第2部(モジュール2)ctdの概要 2.6.2. 薬理試験の …...2.6.2....
TRANSCRIPT

2.6.2. 薬理試験の概要文
スイニー錠 100 mg
ベスコア錠 100 mg
第 2部(モジュール 2)CTD の概要
2.6.2. 薬理試験の概要文
株式会社三和化学研究所/興和株式会社

2.6.2. 薬理試験の概要文
目 次
2.6.2. 薬理試験の概要文 ···············································7 2.6.2.1. まとめ ······························································ 7 2.6.2.1.1. DPP-4 阻害作用(in vitro試験) ··································· 7 2.6.2.1.2. 酵素選択性 ······················································ 7 2.6.2.1.3. DPP-4 阻害作用(in vivo試験) ···································· 8 2.6.2.1.4. 糖質負荷後のGLP-1 濃度に対する作用 ······························· 8 2.6.2.1.5. 絶食時血糖値に対する作用 ········································ 8 2.6.2.1.6. 耐糖能改善作用 ·················································· 8 2.6.2.1.7. 膵β細胞に対する作用 ············································ 8 2.6.2.1.8. 代謝物及び類縁物質のDPP-4 阻害作用(in vitro試験) ··············· 9 2.6.2.1.9. 副次的薬理試験 ·················································· 9 2.6.2.1.10. 安全性薬理試験 ················································· 9 2.6.2.1.11. 薬力学的薬物相互作用試験 ······································ 11
2.6.2.2. 効力を裏付ける試験 ················································· 12 2.6.2.2.1. DPP-4 に対する阻害作用·········································· 12 2.6.2.2.2. DPP-4 に対する阻害様式·········································· 13 2.6.2.2.3. 酵素選択性 ····················································· 16 2.6.2.2.4. 正常ラットの血漿DPP-4 に対する作用 ······························ 17 2.6.2.2.5. イヌの血漿DPP-4 に対する作用···································· 19 2.6.2.2.6. 正常ラットの糖質負荷後のGLP-1 濃度に対する作用 ·················· 21 2.6.2.2.7. 正常ラットの絶食時血糖に対する作用······························ 22 2.6.2.2.8. 正常ラットのOGTTに対する作用 ··································· 23 2.6.2.2.9. Zucker fattyラットのOGTTに対する作用···························· 25 2.6.2.2.10. GKラットのOGTTに対する作用 ···································· 26 2.6.2.2.11. 膵β細胞に対する作用 ·········································· 28 2.6.2.2.12. 代謝物及び類縁物質のDPP-4 及び類縁酵素に対する阻害作用 ········· 34
2.6.2.3. 副次的薬理試験 ····················································· 36 2.6.2.3.1. 各種プロテアーゼに対する作用 ··································· 36 2.6.2.3.2. 各種受容体及びイオンチャネルに対する作用························ 36 2.6.2.3.3. T細胞及びB細胞の活性化に対する作用······························ 36
2.6.2.4. 安全性薬理試験 ····················································· 38 2.6.2.4.1. 中枢神経系に及ぼす作用 ········································· 38 2.6.2.4.2. 呼吸器系に及ぼす作用 ··········································· 38 2.6.2.4.3. 心血管系に及ぼす作用 ··········································· 39 2.6.2.4.4. 尿量,尿中電解質及び尿浸透圧に及ぼす作用························ 42 2.6.2.4.5. 腎機能に及ぼす作用 ············································· 43 2.6.2.4.6. 胃排出能に及ぼす作用 ··········································· 43
- 2 -

2.6.2. 薬理試験の概要文
2.6.2.4.7. 小腸輸送能に及ぼす作用 ········································· 43 2.6.2.4.8. 胃液分泌に及ぼす作用 ··········································· 43 2.6.2.4.9. 自律神経系に及ぼす作用 ········································· 44
2.6.2.5. 薬力学的薬物相互作用試験 ··········································· 45 2.6.2.5.1. GKラットにおけるミグリトールとの併用効果························ 45 2.6.2.5.2. ZDFラットにおけるメトホルミンとの併用効果······················· 47
2.6.2.6. 考察及び結論 ······················································· 49 2.6.2.7. 図表 ······························································· 53 2.6.2.8. 参考文献 ··························································· 54
- 3 -
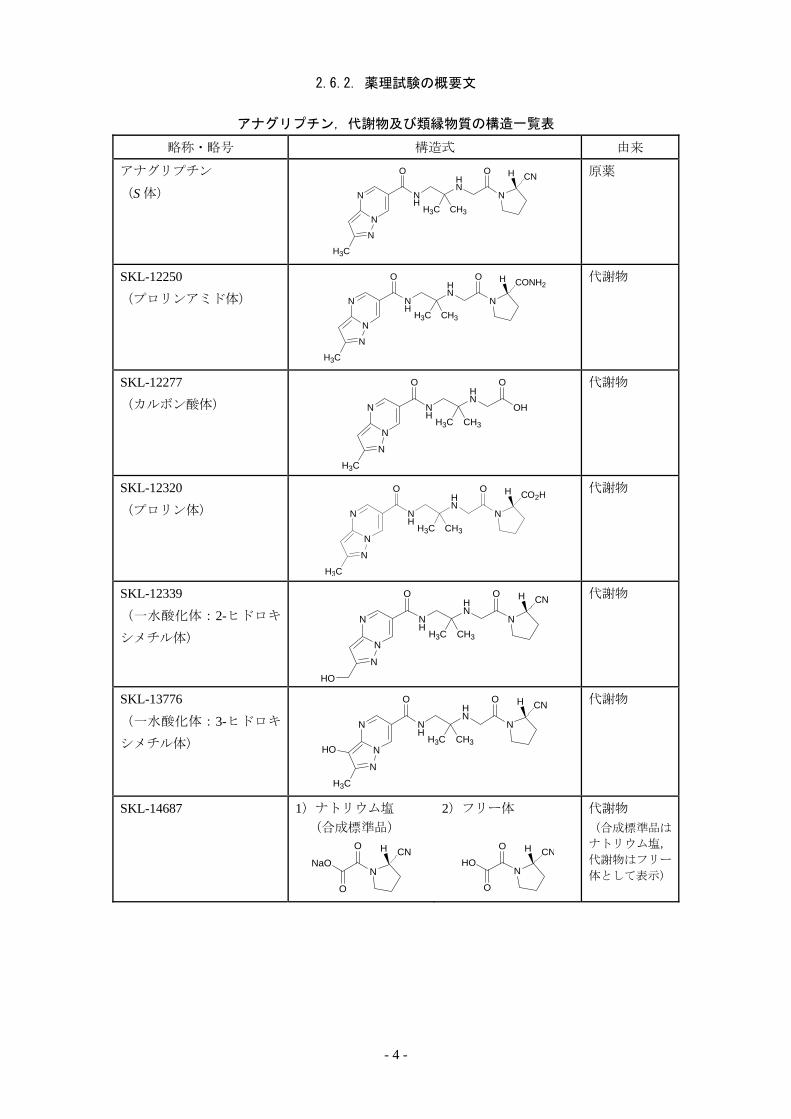
2.6.2. 薬理試験の概要文
アナグリプチン,代謝物及び類縁物質の構造一覧表
略称・略号 構造式 由来
アナグリプチン
(S 体)
原薬
SKL-12250
(プロリンアミド体)
代謝物
SKL-12277
(カルボン酸体)
代謝物
SKL-12320
(プロリン体)
代謝物
SKL-12339
(一水酸化体:2-ヒドロキ
シメチル体)
代謝物
SKL-13776
(一水酸化体:3-ヒドロキ
シメチル体)
代謝物
SKL-14687
1)ナトリウム塩 (合成標準品)
2)フリー体 代謝物 (合成標準品は
ナトリウム塩,
代謝物はフリー
体として表示)
N
HN
NH
O
CH3H3C
O
N
N
CNH
N
H3C
N
HN
NH
O
CH3H3C
O
N
N
CONH2H
N
H3C
OH
HN
NH
O
CH3H3C
O
N
N
N
H3C
N
HN
NH
O
CH3H3C
O
N
N
CO2HH
N
H3C
N
HN
NH
O
CH3H3C
O
N
N
CNH
N
HO
N
HN
NH
O
CH3H3C
O
N
N
CNH
N
H3C
HO
NHO
O CNH
O
NNaO
O CNH
O
- 4 -

2.6.2. 薬理試験の概要文
略称・略号 構造式 由来
SKL-06327 類縁物質
SKL-12309 類縁物質
SKL-13775
(エナンチオマー,R 体)
類縁物質
原薬のエナン
チオマー N
HN
NH
O
CH3H3C
O
N
N
CNH
N
H3C
- 5 -

2.6.2. 薬理試験の概要文
略号一覧表
略号 略号内容 ACE アンジオテンシン I 変換酵素 AFC 7-amino-4-trifluoromethylcoumarin α-GI 剤 α-グルコシダーゼ阻害剤 AMC 7-amino-4-methylcoumarin APA 活動電位振幅 APD 活動電位持続時間 APD30-90 APD90とAPD30との差 APDx x%再分極時活動電位持続時間 APP アミノペプチダーゼ P AUC0-t (0 から t 時間までの)濃度-時間曲線下面積 BG 剤 ビグアナイド剤 BrdU ブロモデオキシウリジン DPP ジペプチジルペプチダーゼ EDTA エチレンジアミン四酢酸 EI half-life 酵素-阻害剤複合体の半減期 ELISA Enzyme-linked immunosorbent assay FAP Fibroblast activation protein α FBS ウシ胎児血清 GIP グルコース依存性インスリン分泌刺激ポリペプチド GK ラット Goto-Kakizaki ラット GLP Good Laboratory Practice GLP-1 グルカゴン様ペプチド-1 Gly-Pro-MCA Glycyl-proline 4-methyl-coumaryl-7-amide HEK293 細胞 Human embryo kidney 293 細胞 hERG Human ether-a-go-go related gene HIT-T15 細胞 Hamster insulinoma tumor-T15 細胞 HPLC 高速液体クロマトグラフ IC50 50%阻害濃度 IC80 80%阻害濃度 ICH 日米 EU 医薬品規制調和国際会議 IK 遅延整流カリウムイオン電流 IKr 急速活性化遅延整流カリウムイオン電流 kon 結合速度定数 koff 解離速度定数 Ki 阻害定数 LAP ロイシルアミノペプチダーゼ LC/MS/MS 液体クロマトグラフ/タンデム質量分析 LPS Lipopolysaccharide OGTT 経口グルコース負荷試験 POP プロリルオリゴペプチダーゼ PQ 間隔 心電図上の P 波の始めから Q 波の始めまでの間隔 QRS 時間 Q 波の始まりから S 波の終わりまでの時間 QT 間隔 心電図上の QRS 波の始めから T 波の終わりまでの間隔 QTc 間隔 心拍数で補正した心電図 QT 間隔 RMP 静止膜電位 STZ ストレプトゾトシン Vmax 最大立ち上がり速度 ZDF ラット Zucker Diabetic Fatty ラット
- 6 -

2.6.2. 薬理試験の概要文
2.6.2. 薬理試験の概要文
2.6.2.1. まとめ
アナグリプチンの効力を裏付ける試験として,種々の in vitro 及び in vivo 試験を実施した。 In vitro 試験では,各種 DPP-4 に対する阻害作用,阻害様式及び酵素選択性を評価し,既存
の DPP-4 阻害剤の DPP-4 阻害作用及び酵素選択性と比較した。また,代謝物及びエナンチオ
マーを含む,原薬の製造工程中に混入する可能性がある類縁物質のDPP-4に対する阻害作用,
並びに主要代謝物の DPP-4 類縁酵素に対する阻害作用も評価した。In vivo 試験では正常動物
を用いて,血漿 DPP-4 活性阻害作用及び血漿 GLP-1 濃度に対する作用を評価し,絶食時血糖
値に対する作用を既存のインスリン分泌促進剤と比較した。また,正常動物及び種々の糖尿
病モデル動物を用いて耐糖能改善作用を評価した。更に,糖尿病モデル動物及びインスリノ
ーマ細胞株を用いて膵 β細胞に対する作用を評価した。 副次的薬理試験として,各種プロテアーゼ,受容体及びイオンチャネルに対する作用,並
びに T 細胞及び B 細胞の活性化に対する作用を検討した。 安全性薬理試験として,中枢神経系,呼吸器系,心血管系,腎/泌尿器系,胃腸管系及び自
律神経系に及ぼす作用を検討した。 薬力学的薬物相互作用試験として,既存の経口血糖降下剤との併用効果を評価した。
2.6.2.1.1. DPP-4 阻害作用(in vitro 試験)
アナグリプチンは,ラット,イヌ及びヒト由来のDPP-4 活性を濃度に依存して阻害した。
ラット及びイヌ血漿DPP-4 に対するIC50値は,それぞれ 5.8 及び 6.4 nmol/Lを示し,ヒト血漿,
ヒト腸管上皮細胞株Caco-2 細胞分画DPP-4 及びヒト組換えDPP-4 に対するIC50値はそれぞれ
5.4,3.5 及び 3.3 nmol/Lを示した。アナグリプチンは,いずれのDPP-4 に対してもほぼ同程度
の阻害作用を示した。アナグリプチンのDPP-4 阻害作用(IC50値=3.3~6.4 nmol/L)は類薬の
ビルダグリプチン(IC50値=2.5~4.2 nmol/L)とほぼ同程度であり,シタグリプチン(IC50値
=14.8~40.3 nmol/L)に比べて強かった。 アナグリプチンは,DPP-4 を時間及び濃度に依存して阻害し,slow-binding inhibitor であっ
た。また,DPP-4 とは可逆的に結合し,DPP-4 活性を競合的に阻害した。
2.6.2.1.2. 酵素選択性
アナグリプチンのDPP-4 類縁酵素に対するIC50値は,DPP-8 で 84.7 μmol/L,DPP-9 で 56.1 μmol/L,FAPで 72.7 μmol/L,DPP-2 で 176.7 μmol/L及びPOPで 229.2 μmol/Lであった。また, APP,prolidase,ACE及びLAPに対して阻害作用を示さなかった(IC50値>500 μmol/L)。これ
らのIC50値をヒト組換えDPP-4 に対するIC50値と比較すると,最も低い値を示したDPP-9 に対
するIC50値でも 17,000 倍となり,アナグリプチンはDPP-4 に対して高い選択性を示した。同
様に,類薬のビルダグリプチン及びシタグリプチンのDPP-4 類縁酵素に対するIC50値をヒト
組換えDPP-4 に対するIC50値と比較すると,それぞれ 440 倍(DPP-9)以上及び 5,730 倍(DPP-8)以上であった。 アナグリプチンは,ビルダグリプチン及びシタグリプチンよりも酵素選択性の高い DPP-4
阻害剤であった。
- 7 -

2.6.2. 薬理試験の概要文
2.6.2.1.3. DPP-4 阻害作用(in vivo 試験)
一晩絶食したラット及びイヌにアナグリプチン(0.3~100 mg/kg 及び 0.3~30 mg/kg)を単
回経口投与したところ,用量に依存して血漿 DPP-4 活性を阻害し,その作用は血漿中アナグ
リプチン濃度の増加に伴い増大した。 ラットにアナグリプチン(3,30 及び 100 mg/kg/日)を 1 日 1 回 6 週間反復経口投与した
ところ,用量に依存して血漿 DPP-4 活性を阻害し,投与後 2,4 及び 6 週間に初回投与時と
比べて作用の減弱は認められなかった。
2.6.2.1.4. 糖質負荷後の GLP-1 濃度に対する作用
一晩絶食したラットにアナグリプチン(3 mg/kg)を単回経口投与したところ,血漿活性型
GLP-1 濃度は経口スターチ負荷後 30 分で最高濃度に達し,対照群では負荷直前に比べて 2倍に上昇したのに対し,アナグリプチン群では 12 倍に上昇した。
2.6.2.1.5. 絶食時血糖値に対する作用
一晩絶食したラットにアナグリプチン(3 及び 30 mg/kg),ナテグリニド(50 及び 100 mg/kg)及びグリベンクラミド(1 及び 3 mg/kg)をそれぞれ単回経口投与したところ,既存のインス
リン分泌促進剤のナテグリニド及びグリベンクラミドは絶食時血漿グルコース濃度を対照群
に比べて更に低下させたが,アナグリプチンは 30 mg/kg においてもほとんど影響を及ぼさな
かった。
2.6.2.1.6. 耐糖能改善作用
正常ラットにおけるアナグリプチン(0.3~30 mg/kg)の単回経口投与は,経口グルコース
負荷後 15 分から 60 分の血漿グルコース濃度上昇を抑制し,3 mg/kg 以上で明らかな作用を
示した。また,負荷後 0 分から 120 分までの血漿グルコース濃度の AUC を減少させ,対照
群に比べて 3 mg/kg 以上で有意に抑制した。 肥満・インスリン抵抗性モデル動物 Zucker fatty ラットにおけるアナグリプチン(1,3 及
び 10 mg/kg)の単回経口投与は,経口グルコース負荷後 0 分から 120 分までの血漿インスリ
ン濃度の AUC を増加させた。また,負荷後 0 分から 120 分までの血漿グルコース濃度変化
量の AUC を用量に依存して減少させ,対照群に比べて 10 mg/kg で有意な作用を示した。 糖尿病モデル動物 GK ラットにおけるアナグリプチン(1,3 及び 10 mg/kg)の単回経口投
与は,経口グルコース負荷後 0 分から 120 分までの血漿インスリン濃度の AUC を用量に依
存して増加させ,10 mg/kg で有意な作用を示した。また,負荷後 0 分から 120 分までの血漿
グルコース濃度変化量の AUC を減少させ,対照群に比べて 3 mg/kg 以上で有意な作用を示し
た。
2.6.2.1.7. 膵β細胞に対する作用
STZ 誘発糖尿病ラットにおけるアナグリプチン(75 及び 500 μg/h)の 4 週間持続的皮下投
与は,膵臓インスリン含量及び膵 β-cell mass を用量に依存して増加させ,対照群に比べて 500 μg/h で有意な作用を示した。 アロキサン誘発糖尿病マウスにおけるアナグリプチン(0.5 又は 1.0 mg/mL)の 4 週間反復
- 8 -

2.6.2. 薬理試験の概要文
飲水投与は,0.5 mg/mL で膵臓インスリン含量を対照群に比べて有意に増加させ,1.0 mg/mLで膵 β-cell mass を対照群に比べて有意に増加させた。 アナグリプチン(0.1~10 nmol/L)は GLP-1(10 pmol/L)共存下で,ハムスター膵 β細胞
由来インスリノーマ細胞株 HIT-T15 の増殖を対照群に比べて有意に増加させた。
2.6.2.1.8. 代謝物及び類縁物質の DPP-4 阻害作用(in vitro 試験)
アナグリプチンのヒトでの主代謝物であるSKL-12320 及び動物で比較的多く認められた代
謝物のSKL-14687 は,ヒト組換えDPP-4 及び類縁酵素に対して阻害作用を示さなかった(IC50
値>500 μmol/L)。その他の代謝物のヒト組換えDPP-4 に対するIC50値は,SKL-12250 で 1,700 nmol/L,SKL-12339 で 3.1 nmol/L及びSKL-13776 で 3.4 nmol/Lを示し,SKL-12277 はDPP-4 阻
害作用を示さなかった(IC50値>500 μmol/L)。 原薬の製造工程中に混入する可能性がある類縁物質のSKL-06327, SKL-12309 及びアナグ
リプチン(S体)のエナンチオマーであるSKL-13775(R体)は,ヒト組換えDPP-4 に対して
阻害作用を示さなかった(IC50値>500 μmol/L)。
2.6.2.1.9. 副次的薬理試験
アナグリプチン及びヒトでの主代謝物である SKL-12320 は,100 及び 500 μmol/L で評価し
たいずれのプロテアーゼ(32 種類)に対しても 50%以上の阻害作用を示さなかった。また,
アナグリプチン及び SKL-12320 は,10 μmol/L で評価したいずれの受容体及びイオンチャネ
ルと特異的リガンドとの結合(60 種類)に対しても 50%以上の阻害作用を示さなかった。 アナグリプチンは,50 μmol/L で抗 CD3ε抗体刺激によるマウス脾臓由来 T 細胞の増殖及び
LPS 刺激によるマウス脾臓由来 B 細胞の増殖に対して 50%以上の阻害作用を示さなかった。
2.6.2.1.10. 安全性薬理試験
(1) ラットを用いた中枢神経系に及ぼす作用
アナグリプチン(500,1000 及び 2000 mg/kg)は,2000 mg/kg までラットの中枢神経系に
作用を及ぼさなかった。しかし,縮瞳が 1000 mg/kg 以上で認められ,末梢神経系への作用が
示唆された。
(2) ラットを用いた呼吸器系に及ぼす作用
アナグリプチン(500,1000 及び 2000 mg/kg)は,2000 mg/kg までラットの呼吸数,1 回
換気量及び分時換気量に問題となる変化を認めなかった。したがって,アナグリプチンは
2000 mg/kg までラットの呼吸機能に作用を及ぼさないと考えられた。
(3) 心血管系に及ぼす作用
1) hERG チャネル発現細胞を用いた hERG 電流に及ぼす作用
アナグリプチン(30,100 及び 300 μg/mL)は,hERG 電流に対して 100 μg/mL 以上で有意
な抑制作用を示した。30,100 及び 300 μg/mL の hERG 電流に対する阻害率は,それぞれ 2.6,12.6 及び 35.8%であった。したがって,アナグリプチンは 30 μg/mL まで hERG 電流に対して
作用を及ぼさないと考えられた。
- 9 -

2.6.2. 薬理試験の概要文
2) モルモット摘出乳頭筋を用いた心筋活動電位持続時間に及ぼす作用
アナグリプチン(30,100 及び 300 μg/mL)は,モルモット摘出乳頭筋のAPDに対して 100 μg/mLでAPD30-90相対値の高値を認め,300 μg/mLでAPA,Vmax,APD30,APD50及びAPD90相対
値の低値,並びにAPD30-90相対値の高値を認めた。したがって,アナグリプチンは 30 μg/mLまでモルモット摘出乳頭筋の活動電位に作用を及ぼさないと考えられた。
3) 無麻酔イヌを用いた心血管系に及ぼす作用
アナグリプチン(30,100 及び 300 mg/kg)は,300 mg/kg までイヌの血圧及び心拍数に作
用を及ぼさなかった。30 及び 100 mg/kg は QRS 時間に作用を及ぼさなかったが,300 mg/kgは可逆的な QRS 時間の延長を認めた。また,いずれの用量も PQ 間隔,QT 間隔及び QTc 間隔に作用を及ぼさなかった。したがって,アナグリプチンは 100 mg/kg まで無麻酔イヌの心
血管系に作用を及ぼさないと考えられた。
(4) ラットを用いた腎/泌尿器系に及ぼす作用
アナグリプチン(200,600 及び 2000 mg/kg)は,600 及び 2000 mg/kg でラットの尿量の増
加,並びに尿中ナトリウム,カリウム及び塩素排泄量の増加を認めた。200 mg/kg において
も,軽度ながら尿中ナトリウム及びカリウム排泄量の増加を認めた。いずれの用量も尿浸透
圧に変化を認めなかった。 アナグリプチン(200,600 及び 2000 mg/kg)は,2000 mg/kg までラットの濾過率に影響を
及ぼさなかったが,600 mg/kg 以上で糸球体濾過量の低下を認め,2000 mg/kg で腎血漿流量
の低下を認めた。
(5) ラットを用いた胃腸管系に及ぼす作用
アナグリプチン(200,600 及び 2000 mg/kg)は,600 及び 2000 mg/kg でラットの胃排出率
の低下を認めた。また,2000 mg/kg で小腸輸送能の抑制及び pH の上昇に伴うペプシン活性
の低下傾向を認めたが,胃液分泌量及び総酸排出量には作用を及ぼさなかった。
(6) モルモット摘出回腸を用いた自律神経系に及ぼす作用
アナグリプチン(38.3,115 及び 383 μg/mL)は,モルモット摘出回腸のアゴニストによる
収縮反応に対して,115 及び 383 μg/mL でヒスタミン収縮高比率の低下作用を示した。アセ
チルコリン収縮高比率の低下作用も 383 μg/mL で認められたが,塩化バリウム及びセロトニ
ン収縮に対する作用,並びにアナグリプチン単独での作用は認められなかった。 (7) 代謝物の安全性薬理試験(hERG 電流に及ぼす作用)
ヒトでの主代謝物である SKL-12320(31.5,105 及び 315 μg/mL)は,いずれの濃度におい
ても hERG 電流に有意な作用を及ぼさなかった。また,31.5,105 及び 315 μg/mL の hERG 電
流に対する阻害率は,それぞれ 1.4,0.0 及び 4.4%であった。したがって,SKL-12320 は 315 μg/mL まで hERG 電流に対して作用を及ぼさず,その作用はアナグリプチンよりも弱いもの
と考えられた。
- 10 -

2.6.2. 薬理試験の概要文
2.6.2.1.11. 薬力学的薬物相互作用試験
GK ラットにおいて,アナグリプチン(3 mg/kg)と α-GI 剤であるミグリトール(3 mg/kg)との併用投与は,経口液体流動食負荷後 0 分から 120 分までの血漿活性型 GLP-1 濃度変化量
の AUC をそれぞれの単独投与に比べて増加させ,アナグリプチンの単独投与の約 2.4 倍高値
を示した。一方,負荷後 0 分から 120 分までの血漿グルコース濃度及び血漿インスリン濃度
の AUC には,それぞれの単独投与との差は認められなかった。 肥満・糖尿病モデル動物の ZDF ラットにおいて,アナグリプチン(3 mg/kg)と BG 剤で
あるメトホルミン(300 mg/kg)との併用投与は,経口スターチ負荷後 0 分から 180 分までの
血漿活性型 GLP-1 濃度変化量の AUC は,それぞれの単独投与の約 4~5 倍高値を示したもの
の,有意な増加ではなかった。また,負荷後 0 分から 180 分までの血漿 GIP 濃度の AUC を
メトホルミン単独投与に比べて減少させたが,アナグリプチンの単独投与との差は認められ
なかった。更に,負荷後 0 分から 180 分までの血漿グルコース濃度の AUC を血漿インスリ
ン濃度の AUC の増加を伴うことなく,アナグリプチンの単独投与に比べて減少させた。
- 11 -

2.6.2. 薬理試験の概要文
2.6.2.2. 効力を裏付ける試験
2.6.2.2.1. DPP-4 に対する阻害作用[資料 4.2.1.1-1,4.2.1.1-2,4.2.1.1-3]
アナグリプチンの DPP-4 阻害作用について,ラット,イヌ及びヒト血漿 DPP-4,ヒト腸管
上皮細胞株 Caco-2 細胞分画 DPP-4,並びにヒト組換え DPP-4 を用いて評価した。また,類薬
のビルダグリプチン( )及びシタグリプチン ( ,以下,シタグリ
プチンとする)の DPP-4 阻害作用と比較した。 DPP-4 活性は,合成基質であるGly-Pro-MCAを用い,DPP-4 により切断され遊離した蛍光
物質AMCの濃度を定量することにより測定した。アナグリプチン,ビルダグリプチン又はシ
タグリプチンに各種DPP-4を加えて室温で30分間反応させた。Gly-Pro-MCAを加えて混和し,
室温で 20 分間反応させた後,酢酸溶液を添加して反応を停止し,励起波長 390 nm及び蛍光
波長 460 nmにおける蛍光強度を測定した。IC50値は,アナグリプチン,ビルダグリプチン及
びシタグリプチン濃度に対してDPP-4 活性をプロットした阻害曲線から,ロジスティック回
帰分析を行って算出した。 アナグリプチンは,ラット,イヌ及びヒト血漿DPP-4,Caco-2 細胞分画DPP-4,並びにヒト
組換えDPP-4 活性を濃度に依存して阻害した。算出したIC50値を表 2.6.2.2-1に示した。 アナグリプチンのラット及びイヌ血漿DPP-4 に対するIC50値は,それぞれ 5.8 nmol/L及び
6.4 nmol/Lであった。また,ヒト血漿,Caco-2 細胞分画DPP-4 及びヒト組換えDPP-4 に対する
IC50値は,それぞれ 5.4 nmol/L,3.5 nmol/L及び 3.3 nmol/Lであった。アナグリプチンは,いず
れのDPP-4 に対してもほぼ同程度の阻害作用を示した。 アナグリプチンのDPP-4 阻害作用(IC50値=3.3~6.4 nmol/L)はビルダグリプチン(IC50値
=2.5~4.2 nmol/L)とほぼ同程度であり,シタグリプチン(IC50値=14.8~40.3 nmol/L)に比べ
て強かった。 表 2.6.2.2-1 ラット,イヌ及びヒト由来 DPP-4 に対するアナグリプチン,ビルダ
グリプチン並びにシタグリプチンの阻害作用
IC50 (nmol/L) DPP-4 由来
アナグリプチン ビルダグリプチン シタグリプチン ラット血漿 5.8 ± 0.4 3.0 ± 0.7 40.3 ± 5.3 イヌ血漿 6.4 ± 0.1 4.2 ± 0.1 17.9 ± 1.2 ヒト血漿 5.4 ± 0.2 3.7 ± 0.1 21.5 ± 1.1
Caco-2 細胞分画 3.5 ± 0.3 2.6 ± 0.3 14.9 ± 0.9 ヒト組換え 3.3 ± 0.3 2.5 ± 0.3 14.8 ± 0.6
平均値±標準偏差(n=3) [資料 4.2.1.1-1,4.2.1.1-2,4.2.1.1-3 より引用,改変]
- 12 -
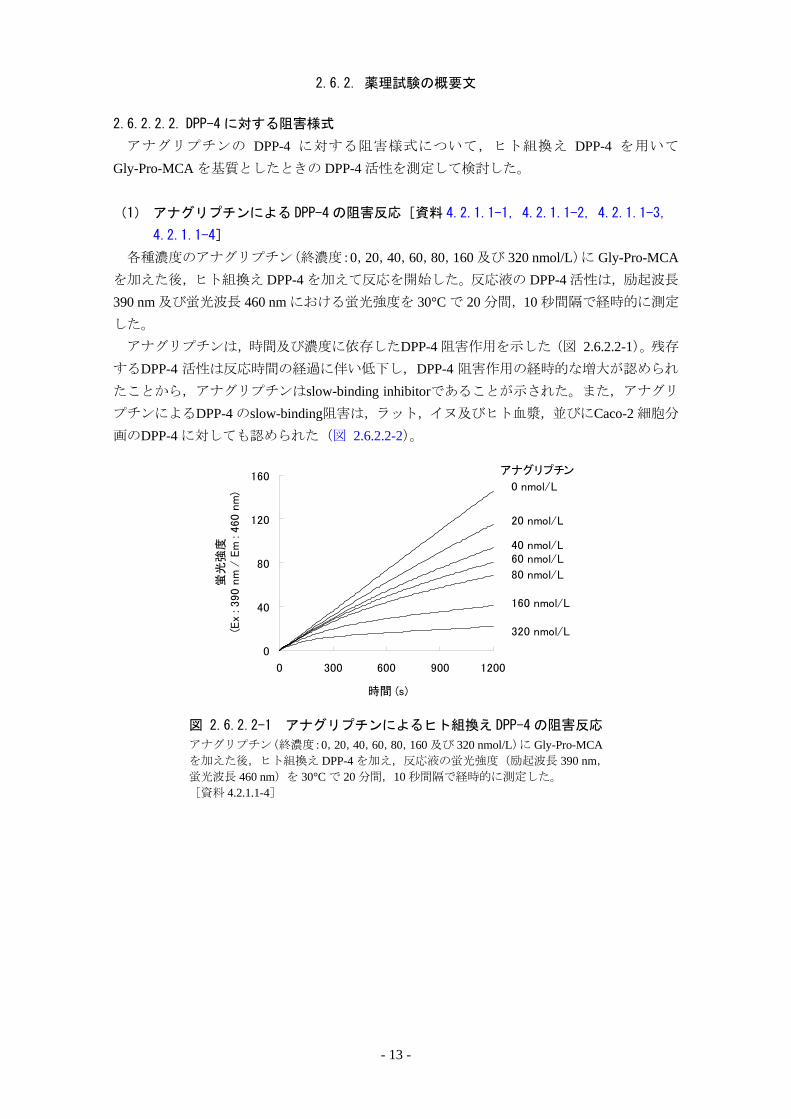
2.6.2. 薬理試験の概要文
2.6.2.2.2. DPP-4 に対する阻害様式
アナグリプチンの DPP-4 に対する阻害様式について,ヒト組換え DPP-4 を用いて
Gly-Pro-MCA を基質としたときの DPP-4 活性を測定して検討した。
(1) アナグリプチンによる DPP-4 の阻害反応[資料 4.2.1.1-1,4.2.1.1-2,4.2.1.1-3,
4.2.1.1-4]
各種濃度のアナグリプチン(終濃度:0,20,40,60,80,160 及び 320 nmol/L)に Gly-Pro-MCAを加えた後,ヒト組換え DPP-4 を加えて反応を開始した。反応液の DPP-4 活性は,励起波長
390 nm 及び蛍光波長 460 nm における蛍光強度を 30°C で 20 分間,10 秒間隔で経時的に測定
した。 アナグリプチンは,時間及び濃度に依存したDPP-4 阻害作用を示した(図 2.6.2.2-1)。残存
するDPP-4 活性は反応時間の経過に伴い低下し,DPP-4 阻害作用の経時的な増大が認められ
たことから,アナグリプチンはslow-binding inhibitorであることが示された。また,アナグリ
プチンによるDPP-4 のslow-binding阻害は,ラット,イヌ及びヒト血漿,並びにCaco-2 細胞分
画のDPP-4 に対しても認められた(図 2.6.2.2-2)。
0
40
80
120
160
0 300 600 900 1200
時間 (s)
蛍光
強度
(Ex
: 390 n
m /
Em
: 460 n
m)
80 nmol/L
160 nmol/L
320 nmol/L
60 nmol/L
アナグリプチン
40 nmol/L
20 nmol/L
0 nmol/L
図 2.6.2.2-1 アナグリプチンによるヒト組換え DPP-4 の阻害反応
アナグリプチン(終濃度:0,20,40,60,80,160 及び 320 nmol/L)に Gly-Pro-MCAを加えた後,ヒト組換え DPP-4 を加え,反応液の蛍光強度(励起波長 390 nm,
蛍光波長 460 nm)を 30°C で 20 分間,10 秒間隔で経時的に測定した。 [資料 4.2.1.1-4]
- 13 -

2.6.2. 薬理試験の概要文
a) ラット血漿 b) イヌ血漿
c) ヒト血漿 d) Caco-2細胞分画
0
50000
100000
150000
200000
250000
300000
350000
0 20 40 60
時間 (min)
蛍光
強度
(Ex
: 390 n
m /
Em
: 4
60 n
m) 0 nmol/L
30 nmol/L100 nmol/L300 nmol/L
アナグリプチン
0
50000
100000
150000
200000
250000
300000
350000
0 20 40
時間 (min)
蛍光
強度
(Ex
: 390
nm
/ E
m : 4
60 n
m)
60
0 nmol/L30 nmol/L100 nmol/L300 nmol/L
アナグリプチン
0
50000
100000
150000
200000
250000
300000
350000
0 20 40
時間 (min)
蛍光
強度
(Ex
: :3
90 n
m /
Em
: 4
60 n
m)
60
0 nmol/L30 nmol/L100 nmol/L300 nmol/L
アナグリプチン
0
50000
100000
150000
200000
250000
300000
350000
0 20 40 60
時間 (min)
蛍光
強度
(Ex
: 390 n
m /
Em
: 4
60 n
m) 0 nmol/L
30 nmol/L100 nmol/L300 nmol/L
アナグリプチン
図 2.6.2.2-2 アナグリプチンによるラット,イヌ,ヒト血漿及び Caco-2 細胞
分画 DPP-4 の阻害反応
アナグリプチン(終濃度:0,30,100 及び 300 nmol/L)に Gly-Pro-MCA を加えた後,血漿(ラ
ット,イヌ,ヒト)又は Caco-2 細胞分画 DPP-4 を加え,反応液の蛍光強度(励起波長 390 nm,
蛍光波長 460 nm)を 30°C で 60 分間,5 分間隔で経時的に測定した。 [資料 4.2.1.1-1,4.2.1.1-2,4.2.1.1-3 より引用,改変]
(2) アナグリプチンと DPP-4 との酵素-阻害剤複合体の解離[資料 4.2.1.1-4]
アナグリプチンと DPP-4 との酵素-阻害剤複合体は,アナグリプチンの終濃度が 300 nmol/Lとなるようにアナグリプチンにヒト組換え DPP-4 を加え,30°C で 30 分間反応させて調製し
た。この酵素-阻害剤複合体を,アナグリプチン濃度が 1 nmol/L となるように Gly-Pro-MCAを含む緩衝液に加えて 301 倍希釈し,複合体の解離反応を開始した。反応液の DPP-4 活性は,
励起波長 390 nm 及び蛍光波長 460 nm における蛍光強度を 30°C で 2 時間,20 秒間隔で経時
的に測定した。対照として,アナグリプチンの代わりに精製水を用いて同様に操作し,反応
液の DPP-4 活性を測定した。 希釈した酵素-阻害剤複合体のDPP-4 活性は,時間の経過に伴い回復したことから,アナグ
リプチンとDPP-4 との結合は可逆的であることが示された(図 2.6.2.2-3)。
- 14 -

2.6.2. 薬理試験の概要文
0
100
200
300
400
0 1200 2400 3600 4800 6000 7200
時間 (s)
蛍光
強度
(Ex
: 390 n
m /
Em
: 460 n
m)
アナグリプチン
1 nmol/L
0 nmol/L
図 2.6.2.2-3 アナグリプチンとヒト組換え DPP-4 との複合体の解離
アナグリプチンとヒト組換えDPP-4との複合体は,アナグリプチン濃度が1 nmol/Lとなるように Gly-Pro-MCA を含む緩衝液で 301 倍希釈し,反応液の蛍光強度(励
起波長 390 nm,蛍光波長 460 nm)を 30°C で 2 時間,20 秒間隔で経時的に測定し
た。 また,定常状態にある DPP-4 とアナグリプチンとの酵素-阻害剤複合体の DPP-4 活性を測
定して,阻害様式を検討した。各種濃度のアナグリプチン(終濃度:0,2.5,5,10 及び 40 nmol/L)にヒト組換え DPP-4 を加え,30°C で 30 分間加温した後,各種濃度の Gly-Pro-MCA(終濃度:
25,50,100,200,400 及び 600 μmol/L)を加えて混和した。室温で 20 分間反応させた後,
酢酸溶液を添加して反応を停止し,励起波長 390 nm 及び蛍光波長 460 nm の蛍光強度を測定
した。 酵素-阻害剤複合体のDPP-4 活性は,いずれのアナグリプチン濃度においても基質濃度の増
加に応じて上昇した(表 2.6.2.2-2)。DPP-4 の活性部位において,アナグリプチンとDPP-4との結合が,基質であるGly-Pro-MCAによって置換されたものと考えられたことから,アナ
グリプチンはDPP-4 を競合的に阻害することが示された。
表 2.6.2.2-2 アナグリプチンと DPP-4 の結合に対する Gly-Pro-MCA 濃度の影響
DPP-4 活性(%) アナグリプチン濃度(nmol/L) 基質濃度
(μmol/L) 2.5 5 10 40
25 84.7 71.1 53.9 24.0 50 84.6 71.8 56.2 27.5 100 86.0 73.6 59.2 33.0 200 86.7 75.2 62.5 39.0 400 87.2 76.8 64.7 44.5 600 87.7 77.1 65.8 47.0
平均値(n=3) DPP-4 活性(%)は,各基質濃度におけるアナグリプチンを含まない場合の DPP-4 活性に対する
百分率(残存率%)として算出した。
- 15 -

2.6.2. 薬理試験の概要文
(3) アナグリプチンによる DPP-4 阻害の反応速度論的解析[資料 4.2.1.1-4]
アナグリプチンによるDPP-4 阻害反応について,競合的なslow-binding inhibitorの反応速度
論モデルに関する反応速度式[1]を用いて解析を行った。アナグリプチンとDPP-4 の結合及び
解離反応における時間‐反応曲線(図 2.6.2.2-1, 図 2.6.2.2-3)の非線形回帰分析を行って,
反応速度論パラメータとして結合速度定数(kon),解離速度定数(koff),阻害定数(Ki)及び
DPP-4 とアナグリプチンの酵素-阻害剤複合体の半減期(EI half-life)を算出した。 アナグリプチンのDPP-4 に対するKiは 7.1 nmol/L,konは 1.04 × 105 (mol/L-1)s-1,koffは 7.36 ×
10-4 s-1及びEI half-lifeは 15.7 minであった(表 2.6.2.2-3)。
表 2.6.2.2-3 アナグリプチンによる DPP-4 阻害の反応速度論パラメータ
kon((mol/L-1)s-1)
koff(s-1)
Ki(nmol/L)
EI half-life (min)
アナグリプチン 1.04 × 105 7.36 × 10-4 7.1 15.7 平均値(n=2) Ki及びEI half-lifeは次式に従って算出した: Ki = koff/kon,EI half-life=0.693/koff
2.6.2.2.3. 酵素選択性[資料 4.2.1.1-5,4.2.1.1-6]
DPP-4 と類似した基質特異性を有するプロリン特異性ペプチダーゼに対するアナグリプチ
ンの阻害作用を評価した。また,ビルダグリプチン及びシタグリプチンの阻害作用と比較し
た。 DPP-4 類縁酵素として,ヒト組換えDPP-8,DPP-9 及びFAP,Caco-2 細胞分画DPP-2,微生
物由来POP,ヒト血漿APP,並びにブタ腎臓由来prolidase,ACE及びLAPを用いた。基質とし
て,DPP-8 及びDPP-9 活性はGly-Pro-MCA,FAP活性はAla-Pro-AFC,DPP-2 活性はLys-Ala-MCA,
POP活性はZ-Gly-Pro-MCA,APP活性はLys(Abz)-Pro-Pro-pNA,prolidase活性はGly-Pro,ACE活性はFA-Phe-Gly-Gly(FAPGG)及びLAP活性はLeu-MCAを用いた。アナグリプチン,ビル
ダグリプチン又はシタグリプチンに各基質を加えて混和し,各酵素を添加した。DPP-8,DPP-9,DPP-2,POP及びLAPは室温で 20 分間,FAPは室温で 60 分間反応させた後に反応を停止し,
生成したAMC量又はAFC量を蛍光法により測定した。APPは室温で 10 分間及び 130 分間反
応させた後,生成したLys(Abz)-Pro-Pro量を蛍光法により測定した。prolidaseは 37°Cで 30 分
間反応させた後,生成したプロリン量を吸光度法により測定した。ACEは 37°Cで 1 分間及び
15 分間反応させた後,分解されたFAPGG量を吸光度法により測定した。IC50値は,各被験物
質濃度及び酵素活性をプロットした阻害曲線から,ロジスティック回帰分析を行って算出し
た。なお,被験物質の濃度を 500 μmol/Lとした場合の酵素活性値が 50%以上のとき,IC50値
は 500 μmol/Lを超える(>500 μmol/L)と判断した。 アナグリプチンのDPP-8 に対するIC50値は 84.7 μmol/L,DPP-9 では 56.1 μmol/L,FAPでは
72.7 μmol/L,DPP-2 では 176.7 μmol/L及びPOPでは 229.2 μmol/Lであった(表 2.6.2.2-4)。ま
た,アナグリプチンはAPP,prolidase,ACE及びLAPに対して阻害作用を示さなかった(IC50
値>500 μmol/L)。 DPP-4 類縁酵素に対するアナグリプチンのIC50値は,いずれも 50 μmol/L以上であった。各
酵素に対するIC50値をヒト組換えDPP-4 に対するIC50値(3.3 nmol/L,表 2.6.2.2-1)を 1 とし
て比較すると,最も低い値を示したDPP-9 に対するIC50値でも 17,000 倍であった。同様に,
- 16 -

2.6.2. 薬理試験の概要文
ビルダグリプチン及びシタグリプチンのDPP-4 類縁酵素に対するIC50値をヒト組換えDPP-4に対するIC50値(それぞれ 2.5 nmol/L及び 14.8 nmol/L,表 2.6.2.2-1)と比較すると,ビルダ
グリプチンではDPP-9 に対するIC50値が 440 倍,シタグリプチンではDPP-8 に対するIC50値が
5,730 倍であった。アナグリプチンは酵素選択性が高いDPP-4 阻害剤であった。
表 2.6.2.2-4 DPP-4 類縁酵素に対するアナグリプチン,ビルダグリプチン並びに
シタグリプチンの阻害作用
IC50 (μmol/L)
アナグリプチン ビルダグリプチン シタグリプチン DPP-8 84.7 ± 9.8 [25,700] 6.4 ± 0.8 [2,560] 84.8 ± 11.6 [5,730] DPP-9 56.1 ± 5.3 [17,000] 1.1 ± 0.1 [440] 299.4 ± 17.1 [20,200]FAP 72.7 ± 1.2 [22,000] 54.6 ± 0.5 [21,800] >500
DPP-2 176.7 ± 16.1 [53,500] >500 129.3 ± 4.5 [8,740] POP 229.2 ± 31.8 [69,500] >500 >500 APP >500 >500 >500
Prolidase >500 >500 >500 ACE >500 >500 >500 LAP >500 >500 >500
平均値±標準偏差(n=3,ただしIC50>500 μmol/Lの場合はn=1) [ ]内の値は各IC50値をヒト組換えDPP-4 に対するIC50値を 1 として比較した相対強度とし,次式に
従って算出した:相対強度=DPP-4 類縁酵素に対するIC50 /ヒト組換えDPP-4 に対するIC50
各酵素活性はそれぞれ以下の基質を用いて測定した。 ・ DPP-8 及び DPP-9: Glycyl-proline 4-methyl-coumaryl-7-amide(Gly-Pro-MCA) ・ FAP: Alanyl-proline 7-amido-4-trifluoromethylcoumarin(Ala-Pro-AFC) ・ DPP-2: Lysyl-alanine 4-methyl-coumaryl-7-amide(Lys-Ala-MCA) ・ POP: N-Benzyloxycarbonyl-glycyl-proline 4-methyl-coumaryl-7-amide(Z-Gly-Pro-MCA) ・ APP: H-Lysyl(2-aminobenzoyl)-prolyl-proline p-nitroanilide(Lys(Abz)-Pro-Pro-pNA) ・ Prolidase: Glycyl-proline(Gly-Pro) ・ ACE: 3-(2-Furyl)acryloyl-phenylalanyl-glycyl-glycine(FA-Phe-Gly-Gly = FAPGG) ・ LAP: Leucine 4-methyl-coumaryl-7-amide(Leu-MCA) [資料 4.2.1.1-5,4.2.1.1-6 より引用,改変]
2.6.2.2.4. 正常ラットの血漿 DPP-4 に対する作用
(1) 単回投与後の DPP-4 活性に対する作用[資料 4.2.1.1-7]
頸静脈に採血用のカテーテルを留置した雄性 Wistar 系ラットを用いた。一晩絶食したラッ
ト(各群 7 例)にアナグリプチン(0.3~100 mg/kg)又は対照として精製水を単回経口投与
し,薬物投与前並びに投与後 0.25,0.5,1,2,4,6,9,12,16 及び 24 時間にカテーテルよ
り経時的に採血した。血漿 DPP-4 活性は合成基質(Gly-Pro-MCA)を用いた蛍光法にて測定
した。 アナグリプチンは,ラット血漿DPP-4 活性を投与後 15 分から 24 時間まで用量に依存して
阻害した(図 2.6.2.2-4)。また,血漿DPP-4 活性抑制効果の持続性も用量に依存して認められ,
3 mg/kgで投与後 2 時間まで 80%以上,10 及び 30 mg/kgで 12 時間まで 50%以上,100 mg/kgで 24 時間まで 50%以上それぞれ抑制した。
- 17 -

2.6.2. 薬理試験の概要文
0
20
40
60
80
100
120
140
160
0 4 8 12 16 20 24
投与後の時間(h)
血漿
DP
P-4活
性(%
)
対照群
0.3 mg/kg
1 mg/kg
3 mg/kg
10 mg/kg
30 mg/kg
100 mg/kg
図 2.6.2.2-4 アナグリプチン投与後のラット血漿 DPP-4 活性の経時的推移
平均値±標準偏差(n=7) (2) DPP-4 活性阻害と薬物濃度との関係[資料 4.2.1.1-8,4.2.1.1-9]
頸静脈に採血用のカテーテルを留置した雄性 Wistar 系ラットを用いた。一晩絶食したラッ
ト(各群 4 又は 5 例)にアナグリプチン(1~100 mg/kg)又は対照として精製水を単回経口
投与し,薬物投与前並びに投与後 0.25,0.5,1,2,4,6,9,12,16 及び 24 時間にカテーテ
ルより経時的に採血した。血漿 DPP-4 活性は合成基質を用いた蛍光法で,血漿中アナグリプ
チン濃度は LC/MS/MS を用いてそれぞれ測定し,血漿 DPP-4 活性と血漿中アナグリプチン濃
度との関係を検討した。 アナグリプチン投与後のラット血漿DPP-4 活性は,血漿中アナグリプチン濃度の上昇に伴
い低下する傾向が認められ,血漿DPP-4 活性に対する血漿中アナグリプチン濃度のIC50値及
びIC80値は,それぞれ 25.7 nmol/L(9.85 ng/mL)及び 81.4 nmol/L(31.2 ng/mL)であった(図 2.6.2.2-5)。
0
50
100
150
200
0.1 1 10 100 1000 10000 100000 1000000
血漿中アナグリプチン濃度(nmol/L)
血漿
DP
P-4活
性(%
)_
実測値
回帰
IC50(nmol/L) = 25.7 [23.0-28.7]
IC80(nmol/L) = 81.4 [68.8-96.3]
図 2.6.2.2-5 血漿 DPP-4 活性と血漿中アナグリプチン濃度との関係(ラット)
図中の[ ]内の数字は 95%信頼区間を示す。[資料 4.2.1.1-8]
- 18 -

2.6.2. 薬理試験の概要文
(3) 反復投与による血漿 DPP-4 活性阻害作用[資料 4.2.1.1-10]
雄性 Wistar 系ラット(各群 8 例)にアナグリプチン(3,30 及び 100 mg/kg/日)又は対照
として精製水を 1 日 1 回 6 週間反復経口投与した。投与初日より 2 週間ごとに一晩絶食した
ラットの尾部より薬物投与直前,並びに投与後 0.5,1,2 及び 6 時間に経時的に採血し,血
漿 DPP-4 活性を,合成基質を用いた蛍光法にて測定した。 アナグリプチンは,初回の経口投与において,3 mg/kgからラット血漿DPP-4 活性を 80%以
上阻害し,投与後 30 分から 6 時間まで用量に依存して阻害した。また,反復投与 2,4 及び
6 週後においてもほぼ同等の血漿DPP-4 活性阻害作用を示し,すべての用量において反復投
与による阻害作用の減弱は認められなかった(図 2.6.2.2-6)。
対照群
0
20
40
60
80
100
120
140
0 2 4 6
投与後の時間(h)
血漿
DP
P-4活
性(%
)
0週2週4週6週
アナグリプチン 3 mg/kg
0
20
40
60
80
100
120
140
0 2 4 6
投与後の時間(h)
血漿
DP
P-4活
性(%
)
0週2週4週6週
アナグリプチン 30 mg/kg
0
20
40
60
80
100
120
140
0 2 4 6
投与後の時間(h)
血漿
DP
P-4活
性(%
)
0週2週4週6週
アナグリプチン 100 mg/kg
0
20
40
60
80
100
120
140
0 2 4 6
投与後の時間(h)
血漿
DP
P-4活
性(%
)
0週2週4週6週
図 2.6.2.2-6 反復経口投与後のラット血漿 DPP-4 活性の経時的推移
平均値±標準偏差(n=8) 2.6.2.2.5. イヌの血漿 DPP-4 に対する作用[資料 4.2.1.1-11,4.2.1.1-12]
一晩絶食した雄性ビーグル犬(各群 4 例)にアナグリプチン(0.3~30 mg/kg)又は対照と
して注射用蒸留水を単回経口投与し,薬物投与前,並びに投与後 0.25,0.5,1,2,4,6,9,12,16,24,36 及び 48 時間に橈側皮静脈より経時的に採血した。血漿 DPP-4 活性は合成基
質を用いた蛍光法で,血漿中アナグリプチン濃度は LC/MS/MS を用いてそれぞれ測定した。 アナグリプチンは,イヌ血漿DPP-4 活性を投与後 15 分から 48 時間まで用量に依存して阻
- 19 -
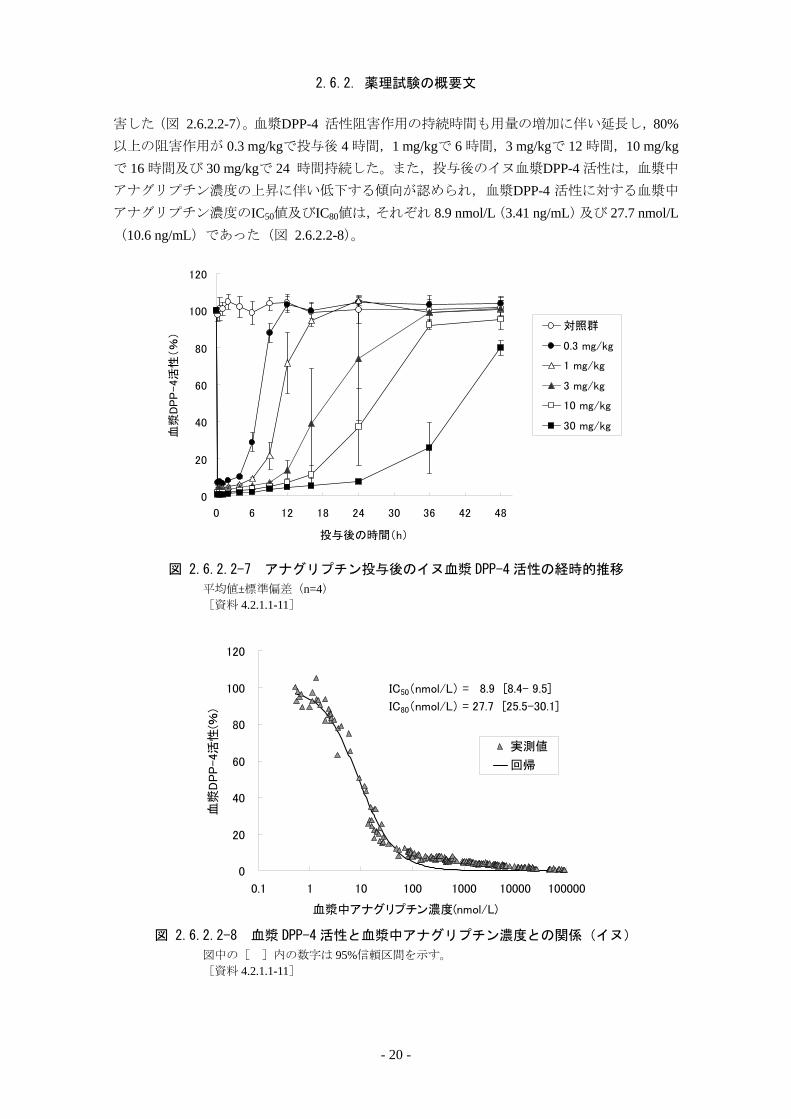
2.6.2. 薬理試験の概要文
害した(図 2.6.2.2-7)。血漿DPP-4 活性阻害作用の持続時間も用量の増加に伴い延長し,80%以上の阻害作用が 0.3 mg/kgで投与後 4 時間,1 mg/kgで 6 時間,3 mg/kgで 12 時間,10 mg/kgで 16 時間及び 30 mg/kgで 24 時間持続した。また,投与後のイヌ血漿DPP-4 活性は,血漿中
アナグリプチン濃度の上昇に伴い低下する傾向が認められ,血漿DPP-4 活性に対する血漿中
アナグリプチン濃度のIC50値及びIC80値は,それぞれ 8.9 nmol/L(3.41 ng/mL)及び 27.7 nmol/L(10.6 ng/mL)であった(図 2.6.2.2-8)。
0
20
40
60
80
100
120
0 6 12 18 24 30 36 42 48
投与後の時間(h)
血漿
DP
P-4活
性(%
)
対照群
0.3 mg/kg
1 mg/kg
3 mg/kg
10 mg/kg
30 mg/kg
図 2.6.2.2-7 アナグリプチン投与後のイヌ血漿 DPP-4 活性の経時的推移
平均値±標準偏差(n=4) [資料 4.2.1.1-11]
0
20
40
60
80
100
120
0.1 1 10 100 1000 10000 100000
血漿中アナグリプチン濃度(nmol/L)
血漿
DP
P-4活
性(%
)_
実測値
回帰
IC50(nmol/L) = 8.9 [8.4- 9.5]
IC80(nmol/L) = 27.7 [25.5-30.1]
図 2.6.2.2-8 血漿 DPP-4 活性と血漿中アナグリプチン濃度との関係(イヌ)
図中の[ ]内の数字は 95%信頼区間を示す。 [資料 4.2.1.1-11]
- 20 -

2.6.2. 薬理試験の概要文
2.6.2.2.6. 正常ラットの糖質負荷後の GLP-1 濃度に対する作用[資料 4.2.1.1-13]
頸静脈に採血用のカテーテルを留置した雄性 Wistar 系ラットを用いた。一晩絶食したラッ
ト(各群 8 例)にアナグリプチン(3 mg/kg)又は対照として精製水を単回経口投与し,30分後に精製水で懸濁させた 2 g/kg のスターチを経口負荷した。薬物投与直前(スターチ負荷
前 30 分),スターチ負荷直前(0 分),並びにスターチ負荷後 10,20,30,60,90 及び 120分にカテーテルより経時的に採血した。血漿活性型 GLP-1 濃度は ELISA 法で,血漿グルコ
ース濃度は酵素法にて,血漿 DPP-4 活性は合成基質を用いた蛍光法にてそれぞれ測定した。 スターチ負荷直前(0 分,薬物投与後 30 分)の対照群及びアナグリプチン群の血漿活性型
GLP-1 濃度は,それぞれ 2.7 ± 3.3 pmol/L及び 1.4 ± 1.6 pmol/Lであり,2 群間に差は認められ
なかった。スターチ負荷後の血漿活性型GLP-1 濃度は,対照群及びアナグリプチン群とも負
荷後 30 分で最高濃度に達し,その後低下した。スターチ負荷後 30 分の血漿活性型GLP-1 濃
度は対照群で 5.6 ± 5.6 pmol/L,アナグリプチンで 16.1 ± 9.4 pmol/Lであり,負荷直前の濃度と
比較すると,対照群では 2 倍とわずかな増加しか見られなかったのに対して,アナグリプチ
ンでは 12 倍に増加した(図 2.6.2.2-9)。 アナグリプチンは,スターチ負荷直前の血漿グルコース濃度に影響を与えなかったが,負
荷後 20,30,60 及び 120 分の血漿グルコース濃度上昇を対照群に比べて有意に抑制した(図 2.6.2.2-10)。また,スターチ負荷後 0 分から 120 分(薬物投与後 30 分から 150 分)の血漿DPP-4活性を 80%以上阻害した。
-5
0
5
10
15
20
25
30
-30 0 30 60 90 120
スターチ負荷後の時間(min)
血漿
活性
型G
LP
-1濃
度(pm
ol/
L)
対照群
アナグリプチン
*
図 2.6.2.2-9 経口スターチ負荷後のラット血漿活性型 GLP-1 濃度の経時的推移
平均値±標準偏差(n=8) *:p<0.05;対照群との有意差(t 検定)
- 21 -

2.6.2. 薬理試験の概要文
50
100
150
200
250
-30 0 30 60 90 120
スターチ負荷後の時間(min)
グル
コー
ス濃
度(m
g/dL
)
対照群
アナグリプチン
* ***
*
図 2.6.2.2-10 経口スターチ負荷後のラット血漿グルコース濃度の経時的推移
平均値±標準偏差(n=8) *:p<0.05,**:p<0.01;対照群との有意差(t 検定)
2.6.2.2.7. 正常ラットの絶食時血糖に対する作用[資料 4.2.1.1-14]
一晩絶食した雄性 Wistar 系ラット(各群 8 例)にアナグリプチン(3,30 mg/kg),ナテグ
リニド(50,100 mg/kg),グリベンクラミド(1,3 mg/kg)又は対照として 0.5%カルボキシ
メチルセルロースナトリウム溶液を単回経口投与した。薬物投与前,並びに投与後 15,30,60,90,120 及び 180 分に尾部より経時的に採血し,血漿グルコース濃度を酵素法にて測定
した。 アナグリプチンは,30 mg/kgで対照群に比べて投与後 15 分にわずかな低下が認められた以
外,絶食時血漿グルコース濃度にほとんど影響を及ぼさなかった。一方,ナテグリニドは,
50及び 100 mg/kgで投与後 15分から絶食時血漿グルコース濃度の有意な一過性の低下が用量
に依存して認められ,投与後 60 分にそれぞれ最低値(83 及び 65 mg/dL)を示した。また,
グリベンクラミドは,1 及び 3 mg/kgで投与後 60 分まで絶食時血漿グルコース濃度にほとん
ど影響を及ぼさなかったが,投与後 90 分以降に用量に依存した有意な低下が認められ,投与
後 120 分にそれぞれ最低値(87 及び 79 mg/dL)を示した(図 2.6.2.2-11)。
- 22 -

2.6.2. 薬理試験の概要文
アナグリプチン
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180
投与後の時間(min)
グル
コー
ス濃
度(m
g/dL)
対照群
3 mg/kg
30 mg/kg
*
ナテグリニド
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180
投与後の時間(min)
グル
コー
ス濃
度(m
g/dL)
対照群
50 mg/kg
100 mg/kg
*
**
**
*********
***
***
グリベンクラミド
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180
投与後の時間(min)
グル
コー
ス濃
度(m
g/dL)
対照群
1 mg/kg
3 mg/kg
**
*****
***
図 2.6.2.2-11 正常ラットの絶食時血漿グルコース濃度の経時的推移
平均値±標準偏差(n=8) *:p<0.05,**:p<0.01,***:p<0.001;対照群との有意差(Dunnett の多重比較) [資料 4.2.1.1-14 より引用,改変]
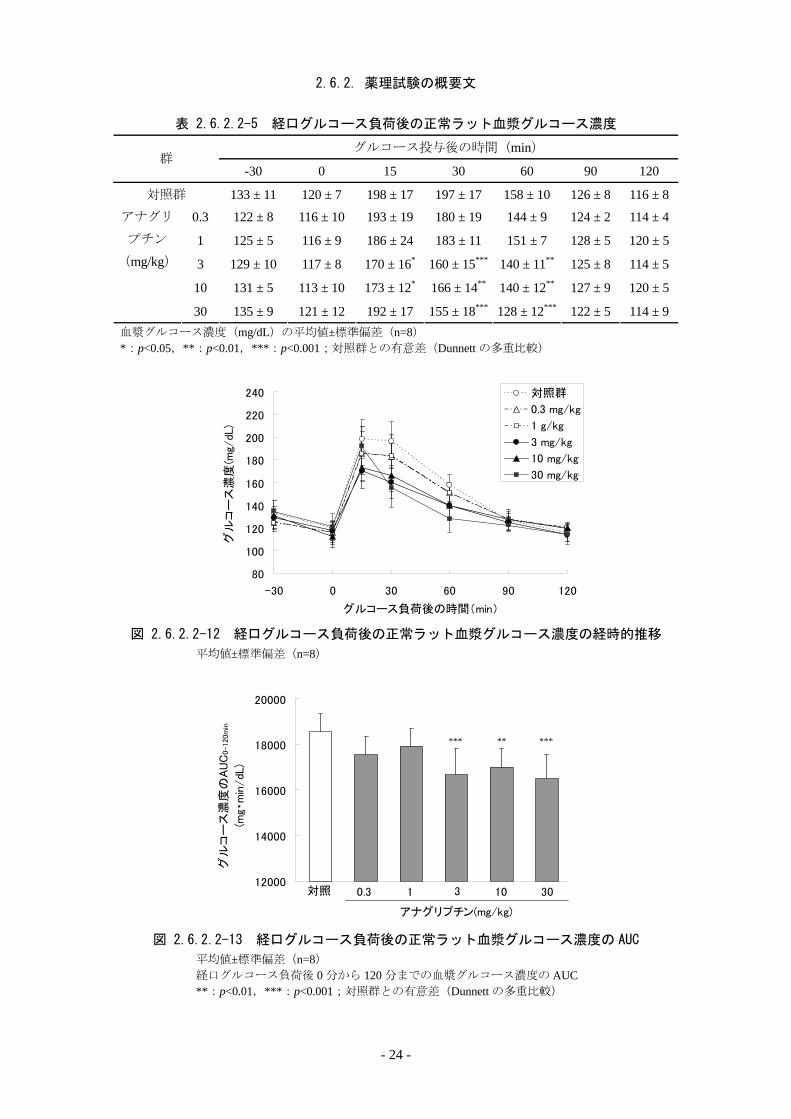
2.6.2.2.8. 正常ラットの OGTT に対する作用[資料 4.2.1.1-15]
一晩絶食した雄性 Wistar 系ラット(各群 8 例)にアナグリプチン(0.3~30 mg/kg)又は対
照として精製水を単回経口投与し,30 分後に 1 g/kg の経口グルコース負荷試験を行った。薬
物投与直前(グルコース負荷前 30 分),グルコース負荷直前(0 分),並びにグルコース負荷
後 15,30,60,90 及び 120 分に尾部より経時的に採血し,血漿グルコース濃度を酵素法にて
測定した。 アナグリプチンは,グルコース負荷前の血漿グルコース濃度に影響を及ぼさなかったが,
負荷後 15 分から 60 分の血漿グルコース濃度の上昇を抑制し,3 mg/kg以上で明らかな抑制作
用を示した(表 2.6.2.2-5,図 2.6.2.2-12)。また,グルコース負荷後 0 分から 120 分までの血
漿グルコース濃度のAUCの増加を抑制し,対照群に比べて 3 mg/kg以上で有意な抑制作用を
示した(図 2.6.2.2-13)。
- 23 -
2.6.2. 薬理試験の概要文
表 2.6.2.2-5 経口グルコース負荷後の正常ラット血漿グルコース濃度
グルコース投与後の時間(min) 群
-30 0 15 30 60 90 120
対照群 133 ± 11 120 ± 7 198 ± 17 197 ± 17 158 ± 10 126 ± 8 116 ± 8
0.3 122 ± 8 116 ± 10 193 ± 19 180 ± 19 144 ± 9 124 ± 2 114 ± 4
1 125 ± 5 116 ± 9 186 ± 24 183 ± 11 151 ± 7 128 ± 5 120 ± 5
3 129 ± 10 117 ± 8 170 ± 16* 160 ± 15*** 140 ± 11** 125 ± 8 114 ± 5
10 131 ± 5 113 ± 10 173 ± 12* 166 ± 14** 140 ± 12** 127 ± 9 120 ± 5
アナグリ
プチン
(mg/kg)
30 135 ± 9 121 ± 12 192 ± 17 155 ± 18*** 128 ± 12*** 122 ± 5 114 ± 9血漿グルコース濃度(mg/dL)の平均値±標準偏差(n=8) *:p<0.05,**:p<0.01,***:p<0.001;対照群との有意差(Dunnett の多重比較)
80
100
120
140
160
180
200
220
240
-30 0 30 60 90 120
グルコース負荷後の時間(min)
グル
コー
ス濃
度(m
g/dL
)_
対照群
0.3 mg/kg
1 g/kg
3 mg/kg
10 mg/kg
30 mg/kg
図 2.6.2.2-12 経口グルコース負荷後の正常ラット血漿グルコース濃度の経時的推移
平均値±標準偏差(n=8)
12000
14000
16000
18000
20000
グル
コー
ス濃
度の
AU
C0-120m
in
(mg・
min
/dL
)
対照
アナグリプチン(mg/kg)
0.3 1 3 10 30
*** ** ***
図 2.6.2.2-13 経口グルコース負荷後の正常ラット血漿グルコース濃度の AUC
平均値±標準偏差(n=8) 経口グルコース負荷後 0 分から 120 分までの血漿グルコース濃度の AUC **:p<0.01,***:p<0.001;対照群との有意差(Dunnett の多重比較)
- 24 -

2.6.2. 薬理試験の概要文
2.6.2.2.9. Zucker fatty ラットの OGTT に対する作用[資料 4.2.1.1-16]
肥満インスリン抵抗性モデル動物である雄性 Zucker fatty ラットを用いた。一晩絶食した
Zucker fatty ラット(12 週齢,各群 6 又は 7 例)にアナグリプチン(1,3 及び 10 mg/kg),ビ
ルダグリプチン( ,10 mg/kg)又は対照(Fatty 対照群)として精製水を単回経口
投与し,30 分後に 2 g/kg の経口グルコース負荷試験を行った。正常対照群(Lean 対照群)
として,雄性 Zucker Lean ラット(12 週齢,7 例)を使用した。薬物投与直前(グルコース
負荷前 30 分),グルコース負荷直前(0 分),並びにグルコース負荷後 10,20,30,60,90及び 120 分に尾部より経時的に採血した。血漿 DPP-4 活性は合成基質を用いた蛍光法で,血
漿インスリン濃度は ELISA 法にて,血漿グルコース濃度は酵素法にてそれぞれ測定した。 アナグリプチンは,Zucker fattyラットのグルコース負荷 0 分(薬物投与後 30 分)における
血漿DPP-4 活性を用量に依存して阻害し,Fatty対照群に比べて 3 mg/kg以上で有意な作用を
示した(図 2.6.2.2-14-a, 2.6.3.2.項)。また,グルコース負荷後 0 分から 120 分までの血漿イン
スリン濃度のAUCをFatty対照群に比べて,いずれの用量においても有意に増加させた(図 2.6.2.2-14-b)。更に,グルコース負荷後 0 分から 120 分までの血漿グルコース濃度変化量の
AUC[⊿AUC(0 分の濃度を基準)]を用量に依存して減少させ,Fatty対照群に比べて 10 mg/kgで有意な作用を示した(図 2.6.2.2-14-c)。 ビルダグリプチンは,10 mg/kgでグルコース負荷 0 分(薬物投与後 30 分)における血漿
DPP-4 活性をFatty対照群に比べて有意に阻害し(図 2.6.2.2-14-a, 2.6.3.2.項),グルコース負荷
後 0 分から 120 分までの血漿インスリン濃度のAUCを有意に増加させた(図 2.6.2.2-14-b)。また,グルコース負荷後 0 分から 120 分までの血漿グルコース濃度の⊿AUC(0 分の濃度を
基準)が減少する傾向を示した(図 2.6.2.2-14-c)。
- 25 -

2.6.2. 薬理試験の概要文
0
4000
8000
12000
16000
グル
コー
ス濃
度の
⊿A
UC
0-120m
in(m
g・m
in/dL)
###
**
c)
Lean対照群
Fatty対照群
1
アナグリプチン (mg/kg)
(n=7) (n=7) (n=7) (n=7) (n=6)
3 10
(n=6)
ビルダグリプチン
(10mg/kg)
0
1000
2000
3000
4000
5000
血漿
イン
スリ
ン濃
度の
AU
C0-120m
in(ng
・min
/m
L)
###
*
*
*
b)
Lean対照群
Fatty対照群
1
アナグリプチン (mg/kg)
(n=7) (n=7) (n=7) (n=7) (n=6)
3 10
(n=6)
ビルダグリプチン
(10mg/kg)
††
0
20
40
60
80
100
120
血漿
DP
P-4活
性(%
)
Lean対照群
Fatty対照群
1
アナグリプチン (mg/kg)
*****
(n=7)
a)
(n=7) (n=7) (n=7) (n=6)
3 10
(n=6)
†††
ビルダグリプチン
(10mg/kg)
図 2.6.2.2-14 経口グルコース負荷後の Zucker fatty ラットにおける血漿 DPP-4 活性,血
漿インスリン濃度の AUC 及び血漿グルコース濃度の⊿AUC
a) 経口グルコース負荷 0 分(薬物投与後 30 分)の血漿 DPP-4 活性 b) 経口グルコース負荷後 0 分から 120 分までの血漿インスリン濃度の AUC c) 経口グルコース負荷後 0 分から 120 分までの血漿グルコース濃度の⊿AUC 平均値±標準偏差(n=6~7) ###:p<0.001;Lean 対照群との有意差(t 検定) *:p<0.05,**:p<0.01,***:p<0.001;Fatty 対照群との有意差(Dunnett の多重比較) ††:p<0.01,†††:p<0.001;Fatty 対照群との有意差(t 検定)
2.6.2.2.10. GK ラットの OGTT に対する作用[資料 4.2.1.1-17]
自然発症糖尿病モデル動物である雄性 GK ラットを用い,頸静脈に採血用のカテーテルを
留置した。一晩絶食したGKラット(10週齢,各群 8例)にアナグリプチン(1,3及び 10 mg/kg),ビルダグリプチン( ,10 mg/kg)又は対照(GK 対照群)として精製水を単回経口
投与し,30 分後に 2 g/kg の経口グルコース負荷試験を行った。正常対照群(Wistar 対照群)
として雄性 Wistar 系ラット(10 週齢,8 例)を使用した。薬物投与直前(グルコース負荷前
30 分),グルコース負荷直前(0 分),並びにグルコース負荷後 15,30,60,90 及び 120 分に
カテーテルより経時的に採血した。血漿 DPP-4 活性は合成基質を用いた蛍光法で,血漿イン
スリン濃度は ELISA 法にて,血漿グルコース濃度は酵素法にてそれぞれ測定した。
- 26 -

2.6.2. 薬理試験の概要文
アナグリプチンは,GKラットのグルコース負荷 0 分及び負荷後 120 分(薬物投与後 30 分
及び 150 分)における血漿DPP-4 活性をそれぞれ用量に依存して阻害し,GK対照群に比べて
3 mg/kg以上で有意な抑制作用を示した(図 2.6.2.2-15-a, 2.6.3.2.項)。また,グルコース負荷
後 0 分から 120 分までの血漿インスリン濃度のAUCを用量依存的に増加させ,GK対照群に
比べて 10 mg/kgで有意な作用を示した(図 2.6.2.2-15-b)。更に,グルコース負荷後 0 分から
120 分までの血漿グルコース濃度の⊿AUC(0 分の濃度を基準)を 3 mg/kg以上でGK対照群
に比べて有意に減少させた(図 2.6.2.2-15-c)。 ビルダグリプチンは,GKラットのグルコース負荷 0 分及び負荷後 120 分(薬物投与後 30
分及び 150 分)の血漿DPP-4 活性をGK対照群に比べて有意に阻害し(図 2.6.2.2-15-a, 2.6.3.2.項),グルコース負荷後 0 分から 120 分までの血漿インスリン濃度のAUCを有意に増加させ
た(図 2.6.2.2-15-b)。また,グルコース負荷後 0 分から 120 分までの血漿グルコース濃度の
⊿AUC(0 分の濃度を基準)をGK対照群に比べて有意に減少させた(図 2.6.2.2-15-c)。
0
5000
10000
15000
20000
25000
グル
コー
ス濃
度の
⊿A
UC
0-120m
in(m
g・m
in/dL
)
###
*
*
c)
††
Wistar対照群
GK対照群
1
アナグリプチン (mg/kg)
103 ビルダグリプチン
(10mg/kg)
0
100
200
300
400イ
ンス
リン
濃度
のA
UC
0-120m
in(ng・
min
/m
L)
#
*
b)
†††
Wistar対照群
GK対照群
1
アナグリプチン (mg/kg)
103 ビルダリプチン
(10mg/kg)
グ
0
20
40
60
80
100
120
血漿
DP
P-4活
性(%
)
0min
120min
Wistar対照群
GK対照群
1
アナグリプチン (mg/kg)
**
***
*****
103
a)
ビルダグリプチン
(10mg/kg)
†††
†††
図 2.6.2.2-15 経口グルコース負荷後の GKラットにおける血漿 DPP-4 活性,血漿インスリ
ン濃度の AUC 及び血漿グルコース濃度の⊿AUC
a) 経口グルコース負荷 0 分及び負荷後 120 分(薬物投与後 30 分及び 150 分)の血漿 DPP-4 活性 b) 経口グルコース負荷後 0 分から 120 分までの血漿インスリン濃度の AUC c) 経口グルコース負荷後 0 分から 120 分までの血漿グルコース濃度の⊿AUC 平均値±標準偏差(n=8) #:p<0.05,###:p<0.001;Wistar 対照群との有意差(t 検定) *:p<0.05,**:p<0.01,***:p<0.001;GK 対照群との有意差(Dunnett の多重比較) ††:p<0.01,†††:p<0.001;GK 対照群との有意差(t 検定)
- 27 -

2.6.2. 薬理試験の概要文
2.6.2.2.11. 膵β細胞に対する作用
(1) STZ 誘発糖尿病ラットにおける持続投与の効果[資料 4.2.1.1-18]
STZ によって糖尿病を惹起させたラットを用いて,アナグリプチンを 4 週間持続的皮下投
与したときの膵 β細胞に対する作用を評価した。 雄性 SD 系ラット(8 週齢)の尾静脈に STZ(30 mg/kg)を静脈内投与して糖尿病を惹起し
た。STZ 投与後 3 日に,血漿グルコース濃度が mg/dL 以上 mg/dL 未満のラットを用
いた。 STZ 誘発糖尿病ラット(8 週齢,各群 10 例)に浸透圧ポンプを用いてアナグリプチン(75
及び 500 μg/h)又は対照として生理食塩水を 4 週間持続的皮下投与した。血漿グルコース濃
度,血漿 DPP-4 活性及び糖化ヘモグロビン値を薬物投与開始前及び投与開始後 2 週間は尾部,
4 週間は鎖骨下静脈より採血して測定した。また,血漿中アナグリプチン濃度及び血漿活性
型 GLP-1 濃度を投与開始後 4 週間に鎖骨下静脈より採血して測定した。血漿グルコース濃度
は酵素法,血漿 DPP-4 活性は蛍光法,及び糖化ヘモグロビン値はグリコヘモグロビン分析計
を用いた HPLC 法にてそれぞれ測定し,血漿中アナグリプチン濃度は LC/MS/MS 及び血漿活
性型 GLP-1 濃度は ELISA 法にてそれぞれ測定した。 投与開始後 4 週間のラットより膵臓を採取し,膵臓インスリン含量の測定試料として尾部,
病理標本作製試料として体部を用いた。膵臓インスリン含量は,膵臓を破砕して調製した膵
臓抽出液のインスリン濃度を ELISA 法にて測定した。膵 β-cell mass は,抗インスリン抗体を
用いて免疫染色を行った病理標本の膵臓総面積に対するインスリン陽性面積比(%)を測定
した。 アナグリプチンは,血漿DPP-4 活性を用量に依存して阻害し,投与開始後 4 週間において,
75 μg/hで約 80%,500 μg/hで約 85%阻害した。このときの血漿中アナグリプチン濃度は,そ
れぞれ 109 ± 47 ng/mL及び 616 ± 133 ng/mLであり,500 μg/hでは 75 μg/hに比べて約 6 倍高値
を示した。また,投与開始後 4 週間の血漿活性型GLP-1 濃度は,対照群で 11.1 ± 5.9 pmol/L,75 μg/hで 21.7 ± 6.3 pmol/L及び 500 μg/hで 26.4 ± 10.7 pmol/Lであり,対照群に比べて用量に依
存した有意な上昇が認められた(表 2.6.2.2-6)。 膵臓インスリン含量は,投与開始後 4 週間において,対照群で 2.89 ± 1.10 μg,アナグリプ
チンの 75 μg/hで 3.51 ± 0.83 μg及び 500 μg/hで 4.65 ± 1.22 μgであり,用量に依存して増加し,
対照群に比べて 500 μg/hで有意な作用を示した(図 2.6.2.2-16-a)。また,膵β-cell massは,対
照群で 0.018 ± 0.005%,75 μg/hで 0.024 ± 0.010%及び 500 μg/hで 0.026 ± 0.008%であり,用量
に依存して増加し,対照群に比べて 500 μg/hで有意な作用を示した(図 2.6.2.2-16-b)。 血漿グルコース濃度は,対照群では溶媒(生理食塩水)投与開始後に上昇し,2 週間で最
大濃度を示した。アナグリプチンでは,投与開始後 2 週間において,いずれの用量も対照群
との差は認められなかった。投与開始後 4 週間において,500 μg/hで対照群に比べてわずか
に低値を示したが,有意な作用ではなかった(図 2.6.2.2-16-c)。 糖化ヘモグロビン値は,対照群では経時的に上昇し,溶媒投与開始後 4 週間で最大値を示
した。アナグリプチンでは,投与開始後 2 及び 4 週間において,用量に依存して低下する傾
向がわずかに認められたが,対照群に比べて有意な差はなく,明らかな作用は認められなか
った(図 2.6.2.2-16-d)。
- 28 -

2.6.2. 薬理試験の概要文
アナグリプチンの持続投与は,STZ 誘発糖尿病ラットにおいて血漿 DPP-4 活性を阻害して
活性型 GLP-1 濃度を上昇させることにより,膵 β細胞量を増大したと考えられた。 表 2.6.2.2-6 STZ 誘発糖尿病ラットにおけるアナグリプチンの持続的皮下投与による
血漿 DPP-4 活性,血漿中薬物濃度及び血漿活性型 GLP-1 濃度
DPP-4 活性 (%)
アナグリプチン濃度 (ng/mL)
活性型 GLP-1 (pmol/L) 群 (n)
2 w 4 w 4 w 4 w 対照 (10) 100.8 ± 13.9 83.0 ± 13.5 - 11.1 ± 5.9
アナグリプチン 75 μg/h (10) 26.2 ± 4.5 * 19.1 ± 3.2 ** 109 ± 47 21.7 ± 6.3 *
500 μg/h (10) 18.2 ± 3.1 *** 14.4 ± 3.1 *** 616 ± 133 26.4 ± 10.7 ***
平均値±標準偏差(n=10) *:p<0.05,**:p<0.01,***:p<0.001;対照群との有意差(Dunnett の多重比較)
200
300
400
500
600
0 2 4
投与後の時間 (w)
グル
コー
ス濃
度 (m
g/dL
)
対照群
75μg/h
500μg/h
c)
3
4
5
6
7
8
9
10
0 2 4
投与後の時間 (w)
糖化
ヘモ
グロ
ビン
値 (%
)
対照群
75μg/h
500μg/h
d)
アナグリプチン (μg/h)
0
2
4
6
対照群 75 500
膵臓
イン
スリ
ン含
量 (μ
g)
**
a)
アナグリプチン (μg/h)
0
0.01
0.02
0.03
0.04
対照群 75 500
膵β
-cell
mas
s (%
)
*
b)
図 2.6.2.2-16 STZ 誘発糖尿病ラットにおけるアナグリプチンの持続的皮下投与による
膵臓インスリン含量,膵β-cell mass 及び血糖パラメータ
a) 4 週間持続的皮下投与後の膵臓インスリン含量 b) 4 週間持続的皮下投与後の膵 β-cell mass c) 持続的皮下投与後 0,2 及び 4 週間の血漿グルコース濃度の経時的推移 d) 持続的皮下投与後 0,2 及び 4 週間の糖化ヘモグロビン値の経時的推移 平均値±標準偏差(n=10) *:p<0.05,**:p<0.01;対照群との有意差(Dunnett の多重比較)
- 29 -

2.6.2. 薬理試験の概要文
(2) アロキサン誘発糖尿病マウスにおける反復投与の効果[資料 4.2.1.1-19(参考資料)]
アロキサンによって糖尿病を惹起させたマウスを用いて,アナグリプチンを 4 週間反復飲
水投与したときの膵 β細胞に対する作用を評価した。 雄性 C57BL/6J マウス(9 週齢)の尾静脈にアロキサン(60 mg/kg)を静脈内投与して糖尿
病を惹起した。アロキサン投与後 3 日に血漿グルコース濃度が mg/dL 以上 mg/dL 未
満のマウスを糖尿病マウスとした。 1) 膵臓インスリン含量に対する作用
アロキサン誘発糖尿病マウス(9 週齢,各群 10 例)にアナグリプチン(0.5 mg/mL),シタ
グリプチン ( ,以下,シタグリプチンとする,0.5 mg/mL)又は対照とし
て精製水を 4 週間反復飲水投与(自由摂取)した。体重及び飲水量から換算した平均 1 日投
与量は,アナグリプチンが 219 ± 51 mg/kg/日,シタグリプチンが 359 ± 29 mg/kg/日であった。 膵臓インスリン含量は,投与開始後 4 週間のマウスより膵臓を採取し,その後破砕して調
製した膵臓抽出液のインスリン濃度を ELISA 法にて測定した。また,血漿グルコース濃度(随
時及び食後)を酵素法,糖化ヘモグロビン値はグリコヘモグロビン分析計を用いた HPLC 法
及び血漿 DPP-4 活性を蛍光法にてそれぞれ測定した。 アナグリプチン及びシタグリプチンは,投与開始後 4 週間の血漿 DPP-4 活性を対照群に比
べて有意に阻害し,対照群に対する阻害率は,それぞれ約 76%及び約 69%であった(アナグ
リプチンは p<0.001,シタグリプチンは p<0.01;対照群との有意差,Dunnett の多重比較)。 膵臓インスリン含量は,投与開始後 4 週間において,対照群で 196 ± 137 ng/g体重,アナグ
リプチンで 434 ± 231 ng/g体重及びシタグリプチンで 239 ± 124 ng/g体重であり,アナグリプ
チン投与後の膵臓インスリン含量が対照群に比べて有意に増加した(図 2.6.2.2-17-a)。 随時血漿グルコース濃度は,対照群ではいずれの時点も 400 mg/dL以上の高値を示した。
アナグリプチンでは,投与開始以降に対照群に比べて低下する傾向を示したが,いずれの時
点も有意な差は認められなかった。シタグリプチンでは,投与開始後 1 週間で対照群に比べ
て低下する傾向を示したが,2 週間以降は作用が認められなかった(図 2.6.2.2-17-b)。 糖化ヘモグロビン値は,対照群では経時的に上昇し,投与開始後 4 週間で最大値を示した。
アナグリプチンでは,投与開始以降に対照群に比べて低下したが,シタグリプチンでは作用
が認められなかった。投与開始後 4 週間における糖化ヘモグロビン値は,対照群で 7.4 ± 1.5%,
アナグリプチン群で 5.7 ± 1.5%及びシタグリプチン群で 6.9 ± 1.3%であり,アナグリプチンで
は対照群に比べて有意に低下した(図 2.6.2.2-17-c)。 食後血漿グルコース濃度は,対照群で 512 ± 129 mg/dLであったのに対し,アナグリプチン
群で 366 ± 138 mg/dL,シタグリプチン群で 452 ± 157 mg/dLであり,いずれも対照群に比べて
有意な作用は認められなかった(図 2.6.2.2-17-d)。
- 30 -

2.6.2. 薬理試験の概要文
0
100
200
300
400
500
600
700
対照群 アナグリプチン(0.5 mg/mL)
シタグリプチン(0.5 mg/mL)
膵臓
イン
スリ
ン含
量 (
ng/
g体重
)
**
a)
0
100
200
300
400
500
600
700
0 2
投与後の時間 (w)
随時
血漿
グル
コー
ス濃
度 (
mg/
dL)
4
対照群
アナグリプチン (0.5 mg/mL)
シタグリプチン (0.5 mg/mL)
b)
0
100
200
300
400
500
600
700
対照群 アナグリプチン(0.5 mg/mL)
シタグリプチン(0.5 mg/mL)
食後
血漿
グル
コー
ス濃
度 (
mg/
dL)
d)
0
2
4
6
8
10
0 2 4
投与後の時間 (w)
糖化
ヘモ
グロ
ビン
値 (
%)
対照群
アナグリプチン (0.5 mg/mL)
シタグリプチン (0.5 mg/mL)
c)
*
図 2.6.2.2-17 アロキサン誘発糖尿病マウスにおけるアナグリプチンの反復投与による
膵臓インスリン含量及び血糖パラメータ
a) 4 週間反復飲水投与後の膵臓インスリン含量 b) 反復飲水投与後 0,1,2,3 及び 4 週間の随時血漿グルコース濃度の経時的推移 c) 反復飲水投与後 0,2 及び 4 週間の糖化ヘモグロビン値の経時的推移 d) 4 週間反復飲水投与後の食後血漿グルコース濃度(絶食 2 時間後に再給餌,1 時間後に採血) 平均値±標準偏差(n=10) *:p<0.05,**:p<0.01;対照群との有意差(Dunnett の多重比較)
2) 膵 β-cell mass に対する作用
アロキサン誘発糖尿病マウス(9 週齢,各群 10 例)にアナグリプチン(1.0 mg/mL)又は
対照として精製水を 4 週間反復飲水投与(自由摂取)した。体重及び飲水量から換算したア
ナグリプチンの平均 1 日投与量は,384 ± 167 mg/kg/日であった。 膵 β-cell mass は,投与開始後 4 週間のマウスより膵臓を採取し,抗インスリン抗体を用い
て免疫染色を行った病理標本の膵臓総面積に対するインスリン陽性面積比(%)を測定した。
また,血漿グルコース濃度(随時及び食後),糖化ヘモグロビン値及び血漿 DPP-4 活性を測
定した。 アナグリプチンは,投与開始後 4 週間の血漿 DPP-4 活性を対照群に比べて有意に阻害し,
対照群に対する阻害率は約 80%であった(p<0.001;対照群との有意差,Dunnett の多重比較)。 膵β-cell massは,対照群で 0.083 ± 0.034%であったのに対し,アナグリプチンで 0.111 ±
- 31 -

2.6.2. 薬理試験の概要文
0.019%と対照群の 1.3 倍に増加し,有意な差が認められた(図 2.6.2.2-18-a)。 随時血漿グルコース濃度は,対照群ではいずれの時点も 400 mg/dL以上の高値を示した。
アナグリプチンでは,投与開始以降に対照群に比べて低下し,2,3 及び 4 週間で有意に低値
を示した(図 2.6.2.2-18-b)。 糖化ヘモグロビン値は,対照群では経時的な上昇が認められ,投与開始後 4 週間で最大値
を示した。投与開始後 2 及び 4 週間における糖化ヘモグロビン値は,対照群ではそれぞれ 6.5 ± 0.7%及び 7.2 ± 1.1%であったのに対し,アナグリプチンではそれぞれ 5.3 ± 0.8%及び 5.1 ± 1.3%であった。アナグリプチンでは,投与開始後 2 及び 4 週間のいずれの時点も対照群に比
べて有意に低下した(図 2.6.2.2-18-c)。 食後血漿グルコース濃度は,対照群で 451 ± 121 mg/dLであったのに対し,アナグリプチン
では 300 ± 104 mg/dLであり,対照群に比べて有意に低下した(図 2.6.2.2-18-d)。
0
100
200
300
400
500
600
700
0 2
投与後の時間 (w)
随時
血漿
グル
コー
ス濃
度 (
mg/
dL)
4
対照群
アナグリプチン (1 mg/mL)
b)
** ** *
0
2
4
6
8
10
0 2 4
投与後の時間 (w)
糖化
ヘモ
グロ
ビン
値 (
%)
対照群
アナグリプチン (1 mg/mL)
c)
*****
0
0.04
0.08
0.12
0.16
対照群 アナグリプチン(1 mg/mL)
膵β
-cell
mas
s (%
)
*
a)
0
100
200
300
400
500
600
700
対照群 アナグリプチン(1 mg/mL)
食後
血糖
グル
コー
ス濃
度 (
mg/
dL)
**
d)
図 2.6.2.2-18 アロキサン誘発糖尿病マウスにおけるアナグリプチンの反復投与による
膵β-cell mass 及び血糖パラメータ
a) 4 週間反復飲水投与後の膵 β-cell mass b) 反復飲水投与後 0,1,2,3 及び 4 週間の随時血漿グルコース濃度の経時的推移 c) 反復飲水投与後 0,2 及び 4 週間の糖化ヘモグロビン値の経時的推移 d) 4 週間反復飲水投与後の食後血漿グルコース濃度(絶食 2 時間後に再給餌,1 時間後に採血) 平均値±標準偏差(n=10) *:p<0.05,**:p<0.01,***:p<0.001;対照群との有意差(Dunnett の多重比較)
- 32 -

2.6.2. 薬理試験の概要文
(3) インスリノーマ細胞の増殖に対する作用[資料 4.2.1.1-19(参考資料)]
ハムスター膵 β細胞由来のインスリノーマ細胞株 HIT-T15 を用いて, GLP-1 とアナグリプ
チンを共存させたときの細胞増殖に対する作用を評価した。 維持培養にて 70~90%コンフルエントとなった HIT-T5 細胞をトリプシン-EDTA で剥離し
た後,10%FBS 及び 25 mmol/L グルコースを含む RPMI1640 培地で再懸濁した。細胞懸濁液
を ウェル細胞培養用マイクロプレートに播種し, 間培養した。細胞を PBS(−)で洗浄
した後,10 mmol/L グルコースを含む RPMI1640 培地 ,GLP-1(培地中での終
濃度:10 pmol/L,以下同じ)とアナグリプチン(0.1,1 及び 10 nmol/L),GLP-1(10 pmol/L)又はアナグリプチン(10 nmol/L)単独,並びに対照として PBS(−)及び陽性対照としてエキセ
ンディン-4(100 pmol/L) 培養した。細胞増
殖の評価は,生細胞量を酵素法で,BrdU 取込み量を ELISA 法にてそれぞれ計測して行った。
測定は quadruplicate で行い, は として ,n=8 で評価した。 アナグリプチンは,10 pmol/LのGLP-1 と共存させたとき,0.1 nmol/L以上でHIT-T5 細胞の
生細胞量及びBrdU取込み量をそれぞれ対照群に比べて有意に増加させた。同様に,陽性対照
であるエキセンディン-4 も,それぞれ対照群に比べて有意に増加させた。一方,GLP-1 又は
アナグリプチン単独は,いずれにも影響を及ぼさなかった(図 2.6.2.2-19)。
0.0
0.5
1.0
1.5
2.0
2.5
対照群 GLP-1
10 pmol/L
アナグリプチン
10 nmol/L 0.1 nmol/L
アナグリプチン
1 nmol/L 10 nmol/L
エキセンディン-4
100 pmol/L
+ GLP-1 10 pmol/L
Brd
U 取
込み
量(相
対値
)
***** ***
***
b)
0.0
0.2
0.4
0.6
0.8
1.0
対照群 GLP-1
10 pmol/L
アナグリプチン
10 nmol/L 0.1 nmol/L
アナグリプチン
1 nmol/L 10 nmol/L
エキセンディン-4
100 pmol/L
+ GLP-1 10 pmol/L
生細
胞量
(相
対値
)
** ** ****
a)a)
図 2.6.2.2-19 ハムスター膵β細胞由来インスリノーマ細胞の増殖に対する作用
a) 生細胞量( ブランクにおける生細胞量を 1 としたときの相対値) b) BrdU 取込み量( ブランクにおける BrdU 取込み量を 1 としたときの相対値) 平均値±標準偏差(n=8) *:p<0.05,**:p<0.01,***:p<0.001;対照群との有意差(Dunnett の多重比較)
- 33 -

2.6.2. 薬理試験の概要文
2.6.2.2.12. 代謝物及び類縁物質の DPP-4 及び類縁酵素に対する阻害作用
ヒトでの主代謝物である SKL-12320,動物で比較的多く認められた SKL-14687 を含む 6 種
類の代謝物,及びアナグリプチンのエナンチオマーを含む原薬の製造工程中に混入する可能
性がある 3 種類の類縁物質の DPP-4 に対する阻害作用を検討した。また,SKL-12320 及び
SKL-14687 については,DPP-4 類縁酵素に対する阻害作用も検討した。
(1) SKL-12320 及び SKL-14687 の DPP-4 及び類縁酵素に対する阻害作用[資料 4.2.1.1-20]
アナグリプチンのニトリルが加水分解された SKL-12320(プロリン体)及びアミンに隣接
するメチレンが酸化された代謝物のナトリウム塩である SKL-14687 を評価した。 DPP-4 活性はヒト組換えDPP-4 を用いてGly-Pro-MCAを基質とし,DPP-4 に対するアナグ
リプチンの阻害活性を評価した方法(2.6.2.2.1.項)と同じ方法により測定した。DPP-4 類縁
酵素として,ヒト組換えDPP-8,DPP-9 及びFAP,ヒトCaco-2 細胞分画DPP-2,微生物由来POP,ヒト血漿APP,並びにブタ腎臓由来prolidase,ACE及びLAPを用いて,アナグリプチンの作用
を評価した方法(2.6.2.2.3.項)と同じ方法により酵素活性を測定した。IC50値は,各被験物質
濃度及び酵素活性をプロットした阻害曲線からロジスティック回帰分析を行って算出した。
なお,被験物質の濃度を 500 μmol/Lとした場合の酵素活性値が 50%以上のとき,IC50値は 500 μmol/Lを超える(>500 μmol/L)と判断した。
SKL-12320 及びSKL-14687 は,DPP-4 及び類縁酵素に対して阻害作用を示さなかった(IC50
値>500 μmol/L)(表 2.6.2.2-7)。
表 2.6.2.2-7 アナグリプチンの代謝物 SKL-12320 及び SKL-14687 の DPP-4 及び
類縁酵素阻害作用
IC50 ( μmol/L) SKL-12320 SKL-14687
DPP-4 >500 >500 DPP-8 >500 >500 DPP-9 >500 >500 FAP >500 >500
DPP-2 >500 >500 POP >500 >500 APP >500 >500
Prolidase >500 >500 ACE >500 >500 LAP >500 >500
n=1,各酵素活性はそれぞれ以下の基質を用いて測定した。 ・ DPP-4,DPP-8 及び DPP-9: Gly-Pro-MCA ・ FAP: Ala-Pro-AFC ・ DPP-2: Lys-Ala-MCA ・ POP: Z-Gly-Pro-MCA ・ APP: Lys(Abz)-Pro-Pro-pNA ・ Prolidase: Gly-Pro ・ ACE: FA-Phe-Gly-Gly(FAPGG) ・ LAP: Leu-MCA
- 34 -

2.6.2. 薬理試験の概要文
(2) その他の代謝物及び類縁物質の DPP-4 に対する阻害作用[資料 4.2.1.1-21]
その他の代謝物として SKL-12250(プロリンアミド体),SKL-12277(カルボン酸体),
SKL-12339(一水酸化体:2-ヒドロキシメチル体)及び SKL-13776(一水酸化体:3-ヒドロキ
シメチル体)を評価した。類縁物質として SKL-06327, SKL-12309 及びアナグリプチンのエナ
ンチオマー(R 体)である SKL-13775 を評価した。 DPP-4 活性はヒト組換えDPP-4 を用いてGly-Pro-MCAを基質とし,アナグリプチンの阻害
活性を評価した方法(2.6.2.2.1.項)と同じ方法により測定した。IC50値は,各被験物質濃度及
びDPP-4 活性をプロットした阻害曲線からロジスティック回帰分析を行って算出した。なお,
被験物質の濃度を 500 μmol/Lとした場合のDPP-4 活性値が 50%以上のとき,IC50値は 500 μmol/Lを超える(>500 μmol/L)と判断した。 代謝物のSKL-12250,SKL-12339 及びSKL-13776 は,ヒト組換えDPP-4 活性を濃度に依存
して阻害した。SKL-12250 のIC50値は 1,700 nmol/Lであり,アナグリプチンのIC50値(3.3 nmol/L,表 2.6.2.2-1)に比較して 515 倍の高値であった(表 2.6.2.2-8)。SKL-12339 及びSKL-13776のIC50値はそれぞれ 3.1 nmol/L及び 3.4 nmol/Lであり,アナグリプチンのIC50値と同等であっ
た。一方,SKL-12277 はヒト組換えDPP-4 に対して阻害作用を示さなかった(IC50値>500 μmol/L)。 類縁物質のSKL-06327, SKL-12309 及びエナンチオマーのSKL-13775 は,ヒト組換えDPP-4
に対して阻害作用を示さなかった(IC50値>500 μmol/L)。
表 2.6.2.2-8 アナグリプチンの代謝物及び類縁物質の DPP-4 に対する阻害作用
IC50 (nmol/L) 相対強度
代謝物 SKL-12250 1,700 ± 100 515 SKL-12277 >500,000 >150,000 SKL-12339 3.1 ± 0.9 0.94 SKL-13776 3.4 ± 0.0 1.03
類縁物質 SKL-06327 >500,000 >150,000 SKL-12309 >500,000 >150,000
エナンチオマー SKL-13775 >500,000 >150,000 平均値±標準偏差(n=3,ただしIC50>500 μmol/Lの場合はn=1) 相対強度は次式に従って算出した:相対強度=各化合物のIC50 /アナグリプチンのIC50
- 35 -

2.6.2. 薬理試験の概要文
2.6.2.3. 副次的薬理試験
2.6.2.3.1. 各種プロテアーゼに対する作用[資料 4.2.1.2-1]
アナグリプチン及びヒトでの主代謝物であるSKL-12320の各種プロテアーゼに対する阻害
作用を確認した。プロテアーゼとして,DPP-4 類縁酵素である DPP-8,DPP-9,FAP などの
プロリン特異性ペプチダーゼを除くセリンプロテアーゼ,システインプロテアーゼ,アスパ
ラギン酸プロテアーゼ及びメタロプロテアーゼを含む 32 種類を用いた。アナグリプチン及び
SKL-12320 の適用濃度は 100 及び 500 μmol/L とし,それぞれの酵素に特異的な合成基質を用
いて酵素活性を測定し,阻害率を算出した。なお,50%以上の阻害作用が認められた場合に
作用ありとした。 アナグリプチン及びSKL-12320 は,いずれのプロテアーゼに対しても阻害作用を示さなか
った(IC50値>500 μmol/L)。
2.6.2.3.2. 各種受容体及びイオンチャネルに対する作用[資料 4.2.1.2-2,4.2.1.2-3]
アナグリプチン及びSKL-12320の各種受容体及びイオンチャネルとリガンドとの結合に対
する阻害作用を確認した。アナグリプチン及び SKL-12320 の適用濃度は 10 μmol/L とし,放
射標識した特異的リガンドを用いて,60 種類の受容体及びイオンチャネルとの結合に対する
阻害率を算出し,50%以上の阻害作用が認められた場合に作用ありとした。 アナグリプチン及びSKL-12320 は,いずれの受容体及びイオンチャネルとリガンドとの結
合に対しても阻害作用を示さなかった(IC50値>10 μmol/L)。 2.6.2.3.3. T 細胞及び B細胞の活性化に対する作用
(1) T 細胞の活性化に対する作用[資料 4.2.1.2-4]
マウス脾細胞を用いた抗 CD3ε抗体刺激による T 細胞の増殖に対するアナグリプチンの作
用を評価した。また,ビルダグリプチン及びシタグリプチンの作用と比較した。 雄性 BALB/c マウスの脾臓よりリンパ球を採取し,50 μmol/L のアナグリプチン,ビルダグ
リプチン及びシタグリプチンとそれぞれ 1 時間プレインキュベーションした。抗 CD3ε 抗体
を固相化したマイクロプレートで 44 時間培養した後,BrdU 取込み量を ELISA 法にて測定し
た。なお,陽性対照物質として,0.5 μmol/L のシクロスポリン A の作用も同時に評価した。 陽性対照物質のシクロスポリンAは,3 回実施した実験のいずれにおいても,T細胞の増殖
を 95%以上抑制した。一方,アナグリプチンの阻害率は 7~13%,ビルダグリプチン及びシ
タグリプチンの阻害率は,それぞれ 6~9%及び 13~34%であり,いずれのDPP-4 阻害剤も,
50 μmol/Lの濃度では,T細胞の活性化に対して 50%以上の阻害作用を示さなかった(IC50値
>50 μmol/L;表 2.6.2.3-1)。
- 36 -

2.6.2. 薬理試験の概要文
表 2.6.2.3-1 マウス脾臓由来 T細胞の増殖に対する作用
阻害率(%) 適用物質
適用濃度 (μmol/L) 1 回目 2 回目 3 回目
媒体 - 0 0 0 アナグリプチン 50 7 13 8 ビルダグリプチン 50 9 6 6 シタグリプチン 50 13 33 34 シクロスポリン A 0.5 95 98 98
(2) B 細胞の活性化に対する作用[資料 4.2.1.2-5]
マウス脾細胞を用いたLPS刺激によるB細胞の増殖に対するアナグリプチンの作用を評価
した。また,ビルダグリプチン及びシタグリプチンの作用と比較した。 雄性 BALB/c マウスの脾臓よりリンパ球を採取し,B 細胞の粗精製を行ったところ,純度
約 80%の B 細胞を得た。B 細胞を 50 μmol/L のアナグリプチン,ビルダグリプチン及びシタ
グリプチンとそれぞれ 1 時間プレインキュベーションした。LPS を 0.25 μg/mL の濃度で添加
し,マイクロプレートで 42 時間培養した後の BrdU 取込み量を ELISA 法にて測定した。な
お,陽性対照物質として,0.01 μmol/L のラパマイシンの作用も同時に評価した。 陽性対照物質のラパマイシンは,3 回実施した実験のいずれにおいても,B細胞の増殖を約
80%抑制した。一方,アナグリプチンの阻害率は 3.3~10.6%,ビルダグリプチン及びシタグ
リプチンの阻害率は,それぞれ 6.7~7.1%及び 5.1~17.9%であり,いずれのDPP-4 阻害剤も,
50 μmol/Lの濃度では,B細胞の活性化に対して 50%以上の阻害作用を示さなかった(IC50値
>50 μmol/L;表 2.6.2.3-2)。
表 2.6.2.3-2 マウス脾臓由来 B細胞の増殖に対する作用
阻害率(%) 適用物質
適用濃度 (μmol/L) 1 回目 2 回目 3 回目
媒体 - 0.0 0.0 0.0 アナグリプチン 50 5.9 10.6 3.3 ビルダグリプチン 50 7.0 6.7 7.1 シタグリプチン 50 17.9 5.1 12.7 ラパマイシン 0.01 78.8 79.2 78.3
- 37 -

2.6.2. 薬理試験の概要文
2.6.2.4. 安全性薬理試験
コアバッテリー試験として,中枢神経系,呼吸器系及び心血管系に対する作用を評価した。
また,補足的安全性薬理試験として,毒性試験で認められた腎/泌尿器系,胃腸管系及び自律
神経系に及ぼす影響を検討するため,これらの組織及び器官に及ぼす作用を評価した。 安全性薬理試験は,ICH ガイドラインの S7A;「安全性薬理試験ガイドラインについて」(医
薬審発第 902 号,平成 13 年 6 月 21 日)及び S7B;”The Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals (2005 年 5 月 12 日)”に従い,「医薬品の安全性に関する非臨床試験の実施の基準に関する
省令」(厚生省令第 21 号,平成 9 年 3 月 26 日)及び関連省令に定められた基準(GLP)に準
拠して実施した。
2.6.2.4.1. 中枢神経系に及ぼす作用[資料 4.2.1.3-1]
1 群 6 匹の雄性Crj:CD(SD)IGS[現行系統名Crl:CD(SD)]ラットにアナグリプチンの 500,1000 及び 2000 mg/kgを単回経口投与し,Irwinの変法[2]に準じて,投与後 0.5,1,3,6 及び
24 時間の一般症状及び行動観察によって中枢神経系に及ぼす作用を検討した。 アナグリプチンの1000 mg/kgでは,投与後6時間に縮瞳が6匹中2匹のラットに認められ,
2000 mg/kg では,投与後 3 時間及び 6 時間に縮瞳がそれぞれ 6 匹中 2 匹及び 3 匹のラットに
認められた。1000 及び 2000 mg/kg において,縮瞳以外の一般症状及び行動に異常及び変化
は認められなかった。また,500 mg/kg では,すべての観察項目で異常及び変化は認められ
なかった。 したがって,アナグリプチンは 2000 mg/kg までラットの中枢神経系に作用を及ぼさないと
考えられた。なお,縮瞳が 1000 mg/kg 以上で認められたが,瞳孔反射は消失していなかった
ことから,末梢神経系への作用が示唆された。 2000 mg/kgにおけるアナグリプチン及びヒトでの主代謝物であるSKL-12320 の推定曝露量
は,同用量におけるラット 4 週間反復経口投与毒性試験及びラット 104 週間反復経口投与が
ん原性試験の初回投与後の血漿中最高濃度(Cmax)がそれぞれ 179 μg/mL及び 6.18 μg/mLであ
ることから(2.6.7.7A.項,2.6.7.10D.項),最高臨床用量である 1 回 200 mgを 1 日 2 回反復経
口投与したときのアナグリプチン及びSKL-12320のCmax:1.2 μg/mL及び 0.162 μg/mL(2.7.6.3.2. 表 3.2-4)と比較すると,それぞれ約 149 倍及び 38 倍の開きがあった。また,縮瞳が認めら
れなかった 500 mg/kgにおけるアナグリプチン及びSKL-12320 の推定曝露量は,ラット 4 週
間反復経口投与毒性試験及びラット 104 週間反復経口投与がん原性試験における 600 mg/kg/日初回投与後のCmax(アナグリプチン:143 μg/mL,SKL-12320:4.00 μg/mL)に近似すると
考えられることから,最高臨床用量における推定曝露量と比較すると,それぞれ約 119 倍及
び 25 倍の開きがあった。
2.6.2.4.2. 呼吸器系に及ぼす作用[資料 4.2.1.3-2]
1 群 6 匹の雄性 Crj:CD(SD)IGS[現行系統名 Crl:CD(SD)]ラットにアナグリプチンの 500,1000 及び 2000 mg/kg を単回経口投与し,呼吸数,1 回換気量及び分時換気量を測定した。 アナグリプチンの 500,1000 及び 2000 mg/kg では,投与後の呼吸数,1 回換気量及び分時
換気量にアナグリプチンの作用に基づくと考えられる変化は認められなかった。
- 38 -

2.6.2. 薬理試験の概要文
したがって,アナグリプチンは 2000 mg/kg までラットの呼吸機能に作用を及ぼさないと考
えられた。 作用が認められなかった 2000 mg/kg と最高臨床用量における推定曝露量を比較すると,ア
ナグリプチン及び SKL-12320 の安全域は,それぞれ約 149 倍及び約 38 倍の開きがあった。
2.6.2.4.3. 心血管系に及ぼす作用
(1) hERG チャネル発現細胞を用いた hERG 電流に及ぼす作用[資料 4.2.1.3-3]
アナグリプチンの心血管系に及ぼす影響を明らかにする試験の一つとして,ヒト胎児腎細
胞株HEK293 に安定発現させたhERGチャネルの電流に及ぼす作用を検討した。hERG電流は
パッチクランプ法により測定した。アナグリプチンは 30,100 及び 300 μg/mLを適用した。
陽性対照物質として設定したクラスIII抗不整脈薬であるE-4031 は 1×10-7 mol/Lを適用した。 E-4031 のhERG電流相対値は 7.8%であり,媒体(92.7%)に比べて有意な低下が認められ,
阻害効果が確認された(表 2.6.2.4-1)。 アナグリプチンの 30,100 及び 300 μg/mLにおけるhERG電流相対値は,それぞれ 90.3,81.0
及び 59.5%であり,100 及び 300 μg/mLでは媒体に比べて有意な低下が認められた(表 2.6.2.4-1)。30,100 及び 300 μg/mLにおけるhERG電流に対する阻害率は,それぞれ 2.6,12.6及び 35.8%であった。 以上の結果から,アナグリプチンは 30 μg/mL まで hERG 電流に作用を及ぼさないと考えら
れた。 作用が認められなかった 30 μg/mL と最高臨床用量における推定曝露量を比較すると,約
25 倍の開きがあった。
表 2.6.2.4-1 hERG 電流に及ぼす作用
適用物質 適用濃度 細胞数 相対値(%) 阻害率(%)
媒体 - 5 92.7±6.7 -
アナグリプチン 30 μg/mL 5 90.3±5.6 2.6
アナグリプチン 100 μg/mL 5 81.0±5.0 * 12.6
アナグリプチン 300 μg/mL 5 59.5±4.6 ** 35.8
E-4031 1×10-7 mol/L 5 7.8±3.6 ## 91.6 相対値=(適用 10 分後の値/適用前の値)×100,平均値±標準偏差 阻害率=100 −[(アナグリプチン又は E-4031 の相対値)/(媒体の相対値)]×100 *:p<0.05,**:p<0.01;媒体との有意差(Dunnett の多重比較) ##:p<0.01;媒体との有意差(t 検定)
(2) モルモット摘出乳頭筋を用いた心筋活動電位持続時間に及ぼす作用[資料 4.2.1.3-4]
モルモット摘出乳頭筋標本を用いて,静止膜電位(RMP),活動電位振幅(APA),最大立
ち上がり速度(Vmax),30%,50%及び 90%再分極時活動電位持続時間(APD30,APD50及び
APD90),並びにAPD90とAPD30との差(APD30-90)に及ぼす作用を検討した。活動電位は微小
電極法により測定した。アナグリプチンは 30,100 及び 300 μg/mLを適用した。陽性対照物
質として設定したクラスIII抗不整脈薬である塩酸ソタロールは 3×10-5 mol/Lを適用した。 塩酸ソタロールでは,適用後のAPD30,APD50及びAPD90が媒体に比べて高値を示し,活動
- 39 -

2.6.2. 薬理試験の概要文
電位持続時間の延長が認められた(表 2.6.2.4-2)。また,適用後のAPD30-90相対値が媒体に比
較して高値を示した。 アナグリプチンの 100 μg/mLでは,適用後のAPD30-90相対値が媒体に比べて高値を示したが,
RMP,APA,Vmax,APD30,APD50 及びAPD90の相対値に媒体との差は認められなかった(表 2.6.2.4-2)。300 μg/mLでは,適用後のAPA及びVmaxの相対値が低値を示した。また,適用 30分後のAPD30,APD50 及びAPD90の相対値が低値を示し,適用 10 分後のAPD30-90相対値が高
値を示した。30 μg/mLでは,すべての値に媒体との差は認められなかった。 以上の結果から,アナグリプチンは 30 μg/mL までモルモット摘出乳頭筋の活動電位に作用
を及ぼさないと考えられた。 作用が認められなかった 30 μg/mL と最高臨床用量における推定曝露量を比較すると,約
25 倍の開きがあった。
表 2.6.2.4-2 モルモット摘出乳頭筋の活動電位パラメータに及ぼす作用
適用物質 媒体 アナグリプチン 塩酸ソタロール
適用濃度
適用後の 時間 - 30 μg/mL 100 μg/mL 300 μg/mL 3×10-5 mol/L
10 min 100.2 ± 0.9 99.3 ± 1.0 99.8 ± 0.5 99.8 ± 0.9 100.0 ± 0.8 RMP
30 min 100.4 ± 0.5 99.6 ± 0.6 99.3 ± 0.6 99.8 ± 0.9 100.0 ± 0.8
10 min 99.9 ± 1.2 99.5 ± 1.2 99.1 ± 0.3 98.3 ± 0.6* 99.7 ± 0.4 APA
30 min 99.9 ± 1.5 99.7 ± 0.9 98.8 ± 1.2 98.0 ± 0.7* 99.7 ± 0.4
10 min 99.5 ± 2.1 99.5 ± 2.4 95.1 ± 2.3 87.7 ± 4.5** 99.4 ± 2.2 Vmax
30 min 98.1 ± 5.5 96.4 ± 6.1 93.5 ± 2.9 84.2 ± 4.2** 99.1 ± 3.6
10 min 102.3 ± 2.2 102.4 ± 1.7 100.6 ± 1.4 101.2 ± 3.2 106.3 ± 1.0## APD30
30 min 104.6 ± 3.3 107.3 ± 2.6 100.5 ± 4.1 92.4 ± 4.2** 109.1 ± 1.5#
10 min 102.8 ± 1.8 103.1 ± 1.7 102.2 ± 1.5 101.9 ± 1.6 110.0 ± 2.3## APD50
30 min 105.2 ± 2.6 105.9 ± 2.0 102.7 ± 3.7 94.5 ± 3.7** 116.7 ± 2.8##
10 min 102.1 ± 1.8 102.8 ± 1.2 103.2 ± 1.9 103.2 ± 0.9 111.4 ± 2.6## APD90
30 min 104.1 ± 2.7 104.6 ± 1.3 103.9 ± 3.3 97.1 ± 3.0** 119.3 ± 4.0##
10 min 1.5 ± 1.6 3.2 ± 1.6 9.3 ± 3.6** 7.9 ± 4.5* 23.4 ± 6.9## APD30-90
30 min 3.0 ± 3.5 0.3 ± 3.8 11.6 ± 2.9** 7.0 ± 3.4 43.7 ± 11.6##
相対値(%)=(post / pre)×100,(pre:適用前の値,post:適用後の値),平均値±標準偏差(n=5) APD30-90相対値(%)=[(APD90post-APD30post)-(APD90pre-APD30pre)]/(APD90pre-APD30pre)×100 *:p<0.05,**:p<0.01;媒体との有意差(Dunnett の多重比較) #:p<0.05,##:p<0.01;媒体との有意差(t 検定)
(3) 無麻酔イヌを用いた心血管系に及ぼす作用[資料 4.2.1.3-5]
無麻酔雄性ビーグル犬にアナグリプチンの 30,100 及び 300 mg/kg を単回経口投与し,投
与後 0.5,1,2,3,6 及び 24 時間の血圧(収縮期血圧,拡張期血圧及び平均血圧),心拍数
及び心電図パラメータ(PQ 間隔,QRS 時間,QT 間隔及び QTc 間隔)に及ぼす作用を検討
した。血圧,心拍数及び心電図パラメータはテレメトリー法により測定した。 アナグリプチンの 30 及び 100 mg/kgは,血圧,心拍数及び心電図パラメータに作用を及ぼ
さなかった。300 mg/kgでは,投与後 1 時間から 6 時間のQRS時間に媒体に比べて有意な延長
- 40 -

2.6.2. 薬理試験の概要文
が認められたが,投与後 24 時間には投与前の値まで回復した(表 2.6.2.4-3)。一方,その他
の心電図パラメータ,血圧及び心拍数には 300 mg/kgでも作用は認められなかった。 したがって,アナグリプチンは 100 mg/kg までイヌの心血管系に作用を及ぼさないと考え
られた。 100 mg/kgにおけるアナグリプチンの推定曝露量は,同用量におけるイヌ 4 週間反復経口投
与毒性試験の初回投与後のCmaxが 83.9 μg/mLであることから(2.6.7.7D.項),最高臨床用量に
おける推定曝露量と比較すると,約 70 倍の開きがあった。
表 2.6.2.4-3 無麻酔イヌの心電図検査に及ぼすアナグリプチンの作用
投与後(時間)
投与量 (mg/kg)
n 投与前 0.5 1 2 3 6 24
媒体 4 97 ± 9 98 ± 5 102 ± 12 94 ± 9 98 ± 8 98 ± 12 89 ± 8
30 4 105 ± 11 99 ± 12 107 ± 11 115 ± 17 107 ± 10 104 ± 19 106 ± 12
100 4 108 ± 11 107 ± 12 114 ± 17 110 ± 10 110 ± 17 113 ± 22 94 ± 11
PQ 間隔
(msec)
300 4 109 ± 4 104 ± 8 110 ± 8 119 ± 17 113 ± 13 111 ± 18 101 ± 7
媒体 4 38 ± 1 38 ± 1 38 ± 2 38 ± 1 38 ± 1 37 ± 1 38 ± 1
30 4 39 ± 2 39 ± 2 39 ± 2 40 ± 2 41 ± 2 38 ± 2 39 ± 1
100 4 38 ± 2 37 ± 1 40 ± 1 41 ± 2 41 ± 2 40 ± 1 38 ± 2
QRS 時間
(msec)
300 4 38 ± 1 39 ± 1 43 ± 3** 44 ± 2** 42 ± 3* 41 ± 2* 38 ± 1
媒体 4 217 ± 14 206 ± 14 219 ± 10 210 ± 18 218 ± 11 221 ± 8 206 ± 6
30 4 218 ± 7 209 ± 10 211 ± 3 222 ± 11 221 ± 10 225 ± 8 225 ± 18
100 4 226 ± 12 213 ± 14 217 ± 8 220 ± 10 217 ± 14 235 ± 3 206 ± 9
QT 間隔
(msec)
300 4 227 ± 14 217 ± 7 210 ± 17 214 ± 12 213 ± 5 217 ± 9 213 ± 14
媒体 4 229 ± 12 221 ± 5 230 ± 14 228 ± 15 234 ± 11 232 ± 8 239 ± 3
30 4 234 ± 3 226 ± 15 232 ± 12 228 ± 19 227 ± 9 239 ± 9 233 ± 12
100 4 237 ± 13 229 ± 7 230 ± 6 241 ± 21 235 ± 8 230 ± 6 228 ± 5
QTc 間隔
(msec)
300 4 229 ± 6 237 ± 13 233 ± 6 235 ± 10 244 ± 8 243 ± 5 234 ± 15
平均値±標準偏差 QTc 間隔は Fridericia の補正式を用いて算出 *:p<0.05,**:p<0.01;媒体との有意差(Dunnett の多重比較)
(4) 心血管系に及ぼす作用について
hERG電流に及ぼす作用試験では,アナグリプチンの 100 及び 300 μg/mLでhERG電流相対
値が低値を示し,急速活性化遅延整流カリウムイオン電流(IKr)の抑制作用が示唆された。
また,モルモット摘出乳頭筋を用いた心筋活動電位持続時間に及ぼす作用試験では,300 μg/mLでAPA及びVmaxの減少が認められ,心筋ナトリウムイオン電流の抑制作用が示唆された。
更に 300 μg/mLでは,APD30,APD50及びAPD90相対値が低値を示し,電位依存性カルシウム
イオン電流及び遅延整流カリウムイオン電流(IK)の抑制作用が示唆された。100 及び 300 μg/mLでAPD30-90相対値が高値を示し,IKの抑制作用が示唆されたが,300 μg/mLでは活動電
位持続時間が短縮したことから,ナトリウムチャネル及びカルシウムチャネルの抑制が,カ
リウムチャネルの抑制より優位に作用すると考えられた。 以上の結果から,アナグリプチンは 30 μg/mL まで,ナトリウムチャネル,カルシウムチャ
ネル及びカリウムチャネルに作用を及ぼさないと考えられた。
- 41 -

2.6.2. 薬理試験の概要文
また,無麻酔イヌを用いた心血管系に及ぼす作用試験において,アナグリプチンの 300 mg/kg で認められた QRS 時間の延長は,ナトリウムチャネルの抑制作用に基づく伝導遅延と
考えられた。なお,本変化は投与後 24 時間には回復する可逆性変化であった。
(5) 代謝物の hERG 電流に及ぼす作用[資料 4.2.1.3-6]
ヒトでの主代謝物SKL-12320 の心血管系に及ぼす影響を明らかにする試験として,HEK293細胞に安定発現させたhERGチャネルの電流に及ぼす作用を検討した。hERG電流はパッチク
ランプ法により測定した。SKL-12320 は 31.5,105 及び 315 μg/mLを適用した。陽性対照物質
として設定したクラスIII抗不整脈薬であるE-4031 は 1×10-7 mol/Lを適用した。 E-4031 のhERG電流相対値は 7.7%であり,媒体(93.5%)に比べて有意な低下が認められ,
阻害効果が確認された(表 2.6.2.4-4)。 SKL-12320 の 31.5,105 及び 315 μg/mLにおけるhERG電流相対値は,それぞれ 92.2,93.5
及び 89.4%であり,媒体に比べて有意な差は認められなかった(表 2.6.2.4-4)。また,31.5,105 及び 315 μg/mLにおけるhERG電流に対する阻害率は,それぞれ 1.4,0.0 及び 4.4%であっ
た。 以上の結果から,SKL-12320は315 μg/mLまで hERG電流に作用を及ぼさないと考えられ,
hERG 電流に対する作用はアナグリプチンよりも弱いものと考えられた。 作用が認められなかった 315 μg/mL と最高臨床用量における推定曝露量を比較すると,約
1944 倍の開きがあった。
表 2.6.2.4-4 hERG 電流に及ぼす SKL-12320 の作用
適用物質 適用濃度 細胞数 相対値(%) 阻害率(%)
媒体 - 5 93.5±3.3 -
SKL-12320 31.5 μg/mL 5 92.2±4.0 1.4
SKL-12320 105 μg/mL 5 93.5±3.7 0.0
SKL-12320 315 μg/mL 5 89.4±2.1 4.4
E-4031 1×10-7 mol/L 5 7.7±2.8 ## 91.8 相対値=(適用 10 分後の値/適用前の値)×100,平均値±標準偏差 阻害率=100-[(SKL-12320 又は E-4031 の相対値)/(媒体の相対値)]×100 ##:p<0.01;媒体との有意差(t 検定)
2.6.2.4.4. 尿量,尿中電解質及び尿浸透圧に及ぼす作用[資料 4.2.1.3-7]
1 群 8 匹の雄性 Crl:CD(SD)ラットにアナグリプチンの 200,600 及び 2000 mg/kg を単回経
口投与し,尿量,尿中電解質及び尿浸透圧に及ぼす作用を検討した。 アナグリプチンの 600 及び 2000 mg/kg では,尿量の増加,並びに尿中ナトリウム,カリウ
ム及び塩素排泄量の増加が認められた。また,200 mg/kg でも,軽度ながら尿中ナトリウム
及びカリウム排泄量の増加が認められた。しかし,いずれの用量においても,尿浸透圧に変
化は認められなかった。
- 42 -

2.6.2. 薬理試験の概要文
2.6.2.4.5. 腎機能に及ぼす作用[資料 4.2.1.3-8]
1 群 8 匹の雄性 Crl:CD(SD)ラットにアナグリプチンの 200,600 及び 2000 mg/kg を単回経
口投与し,糸球体濾過量,腎血漿流量及び濾過率に及ぼす作用を検討した。 アナグリプチンの 2000 mg/kg では,糸球体濾過量の低下が投与後 60 及び 90 分に認められ,
腎血漿流量の低下が投与後 30,60 及び 90 分に認められたが,濾過率に影響は認められなか
った。600 mg/kg でも,糸球体濾過量の低下が投与後 60 分に認められたが,腎血漿流量及び
濾過率には作用を及ぼさなかった。200 mg/kg では,糸球体濾過量,腎血漿流量及び濾過率
に作用を及ぼさなかった。 腎/泌尿器系における安全域は,最低投与量の 200 mg/kgで尿中電解質に対して軽度ではあ
るが有意な増加が認められたため,明確となっていない。なお,200 mg/kgと最高臨床用量に
おける推定曝露量を比較すると,本薬未変化体で約 54 倍及びSKL-12320 で約 13 倍の開きが
あった。しかし,尿中電解質以外に 200 mg/kgで有意な変動は認められていないこと,ラッ
ト 26 週間反復経口投与毒性試験において,腎臓に対する無毒性量は 300 mg/kg/日であり,無
毒性量における曝露量は最高臨床用量における推定曝露量と比較して,Cmaxで 95 倍及び
AUC0-24hで 32 倍の開きがあることが確認されている(2.6.6. 表 2.6.6.9-1)。したがって,本薬
がヒトの腎/泌尿器系に対して問題となるような影響を及ぼす懸念は少ないものと考えられ
た。
2.6.2.4.6. 胃排出能に及ぼす作用[資料 4.2.1.3-9]
1 群 8 匹の雄性 Crl:CD(SD)ラットにアナグリプチンの 200,600 及び 2000 mg/kg を単回経
口投与し,胃排出能に及ぼす作用を検討した。 アナグリプチンの 600 及び 2000 mg/kg では,胃排出率の低下が認められた。200 mg/kg で
は,胃排出率に作用を及ぼさなかった。
2.6.2.4.7. 小腸輸送能に及ぼす作用[資料 4.2.1.3-10]
1 群 8 匹の雄性 Crl:CD(SD)ラットにアナグリプチンの 200,600 及び 2000 mg/kg を単回経
口投与し,小腸輸送能に及ぼす作用を検討した。 アナグリプチンの 2000 mg/kg では,炭末移行率の低下が認められ,小腸輸送能に対する抑
制作用が認められた。200 及び 600 mg/kg では,小腸輸送能に作用を及ぼさなかった。
2.6.2.4.8. 胃液分泌に及ぼす作用[資料 4.2.1.3-11]
1 群 8 匹の雄性 Crl:CD(SD)ラットにアナグリプチンの 200,600 及び 2000 mg/kg を単回経
口投与し,胃液分泌(胃液分泌量,pH,総酸排出量及びペプシン活性)に及ぼす作用を検討
した。 アナグリプチンの 2000 mg/kg では,pH の有意な上昇が認められ,これに伴う変化として
ペプシン活性の低下傾向が認められた。しかし,胃液分泌量及び総酸排出量には作用を及ぼ
さなかった。200 及び 600 mg/kg では,胃液分泌量,pH,総酸排出量及びペプシン活性に作
用を及ぼさなかった。
- 43 -

2.6.2. 薬理試験の概要文
胃腸管系に対する作用が認められなかった用量と最高臨床用量における推定曝露量を比較
すると,本薬未変化体及び SKL-12320 の安全域は,胃排出能で約 54 倍及び 13 倍,小腸輸送
能,胃液分泌で約 119 倍及び 25 倍の開きがあった。
2.6.2.4.9. 自律神経系に及ぼす作用[資料 4.2.1.3-12]
自律神経系に及ぼすアナグリプチンの作用を評価する目的で,1 群 5 標本のモルモット摘
出回腸標本にアナグリプチンを 38.3,115 及び 383 μg/mL(100,300 及び 1000 μmol/L)で適
用し,アセチルコリン,ヒスタミン,塩化バリウム及びセロトニンによる収縮反応に対する
作用を検討した。 アナグリプチンは 383 μg/mL まで,モルモット摘出回腸に対して単独作用を示さなかった。
また,塩化バリウム及びセロトニンによる収縮に対しても,383 μg/mL まで作用を及ぼさな
かった。一方,ヒスタミン収縮高比率の低下が,115 及び 383 μg/mL で認められた。更に,
383 μg/mL では,アセチルコリン収縮高比率の低下も認められた。 それぞれ作用が認められなかった濃度と最高臨床用量における推定曝露量を比較すると,
ヒスタミンで約 32 倍,アセチルコリンで約 96 倍,並びに塩化バリウム及びセロトニンで約
319 倍の開きがあった。
- 44 -

2.6.2. 薬理試験の概要文
2.6.2.5. 薬力学的薬物相互作用試験
2.6.2.5.1. GK ラットにおけるミグリトールとの併用効果[資料 4.2.1.4-1]
雄性 GK ラットの頸静脈に採血用のカテーテルを留置した。一晩絶食した GK ラット(8週齢,各群 7 又は 8 例)に対し,単独投与群には 3 mg/kg のアナグリプチン及び 3 mg/kg の
ミグリトール,併用投与群には 3 mg/kg のアナグリプチンと 3 mg/kg のミグリトール,対照
群(GK 対照群)には精製水をそれぞれ単回経口投与し,15 分後に液体流動食を 10 mL/kg の
容量で経口負荷した。なお,正常対照群(Wistar 対照群)として,雄性 Wistar 系ラット(8週齢,7 例)を使用した。液体流動食負荷前,並びに負荷後 15,30,60,90 及び 120 分にカ
テーテルより経時的に採血した。血漿 DPP-4 活性は蛍光法で,血漿活性型 GLP-1 濃度及び血
漿インスリン濃度は ELISA 法にて,血漿グルコース濃度は酵素法にてそれぞれ測定した。 アナグリプチンは,GK ラットの液体流動食負荷後 30 分(薬物投与後 45 分)における血
漿 DPP-4 活性を,単独投与では約 80%阻害し,ミグリトールとの併用投与では約 75%阻害し
た。一方,ミグリトールの単独投与は,血漿 DPP-4 活性に対して阻害作用を示さなかった
(2.6.3.5.項)。 アナグリプチンとミグリトールの併用投与では,液体流動食負荷後 0 分から 120 分までの
血漿活性型GLP-1 濃度変化量のAUC[⊿AUC(0 分の濃度を基準),253 pmol·min/L]が増加
し,GK対照群(16 pmol·min/L)及びそれぞれの単独投与(アナグリプチン:106 pmol·min/L,ミグリトール:−10 pmol·min/L)との有意な差が認められ,アナグリプチンの単独投与と比
較して約 2.4 倍高値を示した(図 2.6.2.5-1-a)。 液体流動食負荷後 0 分から 120 分までの血漿インスリン濃度及び血漿グルコース濃度の
AUCは,それぞれの単独投与ではGK対照群との有意な差は認められず,また,併用投与で
はそれぞれの単独投与との有意な差は認められなかった(図 2.6.2.5-1-b,図 2.6.2.5-1-c)。
- 45 -

2.6.2. 薬理試験の概要文
0
10000
20000
30000
40000
50000
血漿
グル
コー
ス濃
度の
AU
C0-120m
in_
(m
g ・m
in/dL
)_
§§§
c)
Wistar対照群(n=7)
GK対照群(n=8)
アナグリプチン(3 mg/kg)
(n=7)
ミグリトール(3 mg/kg)
(n=8)
併用
(n=7)
0
100
200
300
400
500
血漿
イン
スリ
ン濃
度の
AU
C0-120m
in_
(ng
・m
in/m
L)_
b)
Wistar対照群(n=7)
GK対照群(n=8)
アナグリプチン(3 mg/kg)
(n=7)
ミグリトール(3 mg/kg)
(n=8)
併用
(n=7)-100
0
100
200
300
400
血漿
活性
型G
LP
-1濃
度の
⊿A
UC
0-120m
in_
(pm
ol・
min
/L)__
Wistar対照群(n=7)
GK対照群(n=8)
アナグリプチン(3 mg/kg)
(n=7)
ミグリトール(3 mg/kg)
(n=8)
併用
(n=7)
***, ##, †††
a)
#
図 2.6.2.5-1 GK ラットにおける液体流動食負荷後の血漿活性型 GLP-1 濃度の⊿AUC,
並びに血漿インスリン濃度及び血漿グルコース濃度の AUC
a) 液体流動食負荷後 0 分から 120 分までの血漿活性型 GLP-1 濃度の⊿AUC b) 液体流動食負荷後 0 分から 120 分までの血漿インスリン濃度の AUC c) 液体流動食負荷後 0 分から 120 分までの血漿グルコース濃度の AUC 平均値±標準偏差 ***:p<0.001;GK ラット対照群との有意差(Tukey の多重比較) #:p<0.05,##:p<0.01;アナグリプチン投与群との有意差(Tukey の多重比較) †††:p<0.001;ミグリトール投与群との有意差(Tukey の多重比較) §§§:p<0.001;Wistar 対照群との有意差(t 検定)
- 46 -

2.6.2. 薬理試験の概要文
2.6.2.5.2. ZDF ラットにおけるメトホルミンとの併用効果[資料 4.2.1.4-2]
肥満・糖尿病モデル動物である雄性 ZDF ラットを用い,頸静脈に採血用のカテーテルを留
置した。一晩絶食した ZDF ラット(10 週齢,各群 8 例)の単独投与群には 3 mg/kg のアナグ
リプチン及び 300 mg/kg のメトホルミン,併用投与群には 3 mg/kg のアナグリプチンと 300 mg/kg のメトホルミン,対照群には精製水をそれぞれ単回経口投与し,15 分後に精製水に懸
濁させた 2 g/kg のスターチを経口負荷した。薬物投与前,スターチ負荷前,並びに負荷後 30,60,120 及び 180 分にカテーテルより経時的に採血した。血漿 DPP-4 活性は蛍光法で,血漿
活性型 GLP-1 濃度,血漿 GIP 濃度及び血漿インスリン濃度は ELISA 法にて,血漿グルコー
ス濃度は酵素法にてそれぞれ測定した。 アナグリプチンの単独投与及びメトホルミンとの併用投与は,ZDF ラットのスターチ負荷
後 30 分(薬物投与後 45 分)における血漿 DPP-4 活性をそれぞれ約 90%阻害した。一方,メ
トホルミンの単独投与は,血漿 DPP-4 活性に対して阻害作用を示さなかった(2.6.3.5.項)。 アナグリプチンとメトホルミンの併用投与では,スターチ負荷後 0 分から 180 分までの血
漿活性型GLP-1 濃度の⊿AUC(0 分の濃度を基準,724 pmol·min/L)がそれぞれの単独投与に
比べて増加し,アナグリプチンの単独投与(187 pmol·min/L)と比較して約 4 倍及びメトホル
ミンの単独投与(154 pmol·min/L)と比較して約 5 倍高値を示したが,統計学的に有意ではな
かった(図 2.6.2.5-2-a)。また,併用投与では,負荷後 0 分から 180 分までの血漿GIP 濃度の
AUC がメトホルミンの単独投与に比べて有意に減少したが,アナグリプチンの単独投与と
の差は認められなかった(図 2.6.2.5-2-b)。 スターチ負荷後 0 分から 180 分までの血漿インスリン濃度のAUCは,アナグリプチンの単
独投与では対照群に比べて有意に増加したが,併用投与では増加は認められず,アナグリプ
チンの単独投与に比べて有意に減少した(図 2.6.2.5-2-c)。また,スターチ負荷後 0 分から
180 分までの血漿グルコース濃度のAUCは,併用投与では対照群及びアナグリプチンの単独
投与に比べて有意に減少したが,メトホルミンの単独投与との差は認められなかった(図 2.6.2.5-2-d)。
- 47 -

2.6.2. 薬理試験の概要文
0
5000
10000
15000
20000
25000
30000
35000血
漿グ
ルコ
ース
濃度
のA
UC
0-180m
in
(m
g ・m
in/dL
)_ ***, †
d)
対照群 アナグリプチン(3 mg/kg)
メトホルミン(300 mg/kg)
併用0
500
1000
1500
2000
2500
血漿
イン
スリ
ン濃
度の
AU
C0-180m
in
(ng
・m
in/m
L)_
*
c)
††
対照群 アナグリプチン(3 mg/kg)
メトホルミン(300 mg/kg)
併用
0
200
400
600
800
1000
1200
血漿
活性
型G
LP
-1濃
度の
⊿A
UC
0-180m
in
(pm
ol・
min
/L)__
**
a)
対照群 アナグリプチン(3 mg/kg)
メトホルミン(300 mg/kg)
併用0
5000
10000
15000
20000
25000
30000
35000
血漿
GIP
濃度
のA
UC
0-180m
in
(pg
・m
in/m
L)__
b)
対照群 アナグリプチン(3 mg/kg)
メトホルミン(300 mg/kg)
併用
#
図 2.6.2.5-2 ZDF ラットにおける経口スターチ負荷後の血漿活性型 GLP-1 濃度の⊿AUC,
並びに血漿 GIP 濃度,血漿インスリン濃度及び血漿グルコース濃度の AUC
a) スターチ負荷後 0 分から 180 分までの血漿活性型 GLP-1 濃度の⊿AUC b) スターチ負荷後 0 分から 180 分までの血漿 GIP 濃度の AUC c) スターチ負荷後 0 分から 180 分までの血漿インスリン濃度の AUC d) スターチ負荷後 0 分から 180 分までの血漿グルコース濃度の AUC 平均値±標準偏差(n=8) *:p<0.05,**:p<0.01,***:p<0.001;対照群との有意差(Tukey の多重比較) #:p<0.05;メトホルミン投与群との有意差(Tukey の多重比較) †:p<0.05,††:p<0.01;アナグリプチン投与群との有意差(Tukey の多重比較)
- 48 -

2.6.2. 薬理試験の概要文
2.6.2.6. 考察及び結論
アナグリプチンは,ラット,イヌ及びヒト由来の各種DPP-4 をIC50値 3.3~6.4 nmol/Lで競
合的に阻害し,いずれのDPP-4 に対してもほぼ同程度の阻害作用を示した。アナグリプチン
のDPP-4 阻害作用は,ビルダグリプチンとほぼ同程度であり,シタグリプチンに比べて強か
った。また,DPP-8 及びDPP-9 を含むDPP-4 類縁酵素に対するIC50値を,ヒト組換えDPP-4 に
対するIC50値と比較すると,最も低い値を示したDPP-9 においても 17,000 倍となり,アナグ
リプチンはDPP-4 に対して高い選択性を示した。DPP-8 及びDPP-9 の阻害がラット及びイヌ
の毒性発現に関与することが,DPP-8/9 選択的阻害剤を用いた検討で報告されていることか
ら,DPP-4 阻害剤の酵素選択性は,薬剤の安全性に影響を及ぼす可能性が示唆される[3]。ア
ナグリプチンの酵素選択性を,類薬のビルダグリプチン及びシタグリプチンと比較した結果,
アナグリプチンでは 17,000 倍(DPP-9)以上であったのに対し,ビルダグリプチン及びシタ
グリプチンでは,それぞれ 440 倍(DPP-9)以上,及び 5,730 倍(DPP-8)以上であった。し
たがって,アナグリプチンは,DPP-4 に対する強力な阻害作用と優れた選択性を有するDPP-4阻害剤であると考えられた。 正常ラット及びイヌにアナグリプチンを単回経口投与した結果,用量に依存して血漿
DPP-4 活性を阻害し,その阻害強度は血漿中のアナグリプチン濃度に依存していた。ラット
への 6 週間反復経口投与において,DPP-4 阻害作用の減弱は認められなかった。また,正常
ラットを用いて,糖質負荷後の GLP-1 濃度に対する作用を評価した結果,経口スターチ負荷
後の血漿活性型 GLP-1 濃度が対照群に比べて著明に上昇した。したがって,アナグリプチン
は,経口投与で強力な血漿 DPP-4 活性阻害作用及び活性型 GLP-1 濃度上昇作用を示すことが
明らかとなった。 DPP-4 阻害剤は,血中に存在する GLP-1 をはじめとするインクレチンの分解を阻害するこ
とで,グルコース依存的なインスリン分泌促進作用を発揮するため,理論的には低血糖は発
現しないものと考えられている。アナグリプチンのラット絶食時血糖に対する作用を既存の
インスリン分泌促進剤と比較した結果,ナテグリニド及びグリベンクラミドで認められた絶
食時血糖値の低下は,アナグリプチンではほとんど認められなかった。したがって,アナグ
リプチンの単独療法では,低血糖の発現リスクは低いと考えられた。 ラットを用いて耐糖能に対する作用を評価した結果,アナグリプチンの単回経口投与は,
経口グルコース負荷後の正常ラットの血漿グルコース濃度の上昇を用量に依存して抑制した。
また,肥満・インスリン抵抗性モデル動物の Zucker fatty ラット及び非肥満 2 型糖尿病モデル
動物の GK ラットにおいて,経口グルコース負荷後の血漿インスリン濃度を増加させ,血漿
グルコース濃度上昇を抑制した。いずれのモデルにおいても,同様に耐糖能に対する作用が
確認されたことから,アナグリプチンは,肥満やインスリン抵抗性の程度に関わらず,広く
2 型糖尿病患者に対して有効性を示す可能性が示唆された。 糖尿病ラット及びマウスを用いたin vivo持続的皮下投与又は反復飲水投与試験の結果,ア
ナグリプチンは,膵臓インスリン含量及び膵β細胞量を増加させることが明らかとなった。
GLP-1 は多様な生理作用を示し,膵β細胞に対してはインスリン分泌及び生合成促進作用のほ
かに,細胞増殖促進作用及び細胞死抑制作用を有することが報告されている[4][5]。したがっ
て,アナグリプチンは,既存のDPP-4 阻害剤及びGLP-1 受容体作動薬と同様に,膵β細胞に対
して保護作用を発揮する可能性が示唆された。
- 49 -

2.6.2. 薬理試験の概要文
また,ハムスター膵 β細胞由来インスリノーマ細胞株HIT-T15を用いた in vitro試験の結果,
アナグリプチンは,GLP-1 共存下において HIT-T15 細胞の増殖を対照群に比べて有意に増加
させた。 アナグリプチンのヒトでの主代謝物であるSKL-12320 は,DPP-4 及び類縁酵素に対して阻
害作用を示さなかった。その他の代謝物では,SKL-14687 及びSKL-12277 は,DPP-4 阻害作
用を示さなかったが,SKL-12250,SKL-12339 及びSKL-13776 にDPP-4 阻害作用が認められ
た。[14C]アナグリプチンをヒトに経口投与したとき,主代謝物はSKL-12320 であり,その
他の代謝物の生成量はいずれも投与量の 1%未満であった(2.7.6.3.3.項)。したがって,代謝
物によるDPP-4 阻害作用は,臨床上特に問題とはならないと考えられた。また,類縁物質で
あるSKL-06327, SKL-12309 及びSKL-13775 は,DPP-4 阻害作用を示さなかった。 以上のことから,アナグリプチンの代謝物及び類縁物質が,DPP-4 及び類縁酵素に対して
作用を及ぼす可能性は極めて低いと考えられた。 副次的薬理試験として,アナグリプチン及び SKL-12320 の各種プロテアーゼ,受容体及び
イオンチャネルに対する阻害作用,並びにアナグリプチンのリンパ球の活性化に対する阻害
作用を検討した。アナグリプチン及び SKL-12320 は,500 μmol/L でいずれのプロテアーゼの
酵素活性に対して阻害作用を示さず,10 μmol/L でいずれの受容体及びイオンチャネルと特異
的リガンドとの結合に対して阻害作用を示さなかった。また,アナグリプチンは 50 μmol/Lで,マウス脾細胞を用いて評価した抗 CD3ε抗体刺激による T 細胞の増殖,及び LPS 刺激に
よる B 細胞の増殖に対して阻害作用を示さなかった。 DPP-4 はリンパ球の表面抗原であるCD26 と同一であり,T細胞活性化の共刺激に関与して
いることが示唆されているが,CD26 の表面抗原としての役割と,DPP-4 の酵素活性との間
に相関がないとの報告もある[6]。本検討により,少なくともアナグリプチンによるDPP-4 酵
素活性阻害は,リンパ球表面に存在するCD26 の表面抗原としての作用に影響を及ぼさない
ことが示唆された。 以上のことから,アナグリプチンの作用はDPP-4 への高い選択性を特徴とし,他の標的分
子に対して薬理作用を示す可能性は極めて低いと考えられた。なお,アナグリプチンの 500,10 及び 50 μmol/Lは,第I相反復投与試験の最終投与時の結果より,最高臨床用量である 1 回
200 mgを 1 日 2 回反復経口投与したときのCmax[1,200 ng/mL(3.1 μmol/L),2.7.6.3.2. 表 3.2-4]のそれぞれ約 161 倍,3 倍及び 16 倍である。同様に,SKL-12320(分子量 402.45)の 500 及
び 10 μmol/Lは,最高臨床用量におけるCmax[162 ng/mL(0.40 μmol/L)]のそれぞれ約 1,250倍及び 25 倍である。
安全性薬理試験において,アナグリプチンはラットの中枢神経系及び呼吸器系に対して作
用を及ぼさなかった。しかし,末梢神経系に対する作用と考えられる縮瞳が,500 mg/kgでは
認められなかったが,1000 mg/kg以上の単回経口投与で認められた。また,縮瞳はラット及
びイヌ反復投与毒性試験においても見られ,ラット 4 週間及び 13 週間反復経口投与毒性試験
での 2000 mg/kg/日投与(2.6.6.3.1.項, 2.6.6.3.2.項),イヌ単回経口投与毒性試験での 300 及び
1000 mg/kg投与(2.6.6.2.2.項),並びにイヌ 4 週間反復経口投与毒性試験での 300 mg/kg/日投
- 50 -

2.6.2. 薬理試験の概要文
与(2.6.6.3.4.項)において,いずれも雌雄に認められた。第I相反復投与試験の最終投与時の
結果より,最高臨床用量である 1 回 200 mgを 1 日 2 回反復経口投与したときのCmax及び
AUC0-24hは,それぞれ 1,200 ng/mL及び 9.46 μg·h/mLと推定された(2.7.6.3.2. 表 3.2-4,ただ
し,AUC0-24 hは最終投与が単回のため 2 倍値とした)。一方,ラット 4 週間反復経口投与毒性
試験の初回投与時の結果より,雄性ラットに 600 mg/kg/日を投与したときの血漿中アナグリ
プチンのCmax及びAUC0-24hは,それぞれ 143 μg/mL及び 376 μg·h/mLであった(2.6.7.7A.項)。
したがって,本試験で縮瞳が発現しなかったラットの 500 mg/kg単回経口投与における曝露
量は,600 mg/kg/日初回投与時の曝露量を下回るものと推定されることから,最高臨床用量
における推定曝露量と比較したところ,Cmaxで 119 倍以上,AUC0-24hで 40 倍以上の開きがあ
った。更に,第I相単回及び反復投与試験において瞳孔径検査を実施したところ,臨床上問題
となる所見は認められていない(2.7.6.3.1.項, 2.7.6.3.2.項)。 また,心血管系に及ぼす作用として,in vitro電気生理学的試験において,30 μg/mL(78.2
μmol/L)まで影響は認められなかったが,100 μg/mL(261 μmol/L)以上で急速活性化遅延整
流カリウムイオン電流の抑制作用が認められ,300 μg/mL(782 μmol/L)でモルモット心筋の
ナトリウムイオン電流の抑制,電位依存性カルシウムイオン電流の抑制及び遅延整流カリウ
ムイオン電流の抑制を示唆する作用が認められた。また,イヌを用いたin vivo試験において,
300 mg/kgでナトリウムチャネルの抑制作用に基づく伝導遅延が示唆されたが,投与後 24 時
間には回復する可逆性の変化であった。イヌ心血管系に対する無作用量は 100 mg/kgと考え
られ,イヌ 4 週間反復経口投与毒性試験の初回投与時の結果より,雄性イヌに 100 mg/kg/日を投与したときの血漿中アナグリプチンのCmax及びAUC0-24hは,それぞれ 83.9 μg/mL及び 376 μg·h/mLであった(2.6.7.7D.項)。このときの曝露量と最高臨床用量における推定曝露量を比
較すると,心血管系に対する安全係数はhERG電流及びモルモット心筋活動電位に対して約
25 倍,イヌ心血管系に対して 40~70 倍と算出された。また,イヌ及びサルの反復経口投与
毒性試験で行った心電図検査において,イヌでは 300 mg/kg/日で(2.6.6.3.4.項),サルでは 200 mg/kg/日以上で(2.6.6.3.5.~7.項),QTc間隔の延長を含む変化が認められたが,いずれも休
薬による回復性が確認された。4 週間反復投与毒性試験の成績から,イヌでは 100 mg/kg/日(28 回投与後の血漿中アナグリプチンのCmax:78.9~84.9 μg/mL,2.6.7.7D.項)まで,サルで
は 60 mg/kg/日(同:26.5~30.5 μg/mL,2.6.7.7E.項)まで,影響が認められなかった。このと
きのCmaxと最高臨床用量におけるCmaxを比較すると,イヌでは 66~71 倍,サルでは 22~25倍の開きがあった。なお,国内で実施された臨床試験において安静時 12 誘導心電図検査を行
った結果,臨床上問題となる所見は認められていない(2.7.6.1.1.~3.項,2.7.6.3.1.~2.項,
2.7.6.3.7.~9.項,2.7.6.4.2.項,2.7.6.5.1.~5.項)。また,海外で実施された外国人健康被験者を
対象としたQTc間隔への影響試験において,単回経口投与で最高投与量の 1600 mgまで,アナ
グリプチンの投与に起因したQTc間隔への影響は認められていない(2.7.6.4.1.項)。 その他,補足的安全性薬理試験として実施したラットの腎/泌尿器系に及ぼす作用試験にお
いて,200 mg/kg以上で尿中電解質排泄量の増加,600 mg/kg以上で尿量の増加及び糸球体濾
過量の低下,2000 mg/kgで腎血漿流量の低下がそれぞれ認められた。また,ラットの胃腸管
系に及ぼす作用試験において,600 mg/kg以上で胃排出能の抑制,2000 mg/kgで小腸輸送能の
抑制及びpHの上昇とこれに伴うペプシン活性の低下傾向が見られ,胃液分泌に対する影響が
認められた。ラット 4 週間反復経口投与毒性試験の初回投与時の結果より,ラットに 200
- 51 -

2.6.2. 薬理試験の概要文
mg/kg/日を投与したときの血漿中アナグリプチンのCmax及びAUC0-24hは,それぞれ 65.3 μg/mL及び 116 μg·h/mLであった(2.6.7.7A.項)。腎/泌尿器系に対して,200 mg/kgでラット尿中電解
質排泄量の増加が認められたが軽度な変化であり,その他には影響が見られていない。同用
量での曝露量(Cmax:65.3 μg/mL)は,最高臨床用量における推定曝露量と比較して約 54 倍
の開きがあった。また,ラット 26 週間反復経口投与毒性試験において,腎臓に対する無毒性
量は 300 mg/kg/日であり,この無毒性量における曝露量は最高臨床用量における推定曝露量
と比較して,Cmaxで 95 倍及びAUC0-24hで 32 倍の開きがあることが確認されている(2.6.6. 表 2.6.6.9-1)。したがって,本薬がヒトの腎/泌尿器系に対して問題となるような影響を及ぼす懸
念は少ないものと考えられた。胃腸管系に対して,作用が認められなかった 200 mg/kgにお
ける推定曝露量と最高臨床用量における推定曝露量の間には,Cmaxで約 54 倍,AUC0-24hで約
12 倍の開きがあった。また,モルモット摘出回腸を用いた自律神経系に及ぼす作用試験にお
いて,115 μg/mL以上でヒスタミン及び 383 μg/mLでアセチルコリンによる収縮反応を抑制し
たが,38.3 μg/mLでは作用を及ぼさなかった。作用が認められなかった 38.3 μg/mLと最高臨
床用量におけるCmaxの間には,約 32 倍の開きがあった。 代謝物の安全性薬理試験として,ヒトでの主代謝物であるSKL-12320 のhERG電流に及ぼす
作用を検討した。SKL-12320 は,315 μg/mLまでhERG電流に作用を及ぼさなかった。なお,
SKL-12320 の 315 μg/mLは,第I相反復投与試験の最終投与時の結果より,最高臨床用量であ
る1回200 mgを1日2回反復経口投与したときの血漿中SKL-12320のCmax(162 ng/mL,2.7.6.3.2. 表 3.2-4)の 1,900 倍以上である。 安全性薬理試験で認められた作用は,効力を裏付ける試験で有効性を示した用量に比べて
高い用量でのみ認められ,最高臨床用量における推定曝露量と比較して十分な開きがあると
推察されたことから,これらの作用が臨床上問題となる可能性は低いものと考えられた。 アナグリプチンと α-GI 剤であるミグリトールとの併用効果は,GK ラットにおける経口液
体流動食負荷試験を行い,また,BG 剤であるメトホルミンとの併用効果は,ZDF ラットに
おける経口スターチ負荷試験を行って評価した。アナグリプチンとミグリトールの併用投与
は,負荷後の血漿活性型GLP-1濃度のAUCをそれぞれの単独投与に比べて有意に増加させ,
アナグリプチンとメトホルミンの併用投与は,負荷後の血漿活性型 GLP-1 濃度の AUC をそ
れぞれの単独投与に比べて増加させたものの,有意な変化ではなかった。 以上より,アナグリプチンは選択的かつ強力に DPP-4 を阻害し,内因性の活性型 GLP-1
濃度上昇を介したグルコース依存的なインスリン分泌を促進して血糖上昇を抑制し,血糖コ
ントロールを改善した。更に,膵 β 細胞に対する保護作用を発揮する可能性が示唆された。
また,作用機序が異なる経口血糖降下剤であるミグリトールとの併用では,単独投与に比べ
て GLP-1 濃度の上昇が期待された。副次的薬理及び安全性薬理評価から,臨床上特に問題と
なる作用は認められなかった。 したがって,アナグリプチンは,2 型糖尿病治療において高い有用性を持つ薬剤として期
待される。
- 52 -

2.6.2. 薬理試験の概要文
2.6.2.7. 図表
図表は本文中の適切な場所に記載した。
- 53 -

2.6.2. 薬理試験の概要文
2.6.2.8. 参考文献
[1] Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv
Enzymol Relat Areas Mol Biol. 1988;61:201-301.
[2] Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for
assessing the behavioral and physiologic state of the mouse. Psychopharmacologia.
1968;13(3):222-57.
[3] Lankas GR, Leiting B, Roy RS, Eiermann GJ, Beconi MG, Biftu T, et al. Dipeptidyl peptidase IV
inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl
peptidases 8 and 9. Diabetes. 2005;54(10):2988-94.
[4] Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology.
2007;132(6):2131-57.
[5] Buteau J. GLP-1 receptor signaling: effects on pancreatic β-cell proliferation and survival. Diabetes
Metab. 2008;34 Suppl 2:S73-7.
[6] Vora KA, Porter G, Peng R, Cui Y, Pryor K, Eiermann G, et al. Genetic ablation or pharmacological
blockade of dipeptidyl peptidase IV does not impact T cell-dependent immune responses. BMC
Immunol. 2009;10:19.
- 54 -