24 - binde- und stützgewebe
TRANSCRIPT

24
24 Binde- und Stützgewebe Rainer Deutzmann, Leena Bruckner-Tuderman, Peter Bruckner
24.1 Zusammensetzung der extrazellulären Matrix (ECM) – 716
24.2 Kollagene – 71624.2.1 Fibrilläre Kollagene – 71624.2.2 Nichtfibrilläre Kollagene – 72224.2.3 Angeborene Erkrankungen des Kollagen-Stoffwechsels – 723
24.3 Elastische Fasern – 724
24.4 Proteoglykane – 72724.4.1 Aggrecan – 72824.4.2 Kleine leucinreiche Proteoglykane – 72824.4.3 Membrangebundene Proteoglykane – 729
24.5 Nichtkollagene, zelladhäsive Glycoproteine – 73024.5.1 Fibronectin – 73124.5.2 Laminine – 73224.5.3 Integrine – 733
24.6 Abbau der extrazellulären Matrix – 736
24.7 Biochemie und Pathobiochemie des Skelettsystems – 73724.7.1 Die extrazelluläre Matrix von Knorpel und Knochen – 73724.7.2 Synthese von Knochen und Knorpel durch Chondrozyten
und Osteoblasten – 73824.7.3 Differenzierung von Osteoklasten und Abbau und Umbau von Knochen
und Knorpel – 74224.7.4 Regulation des Knochenwachstums bis zur Pubertät – 74424.7.5 Homöostase des Skelettsystems – 74424.7.6 Osteoporose und Regulation des Knochenumbaus durch Cytokine
und Steroidhormone – 74524.7.7 Knochenerkrankungen – 746
24.8 Biochemie der Haut – 74724.8.1 Aufbau und Funktionen der Haut – 74724.8.2 Die Epidermis – 74824.8.3 Die dermo-epidermale Junktionszone – 74924.8.4 Die Dermis – 74924.8.5 Pathobiochemie der Haut – 751
Literatur – 754

716 Kapitel 24 · Binde- und Stützgewebe
24
>> Einleitung
Das Bindegewebe durchzieht den gesamten Organismus. Um den vielfältigen Aufgaben als Gerüst- und Stützsubstanz gerecht zu werden, kommt es in den unterschiedlichsten Ausprägungen vor. Dazu gehören feste Strukturen wie Knorpel, Sehnen und Bänder, aber auch das aus locker gepackten fibrillären Strukturen bestehende interstitielle Bindegewebe, das den Extrazellulär-raum ausfüllt und Organe umgibt. Das Bindegewebe ist aber weit mehr als nur das strukturgebende Element des Körpers. Eine Vielzahl von extrazellulären Matrix-Molekülen binden über spezifische Rezeptoren an Zellen und beeinflussen Wachstum, Dif-ferenzierung und Funktion fast aller Zellen des Körpers. Eindrucksvoll konnte dies an Mäusen gezeigt werden, bei denen Rezep-toren oder deren extrazelluläre Liganden inaktiviert waren. Im Extremfall kam es nicht einmal zur Ausbildung des zweiblättrigen Keimblatts.
Aufgrund des komplexen Aufbaus und der ubiquitären Verteilung ist das Bindegewebe an zahlreichen Krankheiten beteiligt. Dazu gehören angeborene Störungen, die auf Defekten in Genen für Strukturproteine oder Zelladhäsionsmoleküle beruhen. Darüber hinaus spielt das Bindegewebe eine entscheidende Rolle bei vielen atrophisierenden und fibrosierenden Prozessen. Pathologische Veränderungen treten bei entzündlichen Reaktionen wie den rheumatischen Erkrankungen auf. Auch die Metas-tasierung von Tumoren wird vom Bindegewebe beeinflusst.
24.1 Zusammensetzung der extra-zellulären Matrix (ECM)
Die verschiedenen Formen des Bindegewebes leiten sich vom embryonalen Bindegewebe, dem Mesenchym ab. Die Bindegewebszellen im engeren Sinne sind die Fibroblas-ten/ Fibrozyten. Abkömmlinge sind die Chondroblasten/Chondrozyten des Knorpels und die Osteoblasten/Osteo-zyten des Knochens. Diese Zelltypen synthetisieren den größten Teil der extrazellulären Matrix, jedoch produzieren auch Epithel- und Endothelzellen sowie glatte Muskelzellen eine Reihe von extrazellulären Matrix-Molekülen, insbe-sondere die meisten Bestandteile der Basalmembran. Die von den verschiedenen Zelltypen sezernierten Proteine können in vier Gruppen eingeteilt werden:4 Kollagene. Sie stellen quantitativ die bedeutendsten
Proteine des Organismus dar und sind die strukturge-benden Proteine von Haut, Sehnen, Bändern sowie der organischen Grundsubstanz von Hartgeweben und der Basalmembranen
4 Elastin. Dieses Protein verleiht Strukturen elastische Eigenschaften, z.B. in der Aortenwand
4 Proteoglykane. Diese Proteinklasse ist wesentlich durch ihren Kohlenhydratanteil definiert. Durch An-heften von sauren repetitiven Disaccharid-Einheiten (7 Kap. 2.1.4) stellen sie Polyanionen dar und sind sowohl für die Schaffung von elastischen wasserge-füllten Kompartimenten (z.B. Knorpel) notwendig als auch für die Regulation der Kollagenfibrillen-Bildung. Proteoglykane sind als Zelloberflächen-assoziierte Co-Rezeptoren essentiell (z.B. Kooperation von Syn-decanen mit Integrinen, Beteiligung verschiedener Heparansulfatproteoglykane an Bindung/Rezeptor-Präsentation von Wnt und Hedgehog, FGF-α TGF -Proteinen)
4 nichtkollagene Glycoproteine. Die Gruppe der nicht-kollagenen Glycoproteine umfasst eine Vielzahl von Molekülen, die für die Zellfunktion essentiell sind. Die
bekanntesten Moleküle dieser Gruppe sind die Lami-nine und das Fibronectin
24.2 Kollagene
! Kollagene sind Moleküle, die aus Polypeptidketten mit vielfach wiederholten Gly-X-Y-Sequenzmotiven beste-hen, die sich zu tripelhelicalen Strukturen zusammen-lagern. Durch Kombination von tripelhelicalen und globulären Domänen entsteht eine Vielzahl von Pro-teinen mit unterschiedlichen Eigenschaften.
Das gemeinsame Strukturmerkmal aller Kollagene sind starre stabförmige Abschnitte, die eine tripelhelicale Kon-formation (7 Kap. 3.3.2) besitzen. Diese Konformation ist durch monoton wiederholte Triplet Sequenzen aus Gly-X-Y bedingt, wobei X und Y häufig Prolin und Hydroxyprolin sind. Ferner tritt in Y-Position bisweilen das für Querver-netzungen wichtige Hydroxylysin auf (7 u.).
Zur Zeit sind wenigstens 40 verschiedene Kollagen-ketten sequenziert, die zu 27 distinkten trimeren Kollagen-molekülen assemblieren können. Dabei unterscheiden sich die einzelnen Kollagentypen strukturell in der Länge der tripelhelicalen Abschnitte, kurzen Unterbrechungen in der Tripelhelix und/oder in der Existenz zusätzlicher globulärer Domänen. Durch die Kombination dieser Module und Merkmale erhalten die Kollagene ihre spezifischen bio-logischen Eigenschaften. Einige Vertreter der Familie sind in . Tab. 24.1 aufgelistet.
24.2.1 Fibrilläre Kollagene
! Die fibrillären Kollagene sind die Hauptkollagene des Bindegewebes von Haut, Knochen, Knorpel, Sehnen und Bändern und besitzen charakteristische Faserstruk-turen.

24717
Die wichtigsten fibrillären Kollagene sind die in . Tab. 24.1 aufgelisteten »klassischen« Typen I, II, III, V, und XI. Die Aufgabe der fibrillären Kollagene ist die Bildung von festen Fasern, die Druck- oder Zugbelastungen aushalten. Im Elektronenmikroskop lassen sie einen Aufbau aus quer gestreift erscheinenden Fibrillen mit einer Periodizität von 67 nm erkennen (. Abb. 24.1, . Abb. 24.4), jedoch sind Anordnung und Dicke in verschiedenen Geweben recht unterschiedlich (. Abb. 24.1). Die dreidimensionalen Strukturen sind den Anforderungen entsprechend opti-miert. In Sehnen z.B., die überwiegend Typ-I-Kollagen ent-halten, sind alle Fibrillen parallel angeordnet, sodass sie maximale Stabilität in Richtung einer Zugbelastung besit-
zen, während sie in der Haut (vor allem Typ-I- und Typ-III-Kollagen) kreuz und quer liegen, um eine Dehnung in alle Richtungen zu ermöglichen. Knorpel hingegen besitzt dünnere Fasern (überwiegend Typ-II-Kollagen), die ein dreidimensionales auf Druckbelastung ausgelegtes Netz-werk ausbilden. Diese morphologischen Befunde beruhen auf unterschiedlichen Eigenschaften der Polypetidketten und Zusammenwirken mit Fibrillendicke-regulierenden Proteinen.
Die Polypeptidketten der fibrillären Kollagene besitzen homologe Aminosäuresequenzen und nahezu identische Domänen-Strukturen (. Abb. 24.2). Das charakteristische Strukturmerkmal ist eine große zentrale Domäne. Sie be-
. Tabelle 24.1. Kollagen-Typen (Auswahl) und repräsentative Expressionsorte
Typ typische Molekül-Zusammensetzung
typisches Vorkommen charakteristische Merkmale
Fibrillen-bildende Kollagene
I [ 1(I)]2 2(I) Haut, Knochen, Sehnen, Bänder häufigstes Kollagen, bildet besonders zugfeste Fibrillen
II [ 1(II)]3 Knorpel, Glaskörper häufigstes Knorpelkollagen
III [ 1(III)]3 dehnbare Gewebe wie Haut, Gefäße
Vorkommen zumeist zusammen mit Typ-I-Kollagen
V [ 1(V)]2 3(V) Haut, Cornea, Nebenkomponente, zusammen mit Typ-I-Kollagen exprimiert
XI 1(XI) 2(XI) 2(XI),[ 1(XI)]2 2(V)
Knorpel, Glaskörper Nebenkomponente, zusammen mit Typ-II-Kollagen exprimiert
Basalmembrankollagene
IV [ 1(IV)]2 2(IV) fast alle Basalmembranen flächiges Netzwerk,Hauptstrukturprotein der Basalmembran3(IV) 4(IV) V) Nierenglomeruli
[ 5(IV)]2 6(IV) + [ 1(IV)]2 2(IV)
Bowmannsche Kapsel
Multiplexine, Basalmembran-assoziierte Kollagene
XV [ 1(XV)]3 vor allem vaskuläre und epitheliale Basalmembranen
C-terminales Fragment hemmt Angiogenese
XVIII [ 1(XVIII)]3 C-terminales Fragment (=Endostatin) hemmt Angiogenese, Mutationen führen zur Erblindung
Fibrillen-assoziierte(FACIT) Kollagene
IX 1(IX) 2(IX) 3(IX) mit Typ II-Kollagen assoziiert knock-out-Mäuse entwickeln Arthrose
XII [ 1(XII)]3 hauptsächlich mit Typ I-Kollagen assoziiert
Bindung an ECM-Moleküle (Proteogykane), Regulation der Fibrillenbildung / Modifikation der Interaktion zwischen Fibrillen (?)
XIV [ 1(XIV)]3
Netzwerk-bildende Kollagene
VIII [ 1(VIII)2] 2(VIII) Endothel, Descemet Membran (Auge) Bildung hexagonaler Netzwerke
X [ 1(X)]3 hypertrophierender Knorpel
Transmembrankollagene
XIII [ 1(XIII)]3 breite Gewebsverteilung Zell-Zell- und Zell-Matrix-Verankerung
XVII [ 1(XVII)]3 Hemidesmosomen der Haut Adhäsion von Keratinozyten an die Basalmembran,Mutationen führen zur Epidermolysis bullosa junctionalis
Sonstige
VI 1(VI) 2(VI) 3(VI) ubiquitär bildet Mikrofibrillen
VII [ 1(VII)]3 Anker-Fibrillen exprimiert an der Dermis-Epidermis-Grenze, Verankerung der Ba-salmembran im interstitiellen Bindegewebe
24.2 · Kollagene

718 Kapitel 24 · Binde- und Stützgewebe
24
steht aus einer tripelhelicalen Domäne von 340 Gly-X-Y-repeats in ununterbrochener Reihenfolge, welche von zwei etwa 20 Aminosäuren langen nichttripelhelicalen Telopep-tiden flankiert ist. Diese Telopeptide sind für die Ausbil-dung von Quervernetzungen essentiell. Neu synthetisierte Kollagenmoleküle enthalten noch zusätzliche Domänen an den Enden, das N-Propetid bzw. C-Propeptid, die je nach Kollagentyp im reifen Molekül jedoch nicht mehr oder
nur noch teilweise vorhanden sind. Zusätzlich besitzen die Ketten noch eine Prä-Sequenz zur Translokation ins endo-plasmatische Retikulum.
Biosynthese der fibrillären Kollagene am Beispiel von Typ-I-Kollagen
! In der Zelle wird Prokollagen gebildet, das charakteris-tische Hydroxylierungen von Prolinen und Lysin auf-weist.
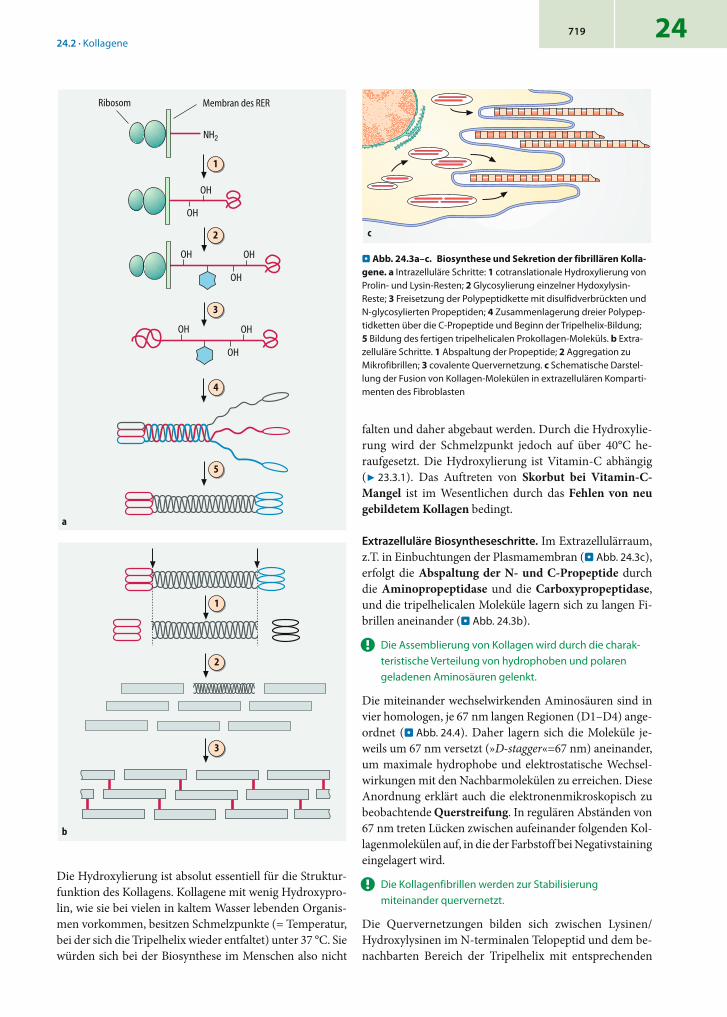
Intrazelluläre Biosyntheseschritte. Die Kollagenketten werden am rauen endoplasmatischen Retikulum (RER) ge-bildet. Die Synthese erfolgt in das Lumen des RER, wobei die Signalsequenz (Präsequenz) abgespalten wird. Etwa 50% der Proline und einige Prozent der Lysine werden in der Y-Position des Gly-X-Y-Triplets hydroxyliert (. Abb. 24.3a). Untersuchungen haben gezeigt, dass tripelhelicale Abschnitte keine Substrate der Prolyl-Hydroxylase oder Lysyl-Hydroxylase darstellen, sodass diese Modifikationen vor der Faltung zum Prokollagen abgeschlossen sein müs-sen. Die beiden Propeptide bilden im ER intramolekulare Disulfidbrücken, das C-Propeptid wird zusätzlich noch mit N-glycosidisch gebundenen Zuckerresten derivatisiert. An die hydroxylierten Lysine werden häufig noch O-glycosi-disch verknüpfte Disaccharide aus Galactose und Glucose (-O-Gal-Glc) angehängt.
Anschließend assemblieren drei Polypeptidketten zum Prokollagen-Molekül. Dieser Prozess wird durch Aneinan-derlagerung der C-terminalen Pro-Domänen eingeleitet. Die Struktur dieser Domänen legt fest, ob Homo- oder Heterotrimere gebildet werden, da nur bestimmte Assozia-tionen stabil sind. Danach faltet sich die Tripel-Helix vom C-terminalen zum N-terminalen Ende hin (. Abb. 24.3a). Das entstehende Prokollagen ist das Endprodukt der intra-zellulären Biosyntheseabschnitte.
! Die Hydroxylgruppen der Hydroxyproline bilden Was-serstoffbrücken zwischen den benachbarten Polypep-tidketten, sodass die Tripelhelix bei physiologischen Temperaturen stabil ist.
. Abb. 24.1a,b. Transmissionselektronenmikroskopische Aufnah-men. a Kollagen-Mikrofibrillen aus Rattenschwanzsehnen. b Epiphysen-knorpel des Hühnerembryos
. Abb. 24.2. Schematische Darstellung der Struktur der Polypep-tidkette von fibrillären Kollagenen am Beispiel der α1(III)-Kette. Die »tripelhelicalen« Bereiche, d.h. die Bereiche, die mit zwei weiteren Kollagenketten eine Tripelhelix ausbilden, sind hellblau dargestellt,
die Spaltstellen für die N- und C-Propeptidasen sind durch Pfeile gekennzeichnet. Die Zahlen geben die Anzahl der Aminosäuren in den einzelnen Domänen an
24719
Die Hydroxylierung ist absolut essentiell für die Struktur-funktion des Kollagens. Kollagene mit wenig Hydroxypro-lin, wie sie bei vielen in kaltem Wasser lebenden Organis-men vorkommen, besitzen Schmelzpunkte (= Temperatur, bei der sich die Tripelhelix wieder entfaltet) unter 37 °C. Sie würden sich bei der Biosynthese im Menschen also nicht
falten und daher abgebaut werden. Durch die Hydroxylie-rung wird der Schmelzpunkt jedoch auf über 40°C he-raufgesetzt. Die Hydroxylierung ist Vitamin-C abhängig (7 23.3.1). Das Auftreten von Skorbut bei Vitamin-C-Mangel ist im Wesentlichen durch das Fehlen von neu gebildetem Kollagen bedingt.
Extrazelluläre Biosyntheseschritte. Im Extrazellulärraum, z.T. in Einbuchtungen der Plasmamembran (. Abb. 24.3c), erfolgt die Abspaltung der N- und C-Propeptide durch die Aminopropeptidase und die Carboxypropep tidase, und die tripelhelicalen Moleküle lagern sich zu langen Fi-brillen aneinander (. Abb. 24.3b).
! Die Assemblierung von Kollagen wird durch die charak-teristische Verteilung von hydrophoben und polaren geladenen Aminosäuren gelenkt.
Die miteinander wechselwirkenden Aminosäuren sind in vier homologen, je 67 nm langen Regionen (D1–D4) ange-ordnet (. Abb. 24.4). Daher lagern sich die Moleküle je-weils um 67 nm versetzt (»D-stagger«=67 nm) aneinander, um maximale hydrophobe und elektrostatische Wechsel-wirkungen mit den Nachbarmolekülen zu erreichen. Diese Anordnung erklärt auch die elektronenmikroskopisch zu beobachtende Querstreifung. In regulären Abständen von 67 nm treten Lücken zwischen aufeinander folgenden Kol-lagenmolekülen auf, in die der Farbstoff bei Negativstaining eingelagert wird.
! Die Kollagenfibrillen werden zur Stabilisierung miteinan der quervernetzt.
Die Quervernetzungen bilden sich zwischen Lysinen/Hydroxylysinen im N-terminalen Telopeptid und dem be-nachbarten Bereich der Tripelhelix mit entsprechenden
. Abb. 24.3a–c. Biosynthese und Sekretion der fibrillären Kolla-gene. a Intrazelluläre Schritte: 1 cotranslationale Hydroxylierung von Prolin- und Lysin-Resten; 2 Glycosylierung einzelner Hydoxylysin-Reste; 3 Freisetzung der Polypeptidkette mit disulfidverbrückten und N-glycosylierten Propeptiden; 4 Zusammenlagerung dreier Polypep-tidketten über die C-Propeptide und Beginn der Tripelhelix-Bildung; 5 Bildung des fertigen tripelhelicalen Prokollagen-Moleküls. b Extra-zelluläre Schritte. 1 Abspaltung der Propeptide; 2 Aggregation zu Mikrofibrillen; 3 covalente Quervernetzung. c Schematische Darstel-lung der Fusion von Kollagen-Molekülen in extrazellulären Komparti-menten des Fibroblasten
24.2 · Kollagene
720 Kapitel 24 · Binde- und Stützgewebe
24
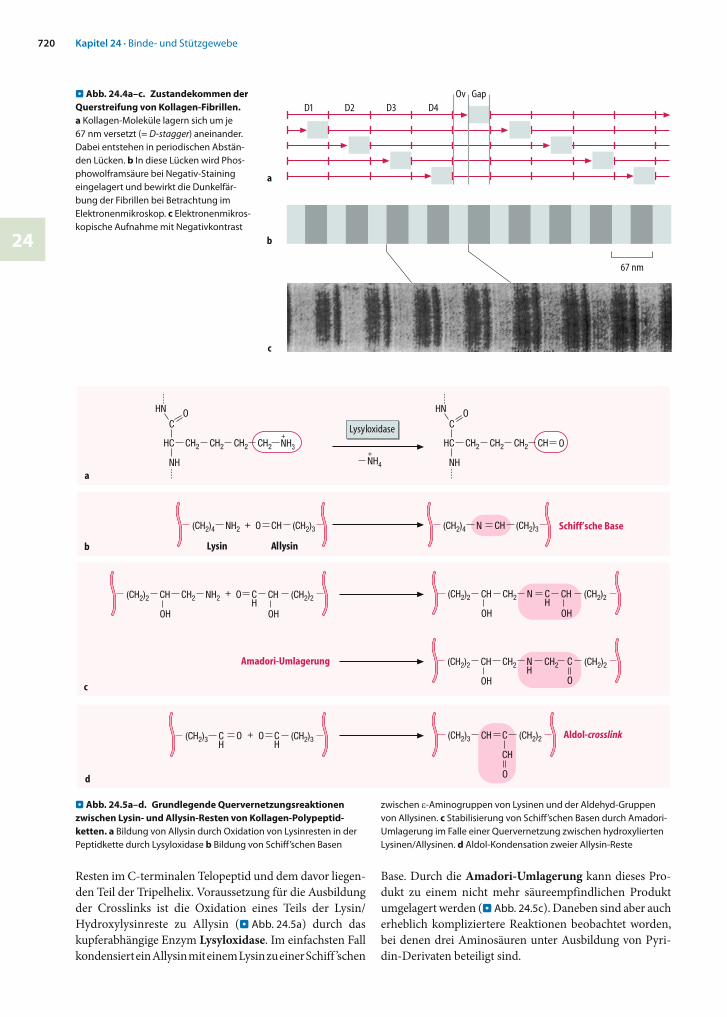
Resten im C-terminalen Telopeptid und dem davor liegen-den Teil der Tripelhelix. Voraussetzung für die Ausbildung der Crosslinks ist die Oxidation eines Teils der Lysin/Hydroxylysinreste zu Allysin (. Abb. 24.5a) durch das kupferabhängige Enzym Lysyloxidase. Im einfachsten Fall kondensiert ein Allysin mit einem Lysin zu einer Schiff ’schen
Base. Durch die Amadori-Umlagerung kann dieses Pro-dukt zu einem nicht mehr säureempfindlichen Produkt umgelagert werden (. Abb. 24.5c). Daneben sind aber auch erheblich kompliziertere Reaktionen beobachtet worden, bei denen drei Aminosäuren unter Ausbildung von Pyri-din-Derivaten beteiligt sind.
. Abb. 24.4a–c. Zustandekommen der Querstreifung von Kollagen-Fibrillen. a Kollagen-Moleküle lagern sich um je 67 nm versetzt (= D-stagger) aneinander. Dabei entstehen in periodischen Abstän-den Lücken. b In diese Lücken wird Phos-phowolframsäure bei Negativ-Staining eingelagert und bewirkt die Dunkelfär-bung der Fibrillen bei Betrachtung im Elektronenmikroskop. c Elektronenmikros-kopi sche Aufnahme mit Negativkontrast
. Abb. 24.5a–d. Grundlegende Quervernetzungsreaktionen zwischen Lysin- und Allysin-Resten von Kollagen-Polypeptid-ketten. a Bildung von Allysin durch Oxidation von Lysinresten in der Peptidkette durch Lysyloxidase b Bildung von Schiff’schen Basen
zwischen -Aminogruppen von Lysinen und der Aldehyd-Gruppen von Allysinen. c Stabilisierung von Schiff’schen Basen durch Amadori-Umlagerung im Falle einer Quervernetzung zwischen hydroxylierten Lysinen/Allysinen. d Aldol-Kondensation zweier Allysin-Reste
24721
! Prokollagen wird in extrazellulären Kompartimenten von Fibroblasten prozessiert und zu Fibrillen aneinan-der gelagert.
Die intrazellulär synthetisierten Prokollagen-Moleküle werden nicht einfach in den Extrazellulärraum sezerniert, vielmehr findet die Prozessierung und Bildung fibrillärer Segmente in extrazellulären Kompartimenten des Fibro-blasten statt. Wie in . Abb. 24.3c angedeutet, reichen Ein-buchtungen bis tief in den Fibroblasten hinein. Sekreto-rische Vakuolen mit Prokollagen fusionieren miteinander und mit der Zellmembran, sodass lange, enge Kanäle ent-stehen, in denen die Fibrillenbildung abläuft. Im Extra-zellulärraum erfolgt dann eine Aneinanderlagerung der fibrillären Segmente, begleitet von Zusammenlagerung zu Bündeln, die gewebsspezifisch zu weiteren Lagen organi-siert werden können (. Abb. 24.1).
Dicke und Zusammensetzung von Kollagen-fibrillen
! Natürlich vorkommende Fibrillen sind in der Regel Mischfibrillen.
Da die tripelhelicalen Abschnitte der verschiedenen Kol-lagentypen die gleiche Länge besitzen, können sie in die gleiche Fibrille eingebaut werden. So bestehen Fibrillen der Cornea z.B. aus einer Mischung von Typ-I- und Typ-V-Kollagen. Sehnen enthalten fast ausschließlich Typ-I-Kol-lagen, mit geringen Beimischungen der Typen-III und –V,
während die Fibrillen der Haut aus den Kollagen-Typen-I und –III aufgebaut sind. Knorpel hingegen enthält Misch-fibrillen aus Typ–II und Typ-XI. Typ-XI-Kollagen ist zwar nur eine Nebenkomponente (5–10%), aber notwendig, da-mit sich überhaupt Fibrillen bilden können.
! Wichtige, die Fibrillendicke regulierende Faktoren sind Mischung verschiedener Kollagentypen, Interaktion mit Proteoglykanen und Fibrillen assoziierten (FACIT)-Kollagenen.
Die verschiedenen Kollagentypen unterscheiden sich von Hause aus in der maximal erreichbaren Fibrillendicke. Wenigstens teilweise beruht dies darauf, dass bei den Kol-lagenen Typ-III, Typ-V und Typ-XI die N-terminalen Propeptide nur zum Teil abgespalten werden, und so die Fibrillenbildung sterisch inhibieren. Es konnte außerdem gezeigt werden, dass die Fibrillenbildung durch das kleine Proteoglykan Decorin (7 Kap. 24.4.2) reguliert wird. Die Be-deutung der FACIT-Kollagene wird im nächsten Abschnitt be handelt.
! Fibroblasten können Kollagenfibrillen organisieren.
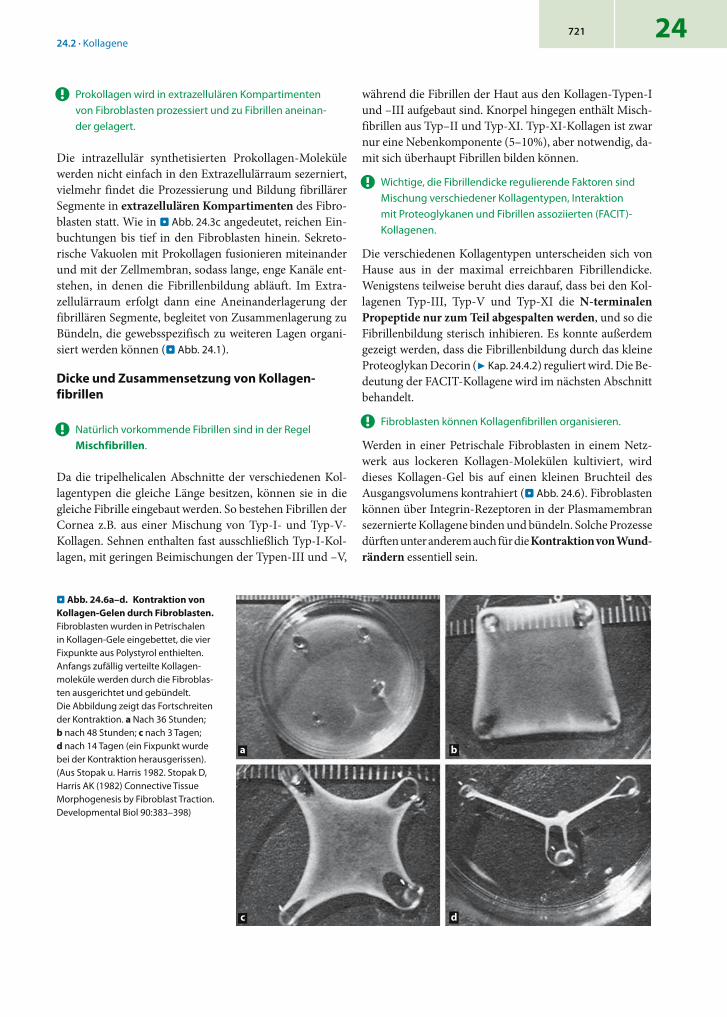
Werden in einer Petrischale Fibroblasten in einem Netz-werk aus lockeren Kollagen-Molekülen kultiviert, wird dieses Kollagen-Gel bis auf einen kleinen Bruchteil des Ausgangsvolumens kontrahiert (. Abb. 24.6). Fibroblasten können über Integrin-Rezeptoren in der Plasmamembran sezernierte Kollagene binden und bündeln. Solche Prozesse dürften unter anderem auch für die Kontraktion von Wund-rändern essentiell sein.
a
c
b
d
. Abb. 24.6a–d. Kontraktion von Kollagen-Gelen durch Fibroblasten. Fibroblasten wurden in Petrischalen in Kollagen-Gele eingebettet, die vier Fixpunkte aus Polystyrol enthielten. Anfangs zufällig verteilte Kollagen-moleküle werden durch die Fibroblas-ten ausgerichtet und gebündelt. Die Abbildung zeigt das Fortschreiten der Kon traktion. a Nach 36 Stunden; b nach 48 Stunden; c nach 3 Tagen; d nach 14 Tagen (ein Fixpunkt wurde bei der Kontraktion herausge rissen). (Aus Stopak u. Harris 1982. Stopak D, Harris AK (1982) Connective Tissue Morphogenesis by Fibroblast Traction. Developmental Biol 90:383–398)
24.2 · Kollagene

722 Kapitel 24 · Binde- und Stützgewebe
24
24.2.2 Nichtfibrilläre Kollagene
! Die nichtfibrillären Kollagene bilden eine Vielzahl ver-schiedener Strukturen mit unterschiedlichen Aufgaben.
Im Gegensatz zu den fibrillären Kollagenen bilden die üb-rigen Kollagene eine sehr heterogene Gruppe. Einige Struk-turen, die weiter unten noch diskutiert werden, sind in . Abb. 24.7 dargestellt. Die tripelhelicalen Segmente sind von unterschiedlicher Länge und z.T. durch längere nicht-tripelhelicale Segmente unterbrochen, die die Moleküle flexibler machen. Ein weiteres Kennzeichen ist die Gegen-wart von zahlreichen nichtkollagenen Domänen. z.B. ent-hält das Kollagen-Typ-VI zahlreiche Kopien von Domänen, die Sequenzabschnitten des von-Willebrand-Faktors ho-molog sind, während Kollagen-Typ-VII eine Anzahl von Fibronectin-Typ-III-Domänen enthält. Die für die fibril-lären Kollagene typische Prozessierung von Propeptiden findet meist nicht statt. Wichtige nichtfibrilläre Kollagene sind:4 FACIT-Kollagene (fibril associated collagens with inter-
rupted triple helices). Diese Gruppe umfasst neben den in . Tab. 24.1 aufgeführten Kollagenen Typ-IX, -XII und -XIV noch eine Reihe weiterer, z.T. noch nicht nä-her charakterisierter Kollagene (XVI, XIX bis XXIII). Für Kollagen Typ-IX wurde gezeigt, dass es in regelmä-ßigen Abständen als monomeres Protein an die Ober-fläche von Kollagen-Typ-II-Fibrillen bindet (. Abb. 24.7e). Der größte Teil des Moleküls ist an der Typ-II-Fibrille fixiert, während der N-terminale Teil, der in einer nichtkollagenen globulären Domäne endet, von der Fibrille wegzeigt. Zusätzlich trägt Kollagen-Typ-IX häufig noch eine Chondroitinsulfat-Proteoglykan-Seitenkette, sodass die Oberfläche der Kollagen-II-Fibrille einen hydrophilen Charakter aufweist. Inte-ressanterweise erkrankten Mäuse mit inaktiviertem Kollagen-IX-Gen im Alter von einigen Monaten an Ar-throse, sodass Kollagen IX dem Gelenkknorpel sozu-sagen als Gelenkschmiermittel eine hydrophile Ober-fläche verleiht, die ihn vor mechanischer Abnutzung schützt
4 Die beiden Kollagene Typ-XII und Typ-XIV hingegen sind überwiegend mit Typ-I-Kollagen-Fibrillen assozi-iert. Sie besitzen eine ähnliche Struktur, bestehend aus einer C-terminalen, Kollagen-bindenden Domäne, und eine große nichtkollagene N-terminale Domäne mit der Fähigkeit an Proteoglykan-Seitenketten und andere ECM-Moleküle zu binden. Man nimmt an, dass die Funktion der beiden Kollagene darin besteht, Wechsel-wirkungen mit anderen extrazellulären Matrix-Mole-külen einzugehen um so mitzuhelfen, den Aufbau der ECM-Struktur zu regulieren. Weiterhin wird eine Rolle bei der Regulation der Fibrillenbildung diskutiert, da Anlagerung an die Oberfläche einer Fibrille ein weiteres Dickenwachstum inhibieren könnte
4 Kollagen-Typ-IV. stellt die Hauptstrukturkomponente der Basalmembran dar und ist für alle tierischen Viel-zeller lebenswichtig. Es besitzt eine fast 400 nm lange tripelhelicale Domäne und zusätzlich eine globuläre Domäne (NC1) am C-terminalen Ende. Durch zahl-reiche, nur wenige Aminosäuren lange Unterbre-chungen der Tripelhelix ist das Molekül flexibler als die fibrillären Kollagene. Kollagen Typ IV bildet ein flächiges Netzwerk: Durch covalente Verknüpfungen über die C-terminalen globulären Domänen werden Dimere gebildet, die lateral miteinander aggregieren können. Zusätzlich können vier Moleküle über die N-terminalen Abschnitte der Tripelhelix (sog. 7S-Do-mäne) aggregieren, sodass eine Struktur entsteht, wie
. Abb. 24.7a–e. Schematische Darstellung der Struktur einiger ausgewählter nichtfibrillärer Kollagene. a Kollagen Typ IV (NC1 =C-terminale nicht-kollagene Domäne; 7 S = N-terminale Quervernet-zungsdomäne); b Kollagen Typ VI; c Kollagen Typ VII; d Kollagen Typ X, e Kollagen Typ IX

24723
sie in . Abb. 24.7a dargestellt ist. Insgesamt existieren sechs verschiedene -Ketten. In den meisten Basal-membranen hat Kollagen IV die Zusammensetzung [ 1(IV)2 2(IV)], in einigen Basalmembranen findet man jedoch auch andere Kombinationen (. Tab. 24.1). So enthalten die Basalmembranen der Glomeruli über-wiegend Moleküle der Zusammensetzung [ 3(IV)
4(IV) 5(IV)], die funktionell nicht durch die 1 und 2-Ketten ersetzt werden können, wie die Analyse von
Gendefekten zeigt (7 u.)4 Mit Basalmembranen assoziiert sind die beiden homo-
logen Kollagene Typ-XV und Typ-XVIII. Sie kommen in den epithelialen und endothelialen Basalmembranen einer Reihe von Geweben vor. Ca. 20 kDa große Spalt-produkte der C-terminalen Domäne (Endostatin im Falle von Col-XVIII) hemmen die Angiogenese. Mu-tationen im Gen für Col-XVIII führen aus noch unbe-kannten Gründen zur Makuladegeneration im Auge
4 Kollagen-Typ-VI ist ein in den meisten interstitiellen Bindegeweben vorkommendes Kollagen. Es ist der Hauptbestandteil der gebänderten Mikrofibrillen (. Abb. 24.7b). Diese Strukturen werden in völlig ande-rer Art und Weise als die Fibrillen der Kollagene I-III und V gebildet. Zunächst lagern sich zwei monomere Moleküle antiparallel zu Dimeren zusammen, die wei-ter zu Tetrameren aggregieren. Diese Tetramere poly-merisieren schließlich über die globulären Enden
4 Kollagen-Typ-VII verankert die Basalmembran von Plattenepithelien mit Ankerplatten im darunter liegen-den Gewebestroma. Nach Abspaltung einer C-termi-nalen, 30 kDa großen globulären Domäne bildet Kol-lagen VII antiparallele Dimere, die zu Bündeln aggre-gieren
4 Kollagen-Typ-X wird nur in hypertrophierendem Knorpel exprimiert und stellt daher ein wichtiges Markerprotein dar. Das monomere Molekül besitzt die Form einer Hantel und polymerisiert zu einem hexa-gonalen Netzwerk. Ähnlich aufgebaut ist Kollagen VIII, das aber eine breitere Verteilung besitzt, und vor allem in der Descemet-Membran und in der subendo-thelialen Matrix prominent ist
4 Transmembrankollagene. Nicht alle Kollagene werden sezerniert, einige besitzen Transmembrandomänen (Typen XIII, XVII, XXIII, XXV). Eine Funktion der Transmembrankollagene ist die Stabilisierung von Zell-Zell- und Zell-Matrix-Interaktionen. Kollagen-Typ-XVII ist ein Bestandteil der Hemidesmosomen und bindet an Laminin und 6-Integrin, Mutationen führen zu schweren Erkrankungen (Epidermolysis bul-losa junctionalis, 7 u.). Eine interessante Eigenschaft von Kollagen Typ-XXV ist die Bindung an Alzheimer Amyloid-Plaques
24.2.3 Angeborene Erkrankungen des Kollagen-Stoffwechsels
Störungen der Kollagen-Expression können auf unter-schiedlichen Ebenen auftreten:4 Störung der Regulation einzelner Gene, wodurch die
ge websspezifische Zusammensetzung der einzelnen Kollagen-Typen verändert wird
4 Mutationen von Kollagenen und Enzymen für die posttranslationalen Modifikationen. Dies führt meist zu Defekten in der makromolekularen Organisation des Kollagens und somit zur Veränderung der biomecha-nischen Eigenschaften, die letztlich für die klinischen Symptome verantwortlich sind
Osteogenesis imperfecta (OI). Die Erkrankung beruht auf einer Synthesestörung von Kollagen I. Betroffen sind alle Kollagen-reichen Organe, dominant ist jedoch der Kno-chen-Phänotyp (wiederholte, zu schweren Skelettdeforma-tionen führende Knochenbrüche bei Belastung). Man un-terscheidet verschiedene Formen der OI, die schwerste Form führt zum Tod im Mutterleib oder kurz nach der Ge-burt. Über 200 verschiedene Mutationen sind beschrieben, darunter Deletionen, Insertionen und Spleiß-Variationen. Die meisten Mutationen bestehen in einer Substitution von Glycinen im Gly-X-Y-Triplet. Dies kann zur Folge haben, dass die Tripelhelix-Bildung verlangsamt wird oder sich überhaupt nicht mehr falten kann, sodass die Ketten im Fibroblasten abgebaut werden. Andere Mutationen verur-sachen Knicke in der Tripelhelix und interferieren so mit dem Wachstum der Fibrillen.
Ehlers-Danlos-Syndrom (EDS). Das Ehlers-Danlos-Syn-drom ist durch Überdehnbarkeit der Haut und Überstreck-barkeit der Gelenke charakterisiert. Trotz der relativ ein-heitlichen Symptomatik liegen der Erkrankung sehr hete-rogene Ursachen zugrunde, und man unterscheidet neun verschiedene Typen, z.B.:4 Typ-IV beruht auf Defekten in der Kollagen-III-Syn-
these. Da Blutgefäße, besonders die großen Arterien, einen hohen Anteil an Kollagen-III besitzen, besteht Neigung zu Gefäßrupturen
4 Typ-V beruht auf einer defekten Lysyloxidase, sodass die Quervernetzung von Kollagen gestört ist. Da dieses Enzym Kupfer-abhängig ist, treten ähnliche Effekte auch beim Mencke-Syndrom, einer Resorptionsstörung von Kupfer, auf
4 Typ-VI beruht ebenfalls auf einer Störung der Querver-netzung von Kollagenfibrillen, in diesem Fall jedoch aufgrund einer defekten Lysylhydroxylase
4 Typ-VII beruht auf einer gestörten Abspaltung der Propeptide, indem entweder die Peptidasen inaktiv oder wenig aktiv sind oder die Erkennungsstelle für die Proteasen mutiert ist
24.2 · Kollagene

724 Kapitel 24 · Binde- und Stützgewebe
24
Alports-Syndrom. Das Alports-Syndrom stellt eine pro-gressive Erbkrankheit dar, die durch Mutationen in den 3-,
4-, besonders aber der -Kette des Typ-IV-Kollagens verursacht wird. Die Folge ist eine Verschlechterung der Nie-renfunktion mit Hämaturie und Proteinurie aufgrund von Strukturveränderungen der glomerulären Basalmembran. Zusätzlich ist die Erkrankung durch Innen ohrschwerhörigkeit und Augenveränderungen gekennzeichnet.
Chondrodysplasien. Eine Vielzahl von Mutationen im Kollagen-II-Gen korreliert mit einer Reihe von Chondro-
dysplasien, die zu Zwergwuchs, Gelenkdeformationen oder anderen Skelettfehlbildungen führen können, da es aufgrund von Kollagen-II-Synthesestörungen zu Stö-rungen in der Knorpelbildung und damit zu Störungen der enchondralen Ossifikation kommt. Mutationen des Typ X-Kol lagens sind die Ursache für die Chondrodys-plasia metaphysaria vom Typ Schmid, klinische Symp-tome sind Verkürzung der Gliedmaßen und verkrümmte Beine.
Epidermolysis bullosa dystrophica beruht auf De-fekten des Kollagens Typ VII (7 Kap. 24.8.5)
In Kürze
Kollagene sind die wichtigsten Strukturproteine des Kör-pers, inzwischen sind wenigstens 27 verschiedene Typen bekannt. Alle Kollagen-Moleküle bestehen aus drei Polypeptid-ketten. Gemeinsames Strukturmerkmal sind vielfach wie-derholte Gly-X-Y-Sequenzen, wobei X und Y häufig Prolin und Hydroxyprolin darstellen. Hydroyxprolin erhöht den Schmelzpunkt der Tripelhelix auf über 40°C. Die Hydroxy-lierung von Prolin und Lysin ist Vitamin-C abhängig. Wei-tere für Kollagen typische modifizierte Aminosäuren sind Hydroxylysin und Allysin.4 Die größte Gruppe innerhalb der Kollagene sind die
fibrillären Kollagene (Typ-I, -II, -III, -V, und -XI), die gebänderte Fibrillen bilden (z.B. in Sehnen, Bändern, Haut und Knorpel). Sie kommen überwiegend in Form von Mischfibrillen vor.
Die komplexe Biosynthese der fibrillären Kollagene lässt sich in mehrere Abschnitte unterteilen:– intrazelluläre Schritte: Biosynthese am RER, cotrans-
lationale Hydroxylierung, Assemblierung der drei Polypeptidketten zu Prokollagen
– Abspaltung der N- und C-Propeptide in extra zellu lä-ren Kompartimenten und Assemblierung zu größeren Einheiten
– Bildung von Fibrillen und Stabilisierung durch Quer-vernetzung (zwischen Lysin und Allysin) im Extrazel-lulärraum
– Organisation der Fibrillen durch Fibroblasten4 Fibrillen-assoziierte Kollagene modifizieren die Oberflä-
chen von Fibrillen. So verleiht das Kollagen Typ IX den Typ-II-Fibrillen des Knorpels den hydrophilen Charakter (wichtig für die Gelenkfunktion)
4 Viele Kollagene bilden keine Fibrillen und sind nicht mit Fibrillen assoziiert. Ein besonders wichtiger Vertreter dieser Gruppe ist das Typ-IV-Kollagen, ein essentieller Bestandteil aller Basalmembranen
Defekte in den Kollagen-Genen führen zu einer Reihe von Erkrankungen wie Osteogenesis imperfecta und Ehlers-Danlos-Syndrom (Mutationen bzw. Defekte in der Prozes-sierung von Typ-I-Kollagen), Alports Syndrom (Defekte im Typ-IV-Kollagen), Chondrodysplasien (Defekte im Typ-II-Kollagen).
24.3 Elastische Fasern
Aufgrund ihrer starren Tripelhelix sind die Kollagene nur bedingt geeignet, Strukturen (z.B. Wände der großen Arte-rien, Lunge) elastische Eigenschaften zu verleihen. Dafür haben die Vertebraten spezielle elastische Fasern entwickelt, die je nach Gewebetyp in morphologisch unterscheidbaren Netzwerken vorkommen.
Die elastischen Fasern bestehen aus einem Kern aus Elastin, der in elektronenmikroskopischen Aufnahmen keine Struktur aufweist und daher »amorph« genannt wird. Der Elastin-Kern wird von einem Mantel aus Mikrofibril-len umgeben. Letztere bestehen aus Fibrillin-Molekülen, an denen Elastin während der Biosynthese der elastischen Fasern polymerisiert (7 u. und . Abb. 24.8). Zusätzlich enthalten die elastischen Fasern noch eine Reihe weiterer Proteine, wie z.B. Mikrofibrillen-assoziierte Glycoproteine
(MAGPs), Emiline und Fibuline. Diese Proteine sind für die Bildung der korrekten Architektur der elastischen Fasern wichtig (7 u.).
! Elastin verleiht den Geweben elastische Eigenschaften.
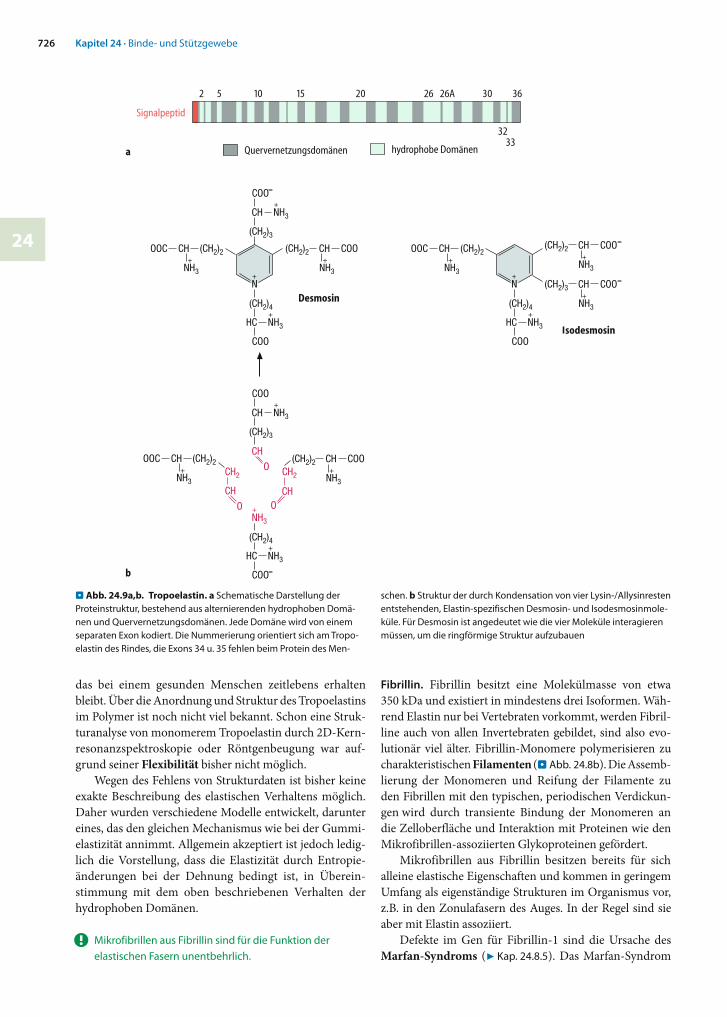
Elastin ist ein unlösliches, quervernetztes Polymer aus mo-nomeren Tropoelastin-Untereinheiten. Dieses 70 kDa große Protein setzt sich zum großen Teil aus alternierenden hydrophoben Bereichen und -helicalen Quervernet-zungs-Domänen zusammen (. Abb. 24.9a).
Die hydrophoben Bereiche sind reich an Glycin, Ala-nin, Valin und Prolin. Sie besitzen einen hohen Anteil an
-Faltblatt-Strukturen und -Turns. Diese Strukturen kön-nen in mehreren, leicht ineinander überführbaren Konfor-mationen vorliegen, besitzen also eine hohe Flexibilität. In dieser Hinsicht ist Tropoelastin ein atypisches Protein, denn normalerweise liegen die hydrophoben Domänen im Inne-

24725
ren des Proteins verborgen und besitzen eine feste, kom-pakte Struktur. Eine zweite atypische Eigenschaft besteht darin, dass die hydrophoben Bereiche des Tropoelastins von einem Wassermantel umgeben sind. Diese Hydrati-sierung wird als wesentlich für das elastische Verhalten erachtet: Bei Dehnung der Peptidkette werden mehr hy-drophobe Seitenketten der Aminosäuren dem Wasser ausgesetzt, sodass das Wasser eine geordnetere Struktur (= Entropieabnahme) einnehmen muss (wie bei der Grenz-fläche zu einem Öltröpfchen). Wenn die Zugbelastung nachlässt, können die hydrophoben Seitenketten wieder stärker miteinander interagieren, die geordnete Hydrat-
hülle kann sich wieder statistisch orientieren (= Entropie-zunahme), sodass eine elastische Rückstellkraft resultiert.
Die Quervernetzungsdomänen enthalten 40 Lysin-Reste, von denen etwa 35 durch Lysyloxidase zu Allysin oxidiert werden, nach dem gleichen Mechanismus wie bei Kollagen (s.o). Die Mehrzahl der Allysinreste bildet Cross-links mit verbliebenen Lysinresten. Dabei entstehen z.T. die gleichen Produkte wie beim Kollagen, zusätzlich werden aber auch elastinspezifische ringförmige Moleküle wie Desmosin und Isodesmosin aufgebaut (. Abb. 24.9b).
Die Quervernetzungen erfolgen intra- und intermole-kular. Dabei entsteht ein hochelastisches, inertes Polymer,
. Abb. 24.8a,b. Architektur und Biosynthese elastischer Fasern. a Fibrillin-Moleküle (rot) assemblieren an der Zelloberfläche und reifen zu Mikrofibrillen (1), danach werden Tropelastinmoleküle (grün) an die Mikrofibrillen angelagert und durch Lysyloxidase quervernetzt (2). Die Polymere aus Elastin wachsen später zusammen und drängen die
Mikrofibrillen zum Rand (3). b Elektronenmikroskopische Aufnahme nach Rotations-Kegelbedampfung von Fibrillen aus Fibrillin. Der Strich entspricht 100 nm. (Aus Ren ZX 1991. Ren ZX et al. (1991) An Analysis by Rotary Shadowing of the Structure of the Mammalian Vitreous Humor and Zonular Apparatus. J Struct Biol 106:57–63)
24.3 · Elastische Fasern
726 Kapitel 24 · Binde- und Stützgewebe
24
das bei einem gesunden Menschen zeitlebens erhalten bleibt. Über die Anordnung und Struktur des Tropoelastins im Polymer ist noch nicht viel bekannt. Schon eine Struk-turanalyse von monomerem Tropoelastin durch 2D-Kern-resonanzspektroskopie oder Röntgenbeugung war auf-grund seiner Flexibilität bisher nicht möglich.
Wegen des Fehlens von Strukturdaten ist bisher keine exakte Beschreibung des elastischen Verhaltens möglich. Daher wurden verschiedene Modelle entwickelt, darunter eines, das den gleichen Mechanismus wie bei der Gummi-elastizität annimmt. Allgemein akzeptiert ist jedoch ledig-lich die Vorstellung, dass die Elastizität durch Entropie-änderungen bei der Dehnung bedingt ist, in Überein-stimmung mit dem oben beschriebenen Verhalten der hydrophoben Domänen.
! Mikrofibrillen aus Fibrillin sind für die Funktion der elastischen Fasern unentbehrlich.
Fibrillin. Fibrillin besitzt eine Molekülmasse von etwa 350 kDa und existiert in mindestens drei Isoformen. Wäh-rend Elastin nur bei Vertebraten vorkommt, werden Fibril-line auch von allen Invertebraten gebildet, sind also evo-lutionär viel älter. Fibrillin-Monomere polymerisieren zu charakteristischen Filamenten (. Abb. 24.8b). Die Assemb-lierung der Monomeren und Reifung der Filamente zu den Fibrillen mit den typischen, periodischen Verdickun-gen wird durch transiente Bindung der Monomeren an die Zelloberfläche und Interaktion mit Proteinen wie den Mikrofibrillen-assoziierten Glykoproteinen gefördert.
Mikrofibrillen aus Fibrillin besitzen bereits für sich alleine elastische Eigenschaften und kommen in geringem Umfang als eigenständige Strukturen im Organismus vor, z.B. in den Zonulafasern des Auges. In der Regel sind sie aber mit Elastin assoziiert.
Defekte im Gen für Fibrillin-1 sind die Ursache des Marfan-Syndroms (7 Kap. 24.8.5). Das Marfan-Syndrom
. Abb. 24.9a,b. Tropoelastin. a Schematische Darstellung der Pro teinstruktur, bestehend aus alternierenden hydrophoben Domä-nen und Quervernetzungsdomänen. Jede Domäne wird von einem separaten Exon kodiert. Die Nummerierung orientiert sich am Tropo-elastin des Rindes, die Exons 34 u. 35 fehlen beim Protein des Men-
schen. b Struktur der durch Kondensation von vier Lysin-/Allysinresten entstehenden, Elastin-spezifischen Desmosin- und Isodesmosinmole-küle. Für Desmosin ist angedeutet wie die vier Moleküle interagieren müssen, um die ringförmige Struktur aufzubauen

24727
ist durch Hochwuchs, Arachnodaktylie und Linsenverän-derungen gekennzeichnet. Entscheidend für den Verlauf der Krankheit sind jedoch Störungen in den Gefäßwänden. Es kommt zur Bildung von Aneurysmen und zu Aortenrup-turen.
Eine der historischen Persönlichkeiten, die an Marfan-Syndrom litten, war wahrscheinlich der amerikanische Prä-sident Abraham Lincoln.
Biosynthese der elastischen Fasern. Zunächst entstehen Mikrofibrillen, die später mit Elastin zusammenwachsen, das die Hauptkomponente in den ausgereiften elastischen Fasern darstellt (. Abb. 24.8), der Elastinanteil ist allerdings von Gewebe zu Gewebe unterschiedlich. Man nimmt an,
dass die Tropoelastinmoleküle durch Bindung an Mikro-fibrillen eine Konformation einnehmen, in der die zu ver-netzenden Lysine/Allysine in der richtigen Position für die Quervernetzung durch Lysyloxidase liegen. Wie neueste Untersuchungen gezeigt haben sind für die Assemblierung des Elastins noch weitere Proteine wie die Emiline und Fibuline erforderlich. Auf molekularer Ebene sind diese Prozesse allerdings erst unzureichend charakterisiert.
Elastische Fasern werden hauptsächlich während der Wachstumsphase der Organe angelegt, später nur noch in begrenztem Umfang. Dies erklärt, warum bei degenerativen oder entzündlichen Reaktionen, bei denen Elastin durch z.B. von Leukozyten gebildete Elastasen degradiert wird, die elastischen Eigenschaften weitgehend verloren gehen.
In Kürze
Elastische Fasern erlauben eine reversible Dehnung und Kontraktion. Elastische Fasern sind im Wesentlichen aus Elastin und Fibrillin aufgebaut:4 Elastin ist ein Polymer, das durch Quervernetzung
von monomeren Tropoelastin-Einheiten entsteht. Die Quervernetzung erfolgt wie beim Kollagen über
Lysin/Allysin, dabei entstehen u.a. die Elastin-spezifi-schen Produkte Desmosin und Isodesmosin
4 Fibrillin bildet Mikrofibrillen, die für die Organisation des Elastins notwendig sind
Defekte im Gen für Fibrillin-1 führen zum Marfan-Syn-drom.
24.4 Proteoglykane
! Proteoglykane sind eine heterogene Gruppe von Pro-teinen, die durch Glycosaminoglykan(GAG)-Seitenket-ten, modifiziert sind.
Proteoglykane sind ubiquitäre Zelloberflächen- und Extra-zelluläre-Matrix-Proteine. Im Unterschied zu den meisten Proteinen, die aufgrund ihrer Aminosäuresequenz in ver-schiedene Familien eingeteilt werden, sind die Proteo glyka-ne durch covalent mit dem Proteingerüst verknüpfte Gly-cosaminoglykan-Seitenketten (GAG) definiert. Diese stel len nichtverzweigte Polymere aus repetitiven Disaccharid-einheiten dar und werden entsprechend der Struktur der Grundbausteine in Chondroitinsulfat, Keratansulfat, Dermatansulfat und Heparansulfat unterteilt. Zur Dar-stellung der Strukturen und Biosynthese dieser Kohlen-hydrate sei auf 7 Kap. 2.14, und 7 Kap. 17.3.5, verwiesen, wo diese Themen bereits diskutiert worden sind. Die Gly-cosaminoglykan-Seitenketten können Größen von einigen 10 kDa besitzen und so die Eigenschaften des Proteins be-stimmen.
Proteoglykane zeigen eine fast unüberschaubare Struk-tur-Vielfalt (. Tabelle 24.2). Diese ist durch zwei Faktoren bedingt:4 Modifikation der Glykane. Die Modifikationen um-
fassen:5 Anheftung von O-Sulfat-Resten an verschiedenen
Positionen
5 Umwandlung von N-Acetylglucosamin in Glucose-N-Sulfat und
5 Isomerisierung von Glucuronsäure zu Iduronsäure
Da diese Modifikationen nur sporadisch innerhalb der Ketten erfolgen, ergibt sich ein komplexes Produktspek-trum.
4 Vielzahl von Proteingerüsten. Die Core-Proteine der Proteoglykane werden von mehr als einhundert Genen kodiert. Die Größe der Polypeptidketten reicht von etwa 10 kDa bis über 400 kDa, sie besitzen daher eine große Strukturvielfalt
Viele Funktionen der Proteoglykane lassen sich mit den biophysikalischen Eigenschaften, dem polaren Charakter und der negativen Ladungen durch Uron- und Sulfon säuren der Glykanketten, erklären. Aufgrund der negativen Ladun-gen stellen Proteoglykane eine Filtrationsbarriere in den Glomeruli der Niere dar. Das Heparansulfat-Proteoglykan Perlecan verhindert, vermutlich zusammen mit anderen Proteoglykanen, den Durchtritt anionischer Serumproteine in den Urin. Eine weitere Funktion ist die Bildung wasser-gefüllter Kompartimente, z.B. im Knorpel. Die GAG-Ket-ten besitzen je nach Typ eine bis vier Sulfatgruppen pro Disaccharid-Einheit, was eine Ladungsdichte von 1–5 La-dungen pro nm ergibt. Die Sulfatreste sind bei physio-logischen pH-Werten voll ionisiert, die fixierten negativen Ladungen ziehen Gegenionen an, vor allem Na+- und Ca2+-Ionen. Die hohe lokale Ionenkonzentration verursacht
24.4 · Proteoglykane

728 Kapitel 24 · Binde- und Stützgewebe
24
einen osmotisch bedingten Wassereinstrom aus den um-liegenden Regionen. Das Ausmaß hängt stark von der Konzentration ab, die in verschiedenen Kompartimenten sehr unterschiedlich ist. Im Knorpel etwa beträgt die extra-zelluläre Na+ -Konzentration 250–350 mM und die Os-molalität 350–450 mosm, verglichen mit etwa 290 mosm der normalen Extrazellulärflüssigkeit.
Mit solchen Funktionen sind die Proteoglykane aber nur unzureichend charakterisiert. Sie sind darüber hinaus von Bedeutung als Corezeptoren für Wachstumsfakto-ren, als Modulatoren der Zell-Zell- und Zell-Matrix-Inter aktion und bei der Regulation der Aktivität eini-ger Proteasen (Heparin-Thrombin-Antithrombin III) (7 Kap. 29.5.3).
24.4.1 Aggrecan
! Aggrecan ist ein lebensnotwendiger Knorpelbaustein. Es ist essentiell für die Funktion des Knorpels als druck-elastische Struktur.
Aggrecan ist das wichtigste Proteoglykan des Knorpels. Das Core-Protein hat eine Größe von etwa 250 kDa. Die N- und C-terminalen Bereiche bilden globuläre Domänen, der Mittelteil hingegen besitzt eine elongierte Struktur, die mit etwa 30 Keratansulfat- und ungefähr 100 (!) Chon-droitinsulfat-Seitenketten substituiert ist, sodass das kom-plette Protein eine Molekülmasse von etwa 3 MDa besitzt und eine hohe Konzentration an negativen Ladungen auf-
weist (. Abb. 24.10b). Aggrecan kommt im Knorpel nicht isoliert vor, sondern bildet riesige Aggregate mit Hyalu-ronsäure (. Abb. 24.10a), die selbst schon ein ungewöhn-lich großes lineares Polymer aus bis zu 25000 Disaccharid-einheiten von Glucuronsäure und N-Acetyl-Glucosamin darstellt (7 Kap. 2.1.4). Die nichtcovalente Bindung an Hyaluronsäure wird durch die N-terminale globuläre Do mäne des Aggrecans vermittelt und durch ein kleines Protein, das so genannte Link-Protein, stabilisiert. Muta-tionen, die zu einem nichtfunktionellen Protein führen, sind letal. Embryonen von Hühnchen und Maus zeigen eine stark verminderte Knorpelbildung und damit auch eine gestörte Knochenbildung. Unmittelbar letal ist vermutlich der Kollaps der Luftröhre. Ein ähnlicher Phänotyp tritt bei Mangel an Sulfattransferasen auf, weil durch Fehlen der sulfatierten Zucker die Ladungskonzentration erniedrigt ist und so die Bildung eines hyperosmotischen (7 o.), rever-sibel deformierbaren Gels verhindert wird.
Ein strukturell mit Aggrecan verwandtes großes Pro-teoglykan ist das in vielen extrazellulären Matrices vorkom-mende Versican. Dieses Proteoglykan schafft ebenfalls eine lockere hydratisierte Matrix. Darüber hinaus kann es auch Prozesse wie Zellwanderung und -Proliferation be-einflussen.
24.4.2 Kleine leucinreiche Proteoglykane
! Proteoglykane wie Decorin, Biglykan und Fibromodulin regulieren die Kollagen-Fibrillenbildung.
. Tabelle 24.2. Übersicht über wichtige Proteoglykane (Auswahl)
Proteoglykan Core-Protein (kDa)a
Typ u. Anzahl der GAG-Kettenb
Vorkommen/Funktion
Basalmembran-Proteoglykane
Perlecan 400–470 HS/CS (3) Integraler Bestandteil der Basalmembran, Filtrationsbarriere
Agrin 225 HS (3) Aggregation von Acetylcholin-Rezeptoren
Hyalectanec
Aggrecan 220–250 CS/KS (~130) Knorpel, Bildung eines hydratisierten Gels
Versican 180–370 CS (~20) Stroma, hydratisiertes Gel, Modulation v. Zell-Matrix-Interaktionen
Kleine leuzinreiche Proteoglykane
Decorin 36 CS/DS (1) Bindung an Kollagen I, Rolle in der Fibrillen-Bildung, Bindung an TGFβ
Fibromodulin 42 KS (4) Bindung an Kollagen I,Rolle in der Fibrillen-Bildung
Membrangebundene Proteoglykane
Betaglykan 100–110 HS/CS (2) Bindung von TGFβ
Syndecane 20–45 HS/CS (3) FGF-Bindung, Zelladhäsiona Die Variationen in den Größen ergeben sich durch Spezies-Unterschiede und alternatives Spleißen.b Art und Anzahl der angehefteten Glycosaminoglykan (GAG)-Seitenketten können von Zelltyp zu Zelltyp variieren. HS = Heparansulfat;
CS = Chondroitinsulfat; KS = Keratansulfat; Hya = Hyaluronsäure.c Komplexe mit Hyaluronsäure und Link-Proteinen.

24729
Die kleinen leucinreichen Proteoglykane besitzen etwa 40 kDa große core-Proteine mit einer N-terminalen, die Glycosaminoglykan-Seitenketten tragenden Domäne, gefolgt von einer Domäne, die aus leucinreichen repeats besteht. Eine wichtige Funktion dieser Proteine ist die Or ganisation von Kollagenfibrillen. Decorin bindet mit seinem core-Protein an die Gap-Region (. Abb. 24.4) von Kollagen-Fibrillen, während die Glycosaminoglykan-Sei-tenkette nach außen gerichtet ist. Dies erschwert die wei-tere Anlagerung von Kollagenmolekülen, verhindert die laterale Fusion von Fibrillen und fördert so die korrekte Fibrillenbildung. Entsprechend besitzen Decorin-Knock-out-Mäuse eine gestörte Kollagen-Fibrillen-Morphologie und die Haut zeigt eine deutlich reduzierte mechanische Belastbarkeit.
24.4.3 Membrangebundene Proteo-glykane
! Viele membrangebundene Proteoglykane sind Corezep-toren für Wachstumsfaktoren.
Zellkulturuntersuchungen aus den frühen 90er Jahren hatten gezeigt, dass die Wirkung von FGF (Fibroblasten-Wachstumsfaktor, 7 Kap. 25.1.3) in Abwesenheit von He-paransulfat drastisch reduziert ist. Röntgenstruktur analysen haben inzwischen die molekulare Ursache geklärt. Eine
Heparansulfat-Seitenkette, die für eine hochaffine Bindung noch ein spezielles Sulfatierungsmuster besitzt, bindet zwei Ligand-Rezeptor-Komplexe und hält sie so in der richtigen Konformation, dass die intrazelluläre Tyrosin-kinase-Aktivität des FGF-Rezeptors, eines typischen Tyro-sinkinase-Rezeptors (7 25.7) durch Autophosphorylierung aktiviert werden kann. Neben FGF sind eine Reihe anderer Wachstum und Differenzierung regulierender Faktoren (TGF , Wnt, Hedgehog) auf zellgebundene Proteoglykane als Cofaktoren angewiesen. Dies können entweder Mem-branproteine sein wie Betaglykan und die Syndecane, oder über einen Lipidanker befestigte Moleküle wie Gly-pican. Betaglykan bildet über sein core-Protein mit TGF (7 Kap. 25.1.3, 25.7.2) einen Komplex, der mit dem TGF-Rezeptor interagiert. Die Aktivierung von Wachstumsfak-toren durch Syndecane und Glypicane hingegen erfordert die Interaktion mit den Heparansulfatketten.
Nicht nur Zelloberflächenproteine binden Wachs-tumsfaktoren, sondern auch einige sezernierte Proteo-glykane. Seit langem ist z.B. der wachstumshemmende Effekt von Heparin bekannt. Dieser dürfte darauf be-ruhen, dass Proteoglykane in der extrazellulären Matrix mit Heparin um die Bindung der Wachstumsfaktoren kon-kurrieren und so die Konzentration an freien Cytokinen regulieren. Andererseits werden auf diese Weise Wachs-tumsfaktoren in der ECM gespeichert und können bei Be-darf durch Hydrolyse der Bindungspartner wieder freige-setzt werden.
. Abb. 24.10a,b. Schematische Darstellung der Aggregate des Proteoglykans Aggrecan mit Hyaluronsäure. a Aggrecan bindet in vielen Kopien entlang des Hyaluronsäure-Fadens und schafft so ein wässriges, elastisches Kompartiment. b Aggrecan-Moleküle binden
über ihre N-terminale globuläre Domäne an Hyaluronsäure. Die Bin-dung wird durch ein sog. Link-Protein verstärkt. KS = Keratansulfat; CS = Chondroitinsulfat
24.4 · Proteoglykane

730 Kapitel 24 · Binde- und Stützgewebe
24
In Kürze
Proteoglykane sind eine heterogene Gruppe von Pro-teinen, die mit Glycosaminoglykan-Seitenketten substitu-iert sind. Glycosaminoglykane sind aufgrund von sulfatier ten Zuckern und Uronsäuren stark negativ geladen. Man unterscheidet 4 Klassen:4 Chondroitinsulfat4 Keratansulfat4 Dermatansulfat4 Heparansulfat/Heparin
Viele Funktionen der Proteoglykane ergeben sich aus den biophysikalischen Eigenschaften der negativ geladenen Zuckerketten, z.B.:4 Filtrationsbarriere in den Glomeruli (z.B. durch Perlecan)4 wassergefüllte hydrophile Kompartimente (z.B. durch
Hyaluronsäure und Aggrecan im Knorpel)
Einige Proteoglykane (wie Decorin) sind wichtig bei der Regulation der Kollagen-Fibrillenbildung. Einige membrangebundene Proteoglykane (z.B. Syn-decane) sind Corezeptoren für Wachstumsfaktoren (FGF, TGFβ) oder für die Zelladhäsion.
24.5 Nichtkollagene, zelladhäsive Glycoproteine
Die extrazelluläre Matrix besitzt nicht nur Strukturfunkti-onen, sondern sie reguliert auch zelluläre Funktionen.
! Die extrazelluläre Matrix dient als Substrat für Zell-Adhäsion und -Wanderung.
Mit Ausnahme einiger hämatopoetischer Zellen sind alle Zellen des Organismus ständig an Substrate wie die Basal-membran oder das lockere Bindegewebe gebunden, die Bindung erfolgt über spezifische zelluläre Rezeptoren, vor
allem die Integrine (7 u.). Je nach Substrat werden die Zel-len zur Wanderung angeregt, haften fest oder versuchen sogar, das Substrat zu meiden. Bei der Gastrulation z.B. wandern die Mesoderm-Zellen über eine Schicht aus Fibro-nectin (7 u.), das gleichsam als Leitschiene für die Zellen dient. Zellwanderungen sind auch im adulten Organismus z.B. im Falle der Fibroblasten und in pathologischen Situa-tionen wie der Wundheilung oder Rekrutierungen von Leukozyten zu Entzündungsherden erforderlich.
! Die extrazelluläre Matrix beeinflusst Zellfunktion, Zell-differenzierung, Zellproliferation und Apoptose.
. Tabelle 24.3. Matrix-Glycoproteine, die Zellfunktionen beeinflussen (Auswahl)
Protein(-Typ) biologische Wirkungen (Auswahl)
matricellular proteinsProteine, die Zellrezeptoren, ECM-Moleküle, Zytokine und Proteasen binden; Modulatoren der Zell-Matrix-Interaktion; wegen der Vielzahl möglicher Interaktionen unterschiedliche Wirkung an verschiedenen Geweben möglich, Beispiele:
SPARC* (= BM40, Osteonectin)
knock-out-Phänotyp: Linsentrübung, erhöhte Adipogenese, Störung der Kollagen-Fibrillenbildung.
Thrombospondine Aktivierung von TGF , Regulation der Kollagenfibrillenbildung, Regulation der Angiogenese, Plättchenaggregation
Tenascin-C, -X, -R, -Y, -W hohe Expression während der Gewebsbildung in der Embryonalentwicklung, Reexpression während Wund-heilung und Tumorbildung. Bei Ausfall: geringe neurologische Defekte (TN-C, -R), erhöhte Aktivität an MMPs und verstärkte Tumor-Metastasierung (TN-X)
Osteopontin Regulation der Calcifizierung, Bindung an Hydroxyapatit
bone sialoprotein Regulation der Calcifizierung, Bindung an Hydroxyapatit
zelladhäsive Proteine
Vitronectin Vorkommen im Blut und in der ECM, beteiligt an der Regulation der Blutgerinnung und dem proteoly tischen Ab-bau der ECM, bindet mehrere Integrine, vor allem v 3 (Modulation von Zellwanderung, Angiogenese)
Fibronectin wichtigstes Zelladhäsionsmolekül in der ECM, Einzelheiten s. Text
Laminine wichtigstes Zelladhäsionsmolekül der Basalmembran, Einzelheiten s. Text
Agrin Basalmembranprotein, verschiedene Spleißvarianten mit unterschiedlichen Aktivitäten; notwendig für die Differenzierung der neuromuskulären Synapse (Aggregation der Acetylcholin- Rezeptoren), knock-out letal
* SPARC = secreted protein acidic and rich in cysteine

24731
Zellen müssen in der richtigen Umgebung angesiedelt sein und die richtige Polarität (apikal-basale Polarität bei Epithelzellen) besitzen, um ihre korrekten Funktionen aus-zuüben. Dies erfordert die Ausbildung spezifischer Zell-Zell- und Zell-Matrix-Kontakte. Die wichtigsten Rezep-toren für extrazelluläre Matrixmoleküle sind die Integrine, ihre Aktivierung ermöglicht in vielen Fällen erst, dass Wachstumsfaktoren/Hormone ihre Wirkung entfalten (Sy-nergismus). Darüber hinaus beeinflussen ECM-Moleküle über Rezeptoren wie die Integrine aber auch direkt die Gen-expression.
Man kennt inzwischen eine große Anzahl von ECM-Molekülen, die eine regulatorische Funktion ausüben, eini-ge sind in . Tab. 24.3 aufgelistet. Aus der Vielfalt an Fak-toren sollen im Folgenden die beiden wichtigsten Zell-adhäsionsproteine, das Fibronectin und die Laminine, vorgestellt werden. Im Anschluss werden die Integrine, die wichtigste ECM-Rezeptor-Familie besprochen.
24.5.1 Fibronectin
Fibronectin ist ein Heterodimer aus zwei etwa 230 kDa großen Polypeptidketten, die nahe am C-terminalen Ende durch Disulfidbrücken zusammen gehalten werden (. Abb. 24.11). Durch alternatives »Spleißen« der RNA eines einzigen Gens entstehen Moleküle mit unterschied-lichen Eigenschaften.4 In der Leber wird die als lösliches Plasma-Fibronectin
bezeichnete Spleißvariante synthetisiert. Das Protein liegt im Plasma in einer Konzentration von ca. 300 mg/l vor und spielt eine wichtige Rolle bei der Wundheilung. Bei der Blutgerinnung wird Fibronectin in das Fibrin-Gerinnsel eingebaut, sodass der Blutpfropf nicht nur die defekte Stelle verschließt, sondern gleichzeitig auch ein Zelladhäsionsmolekül enthält, das von Keratinozyten, Fibroblasten und Zellen des Immunsystems erkannt wird, umso gleichzeitig die Regeneration zu stimulieren
4 Fibroblasten hingegen bilden eine andere Spleiß variante (unlösliches Fibronectin), die in die extrazelluläre
. Abb. 24.11a,b. Struktur von Fibronectin. a Schematische Dar-stellung des modularen Aufbaus einer Fibronectin Untereinheit. Die Polypeptidkette (MG ca. 230 kDa) besteht aus einer Vielzahl von 40–90 Aminosäuren langen Domänen, die aufgrund ihrer Homologie in drei verschiedene Strukturtypen (Typ-I, Typ-II, Typ-III) eingeteilt werden. EDA, EDB und IIICS sind Module, die alternativ gespleißt
werden können. Entlang der Polypeptidkette befinden sich verschie-dene Bindungsregionen für weitere ECM- und Zelloberflächen-Mole-küle. b Elektronenmikroskopische Aufnahme (Rotary Shadowing Verfahren) einzelner Fibronectin-Moleküle. Zwei Polypeptidketten werden über C-terminale Disulfidbrücken verknüpft. (Aufnahme von J. Engel, Basel)
24.5 · Nichtkollagene, zelladhäsive Glycoproteine

732 Kapitel 24 · Binde- und Stützgewebe
24
Matrix in Form von unlöslichen Fibrillen eingelagert wird. Dieses Fibronectin besitzt eine Brückenfunktion zwischen Kollagenfibrillen und anderen ECM-Mole-külen, es dient als Adhäsionsmolekül für verschiedene Zellen, und reguliert dadurch Wanderung und Diffe-renzierung
Den verschiedenen biologischen Funktionen entsprechend besitzt Fibronectin eine Reihe von spezifischen Bindungs-stellen für extrazelluläre Proteine (Fibrin, Heparin, Kolla-gen) und je nach Spleißvariante eine oder mehrere Zellbin-dungsregionen. Letztere binden an Integrine in der Plas-mamembran der adhärierenden Zellen und lösen so eine Signaltransduktionkette aus. Der wichtigste Fibronectin-Rezeptor ist das 5 1-Integrin (7 Kap. 24.5.3).
Die Bedeutung der Fibronectin-vermittelten Zell-Matrix-Interaktion für den Organismus zeigt am deut-lichsten die Tatsache, dass die Inaktivierung des Fibro-nectin-Gens embryonal letal ist (. Abb. 24.12): Die Gastru-lation ist gehemmt (bei der normale Gastrulation wandern
die Mesodermzellen über eine Matrix aus Fibronectin-molekülen) und die mutanten Embryonen zeigen drama-tische Störungen in der Morphogenese mesodermaler Organe (Fehlen stimulierender Signale durch Integrin-ver-mittelte Signaltransduktion).
24.5.2 Laminine
! Multiple Isoformen verleihen Basalmembranen organ-spezifische Funktionen.
Während sich die Fibronectine aus einem Gen durch alter-natives Spleißen ableiten und in der ECM deponiert wer-den, stellen die Laminine eine Multigen-Familie dar und werden in Basalmembranen eingebaut. Die Laminine sind Heterotrimere, bestehend aus je einer -, - und -Kette. Bisher sind fünf -, vier - und drei -Ketten bekannt, die zu mehr als einem Dutzend verschiedener Lamininvarian-ten assembliert werden können. Die Mehrzahl der Mole-küle besitzt eine asymmetrische kreuzförmige Struktur aus drei kurzen und einem langen Arm (. Abb. 24.13). Einige Moleküle besitzen jedoch verkürzte Ketten und zeigen da-her abweichende Formen. Laminine stellen die nichtkolla-gene Hauptkomponente in allen Basalmembranen dar. Sie polymerisieren über die drei terminalen Domänen der drei kurzen Arme zu einem Netzwerk, das über linker-Proteine wie das Nidogen mit dem Gerüst des Kollagens Typ IV ver-knüpft ist. Zusätzlich sind in die Basalmembran noch das Heparansulfatproteoglykan Perlecan und andere Glycos-aminoglykane eingelagert, an die Laminin mit seiner Hepa-rinbindungsstelle am Ende des langen Arms binden kann.
Die Laminine werden entwicklungs- und organspezi-fisch exprimiert. Laminin-1 ( 1 1 1) ist vorwiegend in em-bryonalen Basalmembranen zu finden. Laminin-2 ( 2 1 1) ist die dominierende Isoform in Muskel und peripheren Nerven, neuromuskuläre Synapsen hingegen enthalten La-minin-4 ( 1 2 1). In den Basalmembranen der Haut findet sich Laminin-5 ( 3 3 3), in vielen epithelialen Basalmem-branen ist Laminin-10 ( 5 1 1) die dominierende Isoform.
! Die einzelnen Laminin-Isoformen können sich funktio-nell nicht gegenseitig ersetzen. Der Ausfall jeder der bisher untersuchten Lamininketten erzeugt schwerste Defekte oder ist embryonal letal.
So führt z.B. das Fehlen der 2-Kette zur Muskeldystrophie, das Fehlen der 2-Kette zur Blockierung der neuromus-kulären Signalübertragung, da die Schwannschen Zellen entlang der veränderten Basalmembran in den synaptischen Spalt hineinwachsen. Inaktivierung des Gens der in fast allen Isoformen vorkommenden 1-Kette ist bereits im frühen Blastocysten-Stadium letal. Diese Ergebnisse zeigen, wie wichtig die korrekte Proteinzusammensetzung der Basalmembran für die Entwicklung und die Funktion der adhärierenden Zellen ist.
. Abb. 24.12. Auswirkung der Inaktivierung des Fibronectin-Gens auf die Embryonalentwicklung der Maus. Homozygote (–/–) Mäuse zeigen im Vergleich zu normal aussehenden heterozygoten (+/–) Mäusen eine stark verkürzte anteriore/posteriore Achse. Somiten fehlen sowie differenzierte Strukturen im kaudalen Bereich, die Kopf-falte ist fehlgebildet (Pfeil). Die Aufnahme wurde im Entwicklungssta-dium E8.5 (Tag 8.5 nach Coitus) gemacht. H Herz; Al Allantois; S Somit. (George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO (1993) Defects in mesoderm, neural tube and vascular develop-ment in mouse embryos lacking fibronectin. Development 119:1079–1091)

24733
24.5.3 Integrine
Man kennt eine Reihe von zellulären Rezeptoren, die ECM-Proteine binden, darunter die vor allem als Corezeptoren agierenden membrangebundenen Proteoglykane und das Dystroglykan (7 Kap. 30.2.3). Die am besten untersuchten und universellsten Rezeptoren stellen jedoch die Integrine dar (. Abb. 24.14).
! Integrine sind die größte Rezeptorfamilie für ECM-Moleküle.
Alle Integrine sind heterodimere, aus einer - und einer -Untereinheit bestehende Transmembranproteine. Beide
Ketten sind nichtcovalent miteinander assoziiert und be-sitzen Molmassen im Bereich zwischen 100 und 200 kDa. Es gibt wenigstens 18 -Ketten und 8 -Ketten. Aus der Vielzahl der theoretisch möglichen Kombinationen werden jedoch nur 24 verschiedene Integrine gebildet. Die Rezep-toren für ECM-Moleküle sind meist Heterodimere aus der
1-Kette mit verschiedenen -Ketten. So ist α2β1 ein Kol-lagen-Rezeptor, α5β1 ein Fibronectin-Rezeptor und α6β1 ein typischer Laminin-Rezeptor.
Die Integrine müssen eine Vielzahl von Liganden er-kennen. Eine Gemeinsamkeit ist, dass die Liganden einen Aspartat-Rest in der Bindungsregion enthalten müssen. Die
-Ketten besitzen nämlich ein unvollständig koordiniertes Kation, in der Regel Mg2+, die Koordination wird durch Bindung eines Aspartat-Rests des Liganden vervollständigt (. Abb. 24.14a). Für einige Integrine ist die Erkennung der Tripeptidsequenz Arg-Gly-Asp (so genannte RGD-Se-quenz) für die Bindung des Liganden ausreichend. Meist werden aber komplexere Proteinstrukturen des Liganden erkannt, die mit beiden Integrin-Untereinheiten wechsel-wirken.
Nicht alle Integrine sind Rezeptoren für ECM-Mole-küle. So ist zum Beispiel IIb/ 3 der wichtigste Rezeptor auf Blutplättchen für Fibrinogen (7 Kap. 29.5.2) und 2-In-tegrine vermitteln die Interaktion von Lymphozyten mit Endothelzellen, z.B. bei der Extravasation an Entzündungs-herden (7 Kap. 34.6.1).
Integrine sind bidirektionale Rezeptoren. Viele Inte-grin-Rezeptoren liegen normalerweise in einer inaktiven Konformation vor, wie z.B. der Plättchenrezeptor IIb/ 3. Erst bei Aktivierung der Thrombozyten nach Gefäßver-letzungen (u.a. durch Bindung von subendothelialem Kol-lagen an das ständig aktive 2 1-Integrin) wird der Rezep-tor über eine intrazelluläre Signaltransduktionskette in die bindungskompetente Konformation überführt (inside-out-signaling). Die bedarfsabhängige Aktivierung von IIb/ 3 verhindert in diesem Falle eine unkontrollierte Blutgerin-
. Abb. 24.13a,b. Typische Struktur von Lamininen, dargestellt am Beispiel von Laminin-1. a Elektronenmikroskopische Aufnahme (nach Rotary Shadowing) von Laminin-1 aus einem Maus-Tumor. b Schematische Darstellung des Aufbaus der kreuzförmigen Struktur aus drei unterschiedlichen Polypeptidketten mit Molekulargewichten zwischen 220 und 440 kDa. Alle drei Ketten zeigen einen homologen Aufbau aus sechs unterschiedlichen globulären und stäbchenför-
migen Domänen (I–VI). Die stäbchenförmigen Domänen der kurzen Arme sind aus einer Vielzahl je acht Cystein-Reste enthaltender LE-Module (EGF-ähnliche Lamininmodule) aufgebaut. Der Stab des langen Arms besitzt eine coiled-coil Struktur. Zusätzlich enthält die
1-Kette noch eine große C-terminale globuläre Domäne. Einige wichtige biologisch aktive Regionen sind in der Abbildung gekenn-zeichnet. G-Domäne = globuläre, C-terminale Domäne)
24.5 · Nichtkollagene, zelladhäsive Glycoproteine

734 Kapitel 24 · Binde- und Stützgewebe
24
. Tabelle 24.4. Effekte von Mutationen in Ingegrin-Genen auf die Mausentwicklung (Beispiele)
Integrin-Untereinheit
Phänotyp
α3 Perinatal letal, Defekte in der Nierenentwicklung
α4 Embryonal letal, Defekte in der Plazenta- und Herzentwicklung
α5 Embryonal letal, Defekte in der Mesoderm-Bil-dung
α6 Perinatal letal, Hautablösung
α7 Lebensfähig, Entwicklung einer Muskeldystro-phie
α8 Perinatal letal, Fehlbildung der Nieren
αv Perinatal letal, Rupturen von Gefäßen
β1 Letal, Degeneration der Inneren Zellmasse der Blastocyste
β7 Fehlen von Lymphozyten im Darmbereich
C
. Abb. 24.14a,b. Struktur und Signaltransduktion der Integrine. a Schematische Darstellung der Struktur eines Integrins, bestehend aus einer - und einer -Untereinheit. An der Ligandenbindung sind die N-terminalen Domänen beider Untereinheiten beteiligt, die
Ligan denbindungsregion der -Untereinheit enthält ein zweiwertiges Kation (Mg2+), das einen Aspartatrest in der Bindungsregion des Liganden bindet. b Schematische Darstellung der durch Aktivierung der Integrine ausgelösten Signalwege. (Einzelheiten 7 Text)
nung. Demgegenüber ist das outside-in-signaling der nor-male Signalübertragungsweg von außen in die Zelle.
! Integrine erfüllen lebensnotwendige Aufgaben.
Inaktivierung der Gene für einzelne Integrin-Unterein-heiten verursacht in den meisten Fällen einen charakteris-tischen Phänotyp (. Tabelle 24.4). Die Befunde reichen von Letalität im Blastocystenstadium (7 u.) bis hin zu relativ milden Effekten wie dem Fehlen der Peyerschen Plaques.
! Integrine organisieren das Cytoskelett und aktivieren eine Vielzahl von Signaltransduktionswegen.
Die Integrine besitzen nur kurze cytoplasmatische Domä-nen, die keinerlei eigene enzymatische Aktivitäten aufwei-sen. Entsprechend läuft die Signaltransduktion auch anders ab als bei »konventionellen« Rezeptoren. Die cytoplas ma-tischen Domänen dienen als Anker für die Assemblierung von Multiprotein-Komplexen. Die wichtigsten Reaktionen sind in . Abb. 26.14b zusammengefasst:4 Adhärieren Zellen an immobilisierte Moleküle in der
ECM, lagern sich die vorher mehr oder weniger frei in der Membran diffusiblen Integrine an den Kontaktstel-len zusammen. Über Linker-Proteine wie Vinculin und Talin werden Verbindungen zu Aktin-Filamenten her-
24735
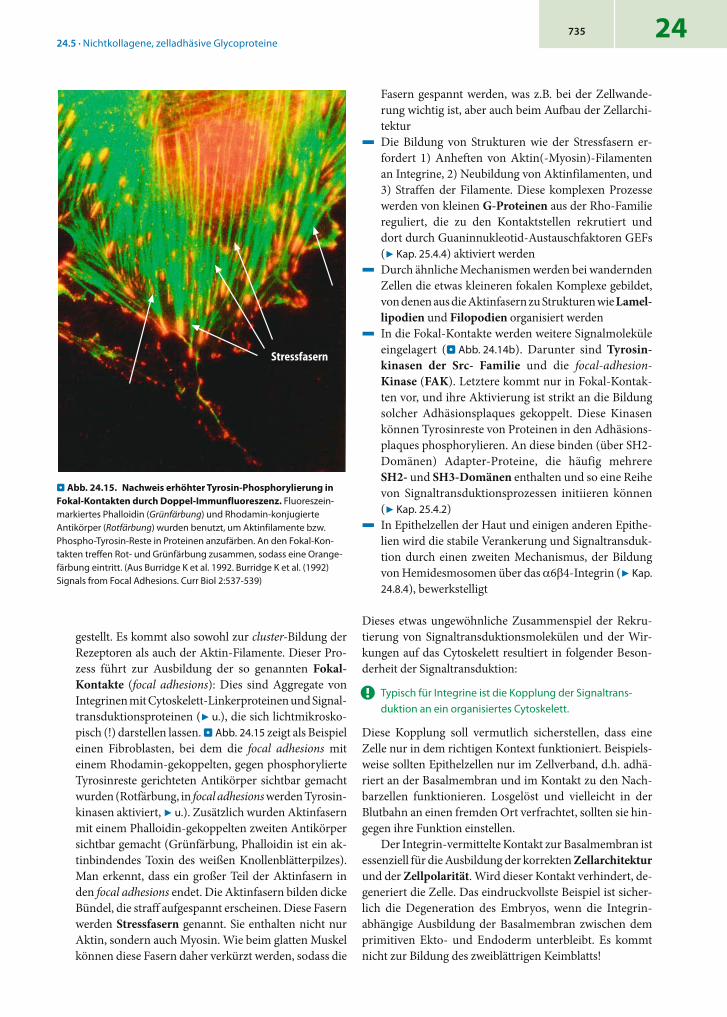
gestellt. Es kommt also sowohl zur cluster-Bildung der Rezeptoren als auch der Aktin-Filamente. Dieser Pro-zess führt zur Ausbildung der so genannten Fokal-Kontakte (focal adhesions): Dies sind Aggregate von Integrinen mit Cytoskelett-Linkerproteinen und Signal-transduktionsproteinen (7 u.), die sich lichtmikrosko-pisch (!) darstellen lassen. . Abb. 24.15 zeigt als Beispiel einen Fibroblasten, bei dem die focal adhesions mit einem Rhodamin-gekoppelten, gegen phosphorylierte Tyrosinreste gerichteten Antikörper sichtbar gemacht wurden (Rotfärbung, in focal adhesions werden Tyrosin-kinasen aktiviert, 7 u.). Zusätzlich wurden Aktinfasern mit einem Phalloidin-gekoppelten zweiten Antikörper sichtbar gemacht (Grünfärbung, Phalloidin ist ein ak-tinbindendes Toxin des weißen Knollenblätterpilzes). Man erkennt, dass ein großer Teil der Aktinfasern in den focal adhesions endet. Die Aktinfasern bilden dicke Bündel, die straff aufgespannt erscheinen. Diese Fasern werden Stressfasern genannt. Sie enthalten nicht nur Aktin, sondern auch Myosin. Wie beim glatten Muskel können diese Fasern daher verkürzt werden, sodass die
Fasern gespannt werden, was z.B. bei der Zellwande-rung wichtig ist, aber auch beim Aufbau der Zellarchi-tektur
4 Die Bildung von Strukturen wie der Stressfasern er-fordert 1) Anheften von Aktin(-Myosin)-Filamenten an Integrine, 2) Neubildung von Aktinfilamenten, und 3) Straffen der Filamente. Diese komplexen Prozesse werden von kleinen G-Proteinen aus der Rho-Familie reguliert, die zu den Kontaktstellen rekrutiert und dort durch Guaninnukleotid-Austauschfaktoren GEFs (7 Kap. 25.4.4) aktiviert werden
4 Durch ähnliche Mechanismen werden bei wandernden Zellen die etwas kleineren fokalen Komplexe gebildet, von denen aus die Aktinfasern zu Strukturen wie Lamel-lipodien und Filopodien organisiert werden
4 In die Fokal-Kontakte werden weitere Signalmoleküle eingelagert (. Abb. 24.14b). Darunter sind Tyrosin-kinasen der Src- Familie und die focal-adhesion-Kinase (FAK). Letztere kommt nur in Fokal-Kontak-ten vor, und ihre Aktivierung ist strikt an die Bildung solcher Adhäsionsplaques gekoppelt. Diese Kinasen können Tyrosinreste von Proteinen in den Adhäsions-plaques phosphorylieren. An diese binden (über SH2-Domänen) Adapter-Proteine, die häufig mehrere SH2- und SH3-Domänen enthalten und so eine Reihe von Signaltransduktionsprozessen initiieren können (7 Kap. 25.4.2)
4 In Epithelzellen der Haut und einigen anderen Epithe-lien wird die stabile Verankerung und Signaltransduk-tion durch einen zweiten Mechanismus, der Bildung von Hemidesmosomen über das 6 4-Integrin (7 Kap. 24.8.4), bewerkstelligt
Dieses etwas ungewöhnliche Zusammenspiel der Rekru-tierung von Signaltransduktionsmolekülen und der Wir-kungen auf das Cytoskelett resultiert in folgender Beson-derheit der Signaltransduktion:
! Typisch für Integrine ist die Kopplung der Signaltrans-duktion an ein organisiertes Cytoskelett.
Diese Kopplung soll vermutlich sicherstellen, dass eine Zelle nur in dem richtigen Kontext funktioniert. Beispiels-weise sollten Epithelzellen nur im Zellverband, d.h. adhä-riert an der Basalmembran und im Kontakt zu den Nach-barzellen funktionieren. Losgelöst und vielleicht in der Blutbahn an einen fremden Ort verfrachtet, sollten sie hin-gegen ihre Funktion einstellen.
Der Integrin-vermittelte Kontakt zur Basalmembran ist essenziell für die Ausbildung der korrekten Zellarchitektur und der Zellpolarität. Wird dieser Kontakt verhindert, de-generiert die Zelle. Das eindruckvollste Beispiel ist sicher-lich die Degeneration des Embryos, wenn die Integrin-abhängige Ausbildung der Basalmembran zwischen dem primitiven Ekto- und Endoderm unterbleibt. Es kommt nicht zur Bildung des zweiblättrigen Keimblatts!
. Abb. 24.15. Nachweis erhöhter Tyrosin-Phosphorylierung in Fokal-Kontakten durch Doppel-Immunfluoreszenz. Fluoreszein-markiertes Phalloidin (Grünfärbung) und Rhodamin-konjugierte Antikörper (Rotfärbung) wurden benutzt, um Aktinfilamente bzw. Phospho-Tyrosin-Reste in Proteinen anzufärben. An den Fokal-Kon-takten treffen Rot- und Grünfärbung zusammen, sodass eine Orange-färbung eintritt. (Aus Burridge K et al. 1992. Burridge K et al. (1992) Signals from Focal Adhesions. Curr Biol 2:537-539)
24.5 · Nichtkollagene, zelladhäsive Glycoproteine

736 Kapitel 24 · Binde- und Stützgewebe
24
Auch weitere durch Integrine stimulierte Reaktion lassen sich mit der Annahme, dass ein priming der Zelle durch Integrin-abhängige Zelladhäsion erforderlich ist, erklären:4 Integrine wirken synergistisch mit Wachstumsfak-
toren. Einige Integrine können ebenso wie Wachs-tumsfaktoren (7 Kap. 25.4.3) über SH2/SH3-Adapter-
proteine Ras aktivieren. Die Zellen müssen adhärent sein, damit die Wachstumsfaktoren wirken können. In Suspension teilen sich nichttransformierte Zellen nicht
4 Integrine sind permissiv für die Zelldifferenzierung. In einigen Zelltypen bewirkt die Adhäsion Austritt aus dem Zellzyklus und Differenzierung
In Kürze
Nichtkollagene Glycoproteine der ECM wie Fibronectin und Laminin sind wichtige Mediatoren4 von Zelladhäsion und -wanderung und4 beeinflussen Zellfunktion, -differenzierung und
-proliferation
Fibronectin kommt in zahlreichen Spleiß-Varianten vor.Es bindet4 sowohl an andere ECM-Moleküle wie Kollagen und
Heparin als auch4 über Integrine an Zellen
Im adulten Organismus ist Fibronectin von Bedeutung4 bei der Wundheilung und4 der Regulation der Aktivität von (Bindegewebs-)-
Zellen
Während der Embryonalentwicklung ist Fibronectin u.a. essentiell für die Bildung mesodermaler Strukturen.Laminine stellen eine große Molekülfamilie aus drei Polypeptidketten dar, die meisten Moleküle besitzen eine typische kreuzförmige Struktur.
Laminine sind4 obligatorische Bestandteile der Basalmembran und4 werden gewebs- und entwicklungsspezifisch ex-
primiert
Der Ausfall der verschiedenen Iso-Formen hat einen charakteristischen, oft letalen Phänotyp.Integrine4 sind die wichtigsten Zelloberflächen-Rezeptoren
für ECM-Moleküle4 bilden eine große Familie von heterodimeren Mole-
külen aus je einer - und einer -Kette4 können in verschiedenen Kombinationen unter-
schiedliche Liganden binden4 binden an die ECM, was für die Ausbildung der Fokal-
Kontakte wichtig ist. Dabei organisieren Integrine das Aktin-Cytoskelett, führen zur Aktivierung von Protein-kinasen und beeinflussen dadurch Signaltransduk-tionswege und den Funktionszustand der Zellen
Inaktivierung von Integrin-Genen verursacht schwerste Schäden.
24.6 Abbau der extrazellulären Matrix
Die extrazelluläre Matrix des Erwachsenen besitzt in der Regel einen recht geringen Stoffumsatz. So werden ca. 2–3% des Hautkollagens täglich erneuert, im gesunden Gelenk-knorpel beträgt die Halbwertszeit viele Jahre. In bestimmten Situationen sind jedoch ein schneller Abbau bzw. Umbau erforderlich. Großflächige Umstrukturierungen finden z.B. bei der Rückbildung des Uterus nach der Geburt und bei der Wundheilung statt, während lokal eng begrenzte Ab-bauprozesse beim Durchtritt von weißen Blutkörperchen aus der Blutbahn ins Interstitium erfolgen. Der Abbau von ECM-Molekülen wird durch spezifische Proteasen vermit-telt. Einige davon sind Serinproteasen, die meisten gehören jedoch zur Familie der Metalloproteinasen:4 Zwei für den Abbau der ECM wichtige Serinproteasen
sind tPA (tissue-type-plasminogen activator) und uPA(urokinase-type-plasminogen-activator). Beide wandeln durch Spaltung einer einzigen Peptidbindung Plas-minogen in Plasmin um. Plasminogen/Plasmin sind wegen ihrer Rolle bei der Auflösung von Blutgerinnseln
durch Spaltung von Fibrin bekannt (7 Kap. 29.5.3), sind aber auch an Umbauprozessen der ECM beteiligt. Plasmin ist in der Lage, verschiedene ECM-Proteine wie Fibronectin und Laminin zu verdauen. Eine Beson-derheit von uPA besteht darin, dass durch Bindung an den Zelloberflächen-Rezeptor uPAR gezielt die ECM in der direkten Umgebung dieser Zelle verdaut werden kann, z.B. bei der Extravasation von Leukozyten.
4 Die Matrix-Metallo-Proteinasen (MMPs) stellen eine Familie von inzwischen über 20 Zink-abhängigen Proteasen dar (. Tabelle 24.5), die entweder sezerniert werden oder in der Membran verankert sind. Alle En-zyme besitzen eine konservierte Protease-Domäne, in der drei Histidin-Reste das Zinkatom im katalytischen Zentrum komplexieren. Zusätzlich finden sich bei den meisten Mitgliedern noch eine oder mehrere C-termi-nale Domänen, die für Substraterkennung und -bin-dung wichtig sind.
! Die Matrix-Metallo-Proteinasen werden als inaktive Pro-Formen (Zymogene) gebildet.

24737
Die Pro-Form enthält eine N-terminale Domäne, die Pro-Domäne, die das katalytische Zentrum blockiert. Die Akti-vierung der Protease erfolgt durch proteolytische Ab-spaltung des Propeptids im Extrazellulärraum. Lediglich Stromelysin-3 und die membrangebundenen MMPs (MT-MMPs) werden bereits im Golgi-Apparat aktiviert.
! Die Matrix-Metallo-Proteinasen besitzen zum Teil eine sehr hohe Substratspezifität.
Die Kollagenasen MMP-1, -8 und -13 spalten die fibril-lären Kollagene I–III, nicht aber Kollagen-Typ-IV. Die Spal-tung erfolgt an einer einzigen Gly-Ile/Leu-Bindung. Die Tripelhelix der Fragmente ist thermisch nicht mehr so stabil und kann von anderen Proteasen, darunter die eben-falls zu den MMPs gehörenden Gelatinasen (MMP-2 und MMP-9), weiter verdaut werden. Letztere vermögen auch das Typ-IV-Kollagen in zwei Fragmente zu zerlegen.
Die Bedeutung der MMPs wurde in jüngerer Zeit auch durch Ausschalten der entsprechenden Gene in Mäusen offenbar. So können Makrophagen von Mäusen, denen MMP-12 fehlt, weder in vitro noch in vivo durch die Basal-membran penetrieren. MMP-9 defiziente Mäuse zeigen verzögertes Wachstum der langen Knochen mit abnorm verdickter Wachstumszone, was auf eine Verzögerung des Abbaus der knorpeligen ECM bei der indirekten Ossifika-tion zurückzuführen ist (s.u.).
! Die Aktivität von MMPs und Serinproteasen wird strikt reguliert.
Eine überschießende Reaktion würde leicht zu einer Ge-webszerstörung führen. Daher erfolgt die Aktivierung lokal begrenzt an der Zelloberfläche von Fibroblasten. Gelatinase wird bei Bedarf durch die membranständige MT1-MMP aktiviert, die gleichzeitig einen Rezeptor für das aktive Enzym darstellt. Darüber hinaus gibt es eine Reihe von Inhibitoren der MMPs, die TIMP (tissue inhibitors of metal-loproteinases) genannt werden. Normalerweise herrscht ein fein reguliertes Gleichgewicht zwischen den MMPs und TIMPs. Störungen des Gleichgewichts führen zu einem er-höhten Kollagenabbau (z.B. bei Metastasierungen) oder zu erniedrigtem Kollagenabbau (Fibrose).
24.7 Biochemie und Pathobiochemie des Skelettsystems
24.7.1 Die extrazelluläre Matrix von Knorpel und Knochen
Knorpel. Knorpel findet sich an den Stellen, wo flexible, druckresistente Strukturen benötigt werden. Er hat als Ge-lenkknorpel und Zwischenwirbelscheibe mechanische Auf-gaben. Darüber hinaus ist Knorpel die Vorstufe für die durch indirekte Ossifikation entstehenden Knochen. Die extrazelluläre Matrix des Knorpels wird von Chondrozyten gebildet, die von ihren eigenen Syntheseprodukten einge-schlossen werden, sodass sie nur noch durch Diffusion von außen ernährt werden können, denn Knorpel ist nicht
. Tabelle 24.5. Matrix-Metalloproteinasen und ihre Substrate (Auswahl) Col = Kollagen
Enzym Alternative Namen Typische Substrate
Kollagenasen
MMP-1 Kollagenase-1, Fibroblasten-Kollagenase Col I, II, III, VII, X, Pro-MMP-2 u.-9
MMP-8 Kollagenase-2, Neutrophilen-Kollagenase Col I, II, III, Aggrecan
MMP-13 Kollagenase-3 Col I, II, III, Aggrecan
Gelatinasen
MMP-2 Gelatinase A Gelatine, Col IV, Col I, V, X, Elastin, Aggrecan, Link-Protein
MMP-9 Gelatinase B Gelatine, Col IV, Col V, XI, Elastin, Aggrecan, Link-Protein
Stromelysine
MMP-3 Sromelysin-1, Proteo-glykanase Kollagene u. nichtkollagene ECM-Proteine,Pro-MMP-1,-8,-9,-13, E-Cadherin, L-Selektin,Inaktivierung v. Protease-Inhibitoren
MMP-10 Stromelysin-2 ähnlich wie MMP-3, schwächer aktiv als MMP-3
Membran-MMPs
MMP-14 MT1-MMP Col I, II, III, Fibronectin, Laminin, Proteoglykane
MMP-16 MT3-MMP Pro-MMP-2
Sonstige
MMP-7 Matrilysin, PUMP-1 ECM-Proteine, Pro-MMP-1,-2 u.-9
MMP-12 Metalloelastase Elastin und weitere ECM-Moleküle
MMP-20 Enamelysin Amelogenin
24.7 · Biochemie und Pathobiochemie des Skelettsystems

738 Kapitel 24 · Binde- und Stützgewebe
24
vaskularisiert. Die Knorpelmatrix stellt im Wesentlichen ein faserverstärktes Gel dar. Der Gelcharakter ist durch polyanionische Aggregate aus Hyaluronsäure mit Proteo-glykanen (Aggrecan) bedingt (7 Kap. 2.1.4, 24.4.1), die zu hohen osmotischen Drücken führen. Daher besitzt Knorpel die Fähigkeit anzuschwellen, der Wasseranteil beträgt etwa 70–80%. Dieses Gel wird durch quervernetzte Fibrillen aus Kollagen-Typ-II in Form gehalten. Dieses Kollagen ist das mengenmäßig dominierende Knorpelkollagen. Daneben findet man noch geringere Mengen der physiologisch eben-falls wichtigen Kollagene Typ-XI und Typ-IX (7 Kap. 24.2.1). Knorpel ist meist von einem Perichondrium umgeben, dies trifft jedoch nicht zu für die Gelenkflächen, die Knorpel-Knochen-Grenze der Gelenke und die Wachstumszone. Im Perichondrium finden sich Kollagene Typ-I, -II und -V. Zusätzlich zu den Kollagenen und Proteoglykanen enthält die Korpel-Matrix noch weitere Proteine, darunter Protease-inhibitoren und die in drei Isoformen bekannten Matri-line (auch cartilage matrix protein genannt). Die mit den Thrombospondinen (. Tabelle 24.3) verwandten Matriline besitzen möglicherweise eine Bedeutung in der Assemblie-rung der Knorpelmatrix oder vermitteln die Adhäsion der Chondrozyten an Knorpel-Matrix-Proteine.
Knochen. Im Gegensatz zum Knorpel ist der Knochen gut durchblutet und innerviert. Die eigentlichen Knochenzel-len sind die Knochenmatrix synthetisierenden Osteoblas-ten/Osteozyten und die Knochenmatrix ab-/umbauenden Osteoklasten. Der Knochen erfüllt mehrere Funktionen. Zum einen ist er ein hochdifferenziertes Stützgewebe, das für die Bewegungen des Körpers und zum Schutz von Organen wie dem Gehirn essentiell ist, zum anderen dient er als Speicher für Calcium- und Phosphationen. Darüber hinaus beherbergt der Knochen das Knochenmark als Stätte der Blutbildung. Das anorganische Knochenmaterial setzt sich vorwiegend aus Calciumphosphaten zusammen, die sehr dem Hydroxylapatit [3Ca3(PO4)2 Ca(OH)2] äh-neln. Zusammen mit 10% Wasser machen die anorga-nischen Bestandteile des Knochens etwa 80% aus. Die rest-lichen 20% entfallen auf organisches Material. Die Haupt-komponente stellt das Kollagen-Typ-I dar, das dem Knochen die mechanische Festigkeit verleiht. Die anorga-nische Matrix alleine ist zu brüchig, um Belastungen Stand zu halten, sie bildet mit dem Kollagen zusammen eine Art »Verbundwerkstoff«. Nichtkollagene Proteine machen nur etwa 10% der organischen Matrix aus, sind aber dennoch für die Knochenentwicklung und Homöostase wichtig. Zu den nichtkollagenen Proteinen gehören Proteoglykane wie Decorin und Biglykan und Zelladhäsionsproteine wie Fibronectin. Weiterhin enthält Knochen eine Reihe von matrizellulären Proteinen (. Tab. 24.3) wie Tenascin, Thrombospondin, Osteopontin und bone sialoprotein.Diese auch in der extrazellulären Matrix von Nicht-Kno-chen-Gewebe weit verbreiteten Proteine unterstützen u.a. die Differenzierung/Funktion von Osteoblasten und Osteo-
klasten. Fehlen dieser Proteine äußert sich u.a. in einer erhöhten Knochenbrüchigkeit und einer verlangsamten Heilung von Knochenbrüchen. Osteopontin und bone-sialoprotein sind integrinbindende, phosphorylierte und sulfatierte Glycoproteine, denen aufgrund ihrer Fähigkeit Hydroxylapatit zu binden eine Funktion in der Mineralisie-rung zugeschrieben wird. Wichtig für die Ossifikation sind auch Proteine mit Vitamin-K abhängig gebildeten -Car-boxy-Glutamat-Resten (= Gla), z.B. Matrix-GLA-Protein und Osteocalcin (7 Kap. 23.2.4). Mäuse mit inaktivierten Genen für Matrix-GLA-Protein und Osteocalcin zeigen überraschenderweise eine verstärkte (!) Knochenbildung. Mäuse ohne GLA-Protein sterben in den ersten beiden Monaten nach der Geburt aufgrund einer exzessiven, ab-normalen Calzifizierung der Arterien. Trotz ihrer Fähigkeit an Apatit zu binden, scheint also die Funktion dieser Pro-teine eher in der kontrollierten Abscheidung von Hydro-xylapatit und der Verhinderung einer überschießenden Ossifikation zu liegen.
Ein wichtiges Markerenzym für die Osteoblastenakti-vität ist eine knochenspezifische alkalische Phosphatase, deren physiologische Funktion allerdings noch nicht genau bekannt ist.
24.7.2 Synthese von Knochen und Knorpel durch Chondrozyten und Osteoblasten
Chondroblasten und Osteoblasten leiten sich von me-senchymalen Stammzellen ab, aus denen in anderen Dif-ferenzierungsprogrammen auch Fibroblasten, Myoblasten und Adipozyten entstehen. Sie sind die beiden zentralen Zelltypen, die die Knochen- und Knorpelsubstanz auf-bauen.
Knorpelbildung. An der Bildung der Knorpelsubstanz sind lediglich die Chondroblasten und Chondrozyten (in Knor-pelmatrix eingebettete Chondroblasten) beteiligt. Sie synthe-tisieren die charakteristischen Knorpelbestandteile wie das Typ-II-Kollagen und Aggrecan.
Knochenbildung. Die Bildung des Knochens verläuft we-sentlich komplexer. Bei der direkten oder desmalen Ossi-fikation differenzieren mesenchymale Zellen direkt zu Osteoblasten. Zunächst wird eine Osteoid genannte orga-nische Matrix aus Kollagen Typ I, Proteoglykanen, Muci-nen und weiteren nichtkollagenen Proteinen wie Osteo-calcin und Osteopontin abgelagert, die anschließend ver-kalkt. Die Osteoblasten werden in diese verkalkte Substanz eingemauert, bleiben aber über Zellfortsätze miteinander in Kontakt. Diese Art der Knochenbildung ist auf relativ wenige Bereiche wie z.B. die Bildung des knöchernen Schä-deldachs beschränkt. Die häufigste Art der Knochen bildung ist die indirekte oder enchondrale Ossifikation. Einen

24739
Überblick gibt . Abb. 24.16. Eingeleitet wird sie durch Kon-densation von Mesenchymzellen. Während die Zellen an der Peripherie das Perichondrium bilden und sich direkt zu Osteoblasten entwickeln, differenzieren die Zellen im Zentrum zu Knorpelzellen. Im Gegensatz zur Chondro-genese bleibt die Differenzierung aber nicht auf der Stufe der Chondrozyten stehen, sondern sie differenzieren über verschiedene Zwischenstadien zu hypertrophen Zellen, die durch Apoptose zugrunde gehen. Die Hypertrophierung ist durch Umschalten der Synthese von Kollagen-Typ-II auf Typ-X und schließlich die Mineralisierung der Knor-pelmatrix gekennzeichnet. Von den hypertrophen Knor-pelzellen sezerniertes VEGF (= vascular endothelial growth factor) führt zur Vaskularisierung der mineralisierten Knorpelmatrix. Mit den Blutgefäßen dringen Zellen ein, die zu Chondroklasten werden und die Knorpelreste abbauen, und schließlich Osteoblasten-Progenitorzellen, die die endgültige Ossifikation einleiten.
Die Knochenmatrix-synthetisierenden Osteoblasten sind spezialisierte, aber noch nicht terminal differen-zierte Zellen. Einige werden schließlich in die Knochen-
matrix eingebettet und differenzieren zu den nicht mehr teilungsfähigen Osteozyten, die über ein Netzwerk von Zellfortsätzen miteinander verbunden bleiben, aber kaum noch Osteoid synthetisieren. Andere sterben durch Apop-tose ab.
Auf molekularer Ebene werden Chondrogenese und Osteo genese durch eine Reihe von Faktoren reguliert, die man in zwei Gruppen unterteilen kann:4 Schlüsseltranskriptionsfaktoren. Sie bewirken die Dif-
ferenzierung von mesenchymalen Vorläuferzellen zu Chondrozyten bzw. Osteoblasten. Diese Faktoren wer-den im Laufe der Embryonalentwicklung an entschei-denden Weichenstellungen exprimiert. Sind sie nicht vorhanden, wird die ganze Zell-Linie (z.B. Chondro-blasten) nicht gebildet. Welche Faktoren die Induktion dieser Transkriptionsfaktoren zum richtigen Zeitpunkt an der richtigen Position bewirken, ist meist erst un-vollständig aufgeklärt
4 Parakrine Wachstumsfaktoren und Hormone. Eine Übersicht ist in . Tab. 24.6 dargestellt. Im Folgenden sind nur die wichtigsten Faktoren dargestellt
. Abb. 24.16. Schematische Darstellung der enchondralen Ossifikation von Röhrenknochen. Mesenchymale Zellen (1) konden-sieren zu einer Knorpelanlage (Knorpelblastem) (2), das Knorpelmo-dell erhält eine perichondrale Knochenmanschette (3), die umschlos-senen Chondrozyten hypertrophieren, verkalken und gehen durch Apoptose zugrunde (4), Blutgefäße wandern ein (5) und mit ihnen Chondroklasten, die die Reste der Knorpelzellen abbauen. In die ent-
standene Markhöhle wandern Osteoblasten-Vorläuferzellen ein und leiten die primäre Ossifikation ein (5). Das Längenwachstum erfolgt an der Wachstumszone zu beiden Seiten der Markhöhle (7 auch . Abb. 26.17a). In späteren Entwicklungsstadien, vor allem nach der Ge burt entsteht in den Epiphysen das sekundäre Ossifikationszen-trum (6, dargestellt sind zwei sekundäre Ossifikationszentren in unter-schiedlichen Entwicklungsstadien)
24.7 · Biochemie und Pathobiochemie des Skelettsystems

740 Kapitel 24 · Binde- und Stützgewebe
24
! Die wichtigsten Transkriptionsfaktoren für die Bildung von Chondrozyten und Osteoblasten sind SOX9 und CBFA-1 (Runx2).
SOX-9 reguliert die frühe Differenzierung der kondensie-renden Mesenchymzellen zu Knorpelzellen und die fol-genden Differenzierungsstadien. SOX-9 stimuliert zu-sammen mit weiteren Mitgliedern der SOX-Familie die Transkription einer Reihe von Matrix-Genen, darunter Typ-II-Kollagen und Aggrecan.
Für die späte Differenzierung von Chondrozyten zu hypertrophen Chondrozyten übernimmt ein zweiter Trans-kriptionsfaktor, CBFA-1 (= core binding factor-1), Syno-nym: Runx-2 (= runt related transcription factor-2), eine zentrale Rolle.
CBFA-1 ist gleichzeitig der Schlüsseltranskriptions-faktor für die Differenzierung von Mesenchymzellen zu Osteoblasten. Ohne CBFA-1 bilden sich zwar noch die knorpeligen Vorläufer der Knochen, aber die Osteoblasten-Differenzierung und somit die Ossifikation unterbleiben. CBFA-1 ist auch für die Expression von Osteoblasten-spe-zifischen Genen erforderlich und wurde in der Tat erstmals als ein die Transkription von Osteocalcin stimulierender Faktor isoliert.
! Parakrin sezernierte Faktoren aus den Familien der BMPs und FGFs besitzen eine Schlüsselfunktion bei allen Stadien der Chondrozyten-Differenzierung.
Die bone morphogenetic proteins (BMPs) aus der TGF -Superfamilie wurden aufgrund ihrer Fähigkeit entdeckt, enchondrale Knochenbildung zu induzieren, wenn sie sub-kutan in Mäuse injiziert wurden. Die BMP-Proteine kom-men in zahlreichen entwicklungsspezifisch exprimierten Isoformen vor und binden an mehrere unterschiedliche Rezeptoren. Einige Vertreter (BMP-4) sind für die Konden-sation der Mesenchymzellen wichtig, für jeden Knochen muss eine Anlage von aggregierten Mesenchymzellen ge-bildet werden. Die Aggregation der Mesenchymzellen wird u.a. durch Expression von Zelladhäsionsmolekülen aus der Cadherin-Familie bewirkt. Außerdem halten BMPs die SOX-Expression aufrecht. BMPs sind auch an den weiteren Schritten der Chondrogenese beteiligt, wobei in jedem Stadium ein charakteristisches Muster an BMPs exprimiert wird.
Darüber hinaus sind BMP-Proteine auch für die Osteo-blastenentwicklung essenziell, von der Differenzierung der Mesenchymzellen über die Reifung der Osteoblasten bis hin zur Apoptose von Osteozyten.
Die Fibroblastenwachstumsfaktoren (FGFs) stellen eine Gruppe von Cytokinen dar, die mit Tyrosinkinasere-zeptoren (FGFRs) der Targetzelle interagieren. Viele der insgesamt 22 FGF-Gene und der vier FGF-Rezeptorgene werden in allen Stadien der Knorpel- und Knochenent-wicklung exprimiert. Eine der bekanntesten Wirkungen ist die Induktion der Extremitätenknospen und die Regula-
tion des proximal-distalen Wachstums durch FGF8, und legt so die Grundlagen für die Arm- und Beinentwicklung. In der Wachstumsfuge wirken sie antagonistisch zu den BMPs (7 u.).
Störungen der FGF-Signaltransduktion sind verant-wortlich für häufige erblich bedingte Fehlregulationen der Knochenbildung: Die Achondrodysplasie, die häufigste Form von Zwergwuchs, wird durch aktivierende Muta-tionen des FGFR-3-Rezeptors verursacht, was zu einer ver-frühten terminalen Differenzierung der Chondroblasten führt.
Die Kraniosynostose ist die vorzeitige Verknöcherung der Schädelnähte. Sie wird meist durch aktivierende Mu-tationen des FGFR-2-Rezeptors verursacht, übermäßig starkes signaling durch FGF stimuliert die Differenzierung von Osteoblasten.
! Ein Regelkreis aus Ihh und PTHrP kontrolliert die Länge der Zone der proliferierenden Chondrozyten in der Wachstumsfuge.
Das Längenwachstum der Röhrenknochen erfolgt an den Wachstumsfugen und ist durch eine charakteristische Ab-folge ruhender, proliferierender und hypertrophierender Chondrozyten gekennzeichnet (. Abb. 24.17a). Die ver-schiedenen Differenzierungsschritte müssen strikt reguliert und koordiniert werden. Werden z.B. unter pathologischen Bedingungen Chondrozyten zu einem vorzeitigen Über-gang von der proliferativen Phase zum hypertrophierten Zustand stimuliert, wird das Längenwachstum des Kno-chens verlangsamt, mit Zwergwuchs als Folge.
Die kontrollierte Abfolge von Reifungsprozessen wird durch komplexe Regelkreise gesteuert, an denen Hormone (Wachstumshormon, IGFs, Schilddrüsenhormone, 7 u.) und Wachstumsfaktoren (BMPs, FGFs, . Abb. 24.17b) beteiligt sind. Ein wichtiger Regelkreis zur Kontrolle der Chondrozyten-Proliferation und -Reifung wird durch in-dian hedgehog (Ihh) und parathormon-related-peptide (PTHrP) gebildet. (. Abb. 24.17a,b).
Indian hedgehog (Ihh) ist ein Mitglied der Hedgehog Familie von lokal wirkenden, sezernierten Proteinen, die nach Bindung an den Rezeptor patched den Transkriptions-faktor Gli (so benannt nach Mutationen die zu Glioblasto-men führen) aktivieren. Ihh wird in prähypertrophen und frühen hypertrophen Chondrozyten exprimiert. Es stimu-liert zum Einen die Proliferation der zum Gelenkende hin gelegenen Chondrozyten (die zur späteren Markhöhle ge-legenen Chondrozyten sind nicht mehr teilungsfähig). Zum Anderen verhindert Ihh die vorzeitige Hypertrophierung der Chondrozyten. Dieser Effekt wird indirekt über Sti-mulierung der PTHrP-Expression vermittelt (7 u.). Eine weitere wichtige Funktion von Ihh ist die Förderung der Bildung von Osteoblasten in der primären Spongiosa und der Knochenmanschette.
Parathormon-related-peptide (PTHrP) bindet an den gleichen Rezeptor wie PTH und wurde in der Tat auch als

24741
Faktor entdeckt, der die Hypercalcämie bei bestimmten Tumoren bewirkt. Erst später wurde seine Bedeutung bei der Knochenentwicklung erkannt. PTHrP wird unter der Wirkung von Ihh von Perichondrium-Zellen und den frü-hen proliferativen Chondrozyten an den periartikulären Enden der langen Knochen exprimiert, der Mechanismus ist noch nicht bekannt. PTHrP wirkt primär, indem es die Chondrozyten im proliferierenden Zustand hält. Wenn Chondrozyten nicht mehr genügend durch durch PTHrP stimuliert werden, können sie nicht mehr zur Proliferation angeregt werden und beginnen, Ihh zu synthetisieren. Durch diesen Regelkreis wird die Länge der proliferativen Zone vorgeben (. Abb. 24.17a). Die Bedeutung des Ihh/PTHrP-Systems erkennt man am besten aus der Tatsache, dass Fehlen von Ihh zu schwersten Wachstumsstörungen führt: Das Wachstum der Röhrenknochen ist praktisch komplett inhibiert, und es werden keine Osteoblasten ge-bildet.
An der Wachstumsfuge müssen noch weitere Faktoren aktiv werden, damit eine normale Knochenentwicklung erfolgt:
! BMPs und FGFs sind Antagonisten bei der Regulation der Chondrozyten-Differenzierung in der Epiphysen-fuge.
BMPs werden in proliferierenden und hypertrophierenden Chondrozyten sowie im Perichondrium exprimiert. Sie fördern die Proliferation von Chondrozyten und verzö-gern/inhibieren ihre terminale Differenzierung. Sie sti-mulieren die Expression von Ihh (und umgekehrt). FGFs hingegen vermindern die Proliferation von Chondro-zyten und beschleunigen die Differenzierung von hyper-
trophen Chondrozyten zu terminal differenzierten Chon-drozyten. Daher ist das Knochenwachstum vom richtigen Verhältnis der beiden Cytokine abhängig (. Abb. 24.17b).
! Parakrine Faktoren regulieren Chondro- und Osteo-genese zusammen mit Hormonen.
An der Entwicklung und Homöostase von Knorpel/Kno-chengewebe sind eine Reihe von Hormonen beteiligt (. Tab. 24.6). PTH, Calcitonin und Dihydroxycalciferol regulieren den Calcium-Haushalt. Die wichtigsten Hormone, die das Längenwachstum während der Embryonalentwicklung und der Kindheit regulieren, sind das Wachstumshormon (GH) und die insulin-like growth factors-1, -2 (7 u.), die Schilddrüsenhormone und die Glucocorticoide, während des Heranwachsens gewinnen die Androgene und Östro-gene an Bedeutung. Sowohl Chondrozyten als auch Osteo-blasten besitzen Rezeptoren für diese Hormone. Die Bio-chemie dieser Hormone wird im Kap. 27 besprochen. Hier soll nur kurz die Beziehung zu den parakrinen Faktoren abgegrenzt werden.
Auf den ersten Blick scheinen Hormone und parakrine Faktoren redundante Funktionen in der Embryonalent-wicklung und der Wachstumsphase zu besitzen, da beide Proliferation und Differenzierung unterstützen. Aber da sowohl Störungen des Hormon-Haushalts als auch Defekte in der Signaltransduktion durch parakrine Wachstums-faktoren zu definierten Defekten im Skelettsystem führen, können sie sich offensichtlich nicht gegenseitig ersetzen. Beide Komponenten sind essenziell.
Einige Funktionen lassen sich klar voneinander abgren-zen. In der Embryonalentwicklung sind morphogenetische Prozesse wie Kondensation der Mesenchymzellen zu Knor-
. Abb. 24.17a,b. Interaktion von PTHrP, Ihh, BMPs und FGFs bei der Regulation der Chondrozytenproliferation und Reifung in der Wachstumszone (spätere Epiphysenfuge, vgl. Abb. 26.16, 5 u. 6).
a biologische Wirkungen von Ihh und negative Rückkopplung zwi-schen Ihh und PTHrP, b Darstellung des Antagonismus zwischen FGFs und BPMs und Beziehung zu Ihh und PTHrP. (Einzelheiten 7 Text)
24.7 · Biochemie und Pathobiochemie des Skelettsystems

742 Kapitel 24 · Binde- und Stützgewebe
24
pelanlagen, Induktion der Extremitätenknospe primär die Domäne der parakrinen Faktoren (BMPs und FGFs). Auf der anderen Seite sind Koordination von Wachstumspro-zessen primär die Aufgabe von Hormonen, z.B. kontrolliert das GH/IGF-System das proportionales Wachstum aller Knochen und die Koordination mit dem Muskelwachstum. Hormone koppeln auch das Wachstum an die Stoffwech-sellage und Energieversorgung, hierbei sind die Schild-drüsenhormone und Glucocorticoide involviert.
An vielen Prozessen sind aber Hormone und parakrine Faktoren gleichermaßen involviert (. Tab. 24.6). So regulie-ren T3/4, GH/IGF-1,2 auf der einen Seite und PTHrP/Ihh, BMP und FGF auf der anderen Seite zusammen die Pro-liferation und Differenzierung von Chondroblasten und Osteoblasten. Man weiß inzwischen, dass die Biosynthese von PTHrP und dessen Rezeptor zumindest teilweise unter Kontrolle der Schilddrüsenhormone steht, sodass sie den Regelkreis zwischen Ihh und PTHrP und somit auch die Länge des Zone zwischen Ruheknorpel und hypertrophie-rendem Knorpel (. Abb. 24.17a) beeinflussen. Auch stimu-lieren Schilddrüsenhormone die Biosynthese von Heparan-
sulfatproteoglykan, einem essenziellen Cofaktor der FGF-Rezeptors (7 o.). In den meisten Fällen sind die molekularen Mechanismen aber noch unbekannt.
Die Wirkung der parakrinen Faktoren ist nicht auf die Wachstumsphase begrenzt. Einige Wachstumsfaktoren wie TGFβ, BMPs und FGFs werden auch im reifen Knochen ge-bildet und in der Knochenmatrix gespeichert. Sie spielen eine wichtige Rolle bei Umbau- und Regenerationspro zessen.
24.7.3 Differenzierung von Osteoklasten und Abbau und Umbau von Knochen und Knorpel
Osteoklasten sind die Zellen, die die Knochensubstanz auf-lösen und so für den Umbau des Skelettsystems unentbehr-lich sind (7 u.).
! Im Gegensatz zu den Chondroblasten und Osteoblas-ten leiten sich die Osteoklasten von Vorläuferzellen der Makrophagen ab.
. Tabelle 24.6. Faktoren, die das Knochenwachstum beeinflussen (Auswahl)
Faktor Effekte
Hormone
T3 erforderlich für die normale Knochenentwicklung, bei Mangel Wachstumsverlangsamung, bei Überschuss beschleu-nigtes Knochenwachstum, fördern hypertrophe Differenzierung von Chondrozyten, Stimulierung der Transkription von IGF und GH
Glucocorticoide Rezeptoren in proliferierenden und hypertrophierenden Chondrozyten und Osteoblasten, Osteoblastenproliferation , Osteoblastendifferenzierung , Knochenmatrixproduktion , Knochenresorption
GH IGF-Produktion (Leber) , Wirkung an der Wachstumsfuge (Epiphysenwachstum): IGF-abhängige und unabhängige Mechanismen, GH postnatal erforderlich, pränatal nicht essentiell
IGF’s Epiphysenwachstum
Vitamin D Stimulierung der Knochenbildung (vor allem durch Erhöhung des Ca2+ -Spiegels), Stimulierung der Osteoblasten (we-niger wichtig), Stimulierung von Osteoklasten
PTH Ca2+ -Mobilisierung aus dem Knochen, kurze Pulse von PTH wirken Osteoporose entgegen
Prostaglandin E2 Osteoklastengenese
Wachstumsfaktoren
Ihh produziert von postmitotischen Chondrozyten, Chondrozyten-Proliferation und -Differenzierung , Osteoblasten- Differenzierung , PTHrP-Bildung , Osteoblasten-Differenzierung im Perichondrium , Ihh-/- Mäuse: kleine Knorpel-Elemente, vermehrt hypertrophierender Knorpel
PTHrP Bindet an PTH-Rezeptor, Proliferation von Chondrozyten, verzögerte Hypertrophierung,
FGFs Chondrozyten: Proliferation , terminale Differenzierung , Osteoblasten: Regulation von Proliferation und Differen-zierung; Effekte hängen vom Entwicklungsstadium ab
BMPs Mesenchym-Kondensationen , Chondrozyten-Proliferation , terminale Differenzierung , Antagonismus zu FGF
Interleukine Osteoklastogenese
Transkriptionsfaktoren
SOX9 Differenzierung von Vorläuferzellen zu Chondrozyten , alle Stadien der Chondrozytendifferenzierung , Transkription von ECM-Proteinen
Cbfa1 (Runx2) Differenzierung von Vorläuferzellen zu Osteoblasten , bei Mangel abnorme Chondrozytenreifung
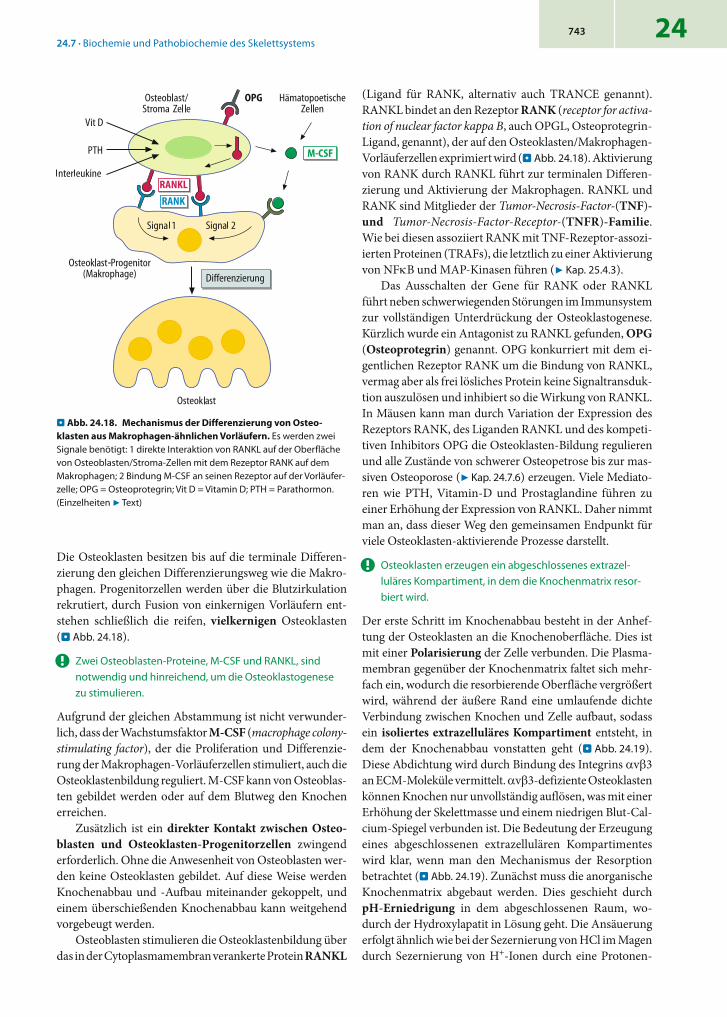
24743
Die Osteoklasten besitzen bis auf die terminale Differen-zierung den gleichen Differenzierungsweg wie die Makro-phagen. Progenitorzellen werden über die Blutzirkulation rekrutiert, durch Fusion von einkernigen Vorläufern ent-stehen schließlich die reifen, vielkernigen Osteoklasten (. Abb. 24.18).
! Zwei Osteoblasten-Proteine, M-CSF und RANKL, sind notwendig und hinreichend, um die Osteoklastogenese zu stimulieren.
Aufgrund der gleichen Abstammung ist nicht verwunder-lich, dass der Wachstumsfaktor M-CSF (macrophage colony-stimulating factor), der die Proliferation und Differenzie-rung der Makrophagen-Vorläuferzellen stimuliert, auch die Osteoklastenbildung reguliert. M-CSF kann von Osteoblas-ten gebildet werden oder auf dem Blutweg den Knochen erreichen.
Zusätzlich ist ein direkter Kontakt zwischen Osteo-blasten und Osteoklasten-Progenitorzellen zwingend erforderlich. Ohne die Anwesenheit von Osteoblasten wer-den keine Osteoklasten gebildet. Auf diese Weise werden Knochenabbau und -Aufbau miteinander gekoppelt, und einem überschießenden Knochenabbau kann weitgehend vorgebeugt werden.
Osteoblasten stimulieren die Osteoklastenbildung über das in der Cytoplasmamembran verankerte Protein RANKL
(Ligand für RANK, alternativ auch TRANCE genannt). RANKL bindet an den Rezeptor RANK (receptor for activa-tion of nuclear factor kappa B, auch OPGL, Osteoprotegrin-Ligand, genannt), der auf den Osteoklasten/Makrophagen-Vorläuferzellen exprimiert wird (. Abb. 24.18). Aktivierung von RANK durch RANKL führt zur terminalen Differen-zierung und Aktivierung der Makrophagen. RANKL und RANK sind Mitglieder der Tumor-Necrosis-Factor-(TNF)- und Tumor-Necrosis-Factor-Receptor-(TNFR)-Familie. Wie bei diesen assoziiert RANK mit TNF-Rezeptor-assozi-ierten Proteinen (TRAFs), die letztlich zu einer Aktivierung von NF B und MAP-Kinasen führen (7 Kap. 25.4.3).
Das Ausschalten der Gene für RANK oder RANKL führt neben schwerwiegenden Störungen im Immunsystem zur vollständigen Unterdrückung der Osteoklastogenese. Kürzlich wurde ein Antagonist zu RANKL gefunden, OPG (Osteoprotegrin) genannt. OPG konkurriert mit dem ei-gentlichen Rezeptor RANK um die Bindung von RANKL, vermag aber als frei lösliches Protein keine Signaltransduk-tion auszulösen und inhibiert so die Wirkung von RANKL. In Mäusen kann man durch Variation der Expression des Rezeptors RANK, des Liganden RANKL und des kompeti-tiven Inhibitors OPG die Osteoklasten-Bildung regulieren und alle Zustände von schwerer Osteopetrose bis zur mas-siven Osteoporose (7 Kap. 24.7.6) erzeugen. Viele Mediato-ren wie PTH, Vitamin-D und Prostaglandine führen zu einer Erhöhung der Expression von RANKL. Daher nimmt man an, dass dieser Weg den gemeinsamen Endpunkt für viele Osteoklasten-aktivierende Prozesse darstellt.
! Osteoklasten erzeugen ein abgeschlossenes extrazel-luläres Kompartiment, in dem die Knochenmatrix resor-biert wird.
Der erste Schritt im Knochenabbau besteht in der Anhef-tung der Osteoklasten an die Knochenoberfläche. Dies ist mit einer Polarisierung der Zelle verbunden. Die Plasma-membran gegenüber der Knochenmatrix faltet sich mehr-fach ein, wodurch die resorbierende Oberfläche vergrößert wird, während der äußere Rand eine umlaufende dichte Verbindung zwischen Knochen und Zelle aufbaut, sodass ein isoliertes extrazelluläres Kompartiment entsteht, in dem der Knochenabbau vonstatten geht (. Abb. 24.19). Diese Abdichtung wird durch Bindung des Integrins v 3 an ECM-Moleküle vermittelt. v 3-defiziente Osteoklasten können Knochen nur unvollständig auflösen, was mit einer Erhöhung der Skelettmasse und einem niedrigen Blut-Cal-cium-Spiegel verbunden ist. Die Bedeutung der Erzeugung eines abgeschlossenen extrazellulären Kompartimentes wird klar, wenn man den Mechanismus der Resorption betrachtet (. Abb. 24.19). Zunächst muss die anorganische Knochenmatrix abgebaut werden. Dies geschieht durch pH-Erniedrigung in dem abgeschlossenen Raum, wo-durch der Hydroxylapatit in Lösung geht. Die Ansäuerung erfolgt ähnlich wie bei der Sezernierung von HCl im Magen durch Sezernierung von H+-Ionen durch eine Protonen-
. Abb. 24.18. Mechanismus der Differenzierung von Osteo-klasten aus Makrophagen-ähnlichen Vorläufern. Es werden zwei Signale benötigt: 1 direkte Interaktion von RANKL auf der Oberfläche von Osteoblasten/Stroma-Zellen mit dem Rezeptor RANK auf dem Makrophagen; 2 Bindung M-CSF an seinen Rezeptor auf der Vorläufer-zelle; OPG = Osteoprotegrin; Vit D = Vitamin D; PTH = Parathormon. (Einzelheiten 7 Text)
24.7 · Biochemie und Pathobiochemie des Skelettsystems

744 Kapitel 24 · Binde- und Stützgewebe
24
ATPase, wobei die Elektroneutralität durch einen ladungs-gekoppelten Chlorid-Kanal gewahrt bleibt. Der intrazel-luläre pH wird durch HCO3
–/Cl– Austausch auf der dem Knochen abgewandten Seite des Osteoklasten aufrechter-halten. Daraufhin wird die entmineralisierte Knochen-matrix durch lysosomale Proteasen abgebaut, eine Schlüs-selstellung nimmt dabei Kathepsin K ein.
24.7.4 Regulation des Knochenwachstums bis zur Pubertät
! Die Geschwindigkeit des postnatalen Skelettwachs-tums wird durch das Wachstumshormon aus der Hypo-physe (GH) reguliert.
Aktivierung von GH-Rezeptoren auf Chondrozyten der Wachstumsfuge führt zur lokalen Produktion von IGF-1 (7 Kap. 27.7.2), das an den IGF-Rezeptor, eine Rezeptor-tyrosinkinase, der Chondrozten bindet. In wie weit syste-misch in der Leber unter Einfluss von GH gebildetes IGF-1 am Knochen eine Rolle spielt, ist noch umstritten. IGF-1 stimuliert die Proliferation von Chondrozyten und deren Größenwachstum (= Zunahme des Zellvolumens). Zusätz-lich scheint GH auch direkte Effekte zu besitzen, indem es Chondrozyten der Ruhezone zur Proliferation anregt. Ein Fehlen von IGF-1 führt zu Zwergwuchs, die Proportionen des Skeletts bleiben dabei jedoch erhalten.
Im Gegensatz zum postnatalen Wachstum wird in der Embryonalentwicklung die Chondrozytenproliferation vor allem durch unabhängig von GH gebildetes IGF-1 und IGF-2 vermittelt.
Für den Wachstumsschub während der Pubertät und die geschlechtsspezifische Ausprägung des Körperbaus sind die Sexualhormone (Testosteron und Östrogen, 7 Kap. 27.5.5, 27.6.5) verantwortlich. Das Längenwachstum der Knochen endet mit Schließung der Epiphysenfuge
während der Pubertät, ebenfalls unter dem Einfluss von Sexualhormonen.
! Die Schließung der Epiphysenfuge wird beim Mann und der Frau durch Östrogen vermittelt.
Indiz dafür ist z.B., dass bei einem 28-jährigen Mann mit defektem Östrogenrezeptor (ER) aber normalem Testos-teronspiegel die Epiphysenfugen noch nicht vollständig geschlossen waren. Ähnliche Effekte waren bei Mäusen mit inaktiviertem ER- -Östrogenrezeptor-Gen sowie bei männlichen Ratten, bei denen durch Gabe von Aromatase-Inhibitoren die Bildung von Östrogenen inhibiert worden war, zu beobachten.
24.7.5 Homöostase des Skelettsystems
In der Wachstumsphase steht durch die Aktivität der Hor-mone und der verschiedenen Wachstums-/Differenzie-rungsfaktoren die Bildung neuer Knochensubstanz im Vordergrund. Nach Beendigung des Wachstums geht das Skelettsystem nicht in einen ruhenden Zustand über, son-dern bleibt nach wie vor sehr aktiv, denn es erfolgt ein steter Umbau des Knochens.
Dies ist erforderlich, da die Knochen einerseits als Cal-cium-Speicher fungieren und bei Bedarf Calcium freige-setzt werden muss. Zum anderen wird das Skelettsystem den mechanischen Erfordernissen angepasst, die Knochen-bälkchen werden in Richtung der maximalen Belastung angeordnet, ein Prozess der noch weitestgehend unverstan-den ist, aber immense physiologische Bedeutung besitzt, da sonst der Knochen bei mechanischer Belastung leicht brechen würde. Ständig wird Knochensubstanz durch die Osteoklasten abgebaut und gleichzeitig neue Knochen-substanz durch die Osteoblasten hergestellt. Man schätzt, dass dies im menschlichen Skelett an etwa 1 bis 2 Millionen Stellen gleichzeitig geschieht. Etwa alle zehn Jahre ist das Skelettsystem einmal erneuert. Beim jungen Erwachsenen sind Aufbau und Abbau exakt ausbalanciert, mit zuneh-mendem Alter verschiebt sich das Gleichgewicht jedoch zugunsten der Osteoklasten, es kommt zur Osteoporose, die ein großes gesundheitliches Problem darstellt, insbe-sondere bei Frauen nach der Menopause.
Die Osteoblasten- und Osteoklastenaktivität kann auf zwei Wegen reguliert werden. Zum einen können reife Zel-len zu Knochenaufbau bzw. Knochenabbau aktiviert wer-den. Zum anderen können Vorläuferzellen zur Proliferation und Differenzierung stimuliert werden, umso mehr Zellen zur Verfügung zu stellen. Beide Wege werden durch die verschiedenen lokalen und systemischen Faktoren unter-stützt. Es ist in vielen Fällen jedoch noch unklar, auf wel-chen Mechanismen die Wirkungen beruhen.
. Abb. 24.19. Mechanismus der Knochenresorption durch Osteo-klasten. (Einzelheiten 7 Text)

24745
24.7.6 Osteoporose und Regulation des Knochenumbaus durch Cytokine und Steroidhormone
Regulation durch Cytokine. Der Einfluss von Cytokinen auf die Knochenbildung wurde aufgrund der Beobachtung entdeckt, dass stimulierte Monozyten Faktoren ins Blut aus-schütten, die die Knochenresorption fördern.
! Cytokine stimulieren die Knochenresorption und för-dern so die Osteoporose.
Diese Aktivität wurde Osteoklasten-aktivierender Faktor (OAF) genannt und später als Interleukin 1 (IL-1) identi-fiziert. In der Folgezeit wurde eine Vielzahl von weiteren Cytokinen entdeckt, die die Knochenresorption stimulie-ren (darunter TNF- , IL-6, IL-11, IL-15 und IL-17). Es wurden aber auch andere Interleukine gefunden, die die Knochenresorption inhibieren (IL-4, IL-10, IL-13, IL-18, IFN- ). TGF- und Prostaglandine können sowohl inhi-bieren als auch stimulieren, je nach Funktionszustand der Zellen. Die Wirkung am Knochen von Interleukinen und anderen Cytokinen, die eigentlich aufgrund ihrer Funktion bei der Immunabwehr bekannt sind, wird leichter ver-ständlich, seit man weiß, dass die Osteoklasten sich von den Makrophagen ableiten.
Die Cytokine werden teils in den Stromazellen/Osteo-blasten, teils in den hämatopoetischen Zellen/Osteoklasten exprimiert und bilden im Knochen ein komplexes Netz-werk. IL-1 kann zum Beispiel durch autokrine und para-krine Mechanismen seine eigene Biosynthese und die von weiteren Interleukinen wie IL-6 stimulieren. Als Resultat der Cytokin-Wirkungen kommt es zu einer Stimulierung der Proliferation, Differenzierung und Aktivierung von Osteoklasten. Die Aktivierung der Osteoklasten kann auf zwei verschiedenen Wegen erfolgen:4 Cytokine können direkt an Rezeptoren auf Osteo-
klasten binden und intrazelluläre Signale auslösen4 Sie können auf indirektem Wege wirken, indem die
verschiedenen Interleukin-vermittelten Reaktionen in der Bildung von M-CSF und RANKL einmünden, die dann wie bereits diskutiert die Osteoklasten aktivieren
Regulation durch Steroidhormone. Steroidhormone be-sitzen einen großen Einfluss auf die Homöostase des Ske-lettsystems. Von großer Bedeutung sind vor allem zwei Gruppen von Hormonen, die Sexualhormone und die Glucocorticoide.
! Östrogene und Androgene bewirken bei Mann und Frau eine positive Bilanz der Knochendichte mit verbes-serten biomechanischen Eigenschaften.
Die Sexualhormone wirken wahrscheinlich primär an den Osteoblasten, obwohl auch Osteoklasten Rezeptoren für Östrogene bzw. Androgene besitzen. Die Wirkungen auf Proliferation und Differenzierung der Osteoblasten sind
komplex und molekular noch nicht gut verstanden, da Osteoblasten bzw. deren Vorläuferzellen je nach Differenzie-rungszustand und Umgebung unterschiedlich auf Sexual-hormone ansprechen. Neben der positiven Wirkung auf die Knochendichte sind die Sexualhormone, wie in Kap. 24.7.4 dargelegt, auch für die geschlechtsspezifische Ausprägung des Knochenbaus und für die Schließung der Epiphysen-fugen verantwortlich.
! Östrogene und Androgene verhindern ein Überschie-ßen der Osteoklastentätigkeit und wirken so antiosteo-porotisch.
Östrogene und Androgene inhibieren die Transkription einer Reihe von den Knochenabbau stimulierenden Cytoki-nen (7 o.) wie IL-1, IL-6, TNF- , CSF und Prostaglandin E2. Die Effekte werden durch die gleichzeitige Inhibition der Expression von Cytokin-Rezeptoren noch verstärkt, wie im Falle des IL-6 Rezeptors gezeigt worden ist. Zusätzlich kön-nen Östrogene die durch RANKL vermittelte Aktivierung von Osteoklasten blockieren. Darüber hinaus kann Östro-gen die Apoptose von Osteoklasten fördern. Es wird ange-nommen, dass an diesem Prozess TGF beteiligt ist, dessen Transkription im Gegensatz zu den meisten Cyto kinen durch Sexualhormone erhöht wird.
In der Summe führen die Effekte der Sexualhormone daher zu einer Verminderung der Osteoklastentätigkeit und wirken so einer Osteoporose entgegen.
! Hohe Spiegel an Glucocorticoiden führen zur Entmine-ralisierung des Knochens.
Über die physiologische Rolle der Glucocorticoide im Kno-chenstoffwechsel weiß man noch wenig, dagegen sind die durch Hormonüberschuss verursachten Effekte gut unter-sucht, wie im Falle des Cushing-Syndroms oder heute häu-figer bei Einsatz von Cortisolderivaten als Immunsuppres-siva. Zielzelle der Hormonwirkung ist der Osteoblast. Die-ser besitzt Rezeptoren für Glucocorticoide, in Osteoklasten hingegen sind bisher noch keine Glucocorticoid-Rezep-toren nachgewiesen worden. Glucocorticoide hemmen die Bildung und Aktivierung von Osteoblasten und damit auch die Neubildung von Osteoklasten (7 Kap. 24.7.3). Zusätz-lich ist die Apoptoserate der Osteoblasten und Osteozyten erhöht. Als Teil der entzündungshemmenden Wirkung der Glucocorticoide wird die Expression von Kollagenase und Interleukin 6 inhibiert. Diese Effekte sollte zu einer Ver-minderung des Knochenabbaus führen. Dennoch kommt es wegen einer verstärkten Aktivierung der vorhandenen Osteoklasten letztlich zur Osteoporose. Ein Effekt, der da-bei eine Rolle spielt, ist eine verminderte Transkription der RNA für Osteoprotegrin bei gleichzeitiger Erhöhung der Transkription seines Liganden (7 Kap. 24.7.3).
24.7 · Biochemie und Pathobiochemie des Skelettsystems

746 Kapitel 24 · Binde- und Stützgewebe
24
24.7.7 Knochenerkrankungen
Das Skelettsystem ist einer Reihe von pathophysiologischen Veränderungen unterworfen, beginnend von angeborenen Fehlbildungen bis hin zu erworbenen Schädigungen durch Fehlhaltung und falsche Belastung und Störungen der Kno-chenhomöostase. Aufgrund der damit verbundenen lang-fristigen gesundheitlichen Beeinträchtigung sind Kno-chenerkrankungen daher von großer volkswirtschaftlicher Bedeutung. Im Folgenden sollen einige Krankheiten im Zu-sammenhang mit Störungen der Knochenhomöostase dar-gestellt werden.
Osteoporose. Die Verringerung der Knochenmasse ver-bunden mit einer Verschlechterung der Knochenarchitek-tur nach dem 40sten Lebensjahr stellt ein großes gesund-heitliches Problem dar, da bereits ein 10%iger Verlust an Knochenmasse das Risiko eines Knochenbruchs verdop-pelt. Man hat abgeschätzt, dass allein in den Vereinigten Staaten mehr als 25% der Frauen in der Postmenopause eine um 10 bis 25% reduzierte Knochendichte besitzen und etwa 5 Millionen Frauen bereits einen Knochenbruch er-litten haben. Die häufigste Ursache der Osteoporose bei Frauen ist der Abfall an Östrogen in der Menopause. Da Östrogene den Spiegel an Cytokinen wie IL-1, IL-6 und TNF- senken, führt ein Abfall des Östrogenspiegels na-türlich zu einer Erhöhung der Cytokinspiegel und damit zu verstärktem Knochenabbau. Nicht nur Frauen, sondern auch Männer im fortgeschrittenen Alter leiden unter fort-schreitender Osteoporose, vermutlich bedingt durch einen Abfall an Sexualhormonen. Aber auch die Tatsache, dass mit fortschreitendem Alter immer weniger teilungsfähige fibroblastenartige Zellen vorhanden sind, dürfte eine wesentliche Rolle spielen. Die dritthäufigste Ursache ist inzwischen die Behandlung mit Cortison-Präparaten. Wei-tere Ursachen sind Multiple Myelomatose, Hyperparathy-reoidismus und Hyperthyreoidismus.
Paget-Krankheit. Diese Erkrankung betrifft etwa 3% der über 40-jährigen Nordeuropäer und der weißen Bevölke-rung Nordamerikas. Die Erkrankung ist durch Verkrüm-mung und Verdickung einzelner Röhrenknochen mit Nei-gung zu Spontanfrakturen gekennzeichnet. Die Ursache ist noch nicht vollends geklärt, beruht aber vermutlich auf einer Infektion mit Paramyxoviren, zu denen auch das Masernvirus gehört. In vitro konnte durch Transfektion von Osteoklasten-Vorläuferzellen mit retroviralen Vektoren, die das Gen für Masern-Nukleocapsidprotein trugen, eine Aktivierung der Expression von RANK, Aktivierung von NF B und erhöhte Empfindlichkeit gegenüber Vitamin D beobachtet werden. Dafür spricht auch, dass bei einer auto-somal dominanten juvenilen Variante der Paget-Krankheit aktivierende Mutationen des RANK-Gens nachgewiesen wurden.
Entzündliche Knochenerkrankungen. Rheumatische Er-krankungen sind durch Zerstörung des Gelenkknorpels und eine exzessive subchondrale Knochenresorption ge-kennzeichnet. In den entzündeten Regionen erhöht sich die Konzentration verschiedener Cytokine, darunter IL-1, On-costatin M, IL-6, IL-13, IL-17 und PTHrP. In der rheuma-tischen Synovialmembran finden sich vermehrt Makro-phagen sowie aktivierte Fibroblasten und T-Zellen. Die beiden letzteren Zelltypen exprimieren RANKL, das die Osteoklastenbildung ohne Beteiligung von weiteren Zell-typen stimulieren kann.
InfoboxKnochenstoffwechselSeit seinem 14. Lebensjahr war der großartige Maler Henri Toulouse-Lautrec (1864–1901) nicht mehr ge-wachsen. Er blieb 1,52 m groß und wurde zu einem deformierten Zwerg. Seine Beine waren die eines Kin-des, beim Gehen war er behindert und musste einen Stock benutzen. Er sagte: »Ich gehe schlecht wie ein Enterich.« Im Zoo stellte er vor den Pinguinen fest: »Die watscheln ja genau wie ich!« Sein Leiden war eine seltene Erbkrankheit der Skelettentwicklung mit der Bezeichnung Pyknodysos-tose. Man weiß inzwischen, dass diese Erkrankung auf Mutationen im Gen für Kathepsin K beruhen. Dies führt zu einem gestörten Abbau der organischen Knochen-matrix. Die Resorptionslakunen der Osteoblasten ent-halten große Mengen unverdauter Kollagenfibrillen. Es resultiert zwar eine röntgenologisch vermehrte Kno-chendichte, jedoch kommt es zur abnorm gesteigerten Brüchigkeit. Die Ursache von Lautrecs zwergenhafter Gestalt war also nicht, wie oft behauptet, schlecht verheilte Knochenbrüche, sondern eine rezessiv-autosomal erbliche Skelettdysplasie. Der später weltberühmte Maler ist im 37. Lebensjahr gestorben.
Therapiemöglichkeiten. Zur Behandlung der nach der Menopause auftretenden Osteoporose werden in erster Linie Östrogene gegeben, um den Abfall des Hormonspie-gels zu kompensieren. Da eine Östrogenbehandlung mit einem erhöhten Risiko für Unterleibskrebs verbunden ist, wird es in Verbindung mit einem Gestagen verabreicht. In den letzten Jahren wurden Agenzien entwickelt, die die Wirkung von Östrogen ganz oder teilweise nachahmen können, so genannte selektive Östrogen Rezeptor Modu-latoren (SERMs). Der erste Vertreter dieser Substanzklasse war das Triphenylethylen-Derivat Tamoxifen. Mit derar-tigen Wirkstoffen wird es in der Zukunft wahrscheinlich gelingen, die positive Wirkung von Östrogen am Knochen zu substituieren, ohne die negativen Effekte in Kauf neh-men zu müssen. Eine andere Klasse von antiosteoporotisch

24747
wirkenden Substanzen sind die Bisphosphonate, in denen der Sauerstoff von Pyrophosphat (-O3P-O-PO3-) durch eine Methylengruppe (-O3P-CH2-PO3-) mit verschiedenen Seitenketten ersetzt ist. Diese Substanzen werden von Os-teoklasten aufgenommen und wirken wahrscheinlich durch Inhibition der Farnesylpyrophosphatsynthase (7 Kap. 18.3.1)
und hemmen so die Prenylierung und damit die Mem-branverankerung von kleinen G-Proteinen wie Rho, Rab und Cdc42, die für die Aktivierung und das Überleben der Osteoklasten wichtig sind. Aufgrund ihres Wirkungsme-chanismus können die Bisphosphonate Knochenresorption unabhängig von der Ursache verhindern.
In Kürze
Knorpel und Knochen besitzen eine sehr spezialisierte ECM:4 Knorpel stellt im Wesentlichen ein elastisches
hydratisiertes Gel dar, das aus riesigen Komplexen zwischen Hyaluronsäure und dem Proteoglykan Aggrecan besteht und durch Typ II Kollagen-Fasern in Form gehalten wird
4 Knochen besitzt eine aus Hydroxyapatit aufgebaute ECM, die durch Kollagen Typ I-Fasern ihre Stabilität erlangt
4 Darüber hinaus enthalten Knochen und Knorpel noch eine Reihe von anderen, nichtkollagenen Proteinen, die z.T. für die Interaktion der Zellen mit der ECM von Bedeutung sind
Knochen- und Knorpel-spezifische Zellen sind die Chon-drozyten, Osteoblasten/Osteozyten und die Osteo klasten.4 Chondrozyten und Osteoblasten leiten sich von Fi-
broblasten ab. Die Differenzierung ist von Trans-kriptionsfaktoren wie SOX-9 und CBFA-1 und Wachs-tums-/Differenzierungsfaktoren wie Ihh und PTHrP und verschiedenen Isoformen aus den TGF /BMP- und FGF-Familien abhängig. Zusätzlich ist eine Reihe von Hormonen (GH, IGF, T3, Glucocorticoide) invol-viert
4 Osteoklasten leiten sich von der Monozyten/Makro-phagen-Linie ab. Die Differenzierung erfordert das Zusammenwirken zweier unterschiedlicher Signale:
– sezernierte Faktoren: Bindung von M-CSF an Rezep to-ren auf den Vorläuferzellen
– direkte Zell-Zell-Kontakte mit Osteoblasten: Inter-aktion des Zellmembranproteins RANKL auf Osteo-blasten mit RANK auf den Präkursorzellen
Osteoklasten bauen Knochen durch Erzeugung eines abge-schlossenen extrazellulären Kompartiments ab, in das HCl und lysosomale Enzyme sezerniert werden. Die Homöostase des Skelettsystems erfordert eine komplexe hormonelle Regulation:4 Knochenwachstum unter Einfluss von Wachstumshor-
monen (GH und IGF), Schließung der Epiphysen fuge in der Pubertät unter Einfluss von Östrogen
4 Hormone des Calcium-Stoffwechsels (PTH, Vitamin D, Calcitonin): Mineralisation und Demineralisation
4 Androgene und Östrogene: positive Bilanz, geschlechts-spezifische Ausprägung
4 Glucocorticoide: hohe Spiegel bewirken Entmineralisie-rung
4 Cytokine (Interleukine): stimulieren Knochenabbau
Das Skelettsystem ist einer Reihe von pathophysio logi schen Veränderungen unterworfen. Am bedeutendsten sind:4 Osteoporose, besonders in der Postmenopause bei
Frauen4 entzündliche Knochenerkrankungen wie Rheuma mit
erhöhten Cytokin-Spiegeln
24.8 Biochemie der Haut
24.8.1 Aufbau und Funktionen der Haut
Die Haut ist das Grenzorgan zur Umwelt. Sie besteht von außen nach innen aus drei Schichten:4 Die Epidermis ist ein vielschichtiges Plattenepithel und
ist Träger der Hornschicht, der äußersten, nicht mehr vitalen Hautschicht. Sie enthält drei Hauptzelltypen, die Keratinozyten, die Melanozyten und die immunkom-petenten Langerhans-Zellen
4 Durch eine Basalmembran von der Epidermis getrennt, ist die Dermis das bindegewebige Gerüst der Haut und gleichzeitig der vaskularisierte Versorgungsteil
4 Die Subkutis ist ein Fettgewebspolster, das auf den Fas-zien der darunter liegenden Muskulatur aufliegt
Die Haut erfüllt folgende Funktionen:4 Barrierefunktion gegen die Umwelt, einschließlich
Mikroorganismen4 Schutz vor Wasserverlust4 mechanischer Schutz gegen Traumen4 Schutz vor physikalischen Einwirkungen, wie UV-Licht,
Hitze oder Kälte4 Regulation der Körpertemperatur4 immunologische Abwehr4 Sinnesfunktionen wie Tast- und Schmerzreize
Zur Erfüllung spezifischer Aufgaben weist die Haut regionale Unterschiede auf, die sich in der Beschaffen-heit und Dimensionierung der einzelnen Schichten und der makromolekularen Zusammensetzung manifestie-ren.
24.8 · Biochemie der Haut
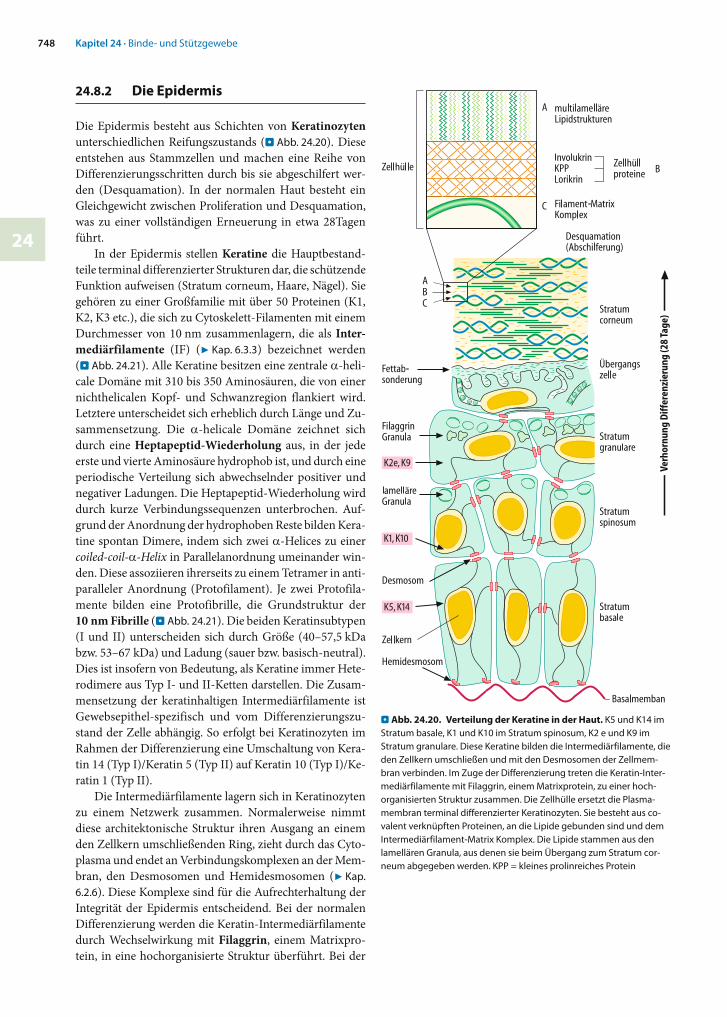
748 Kapitel 24 · Binde- und Stützgewebe
24
24.8.2 Die Epidermis
Die Epidermis besteht aus Schichten von Keratinozyten unterschiedlichen Reifungszustands (. Abb. 24.20). Diese entstehen aus Stammzellen und machen eine Reihe von Differenzierungsschritten durch bis sie abgeschilfert wer-den (Desquamation). In der normalen Haut besteht ein Gleichgewicht zwischen Proliferation und Desquamation, was zu einer vollständigen Erneuerung in etwa 28Tagen führt.
In der Epidermis stellen Keratine die Hauptbestand-teile terminal differenzierter Strukturen dar, die schützende Funktion aufweisen (Stratum corneum, Haare, Nägel). Sie gehören zu einer Großfamilie mit über 50 Proteinen (K1, K2, K3 etc.), die sich zu Cytoskelett-Filamenten mit einem Durchmesser von 10 nm zusammenlagern, die als Inter-mediärfilamente (IF) (7 Kap. 6.3.3) bezeichnet werden (. Abb. 24.21). Alle Keratine besitzen eine zentrale -heli-cale Domäne mit 310 bis 350 Aminosäuren, die von einer nichthelicalen Kopf- und Schwanzregion flankiert wird. Letztere unterscheidet sich erheblich durch Länge und Zu-sammensetzung. Die -helicale Domäne zeichnet sich durch eine Heptapeptid-Wiederholung aus, in der jede erste und vierte Aminosäure hydrophob ist, und durch eine periodische Verteilung sich abwechselnder positiver und negativer Ladungen. Die Heptapeptid-Wiederholung wird durch kurze Verbindungssequenzen unterbrochen. Auf-grund der Anordnung der hydrophoben Reste bilden Kera-tine spontan Dimere, indem sich zwei -Helices zu einer coiled-coil- -Helix in Parallelanordnung umeinander win-den. Diese assoziieren ihrerseits zu einem Tetramer in anti-paralleler Anordnung (Protofilament). Je zwei Protofila-mente bilden eine Protofibrille, die Grundstruktur der 10 nm Fibrille (. Abb. 24.21). Die beiden Keratinsubtypen (I und II) unterscheiden sich durch Größe (40–57,5 kDa bzw. 53–67 kDa) und Ladung (sauer bzw. basisch-neutral). Dies ist insofern von Bedeutung, als Keratine immer Hete-rodimere aus Typ I- und II-Ketten darstellen. Die Zusam-mensetzung der keratinhaltigen Intermediärfilamente ist Gewebsepithel-spezifisch und vom Differenzierungszu-stand der Zelle abhängig. So erfolgt bei Keratinozyten im Rahmen der Differenzierung eine Umschaltung von Kera-tin 14 (Typ I)/Keratin 5 (Typ II) auf Keratin 10 (Typ I)/Ke-ratin 1 (Typ II).
Die Intermediärfilamente lagern sich in Keratinozyten zu einem Netzwerk zusammen. Normalerweise nimmt diese architektonische Struktur ihren Ausgang an einem den Zellkern umschließenden Ring, zieht durch das Cyto-plasma und endet an Verbindungskomplexen an der Mem-bran, den Desmosomen und Hemidesmosomen (7 Kap. 6.2.6). Diese Komplexe sind für die Aufrechterhaltung der Integrität der Epidermis entscheidend. Bei der normalen Differenzierung werden die Keratin-Intermediärfilamente durch Wechselwirkung mit Filaggrin, einem Matrixpro-tein, in eine hochorganisierte Struktur überführt. Bei der
. Abb. 24.20. Verteilung der Keratine in der Haut. K5 und K14 im Stratum basale, K1 und K10 im Stratum spinosum, K2 e und K9 im Stratum granulare. Diese Keratine bilden die Intermediärfilamente, die den Zellkern umschließen und mit den Desmosomen der Zellmem-bran verbinden. Im Zuge der Differenzierung treten die Keratin-Inter-mediärfilamente mit Filaggrin, einem Matrixprotein, zu einer hoch-organisierten Struktur zusammen. Die Zellhülle ersetzt die Plasma-membran terminal differenzierter Keratinozyten. Sie besteht aus co-valent verknüpften Proteinen, an die Lipide gebunden sind und dem Intermediärfilament-Matrix Komplex. Die Lipide stammen aus den lamellären Granula, aus denen sie beim Übergang zum Stratum cor-neum abgegeben werden. KPP = kleines prolinreiches Protein
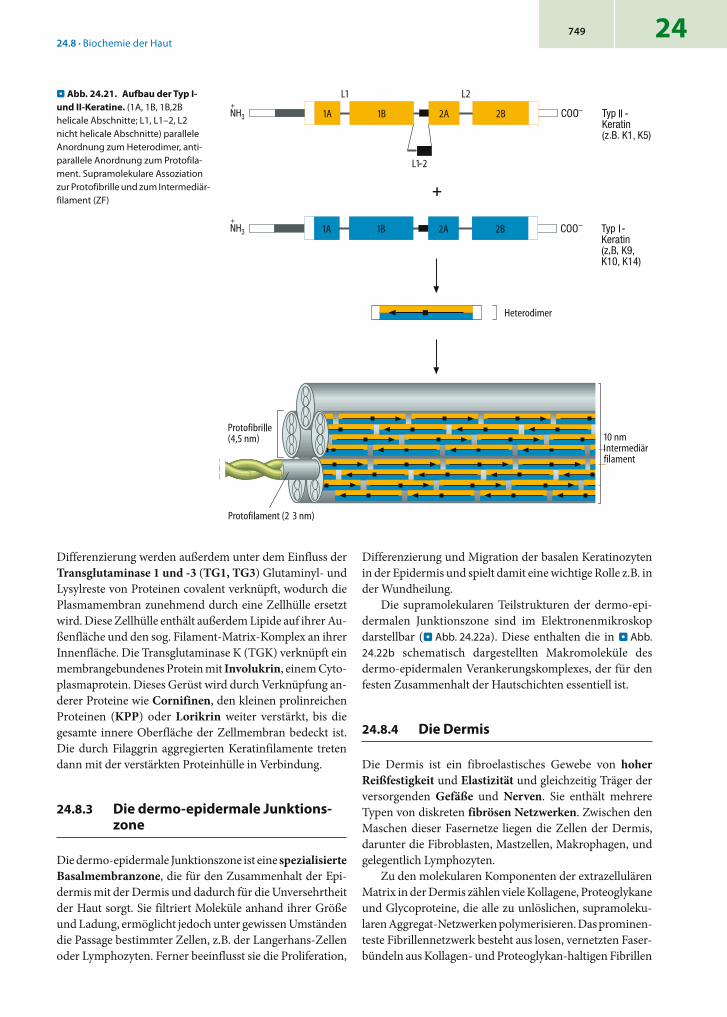
24749
Differenzierung werden außerdem unter dem Einfluss der Transglutaminase 1 und -3 (TG1, TG3) Glutaminyl- und Lysylreste von Proteinen covalent verknüpft, wodurch die Plasmamembran zunehmend durch eine Zellhülle ersetzt wird. Diese Zellhülle enthält außerdem Lipide auf ihrer Au-ßenfläche und den sog. Filament-Matrix-Komplex an ihrer Innenfläche. Die Transglutaminase K (TGK) verknüpft ein membrangebundenes Protein mit Involukrin, einem Cyto-plasmaprotein. Dieses Gerüst wird durch Verknüpfung an-derer Proteine wie Cornifinen, den kleinen prolinreichen Proteinen (KPP) oder Lorikrin weiter verstärkt, bis die gesamte innere Oberfläche der Zellmembran bedeckt ist. Die durch Filaggrin aggregierten Keratinfilamente treten dann mit der verstärkten Proteinhülle in Verbindung.
24.8.3 Die dermo-epidermale Junktions-zone
Die dermo-epidermale Junktionszone ist eine spezialisierte Basalmembranzone, die für den Zusammenhalt der Epi-dermis mit der Dermis und dadurch für die Unversehrtheit der Haut sorgt. Sie filtriert Moleküle anhand ihrer Größe und Ladung, ermöglicht jedoch unter gewissen Umständen die Passage bestimmter Zellen, z.B. der Langerhans-Zellen oder Lymphozyten. Ferner beeinflusst sie die Proliferation,
Differenzierung und Migration der basalen Keratinozyten in der Epidermis und spielt damit eine wichtige Rolle z.B. in der Wundheilung.
Die supramolekularen Teilstrukturen der dermo-epi-dermalen Junktionszone sind im Elektronenmikroskop darstellbar (. Abb. 24.22a). Diese enthalten die in . Abb. 24.22b schematisch dargestellten Makromoleküle des dermo-epidermalen Verankerungskomplexes, der für den festen Zusammenhalt der Hautschichten essentiell ist.
24.8.4 Die Dermis
Die Dermis ist ein fibroelastisches Gewebe von hoher Reißfestigkeit und Elastizität und gleichzeitig Träger der versorgenden Gefäße und Nerven. Sie enthält mehrere Typen von diskreten fibrösen Netzwerken. Zwischen den Maschen dieser Fasernetze liegen die Zellen der Dermis, darunter die Fibroblasten, Mastzellen, Makrophagen, und gelegentlich Lymphozyten.
Zu den molekularen Komponenten der extrazellulären Matrix in der Dermis zählen viele Kollagene, Proteoglykane und Glycoproteine, die alle zu unlöslichen, supramoleku-laren Aggregat-Netzwerken polymerisieren. Das prominen-teste Fibrillennetzwerk besteht aus losen, vernetzten Faser-bündeln aus Kollagen- und Proteoglykan-haltigen Fibrillen
. Abb. 24.21. Aufbau der Typ I-und II-Keratine. (1A, 1B, 1B,2B helicale Abschnitte; L1, L1–2, L2 nicht helicale Abschnitte) parallele Anordnung zum Heterodimer, anti-parallele Anordnung zum Proto fila-ment. Supramolekulare Assoziation zur Protofibrille und zum Intermediär-filament (ZF)
24.8 · Biochemie der Haut

750 Kapitel 24 · Binde- und Stützgewebe
24
. Abb. 24.22a,b. Elektronenmikroskopische Aufnahme der dermo-epidermalen Junktion. a Der basale Keratinozyt (K) sitzt auf einer zweischichtigen Basalmembran bestehend aus einer elektronen-optisch hellen Zone, Lamina lucida (LL), und aus einer elektronen-optisch dunklen Zone, Lamina densa (LD). Die intrazellulären, dunkel gefärbten feinen Intermediärfilamente docken an den knopfförmigen Hemidesmosomen (HD) ein. Die feinen Verankerungsfilamente zwi-schen den Hemidesmosomen und der Lamina densa sind kaum sicht-bar. Dagegen sind die quer gestreiften Verankerungsfibrillen (AF = anchoring fibrils) unterhalb der Lamina densa prominent. b Schema-tische Darstellung der molekularen Komponenten der dermo-epider-malen Junktion. Die Hemidesmosomen an der Plasmamembran der basalen Zelle sind Proteinaggregate, die eine Verbindung zwischen den Keratinfilamenten und der Basalmembran sichern. Auf der intra-zellulären Seite vermitteln BP230 und Plectin als Linker-Proteine die Bindung zwischen den Keratinfilamenten und dem Hemidesmosom, während auf der extrazellulären Seite die Laminin 5- und Kollagen
Typ XVII-haltigen Verankerungsfilamente die Lamina lucida durch-spannen. Dabei funktionieren die Transmembranproteine Integrin
6 4 und Kollagen Typ XVII auch als Zelladhäsionsrezeptoren. Die Lamina densa, eine ultrastrukturell amorphe Schicht, ist durch Kol-lagen Typ VII-haltige Verankerungsfibrillen mit dem unterliegenden Bindegewebe verankert. Ein Verankerungskomplex mit Hemidesmo-somen, Verankerungsfilamenten und Verankerungsfibrillen ist charak-teristisch für die Haut und kommt in anderen Basalmembranen nicht vor. Mutationen in den erwähnten Proteinen führen zum vermin-derten Zusammenhalt der Hautschichten und zur Blasenbildung als Symptom (Epidermolysis-bullosa-Gruppe). Rechts ist die Blasenbil-dungsebene bei den verschiedenen Epidermolysis-bullosa-Formen markiert (Pfeile). Die Blasenbildungsebene ist von der Lokalisation des betroffenen Proteins abhängig. Die entsprechenden Gendefekte sind in Tabelle 26.8 aufgelistet. EBS = Epidermolysis bullosa simplex; EBJ = Epidermolysis bullosa junctionalis; EBD = Epidermolysis bullosa dystrophica

24751
(D-Periodizität: 64 nm). Außerdem finden sich elastische Fasern, die mit perlenkettenartigen Mikrofibrillen verge-sellschaftet sind (7 Kap. 24.2.1, 24.3). Andere Netzwerke werden z.B. durch Fibronectin gebildet. Schließlich kom-men auch Filamente mit einer Periodizität von 100 nm vor, deren Hauptkomponente das Kollagen Typ VI ist. Die In-forma tion für den Aufbau einer gegebenen Aggregatstruk-tur ist in der Zusammensetzung der makromolekularen Bestandteile, sowie in deren Primärstruktur enthalten und die Poly merisierung der Fibrillen-Netzwerke ist auf mole-kularer Ebene hierarchisch und streng reguliert.
Die Dermis enthält mindestens 12 verschiedene Kol-lagentypen (. Tabelle 24.1). Typische dermale Fibrillen sind immer Mischfibrillen, die mehrere Kollagentypen sowie nichtkollagene Komponenten enthalten. Die charak-teristischen, im Elektronenmikroskop sichtbaren quer ge-streiften Fibrillen in der Dermis enthalten als Hauptkom-ponente das ubiquitäre Kollagen Typ I, gemischt mit etwa 10–15% Typ III. Ferner finden sich geringe Mengen der Kollagene Typ V, Typ XII und Typ XIV, die jedoch für die Fibrillenorganisation essentiell sind. Der Kollagen Typ III-Gehalt nimmt mit dem Alter ab.
Die Kollagen Typen IV, VII, VIII, XVII und XVIII kom-men in den verschiedenen epithelialen und vaskulären Ba-salmembranzonen in der Haut vor und sind dort Bestand-teile von diskreten Suprastrukturen.
Die Elastizität der Haut beruht auf der Struktur der elas tischen Fasern, die in verschiedenen Dermis-Schichten in unterschiedlicher Zusammensetzung vorkommen. Die Fasern sind unterschiedlich dick und zum Teil verzweigt. Ultrastrukturell sind zwei Komponenten erkennbar, eine amorphe Masse, die die dehnbare Komponente darstellt, und 10–12 nm dicke Mikrofibrillen, die die elastischen Fa-sern mit dem angrenzenden Gewebe verbinden. Das amor-phe Material besteht vorwiegend aus Elastin, einem stark quervernetzten Protein, das -Faltblatt-Elemente enthält. Die mikrofibrilläre Komponente der Fasern sorgt für deren Festigkeit und Begrenzung der Dehnbarkeit, ihr Hauptbe-standteil ist Fibrillin-1, ein Makromolekül, das mit anderen Proteinen wie Fibrillin-2 zu Mikrofibrillen polymerisiert.
Zwischen den Fibrillennetzwerken ist eine amorphe extrafibrilläre Matrix eingelagert. Deren Hauptkompo-nenten sind neben Wasser Proteoglykane (z.B. Versican, Decorin), Hyaluronan und Glycoproteine. Die extrafibril-läre Matrix hat in der Haut nicht nur eine wichtige Füll- und Stützfunktion, sondern ist auch biologisch aktiv. Viele Glycoproteine und Proteoglykane sind wesentlich für den korrekten Aufbau der Gewebearchitektur und liefern spe-zifische Informationen an die Zellen. Heparansulfatsei-tenketten der Proteoglykane und kleine Proteoglykane, z.B. Decorin und Biglykan, funktionieren auch als Speicher oder Modulatoren von Wachstumsfaktoren der FGF- und TGF -Familien.
Die dermalen Glycoproteine besitzen adhäsive Eigen-schaften und sind an der Regulation der Fibrillenbildung
und bestimmter zellulärer Funktionen beteiligt. Ein wich-tiges multifunktionelles Glycoprotein ist Fibronectin (7 Kap. 24.5.1). Das im Plasma in einer löslichen, globulären Form vorkommende Protein polymerisiert in der extrazel-lulären Matrix vieler Gewebe, u.a. in der Dermis, zu Fibril-len, welche die Adhäsion, die Migration und die Differen-zierung von vielen verschiedenen Zellen regulieren.
24.8.5 Pathobiochemie der Haut
Verhornungsstörungen. Vielfältige genetische Defekte mit Hautmanifestationen sind bekannt, betroffen können alle Hautschichten sein. Verhornungsstörungen sind die Kon-sequenz von Mutationen in den Genen für epidermale Strukturproteine. Zum Beispiel führen Mutationen in den Genen für Keratin 1, Keratin 10 oder Transglutaminase-1 (. Tabelle 24.7) zu erblichen Ichthyosen (Fischschuppen-krankheit). Die durch die Transglutaminase vernetzten Keratinfilament-Aggregate des Stratum Corneum werden unter diesen Umständen nicht mehr normal gebildet und mit der Zellhülle verknüpft, was zu einer charakteristischen, grob lamellären Schuppenbildung führt.
Bindegewebskrankheiten. Mutationen in den Genen für die Hautkollagene, für das Elastin sowie für die Fibrilline führen zu einem breiten Spektrum von erblichen Haut-krankheiten oder Syndromen mit Hautbeteiligung . Tabel-le 24.7). Die durch Kollagenmutationen herbeigeführten molekularen Defekte zeichnen sich durch eine abnormale Struktur oder Stabilität der kollagenhaltigen Fasern und damit eine stark erhöhte Dehnbarkeit oder verminderte Reißfestigkeit der Dermis aus. Diese Situation findet sich z.B. bei verschiedenen Formen des Ehlers-Danlos-Syn-droms (. Abb. 24.23a). In ähnlicher Weise sind Mutationen im Fibrillin-1-Gen die Ursache für das Marfan-Syndrom und im Fibrillin-2-Gen für die kongenitale kontraktu-rale Arachnodaktylie. Elastin-Mutationen können auto-somal dominante Formen von Cutis laxa verursachen (. Abb. 24.23b).
Blasen bildende Hautkrankheiten. Ein illustratives Beispiel für die unterschiedlichen Ursachen von Hautkrankheiten stellen die Defekte der dermo-epidermalen Junktionszone dar (. Abb. 24.23c). Daran lassen sich die genetischen und erworbenen molekularen Abnormitäten einander gegen-überstellen, bzw. miteinander korrelieren.
Die Epidermolysis bullosa (EB) gehört zu einer Grup-pe von erblichen Krankheiten, bei denen durch minimale Verletzungen Blasenbildung hervorrufen wird. Ursachen sind Mutationen in den Genen für Proteine des Veranke-rungskomplexes und dadurch Verminderung des dermo-epidermalen Zusammenhalts. Es werden drei EB-Haupt-gruppen unterschieden: EB simplex, EB junctionalis und EB dystrophica.
24.8 · Biochemie der Haut

752 Kapitel 24 · Binde- und Stützgewebe
24
Im Fall der EB simplex (EBS) erfolgt die Blasenbildung innerhalb des Stratum basale. Abnormale Intermediärfila-mente sind die Folge von Mutationen in den Genen für die Keratine 5 und 14 oder Plectin. Diese Makromoleküle sind für den Aufbau und die Verankerung des Intermediärfila-ment-Netzwerks der Zelle essentiell. Bei der EB junctionalis (EBJ) tritt die Spaltbildung entlang der Lamina lucida aufgrund von abnormalen Verankerungsfilamenten auf. Hier sind Mutationen in den Genen für Laminin 5, 6 4-Integrin und Kollagen Typ XVII ursächlich beteiligt (. Abb. 24.23c). Die Mutationen führen zu abnormalen
Proteinstrukturen und zu ultrastrukturell rudimentären Hemidesmosomen, die nicht funktionell sind. Bei der Epi-dermolysis bullosa dystrophica (EBD) findet die Spaltung unterhalb der Lamina densa auf der Ebene der Veranke-rungsfibrillen statt. Bei der EBD sind die Verankerungs-fibrillen entweder abnormal oder in ihrer Anzahl vermin-dert. Für alle molekular identifizierten EBD-Varianten sind Mutationen im Gen für Kollagen Typ VII verantwortlich.
Bis heute sind zahlreiche Mutationen in den Genen für die erwähnten Proteine bekannt (. Tabelle 24.8). Null-Mutationen, die zum vollständigen Fehlen eines
. Abb. 24.23a–c. Klinische Symptome von genetischen Haut-krankheiten. Mutationen in den Genen für Strukturproteine der Haut können vielfältige Symptome verursachen. Typische Beispiele sind in a–c dargestellt. a Dünne und stark dehnbare Haut beim Ehlers-Danlos-
Syndrom (Kollagen-Typ-I-Mutation). b Schlaffe, »zu große« Haut bei Cutis laxa (Elastin Mutation). c Blasenbildung an der Hand nach mini-maler mechanischer Belastung (Kollagen Typ XVII Mutation)
. Tabelle 24.7. Erbliche Hautkrankheiten oder -symptome durch mutierte Strukturproteine. EB Epidermolysis bullosa
Protein Gen Krankheit Hautsymptome
Keratin 1 KRT1 Ichthyose(epidermolytische Hyperkeratose)
Starke Schuppung und Rötung der Haut
Keratin 10 KRT10 Ichthyose(epidermolytische Hyperkeratose)
Starke Schuppung und Rötung der Haut
Kollagen Typ I COL1A1 Ehlers-Danlos-Syndrom Typ I Schwere Form; dünne, dehnbare Haut
Ehlers-Danlos-Syndrom Typ VII A & VII B dünne, dehnbare Haut
Kollagen Typ III COL3A1 Ehlers-Danlos-Syndrom Typ IV Fragilität, Hämatombildung
Kollagen Typ V COL5A1 Ehlers-Danlos-Syndrom Typ I Schwere Form; dünne, dehnbare Haut
COL5A2 Ehlers-Danlos-Syndrom Typ I Schwere Form; dünne, dehnbare Haut
Kollagen Typ VI COL6A1 Ullrich-Syndrom Muskeldystrophie und teigige Haut
Kollagen Typ VII COL7A1 EB dystrophica Blasenbildung
Kollagen Typ XVII COL17A1 EB junctionalis Blasenbildung
Laminin 5 LAMA3LAMB3LAMC2
EB junctionalis Blasenbildung
Fibrillin 1 FBN1 Marfan-Syndrom Dünne, leicht dehnbare Haut
Elastin ELN Cutis laxa Schlaffe »zu große« Haut

24753
dieser Proteine führen, sind in der Regel mit schweren Krank heitsbildern assoziiert. Andere Mutationen, z.B. Amino säuresubstitutionen oder kleine Deletionen oder Inser tionen, verursachen meistens mildere Symptome (. Abb. 24.23c).
Autoimmunkrankheiten. Erworbene Blasen bildende Haut-krankheiten können durch Autoantikörper verursacht werden, die gegen Strukturproteine der dermo-epiderma-len Junktionszone gerichtet sind. Man vermutet, dass die Bindung der Autoantikörper zu einer Funktionsverminde-rung der Proteine und damit zum geschwächten dermo-epidermalen Zusammenhalt führen. Zum Beispiel kommen beim bullösen Pemphigoid sowohl zirkulierende als auch gewebeständige Autoantikörper gegen Kollagen Typ XVII oder Laminin 5 vor, oder bei der Epidermolysis bullosa acquisita Autoantikörper gegen Kollagen Typ VII. Die Auto-antikörper können durch Immunfluoreszenzfärbung in der Haut nachgewiesen werden (. Abb. 24.24).
. Abb. 24.24. Immunfluoreszenz-Färbung der Haut beim bullö-sen Pemphigoid. Die Epidermis ist durch eine Blase von der Dermis getrennt (Stern). Autoantikörper gegen Kollagen Typ XVII (grüne Fluoreszenzfärbung, Pfeile) sind am Blasendach sichtbar. E = Epidermis; D = Dermis
. Tabelle 24.8. Molekulare Grundlagen der Epidermolysis bullosa (EB)
EB-Subtyp Gen Protein Abnormale Struktur
EB simplex KRT5 Keratrin 5 Keratinfilamente
EB simplex KRT14 Keratin 14 Keratinfilamente
EB simplex-MDa PLEC1 Plectin Hemidesmosom
EB junctionalis LAMA3 Laminin 5 (α3-Kette) Verankerungsfilamente
EB junctionalis LAMB3 Laminin 5 (β3-Kette) Verankerungsfilamente
EB junctionalis LAMC2 Laminin 5 (γ2-Kette) Verankerungsfilamente
EB junctionalis COL17A1 Kollagen Typ XVII Verankerungsfilamente
EB junctionalis ITGA6 α6β4-Integrin Hemidesmosom
EB junctionalis ITGB4 α6β4-Integrin Hemidesmosom
EB dystrophica COL7A1 Kollagen Typ VII Verankerungsfibrillen
a MD mit Muskeldystrophie. Plectin kommt in der Haut und im Muskel vor.
In Kürze
Die Haut ist das Grenzorgan zur Umwelt. Sie besteht aus drei Schichten:4 Epidermis4 Dermis und4 Subkutis
Dermis und Epidermis sind durch die dermo-epidermale Junktionszone verbunden. Hauptbestandteil der terminal differenzierten Struk-turen (Corneum, Haare, Nägel) sind die Keratine, eine Großfamilie von etwa 40 verschiedenen Proteinen. Die
Keratine bilden 10 nm-dicke Keratin-Intermediär filamente: Die Assemblierung erfolgt über Dimere mit einer coiled-coil
-helicalen Struktur, die über Proteo filamente und Proteo-fibrillen zu den reifen 10 nm-Fibrillen polymerisieren. Das Stratum corneum bildet eine hochorganisierte Struktur aus Keratin-Fibrillen, zusammen mit Filaggrin und weiteren Proteinen und Lipiden. Durch Transglut aminase 1 und 3 wird das Netzwerk zur Stabilisierung extensiv quer-vernetzt. Der Zusammenhalt zwischen Dermis und Epidermis wird durch eine spezialisierte Basalmembran-Region, die
6
24.8 · Biochemie der Haut

754 Kapitel 24 · Binde- und Stützgewebe
24
dermo-epidermale Junktionszone, vermittelt: Hemi-desmosomen an der basalen Seite der Keratinozyten stehen über das Integrin 6 4, Laminin-5 und das Trans-membran-Kollagen XVII mit Ankerfibrillen aus Kollagen VII und dem interstitiellen Bindegewebe in Verbindung. Die Dermis ist ein fibroelastisches Gewebe mit mehre-ren Typen von fibrösen Netzwerken aus mindes tens 12 verschiedenen Kollagentypen, Proteoglykanen und Fibro-nectin. Die Elastizität der Haut wird durch elastische Fa-sern aus Elastin und Fibrillin gewährleistet. Die fibrillären Strukturen sind in eine amorphe Matrix aus Hyaluronäure und Proteoglykanen wie Ver sican ein-gebettet.
Alle Hautschichten können von vielfältigen geneti schen Defekten betroffen sein. Die bekanntesten sind:4 Verhornungsstörungen (Ichthyosen)4 Mutationen in Bindegewebsproteinen, die zu einer
erhöhten Dehnbarkeit/verminderten Reißfestigkeit führen (z.B. beim Marfan-Syndrom)
4 Blasen-bildende Krankheiten aufgrund von Defekten in der dermo-epidermalen Junktionszone (verschiedene Formen der Epidermolysis bullosa)
Eine der bekanntesten Autoimmunerkrankung ist das bullöse Pemphigoid, bei der Autoantikörper gegen das Kollagen Typ XVII gebildet werden.
Literatur
Monographien und LehrbücherKreis T, Vale R (1999) Guidebook to the Extracellular Matrix, Anchor,
and Adhesion Proteins, 2nd edn. A Sambrook and Tooze publica-tion at Oxford University Press, New York
Iozzo R (2000) Proteoglycans: Structure, Biology and Molecular Inter-actions. Marcel Dekker Press
Bilezikian JP, Raisz LG, Rodan GA (2002) Principles of Bone Biology. Academic Press, San Diego, USA
Original- und ÜbersichtsarbeitenMyllyharju J, Kivirikko KI (2001) Collagens and Collagen-related di seases.
Ann Med 33:7–21Prockop DJ,Kivirikko KI (1995) Collagens: Molecular Biology, Diseases,
and Potentials for Therapy. Annu Rev Biochem 64:403–434Kielty CM, Sheratt MJ, Shuttleworth A (2002) Elastic fibres. J Cell Sci
115:2817–2828Kornblihtt AR, Pesce CG, Alonso CR, Cramer P, Srebrow A, Werbajh S,
Muro AF (1996) The fibronectin gene as a model for splicing and transcription FASEB J 10:248–257
Nature Insight Reviews: Bone and Cartilage (2003) Nature 423:315–361
a) Kronenberg HM. Developmental regulation of the growth plate, pp 423:332–336
b) Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. pp 337–342
c) Harada S, Rodan GA. Control of osteoblast function and regula-tion of bone mass. pp 349–355
Rodan GA, Martin TJ (2000) Therapeutic Approaches to Bone Diseases. Science 289:1508–1514
Jilka RL (1998) Cytokines, bone remodelling, and estrogen deficiency: a 1998 update. Bone 23:75–81
Yurchenco PD, Amenta PS (2004) Basement membrane assembly, stabil-ity and activities observed through a developmental lens. Matrix Biol 22:521–538
Hynes RO (2002) Integrins: Bidirectional, Allosteric Signaling Machines. Cell 110:673–687
Miner JH, Yurchenco PD (2004) Laminin Functions in Tissue Morpho-genesis. Annu Rev Cell Dev Biol 20:255–284
Bruckner-Tuderman L, Bruckner P (1998) Genetic diseases of the extra-cellular matrix: more than just connective tissue disorders. J Mol Med 76:226–237
Nievers MG, Schaapveld RQ, Sonnenberg A (1999) Biology and function of hemidesmosomes. Matrix Biol 18:5–17
Schumann H, Beljan G, Bruckner-Tuderman L (2001) Epidermolysis bul-losa: eine interdisziplinäre Herausforderung. Neues über Genetik, Pathophysiologie und Management. Deutsches Ärzteblatt 98:A1559–1563
Jainta S, Schmidt E, Bröcker, E-B, Zillikens D (2001) Diagnostik und Therapie bullöser Autoimmunerkrankungen der Haut. Deutsches Ärzteblatt 98:A1320–1325
Alford AI, Hankenson KD (2006) Matricellular proteins: Extracellular modulators of bone development, remodeling, and regeneration. Bone, 38:749–757
Links im Netz7 www.lehrbuch-medizin.de/biochemie