2 トランスクリプトミクス...- 38 - トランスクリプトミクス 鲛...
TRANSCRIPT

トランスクリプトームシーケンス解析
RNA-Seq (Quantification)解析
真核生物 Small RNA解析
MicroRNAおよびmRNAの統合解析
FFPE RNA-Seqトランスクリプトーム解析
Long Non-Coding RNAシーケンス解析
メタトランスクリプトーム解析
RIP-Seq解析
トランスクリプトミクス2

- 38 -
トランスクリプトミクス
Ⅱ
トランスクリプトームシーケンス解析
製品概要 トランスクリプトームは次世代ハイスループットシーケンシングで、特定の状況下において、ある生物種の特定
組織や器官のすべての転写配列情報を網羅的かつ迅速に得ることができます。既に基礎研究・臨床診断・創薬など
の分野で利用されています。
TruSeq トランスクリプトーム解析は、微量のサンプルで解析を行なうことが出来ます。例えば Illumina TruSeqTM RNA Sample Preparation Kit を用いた場合、ヒト・マウス・ラットは 200ng のサンプルで解析することができます。
さらに、SMARTerTM PCR cDNA Synthesis Kit を用いると、ヒトはわずか 30ng のサンプルで解析が可能です。これ
により、トランスクリプトーム解析の応用範囲が大きく広がることが期待されています。
技術特長 • 生物種制限なしの全トランスクリプトームの解析:特異的なプローブの事前のデザインが不要。生物種の遺伝子
やゲノム情報なしで生物種を直接かつ網羅的に解析可能
• 高カバー率:デジタル信号・生体内の全転写物のほぼ全てを直接解析可能
• 高解像度:遺伝子ファミリーの中で似ている遺伝子や、選択的スプライシングが起きる SNP を検出可能
• 広検出範囲:コピーしたリードを数個から数十万個まで精確に計算可能
• Illumina® TruSeqTM RNA Sample Preparation Kit と SMARTerTM PCR cDNA Synthesis Kit を用いた、高品質なシー
ケンス解析データ
• 微量のサンプルでライブラリーの作製が可能
• ビーズ精製および試薬のミックスを使用し、短時間でライブラリー作製が可能
ワークフロー
真核生物Oligo (dT) を利用して
mRNA を単離
原核生物 rRNA の除去トータル RNA
ライブラリー作製
シーケンシング
生データ
品質管理
クリーンデータ• コンティグの長さの分布
• Unigene の長さの分布
Unigene アセンブリー
Differentially expressed genes(DEGs) スクリーニング
Unigene 機能
アノテーション
SNP 解析と
SSR 解析
• GO/KEGG/COG アノテーション
• CDS 予測
生物学解析• Gene Ontology (GO) アノテーション
• Pathway enrichment 解析
• 主成分分析
• Conditional-specific • expressed 解析
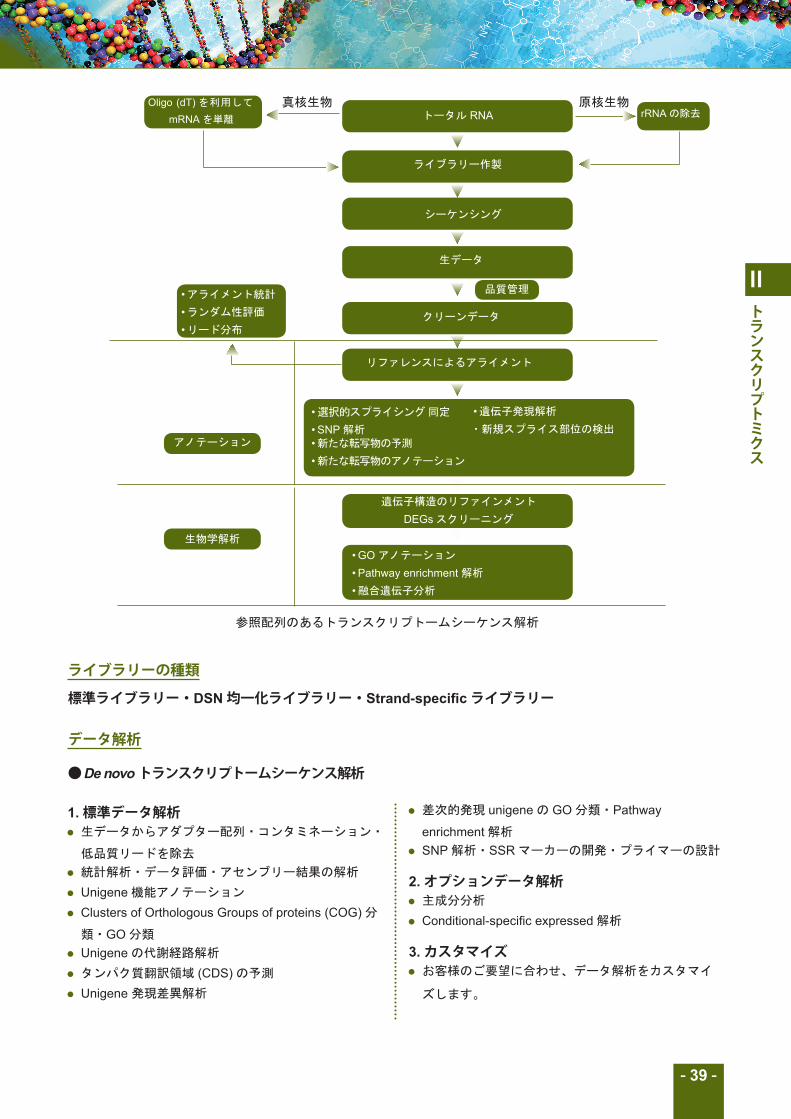
De novo トランスクリプトームシーケンス解析
- 39 -
トランスクリプトミクス
Ⅱ
トータル RNA
ライブラリー作製
シーケンシング
生データ
クリーンデータ
リファレンスによるアライメント
• 選択的スプライシング 同定
• SNP 解析• 新たな転写物の予測
• 新たな転写物のアノテーション
遺伝子構造のリファインメント
DEGs スクリーニング
• GO アノテーション
• Pathway enrichment 解析
• 融合遺伝子分析
• アライメント統計
• ランダム性評価
• リード分布
アノテーション
生物学解析
品質管理
• 遺伝子発現解析
・新規スプライス部位の検出
参照配列のあるトランスクリプトームシーケンス解析
1.標準データ解析 • 生データからアダプター配列・コンタミネーション・
低品質リードを除去
• 統計解析・データ評価・アセンブリー結果の解析
• Unigene 機能アノテーション
• Clusters of Orthologous Groups of proteins (COG) 分
類・GO 分類
• Unigene の代謝経路解析
• タンパク質翻訳領域 (CDS) の予測
• Unigene 発現差異解析
• 差次的発現 unigene の GO 分類・Pathway
enrichment 解析
• SNP 解析・SSR マーカーの開発・プライマーの設計
2.オプションデータ解析 • 主成分分析
• Conditional-specific expressed 解析
3.カスタマイズ • お客様のご要望に合わせ、データ解析をカスタマイ
ズします。
ライブラリーの種類
標準ライブラリー・DSN均一化ライブラリー・Strand-specificライブラリー
データ解析
●De novoトランスクリプトームシーケンス解析
真核生物Oligo (dT) を利用して
mRNA を単離
原核生物rRNA の除去

- 40 -
トランスクリプトミクス
Ⅱ
技術パラメーター
1.サンプル要件植物と真菌 ヒト・ラット・マウス その他
サンプル DNase 処理後の RNA(タンパク質の混入なし)
サンプル量 ≧ 1µg≧ 200ng(SMARTerTM PCR cDNA Synthesis Kit を利用の場合は、30ng でも解析可能 )
≧ 1µg
サンプル濃度 ≧ 20ng/µL ≧ 5ng/µL ≧ 20ng/µL
サンプル純度 28S:18S ≧ 1.0、RIN ≧ 6.5 28S:18S ≧ 1.0、RIN ≧ 7.023S:16S ≧ 1.0、 RIN ≧ 7.0(昆虫の場合は不要)
※ライブラリー作製が困難と判断される場合がありますので、予備のサンプルの同梱を推奨しています。
2.シーケンス解析:101PE (解析内容により 51PE の選択も可能)
3.推奨ライブラリーサイズ:160bp
納品物1. シーケンシング結果 (clean data) は FASTQ ファイルで納品
2. 納品データ例
●De novo トランスクリプトームシーケンス解析
1.標準データ解析 • 生データからアダプター配列・コンタミネーション・
低品質リードを除去
• シーケンシングの評価
• 遺伝子の発現解析・アノテーション
• 遺伝子発現差異解析
• DEG の発現パターン解析・GO 解析・Pathway
enrichment 解析
• 遺伝子構造のリファインメント(真核生物のみ)
• 選択的スプライシングの同定(真核生物のみ)
• 新たな転写物の予測とアノテーション
• SNP 解析
2.オプションデータ解析 • 融合遺伝子分析(ヒトのみ)
3.カスタマイズ • お客様のご要望に合わせ、データ解析をカスタマイ
ズします。
●リファレンスゲノム配列が存在するトランスクリプトーム
図 1 Expression level in paired samples 図 2 COG分類

- 41 -
トランスクリプトミクス
Ⅱ
~62%
~2%~4%
~4%~4%
~4%~4%
~4%~5%~8%
90%-100% (18743)80%-90% (2510)70%-80% (1519)60%-70% (1284)50%-60% (1125)40%-50% (1148)30%-40% (1134)20%-30% (1182)10%-20% (1171)0%-10% (523)
図 3 遺伝子カバー率の統計
2095 2504 34424864 4531
6049 6724
10616
20000 20000
10000 10000
0 0
Exon Skipping
Intron Retention
Alternative 5' Splicing
Alternative 3' Splicing
Num
ber o
f Gen
e
Num
ber o
f AS
Eve
nt
図 4 選択的スプライシング
Stat. of Map to Genome
Map to Genome Reads number Percentage (%)
Total Reads 50723472 100.00
Total BasePairs 4565112480 100.00
Total Mapped Reads 34741646 68.49
Perfect match 13039750 25.71
≦ 5bp mismatch 21701896 42.78
Unique match 32662568 64.39
Multi-position match 2079078 4.10
Total Unmapped Reads 15981826 31.51
Stat. of Map to Gene
Map to Gene Reads number Percentage (%)
Total Reads 5723472 100.00
Total BasePairs 4565112480 100.00
Total Mapped Reads 28039137 55.28
Perfect match 11432658 22.54
≦ 5bp mismatch 16606479 32.74
Unique match 23144279 45.63
Multi-position match 4894858 9.65
Total Unmapped Reads 22684335 44.72
納期ライブラリー作製・シーケンシング・標準データ解析:約 8 週間
原核 Strand-specific サンプル:約 10 週間
表 1 遺伝子発現差異解析 (DEGs)
Gene ID 99152 28015
Symbol Anapc2 Grinl1a
Gene Length 3021 2265
Sample1-Exp 1211 1822
Sample2-Exp 1471 2213
Sample1-RPKM 35.40935 71.0566512
Sample2-RPKM 43.45366 87.192198
log2Ratio 0.295434 0.295229
Up/Down Up Up
P-value 1.23722E-07 8.92E-11
FDR 1.21E-06 1.293144225
●参照配列のあるトランスクリプトームシーケンス解析
表 2 アライメントの統計

- 42 -
トランスクリプトミクス
Ⅱ
RNA-Seq (Quantification)解析
製品概要遺伝子のトランスクリプトームレベルの研究は、機能ゲノミクスと医学研究の基本です。RNA-Seq は、特定の生物
過程の中の遺伝子発見差異を研究する技術です。
技術特長 • バックグラウンドノイズのないデジタル信号
• ハイスループット・高再現性
• 広範囲の遺伝子発現
• 少量の RNA(200ng)で解析可能
• Reads per kilobase of exon model per million mapped reads (RPKM) 方法による高精度な遺伝子発現レベルの計算
ワークフロー
トータル RNA
ライブラリー作製
シーケンシング
生データ
クリーンデータ実験の再現性解析
参照配列とのアライメント
可視化
DEGs スクリーニング
• グループ間 DEG の発現パターン解析• DEG の発現パターン解析
• GO 解析
• Pathway enrichment 解析
• 差異発現パターンのクラスター分析
• タンパク質間相互作用の解析
• シーケンシングの飽和度解析
• カバー率の解析
• リードの分布解析品質管理
アノテーション
生物学解析
遺伝子発現量の統計
真核生物Oligo (dT) を利用して
mRNA を単離
原核生物rRNA の除去

- 43 -
トランスクリプトミクス
Ⅱ
データ解析1.標準データ解析 • 生データからアダプター配列・コンタミネーション・
低品質リードを除去
• シーケンシングの評価
• 遺伝子発現アノテーション
• 遺伝子発現差異の解析
• グループの DEG 発現解析(グループ数 n ≧ 2、
biological replicates/ グループ≧ 3) • DEG の発現パターン解析
• DEG の GO enrichment 解析
• GO 機能解析(Web Gene Ontology Annotation Plot:
WEGO 解析)
• DEG の Pathway enrichment 解析
• GO 分類(WEGO 解析)
• タンパク質間相互作用の解析
2.カスタマイズ • お客様のご要望に合わせ、データ解析をカスタマイ
ズします。
技術パラメーター
1.サンプル要件サンプル DNase 処理後の RNA(タンパク質の混入無し)
サンプル量
ヒト、ラット、マウス その他の真核生物
トータル RNA ≧ 2μg トータル RNA ≧ 5µg
200ng でも解析可能
サンプル 濃度
≧ 80ng/μL ≧ 200ng/μL
サンプル
純度
動物 植物と真菌
RIN ≧ 7.0(昆虫の場合は不要)
RIN ≧ 6.5
OD260/280 ≧ 1.8、OD260/230 ≧ 1.8、28S:18S > 1.0
※ライブラリー作製が困難と判断される場合がありますので、
予備のサンプルの同梱を推奨しています。
2.シーケンス解析:50SE
3. 推奨ライブラリーサイズ:約 200bp
納品物1. シーケンシング結果 (clean data) は FASTQ ファイル
で納品
2. 納品データ例
表 1 遺伝子アライメントの統計図
Map to Gene Reads number Percentage (%)
Total Reads 6246382 100.00
Total BasePairs 306072718 100.00
Total Mapped Reads 4957931 79.37
lperfect match 4064918 65.08
≦ 2bp mismatch 893013 14.30
Unique match 4000400 64.04
Multi-position match 957531 15.33
Total Unmapped Reads 1288451 20.63
2.0
0
0
0
27.0
chr7 (317642 nt/window, 500 windows)
0.3
Gen
eNum
ber
Cov
erag
elo
g 2(R
eads
Num
ber)
図 1 リファレンスゲノムにおけるリードの分布
90%-100% (9978)
80%-90% (7507)70%-80% (5775)
60%-70% (4820)
50%-60% (3857)
40%-50% (3205)30%-40% (3081)
20%-30% (3241)
10%-20% (1748)0%-10% (996)
~17%
~23%
~2%
~4%
~7%
~7%~7%
~9%
~11%
~13%
図 2 遺伝子カバレッジの統計
納期ライブラリー作製・シーケンシング・標準データ解析:約 6 週間

- 44 -
トランスクリプトミクス
Ⅱ
真核生物 Small RNA解析
製品概要次世代シーケンシング技術に基づいた Small RNA シーケンシングは、microRNA・siRNA・piRNA などの small RNA の同定と検出において最も信頼できる方法です。特に microRNA は細胞の成長・発展・分化・死亡に決定的な
役割を果たすので、microRNA のシーケンシングにより制御因子を全般的に見られ、疾患メカニズムをよりよく理解
できます。生体分子研究分野において、Small RNA シーケンシングはバイオマーカーと薬物ターゲット研究のため
によく使われる手段です。
技術特長 • 単一塩基解像度で microRNA の SNP を検出
• コピー数が 2 ~数十万までの small RNA を幅広く検出可能
• 既知と未知の microRNAs を検出
• 即時更新のデータベース
ワークフロー
トータル RNA
ライブラリー作製
50SE シーケンシング
生データ
クリーンデータ
• アライメント
• ランダム検査
• リードの分布
既知 piRNA・siRNA・rRNA など
既知 microRNAエクソンでまだアノテー
ションを行わないリード
新たな microRNA の予測
ファマリー解析 • 差異発現解析
• クラスター分析
• 標的遺伝子の予測
• 標的遺伝子のアノテーション
• Small RNA を元にした情報編集
可視化
アノテーション
生物学解析
品質管理
1.標準データ解析 • 生データからアダプター配列・コンタミネーション・
低品質リードを除去
• small RNA の長さの分布の統計 (18~30 nt)
• サンプル間にある共通配列と特異配列の分析
• 選ばれたリファレンスゲノムで small RNA 分布の探索
• Rfam と Genbank データベースをアライメントし、
rRNAs・tRNAs・snRNAs・snoRNAs などを同定
• small RNAs と反復配列を同定
• small RNAs とエクソン・イントロンを同定
• small RNAs と miRBase で指定した範囲の既知
miRNAs を同定
• small RNAs の分類アノテーション
• Mireap プログラムでの新規 miRNAs の予測とその二
次構造の作成
• 既知 miRNAs の発現パターンの解析
データ解析
- 45 -
トランスクリプトミクス
Ⅱ技術パラメーター
1.サンプル要件
※ライブラリー作製が困難と判断される場合がありますので、予備のサンプルの同梱を推奨しています。
• 既知 miRNAs の家系解析
2.オプションデータ解析 • 既知・新規 miRNA の標的遺伝子の予測
• miRNA の標的遺伝子の GO アノテーションと KEGG
経路解析
• 既知 miRNA-editing 解析
• miRNA の発現差異解析とクラスター分析
• 発現変動 miRNA の標的遺伝子の予測
• 発現変動 miRNA の標的遺伝子解析・GO アノテーショ
ンと KEGG 経路解析
3.カスタマイズ • お客様のご要望に合わせ、データ解析をカスタマイ
ズします。
Length (nt)
Freq
uenc
e pe
rcen
t
0.01 0.02 0.08 0.45 0.68 1.17 1.91 2.37 2.654.92
1.01 0.14 0.03 0.01
10.33
16.29
10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28
57.9260%
50%
40%
30%
20%
10%
0
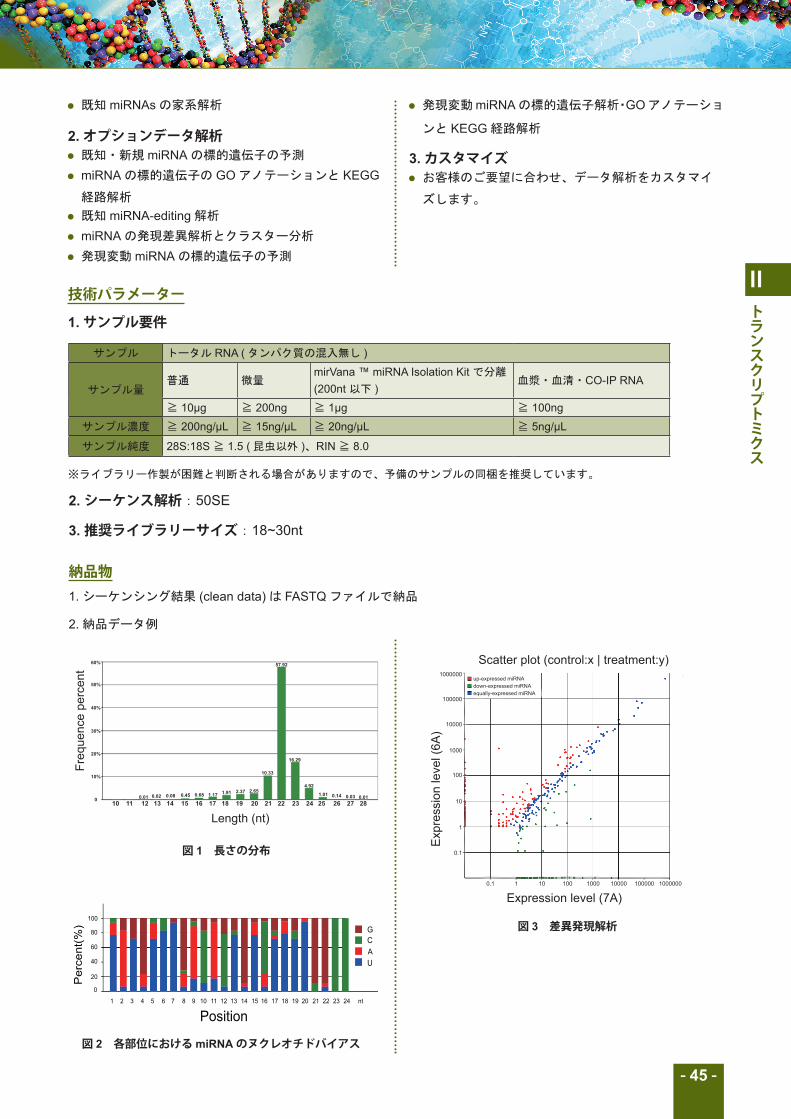
図 1 長さの分布
Per
cent
(%)
Position
100
80
60
40
20
0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 nt
GCAU
Scatter plot (control:x | treatment:y)1000000
1000000
100000
100000
10000
10000
1000
1000
100
100
10
10
Expression level (7A)
1
1
0.1
0.1
up-expressed miRNAdown-expressed miRNAequally-expressed miRNA
Exp
ress
ion
leve
l (6A
)
図 3 差異発現解析
サンプル トータル RNA ( タンパク質の混入無し )
サンプル量普通 微量
mirVana ™ miRNA Isolation Kit で分離
(200nt 以下 )血漿・血清・CO-IP RNA
≧ 10μg ≧ 200ng ≧ 1μg ≧ 100ng
サンプル濃度 ≧ 200ng/μL ≧ 15ng/μL ≧ 20ng/μL ≧ 5ng/μL
サンプル純度 28S:18S ≧ 1.5 ( 昆虫以外 )、RIN ≧ 8.0
2. シーケンス解析:50SE
3. 推奨ライブラリーサイズ:18~30nt
納品物1. シーケンシング結果 (clean data) は FASTQ ファイルで納品
2. 納品データ例
図 2 各部位におけるmiRNAのヌクレオチドバイアス
- 46 -
トランスクリプトミクス
Ⅱ
表 1 発現差異比較解析(2サンプル間)
pairwise miR-name S1-std S2-std fold-change(log2 S2/S1) p-value
S1-S2 mmu-let-7a-5p 4847.7382 2821.0664 -0.78107117 0
S1-S2 mmu-let-7b-5p 2196.2147 1482.2416 -0.56723848
S1-S2 mmu-let-7c-5p 8002.0895 5311.7130 -0.59119957 0
S1-S2 mmu-let-7d-3p 7.4349 4.0061 -0.89211490
S1-S2 mmu-let-7d-5p 540.0024 372.2430 -0.53672110
S1-S2 mmu-let-7e-5p 358.9780 244.6900 -0.55294029
S1-S2 mmu-let-7f-5p 7119.7569 4502.6494 -0.66105384 0
S1-S2 mmu-let-7g-5p 1452.7213 904.0873 -0.68422396
S1-S2 mmu-let-7i-5p 285.5984 179.4714 -0.67023394
S1-S2 mmu-miR-100-5p 6.9500 5.6085 -0.30939801
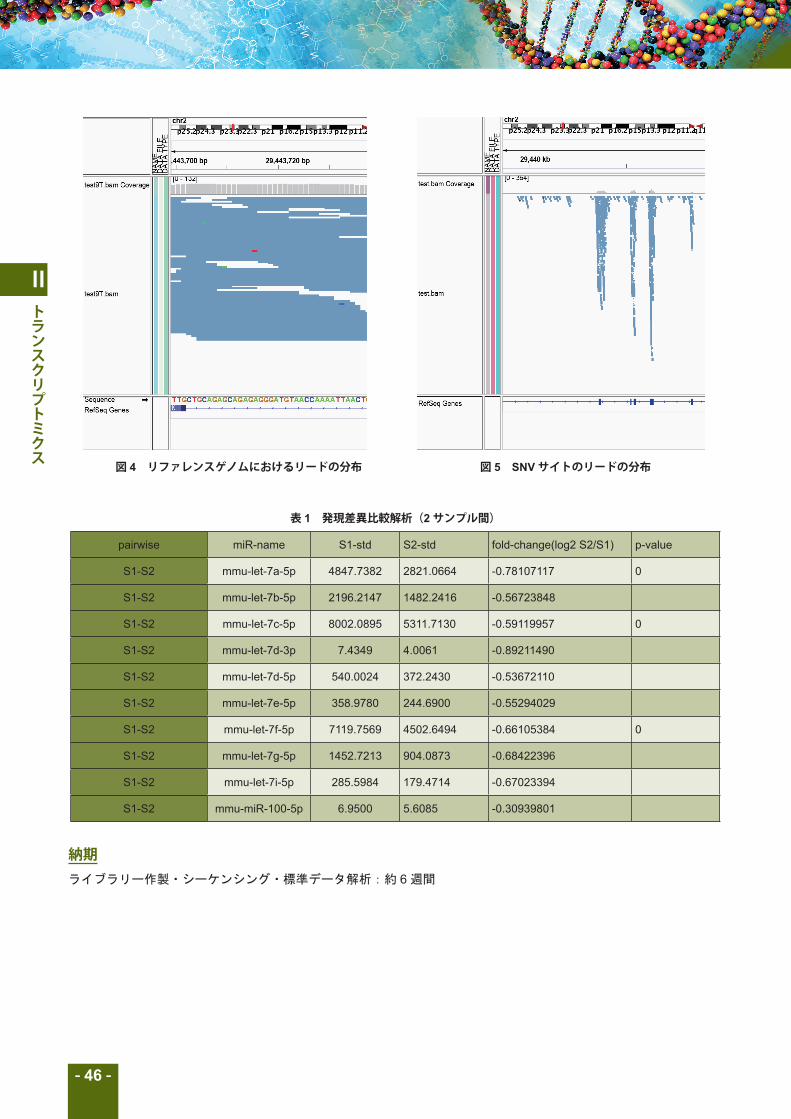
図 4 リファレンスゲノムにおけるリードの分布 図 5 SNVサイトのリードの分布
納期ライブラリー作製・シーケンシング・標準データ解析:約 6 週間

- 47 -
トランスクリプトミクス
Ⅱ
MicroRNAおよびmRNAの統合解析
製品概要 現在、機能ゲノミクス分野において、マイクロ RNA(miRNA)とメッセンジャー RNA(mRNA)の研究が注目さ
れています。Small RNA シーケンシング、RNA-Seq(トランスクリプトーム)と RNA-Seq(定量)の登場後、BGIは先駆者として、microRNA および mRNA の統合解析の発展に注力しています。
MicroRNA および mRNA の統合解析は発生生物学、機能ゲノミクスと疾患進行などの領域で応用することができ
ます。BGI は異なる RNA シーケンス解析法を効果的に融合し、より多くのハイスループットデータを簡単に処理・
探索できるようになりました。
技術特長 • 次世代シーケンシング(NGS)データとマイクロアレイデータを含むハイスルプットデータの統合解析を実施
• データの利用率の向上、標的遺伝子予測の改善
ワークフロー
miRNA の発現情報
miRNA 標的遺伝子の予測miRNA 差異解析 遺伝子差異解析
相関解析
標的遺伝子のスクリーニング
遺伝子の機能アノテーション
Pathway ネットワークの作成
miRNA と遺伝子の共発現解析
制御ネットワークの作成
遺伝子の発現情報
MicroRNAとmRNA統合解析内容 • miRNA と遺伝子の発現差異解析
• miRNA 標的遺伝子の予測
• miRNA と遺伝子の共発現解析
• miRNA と遺伝子の相関解析
• 発現相違の miRNA および遺伝子の調製ネットワーク
• KEGG と GO 解析
• Gene-miRNA-GO と Gene-miRNA-KEGG の結果分類

- 48 -
トランスクリプトミクス
Ⅱ
BGIの実績患者の microRNA-mRNA の統合解析は、疾患診断および予後のバイオマーカーのスクリーニングに多大な影響を与
えました。
・疾病の診断
・疾病の予測
研究目的
・血清
・組織
サンプル選択 実験手順
・ライブラリー作製
・シーケンシング
・発現差異
・標的予測
データ解析
・有病率
・生物機能解析
フォローアップ検証
A. 健常者と比較し、淡明型腎細胞癌(ccRCC)患者の特異的
に発現する miRNA および mRNA の数(P<0.01,FDR ≦ 0.001)※ K1 ~ K55 は患者の No.
B. 異なる組織で観察された新規 miRNA 候補
C. 異なる組織で観察された新規 mRNA 候補
4 48 4(7.14%) (7.14%)
ccRCCNormal adjacent tissues
18 562 6(3.07%) (1.02%)
ccRCCNormal adjacent tissues
図 1 淡明型腎細胞癌(ccRCC)患者のmiRNAおよびmRNAの発現情報
Zhou L, Chen J, Li Z, Li X, Hu X, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE. 2010, 5(12): e15224.
納期約 3 週間(サンプル数が 20 以下の場合)
miRNAs up-regulated miRNAs down-regulatedmRNAs up-regulated mRNAs down-regulated
50
0
8000
K1 K2
miR
NA
s di
ffere
ntia
lly e
xpre
ssed
mR
NA
s di
ffere
ntia
lly e
spre
ssed
100
150
200
K39 K6K3 K27K7 K38 K44 K55
6000
4000
2000
0

- 49 -
トランスクリプトミクス
Ⅱ
ワークフロー
精製したトータル RNA
RNA 断片化
cDNA 合成
アダプターのライゲーション &PCR
DSN 均一化
FFPE RNA-Seqトランスクリプトーム解析
製品概要 現在、腫瘍サンプルの約 90%はホルマリンで固定し、パラフィンで包埋 (FFPE) されたサンプルであり、バイオマー
カーの発見、新薬の開発、疾病の診断および治療などについて豊富な情報を持っています。
しかし、FFPE サンプルは、希に核酸の分解と塩基変化を起こすため、組織から完全な情報を取得するのは困難で
す。現在のところ、FFPE サンプルの核酸情報の取得はマイクロアレイやプロテオミクス技術が利用されていますが、
次世代シーケンス解析の利用はごくわずかです。
BGI では FFPE サンプルの完全なトランスクリプトーム情報をシーケンス解析する新しい方法を開発しました。
この方法を評価するため、同じ腫瘍組織の FFPE サンプルと新鮮凍結 (FF) サンプルによるシーケンス解析のデータ
を比較しました。その結果、新しいシーケンス解析方法は FFPE サンプルの遺伝子発現のプロファイリング研究に
適応することが明らかになりました。
技術特長 • DSN 均一化ライブラリー
• サンプルの量が少ない場合(最低 200ng)でも解析可能
• ハイスループット
• FFPE サンプルの専門的なバイオインフォマティクス解析
• マルチオミクス技術を利用した全面的・系統的な解析

- 50 -
トランスクリプトミクス
Ⅱ
技術パラメーター
1.サンプル要件
2. RNA抽出キット:Ambion® RecoverAllTM Total Nucleic Acid Isolation Kit
3.シーケンス解析:Illumina HiSeq
4.データ量:推薦 5Gb clean data 以上
BGIの実績 • サンプル情報
表 1 同じ腫瘍組織からの FFPE腫瘍サンプルと新鮮凍結 (FF)サンプルの情報
Donor IDFF FFPE
T1 T2 T3 N T10 T20 T30 N0
Block Age (Years)
3.0 1.5 1.0 0.2 3.0 1.5 1.0 0.2
RIN 7.5 4.8 6.4 2.6 2.4 2.4 2.4 2.4
28S:18S 1.0 1.0 0.8 1.4 0.5 0 0 0
T:腫瘍サンプル、N:コントロール、1-3:癌のサブタイプ
図 1 T30 サンプルの RNA分析結果(Agilent 2100)
RNA は顕著に分解されていました。
サンプル トータル RNA
サンプル量 ≧ 200ng
サンプル濃度 ≧ 20ng/μL
サンプル純度 RIN ≧ 2.3
- 51 -
トランスクリプトミクス
Ⅱ
• 納品データ例
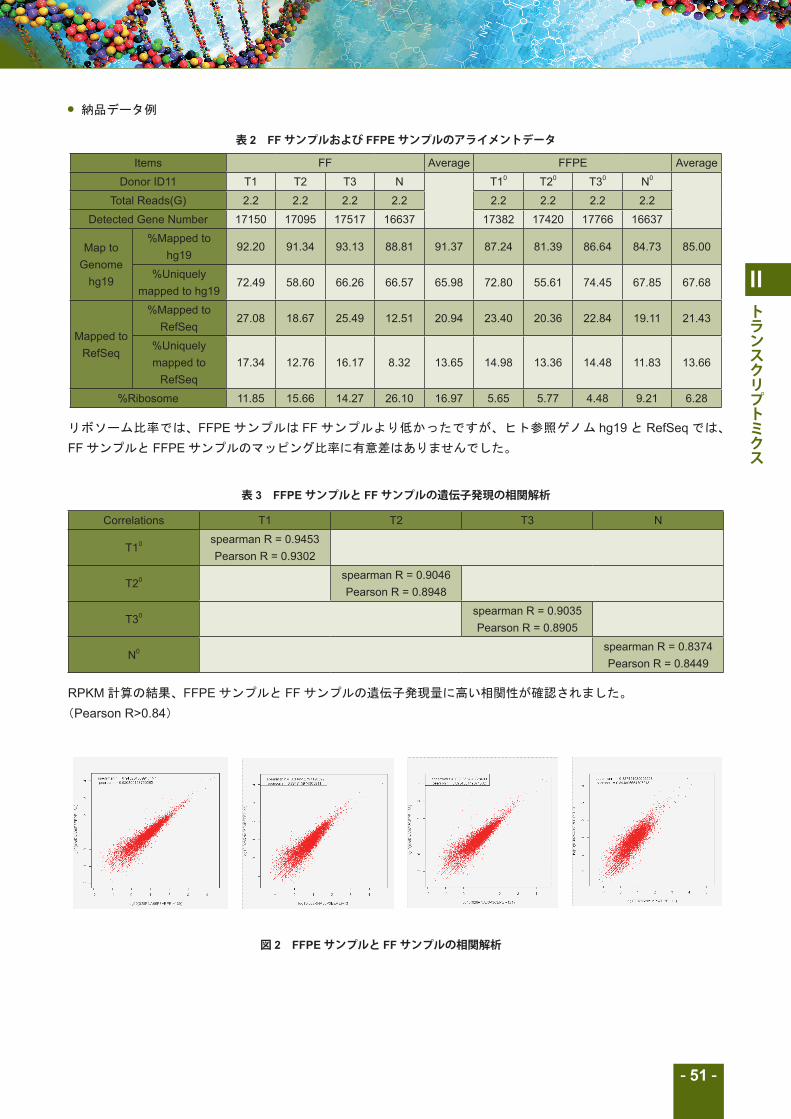
表 2 FFサンプルおよび FFPEサンプルのアライメントデータ
Items FF Average FFPE Average
Donor ID11 T1 T2 T3 N T10 T20 T30 N0
Total Reads(G) 2.2 2.2 2.2 2.2 2.2 2.2 2.2 2.2
Detected Gene Number 17150 17095 17517 16637 17382 17420 17766 16637
Map to Genome
hg19
%Mapped to hg19
92.20 91.34 93.13 88.81 91.37 87.24 81.39 86.64 84.73 85.00
%Uniquely mapped to hg19
72.49 58.60 66.26 66.57 65.98 72.80 55.61 74.45 67.85 67.68
Mapped to RefSeq
%Mapped to RefSeq
27.08 18.67 25.49 12.51 20.94 23.40 20.36 22.84 19.11 21.43
%Uniquely mapped to
RefSeq17.34 12.76 16.17 8.32 13.65 14.98 13.36 14.48 11.83 13.66
%Ribosome 11.85 15.66 14.27 26.10 16.97 5.65 5.77 4.48 9.21 6.28
リボソーム比率では、FFPE サンプルは FF サンプルより低かったですが、ヒト参照ゲノム hg19 と RefSeq では、
FF サンプルと FFPE サンプルのマッピング比率に有意差はありませんでした。
表 3 FFPEサンプルと FFサンプルの遺伝子発現の相関解析
Correlations T1 T2 T3 N
T10 spearman R = 0.9453 Pearson R = 0.9302
T20 spearman R = 0.9046 Pearson R = 0.8948
T30 spearman R = 0.9035 Pearson R = 0.8905
N0 spearman R = 0.8374 Pearson R = 0.8449
RPKM 計算の結果、FFPE サンプルと FF サンプルの遺伝子発現量に高い相関性が確認されました。
(Pearson R>0.84)
図 2 FFPEサンプルと FFサンプルの相関解析

- 52 -
トランスクリプトミクス
Ⅱ
Long Non-Coding RNAシーケンス解析
製品概要 長鎖非コード RNAs(Long non-coding RNAs, Lnc RNAs)はタンパク質へ翻訳されていない 200nt 以上の長さの
RNAs (rRNA を含まない ) を言い、ポリ A の有無を問わず、非コード RNAs(non-coding RNAs, ncRNAs)内で大き
な割合を占め、エピジェネティック制御、転写調節と転写後調節などで遺伝子発現を調節し、生命活動に重要な役
割を果たしています。
Lnc RNAs シーケンス解析(LncRNA-seq)は特定方法(例えば、Ribo-ZeroTM キット)を利用してサンプル中の
rRNA の存在量を減らし、濃縮された RNA を Illumina HiSeq2000 プラットフォームでシーケンシングし、LncRNAの鑑定、予測と差異分析を行います。迅速で精確な、特定生物学過程或いは疾患関連の LncRNA 情報の研究方法です。
技術特長 • シーケンスカバー率が高く、データ量を変更でき、ほぼすべての転写物をカバー
• 高精度、大量の配列情報を獲得
• 既知 LncRNA の鑑定と未知 LncRNA の予測のほか、mRNA データと組み合わせての共発現のアノテーション
ワークフロー
新しい LncRNA予測
LncRNA機能予測
トータル RNA
生データ
ライブラリー作製
rRNA データ除去
rRNA 除去
クリーンデータ
シーケンシング
転写物再構成
発現差異解析
品質管理、データの
フィルタリング
データ解析
1.標準データ解析 • 既知 LncRNA の鑑定
• 未知 LncRNA の発見
• LncRNA の機能予測
• LncRNA ファミリーの予測分類
• LncRNA 定量と差異分析
• 疾患マーカーの探索
2.オプションデータ解析 • CNC(coding-non-coding)共発現アノテーション
• Pathway enrichment 解析
技術パラメーター
1.サンプル要件
※ライブラリー作製が困難と判断される場合がありますので、予備のサンプルの同梱を推奨しています。
サンプル DNase 処理後のトータル RNA(タンパク質の混入なし)
サンプル量ヒト・ラット・マウス
≧ 5μg
サンプル濃度 ≧ 200ng/μL
サンプル純度 RIN ≧ 7.0、28S/18S ≧ 1.0
- 53 -
トランスクリプトミクス
Ⅱ
2.シーケンス解析:Illumina HiSeq
3. 推奨ライブラリーサイズ:140 ~ 160bp
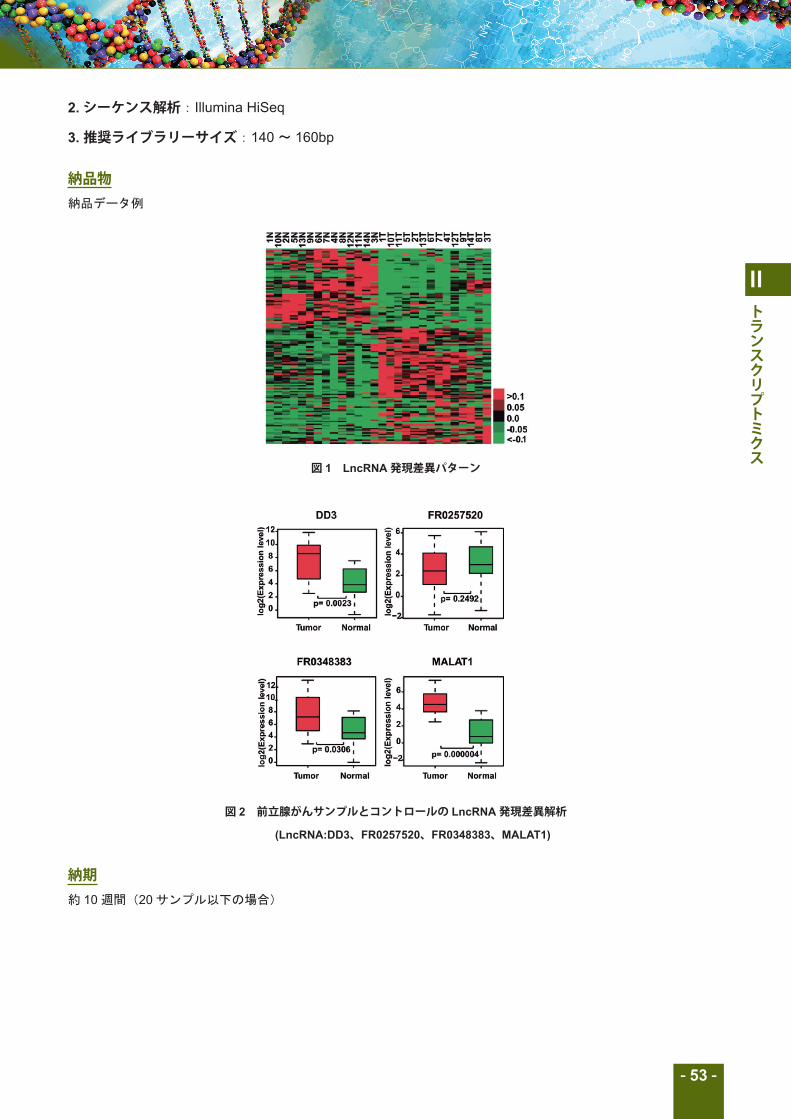
納品物納品データ例
図 1 LncRNA発現差異パターン
図 2 前立腺がんサンプルとコントロールの LncRNA発現差異解析
(LncRNA:DD3、FR0257520、FR0348383、MALAT1)
納期約 10 週間(20 サンプル以下の場合)

- 54 -
トランスクリプトミクス
Ⅱ
メタトランスクリプトーム解析
製品概要 メタトランスクリプトーム解析では、環境微生物の群集の動態構造と機能を調べます。環境サンプルの中のすべ
ての微生物のトランスクリプト産物に対してハイスループットシーケンシングを行い、参照配列が無くてもトラン
スクリプト産物の種の多様性を分析できます。
ソフトウェアを使って RNA レベルで機能遺伝子を発見し、異なる環境の中の微生物遺伝子のコントロールと表現
の相違性を示したり、未知遺伝子の機能や、遺伝子のコントロール原理、微生物群集・環境の相互作用などの研究
が可能です。
技術特長 • 環境中の微生物サンプルの Total RNA を直接抽出してライブラリーを作製し、ハイスループットシーケンシング
をすることができます。従来の分離培養された微生物に比べ、自然環境の微生物の構造と機能をより正確に反映
できます。
• 微生物分離培養技術や、クローン技術など不要
• 環境微生物の動態構造と機能の情報を取得可能
• データ解析のカスタマイズが可能
ワークフロー
データ解析
1.標準データ解析
Total RNA
ライブラリー作製
シーケンス解析(101PE)
データ解析
• データの処理
アダプター、rRNA と低品質データを除去
ホストのコンタミネーションを除去
• 配列のアセンブリーと統計
コンティグの構築
コンティグの長さの統計
cDNA リード利用率の統計
• ゲノムの構成解析
遺伝子構成
• 遺伝子発現解析
遺伝子カバレッジ、深度の統計
遺伝子発現量の計算
• 遺伝子機能アノテーション
KEGG データベースに基づく機能アノテーション
CAZy(carbohybrate-Active Enzymes)データベース
に基づく機能アノテーション
eggNOG(evolut ionary genealogy of genes: Nonsupervised Orthologous Groups)データベース
に基づく機能アノテーション
GO データベースに基づく機能アノテーション
抗生物質の抗性因子アノテーション
各アノテーション結果の発現量統計

- 55 -
トランスクリプトミクス
Ⅱ
• 発現差異解析(サンプル> 2)
DEG (Differentially expressed genes) のスクリーニング
遺伝子発現差異パターンのクラスタリング
種の DEG のアノテーション
DEG の KEGG、CAZy、eggNOG、GO 解析
2.オプションデータ解析 • お客様のご要望に合わせ、データ解析をカスタマイズします。
技術パラメーター
1.サンプル要件
サンプル量 ≧ 5μg
サンプル濃度 ≧ 150ng/μL
サンプル純度23S/16S ≧ 1.0、RIN ≧ 7.0、OD260/280 ≧ 1.8、OD260/230 ≧ 1.8 ベースラインがスムーズで5Sピークがノーマルであること。
※土壌・海水などの複雑環境のサンプルの場合、サンプル要件は異なりますので、別途お問い合わせください。
2.シーケンス解析:Illumina HiSeq・MiSeq
納品物 • シーケンシング結果(clean data)は FASTQ ファイルで納品
• 納品データ例
図 1 SEED サブシステムの相対量に基づいた、全 cDNA と DNA データセットのクラスタリング
納期お問い合わせください。

- 56 -
トランスクリプトミクス
Ⅱ
RIP-Seq解析
製品概要RIP(RNA Binding Protein Immunoprecipitation)は、特定の RNA とタンパク質の相互作用を同定するための、優れ
た技術です。RIP では、特定のタンパク質の抗体によって免疫沈降させたタンパク質 -RNA 複合体を、標的 RNA の
抽出後、転写後調節ネットワークのダイナミックプロセスを調べます。RIP と次世代シーケンス解析を組み合わせ
た RIP-Seq 解析では、全ゲノムレベルで精確にタンパク質と RNA の相互作用と調節を識別することが可能です。こ
の技術により、がんや他の疾病の総合的な RNA 変化のハイスループット分析が可能になります。
技術特長 • 高コストパフォーマンス
• 広い研究範囲
• ハイスループット
ワークフロー
RIP-ed RNA
ライブラリー作製
クリーンデータ
シーケンシング
ゲノムマッピング
生データ
品質管理
ピークコール
ピークの長さ ピークのカバー率 ピーク関連遺伝子ピークのアノテーション
差異解析
ピーク差異
GO 解析
遺伝子差異
Pathway 解析
Motif 解析
アライメント統計
カバー率 / シーケンスの
リード深度統計

- 57 -
トランスクリプトミクス
Ⅱ
技術パラメーター1.サンプル要件
※ RNA は分解を防ぐため、抽出後 72 時間以内に、ドライアイスを同梱して輸送してください。
2.シーケンス解析
ライブラリーサイズが 150nt 未満:50SE
ライブラリーサイズが 150nt 以上:101PE
納品物1. 参考データ量:20Mb
2. 納品データ例
図 1 結合 Motif のリファイン
納期ライブラリー作製・シーケンシング・標準データ解析:約 10 週間
サンプル RNA(ヒト・マウス・ラット)
サンプル量 ≧ 100ng
サンプル濃度 ≧ 5ng/μL
データ解析
1.標準データ分析 • アダプター配列・コンタミネーション・低品質リー
ドを除去し、産出量を統計
• リファレンスゲノム配列とアライメント
• 全ゲノムのシーケンシングリードの深度分析
• ピーク解析(データ豊富区の情報統計)
• ピークアノテーションとその関連遺伝子の GO、
Pathway enrichment 解析
• サンプル間の差異解析
2.オプションデータ解析 • Motif 解析
3.カスタマイズ • お客様のご要望に合わせ、データ解析をカスタマイ
ズします。