
i
NEY CARTER DO CARMO BORGES
APLICAÇÃO DOS ESTUDOS DE
BIOEQUIVALÊNCIA AOS FÁRMACOS DE
AÇÃO CARDIOVASCULAR
CAMPINAS 2005

iii
NEY CARTER DO CARMO BORGES
APLICAÇÃO DOS ESTUDOS DE
BIOEQUIVALÊNCIA AOS FÁRMACOS DE
AÇÃO CARDIOVASCULAR Tese de doutorado apresentada à Pós-Graduação da Faculdade de Ciências Médicas da Universidade Estadual de Campinas para a obtenção do título de Doutor em Farmacologia.
Orientador: Prof. Dr. Glberto de Nucci
CAMPINAS 2005

iv
FICHA CATALOGRÁFICA ELABORADA PELA BIBLIOTECA DA FACULDADE DE CIÊNCIAS MÉDICAS DA UNICAMP
Bibliotecário: Sandra Lúcia Pereira – CRB-8ª / 6044
Borges, Ney Carter do Carmo B644a Aplicação de estudos de bioequivalência às drogas de ação
cardiovascular. / Ney Carter do Carmo Borges. Campinas, SP : [s.n.], 2005.
Orientador : Gilberto de Nucci Tese ( Doutorado ) Universidade Estadual de Campinas. Faculdade
de Ciências Médicas. 1. Medicamentos. 2. Plasma. 3. Medicamentos genéricos-
farmacocinética. 4. Espectometria de massa. I. Nucci, Gilberto de. II. Universidade Estadual de Campinas. Faculdade de Ciências Médicas. III. Título.
(slp/fcm)

v
UNICAMP
Banca Examinadora da Tese de Doutorado
Orientador:
Prof. Dr. Gilberto de Nucci Membros:
Prof. Dr. Gilberto de Nucci
Prof. Dr. Eduardo Arantes Nogueira
Prof. Dr. José Roberto Maiello
Prof. Dr. Francisco Hideo Aoki
Prof. Dr. José Francisco Kerr Saraiva Programa de Pós-Graduação em Farmacologia da Faculdade de Ciências Médicas da Universidade Estadual de Campinas.
Data: 14/10/2005

vii
AGRADECIMENTOS
Agradeço a Deus pela chance de vida.
A minha esposa Márcia e aos meus filhos Bruno, Diego e Ney Jr., pela
tolerância e amor dedicados a mim.
Em especial ao Prof. Dr. Gilberto De Nucci - meu orientador e amigo, pela
paciência e confiança, mesmo após 10 longos anos de ausência.
Ao Dr. Gustavo Duarte Mendes pelo apoio e colaboração constante.
A todos que de forma direta e indireta contribuíram para realização deste
trabalho.
A todos o meu mais sincero obrigado.

ix
Trabalho, solidariedade e tolerância. León Hippolyte Denizart Rivail

xi
SUMÁRIO
LISTA DE ABREVIATURAS...............................................................................................xv
RESUMO.......................................................................................................................... xvii
SUMMARY ........................................................................................................................ xix
1. INTRODUÇÃO .............................................................................................................. 21
2. OBJETIVO..................................................................................................................... 29
CAPÍTULO 1 - Ticlopidine quantification in human plasma by high-performance liquid chromatography coupled to electrospray tandem mass spectrometry.application to bioequivalence study.................................................... 33
CAPÍTULO 2 - Comparative bioavailability study with two gemfibrozil tablets formulations in healthy volunteers ....................................................... 43
CAPÍTULO 3 - Quantification of carvedilol in human plasma by high-performance liquid chromatography coupled to electrospray tandem mass spectrometry application to bioequivalence study....................................... 51
CAPÍTULO 4 - Verapamil quantification in human plasma by liquid chromatography coupled to tandem mass spectrometry an application for bioequivalence study.................................................................................. 63
CAPÍTULO 5 - Comparative bioavailability of two ramipril formulations in healthy volunteers after a single dose administration ............................... 73
CAPÍTULO 6 - A bioequivalence study of citalopram based on quantification by liquid high-performance liquid chromatography coupled to electrospray tandem mass spectrometry ......................................................... 93
3. DISCUSSÃO ............................................................................................................... 105
4. CONCLUSÃO.............................................................................................................. 115
5. REFERÊNCIAS BIBLIOGRÁFICAS ........................................................................... 119

xiii
LISTA DE TABELAS
TABELA 1 - Anamnese e Exames Laboratoriais vinculados ao Processo de Seleção dos Voluntários ................................................................................................. 112
TABELA 2 - Características gerais da metodologia empregada através de cromatografia liquida de alta eficiência acoplada a espectrômetro de massa (LC-MS-MS) para quantificação dos diferentes medicamentos ...................................... 113

xv
LISTA DE ABREVIATURAS
AHA - American Heart Association
ANVISA - Agência Nacional de Vigilância Sanitária
ASC - Área sob a curva farmacocinética da substância analisada
BPFC - Boas Práticas de Fabricação e Controle de Qualidade
BPL - Boas Práticas de Laboratório
CB - Ciências Biomédicas da Universidade de São Paulo
CEP - Comitê de Ética em Pesquisa
Cmax - Pico de concentração plasmática máxima do medicamento
CONEP - Conselho Nacional de Ética em Pesquisa
FDA - Food and Drug Administration -Agência de Controle de
Medicamentos e Alimentos dos Estados Unidos da América
FUNCOR - Fundo de Aperfeiçoamento e Pesquisa Cardiológica
BPC - Boas Práticas Clínicas
HU - Hospital Universitário da Universidade de São Paulo
ICB - Instituto de Ciências Biomédicas
ICH - Conferência Internacional de Harmonização
LC-MS-MS - Cromatografia líquida de alta eficiência, acoplada a espectrômetro de
massa
LOQ - Limite de Quantificação
OMS - Organização Mundial da Saúde
RDC - Resolução da Diretoria Colegiada
SBC - Sociedade Brasileira de Cardiologia
Tmax - Tempo do pico de concentração máxima da droga
UNICAMP - Universidade Estadual de Campinas
USP - Universidade de São Paulo

xvii
RESUMO
A quantificação de substâncias farmacologicamente ativas em matrizes biológicas
pode ser obtida através de estudos de bioequivalência. Hoje em dia, os estudos
de bioequivalência com drogas de ação cardiovascular são pontos crítico na
comunidade científica, usando detectores tais como a espectrometria de massa
acoplada com cromatografia líquida de alta eficiência; o HPLC-MS-MS vem sendo
aplicado progressivamente para quantificação de fármacos em várias partes do
mundo inclusive no Brasil. Muitos aspectos diferentes, tais como uma
sensibilidade mais elevada, precisão, explicam certamente este fato: a
substituição com fármacos genéricos dos fármacos de referência, transformou-se
uma prática difundida em nosso país. Após aprovação pela ANVISA estas drogas
sendo bioequivalentes, são uma opção aos pacientes e as seguradoras de saúde
as quais procuram maneiras para baixar custos do sistema de saúde. Os
interesses foram levantados, entretanto, com o uso descontrolado de tal
substituição pode certamente ser prejudicial em determinados casos. Neste
estudo, nós analisamos 6 (seis) diferentes estudos de bioequivalência. As
amostras do plasma foram extraídas e quantificadas pelo HPLC-MS-MS na
unidade analítica de Cartesius (SP de São Paulo I). Os estudos aqui apresentados
são práticos, precisos com elevada significância e exatidão na avaliação de tais
drogas de ação cardiovascular, podendo contribuir desta forma com a comunidade
de médicos, profissionais da saúde e com os pacientes portadores de doença
cardiovascular na determinação se tais são bioequivalentes.
Palavras chave: Medicamentos, Plasma, Medicamentos genéricos,
Farmacocinética, Espectometria de massa.

xix
SUMMARY
The quantification of pharmacologically active substances in biological materials
can be performed through bioequivalence studies. Now-a-days, bioequivalence
study of cardiovascular drugs is critical point in scientific community, using
detectors such as mass spectrometry with HPLC; HPLC-MS-MS have been
progressively applied to this drugs quantifications in worldwide, including Brazil.
Many different aspects, such as higher sensitivity, precision, surely explain this
fact, specially generic substitution for drugs has become a widespread practice as
ANVISA approves bioequivalent drugs, and patients and payors seek ways to trim
healthcare costs. Concerns have been raised, however, that uncontrolled use of
such substitution may indeed be harmful in certain cases. In this study, we analyze
6 different bioequivalence studies. Plasma samples were extracted and quantified
by HPLC-MS-MS at the Cartesius Analytical Unit (ICB/USP - São Paulo - SP). The
herein presented studies practical with significantly high precision and accuracy in
evaluation of such drugs to contribute of physician’s community with cardiovascular
patients in determination if all of them are bioequivalence.
Key words: Drugs, Plasma, Generic drugs, Pharmacokinetics, Mass spectrometry .

1
Introdução

Introdução
23
1. Aplicações dos estudos farmacocinéticos.
O comportamento farmacocinético de uma nova droga é, normalmente,
determinado em animais antes de ser utilizado em humanos. Os testes em
animais de laboratório seguem até o ponto em que se faz necessário obter
resultados em experimentos clínicos envolvendo seres humanos, pois existem
variações nos parâmetros farmacocinéticos quando comparamos resultados
obtidos em animais e em humanos (ROBERTS et aI.,1988). Um número cada vez
maior de novas substâncias com potencial terapêutico, bem como a
comercialização de produtos similares por diferentes empresas, tem estimulado o
desenvolvimento de novos métodos para o controle de qualidade. Os ensaios
clínicos tomaram-se importantes ferramentas para avaliar isolada ou de forma
comparativa as propriedades farmacocinéticas e farmacodinâmicas (ATKINSON et
aI., 2001).O desenvolvimento e a regulamentação destes ensaios clínicos
tornaram-se reconhecidamente mais importantes a partir da década de 50, quando
passaram a ser conduzidos de acordo com os interesses científicos e éticos. Em
1959 BULL introduz o conceito de estudo duplo cego, onde nem médico nem
paciente tinham conhecimento prévio da formulação administrada. Sir Austin
Bradford Hill foi o principal pesquisador envolvido com ensaios clínicos no
"Medical Research Council" tendo organizado vários estudos clínicos e publicado
diversos artigos sobre condução destes protocolos, demonstrando a importância
da escolha dos pacientes, randomização, avaliação objetiva e análise estatística
(ATKINSON et al., 2001). Os motivos que retardaram o progresso dos ensaios
clínicos foram "reverência à autoridade, relacionamento entre médico e paciente,
poucos registros, falta de facilidades para investigação e falta de remédios ativos".
A razão mais importante para o desenvolvimento de novos estudos foi a crescente
preocupação com o tratamento de doenças não transmissíveis (BULL et al., 1959).
A indústria farmacêutica sofreu uma enorme expansão nos últimos 40 anos, e com
isso, novas drogas foram descobertas e sintetizadas. Antes da II Guerra Mundial
não havia qualquer controle para que uma droga fosse livremente comercializada.
Contudo nos Estados Unidos já existia, desde 1938, legislação para uso de drogas

Introdução
24
em animais. Foi apenas na década de 60, com a catástrofe da talidomida, que
tanto os Estados Unidos como a Inglaterra, intensificaram as regulamentações
para execução de ensaios em humanos. A partir de 1963 é obrigatória uma
aprovação oficial para que a droga inicie um ensaio clínico e posteriormente, nova
aprovação para ser comercializada no Reino Unido. Nos Estados Unidos, o FDA
(FOOD AND DRUG ADMINISTRATION) desenvolveu e aplicou diretriz (a partir da
década de 70) para a elaboração de um modelo seguro de pesquisa clínica,
seguido por muitos outros países, diferindo do modelo britânico, principalmente
por valorizar sobremaneira a documentação dos dados obtidos (ATKINSON at al.,
2001).
Em 1964, a Associação Médica Mundial aprova em Helsinque um
documento com princípios para proteção de indivíduos em pesquisa biomédica.
São introduzidos conceitos de responsabilidades do investigador, comitês de ética
e consentimento livre e esclarecido. Com revisões periódicas posteriores (1975,
1983, 1989 e 2000), a Declaração de Helsinque constitui-se, atualmente, no
documento universal que rege os parâmetros para o desenvolvimento científico e
tecnológico envolvendo seres humanos.
Em 1977, o FDA publica as primeiras diretrizes para pesquisas clínicas
com o objetivo de garantir qualidade dos dados e proteger os participantes das
mesmas. Ente 1977 e 1981, novas diretrizes sobre Boas Práticas Clínicas são
publicadas. Em 1988, uma consolidação de um código de Boas Práticas Clínicas
(BPC) é publicada pelo FDA.
As Boas Práticas Clínicas também foram adotadas em outros países:
• 1985 – Japão e Canadá
• 1986 – Inglaterra
• 1991 – Austrália e Comunidade Européia
• 1995 – Publicação do código de Boas Práticas Clínicas (GCP) pela
OMS.

Introdução
25
Em 1996, a realização da Conferência Internacional de Harmonização
(ICH) e posterior introdução das alterações propostas serviram de alicerce para
que os estudos clínicos pudessem ser conduzidos de acordo com as normas e
regulamentos similares em diferentes países e em conformidade com elevados
padrões éticos e científicos.
No Brasil, a implantação de normas definindo a pesquisa em seres
humanos deu-se com a Resolução nº 01/88, através de iniciativa do Professor
Elisaldo Carlini. Em outubro de 1996, esta foi revogada pela resolução nº
196/MS/CNS, sendo posteriormente completada pela Resolução nº 251/97. Por
meio destas Resoluções, o Ministério da Saúde define diretriz e normas
objetivando promover a proteção de sujeito de pesquisas envolvendo seres
humanos.
A Resolução 196/96, baseada nos quatro referenciais básicos da
bioética, autonomia, não maleficência, beneficência e justiça, traz à comunidade
científica, bem como à sociedade brasileira, reflexões sobre os aspectos éticos da
pesquisa envolvendo seres humanos e estabelece as diretrizes para a
implantação de um sistema de revisão ética em pesquisa, composto por Comitês
de Ética em Pesquisa (CEP) e pela Comissão Nacional de Ética em Pesquisa
(CONEP), do Conselho Nacional de Saúde.
Assim, a Resolução 196/96, bem como as demais que complementam,
traz ao pesquisador as orientações para que o mesmo possa desenvolver, de
forma ética, pesquisas envolvendo seres humanos.
Esta Resolução estabelece que todas as pesquisas desenvolvidas com
seres humanos devem ser submetidas à apreciação de um Comitê de Ética em
Pesquisa (CEP), credenciado pela CONEP (Comissão Nacional de Ética em
Pesquisa). A resolução também preconiza que todas as instituições que realizam
pesquisas implantem o CEP, a fim de se promover em toda a rede o
desenvolvimento científico e tecnológico fundamentado nos princípios de ética e
do respeito à cidadania. Na impossibilidade de constituir um CEP de outra

Introdução
26
instituição ou o pesquisador principal deverá submeter o projeto a apreciação do
CEP de outra instituição, dentre os indicados pelo CONEP.
2. Genéricos
Medicamento genérico é um medicamento similar a um produto de
referência ou inovador, que pretende ser com este intercambiável, geralmente
produzido após expiração ou renúncia da proteção patenteária ou de outros
direitos de exclusividade, comprovada a sua eficácia, segurança e qualidade, e
designado pela Denominação Comum Brasileira ou, na sua ausência, pela
Denominação Comum Internacional.
No Brasil, a questão dos genéricos começa a tomar fôlego, a partir da
Lei 9.787, sancionada pelo presidente da República Fernando Henrique Cardoso,
em 10 de fevereiro de 1999, que estabelece o que é medicamento genérico e
dispõe sobre a utilização de nomes genéricos em produtos farmacêuticos, sob
fiscalização da ANVISA (Agência Nacional de Vigilância Sanitária). Essa
legislação foi revista e atualizada com o objetivo de melhor esclarecer as
exigências, em relação aos medicamentos genéricos através da RDC 10 de
janeiro de 2001. Posteriormente, foi editada a RDC 84, de março de 2002, com o
intuito de detalhar as regulamentações já previstas na RDC 10. Esta nova
resolução transforma os anexos da RDC 10 em Guias publicados na forma de
resoluções (RE), como por exemplo, a RE 478-Guia para provas de
bioequivalência de medicamentos Genéricos (RIBEIRO, 2000). O objetivo dessa
Legislação é aumentar a oferta de medicamentos, o que resultará numa maior
concorrência entre os fabricantes e na diminuição dos gastos da população com
os mesmos (ABIFAM, 2001). O surgimento dos produtos denominados genéricos
aparece como uma alternativa aos produtos tradicionais e podem revolucionar as
questões inerentes à falta de recursos dos governos para subsidiar a população
de baixa renda (ABIFAM,2001).

Introdução
27
Do ponto de vista econômico, fala-se de um bem substituto à altura dos
concorrentes, tendo sua intercambialidade garantida pelos estudos de
bioequivalência, com preços competitivos e que podem mudar o cenário atual de
consumo da população brasileira, principalmente referindo-se aos menos
favorecidos (RIBEIRO, 2000).
A adoção de uma política de medicamentos genéricos, envolvendo a
produção, a garantia de qualidade, a prescrição, a dispensação e o uso dos
mesmos é parte fundamental de uma diretriz para promoção e o uso racional de
medicamentos em nosso País. A promoção do uso racional de medicamentos é,
também, uma das principais diretrizes preconizadas pela Organização Mundial da
Saúde - OMS. A fim de alcançar esse objetivo, é fundamental a realização de
estudos de farmacocinética comparada de fármacos, para verificar se existe
bioequivalência entre o medicamento teste e o medicamento referência adotado
como padrão pelo Ministério da Saúde. Também se torna necessário o
desenvolvimento de metodologias de baixo custo e fácil aplicação, com
sensibilidade e seletividade adequadas para realização desde tipo de estudo.
Considerando a importância dos estudos de biodisponibilidade/
bioequivalência no contexto da estratégia da política de implantação de
medicamentos genéricos no Brasil, os pesquisadores deverão estar atentos às
diretrizes estabelecidas pelo Ministério da Saúde na área de ética em pesquisas.
Pode-se afirmar que a cerca de estudos de biodisponibilidade iniciou-se
a partir de 1945, com a primeira publicação do conceito de disponibilidade
biológica. O desenvolvimento, durante a década de 1960, de técnicas analíticas
possibilitou o desenvolvimento de métodos sensíveis o suficiente para permitir a
quantificação de drogas ou metabólitos, inicialmente a urina, e posteriormente no
plasma, o que possibilitou a avaliação e comparação de biodisponibilidade de
diferentes formulações em voluntários, bem como a demonstração de que
diferenças significativas entre estas podem ocorrer.
Após a legislação de registro compulsório de medicamentos em 1969,
que facilitou a entrada de medicamentos genéricos no mercado canadense, o

Introdução
28
“Drugs Directorate” do “Canadian Federal Department of Health and Welfare”
começou a utilizar bioequivalencia como uma medida para aprovar o registro de
um medicamento durante a década de 1970. Este programa, juntamente com
outras informações, foi analisado pelo FDA, órgão a editar as primeiras diretrizes
para a realização de estudos de bioequivalência em 1977 A aplicação destas
diretrizes foi ampliada no “Drug Price Competition na Patent Term Restoration Act”
de 1984, o qual concedia ao FDA poderes para autorizar a aprovação de drogas
genéricas sem evidências clínicas de segurança ou eficácia, desde que a droga
seja bioequivalente ao produto inovador.

2
Objetivo

Objetivo
31
O presente trabalho teve por objetivo principal avaliar o desempenho de
6 (seis) fármacos com ação cardiovascular através de estudos de bioequivalência
realizado em voluntários sadios de ambos os sexos e em hospital brasileiro, pela
Unidade Analítica Cartesius - do Instituto de Ciências Biomédicas (ICB) da
Universidade de São Paulo (USP). Os estudos apresentados são os dos seguintes
medicamentos: (ticlopidina, gemfibrozil, carvedilol, verapamil, ramipril e
citalopram).

CAPÍTULO 1
Artigo publicado
“Ticlopidine quantification in human plasma by high-performance
liquid chromatography coupled to electrospray tandem mass
spectrometry. Appilcation to bioequivalence study”
Ney Carter do Carmo Borges,1 Gustavo Duarte Mendes,2,3 André Borges,2,3
Sandro Evandir de Oliveira,2,3 Rafael Eliseo Barrientos-Astigarraga2 and
Gilberto De Nucci1,2,3* - J.Mass.Spectrom- 39: 1562 -1569 , 2004

Capítulo 1
35

Capítulo 1
36

Capítulo 1
37

Capítulo 1
38

Capítulo 1
39

Capítulo 1
40

Capítulo 1
41

Capítulo 1
42

CAPÍTULO 2
Artigo publicado
“Comparative bioavailability study with two gemfibrozil tablet
formulations in healthy volunteers”
Ney Carter do C. Borges a ,Gustavo Duarte Mendesbc ,Rafael E.Barrientos-
Astigarragab ,Eduardo Zappib ,Fabiana Duarte Mendesb,and Gilberto De
Nuccia,b,c
Arzneim. Forch. Drug. Res. 55: 382 – 386, 2005

Capítulo 2
45

Capítulo 2
46

Capítulo 2
47

Capítulo 2
48

Capítulo 2
49

CAPÍTULO 3
Artigo publicado
“Quantification of carvedilol un human plasma by high-
performance liquid chromatography coupled to electrospray
tandem mass spectrometry application to bioequivalence study”
Ney Carter do Carmo Borgesa , Gustavo Duarte Mendesb,c, Diogo de Oliveira
Silvac ,Vinicius Marcondes Rezendec , Rafael Eliseo Barrientos-astigarragab ,
Gilberto De Nuccia,b,c,* J.Chromatogr B. 822: 253 – 262, 2005

Capítulo 3
53

Capítulo 3
54

Capítulo 3
55

Capítulo 3
56

Capítulo 3
57

Capítulo 3
58

Capítulo 3
59

Capítulo 3
60

Capítulo 3
61

CAPÍTULO 4
Artigo Publicado
“Verapramil quantification in human plasma by liquid
chromatography coupled to tandem mass spectrometry an
application for bioequivalence study”
Ney Carter do C.Borgesa ,Gustavo Duarte Mendesb,c , Rafael E.Barrientos-
Astigarragab , PauloGalvinasb ,Celso H.Oliveirab ,Gilberto De Nuccia,b,c,*
J.Chromatogr B. 827 ( 2005 ) - 165-172

Capítulo 4
65

Capítulo 4
66

Capítulo 4
67
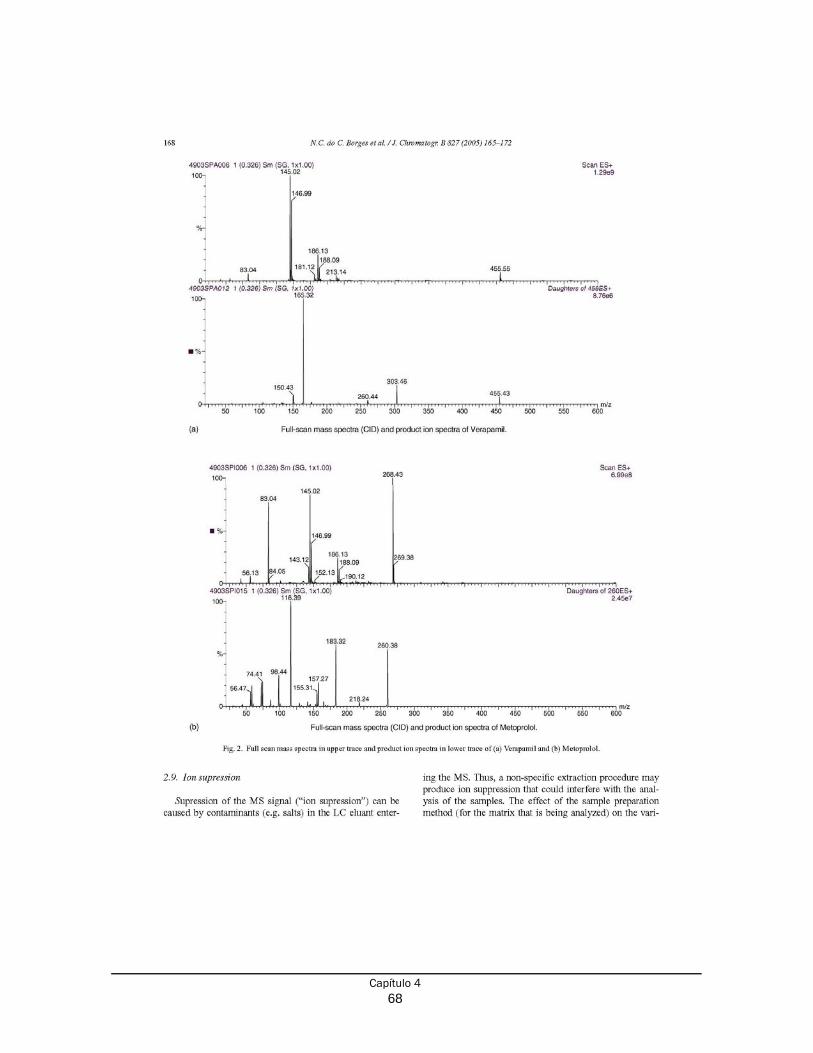
Capítulo 4
68

Capítulo 4
69

Capítulo 4
70

Capítulo 4
71

Capítulo 4
72

CAPÍTULO 5
Artigo Submetido
“Comparative bioavailability of two ramipril formulations in
healthy human volunteers after a single dose administration”
Gustavo Duarte Mendes1,3 ,André Tasso Dantas Sanfelice1 ,Ney Carter do
Carmo Borges2 ,Luiz Eduardo Cavedal2 ,João Henrique Módulo3 and Gilberto
De Nucci1,2,3*
Int.J.Clin.Pharmacol.Ther. 2005- IN PRESS


Capítulo 5
75

Capítulo 5
76
Comparative bioavailability of two Ramipril formulations in healthy human
volunteers after a single dose administration.
Gustavo Duarte Mendes1,3, André Tasso Dantas Sanfelice1, Ney Carter do Carmo
Borges2, Luiz Eduardo Cavedal2, João Henrique Módolo3 & Gilberto De Nucci1,2,3 *
1 Department of Internal Medicine, State University of Campinas, Campinas/SP,
Brazil
2 Department of Pharmacology, State University of Campinas, PO Box 6111
Campinas/SP, Brazil
3 Cartesius Development of Clinical Research, Jesuino Marcondes Machado Ave,
415, 13092-320 Campinas/SP, Brazil
Running title: ramipril and ramiprilat bioequivalence study
Key words: tandem mass spectrometry, LC/MS/MS, bioavailability,
pharmacokinetics
* Author for correspondence:
Gilberto De Nucci, MD, PhD.
415 Jesuino Marcondes Machado Ave
Campinas, SP, Brazil
Postal Code: 13092-320
Fax: 55 19 3252-1516
E-mail: [email protected]

Capítulo 5
77
Summary
Objective: To assess the bioequivalence of one ramipril 5 mg tablet formulation
(ramipril test formulation from Laboratórios Biosintética Ltda and Triatec® from
Aventis Pharma standard reference formulation) in 26 healthy volunteers of both
sexes. Methods: The study was conducted using an open, randomized, two-period
crossover design with a 2-week washout interval. Plasma samples were obtained
over a 36 h period. Plasma ramipril and ramiprilat concentrations were analyzed by
liquid chromatography coupled to tandem mass spectrometry (LC-MS-MS) with
positive ion electrospray ionization using multiple reaction monitoring (MRM). From
the ramipril and ramiprilat plasma concentration vs time curves, the following
pharmacokinetic parameters were obtained: AUC0-36h AUClast, AUC0–inf and Cmax.
Results: The limit of quantification was 0.2 ng.mL-1 and 1.0 ng.mL-1 for ramipril and
ramiprilat, respectively. The geometric mean and respective 90% confidence
interval (CI) Ramipril/Triatec® and Ramiprilat/ Triatec® percent ratios were
respectively: 104.69% (90% CI= 93.21%-117.59%) for Cmax,102.49% (90% CI=
92.76%-113.24%) for AUClast,102.97% (90% CI= 93.21%-113.74%) for AUC0-
36h,103.60% (90% CI= 93.56%-114.73%) for AUCinf,108.48% (90% CI= 98.86%-
119.03%) for Cmax,105.88% (90% CI= 101.55%-110.39%) for AUClast,105.81%
(90% CI= 101.53%-110.27%) for AUC0-36h, 97.30% (90% CI= 90.17%-104.99%) for
AUCinf. Conclusion: Since the 90% CI for AUClast, AUC0-36h, AUC0–inf and Cmax ratios
were within the 80–125% interval proposed by the US FDA, it was concluded that
ramipril formulation elaborated by Laboratórios Biosintética Ltda is bioequivalent to
Triatec® formulation for both the rate and the extent of absorption.
Introduction
The ramipril, 2-[N-[(S)-1-(ethoxycarbonyl)-3-phenylpropyl)]-L-alanyl]-
(1S,3S,5S) 2-azabicylclo(3-3-0) octane carboxylic acid, is an angiotensin-
converting enzyme inhibitor (ACEI) [1], that regulates the hypertension and
treatment of congestive heart failure [2,3,4]. It is a prodrug [1,2], that acts on the

Capítulo 5
78
renin-angiotensin aldosterone system by inhibiting the conversion of the inactive
angiotensin I to the highly potent vasoconstrictor, angiotensin II.
The aim of this study was to compare in healthy volunteers of both sexes,
the pharmacokinetic profiles and to evaluate the bioequivalence of two ramipril 5
mg tablet formulations. Ramipril and ramiprilat plasma levels were measured by
high performance liquid chromatography coupled to tandem mass spectrometry
(LC-MS-MS) in human plasma using enalapril and enalaprilat as the internal
standards.
Materials and methods
Clinical protocol Twenty six healthy volunteers of both sexes aged between 18 and 50 years
and within 15% of the ideal body weight, were selected for the study after
assessment of their health status by clinical evaluation (physical examination,
ECG) and the following laboratory tests: blood glucose, urea, creatinine, AST, ALT,
alkaline phosphatase, γ-GT, total bilirubin, albumin and total protein, triglyceride,
total cholesterol, hemoglobin, hematocrit, total and differential white cell counts,
and routine urinalysis. All subjects were negative for HIV, HCV and HBV (except
for serological scar).
The study was begun with 26 healthy volunteers and finished with 24
healthy volunteers. Two volunteers dropped out of the study for personal reasons.
The volunteers had the following clinical characteristics (according to gender and
expressed as mean ± SD [range]): male: age: 30.7 ± 8.8 yr. [20 – 44], height: 172.0
± 8.0 cm [160.0 – 185.0], body weight: 68.1 ± 7.8 kg [55.0 – 81.5]; female: age:
32.4 ± 6.2 yr. [22 – 39], height: 157.0 ± 6.0 cm [144.0 – 165.0], body weight: 58.7 ±
5.7 kg [49.5 – 68.2]. All subjects gave written informed consent and the State
University of Campinas - Unicamp approved the clinical protocol. The study was
conducted in accordance with the provisions of the Declaration of Helsinki (1964),
Tokyo (1975), Venice (1983), Hong Kong (1989), Somerset West (1996) and

Capítulo 5
79
Edinburgh (2000) revisions. After screening and wash-out period (of at least 2
weeks), the individuals who qualified were confined for 2 periods of approximately
48 hours.
The study was a single dose, two-way randomized crossover design with a
two-week washout period between the doses. During each period, the volunteers
were hospitalized at 03:00 p.m. and after an overnight fast they received (at
approximately 7:00 a.m.) a single dose of ramipril (5 mg of either tablet
formulation). Water (200 mL) was given immediately after the drug administration
and the volunteers were then fasted for 2 h, after which period, a standard lunch
was served; an evening meal was provided 10 h after dosing. Standard meal was
constituted by rice, beans, vegetables, and fried chicken meat plus a fruit as
dessert. No other food was permitted during the "in-house" period and liquid
consumption was allowed ad libitum after lunch (with the exception of xanthine-
containing drinks, including tea, coffee, and cola). At each time 0, 2, 4, 6, 8, 12, 18,
24 and 36 hours, systolic and diastolic arterial pressure (measured non-invasively
with a sphygmomanometer), heart rate and temperature were recorded.
Formulations
The following formulations were employed: ramipril (test formulation from
Laboratórios Biosintética Ltda, Brazil; lot N° 273/03, expiry 07/2005) and Triatec®
(standard reference formulation from Aventis Pharma, lot N° 204971, expiry date
06/2005.)
Drug analysis Blood samples (4 mL) from a suitable antecubital vein were collected by
indwelling catheter into EDTA containing tubes before and 0.33h, 0.66h, 1h, 1.33h,
1.66h, 2h, 2.5h, 3h, 3.5h, 4h, 4.5h, 5h, 5.5h, 6h, 7h, 8h, 10h, 12h, 14h, 16h, 18h,
24h, 32h and 36h post-dosing. The blood samples were centrifuged at
approximately 2000 x g for 1 min at 4°C and the plasma decanted stored at -20°C
until assayed for ramipril and ramiprilat contents.

Capítulo 5
80
Drug plasma extraction was performed using a solid-phase extraction.
Briefly, 500 µL of each plasma sample were introduced into glass tube following by
the internal standards (50 µL of 0.1 µg/mL of Enalapril/Enalaprilat in
methanol/water 50/50; v/v solution) and vortex-mixed the samples for
approximately 10s. The HLB Oasis SPE cartridges for the assay were pre-
conditioned by washing with 1 mL methanol and with 1 mL of deionized water. All
samples were applied to individual HLB Oasis SPE cartridges, then washed (5
times) the cartridges with 1 mL of washing solution (aqueous hydrochloric acid
solution (10 mM)). Cartridges were then eluted with 0.5 mL of the methanol and the
solvent was evaporated using a flow of nitrogen. The residue was dissolved with
0.2 mL of pure deionized water and vortex-mixed for 15 s. All solutions were
transferred to 96-well plates and placed them into auto injector racks. Typical
standard retention times were 2.1, 1.6, 1.7 and 1.3 min for ramipril, ramiprilat,
enalapril and enalaprilat, respectively. The mass spectrometer (Micromass model
Quattro Ultima) equipped with an electrospray source using a crossflow counter
electrode was run in positive mode (ES+), was set up in Multiple Reaction
Monitoring (MRM), monitoring the transitions 417.00 > 234.0, 389.00 > 206.00,
377.00 > 234.0 and 349.00 > 206.0 for ramipril, ramiprilat, enalapril maleate and
enalaprilat, respectively as the precursor ions m/z and the respective product ions.
Stability
Stability quality control plasma samples (0.6, 4.0 and 40.0 ng/mL for ramipril
and 3.0, 9.0 and 16.0 ng/mL for ramiprilat) were subjected to short-term (6h) room
temperature, three freeze/thaw (-20 to 25°C) cycles, 73h autosampler stability
(room temperature) and long-term stability (116 days) tests. Subsequently, the
ramipril and ramiprilat concentrations were measured compared to freshly
prepared samples. The significance of the results obtained was analyzed by
Student’s t-test (p<0.05).

Capítulo 5
81
Pharmacokinetics and statistical analysis
Bioequivalence between the two formulations was assessed by calculating
individual test/reference ratios for the peak of concentration (Cmax), area under
curve (AUC) of plasma concentration until the last concentration observed
(AUClast), and the area under curve between the first sample (pre-dosage) and
infinite (AUC0-inf). The Cmax and the time taken to achieve this concentration (Tmax)
were obtained directly from the curves. The areas under the ramipril and ramiprilat
plasma concentration vs. time curves from 0-to the last detectable concentration
(AUClast) were calculated by applying the linear trapezoid rule. Extrapolation of
these areas to infinity (AUC0-inf) was done by adding the value Clast/ke to the
calculated AUClast (where Clast = the last detectable concentration). The AUC and
Cmax data for the two formulations were analyzed by ANOVA to establish whether
the 90% CI of the ratios was within the 80-125% interval indicating bioequivalence
as proposed by the US Food and Drug Administration. Parametric and non-
parametric analyses of ln-transformed arithmetic means and individual Tmax
differences between test and reference formulations were also performed.
Results
The ramipril was well tolerated at the administered doses and no significant adverse
reactions were observed or reported. All biochemical parameters did not present any
clinical relevant alterations.
The mass chromatograms of a sample are shown in Figure 1, in which both the
retention times of Ramipril, Ramiprilat, Enalapril and Enalaprillat were 2.1, 1.6, 1.7 and
1.3 min respectively. Table 1 shows the precision and accuracy for the LOQ and QCs.
The mean ramipril plasma concentrations vs time profiles after a single oral dose of
each 5mg tablet formulation of ramipril is shown in Figure 2. Table 2 shows the
mean pharmacokinetics parameters obtained from 26 volunteers after the
administration of 5 mg ramipril tablet. Table 3 shows geometric mean of the
individual Cmax, AUClast, AUC0-36h, AUCinf, (test/reference formulation), the
respective 90% confidence intervals (CI), Intra-subject CV for the 26 volunteers
and separated gender.
Discussion

Capítulo 5
82
Ramipril has been determined in plasma and other biological fluids by
several methods such as reversed-phase high-pressure liquid chromatography
quantified by radioimmunoassay (LOQ 0.25 - 0.4 fmol/mL RT 6 min) [2,3], gas
chromatography-mass spectrometry (LOQ 10 ng/mL; RT 10 min) [6], UV
spectrophotometry (LOQ 23000 ng/mL) [8], reversed-phase high-pressure liquid
chromatography (LOQ 500 ng/mL; RT 8.5-30 min) [9], polarography (LOQ 20
ng/mL) [7], high-pressure liquid chromatography coupled with UV detection (LOQ
180 ng/mL RT 7.36 min) [10] and liquid chromatography coupled to tandem mass
spectrometry (LOQ 0.5ng/mL RT 1.0-1.5 min for both ramipril and ramiprilat) [11].
Our method has a suitable LOQ (0.2 ng/mL,1 ng/mL ramipril and ramiprilat) is
sufficient for bioequivalence study and can be performed in a short time (RT 2.1,
1.6 min for ramipril and ramiprilat, respectively), employing a high yield solid-phase
extraction (mean recovery 93.4%).
After the oral administration of the ramipril tablets, the observed ramapril
and ramiprilat peak plasma concentration (Cmax) values and the time values (Tmax)
were similar to those reported in the literature [12,13] and equivalent between the
formulations (Table 2 and 3).
In addition, the calculated 90% CI for mean Cmax, AUC0-36h, AUClast and
AUC0-inf Ramipril/Triatec® and Ramiprilat/Triatec® individual ratios were within the
80–125% interval defined by the US Food and Drug Administration [14,15], thus
establishing the bioequivalence of the two formulations.
For future studies, we proposed the quantification of the ramipril only, since the
intra-subject variability of the metabolite is much lower than of the parent, making it
less discriminative for bioequivalence.
Conclusion
Since the 90% CI for Cmax and AUClast ratios were all inside the 80-125%
interval proposed by the US Food and Drug Administration Agency, it was concluded
that ramipril formulation elaborated by Laboratórios Biosintética, Brazil is bioequivalent
to Triatec® formulation for both the rate and the extent of absorption [14,15].

Capítulo 5
83
References
1. Becker, R.H., Scholkens, B. A., Metzger, M., Schulze,
K.J.,Pharmacological properties of the new orally active angiotensin
converting enzyme inhibitor 2-[N- [ (S) – 1 - ethoxycarbonyl- 3- phenylpropyl]
- L- alanyl] - (1S,3S,5S) -2-azabicyclo[3.3.0]octane-3-carboxylic acid (Hoe
498). Arzneimittelforschung. 34:1411- 6 (1984)
2. Nussberger, J., Brunner, D.B., Waeber, B., Brunner, H.R., True versus
immunoreactive angiotensin II in human plasma. Hypertension. :7:I1-7 (1985)
3. Campbell, D.J., Kladis, A., Duncan, A,M.,Nephrectomy, converting enzyme
inhibition, and angiotensin peptides., Hypertension. 2:513-22 (1993)
4. Unger, T., Moursi, M., Ganten, D., Hermann, K., Lang, R.E.,Antihypertensive
action of the converting enzyme inhibitor perindopril (S9490-3) in
spontaneously hypertensive rats: comparison with enalapril (MK421) and
ramipril (Hoe498). J Cardiovasc Pharmacol. 8:276-85 (1986)
5. Verho,M., Luck, C., Stelter, W.J., Rangoonwala, B.,Bender,N.,
Pharmacokinetics metabolism and biliary and urinary excretion of oral
ramipril in man .Curr Med Res Opin.13:264-73 (1995)
7. Al-Majed, A,A., Belal, F., Abadi, A., Al-Obaid, A,M..The voltammetric study
and determination of ramipril in dosage forms and biological fluids. Farmaco.
55:233-8 2000

Capítulo 5
84
8. Bonazzi, D., Gotti, R., Andrisano, V., Cavrini, V,. Analysis of ACE inhibitors in
pharmaceutical dosage forms by derivative UV spectroscopy and liquid
chromatography (HPLC). J Pharm Biomed Anal. 16:431-8 (1997)
9. Hogan, B,L., Williams, M., Idiculla, A., Veysoglu, T.,Parente, E.,Development
and validation of a liquid chromatographic method for the determination of
the related substances of ramipril in Altace capsules. J Pharm Biomed Anal.
23:637-51 ( 2000)
10. Belal, F., Al-Zaagi, I,A,. Gadkariem, E,A., Abounassif, M,A., A stability-
indicating LC method for the simultaneous determination of ramipril and
hydrochlorothiazide in dosage forms. J Pharm Biomed Anal. 24:335-42 2001
11. Zhu, Z., Vachareau, A., Neirinck, L., Liquid chromatography-mass
spectrometry method for determination of ramipril and its active metabolite
ramiprilat in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci.
779:297- 306 (2002)
12. Lee, I.D., Hunt, T.L., Bradley, C.R, Copp, C., Griffiths, L., Brobst-Kromer.,
Effects on the pharmacokinetics and pharmacodynamics in the elderly of
coadministering ramiprilwith water, apple juice, and applesauce.
J. Pharm Res.13:639-42 (1996)
13.Van, Griensven, J.M., Schoemaker, R.C., Cohen, A.F., Luus, H.G, Seibert-
Grafe, M.,Rothig, H,J., Pharmacokinetics, pharmacodynamics and

Capítulo 5
85
bioavailability of the ACE inhibitor ramipril. Eur J Clin Pharmacol. 47:513-8
(1995)
14. Food and Drug Administration. In vivo bioequivalence guidances.
Pharmacopeial 19:6501-6508 (1993)
15. Food and Drug Administration. Bioavailability and bioequivalence
requeriments; abbreviated applications; proposed revisions-FDA. Proposed
rule. Fed. Regist. 63: 64 222–64 228 (1998)

Capítulo 5
86
Figure 1 - Selected ion chromatograms (MRM) of 2 channels ES+. 1A: blank
human plasma for monitoring the transitions 417.00 > 234.0, 389.00 > 206.00,
377.00 > 234.0 and 349.00 > 206.0 for ramipril, ramiprilat, enalapril maleate and
enalaprilat, respectively. The peaks illustrate the retention time and integrated
area.
Figure 2 - Mean concentrations vs. time curve for ramipril and ramiprilat.
Table 1 - Quantified concentration (µg/mL) of individual samples for intra-batch
and inter-batch validation.
Table 2 - Mean pharmacokinetic parameters obtained from volunteers after oral
administration of 5 mg Ramipril tablets.
Table 3 - Geometric mean of the individual Cmax, AUClast, AUCinf, (test/reference
formulation), the respective 90% confidence intervals (CI) and Intra-subject CV and
for each gender.

Capítulo 5
87
Figure 1: MRM chromatograms of blank normal human plasma: (A) ramipril, (B).
ramiprilat, (C) enalapril and (D) enalaprilat; MRM chromatogram of the LOQ: (E)
ramipril, (F). ramiprilat, (G) enalapril and (H) enalaprilat.

Capítulo 5
88
0 1 2 3 4 5 6 7 8 9 10
0.0
2.5
5.0
7.5
10.0
12.5 Ramipril - referenceRamipril - test
12 18 24 30 36
Ramiprilat - referenceRamiprilat - test
Time (h)
Con
cent
ratio
n M
ean
(ng/
mL)
Figure 2: Ramipril and ramiprilat plasma means concentrations vs. time profile
obtained after the single oral administration of 5 mg of ramipril formulation.

Capítulo 5
89
Table 1: Accuracy and precision data for ramipril and ramiprilat from the pre-study validation in human plasma.
Intra-batch - Ramipril Nominal
concentration (ng.mL-1)
0.2 0.6 4.0 40.0
Mean Range
0.22 0.20 – 0.24
0.63 0.57 – 0.68
4.32 4.14 – 4.41
42.54 40.50 – 45.00
Accuracy (%) 111.69 104.96 107.91 106.34 Precision (%) 5.29 5.51 2.08 3.79
Inter-batch – Ramipril Nominal
concentration (ng.mL-1)
0.2 0.6 4.0 40.0
Mean Range
0.21 0.18 – 0.24
0.61 0.52 – 0.68
3.95 3.45 – 4.41
39.57 32.60 – 45.00
Accuracy (%) 105.56 100.86 98.67 98.92 Precision (%) 8.33 7.00 7.48 7.30
Intra-batch – Ramiprilat Nominal
concentration (ng.mL-1)
1.0 3.0 9.0 16.0
Mean Range
1.20 1.15 – 1.25
3.04 2.85 – 3.21
9.31 8.83 – 9.76
16.8 16.10 – 17.50
Accuracy (%) 119.50 101.42 103.40 105.08 Precision (%) 2.79 4.22 3.24 2.47
Inter-batch - Ramiprilat Nominal
concentration (ng.mL-1)
1.0 3.0 9.0 16.0
Mean Range
1.14 1.02 – 1.25
3.02 2.82 – 3.21
9.24 8.52 – 9.76
16.7 15.50 – 17.60
Accuracy (%) 113.96 100.58 102.63 104.48 Precision (%) 5.38 3.78 2.72 3.56

Capítulo 5
90
Ramipril Ramiprilat
Pharmacokinetic Parameter Triatec®
5 mg Ramipril
5 mg Triatec®
5 mg Ramipril
5 mg AUClast (ng*h/mL)
Geom. Mean SD
12.77 5.09
12.84 4.58
105.95 25.22
112.18 21.70
AUCinf (ng*h/mL) Geom. Mean
SD
13.33 5.15
13.59 4.72
187.02 42.78
184.48 39.56
Cmax (ng/mL) Geom. Mean
SD
12.82 5.87
13.00 4.99
9.81 6.34
10.45 5.80
K (1/h) Median Range
0.52 0.03 – 1.29
0.57 0.01 – 2.40
0.02 0.01 – 0.05
0.03 0.01 – 0.06
T1/2 (h) Median Range
1.33 0.54 – 23.52
1.22 0.29 – 61.69
35.08 13.30 – 80.64
26.73 11.65 – 75.66
Tmax (h) Median Range
0.33 0.33 – 1.00
0.67 0.33 – 1.00
3.00 1.33 – 10.00
3.00 1.33 – 6.00
Table 2: Mean pharmacokinetic parameters obtained from 26 volunteers after administration of each 5 mg ramipril tablet formulation.

Capítulo 5
91
n = 24 Ramipril Ramiprilat Parametric Parametric
Triatec® / Ramipril
5 mg Geom. Mean 90% CI
Intra subject
CV% Geom. Mean 90% CI
Intra subject
CV% AUClast 102.49 92.76 – 113.25 19.57 105.88 101.56 – 110.39 7.92 AUCinf 103.60 93.56 – 114.73 19.79 97.30 90.17 – 104.99 15.71 Cmax 104.69 93.21 – 117.59 22.31 108.48 98.86 – 119.04 17.21
Man - n = 11 Ramipril Ramiprilat Parametric Parametric
Triatec® / Ramipril
5 mg Geom. Mean 90% CI
Intra subject
CV% Geom. Mean 90% CI
Intra subject
CV% AUClast 114.39 95.43 - 137.12 19.50 104.39 98.85 - 110.26 6.43 AUCinf 116.44 96.85 - 139.99 19.47 96.17 84.17 - 109.89 17.05 Cmax 116.55 97.75 - 138.98 18.58 110.90 98.72 - 124.58 12.91
Woman - n = 13 Ramipril Ramiprilat Parametric Parametric
Triatec® / Ramipril
5 mg Geom. Mean 90% CI
Intra subject
CV% Geom. Mean 90% CI
Intra subject
CV% AUClast 93.06 82.74 - 104.67 17.88 105.63 98.99 - 112.7 8.69 AUCinf 93.60 83.04 - 105.50 18.10 98.52 88.21 - 110.04 15.88 Cmax 96.22 80.79 - 114.61 25.71 103.86 89.48 - 120.54 20.30
Table 3: Geometric mean of the individual AUClast, AUC0-∞, and Cmax ratios (test/reference formulation), the respective 90% confidence intervals (CI) and CV.

CAPÍTULO 6
Artigo Publicado
“A bioequivalence study of citalopram base don quantification by
high-performance liquid chromatography coupled to electrospray
tandem mass spectrometry”
G Duarte Mendes1,2, N Cater do Carmo Borges2, A. dos Santos Pereira2, F.
Duarte Mendes2, R. Eliseo Barrientos-Astigarraga2 and G. De Nucci1,2
Int.J.Clin.Pharmacol.Ther. v.43 - n. 8/2005 (389-398)

Capítulo 6
95

Capítulo 6
96

Capítulo 6
97

Capítulo 6
98

Capítulo 6
99

Capítulo 6
100

Capítulo 6
101

Capítulo 6
102

Capítulo 6
103

Capítulo 6
104

4
Discussão

Discussão
107
O objetivo final dos estudos de bioequivalência é o tratamento ou
prevenção de doenças com fármacos de padrões farmacocinéticos e qualidades
comprovados. No presente trabalhos foram utilizados como matriz biológica o
plasma humano para quantificação farmacocinética dos fármacos através do
método de cromatografia líquida acoplada a espectrômetro de massa (LC-MS-
MS). A quantificação de fármacos através de LC-MS-MS é geralmente baseada na
razão de áreas de picos do analito e do padrão interno (resposta). A utilização do
padrão interno determina uma compensação de diferenças no comportamento do
analito na extração, eventuais falhas no processo de injeção e em diferenças na
eficiência da ionização. Padrões internos mais utilizados são fármacos de
estrutura química semelhante ao analito ou o próprio analito que contenha alguns
dos seguintes isótopos estáveis: 2H, 13C, 180 ou 15N. A Anamnese e Exames
Laboratoriais vinculados ao Processo de Seleção dos Voluntários incluindo
avaliação clínica, eletrocardiográfica e laboratorial são demonstrados na Tabela 1. A quantificação das amostras de medicamentos foi realizada de acordo com os
parâmetros apresentados na Tabela 2 e Capítulos 1 a 6. Nesses capítulos
encontram-se os dados e resultados, gráficos, tabelas e análise estatística
correspondentes. Foi apresentado no Capítulo 1 e 3 pela primeira vez na literatura
mundial, o limite de quantificação (LOQ) no LCMSMS de 1 ng/mL e 0.1 ng/mL,
garantindo maior velocidade no processamento das amostras de plasma, maior
acurácia , maior sensibilidade e especifidade associado com menor custo por
estudo realizado. A biodisponibilidade e o bioequivalência entre duas formulações,
e a seleção das formulações, emergiram como temas críticos na medicina durante
os últimos anos em nosso país. O interesse em baixar custos do cuidado de saúde
resultou em um aumento tremendo no uso de genéricos; atualmente
aproximadamente mais da metade de todas as prescrições escritas é para as
drogas que podem ser substituídas com um produto genérico (MILLER e
STRONG 1990). Sobre 80% das aproximadamente 10.000 drogas da prescrição
disponíveis em 1990 (U.S.A.) já estavam disponível em mais de uma fonte (The
Food and Drug Letter 1990). Com a disponibilidade e o uso crescentes de
produtos genéricos, os profissionais de saúde são confrontados com uma

Discussão
108
disposição sempre maior dos genéricos produzidos por vários laboratórios
nacionais e internacionais, tendo como principal tarefa a de selecionar aqueles
que são terapeuticamente equivalentes. Este crescimento fenomenal da indústria
farmacêutica dos genéricos e da abundância de produtos oriundos de diferentes
laboratórios chamou-nos a atenção para algumas perguntas que muitos
profissionais e cientistas de saúde têm a respeito da equivalência terapêutica
destes produtos, particularmente daqueles em determinadas categorias
terapêuticas, críticas tais como os genéricos como os de ação cardiovascular
(MILLER e STROM, 1990; LAMY, 1985; FOSTER, 1991). Inerente aos guidelines
nacionais e internacionais, atualmente aceitos, para a substituição de uma
formulação inovadora, é a suposição de que a droga genérica seja considerada
bioequivalente a formulação inovadora ou de marca, produzirá o mesmo efeito
clínico. Tão direto como esta indicação a respeito da bioequivalência, parece ser
que gerou a controvérsia de muitos entre os cientistas e profissionais no campo do
cuidado a saúde. As numerosas publicações, indicam que há um interesse de que
os padrões atuais para a aprovação de drogas genéricas não podem sempre
assegurar a equivalência terapêutica (MEYER, 1985; LEVY,1960; LAMY, 1986;
DETTELBACH, 1986; SCHWARTZ, 1985; BERGER, 1980; LAMY, 1986;
HORWITZ, 1985; GOTTSCHALK, 1986;BARONE e COLAIZZI, 1985; STROM,
1987; WEAVER, 1987; NUWER, 1990).
Estudos de bioequivalência são realizados como substitutos da
equivalência terapêutica, com o objetivo de demonstrar que a eficácia, segurança
e qualidade de um determinado medicamento não apresentam diferenças
clinicamente significativas em relação a um medicamento referência no mercado
nacional. Segundo a legislação brasileira em vigor (RDC no135, de 29 de maio de
2003), dois medicamentos serão considerados bioequivalentes, quando eles forem
equivalentes farmacêuticos e, ao serem administrados, na mesma dose molar, nas
mesmas condições experimentais, não apresentem diferenças estatisticamente
significativas em relação a biodisponibilidade, a qual é determinada pela
quantificação dos seguintes parâmetros farmacocinéticos: área sob a curva de

Discussão
109
concentração plasmática versus tempo (ASC), pico de concentração plasmática
(Cmax) e, eventualmente, tempo para se atingir Cmax (Tmax).
A equivalência farmacêutica entre dois medicamentos relaciona-se à
comprovação de que ambos contêm o mesmo fármaco (mesma base, sal ou éster
da mesma molécula terapeuticamente ativa), na mesma dosagem e forma
farmacêutica, o que pode ser avaliado por meio de testes in vitro (SHARGEL e
YU, 1999; WHO, 1999). Portanto, pode ser considerada como um indicativo da
bioequivalência entre os medicamentos em estudo, entretanto não é o suficente
para garantir a bioequivalência. A legislação brasileira, tendo como base a
regulamentação técnica e a experiência de diversos países na área de
medicamentos genéricos, estabelece que, para um medicamento ser registrado
como genérico, é necessário que se comprove sua equivalência farmacêutica e
bioequivalência (mesma biodisponibilidade) em relação ao medicamento de
referência indicado pela Anvisa (RDC no135, de 29 de maio de 2003). Tal fato,
aliado ao cumprimento das Boas Práticas de Fabricação e Controle de Qualidade
(BPFC), fornece as bases técnicas e científicas para a intercambialidade entre o
genérico e seu medicamento de referência, uma vez que, nesse caso, ambos
podem ser considerados equivalentes terapêuticos, ou seja, medicamentos que
apresentam a mesma eficácia clínica e o mesmo potencial para gerar efeitos
adversos (MARZO e BALANT, 1995; MEREDITH, 1996; WHO, 1996; BENET,
1999; MARZO, 1999; MEYER, 1999). O medicamento de referência é, geralmente,
o inovador cuja biodisponibilidade foi determinada, durante o desenvolvimento do
produto, e que teve sua eficácia e segurança comprovada por meio de ensaios
clínicos, antes da obtenção do registro para comercialização. Nesse caso, a
empresa fabricante desenvolveu a formulação e a forma farmacêutica adequadas
à via de administração e ao objetivo terapêutico do medicamento, estabelecendo e
validando os processos de fabricação, bem como as especificações que deverão
ser reproduzidas posteriormente, lote a lote (STORPIRTIS, 1999). Para o
medicamento genérico, o fabricante deve investir no desenvolvimento
farmacotécnico de um produto que cumpra com as mesmas especificações in
vitro, em relação ao medicamento de referência. Entretanto, aceita-se que a

Discussão
110
formulação e o processo de fabricação não sejam idênticos, o que geralmente
ocorre devido aos diferentes equipamentos e fornecedores de matérias-primas
empregados por distintos fabricantes, desde que essas diferenças não
comprometam a bioequivalência entre os produtos (DIGHE, 1999). Nesse
contexto, é fundamental ressaltar que diferenças em relação a características
físicas e físico-químicas do fármaco e demais componentes da formulação, bem
como nos processos de fabricação, podem gerar diferenças na biodisponibilidade
que, no caso do genérico, podem comprometer a bioequivalência. Portanto, as
preocupações em termos de biodisponibilidade, bioequivalência, recaem sobre
medicamentos apresentados sob formas farmacêuticas para as quais existem
muitos fatores que podem alterar a liberação, a dissolução e a absorção do
fármaco no organismo. Tais fatores devem ser amplamente estudados durante o
desenvolvimento farmacotécnico do produto, o que, no entanto, não exclui a
necessidade da realização do teste de bioequivalência (BANAKAR, 1992;
MARQUES et al., 2002).
Desse modo, o teste de bioequivalência realizado, de acordo com as
Boas Práticas de Clínica (BPC) e de Laboratório (BPL), empregando-se
voluntários sadios, é fundamental para garantir que dois medicamentos que
comprovaram a equivalência farmacêutica apresentarão o mesmo desempenho no
organismo em relação à biodisponibilidade, expressa em termos da quantidade
absorvida do fármaco, a partir da forma farmacêutica administrada, e da
velocidade do processo de absorção (SHARGEL e YU, 1999).
A Sociedade Brasileira de Cardiologia através do FUNCOR criou o Selo
de Medicamentos da SBC/FUNCOR (http://prevencao.cardiol.br/selodeaprovacao/)
que é a certificação para orientar o consumidor que determinado medicamento
apresenta qualidade comprovada e está indicado para o tratamento ou prevenção
de doenças cardiovasculares, segundo evidencias técnico - científicas. A
certificação, dada através do Comitê do Selo de Aprovação da SBC/FUNCOR, é
baseada nos seguintes requisitos: Satisfação das leis e portarias vigentes:

Discussão
111
• Leis que regulamentam a entrada de medicamentos "referência" no
país;
• Leis que regulamentam a produção de "similares" no país;
• Lei 9787/99, publicada em 10/08/99 que trata da regulamentação dos
medicamentos genéricos.
Habitualidade de uso internacional e nacional, fazendo parte do
consenso da Sociedade Brasileira de Cardiologia e da AHA (American Heart
Association).
Aprovação pela comunidade científica brasileira – uso habitual, de
preferência fazendo parte do bulário de serviços universitários e hospitais de
referência.
Atender às exigências da ANVISA – Ministério da Saúde
Exigência de testes de bioequivalencia e biodisponibilidade para
produtos que não são "referência" (realizados em laboratórios nacionais,
credenciados pelo Ministério da Saúde)
O Comitê do Selo de Aprovação da FUNCOR é formado por médicos
cardiologistas, farmacologistas, bioquímicos e se necessário um convidado
especialista no assunto, que se reúnem para deliberação, análise e aprovação do
produto. Os padrões de referência usados são submetidos às revisões periódicas
de acordo com estudos e trabalhos científicos publicados em revistas
especializadas. A SBC/FUNCOR, por meio de seus profissionais especializados,
está apta a fornecer laudo técnico - científico a produtos relacionados à prevenção
de doenças cardiovasculares e promoção ou manutenção da saúde.

Discussão
112
Tabela 1 - Anamnese e Examaes Laboratoriais vinculados ao Processo de Seleção dos Voluntários
História Médica
Alergias; olhos, nariz e garganta; sistemas respiratório, cardiovascular, gastrointestinal, genito urinário, nervoso central, hematopoético-linfático, endócrino; dermatológico, musculoesquelético; estabilidade emocional, história familiar, cirúrgica.
Exame Físico Olhos, orelhas, nariz, garganta, pescoço(incluindo tireóide), coração, pulmões, abdomem (incluindo fígado e baço), pele, linfonodos, urogenital, sistema nervoso, esqueleto e músculos
Dados antropométricos Pressão arterial (medida 5 minutos após descanso), na posição sentada, pulso, altura, peso (roupas leves), índice de massa corpórea, temperatura em 0C
Categoria Exames
ECG ECG padrão com 12 derivações
Análise hematológica hemoglobina, hematócrito, contagem total e diferencial de leucócitos, contagem de glóbulos vermelhos, contagem de plaquetas
Análise Bioquímica Ureia, creatinina, bilirrubina total, proteinas totais, albumina, glicose em jejum, fosfatase alcalina, SGOT, SGTP, colesterol total, triglicérides, ácido úrico, γGT, potássio, sódio .
Urina Sumário de Urina (urina I)
Fezes Protoparasitológico
Sorologia Análise Sorológica para: hepatite B, hepatite C e HIV(1+2)β-HCG para mulheres

Discussão 113
Tabe
la 2
- C
arac
terís
ticas
ger
ais
da m
etod
olog
ia e
mpr
egad
a at
ravé
s de
cro
mat
ogra
fia li
quid
a de
alta
ef
iciê
ncia
ac
opla
da
a es
pect
rôm
etro
de
m
assa
(L
C-M
S-M
S)
para
qu
antif
icaç
ão
dos
dife
rent
es
med
icam
ento
s
Ti
clop
idin
a G
emfib
rozi
l C
arve
dilo
l R
amip
ril/
Ram
ipril
ato
Vera
pam
il C
italo
pram
Nom
e do
pad
rão
inte
rno
(IS)
esco
lhid
o C
lopi
dogr
el
Bez
afib
rato
M
etop
rolo
l E
nala
pril/
E
nala
prila
to
Met
opro
lol
Fluo
xetin
a
Mat
riz b
ioló
gica
P
lasm
a P
lasm
a P
lasm
a P
lasm
a P
lasm
a P
lasm
a
Tipo
de
extr
ação
Lí
quid
a/líq
uida
Lí
quid
a/líq
uida
Lí
quid
a/líq
uida
S
ólid
a/líq
uida
Lí
quid
a/líq
uida
Lí
quid
a/líq
uida
Mod
elo
mic
rom
ass
Qua
ttro
Ulti
ma
Qua
ttro
LC
Qua
ttro
LC
Qua
ttro
Ulti
ma
Qua
ttro
Ulti
ma
Qua
ttro
Mic
ro
Tipo
de
elec
tros
pray
+
_ +
+ +
+
Tran
siçã
o an
alito
(m/z
) 26
4,1>
153,
9 24
8,9>
121,
1 40
7,2>
99,8
41
7,0>
234,
0 38
9,0>
206,
0 45
5,4>
165,
2 32
5,1>
108,
5
Tran
siçã
o pa
drão
inte
rno
(m/z
) 32
2,1>
212,
0 35
9,7>
274,
2 26
8,3>
116,
1 37
7,0>
234,
0 34
9,0>
206,
0 26
8,5>
116,
4 31
0,1>
43,1
Volt.
Con
e an
alito
/IS (V
) 35
/ 35
20
/ 15
35
/ 30
30
/ 30
60
/ 17
35
/ 20
Ener
gia
colis
ão a
nalit
o/IS
(eV)
15
/ 15
30
/ 15
30
/ 18
15
/ 15
25
/ 45
25
/ 10
Dw
ell t
ime
(s)
0,3
/ 0,3
0,
2 / 0
,2
0,3
/ 0,3
0,
5 / 0
,5
0,2
/ 0,2
0,
2 / 0
,2
Tem
po d
e co
rrid
a (m
in)
3,0
4,0
3,5
2,5
2,3
5,0
Tem
po d
e re
tenç
ão (a
nalit
o/IS
; m
in)
1,8
/ 2,1
3,
4 / 2
,1
1,6
/ 1,6
2,
1/1,
6 1,
7/1,
3 2,
3 / 2
,0
1,6
/ 1,8
Lim
ite q
uant
ifica
ção
(LO
Q; n
g/m
l) 1,
0 50
,0
0,1
0,2/
1,0
1,0
0,5
Prec
isão
% (L
OQ
) 5,
5 6,
7 8,
2 5,
2/2,
7 15
,0
18,5
Exat
idão
% (L
OQ
) 11
3,8
95,5
10
3,1
111,
6/11
9,5
95,2
-2
,9

6
Conclusão

Conclusão
117
Os estudos apresentados demonstram uma grande aplicabilidade dos
estudos de bioequivalência com drogas de ação cardiovasculares aplicados com
uso de cromatografia líquida de alta eficiência acoplada a espectrômetro de massa
para a determinação de medicamentos em plasma humano. Essa aplicabilidade
pode ser utilizada com vantagens devido a alta confiabilidade dos métodos em
determinar com precisão e exatidão as concentrações de fármacos aliados com
custo por amostra quantificada menor. Deve-se ressaltar que um país como o
Brasil de dimensões continentais com parcos recurssos destinados à saúde, os
medicamentos genéricos são seguramente uma opção extremamente viável, para
o tratamento de patologias que têm alto índice de morbi-mortalidade.

7
Referências Bibliográficas

Referências Bibliográficas
121
ABIFAM. Perfil mercadológico dos medicamentos genéricos. 2001.
ABSHAGEN, U. A new molecule with vasodilating and beta-adrenoceptor blocking
properties. J Cardiovasc Pharmacol, 10: S23-32, 1987.
ANVISA . RDC n084 de 19 de março de 2002
ANVISA, RE nº 896, 29 de maio de 2003, Guia para Provas de Biodisponibilidade
Relativa/Bioequivalência de Medicamentos.
ANVISA. RDC n.135, de 29 de maio de 2003. Regulamento técnico para medicamentos genéricos. Diário Oficial da União, Brasília, 02 jun. 2003a.
ANVISA. RDC N° 10, de 2 de janeiro de 2001.
ARNOUX, P., SALES, Y., MANDRAY, M., LECHAT, P., BERGER, Y., CANO, J.P.
Quantitative high-performance liquid chromatographic, gas chromatographic, and
gas chromatographic-mass spectrometric analysis of ticlopidine in baboon plasma
after solid-phase extraction. J Pharm Sci, 11:1092-5, 1991.
ATKINSON, H. C.; BEGG, E. J.; DARLOW, B. A. Drugs in human milk;clinical
pharmacokinetic consideratios. Clin Pharmacokinet, 14: 217-240,1988.
ATKINSON, J. A.; DANIELS, C. E.; DECRICK, R. L.; GRUDZINSKAS, C.
V.;MARKEY, S. P. Principles of clinical pharmacology. Academic
Press,London, 2001 .
BANAKAR, U.K. - Pharmaceutical Dissolution Testing. New York: Marcel
Dekker Inc., 1992. 437p.
BARONE, J.A., COLAIZZI, J.L., Critical Evaluation of Thioridazine Bioequivalence.
Drug Intel Clin Pharm, 19:847-858, 1985.

Referências Bibliográficas
122
BARTSCH, W., SPONER, G., STREIN, K., MULLER-BECKMANN, B., KLING, L.,
BOHM, E., MARTIN, U., BORBE, H.O. Pharmacological characteristics of the
stereoisomers of carvedilol. Eur J Clin Pharmacol, 38: S104-7, 1990.
BAUMANN, P., LARSEN, F. The pharmacokinetics of citalopram. Rev Contemp
Pharmacother, 6: 287-295, 1995.
BEHN, F., LAER, S., SCHOLZ, H. Determination of carvedilol in human cardiac
tissue by high-performance liquid chromatography. J Chromatogr Sci, 39: 121-4,
2001.
BENET, L, Z. – Understanding bioequivalence testing. Transplant Proc (New
York), 31, suppl. 3A, :75-95, 1999.
BERGE, B. Drug Product Selection: Are All Drugs Created Equal? M M & M,
Sep:46-53, 1980.
BLUME, H., SCHEIDEL, B., SIEWART, M. Proceedings of Bio-Internacional, 92
(1993) 44.
BORGES, N.C.C., MENDES, G.D., BARRIENTOS- ASTIGARRAGA, R.E.,
GALVINAS, P., OLIVEIRA, C.H., De NUCCI, G. Verapamil quantification in human
plasma by liquid chromatography coupled to tandem mass spectrometry na
application for bioequivalence study. J Chromatogr B Analyt Technol Biomed Life Sci, 822: 2005 – In Press.
BORGES, N.C.C., MENDES, G.D., BARRIENTOS- ASTIGARRAGA, R.E., ZAPPI,
E., MENDES, F.D., DE NUCCI,G. Comparative bioequivalence study with two
gemfibrozil tablet formulations in healthy volunteers. ArzneimForchDrugRes, 55:
382 – 386, 2005.

Referências Bibliográficas
123
BORGES, N.C.C., MENDES, G.D., BARRIENTOS- ASTIGARRAGA, R.E., ZAPPI,
E., MENDES, F.D., DE NUCCI,G. Comparative bioequivalence study with two
gemfibrozil tablet formulations in healthy volunteers. ArzneimForchDrugRes, 55:
382 – 386, 2005.
BORGES, N.C.C., MENDES, G.D., BORGES, A., OLIVEIRA, S.E. DE.,
BARRIENTOS-ASTIGARRAGA, R.E., DE NUCCI,G. Ticlopidine quantification in
human plasma by high- performance liquid chromatography coupled to
electrospray tandem mass spectrometry.application to bioequivalence study. J Mass Spectrom, 39 : 1562 – 1569, 2004.
BORGES, N.C.C., MENDES, G.D., BORGES, A., OLIVEIRA, S.E. DE.,
BARRIENTOS-ASTIGARRAGA, R.E., DE NUCCI,G. Ticlopidine quantification in
human plasma by high-performance liquid chromatography coupled to electrospray
tandem mass spectrometry.application to bioequivalence study. J Mass Spectrom, 39 : 1562 – 1569, 2004.
BORGES, N.C.C., MENDES, G.D.,SILVA,D.O.,REZENDE,V.M., BARRIENTOS-
ASTIGARRAGA, R.E., DE NUCCI,G. J Chromatogr B Analyt Technol Biomed Life Sci, 822: 253 -262, 2005
BORGES, N.C.C., MENDES, G.D.,SILVA,D.O.,REZENDE,V.M., BARRIENTOS-
ASTIGARRAGA, R.E., DE NUCCI,G. J Chromatogr B Analyt Technol Biomed Life Sci, 822: 253 -262, 2005
BRUNO, J.J. The mechanisms of action of ticlopidine. Thromb Res Suppl, 4: 59-
67, 1983
BULL, J. P. The historical development of clinical therapeutics trials. J Chron
DiS,10:218-48, 1959.

Referências Bibliográficas
124
CECCATO, A., CHIAP, P.,HUBERT, P.H., TOUSSAINT, B., CROMMEN, J
.Automated determination of verapamil and norverapamil in human plasma with
on-line coupling of dialysis to high-performance liquid chromatography and
fluorometric detection. J Chromatogr A, 750: 351-60, 1996
CLOHS, L., MCERLANE, K.M.Comparison between capillary electrophoresis and
high-performance liquid chromatography for the stereoselective analysis of
carvedilol in serum. J Pharm Biomed Anal, 31: 407-12, 2003.
COLAIZZI, J., LOWENTHAL, D. Critical Therapeutic Categories: A
Contraindication to Generic Substitution? Clin Therap, 8: 370-379, 1986.
COLLlNS, C.; BRAGA, G. L.; BONATO, P. S. Introdução à métodos cromatográficos. 6a ed. Campinas, Editora da UNICAMP, 1995.
DAL BO, L., VERGA, F., MARZO, A, AMBROSOLI, L., POLI, A. Determination of
ticlopidine in human plasma by high-performance liquid chromatography and
ultraviolet absorbance detection. J Chromatogr B Biomed Appl, 2: 404-9, 1995.
DE MEY, C., BREITHAUPT, K., SCHLOOS, J, NEUGEBAUER, G, PALM, D,
BELZ, G.G. Dose-effect and pharmacokinetic-pharmacodynamic relationships of
the beta 1-adrenergic receptor blocking properties of various doses of carvedilol in
healthy humans. Clin Pharmacol Ther, 55: 329-37, 1994.
DESAGER, J.P., VAN NIEUWENHUYZE, Y., DRICOT, J., BERGER, Y.,
HARVENGT, C. Pharmacokinetic profile and bioavailability of a new galenic
formulation of ticlopidine. Int J Clin Pharmacol Res, 4: 247-50, 1990
DETHY, J.M., ACKERMANN, B.L, DELATOUR, C., HENION, J.D., SCHULTZ,
G.A. Demonstration of direct bioanalysis of drugs in plasma using
nanoelectrospray infusion from a silicon chip coupled with tandem mass
spectrometry. Anal Chem, 75: 805-11, 2003.

Referências Bibliográficas
125
DETTELBACH , H.R. A Time to Speak Out on Bioequivalence and Therapeutic
Equivalence. J Clin Pharmacol,26: 307-308, 1986.
DIGHE, S. V. – A review of the safety of generic drugs. Transplant Proc (New
York), 31, suppl. 3A,: 235-245, 1999.
DIPERRI, T., PASINI, F.L., FRIGERIO, C., BLARDI, P., CENTINI, F., MESSA,
G.L., GHEZZI, A., VOLPI, L. Pharmacodynamics of ticlopidine in man in relation to
plasma and blood cell concentration. Eur J Clin Pharmacol, 5: 429-34, 1991
DRUMMER, O.H, HOROMIDIS, S., KOURTIS, S., SYRJANEN, M.L, TIPPET, P.
Capillary gas chromatographic drug screen for use in forensic toxicology. J Anal Toxicol, 18: 134-8, 1994.
EICHELBAUM, M., ENDE, M., REMBERG, G., SCHOMERUS, M, DENGLER, H.J.
The metabolism of DL-[14C] verapamil in man. Drug Metab Dispos, 7 145-8,
1979.
EISENBERG, E.J., PATTERSON, W.R., KAHN, .GC. High-performance liquid
chromatographic method for the simultaneous determination of the enantiomers of
carvedilol and its O-desmethyl metabolite in human plasma after chiral
derivatization. J Chromatogr, 25: 105-15, 1989.
EVANS, J.R., FORLAND, S.C., CUTLER, R.E. The Effect of renal Function on the
Pharmacokinetics of Gemfibrozil. J Clin Pharmacol, 27: 994-1000, 1987.
FAHLBUSCH, S.A., TSIKAS, D., MEHLS, C., GUTZKI, F.M, BOGER, R.H.,
FROLICH, J.C., STICHTENOTH, D.O. Effects of carvedilol on oxidative stress in
human endothelial cells and healthy volunteers. Eur J Clin Pharmacol, 60: 83-8,
2004.
FDA . Guidance for Industry. U.S. Department of Health and Human Services
- Food and Drug Administration, CDER, march 2003, p. 18.

Referências Bibliográficas
126
FELISTE, R., DELEBASSEE, D., SIMON, M.F., CHAP, H., DEFREYN, G.,
VALLEE, E., DOUSTE-BLAZY, L., MAFFRAND, J.P. Broad spectrum anti-platelet
activity of ticlopidine and PCR 4099 involves the suppression of the effects of
released ADP. Thromb Res. 4: 403-15, 1987
FLECKENSTEIN A. History of calcium antagonists. Circ Res, 52 :I3-16. Review
1983.
FOOD AND DRUG ADMINISTRATION - In vivo bioequivalence guidances.
Pharmacopeial Forum, 19: 6501-6508, 1993.
FOOD AND DRUG ADMINISTRATION Bioavailability and bioequivalence
requeriments; abbreviated applications; proposed revisions-FDA. Proposed rule.
Fed. Regist, 63, 64 222 – 64228, 1998.
FOOD AND DRUG ADMINISTRATION, Fed. Reg. 63 : 64222, 1998
FOOD AND DRUG ADMINISTRATION-Bioavailability and bioequivalence
requeriments. Fed Regist, 320: 154-173, 1985.
FOSTER , T.S. Selecting Therapeutically Equivalent Products: Special Cases. Am Pharm, NS31: 49-54,1991.
FRICK, M.H., ELO, O., HAAPA, K., HEINONEN, O.P., HEINSALMI, P., HELO, P.,
HUTTUNEN, J.K., KAITANIEMI, P., KOSKINEN, P., MANNINEN, V. Helsinki Heart
Study: Primary prevention trial with gemfibrozil in middle-aged men with
dyslipidemia. N Engl J Med, 317:1237-1245, 1987.
FRICK, M.H., HEINONEN, O.P., HUTTUNEN, J.K., KOSKINEN, P., MANTTARI,
M., MANNINEN, V. Efficacy of gemfibrozil in dyslipidemic subjects with suspected
heart disease. An ancillary study in the Helsinki Heart Study frame population.
Annals of Medicine, 25: 1-45, 1993.

Referências Bibliográficas
127
FURLAN, G., MALUSA, N., STROHMAYER, A., KLUGMANN, F.B., DECORTI, G.,
CANDUSSIO, L., KLUGMANN, S. Optimised method for determination of
ticlopidine in serum by high performance liquid chromatography. Farmaco, 11: 747-51, 1996.
GARCIA, M.A, ARAMAYANA, J.J, BREGANTE, M.A, FRAILE, L.J, SOLANS ,C.
Simultaneous determination of verapamil and norverapamil in biological samples
by high-performance liquid chromatography using ultraviolet detection. J Chromatogr B Biomed Sci Appl, 693: 377-82, 1997.
GENT, M., BLAKELY, J.A, EASTON, J.D., ELLIS, D.J., HACHINSKI, V.C.,
HARBISON, J.W., PANAK, E., ROBERTS, R.S., SICURELLA ,J., TURPIE, A.G.
The Canadian American ticlopidine Study (CATS) in thromboembolic stroke.
Lancet, 8649: 215-20, 1989
GERGOV, M., ROBSON, J.N., DUCHOSLAV, E., OJANPERA, I. Automated liquid
chromatographic/tandem mass spectrometric method for screening beta-blocking
drugs in urine. J Mass Spectrom, 35: 912-8, 2000.
GONZALEZ-PENAS, E., AGARRABERES, S., LOPEZ-OCARIZ, A., GARCIA-
QUETGLAS, E., CAMPANERO, M.A., CARBALLAL, J.J., HONORATO, J. A
sensitive method for the determination of gemfibrozil in human plasma samples by
RP-LC. J Pharm Biomed Anal, 26: 7-14, 2001.
GOTTSCHALK, L.A. Clinical Relevance of the Bioavailability/Bioequivalence
Controvers. J Clin Psychiatry,47(9, Suppl): 3-5, 1986.
GUTIERREZ, M.M., ABRAMOWITZ, W. Pharmacokinetic comparison of oral
solution and tablet formulations of citalopram: a single-dose, randomized,
crossover study. Clinical Therapeutics, 22: 1525-1532, 2000.

Referências Bibliográficas
128
HEDELAND, M., FREDRIKSSON, E., LENNERNAS, H., BONDESSON, U.
Simultaneous quantification of the enantiomers of verapamil and its N-
demethylated metabolite in human plasma using liquid chromatography-tandem
mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 804: 303-
11, 2004.
HENGY, H., KOLLE, E.U. Determination of gemfibrozil in plasma by high
performance liquid chromatography. Arzneimittelforschung, 35: 637-9, 1985.
HOKAMA, N., HOBARA, N., KAMEYA, H., OHSHIRO, S., SAKANASHI, M. Rapid
and simple micro-determination of carvedilol in rat plasma by high-performance
liquid chromatography. J Chromatogr B Biomed SciAppl, 732: 233-8, 1999.
HORNE, C., STENZHORN, G., BLUME, H., KNAUF, H., MUSTSCHELER, E.
Bioavailability study of two different verapamil formulations. Arch Pharm (Weinheim), 325: 531-6. 1992.
HORWITZ, N. Generic Bioequivalence Tests are Flawed, Medical Tribune, 26 : 1,
1985.
HYNNING, P.A., ANDERSON, P., BONODESSON, U., BORÉUS, L.O. Liquid-
chromatographic quantification compared with gas-chromatographic-mass-
spectrometric determination of verapamil and norverapamil in plasma. Clin Chem,
34: 2502-3, 1988.
IDBORG-BJORKMAN, H., EDLUND, P.O., KVALHEIM, O.M., SCHUPPE-
KOISTINEN, I., JACOBSSON, S.P. Screening of biomarkers in rat urine using
LC/electrospray ionization-MS and two-way data analysis, Anal Chem, 75: 4784-
4792, 2003.
ITOH, S., SUZUKI, S., ANDO, S., FUNATSU, Y., YAMAZAKI, M., TANABE, K.,
SAWANOI, M. Determination of ticlopidine in rabbit plasma by high-performance
liquid chromatography. Chem Pharm Bull, 3:1304-7, 1987.

Referências Bibliográficas
129
KANG, D., VEROTTA, D., KRECIC-SHEPARD, M.E., MODI, N.B., GRUPTA, S.K.,
SCHWARTAZ, J.B. Population analyses of sustained-release verapamil in
patients: effects of sex, race, and smoking. Clin Pharmacol Ther, 73 : 31-40, 2003
KERSTEN, S., DESVERGNE, B., WAHLI, W. Roles of PPARs in health and
disease. Nature, 405: 421-4, 2000.
KNUDSEN, J.B., BASTAIN, W., SEFTON, C.M., ALLEN, J.G., DICKINSON, J.P.
Pharmacokinetics of ticlopidine during chronic oral administration to healthy
volunteers and its effects on antipyrine pharmacokinetics. Xenobiotica, 5: 579-89,
1992
KNUDSEN, J.B., GORMSEN,J. The effect of ticlopidine on platelet function in
normal volunteers and in patients with platelet hyperaggregability in vitro. Thromb Res. 16: 663-71, 1979
KOLBAH, T.A, ZAVITSANOS, A.P. Chiral bioanalysis by normal phase high-
perfomance liquid Chromatography - atmospheric pressure ionization tandem
mass spectrometry . J Chromatogr A, 759: 65 – 67, 1997
KONISHI, H., NISHIO, S., TSUTAMOTO, T., MINOUCHI, T., YAMAJI, A. Serum
carvedilol concentration and its relation to change in plasma brain natriuretic
peptide level in the treatment of heart failure: a preliminary study. Int J Clin
Pharmacol Ther, 41: 578-86, 2003.
KOSEL, M., EAP, C.B, AMEY, M., BAUMANN, P. Analysis of the enantiomers of
citalopram and its demethylated metabolites using chiral liquid chromatography. J Chromatogr B, 719: 234-238, 1998.
LAGANA, A., BELLAGAMBA, G., ASCENZO, D. GENTILI, A., MARINO, A.
Evaluation of ticlopidine in human serum and plaque by liquid
chromatography/atmospheric pressure chemical ionization mass espectrometry.
Analytica Chimica Acta, 354: 87-95, 1997.

Referências Bibliográficas
130
LAMY , P. Generic Equivalents: Issues and Concerns. J Clin Pharmacol, 26: 309-
316, 1986.
LAMY, P. Critical Patients, Critical Drugs, Critical Diseases. Maryland Pharmacist, 61: 22-25, 1985.
LAMY, P. What Should We Know about Generics? Geriatric Medicine Today, 5:
25-27, 1986.
LEVY,G. The Therapeutic Implications of Brand Interchange. Am J Hosp Pharm,
17: 756-760, 1960.
LONG, Y., FENG, J., Spectrophotometry of verapamil tablests with charge transfer
reaction TONG, S. Zhongguo Yiyao Gongye Zazhi, 24 : 267 – 270, 1993.
MACEK, J., PTACEK, P., KLIMA, J. Rapid determination of citalopram in human
plasma by high-performance liquid chromatography. J Chromatogr B, 755: 279-
285, 2001.
MACHIDA, M., WATANABE, M., TAKECHI, S., KAKINOKI, S., NOMURA, A.
Measurement of carvedilol in plasma by high-performance liquid chromatography
with electrochemical detection. J Chromatogr B Analyt Technol Biomed Life Sci, 798: 187-91, 2003.
MANNINEN, V., ELO, M.O., FRICK, M.H., HAAPA, K., HEINONEN, O.P.,
HEINSALMI, P., HELO, P., HUTTUNEN, J.K., KAITANIEMI, P., KOSKINEN, P.
Lipid alterations and decline in the incidence of coronary heart disease in the
Helsinki Heart Study. JAMA, 260: 641-651, 1998.
MANZOORI, J.L., AMJADI, M. Spectrofluorimetric and micelle-enhanced
spectrofluorimetric methods for the determination of gemfibrozil in pharmaceutical
preparations. J Pharm Biomed Anal, 31: 507-13, 2003.

Referências Bibliográficas
131
MARQUES, M.R.C. & BROWN, W. – Desenvolvimento e validação de métodos de
dissolução para formas farmacêuticas sólidas orais. Rev Analytica, n.1, p.48-51,
2002.
MARZO, A. – Open questions on bioequivalence: some problems and some
solutions. Pharm Res ( New York), 40, : 357-368, 1999.
MARZO, A., BALANT, L.P. – Bioequivalence: an updated reappraisal addressed to
applications o interchangeable multisorce pharmaceutical products. Arzneim–
Forsch/Drug Res, (Aulendorf), 45, : 109-115, 1995.
MASCHER, H. J., KIKUTA, C., MILLENDORFER, A. & LUDWIG, G. Int J Clin
pharmacol Ther, 35: 9-13, 1997.
MATSUI, E., HOSHINO, M., MATSUI, A., OKAHIRA, A. Simultaneous
determination of citalopram and its metabolites by high-performance liquid
chromatography with column switching and fluorescence detection by direct
plasma injection. J Chromatogr B, 668: 299-307, 1995.
MENDES, G.D., BORGES, N.C.C., PEREIRA, A.S., MENDES, F.D.,
BARRIENTOS-ASTIGARRAGA, R.E., De NUCCI,G. A bioequivalence study of
citalopram base don quantification by high-performance liquid chromatography
coupled to electrospray tandem mass spectrometry. Int J Clin Pharmacol ther, 2005 In Press.
MENDES, G.D., SANFELICE, A.T.D., BORGES,N.C.C., CAVEDAL, L.E.,
MODOLO,J.H., De NUCCI,G. Comparative bioavailability of two ramipril
formulations in healthy human volunteers after a single dose administration. Int J Clin Pharmacol ther, 2005. In Press.
MEREDITH, P.A. – Generic drugs: therapeutic equivalence. Drug Saf, (Auckland), 15, : 233-242, 1996.

Referências Bibliográficas
132
MEYER, M. The Therapeutic Equivalence of Drug Products. A Second Look, The
University of Tennessee Center for the Health Sciences. Memphis, 1985.
MEYER,G. F. – History and regulatory issues of generic drugs. Transplant Proc (New York), 31, suppl. 3A, .105-125, 1999.
MILLER, S.W., STROM, J.G. Drug Product Selection: Implications for the Geriatric
Patient. The Consultant Pharmacist,5: 30-37, 1990.
MILNE, R.J, GOA, K.L Citalopram. A review of its pharmacodynamic and
pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs,
41: 450-477, 1991
MORGAN, T. Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin Pharmacokinet, 26: 335-46, 1994.
MULLER, C., VOGT, S., GOERKE, R., KORDON, A., WEINMANN, W.
Identification of selected psycho pharmaceuticals and their metabolites in hair by
LC/ESI-CID/MS and LC/MS/MS. Forensic Science International, 113: 415-421,
2000.
MULLETT, W.M., WALLES,M., LEVSEN, K., BORLAK, J., PAWLISZYN, J.
Multidimensional on-line sample preparation of verapamil and its metabolites by a
molecularly imprinted polymer coupled to liquid chromatography-mass
spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 801: 297-306,
2004.
MUSCARA, M.N., De NUCCI, G. Measurement of plasma verapamil levels by
high-performance liquid chromatography. Braz J Med Biol Res, 26: 753-63, 1993.
NAKAGAWA, A., SHIGETA, A., IWABUCHI, H., HORIGUCHI, M., NAKAMURA, K.,
TAKAHAGI, H. Simultaneous determination of gemfibrozil and its metabolites in
plasma and urine by a fully automated high performance liquid chromatographic
system. Biomed Chromatogr, 5: 68-73, 1991

Referências Bibliográficas
133
NASH, D.T. Gemfibrozil - a new lipid lowering agent. J Med, 11:107-16, 1980.
NEUGEBAUER, G., AKPAN, W., KAUFMANN, B., REIFF, K. Stereoselective
disposition of carvedilol in man after intravenous and oral administration of the
racemic compound. Eur J Clin Pharmacol, 38: S108-11, 1990.
NISSEN, D . Mosby’s Drug Consult III, 617, 2002.
NUWER, M.R., et al. Generic Substitutions for Antiepileptic Drugs. Neurology, 40:
1647-1651, 1990.
ORAVCOVA, J., SOJKOVA, D., LINDNER, W. Comparison of the Hummel-Dreyer
method in high-performance liquid chromatography and capillary electrophoresis
conditions for study of the interaction of (RS)-, (R)- and (S)-carvedilol with isolated
plasma proteins. J Chromatogr B Biomed Appl, 682: 349-57, 1996.
OVERO, K.F. Fluorescence assay of citalopram and its metabolites in plasma by
scanning densitometry of thin-layer chromatograms. J Chromatogr, 224: 526-31,
1981.
OVERO, K.F. Preliminary studies of the kinetics of citalopram in man. Eur J Clin Pharmacol, 14: 69-73, 1978.
OYEHAUG, E., OSTENSEN, E.T., SALVESEN, B. Determination of the
antidepressant agent citalopram and metabolites in plasma by liquid
chromatography with fluorescence detection. J Chromatogr, 227: 129-135, 1982.
OYEHAUG,E.,OSTENSEN, E.T.,SALVESEN,B. High-performance liquid
chromatographic determination of citalopram and four of its metabolites in plasma
and urine samples from psychiatric patients. J Chromatogr, 308: 199-208, 1984.
PACKER, M., BRISTOW, M.R, COHN, J.N., COLUCCI, W.S., FOWLER, M.B.,
GILBERT, E.M., SHUSTERMAN, N.H. The effect of carvedilol on morbidity and
mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study
Group. N Engl J Med, 334: 1349-55, 1996.

Referências Bibliográficas
134
PFEFFER, M.A., STEVENSON, L.W. Beta-adrenergic blockers and survival in
heart. N Engl J Med, 334: 1396-7, 1996.
PTACEK, P., MACEK, J., KLIMA, J.Liquid chromatographic determination of
carvedilol in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci, 789: 405-410, 2003.
RANDINITIS, E.J., KINKEL, A.W., NELSON, C., PARKER, T.D. 3rd Gas
chromatographic determination of gemfibrozil and its metabolites in plasma and
urine. J Chromatogr, 307: 210-5, 1984.
RANDINITIS, E.J., PARKER, T.D., 3rd, KINKEL, A.W. Liquid chromatographic
determination of gemfibrozil and its metabolite in plasma. J Chromatogr, 383:
444-8, 1986.
REYMOND P, AMEY M, SOUCHE A, LAMBERT S, KONRAT H, EAP CB,
BAUMANN P. Determination of plasma levels of citalopram and its demethylated
and deaminated metabolites by gas chromatography and gas chromatography-
mass spectrometry. J Chromatogr, 616: 221-228, 1993.
REYNOLDS J.E.F. The Extra Pharmacopeia, 29 th ed. 1989.
RIBEIRO, A. N. F. Medicamentos Genéricos - Informações para Farmacêuticos ou profissionais da Saúde. Maio, 2000.
Richter O.V, Eichelbaum M, Schönberg F, Hofmann U. Rapid and highly sensitive
method for the determination of verapamil, [2H7]verapamil and metabolites in
biological fluids by liquid chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl, 738: 137-47, 2000.
ROADCAP, B.A., MUSSON, D.G., ROGERS, J.D., ZHAO, J.J. Sensitive method
for the quantitative determination of gemfibrozil in dog plasma by liquid-liquid
cartridge extraction and liquid chromatography-tandem mass spectrometry. J
Chromatogr B Analyt Technol Biomed Life Sci, 791: 161-70, 2003.

Referências Bibliográficas
135
ROBERTS, M. S.; DONALDSON, J. D.; ROWLAND, M. Models of hepatic
elimination: Comparison of stochastic models to describe residence time
distributions and to predict the influence of drug distribution, enzyme heterogeniety, and systemic recycling on hepatic elimination. J Pharmacokinetic Biopharm, 16: 1-83, 1988.
ROCHAT B, AMEY M, BAUMANN E. Analysis of enantiomers of citalopram and its
demethylated metabolites in plasma of depressive patients using chiral reverse-
phase liquid chromatography. Ther Drug Monit, 17: 273-279, 1995.
ROCHAT B, AMEY M, GELDEREN HV, TESTA B, BAUMANN E. Determination of
the enantiomers of citalopram, its demethylated and propionic acid metabolites in
human plasma by chiral HPLC. Chirality, 7: 389-395, 1995.
RÓNA K, ARY K, GACHÁLYI B, KLEBOVICH I. Liquid chromatographic method
for the determination os ticlopidine in human plasma. Journal of Chromatography, 693:393-398, 1996.
ROP, P.P., DURAND, A., VIALA, A., JORGENSEN, A. Simultaneous determination
of citalopram, monodesmethylcitalopram and didesmethylcitalopram in plasma by
high-performance liquid chromatography after column extraction. J Chromatog,
527: 226-232, 1995.
ROP, P.P., VIALA, A., DURAND, A., CONQUY, T. Determination of citalopram,
amitriptyline and clomipramine in plasma by reversed-phase high-performance
liquid chromatography. J Chromatogr, 338: 171-178, 1985.
RUFFOLO, R.R., Jr., GELLAI, M., HIEBLE, J.P., WILLETTE, R.N., NICHOLS, A.J.
The pharmacology of carvedilol. Eur J Clin Pharmacol, 38: S82-8, 1990.
SALAMA, Z.B., DILGER, C., CZOGALLA, W., OTTO, R., JAEGER, H. Quantitative
determination of verapamil and metabolites in human serum by high-performance
liquid chromatography and its application to biopharmaceutic investigations.
Arzneimittelforschung, 39: 210-5, 1989.

Referências Bibliográficas
136
SALTIEL, E., WARD, A. Ticlopidine. A review of its pharmacodynamic and
pharmacokinetic properties, and therapeutic efficacy in platelet-dependent disease
states. Drugs, 2: 222-62, 1987
SBC/FUNCOR, Sociedade Brasileira de Cardiologia/ Fundo de Aperfeiçoamento e
Pesquisa Cardiológica. Disponível em: http://prevencao.cardiol.br/selodeaprovacao/.
Acesso em 18 ago.2005.
SCHAEFER, W.H., POLITOWSKI, J., HWANG, B, DIXON, F. Jr., GOALWIN, A.,
GUTZAIT, L., ANDERSON, K., DEBROSSE, C., BEAN, M., RHODES, G.R.
Metabolism of carvedilol in dogs, rats, and mice. Drug Metab Dispos, 26: 958-69,
1998.
SCHWARTZ, L., The Debate Over Substitution Policy. Am J Med, 79: 38-44,
1985.
SHAH, J., FRATIS, A., ELLIS, D., MURAKAMI, S., TEITELBAUM, P. Effect of food
and antacid on absorption of orally administered ticlopidine hydrochloride. J Clin
Pharmacol, 8: 733-6, 1990
SHAH, J., TEITELBAUM, P., MOLONY, B., GABUZDA, T., MASSEY, I. Single and
multiple dose pharmacokinetics of ticlopidine in young and elderly subjects. Br J
Clin Pharmacol, 6: 761-4, 1991.
SHARGEL L. & YU A.B.C. – Applied biopharmaceutics and pharmacokinetics.
4a. ed. Stamford: Appleton & Lange, 1999. 768p.
SOINI, H., RIEKKALA, M.L, NOVOTNY, M.V. Chiral separations of basic drugs
and quantitation of bupivacaine enantiomers in serum by capillary electrophoresis
with modified cyclodextrin buffers. J Chromatogr, 608: 265-74, 1992.

Referências Bibliográficas
137
SPAHN, H., HENKE, W., LANGGUTH, P., SCHLOOS, J., MUTSCHLER, E.
Measurement of carvedilol enantiomers in human plasma and urine using S-
naproxen chloride for chiral derivatization. Arch Pharm (Weinheim) 323: 465-9,
1990.
SPIEGELHALDER, B, EICHELBAUM, M. Determination of verapamil in human
plasma by mass fragmentography using stable isotope-labelled verapamil as
internal standard.
Arzneimittelforschung, 27: 94-7, 1977.
SPONER, G., STREIN, K., MULLER-BECKMANN, B., BARTSCH, W. Studies on
the mode of vasodilating action of carvedilol. J Cardiovasc Pharmacol, 10 :S42-8,
1987.
STORPIRTS, S., Biofarmacocinética. Fundamentos de biodisponibilidade,
bioequivalência, dissolução e intercambialidade de medicamentos genéricos.
Apostila do curso Biofarmacotécnica. São Paulo: XI Congresso Paulista
Farmacêuticos e III Seminário internacional de Farmacêuticos , 1999.
STREIN, K., SPONER, G., MULLER-BECKMANN, B., BARTSCH, W.
Pharmacological profile of carvedilol, a compound with beta-blocking and
vasodilating properties. J Cardiovasc Pharmacol, 10: S33-41, 1987.
STROM B.L. Generic Drug Substitution Revisited. N Eng J Med, 316: 1456-1462,
1987.
TASSIN ,J.P., DUBOIS, J., ATASSI, G., HANOCQ, M., Simultaneous
determination of cytotoxic (adriamycin, vincristine) and modulator of resistance
(verapamil, S 9788) drugs in human cells by high-performance liquid
chromatography and ultraviolet detection. J Chromatogr B Biomed Sci Appl, 691: 449-56, 1997.

Referências Bibliográficas
138
TENERO, D., BOIKE, S., BOYLE, D., ILSON, B., FESNIAK, H.F., BROZENA, S.,
JORKASKY, D..Steady-state pharmacokinetics of carvedilol and its enantiomers in
patients with congestive heart failure. J Clin Pharmacol, 40: 844-53, 2000.
THOMAS, H., COLEY, H.M. Overcoming multidrug resistance in cancer: an update
on the clinical strategy of inhibiting p-glycoprotein. Cancer Control, 10:159-65.
Review, 2003.
TODD, P.A., WARD, A. Gemfibrozil. A review of its pharmacodynamic and
pharmacokinetic properties, and therapeutic use in dyslipidaemia. Drugs, 36:314-
39, 1988.
TSANG, Y.C., POP, R., GORDON, P., HEMS, J., SPINO, M. High variability in
drug pharmacokinetics complicates determination of bioequivalence: experience
with verapamil. Pharm Res,13: 846-50, 1996.
TUOMILEHTO, J., SALONEN, J., KUUISTO, P., VIRTAMO, J., MANNINEN V.,
MALKONEN M. A clofibrate controlled trial of gemfibrozil in the treatment of
hyperlipidaemias. Proc R Soc Med, 69: 38-40, 1976.
VARIN, F., CUBEDDU, L.X., POWELL, JR. Liquid chromatographic assay and
disposition of carvedilol in healthy volunteers. J Pharm Sci, 75: 1195-7, 1986.
WALLES, M, MULLETT, W.M, LEVSEN, K, BORLAK, J., WUNSCH, G.,
PAWLISZYN, J. Verapamil drug metabolism studies by automated in-tube solid
phase microextraction. J Pharm Biomed Anal, 30 307-19, 2002.
WALLES, M., BORLAK, J, LEVSEN, K . Application of restricted access material
(RAM) with precolumn-switching and matrix solid-phase dispersion (MSPD) to the
study of the metabolism and pharmacokinetics of Verapamil. Anal Bioanal Chem,
374: 1179-86, 2002.

Referências Bibliográficas
139
WALLES, M., THUM, T., LEVSEN, K., BORLAK, J. Metabolism of verapamil: 24
new phase I and phase II metabolites identified in cell cultures of rat hepatocytes
by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci, 798: 265-74, 2003.
WEAVER L.C. Drug Cost Containment and the Case for Generics. IPU Review,
12: 320-324, 1987.
WORLD HEALTH ORGANIZATION – Expert Committee on Specification for Pharmaceutical Preparation. 34 report. Geneva: WHO, 1996. 46p.
WORLD HEALTH ORGANIZATION – Marketing authorization of pharmaceutical products with special reference to multisource (generic) products: a manual for a drug regulatory authority. Geneva: [s.n.], 1999.







