
104
Aromatische Kohlenwasserstoffe Aromatizität Das Benzolproblem 1825 Faraday Im Gasöl: Kohlenwasserstoff C6H6, Sdp. 80 °C, Schmp. 5.5 °C 1834 Mitscherlich
Benzoesäure liefert beim erhitzen den selben Kohlenwasserstoff C6H6
Welche Strukturisomere kommen in Frage ?
Org09_6a.cw2

105
Historisch folgende Vorschläge:
Kekulé Dewar Baeyer/Armstrong Ladenburg
1859 1867 1887 1879 Formeln in vieler Hinsicht unbefriedigend, da Benzol keinen olefinischen Charakter und keine Ringspannung aufweist
C6H6
Br2
Br2, FeBr3
HBr
KMnO4
C6H5Br + HBr
Kekulé
Oszillation
Armstrong/Baeyer
innere
Absättigung Die Erklärung liefert erst die moderne Elektronentheorie
HHH H
HH
Erwartung:
H
H H
H
HH
kurz kurz
kurz
lang
lang
lang
Realität:In der Kristallstrukturanalysealle Bindungen gleich lang: 1.39 AWie erklärt man das ?
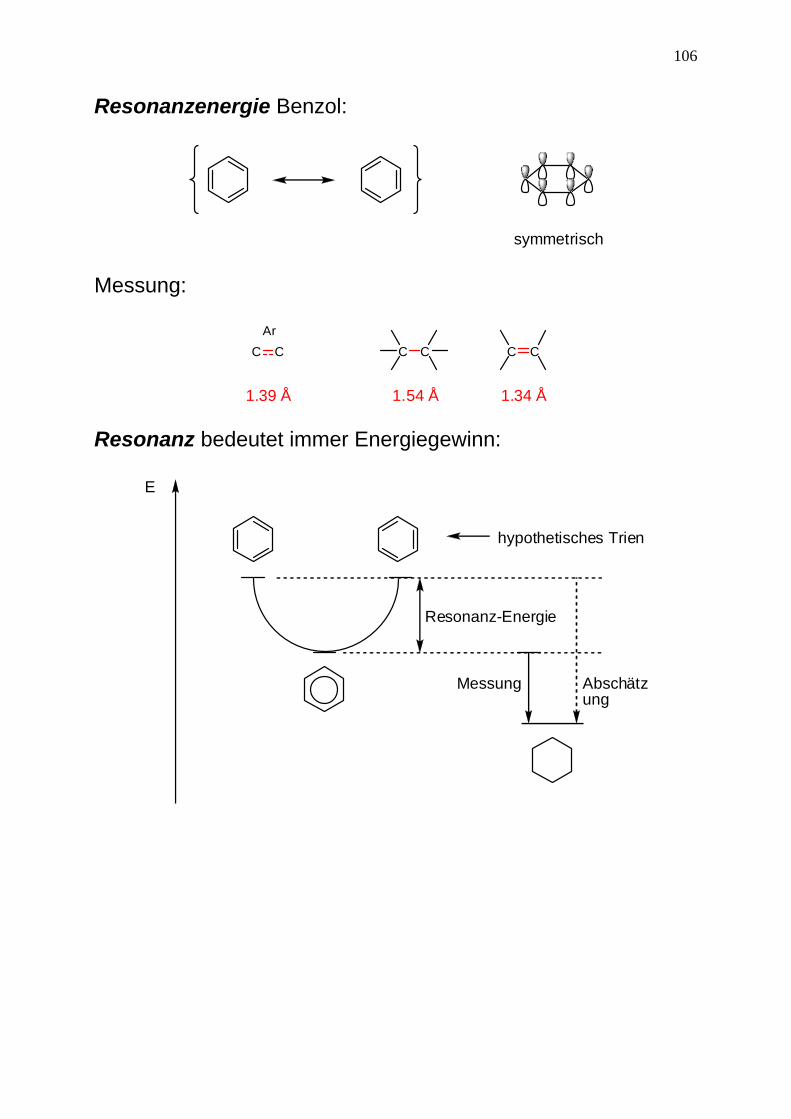
106
Resonanzenergie Benzol:
symmetrisch Messung:
C C C CC CAr
1.39 Å 1.54 Å 1.34 Å Resonanz bedeutet immer Energiegewinn:
E
Resonanz-Energie
Abschätzung
Messung
hypothetisches Trien

107
-103
-111
-119
+23
-207
RE = -126
RE = 8 kJ/mol
Methode Hydrierwärme RE = -126 kJ/mol Grenzen der Resonanztheorie: Willstätter 1908
N CH3
O
Pseudopelletierin gelb / Olefin
13 Stufen
Synthese von COT heute? Resonanztheorie keine Erklärung für Unterschied zu Benzol Erklärung durch MO-Theorie, Hückel-Regel 1938 E. Hückel (Anerkennung erst nach 2. Weltkrieg)

108
x p-Orbitale
n π-Elektronen Hückel-Regel cyclische, planare, durchgehend konjugierte π-Systeme (4n+2) π-Elektronen: aromatisch stabilisiert 4n π-Elektronen: olefinisch oder destabilisiert n = 0,1,2 aromatisch:
Ph
Ph Ph
planar Tropylium-ion
Cyclopropenylium-ion
C leeres p
6 π-El. 6 π-El. 2 π-El.
nicht aromatisch:
4 π-Elektronen
8 π-Elektronennicht planar
4 π-Elektronen
nicht durchgehendkonjugiert
H

109
Weitere aromatische Verbindungen Mehrkernige aromatische Kohlenwasserstoffe
Naphthalin Anthracen
Phenanthren
Benzol
Pyren
Perylen
Chrysen
Benzpyren
Cyclopentadienyl-Anion
pKs = 15C
H H
....
H
- H
Aromatische Heterocyclen
NN
NH
N N H
Pyridinbasisch
Pyrrolnicht basisch
Chinolinbasisch
O
Furan
S
Thiophen
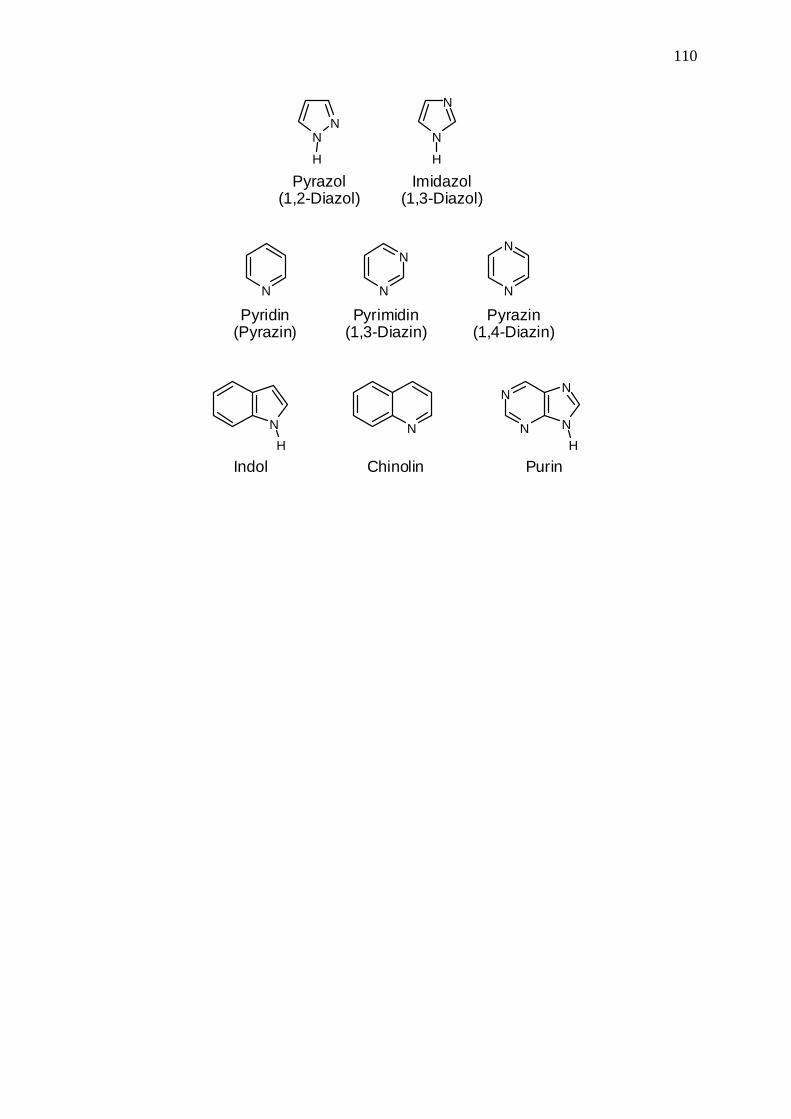
110
NN
N
N
N N
N
N
N
N
N
N N
N
N
H H
H H
Pyrazol(1,2-Diazol)
Imidazol(1,3-Diazol)
Pyridin(Pyrazin)
Pyrimidin(1,3-Diazin)
Pyrazin(1,4-Diazin)
Indol Chinolin Purin

111
Erdöl und Kohle Erdöl Hauptsächlich aliphatische Verbindungen a) Entstehung Durch Absterben von Organismen in küstennahen Regionen, reduzierende Zersetzung:
Organische Materie100-200°C
Druck,Reduktion,
Mikroorganismen
Erdöl +Erdgas
Biologischer Ursprung abgesichert (Porphyrine und Steroide) Hauptfundorte, Bedeutung siehe erstes Kapitel b) Zusammensetzung Je nach Fundort sehr verschieden 81-87 % C, 10-14 % H, 0-7 % O, 0-7 % S, wenig N
Alkane Cycloalkane (Naphthene)
Alkene Aromaten
Regelfall ehemalige UDSSR,
Rumänien bis 80%
Cyclopentan, Cyclohexan
- Indonesien bis 40 % Aromaten

112
c) Verarbeitung Auftrennung durch Destillation, siehe Beginn
Fraktionen aus Erdöl
Fraktion
Erdgas (C1 - C4)
Rohbezin
Leuchtöl
Gasöl
Schmieröl /Paraffine
Rückstand(Asphalt / Pech)
Siedepunkt [°C] Ausbeute [%]
< 150
150 - 300
300 - 350
> 350
20
6
40
30
Destillation
Fraktion
Petrolether
Benzin
Ligroin
Schwerbenzin
Siedepunkt [°C]
40 - 70
70 - 90
90 -120
120 - 150
d) Treibstoffe Zwei Probleme: - Direkte Fraktionierung Erdöl zu wenig - Klopfen Was ist Klopfen? n-Alkane haben schlechte Verbrennungseigenschaften, zünden im Motor schon bei Kompression von Gas/Luft-Gemisch bevor Zündfunke kommt. Verzweigte Alkane und Aromaten sind besser. Qualitätsmaß für Treibstoff: Octanzahl (OZ)
H3C(CH2)6CH3 CCH3
CH3H3C CH
n-Heptan IsooctanOZ = 0 OZ = 100
CH3
CH3CH2
H2
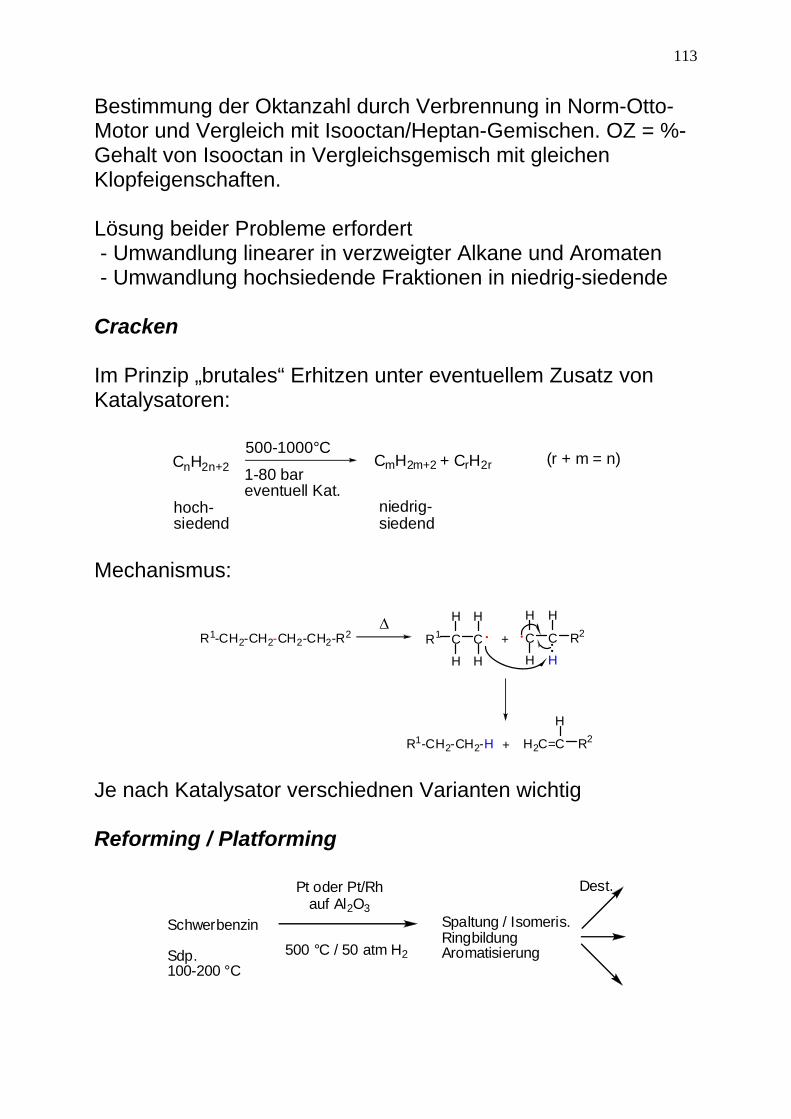
113
Bestimmung der Oktanzahl durch Verbrennung in Norm-Otto-Motor und Vergleich mit Isooctan/Heptan-Gemischen. OZ = %-Gehalt von Isooctan in Vergleichsgemisch mit gleichen Klopfeigenschaften. Lösung beider Probleme erfordert - Umwandlung linearer in verzweigter Alkane und Aromaten - Umwandlung hochsiedende Fraktionen in niedrig-siedende Cracken Im Prinzip „brutales“ Erhitzen unter eventuellem Zusatz von Katalysatoren:
CnH2n+2 CmH2m+2 + CrH2r500-1000°C
1-80 bareventuell Kat.
hoch-siedend
niedrig-siedend
(r + m = n)
Mechanismus:
R1 C
H
H
C
H
H
+ C
H
C
H H
H
R2
R1-CH2-CH2-H + H2C=C R2
H
ΔR1-CH2-CH2-CH2-CH2-R2
Je nach Katalysator verschiednen Varianten wichtig Reforming / Platforming
Schwerbenzin
Sdp.100-200 °C
Pt oder Pt/Rhauf Al2O3
500 °C / 50 atm H2
Spaltung / Isomeris.RingbildungAromatisierung
Dest.

114
Z. B. aus
z. B. aus
+ 2
+ +
Aus Crackprozessen hauptsächlich Benzol, Toluol, Xylole Andere Aromaten vorwiegend aus Kohle Kohle entstand aus Pflanzen unter hohem Druck und hoher Temperatur unter Mitwirkung von Mikroorganismen durch Reduktion: >90% C, daneben viel S, manchmal P! Insgesamt viel mehr Kohle als Erdöl vorhanden! Kohle entweder direkt verbrannt oder
Kohle> 1000°C
Koks + Teer + CH4 + H2 + CO
Kohlen-stoff
aromatischeKohlenwasserstoffe
Für chemische Industrie Teer wichtig: fraktionierte Destillation und dann andere Trennprozesse

115
Produkt Ausbeute [%]
Koks
Kokereigas (H2, CH4, CO)
Teer
Benzol
Ammoniak
Schwefelwasserstof f
Wasser, Sonstiges
70
15
5
1
0.25
0.2
8.5
Rückstand (Teerpech) 50 - 60
N
Fraktion Siedepunkt [°C] Ausbeute [%] Strukturen
Leichtöl
Mittelöl
Schweröl
Anthracenöl
80 - 170
170 - 250
250 - 300
300 - 400
2.5 - 6
10 - 12
8 - 10
18 - 20
OH
N
Destillation
Elektrophile aromatische Substitution Substitution am Benzol a) Bromierung und Chlorierung
Raumtemperatur
Br2
Bruttoreaktion:
H
Br2
(FeBr3)Kat
Br
+ HBr
Chlorierung analog mit Cl2/AlCl3

116
Mechanismus: Erzeugung Elektrophil
Br Br + FeBr3 Br Br FeBr3
δ+ δ-
Reaktion via σ-Komplex
Br Br FeBr3
δ+ δ-Br Br FeBr3
+ -
H Br H Br H Br
Br
H
+ HBr + FeBr3 Reaktionsprofil
EnergieE
E+ +
aromatisch
Zwischenstufe:nicht aromatisch
Ea1geschwindigkeits-bestimmenderSchritt
HE
+
++
Ea2kleine Aktivierungnotwendig=> schneller Schritt
aromatisch
EH+ +
HE
+
++
HE
+
HE
+
HE
+

117
regeneratives Verhalten des π-Systems, die Rückbildung eines aromatischen Systems ist energetisch günstiger als die Addition zum Sechsring mit zwei Doppelbindungen => Substitution am planaren sp2-Kohlenstoffatom nach einem Additions-Eliminierungs-Mechanismus über eine Zwischenstufe mit tetraedrischem, sp3-hybridisiertem Kohlenstoffatom b) Nitrierung
N NH2O O
NOO
HNO3 konz./H2SO4 konz.
10°C
Zn / H+
Nitrobenzol Anilin Bezeichnungen: NO2 Nitrogruppe ; NH2 Aminogruppe Zum Mechanismus: 2 Typen von Reaktionen werden unterschieden Erzeugung Elektrophil: Erzeugung Elektrophil: Nitronium-Ion NO O
HO NO
O+ H HSO4
raschO N
O
OH
H
langsamO
H
H
O
N
O
HSO4 HSO4 Aromatische Substitution:
NHO
O
weiter analog Bromierung
O N OH

118
c) Sulfonierung
SO3HHH2SO4
10% SO3
35°C30 min
Gemisch aus H2SO4 und SO3 = Oleum Elektrophil:
SO
OO SO
OO
Wichtig Sulfonierung ist reversibel: deshalb Sulfonylgruppe abspaltbar mit starker Säure (H+).
SO3HH H SOO
OO S O
O
Wegen Reversibilität ist SO3H-Gruppe abspaltbar mit H+ , kann also als dirigierender Hilfs-Substituent verwendet werden !
in der Medizin werden antibakterielle Sulfonamide verwendet:
HNR
S NHO
O
R'
z.B. Sulfadiazin (gegen Malaria)
H2N S NO
O
H
NN

119
Besonders interessanter Fall ist Naphthalin
H2SO480°C
H2SO4160°C
H
H
SO3H
SO3H
kinetischeKontrolle
thermodynamischeKontrolle
Reaktionsprofil:

120
Zweitsubstitution Ein Benzolderivat kann in der Regel nochmals substituiert werden. Dies bringt zwei Probleme mit sich: Reaktivität Regioselektivität
R
Wo greift das Elektrophil an ?
E+
ipso
orthometa
para
E
RE
R
E
R
E
völlig andere Verbindung,kein Isomer !!
Konstitutionsisomere

121
Isomerie von Benzolderivaten mit gleichen Substituenten
X
X
X
X
X
X
X
X
X
XX
XX
X
X X
X
X
X X
XX
X X X
X
X
X
ortho (o) meta (m) para (p)
vicinal(vic.)
asymmetrisch(asym.)
symmetrisch(sym.)
X
X
X
X
X
X
X
X
X
X
X
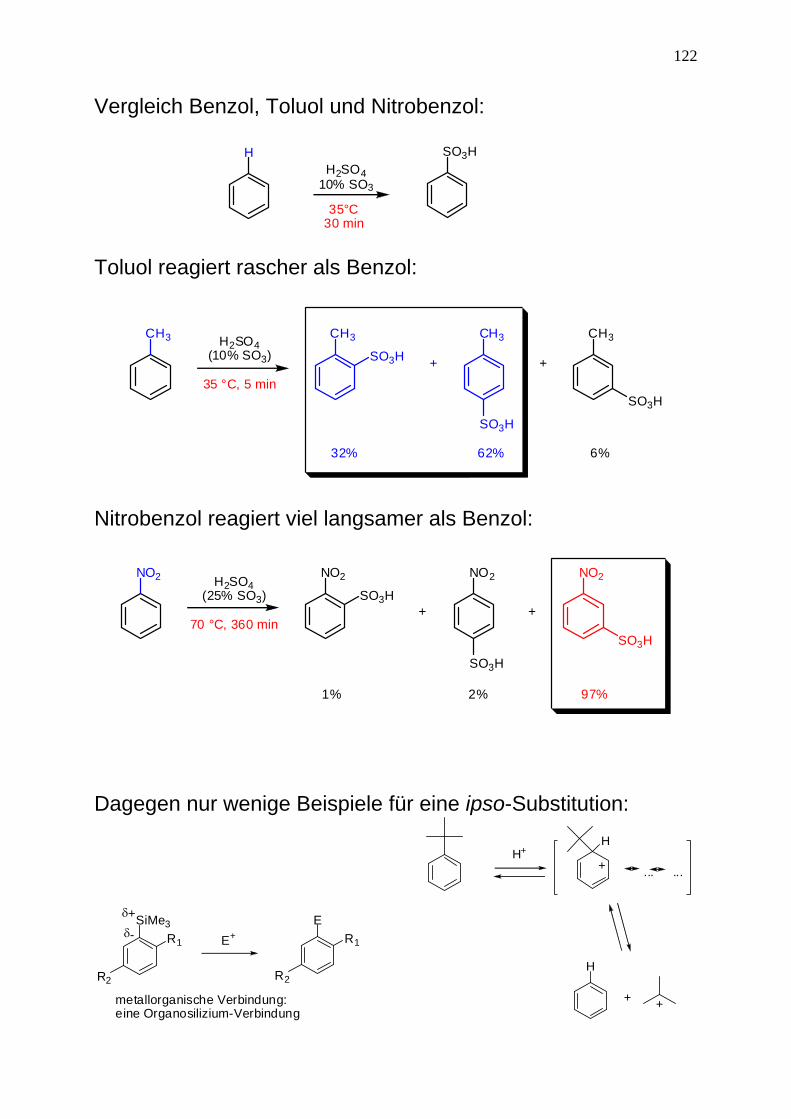
122
Vergleich Benzol, Toluol und Nitrobenzol:
SO3HHH2SO4
10% SO3
35°C30 min
Toluol reagiert rascher als Benzol:
CH3 CH3 CH3 CH3
SO3H + +
H2SO4(10% SO3)
35 °C, 5 min
32% 62% 6%
SO3H
SO3H
Nitrobenzol reagiert viel langsamer als Benzol:
NO2 NO2 NO2 NO2
SO3H+ +
H2SO4(25% SO3)
70 °C, 360 min
1% 2% 97%
SO3H
SO3H
Dagegen nur wenige Beispiele für eine ipso-Substitution:
SiMe3R1
R2
δ+δ- E+
ER1
R2
metallorganische Verbindung:eine Organosilizium-Verbindung
H+H
+ ......
H
+ +

123
Allgemein: Substituenten 1. Ordnung dirigieren o/p aktivieren NR2 OR Alkyl Aryl Substituenten 2. Ordnung dirigieren m desaktivieren NO2 (C=O)R NR3
+ SO3H Substituenteneffekte Ein Substituent wirkt auf ein C-Gerüst über das σ- und das π-System und über den Raum direkt. Die Anteile unterscheidet man als I- und M-Effekt sowie sterischen Effekt. Zwei Grundsysteme analog
R R
RR
R +I σ-Donor +M π-Donor R -I σ-Akzeptor -M π-Akzeptor

124
Zweitsubstitution Die Regioselektivität der Zweitsubstitution wird durch die
Substituenteneffekte des Erstsubstituenten im σ-Komplex bestimmt:
R R
E
RE
R
E
R Donor: günstigR Akzeptor: ungünstig
R Donor: günstigR Akzeptor: ungünstig
R unwichtig
E+
R
HE
= 1/3
R
EH
R
EH
σ-Komplex
Konsequenz: +M/+I Stabilisierung der positiven Ladung im σ-Komplex,
wenn Substituent relativ zu E in o/p-Stellung → o/p-dirigierend, aktivierend -M/-I stark destabilisierend in der o/p-Stellung, schwächer
in der m-Stellung → m-dirigierend, desaktivierend Antagonismus: +M/-I, z.B. bei OR und NR2 dominiert +M, aber Sonderfall Halogene: Regioselektivität o/p (wg. +M), aber
desaktivierend (wg. -I)
125
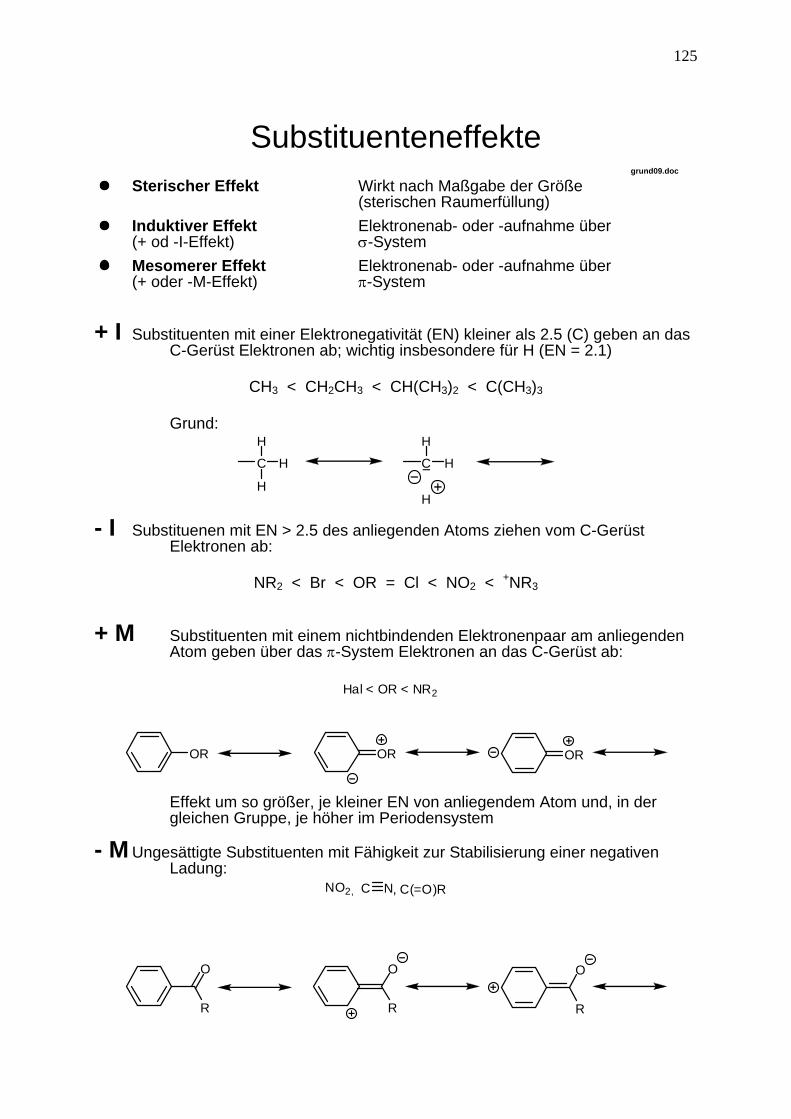
Substituenteneffekte
grund09.doc Sterischer Effekt Wirkt nach Maßgabe der Größe
(sterischen Raumerfüllung) Induktiver Effekt
(+ od -I-Effekt) Elektronenab- oder -aufnahme über σ-System
Mesomerer Effekt (+ oder -M-Effekt)
Elektronenab- oder -aufnahme über π-System
+ I Substituenten mit einer Elektronegativität (EN) kleiner als 2.5 (C) geben an das
C-Gerüst Elektronen ab; wichtig insbesondere für H (EN = 2.1)
CH3 < CH2CH3 < CH(CH3)2 < C(CH3)3
Grund:
C
H
H
H C
H
H
H - I Substituenen mit EN > 2.5 des anliegenden Atoms ziehen vom C-Gerüst
Elektronen ab:
NR2 < Br < OR = Cl < NO2 < +NR3
+ M Substituenten mit einem nichtbindenden Elektronenpaar am anliegenden Atom geben über das π-System Elektronen an das C-Gerüst ab:
OR OR OR
Hal < OR < NR2
Effekt um so größer, je kleiner EN von anliegendem Atom und, in der
gleichen Gruppe, je höher im Periodensystem - M Ungesättigte Substituenten mit Fähigkeit zur Stabilisierung einer negativen
Ladung:
O
R
O
R
O
R
NO2, C N, C(=O)R
126
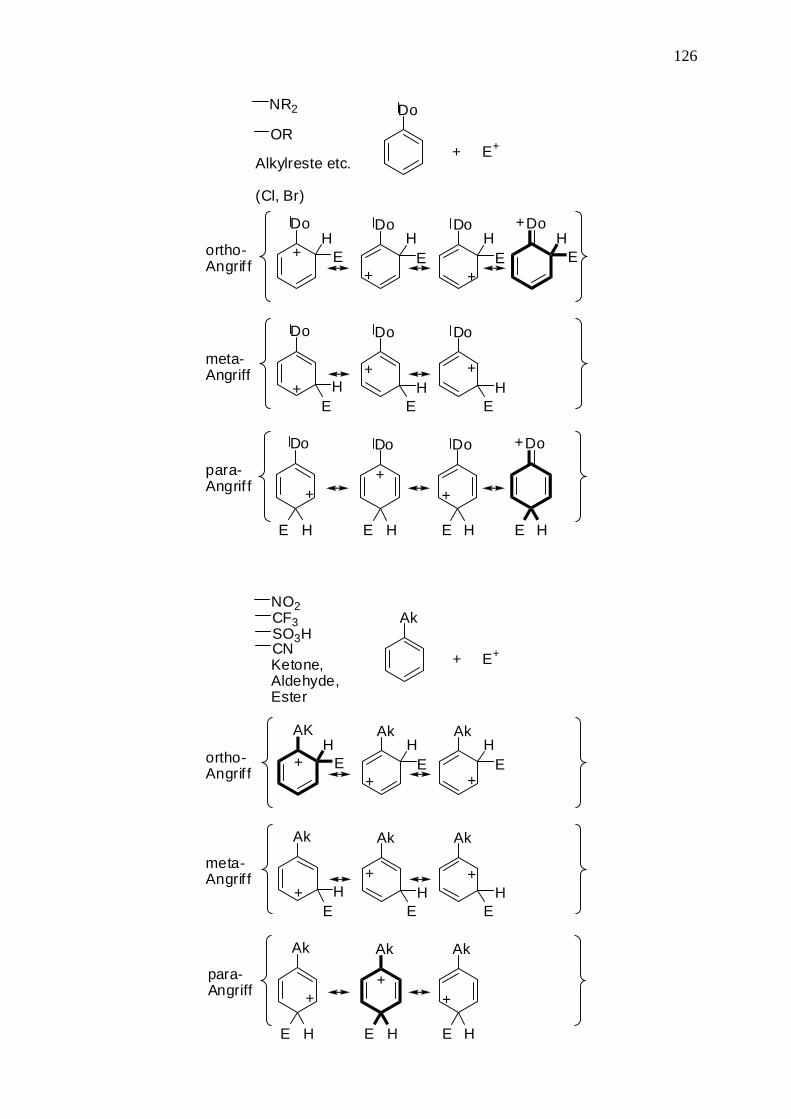
NR2
OR
Alkylreste etc.
(Cl, Br)
Do
+ E+
ortho-Angrif f
DoH
E
Do
EH
Do
EH
Do
EH
++ +
+
meta-Angriff
Do Do Do
++ +
HE
HE
HE
para-Angrif f
Do Do Do Do
++
+
+
HE E H E H E H
NO2CF3 Ak
+ E+
ortho-Angrif f
AKH
E EH
Ak
EH
++ +
meta-Angrif f
Ak Ak Ak
++ +
HE
HE
HE
para-Angriff
Ak Ak Ak
++
+
HE E H E H
SO3HCNKetone,Aldehyde,Ester
Ak

127
Alkylaromaten, Friedel-Crafts-Reaktion Äußerst wichtige Verbindungen, vor allem als Lösungsmittel und sog. Zwischenprodukte. Aus Crack-Verfahren billig anfallend. Wichtige Methylderivate des Benzols
Sdp. [°C] Schmp. [°C]
Benzol
80
6
CH3
Toluol
111
-95
CH3CH3
ortho-Xylol
144
-25
CH3
CH3
meta-Xylol
139
-48
CH3
CH3
para-Xylol
138
13

128
Herstellung der Alkylaromaten a) Friedel-Crafts-Alkylierung
H3C ClAlCl3 CH3
AlCl4
HAlCl4
HCl
CH3 CH3
CH3
rascher alsBenzol !
Xylole Katalysecyclus Toluol reagiert rascher als Benzol. Ferner alle Reaktionen reversibel, deshalb Gemische, die durch Destillation aufgetrennt werden. Allgemein: Lewis-Säure = Friedel-Crafts-Katalysator: AlCl3, FeBr3, BF3•OEt2, TiCl4 Reaktion auch mehrfach möglich:
AlCl3+ CCl4
- 3 HClC Cl Triphenylchlormethan
"Tritylchlorid"3

129
Beispiel für eine elektrophile aromatische Alkylierung:Cumol
++ [H+]
+H
+
H
H+
- [H+]
Cumol
b) Friedel-Crafts-Acylierung Präparativ sauberer als Alkylierung, da Produkt deaktiviert.
AlCl3
RCl
O RO RHH
Zn/HgHCl
Clemmenson-Reduktion
Clemmensen-Reduktion besonders gut mit Arylketonen! "Katalysator" AlCl3 wird in der Regel in molarer Menge benötigt, wegen Produktinhibition:
RO
Cl
AlCl3R
O
Ar
AlCl3R
O
Ar+ R
O
Cl+

130
Reaktionen, Benzyl-Resonanz Toluol reagiert mit Elektrophilen schneller als Benzol.
HNO3H2SO4 Gemisch
auso-, p-Nitrotoluol
NO2
NO2 Halogenierung zeigt interessante Besoderheiten:
KKK
CH3 CH3Cl
CH3
Cl
+Cl2 / AlCl3
0°C
SSS
CH3 CH2ClCl2 / hv
Siedehitze- HCl
Grund Radikalkettenreaktion:
Start
Kette
Abbruch
Cl2 2 Cl
ArCH3 ArCH2
ArCH2Cl
Dibenzyl
Cl HCl
Cl+ Cl2
+
+
+
ArCH4
h . ν
2 ArCH2 ArH2C CH2Ar

131
Allgemein: Radikal in benzylischer oder allylischer Stellung besonders stabilisiert: Ph-CH2 Benzyl H2C=CH-CH2 Allyl
HH
RE = 50 KJ/mol
Das Triphenylmethyl-Radikal Versuch Mit drei Phenylgruppen besonders große Stabilisierung (Gomberg 1900)
CC Cl2Zn
- ZnCl2
CH
O2
C O O C
gelb-orangelanglebigesRadikal
(10 Grenzstrukturen)
farblosPeroxid früher Hexaphenyläthan







