蛋白质纯化技术及应用 -...
TRANSCRIPT

书书书
蛋白质纯化技术及应用
陆 健 等编著

(京)新登字039号
图书在版编目 (CIP)数据
蛋白质纯化技术及应用/陆健等编著—北京:化学工
业出版社,20056 ISBN7502573828
Ⅰ蛋… Ⅱ陆… Ⅲ蛋白质提纯技术 ⅣQ51
中国版本图书馆CIP数据核字 (2005)第070811号
蛋白质纯化技术及应用陆 健 等编著
责任编辑:侯玉周
文字编辑:焦欣渝
责任校对:边 涛
封面设计:鲍 萌
化学工业出版社出版发行
(北京市朝阳区惠新里3号 邮政编码100029)购书咨询:(010)64982530 (010)64918013购书传真:(010)64982630http://www.cip.com.cn
新华书店北京发行所经销
大厂聚鑫印刷有限责任公司印刷
三河市东柳装订厂装订
开本720mm×1000mm 1/16 印张22 字数402千字
2005年9月第1版 2005年9月北京第1次印刷
ISBN7502573828定 价:4900元
版权所有 违者必究
该书如有缺页、倒页、脱页者,本社发行部负责退换

前 言蛋白质是一切生命活动的主要体 现 者 和 物 质 基 础。随 着 分 子 生 物 学、结 构 生 物 学、基 因
组学等研究的不断深入,许多学者将生命 科 学 领 域 的 研 究 焦 点 从 基 因 转 向 蛋 白 质,这 进 一 步
促进了蛋白质组学和蛋白质工程研究的发 展。蛋 白 质 研 究 已 成 为 当 前 生 物 化 学 和 分 子 生 物 学
领域内最为活跃的研究之一。
要研究蛋白质的结构和功能,首先要 得 到 高 纯 度 的 具 有 生 物 学 活 性 的 目 的 蛋 白。而 蛋 白
质在组织或细胞中一般都是以复杂的混合 物 的 形 式 存 在,每 种 类 型 的 细 胞 都 含 有 很 多 不 同 结
构和功能的蛋白质,这使蛋白质的分离纯 化 成 为 一 项 精 细 而 复 杂 的 任 务。虽 然 通 过 分 子 生 物
学手段可推导出蛋白质一级结构链的氨基 酸 排 列 顺 序 和 组 成,但 仍 有 许 多 问 题 需 要 通 过 对 蛋
白质本身的空间结构和表达模式的研究 才 能 得 以 更 好 的 解 释 和 完 善。此 外,利 用 蛋 白 质 工 程
对蛋白质的结构进行改造,也必须先要了 解 蛋 白 质 的 基 本 结 构。蛋 白 质 的 分 离 纯 化 是 广 大 生
物学、化学和医学工作者十分关心并且 长 期 深 入 研 究 的 领 域,在 获 得 高 纯 度 目 的 蛋 白 后,才
能够进行其性质的研究,从而有可能 大 规 模 生 产 应 用,以 造 福 人 类。因 此,蛋 白 质 纯 化 不 论
是在基础理论研究、揭示生命现象的本 质 方 面,还 是 在 蛋 白 质 工 程、基 因 工 程 和 细 胞 工 程 等
产品的工业化生产方面,都是非常基础、非常重要、非常关键的。
本书全面系统而深入地介绍了蛋白 质 纯 化 理 论,并 且 将 理 论 和 实 践 联 系,便 于 读 者 系 统
了解蛋白质纯化技术理论和具体实践应 用 过 程。书 中 既 介 绍 了 基 本 实 验 步 骤,也 介 绍 了 综 合
性蛋白质纯化研究的实例。全书主要 内 容 包 括:蛋 白 质 样 品 的 预 处 理,凝 胶 过 滤 色 谱,离 子
交换色谱,亲和色谱,共价色谱,电泳技术,特殊用 途 蛋 白 质 的 纯 化,不 同 来 源 蛋 白 质 的 纯
化,蛋白质的常用指标分析以及实验部分。
本书由陆健、周楠迪、史锋、曹 钰、王 栋、孔 维 宝 编 写。几 位 年 轻 老 师,充 分 利 用 在 国
外学习、研究的时间,结合亲 身 研 究 经 历,收 集 了 很 多 资 料,并 进 行 了 归 纳 整 理。李 胤、赵
海峰、李旺军、蔡国林、冯凌蕾、刘军、王璐等参与了 资 料 整 理 以 及 部 分 计 算 机 文 字 处 理 工
作,全书由陆健统稿、定稿。
由于作者水平所限,加之时间仓促,书中可能存在许多不足,真诚期待广大读者赐教。
衷心感谢化学工业出版社对本书的大力支持。
编著者
2005年4月于无锡

目 录1 绪论 1!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
11 蛋白质纯化的意义 1!!!!!!!!!!!!!!!!!!!!!!
12 纯化蛋白质常用的方法 1!!!!!!!!!!!!!!!!!!!!
13 蛋白质纯化的设计 2!!!!!!!!!!!!!!!!!!!!!!
131 基本原则 2!!!!!!!!!!!!!!!!!!!!!!!!
132 过程优化 3!!!!!!!!!!!!!!!!!!!!!!!!
133 经济性 4!!!!!!!!!!!!!!!!!!!!!!!!!
14 规模化蛋白质纯化 5!!!!!!!!!!!!!!!!!!!!!!
141 基本原则 5!!!!!!!!!!!!!!!!!!!!!!!!
142 主要操作过程 6!!!!!!!!!!!!!!!!!!!!!!
2 蛋白质样品的预处理 11!!!!!!!!!!!!!!!!!!!!!!!
21 蛋白质样品的提取和分离 11!!!!!!!!!!!!!!!!!!!
211 细胞的破碎 12!!!!!!!!!!!!!!!!!!!!!!!
212 蛋白质样品的分离 15!!!!!!!!!!!!!!!!!!!!
22 蛋白质样品的浓缩 17!!!!!!!!!!!!!!!!!!!!!!
221 吸附法 18!!!!!!!!!!!!!!!!!!!!!!!!!
222 超滤法 18!!!!!!!!!!!!!!!!!!!!!!!!!
223 沉淀法 20!!!!!!!!!!!!!!!!!!!!!!!!!
224 透析法 21!!!!!!!!!!!!!!!!!!!!!!!!!
225 冷冻干燥法 21!!!!!!!!!!!!!!!!!!!!!!!
226 双水相分离法 21!!!!!!!!!!!!!!!!!!!!!!
23 蛋白质样品的沉淀 22!!!!!!!!!!!!!!!!!!!!!!
231 盐析法 23!!!!!!!!!!!!!!!!!!!!!!!!!
232 有机溶剂沉淀法 25!!!!!!!!!!!!!!!!!!!!!
233 等电点沉淀法 26!!!!!!!!!!!!!!!!!!!!!!
234 聚乙二醇沉淀法 26!!!!!!!!!!!!!!!!!!!!!
235 选择性沉淀法 27!!!!!!!!!!!!!!!!!!!!!!
24 蛋白质样品的透析 27!!!!!!!!!!!!!!!!!!!!!!
241 基本原理 27!!!!!!!!!!!!!!!!!!!!!!!!
242 应用 28!!!!!!!!!!!!!!!!!!!!!!!!!!
3 凝胶过滤色谱 29!!!!!!!!!!!!!!!!!!!!!!!!!!
31 概述 29!!!!!!!!!!!!!!!!!!!!!!!!!!!!

311 基本原理 29!!!!!!!!!!!!!!!!!!!!!!!!
312 凝胶的性质和种类 36!!!!!!!!!!!!!!!!!!!!
313 凝胶介质的选择 41!!!!!!!!!!!!!!!!!!!!!
32 凝胶柱的操作 42!!!!!!!!!!!!!!!!!!!!!!!!
321 装柱 42!!!!!!!!!!!!!!!!!!!!!!!!!!
322 样品和缓冲液的准备 46!!!!!!!!!!!!!!!!!!!
323 色谱条件 47!!!!!!!!!!!!!!!!!!!!!!!!
324 加样和洗脱 48!!!!!!!!!!!!!!!!!!!!!!!
325 凝胶柱的再生及保存 49!!!!!!!!!!!!!!!!!!!
33 应用 49!!!!!!!!!!!!!!!!!!!!!!!!!!!!
331 脱盐 49!!!!!!!!!!!!!!!!!!!!!!!!!!
332 蛋白质分离 50!!!!!!!!!!!!!!!!!!!!!!!
333 测定蛋白质分子量 51!!!!!!!!!!!!!!!!!!!!
334 蛋白质复性的研究 51!!!!!!!!!!!!!!!!!!!!
335 其他 52!!!!!!!!!!!!!!!!!!!!!!!!!!
4 离子交换色谱 53!!!!!!!!!!!!!!!!!!!!!!!!!!
41 基本概念 54!!!!!!!!!!!!!!!!!!!!!!!!!!
411 蛋白质的电性质 55!!!!!!!!!!!!!!!!!!!!!
412 离子交换理论 60!!!!!!!!!!!!!!!!!!!!!!
413 离子交换的分辨率 62!!!!!!!!!!!!!!!!!!!!
42 固定相:离子交换剂 66!!!!!!!!!!!!!!!!!!!!!
421 基本结构 66!!!!!!!!!!!!!!!!!!!!!!!!
422 功能基团和酸碱性质 67!!!!!!!!!!!!!!!!!!!
423 离子交换剂的部分性质 68!!!!!!!!!!!!!!!!!!
424 离子交换剂的类型 74!!!!!!!!!!!!!!!!!!!!
43 流动相:缓冲液和盐 86!!!!!!!!!!!!!!!!!!!!!
431 缓冲液的类型 87!!!!!!!!!!!!!!!!!!!!!!
432 缓冲液的pH和浓度 88!!!!!!!!!!!!!!!!!!!
433 离子对色谱行为的影响 89!!!!!!!!!!!!!!!!!!
434 添加剂 90!!!!!!!!!!!!!!!!!!!!!!!!!
44 实验方案设计 91!!!!!!!!!!!!!!!!!!!!!!!!
441 离子交换剂的选择 91!!!!!!!!!!!!!!!!!!!!
442 缓冲液的选择和色谱条件的确定 94!!!!!!!!!!!!!!
443 色谱柱的尺寸 98!!!!!!!!!!!!!!!!!!!!!!

444 分批分离 99!!!!!!!!!!!!!!!!!!!!!!!!
45 色谱技术 100!!!!!!!!!!!!!!!!!!!!!!!!!
451 离子交换剂的准备 100!!!!!!!!!!!!!!!!!!!
452 样品的准备 102!!!!!!!!!!!!!!!!!!!!!!
453 加样 103!!!!!!!!!!!!!!!!!!!!!!!!!
454 洗脱技术 105!!!!!!!!!!!!!!!!!!!!!!!
455 样品的收集和处理 113!!!!!!!!!!!!!!!!!!!
456 离子交换剂的再生、清洗、消毒和储存 114!!!!!!!!!!
46 应用实例 115!!!!!!!!!!!!!!!!!!!!!!!!!
461 用DEAESepharose色谱分离真菌纤维素酶 115!!!!!!!!
462 用 MonoS和 MonoQ连续色谱法从鸡肌肉组织分级分离
糖酵解酶类 116!!!!!!!!!!!!!!!!!!!!!!
463 用 MonoQ阴离子交换分离白血病细胞N乙酰βD氨基
葡萄糖苷酶的同工酶 117!!!!!!!!!!!!!!!!!!
464 用BioRex70色谱分离眼镜蛇神经毒素的六种单乙酰基衍
生物 118!!!!!!!!!!!!!!!!!!!!!!!!!
465 用阶段洗脱法大规模纯化人血清白蛋白和免疫球蛋白G 119!!!
5 亲和色谱 120!!!!!!!!!!!!!!!!!!!!!!!!!!!
51 亲和吸附剂 121!!!!!!!!!!!!!!!!!!!!!!!!
511 配体和载体的选择 121!!!!!!!!!!!!!!!!!!!
512 亲和吸附剂的制备方法 123!!!!!!!!!!!!!!!!!
513 商品化基团特异性吸附剂 128!!!!!!!!!!!!!!!!
52 色谱技术 130!!!!!!!!!!!!!!!!!!!!!!!!!
521 亲和色谱操作过程 130!!!!!!!!!!!!!!!!!!!
522 亲和色谱的操作注意事项 132!!!!!!!!!!!!!!!!
523 亲和色谱的吸附容量与吸附效率 132!!!!!!!!!!!!!
53 亲和色谱的特殊类型 133!!!!!!!!!!!!!!!!!!!!
531 凝集素亲和色谱 133!!!!!!!!!!!!!!!!!!!!
532 免疫亲和色谱 137!!!!!!!!!!!!!!!!!!!!!
533 环氧乙烷丙烯酰胺珠亲和色谱 140!!!!!!!!!!!!!!
534 金属螯合亲和色谱 142!!!!!!!!!!!!!!!!!!!
535 疏水作用色谱 144!!!!!!!!!!!!!!!!!!!!!
54 应用 145!!!!!!!!!!!!!!!!!!!!!!!!!!!
541 抗体和抗原的纯化 145!!!!!!!!!!!!!!!!!!!

542 酶的纯化 145!!!!!!!!!!!!!!!!!!!!!!!
543 糖蛋白的纯化 145!!!!!!!!!!!!!!!!!!!!!
544 脂蛋白的纯化 145!!!!!!!!!!!!!!!!!!!!!
6 共价色谱 146!!!!!!!!!!!!!!!!!!!!!!!!!!!
61 巯基的化学性质 147!!!!!!!!!!!!!!!!!!!!!!
611 离子化、氧化、金属联结、烷基化 147!!!!!!!!!!!!
612 巯基二硫化物互换反应 148!!!!!!!!!!!!!!!!!
613 与活性二硫化物的反应 149!!!!!!!!!!!!!!!!!
614 巯基与二硫氧化物的反应 150!!!!!!!!!!!!!!!!
62 含巯基的蛋白质 151!!!!!!!!!!!!!!!!!!!!!!
621 生物体中的氧化还原态 151!!!!!!!!!!!!!!!!!
622 胞内与胞外的蛋白质 151!!!!!!!!!!!!!!!!!!
623 蛋白质巯基基团的解蔽 151!!!!!!!!!!!!!!!!!
624 蛋白质二硫键的还原 152!!!!!!!!!!!!!!!!!!
625 巯基的引入 152!!!!!!!!!!!!!!!!!!!!!!
626 巯基基团的衍生化作用 152!!!!!!!!!!!!!!!!!
63 共价色谱的凝胶 152!!!!!!!!!!!!!!!!!!!!!!
631 原理 152!!!!!!!!!!!!!!!!!!!!!!!!!
632 凝胶的基质 153!!!!!!!!!!!!!!!!!!!!!!
633 凝胶的固定相 153!!!!!!!!!!!!!!!!!!!!!
634 固定相活性基团的比较 153!!!!!!!!!!!!!!!!!
635 凝胶固定相的引入 155!!!!!!!!!!!!!!!!!!!
636 凝胶的取代程度和实际容量 157!!!!!!!!!!!!!!!
64 色谱技术 158!!!!!!!!!!!!!!!!!!!!!!!!!
641 预备 158!!!!!!!!!!!!!!!!!!!!!!!!!
642 蛋白样品的结合 158!!!!!!!!!!!!!!!!!!!!
643 洗涤 159!!!!!!!!!!!!!!!!!!!!!!!!!
644 还原洗脱 159!!!!!!!!!!!!!!!!!!!!!!!
645 巯基蛋白的回收 160!!!!!!!!!!!!!!!!!!!!
646 巯基凝胶的再生 161!!!!!!!!!!!!!!!!!!!!
647 反向共价色谱 162!!!!!!!!!!!!!!!!!!!!!
65 应用实例 162!!!!!!!!!!!!!!!!!!!!!!!!!
651 从刀豆中分离脲酶 162!!!!!!!!!!!!!!!!!!!
652 木瓜蛋白酶的纯化 163!!!!!!!!!!!!!!!!!!!

653 共价色谱的顺序洗脱 164!!!!!!!!!!!!!!!!!!
654 巯基多肽的纯化 165!!!!!!!!!!!!!!!!!!!!
655 大肠杆菌β半乳糖苷酶的可逆固定 165!!!!!!!!!!!!
656 蛋白质亚基的鉴定 166!!!!!!!!!!!!!!!!!!!
7 电泳技术 167!!!!!!!!!!!!!!!!!!!!!!!!!!!
71 概述 167!!!!!!!!!!!!!!!!!!!!!!!!!!!
711 原理 167!!!!!!!!!!!!!!!!!!!!!!!!!
712 分类 169!!!!!!!!!!!!!!!!!!!!!!!!!
713 介质 170!!!!!!!!!!!!!!!!!!!!!!!!!
714 电泳仪器 172!!!!!!!!!!!!!!!!!!!!!!!
715 染色方法 173!!!!!!!!!!!!!!!!!!!!!!!
72 常规聚丙烯酰胺凝胶电泳 174!!!!!!!!!!!!!!!!!!
721 原理 174!!!!!!!!!!!!!!!!!!!!!!!!!
722 常规PAGE的类型 175!!!!!!!!!!!!!!!!!!!
723 影响分离结果的外在因素 175!!!!!!!!!!!!!!!!
724 蛋白质分子量的测定 176!!!!!!!!!!!!!!!!!!
725 常规PAGE的实验方法 177!!!!!!!!!!!!!!!!!
726 PAGE的应用范围 181!!!!!!!!!!!!!!!!!!!
73 SDS聚丙烯酰胺凝胶电泳 181!!!!!!!!!!!!!!!!!!
731 原理 181!!!!!!!!!!!!!!!!!!!!!!!!!
732 SDSPAGE的类型 182!!!!!!!!!!!!!!!!!!!
733 影响分离结果的外在因素 182!!!!!!!!!!!!!!!!
734 蛋白质分子量的测定 183!!!!!!!!!!!!!!!!!!
735 SDS聚丙烯酰胺凝胶电泳的实验方法 183!!!!!!!!!!!
736 SDSPAGE的应用范围 184!!!!!!!!!!!!!!!!!
74 等电聚焦 184!!!!!!!!!!!!!!!!!!!!!!!!!
741 原理 185!!!!!!!!!!!!!!!!!!!!!!!!!
742 载体两性电解质pH梯度的形成 186!!!!!!!!!!!!!
743 载体两性电解质等电聚焦方法 187!!!!!!!!!!!!!!
744 固相pH梯度的介质及其pH梯度的形成 189!!!!!!!!!
745 固相pH梯度等电聚焦方法 192!!!!!!!!!!!!!!!
746 等电聚焦的应用范围 194!!!!!!!!!!!!!!!!!!
75 双向电泳 194!!!!!!!!!!!!!!!!!!!!!!!!!
751 原理 194!!!!!!!!!!!!!!!!!!!!!!!!!

752 双向电泳实验方法 194!!!!!!!!!!!!!!!!!!!
753 应用 196!!!!!!!!!!!!!!!!!!!!!!!!!
76 免疫电泳 197!!!!!!!!!!!!!!!!!!!!!!!!!
761 原理 197!!!!!!!!!!!!!!!!!!!!!!!!!
762 电泳方法 199!!!!!!!!!!!!!!!!!!!!!!!
763 应用 200!!!!!!!!!!!!!!!!!!!!!!!!!
77 蛋白质印迹 200!!!!!!!!!!!!!!!!!!!!!!!!
771 原理 200!!!!!!!!!!!!!!!!!!!!!!!!!
772 试验材料的选择 201!!!!!!!!!!!!!!!!!!!!
773 蛋白质印迹试验方法 202!!!!!!!!!!!!!!!!!!
774 应用 203!!!!!!!!!!!!!!!!!!!!!!!!!
78 毛细管电泳 203!!!!!!!!!!!!!!!!!!!!!!!!
8 特殊用途蛋白质的纯化 204!!!!!!!!!!!!!!!!!!!!!
81 测序用蛋白质的纯化 204!!!!!!!!!!!!!!!!!!!!
811 测序蛋白纯化技术 205!!!!!!!!!!!!!!!!!!!
812 多肽的制备和纯化 207!!!!!!!!!!!!!!!!!!!
82 用于晶体学研究的蛋白质纯化 208!!!!!!!!!!!!!!!!
821 蛋白质结晶方法 211!!!!!!!!!!!!!!!!!!!!
822 不均一性对蛋白质结晶的影响 214!!!!!!!!!!!!!!
83 融合蛋白的纯化 218!!!!!!!!!!!!!!!!!!!!!!
831 融合蛋白的纯化过程 219!!!!!!!!!!!!!!!!!!
832 蛋白质的水解保护 225!!!!!!!!!!!!!!!!!!!
833 常用融合技术 227!!!!!!!!!!!!!!!!!!!!!
834 融合蛋白纯化实例 231!!!!!!!!!!!!!!!!!!!
84 包含体的初步纯化 233!!!!!!!!!!!!!!!!!!!!!
841 影响包含体形成的因素 233!!!!!!!!!!!!!!!!!
842 细胞匀浆中包含体的分离 233!!!!!!!!!!!!!!!!
843 包含体的洗涤 233!!!!!!!!!!!!!!!!!!!!!
85 膜蛋白的纯化 234!!!!!!!!!!!!!!!!!!!!!!!
851 蛋白质的溶解性 235!!!!!!!!!!!!!!!!!!!!
852 膜蛋白的纯化 237!!!!!!!!!!!!!!!!!!!!!
86 治疗用蛋白质的纯化 238!!!!!!!!!!!!!!!!!!!!
861 治疗用蛋白纯化的关键问题 239!!!!!!!!!!!!!!!
862 工艺确认 242!!!!!!!!!!!!!!!!!!!!!!!

9 不同来源蛋白质的纯化 244!!!!!!!!!!!!!!!!!!!!!
91 微生物细胞培养蛋白质的纯化 244!!!!!!!!!!!!!!!!
911 色谱纯化方法 244!!!!!!!!!!!!!!!!!!!!!
912 蛋白质离子交换纯化 245!!!!!!!!!!!!!!!!!!
92 哺乳动物细胞培养蛋白质的纯化 250!!!!!!!!!!!!!!!
921 工艺设计 251!!!!!!!!!!!!!!!!!!!!!!!
922 细胞分离 252!!!!!!!!!!!!!!!!!!!!!!!
923 产品的初步回收和分离 252!!!!!!!!!!!!!!!!!
924 主要纯化方法 252!!!!!!!!!!!!!!!!!!!!!
925 样品纯化举例———单克隆抗体的纯化 253!!!!!!!!!!!
926 消除污染 254!!!!!!!!!!!!!!!!!!!!!!!
93 动物组织蛋白质的纯化 256!!!!!!!!!!!!!!!!!!!
931 组织的选择 256!!!!!!!!!!!!!!!!!!!!!!
932 组织破碎 257!!!!!!!!!!!!!!!!!!!!!!!
933 防止蛋白质的有害水解 257!!!!!!!!!!!!!!!!!
934 亚细胞分级分离 257!!!!!!!!!!!!!!!!!!!!
935 膜蛋白的溶解 259!!!!!!!!!!!!!!!!!!!!!
936 动物蛋白质纯化举例 260!!!!!!!!!!!!!!!!!!
94 植物组织蛋白质的纯化 263!!!!!!!!!!!!!!!!!!!
941 影响植物组织蛋白质纯化的因素 263!!!!!!!!!!!!!
942 材料来源 263!!!!!!!!!!!!!!!!!!!!!!!
943 确定植物蛋白种类的直接和间接策略 267!!!!!!!!!!!
944 样品纯化举例 268!!!!!!!!!!!!!!!!!!!!!
10 蛋白质的常用指标分析 271!!!!!!!!!!!!!!!!!!!!!
101 蛋白质的含量分析 271!!!!!!!!!!!!!!!!!!!!!
1011 测定蛋白质浓度的方法 271!!!!!!!!!!!!!!!!!
1012 紫外分光光度法 271!!!!!!!!!!!!!!!!!!!!
1013 Lowry法 274!!!!!!!!!!!!!!!!!!!!!!
1014 二喹啉甲酸检测法 277!!!!!!!!!!!!!!!!!!!
1015 考马斯亮蓝染色法 278!!!!!!!!!!!!!!!!!!!
102 蛋白质的分子量测定 281!!!!!!!!!!!!!!!!!!!!
1021 SDSPAGE凝胶电泳法测定蛋白质的分子量 282!!!!!!!
1022 凝胶过滤色谱法测定天然蛋白质的分子量 286!!!!!!!!!
103 蛋白质的等电点测定 288!!!!!!!!!!!!!!!!!!!!

1031 蛋白质的等电点 288!!!!!!!!!!!!!!!!!!!!
1032 等电聚焦技术 289!!!!!!!!!!!!!!!!!!!!!
1033 超薄聚丙烯酰胺凝胶等电聚焦法测定蛋白质的等电点 289!!!!
104 蛋白质的纯度分析 294!!!!!!!!!!!!!!!!!!!!!
1041 纯度标准 294!!!!!!!!!!!!!!!!!!!!!!!
1042 常用的纯度检测方法 294!!!!!!!!!!!!!!!!!!
105 糖蛋白质中的糖含量分析 296!!!!!!!!!!!!!!!!!!
1051 糖蛋白的化学分析 296!!!!!!!!!!!!!!!!!!!
1052 SDS凝胶中的糖蛋白染色法 297!!!!!!!!!!!!!!
106 蛋白质和肽的N末端氨基酸测定 299!!!!!!!!!!!!!!
1061 FDNB法 300!!!!!!!!!!!!!!!!!!!!!!!
1062 DNS分析法 301!!!!!!!!!!!!!!!!!!!!!
1063 Edman降解法 304!!!!!!!!!!!!!!!!!!!!
107 蛋白质N末端序列分析 306!!!!!!!!!!!!!!!!!!
1071 DNSEdman法 308!!!!!!!!!!!!!!!!!!!!
1072 DABITC/PITC双偶合法 309!!!!!!!!!!!!!!!!
108 蛋白质和肽的C末端分析 313!!!!!!!!!!!!!!!!!
1081 概述 313!!!!!!!!!!!!!!!!!!!!!!!!!
1082 C末端序列分析的羧肽酶Y法 314!!!!!!!!!!!!!
11 实验部分 317!!!!!!!!!!!!!!!!!!!!!!!!!!!
111 用于蛋白质纯化的仪器 (工作站) 317!!!!!!!!!!!!!!
1111 蛋白质纯化系统的主要部件 317!!!!!!!!!!!!!!!
1112 几种常见的蛋白质色谱系统 320!!!!!!!!!!!!!!!
112 蛋白质分离纯化实例 322!!!!!!!!!!!!!!!!!!!!
1121 蔗糖酶的分离纯化 322!!!!!!!!!!!!!!!!!!!
1122 转谷氨酰胺酶的分离纯化 323!!!!!!!!!!!!!!!!
1123 纯化重组蛋白Pseudomonasaeruginosa外毒素A 325!!!!!
1124 Ecoli中RNA聚合酶的提取和纯化 329!!!!!!!!!!!
1125 从Ecoli中分离制备高纯度的去乙酰头孢菌素C合成酶 330!!参考文献 334!!!!!!!!!!!!!!!!!!!!!!!!!!!!!

书书书
1
1 绪 论
11 蛋白质纯化的意义
蛋白质是包括人类在内的各种生物有机体的重要组成成分,是生命的物质基
础之一。生物体的生长、发育、遗传和繁殖等一切生命活动都离不开蛋白质。机
体内的一些生理活性物质,如多肽类激素、抗体、酶、核蛋白等都是蛋白质,它
们对调节生理功能、维持新陈代谢起着极其重要的作用。在生命活动中蛋白质无
处不在,而且不同的蛋白质有不同的结构和功能。如果生物体内的蛋白质发生不
正常变化或失去,那么生命活动将会出现病变或停止。因此,研究蛋白质有着极
为重要的生物学意义。随着分子生物学、结构生物学、基因组学等研究的不断深入,人们意识到仅
仅依靠基因组的序列分析来试图阐明生命活动的现象和本质是远远不够的。只有
从蛋白质组学的角度对所有蛋白质的总和进行研究,才能更科学地掌握生命现象
和活动规律,更完善地揭示生命的本质。由此许多学者将生命科学领域的研究焦
点从基因转向蛋白质,使蛋白质成为揭示生命活动现象和分子生物学机理的重要
研究对象。研究蛋白质首要的步骤是将目的蛋白从复杂的大分子混合物中分离纯
化出来,得到高纯度具有生物学活性的目的物。因此,高效的纯化技术和手段是
蛋白质研究的重要基础和关键之一。
12 纯化蛋白质常用的方法
蛋白质纯化的总体目标是设法在高效率、高得率的条件下获得高纯度、高活
性和完整的目的蛋白。采用一种或一套现成的方法把任何一种蛋白质从复杂的混
合体系中分离纯化出来显得比较困难,但对于任何一种蛋白质都有可能选择适当
的纯化方法来获得高纯度的活性蛋白质。因此,选择科学合理的纯化方法对于分
离纯化目的蛋白非常重要。纯化方法的选择和确定要根据不同蛋白质样品的性质和具体的研究目的来决
定。常用于初步提取和浓缩蛋白质的方法主要有吸附法、超滤法、沉淀法 (如盐
析、有机溶剂沉淀、等电点沉淀和选择性沉淀等)、透析法等。在要求高分辨率
的条件下,通常采用色谱法 (如凝胶过滤、离子交换、亲和色谱和共价色谱等)和电泳法 (如等电聚焦、双向电泳、毛细管电泳和免疫电泳等)。这些分离纯化

2
方法的原理主要是基于蛋白质在溶解性、带电荷性、分子量大小或亲和特异性等
方面的差异。色谱技术和电泳技术在纯化蛋白质方面的研究和应用比较广泛、深
入,详细内容见书中的相关章节。本节主要介绍蛋白质纯化设计的一些基本原则,对纯化过程的优化,以及经
济性方面的考虑,同时介绍规模化纯化蛋白质所需考虑的一些原则和操作过程。
13 蛋白质纯化的设计
131 基本原则
1311 原则
纯化蛋白质通常是为了获得纯蛋白质以便深入研究蛋白质的活性、结构、结
构与功能之间的关系。首先,必须了解待纯化样品中目的蛋白及主要杂质的性质,尽可能多收集有
关蛋白质的来源、性质 (分 子 大 小、等 电 点)和 稳 定 性 (蛋 白 质 对 温 度、极 端
pH、蛋白酶、氧和 金 属 离 子 等 的 耐 受 性)等 信 息,这 有 助 于 设 计 蛋 白 质 纯 化。比如目的蛋白是糖蛋白或脂蛋白,它们就明显区别于大多数的杂蛋白;蛋白质是
细胞内还是细胞外,可溶还是不可溶等都将影响到 蛋 白 质 提 取 方 法 和 缓 冲 液 组
成,对于细胞外蛋白质,除去细胞可以有助于纯化过程,与膜结合的蛋白质需要
用有机溶剂先溶解。其次,纯化开始之前必须了解最终产品的用途,从而设计蛋白质纯化过程,
同时要综合考虑纯化产品的质量、数量和经济性等三个方面的要求。纯化后的蛋
白质纯度要多高,纯化过程中能允许损失多少活性以及纯化过程需多少时间和成
本等都受到目的蛋白用途的影响。目的蛋白纯度如果要求越高,往往所需要的操
作时间越长,成本越高。最后,充分了解各分离纯化技术操作单元的大量信息也很重要,比如在细胞
破碎时,需要了解包括流速、搅拌器类型、操作压力、细胞浓度和种类、产品释
放的碎片和大小等;设计分析吸附色谱时,要了解包括色谱柱特征、凝胶或其他
吸附剂的性能 (结合能力、解离常数、流速等)。
1312 蛋白质的纯度
对目的蛋白纯度的要求取决于纯蛋白质的用途,如果是工业化应用 (在食品
工业或日用化学工业),则需要大量的产品,此时纯度是次要的。如果纯蛋白质
被用于研究,所需数量比较少,在酶学研究中,80%~90%的纯度就足够了,蛋
白质结构研究中,纯度要在95%以上,而用于医疗的蛋白质,必须考虑到所有
的杂质,对最终产物必须分析污染蛋白质、DNA和在纯化过程中的添加物。可
以采取一些特别的步骤除去这些杂质,但是额外的纯化步骤虽然可以提高纯度,

3
却由于除掉最终的百分之几杂质比最初的纯化要困难得多,会导致收率下降,增
加成本,延长了操作时间。为应用于研究而纯化蛋白质,规模小,额外的纯化步
骤所需要的花费并不重要,但工业化纯化蛋白质则需要综合考虑纯化的经济性。
1313 蛋白质活性的保持
在大多数情况下,纯化后蛋白质应尽可能保持活性,要采取尽可能少的纯化
步骤以减少蛋白质变性和蛋白质水解。尽量避免比较粗放的条件 (如极端pH、有机溶剂等),选用合适的缓冲液,在纯化的整个过程中,需 要 控 制pH,以 减
少蛋白质变性,通常采用20~50mmol/L的缓冲液浓度。蛋白质样品的贮存温度
宜在4℃或更低,如果贮存时间长或蛋白质易水解,可采用冷冻方法,用液氮、干冰或甲醇进行快速冷冻比较好,但是冷冻法对有些酶也不适用。在低温 (4℃)操作,加快纯化过程而缩短操作时间,或加入蛋白酶抑制剂,都可以降低蛋白质
水解程度。溶酶体是蛋白酶主要来源,尽量避免其受到破坏而释放出蛋白酶,在
酶提取液中加入蔗糖或麦芽糖可降低破坏程度。然而,即使是微量蛋白酶,也能
水解大量蛋白质,因 此 在 纯 化 后 期 也 要 仔 细 操 作。由 于 许 多 蛋 白 酶 分 子 量?在
20000~30000之间,凝胶过滤可用于蛋白质初步纯化,以分离大多数蛋白酶污
染物。选择简单、快速、高专一性的酶分析方法,可以缩短样品在纯化各步骤间
的贮存时间,也可降低蛋白质水解程度。
1314 蛋白质的数量
纯蛋白质的数量不仅仅受原材 料 数 量 的 影 响,还 受 到 蛋 白 质 纯 化 收 率 的 影
响。在有不同原料可供选择时,一般选用目的蛋白最稳定、含量最丰富的,同时
也考虑获得原料是否容易,数量是否足够多,成本是否比较低。在纯化过程,每
一步骤中都会损失蛋白质,所以纯化步骤应尽量少,以得到高蛋白质收率,但是
纯化步骤的减少会降低目的蛋白的最终纯度,需要恰当选择各种纯化方法以提高
蛋白质收率。
132 过程优化
蛋白质纯化的过程优化就是使蛋白质纯化后收率、纯度最高,成本最低。在有纯化方案可供实施时,需要对纯化过程进行优化,此时已经了解了纯化
过程条件和目的蛋白产品的特性。过程优化必须解决两类问题:一是在可替换的
操作中作出选择 (如离心或超滤等);二是设计理想的色谱顺序,以最少的步骤
(一、二或三步)获得最大的产量。每一分离纯化方法都要评估其处理样品能力、蛋白质收率和纯化成本。在纯
? 本书中 “分子量”一词即指 “相对分子质量”。———编辑注

4
化初期阶段,高处理量和低成本十分重要,而在后期高分辨率很重要。纯化分离
初期要求减小样品体积,常常使用高处理量的沉淀技术,色谱中吸附法处理量最
大,一般不采用凝胶过滤作为纯化初期操作步骤,因为它处理量小。使用高分辨
率技术可以减少纯化步骤,也使蛋白质更纯。沉淀步骤分辨率低,而色谱法高,亲和色谱有很高分辨率。各纯化技术都有一个平均收率范围,硫酸铵沉淀法和双
水相提取法的收率通常大于80%。亲和色谱法收率较低 (在60%以下),且成本
高,因此一般用在纯化的后期。通常先用低成本技术以除去大部分杂质,避免损
坏价格高的色谱柱。纯化技术顺序的选择上,一般先使用沉淀技术,然后依次是离子交换、亲和
色谱,最后是凝胶过滤,这样,每一种技术利用了蛋白质的不同性质,沉淀技术
能处理大量的高浓度蛋白质溶液,离子交换除去大部分杂蛋白以便进入价格高的
亲和色谱柱,凝胶过滤用于最后纯化环节,这时处理量已不是问题。但是,这并
非是固定不变的纯化蛋白质的最佳方案,比如有时凝胶过滤就用作最早的分离步
骤,以便获得多组分酶中的高或低分子量部分。总之,纯化方案的确定,除考虑
利用蛋白质不同的特性外,步骤应尽可能少,尽量使分离产物不需进一步处理就
可以直接进行下一步操作。对所有蛋白质适用的纯化方案是不存在的,因为原料来源和各种蛋白质纯化
的要求不同。可以通过试用多种方法先找出大致途径,或使用专家系统,以计算
机为基础的专家系统在人工智能领域已经成了非常重要的工具,现有的人工智能
可以通过相互作用的逻辑推理来找出蛋白质纯化的最理想方案。对100种纯化过
程进行分析,平 均 分 离 纯 化 步 骤 是 四 步,总 收 率28%,平 均 每 一 步 纯 化 倍 数
为8。
133 经济性
在实验室进行蛋白质 纯 化 时,大 多 是 为 了 进 行 研 究,一 般 不 太 考 虑 成 本 问
题,而规模化纯化蛋白质时,除了提高产量,获得更多的纯蛋白质产品,还有一
个目的就是尽可能降低成本。实验室规模和生产规模的纯化 成 本 主 要 包 括 以 下 几 个 方 面:固 定 资 产 投 入
(设备仪器、泵、盛液罐等)、化学材料和消耗品 (塑料样品管、样品罐、无菌移
液管、色谱柱等)、运 行 费 用 (包 括 人 员 工 资、设 备 运 行 费 用、供 热 空 调 费 用
等)。随规模增 大,设 备 耗 费 明 显 增 加。实 验 室 里 的 设 备 一 般 有pH 计,天 平,磁力 搅 拌 器,4℃冷 柜 或 冷 藏 室,制 冰 机,低 温 冰 柜 (-4~8℃,-15~-20℃),分光光度计 (205~850nm),电泳,破碎设备,带制冷 (4℃)的离心
机,以及柱色谱 (包括分部 收 集 器,UV监 测 仪 及 记 录 仪,蠕 动 泵 和 离 子 交 换
柱、亲和色谱柱、凝胶过滤 柱 等 各 种 色 谱 柱)。有 时 也 需 要 用 于 结 构 研 究 的 氨

5
基酸分析仪和蛋白质测序仪等。在实验室中,为了方便,相当一部分的 化 学 溶
液被浪费,使得化学药品的支出成本与成批处理量并不成绝对正比。中 试 规 模
中,不锈钢容器和现代化的自动装置使投入有所增长,除了提纯车间,还 需 要
办公室、仓库、实验室等。生产规模进行蛋白质纯化比较直接反应纯化 过 程 的
实际成本。
14 规模化蛋白质纯化
商业化的多肽和蛋白质生产至今还仅仅限于一些由微生物产生的或是从动植
物组织中提取出的酶。重组DNA和组织培养技术的进展使得规模化生产蛋白质
和多肽成为可能,其中不少产品都是和人类健康有关的,比如干扰素、抗生素和
疫苗等。规模化纯化蛋白质,目的是尽可能提高产量,并降低成本,同时也尽可能避
免人为因素干扰,纯化过程还应尽可能精简。进行规模化纯化蛋白质时,已经基
本了解目的蛋白、原料、生产方法、产品纯度要求和最终数量等,但是纯化过程
中很多方面可能要重新评估并作出适当的改变。从实验室规模纯化蛋白质向工业
化规模纯化生产蛋白质的扩大,属于工程学的范畴。工厂规模是一次性的,它必
须成功。以下介绍规模化纯化蛋白质所需遵循的原则,规模化操作过程和优化控制,
以及规模化纯化中遇到的在实验室操作中可能被忽略的问题。
141 基本原则
合理设计纯化方案。在规模化纯化蛋白质过程中,主要步骤通常不超过4~5个:细胞分离,细胞破碎和碎片分离 (仅对胞内蛋白而言),浓缩,预处理或
初步分离纯化,高效分离纯化。要尽可能了解目的蛋白和杂质的物化性质,根据
被分离物质不同的物理化学性质来进行纯化;尽可能早使用高选择性的步骤;尽
可能早分离除去数量最多的杂质,在分离纯化的早 期 一 般 是 浓 缩 蛋 白 质 并 除 去
水;最后执行特别费时间或成本很高的步骤。选择适当的发酵系统和发酵条件。发酵过程和蛋白质分离纯化过程是相互影
响的,合适的发酵将有助于发酵产品的分离及蛋白质的纯化。要严格控制、防止
蛋白酶和细菌的污染。规模化纯化蛋白质时,要保持蛋白质活性,减少蛋白质变性及失活。选择适
当的操作温度 (尽量在低温下进行操作)、pH 和泵,可以有效地控制蛋白质的
变性。为了避免因为肽酶的作用等因素造成蛋白质降解,要保持比较低的浓度,尽量缩短操作时间。随着规模的扩大,样品溶液浓度要逐步递增,以减少对色谱
过程的影响。

6
对泵的选择要考虑到泵的耐压、流量、稳定性等性能,泵的材料以及外罩的
绝缘、防火和抗腐蚀,另外还必须综合考虑空气、清洁和卫生因素等。考虑到价
格及其适应性,离心叶轮泵适用于缓冲液,而产物的输送可以用瓣轮泵、旋转活
塞泵或推进式真空泵。比较大直径的管道用于规模化纯化蛋白质时,最好采用不
锈钢材料,且必须保证管道内部的光洁和焊缝质量,以防止微生物污染。管道和
阀门的选择,可以像泵一样按照相同的标准进行。保持清洁和卫生。即使 是 低 水 平 的 微 生 物 污 染 也 会 造 成 很 大 的 麻 烦:过 滤
机、色谱柱寿命的缩短,产品稳定性的降低等。因此,所有设备都要定期进行严
格的清洁和卫生工作。
142 主要操作过程
1421 细胞分离
分离微生物细胞的主要措施为离心、传统过滤和膜过滤。传统过滤用于生物
悬浮物的分离是困难的,选择离 心 或 膜 过 滤,需 要 综 合 考 虑 资 金、操 作 维 修 费
用、回收率和过程时间等因素。影响分离过程经济效益的一个主要因素是被分离
颗粒的大小。工业用离心机通常设计成连续进料。细胞在离心机中沉淀越快,在转子中的
保留时间就越短,相应地,离心处理量越大。决定离心机容量的一个主要因素是
相关粒子的大小。通过离心操作回收酵母已有很多年,而动物细胞的处理更加复
杂,因为它们很容易破裂,当通过离心系统时,可能会分解产生微小的碎片。大
规模分离细菌和细胞碎片最常用的离心机是多层转盘离心机。在低流速 (300~500L/h)时,可以将05μm 以 上 的 颗 粒 分 离,而 在 高 流 速 (3000~5000L/h)时,可以将整个细胞分离出来。但是,离心操作费用高,也难于无菌操作,产生
热量,形成噪声。过滤技术 (深层过滤,微孔过滤,超滤)常用于固液分离。旋转式真空过滤
机是比较常见的一种,已应用于大规模生产酶制剂过程,从发酵液中除去细菌菌
体或丝状微生物。膜过滤系统越来越多应用于规模化从发酵液中分离细胞和细胞
碎片,在无菌分离的效果方面远好于价格昂贵的离心机。然而,膜过滤的主要缺
点是不能处理高浓度细胞,高浓度细胞会明显降低过滤速度。微滤能够截留溶液
中非蛋白质的微小物质,而超滤能够截留溶液中的蛋白质。
1422 细胞破碎和碎片分离
对于细胞内蛋白质的分离,需要先有效破碎细胞。通常先收集细胞,然后进
行洗涤以除去残留的培养基,对于仅仅由细胞膜包围着的细胞,比如哺乳动物细
胞,破碎比较容易,如果存在聚合细胞壁结构 (比如微生物细胞),破碎时就会
困难些。

7
实验室中常用的超声波法由于较难以将足够能量输送到大量体积的细胞悬浮
液中,不适于大规模破碎。适用于规模化操作的细胞破碎方法主要有均质法和高
速玻璃珠研磨法,需要注意的是,应尽量缩短操作时间。均质法是当前最广泛使用的破碎技术,产量可达6000L/h,但它不太适用于
高等真菌。装置如果没有制冷,由于释放大量的机械能,温度上升比较高,有些
蛋白质会因此而变性,所以样品溶液在进入设备前及离开设备后都要尽量冷却。研磨法采用无铅玻璃珠 (04~15mm),细 胞 悬 浮 液 在 有 小 玻 璃 珠 的 情 况
下高速搅拌,最大处理量可以达到2000L/h。最佳玻璃珠尺寸的选择取决于破碎
对象的大小和所释放产物所处的位置,小的玻璃珠对破碎细菌非常有效,而大的
玻璃珠对酵母细胞很有效。该法的破碎效率受破碎对象、细胞浓度、搅拌速度、玻璃珠大小、温度和进料速度等因素的影响,细胞破碎效率随搅拌速度增加而达
到一最高值,然后随速度增加而下降。相对于均质法,在搅拌速度不高时,细胞
浓度会影响到研磨法的破碎效率。最合适的细胞浓度随微生物种类不同而变化,一般为细胞湿重的30%~60%。操作温度通常限制在很窄的范围内,通常在5~15℃,以免造成蛋白质热变性,一般需要采用夹套方式进行冷却。
尽管规模化操作通常采用机械破碎法,但由于细胞要彻底破碎以获得较高的
收率,细胞内所有物质都被释放,因此,必须从复杂混合物 (包括蛋白质、核酸
及细胞壁碎片)中分离出目的产物,目的蛋白可能 会 受 到 释 放 出 的 蛋 白 酶 的 降
解。另外,核酸不仅难以分离,而且大大提高了悬浮液的黏度。除去核酸的技术
包括使用核酸酶和沉淀。核酸污染虽然含量很低,但是对于医药级蛋白质还是有
影响。酶解法条件温和,对降解细胞壁结构有特异性,是比较理想的方法。但是不
同细胞组织需要不同的酶,而且因所处的不同生理状态,细胞对酶解的敏感程度
会有明显变化,因而酶解法还没有被广泛用于规模化破碎微生物细胞,但对于价
值较高、采用机械法会造成损失的产品,酶解法已经显示出了良好的应用前景。细胞破碎过程中产生的细胞碎片,必须除去,以免污染或者堵塞纯化蛋白质
所用的色谱柱,可以先进行絮凝,再通过离心法将其碎片分离出来,但对直径小
于05μm颗粒的处理效果不太理想。双水相提取法也常用于分离碎细胞和细胞
碎片。
1423 浓缩
早期纯化阶段需要将蛋白质溶液浓缩10~50倍,以减小过程体积并使随后
的操作更容易处理,这在规模化纯化蛋白质时是个重要的必不可少的步骤,超滤
是比较合适的技术,一般采用错流系统。沉淀法在大规模工业生产中并不可取,因为这种方法处理量很小;引入的外

8
来试剂必须在随后去除或者回收;对于很稀浓度蛋白质溶液,该方法处理效率不
高,且收率很低;使用有机溶剂会造成维持设备的成本,生产区域的防火、防火
花的成本较高。双水相萃取技术可以用于实验室规模或中试规模,但工业化规模
还不多见。
1424 色谱分离
采用色谱技术从粗产品中分离纯化蛋白质,在生产中始终都要慎重操作。经
过浓缩后,得到了蛋白质、其他成分 (比如类脂物、细胞壁或其他多糖类)、盐
和水的混合液,随后一般经2~4步色谱纯化才能达到所要求的蛋白质纯度。可
以从不同的色谱方法中进行选择,实验室实验结果有参考作用,专家系统也有助
于优化蛋白质纯化的顺序。首先采用典型操作 (如吸附、双水相萃取或盐析沉淀
等)来提高收率,减少纯化步骤,然后采用高效液相离子交换色谱或亲和色谱纯
化蛋白质,得到99% (通常情况下95%~98%)纯度的产品。经过上述步骤之
后,如果有需要,可进一步采取其他方法以获得超高纯度。在设计规模化色谱操作时,要保证色谱过程可以重复,安全性好,能满足卫
生要求,色谱操作过程、设备、环境、厂房的设计也能保障整个操作环节的卫生
要求及安全性,同时方便于色谱过程数据的收集。通常需要综合考虑以下几个因
素:操作过程和装置的控制及可靠性,卫生条件的维持,固定投资费用及操作成
本等。典型的大规模柱色谱系统包含了一系列的用泵连接到色谱柱的容器及相关的
仪器,比如:色谱柱,装缓冲液原料、产物及废物的容器,泵,过滤器,空气传
感器,压力传感器,压力仪表,流量传感器和原位清洗设备等。一般色谱系统都
是自动化控制,自动化可以改善色谱过程的控制及重复性,可遥控操作,更加可
靠,并可以提高生产效率。操作过程中需要注意消毒,还需要防止灰尘的积聚及
污染物进入到色谱系统中,以保持卫生状况良好。通常在使用系统前,对所有的
设备、管道和阀门进行原位清洗。对进入色谱系统的缓冲液及样品溶液,一般都
先过滤,以减轻对色谱系统的生物负担。纯化环节的费用通常是蛋白质产品总成
本中的主要部分,因此,需要严格控制纯化成本,尽可能提高对仪器设备的利用
率。在规模化商业生产中,根据生产规模的大小、预期产品的售价、原材料来源
等因素,选择使用不同等级的化学试剂原料。最好预先经过试验,以保证所用材
料的可靠性。如果食品级试剂可行,则可有效降低生产成本。在大规模色谱分离时,由于样品的体积很大,加样时间常常很长,成为分离
过程的限制性因素,此时往往可以采用分批分离的方法,即吸附过程甚至吸附和
解吸过程都不在色谱柱中进行,样品在容器中与离子交换剂充分混合完成吸附,将上清液除去,然后将吸附了蛋白质的离子交换剂装柱并进行洗脱,或者直接在

9
容器中用阶段洗脱的方法进行洗脱,此方法可以大大提高分离速度,当然也会使
分辨率变低,适合在规模化分离的前期使用。离子交换因其具有很高的样品吸附容量,不但在实验室分离中广泛被使用,
而且适合于规模化的蛋白质纯化。离子交换剂具有 从 稀 溶 液 中 浓 缩 蛋 白 质 的 能
力,也适合于从大体积的样品 (如发酵液)中纯化所需蛋白质。对于实验室小规
模的制备,体积为500ml的色谱柱基本能 够 满 足 需 要,而 在 工 业 化 应 用 中,色
谱柱的体积可以 被 放 大 到100~200L。由 于 分 离 的 原 理 和 过 程 是 一 致 的,很 明
显,根据已经优化好的小规模分离条件,结合放大时的容量因子,可以推测出大
规模分离时所需的操作条件。放大过程中的原则是,保持色谱操作上样、吸附和洗脱的条件不变,这里包
括样品的组成、离子交换剂的种类、样品介质比 (mg蛋白/ml离子交换剂)、起
始和洗脱缓冲 液 的 组 成、pH 和 离 子 强 度 等,在 此 基 础 上 根 据 样 品 量 放 大 柱 体
积、洗脱剂的体积、流速等参数。其 中 柱 体 积 是 随 样 品 体 积 的 增 大 而 线 性 增 大
的,并且柱体积的增大主要通过选用内径大的色谱柱来实现,柱高一般不宜有太
大的增加,否则会造成分离时间的增加和样品峰宽的加大。至于洗脱剂的体积,无论采用哪种洗脱方法,也都是随样品体积增加而增加的。色谱操作时的流速有
两种常用的表示方法,即线性流速 (cm/h)和体积流速 (ml/min)。一般在讨论
离子交换剂的流速特性和动力学时采用线性流速,而在实际操作过程中通过泵直
接调节控制的是体积流速。它们之间的换算关系为:体积流速 (ml/min)=线性
流速(cm/h)×色谱柱截面积(cm2)/60。在放大过程中应保持线性流速不变,而
体积流速将随色谱柱截面积的增大而增加。以上是基于很多参数不变的前提,但实际上,大规模纯化中有些参数很难做
到完全不变,比如样品的成分和样品介质比。通过发酵、动植物细胞培养或者从
生物原料提取目的蛋白时,每一批次的样品在组成、总蛋白含量、目的蛋白所占
比例等很多方面总是有一定的波动,这会造成色谱柱内离子交换剂上实际吸附的
蛋白数量和组成每次都有所不同,这往往会导致分离的效果发生改变。特别是大
规模分离时样品介质比往往较大,就是说,离子交换剂的交换容量已经被利用到
很高的程度,此时样品中蛋白质组成和含量发生变化,可能会造成目的蛋白不能
被完全吸附,因此放大过程中,在有效交换容量上应留有一定的余地。大规模分
离中使用的离子交换剂应当具有良好的物理和化学稳定性,基质颗粒通常不宜太
小,因为基质粒度的大小与色谱柱内的背景压力直接相关,相同的粒度情况下,颗粒直径分布均匀有利于保持相对较低的背景压力。另外,由于大规模分离时交
换剂用量很大,必须考虑介质的成本。在大规模分离中采用的是直径很大的色谱柱,此时如果将样品或缓冲液直接

10
加到交换剂的床面上,是很难保证液流能均匀进入柱床的,所以在柱床的上方设
计一个液流分配器,以便采用多口注入方式。还要注意上样前必须保证样品溶液和起始缓冲液在pH和离子强度方面保持
完全一致,这可以通过缓冲液交换或者透析等方法来实现。但体积很大的样品显
然无法进行上述操作,此时可以通过对样品进行稀释,直至其与起始缓冲液离子
强度一致,然后再用酸碱调节样品溶液pH,使之与起始pH一致。这样,虽然
样品溶液在缓冲物质和盐的组成上与起始缓冲液并不相同,但由于pH和离子强
度方面的一致性,也能确保目的蛋白吸附在色谱柱上。新发展起来的 适 用 于 规 模 化 的 是 快 速 蛋 白 质 液 相 色 谱 (fastproteinliquid
chromatograph,FPLC)。FPLC在中等压力 条 件 下 工 作,比 传 统 色 谱 分 离 速 度
更快,分离效果更好。FPLC在实验室中被广泛接受,在中试和大规模蛋白质纯
化中也显示出了强大的潜力。相比于高压系统 (如 HPLC),它的一个优点就是
其设备用塑料而非不锈钢制成,可耐任何缓冲液,因而适于分离纯化蛋白质。

11
2 蛋白质样品的预处理
21 蛋白质样品的提取和分离
在蛋白质生产、分离和纯化中,占总生产费用中绝大部分的是分离和纯化蛋
白质过程。蛋白质生产过程影响到其分离纯化过程的难易程度及费用的多少。以
发酵法生产蛋白质为例,一般在发酵液中目的蛋 白 的 浓 度 比 较 低,而 且 常 含 有
性质相近的杂蛋白,分离纯化蛋白质需要一系列的步骤,步骤的多少及 对 目 的
蛋白纯度高低的要求将影响 到 最 终 的 成 本。提 高 在 发 酵 液 中 目 的 蛋 白 的 浓 度,或采用适当的从发 酵 液 中 提 取 分 离 目 的 蛋 白 的 方 法,都 可 以 降 低 最 终 产 品 的
成本。蛋白质样品的提取和分离是整个蛋白质纯化过程的第一步,在蛋白质纯化中
首先要获得蛋白质的提取液,恰当地选择蛋白质提取及分离的方法将有助于随后
的纯化过程。对于固态样品材料,一般先进行目的蛋白的提取,然后分离纯化。对于微生物发酵或动植物细胞培养得到的悬浮液,具体的操作方法视蛋白质在细
胞内外的位置而不同。对于细胞内蛋白质,必须先破碎细胞。在发酵罐中生长的
细胞要先浓缩,从而提高细胞破碎的效率。在细胞破碎过程中加入蛋白酶的抑制
剂,可避免因细胞破碎而释放出的蛋白酶的作用,应尽量缩短操作时间并且在较
低温度下 (比如4℃)操作。细菌细胞和细胞碎片难于离心或过滤除 去,因 为 它
们太小、可压缩。采用絮凝方法有助于除去这些颗粒。对于细胞外蛋 白 质,目
的蛋白存在于悬浮液中,可直接采用离心、过滤等操作来进行固液分 离,除 去
细胞和其他颗粒物质,以排除它们对随后的纯化过程的干扰。提取液若 准 备 进
入色谱柱,则必须先除去所有颗粒物质,以免污染和堵塞色谱柱。如果 提 取 液
随后要进行沉淀,对少量颗粒物质可以忽略不处理,它们会在沉淀过程 中 聚 沉
而被除去。对蛋白质样品的提取应尽量多 地 提 取 出 目 的 蛋 白,并 且 保 持 其 活 性 少 受 损
失,避免目的蛋白因热、pH或存在有机溶剂、金属离子、蛋白酶等因素而损失
活性。蛋白质的提取常用各种水溶液,一般在偏离蛋白质等电点05pH单位以
上,此时蛋白质的溶解度增加。提取等电点在碱性范围的蛋白质,可以用稀酸;对等电点在酸性范围的蛋白质,可以用稀碱提取,以尽可能提高目的蛋白在提取
液中的溶解度。但是也要注意提取液的pH应该在目的蛋白稳定的范围内,避免

12
极端pH。提取液的离子强度一般不能太高,以免引起盐析。提取液与提取物的
比例通常为5∶1,比例如果过大,会影响到随后纯化过程所用时间及纯化效率。比如,研究固体培养的麦曲中酶系统,可以用5倍于麦曲重量的适当pH的缓冲
液,在4℃浸泡麦曲过夜,然后进行过滤即可。细胞破碎后进行目的蛋白的提取。大部分目的蛋白保留在液相中,为了减少
损失,常用大量的缓冲液。对于动物组织,破碎用缓冲液体积为动物组织的2~25倍;对于植物细胞,可以用较少数量的缓冲液来提取。选择破碎细胞用的缓
冲液受到目的蛋白的稳定性和可溶性的影响,并且受到随后的纯化过程的影响,可以用水,但是会造成离子强度和pH降低。在低离子强度和pH下,很多蛋白
质不可溶,或者失去活性。接近细胞生理条件 (比如pH≈7、015mol/LNaCl)对于提取大多数 蛋 白 质 都 很 有 效。常 用pH=7~75、20~50mmol/L磷 酸 钠,或pH75、01mol/LTrisHCl(含0~015mol/LNaCl)缓冲液。
双水相萃取法 (twoaqueousphaseextraction)是近年发展起来、目前已较
广泛用于蛋白质分离纯化的方法,利用目的蛋白和杂质在两相中不同的分配,进
行目的蛋白的分离。该法能够保留蛋白质的活性,在提取蛋白质的同时还能起到
纯化作用,收率较高。使用较多的是聚乙二醇 (polyethyleneglycol,PEG)/葡
聚糖体系和聚乙二醇/无机盐体系。比如,采 用PEG4000/葡 聚 糖 从 破 碎 的 细 胞
中分离甲酸脱氢酶,收率在90%以上;采用PEG1550/磷酸 钾 分 离β葡 萄 糖 苷
酶,收率达到98%。采用 双 水 相 分 离 法 可 以 直 接 在 细 胞 破 碎 后 进 行 目 的 蛋 白
的提取,目的蛋 白 常 分 布 在 上 相 并 得 到 浓 缩,细 胞 碎 片 等 固 体 物 分 布 在 下
相中。
211 细胞的破碎
很多情况下,微生物分泌蛋白质到培养基中,提取分离蛋白质就比较容易,而对于细胞内蛋白质 (比如酵母产生的葡萄糖6磷酸脱氢酶,黑曲霉产生的过
氧化氢酶及葡萄糖氧化酶等),则需要采用机械方法或非机械方法对细胞进行有
效的破碎。在实验室内已经有不少破碎细胞的方法,常用的机械方法包括超声波
法、高压匀浆法、高速珠磨 法 等,非 机 械 方 法 有 干 燥 法、冻 融 法、渗 透 压 冲 击
法、酶解法、溶剂法。大多数动物蛋白质处于特别的组织或肌肉内,脂肪包围在外部,影响到组织
破碎后的蛋白质分离,必须仔细地先除去这部分,然后再破碎以释放出蛋白质。动物细胞比细菌和酵母细胞易于破碎。硬的植物组织 (种子)可以通过研磨来均
质,同样需除去影响到随后纯化过程的物质,绿叶和根组织可以进行均质化,纤
维状组织可以切割成块状。植物细胞中的酚类化合 物 可 以 结 合 蛋 白 质 而 使 之 失
活,其在破碎过程中也被释放出,因此需要快速提取破碎液体中的目的蛋白。

13
细胞破碎是获得微生物细胞内产物的中间一步,因此在设计方案时需要综合
考虑上游和下游过程的影响,上游过程中细胞组织所处的生理活性状态将影响到
细胞的破碎,下游过程中须考虑到破碎细胞时所产生的细胞碎片,防止其他蛋白
质对产物的污染及产物的失活。选择细胞的破碎方法要考虑破碎的目的和破碎对
象的类型。如果为了定量获得细胞内蛋白质以进一步研究,破碎收率就很重要。细胞的类型、大小、形态、生长条件、细胞壁结构及环境温度、pH等因素都会
造成细胞对不同破碎方法的敏感度也不同。可以根据实验及以往的经验来选择破
碎方法,但要注意破碎不能影响到目的蛋白产物的活性,在细胞的破碎过程中尽
量避免将产物暴露在不利的条件下。破碎过程中,常常产生比较多的热量,需要
预先冷却样品 (最好到4℃),在 破 碎 过 程 中,应 尽 可 能 保 持 低 温。另 外,一 旦
细胞被破碎,就失去了代谢调节机制的控制,目的蛋白就会受蛋白酶的作用,因
此需要迅速提取目的蛋白,或加入抑制剂,或降温以减小蛋白酶的作用。此外,加入适当的核酸酶或调节pH,可以降低破碎后的黏度。破碎后蛋白质还会遇到
氧化问题,加入5~20mmol/L2巯基乙醇或1~5mmol/LEDTA可以减小氧化
程度。对于细胞破碎效果的考察,可以测定细胞破碎后释放出的蛋白质数量,或者
利用显微镜直接进行观察。以下介绍几种主要的细胞破碎的方法。
2111 超声波法
强烈的超声波造成细胞悬浮液中气泡的增大和爆裂,通过机械剪切力使细菌
破裂,可利用15~25kHz频率的超声波进行细胞破碎,该方法因效果好、所需
样品数量少而在实验室广泛使用。破碎效率受到声频、声能、处理时间长短、细
胞类型及浓度等的影响。在高黏度溶液中或者产生泡沫的话,效率会降低。该过
程会产生相当的热量,可能会使蛋白质变性,加速蛋白质水解。超声破碎法应以
短脉冲的方式,间隔冷却。具 体 操 作 中,用 于 分 析 时,将 不 超 过03ml的 细 胞
泥悬浮于3ml的冰冻的溶解缓冲液中,将5mm直径的探头恰好置于液面下,在
不起泡的情况下尽可能增加功率。于冰浴中超声破碎3次,每次1min,中间冷
却1min。用于制备时,将5ml细 胞 泥 悬 浮 于50ml溶 解 缓 冲 液 中,以10mm探
头,于冰上50W 超声破碎2次,每次4min,中间冷却4min。但是超声波法不适宜于大规模的细胞破碎。在破碎过程中,最好要有冷却夹
套以避免温度的升高。
2112 高速珠磨法
高速珠磨法是很有效的破碎细 胞 的 方 法,少 量 样 品 可 以 在 聚 乙 烯 试 管 内 进
行,较多的样品需高速珠磨机,该法较多用于酵母细胞的破碎。影响破碎效果的

14
因素有细胞浓度、珠磨机的搅拌速度、珠大小及操作温度等。最佳的细胞浓度一
般由实验确定,细胞浓度较低时,产生的热量较少,但是单位细胞重量的能耗增
加。珠磨机的搅拌速度须限制在合适的范围内以减少产生的热量,避免造成蛋白
质的失活。实验室内使用的玻璃珠或钢珠一般为02mm,但是也要考虑细胞破
碎对象中目的蛋白及其在细胞中处的位置。由于在操作过程中产生热量,因而采
用冷却夹套方式调节操作温度。对于破碎 对 象,最 好 先 预 冷 却 后 再 进 行 细 胞 破
碎。玻璃珠在使用前,通常也需清洗和预冷却。
2113 高压匀浆法
高压匀浆过程中,细胞受到剪切、碰撞、压力骤变等作用而被破碎,释放出
目的蛋白,这是一种常用的破碎细胞的方法,比较适合于破碎酵母和细菌,也用
于大规模破碎细胞。影响破碎效果的因素主要是操作压力、温度、破碎细胞的种
类及其生理状态等。对于细胞膜结合的蛋白质,一般需要多次破碎才行。通常待
破碎的细胞湿重与缓冲液体积之比为1∶2~1∶5。操作应尽可能在低温下进行,以免因为 产 生 太 多 的 热 量 而 导 致 目 的 蛋 白 的 失 活。常 用 的 匀 浆 机 有 MantonGaulinAPV型。
2114 冻融法
将待破碎的细胞置于低温下 (-15℃)冷冻,再在室温下融化,反复多次进
行从而达到破壁的效果。该方法比较适合于细胞壁较脆弱易破的微生物菌体。但
是该法也存在破碎率低的问题,有时还会造成部分蛋白质的失活。
2115 渗透压冲击法
将待破碎的细胞先置于高渗溶液中,然后再转入低渗缓冲液或水中,由于渗
透压发生变化,使得细胞壁破裂,释放出胞内蛋白质产物到溶液中。该方法对于
分析制 备 较 为 有 用。将 收 集 的05ml细 胞 悬 浮 于4ml20% (200g/L)蔗 糖,
30mmol/LTrisHCL (pH=80,1mmol/LEDTA,01%TritonX100)溶 液
中,室温下保持5min。10000g离心3min,收集细胞。再悬浮于2ml冷水。冰浴
中温和搅拌10min,4℃下5000g离心3min,收集含有溶解蛋白的上清液。
2116 酶解法
该法专一性强,操作条件温和,收率高,只需要选择合适的酶及反应条件,就可以有效地进行细胞的破碎。溶菌酶是应用最广泛的酶,可以有效地降解革兰
阳性菌细胞壁,但是对革兰阴性菌效果不太好 (因为其存在脂多糖层)。对于酵
母细胞,可以采用蜗牛酶。以Micrococcuslysodeikticus为例,收获并且重新悬浮Mlysodeikticus细胞
于05% NaCl溶液中,达到5% (50g/L)的浓度,加入干的鸡蛋白00125g/g

15
干细胞,在室温下温和搅拌20min,加入等体积的95%乙醇,除去细胞碎片,离
心得到蛋白质,将乙醇浓度提高到75% (体积分数)以沉淀过氧化氢酶。来源于CorynebacteriumspOZ21的丝状真菌细胞壁溶解酶Yatalase(日本
Takara公司)已用于实验室的研究中,Yatalase含 有 壳 多 糖 酶、壳 二 糖 酶 和 葡
聚糖酶活性,是一种很有效的溶解丝状真菌细胞壁的酶,可在室温下保存,最适
作用温度为40℃,在pH=30~100范围内稳定。利用Yatalase在37℃作用于
Aspergilluskawachii经清洗过的菌丝体3h,可以有效释放出与细胞壁结合的葡
萄糖苷酶。利用Yatalase作用于Aspergillusoryzae菌丝体4h,然后3000r/min离心,可以得到与细胞壁结合的木聚糖酶,Yatalase的用量一般为菌丝体重量的
25%~3%即可。使用酶解法需要先试验,以确定合适的酶解温度、pH及酶的用量。由于酶
的成本比较高,该法目前还只限于在实验室使用。
212 蛋白质样品的分离
2121 聚沉和絮凝
聚沉 (coagulation)是 在 聚 沉 剂 作 用 下,使 颗 粒 物 质 相 互 聚 集 为 较 大
(1mm)的聚沉物。为了促进聚沉的产生,可以降温至20℃以下 (对于酵母细胞
很有效)进行;调整pH 至3~6;提高离子强度;增加颗粒数 量;增 加 多 价 金
属离子 (比如Fe3+或Al3+)或添加聚合高分子电解质。聚沉剂主要是无机盐类 (比如氯化锌、氯化铁、氯化铝、硫酸锌、硫酸铝和
硫酸铁等)和聚合无机盐 (聚合铝和聚合铁等),通常在高搅拌速度时加入聚沉
剂,然后缓慢搅拌以促进聚沉,控制搅拌速度非常重要。聚沉剂贮藏液必须定期
配制,最好是每周配制一次。可以在600ml以 上 的 烧 杯 中 以 一 定 的 速 度 搅 拌 溶 液,以 检 测 聚 沉 的 效 率,
具体有以下几种方法:①测定沉降速度 (比如测定悬浮液浊度的降低程度或测定
颗粒沉降速度);②测定通过滤纸的速度;③测定滤液的澄清度;④测量聚沉前
后颗粒的大小变化;⑤测定悬浮液中残留的主要颗粒的数目。方法①、④和⑤适
用于离心操作,方法②、③、④和⑤也适合于过滤操作。通过测定沉降速度来研
究聚沉条件,以得到最适合的聚沉剂用量或最适的溶液pH等。可以按照以下方法对聚沉条件进行优化:准备并且配制所需聚沉剂和化学溶
液,加入500~1000ml的悬浮液样品至带搅拌的烧杯中,以300r/min速度搅拌
样品,通过加入酸或碱调整各样品的pH (比如按6个样品计,调节至pH=4,
5,6,7,8,9),同时加入聚 沉 剂 溶 液,并 用 秒 表 计 时,保 持 固 定 时 间 (比 如
1min),降低搅拌速度至维持在悬浮液中凝聚即可 (比如60r/min),可找出最合
适的聚沉条件。

16
絮凝 (flocculation)是通过絮凝剂的作用,将一些不稳定颗粒诱导结合到一
起,逐渐紧密,随后形成更大的凝聚物。常用絮凝剂有淀粉、树脂、单 宁、离
子交换树脂和纤维素衍生物等,粉末状多聚物要小心溶解,先加入少量 甲 醇 或
乙醇,然后快速加入水,同时充分搅拌。絮凝剂一般在聚沉剂后使用,选 择 时
主要考虑成本、毒性等,通过试验可以获得最适合的絮凝剂类型、用量 及 处 理
条件。对絮凝条件进行优化时,准备并且配制所需絮凝剂和化学溶液,加入500~
1000ml的悬浮液样品至带搅拌的烧杯中,以300r/min速度搅拌样品,通过加入
酸或碱调整各样品的pH,同时加入絮凝剂溶液,并用秒表计时,保持快速搅拌
5min,加入絮凝剂溶液,快速搅拌1min,降低搅拌速度 (比如50r/min),维持
15min,分析絮凝情况。可找出最合适的絮凝条件。
2122 离心
离心是有效的进行固液分离的手段,离心沉降,在不很稠的液体中符合斯托
克斯 (Stokes)规则:
vg=D2(γ1-γ2)g
18μ
g=ω2r
ω=πn30
式中 vg———沉降速度,m/s;
D———颗粒直径,m;
γ1———颗粒密度,kg/m3;
γ2———液体密度,kg/m3;
g———重力加速度,m/s2;
μ———液体黏度,kg/(m·s);
ω———角速度,rad/s;
r———颗粒至旋转中心的距离,m;
n———每分钟转子的转速,r/min。离心操作中沉降速度受到颗粒大小、颗粒与溶液的密度差、液体黏度、温度
等因素的影响,通过增大固体颗粒直径或颗粒与液体的密度差,或降低液体黏度
都可提高沉降速度。在实验室规模可以使用管式离心机,管式离心机有台式和立
式的,大部分带冷却,有不同类型转子可供选择,在离心管的材料、运转方式、转子的材料等方面有所不同。离心管使用玻璃、聚丙烯和聚碳酸酯等材料,玻璃

17
离心管因能够适应绝大多数的试剂和消毒剂而被 广 泛 使 用。塑 料 离 心 管 价 格 便
宜,但需要注意生产厂商提供的最大离心力限制以及塑料离心管对化学试剂和热
灭菌的耐受能力。低速 (≤10000g)转子常采用钢、铝或铜材料,中速 (10000~100000g)转子用铝或钛合金材料,高 速 (100000~600000g)转 子 用 钛 合 金 材
料。离心机和转子应定期 (最好是每次使用后)用水进行清洗,并擦干。离心分
离速度快,效率高,操作时卫生条件易于控制,但是设备投资较大。对于低黏度介质中的细菌,2000~3000g、10~15min就可以离心沉降下来;
在高黏度溶液中,需要高的离心力和比较 长 的 时 间,比 如 对 细 胞 碎 片 或 真 菌 孢
子,需要高的离心力 (12000g)和比较长的时间 (30~45min);对于蛋 白 质 沉
淀,15000g、10min或5000g、30min就足够了。
2123 过滤
在经过离心操作后,样品一般还需过滤才进入下一个阶段,即样品的浓缩。过滤是为了除去蛋白质样品溶液中细小 的 颗 粒,在 实 验 室 内,通 常 先 用 滤 纸 过
滤,再进行膜过滤,一般选用孔径为02μm和045μm的 塑 料 膜。过 滤 操 作 可
以在常压或减压状态下进行。膜过滤可以除去细胞和细胞碎片,分离效率高,流速高,所需时间短,处理
量大,设备可靠,简单。膜的材料有聚丙烯、醋酸纤维和硝酸纤维等。对于几毫
升的少量样品,可以选用GelmanLaboratory的注射型、045μm膜过滤器,非
常方便。影响膜过滤的因素主要有:压力差,颗粒浓度和操作温度。膜两侧的压力差
最好保持在较小且可以提供足够的驱动力水平,以保证流量在合适的范围内,压
力差增加,流量也增加。但是超过一定的压力差,流量就不会增加了,过高的压
力差反而会降低流量。进料中固体浓度增加会导致流量的下降。升高操作温度可
以降低溶液的黏度,从而提高流量,但是从蛋白质纯化的总体来考虑,还是在低
温 (4℃)进行膜过滤比较合适。为了能够重复使用膜或在长时 间 操 作 过 程 中 保 证 高 的 流 量,需 要 定 时 清 洗
膜,具体有物理方法、化学方法、生物方法等。物理方法有水洗,反冲,超声波
处理等,无机膜可以用热处理,有许多化学试剂可以用于清洗膜,比如01mol/LNaOH、1~2mol/LNaCl、01mol/LHCl、025%~03% HNO3 或1%胰蛋白
酶等。需要注意膜生产厂家对清洗试剂的要求。
22 蛋白质样品的浓缩
澄清后的蛋白质溶液 常 常 需 进 行 浓 缩,以 提 高 蛋 白 质 的 浓 度,减 少 样 品 体
积,这样有利于随后进一步进行 蛋 白 质 的 色 谱 纯 化。浓 缩 方 法 有 吸 附 法、超 滤

18
法、沉淀法、透析法、冷冻干燥法和双水相分离法等。
221 吸附法
这是最简单、快速的浓缩蛋白质溶液的方法,所需仪器简单,适用于稳定性
比较差的蛋白质。将干的 惰 性 多 孔 基 质 聚 合 物,比 如 葡 聚 糖 凝 胶 (Sephadex),加入到蛋白质溶液中吸收水和其他小分子,当凝胶完全膨胀后,用过滤或离心方
法除去凝胶,分离出蛋白质。常用的凝胶有SephadexG系 列 及BioGelP系 列
凝胶。但是该方法选择性比较差,不能连续操作,浓缩倍数较低,蛋白质收率较
低,一般只有80%~90%。具 体 操 作 举 例:将20g干 的SephadexG25加 入 到
100ml蛋 白 质 溶 液 中,放 置15~20min后,2000g 离 心10min或 用 滤 纸 过 滤
即可。
222 超滤法
超滤是在离心力或较高的压力下,选择适当孔径的半透膜 (膜的孔径从1~20nm),从而使水和其他小分子物质通过半透膜,而所需蛋白质不能通过膜。超
滤浓缩与其他方法相比有其优点,沉淀作用对样品中蛋白质浓度要求在100mg/
L以上,并且由于相变导致收率较低;用透析法浓缩需要比较长的时间,体积也
受到一定的限制;冷冻干燥所需时间也长,收率也低,而且在浓缩蛋白质的同时
也浓缩了盐。截留分子量 (molecularweightcutoff,MWCO)被定义为不能通过膜的最
小分子量。理想状态下,MWCO为10000的膜可以完全截留分子 量 超 过10000的分子,而分子量小于10000的分子可以完全通过该膜,但是膜孔径分布是不均
一的,在平均 MWCO附近具有一定的分布,分布范围同膜的生产方法与生产厂
家有关,因此选择膜的 MWCO通常要比所需要的蛋白质分子量低20%以上。如
果选择的 MWCO过小,那么通过膜的速度将会减慢,造成操作时间延长,或者
需要更高的操作压力。传统的膜由纤维质材料组成 (比如醋酸纤维等),这些材料来源丰富,但是
它们的化学稳定性 和 热 稳 定 性 有 限,一 般 要 求 在pH=3~6和30℃以 下 使 用,限制了其使用范围。一些塑料膜已经广泛使用,比如聚碳酸酯、聚酰胺、聚砜、聚氯乙烯和聚丙烯腈等。这些膜可以在广泛的化学条件下使用,在pH=1~13和许多有机溶剂存在时都可以使用,耐热稳定性可以达到80℃,对其中的一些
膜还可以进行自动灭菌。影响超滤的因素主要是流量,尤其对于大规模的浓缩。操作压力、样品溶液
的组成及黏度、流速、膜以及超滤系统的选择都影响到流量。高的压力能够提高
流量,但是如果超过一定的限制值后,极化作用就会限制流量,而且会增加膜的
污染和泵的运行费用;高的流速可以减轻膜的污染和极化作用,但是过高的剪切

19
力会使蛋白质变性;溶液黏度升高会使流速减慢,提高温度虽然可以降低黏度,但一般不考虑此措施,以免因为热而使蛋白质变性;通常在浓缩前使用离心、絮
凝、透析、粗滤或微滤来除去溶液中的杂质,以减少对膜的污染;缓冲液的pH不要接近或等于目的蛋白的等电点。
超滤搅拌装置 适 用 的 膜 主 要 有 Amicon公 司 的PM、XM、YM、YC系 列,
Millipore公司的聚砜和纤维素系列膜。表21介绍了Amicon公司生产的几种常
用膜。
表21 Amicon公司生产的几种常用膜
膜 MWCO 灭 菌 温 度 限 制 不 适 用 对 象
PM 10000,30000 5%福尔马林,2%戊二醛 100℃ 有机溶剂,强酸,部分洗涤剂
XM 50000,300000 5%福 尔 马 林,2%戊 二
醛,70%乙醇
70℃ 有机溶剂,强酸
YM 1000, 3000,
10000, 30000,
100000
2%戊二醛,70%乙醇,置
于 缓 冲 液 中 121℃ 灭 菌
20min
121℃(置 于 缓
冲液中) 部 分 有 机 溶 剂,福 尔 马 林,胺,
pH<3或pH>13,>10%磷 酸,
>10mol/L盐酸,>05%苯酚
YC 500 5%福尔马林,2%戊二醛 50℃ 有 机 溶 剂,强 碱,>10%乙 醇,
pH<4或pH>8ZM 500000 5%福 尔 马 林,2%戊 二
醛,置于缓 冲 液 中121℃灭
菌20min
121℃(置 于 缓
冲液中) 有机溶剂,强酸,强碱,部分洗涤
剂,>20%乙醇
图21 Omegacell装置
1—样品口;2—减压阀;3—搅拌;4—储存盖子;
5—出口帽;6—容器;7—底座;8—滤液管;
9—固定容器底座
超滤浓缩的具体操作可以根据所需处理的样品体积来选择,一般用于超滤浓
缩的装置 可 以 从 Millipore、Filtron、Whatman、Amicon和Sartorius等 公 司 获
得,样 品 处 理 量 可 从 几 毫 升 到 几 百 毫
升,通常用于蛋白质浓缩的膜的MWCO从1000~1000000。PALL公 司 的 仪 器
可以浓缩5~150ml的样品,其中150ml的有Navacell或Omegacell(图21)两
种类型装置。样 品 体 积 比 较 少 时,可 以
利用离心力作用驱动小分子物 质 通 过 半
透 膜。 以 PALLFILTRON 公 司 的
MACROSEPTM CentrifugalConcentrator为例 (图22),有不同 MWCO的膜
可供选择:1000、3000、10000、30000、
50000、100000、300000和1000000等,适用于5~15ml的 样 品,一 般 在3000g离心10~30min即 可;如 果 处 理 的 样 品

20
很少,可 以 使 用 NANOSEPTM Microconcentrator (图23),有3000、10000、
30000、100000和300000不 同 MWCO的 膜 可 供 选 择,每 次 处 理 样 品005~05ml。
图22 MACROSEPTMCentrifugal
Concentrator1—盖子;2—膜板;3—样品室;
4—滤液室;5—浓缩液室
图23 NANOSEPTM
Microconcentrator1—滤液室;2—样品室;
3—OMEGA膜
使用超滤搅拌装置时,按照生产厂家的说明先处理超滤膜,一 般 是 在 纯 水
中浸泡1h (换水3~4次),将 膜 正 确 放 置 好,安 装 好 超 滤 搅 拌 装 置,调 节 氮
气压力至合适 (压力不要超过生产厂家 推 荐 的 压 力 上 限 值),泵 送 纯 水 以 检 查
装置的密封性,在需要的情况下可以用 泵 送1%纯 蛋 白 质 溶 液 (其 分 子 量 应 远
大于膜的 MWCO)来检查膜 的 完 整 性。采 用 生 产 厂 家 推 荐 的 方 法 清 洗 膜,然
后以合适的流速和 搅 拌 速 度 进 行 超 滤 浓 缩 样 品 溶 液,达 到 所 需 浓 度 后 关 掉 氮
气,将减压安全阀打开,停止搅拌,小心取出浓缩液即可。使用后的膜 可 以 用
纯水、1mol/L氢氧 化 钠 或5%氯 化 钠 来 清 洗,清 洗 后 的 膜 还 要 用 水 浸 泡,以
除去清洗剂的残 留。正 常 情 况 下,清 洗 后 的 膜 应 能 达 到 膜 最 初 流 量 的95%以
上。所有塑料膜都要避免干燥,一般放在10%乙醇中于4℃保存,同时还 可 以
防止微生物污染。有时,超滤系统和膜需要进行热灭菌,要仔细阅读生 产 厂 家
的说明书以进行 选 择。也 可 以 用 化 学 方 法 进 行,1%~5%甲 醛 或25%~75%乙醇都可以。
223 沉淀法
在早期的蛋白质纯化过程中,沉淀法作为分离蛋白质的主要方法之一,但是
现在该法只用于蛋白质的初步分离,然后采用色谱方法进一步纯化。沉淀法也用
于蛋白质的浓缩。蛋白质分子在水溶液中的溶解性受到蛋白质分子表面亲水性和
疏水性带电基团分布的影响,这些基团与水溶液中离子基团互相作用,通过改变

21
pH或离子强度,加入有机溶剂或多聚物,可以促进蛋白质分子凝聚,形成蛋白
质沉淀,通过离心或过滤可以获得沉淀物,然后利用合适的缓冲液清洗、溶解沉
淀物,再经过透析或凝胶过滤,除去残留的溶剂成分。用于浓缩蛋白质的沉淀法有盐析法、有机溶剂沉淀法、等电点沉淀法、聚乙
二醇沉淀法等,具体可以参见本章第23节内容。
224 透析法
用透析 (dialysis,又 称 渗 析)来 进 行 浓 缩 比 较 适 用 于50ml以 下 的 样 品 体
积,但操作时间稍长,常使用20%的分子量大于20000的聚乙二醇或干的葡聚
糖凝胶。具体操作过程如下:将商品化的透析袋适当处理后,封住一端 (打结或
使用透析夹),可先用纯水检查透析袋的完整性,然后将需要浓缩的蛋白质溶液
倒入透析袋中,赶出袋中的空气,封住袋子的另外一端,将透析袋放入10倍于
蛋白质样品体积的装有20% (200g/L)聚乙二醇溶液的容器中,用磁力搅拌器
缓慢搅拌,至达 到 所 需 浓 度 为 止。若 使 用 干 的 葡 聚 糖 凝 胶 G25,用 量 为25g/
100ml蛋白质样品,需注意不能够浓缩到完全脱水。
225 冷冻干燥法
冷冻干燥技术在热不 稳 定 的 蛋 白 质、多 肽 等 的 浓 缩、保 存 方 面 有 着 重 要 作
用,利用低温、低压来去除可溶性的水,它对于超滤膜不能够截留的低分子量多
肽浓缩效果好。冷冻干燥系统由干燥箱、真空系统、制冷系统组成,近年来还发展了自动整
理设备、自动装瓶和倒瓶设备。干燥箱通常由不锈钢制成,是压力容器,其内表
面光洁性要好,以便于清洁,提高抗腐蚀性。冷冻干燥系统中最关键的是旋转式
油泵。具体操作时,将需冻干的样品装入冷冻干燥烧瓶中,小玻璃瓶常用一个橡
皮塞部分地塞住,在塞子上开一道槽使水蒸气逸出。小玻璃瓶放在金属托盘中,放入冻干机进行浓缩。
对蛋白质,干燥后残余水的多少可能影响其活性的稳定性。将样品干燥到最
低残余水的水平,再细分小瓶,在几个相对不同的湿度得到不同残余水含量的样
品,进行稳定性研究,以确定合适的残余水分含量。
226 双水相分离法
利用两种多聚物,或多聚物与盐在水相中的不相溶性,可以从细胞破碎后的
细胞碎片中直接分离、纯化蛋白质,同时起到浓缩蛋白质的作用。该方法比较温
和,一般不造成蛋白质的变性失活,可在室温下进行,双水相中聚合物还可提高
蛋白质的稳定性。最常用的多聚物是聚乙二醇和葡聚糖。表22列出了常用的双
水相系统。葡聚糖的使用由于其成本较高及其造成溶液黏度较高而受到限制,可
以使用价格较低,黏度也不高的变性淀粉替代葡聚糖。

22
表22 常用双水相系统
聚合物Ⅰ 聚合物Ⅱ或无机盐 聚合物Ⅰ 聚合物Ⅱ或无机盐
聚乙二醇 葡聚糖
聚乙烯醇
聚乙烯吡咯烷酮
磷酸钾
硫酸铵
聚丙二醇 聚乙二醇
葡聚糖
聚乙烯醇
聚乙烯吡咯烷酮
甘油
聚乙烯吡咯烷酮 磷酸钾
DEAE葡聚糖盐酸 聚乙烯醇
聚乙二醇Li2SO4聚丙二醇NaCl
羧甲基葡聚糖钠 聚乙二醇NaCl聚乙烯醇NaCl
羧甲基纤维素钠
葡聚糖硫酸钠 聚乙二醇NaCl聚乙烯醇NaCl
甲基纤维素NaCl葡聚糖NaCl
采用双水相系统浓缩目的蛋白,受到聚合物分子量及浓度、溶液pH、离子
强度、盐类型及浓度等因素的影响,各因素之间也相互影响。以PEG/葡聚糖系
统为例,通过降低PEG分子量、增加葡聚糖分子量或 提 高pH,都 可 以 提 高 目
的蛋白在PEG相中的分配系数。在实验室中,可采用5~10ml塑料离心管试验比较合适的条件。先 配 制 较
高浓度的溶液 (40%PEG和30%葡聚糖),使用时按照一定比例 添 加,以 达 到
在双水相系统中的预期浓度。操作 时 先 混 合 蛋 白 质 样 品、PEG和 葡 聚 糖 溶 液
1min,然 后3000g离 心5min,富 集 在 聚 合 物 相 中 的 目 的 蛋 白 经 过 滤 得 到。分 别 测 定 两 相 的 体 积 及 目 的 蛋 白 在 两 相 中 的 浓 度,最 后 确 定PEG和 葡 聚 糖
各 自 合 适 的 使 用 浓 度。一 般 采 用Bradford方 法 测 定 蛋 白 质,以 避 免PEG的
影 响。
23 蛋白质样品的沉淀
沉淀法是比较传统的分离纯化 蛋 白 质 的 方 法,目 前 仍 然 在 实 验 室 内 广 泛 使
用。该法所需设备简单,操作方便,在蛋白质纯化的初期,可以迅速减少样品体
积,起到浓缩的作用,便于后续的纯化,降低纯化成本;还可以尽快将目的蛋白
与杂质分开,提高目的蛋白的稳定性;通过该方法,目的蛋白的收率比较高。但
由于沉淀法对提高 蛋 白 质 纯 度 的 幅 度 比 较 有 限,该 法 常 只 用 于 蛋 白 质 的 初 步
纯化。以下几种沉淀方法比较常用:盐析法,有机溶剂沉淀法,等电点沉淀法,聚
乙二醇沉淀法等。选择沉淀方法时,需要考虑沉淀剂对目的蛋白稳定性的影响、沉淀剂的成本以及操作难易程度、沉淀剂的去除及残留、目的蛋白的纯度要求及
收率要求等。

23
231 盐析法
沉淀法中常用的是盐析法,该方法条件温和、操作简便。加入中性盐到蛋白
质溶液中,蛋白质的溶解度开始增大,但随着继续加入盐,蛋白质的溶解度却逐
步减小,并形成沉淀,这就是盐析。不同蛋白质在不同的中性盐浓度下析出,利
用此原理,可对溶液中的杂蛋白及目的蛋白进行分级沉淀。通常蛋白质受到中性
盐的保护作用,不会因为盐析而变性失活。盐析受到蛋白质表面疏水性的影响。疏水基团主要存在于蛋白质内部,也有一些分布在蛋白质的表面,水分子与这些
基团接触,加入盐到溶液中后,随着盐浓度的提高,水分子被从蛋白质分子周围
移开,蛋白质分子表面的疏水基团互相作用,导致了蛋白质的凝聚、沉淀。蛋白质样品的组成、浓度、pH、操作温度、添加硫酸铵的速度、搅拌速度
等都影响到沉淀效果。盐析操作最好在4℃进行,操作时一边搅拌一边缓慢加入
沉淀剂,以免造成局部沉淀剂浓度过高。通过离心 (10000g、10min)或过滤可
得到沉淀物。对于目的蛋白的沉淀物,可用1~2倍体积的缓冲液溶解,不溶解
的部分可离心除去。采用盐析法沉淀目的蛋白,起到了浓缩及初步分离纯化蛋白质的作用,盐析
条件的确定必须综合考虑原材料的来源、纯化目的、盐析后目的蛋白的收率及纯
化倍数等因素。要选择好合适的盐,可以优先考虑价格低、溶解性好、溶解过程
中产生热量少、溶解后形成的溶液密度与沉淀的密度有明显差别的盐。高价阴离
子的沉淀效果比较好,磷酸盐的效果好于硫酸盐、醋酸盐和硝酸盐,单价阳离子
中,铵离子效果要好于钾离子和钠离子。磷酸钾、磷酸钠、硫酸钠和硫酸铵都可
以采用,磷酸钾和磷酸钠的价格比较高,硫酸钠在40℃以下的溶解度较低,只
适合于热稳定性好的目的蛋白。硫酸铵是最常用的,因为它便宜,溶解度高,对
温度 的 敏 感 性 低,在 纯 水 中 的 饱 和 浓 度 接 近4mol/L,饱 和 溶 液 的 密 度 为
1235g/ml。由于蛋白质沉淀的密度与硫酸铵饱和溶液的密度比较接近,因此离
心分离沉淀物时一般需要高速离心机。另外,硫酸铵会使溶液略微酸化,可用氨
水调节pH在60~75之间,在盐析除去杂蛋白时,可使溶液的pH 远离目的
蛋白的等电点,而在沉淀目的蛋白时,溶液的pH应接近目的蛋白的等电点。如
果需要更高的pH,可以用柠檬酸钠替代硫酸铵。如果目的蛋白对重金 属 敏 感,需加入EDTA至溶液中。
硫酸铵的浓度多用 “饱和度”表示。实际所需的硫酸铵饱和度,应通过试验
来确定,可以先使蛋白质溶液达到某一硫 酸 铵 饱 和 度,此 时 目 的 蛋 白 不 发 生 沉
淀,离心除去杂蛋白沉淀,再加入硫酸铵至更高的另一饱和度,尽量多地沉淀目
的蛋白,离心后获得目的蛋白沉淀。具体操作可在4℃进行:各取50ml的等量
蛋白质样品溶液,放入烧杯中,预先冷却到4℃,计算出至20%、30%、40%、

24
50%、60%、70%、80%、90%和100%饱和度时所需要的硫酸铵数量,准确称
取所需的硫酸铵。将硫酸铵缓慢加入到各蛋白质溶液中,同时使用磁力搅拌器缓
慢搅拌,当溶液出现浑浊时,静置1h以上,3000g离心30~60min,移 去 上 清
液,取出沉淀物。用2倍于沉淀物体积的等量缓冲液溶解沉淀物 (如果还有不溶
性物质,可以3000g离心10~20min,除去不溶性物质)。对于溶解液中的总蛋
白质含量以及目的蛋白的含量进行分析,采用Bradford方法为宜,以免受到硫
酸铵的影响;或者先对蛋白质沉淀的溶解液脱盐,再分析蛋白质。以总蛋白质和
目的蛋白浓度对硫酸铵饱和度作图,可以确定不沉淀目的蛋白的最高饱和度和沉
淀目的蛋白的最适饱和度。盐析时,达到不同饱和度所需要添加硫酸铵的质量可通过表23查得。也可
利用下列公式计算出在20℃时达到所需硫酸铵饱和度应在每升溶液中加入的硫
酸铵质量:
m(g)=533S2-S1
100-03S2式中 S1———溶液起始硫酸铵饱和度,%;
S2———溶液达到的硫酸铵饱和度,%。
表23 达到不同饱和度每升溶液中加入固体硫酸铵的质量/g
硫酸铵起始
饱和度/%
硫酸铵最终饱和度/%
10 20 25 30 35 40 45 50 55 60 65 70 75 80 90 100
0 56 114 144 176 209 243 277 313 351 390 430 472 516 561 662 76710 57 86 118 150 183 216 251 288 326 365 406 449 494 592 69420 29 59 91 123 155 189 225 262 300 340 382 424 520 61925 30 61 93 125 158 193 230 267 307 348 390 485 58330 30 62 94 127 162 198 235 273 314 356 449 54635 31 63 94 129 164 200 238 278 319 411 50640 31 63 97 132 168 205 245 285 375 46945 32 65 99 134 171 210 250 339 43150 33 66 101 137 176 214 302 39255 33 67 103 141 179 264 35360 34 69 105 143 227 31465 34 70 107 190 27570 35 72 153 23775 36 115 19880 77 15790 79
以Aspergilluskawachii木聚糖酶的纯化为例,取100ml的发酵上清液,放
入烧杯中,预冷到4℃,计算出至60%饱和度时所需硫酸铵数量,准确称取,将

25
硫酸铵慢慢加入到溶液中,磁力搅拌器同时进行缓慢搅拌,使硫酸铵溶解充分,在4℃静置1h,7600g离心20min,取出沉淀物,用pH=50的醋酸盐缓冲液溶
解沉淀物,经过凝胶柱脱盐后,进入下一纯化步骤。
232 有机溶剂沉淀法
采用有机溶剂沉淀蛋白质的技术已经有较长的历史了。该法的优点是有机溶
剂一般不会残留在产品中,容易蒸发除去;有机溶剂密度低,与沉淀物质的密度
差大,便于离心分离。有机溶剂沉淀蛋白质被认为是有机溶剂破坏了蛋白质分子之间的某些键,使
蛋白质分子的空间结构发生了变化,一些原先存在于蛋白质分子内部的疏水基团
被暴露于 表 面,并 与 有 机 溶 剂 的 疏 水 基 团 结 合,形 成 了 疏 水 层,使 得 蛋 白 质
沉淀。在操作过程中应保持低温 (0℃或以下)、快速,选择低毒性、不与目的蛋白
发生作用的水溶性有机溶剂,常用的有机溶剂是丙酮和乙醇,有时也用到甲醇和
丙醇。使用有机溶剂时,需小心操作,注意安全。大多数蛋白质通过加入等体积
的丙酮或4倍体积的乙醇就可以沉淀下来,但也造成了蛋白质溶液被稀释,所以
蛋白质的浓度一般要在1mg/ml以上。有机溶剂沉淀 蛋 白 质 的 效 率 除 了 有 机 溶 剂 种 类 外,还 受 到 操 作 温 度、pH、
蛋白质分子大小等因素的影响。温度低,沉淀比较完全,在整个沉淀蛋白质及分
离沉淀的过程中,可在盐水冰浴中进行,以保持低温操作 (4℃),将预先冷却过
的有机溶剂缓慢加入到冷却了的蛋白质溶液中,同时不断搅拌,以避免局部有机
溶剂浓度过高或升温过大而造成蛋白质失活。蛋白质溶液的pH应该选择使目的
蛋白性质稳定的区域,并尽可能接近其等电点。分子量大的蛋白质可以在较低的
有机溶剂浓度下沉淀。可以通过在10000g离心5~15min后获 得 沉 淀,对 于 离
心机的转子要预冷却,离心机应在0~4℃进行操作,以防止部分沉淀重新溶解,或者蛋白质发生变性。沉淀物可以用1~2倍体积的缓冲液溶解,不溶解的部分
是已经变性了的蛋白质,通过离心除去。以丙酮为例,将等体积的丙酮 (已经冷却到-20℃)缓慢加入到蛋白质溶液
中,同时 利 用 冰 浴 进 行 冷 却,不 断 地 缓 慢 搅 拌10~20min,在10000g 离 心
15min后获得沉淀,用2倍体积的缓冲液溶解沉淀。有机溶剂 沉 淀 蛋 白 质 时,需 加 入 的 有 机 溶 剂 数 量,可 以 由 下 列 公 式 计 算
得到:
V=V0S2-S1S0-S2
式中 V———加入的有机溶剂体积,L;

26
V0———蛋白质样品的起始体积,L;
S0———加入的有机溶剂浓度,%;
S1———蛋白质样品中的有机溶剂浓度,%;
S2———蛋白质样品中需达到的有机溶剂浓度,%。有机溶剂沉淀法比盐析法,更容易使蛋白质变性,这是不足的方面。对于一
些疏水性蛋白质,尤其是位于细胞膜的蛋白质,不采用此法,因为这些蛋白质不
被有机溶剂沉淀,反而会被有机溶剂从细胞膜溶解下来。
233 等电点沉淀法
该方法是最早使用的沉淀蛋白质的方法之一。蛋白质分子表面存在带正电荷
和负电荷的基团,在等电点时,蛋白质分子的正、负电荷相等,净电荷为零,分
子间不再发生静电排斥,而是产生静电引力,蛋白质的溶解度最低,可以被沉淀
出来。利用此原理,将蛋白质样品溶液的pH调节至其等电点,大大降低其溶解
度,从而沉淀得到目的蛋白,再将 沉 淀 溶 解 于 适 当 的 缓 冲 液 中,用 于 随 后 的 纯
化。利用不 同 蛋 白 质 具 有 不 同 的 等 电 点,也 可 以 采 用 该 法,依 次 改 变 溶 液 的
pH,分别沉淀、除去杂蛋白,从而获得目的蛋白,这比较适合于沉淀过程中容
易发生变性失活的目的蛋白。该方法的优点是很多蛋白质的等电点在偏酸性范围内,比如血清蛋白的等电
点在49,脲酶的等电点在50,来源于米曲霉RIB128的木聚糖酶B的等电点
为44。调节pH所需的无机酸价格低,操作也比较方便。但是,对低pH敏感
的目的蛋白,应避免使用此法。
234 聚乙二醇沉淀法
许多分子量高的非离子聚合物,比如聚乙二醇、葡聚糖等,都可以沉淀蛋白
质,常用的是PEG。一 般 使 用 分 子 量4000以 上 的PEG,常 用 分 子 量6000和
20000的。PEG无毒,不可燃,操作条件温和、简便,沉淀比较完全,而且对蛋
白质有一定的保护作用。
PEG沉淀蛋白质的机理被认为是:PEG分子在溶液中形成了网状结构,与
溶液中的蛋白质分子发生空间排斥作用,使蛋白质分子凝聚、沉淀。该方法受到
蛋白质的分子量、浓度、溶液的pH、温度以及PEG的平 均 分 子 量 等 因 素 的 影
响:蛋白质的分子量越大,将蛋白质沉淀所需的PEG浓度越小;蛋白质浓度越
高,越易于沉淀,但是蛋白质浓度也不能太高,一般要小于10mg/ml;pH越接
近蛋白质的等电点,所需PEG浓度也越低;在30℃以下一般都可以使用此法,只是操作时需要考虑到目的蛋白对温度的敏感性;PEG的分子量越高,沉淀蛋
白质所需的浓 度 越 低,但 是PEG的 分 子 量 过 高,会 造 成 溶 液 黏 度 大,不 利 于
操作。

27
具体操作:将100~150ml的50% (500g/L)PEG6000溶液加入到100ml蛋白质样品溶液中,缓慢搅拌60min,至沉淀完全,然后在5000g离 心30min,将所获得的沉淀部分溶解于2倍体积的缓冲液中。
235 选择性沉淀法
不同蛋白质在不同的条件下有不同的稳定性,改变温度、pH或加入有机溶
剂,许多杂蛋白发生变性沉淀,而目的蛋白不形成沉淀,就可以分离出比较纯的
目的蛋白。利用杂蛋白在不同温度下产生沉淀,将样品溶液升温至45~65℃,保温一
定时间,使杂蛋白形成最大程度的沉淀,同时,目的蛋白的活性损失最少。由于
不少蛋白酶在此温度范围内比较稳定,为了避免样品中发生酶解而造成目的蛋白
的活性损失,操作前可适当加入蛋白酶抑制剂。很多蛋白质在pH=50或以下时被沉淀,只有少数蛋白质在中性或碱性条
件下形成沉淀。如 果 目 的 蛋 白 在 此pH 范 围 内 能 够 保 持 稳 定,就 可 以 通 过 调 节
pH来除去杂蛋白。该方法尤适用于初步纯化原核生物中表达的重组蛋白质,因
为很多细菌蛋白质的等电点在pH=5左右,通过调节pH 至其等电点,可以先
除去这部分蛋 白 质。调 节pH 时,常 用 醋 酸、柠 檬 酸 或 碳 酸 钠,也 可 采 用 高 氯
酸、三氯醋酸等强酸,但是需要注意安全。加入10%的三氯醋酸可以沉淀大部
分的蛋白质,20%的三氯醋酸可以沉淀分子量低于20000的蛋白质,操作时需在
冰水浴中进行,沉淀物可以10000g离心10min,获得的沉淀部分可用乙醇和乙
醚 (1∶1)清洗,然后溶解于适当的缓冲液中。杂蛋白在有机溶剂 (丙酮或乙醇)作用下,能够形成沉淀,一般可在20~
30℃加入有机溶剂,以促进变性,然后冷却到-20℃,保持1h以上,尽可能完
全沉淀杂蛋白,从而获得目的蛋白。这比较适合于沉淀过程中易发生变性的目的
蛋白。
24 蛋白质样品的透析
241 基本原理
为了提高随后纯化环节的效率,在纯化过程中通过透析进行除盐或更换缓冲
液。将蛋白质样品放入半透膜袋中,置于所需的缓冲液中,由于袋内小分子的渗
透压高于袋外的缓冲液,根据渗透压和分子自由扩散的原理,小分子物质可以自
由通过半透膜,大分子物质被保留在袋内,随时间的延长,小分子向袋外扩散的
速度逐渐减慢,最后半透膜内外的分子进出速度达到平衡。半透膜用醋酸纤维制成,孔径在1~20nm,它决定了膜的 MWCO。有些透
析袋需先进行预处理以除去金属物质,并保证膜孔的一致性。现在商品化的透析

28
膜 (比如日本三光纯药公司生产的无缝纤维透析膜)一般不需要预处理了,只要
使用前在纯水或适 当 的 缓 冲 液 中 浸 泡 几 分 钟 即 可。透 析 操 作 可 在4℃下 过 夜 进
行,以充分利用时间。
242 应用
在米曲霉RIB128 (AspergillusoryzaeRIB128)木 聚 糖 酶 的 纯 化 过 程 中,多次用到透析。选择合适的商品透析袋 (日本三光纯药公司生产的 UC2432100型),截取足够的 长 度,浸 入 纯 水 中 浸 泡10min,用 透 析 夹 封 住 透 析 袋 的 一 端,先用纯水检查透析袋的完整性,然后将样品蛋白质溶液注入透析袋中,赶出袋中
的空气,封住袋子的另外一端,将透析袋放入足够体积的pH=50的醋酸盐缓
冲液中,缓慢搅拌,在4℃过夜。透析袋要足够长,是因为样品在透析过程中体积会有不同程度的增加,如果
透析袋内预留的空间太小,有可能会因为样品体积增加过多而造成透析袋破裂。缓冲液的体积在条件允许时,可以是样品体积的几十倍至100倍,以免为了达到
平衡而中途再更换缓冲液。在操作过程中最好戴手套,以免造成污染。

29
3 凝胶过滤色谱
31 概述
凝胶过滤色谱 (gelfiltrationchromatography,GFC)是利用具有多孔网状
结构的凝胶颗粒的分子筛作用,根据被分离样品中各组分分子量大小的差异进行
洗脱分离的一项技术。Porath等首次用 一 种 多 孔 网 状 高 聚 物———交 联 葡 聚 糖 凝
胶作为固定相,在水系流动相 (或称洗脱液)中分离了不同分子量的物质,被称
为凝胶过滤色 谱。1964年,Moore制 备 了 具 有 不 同 孔 径 的 交 联 聚 苯 乙 烯 凝 胶,在有机流动相中进行了样 品 分 离,称 为 凝 胶 渗 透 色 谱 (gelpermeationchromatography,GPC)。它们的分离原理相同,但 通 常 将 流 动 相 为 水 溶 液 的 称 凝 胶 过
滤色谱,流动相为有机溶剂的称凝胶渗透色谱,后来将这两种统称为尺寸排阻色
谱 (sizeexclusionchromatography,SEC)。本书中将该纯化技术统称为凝胶过
滤色谱。凝胶过滤色谱技术是蛋白质研究领域内一种高效的分离纯化手段。一方面从
理论上讲,蛋白质样品在凝胶柱内几乎和固定相基质不发生任何作用,另一方面
样品洗脱液均为含盐或纯水系缓冲液,而且整个操作过程中洗脱液组成不发生变
化,所以该技术在分离纯化蛋白质时具有操作方便、洗脱条件温和、重复性好、不需要有机溶剂、样品不易变性和回收率高等诸多优点。该技术主要可用于蛋白
质的浓缩纯化,分子量及其分布范围测定,样品脱盐以及更换蛋白质缓冲液等。近年来也在蛋白质复性研究中得到了广泛应用。在研究中为了得到较高纯度的蛋
白质样品,通常将凝胶过滤色谱和离子交换色谱结合使用,而凝胶过滤色谱可在
蛋白质纯化的任何一个阶段使用。
311 基本原理
3111 凝胶的分子筛效应
采用凝胶过滤色谱可将不同分子大小的样品分离开来,这主要归功于固定相
基质———凝胶。凝胶是一种具有三维多空网状结构的珠状颗粒物,具有一定的孔
径和交联度,它们不溶于水,但具有吸水膨胀的特性,因此具有一定的膨胀度。同一型号的凝胶颗粒的细微结构和筛孔直径均匀一致,小分子物质可以 进 入 凝
胶内部网孔,而大分子物质则排阻于颗粒之外,显示了良好的分子筛功 能 (见
图31)。

30
图31 凝胶颗粒结构示意
由于凝胶对不同分子量大小的物质的排阻和扩散作用的程度不同,最终导致
大分子物质先流出,中等分子物质后流出,最小的分子最后流出,这种现象叫做
凝胶的分子筛效应。具有多孔网状结构的凝胶就是一种很好的分子筛。当不同分
子大小的物质通过由这种凝胶颗粒填装成的色谱柱时,分子直径比凝胶最大孔隙
直径大的物质,就会被全部排阻在凝胶颗粒之外,称为全排阻;直径比凝胶最小
孔径小的小分子溶质和洗脱液分子能自由进出凝 胶 内 部 的 全 部 孔 隙,为 全 部 渗
透;而分子大小介于两者之间的溶质分子在其中被部分排阻 (或部分渗透)。所
以在凝胶过滤色谱分离样品的过程中,分子有三种状况:第一类为大分子,完全
不能进入凝胶的任何内部孔隙;第二类为适中大小分子,能部分进入与其孔径大
小相应的凝胶内部孔隙中;第三类为小分子,能进入凝胶内部的全部孔隙。若样
品中主要组分为大、中、小三类分子,则彼此间比较容易分开,这种分离称为组
别分离 (groupseparations),而 第 二 类 样 品 的 分 离 称 为 分 级 分 离 (fractionations)。分级分离对凝胶和各种色谱条件的要求比组别分离要高。但是每种凝胶
都有一定的分离范围,在其分离范围之外的混合分子是很难分离的。即使两种全
排阻的有不同大小的分子,也不能进行有效分离。同样,如果两种分子都能全部
渗入凝胶内部孔隙,它们即使在分子大小上存在差异,也不能得到有效分离。采用凝胶过滤色谱技术分离纯化蛋白质时,主要是根据蛋白质样品分子量大
小差异这一物理特性。在凝胶的分子量分离范围之内,几种不同分子量的蛋白质
都能不同程度地进入凝胶网孔,但由于它们被排阻和扩散的程度不同,在凝胶柱
中所经过的路程不同,最终造成被洗出柱子的时间也不同,从而可以达到分离的
目的。当含有不同分子量大小的混合蛋白质样品加到用凝胶颗粒填装而成的色谱柱

31
上时,这些物质随洗脱液的流动而发生移动,在柱内存在垂直向下的因重力引起
的移动和无规则的扩散两种运动方式。大分子蛋白由于直径较大,不能进入凝胶
颗粒内的微孔,只能在颗粒之间的空隙流动,所以在洗脱时下移速度较快,最先
被洗出凝胶柱;小分子蛋白除了可在凝胶颗粒间的空隙中扩散外,还可以渗透到
凝胶内孔中,下移过程中在凝胶内部和颗粒间隙之 间 不 断 往 复 运 动 使 其 行 程 较
长,最后被洗脱出柱;中等分子蛋白流出的时间介于大小分子之间,分子越大流
出的时间越早,最终使样品中不同分子大 小 的 蛋 白 质 彼 此 分 离。其 分 离 原 理 见
图32。
图32 凝胶过滤色谱的原理
3112 凝胶过滤色谱中的重要参数
(1)凝胶的参数
① 凝胶颗粒大小 颗粒的大小通常用目 (mesh)或直径 (μm)来表示。色
谱分离用的凝胶一般都制成球形颗粒。凝胶颗粒的大小直接关系到色谱柱的流速
和分辨率,颗粒越大,流速越快,但分辨率较差;相反,颗粒越小,流速越慢,但分辨率提高。
② 排阻极限 指不能进入凝胶颗粒内部孔隙的最小分子的分子量。排阻极
限反应某种凝胶能有效分离的最大分子量,所有大于排阻极限的分子均被完全排
阻,不能分离。如SephadexG75的排阻极限为80000Da?,它表明分子量大于
80000Da的分子为全排阻。
? Da:即dalton,道尔顿,非法定质 量 单 位,等 于 一 个 氢 原 子 的 质 量 (167×10-24g)。表 示 分 子
质量的大小,本书中按照行业习惯将其称为分子量,但应与相对分子质量有所区别。———编辑注

32
③ 分级范围 (或工作范围) 所能进入凝胶内部孔隙的最大分子和最小分子
的分子量范围。它表示一种凝胶适用的分离范围,如SephadexG50对球形蛋白
的分级分离范围为1500~30000Da,它表示通过SephadexG50可有效分离分子
量在这个范围内的球形蛋白 (不同分子形状对分离效果的影响在后面讨论)。
④ 吸水量 1g干胶吸收水的体积或质量。凝胶型号越大,吸水量越大。例
如SephadexG50的吸水量为5ml(或g),而SephadexG150的为15ml(或g)。
⑤ 床体积 1g干胶吸水后的最终体积。(2)体积参数
① 外水体积 柱内凝胶颗粒之间的空隙体积的总和,用Vo表示。
② 基质体积 干胶溶胀后颗粒本身所固有的体积,用Vg表示。
③ 内水体积 凝胶颗粒内部孔隙体积的总和,或者为小分子溶质可渗透进
入凝胶内部体积的总和,又称固定相体积,用Vi表示。
④ 柱床体积 凝胶柱装成后从柱底板到凝胶沉积层表面的总体积,又称床
体积,用Vt表示。见图33。其计算公式为:
Vt=Vo+Vi+Vg (31)由于Vg相对较小,可忽略不计,故有:
Vt=Vo+Vi (32)
图33 凝胶柱各种体积示意 (阴影部分)
⑤ 洗脱体积 某一被分离样品从凝胶柱内完全被洗脱出来时所需洗脱液的
体积,用Ve表示。其计算公式为:
Ve=Vo+KdVi (33)
式中,Kd是分配系数;Ve一般介于Vo和Vo+Vi之 间。对 于 完 全 排 阻 的 大
分子,由于其不进入凝胶内部,只能在颗粒间隙中流动,故其洗脱体积Ve=Vo;
而对于完全渗透的小分子,由于它可在整个凝胶柱内流动 (忽略Vg),故其洗脱

33
体积Ve=Vo+Vi;分子量介于两者之间的分子,它们的洗脱体积也介于两者之
间;一些吸附蛋白在最后洗出。蛋白质分子大小与洗脱曲线的关系见图34。
图34 凝胶过滤色谱洗脱曲线示意
1—完全排阻的大分子;2—中等分子;3—完全渗透的小分子;4—吸附分子
(3)分配系数和有效分配系数 凝胶过滤色谱中,某一蛋白组分在流动相和
固定相之间的分配关系叫做分配系数,常用Kd表示,其值介于0~1之间。由式
(33)可得出Kd的计算公式为:
Kd=Ve-VoVi
(34)
对于一恒定的凝胶色谱系统,其Vi和Vo都是恒定的,而且Vi和Vo都可测
定 (具体方法在后面介绍),所 以 测 定 了 某 一 组 分 的Ve就 可 得 到 该 组 分 的Kd。Kd与Ve成正比关系,Ve越大,Kd越大。从理论上讲,所有分离的蛋白组分的洗
脱体积只能在Vo和Vo+Vi之 间。全 部 排 阻 的 大 分 子 蛋 白,因 其Ve=Vo,故
Kd=0;而完全能渗入凝胶内部孔隙的小分子蛋白,其Ve=Vo+Vi,故Kd=1;分子量介于两者之间的0<Kd<1。但是当蛋白质在凝胶柱 内 有 吸 附 时Kd>1;当凝胶床有裂缝而发生洗脱短路时Kd<0。
由于在实验中准确测定Vi的值比较困难,所以提出了有效分配系数的概念,常用Kav表示,它将基质体积和流动相在凝胶颗粒外的体积总体看作外水体积。
Kav实则为蛋白组分在内水体积和外水体积中的浓度分配比。测出Vt、Vo和Ve即可得出Kav。对于一些基质体积不能忽略或精确度要求很高的,用Kav表示分
配系数较为准确,其计算公式如下:
Kav=Ve-VoVt-Vo
(35)
3113 影响分离效果的主要因素
(1)流速 流速对分离效果的影响较大,所以在洗脱分离蛋白质样品时保持

34
合适而恒定的流速非常重要。凝胶色谱系统中洗脱流速主要取决于凝胶柱的内径
和高度、凝胶种类、颗粒大小以及分离类型等。一般在开始正式洗脱之前,先要
进行预备试验以确定合适的流速。较缓的流速可使 蛋 白 样 品 与 凝 胶 基 质 充 分 平
衡,达到理想的分离效果,但是流速过低会 造 成 样 品 在 凝 胶 床 内 的 横 向 扩 散 增
大,使峰宽变宽,降低分辨率。此外,还延长了洗脱时间,降低工作效率。高流
速易引起洗脱峰的重叠,使本来可分开的组分重合,而且会增大柱压,影响分离
效果。一般凝胶的线性流速控制在2~10cm/h。商品凝胶出厂时,商家提供的一些
流速参数可供使用者参考。在一般实验中通常用蠕动泵调节流速,没有条件的可通
过控制一定的高度压差来控制流速。在实验中通常使用体积流速 (ml/min),有时
为了实验需要,还需将体积流速转换成线性流速 (cm/h),其转换公式如下:
线性流速(cm/h)=体积流速(ml/min)×60
柱子横切面积(cm2)(36)
图35 Vs对洗脱曲线的影响及相应Ve的计算
(a)Vs可忽略;(b)Vs不能忽略;(c)Vs过量
(2)加样体积 加样体积 (Vs)也可对蛋白样品的分离效果造成较大 的 影
响。体积过大,会造成平台洗脱峰或相邻峰的重叠,影响分离效果;加样过少,目的蛋白组分的收集量少、稀释倍数增大、浓度低,降低实验效率。而且不同的
加样体积对洗脱曲线及Ve的计算均有不同程度的影响 (见图35)。当加样体积
很少时 (与Ve 相比可忽略),所出峰尖锐,峰宽较小,其Ve可计为从加样体积
起始到洗脱峰顶的体积 [图35 (a)];当加样体积与Ve 相比不能忽略时,Ve可

35
计为从样品 体 积 的 一 半 到 峰 顶 位 置 的 体 积 [图35 (b)];当 加 样 体 积 很 大 时,洗脱峰出现平台,其Ve可计为从加样体积开始到洗脱峰升高时拐点 (或半峰高
处)间的体积 [图35 (c)]。(3)样品浓度 样品进样前应高度浓缩,但是浓度不可过大,否则会影响分
离效果。蛋白质浓度应小于70mg/ml,一般在10~20mg/ml。必 要 时,需 进 行
样品浓度系列试验,以确定最佳值。过低的样品浓度可造成某一蛋白组分的过度
稀释,影响收集;而浓度过高时会使流动相不稳定,造成洗脱峰的变形或重叠。此外,样品浓度还和分离纯化的类型有关,进行蛋白组别分离或脱盐除杂时,由
于目的蛋白和杂质的分子量差异较大,故可适当提高样品浓度,而进行分子量差
异较小的分级分离或分析性实验时,样品浓度尽量要低一些。(4)离子强度 样品洗脱液中含有一定离子强度的盐溶液可防止蛋白质与蛋
白质之间或蛋白质与凝胶介质之间的相互作用,排除蛋白质与流动相或与凝胶介
质相互作用的干扰。一些不带电荷的中性蛋白在纯水中可实现有效分离,而在分
离一些带电荷的蛋白质样品时特别要注意这一影响,一定要调整洗脱液的离子强
度,排除离子吸附等干扰。例如,Sephadex因含有羧基基团,呈弱酸 性,所 以
在用该类凝胶进 行 色 谱 操 作 时 常 使 用 一 定 离 子 强 度 的 盐 溶 液 (一 般 高 于005)作为洗脱液,这样就可以避免Sephadex与碱性蛋白发生吸附。在分离纯化蛋白
质时一般使用20~100mmol/LNaCl作为盐溶液。但应注意如果盐浓度过高,会
引起凝胶柱床体积的变化。(5)pH 由于在凝胶过滤色谱中所用的平衡液和洗脱液一致,故在整个操
作中pH不发生变 化,选 用 具 有 一 定 缓 冲 能 力 的 缓 冲 液 即 可 达 到 控 制pH 的 目
的。维持洗脱液一定的pH,一方面是考虑蛋白质样品的稳定性和溶解性,需尽
量避开靠近蛋白样品等电点的pH范围,以防蛋白质沉淀。另一方面,平衡、洗
脱中pH的变化会造成凝胶床体积的变化,影响分离纯化的效率和效果。所以在
确定pH时要考虑蛋白质样品性质和凝胶所能耐受的pH 范围这两方面的因素。
pH一般控制在6~8,磷酸盐和TrisHCl缓冲液的使用较多。生产商家提供的
一些凝胶所适合使用的pH范围可供参考。(6)分子形状 溶质的大小和分子量主要取决于分子形状,洗脱液中所溶解
的蛋白质具有各种不同的分子形状 (如球蛋白、微不对称球蛋白、含纤维杆状蛋
白和变性后的弯曲结构等),而在理想的 凝 胶 过 滤 色 谱 中,蛋 白 质 为 球 形 分 子,在柱内的保留时间仅与其分子大小和凝胶孔径大小之间的差别有关,所以不同形
状的蛋白质分子对分离也有不同程度的影响。不同形状分子溶质的理论校准曲线
见图36。在测定一些非球形蛋白质分子量的实验过程中,通常在样品中加入高浓
度的变性剂 (如8mol/L尿素),使各蛋白组分发生卷曲后,分子形状趋于一致。

36
图36 不同形状分子溶质的
理论校准曲线
(7)其他因素 除 以 上 因 素 之 外,操
作温度、凝胶颗粒 的 种 类 和 直 径、柱 子 的
长度和内径等都对蛋白质样品的分 离 效 果
有影响。在操作过程中保持 合 适 的 温 度 对
分离非常重要,操作温度要 控 制 在 凝 胶 的
最适用范围内,否则会影响 到 凝 胶 分 级 范
围、排阻极限的准确性和柱床体积的变化。
312 凝胶的性质和种类
作为分离 的 核 心,凝 胶 首 先 要 具 有 三
维多孔网状的分子筛结构 特 性,能 使 各 种
不同分子量的蛋白质分子 得 以 分 离。凝 胶
介质要 具 备 较 大 的 外 水 体 积 和 内 水 体 积,亲水性好且具有一定的机 械 强 度、化 学 惰
性和良好的色谱稳定性。在凝胶过滤色谱固定相载体的选择中被广泛应用的主要
有交联葡聚糖凝胶、琼脂糖凝胶、交联琼脂糖、聚丙烯酰胺凝胶以及聚丙烯酰胺
和琼脂糖的交联物等。近年来许多厂家还开发出了硅胶质的固定相载体,可实现
高通量和高流速分离。下面分有机和无机填料两类介绍。
3121 有机凝胶填料
(1)交联 葡 聚 糖 凝 胶 商 品 种 类 有Sephadex和Sephacryl两 种,主 要 由
PharmaciaBiotech公司生产。Sephadex不溶水,但有较强的亲水性,能迅速在
水和电解质溶液中吸水膨胀,而且在碱性环境中比较稳定,所以用适当浓度的碱
液 (一般为02mol/L)可除去吸附在凝胶上的污染物。SephadexG是由葡聚糖
和3氯1,2环氧丙烷 (交联剂)以醚键交联形成的具有三维多孔网状结构的高
聚物,其交联度 由 交 联 剂 的 百 分 比 决 定。SephadexG的 种 类 主 要 有 G10~G200共八种,G后面的阿拉伯数字表示每克干胶吸水量 (g水/g干胶)的10倍。例如,SephadexG75表示该凝胶在吸水膨胀时每克干胶能吸水75g。G反映凝
胶的洗水量、排阻极限及分离范围。例如:SephadexG10的网孔结构紧密,孔
径 小,吸 水 率 低,排 阻 极 限 小,只 能 分 离 分 子 量 较 小 的 物 质;而 SephadexG200的孔径大,吸水率高,可分离分子量较大的物质。因强氧化剂和强酸可使
Sephadex中起交联作用的糖苷键水解断裂,所以在使用时要防止其与强氧化剂
和强酸接触。在中性条件下,Sephadex悬浮液可进行高温煮沸溶胀和消毒,其
性质不受影响。在SephadexG25和G50中分别加入亲脂性的羟丙基基团,形成烷基化葡
聚糖凝胶———SephadexLH型。它 是 一 种 同 时 具 备 吸 附 性 和 分 子 筛 功 能 的 独 特

37
凝胶介质,主要型号有SephadexLH20和LH60,适用于有机溶剂洗脱,分离
脂溶性物质,具有高处理量 (可达300mg样品/ml凝胶),可分离结构非常相近
的分子,而且分离效果好。各种不同型号的Sephadex见表31。
表31 交联葡聚糖凝胶 (Sephadex)种类及其特性
型 号分子量分离
范围/Da
吸水量
/(ml/g)
最小溶胀时间/h
20~25℃ 100℃
床体积
/(ml/mg干凝胶)
G10 <700 10±01 3 1 2~3G15 <1500 15±02 3 1 25~35G25 1000~5000 25±02 3 1 4~6G50 1500~30000 50±03 3 1 9~11G75 3000~70000 75±05 24 1 12~15G100 4000~150000 100±10 72 15 15~20G150 5000~300000 150±15 72 15 20~30G200 5000~600000 200±20 72 2 30~40
Sephacryl是由葡聚糖和亚甲基双丙烯酰胺 (N,N′methylenebisacrylamide)交联而成的新型葡聚糖凝胶,其优点为分离范围较Sephadex大,适合于分离分
子量较大的蛋白质或糖蛋白,而且物理化学稳定性高,在各种溶剂中不易分解,耐高温高压,也可在较高的流速下实现较好的分辨率。
(2)琼脂糖和交 联 琼 脂 糖 凝 胶 琼 脂 糖 在100℃时 呈 液 态,当 温 度 下 降 至
45℃以下时,多糖链之间相互连接成线性双链单环状,继续降温,即凝聚成琼脂
糖凝胶。琼脂糖凝胶依靠糖链间的次级链维持成网状结构,网状结构的疏密程度
取决于干胶的浓度。因琼脂糖凝胶在40℃以上时开始融化,所以一般只能在低
温环境中操作 (0~30℃为宜),而且在干燥状态下易破裂,因此不能进行高温高
压消毒,只能进行化学灭菌处理,将其悬浮存放于含防腐剂的水溶液中。琼脂糖凝胶是一种大孔凝胶,对样品的吸附作用很小,排阻极限很大,分离
范围很广,适合于分离分子量在400000Da以上的 大 分 子 蛋 白,但 分 辨 率 较 低。常见的琼脂糖凝胶有Sepharose、BioGelA和Gelarose等。Sepharose按凝胶中
干胶的百分含量分为Sepharose2B、Sepharose4B和Sepharose6B。
Sepharose在强碱条件下与2,3二溴丙醇或环氧氯丙烷进行交联形成CL型
交联琼脂糖,分离特性基本没有改变,但抗热抗压性都有所提高,可以在更广泛
的pH范围内使用,稳定工作的pH范围为30~130 (通常,Sepharose的pH范围在45~90)(见表32)。
(3)聚丙烯酰胺凝胶 是 一 种 合 成 的 凝 胶,由 单 体 丙 烯 酰 胺 (acrylamide)和交联剂亚甲基双丙烯酰胺交联聚合而成。其商品名为BioGelP,美国BioRad公司生产,干粉呈颗粒状,在溶剂 中 能 自 动 溶 成 凝 胶。通 过 改 变 丙 烯 酰 胺 的 浓

38
度,可获得不 同 交 联 度 的 产 物。根 据 胶 的 分 离 范 围 不 同,分 成BioGelP2至
BioGelP300,P后面的数字乘以1000相当于其排阻极限,P后面的数字越大,可分离的分子量范围也越大。聚丙烯酰胺的化学性质不活泼,但是在极端pH下
会被水解,产生具有离子交换性质的羧基,因此pH应尽量控制在2~10之间。聚丙烯酰胺凝胶在水溶液、一般有机溶剂和盐溶液中都比较稳定。该类凝胶适合
于测定蛋白质的分子量。表33中各型号的凝胶都是亲水性的,在水和缓冲溶液
中很容易膨胀。
表32 Sepharose系列凝胶特性比较
项 目Sepharose2B
Sepharose4B
Sepharose6B
SepharoseCL2B
SepharoseCL4B
SepharoseCL6B
琼脂糖/% 2 4 6 2 4 6分子量分离范围/kDa 70~40000 70~20000 10~4000 70~40000 60~20000 10~4000颗粒大小/μm 60~200 45~165 45~165 60~200 45~165 45~165推荐流速/(cm/h) 10 115 14 15 26 30
pH范围 4~9 4~9 4~9 3~13 3~13 3~13适用范围
蛋白质、病毒、核酸、多糖
蛋白质、多糖
蛋白质、多糖
蛋白质、病毒、核酸、多糖
蛋白质、多糖
蛋白质、多糖
表33 各种聚丙烯酰胺凝胶的技术参数 (BioRad公司,美国)
型 号 排阻下限/Da分子量分离
范围/Da
溶胀后的床体积
/(ml/g干凝胶)最少溶胀时间
(室温)/h
BioGelP2 1600 200~2000 38 2~4
BioGelP4 3600 500~4000 58 2~4
BioGelP6 4600 1000~5000 88 2~4
BioGelP10 10000 5000~17000 124 2~4
BioGelP30 30000 20000~50000 149 10~12
BioGelP60 60000 30000~70000 190 10~12
BioGelP100 100000 40000~100000 190 24
BioGelP150 150000 50000~150000 240 24
BioGelP200 200000 80000~300000 340 48
BioGelP300 300000 100000~400000 400 48
(4)聚苯乙烯 凝 胶 商 品 名 为Styrogel,具 有 较 大 的 网 孔 结 构,机 械 强 度
高,可用于分离分子量在1600~40000000之间的大分子,适用于蛋白质分子量
测定和分级分离。(5)交联丙烯基葡聚 糖 凝 胶 此 凝 胶 (SephacrylSX00)是 由 亚 甲 基 双 丙
烯酰胺交联的丙烯基葡聚糖,该凝胶质地硬、强度大、较Sephadex耐压,主要
型号有SephacrylS100、S200、S300、S400和S500等,型号愈大,孔径愈大,

39
分子量范围也越大,pH稳定范围在2~11之间。常用凝胶性能的比较见表34。
表34 几种常见凝胶的结构性能比较
凝 胶 种 类 交联葡聚糖 琼脂糖 交联琼脂糖 聚丙烯酰胺 聚苯乙烯
单体 葡聚糖 βD半 乳 糖 和
3,6脱 水L半 乳
糖 交 替 结 合 成 的
多聚糖
βD半 乳 糖 和
3,6脱 水L半 乳
糖交 替 结 合 成 的
多聚糖
丙烯酰胺 苯乙烯
交联剂 环氧氯丙烷 随 温 度 降 低 而
交联
2,3二 溴 丙 醇
或环氧氯丙烷
N,N′亚甲基双
丙烯酰胺
pH稳定工作范围 20~120 45~90 30~130 20~110 20~12物理稳定性 对热较稳定,
可高温灭菌
抗 热 性 较 差,在
0~45℃ 使 用,抗
压性较好
抗 热、抗 压 性
较琼脂糖均高
对 热 较 稳 定,可
高温灭菌
适用于有机
相,稳定性高
分子量范围/Da 102~6×106 103~4×107 103~4×107 102~4×106 103~4×107
吸附性 具离子性 离子强度>002 吸附性下降 离子强度>002
3122 无机填料
(1)多孔硅胶 采用交联葡聚糖、琼脂糖和聚丙烯酰胺凝胶等作为固定相基
质分离纯化蛋白质是凝胶过滤色谱最先发展的技术。然而上述填料均为软胶和半
硬质胶,分离度低,不能长时间耐受高压和高流速,使用寿命比较短,而且在极
端pH、高盐或特殊有机溶剂中的使用受到很大的限制。硅胶基质的刚性亲水性
填料的出现,大大提高了分离纯化生物高分子和有机聚合物的手段。多孔硅胶是
将硅胶制成具有一定直径的多孔网状结构的球状颗粒,它属于硬质无机填料,其
最大优点是物理化学稳定性高,耐热耐压,使用寿命长,分子量分离范围一般在
102~5×106Da。无机填料的柱效一般较低,但用微粒多孔硅胶制成的高效色谱
柱也可达到很高的柱效。例如日本Tosoh公司、美国 Waters公司等开发出的以
完全多孔硅胶和多聚物填料为基质的色谱柱,能够实现大小生物分子和有机聚合
物的快速、高分辨率分离。Tosoh公司的主要产品包括在水相体系中分离时所采
用 的 硅 胶 TSKGelSW 填 料、多 聚 物 TSKGelPW 和 ToyopearlHW 填 料。
TSKGelSW适合分离单 一 分 散 性 生 物 聚 合 物,如 蛋 白 质;而TSKGelPW 则
可用于多元分散性化合物的分离,如多聚糖以及合成聚合物。Waters公司开发
的以10μm二醇键合相的硅胶为填料的ProteinPakTM系列色谱柱可分离分子量
范围相差15%的蛋白质分子,另外还有BiosuiteTM和BiosuiteTMUHR系列。表
35列出一些硅胶填料的商品柱。在实际纯化操作中,可根据蛋白质的不同性质
和纯化目的选择合适填料的成品或预装分离柱。YMCPack商品柱在实际分离纯
化中的应用见图37和图38。

40
表35 部分硅胶填料的商品柱
型 号 规格/mm 球蛋白分离范围/Da 生 产 商
BioSuiteTM125,4μmUHR 46×300 5000~150000BioSuiteTM250,4μmUHR 46×300 10000~500000BioSuiteTM125,5μmHR 78×300 5000~150000BioSuiteTM250,5μmHR 78×300 10000~500000BioSuiteTM450,8μmHR 78×300 20000~7000000BioSuiteTM125,10μm 75×300 5000~100000BioSuiteTM250,13μm 215×300 10000~500000BioSuiteTM450,13μm 75×300 20000~7000000
美国
Waters公司
ProteinPakTM60 78×300 1000~20000ProteinPakTM125 78×300 2000~80000ProteinPakTM300SW 75×300 10000~300000ProteinPakTM200SW 80×300 500~60000
美国
Waters公司
TSKG2000SW,10μm,125nm 75×300 5000~100000TSKG2000SWxl,5μm,125nm 78×300 5000~150000TSKG3000SW,10μm,25nm 75×300 10000~500000TSKG3000SWxl,5μm,25nm 78×300 10000~500000TSKG2000SW,13μm,45nm 75×300 20000~700000TSKG2000SWxl,8μm,45nm 78×300 20000~10000000
日本
Tosoh公司
图37 YMCPackDiol硅胶柱色谱图 (一)样 品:1—glutamatedehydrogenase (谷 氨 酸 脱 氢
酶);2—lactatedehydrogenase (乳 酸 脱 氢 酶);3—enolase(烯醇化酶);4—adenylatekinase(腺
苷酸激酶);5—cytochromeC (细胞色素C)色谱柱:YMCPackDiol,500mm×80mm,孔 径:20nm
洗脱 液:01mol/LKH2PO4K2HPO4 含02mol/LNaCl(pH=70)
流速:07ml/min紫外检测波长:280nm
图38 YMCPackDiol硅胶柱色谱图 (二)样品:1—insulin (牛胰 岛 素);2—neurotensin (神
经降压素);3—angiotensnⅡ (血 管 紧 张 素Ⅱ);4—glycine(甘氨酸)
色谱柱:YMCPackDiol,500mm×80mmid洗脱 液:01mol/LKH2PO4K2HPO4 (pH=70)NaCl/乙腈 (70∶30)
流速:07ml/min紫外检测波长:215nm

41
(2)多孔玻璃 使用多孔玻璃作填料易破碎,在填装时不宜过于紧密,柱效
也相 对 较 低。其 分 子 量 分 离 范 围 一 般 在3~9000kDa之 间。在 分 离 纯 化 蛋 白 质
中,多孔硅胶和多孔玻璃作为无机填料最大的缺点是由于它们表面具有离子性,故对蛋白质有较强的吸附性,而且不能在强碱性环境中使用,一般操作pH要低
于85。可通过表面处理或选择合适的洗脱液来消除和降低其吸附性。
313 凝胶介质的选择
前面已提到凝胶介质要有较强的化学惰性和机械强度,在操作时不易引起降
解或变形,可在较广的pH和温度范围内使用,而且寿命长。除此之外,最理想
的凝胶是与所分离的目的蛋白或流动相不发生任何作用。由于在一些凝胶表面存
在离子交换基团,会不同程度地吸附带电荷蛋白质,产生离子交换的效果,所以
选择凝胶时,表面最好不要有离子交换基团,在必要时,可通过表面活性处理或
调整洗脱液的离子强度来降低吸附性。不同类型的凝胶有各自不同的排阻极限和分子量分离范围,所以在选择凝胶
时要根据所纯化的目的蛋白的分子量大小和性质来确定凝胶的种类。按照组别分
离和分级分离的不同要求,在进行组别分离时要选择能将大分子完全排阻而小分
子完全渗透的凝胶,常选用排阻极限较小的凝胶类型,以获得理想的分离效果。例如,在蛋白质脱盐时,可选用排阻极限较小的SephadexG25。分级分离时一
方面要保证样品的分子量分布在所选凝胶的分级范围之内,如果分离范围过小,一些组分不能得到分离;另一方面,凝胶的分级范围不能过大,否则会降低分辨
率,影响分离效果。在分离纯化已知分子量的蛋白质时,选用分级范围较窄的凝胶可获得较好的
分离效果,而且应尽量使所选凝胶的分级范围的中 间 值 接 近 于 目 的 蛋 白 的 分 子
量。以Sephadex为例,SephadexG50可 用 于 分 离 分 子 量 在1500~30000Da的
球蛋白,而SephadexG75的分离分子量范围在3000~70000Da,所以如果要分
离分子量 为10000Da和20000Da的 蛋 白 质,两 种 均 可 使 用,但 由 于SephadexG50的分离范围较窄,其分辨率也较高。如分离一个分子量为3000Da的小分子
蛋白,就选用SephadexG25。已 知 小 分 子 蛋 白 质 一 般 可 选 用SephadexG≤50或SephacrylS100,大分子蛋白的 分 离 纯 化 可 选 用SephadexG≥100或 琼 脂 糖
凝胶。此外,凝胶颗粒的大小直接关系到蛋白质样品的分离效果。一般来说,颗粒
直径越小,分离效果越好,但流速缓慢,所需分离时间长;较大直径的凝胶颗粒
虽然可提高流速,但会使区带发生扩散,易产生平而宽的洗脱峰,所以要按具体
情况来选择凝胶类型和型号。如果样品中各个组分的分子量差别较大,则可以选
用大粒径的凝胶,实现快速分离的目的;如果组分分子量差别较小,则要使用小

42
粒径的凝胶以提高分辨率。在一些特殊条件下进行分离 (如强酸、强碱或有机溶
剂等)时,则要慎重选择适宜类型的凝胶。
32 凝胶柱的操作
321 装柱
3211 色谱柱的选择
在选择色谱柱时,从外观、质 地 结 构 和 色 谱 性 能 来 讲,所 选 柱 子 要 透 明 光
滑,内径一致,耐压防腐,柱子物料须生物兼容,并有良好的物理化学抗性和稳
定性。一般的柱材料多为玻璃或有机玻璃,在使用钢柱时,特别要注意防腐蚀。另外,柱子终端要有采集器和过滤网,柱底板要求耐压且不容易堵塞。死体积应
小于0001Vt,如果死体积过大,被分离组分可能会重新混合,导致洗脱峰出现
拖尾现象,降低分辨率。底板过滤介质要求新,且带有能与凝胶融合的网孔,避
免接触网孔和过滤介质的表面,因为指纹会使样品的分离程度下降。在装柱或重
新装柱时,所有部位必须彻底清洗干净。色谱柱的长度和内径主要取决于加样体积的多少和对分辨率的要求。色谱柱
的长度对分辨率的影响较大,长柱的分辨率要比短柱的高,但过长会引起柱内不
均一、流速过慢,柱长度一般不超过100cm。同时,也要有适当的柱内径,内径
过粗会产生蛋白样品较为严重的横向扩散现象,内径过细,靠近柱内壁的流速会
大于中心的流速,产生 “器壁效应”,影响分离速度和效果。通常使用的凝胶柱长度在25~70cm,内径在4~16mm,H/D (高径比)一
般在 (25∶1)~(100∶1)之间。用于组别分离的凝胶柱,其 H/D 可相对低一
些;进行分级分离时H/D可在 (30∶1)~(100∶1);而脱盐柱由于对分辨率的
要求较低,一般用短柱,H/D大多在 (5∶1)~(25∶1)之间。如果要分离的蛋
白质分子量相差较大,可选用H/D=(15∶1)~(50∶1)的柱子,且柱体积要大
于4~15倍的 样 品 体 积;如 要 分 离 的 蛋 白 质 分 子 量 差 异 较 小,则 选 用 H/D=(20∶1)~(100∶1)的细柱,且柱体积要大于25~100倍的样品体积。在纯化蛋
白质时,为了得到较好的分辨率,柱子的H/D应在 (20∶1)~(40∶1)。此外,选择柱子时还应考虑凝胶颗粒的大小。如填装小颗粒凝胶,适合使用
较大直径的色谱柱;用粗颗粒填装时,则用较小直径的柱子。
3212 凝胶的预处理
市售凝 胶 一 般 呈 干 粉 颗 粒 状 (如Sephadex)或 水 悬 浮 状 (如 SepharoseCL)。一些不宜在脱水干燥状态下保存的凝胶 (如琼脂糖凝胶),应存放在含防
腐剂的水溶液中,这些凝胶在使用前不需要进行溶胀处理。另外,多孔硅胶和多
孔玻璃也不需要溶胀处理。为了尽量减少不同溶剂对柱床体积的影响,获得理想

43
的分离效果,应将干胶缓慢倒入5~10倍干胶体积的洗脱液中充分溶胀。若溶胀
不够,则达不到有效的分离,而且在使用过程中会引起凝胶柱破裂。根据蛋白质样品的分子量和分辨率要求选择好所用凝胶的类型后,再根据柱
体积和凝胶的吸水性质估算出干胶的用量。计算干胶用量的公式见式 (37)。由
于凝胶在预处理和实验操作过程中有一定量的损失,所以在实际称取时应高于理
论计算值的10%~20%。不同型号的凝胶在不同规格凝胶柱中的用量见表36。
干胶用量(g)=柱床体积(ml)
凝胶的床体积(ml/g)(37)
表36 不同型号的凝胶在不同规格凝胶柱中的用量
内径/mm凝胶柱规格
高度/cm容量/ml
凝胶的规格和用量/g
G25 G50 G100 G200
9 15 95 25 1 06 039 30 19 5 2 12 069 60 38 10 4 25 1216 20 40 10 4 25 1216 40 80 20 8 50 2416 70 140 35 14 90 4416 100 200 50 20 125 626 40 210 50 20 12 726 70 370 90 35 20 1226 100 530 130 50 30 17
不同类型的凝胶所需 的 溶 胀 时 间 不 同。一 般 型 号 较 小、排 阻 极 限 较 低 的 凝
胶,其吸水量较低,溶胀所需时间较短,在20℃条件下需3~4h即可;型号较
大、排阻极限较高的凝胶,其吸 水 量 较 大,故 所 需 的 溶 胀 时 间 也 较 长,在20℃左右需十几小时到几十小时,例如Sephadex型号在G100以上的干胶所需的溶
胀时间在72h以上。加热溶胀是常用的凝胶预处理方法,即把所称量的凝胶干粉溶在洗脱液中逐
渐加热至接近沸腾。这种方法可大大缩短溶胀时间,一般在1~2h内即可完成,而且可起到溶胀、除气和杀菌的三重效果。在凝胶溶胀过程中要不断缓慢搅拌,但不能剧烈搅拌,因为这样容易引起凝胶颗粒的破碎,最终使细小颗粒堵塞凝胶
柱而影响到流速和分离效果。此外,还应尽量避免在酸或碱中进行加热溶胀,以
防破坏凝胶的网孔结构。待充分溶胀后,应将凝胶匀浆悬浮,看上清中是否有杂质或不均一的细小颗
粒,如有则需反复倾倒去除,直至上清液不再浑浊为止。对溶胀后的凝胶进行除
气泡是非常重要的,否则也会影响分离效果,一般可通过真空抽气或加热煮沸的

44
方法排除气泡。由于凝胶价格昂贵,在使用过程中要尽量减少损失和浪费,剩余
凝胶要保存好,以备后用。
3213 具体装柱步骤
凝胶柱装填方法主要有电动法 (见图39)和手动法,实验室一般采用手动法。
图39 电动搅拌装柱示意
下面介绍手动法装柱的具体过程。(1)制备凝胶匀浆 将溶胀好的一定体积
的凝胶制成含75%凝胶和25%洗脱液的匀浆。(2)安装色 谱 柱 取 所 选 的 干 净 玻 璃 柱
(或有机玻璃)垂直固定在铁架台上,保证装
置周围 无 阳 光 照 射 和 空 气 流 动,以 防 造 成 影
响,若有条件,可在专门的装柱柜中进行操作。(3)测定Vt 在距柱上端约5cm处作一
标记,关 闭 柱 出 口,加 入 高 于 标 记 位 置 约
2~3cm的去离子水;打开出口,当液面降至
标记处时 立 即 关 闭 出 水 口;打 开 出 口 并 用 量
筒收集流 出 的 去 离 子 水,待 水 面 降 至 柱 底 筛
板时 关 闭 出 口,所 得 去 离 子 水 的 体 积 即 为
Vt。该方法 所 得 值 不 精 确,另 外 可 通 过 测 定
最终所装柱床的高度来计算Vt。(4)灌胶 平 衡 关 闭 出 水 口,在 柱 中 加
入约1/3柱床体积的洗脱液,边轻轻搅拌边将凝胶匀浆通过内壁一侧或玻璃棒引
流倾入柱中,待凝胶沉积约1~2cm后,打开柱出口让水流出,流速一般为3~6ml/10min;同时不断缓慢加入凝胶悬液,待沉积胶面上升至标记处时,灌胶完
毕;关闭出水口,静置片刻,等凝胶完全沉降后,接上平衡液储液器和蠕动泵,检查密闭性,用3~5倍柱床体积的平衡液平衡柱子,使柱床稳定。
注意事项:灌胶过程尽量一次性完成,否则柱床不均一,进行二次灌胶时须
将表面沉积凝胶层搅拌起,以防柱床出现分层。装柱过程中不能产生气泡,若在
早期产生可通过轻微搅拌去除。在任何时候柱床内都要充满缓冲液,否则不能进
行正常分离。对长期使用的凝胶柱建议使用流动相转换接头 (flowadaptor),这
样不但可以保护柱床、节省凝胶,还可防止洗脱液中的大颗粒物进入凝胶床而发
生堵塞,从而延长柱子的使用寿命。必要时要重新装柱,再次填装前要清洗干净
柱。一般的凝胶过滤色谱装置见图310。(5)其他装柱方法 在填装SephacrylSHR凝胶柱时,要采用两步法按所
推荐流速进行装柱。首先在所推荐的流速下将凝胶在2h内或者到一定的柱床高

45
度时填装完,然后将流速升高到所推荐 的 值,并 在60min内 完 成。装 柱 时 的 流
速见表37。
图310 凝胶过滤色谱装置示意
表37 用SephacrylSHR凝胶填装不同型号空柱时的流速/(ml/min)
柱 子 型 号 第 一 步 第 二 步
XK16/70 10 18XK16/100 10 17XK26/70 25 50XK26/100 25 45XK50/100 83 133
(6)检测柱床及柱效 装填平衡后的凝胶柱用肉眼观察应均匀,无纹路,无
气泡。另外,可通过加样洗脱易于观测的有色物质,如蓝色葡聚糖2000、血红
蛋白和细胞色素等 (浓度为2g/L),来观察色带在柱中的移动变化,检测柱床的
均匀程度和装柱质量。如果在洗脱过程中色带下降均匀平整,则表明凝胶填装理
想;如色带歪曲弥散,则需要重新装柱。如果柱子是用来测定蛋白质分子量的,则检测标定物质应选用与样品有相似性质和形状的已知蛋白,如果没有合适的标
定物质,则可使用葡聚糖、支链淀粉、乙二醇或其他一些性质类似的聚合物来代
替。装柱质量还可通过低分子量的彼此无 作 用 的 溶 质 在 高 流 速 下 来 检 测,如 丙
酮、硝酸钠、葡萄 糖 等。一 般 用<05% (30μm 介 质)或<1% (90μm 介 质)

46
柱体积的1%的丙酮检测柱效和峰形 (见图311)。
图311 一般柱效检测图
理论塔板数:
N=16tR( )W2=554 tR
Wh/( )22
峰对称因子:
As=ba式中,N 表示理论塔板数;tR 为 进 样 点 到 色 谱 峰 极 大 点 的 距 离,mm;W,
Wh/2分别表示峰底宽和半高峰宽,mm;a,b分 别 为 峰 高 的10%时 所 对 应 的 左
右半峰宽,mm (具体见图311中标示)。另外,还可参照生产商提供的检测方法。(7)测定Vo和Vi Vo (或Vi)的测定一般选用完全排阻的大分子物质 (或
完全渗透的小分子物质)来测定,其Ve即为凝胶柱的Vo (或Vi)。Vo的标定物
通常为蓝色葡聚糖2000,它具有 很 大 的 分 子 量 (2000000),为 完 全 排 阻;重 铬
酸钾可作Vi的标定物,为完全渗透。
322 样品和缓冲液的准备
3221 蛋白质样品制备
蛋白质样品的制备是将蛋白质粗品在一定体积的洗脱缓冲液中溶解成一定浓
度的进样液。所制得样品的溶解性和稳定性要好,保证在洗脱过程中不发生变性
和沉淀。对于分析性的蛋白质样品,在进样之前一定要确定其浓度,而且浓度尽
可能要低一些。对于一些特殊实验,如利用凝胶过滤色谱进行蛋白质复性研究,

47
其浓度可提高 (有些可达80mg/ml),制备性的样品浓度也可稍高。此外,所得
样品的黏度不能过大,否则会使色谱区带和流动相不稳定,产生歪曲洗脱峰,样
品黏度与洗脱液黏度之比应小于1∶5。如果样品黏度很高,则可以通过增加洗
脱液的黏度来弥补,比如添加蔗糖或葡聚糖,但这需要低流速以维持窄的峰宽和
较低的压力。在进样之前还可采用045μm滤膜过滤样品,以防止柱入口处被堵
塞而影响分离效果和柱子的使用寿命。
3222 缓冲液准备
因为凝胶色谱的分离依据主要是依靠样品分子量的差异,而不依赖于改变流
动相的组成,所以对洗脱液的要求低于其他几种色谱法。用单一缓冲液或含盐缓
冲液作为洗脱液即可,而对于一些确定无吸附作用的蛋白质样品甚至可单独用去
离子水进行洗脱。准备缓冲液时主要考虑两方面因素:样品的溶解性、稳定性;样品与凝胶介
质可能会发生的吸附。所使用的缓冲液一定要保证蛋白质样品在其中不能变性或
沉淀,pH值范围应选在样品较稳定、溶解性良好的范围之内,为了防止凝胶可
能对带电荷蛋白质的吸附作用,在缓冲液中要含有一定离子强度的盐 (一般使用
NaCl),对蛋白质起稳定和保护作用。普遍使用的柠檬酸、磷酸、硼酸缓冲液的pH范围在2~12,这些缓冲液即
使在离子强度影响 下 变 化 也 很 小。缓 冲 液 中 离 子 的 解 离 度pKa 应 当 与 所 需pH接近,以起到最佳的缓冲作用。如果缓冲液 缓 冲 能 力 不 够 或 缓 冲 液 与 样 品 不 相
溶,则应考虑使用生物缓冲液。一般磷酸缓冲液应用较多,适用于中性环境,在
进行蛋白质制备或脱盐操作时,可考虑使用醋酸铵或碳酸氢铵缓冲液,因为这些
离子在冷冻干燥过程中会被除去。缓冲液在进柱之前,也需要经过045μm滤膜过滤,防止杂质进入柱内。滤
膜的选择要根据样品分子量的大小和流动相的组成,使用较多的为045μm孔径
的,分有机相和水相滤膜两种。用水溶液洗脱时用水相滤膜过滤,用有机溶剂作
流动相时,用有机滤膜过滤。
323 色谱条件
由于在洗脱过程中,流动相的组成不发生变化,所以凝胶过滤色谱中所要确
定的色谱条件较离子交换色谱的简单。色谱条件主要包括柱尺寸、凝胶类型、缓
冲液类型、提供 离 子 强 度 的 盐 类 及 浓 度、洗 脱 液 的pH、流 速 和 紫 外 检 测 波 长
等。在分离结果不理想时,可通过检测柱效、清洗或重装柱子、减少样品体积、降低样品浓度、控制流速、调整或更换缓冲液及盐类型、改变pH或改用其他种
类的凝胶等措施来改善色谱条件。在一些特殊实验中还要保持一定的温度,此时
可将凝胶柱放置在恒温箱中进行操作。

48
324 加样和洗脱
3241 加样体积及方法
(1)加样体积 关于加样体积对分离的影响在前面已介绍。加样体积的多少
主要与柱床体积和所要达到的分离度有关。在高精确度的分析性凝胶色谱中,如
果所使用的检测方法灵敏度高或柱床体积小,加样体积则要尽可能小,必要时先
进行预备试验,尝试不同加样体积对分离效果的影响,从而找到合适的值。对于
含较少组分的蛋白质样品来说,加样体积一般不能超过两组分洗脱体积之差。几
个刚能分开的蛋白质组分,加样 体 积 越 少,其 分 辨 率 越 高,必 要 时 还 需 减 少 体
积;而对于几个分得很开的蛋白质组分来说,为提高实验效率,其加样体积可适
当增大。一般分级分离时加样体积约为柱床体积的1%~5%,当加样体积高于
5%时会影响分离效果,而低于1%时,并不能提高分辨效率。在进行组别分离时
由于样品分子量差异较大,所以加样体积可增大,约为柱床体积的10%~25%。此外,制备性的凝胶柱一般柱床体积较大,所以加样体积也较大。
(2)加样方法 实验室常采用的加样方法有自动加样法、加样器法和手动法
三种。加样时应特别注意不能干扰凝胶床表面,而且柱内的缓冲液在任何时候都
不能低于凝胶床表面,否则会影响分离效果或需要重新装柱。这里主要介绍手动加样法,其基本要求是快速、均匀,对床面无影响,具体
方法如下:填装好的凝胶柱经平衡后,用胶头滴管吸去床上层部分多余洗脱液,待洗脱液下降至近床表面2~3mm时,关闭柱出口 (注意不能使洗 脱 液 全 部 流
干,若流干,需要重新装柱);用胶 头 滴 管 或 加 样 器 吸 取 一 定 体 积 的 样 品 溶 液,贴柱内壁旋转缓缓加入,以防加样过快导致凝胶床表面受损;加样完毕后,打开
柱出口,让样品液缓慢渗入凝胶床内;当样品液面恰与凝胶床表面持平时 (未排
干),按前面同样方法小心加入几毫升洗脱液冲洗内壁,并使洗脱液高出凝胶床
表面2~3cm,待恒流洗脱。
3242 洗脱
加样完毕后,检查柱及管路的密封性,将凝胶柱与洗脱液储瓶、蠕动泵、紫
外检测器、分部收集器及记录仪等连接,开始洗脱。根据预先设定的流速,按每
管2~5ml定量收集洗脱液,并排好顺序,待分析活性。通过紫外检测仪和记录
仪观察,当洗脱曲线的基线不再上升时说明样品已完全洗出,继续洗脱平衡一段
时间 (时间根据柱床体积定)后,用同样步骤进行下一轮实验。由于样品与凝胶
介质不发生作用,所以中间平衡所需时间较短。前面已提到凝胶色谱对洗脱的要求比较低,所以一般采用简单洗脱法,即在
洗脱过程中使用组成和pH不发生变化的流动相,这也是凝胶过滤色谱得以广泛
应用的一个重要原因。始终用同样组成和性质的缓冲液作洗脱液是凝胶色谱技术

49
对流动相的基本要求。洗脱中流速的控制通常采用蠕动泵,也可用控制操作压力
的方法来实现。
325 凝胶柱的再生及保存
3251 清洗和再生
凝胶柱在多次反复使用后,由于床体积变小、流速降低、杂质或微生物污染
等原因,会影响分离效果,此时必须进行柱的清洗和再生处理。一般用洗脱液继
续平衡即可达到清洗目的。被脂肪污染的凝胶柱再生处理可用02mol/LNaOH(除Sepharose)或非离子表面活性去垢剂。一些结合在凝胶上的很难去除的蛋白
质,可通过加入2%的蛋白酶液,进行处理清洗。
3252 保存
凝胶的保存一般有湿法和干法两种。湿法保存是将清洗干净的凝胶悬浮在002%的叠氮化钠或0002%洗 必 泰
中,于4℃下保存;或者先用水冲洗干净,然后用60%~70%的乙醇溶液冲洗,可起到抑菌作用。其他的一些防腐抑菌物质有可乐酮 (001%~002%)、乙基
汞代巯基水杨酸钠 (005%~001%)、苯基汞代盐 (0001%~001%)。若要长时间保存,则需要将凝胶清洗后脱水干燥,再将干胶浸泡在50%的
乙醇中脱水,抽干后再逐步提高乙醇浓度反复浸泡脱水,以95%乙醇脱水抽干
后再经乙醚脱水,置60℃烘箱中烘干,装瓶保存。应该注意的是溶胀后的凝胶
不能直接高温烘干,以避免破坏凝胶的结构。
33 应用
凝胶过滤色谱在蛋白质研究中的应用主要有:(1)脱盐 缓冲液交换,大小分子分离;此时的蛋白质和杂质的分离大小差
异大,故对系统和操作参数要求低。(2)蛋白质分级分离 各组分分子大小差异小,对系统要求高,分离相对困难。(3)分子量测定 应保证蛋白质结构的完整性,对球形蛋白质分子量的测定
比较准确,对分离条件要求高。(4)复性研究 依据蛋白质在复性过程中空间结构的变化,不同时间在固定
相和流动相之间的分配不同。
331 脱盐
从20世纪60年代开始,凝胶过滤色谱技术就被用于蛋白质的脱盐。利用凝
胶过滤色谱进行蛋白质的脱盐或去除小分子杂质是一种快速高效的方法。因为蛋
白质与盐类的分子量差异较大,所以对凝胶和色谱系统的要求不高。蛋白质的分

50
子量比盐类的分子量要大得多,所以蛋白质在外水体积中被洗脱,即使在高流速
下进行脱盐也不会影响目的蛋白的分离效率。脱盐所用的洗脱液和添加剂必须是
挥发性的,否则脱盐操作应在去离子水中进行。凝胶可选择性地将蛋白质排阻而
达到脱盐的目的,一般不会造成蛋白质样品较大倍数的稀释或发生变性。在脱盐
的操作中,在除杂的同时也可达到更换蛋白质缓冲液的目的。
332 蛋白质分离
在蛋白质的分离中,凝胶过滤色谱是一种重要的分离手段,因为它是依据样
品分子量的不同来进行分离的,凝胶介质和蛋白质之间不发生任何作用,所以不
改变样品的生物活性,而且含盐的水系缓冲液对蛋白质还起到一定的保护作用。一般而言,凝胶过滤色谱通常用在离子交换色谱或亲和色谱之后,以进一步纯化
目的蛋白,但有时,在存在同工酶,且含量低、分子量很大或很小时,可以在纯
化的初期先采用凝胶过滤色谱,以获得所需酶蛋白,然后进一步纯化此酶蛋白,并分离纯化其他目的蛋白。
使用凝胶过滤色谱分离蛋白质最理想的情况是全排阻蛋白质而允许杂质渗入
凝胶内部,或反之。通常情况下蛋白质分子量相差两个数量级,需要使用适当的
凝胶才能有效分离。用ProteinPakTM300SW色谱柱分离几种蛋白质的色谱图见图312。
图312 ProteinPakTM300SW色谱柱分离蛋白应用实例
样品:50μl蛋白质标样,5mg/ml色谱柱:ProteinPakTM300SW,75mm×300mm流动相:01mol/LK2HPO4,pH=70流速:10ml/min检测:280nm
1—蓝色葡聚糖;2—铁蛋白;3—牛血清蛋白;4—卵清蛋白;
5—胰蛋白酶抑制剂;6—核糖核酸酶;7—Ruonosine

51
333 测定蛋白质分子量
对于同一型号的凝胶,在一定的分子量范围内,各个组分的Kd与其分子量
的对数成线性关系,见式 (38):
Kd=-blgMw+c (38)式中 b,c———常数;
Mw———蛋白质的分子量;
Kd———分配系数。另外,由于Ve和Kd也成线性关系,所以有式 (39):
Ve=-b′lgMw+c′ (39)式中 b′,c′———常数。
通过将一些已知分子量的蛋白质在同一凝胶柱上以相同条件进行洗脱,测定
它们的Kd (或Ve),根据上述关系式绘出标准曲线,得出回归方程,然后在相
同的洗脱条件下测定目的蛋白的Kd (或Ve),通过回归方程即可求出目的蛋白
的分子量。采用该方法测定蛋白质分子量的准确性受许多因素的影响,如蛋白质与凝胶
介质的吸附、分子形状等。在理想的凝胶过滤色谱中,蛋白质的形状为球状,并
且与凝胶不发生任何作用,所以用来测定分子量的蛋白质样品和分子量标准应该
都是球状蛋白,否则所得值会有较大的误差。对于一些非球状的分子,可以通过
预变性处理 (如用盐酸胍或尿素)使其结构发生卷曲,以减少误差。表38给出
了一些常用蛋白质分子量标准。
表38 常用蛋白质分子量标准
高分子量标准 中分子量标准 低分子量标准
蛋 白 质 分子量/Da 蛋 白 质 分子量/Da 蛋 白 质 分子量/Da
肌球蛋白 212000 磷酸化酶B 97400 碳酸酐酶 31000
β半乳糖苷酶 116000 牛血清蛋白 66200 大豆胰蛋白酶抑制剂 21500磷酸化酶B 97400 谷氨酸脱氢酶 55000 马心肌球蛋白 16900牛血清蛋白 66200 卵清蛋白 42700 溶菌酶 14400过氧化氢酶 57000 醛缩酶 40000 肌球蛋白(F1) 8100醛缩酶 40000 碳酸酐酶 31000 肌球蛋白(F2) 6200
大豆胰蛋白酶抑制剂 21500 肌球蛋白(F3) 2500溶菌酶 14400
334 蛋白质复性的研究
凝胶过滤色谱在蛋白质复性方面的应用主要基于蛋白质在凝胶柱内的Vi和
Vo间形成不同的分配区域。Geng等用凝胶过滤色谱首次对溶菌酶、核糖核酸酶

52
(RNase)和牛血清白蛋白 (BSA)进 行 了 复 性 研 究。不 同 形 态 的 变 性 蛋 白 质 因
分子量差异或与凝胶介质作用力的强弱不同在固定相和流 动 相 之 间 进 行 动 态 分
配。变性蛋白质由于所占体积较大,随流动相 (或称复性液)从凝胶颗粒间的空
隙中下移;而部分折叠或完全折叠复性的蛋白质则进入部分凝胶内孔,移动速度
减慢;变性剂由于分子量小,可进入大部分凝胶内部,下移速度最慢。蛋白质在凝胶柱内的复性机理可初步解释为:变性蛋白在刚加入凝胶柱顶端
时,由于有高浓度的变性剂存在,变 性 蛋 白 呈 无 规 则 卷 曲 状 结 构,所 占 体 积 较
大,而且有较大的动力学水合半径 (又称Stokes半径),所以不能进入凝胶颗粒
内部,只能进入颗粒间隙,迁移速度快;而变性剂由于分子量小,可进入颗粒内
部的小孔,因而迁移速度慢。当用缓冲溶液洗脱变性蛋白时,在流过凝胶柱的过
程中,变性剂的浓度不断降低,最后和流动相平衡,在从变性剂溶液转换成复性
缓冲液时,起到阻止或减小变性蛋白间的相互聚集的作用。变性蛋白开始复性,
Stokes半径不断减小,结构趋于正常,部分复性或 折 叠 的 蛋 白 质 可 进 入 凝 胶 颗
粒的内部,当蛋白完全复性时,最终以天然形态被洗脱出柱。与常用的稀释复性法相比,凝胶色谱复性具有许多优点,例如:能在高浓度
下对蛋白进行复性 (起始浓度可高达80mg/ml);复性效率高,能够抑制复性过
程中聚集体的产生;在复性的同时进行分离等。到目前为止,已有多种蛋白质通
过凝胶过滤色谱获得成功复性,例如重组人尿剂酶纤溶酶原激活因子、重组羊生
长激素和重组人白介素26等。
335 其他
凝胶过滤色谱的应用还包括以下几方面:(1)浓缩蛋白质溶液 利用凝胶颗粒的吸水性可对含量较低的高分子蛋白质
溶液进行浓缩。例如将粗粒径的Sephadex干胶加入溶液中,因其较大的吸水性,小分子物质会渗入凝胶内部,而大分子物质被排阻在颗粒外,起到浓缩样品的效
果。凝胶颗粒可通过离心或过滤去除。这种浓缩方法基本不改变溶液的离子强度
和pH,但由于凝胶的价格昂贵,该方法不常用。(2)去热原物质 利用凝胶色谱的排阻效应可将小分子和大分子的热原物质
(一些微生物产生的含热较多的糖蛋白复合物)分开,例如去除注射液中的热原
物质。(3)更换蛋白质缓冲溶液 在盐析提取或在离 子 交 换 柱 上 高 盐 洗 脱 的 蛋 白
质,可经过凝胶过滤色谱同时达到更换不同离子强度缓冲溶液和除盐的目的。(4)分析蛋白质结构 对于已部分纯化的蛋白质,通过凝胶过滤色谱可分析
其二聚体或三聚体结构。

53
4 离子交换色谱
离子交换色谱 (ionexchangechromatography,IEC)是 发 展 最 早 的 色 谱 技
术之一。古代人们使用沙石来净化饮用水就是利用了离子交换的原理,而第一篇
关于离子交换的研究报告来自1850年英国农业化学家Thompson对土壤中钙离
子和铵离子交换及其当量关系的研究。20世纪初,离子交换开始用于水的软化
和糖类的处理。离子交换剂是离子交换技术的核心和基础,20世纪30年代人工
合成离子交换树脂的出现对于离子交换技术的发展具有重要意义,当时先由Adams和 Holmes合成了苯酚磺酸树脂和聚胺型树脂。美国的DAlelio在此基础上
用苯乙烯和二乙烯苯合成了第一个单功能基团的强酸型树脂,而基于苯乙烯二
乙烯苯的离子交换树脂至今仍是最广泛使用的一类离子交换树脂。离子交换在蛋
白质化学中的首次运用是在果胶聚半乳糖醛酸酶的制备过程中,作为杂质的果胶
甲酯酶被吸附于聚苯乙烯阳离子交换剂 (AmberliteIR100,磺酸型)而得以除
去。数年后异丁烯酸树脂 (AmberliteIRC50)被成功地用于分离碱性蛋白质如
细胞色素C、核 糖 核 酸 酶、溶 菌 酶 等,这 是 由 于 异 丁 烯 酸 树 脂 较 高 的pKa 值
(pKa=65)使它适合于碱性蛋白质的分离。尽管离子交换树脂后来得到了非常广泛的应用,但研究表明该离子交换剂并
不十分适合对生物大分子如蛋白质、核酸、多糖等的分离,这是因为:①离子交
换树脂交联度太大而颗粒内网孔较小,蛋白质分子无法进入颗粒内部,只能吸附
在树脂颗粒的表面,造成有效交换容量很小;②树脂表面电荷密度过大,使蛋白
质在其上吸附得过于牢固,必须使用比较极端的条件才能将其洗脱下来,而这样
的条件往往容易造成蛋白质变性;③树脂的骨架是疏水性,一旦与蛋白质之间发
生疏水相互作用,容易造成蛋白质变性失活。
20世纪50年代中期,Sober和Peterson合成 了 羧 甲 基 (CM)纤 维 素 和 二
乙氨乙基 (DEAE)纤维素,这是两种亲水性和大孔型离子交换剂,其亲水性减
少了离子交换剂与蛋白质之间静电作用以外的作用力,大孔型结构使蛋白质能进
入网孔内部从而大大提高了有效交换容量,而纤维素上较少的离子基团有利于蛋
白质的洗脱,因此这两种离子交换剂得到了非常广泛的应用。此后,多种色谱介
质特别是颗粒型介质被开发和合成,包括交联葡聚糖、交联琼脂糖、聚丙烯酰胺
以及一些人工合成的亲水性聚合物等,以这些介质为骨架结合上带电基团衍生而
成的离子交换剂也层出不穷,并且大大推动了离子交换技术在生化分离中的发展

54
和应用。目前,离子交换色谱已成为蛋白质分离纯化中最常用的手段,统计显示,在
蛋白质的纯化方案中,使用到离子交换色谱的占75%,其次是使用亲和色谱和
凝胶过滤色谱,分别占60%和50%。事实上,蛋白质的纯化过程往往是将若干
纯化技术联合使用而实现的,在此过程中,选择合理的纯化技术固然很重要,然
而如何将这些技术合理的组合和按顺序使用也是进行成功分离所必须考虑的。一
般而言,在分离纯化的初始阶段、中 间 阶 段 和 精 制 阶 段,所 需 解 决 的 侧 重 点 不
同,人们会据此选择不同的纯化技术,图41显示了在纯化的不同阶段,各种常
用的纯化技术被采用的频率分布。
图41 常用纯化技术在纯化的不同阶段被采用的情况
离子交换色谱之所以得到如此广泛的应用,是因为其具有 以 下 特 点:①分
辨率高,随着各种高效色谱介质的出现,选择合适的离子交换剂能够确 保 离 子
交换色谱有着良好的选择性和分辨率;②蛋白交换容量高,有利于放大 分 离 规
模和在工业生产中应用,而 这 一 点 是 凝 胶 过 滤 等 方 法 很 难 达 到 的;③应 用 灵
活,通过选择不同的离子交 换 剂,控 制 缓 冲 液 的 组 成 和pH、离 子 强 度 条 件 可
以优化分离过程;④分离 原 理 比 较 明 确,该 技 术 是 依 据 电 荷 不 同 进 行 分 离 的,不过对于蛋白质这样的大分子,除了静电作用外,疏水相互作用、氢键 等 非 离
子作用以及缓冲离子的性质也会影响到分离行为;⑤操作简单易行,在 大 规 模
分离样品而分辨率要求又并不高时,对蛋白质进行吸附和解吸甚至可以 不 用 在
色谱柱中进行。
41 基本概念
用离子交换色谱分离生物分子的基础是待分离物质在特定条件下与离子交换

55
剂带相反电荷因而能够与之竞争结合,而不同的分子在此条件下带电荷的种类、数量及电荷的分布不同,表现出与离子交换剂在结合强度上的差异,在离子交换
色谱时按结合力由弱到强的顺序被洗脱下来而得以分离。离子交换色谱的原理和
一般步骤如图42所示。
图42 离子交换色谱原理
1—上样阶段,此时离子交换剂与平衡离子结合;2—吸附阶段,混合样品中的分子与离子
交换剂结合;3—开始解吸阶段,杂质分子与离子交换剂之间结合较弱而先被洗脱,目的分
子仍处于吸附状态;4—完全解吸阶段,目的分子被洗脱;5—再生阶段,用起始缓冲液
重新平衡色谱柱,以备下次使用
411 蛋白质的电性质
4111 蛋白质的带电基团
蛋白质之所以能够在离子交换剂上发生吸附是由于其表面带有电荷。蛋白质
分子中的带电基团来源有两种:一种来自于特定的氨基酸;另一种是蛋白质在修
饰过程中引入的。蛋白质由氨基酸组成。组成蛋白质时,氨基酸的α氨基和α羧
基形成肽键而不再发生解离。但很多氨基酸的侧链带有可解离基团,其中有的能
进行酸性解离而带上负电荷,如天冬氨酸 和 谷 氨 酸 的 侧 链 羧 基、酪 氨 酸 的 酚 羟
基、半胱氨酸的巯基;有的能进行 碱 性 解 离 而 带 上 正 电 荷,如 赖 氨 酸 的 侧 链 氨
基、精氨酸的胍基、组氨酸的咪唑基。此外,在肽链的 N末端还有一个游离氨
基,C末端还有一个游离羧基,两者都能发生解离反应。这些基团的pK′值与游
离氨基酸中的pK′值是不完全相同的,一般来说,它们比游离氨基酸中的pK′值
向靠近中性的方向偏移 (表41)。此外,侧链可解离基团在蛋白质三级结构中
的位置在很大程度上也会影响到pK′值。如果是结合蛋白质,则辅基中可能也含
有可解离基团。

56
表41 蛋白质分子中可解离基团的pK′值
可 解 离 基 团 pK′(25℃)
α羧基 幑幐COOH —COO- 3.0~3.2
β羧基(Asp) 幑幐COOH —COO- 3.0~4.7
γ羧基(Glu) 幑幐COOH —COO- 4.4
咪唑基(His) 5.6~7.0
α氨基 N+H 幑幐3 —NH2 7.6~8.4
ε氨基(Lys) N+H 幑幐3 —NH2 9.4~10.6
巯基(Cys) 幑幐SH —S- 9.1~10.8
苯酚基(Tyr師師師師
師師) 幑幐師師師師
師師OH O - 9.8~10.4
胍基(Arg) CNH2
NH+ 幑幐
2
CNH2
NH
11.6~12.6
许多蛋白质在翻译后修饰过程中也会引入可解离基团,这些基团多数是酸性
基团,如:磷蛋白中的磷酸基,凝血因子 中 的γ羧 基,糖 蛋 白 寡 糖 链 上 的 唾 液
酸残基等。相对来说引入碱性基团的机会较少,但有的修饰作用,如组蛋白中赖
氨酸和组氨酸的甲基化作用会增加这些氨基酸侧链的碱性。翻译后修饰也可能消
除原先解离基团的酸碱性质,如:N末端的α氨基发生乙酰化或琥珀酰化修饰,
N末端的谷氨酸环化生成焦谷氨酸,C末端的α羧基发生酰胺化或酯化等。值得
注意的是,有时同一种蛋白质的翻译后修饰作用进行的程度不同,形成一个系列
的不均一的异构体,它们在离子交换色谱中表现出不均一性。例如:胰核糖核酸
酶就存在着糖基化程度不同的 A、B、C、D四种异构体,离子交换法可以将其
分开。由于蛋白质中带有众多可解离基团,在离子交换所使用的pH范围内,蛋白
质既带正电荷又带负电荷,不同蛋白质正是因为电性质不同而得以分离。
4112 pH参数和蛋白质的滴定曲线
蛋白质分子所带电荷的种类和数量并非常数,而是与溶液的pH直接相关。当溶液pH低时,蛋白质结合更多的氢离子,分子内负电荷减少而正电荷增多;而当溶液pH高时,蛋白质解离出更多的氢离子,分子内正电荷减少而负电荷增
多。当pH到达某特定值时,蛋白质所带正负电荷数相等,净电荷为零,此时的
pH即为蛋白质的等电点 (pI)。当pH<pI时蛋白质带净的正电荷,而当pH>

57
pI时蛋白质带净的负电荷。进行离子交换色谱时,选用的起始pH 一般 与 目 的
分子的等电点之间要有一个差值,合理选择这个差值能够保证目的分子带上一定
电荷而发生吸附,同时杂质分子尽可能少地被吸附。由于大多数蛋白质含酸性基团多于碱性基团,因而等电点低于7。因此离子
交换色谱中对于一个背景不明的蛋白质,往往先尝试在略偏碱性的条件下采用阴
离子交换剂 (吸附阴离子)进行分离,由于等电点低于7的蛋白质在此pH下带
负电荷而能被吸附。一些碱性蛋白质中含有较多的碱性基团,如赖氨酸、精氨酸
残基,因而等电点较高。胰核糖核 酸 酶 属 于 较 温 和 的 碱 性 蛋 白 质,其 等 电 点 在
95左右,更 强 的 碱 性 蛋 白 还 有 细 胞 色 素 C(pI=102)、卵 清 溶 菌 酶 (pI=114)等。而一些酸性蛋白质含有较多的酸性基团,如天冬氨酸、谷氨酸残基、翻译后修饰中引入的唾液酸残基等,因而等电点较低。例如胃蛋白酶的等电点在
30左右。在进行离子交换色谱前,了解样品组分的等电点信息对于色谱条件的选择是
很有价值的,如果能够获得各组分分子电荷随pH变化的情况,将有助于选择出
分辨率最高的色谱条件。蛋白质的滴定曲线正是给出了蛋白质的净电荷随pH变
化的情况 (图43)。
图43 蛋白质滴定曲线
从图中可以看出,此蛋白质等电点为7,pH<7时带正电荷,并且pH越小
所带正电荷数越多;pH>7时带负电荷,并且pH越大所带负电荷数越多。蛋白
质的滴定曲线可以通过电泳的方法获得,步骤如下:①制备一块正方形凝胶,其
中含有能够用于生成pH梯度的两性电解质;②在第一向即x轴方向进行电泳聚
焦,就在凝胶中产生了一个稳定的连续的pH梯度 (x轴方向);③在整个x轴
方向加上蛋白样品,并在垂直于第一向的第二向 (即y轴 方 向)进 行 电 泳,由

58
于在x轴的不同部位pH不同,蛋白质所带的电荷的种类和数量就不同,表现为
电泳过程中迁移的方向和速度上的差异,从而得到迁移率 (与蛋白质净电荷数存
在正比关系)与pH的关系曲线,这就是蛋白质的滴定曲线 (图44)。
图44 绘制蛋白质滴定曲线的主要步骤
比较样品中目的蛋白和主要杂蛋白的滴定曲线,有助于离子交换色谱起始条
件的选择,从图45可以看出,在pH为8~9时,目的蛋白和主要杂蛋白之间
电荷差异较大,有利于它们之间的分离,因此可选择该pH作为离子交换的起始
pH。尽管pH进一步升高,这两种蛋白质之间电荷差异可能会更大,但过高的
pH会影响到蛋白质活性而不宜采用。
图45 根据滴定曲线确定起始pH
4113 与色谱相关的区域和Z值
在离子交换色谱中蛋白质的色谱行为不单取决于所带电荷的种类和数量,还
与电荷在蛋白质分子中的分布密切相关。人们在实验中发现,有些蛋白质在处于
pI、净电荷为零时仍能与离子交换剂结合,甚至当蛋白质与离子交换剂带同种电
荷时也能结合。例如:磷酸甘油酸激酶在带有少量净的负电荷时的pH条件下仍

59
能结合到阳离子交换剂 (吸附阳离子)上。事实上,处于蛋白质分子内部的电荷
(它们可能形成盐键维持蛋白质的空间结构)对此蛋白质的色谱行为并不产生影
响,而是由蛋白质表面的电荷决定了色谱行为。并且蛋白质表面电荷的分布也是
不均匀的,电荷在某些表面区域内比较密集,它们往往就是在离子交换时与交换
剂发生接触的区域,也称为色谱相关区域,此区域的电性质决定了蛋白质能与何
种离子交换剂结合。上述磷酸甘油酸激酶之所以在带净的负电荷时仍能与阳离子
交换剂结合,就是因为酶分子表面的特定区域内带有一簇正电荷。色谱相关区域由若干氨基酸残基组成,它们在蛋白质一级结构上可能相距甚
远,但形成空间结构时相互靠近并分布于蛋白质的表面。色谱相关区域的变化将
会直接改变色谱结果。研究表明,来自于眼镜蛇的神经毒素有六种单乙酰基的衍
生物,由于乙酰基是连接到赖氨酸的ε氨基上,乙酰化后会使神经毒素失去一个
正电荷。显然,六种单乙酰基衍生物的净电荷数是相同的,但由于它们分别在分
子内 不 同 的 位 置 丢 失 了 一 个 正 电 荷,造 成 表 面 电 荷 的 分 布 有 所 不 同,采 用
BioRex70离子交换剂可以将它们分离。
Z值是指蛋白质分子上与离子交换剂发生相互作用的带电位点的数目。在进
行离子交换色谱时,Z值越大,蛋白质与交换剂的结合越牢固;Z值越小,结合
越松弛。Boardman和Partridge通 过 实 验 证 明 了 随 着Z值 的 增 大,细 胞 色 素C在AmberliteIRC50上的阻滞作用迅速增强。
蛋白质在特定条件下的Z值可以通过同溶剂洗脱 (用恒定离子强度的缓冲
液进行洗脱)测定出相应的保留值后计算得出。
lgk′=Z·lg1D0+lgI
式中 D0———置换离子的摩尔浓度;
I———常数项;
k′———容量因子,定义为分配平衡时某物质在固定相和流动相 中 物 质 的
量之比,k′=tR-t0t0;
tR———该蛋白质的保留时间;
t0———非滞留组分的保留时间。根据计算得出的Z值并不是个整数。
Z值并不是蛋 白 质 的 特 征 性 常 数,它 的 取 值 与 很 多 因 素 有 关,包 括 溶 液 的
pH、蛋白质的构象、其他离子的存在、结合到吸附剂上的蛋白质数量、离子交
换剂性质等。Z值是受多种因子综合作用得到的一个平均值,研究表明它与蛋白
质的净电荷之间没有直接关系,分子量大的蛋白质与离子交换剂之间的接触位点

60
未必多于分子量小的蛋白质,蛋白质的构象变化将会造成电荷分布情况的改变而
引起Z值变化,而随着分离时加样量的增加Z值会下降。
412 离子交换理论
4121 离子交换作用
离子交换剂由不溶性高分子基质、荷电功能基团和与功能基团电性相反的反
离子组成,在水溶液中,与功能基团带相反电荷的离子 (包括缓冲液中的离子、蛋白质形成的离子)依靠静电引力能够吸附在其表面。这样,各种离子与离子交
换剂结合时存在竞争关系。无机离子与交换剂的结合能力与离子所带电荷成正比,与该离子形成的水合
离子半径成反比。也就是说,离子的价态越高,结合力越强,价态相同时,原子
序数越高,结合力越强。在阳离子交换剂上,常见离子结合力强弱顺序为:
Li+<Na+<K+<Rb+<Cs+
Mg2+<Ca2+<Sr2+<Ba2+
Na+<Ca2+<Al3+< Ti4+
在阴离子交换剂上,结合力强弱顺序为:
F-<Cl-<Br-<I-
对于蛋白质这样带电荷的生物大分子,与离子交换剂的结合能力首先取决于
溶液pH,它决定了蛋白质的带电状态,然后是蛋白质的色谱相关区域,也就是
电荷在蛋白质表面的分布情况,这些在前面都已经提到。此外,还取决于溶液中
离子的种类和离子强度。溶液中的无机离子和蛋白质竞争性的与交换剂结合,在
起始条件下,溶液中离子强度较低,上样后由于蛋白质带电荷数较多,与交换剂
之间结合能力更强,因此能取代离子而吸附到交换剂上;在进行洗脱时,往往通
过提高溶液的离子强度,增加了离子的竞争性结合能力,使得蛋白质样品从交换
剂上解吸,这就是离子交换色谱的本质。
4122 pH和离子强度I的影响
pH和离子强度I是 控 制 蛋 白 质 离 子 交 换 行 为、分 辨 率、回 收 率 等 的 重 要
因素。
pH决定了蛋白质以及离子交换剂的带电荷情况,因而是决定蛋白质是否发
生吸附的最重要参数。在进行分离时,应当控制pH使得蛋白质和离子交换剂带
相反的电荷,这里涉及两个方面。一方面,离子交换剂有一个工作pH范围,在
此范围内能够确保离子交换剂带充足的电荷。通常,阳离子交换剂应用时有一个
pH下限,低于此pH会有很大一部分离子交换基团失去负电荷而不再能结合阳
离子;而阴离子交换剂应用时有一个pH上限,高于此pH会有很大一部分离子

61
交换基团失去正电荷而不能再结合阴离子。另一方面,溶液的pH直接决定了蛋
白质带电荷的种类和数量,选择适当的pH,能够保证目的蛋白分子与离子交换
剂带相反电荷而被吸附,同时如果pH距离蛋白质的等电点过远,则造成蛋白质
与离子交换剂结合过于牢固而不易洗脱。在选择操作pH时还需特别注意目的蛋白的pH稳定范围,若超出此范围会
造成蛋白质活性丧失,导致回收率下降。特别是由于陶南效应,离子交换剂表面
pH与溶液pH是不一致的。在阳离子交换剂表面的微环境中,H+ 被阳 离 子 交
换基团吸引而OH-离子被排斥,造成交换剂表面pH比周围缓冲液中低1个pH单位;而阴离子交换剂表面的微环境中,OH- 被阴离子交换基团吸引而 H+ 离
子被排斥,造成交换剂表面pH比周围缓冲液中高1个pH单位。例如:某种蛋
白质在pH=5时 被 阳 离 子 交 换 剂 吸 附,实 际 上 该 蛋 白 质 在 交 换 剂 表 面 是 处 在
pH=4的环境中,若此蛋白质在此pH条件下不稳定就会失活。大多数蛋白质在
pH=4以下稳定性都会下降而使回收率降低。由于溶液中的其他离子与蛋白质竞争结合到离子交换剂上,因此离子种类和
离子强度I是影响蛋白质结合和洗 脱 的 另 一 重 要 因 素。在 低 的 离 子 强 度I条 件
下,蛋白质通过荷电基团结合至离子交换剂上带相反电荷的功能基团上;当竞争
离子浓度即离子强度I逐渐升高时,蛋白质逐渐被置换下来。对于带有特定电荷
数的蛋白质,需要多高的盐浓度才能将其从离子交换剂上洗脱下来,并没有一个
固定的规律,需要从实验中摸索。绝大多数蛋白质在1mol/L的盐浓度下能够被
洗脱,因此在探索条件阶段,人们常把洗脱盐的终浓度定为1mol/L。事实上在
溶液中盐类还常扮演稳定蛋白质结构的角色,为防止蛋白质发生变性或沉淀,离
子强度不宜太低。另外,离子的类型也是一个重要因素,不同离子从交换剂上将
蛋白质置换下来的能力是不同的,并且离子类型还对分辨率和不同蛋白质的洗脱
顺序会产生影响。
4123 其他作用力:疏水相互作用和氢键
虽然蛋白质与离子交换剂发生结合主要依靠相反电荷之间的离子键,但实际
上此过程中还可能存在其他的作用力,常见的就是疏水相互作用和氢键。疏水相互作用主要出现在使用带非极性骨架的离子交换剂时,例如离子交换
树脂,特别是聚苯乙烯树脂,骨架带有较强的疏水性,能与蛋白质分子中的一些
疏水性氨基酸残基通过疏水相互作用结合。虽然前面提到常规的离子交换树脂因
其性质上的缺点在蛋白质分离中并不常用,但在现代 HPLC中还是有部分介质
是以树脂作为骨架的。氢键则主要出现在使用以亲水性高分子为骨架的离子交换
剂时,例如使用较为广泛的以Sephadex或Sepharose为基质的离子交换剂,骨
架糖链中的羟基、羧基等基团能够与蛋白质分子中带亲水侧链的氨基酸残基之间

62
形成氢键。当两种蛋白质在电性质方面很接近时,这些额外的作用力在分离时起
着决定性的作用,据此往往可以实现分离。不过这些作用力对色谱行为产生的影
响通常很难预测,不同的蛋白质往往相差很大,因此不具有通用性。
4124 离子交换动力学
离子交换的发生及进行的程度即离子交换平衡取决于离子作用,而离子交换
动力学则取决于离子交换剂的颗粒结构。离子交换剂的骨架是具有网孔状结构的颗粒状凝胶,而荷电功能基团均匀分
布在凝胶颗粒的表面及网孔内部,蛋白质分子依据分子量的不同,不同程度地进
入凝胶颗粒内部,将荷电功能基团上的反离子置换下来而自身结合到离子交换剂
上。从动力学角度分析,整个过程可分为五个步骤:①蛋白质在溶液中经扩散作
用到达凝胶颗粒表面,亲水性的凝胶和水分子发生氢键作用,从而在凝胶表面束
缚了一层结合水构成水膜,水膜的厚度取决于凝胶的亲水性强弱、色谱时流速的
快慢,亲水性越强,流速越慢,水膜越厚,反之水膜则越薄,蛋白质通过扩散穿
过水膜到达凝胶表面的过程称为膜扩散,速度取决于水膜两侧蛋白质的浓度差;
②蛋白质分子进入凝胶颗粒网孔,并到达 发 生 交 换 的 位 置,此 过 程 称 为 粒 子 扩
散,其速度取决于凝胶颗粒网孔大小 (交联度)、交换剂功能基团种类、蛋白质
分子大小和带电荷数等多种因素;③蛋白质取代交换剂上的反离子而发生离子交
换;④被置换下来的反离子扩散到达凝胶颗粒表面,也即粒子扩散,方向与步骤
②相反;⑤反离子通过扩散穿过 水 膜 到 达 溶 液 中,也 即 膜 扩 散,方 向 与 步 骤①相反。
根据电荷平衡的原则,一定时间内,一个带电蛋白质分子进入凝胶颗粒,就
有与该蛋白质所带净电荷数相当数量的反离子扩散出凝胶颗粒。也就是说,蛋白
质与反离子从电荷数量上看,膜扩散和粒子扩散的速率相同而方向相反。由此,上述五个步骤实际上就是膜扩散、粒子扩散和交换反应三个过程。其中交换反应
通常速度比较快,而膜扩散和粒子扩散速度较慢,当溶液中蛋白质浓度较低时,膜扩散过程往往最慢,成为整个过程的限 制 性 步 骤;当 溶 液 中 蛋 白 质 浓 度 较 高
时,粒子扩散过程往往最慢而成为整个过程的限制性步骤。
413 离子交换的分辨率
这部分内容讨论离子交换色谱中影响分离效果的主要理论参数。同其他色谱分离一样,离子交换的效果常用两个目的峰的分辨率 (RS)来
描述。分辨率定义为两个洗脱峰峰顶对应的洗脱体积之差比上两峰在基线上峰宽
的和的平均值 (图46)。
RS=2(VR2-VR1)Wb1+Wb2
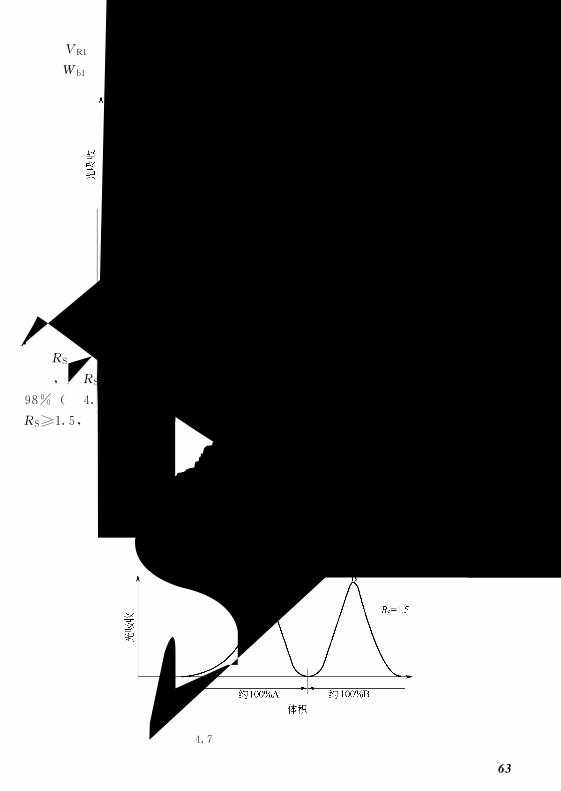
63
式中 VR1和VR2———洗脱峰1和峰2的洗脱体积;
Wb1和Wb2———洗脱峰1和峰2的峰宽。
图46 分辨率RS 的定义
洗脱体积和峰宽应使用相同的体积单位表示。
图47 不同分辨率下的分离效果
RS是相邻两个洗脱峰相对分离程度的衡量标准。假设两个相邻洗脱峰面积
相等,当RS=10时,理论上在回收率为98%时收集得到的洗脱峰纯度可达到
98% (图47),此时两峰实现主体分离;若要达到完全分离 (基线分离),要求
RS≥15,此时收集得到的洗脱峰理论上纯度可达到100%。需要指出的是,一

64
个完全分离的洗脱峰在很多时候并不意味着是单一组分,当分离参数改变后,往
往又能分开成为若干个峰。一个纯化系统能够达到的分辨率取决于该系统的选择性、效率和容量,这是
柱色谱中的三个最重要的参数。RS的表达式为:
RS=14·α-1α·槡N· k′
1+k′式中 α———选择性;
N———柱效率;
k′———容量因子。
4131 容量因子
容量因子k′又称保留因子,是色谱分离中常 用 的 组 分 保 留 值 表 示 法,注 意
不要将其与离子交换剂的吸附容量 (mg样品/ml胶)相混淆。关于容量因子k′的定义和计算方法在前面的4113部分已经讨论过。k′是针对单个洗脱峰进行
计算的,在计算RS时,k′应当取洗脱峰1和2对应的k′1和k′2的平均值。一般来说,容量因子k′的取值与蛋白质在固定相 (离 子 交 换 剂)和 流 动 相
(缓冲液)中的分配性质、操作温度及固定相和流动相的体积比有关,而与柱尺
寸和流速无关。包括离子交换色谱在内的吸附技术都具有很高的容量因子,这是
因为通过选择实验条件可以使得某蛋白质的洗脱时间 (或洗脱体积)大大超过非
滞留组分的洗脱时间 (或洗脱体积),由k′的计算公式看出,某洗脱峰洗脱时间
tR (或洗脱体积VR)越大,则k′的取值越大。而凝胶过滤则完全不同,由于所
有的峰都在一个柱体积 (Vt)被洗脱,即VR≤Vt,其k′的取值有一个上限。
4132 柱效率
柱效率用在特定实验条件下色谱柱的理论塔板数N 来表示,通常表示为每
米色谱床所包含的理论塔板数,其计算公式如下:
N=554· VRW( )h
2
式中 VR———洗脱峰的洗脱体积;
Wh———洗脱峰半峰高 (h1/2)时对应的峰宽。柱效率还可用一个理论塔板的高度H 来表示:
H=L/N式中 L———柱长。
由于N 的取值与流速、上样量等实验条件有关,在进行比较时需要在相同
条件下进行测定。在离子交换色谱中测定柱效率是在同溶剂条件下进行的,并且
使用不与交换剂的基质发生相互作用的物质作为洗脱剂。

65
柱效率下降会导致色谱区带变宽,也就是洗脱峰的峰宽变大。导致区带变宽
的原因之一是蛋白质分子在色谱柱床中 (包括在固定相和流动相中)发生纵向扩
散,实验中采用尺寸较小的凝胶颗粒作为离子交换剂的基质,可以有效减小能够
发生纵向扩散的距离,从而大大提高柱效率。现代离子交换介质中有一些是以很
小的颗粒作为基质的 (目 前 最 小 的 颗 粒 直 径 小 于3μm),它 们 有 着 非 常 高 的 效
率,但是随着基质颗粒直径的减小,也有不利因素出现。很小的颗粒往往是非孔
型的,蛋白质只能结合在其表面,以它作为基质连上功能基团后的离子交换剂对
蛋白质的交换容量会下降。另外,随着基质颗粒的减小,分离时产生的背景压力
会增大,不利于进行快速、大规模的色谱。除了基质颗粒大小外,实验技术是影
响柱效率的另一重要因素,色谱柱床面不平整,柱床内有气泡等都会导致柱效率
下降,因此色谱过程中操作条件的控制对色谱效果影响很大。
4133 选择性
选择性是一个系统分离不同蛋白峰的能力,习惯用相对保留值α来表示,α定义为:
α=k′2k′1=
VR2-V0VR1-V0≈
VR2VR1
式中 k′1,k′2———洗脱峰1和2的容量因子;
VR1,VR2———洗脱峰1和2的洗脱体积;
V0———非滞留组分的洗脱体积。从对分辨 率 的 影 响 上 来 看,好 的 选 择 性 往 往 比 高 的 柱 效 率 更 为 重 要 (图
48)。
图48 选择性和柱效率对分辨率的影响
影响到离子交换色谱选择性的因素包括:离子交换剂上功能基团的性质和数
量,pH和离子强度等实验条件。由于上述实验条件是容易进行调控的,因而人
们更乐于通过控制实验条件来提高选择性,从而获得好的分辨率。

66
根据前面所述RS的解析表达式可知,为了提高一个分离过程的分辨率RS以
达到满意的分离效果,必须满足以下条件:①α>1,当VR1=VR2时α=1,此时
两峰完全重叠,分 辨 率 为0,α的 值 越 大,两 洗 脱 峰 相 距 越 远,分 辨 率 越 高;
②N 尽可能大,理论塔板数越大,柱效率越高,分辨率也越高;③k′≠0,根据
k′的计算公式,当两种蛋白的洗脱体积VR1和VR2均等于非滞留组分的洗脱体积
V0时,k′=0,此时无法实现分离,分辨率为0,选择合适的色谱条件,使得至
少一种蛋白质在离子交换剂上发生吸附,就能满足k′≠0,增加k′的值将提高分
辨率RS,有利于分离。图49表示了通过改变k′的值、增大N、增加α的值对
分离产生的优化效果。
图49 影响相邻两个洗脱峰分离的因素
t0—非滞留组分保留时间
42 固定相:离子交换剂
离子交换剂是离子交换色谱的基础,它们很大程度上决定着分离过程的分辨
率 (通过决定容量因子k′、理论塔板数N 和选择性α来实现),同时还决定着分
离的规模和成本,是离子交换技术中最为关键的因素,现代离子交换技术的进步
得益于优质高效离子交换剂的研制和应用。
421 基本结构
离子交换剂由水不溶性基质和共价结合在基质上的带电功能基团组成,带电
功能基团上还结合有可移动的与功能基团带相反电荷的反离子 (又称平衡离子)。反离子可被带同种电荷的其他离子取代而发生可逆的离子交换,此过程中基质的

67
性质不发生改变。离子交换剂所带功能基团分为酸性基团和碱性基团两类,其中带酸性功能基
团的离子交换剂在工作pH范围内解离出质子而带有负电荷,能够结合溶液中带
正电荷的离子 (阳离子),因此被称为阳离子交换剂;而带碱性功能基团的离子
交换剂在工作pH范围内结合质子而带有正电荷,能够结合溶液中带负电荷的离
子 (阴离子),因此被称为阴离子交换剂。基质是一类水不溶性的化合物,常见的有:①树脂,包括疏水性的聚苯乙烯
树脂和部分疏水性的聚异丁烯酸及聚丙烯酸树脂;②天然的或人工合成的亲水性
大孔型聚合物,如纤维素、葡聚糖、琼脂糖、聚丙烯酰胺等;③各类人工合成的
具有一定硬度、适合于高压色谱的亲水性大孔型聚合物;④人工合成的颗粒很小
的无孔型聚合物。基质的特性不但决定了很多色谱性质如效率、容量和回收率,还决定了离子
交换剂的化学稳定性、机械强度和流体性质,并且会影响到蛋白质生物活性的维
持。理想的基质应当满足以下条件:①有较强的机械稳定性,能适合高流速的需
要;②具有亲水性,至少在颗粒表面应当是亲水性的;③高化学稳定性,在很宽
的pH范围内保持稳定,耐去污剂、有机溶剂,耐高温,从而方便清洗和消毒灭
菌;④高的交换容量,以 满 足 分 离 较 大 规 模 样 品 的 需 要;⑤球 形,颗 粒 直 径 均
匀;⑥价格尽可能便宜以降低分离成本。
422 功能基团和酸碱性质
带电功能基团决定了离子交换剂的基本性质,功能基团的类型决定了离子交
换剂的类型和强弱,它们的总量和有效数量决定了离子交换剂的总交换容量和有
效交换容量。根据功能基团是酸性基团还是碱性基团,离子交换剂分为阴离子交换剂和阳
离子交换剂。此外,根据功能基团的解离性质,离子交换剂还可分为强型离子交
换剂和弱型离子交换剂,强弱并不是指蛋白质与交换剂结合的牢固程度,而是取
决于带电功能基 团 的pKa 值。强 型 离 子 交 换 剂 带 有 强 酸 性 或 强 碱 性 功 能 基 团,其pKa 值很低或很高,远离中性,因此解离程度大,在很 宽 的pH 范 围 内 都 处
于完全解离状态;弱型离子交换剂则带有弱酸性或弱碱性功能基团,其pKa 值
与强型交换剂相比靠近中性,工作pH范围较窄,超出此范围会造成解离程度下
降,带电荷数量明显减少,导致无法充分结合蛋白质。当操作pH远离pKa 时,无论是强型还是弱型离子交换剂都能牢固吸附蛋白质。结合上述两种分类方法,离子交换剂可以分为强酸型 (强阳)、强碱型 (强阴)、弱酸型 (弱阳)和弱碱型
(弱阴)四种。一般来说,阳离子交换剂应用时有一个pH 下限,低于此pH 功
能基团开始失去负电荷,强酸型交换剂的pH工作下限低于弱酸型交换剂;弱碱

68
型交换剂应用时有一个pH上限,高于此pH功能基团开始失去正电荷,以季铵
基为功能基团的强碱型交换剂没有pH工作上限,因为季铵基在任何pH下都不
会失去正电荷。当然,除了离子交换剂的电荷性质外,选择操作pH时还必须考
虑到目的蛋白的pH稳定范围。事实上色谱分析时很少用到低于4或是高于8的
pH条件,因为多数蛋白质在那样的pH下稳定性变差,有时还会造成蛋白质近
乎不可逆地结合到交换剂上,而且陶南效应会使蛋白质在交换剂表面的环境pH更趋极端,直接导致蛋白质的回收率急剧下降。
表42列出了比较常用的离子交换功能基团的名称和种类。强酸型交换剂中
最常用的是磺基,弱酸型最常用的是羧基,强碱型最常用的是季铵基,弱碱型最
常用的是氨基。除了磷酸基,其他功能基团一般都只带单个正电荷或负电荷。
表42 常见的离子交换功能基团
类 型 名 称 英 文 符 号 功 能 基 团
阴离子交换剂 二乙基氨乙基 DEAE —OCH2CH2N+H(C2H5)2季铵基乙基 QAE —OCH2CH2N+(C2H5)2CH2CH(OH)CH3季铵基 Q —OCH2N+(CH3)3三乙基氨乙基 TEAE —OCH2CH2N+(C2H5)3氨乙基 AE —OCH2CH2NH+3
阳离子交换剂 羧甲基 CM —OCH2COO-
磺丙基 SP —OCH2CH2CH2SO-3磺甲基 S —OCH2SO-3磷酸基 P —OPO3H2
423 离子交换剂的部分性质
4231 粒度
粒度是指离子交换剂的基质颗粒的大小,用基质颗粒的直径来表示。用于生
化分离的离 子 交 换 剂 的 颗 粒 直 径 一 般 都 在3~300μm 范 围 内,其 中 直 径 大 于
150μm的被认为是大颗粒,而直径小于30μm的是细颗粒。基质颗粒大小直接关
系到分离效果,颗粒越小,色谱柱的理论塔板数就越大,分离效果越好,但是同
时交换容量会变小,而色谱时背景压力会增大,限制了流速的提高,因此细颗粒
的介质通常适用于分析型分离;相反,基质颗粒越大,柱效率会下降,但是离子
交换剂的交换容量会提高,背景压力会减小,因此大颗粒介质适合于实验室小规
模分离及大规模工业化分离。值得注意的是,除了颗粒大小,基质颗粒的均匀程
度对分离效果的影响也很大,颗粒直径越均一,分离效果越好。
4232 交联度和网孔结构
离子交换剂的基质是通过交联剂将线性大分子交联形成的网孔状颗粒。不同
的离子交换剂使用不同的交联剂,如:聚苯乙烯树脂使用二乙烯苯作为交联剂,葡

69
聚糖和纤维素交换剂使用环氧氯丙烷作为交联剂,琼脂糖交换剂使用二溴丙醇作为
交联剂。交联度用交联剂在基质中所占的百分含量来表示,不同的离子交换剂交联度
相差很大,比如离子交换树脂的交联度通常在2%~20%之间变化。交联度的大小影
响着离子交换剂的很多特性,包括机械强度、膨胀度、网孔大小、交换容量等。离子交换剂的交联度决 定 着 网 孔 大 小,显 然,交 联 度 越 高,网 孔 的 孔 径 越
小;而交联度越低,网孔孔径越大。但琼脂糖交换剂是个例外,这种介质所形成
的网孔结构是由琼脂糖的胶束构成的,其孔径仅与琼脂糖的浓度有关,交联剂的
用量对孔径影响不大。由于离子交换分离中常用的介质具有网孔状结构,进行离子交换色谱时往往
具有分子筛和离子交换双重功效。分子直径小于介质孔径的蛋白质分子能够进入
介质颗粒内部,并与颗粒内部的功能基团发生结合,而分子直径大于介质孔径的
蛋白质被排阻在颗粒外,只能与颗粒表面的功能基团结合,此时交换容量会受到
很大影响。显然,颗粒孔径的均匀程度也会影响到分离效果。
4233 电荷密度
电荷密度是指基质颗粒上单位表面积的取代基 (功能集团)的数量,它决定
着离子交换剂的交换容量、膨胀度等性质。对小分子进行离子交换时,这些离子
与功能基团间按物质的量之比1∶1结合,交换剂上功能基团密度越大,则工作
能力越强,单位数量的交换剂能够结合更多的离子,因此高电荷密度的离子交换
剂具有优势。但是在对蛋白质进行离子交换色谱时情况并不相同。在决定交换容
量方面,交联度比电荷密度重要得多。由于蛋白质是大分子,如果基质颗粒孔径
太小以致蛋白质分子不能进入颗粒网孔,即使颗粒表面电荷密度很大,交换容量
也不会高。并且,由于蛋白质分子带电荷数量往往较多,与交换剂上功能基团结
合时物质的量的关系并不是1∶1,而是多点结合,过高的电荷密度会导致蛋白
质在离子交换剂上结合过于牢固而难以洗脱,易造成蛋白质变性,回收率下降。因此,蛋白质离子交换所用的介质电荷密度往往较低。
4234 膨胀度
膨胀度又称吸水值,是指干态的离子交换剂在水溶液中吸水后造成体积膨胀
的程度,通常用每克干胶吸水膨胀后的体积 (毫升)来表示。影响离子交换剂膨
胀度的因素包括:(1)基质种类 一般来说,具有亲水性基质的离子交换剂膨胀度要大大高于
具有疏水性基质的离子交换剂,前者膨胀度可以达到60ml/g干胶,而后者有时
在溶液中体积变化非常小。(2)交联度 无论是哪一类离子交换剂,其膨胀度都随着交联度的增加而下
降,图410显示了四种基于交联葡聚糖凝胶Sephadex的离子交换剂在不同交联

70
度下的膨胀度情况,其中A50和A25分别是以SephadexG50和G25作为基
质连上碱性功能基团形成的阴离子交换剂 (A代表阴离子交换剂,50和25代表
基质类型),C50和C25分别是以SephadexG50和G25作为基质连上酸性功
能基团形成的阳离子交换剂 (C代表阳离子交换剂)。由于SephadexG25交联
度高于G50,所以相应的A50和C50交换剂膨胀度分别高于A25和C25。
图410 离子交换剂的膨胀度与交换剂类型和溶液离子强度的关系
色谱柱:15mm×30cm操作压力:30cmH2O?
缓冲液:(a)和 (b)为pH=76的TrisHCl缓冲液;
(c)和 (d)为pH=43的醋酸缓冲液
离子强度用NaCl进行调节
(3)带电基团的强弱 在相同 交 联 度 的 情 况 下,SephadexA50和C50系
? 1cmH2O=980665Pa。

71
列的离子交换剂比它们的基质SephadexG50膨胀度大得多,原因在于功能基团
带同种电荷,相互排斥而扩展了凝胶颗粒的体积。同样的道理,强酸型和强碱型
离子交换剂由于带电荷数量多于相应的弱酸型和弱碱型离子交换剂,其膨胀度相
应的也比后者高。比较图410中 (a)、 (b)两图,相同条件下 QAESephadex(强碱型)的膨胀度高于DEAESephadex (弱碱型)。
(4)溶液的离子强度 和pH 对 于 某 种 特 定 的 离 子 交 换 剂,膨 胀 度 不 是 常
数,其数值受到溶液离子强度和pH的影响,特别是交联度较小的交换剂,其结
构强度不如交联度较大的交换剂,其膨胀度更易受上述因素影响。从图410中
可以看出,以SephadexG50为基质的四种离子交换剂随溶液离子强度的增加,膨胀度减小,这是由于低离子强度下,离子交换剂中带同种电荷的功能 基 团 之
间的静电 斥 力 很 大,基 质 颗 粒 体 积 被 胀 大,而 高 的 离 子 强 度 可 以 降 低 静 电 斥
力。正因为如此,交联度小的离子交换剂应避免放置在水中或浓度很低的盐溶液
中,防止颗粒过分膨胀导致破裂。而相对来说,离子强度的变化对交联度高的以
SephadexG25为基质的四种离子交换剂的膨胀度影响不大。
pH对膨胀度产生影响是因为pH 的 变 化 导 致 离 子 交 换 剂 带 电 状 态 的 改 变,从而使存在于交换剂中的静电斥力的大小发生变化所致。这一点在弱型离子交换
剂中表现较为明显,从图411中可以看出,对于以SephadexG50为基质的离
子交换剂,弱碱型的DEAESephadex随pH增加,逐渐失去正电荷,静电斥力
减小,膨胀度随之减小;而弱酸型的CMSephadex随pH降低,逐渐失去负电荷,膨胀度随之减小。强酸型的SPSephadex由于pKa值很低,随pH改变,其膨胀度
变化较小,而强碱型的QAESephadex由于QAE基团在任何pH下都不会失去正
电荷,其膨胀度不随pH而改变。与离子强度的影响相似,pH的改变对于交联度
高的以SephadexG25为基质的四种离子交换剂的膨胀度影响不大。应当指出,让离子交换剂在具有一定离子强度的缓冲液中充分膨胀,可以使
颗粒网孔增大并保持孔径稳定,这对于交换剂的交换容量和分离效果都是有帮助
的。正因为如此,离子交换剂使用前要进行预溶胀。
4235 交换容量
交换容量是指离子交换剂能够结合溶液中可交换离子的能力,通常分为总交
换容量和有效交换容量。总交换容量又称总离子容量,用每克干介质或每毫升湿胶包含的带电功能基
团的数量来表示,其数值的大小取决于离子交换剂的取代程度也就是电荷密度。总交换容量可以通过酸碱滴定的方法来测定,也就是利用交换剂上的酸性或碱性
基团同已知浓度的酸碱发生反应,利用物质的量的关系算出交换剂上功能基团的
数量。也可使离子交换剂和某种小的离子之间发生完全交换,然后通过测定该离

72
图411 离子交换剂的膨胀度与溶液pH的关系
色谱柱:15mm×30cm操作压力:30cmH2O
缓冲液:(a)和 (b)使用咪唑乙二胺缓冲液,(c)和 (d)使用磷酸缓冲液
离子强度均为005
子的含量算出总交换容量,此方法的依据是小的离子与交换剂上功能基团的结合
满足1∶1的物质的量之比。总交换容量是离子交换剂的基础参数,并且对于一
种特定的交换剂其数值是恒定的。总交换容量并不反映交换剂对蛋白质的结合情况,这是因为交换剂所能结合
某蛋白质的数量与该蛋白质的分子大小、交换剂的网孔结构及所选择的实验条件
等多种因素有关。例如,每毫升DEAESephadexA50能够结合250mg血 红 蛋
白,而相同条件下,DEAESephadexA25由于交联度大,网孔孔径小,每毫升
只能结合70mg血红蛋白。因此,人们提出了有效交换容量的概念,表示为蛋白
结合容量,是指每克干介质或每毫升湿胶在一定的实验条件下吸附某种蛋白质的

73
实际容量。由于有效容量是一个变值,比如pH和离子强度,它们会影响蛋白质
及离子交换剂的电荷和静电力的强弱,显然会对有效容量产生影响,因此,在说
明其数值时应当同时指出实验条件。表43列出了几种常用的基于Sephadex的
离子交换剂的总交换容量和对特定蛋白质的有效容量。从表中数据可以看出,一
般情况下,同一种交换剂对于分子量小的蛋白质有效交换容量较大,而对于分子
量大的蛋白质有效容量较小;对于同一种蛋白质,离子交换剂的交联度越小有效
容量越大,而交联度越大有效容量越小。
表43 几种常用离子交换剂的交换容量
离子交换剂名称
总交换容量 有效交换容量/(mg/ml湿胶)
(μmol/mg干胶)
(μmol/ml湿胶)
甲状腺球蛋
白(669kDa)
人血清白蛋
白(68kDa)α乳清蛋白
(14.3kDa)
免疫球蛋白
G(160kDa)
牛碳氧血红
蛋白(69kDa)
DEAESephadexA25 3.5±0.5 500 1.0 30.0 140.0 — —
DEAESephadexA50 3.5±0.5 175 2.0 110.0 50.0 — —
QAESephadexA25 3.0±0.4 500 1.5 10.0 110.0 — —
QAESephadexA50 3.0±0.4 100 1.2 80.0 30.0 — —
CMSephadexC25 4.5±0.5 550 — — — 1.6 70.0CMSephadexC50 4.5±0.5 170 — — — 7.0 140.0SPSephadexC25 2.3±0.3 300 — — — 1.1 70.0SPSephadexC50 2.3±0.3 90 — — — 8.0 110.0
注:测定实验条件:流速75cm/h,阴离子交换剂使用的起始缓冲液为pH=83、005mol/L的Tris
HCl缓冲液,阳离子交换剂 使 用 的 起 始 缓 冲 液 为pH=50、01mol/L的 醋 酸 缓 冲 液,极 限 缓 冲 液 为 含
2mol/L的NaCl的相应的起始缓冲液。
有效交换容量中又分出静态容量和动态容量的概念,这是由 于 色 谱 过 程 中
的流速对有效容量的影响较大。静态容量是指在流速为零,蛋白质有充 足 的 时
间从流动相扩散到固定相并吸附在交换剂上,此时离子交换剂对蛋白质 的 吸 附
可以达到饱和,对应的结合容量即为静态容量。在流速很低时测出的有 效 容 量
近似于静态容量。而当色谱流速增大后,蛋白质来不及使离子交换剂达 到 完 全
饱和,这种在特定流速下测出的有效容量为动态容量,它在数值上总是 小 于 静
态容量。某种离子交换剂对特定蛋白质的静态容量的测定可采用试管小样法。具体操
作如下:取若干支试管,分别往其中加入相同体积的已经用起始缓冲液平衡好的
离子交换剂,再分别加入一系列不同浓度 的 蛋 白 质 溶 液,混 合 振 荡 确 保 充 分 吸
附,然后分析上清液中是否含有蛋白质成分,根据能使上清液中无蛋白质的最大
蛋白质浓度可以计算出每毫升离子交换剂所能吸附的蛋白质的上限,此即为静态
容量。测定动态容量则必须在色谱条件下进行。取一定体积的离子交换剂装柱 (为

74
了缩短测定时间,通常只需装一根体积1ml左右的柱子),用 起 始 缓 冲 液 平 衡,在特定的流速下加样至色谱柱上端,注意样品应当溶于起始缓冲液,浓度在1~5mg/ml。为确保离子交换剂满负荷吸附蛋白质,应不停加样直至紫外检测器测
出有蛋白质从柱下端流出,并且检测器上吸光度的读数达到最大值的一半 (不含蛋
白质的起始缓冲液吸光度为0,样品蛋白质溶液为100%),停止加样,用起始缓冲
液将不能吸附的蛋白质继续洗出,使柱下端流出液的吸光度回到0附近,此时柱内
的离子交换剂处于此流速下的最大吸附状态,而柱内液体中又不含不被吸附的蛋白
质。改变缓冲液条件,使得蛋白质从色谱柱上解吸,收集洗脱液,测定出其中的总
蛋白,除以离子交换剂体积后可算出该交换剂在此流速下的动态容量。
424 离子交换剂的类型
离子交换剂的分类可以依据其基质成分或功能基团来进行,按功能基团分为
强阳、强阴、弱阳、弱阴四种类型,这在前面已经讨论过,这里以基质构成来介
绍一下常用离子交换剂的种类,最为常见 的 包 括 离 子 交 换 树 脂、离 子 交 换 纤 维
素、离子交换葡聚糖和离子交换琼脂糖等。
4241 离子交换树脂
树脂是一类通过化学方法合成的具有特殊网状结构的不溶性高分子化合物,网状结构的形成往往通过单体聚合时加入一定比例的交联剂来实现。树脂本身又
分为好多种类,最常见的包括聚苯乙烯树脂、聚异丁烯酸树脂、聚丙烯酸树脂、酚醛树脂等。再通过化学方法往树脂骨架上引入功能基团就得到离子交换树脂。离子交换树脂种类很多,不同的生产厂家生产的离子交换树脂所标的型号和名称
各不相同,但有的在性能上是非常相似的。表44列出了一些常见离子交换树脂
的型号和特点。
表44 部分常见的离子交换树脂规格
名称及型号 基质类型 功能基团性质 交联度/% 湿交换容量/(mmol/ml) 排阻极限
Dowex1X2 聚苯乙烯 强碱 2 0.6 2700
Dowex1X4 聚苯乙烯 强碱 4 1 1400
Dowex1X8 聚苯乙烯 强碱 8 1.2 1000
Dowex2X8 聚苯乙烯 强碱 8 1.4 1000
Dowex2X10 聚苯乙烯 强碱 10 1.5
Dowex3X4A 聚苯乙烯 弱碱 4 1 1400
Dowex4X4 聚苯乙烯 弱碱 4 0.8 1400
Dowex50WX2 聚苯乙烯 强酸 2 0.7
Dowex50WX4 聚苯乙烯 强酸 4 1.2
Dowex50WX8 聚苯乙烯 强酸 8 1.7
Dowex50WX12 聚苯乙烯 强酸 12 2.3

75
续表
名称及型号 基质类型 功能基团性质 交联度/% 湿交换容量/(mmol/ml) 排阻极限
BioRex5 聚烃基亚胺 强弱碱混合 4 2.8 1400
BioRex9 聚烃基亚胺 强碱 4 1.3 1400
BioRex70 聚异丁烯酸 弱酸 10 3.3
AmberliteIRA401 聚苯乙烯 强碱 4 1.0
AmberliteIRA402 聚苯乙烯 强碱 6 1.25
AmberliteCG400 聚苯乙烯 强碱 8 1.4
AmberliteIRA904 大网络树脂 强碱 4 0.7
AmberliteIRA410 聚苯乙烯 强碱 8 1.4
AmberliteIR45 聚苯乙烯 弱碱 1.9
AmberliteIRA67 聚异丁烯酸 弱碱 1.6
AmberliteIRA93 大网络树脂 弱碱 1.4
AmberliteIR120 聚苯乙烯 强酸 8 1.9
AmberliteGC120 聚苯乙烯 强酸 8 1.9
AmberliteIRC50 聚异丁烯酸 弱酸 5 3.5
AmberliteGC50 聚异丁烯酸 弱酸 5 3.5
Duolite113 聚苯乙烯 强碱 2~9 1.3
DuoliteA161 大孔型树脂 强碱 1.1
DuoliteA116 聚苯乙烯 强碱 1.4
DuoliteA162 大孔型树脂 强碱 1.1
DuoliteA303 聚苯乙烯 强碱 7~9 1.3
DuoliteA378 大孔型树脂 强碱 1.1
Duolite225 聚苯乙烯 强酸 4,8,12 4.5~5.0mmol/g(干)①
DuoliteC255 聚苯乙烯 强酸 10 2.0
DuoliteC26C 大孔型树脂 强酸 高 1.8
① 每克干胶的交换容量,该产品未提供湿交换容量数值。
前面已经提到,由于孔径过小,蛋白质分子无法进入颗粒内部造成有效交换
容量太小,并且电荷密度太高,会使蛋白质结合过于牢固而不易洗脱,尽管离子
交换树脂在脱盐、小分子的氨基酸及短肽的分离中有着非常广泛的应用,但通常
不用于蛋白质的色谱。不过也有例外,比如聚丙烯酸树脂被发现很适合分离小分
子量的碱性蛋白质,并且分辨率很高,另外在高效液相介质中也常有以树脂为骨
架的离子交换剂。
4242 离子交换纤维素
基于纤维 素 的 离 子 交 换 剂 是 最 早 用 于 分 离 蛋 白 质 的 离 子 交 换 剂 之 一,于
1954年由Peterson和Sober最先报道,在当时蛋白质的色谱领域是一个重大突
破,而这类交换剂至今仍在广泛使用。纤维素离子交换剂的特点是表面积大,开
放性的骨架允许大分子自由通过,同时对大分子的交换容量较大。纤维素的亲水

76
性结构使其表面结合着4倍于自身重量的结合水,形成的交换剂与蛋白质之间结
合时离子键以外的作用力 (如疏水相互作用等)很 低,这 使 得 色 谱 时 洗 脱 很 方
便,并且蛋白质活性的回收率高。离子交换纤维素存在着纤维状和微粒状两种不同的物理形态。纤维状的交换
剂是将纤维素直接衍生化,连接上功能基团而形成的。纤维素分子是由葡萄糖通
过β(1→4)糖苷键连接形成的大分子,天然状态下相邻的多糖链之间形成数量
巨大的氢键,从而在这些区域形成微晶结 构,而 微 晶 区 域 周 围 又 散 布 着 非 晶 型
(无定形)区域。而微粒状的交换剂是将纤维素进行部分酸水解,除去了大部分
非晶型区域后经衍生化形成的,这样的处理使交换剂的外水空间增大,结构也更
为牢固,并且电荷分布更均匀。英国的 Whatman公司是最早生产离子交换纤维素的企业之一,产品种类较
多,如表45所示。其中DEAE纤维素和CM纤维素属于常规生化分离中最常
用的介质类型。
表45 Whatman公司的离子交换纤维素
纤维素名称 功能基团性质 基 质 类 型 工作pH范围 标称流速/(cm/h) 蛋白容量/(mg/ml)
DE51 弱碱 微粒状 2~9 40 30
DE52 弱碱 微粒状 2~9 40 120
DE53 弱碱 微粒状 2~9 40 120
QA52 强碱 微粒状 2~12 40 140
DE92 弱碱 纤维状 2~9 75 40
QA92 强碱 纤维状 2~12 75 40
SE52 强酸 微粒状 2~12 40 200
SE53 强酸 微粒状 2~12 40 280
CM52 弱酸 微粒状 4.5~10 40 180
SE92 强酸 纤维状 2~12 75 180
CM92 弱酸 纤维状 4.5~10 75 200
P11 强弱混合 纤维状 3~10 30 40
Pharmacia公司生产的DEAESephacel是另一种常用的纤维素离子交换剂。它是将微晶纤维素的微晶区先解体,然后再重新形成珠状结构的凝胶,颗粒直径
在40~160μm。维持交换剂结构的主要作用力还是纤维 素 分 子 间 的 氢 键,此 外
还通过环 氧 氯 丙 烷 交 联 强 化 凝 胶 结 构,在 此 基 础 上 再 连 上 功 能 基 团。DEAESephacel的一些特点见表46。
此外,BioRad公司生产的Cellex系列也属于常用的离子交换纤维素,其种
类和特性见表47。

77
表46 Pharmacia公司的DEAESephacel离子交换剂
项 目 DEAESephacel
类型 弱阴离子交换剂
总交换容量/(μmol/ml) 95~135排阻极限 1×106
有效容量/(mg/ml)
甲状腺球蛋白 6.4
人血清白蛋白 150
α乳清蛋白 120粒度/μm 40~160稳定pH范围 2~12灭菌条件 Cl-型在pH=7时可在121℃灭菌30min保存条件 20%乙醇中
表47 BioRad公司的Cellex系列离子交换剂
型 号 功能基团类型 基质类型 总交换容量/(mmol/g)
CellexD 碱性 纤维状 0.7±0.1CellexCM 酸性 纤维状 0.7±0.1CellexE 酸性 纤维状 0.3±0.05CellexP 碱性 纤维状 0.85±0.1
4243 离子交换葡聚糖
离子交换葡聚糖是Pharmacia公司以两种交联葡聚糖凝胶SephadexG25和
G50为基质,连接上四种不同的功能基团后形成的离子交换剂。这类交换剂的
型号中分别用字母A和C代表阴离子交换剂和阳离子交换剂,用数字25或50代表其基质是SephadexG25或G50。
Sephadex具有亲水性和很低的非特异性吸附的性质,因而很适合作为离子
交换剂的基质,并且由于这种基质具有分子筛效应,使得这类交换剂在分离蛋白
质时同时具有离子交换作用和分子筛效应。葡聚糖离子交换剂在交换容量方面要
明显高于纤维素离子交换剂。对于分子量小于30000的蛋白质,以SephadexG25为基质的A25和C25比以SephadexG50为基质的A50和C50有着更大
的交换容量;而对于分子量在30000~100000之间的蛋白质,A50和C50由于
凝胶颗粒网孔孔径较大,其交换容量要明显大于A25和C25;若蛋白质的分子
量大于100000,对于25系列和50系列的交换剂都是全排阻的,即蛋白质分子
不能进入凝胶颗粒内部,此时分子的交换与结合仅发生在颗粒表面,25系列的
葡聚糖交换剂的总交换容量高而表现出优势。前面已经提到,交联度较小的50系列的葡聚糖交换剂,其膨胀度大于25系列,并且膨胀度还受离子强 度 和pH

78
的影响。当离子强度由001mol/L增加到05mol/L时,一根DEAESephadexA50或CMSephadexC50色谱柱的体积会缩小至原来的一半。不过实验表明这
种体积变化对色谱结果似乎不会产生什么不利影响,至少在使用连续梯度洗脱时
不会。表48列出了葡聚糖离子交换剂的类型和特性,关于它们交换容量的数据
见表43。
表48 葡聚糖离子交换剂的类型和特性
项 目DEAESephadex QAESephadex CMSephadex SPSephadex
A25 A50 A25 A50 C25 C50 C25 C50
功能基团 二乙基氨乙基二乙基(2羟丙基)
氨乙基羧甲基 磺丙基
种类 弱碱型 强碱型 弱酸型 强酸型
平衡离子 Cl- Cl- Na+ Na+
工作pH范围 2~9 2~9 2~10 2~10 6~13 6~10 2~10 2~10
化学稳定性 在水、盐溶液、有机溶剂、碱和弱酸中稳定,pH<2时不稳定,应避免接触氧化 剂 和 葡
聚糖酶
物理稳定性 在中性pH和以盐形式存在时,可于121℃灭菌30min。A25和C25交联度高,稳定
性好于A50和C50,后两者随溶液pH和离子强度变化体积会发生较大变化
耐压/MPa 011 001 011 001 013 001 013 001
流速限/(cm/h) 475 45 475 45 475 45 475 45
4244 离子交换琼脂糖
这是一类在交联琼脂糖凝胶的基础上连上功能基团形成的离子交换剂,这类
凝胶比葡聚糖凝胶更显多孔结构,由于其孔径稍大,对于分子量在106以内的球
状蛋白质表现出很好的交换容量。琼脂糖离子交换剂的多糖链排列成束,不同程
度的交联使得束状结构进一步强化,因此该介质表现牢固,体积 (膨胀度)随离
子强 度 或pH 的 变 化 改 变 很 小。离 子 交 换 琼 脂 糖 常 用 的 有 Pharmacia公 司 的
Sepharose系列和BioRad公司的BioGelA系列。
Pharmacia公司的Sepharose离子交换剂又包括了SepharoseCL6B、Sepharose HighPerformance (Sepharose HP)、SepharoseFastFlow (SepharoseFF)、SepharoseBigBeads(SepharoseBB)、STREAMLINE等系列。
SepharoseCL6B系列是由交联琼脂糖凝胶SepharoseCL6B引入功能基团
而来的,属于大孔型珠状离子交换剂,主要颗粒直径为90μm。这类离子交换剂
有着较好的分辨率,较高的交换容量,适合于各类大分子的分离。表49列出了
两种主要的SepharoseCL6B系列交换剂的特性。
SepharoseHighPerformance是基于高度交联琼脂糖的颗粒状交 换 剂,凝 胶

79
颗 粒 粒 径 平 均 为34μm,高 的 交 联 度 赋 予 该 介 质 极 好 的 物 理 稳 定 性,其 体 积
不 随pH和 离 子 强 度 而 改 变,小 的 粒 度 使 该 介 质 具 有 很 高 的 效 率 和 选 择 性,因 此 在 操 作 时 有 着 很 高 的 分 辨 率,是Sepharose系 列 中 颗 粒 最 细、分 辨 率 最
高、化 学 稳 定 性 最 好 的 一 类。该 系 列 主 要 有QSepharoseHighPerformance和
SPSepharoseHighPerformance两 种 介 质,都 属 于 强 离 子 交 换 剂,其 特 性 见
表410。
表49 SepharoseCL6B离子交换剂的特性
项 目 DEAESepharoseCL6B CMSepharoseCL6B
功能基团 二乙基氨乙基 羧甲基
功能基团类型 弱碱型 弱酸型
基质结构 6%交联度的琼脂糖
排阻极限 4×106
总交换容量/(μmol/ml) 130~170 100~140
有效交换容量/(mg/ml)①
甲状腺球蛋白(669kDa) 2.0 —
人血清白蛋白(68kDa) 170 —
α乳清蛋白(14.3kDa) 150 —
免疫球蛋白G(160kDa) — 9.5
牛碳氧血红蛋白(69kDa) — 75
核糖核酸酶(13.7kDa) — 120
工作pH范围 2~9 6~10
pH稳定性
短时间 2~14 2~14
长时间 3~12 4~13
粒形与粒度 球状颗粒,湿胶粒径45~165μm,均值90μm
最大流速/(cm/h) 100
建议工作流速/(cm/h) <60
化学稳定性 在常 用 缓 冲 液、1mol/LNaOH 或 乙 酸、8mol/L尿 素 或 盐 酸 胍、乙
醇、甲醇等有机溶剂中均稳定
物理稳定性 在中性pH和以盐形式存在时,可于121℃灭菌30min。颗粒硬 度
大于Sephadex系列的离子交 换 剂,溶 液pH和 离 子 强 度 对 体 积 影 响
很小
① 有效容量测定实验条件:流速75cm/h,阴离子交换剂使用的起始 缓 冲 液 为pH=83,005mol/L的TrisHCl缓冲液,阳离子交换剂使用的起始缓冲液为pH=50,01mol/L的醋酸缓冲液,极限缓冲液
为含2mol/LNaCl的相应起始缓冲液。

80
表410 SepharoseHighPerformance离子交换剂的特性
项 目 QSepharoseHighPerformance SPSepharoseHighPerformance
功能基团 三甲胺基 磺丙基
功能基团类型 强碱型 强酸型
总交换容量/(μmol/ml) 140~200 140~200有效交换容量/(mg/ml)
牛血清白蛋白(67kDa) 70 —
核糖核酸酶(13.7kDa) — 55建议工作流速/(cm/h) <150
基质粒度 粒径24~44μm,平均34μm
排阻极限 4×106
工作pH范围 2~12 4~13
pH稳定性
短时间 1~14 3~14 长时间 2~12 4~13
化学稳定性 在常用缓冲液、8mol/L尿 素、6mol/L盐 酸 胍、70%乙 醇、1mol/L
NaOH中稳定
Pharmacia还将SepharoseHighPerformance介质填充成预装柱出售,商品
名为 HiTrap、HiLoad和BioPilot,其柱尺寸顺序增大,分别满足从实验室到小
规模生产等不同规模分离的要求。
SepharoseFastFlow也是基于高度交联琼脂糖的颗粒状交换剂,其凝胶颗粒粒
径要大于SepharoseHighPerformance系列,平均为90μm,这使得它比后者具有更
好的流 速 性 能,其 线 性 流 速 最 大 可 达750cm/h,适 合 大 规 模 分 离 纯 化 的 要 求。
SepharoseFastFlow系列常见的包括四种介质 (表411),覆盖了强阴、强阳、弱
阴、弱阳四种类型。Pharmacia也将SepharoseFastFlow预装成HiLoad柱出售。
SepharoseBigBeads同样是基于高度交联琼脂糖的颗粒状交换剂,其凝胶颗
粒粒径更大,平均为200μm,大的粒度赋予该介 质 极 好 的 流 速 特 性,即 使 在 分
离黏性样品时仍能保持高的流速,因此非常适合大规模分离使用。该系列主要有
QSepharoseBigBeads和SPSepharoseBigBeads两种介质,都属于强离子交换
剂,其特性见表412。
表411 SepharoseFastFlow离子交换剂的特性
项 目QSepharose
FastFlow
SPSepharose
FastFlow
DEAESepharose
FastFlow
CMSepharose
FastFlow功能基团 三甲胺基 磺丙基 二乙基氨乙基 羧甲基
功能基团类型 强碱型 强酸型 弱碱型 弱酸型
总交换容量/(μmol/ml) 180~250 180~250 110~160 90~130

81
续表
项 目QSepharose
FastFlow
SPSepharose
FastFlow
DEAESepharose
FastFlow
CMSepharose
FastFlow有效交换容量/(mg/ml) 甲状腺球蛋白(669kDa) 3 — 3.1 人血清白蛋白(68kDa) 120 — 110 — α乳清蛋白(14.3kDa) 110 — 100 — 免疫球蛋白G(160kDa) — 50 — 15 牛碳氧血红蛋白(69kDa) — 50 — 30 核糖核酸酶(13.7kDa) — 70 — 50基质粒度 粒径45~165μm,平均90μm排阻极限 4×106
工作pH范围 2~12 4~13 2~9 6~10
pH稳定性
短时间 1~14 3~14 1~14 2~14 长时间 2~12 4~13 2~13 4~13最大流速/(cm/h) 750
建议工作流速/(cm/h) 100~300
化学稳定性 在 常 用 缓 冲 液、8mol/L 尿 素、8mol/L 盐 酸 胍、70%乙 醇、1mol/L
NaOH中均稳定
物理稳定性 在中性pH和以盐形式 存 在 时,可 于121℃灭 菌30min。溶 液pH 和
离子强度对体积影响很小
保存条件 20%乙醇
表412 SepharoseBigBeads离子交换剂的特性
项 目 QSepharoseBigBeads SPSepharoseBigBeads功能基团 三甲胺基 磺丙基
功能基团类型 强碱型 强酸型
总交换容量/(μmol/ml) 180~250 180~250建议工作流速/(cm/h) 1200~1800基质粒度 粒径100~300μm,平均200μm排阻极限 4×106
工作pH范围 2~12 4~13pH稳定性
短时间 2~14 3~14 长时间 2~12 4~13化学稳定性 在常用缓冲液、8mol/L尿素、6mol/L盐酸胍、70%乙醇、1mol/LNaOH中稳定
STREAMLINE系列则 是 专 门 为 膨 胀 床 吸 附 技 术 而 开 发 的 吸 附 剂,其 基 质
类似于其他Sepharose系列的介质,包括STREAMLINESP和STREAMLINEDEAE两种,这里不再展开介绍。
BioRad公司开发的BioGelA系 列 是 基 于4%交 联 琼 脂 糖 的 离 子 交 换 剂,主要有DEAEBioGelA和CMBioGelA两种,它们同样具有物 化 稳 定 性 高、

82
回收率好等特点。这两种介质的特性见表413。
表413 BioGelA离子交换剂的特性
项 目 DEAEBioGelA CMBioGelA
功能基团 二乙基氨乙基 羧甲基
功能基团类型 弱碱型 弱酸型
总交换容量/(μmol/ml) 20±5 20±5
有效交换容量(血红蛋白)/(mg/ml) 45±10 45±10
工作pH范围 2~95 45~10
粒度/μm 80~150 80~300
排阻极限 5×106
线性流速/(cm/h) >20
4245 高效离子交换剂
高效离子交换剂指的是在中压色谱或高压色谱中使用的粒度较小而有一定的
硬度,分辨率很高的一类离子交换剂,其中包含了很多种类。
Pharmacia公司的SOURCE系列离子交换剂是在聚苯乙烯二乙烯苯的表面连
接上亲水基团后再结合功能基团而形成的。聚苯乙烯二乙烯苯的刚性结构确保了
该交换剂有很高的硬度,能够承受非常高的流速,使分离时间大大缩短;亲水的表
面极大地降低了蛋白质和基质之间出现疏水相互作用的可能性,非特异性吸附很
低,因此蛋白质活性的回收率很高。SOURCE系列有两种不同的基质粒度 (15μm和30μm),分别称为SOURCE15和SOURCE30,小的粒度赋予该介质很高的分
辨率。基质大而均一的孔径赋予其很大的表面积,在从小肽、寡核苷酸到大分子蛋
白质的很大的分子范围内都表现出好的交换容量和分辨率。SOURCE系列交换剂
所连接的功能基团主要有Q和S两种,该系列交换剂的特性见表414。Pharmacia公司已将SOURCE系列离子交换剂预装成RESOURCE柱出售。
表414 SOURCE系列离子交换剂的特性
项 目 SOURCE15Q SOURCE30Q SOURCE15S SOURCE30S功能基团 三甲胺基 磺甲基
功能基团类型 强碱型 强酸型
基质 聚苯乙烯二乙烯苯
颗粒特点 刚性、球状、多孔、单分散性
粒度/μm 15 30 15 30有效交换容量/(mg/ml)①
溶菌酶(145kDa) — — 80 80 牛血清白蛋白(67kDa) 45 45 — —工作pH范围 2~12

83
续表
项 目 SOURCE15Q SOURCE30Q SOURCE15S SOURCE30S
pH稳定性
短时间 1~14
长时间 2~12
最大流速/(cm/h) 1800 2000 1800 2000
建议工作流速/(cm/h) 30~600 300~1000 30~600 300~1000
溶剂限制
在醇(C1~C4)/水溶液中稳 定。100%二 甲 亚 砜,二 甲 基 甲 酰 胺,甲 酸 会 改
变色谱行为。应避免氧化剂 和 高 反 应 性 试 剂。可 使 用 非 离 子 型 或 与 交 换 剂
带同种电荷的去污剂
① 测定条件:流速300cm/h,5mg/ml蛋白质溶于20mmol/L的pH=68磷酸钠缓冲液 (溶菌酶)或
20mmol/L的pH=70BisTris丙烷缓冲液 (牛血清白蛋白)。
MonoBeads系列离子交 换 剂 是Pharmacia公 司 开 发 的 又 一 类 高 效 离 子 交 换
介质。其基质成分与SOURCE系列类似,也是聚苯乙烯二乙烯苯的结构连上亲
水基团形成亲水表面。MonoBeads系列的基质粒度比SOURCE系列更小,仅为
10μm,并且粒径分布非常均匀,误差仅±05μm,MonoBeads的名称就由此而
来。高硬度、单分散性的特点使该介质能够在很高的流速下使用,并且不会造成
背景压力过高;小而精细的粒度赋予该介质极高的效率,所有预装的 MonoBeads色谱柱每米的理论塔板数都在25000左右。由于没有非特异性的吸附作用,该介质
对蛋白质总量和活性的回收率都很高。MonoBeads系列离子交换剂主要包括 MonoQ和 MonoS两种类型,其特性见表415,它们都已制成预装柱出售。
表415 MonoBeads系列离子交换剂的特性
项 目 MonoQ MonoS
功能基团 三甲胺基 磺甲基
功能基团类型 强碱型 强酸型
总交换容量/(μmol/ml) 270~370 140~180
有效交换容量/(mg/ml)
甲状腺球蛋白(669kDa) 25 —
人血清白蛋白(68kDa) 65 —
α乳清蛋白(14.3kDa) 80 —
免疫球蛋白G(160kDa) — 75
核糖核酸酶(13.7kDa) — 75
典型的蛋白质总量回收率/% 90~100
典型的活性回收率/% >80
粒度/μm 10±0.5

84
续表
项 目 MonoQ MonoS
排阻极限 >107
工作pH范围 3~11
pH稳定性
短时间 2~14
长时间 2~12
溶剂限制
在醇(C1~C4)/水溶液中稳定。100%二甲亚砜,二甲基
甲酰胺,甲酸会改变色谱行为。应避免氧化剂和高反应 性
试剂。可使用非离子型或与交换剂带同种电荷的去污剂
高效离子 交 换 剂 的 种 类 还 有 很 多,比 如 TosoHaas公 司 的 DEAE3SW 和
CM3SW,它们采用的基质是二氧化硅,外面覆盖带有功能基团的亲水层;BioRad公司的DEAE5PW和CM5PW,采用基质为合成高分子有机聚合物。尽
管适用于中高压色谱的高效离子交换剂种类众多,但 实 验 表 明 有 着 相 同 功 能 基
团、孔结构和颗粒尺寸的交换剂的色谱行为是非常类似的。与常规离子介质相比,高效介质的分辨率有很大的提高,能够取得令人满意
的纯化效果,其缺点是介质的成本更高,处理样品的数量较少,不适合于大规模
的制备。事实上,高效介质一般不用于对粗提取物进行分离纯化,因为其容量十
分有限并且粗提取物中大量的杂质容易污染高效介质。在对粗提取物进行了初步
的纯化操作,去除了大部分杂质后,高效介质由于其高分辨率,在进一步纯化和
最后的精制阶段是十分有用的。
4246 非孔型离子交换剂
在离子交换剂的研制过程中,人们为了获得高的有效交换容 量 趋 向 于 开 发
大孔型的介质,然而在高效 液 相 色 谱 中,现 在 又 开 发 出 一 些 非 孔 型 离 子 交 换
剂,用这类介质进行色谱 时,所 有 的 结 合 都 发 生 在 颗 粒 的 表 面,由 于 蛋 白 质
在 流 动 相 和 固 定 相 间 的 扩 散 距 离 很 小,离 过 程 可 以 很 快 完 成,又 由 于 该 介 质
的 粒 度 非 常 小,因 此 分 辨 率 非 常 高。实 际 上,这 类 介 质 是 在 牺 牲 了 容 量 的 前
提 下,可 以 比 网 孔 型 介 质 分 离 速 度 快5~6倍 的 基 础 上 获 得 比 后 者 更 高 的 分
辨 率,所 以 这 类 介 质 适 合 于 在 纯 化 阶 段 的 后 期 进 行 分 析 型 或 微 量 制 备 型 的 分
离 纯 化。
Pharmacia公司的 MiniBeads系列离子交换剂就属于这类介质,其基质为非
孔型亲水性聚合物,颗粒直 径 很 小,仅 为3μm,MiniBeads的 名 称 也 由 此 而 来,并且颗粒非常均匀,具有单分散性的特点,这使得介质在高流速下仍能保持较低
的背景压力。MiniBeads系列的交换剂有两种:MiniQ和 MiniS,Pharmacia公

85
司将其 制 成 预 装 柱 MiniQ PC32/3和 MiniSPC32/3后 出 售,其 特 性 见
表416。
表416 MiniBeads系列预装柱的特性
项 目 MiniQPC32/3 MiniSPC32/3
功能基团 三甲胺基 磺甲基
功能基团类型 强碱型 强酸型
柱尺寸(内径×床高)/(mm×mm) 3.2×30
柱体积/ml 0.24
基质粒度/μm 3
总交换容量/(μmol/ml) 60~90 16~30
有效交换容量/(mg/柱)
α淀粉酶(49kDa) 1.4 —
胰蛋白酶抑制剂(20.1kDa) 1.4 —
核糖核酸酶(13.7kDa) — 1.3
溶菌酶(14.3kDa) — 1.3
最大上样量/(mg/柱) 1~1.5
实际可上样量/(μg/柱) <200
典型蛋白质回收率/% 70~90
工作pH范围 3~11
pH稳定性
短时间 1~14
长时间 3~11
最大流速/(ml/min) 1.0
操作压力限/(MPa) 10
正常分离时间/min 5~20
4247 其他离子交换剂
TosoHass公司的Toyopearl系 列 离 子 交 换 剂 是 以 聚 乙 烯 醇 Toyopearl凝 胶
作为基 质,连 接 上 不 同 功 能 基 团 后 形 成 的,目 前 商 品 化 的 仅 DEAE和 CMToyopearl两种。Toyopearl为多孔 型 三 维 网 状 结 构,含 有 丰 富 的 羟 基,骨 架 表
现为高度亲水性,衍生而成的离子交换剂结构牢固,稳定性较好,pH和离子强
度对体积影响不大,其部分特性见表417。

86
表417 Toyopearl系列离子交换剂的特性
项 目 DEAEToyopearl650 CMToyopearl650
功能基团 二乙基氨乙基 羧甲基
功能基团类型 弱碱型 弱酸型
粒度/μm 20~40,40~80 20~40,40~80
工作pH范围 2~9 6~10
排阻极限(聚乙二醇) 1000000
总交换容量/(mmol/ml) 108 120
有效交换容量/(mg/ml)
牛血清白蛋白 26 45
铁蛋白 15.0 —
甲状腺球蛋白 11.5 —
流速/[ml/(h·cm2)] >300
Parath等制备了兼性离子交换剂,在基 质 上 连 接 上 如β丙 氨 酸、对 氨 基 苯
磺酸、精氨酸等偶极离子,由 于 在 兼 性 离 子 周 围 电 场 电 压 下 降 快 于 单 位 电 荷,蛋白质从这类介质上解吸更为容易。蛋白质也以兼性离子形式存在,其 所 带 正
负电荷都能与交换剂结合,而缓冲液中的阴、阳离子也都能与蛋白质竞 争 结 合
位点。与通常的交换剂相比,蛋白质在兼性离子交换剂上结合时有着一 定 的 定
向形式,这与蛋白质分子表面的电荷分布又是直接相关的。因此,用通 常 的 离
子交换剂 不 能 得 到 很 好 分 离 效 果 时,采 用 兼 性 离 子 交 换 剂 有 时 能 取 得 良 好
效果。有人还研究了将包含阴、阳离子的磷脂的脂质体固定在凝胶网孔中,形成特
殊的兼性离子交换剂。研究表明在分离核糖核酸酶A、细胞色素C和溶菌酶时,包含磷脂酰丝氨酸脂质体的介质分辨率甚至要高于 MonoS,并且在很低的离子
强度下即可完成洗脱过程。离子交换剂是离子交换技术的关键所在,其本身的研制和开发仍在不断进行
之中,其中有普遍应用价值的介质也在不断被商品化,为生化分离技术的进步打
下基础。
43 流动相:缓冲液和盐
任何分配色谱都是利用不同物质对固定相和流动相的亲和力不同而进行分离
的,离子交换色谱也属于这种类型,当作为固定相离子交换剂选定后,流动相对
于分离效果的影响同样至关重要。流动相的成分有两类:一是用于维持溶液pH恒定,以确保蛋白质和交换剂带相反电荷而发生结合的缓冲物质;二是在洗脱阶

87
段,通过提高离子强度将蛋白质从交换剂上洗脱下来所用的盐,当然,如果采用
的是改变pH的方法进行洗脱,不需用到此成分。
431 缓冲液的类型
在离子交换过程中保持pH 的恒定是十分重要的,正如前面讨论过的,pH的改变会造成蛋白质带电荷数量和分布状况发生变化,从而直接影响到蛋白质是
否能结合在交换剂上以及结合力的强弱。因此,在离子交换色谱中流动相必须使
用缓冲液。缓冲液的种类很多,能 够 起 缓 冲 作 用 的 物 质 可 分 为 两 类:第 一 类 是 由 弱 酸
(或弱碱)及相应的盐构成的系统;第二类是兼性离子化合物。对于第一类缓冲
物质,在进行离子交换时,如果缓冲离子所带的电荷与离子交换剂上的功能基团
相反,将参与离子交换过程,并可能对局部pH产生影响,因此应尽可能采用与
功能基团带同种电荷的缓冲离子,即:使用阴离子交换剂时选择带正电荷的缓冲
离子;使用阳离子交换剂时选择带负电荷的缓冲离子。当然这也并不是绝对的,比如磷酸盐缓冲液也经常在阴离子交换过程中被采用,但在这种情况下应特别注
意在上样前充分平衡,确保色谱系统的pH和离子强度与起始缓冲液一致。第二
类缓冲物质在阴、阳离子交换中均能采用。表418和表419分别列出了阳离子
交换色谱和阴离子交换色谱时常用的缓冲液。
表418 阳离子交换色谱常用的缓冲液
缓 冲 物 质 pKa 建议浓度/(mmol/L) 平 衡 离 子
马来酸 2.00 20 Na+
丙二酸 2.88 20 Na+/Li+
柠檬酸 3.13 20 Na+
乳酸 3.81 50 Na+
甲酸 3.75 50 Na+/Li+
丁二酸 4.21 50 Na+
乙酸 4.76 50 Na+/Li+
丙二酸 5.68 50 Na+/Li+
磷酸盐 7.20 50 Na+
Hepes[4(2羟乙基)哌嗪1乙磺酸] 7.55 50 Na+/Li+
Bicine[N,N双(2羟乙基)甘氨酸] 8.35 50 Na+
在离子交换过程中虽然可以除去很多杂蛋白,起到纯化效果,但目的蛋白的
洗脱峰中必然含有大量缓冲物质和盐的成分,这些成分的引入对于目的蛋白来说
本身也是一种杂质。特别在色谱后需对洗脱峰进行冷冻干燥,以得到纯蛋白样品
时,在冻干后的粉末中往往绝大部分是缓冲物质和盐。如果在冻干前进行脱盐或

88
透析操作,虽然可以基本除去这些杂质,但 也 有 可 能 造 成 蛋 白 活 性 的 回 收 率 下
降。此时应优先考虑采用挥发性的缓冲物质,这样在冻干阶段可以将这部分杂质
除去,常见的挥发性缓冲物质列于表420。
表419 阴离子交换色谱常用的缓冲液
缓 冲 物 质 pKa 建议浓度/(mmol/L) 平衡离子
N甲基哌嗪 4.75 20 Cl-
哌嗪 5.68 20 Cl-/HCOO-
L组氨酸 5.96 20 Cl-
BisTris 6.46 20 Cl-
BisTris丙烷 6.80 20 Cl-
三乙醇胺 7.76 20 Cl-/HCOO-
Tris 8.06 20 Cl-
N甲基二乙醇胺 8.52 50 SO-2/Cl-/HCOO-
二乙醇胺 8.88 20(pH=8.4),50(pH=8.8) Cl-
1,3二氨基丙烷 8.64 20 Cl-
乙醇胺 9.50 20 Cl-
哌嗪 9.73 20 Cl-
1,3二氨基丙烷 10.47 20 Cl-
哌嗪 11.12 20 Cl-
磷酸盐 12.33 20 Cl-
表420 挥发性缓冲系统
缓冲系统(碱/酸) pKa 平衡离子 缓冲系统(碱/酸) pKa 平衡离子
甲酸/氨水 3.75 H+
吡啶/甲酸 3.75,5.25 HCOO-
三甲胺/甲酸 3.75,9.25 HCOO-
吡啶/乙酸 4.76,5.25 CH3COO-
三甲胺/HCl 9.26 Cl-
N乙基吗啉/乙酸 4.76,7.8 CH3COO-
氨/甲酸 3.75,9.25 HCOO-
氨/乙酸 4.76,9.25 CH3COO-
三甲胺/碳酸 6.50,9.25 CO2-3碳酸氢铵/HCl 6.50,9.26 HCO-3乙醇胺/HCl 9.5 Cl-
432 缓冲液的pH和浓度
离子交换色谱通常至少会使用两种不同的缓冲液:一种用于蛋白质上样并洗
去不吸附的杂质,称为起始缓冲液;另一种用于洗脱吸附在柱上的蛋白质,称为
洗脱缓冲液。达到最终pH和盐浓度的洗脱缓冲液称为极限缓冲液。这里又分为
两种情况,在 完 成 吸 附 后 将 目 的 蛋 白 洗 脱 下 来 有 两 种 方 法:一 种 是 通 过 改 变
pH,使蛋白质的带电状态发生变化而不能结合在交换剂上,此方法会用到两种
不同pH的起始和洗脱缓冲液;另一种是通过增加离子强度,虽然不改变蛋白质

89
的带电状况,但高的离子强度会降低蛋白质与离子交换剂之间的静电引力,从而
将蛋白质洗脱下来,此方法会用到两种pH相同而离子强度不同的缓冲液,通常
极限缓冲液是向起始缓冲液中添加特定浓度的盐类而形成。为了保证目的蛋白在吸附阶段能结合到交换剂上,起始缓冲液的浓度一般比
较低,介于001~005mol/L。但过低的起始浓度有可能造成蛋白质在离子交换
剂上吸附过于牢固而给洗脱造成困难,同时也应当控制吸附阶段的时间。研究显
示,将蛋白质吸附在离子交换柱上过夜再进行洗脱,比直接进行洗脱明显困难,这是由于长时间的吸附会引发蛋白质的构象缓慢发生改变,这种改变使之与交换
剂结合更为牢固,并且这种构象变化一般都会造成蛋白质活性下降。合适的起始
缓冲液浓度可以通过小样试验确定。在采用改变pH的方法洗脱时,洗脱缓冲液的缓冲成分种类往往与起始缓冲
液相同,只是控制比例关系不同造成最终pH不同。洗脱缓冲液在浓度方面并没
有特殊的要求,常与起始缓冲液相同。在采用增加离子强度的方法洗脱时,所用
洗脱缓冲液在缓冲物质种类、pH和浓度上往往都与起始缓冲液相同,通常是通
过向起始缓冲液中添加特定浓度的其他种类盐 (如NaCl)来增加离子强度。所以,实际上无论是何种缓冲液,缓冲物质的浓度通常都不大,为了使其具
有足够的缓冲能力,必须满足一定的条件。根据 HendersonHasselbalch公式:
pH=pKa+lg[A-][HA]
当缓冲液的pH接近缓冲物质的pKa 时,才具 有 良 好 的 缓 冲 能 力。最 大 缓
冲能力出现在pKa 处,偏离pKa 值1个pH单位缓冲能力将下降5倍。一般来
说,缓冲液的有效pH范围约为pKa±2pH单位,而要获得足够强的缓冲能力,
pH最好在pKa±05pH单位之间。当弱酸 (或弱碱)与对应的盐的物质的量之
比接近1∶1时,此缓冲体系的缓冲能力最强。所以在选择缓冲液时,首先应根
据起始缓冲液 所 需 要 的pH,选 择pKa 值 与 之 接 近 的 缓 冲 物 质,使 弱 酸 (或 弱
碱)与对应的盐的物质的量之比接近1。需要注意的是,缓冲物质的pKa 值是会
随温度变化的,在温度相差较大时会对溶液pH产生明显的影响。例如,在室温
下配制出的Tris缓冲液比在冷藏室 (4℃)中的低005个pH单位。
433 离子对色谱行为的影响
采用增加离子强度的方法洗脱蛋白质时,通常选用某种非缓冲盐类,最常用
的是NaCl,添加到起始缓冲液中,构成了洗脱缓冲液。在进行色谱分离时,洗
脱缓冲液中的缓冲物质固然会影响到色谱效果,实际上非缓冲盐的种类和性质同
样会影响到色谱时的分辨率和选择性,使用不同的非缓冲盐离子时,可能造成不
同物质被洗脱的先后顺序发生变化,因此非缓冲盐的选择同样具有重要意义。
90
在前面离子交换理论部分讨论过,价态高的离子与交换剂的结合力更强,所
以一般情况下,多价离子是比一价离子更好的置换离子,它们能使蛋白质比较早
被洗脱下来 (保留值小)。所以在阳离子交换剂上,蛋白质的保留值与置换离子
的关系为:Ba2+<Ca2+< Mg2+< NH+4 < K+< Na+<Li+,但也存在例外,
比如对于溶菌酶来说,NH+4 是比 Mg2+更有效的置换离子。在选择置换离子时,应当考虑到多方面的因素,强的置换离子能有效缩短色谱时间,但往往也降低蛋
白质回收率。一般情况下选择置换能力居中的盐离子较为合适,在洗脱与交换剂
牢固结合的蛋白质时,选用强的置换离子具有优势。不但置换离子,洗脱盐中与功能基团带同种电荷而不参与离子交换过程的离
子也会影响到蛋白质的色谱行为,原因也有多种。有些是因为离子与蛋白质发生
作用导致的。例如,用 AmberliteIRC50柱 (弱阳 离 子 交 换 柱)分 离 核 糖 核 酸
酶A,分别采用磷酸钠和氯化钠进行洗脱,前者洗脱速度比后者快得多,这是因
为磷酸根与该酶活性中心的组氨酸残基结合导致的。此外,二价离子如Ca2+ 和
Mg2+能与蛋白质分子中的酰胺基形成复合物而影响到其色谱行为。总之,离子对色谱行为的影响是多方面的,没有一定的规律。在首次进行色
谱分离时,可优先选择比较通用的一些缓 冲 物 质 和 盐 类,在 此 基 础 上 再 进 一 步
优化。
434 添加剂
在有些情况下,在进行离子交换时还会添加一些其他的化合物,其作用主要
有:增加蛋白质的溶解度,提高色谱分辨率,保护待分离物质等。大多数蛋白质是溶于水的,但也有部分蛋白质疏水性比较强,在水中的溶解
度很小,这使得色谱操作难于实现。具体例子是膜蛋白的分离。膜蛋白存在于生
物膜中,其表面是疏水区,与生物膜中疏水性的脂质成分以疏水相互作用结合,这类蛋白在水中是不溶的或溶解度很低。向溶液中添加非离子型或兼性离子型去
污剂可以使膜蛋白溶解并分散在水溶液 中,便 于 色 谱 的 进 行。有 机 溶 剂 (如 氯
仿、甲醇等)也能增加疏水性蛋白质的溶解度,而且由于有机溶剂降低溶液的介
电常数,使得蛋白质与交换剂之间的静电作用力更强,吸附更为牢固。某些情况下,添加特定的物质还能提高色谱时的分辨率。例如,在分离一些
蛋白质时,添加甜菜碱或牛磺酸能减少蛋白聚集体的形成及其与交换剂的结合,从而提高了分辨率。有的中性聚合物如聚乙二醇 (PEG)能与蛋白质竞争溶液中
的水分子,这将增加蛋白质与离子交换剂的相互作用而提高分辨率。分离过程中有时还需添加酶抑制剂。例如在蛋白质分离过程中,样品体系中
如果有蛋白酶存在,会造成目的蛋白被水解,使回收率下降,添加蛋白酶抑制剂
能够有效保护目的蛋白。丝氨酸蛋白酶常用的抑制剂是苯甲磺酰氟,巯基蛋白酶

91
常用的抑制剂是碘乙酰胺,金属蛋白酶可采用金属螯合剂作为抑制剂。
44 实验方案设计
不存在一套方案可以适用于不同的蛋白质的分离纯化,因此对于任何一种蛋
白质,要想获得好的分离效果,实验方案的设计和优化是至关重要的。在选择了
采用离子交换技术后,需要考虑的问题包括:分离的规模有多大,采用何种分离
模式,选用什么样的离子交换剂,何种规格的色谱柱,选用何种缓冲液,起始条
件如何确定,采用何种方法洗脱等。只有在这些问题都得到解决之后,才能设计
出初步的实验方案并付诸实施,然后根据实验结果进一步改进方案。
441 离子交换剂的选择
离子交换剂的种类很多,没有一 种 离 子 交 换 剂 能 够 适 合 各 种 不 同 的 分 离 要
求,所以必须根据分离的实际情况,选择适合的离子交换剂,这中间包含了选择
适合的基质和功能基团。在选择离子交换剂时,主要的依据包括:分离的规模,采用的模式和特殊要求,目的蛋白的分子大小、等电点和化学稳定性等性质。
首先是关于分离的规模和选用的模式。对于分析型分离和实验室小规模制备
型分离,一般采用常规的柱色谱 技 术;而 对 于 大 规 模 工 业 化 分 离,可 以 有 柱 色
谱、膨胀床吸附、分批分离等多种模式可供选择。在大规模分离中采用柱色谱一
般都是到了分离的中后期,因为在前期粗样品中往往含有大量的杂质甚至是颗粒
状物质,将这样的样品直接上色谱柱,不但会对离子交换剂造成不可逆的污染,使之难以清洗和再次使用,而且会降低交换剂的吸附容量,加样时间也会很长,使整个分离过程用时很长。而在分离的中后期,大多数杂质已经被除去,选用柱
色谱可以获得良好的分离效果,此时选择离子交换剂时,介质的流速特性和交换
容量应当作为重要参数。其次是这步离子交换在整个目的蛋白的纯化过程中所处的次序以及所要求的
分辨率。任何一种蛋白质的纯化过程都不是一步能够完成的,通常从原料到最终
的纯品要经历预处理、初分级和细分级等阶段,每个阶段的分离手段和目的也不
完全一致。有些情况下,某种蛋白质的纯化过程中也可能使用到两个离子交换步
骤,但在分离过程中所处的次序不同,所采用的模式、离子交换剂、洗脱技术等
也就不同,在预处理和初分级阶段,离子交换的目的是提取和浓缩目的蛋白,而
在细分级和最后精制阶段,离子交换的目的应当是除去样品中混有的杂质,获得
单一目的蛋白。在预处理和初分级阶段采用离子交换技术,应当选用颗粒尺寸较大,具有大
孔型结构,并且流速特性好的离子交换剂,高 效 离 子 交 换 剂 在 此 阶 段 是 不 适 合
的。粗样品中杂质含量大,其中有的杂质会造成交换剂的污染,甚至清洗后也不

92
能完全恢复,造成使用寿命缩短,因此离子交换剂的成本也是应当考虑的因素。相对来说,离子交换树脂、离子交换纤维素、离子交换葡聚糖等价格比较低,有
时也能获得不错的分离效果。在细分级乃至最后的精制阶段,分辨率的要求就越
来越高,而分离过程中随着样品量逐渐减少,容量和速度等都成为次要的指标。此时若采用离子交换法,应当选用小颗粒、高分辨率的介质,高效离子交换剂、非孔型离子交换剂、基于琼脂糖的离子交换剂等都是比较理想的选择。在分离过
程的任何阶段,回收率始终是一个重要参数,在纯化高价值的产品时更是如此。到了纯化过程的后期,回收率会显得更为重要,因为随着不断的纯化,单位产品
的价值在不断增加。接着应当考虑目的蛋白的分子大小和离子交换剂的有效交换容量。离子交换
剂的取代程度决定了其总交换容量,而对于某种蛋白质的有效交换容量,则取决
于这种蛋白质是否能够达到功能基团的表面并发生结合,在此过程中基质网孔的
孔径起着决定性的作用。如果蛋白质分子不能进入离子交换剂的网孔结构,而只
是与基质颗粒表面的功能基团结合,将会大大降低有效交换容量。因此,在分离
分子量较大的蛋白质时,需要采用孔径尺寸较大的基质,在各种离子交换剂中,基于纤维素和琼脂糖的交换剂所形成的孔径是最大的,天然蛋白质的分子量通常
都低于它们的排阻极限。在分离分子量较小的蛋白质时,网孔较小的介质具有一
定的优势,基于交联葡聚糖的交换剂属于此类。在选择离子交换剂时通常不必考
虑分子筛效应对分离结果产生影响的可能性,在离子交换色谱时,电荷作用是决
定离子交换洗脱顺序的主要因 素。例 如,从 蛇 液 中 分 离 小 分 子 蛋 白 质 时,使 用
SPSephadexC25 (排阻极限30kDa)和SPSephadexC50 (排阻极限200kDa)进行色谱得到的结果是相同的。因此,人们在选择交换剂时只要确保基质孔径能
允许目的蛋白分子进入,进一步增加孔径不会影响结果。无论是常规介质还是高
效介质,在具有类似孔径、容量、粒度和亲水性的介质中选择一种时,往往可以
是随意的。以上考虑主要针对离子交换剂基质的选择,而在确定所选用的基质后,选择
何种功能基 团 是 另 一 个 关 键 所 在,这 主 要 取 决 于 目 的 蛋 白 的 电 性 质 和pH 稳
定性。在最理想的状态下,待分离样品中目的蛋白和主要杂蛋白的等电点pI及电
荷随pH的变化情况已经有所了解 (这可以通过绘制蛋白质滴定曲线来实现,见
4112节),则功能基团比较容易确定。如图45所示,假定图中实线代表目
的蛋白的滴定曲线,虚线代表样品中主要杂蛋白的滴定曲线,那么可以获得如下
信息:目的蛋白的pI=70,pH<70将带净的正电荷,而pH>70将带净的
负电荷;主要杂蛋白pI=82,pH<82将带净的正电荷,而pH>82将带净

93
的负电荷;图中两条曲线在pH=90时纵坐标 (两种蛋白质的电荷)差异最大,当然pH进一步增 大 这 种 差 异 还 可 能 进 一 步 增 大,只 是 图 中 未 表 示 出 来,但 随
pH的增大,蛋白质的稳定性会迅速下降,导致活性的回收率下降,而陶南效应
会进一步增强蛋白质的变性,所以在蛋白质的离子 交 换 色 谱 中 很 少 会 使 用 高 于
90的pH。事实上,当pH>75以后,两种蛋白质的电荷差异已经能够满足分
离的要求,但pH=75距离目的蛋白的pI较近,目的蛋白所带净电荷较少,为
了确保目的蛋白具备充足的电荷与离子交换剂结合,选择pH=80~85是比较
合适的,此时目的蛋白带负电荷,所以应选择一种碱性功能基团,也就是选择阴
离子交换剂。多数情况下,人们对样品的了解程度并没有达到上述要求,此时离子交换剂
功能基团的选择依赖于对目的蛋白的等电点和pH稳定性的了解。根据经验,离
子交换色谱时的pH可以选择在目的蛋白pI上下1~2个pH单位,此时既保证
目的蛋白有充足的电荷结合到交换剂上,又不致结合得过于牢固而难以洗脱。而
pH具体选择比pI高1~2个单位还是低1~2个单位,取决于pI的值和蛋白质
的pH稳 定 范 围。例 如,某 蛋 白pI=40,在pH=36~80范 围 内 保 持 稳 定,即pH稳定范围主要分布在pI之上,那么应当选择pH=50~60作为色谱条
件,此时目的蛋白带负电荷,需选用阴离子交换剂;再如,某蛋白pI=80,在
pH=55~85范围内保持稳定,即pH稳定范围主要分布在pI之下,那么应当
选择pH=60~70作为色谱条件,此时目的蛋白带正电荷,需选用阳离子交换
剂;又如,某蛋白pI=60,在pH=40~85范围内保持稳定,也就是说在pI两侧都有较好的稳定性,那么色谱条件既可以选择pH=45~50,此时需选用
阳离子交换剂,也可以选择pH=70~80,此时需选用阴离子交换剂。当然,还存在这样的情况,那就是对目的蛋白的pI和电性质一无所知。鉴
于大多数的蛋白 质pI<70,可 以 优 先 考 虑 选 用 阴 离 子 交 换 剂,分 离 时 采 用 的
pH可通过试管小样法确定,具体操作见后文 (442节)中起始pH确定部分。在选择好使用阳离子或是阴离子交换剂以及色谱的起始pH后,还必须在强
型或弱型离子交换基团中选择一种。强、弱离子交换剂之间最大的差异在于适用
的pH范围不同,强离子交换剂在较宽的pH范围内能保持带电荷状态,而弱离
子交换剂只能在较窄的pH范围内使用。如果在比较极端的pH下才能获得最好
的分辨率而目的蛋白在此pH下又能保持稳定,那么毫无疑问应当选择强离子交
换剂。通常pH<5时,只有带磺酸基或磷酸基的阳离子交换剂能够保持离子化
状态,而pH>9时,需要使用季铵型阴离子交换剂。多数蛋白质的pI在中性或
略偏酸性的范围内,进行分离时采用的pH也往往靠近中性 (pH=6~8),此时
无论选用强离子交换剂还是弱离子交换剂都能实现分离,习惯上常采用弱型离子

94
交换剂。在某些情况下进行离子交换色谱时,除了离子交换作用外,还有其他机制也
会对分离产生很大的影响。例如,在分离较小的碱性蛋白混合物 (如核糖核酸酶
和毒素蛋白)时,采用聚异丁烯酸树脂 AmberliteIRC50或BioRex70可以获
得很高的分辨率,这是由于氢键在分离过程中扮演着非常重要的角色。又如,在
分离一些以包含磷酸基团的化合物为底物的酶 (如氨酰tRNA合成酶、己糖 激
酶、磷酸葡萄糖异构酶、肌酸激酶 等)时,选 用 磷 酸 纤 维 素 可 以 获 得 理 想 的 效
果,这是因为介质上的磷酸基团和酶分子存在专一性的相互作用,有点类似于亲
和色谱。当然,这些情况往往难以预测,因此很难据此制定分离计划和选择离子
交换介质。另外还有一种方法虽然使用相对较少,但有时也能获得令人满意的效果,那
就是使目的蛋白在起始条件下不发生吸附而直接从色谱柱中流出,形成穿透峰,而多数杂蛋白在此条件下都被吸附在色谱柱上,从而达到分离的目的,该过程也
称为起始相洗脱。此时应当通过选择离子交换剂及控制pH条件使目的蛋白和功
能基团带同种电荷。例如,某蛋白pI=85,pH稳定范围在50~80,样品中
主要杂蛋白的pI都在中性和略偏酸性范围内,此时可以选择一种阴离子交换剂,如DEAE纤维素,在pH=75条件下进行色谱,此时目的蛋白带正电荷,不被
吸附而直接流出,杂蛋白带负电荷而被吸附在色谱柱上,从而实现了分离。
442 缓冲液的选择和色谱条件的确定
流动相缓冲液的确定包括缓冲物质的种类,起始缓冲液的pH和离子强度,极限缓冲液的pH和离子强度等。
一般的原则是,进行阳离子交换时,选用缓冲离子为阴离子的缓冲物质;进
行阴离子交换时,选用缓冲离子为阳离子的缓冲物质 (表418、表419)。当然
这并不是绝对的,但是当选用的缓冲离子与功能基团带相反电荷时,应注意在上
样前确保色谱系统与起始缓冲液之间在pH和离子强度上都达到平衡。在此前提
下,选用何种类型的缓冲物质取决于色谱时的pH条件,后者又与目的蛋白的等
电点有关。前 文 中 已 经 提 及,在 已 经 知 道 目 的 蛋 白 等 电 点 时,起 始 缓 冲 液 的
pH一般选择距离目的 蛋 白 的pI约1~2个pH单 位,对 于 阳 离 子 交 换,起 始
缓冲液pH低于等电点1~2个pH单位;对于阴离子交换,起 始 缓 冲 液pH高
于等电点1~2个pH 单 位。确 定 了 起 始pH 以 后,根 据 缓 冲 物 质 的pKa 值,选用pKa与起始pH很 接 近 的 物 质 作 为 缓 冲 成 分。例 如,某 蛋 白 质pI=65,选用DEAESephadexC50作为色谱介质,起始缓冲 液pH定 为80,则 可 以 选
择Tris缓冲液 (pKa=806),该成分在此pH下有着很好的缓冲能力。至于起
始缓冲液的浓度,一般情况下低一些比较合适,表418和表419中列出了建议

95
浓度,能够保证缓冲能力,而过高的浓度将会带来额外的溶液离子强度的增加,可能对目的蛋白的吸附产生影响。当然,为了获得最好的效果,起始缓冲液的离
子强度可以采用能使目的蛋白发生吸附的最高离子强度,而洗脱时则采用使目的
蛋白解吸所需的最低离子强度,至于这些离子强度分别为多少,可以通过试管小
试法确定。在很多情况下,对目的蛋白的性质还不清楚,甚至有时连目的蛋白到底是何
种蛋白质也是未知的,此时缓冲液及起始pH的确定可以通过试管小试法确定,具体操作如下:①准备10支15ml试管;②每管加入01g基于交联葡聚糖的离
子交 换 剂 或15ml基 于 琼 脂 糖 或 纤 维 素 的 离 子 交 换 剂;③配 制 一 系 列 浓 度 为
05mol/L的不同pH的缓冲液,相邻两种缓冲液的pH相差05个 单 位,如 果
使用阴离子交换剂,这个系列的缓冲液pH分布在5~9,如果使用阳离子交换
剂,这个系列的缓冲液pH分布在4~8,随着pH的不同可能需要选择不同的缓
冲物质,因为任何一种缓冲物质都很难在如此广的pH范围内保持良好的缓冲能
力;④各取10ml上述不同pH的缓冲液分别加入10支试管中用以平衡交换剂,混合一段时间后弃去上清液,再分别加入10ml新鲜缓冲液,反复10次后可以使
试管内的交换剂在pH上完全与缓冲液达到平衡 (之所以采用高浓度的缓冲液是
为了使离子交换剂能迅速在pH方面达到平衡,此过程通常远远慢于离子强度方
面达到平衡);⑤再用10ml低浓度 (交联葡聚糖交换剂可采用005mol/L的浓
度,琼脂糖和纤维素交换剂可采用001mol/L的 浓 度)的 同 一pH 的 缓 冲 液 洗
涤各试管中的交换剂,反复5次可确保试管内的交换剂在离子强度方面与起始缓
冲液保持 一 致;⑥各 支 试 管 中 加 入 相 同 数 量 的 样 品,混 合 后 放 置5~10min;
⑦使离子交换剂沉降,分析上 清 液 中 目 的 蛋 白 含 量,结 果 如 图412 (a)所 示。从图中可以看出,当起始缓冲液pH>70时,目的蛋白可以完全吸附在离子交
换剂上,因此选择pH=70~75作为起始pH。在此pH范围内,如果进行阳
离子交换色谱,可选择磷酸盐缓冲液或 Hepes缓冲液等;如果采用阴离子交换
色谱,可选择Tris缓冲液或三乙醇胺等。如果离子交换色谱过程中,在完成吸
附后采用改变pH的方法来洗脱目的蛋 白,那 么 从 图 中 可 以 看 出 选 择pH=50的洗脱缓冲液即可。
在离子交换色谱中,确定起始缓冲液的离子强度时,多数情况下直接由缓冲
物质提供离子强度,不再向缓冲液中添加 非 缓 冲 盐。缓 冲 物 质 的 浓 度 (一 般 为
002~005mol/L)决定了离子强度,只要起始pH选择合适,在此离子强度下
目的蛋白完全能够与交换剂结合。但有些蛋白质在这种低的离子强度下可能会与
交换剂结合过于牢固而难以洗脱,甚至导致结构破坏,此时在吸附阶段就应当向
起始缓冲液中额外添加非缓冲盐来提供较高的离子强度,减弱蛋白质与交换剂之

96
图412 试管法确定起始pH和离子强度
(a)确定起始pH为70;(b)确定起始盐浓度为015mol/L,洗脱盐浓度为03mol/L
间的作用力。非缓冲盐的浓度应当是多少,可以通过试管小试法确定。另外,多
数情况下在完成吸附后,人们通过增加洗脱液离子强度的方式将目的蛋白从色谱
柱上洗脱下来,洗脱缓冲液就是由起始缓冲液添加特定浓度的非缓冲盐组成,而
非缓冲盐所需的浓度同样可以通过试管小试法确定。试管法确定起始和洗脱缓冲
液所需离子强度的具体操作类似于起始pH的确定,只是10支试管中分别用相
同pH而不同浓度非 缓 冲 盐 的 缓 冲 液 分 别 平 衡,相 邻 两 管 非 缓 冲 盐 的 浓 度 相 差
005mol/L,然后加入等量蛋白质,充分混合后也对上清液目的蛋白含量进行分
析,结果见图412 (b)。从图中可以看出,015mol/L是使得目的蛋白能够吸
附的最高非缓冲盐浓度,而03mol/L是使得目的蛋白发生解吸的最低非缓冲盐
浓度。由此确定,起始缓冲液中应添加非缓冲盐使其终浓度为015mol/L,而洗
脱缓冲液中非缓冲盐浓度增加到03mol/L,这两种缓冲液的缓冲物质组成、浓
度及pH均应相同。在对由少数几种成分组成的混合样品进行分离时,绘制色谱图是确定分离条
件的非常 有 效 的 手 段,通 过 色 谱 图 可 以 很 方 便 确 定 合 适 的 离 子 交 换 剂、起 始
pH、起始和洗脱离子强度。色谱图绘制的方法为:选择阴、阳离子交换剂各一
种,如 MonoQ和 MonoS,分别填充成一系列相同的色谱柱 (当然也可以各填
充一根柱,每次在一种pH条件下进行实验,做完后再平衡到另一种pH条件进
行实验),选择7种具有相同离子强度和不同pH的缓冲液分别充分平衡7根色
谱柱,例如,实验中采用pH=30(A)、pH=50(B)、pH=65(C)的缓冲液
平衡 MonoS柱,采用pH=80(D)、pH=95(E)、pH=110(F)、pH=120(G)的缓冲液平衡 MonoQ柱,然后各柱均加入样品,假设样品中含有Ⅰ、Ⅱ、
Ⅲ三种主要组分,用非缓冲盐的浓度梯度洗脱,即逐步增加洗脱缓冲液中的非缓
冲盐浓度,记录下每根柱中三种组分的洗脱顺序以及洗脱峰对应的离子强度、洗

97
图413 色谱图的绘制
脱时间或洗脱体积。图413 (a)显示了在pH=30、50、110条件下的洗脱
情况,从各峰对应的横坐标可以读出洗脱时的离子强度。然后以Ⅰ、Ⅱ、Ⅲ三种
组分在各pH条件下被洗脱时的离子强度对pH作图,得到图413 (b)所示曲
线。在上述7种pH对应的横坐标处都有三个点,分别对应了此pH条件下三种
组分被洗脱时所需的离子强度。例如在pH=30时,对应的三个点纵坐标由小
到大为Ⅲ、Ⅱ、Ⅰ,说明此pH下各组分的洗脱顺序是Ⅲ、Ⅱ、Ⅰ;在pH=80时,洗脱顺序为Ⅱ、Ⅰ、Ⅲ;而在pH=95时,洗脱顺序为Ⅱ、Ⅲ、Ⅰ……从
图中可以看出,在pH=65时,Ⅰ和Ⅱ几乎在相同的离子强度被洗脱而无法实

98
现分离,在pH=30~50及90~105范 围 内,三 种 组 分 能 够 实 现 很 好 的 分
离。因此,色谱条件可以选择为:采用阳离子交换剂 MonoS,起始pH为50;或采用阴离子交换剂 MonoQ,起始pH为90,均采用增加洗脱液中NaCl浓度
的方法进行洗脱。当然 实 验 中 具 体 采 用 哪 套 方 案 还 应 考 虑 目 的 蛋 白 的pH 稳 定
范围。选择缓冲液时除了考虑缓冲物质成分、pH和离子强度外,有时还需要注意
其他一些问题。如前所述,当离子交换后的洗脱组分需要进行冷冻干燥时,应考
虑选用挥发性的缓冲物质,以尽可能少地将杂质成分带入最终产品中。如果是大
规模工业化分离,缓冲液的用量往往是非常大的,缓冲物质的价格也是必须考虑
的问题,应当在pKa 值 相 近 的 几 种 缓 冲 物 质 中 选 用 最 廉 价 的。在 某 些 情 况 下,有的缓冲物质可能会对分离和检测产生干扰,例如 与 蛋 白 质 上 特 定 基 团 发 生 结
合,或是在紫外区有吸收而干扰蛋白质的A280检测等。以上是离子交换色谱 前 对 交 换 剂、缓 冲 液、起 始 条 件 等 进 行 选 择 的 一 般 原
则,而在实际的实验研究 中,有 时 也 不 完 全 遵 循 上 述 规 则。例 如,在 使 用BioRex70或AmberliteIRC50分离神经毒素及其衍生物时常使用pH=79的碳酸
氢铵缓冲液和pH为中性的乙酸铵缓冲液,在上述pH条件下,这两种缓冲物质
的缓冲能力都是非常弱的,但BioRex70树脂本身是一种强的缓冲剂,洗脱时
的实际pH取决于树脂的平衡条件及缓冲液的离子强度。不仅如此,有些研究中
在使用本身没有强缓冲能力的交换剂如SPSephadexC25或BioGelTSKSP时
也经常使用pH为中性的乙酸铵缓冲液。另外,缓冲离子为阴离子的磷酸盐缓冲
液也经常在阴离子交换过程中被使用。所以,任何一套实验方案都应该通过实践
检验并不断优化。
443 色谱柱的尺寸
色谱柱的尺寸通常用内径 (mm)×高度 (mm)来表示,据此可以计算 出
另外两个常用指标:柱体积 (ml)和高径比。色谱柱的体积直接决定了离子交换剂的用量,而实验中离子交换剂的用量应
取决于待分离样品的量以及离子交换剂对该样品的有效交换容量。但是实践中很
少采用这种方法来计算交换剂的用量,这是因为样品中的蛋白质含量往往不太确
定,而且样品中除了目的蛋白之外还含有大量的杂蛋白,在给定的色谱条件下,有些杂蛋白能够与交换剂结合,这种结合会影响到目的蛋白在交换剂上的有效交
换容量,所以通常情况下柱体积应当足够大。为了在色谱中获得很好的分辨率,建议样品蛋白质的量不要超过有效交换容量的10%~20%,当然,柱体积进一
步加大,除了交换剂的成本会增加,不会对色谱结果产生不利影响。然而受实验
条件的影响,往往是根据已有色谱柱的体积来决定加样量的多少,这些将在后文

99
中介绍。同其他高选择性的吸附技术一样,离子交换色谱通常选用短的色谱柱,即高
径比小的色谱柱,典型的离子交换柱高度在5~20cm,高径比一般小于5。这是
由于离子交换分离蛋白质是依靠吸附强弱的不同,发生吸附时蛋白质会优先结合
在色谱柱的上部,如果柱床过高,当发生解吸时蛋白质需要经过一段较长的色谱
床才能从柱下端流出,这会造成区带扩散,导致洗脱峰变宽,而且使操作时间大
大延长。在大规模的分离过程中,确定分离参数后,可以通过增加色谱柱的直径
来达到扩大分离规模的目的。有一个例外就是同溶剂洗脱,从上样到目的蛋白洗
脱始终使用同一种缓冲液,此时需要高径比大的色谱柱,因为多种蛋白质在柱上
同时发生迁移,色谱柱越长,区带间的距离拉得越大,分辨率就越高。
444 分批分离
严格来说,分批分离过程不属于色谱的范畴,这是一种在色谱柱外的容器中
完成吸附和洗脱过程的离子交换技术,它是由柱分离衍生而来的,但是其本质与
柱色谱时的阶段洗脱并无本质区别。采用分批分离法,起始条件 (pH和离子强度)的确定与柱色谱相同,但是
考虑到该过程中蛋白质与交换剂的结合不如色谱 时 充 分,为 了 获 得 最 大 的 回 收
率,所选择的起始条件应当使目的蛋白在交换剂上吸附得比柱色谱时更为牢固,比如将起始pH定在偏离目的蛋白等电点2~3个pH单位处。进行分批分离时,将样品溶液与已经用起始缓冲液平衡好的离子交换剂混合,轻微搅拌,直至样品
的吸附达到平衡。此过程所需的时间与样品、交换剂的体积以及它们体积之比有
关,可通过测定上清液目的蛋白含量来确定。通常1h的时间对于完成吸附过程
是比较充裕的。然后将混合物过滤,除去上清液,如果发现上清液中有目的蛋白
存在,可适量添加新的离子交换剂再次吸附,过滤,将两次的离子交换剂合并。用起始缓冲液洗涤离子交换剂,除去其表面沾染的杂蛋白,在此条件下目的蛋白
不会被洗脱。然后添加1~2倍于离子交换剂体积的洗脱缓冲液,混合均匀,轻
微搅拌直至目的蛋白充分解吸,这通常需要30min或 更 长 的 时 间。最 后 过 滤 得
到的滤液即为含有分离后的目的蛋白的溶液。还有一种方法将分批分离和柱色谱
结合,即在分批分离操作中,完成吸附,除去上清液并完成对交换剂的洗涤后,将结合着目的蛋白的离子交换剂装入色谱柱中,然后用与柱色谱相同的方法进行
阶段洗脱,此方法的分辨率低于柱色谱而高于一般的分批分离过程。相对于柱色谱,分批分离是一种粗糙的分离手段,介质的选择上首先考虑经
济性,廉价的、能够以固态形式直接添加的介质 (如基于纤维素或交联葡聚糖的
离子交换剂)都是很理想的选择。分批分离过程一般用在分离纯化过程的初期,是一种快速分离技术,尽管其

100
分辨率明显低于柱色谱,但它也具有一些特殊的优势。例如在处理体积很大而蛋
白浓度又很低的样品时,采用柱色谱分离,仅加样就会耗费大量的时间,而分批
法速度会快得多,而且通过控制添加洗脱缓冲液的体积,可以起到很好的浓缩目
的蛋白的作用。此外,当样品溶液黏度较大时,用柱色谱分离柱中会产生很高的
背景压力,使流速大大降低,有时样品中含有脂类杂质甚至会堵塞色谱柱,而采
用分批分离就不存在这样的问题。
45 色谱技术
451 离子交换剂的准备
确定实验方案后,在进行离子交换色谱前,必须先将离子交换剂准备好,这
包括离子交换剂的预溶胀和清洗、装柱以及用起始缓冲液平衡色谱柱,直至流出
液在pH和离子强度方面与起始缓冲液一致。
4511 离子交换剂的预处理
各种不同的离子交换剂在使用前所需进行的预处理是不同的。
Source系列、MonoBeads系列和 MiniBeads系列等细颗粒介质通常 产 品 供
应商已经将其制成预装柱出售,使用前不需预处理,直接用起始缓冲液平衡后即
可加样。基于琼脂糖 的Sepharose系 列 和BioGelA系 列 离 子 交 换 剂,以 及DEAE
Sephacel交换剂是以液态湿胶形式出售的,一般也无需预处理,使用前倾去上清
液,按湿胶∶缓冲液(体积比)=3∶1的比例添加起始缓冲液,搅匀后即可装柱。基于Sephadex的离子交换剂以固态干胶形式出售,使用前需进行溶胀。溶
胀是介质颗粒吸水膨胀的过程,膨胀度与基质种类、交联度、带电基团种类、溶
液的pH和离子强度 等 有 关。溶 胀 过 程 通 常 应 将 交 换 剂 放 置 在 起 始 缓 冲 液 中 进
行,完全溶胀在常温下需1~2天,在沸水浴中需2h左右,在高温下溶胀还能起
到脱气的作用。在溶胀过程中应避免强烈搅拌,例如用磁力搅拌器搅拌可能导致
凝胶颗粒破裂。另外,对干胶进行溶胀时不能使用蒸馏水,因为在溶液离子强度
很低的情况下,交换剂内部功能基团之间的静电斥力会变得很大,容易将凝胶颗
粒胀破。基于纤维素的离子交换剂也是以干态形式出售的,使用前需要进行溶胀。纤
维素的微晶结构是不会被静电斥力破坏的,故可以用蒸馏水进行溶胀,加热能加
快此过程。离子交换纤维素在加工制造过程中会产生一些细颗粒,它们的存在会
影响流速特性,应当将其除去。具体方法是 将 纤 维 素 在 水 中 搅 匀 后 进 行 自 然 沉
降,一段时间后将上清液中的漂浮物除去,然后再加入一定体积的水混合,反复
数次即可。有些纤维素产品制造后未经处理,其中含有很多杂质成分,在使用前

101
必须进行洗涤。洗涤的方法是先用05mol/L的NaOH浸泡半小时,除去碱液,用蒸馏水将交换剂 洗 至 中 性,再 用05mol/L的 HCl浸 泡 半 小 时,然 后 洗 至 中
性,再用05mol/L的NaOH浸泡半小时,最后洗至中性即可。酸碱洗涤顺序也
可反过来,用酸—碱—酸的顺序洗涤。酸碱处理的顺序决定了最终离子交换剂上
平衡离子的类型,对于阴离子交换纤维素,用碱—酸—碱的顺序洗涤,最终平衡
离子是OH-,用酸—碱—酸的顺序洗涤,平衡离子为Cl-;对于阳离子交换纤
维素,用碱—酸—碱的顺序洗 涤,最 终 平 衡 离 子 是 Na+,用 酸—碱—酸 的 顺 序
洗涤,平衡离子为 H+。洗涤以后,还必须用酸或碱将离子交换纤维素的pH调
节到起始pH,再装柱使用。离子交换树脂有的以固态颗粒形式出售,有的以液态形式出售,使用前的处
理方法类似于离子交换纤维素,也要先除去细颗粒,然后用酸碱轮流洗涤,洗涤
时所使用的酸碱浓度大于纤维素交换剂,一般可用2mol/L的NaOH或 HCl。
4512 装柱
装柱是色谱技术的一个重要环节,装柱质量的好坏直接影响到分离效果,填
充得不好的色谱柱会导致柱床内液体流 动 不 均 匀,造 成 区 带 扩 散,大 大 影 响 分
辨率。装柱之前先用缓冲液将色谱柱底部死空间内的空气排空,具体操作可将柱下
端接口连接恒流泵,缓冲液从色谱柱下端 泵 入,直 至 柱 中 可 以 看 到 少 量 液 体 为
止。将预处理好的离子交换剂放 置 在 烧 杯 中,倒 掉 过 多 的 液 体,大 致 使 得 交 换
剂∶上清液 (体积比)=3∶1,轻微搅拌混匀,利用玻棒引流尽可能一次性将介
质倾入色谱柱,注意液体应沿着柱内壁流下,防止有气泡产生,当介质沉降后发
现需要再向柱内补加交换剂时,应当将沉降表面轻轻搅起,然后再次倾入,防止
两次倾注时产生界面。装柱后应当对装柱情况进行检查,可以在柱子后方放置一盏灯,或者将柱子
对着亮光,利用透过光检查柱床是否规整,是否有气泡或界面存在。应慎用染料
物质来检查装柱情况,因为染料分子往往带有大量电荷,例如蓝色葡聚糖2000会牢固结合在阴离子交换剂上而难于洗脱。
4513 选择预装柱
很多公司将自己的色谱介质制成预装柱后出售,由于采用了优良的材料,合
理的色谱柱结构设计和良好的装柱技术,这些色谱柱在耐用性、清洗效果、柱效
率等很多方面要优于自装柱。预装柱有不同的尺寸规格,可适用于不同的分离规
模。在条件许可的情况下,选择合适的预装柱有助于获得理想的分离效果。
4514 柱的平衡
平衡的目的是为了确保离子交换剂上的功能基团与起始缓冲液中的平衡离子

102
间达到吸附平衡。判断色谱柱是否已经达到平衡是通过检验柱中流出的洗脱液与
起始缓冲液在pH、电导和离子强度方面是否达到一致,由于在这几个指标中通
常pH最难达到平衡,所以往往检测洗脱液的pH即可。对于一些弱型离子交换剂,由于本身具有一定的缓冲能力,直接用起始缓冲
液是很难将其平衡至起始pH的,此类交换剂在装柱前就应当用酸或碱将其pH先调节至起始pH,装柱后再用起始缓冲液进行平衡。有时为了加快离子交换剂
达到平衡的速度,先选择与起始缓冲液有着相同pH但是浓度更大 (如05mol/L)的缓冲液平衡色谱柱,由于其缓冲能力更大,较短时间内交换剂在pH方面就达
到了平衡,再换用起始缓冲液进行平衡,最终使洗脱液在离子强度方面也与起始
缓冲液一致。通常在pH预先调节的情况下,至少使用相当于2倍色谱柱体积的
起始缓冲液可以完成平衡过程。
452 样品的准备
想要获得好的分辨率及延长色谱柱的使用寿命,样品中必须无颗粒状物质存
在。通常颗粒状物质主要出现在粗样品中,其成分多为细胞碎片、不溶性蛋白质
聚合物、脂质污染物等,而色谱后获得的样品中一般不会含有颗粒物。因此浑浊
的样品在上柱前先过滤或离心除去颗粒状物质。一般来说,所使用的分离介质颗
粒越小,对样品溶 液 的 澄 清 度 要 求 越 高。在 使 用 平 均 粒 度 在90μm以 上 的 介 质
时,使用孔径 为1μm的 滤 膜 进 行 过 滤 就 能 够 达 到 要 求;在 使 用 平 均 粒 度 小 于
90μm的介质时,应使用孔径为045μm的滤膜进行过滤;需要无菌过滤或澄清
度特别高的样品时,可使用孔径为022μm的滤膜进行过滤。当样品体积非常小
或滤膜对目的 蛋 白 有 吸 附 作 用 时,为 了 减 少 样 品 的 损 失,选 择 在10000g离 心
15min也能除去颗粒物。样品的黏度是影响上样量和分离效果的一个重要方面,高黏性的样品会造成
色谱过程中区带不稳定及不规则的流型。因此样品相对于洗脱剂的黏度有一定的
限制,通常样品黏度最大不超过4×10-3Pa·s,对应的蛋白质浓度不超过5%。如果样品黏度过高是由于浓度过大造成的,那么用起始缓冲液稀释样品可以达到
降低黏度的目的;如果黏度是由样品中存在的核酸等杂质引起的,可以通过添加
大分子聚阳离子化合物如聚乙酰亚胺或鱼精蛋白硫酸盐将其沉淀,或者添加核酸
内切酶来降低黏度,但这些物质的添加无疑会引入额外的杂质,在后续分离中要
将其除去。在离子交换色谱上样前必须确保样品溶液在pH和离子强度方面与起始缓冲
液是一致的,这样才能保证在起始条件下目的蛋白能吸附在离子交换剂上。对于
固体样品,将其溶于起始缓冲液并校对pH即可实现,不过由于样品中含有离子
形式的杂质,以及目的蛋白本身也是离子形式,样品溶液的离子强度会略大于起

103
始缓冲液,这种离子强度的增加一般可忽略不计,当发现增加的程度影响到目的
蛋白的吸附时可进行适当的稀释。对于蛋白质溶液,可以按一定的比例与浓缩形
式的起始缓冲液 (pH相同而浓度为起始缓冲液的2~10倍)混合,具体比例根
据缓冲液浓度及样品体积进行计算,使混合溶液的离子强度与起始缓冲液达到一
致。当然,由于与上述固体样品同样的原因,样品溶液的离子强度也会略大于起
始缓冲液。要想使样品溶液的离子成分与起始缓冲液完全相同,最好的方法是进
行缓冲液交换或透析操作。缓冲液交换一般是通过SephadexG25凝胶过滤来实
现的,将样品加到已经用起始缓冲液平衡好的凝胶柱上,蛋白质等大分子从外水
空间流出,而样品溶液中的小分子进入凝胶颗粒内部,由此大小分子间实现了组
别分离,当蛋白质从凝胶柱下方流出时,已经溶于起始缓冲液体系中了。采用透
析法时,将样品置于透析袋内,放置在盛有起始缓冲液的大烧杯中,缓慢搅拌,使透析袋内外离子达到平衡,更换烧杯内的起始缓冲液,反复数次可使样品溶液
与起始缓冲液在pH和离子强度方面达到一致。当样品的体积很大时,无法采用
凝胶过滤或透析法,此时可以通过稀释样品的方法使其离子强度与起始缓冲液一
致,再用酸碱调节pH至起始pH,同样能够确保目的蛋白发生吸附。
453 加样
当色谱柱和样品都准备好以后,接下来就是将样品加到色谱柱上端,使样品
溶液进入柱床,吸附目的蛋白,并用起始缓冲液将不发生吸附的杂蛋白洗去。
4531 加样量的选择
虽然在设计实验方案时,应当考虑根据所需分离样品的量来确定色谱柱的规
格和离子交换剂的用量,但在很多情况下,人们需要根据已有实验条件,即色谱
柱和离子交换剂的规格,来确定色谱时的最大加样量。理论上,将所使用的离子交换剂的有效交换容量 (mg蛋白/ml胶)乘以色
谱柱柱床的体积 (ml)就可以计算出最大加样量 (mg)。但事实上有效交换容量
并不是常数,它随蛋白质种类的不同而有不同的取值。作为某种离子交换剂的说
明书,只会给出少数几种常见蛋白质的有效交换容量,而目的蛋白在此交换剂上
的有效交换容量往往是未知的。此时可以从已知有效交换容量的蛋白质中选择一
种与目的蛋白分子量比较接近的蛋白质,用它的有效交换容量来近似计算目的蛋
白的最大加样量。实际情况比较复杂,样品溶液中的杂质组分往往也能与离子交
换剂结合,它们的种类和数量会影响到目的蛋白的实际交换容量,因此,在离子
交换色谱中为了获得理想的分离效果,建议样品中目的蛋白的含量不应超过其有
效交换容量的10%~20%。以上是通过理论计算的方法来确定加样量,实践中有时目的蛋白的含量是未
知的,样品中杂质成分对有效交换容量的影响也比较复杂,此时可以通过试管小

104
试法来确定最大加样量。具体的操作类似于用试管小试法确定起始pH和离子强
度。在10支试管中分别加入相同 数 量 的 离 子 交 换 剂,用 起 始 缓 冲 液 充 分 洗 涤、平衡,然后往10支试管中分别加入数量递增的样品溶液,相邻两支试管间加样
量的差值是恒定的,充分混合以完成吸附,分析上清液中目的蛋白的含量,上清
液中不含目的蛋白而加样最多的试管对应的加样量即为试 管 条 件 下 的 最 大 加 样
量,根据试管内以及色谱柱床离子交换剂的体积可以计算出色谱条件下的最大加
样量。考虑到色谱时随流速增加动态交换容量会下降,实际的加样量必须低于最
大加样量。
4532 加样方法和注意事项
加样是将一定体积的样品溶液添加至柱床的床面,依靠重力或泵提供的压力
使样品进入床面的过程。在加样过程中样品溶液应尽可能均匀添加至床面,这样
在柱床的横截面上样品能够均匀进入,另外要防止液流破坏床面的平整性,否则
会造成区带形状变差,洗脱峰变宽。加样的方法有多种,如果采用的是成套液相色谱系统,一般都提供标准的加
样方法,如通过进样器或注射器等,最终利用泵将样品溶液加入柱床。对于预装
柱,柱的上端一般带有液流分配器,能保证样品加入时平均分布在床面上并均匀
进入柱床。加样时流速会影响到样品的吸附,如果起始条件下目的蛋白能够较牢
固地吸附在交换剂上,则加样的流速可以取较大的值;而如果起始条件与目的蛋
白解吸条件相距不远,流速应当慢一些,保证目的蛋白有充足的时间发生吸附。对于黏度相对较大的样品,加样时流速也不能太大。
对于自装柱,常见的加样方法有排干法和液面下加样法。排干法是最常用的,所需设备最少,但对操作的要求较高。加样前先将色谱
柱上口拧开,让床面之上的液体靠重力作用自然排干,当床面刚好暴露时将色谱
柱下端阀门关闭,此时柱内液体不能继续流出,用吸管将样品加到床面上,但一
定要注意不能破坏床面的平整,以免造成区带的扭曲和倾斜。然后打开柱下端阀
门,受重力作用样品溶液进入床面。必要时用少量起始缓冲液洗涤色谱柱上端,打开阀门使洗涤液也进入床面。最后用起始缓冲液充满色谱柱内床面上端空间,拧紧色谱柱上口,即可以用恒流泵泵入起始缓冲液 完 成 吸 附 过 程 并 洗 去 不 吸 附
物质。液面下加样法则是将色谱柱上口拧开,不排干床面之上的液体,而是利用带
有长的针尖的注射器将样品溶液轻轻注入液面下,使其平铺在床面上。注意采用
此方法时样品溶液的密度必须大于起始缓冲液,这可以通过向样品溶液中添加葡
萄糖来实现。打开柱下端阀门后,同样依靠重力,样品会进入床面,然后拧紧柱
上口,用起始缓冲液进行洗涤。

105
采用这两种方法加样时不存在流速过快的问题。样品进入柱床后,接着就是用起始缓冲液洗涤柱床,将起始条件下不能被吸
附的物质从柱中洗去,在色谱图上形成穿透峰。通常在色谱柱下端连接一个紫外
检测器,根据测定出280nm处吸光度来指示出峰情况,洗涤过程一般需进行到
出峰后紫外吸收重新回到初始值附近为止。洗涤所需起始缓冲液的量一般不会超
过2倍柱体积,在理想状态下甚至1倍柱体积的起始缓冲液即可完成洗涤过程。流速对清洗过程不会产生大的影响,因此必要时特别是大规模分离时,可以适当
提高洗涤流速来缩短分离时间。如果色谱的起始条件是预先摸索过的,目的蛋白
此时不会出现在穿透峰中,因此洗涤时柱下端流出的洗脱液不需进行收集。而如
果采用的是起始相洗脱,目的蛋白在起始条件下不发生吸附,而其他杂质成分被
吸附在色谱柱上,那么应当对洗脱液进行分部收集,洗涤完毕将穿透峰对应的分
部合并,可得到经过纯化的目的蛋白溶液。
454 洗脱技术
多数情况下,在完成了加样吸附和洗涤过程后,大部分的杂质已经从色谱柱
中被洗去,形成穿透峰,即样品已经实现部分分离。然后需要改变洗脱条件,使
起始条件下发生吸附的蛋白质从离子交换剂上解吸而被洗脱,如果控制洗脱条件
变化的程度,还可以实现不同的组分在不同时间发生解吸,从而对吸附在柱上的
杂质进一步分离。要想使蛋白质从离子交换剂上被洗脱,可采取的方法有:(1)改变洗脱剂的pH 这会导致蛋白质分子带电荷情况的变化,当pH接
近蛋白质等电点时,蛋白质分子失去净电荷,从交换剂上解吸并被洗脱下来。对
于阴离子交换剂,为了使蛋白质解吸应当降低洗脱剂的pH,使目的蛋白带负电
荷减少;对于阳离子交换剂,洗脱时应当升高洗脱剂的pH,使目的蛋白带正电
荷减少,从而被洗脱下来。(2)增加洗脱剂的离子强度 此时蛋白质与交换剂的带电状态均未改变,但
离子与蛋白质竞争结合交换剂,降低了蛋白质与交 换 剂 之 间 的 相 互 作 用 而 导 致
洗脱。(3)向洗脱剂中添加特定离子 某种特定的蛋白质能与该离子发生特异性相
互作用而被置换下来,这种洗脱方式称为亲和洗脱。(4)向洗脱剂中添加一种置换剂 它能置换离子交换剂上所有蛋白质,蛋白
质先于置换剂从柱中流出,这种方式称为置换色谱。根据洗脱剂发生改变时连续
与否,洗脱技术分为阶段洗脱和梯度洗脱。阶段洗脱是指在一个时间段内用同一
种洗脱剂进行洗脱,而在下一个时间段用另一种改变了pH或离子强度等条件的
洗脱剂进行洗脱的分段式不连续洗脱方式。梯度洗 脱 则 是 一 种 连 续 性 的 洗 脱 方

106
式,在洗脱过程中洗脱剂的pH或离子强度等条件在不断发生改变。
4541 阶段洗脱
如上所述,阶段洗脱是用一系列洗脱剂进行的同溶剂洗脱,在每一阶段,仅
仅需要约1倍柱体积的洗脱剂将此条件下发生解吸的组分洗脱下来。阶段洗脱分
为pH阶段洗脱和离子强度阶段洗脱。(1)pH阶段洗脱 是用一系列具有不同pH的缓冲液进行洗脱,多数情况
下这些缓冲液的缓冲物质是相同的,只是弱酸碱和盐的比例不同。pH的改变造
成蛋白质带电状态的改变,在某种pH下一些蛋白质不能再结合在交换剂上而被
洗脱,再次改变pH 将 使 另 一 些 蛋 白 质 被 洗 脱,这 样,目 的 蛋 白 会 在 某 一 特 定
pH的缓冲液中被洗脱,与在其他阶段被洗脱的杂蛋白分离开。此方法再现性较
好,不同pH的缓冲液也易于配制,但要注意在使用弱型离子交换剂时,在达到
新的pH之前有一段短的重新平衡时间。另外,蛋白质被洗脱时的pH往往靠近
其等电点,因为此时蛋白质带净电荷数逐渐减少,与交换剂之间的结合力减弱,但很多蛋白质在靠近等电点时其溶解度达到最小,须防止蛋白质在色谱柱中发生
等电点沉淀,所以在分离前有必要测试在所使用的pH和离子强度条件下样品组
分的溶解度。(2)离子强度阶段洗脱 是使用具有相同pH而离子强度不同的同一种缓冲
液进行洗脱,不同的离子强度通常通过添加不同比例的非缓冲盐来实现,最常用
的是NaCl,也可通过添加乙酸铵等挥发性盐的方法来增加离子强度,或直接增
加缓冲液中缓冲物质的浓度来增加离子强度。例如,起始缓冲液采用pH=65、
002mol/L磷酸盐缓冲 液,而 洗 脱 缓 冲 液 采 用pH=65、015mol/L磷 酸 盐 缓
冲液。增加离子强度后,减弱了蛋白质与离子交换剂之间的静电作用力,首先使
一些带净电荷较少,吸附得不太牢固的蛋白质解吸而被洗脱下来,再增加离子强
度,使结合得更为牢固的一些蛋白质被洗脱,目的蛋白将在特定离子强度的缓冲
液中被洗脱而与在其他离子强度下被洗脱的杂蛋白实现分离。通常,含有1mol/LNaCl的缓冲液能够将几乎所有蛋白质洗脱下来。
阶段洗脱技术操作简单,对于一些色谱条件已经清楚的蛋白质往往有着高的
分辨率,图414显示了用离子强度的梯度洗脱和阶段洗脱分离牛血清蛋白的效
果,可以看出阶段洗脱要优于梯度洗脱。但阶段洗脱有时效果并不理想,图415显示了用离子强度阶段洗脱和线性
梯度洗脱的分离效果,显然阶段洗脱效果较差,峰2和峰6在线性梯度洗脱时又
分辨出几个组分,而峰3和峰4实际上是同一组分,在阶段洗脱时却被分成两个
洗脱峰。因此采用阶段洗脱时,设计各阶段的洗脱条件和解释分析结果时必须十
分谨慎,若相邻两个阶段间pH或离子强度条件变化过大,会使得吸附强弱较为

107
图414 离子强度梯度洗脱和阶段洗脱分离牛血清蛋白图谱
色谱柱尺寸:15cm×26cm。离子交换剂:QAESephadexA50。
样品:4ml3%冻干牛血清。洗脱缓冲液:pH=65,01mol/L
TrisHCl。流速:02ml/min(a)梯度洗脱,NaCl浓度0~05mol/L;(b)阶段洗脱,
各阶段NaCl浓度以01mol/L递增
接近的不同组分在同一阶段被洗脱,此时典型的洗脱峰具有陡峭的前沿和拖尾的
后沿。而两个阶段间洗脱条件变化过小,则可能会因为洗脱剂的洗脱能力太弱而
形成宽而拖尾的洗脱峰。另外,如果某阶段未完全将解吸蛋白洗下就过早更换洗
脱剂,拖尾现象会造成错误洗脱峰的出现。因此,在初次分离目的蛋白时,人们
往往先采用连续梯度洗脱,待确定了样品的特性和洗脱条件后再考虑采用阶段洗
脱来优化分离效果,缩短操作时间。
4542 梯度洗脱
在进行梯度洗脱时,洗脱缓冲液的离子强度或pH是连续发生变化的,在某
一条件下,吸附最弱的蛋白质先被洗脱,进一步改变洗脱条件后另一种蛋白质被
洗脱。梯度洗脱与阶段洗脱原理上是相同的,但由于洗脱剂的洗脱能力是连续增
加的,洗脱峰的峰宽一般会小于阶段洗脱,拖尾现象也会得到明显的改善甚至完

108
图415 离子强度阶段洗脱和线性梯度洗脱效果对比
(a)分四步阶段洗脱;(b)线性梯度洗脱
图中阶段洗脱由于洗脱条件变化过快,其洗脱峰2和6在梯度洗脱时又分
辨出不同组分,而阶段洗脱时的洗脱峰3和4实际上为同一组分
全消除,而且不会造成同一组分被分离成几个峰的现象,常常能获得较高的分辨
率。梯度洗脱也分为pH梯度洗脱和离子强度梯度洗脱。获得连续线性pH梯度比较困难,它无法通过按照线性体积比混合两种不同
pH的缓冲液来 实 现,且pH 的 改 变 往 往 会 使 得 离 子 强 度 同 步 发 生 变 化,因 此
pH梯度洗脱很少被使用。在确实需进行pH梯度洗脱时,通过混合两种相同缓
冲物质组成的缓冲液,能够在很窄的pH范围内获得线性pH梯度,此范围最大
为2个pH单位,通常是在缓冲物质的pKa 上下各1个pH单位。离子强度梯度洗脱即盐浓度梯 度 洗 脱,是 离 子 交 换 色 谱 中 最 常 用 的 洗 脱 技
术,它再现性好,操作方便,只需将两种不同离子强度的缓冲液 (起始缓冲液和
极限缓冲液)按比例混合即可得到所需的离子强度梯度,此过程中缓冲液的pH始终不变。起始缓冲液是根据实验起始条件而选择的由浓度很低的缓冲物质组成
的,具有特定pH,通常缓冲物质浓度在002~005mol/L。极限缓冲液是向起
始缓冲液中添加了非缓冲盐 (如NaCl)后得到的,也可以是缓冲物质和pH 与

109
起始缓冲液相同但缓冲物质浓度较高的缓冲溶液,其中前一种方法使用较多。例
如,某离子交换色谱的起始缓冲液为pH=65、005mol/L的TrisHCl缓冲液,极限缓冲液为含1mol/LNaCl的pH=65、005mol/L的TrisHCl缓冲液,将
两种缓冲液按一定的比例混合,可以得到NaCl浓度在0~1mol/L之间连续变化
图416 梯度混合器装置
的离子强度梯度。在色谱过程中梯度缓冲液可以 通 过 梯
度混合器来获得。最简单的梯度混合器由
通过 一 根 导 管 连 接 的 两 个 烧 杯 组 成,图
416为市售梯度混合器,它们都是根据连
通器原理来产生连续变化的梯度。容器 A中首先放置的是起始缓冲液,容器B中放
置极限缓 冲 液,容 器 A 和B通 过 管 路 相
通,容器A内置搅拌装置 (常用磁力搅拌
子)并且底部有管路导出。开始 时,装 入
容器A内的起始缓冲液和装入容器B内的
极限 缓 冲 液 的 液 面 高 度 应 保 持 相 同,这
样,打开A、B间的管路,当容器A中液
体作为 洗 脱 缓 冲 液 流 出 时,其 液 面 下 降,根据连通器原理,容器B中的极限缓冲液 会通过管路流到A中,使容器A和B的液面高度始终保持一致,此过程中容器A中的洗脱缓冲液离子强度在不断增加,到两个容器中液体都用完的时刻,容器A中的洗脱缓冲液等价于极限缓冲液。在
洗脱过程中的某一时刻,梯度混合器中流出的梯度缓冲液中的盐浓度 (如NaCl浓
度)可以根据以下公式计算得出:
c=c2-(c2-c1)·(1-V/V0)A2/A1式中 c———容器A中流出洗脱剂体积为V 时洗脱缓冲液的盐浓度;
c1———起始缓冲液中的盐浓度,一般为0;
c2———极限缓冲液中的盐浓度;
V———已经从梯度混合器中流出的洗脱剂的体积;
V0———梯度洗脱剂的总体积,是起始缓冲液和极限缓冲液体积的总和;
A1———容器A的横截面积;
A2———容器B的横截面积。盐浓度梯度曲线可以有不同的形状,当A1=A2时,即盛放起始缓冲液和极
限缓冲液的容器横截面积相同时,产生的是线性梯度 [图417 (a)];当A1<A2时,即盛放起始缓冲液容器的横截面积小于盛放极限缓冲液容器的横截面积

110
时,产生的是凸形梯度 [图417 (b)];当A1>A2时,即盛放起始缓冲液容器
的横截面积大于 盛 放 极 限 缓 冲 液 容 器 的 横 截 面 积 时,产 生 的 是 凹 形 梯 度 [图
417 (c)]。
图417 梯度的形状及产生装置
如果分离采用的是成套液相色谱系统,仪器内都含有梯度产生装置,不需额
外的梯度混合器,通过两个泵或者一个泵联一个转换阀,可以任意地由起始和极
限缓冲液产生线性、凸形、凹形以及更为复杂的混合型梯度,并且这种梯度的产
生可以通过计算机程序进行控制,操作时只需配制好两种缓冲液,即可在计算机
中用软件对所需梯度进行设置,色谱分离时系统会自动调节泵的流速来产生所需
的梯度缓冲液。在选择梯度的形状时,如果是对目的蛋白进行首次离子交换分离,建议优先
采用线性梯度,多数情况下NaCl的浓度范围可选择在0~05mol/L之间变化,梯度洗脱的结果可以作为进一步优化分离的依据,通过改变梯度的形状和斜率来
提高分辨率,也可以根据目的蛋白被洗脱时的盐浓度确定进行阶段洗脱的条件。凸形梯度有利于改善梯度末尾部分的分辨率,对于一份混合样品,如果开始阶段
几个洗脱峰之间能够很好地分离,而末尾部分的几个组分吸附能力比较接近时,采用凸形梯度既有利于末尾部分组分的分离,又可以缩短分离时间,加快速度。凹形梯度有利于改善梯度起始阶段的分辨率,如果混合样品起始部分组分吸附能
力比较接近而末尾部分容易分开,采用凹形梯度能达到改善分辨率和缩短分离时
间的效果。混合型梯度可以在组分吸附能力相近需要改善分辨率的位点提供足够
的分辨率,而在对分辨率不作要求的位点采用陡峭的梯度缩短时间,从而实现分

111
辨率和分离速度的最佳结合。但混合型梯度形状的确定决不是随意的,而是根据
线性梯度洗脱结果优化而来的。除了梯度的形状,梯度的高度和斜率也影响着分离的速度和效果。梯度的高
度是指起始缓冲液与洗脱终止时梯度缓冲液中盐浓度的差值,通常起始缓冲液中
盐浓度为零,而洗脱终止时洗脱缓冲液盐浓度在02~10mol/L,即梯度的高度
在02~10mol/L之间。梯度的斜率是指梯度的高度与完成此梯度时所用洗脱
剂体积 (或时间)的比值。通常较低的斜率有利于改善吸附能力相近组分之间的
分辨率,但会导致洗脱峰峰宽加大和分离时间的延长;相反,较大的斜率可以实
现快速分离,得到尖的洗脱峰,但不同组分之间的分离度差异会减小。图418
图418 梯度斜率对分辨率的影响
色谱柱:MonoQHR5/5。样品:部分纯化的强啡肽转化酶。起始缓冲液:
pH=70,20mmol/LTris缓冲液。极限缓冲液:含10mol/LNaCl的
pH=70,20mmol/LTris缓冲液。流速:1ml/min(a)在20个柱体积内NaCl浓度从零增至10mol/L;
(b)在20个柱体积内NaCl浓度从零增至04mol/L
显示了不同斜率的梯度洗脱在分离效果上的差异,从图中可以看出,斜率降低后
分辨率得到改善。当然,并不是任何情况下都能通过改变斜率来解决分辨率的问
题,有时需要调整pH等其他条件。对于第一次进行离子交换分离的样品,在梯
度洗脱时常用 的 条 件 是:采 用 线 性 梯 度,在10个 柱 体 积 内 盐 浓 度 从 零 提 高 至

112
05mol/L或10mol/L,比如柱体积为20ml,则在洗脱剂总量200ml内完成整
个梯度。此后可以根据分离图谱,在洗脱峰比较靠近,分辨率不理想的部分降低
梯度的斜率,而在没有出现洗脱峰的部分 提 高 梯 度 的 斜 率,从 而 形 成 混 合 型 梯
度,优化分辨率和分离速度。在洗脱过程中,流速会影响到待分离物质的分辨率。蛋白质等大分子物质由
于扩散速度慢,达到吸附和解吸平衡需要一定的时间,这将限制加样和洗脱过程
中的流速。多数情况下适当降低洗脱过程的流速可以提高色谱的分辨率,但会延
长分离所需的时间,图419显示了用DEAESephadexA50色谱柱分离样品时
流速对分辨率的影响。不过也有例外,例如在使用基于琼脂糖的离子交换剂时,采用较快的流速对分辨率影响不大。因此,在初次分离时可以选择适中的流速,根据色谱结果是否达到分辨率的要求,再对流速进行优化。
图419 流速对分辨率的影响
色谱柱:DEAESephadexA50离子交换柱。
起始缓冲液:pH=83,01mol/LTrisHCl。
极限缓冲液:含03mol/LNaCl的pH=83,01mol/LTrisHCl(a)流速8ml/h;(b)流速20ml/h
峰1—碳氧血红蛋白;峰2—血清白蛋白单体;峰3—血清白蛋白二聚体
4543 亲和洗脱
亲和洗脱是向洗脱剂中加入能与蛋白质发生特异性相互作用的离子而将蛋白

113
质从离子交换剂上置换下来的洗脱方式。在亲和洗脱中,蛋白质的吸附是非特异
性的,而解吸却是特异性的。亲和洗脱经常用于酶的离子交换分离,使用离子状态的底物作为洗脱物质,
底物和离子交换剂带同种电荷,利用酶和底物之间的专一性结合可以使酶蛋白解
吸,这种洗脱方式又称底物洗脱。典型的例子是以1,6二磷酸果糖作为洗脱剂,从CM纤维素色谱柱上洗脱果糖1,6二磷酸酶,底物分子完全解离时带四个负
电荷,而果糖1,6二磷酸酶由四个亚基组成,每个亚基都能结合一个底物分子,因此该酶与底物结合后其净电荷的变化达到16个单位,这足以将其从阳离子交
换剂上洗脱下来。并不是任何底物都适合作为洗脱剂将酶蛋白洗脱的。有的底物与酶的亲和力
大,并且结合后能使酶的构象发生有利于 解 吸 的 变 化,这 类 底 物 是 有 效 的 洗 脱
剂。而在其他情况下,为了得到良好的洗脱效果,一个分子量达到10万的蛋白
质应至少结合4个与交换剂功能基团同类的电荷。
4544 置换色谱
在置换色谱中,采用特殊的置换物质来进行洗脱,置换剂与离子交换剂之间
具有很强的亲和性,因此包括蛋白质在内的所有物质都从色谱柱上被置换下来,以相同的速率先于置换剂从柱下端流出,形成一系列矩形的洗脱峰。在置换洗脱
时,不存在一个目的蛋白被洗脱而其他蛋白仍被吸附的条件,被洗脱的不同组分
之间也不存在取代吸附关系。置换剂常见的是一些多聚离子,它们带大量与交换
剂上功能基团相反的电荷,因此与交换剂之间有着非常高的亲和力。在离子交换中较少使用置换色谱,因为很难找到合适的置换剂来分离不同蛋
白组分。不过也有成功应用的报道,Peterson和Liao等研究了用置换色谱的方
法分离β乳球蛋白,将100mgβ乳球蛋白样品加样至35mlTSKDEAE色谱柱
后,用pH=70的磷酸钠缓冲液完成吸附并洗去穿透组分,然后用羧甲基葡聚
糖和硫酸软骨素作为置换剂洗脱,得到两个矩形洗脱峰,该法的优势是其容量为
普通离子交换色谱的2倍。
455 样品的收集和处理
通常色谱柱的下端连接一个紫外检测器,用于检测蛋白质组分的洗脱过程,而紫外检测器后又连着分部收集器,用以收集柱下端流出的洗脱液。人们通过测
定洗脱液在280nm处的吸光度可以判断蛋白质组分何时被洗脱,以及各洗脱峰
分别分部收集在哪几支试管中。但人们很难判断图谱中哪个洗脱峰中存在目的蛋
白,因此在分部收集后需要测定目的蛋白的活性或其他性质来判定目的蛋白峰,如果需要比较精确测定各洗脱峰中蛋白质的浓度,还需要采用其他蛋白质定量测
定方法。

114
有时会出现目的蛋白与其他组分未能完全分开的情况。如 图47所 示,当
RS=15时,两个相邻洗脱峰被完全分开;当RS=1时,两个洗脱峰并未 完 全
分离,但此时两种组分间产生的相互干扰是很小的,必要时弃去与相邻 峰 产 生
干扰的分部,可以得到较纯 的 组 分。但 是 当 目 的 蛋 白 峰 是 一 个 不 对 称 的 洗 脱
峰,或者与另一组分靠得很近时,就需要对收集到的目的蛋白进一步 分 离,这
种分离可仍然采用离子交换色谱,也可采用其他技术。当然,即便是目 的 蛋 白
峰与其他组分的洗脱峰已完全分开,也不能断定已经获得纯的目的蛋 白,还 需
进行纯度鉴定。有时目的蛋白组分在色谱时被分离成多个洗脱峰,导致这种现象的原因是多
方面的,可能是加样量过大造成的,也可能是起始条件过分远离解吸条件,目的
蛋白结合过于牢固而造成部分变性引起的,目的蛋白如果是多亚基的,亚基之间
发生解聚也会导致多洗脱峰的出现。遇到这种现象时应对其产生原因进行分析,通过改变色谱条件来排除。
采用冷冻干燥的方法可以对收集的样品进行浓缩后得到固体粉末,如果色谱
时使用了挥发性缓冲物质作为洗脱剂,此过程将比较方便,并且在固体中没有缓
冲物质残留。乙酸铵是常用的挥发性缓冲物质,由于氨比乙酸更易挥发,在冻干
过程中pH会下降,从乙酸中冻干会使一些蛋白质发生聚集,为了避免这种情况
应将pH调节至7~8。
456 离子交换剂的再生、清洗、消毒和储存
每次色谱完后,往往还有一些结合比较牢固的物质残留在离子交换剂上,如
变性蛋白、脂类等,它们的残留会干扰正常的吸附,影响到蛋白质在色谱时的表
现,并可能对样品造成污染,甚至堵塞色谱柱。因此,每次使用后,应彻底清洗
掉柱中的结合物质,恢复介质的原始功能。再生过程根据介质的稳定性和功能基团不同有着不同的方法,通常离子交换
剂的制造商会提供介质再生和清洗的方法。一般情况下,人们采用最终浓度达到
20mol/L的盐溶液清洗色谱柱可以除去以离子键与交换剂结合的物质,选择盐
的种类时应含有离子交换剂的平衡离子,以使再次使用前的平衡过程容易进行,
NaCl是最常规的选择,其中的Na+是大多数阳离子交换剂的平衡离子,而Cl-
是大多数阴离子交换剂的平衡离子。当色谱柱上结合了以非离子键吸附的污染物
后,用盐溶液无法将其除去,此时应选择更为严格的清洗方案,碱和酸是良好的
清洗剂,但使用时应注意离子交换剂的pH稳定性。通常基于琼脂糖的离子交换
剂可以用盐溶液和05mol/LNaOH进行清洗,基于纤维素的离子交换剂用盐溶
液和01mol/L的NaOH进行清洗,基于葡聚糖和琼脂糖的交换剂应避免pH<3的 酸 性 环 境,它 们 可 以 用10mol/L 的 乙 酸 钠 (用 强 酸 调 节 pH=3)和

115
05mol/LNaOH交替清洗。脂类或脂蛋白污染物可以用非离子型去污剂或乙醇
来清洗。高效离子交换介质和预装柱通常都是原位清洗 (CIP),将清洗剂 直 接
通过泵加入色谱柱,污染物从柱下端被洗出,清洗后柱效率几乎不受影响。对于
自装柱,既可以进行原位清洗 (需注意清洗剂的使用会使柱床的体积发生很大的
变化),也可以将交换剂从色谱柱中取出后进行清洗。消毒过程使得微生物失活,细胞和微生物的生长不仅会严重污染样品,还会
影响离子交换柱的色谱特性,并可能堵塞色谱柱,因此在离子交换剂使用前和储
存前都应当进行消毒操作。在使用色谱前,最常用消毒方法的是以 NaOH溶液
清洗色谱柱,NaOH同时具有清洗和消毒的效果。如果离子交换剂需长期贮存,应将其浸泡在防腐剂中,阴离子交换剂可浸泡在含20%乙醇的02mol/L乙 酸
中,阳离子交换剂可浸泡在含20%乙醇的001mol/LNaOH中,有时也可使用
叠氮化钠等其他防腐剂。对于从未使用过的介质,应放置在密闭容器中在4~25℃保存。不能将介质
冷冻存放,因为冰晶会使凝胶颗粒破碎,产生细的粉末。对于已使用过的介质,在彻底清洗后可浸 泡 在 含 防 腐 剂 的 溶 液 中 于4~8℃保 存,如 果 保 存 时 间 很 长,还需定时更换防腐剂。
46 应用实例
461 用DEAESepharose色谱分离真菌纤维素酶
真菌Trichodermareesei培养液经过滤后作为提取纤维素酶的原料。将滤液
先在SephadexG25凝胶柱上进行缓冲液交换,用pH=50、001mol/L的乙酸
铵平衡并洗脱。收集洗脱峰加样到DEAESepharoseCL6B柱 (26cm×30cm),用pH=50、001mol/L的乙酸铵溶液 完 成 吸 附 并 洗 去 不 发 生 吸 附 的 杂 质,然
后用乙酸铵浓度线性梯度进行洗脱,在1000ml内乙酸铵浓度从001mol/L增加
至05mol/L (pH=50),流速为40ml/h,用280nm紫外线检测洗脱液,分离
出A、B、C三个洗脱峰。纤维素酶活性测定方法有多种,分别对三个洗脱峰测
定了CMC (羧甲基纤维素)水解活性和微晶纤维素水解活性,发现三个洗脱峰
都有纤维素 酶 活 性 [图420 (a)]。收 集 其 中 酸 性 最 强 的 组 分C,冻 干 后 溶 于
pH=37、005mol/L的 乙 酸 铵 中,在 此 条 件 下 再 次 加 样 到 已 平 衡 的 DEAESepharoseCL6B柱 (26cm×30cm)进 行 二 次 色 谱,仍 然 采 用 浓 度 线 性 梯 度
进行 洗 脱,但 梯 度 的 高 度 和 斜 率 作 了 调 整,在 700ml内 乙 酸 铵 浓 度 从
005mol/L增加至03mol/L (pH=37),组分C进 一 步 被 分 离 成 两 个 纯 的 纤
维二糖水解酶 [图420 (b)],这 两 个 组 分 在pH=50时 同 时 洗 脱,然 而 在
pH=37时得到分离。

116
图420 利用DEAESepharoseCL6B从真菌Trichodermareesei中分离纤维素酶
(a)在pH=50条件下进行色谱;(b)在pH=37条件下对组分C二次色谱
462 用 MonoS和 MonoQ连续色谱法从鸡肌肉组织分级分离糖酵
解酶类
从天然来源的复杂样品中获取纯的蛋白质往往要系统利用一 些 具 有 不 同 选
择性的方法,一个典型的例子是顺序使用阳离子和阴离子交换色谱从鸡 肌 肉 组
织中纯化肌酸激 酶 (CK)。在 进 行 阳 离 子 交 换 前,将 鸡 肌 提 取 物 用SephadexG25凝胶柱进行缓冲液 交 换,使 样 品 溶 于 起 始 缓 冲 液 中。样 品 经022μm滤

117
膜过 滤,取 800μl加 样 至 MonoS HR10/10 离 子 交 换 柱,用 pH=60、
005mol/L的 MES (2吗啉代乙磺酸一水合物,是一种兼性离子缓冲液)洗 涤
柱子,然后用0~05mol/L的NaCl进行盐 浓 度 梯 度 洗 脱。肌 酸 激 酶 和 磷 酸 甘
油酸变位酶在穿 透 峰 中 被 洗 脱,而 其 他 酶 类 在 NaCl梯 度 中 被 陆 续 洗 脱 [图
421 (a)]。收集穿透峰分部,再 次 用SephadexG25凝 胶 柱 进 行 缓 冲 液 交 换,使样品溶于阴离子交换所用起始缓冲液。取1ml样品加样至 MonoQHR5/5离子
交换柱,用pH=80、002mol/L的 Tris缓 冲 液 洗 涤,然 后 用0~05mol/L的
NaCl进行盐浓度梯度洗脱,结果见图421(b),肌酸激酶得到了很好的分离。
图421 用 MonoS和 MonoQ顺序色谱从鸡肌肉提取物中分离肌酸激酶
(a)在 MonoSHR10/10上进行分离;(b)在 MonoQHR5/5上进行分离
图中数字表示主要蛋白峰分部;“CK活性”旁边有一阴影,表示色谱图中的肌酸
激酶活性峰分部,应当是 (b)中的峰6
463 用 MonoQ阴离子交换分离白血病细胞N乙酰βD氨基葡萄
糖苷酶的同工酶
同工酶通常有着近似的分子量,用凝胶过滤无法对其分离,而根据同工酶之
间在氨基酸组成上的差异,它们的电性质不完全相同,因而可以用离子交换法分
离同工酶。N乙 酰βD氨 基 葡 萄 糖 苷 酶 是 一 种 在 白 血 病 的 诊 断 中 常 用 的 酶,该酶具 有 若 干 同 工 酶 形 式,选 用 MonoQ阴 离 子 交 换 柱,用 NaCl浓 度 梯 度 洗
脱,可以成功分离这些同工酶 (图422),而在高效离子交换介质出现以前,这

118
种分离仅能通过等电聚焦来实现。
464 用BioRex70色 谱 分 离 眼 镜 蛇 神 经 毒 素 的 六 种 单 乙 酰 基 衍
生物
人们通过化学修饰的方法可以研究蛋白质中氨基酸侧链的功能,在 化 学 反
图422 用 MonoQ分离白血病细胞内
N乙酰βD氨基葡萄糖苷酶的同工酶
应中,通常 若 干 不 同 的 侧 链 会 被 衍 生 化,人们需要对各衍生物进行纯化和 分 析。眼
镜蛇Najanajasiamensis神经 毒 素 是 一 种
小分子 的 碱 性 蛋 白 质,由71个 氨 基 酸 残
基组成 一 条 多 肽 链,链 内 有5个 二 硫 键。该蛋白质包含5个赖氨酸残基的侧 链 氨 基
和1个 游 离 的 N末 端 氨 基,这6个 氨 基
均能与乙酸酐反应生成乙酰基衍 生 物,从
而能形成6种单乙酰基衍生物,它 们 有 着
完全相同的净电荷数,但电荷在蛋 白 质 表
面的分布不同,据此可以用聚异丁 烯 酸 树
树脂BioRex70将它们分开。将毒素衍生
物溶于pH=67、002mol/L的乙 酸 铵 缓
冲液,加 样 至 已 用pH=67、02mol/L
图423 在BioRex70色谱柱上用乙酸铵浓度梯度洗脱6种
眼镜蛇神经毒素单乙酰基衍生物
的乙酸铵平衡好 的BioRex70离 子 交 换 柱 (2cm×305cm),先 用pH=67、
005mol/L的乙酸铵缓冲液洗涤,再用凹形梯度将乙酸铵浓度从005mol/L提
高至14mol/L进行洗脱 (pH=67)。从 图423可 以 看 出,未 衍 生 化 的 毒 素
最晚被洗脱 (洗脱峰1),这是因 为 未 衍 生 化 的 毒 素 蛋 白 正 电 荷 数 量 不 变,超
过六种单衍生物。少量的二乙酰化和三乙酰化衍生物则出现于色谱图谱 的 起 始
位置。

119
465 用阶段洗脱法大规模纯化人血清白蛋白和免疫球蛋白G在科学研究和治疗等多种领域都需要用到高纯度的人血浆蛋白,通常人们采
用Cohn冷冻乙醇法可以大量分离制备人血浆蛋白。新的研究表明采用阴离子和
阳离子交换相结合的方法也能大规模纯化血浆蛋白。离心后用缓冲液交换法将样
品溶于0025mol/L的乙酸钠缓冲液,用乙酸调节pH至52,使优球蛋白沉淀,离心并过滤,加样至已平衡好的DEAESepharoseFastFlow离子交换柱,加样
量为每升离子交换剂35g蛋白质。在起始条件下免疫球蛋白G(IgG)被洗脱,然
后采用阶段洗脱法,用pH=45的缓冲液将白蛋白洗脱下来。将出峰直接加样
至已平衡的CMSepharose柱,用pH=55、011mol/L的乙酸钠缓冲液进行洗
脱,可获得高纯度的白蛋白。
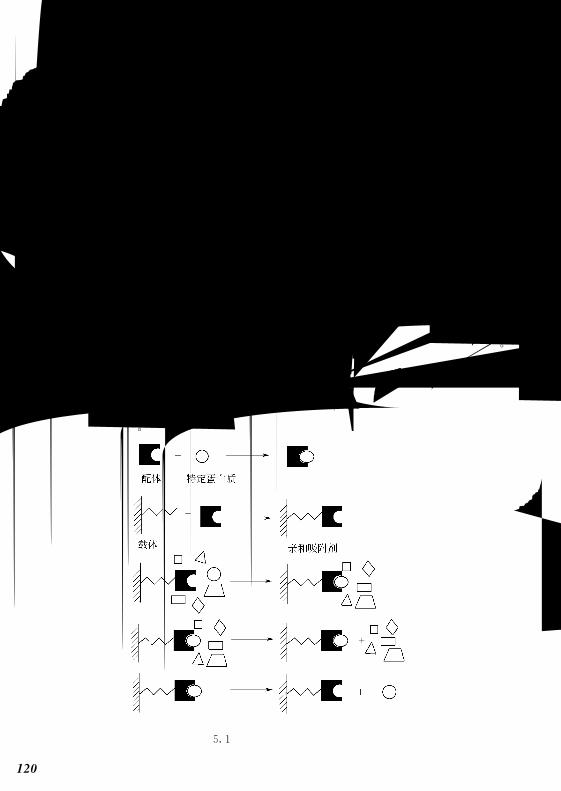
120
5 亲 和 色 谱
亲和色谱是利用蛋白质分子的生物学活性,而不是利用其物化特性来进行分
离,即以蛋白质和配体之间的特异性亲和力作为分离的基础,因而具有高度选择
性,其纯化程度有时可高达1000倍以上,因此亲和色谱是一种非常有效的蛋白
质纯化方法,多用于从大量的复杂溶液中分离少量的特定蛋白质,且这种纯化方
法同时具有浓缩的效果。有时仅用亲和色谱一步分离过程,就能达到快速而且满
意的蛋白质纯化效果,这是其他纯化方法所无法比拟的。亲和色谱中蛋白质与配体之间的结合类型主要有:酶的活性中心或别构中心
通过次级键与专一性底物、辅酶、激活剂或抑制剂结合,抗原与抗体,激素等与其
受体,生物素与抗生物素蛋白/链霉抗生物素蛋白,糖蛋白与凝集素等的结合。这些
蛋白质与其配体之间能够可逆地结合和解离,因而可由此进行蛋白质的分离和纯化。亲和色谱的操作主要分为以下几步 (图51):选择合适的配体;将配体固
图51 亲和色谱操作过程
定在载体上,制成亲和吸附剂,而配体的特异性结合活性不被破坏;将特定蛋白
质与固定化亲和吸附剂相结合;用缓冲液洗掉杂质;将结合在亲和吸附剂上的特
定蛋白质洗脱下来。

121
51 亲和吸附剂
在用亲和色谱进行蛋白质的分离纯化时,首先要制备有效的亲和吸附剂。固
相亲和吸附剂是利用合适的化学偶联方法,将配体和固相载体经连接臂连接而成
的。制备过程包括:选择合适的配体;选择合适的载体和连接臂;用合适的方法
将配体化学偶联到载体上。有两种方法可以获得亲和吸附剂:(1)自行制备 针对待分离蛋白质的性质,选择专一性配体,然后用合适的
化学偶联方法制备亲和吸附剂。由于所选配体对特定蛋白质具有高度特异性,因
此能提高待分离蛋白质的纯化程度,并能有效地浓缩待分离蛋白质。但是由于每
分离一种新蛋白质就要寻找一种新的配体,因此非常费时,成本很高。(2)购买商品化亲和吸附剂 商品化亲和吸附剂都是基团特异性吸附剂,其
配体是针对某个化学基团具有亲和力,因此这类吸附剂的特异性较低,往往用于
纯化一类蛋白质,如用凝集素分离糖蛋白、用蛋白质 A分离抗体等,其纯化程
度可能不如前者,但却非常方便。
511 配体和载体的选择
5111 配体的选择
在进行亲和色谱之前首先要选择合适的配体,配体分为两种主要类型:(1)基团特异性配体 对某个化学基团,即某一类蛋白质具有结合活性。(2)专一性配体 只对一种蛋白质具有结合活性。对于基团特异性配体,目前已有制备好的商品化亲和吸附剂出售,将在513
节中专门对此进行介绍,这里仅讨论专一性配体的选择及其亲和吸附剂的制备。选择配体时需要考虑的因素有:(1)配体对蛋白质的特异性 如果对蛋白质的生物性质有充分的了解,就可
以选择高特异性的、只识别待分离蛋白质的配体;否则的话,可以选择基团特异
性配体。(2)配体与蛋白质结合的可逆性 配体既要能有效地与蛋白质结合,又要能
有效地与蛋白质解离。(3)配体对蛋白质的亲和性 配体对蛋白质的亲和性既不可太低,这样不易
结合;又不可太高,这样结合后不易解离。合适的配体蛋白质间的解离平衡常
数KL 应在10-8~10-4mol/L之间。如果KL 能随着洗 脱 条 件 的 调 整 而 明 显 改
变,则适宜选择高亲和性的配体。(4)配体的大小 配体必须足够大,它既能特异性地与待纯化的蛋白质分子
相结合,又存在另一个与载体相结合的位点。若配体太小,可以在配体和载体之

122
间接入一个连接臂。亲和色谱中配体的选择是实验成败的关键,对不同的待分离蛋白质,应选择
相应的配体,表51简要列出了一些蛋白质纯化时的合适配体。
表51 蛋白质亲和色谱中的合适配体
配 体 待纯化的蛋白质 配 体 待纯化的蛋白质
抗原 特定单克隆抗体或多克隆抗体
单克隆抗体 特定抗原
蛋 白 质 A/蛋
白质G 免疫球蛋白
蛋白酶抑制剂 蛋白酶
磷酸 磷酸酶
三嗪染料 脱氢酶、激 酶、聚 合 酶、限 制 酶、干扰素
抗生物素蛋白 含生物素的酶
肝素 凝 聚 因 子、脂 酶、结 缔 组 织 蛋 白
酶、DNA聚合酶
胆固醇 胆固醇受体、胆固醇结合蛋白
脂肪酸 脂肪酸结合蛋白、白蛋白
核苷酸 核苷酸结合蛋白、需核苷酸的酶
苯基硼酸盐 糖蛋白
凝集素 糖蛋白
糖 凝集素、糖苷酶
5112 载体和连接臂的选择
对于亲和色谱,除了要选择合适的配体外,还要选择一个合适的载体。理想
的载体既要对样品不产生非特异性吸附作用,又要有足够的可同配体相结合的功
能基团;既要高度亲水,又要不溶于水;要具有化学和物理稳定性、一定的机械
强度和均匀性;并且载体必须是孔径较大 的 多 孔 性 材 料,不 会 阻 止 蛋 白 质 的 通
过。亲和色谱的载体多为凝胶,其中琼脂糖是比较理想的载体,另外聚丙烯酰胺
凝胶等也可以作为亲和色谱的载体。当配体比较小,而待分离蛋白质比较大时,由于载体表面的邻近效应会限制
配体与蛋白质的结合,这时通过连接臂连接配体和载体,就不会阻止配体与蛋白
质的结合。连接臂的长度是非常重要的,一般其甲基数目为6~8,并且要求连
接臂亲水,但不会结合蛋白质。目前市场上有接有连接臂的商品化载体出售 (表52)。
表52 接有连接臂的亲和色谱用载体
载 体 连 接 臂 供 应 商
3氨基丙基和琥珀酰氨基丙基 BioRad3,3′二氨基丙基和琥珀酰二氨基丙基 BioRad1,2二氨基乙烷,1二氨基己烷,3,3′二氨基二丙基胺 ICN
琼脂糖1,6二氨基己烷,6氨基己酸 Pharmacia3,3′二氨基丙基胺,对苯甲酰基3,3′二氨基丙基胺 Serva氨基烷基 Miles
聚丙烯酰胺琼脂糖 1,6二氨基己烷,6氨基己酸 IBF
多孔玻璃 氨基丙基,氨基己基 Merck

123
512 亲和吸附剂的制备方法
选择好合适的配体、载体后,就要用适当的化学方法,将载体活化并与配体
相偶联,以得到亲和吸附剂。然后封闭载体上的游离活化基团,并测定偶联配体
的浓度。
5121 载体的活化和配体的偶联方法
有多种方法可以完成载体和配体的偶联过程。偶联过程 (图52)通常分为
两步:将反应基团结合到载体上,即 活 化 载 体;将 活 化 好 的 载 体 与 配 体 共 价 偶
联。有时在活化载体的同时,就可以插入一连接臂。目前市面上有预活化好的载
体供选择 (表53)。除两步偶联过程外,也可以活化和偶联一步完成,但这种
方法很少使用。
图52 亲和吸附剂的两步偶联制备过程
表53 亲和色谱用商品化预活化载体
活 化 方 法 载 体 供 应 商
溴化氰法 Sepharose(琼脂糖/交联琼脂糖) Pharmacia环氧法 EupergitC(聚丙烯酸盐/聚丙烯酰胺) RhomPharma环氧法 SeparonHEMA(聚羟乙基异丁烯酸) Anachem/Tessek环氧法 Memsep(纤维素) DominickHunter羰基二咪唑法 ReactiGel(交联琼脂糖)等 Pierce磺酰氯法 Sepharose(琼脂糖/交联琼脂糖) Pharmacia磺酰氯法Tresyl Selectispher10(硅胶) Pierce
FMP①法 Avidgel(交联琼脂糖) BioProbe戊二醛法 Ultrogel(聚丙烯酰胺琼脂糖) IBF戊二醛法 Magnogel(磁化Ultrogel) IBF戊二醛法 MacrosorbK(硅藻土) SterlingOrganics乙醛法 ActigelA(交联琼脂糖) Sterogene
N羟基琥珀酰亚胺法 AffiGel10,15(交联琼脂糖) BioRad
N羟基琥珀酰亚胺法 AffiPrep10(合成聚合物) BioRad
① 甲苯4磺化2氟1甲基吡啶 。

124
载体和配体的性质不同,则偶联方法也有差异。当某一种偶联方法的结合效
率低或导致配体失活时,应选择其他的偶联方法。表54列出了各种活化及偶联
方法,后面还将对一些主要的方法进行介绍。
表54 亲和色谱的载体活化和配体偶联方法
偶 联 方 法 偶 联 试 剂 载体的活化基团 配体的偶联基团
溴化氰法 溴化氰 多糖的—OH 配体的—NH2双环氧法 双环氧化物 凝胶的—OH或—NH2 配 体 的 亲 核 基 团 (—SH/
—NH2/—OH)羰基二咪唑法 羰基二咪唑 多糖 配体的—NH2磺酰氯法 对甲苯 磺 酰 或 三 氟 代 乙
磺酰
多糖的—OH 配体 的 亲 核 基 团(—NH2/—SH)
高碘酸盐法 高碘酸钠 多糖的连二醇基 配体的—NH2磺化氟甲基吡啶 法 甲苯4磺化2氟1甲基
吡啶
多糖的—OH 配体的—NH2
戊二醛法 戊二醛 载体的氨基或酰胺基 配体的—NH2交联法 碳化二亚胺 载体的—COOH 配体的亲核基团,如—NH2
(1)溴化氰法 溴化氰法是最常用的配体固定化技术,大多数配体都能容易
地用此法偶联 (图53)。
OH
OH
多糖载体
帒帒 →+ Br C N O C N+帒帒 HBr
活化多糖:氰酸酯
O C N+NH帒帒 2 →配体
O C
N
H
NH配体 异脲衍生物
OO
C NH配体 N位取代的亚氨碳酸盐
O
C
O
NH配体 N位取代的氨基甲酸酯
活化多糖 亲和吸附剂
图53 溴化氰法制备亲和吸附剂的原理
① 载体活化 溴化氰和多糖类载体 (如琼脂糖)的羟基反应,使其活化为
氰酸酯。
② 配体偶联 活化的多糖与连接臂或配体上的氨基发生偶联反应,得到亲
和吸附剂。载体活化时溴化氰的用量为50~300mg/ml凝胶,此用量决定于需偶联的配
体的量。由于氰 酸 酯 在 低 温 下 稳 定,因 此 一 般 在 低 温、pH=11下 进 行 活 化 反

125
应,反应后需要 立 即 用 冰 蒸 馏 水 抽 滤 洗 涤。而 偶 联 反 应 的 条 件 为:4℃,pH=8~10碳酸或硼酸缓冲液 (既要使配体的氨基处于非质子化状态,即pH>pKa,又要使氰酸酯稳定,而pH>10氰酸酯的稳定性急剧下降),反应约12h。
溴化氰法制备亲和吸附剂的操作过程如下:琼脂糖凝胶低温下平衡;30℃、
pH=11条件下,将溴化氰加入琼脂糖凝胶中溶解并活化10~12min;快速抽滤
并用冰水洗涤;用pH=8偶联缓冲液洗涤平衡;4℃下将含配体的偶联缓冲液加
入活化琼脂糖中偶联12h;加入乙醇胺或1氨基丙二醇除去未偶联的活化基团;依次用水、2mol/L氯化钾洗涤数次以除去游离配体或乙醇胺。
溴化氰法有三个缺点:此亲和吸附剂对亲核进攻很敏感,在极端pH下会缓
慢 水 解,导 致 配 体 脱 落;溴 化 氰 有 剧 毒,建 议 采 用 预 活 化 好 的 琼 脂 糖,在
5322节(2)中将介绍预活化琼脂糖与抗体的偶联过程;溴化氰法生成的亲和
吸附剂N取代异脲在生理条件下带正电 (pKa≈95),因而具有阴离子交换剂
的作用。(2)双环氧法 双环氧法也是一种较常用的制备亲和吸附剂的方法。
① 载体活化 在碱性条件下,双环氧化物活化含羟基或氨基的凝胶,产生
含一长亲水链的活化环氧琼脂糖。如等量琼脂糖凝胶与1,4丁二醇2缩水甘油
醚混合分散后,再加入等量06mol/LNaOH,25℃旋转反应8h,然后用蒸馏水
洗涤以终止反应,即得到环氧琼脂糖。
② 配体 偶 联 活 化 琼 脂 糖 上 的 环 氧 乙 烷 基 团 与 亲 核 试 剂 (如 含—SH、—NH2 或—OH的有机分子)反应制备亲和吸附剂 (图54)。偶联的温度或pH升高,则偶联效率提高。
CH2OCH2
CH CH 2
+ NH2— 配体O CH2
OCH2
CH CH 2
SH— 配体O
CH2
OCH2
CH CH 2
OH— 配体
→
O
CH2OCH2
CH CH2—NH— 配体OH
CH2
OCH2
CH CH2—S— 配体OH
CH2
OCH2
CH CH2—O— 配体OH
活化环氧琼脂糖 亲核配体 亲核吸附剂
图54 双环氧法制备亲和吸附剂的原理
用活化的环氧乙烷载体制备亲和吸附剂的操作过程将在5332节中介绍。此法的特点:活化的同 时 产 生 一 长 链 亲 水 连 接 臂;活 化 偶 联 时 不 产 生 新 电
荷,从而不会产生非特异性吸附作用;环氧偶联得到的亲和吸附剂很稳定;双环
氧法偶联的配体量要少于溴化氰法偶联的配体量。(3)羰基二咪唑法

126
① 载体活化 无水条件下,羰基化试剂如N,N′羰 基 二 咪 唑 (CDI)活 化
多糖载体,产生活性咪唑碳酸盐衍生物。
② 配体偶联 在碱性条件下,活化多糖与含伯胺基的配体反应生成稳定的
碳酸盐衍生物。偶联条件为室温下,01mol/LpH=85~10的硼酸缓冲液。用CDI活化的琼 脂 糖 凝 胶 制 备 亲 和 吸 附 剂 的 操 作 过 程 将 在5311(2)中
介绍。此法的特点:CDI无毒且此法的活化程度高;此亲和吸附剂不会产生离子交
换剂的作用;CDI活化的琼 脂 糖 在 无 水 条 件 下 稳 定,多 溶 于 丙 酮 中,使 用 (偶
联)前需除去丙酮。(4)磺酰氯法
① 载体活化 有机磺酰卤化物与载体的羟基反应,生成磺酰酯,其磺酰基
团易离去。一般用对甲苯磺酰 (tosyl)或三氟代乙磺酰 (tresyl)在有机相中活
化载体,活化后的载体在1mmol/LHCl或干燥状态下很稳定。
② 配体偶联 配 体 的 亲 核 基 团 (氨 基、巯 基)取 代 磺 酰 基 团,从 而 有 效、快速地偶联。用三氟代乙磺酰进行活化、偶联时,偶 联 反 应 在 中 性pH、4℃下
进行;而用对甲苯磺酰活化、偶联时,载体需在碱性pH (pH=9~105的碳酸
盐缓冲液)、40℃下进行偶联。此法的特点在于当用对甲苯磺酰活化、偶联时,可用分光光度法进行监测。(5)高碘酸盐法
① 载体活化 高碘酸钠使多糖载体的连二醇基氧化形成醛基,NaIO4 溶于
水,很容易从活化凝胶中洗去,活化凝胶在4℃可稳定几天。
② 配体偶 联 活 化 凝 胶 的 醛 基 与 配 体 的 伯 氨 基 在pH=4~6下 反 应 形 成
Schiffs碱,此Schiffs碱经硼氢化钠或硼氢化氰还原而稳定 (图55)。
OH
OH+NaIO →4 CHO
CHO+NH2— →配体 CH N— 配体NaBH
→4
CH 2 NH— 配体
图55 高碘酸盐法制备亲和吸附剂的原理
高碘酸盐法活化偶联的操作过程为:
a活化 凝胶载体与02mol/LNaIO4 等体积混合,置于密闭瓶中,室温下
轻微混合2h。
b洗涤 依次用蒸馏水,pH=60、05mol/L磷酸缓冲液彻底洗胶。
c偶联 将活化凝胶加到含25mmol/L配体和05mol/L硼氢化钠的pH=

127
60、05mol/L磷酸缓冲液中,室温下轻微混合3天。
d去活化 4℃下,用1mol/L硼氢化钠与偶联好的凝胶共浴15h,还原未
偶联的醛基。
e洗涤 依次用蒸馏水、储存缓冲液彻底洗胶。高碘酸盐法使用很方便,特别是当配体对碱性pH敏感时,而且无毒,操作
安全,价格低廉,烷基胺产物很稳定。(6)磺化氟甲基吡啶 法 磺化氟甲基吡啶 活化偶联法是近年来用于亲和
色谱的一个重要方法,值得推广。
① 载 体 活 化 在 极 性 有 机 溶 剂 中,用 甲 苯4磺 化2氟1甲 基 吡 啶 (FMP)活化多糖的羟基,活化反应在叔胺如三乙基胺存在下进行,室温10min内即可完成反应。
② 配体偶联 在pH=8~9的水溶液或有机溶剂中,产生的活化基团2烷
氧基1甲基吡啶 盐与配体的氨基发生偶联反应。此法的特点是:活化和 偶 联 过 程 都 可 以 用 分 光 光 度 法 监 测,因 为 离 去 基 团
1甲基2吡啶在297nm有 强 烈 的 光 吸 收;FMP无 毒,较 便 宜,且 在 高 度 取 代
时,形成稳定的、非离子化的交联。(7)戊二醛法
① 载 体 活 化 在20~40℃下,用25%戊 二 醛 溶 液 (溶 于 pH=75,
05mol/L磷酸缓冲液中)将载体的氨基或酰胺基活化过夜。
② 配体 偶 联 在40℃、同 样 的 缓 冲 液 中,配 体 的 伯 胺 基 与 活 化 载 体
偶联。 此法的特点是:简单,便宜,适宜对碱性pH敏感的配体进行活化偶联;此
亲和吸附剂稳定;在偶联的同时引入一连接臂;在储存时戊二醛会聚合,且戊二
醛有中等毒性。(8)交联法 用碳化二亚胺使载体的羧基和配体的亲核基团如氨基偶联形成
肽键。由于二环己基碳化二亚胺不溶于水,只能溶于有机溶剂中,如二 烷、二
甲亚砜、80% (体积分数)吡啶或乙腈,因此面临着除去不溶性脲和其他副产品
的问题,而水溶性的碳化二亚胺更方便。交联法制备亲和吸附剂的主要过程为:
① 准备载体 用水彻底洗涤含羧基的载体,并调pH至40~45。
② 加入交联剂 将40mg1乙基(3二甲基氨基丙基)碳化二亚胺溶于10mlpH=45的缓冲液中,然后加到4ml载体中。
③ 偶联 加入含氨基的配体,室温下旋转混合1~2h,进行偶联反应。
④ 终止反应 过滤亲和吸附剂,洗去未反应的碳化二亚胺和副产品。

128
5122 活化基团的封阻
将载体活化后,活 化 基 团 的 浓 度 一 般 为5~25μmol/ml,而 偶 联 上 的 配 体
的浓度为1~10μmol/ml,因此偶联后一些残存的 活 化 基 团 还 会 保 留 在 凝 胶 中,必须加入 低 分 子 量 的 化 合 物 封 闭 这 些 基 团。最 常 用 的 封 闭 试 剂 是pH=80的1mol/L乙醇胺,用 它 与 活 化 载 体 在 室 温 下 反 应1h就 可 以 封 闭 活 化 基 团。如果用其他小分子物质,如甘氨酸,则必须检验一下它们是否会导致非 特 异 性
吸附。在 封 闭 的 后 期,应 快 速 洗 涤 吸 附 剂,然 后 将 它 储 存 在002%叠 氮 化
钠中。
5123 配体浓度的估计
将配体固定化到载体上后,需检测一下配体的浓度,以判断偶联过程成功与
否。测定偶联上的配体的浓度有这样几种方法:(1)差示分析法 用加入的配体量减去洗脱液中游离配体量,即可得出偶联
上的配体量。(2)直接测定法 直接测定制备好的亲和吸附剂上配体的浓度,可以根据配
体的性质采用分光光度法、蛋白质测定方法或特定基团测定方法来测定。(3)放射分析法 对于具有放射活性的配体,可以很灵敏地测出固相化配体
的浓度。(4)水解法 当配体的测定不受大量的载体水解产物干扰时,可以用此法测
定亲和吸附剂上配体的浓度。水解的方法可视配体、载体的偶联情况而定,可以
采用酸水解或酶水解。
513 商品化基团特异性吸附剂
常见的商品化基团特异性亲和吸附剂有凝集素、蛋白质A和固定化染料等。
5131 凝集素
凝集素是一类来自于霉菌、植物和动物的蛋白质,它们能可逆地与特定的糖
残基结合,因此对于分离提纯含糖基的生物大分子 (如糖蛋白、血清脂蛋白及膜
蛋白等)非常有用。通过与红细胞表面特异性受体结合,凝集素能凝集各种类型
的红细胞,因此得名。不同的凝集素表现出不同的特异性,如伴刀豆凝集素 A和晶状体凝集素能
够与C3、C4、C5位上含羟基的糖类结合,而小麦胚芽凝集素能与N乙酰葡糖
胺残基结合。吸附后,可以用盐梯度溶液或含游离糖的溶液将待纯化蛋白质从亲
和柱上洗脱下来。凝集素亲和色谱将在531节中详细介绍。
5132 蛋白质A蛋白质A用于纯化IgG型抗体或与抗体有特异性亲和力的抗原。蛋白质A

129
是一种商品化表面蛋白,来自金黄色葡萄球菌,它具有特异的、可同大多数哺乳
动物的IgG型抗体的不变区域Fc相结合的能力,但是它们的相互作用与抗体的
Fab组分无 关,因 此 可 用 于 纯 化 各 种IgG。蛋 白 质 A 与 鼠IgG1,马IgGc,鸡
IgG,以及大多数IgA和IgM结合力很弱,与人IgG3,大鼠IgG2a和IgG2b无结
合力。将IgG抗体通过Fc固定在蛋白质A琼脂糖色谱柱上,其Fab就可以与对
应的抗原相吸附,因此可用于提纯所需抗原。蛋白质A与IgG的结合力随着盐浓度的提高和pH 的增加而增强。在高盐
浓度及高pH下,如:3mol/LNaCl,pH=8~9,常用磷酸盐或Tris作为吸附
缓冲液。洗脱时应采用01mol/L柠檬酸或1mol/L醋酸降低pH 梯度洗脱。由
于配体本身对6mol/L盐酸胍稳定,因此可以用6mol/L盐酸胍不断清洗蛋白质
A亲和柱,它一般储存于70%乙醇中。
5133 蛋白质G蛋白质G可以与那些不能被蛋白质A结合的IgG相结合 (除了鸡IgG外)。一般在生理缓冲液中吸附,然后用01mol/LpH=25甘氨酸HCl缓冲液
洗脱,洗脱后的蛋 白 质 应 立 即 中 和。色 谱 柱 的 清 洗 采 用 低pH 的6mol/L盐 酸
胍,并储存于70%乙醇中。
5134 固定化染料
固定化染料用于纯化许多蛋白质和酶,作为配体的染料是含一氯或二氯三嗪
基团的三嗪染料,其中最广泛应用的染料之一是CibacronBlueF3GA,这种染
料在结构上 (图56)与核苷酸类辅助因子如NAD+和NADP+很相似,因此可用
于纯化含有这些辅助因子的酶以及人血清白蛋白、成纤维细胞干扰素、脂蛋白等
師師師師帪帪
。
師師師師帪帪
O
O NH
2SO3H
師師師師帪帪
NH
SO3
H
師師師師帪帪
NH
N
N
Cl
NH
師師師師帪帪
N
SO3
H
图56 CibacronBlueF3GA的结构
三嗪染料可偶联 含 羟 基 的 载 体。三 嗪 染
料法制备亲和吸附剂的主要过程为:(1)准 备 载 体 用 水 洗 琼 脂 糖,并 将
20g湿重琼脂糖分散于50ml水中。(2)准备染料 将200mg染料溶于20ml
水中。(3)混合 将载体和染料溶液彻底混合。(4)偶联 先加入10ml20% (200g/L)
氯化钠溶液,室温放置30min,再加入20ml5% (50g/L)碳酸钠溶液,于45℃偶联 (对于二氯三嗪染料,偶联约需1h;对于一氯三嗪染料,偶联约需40h)。
(5)洗涤 用温水、6mol/L尿素和1% (10g/L)碳酸钠洗涤亲和吸附剂,直至洗涤液无色。
(6)储存 于含002%叠氮化钠的01%碳酸钠溶液中储存。

130
对于固 定 化 CibacronBlueF3GA,一 般 用 低 摩 尔 浓 度、pH=7~85的
TrisHCl作为吸附缓冲液。降低pH会加强结合力。洗脱时用达1mol/LKCl的
盐梯度洗脱或采用亲和洗脱。亲和柱可以用8mol/L尿素清洗,但是脂蛋白会堵
塞柱表面,这时最好拆柱,将固定化染料浸泡于蒸馏水中,除去脂蛋白颗粒,再
用极端pH的尿素溶液洗几次。染料分子的渗漏可通过颜色方便地观察到。除了固定化CibacronBlueF3GA外,目前有许多其他活性染料用于蛋白质
纯化。染料配体亲和吸附剂能非常有效地用于串联纯化中。由于现在还无法预测
一种特定蛋白质与哪些或哪种染料能发生特异性结合,因此必须对染料配体库进
行系统扫 描 测 定。其 总 体 色 谱 过 程 与 前 面 的 CibacronBlueF3GA 色 谱 过 程
相同。
5135 赖氨酸
赖氨酸可结合纤溶酶原、纤溶酶和纤溶酶原激活剂。一般在生理缓冲液中能
有效吸 附,接 着 用 含05mol/LNaCl的 相 同 缓 冲 液 洗 去 杂 蛋 白,最 后 可 用
02mol/Lε氨基己酸溶液特异洗脱目的蛋白。
52 色谱技术
获得合适的亲和吸附剂后,就可以将亲和吸附剂装入色谱柱进行亲和色谱。亲和色谱的操作过程与凝胶过滤色谱和离子交换 色 谱 大 体 相 似,但 也 有 某 些 差
别。其总体过程依然是:装柱,平衡,制备粗样,加样,洗脱。其中加样过程与
离子交换色谱相似。对于亲和色谱,加样过程实际上是特定蛋白质的吸附过程,因此称之为吸附。加样量可以很大,只要不超过柱的吸附容量,可以采取流动加
样或循环加样方式,而不必担心样品的扩散。洗脱过程分为两阶段,首先是洗去杂
蛋白,然后再将待分离蛋白质洗脱下来。杂蛋白也可以在流动吸附的同时被洗去。
521 亲和色谱操作过程
5211 吸附
吸附的目的是使亲和吸附剂上的配体与待纯化蛋白质间形成紧密结合物。由
于蛋白质与配体之间是通过次级键相互作用发生结合的,因此预先了解配体蛋
白质间的相互作用类型将有助于选择合适的吸附缓冲液。如果其相互作用方式主
要是疏水键,提高离子强度或pH将增强吸附,而低温会弱化疏水作用。另一种
提高目的蛋白对亲和吸附剂亲和力的方法是增加固定化配体的浓度。有时延长吸
附时间也能提高蛋白质与固定化配体的结合程度,如采用二次吸附,或分批式反
应,或使用长柱子、低流速、低样品量加样方式。如果不了解蛋白质与配体之间
的相互作用类型,可预先用小样进行比较实验,以确定最佳吸附条件。

131
5212 洗去杂蛋白
如果配体蛋白质间的亲和力很高,则可以方便、稳定地将非特异吸附的蛋
白质解吸下来,解吸缓冲液应该处于最佳的吸附缓冲液和洗脱缓冲液之间,即如
果某种蛋白质在低摩尔浓度的磷酸缓冲液中吸附,在075mol/LNaCl下 洗 脱,则可以采用04mol/LNaCl洗柱子,以观察是否能提高洗脱后样品的纯度。
5213 洗脱
吸附在亲和柱上的蛋白质分子与周围的固定化配体之间处于平衡状态。通过
改变配体蛋白质复合物间的环境,从而降低蛋白质对配体的亲和性,就能够有
效地将蛋白质洗脱下来。由于配体与蛋白质间的相互作用是基于不同的次级键相
互作用,因此任何能破坏这些次级键的溶剂条件都会使配体蛋白质复合物不稳
定。但要注意防止蛋白质或配体发生不可逆变性。有效的洗脱方法主要有以下几种 (排列越前的洗脱方法越温和):(1)改变缓冲液的离子强度 如果配体与蛋白质间离子键占优势,则增加离
子强度就能减弱蛋白质与配体间的非共价相互作用,从而将蛋白质从配体上解吸
下来。相反,如果他们之间疏水相互作用占优势,则降低离子强度能够有效地将
蛋白质从亲和柱上洗脱下来。(2)改变缓冲液的pH 改变pH,就会改变结合表面带电基团的电性和电
量,从而破坏异性离子间盐桥的形成,并进而降低蛋白质与配体间相互作用的强
度。虽然改变pH与改变离子强度一样有效,但是由于pH梯度难于实现,因此
不如离子强度梯度洗脱那么普遍,且只能用分步洗脱方式。(3)亲和洗脱 亲和洗脱就是使用游离配体,或其他能同配体或待洗脱蛋白
质发生更强特异性亲和作用的分子,将蛋白质从固定化配体上竞争下来。亲和洗
脱的价格可能会比较高,且洗脱下来的蛋白质可能很难与洗脱液分离。亲和洗脱
的例子有:用不同的核苷酸将脱氨酶从染料柱上洗脱下来,用游离糖将糖蛋白从
凝集素柱上洗脱下来等。(4)使用促溶剂 当配体与蛋白质间 的 亲 和 力 很 强,其 他 洗 脱 方 法 都 失 败
时,可以采用促溶剂。促溶剂能破坏水的结构,降低配体蛋白质间相互作用的
强度,从而将蛋白质解吸下来,常用的促溶剂 有3mol/L硫 氰 化 钾、2mol/L碘
化钾、4mol/LMgCl2。(5)使用蛋白质变性剂 8mol/L尿素,6mol/L盐酸胍也可能有效。对于促
溶剂和变性剂,在使用之前,必须测试一下它们对蛋白质活性的影响。(6)使用极性还原剂 可以尝试采用10% (体积分数)二 烷或50% (体
积分数)1,2乙二醇。低浓度洗涤剂也能有效洗脱。另外,有时改变温度也能够
有效洗脱目的蛋白。

132
5214 再生
亲和柱的再生通常采用高浓度盐溶液,如2mol/LKCl。一般不使用极端pH条件或加热灭菌法来清洗、再生亲和柱,特别是对于以蛋白质作配体的亲和吸附
剂。亲和吸附剂在储存时应加抑菌剂,如002% (02g/L)叠氮化钠。
522 亲和色谱的操作注意事项
亲和色谱的操作模式主要有分批式和柱式两种,一般多采用柱式操作。分批
式适用于高亲和系统,特别是当样品体积很大、浓度不稳定时,在分批式操作时
应避免吸附反应时间过长。而低亲和系统应采用柱式操作,它简便、灵活,因而
使用较多。亲和色谱所需亲和柱的总床体积取决于其吸附容量和分离类型。对于某一目
的蛋白,如果配体和洗脱条件有很高的选择性,则蛋白质的分辨率决定于亲和吸
附剂本身,而基本不受柱长的影响,因此对于高亲和系统来说,短粗的柱子比较
合适。亲和色谱的流速决定于载体的孔径和待纯化蛋白质的大小。流速应尽量慢,
如果流速过快,在柱吸附饱和前,目的蛋白就会出现在流出液中,并导致洗脱阶
段峰脱尾,对于以琼脂糖作载体的亲和吸附剂,推荐采用10ml/(cm2·h)的线
性流速。在洗去杂蛋白和再平衡步骤中可以采用快流速。亲和色谱的一个严重缺陷就是配体的渗漏问题。配体渗漏的原因可能在于固
定化配体的不稳定性。配体渗漏后会降低亲和吸附剂的吸附容量。对于高亲和系
统来说,痕量蛋白质的分离尤其会受制于配体的脱落情况。
523 亲和色谱的吸附容量与吸附效率
5231 吸附容量
亲和色谱的吸附容量是指在选定的有效吸附和洗脱条件下,能吸附到特定亲
和柱上的目的蛋白的总量。其测定方法如下:用未使用过的亲和吸附剂制备一根
1ml床体积的亲和柱,以选定缓冲液用6~8ml/h的速度平衡;将待纯化的粗蛋
白溶液以恒定流速进样,溶液中目的蛋白的浓度为P0,收集流出液 (1ml/管);测定流出液的蛋白质吸光度 (A280),并测定各收集管中目的蛋白的浓度P;当
流出液中P 达到P0 时,亲和柱的吸附容量达到饱和;用解吸缓冲液洗去杂蛋
白,直至A280回到基线;用选定的洗脱液洗脱,测定流 出 液 中 目 的 蛋 白 的 浓 度
P,并计算其产量。在图57中:Ve (目的蛋白的吸附体积)表示流出液中P达到P0 的50%
时的体积;V0 表示非吸附蛋白质的浓度达到起始浓度的50%时的体积;吸附容
量=P0(Ve-V0);工作容量是指洗脱后收集到的目的蛋白的产量。

133
理论上讲,吸附容量应该等于工作容量。如果工作容量比吸附容量低很多,则目的蛋白没有被有效洗脱下来,应改进洗脱条件。对于旧的亲和吸附剂,当其
工作容量大于起始值的80%时就可以重复使用。
图57 亲和吸附剂吸附容量测定
5232 配体效率
配体效率定义为工作容量/理论容量×100。理论容量=固定化配体的浓度×单位摩尔配体的结合位点数目。实际上,对于小分子配体而言,配体效率通常等于或小于1%,这表明配体
分子被固定化后难以被目的蛋白接近。而对于大分子配体,如单克隆抗体,则配
体效率通常大于10%,这 一 点 反 映 了 用 大 分 子 配 体 只 能 得 到 低 的 固 定 化 密 度。对于成功的亲和色谱,应最大程度地提高配体效率。
53 亲和色谱的特殊类型
531 凝集素亲和色谱
凝集素是一类能选择性结合碳 水 化 合 物 的 蛋 白 质 或 糖 蛋 白,可 专 门 用 于 分
离、提纯含碳水化合物的生物大分子,如糖蛋白等。不同来源的凝集素其特异性有所不同,但具有共同的结构性质。它们都由一
种或多种亚基组成,一般为四亚基蛋白质,每个亚基都有糖基结合位点。如果所
有的亚基都相同,凝集素的多个结合位点都识别同一种特异性糖基。如果它具有
两种功能上有差别的亚基,凝集素就具有结合两种不同糖基的能力。常用的凝集
素的一些重要的结构性质列于表55。
5311 凝集素色谱材料
(1)凝集素的 选 择 由 于 不 同 的 凝 集 素 对 不 同 的 糖 基 有 特 异 性 (表55),因此针对某一特定的目的糖蛋白,应选择合适的凝集素。例如,许多受体结合于

134
麦芽凝集素,而细胞因子和其他生长因子则优先结合于伴刀豆球蛋白 A或扁豆
凝集素。选择凝集素时需考虑两个因素:糖 蛋 白 上 寡 糖 的 性 质 和 凝 集 素 的 可 获
得性。
表55 亲和色谱中常用的凝集素
凝 集 素 分子量 亚基数目 特 异 性 洗 脱 糖 类 所需离子
伴刀豆球蛋白A 55000 2/4 αD甘露糖
αD葡萄糖
α甲基甘露糖
α甲基葡萄糖
Ca2+,Mn2+
扁豆凝集素 49000 2 αD甘露糖
αD葡萄糖
α甲基甘露糖
α甲基葡萄糖
Ca2+,Mn2+
豌豆凝集素 49000 4(αβ) αD甘露糖 α甲基甘露糖
蓖麻凝集素 120000 4 βD半乳糖 乳糖,半乳糖
蓖麻凝集素 60000 2 αDN乙酰半乳糖胺 αDN乙酰半乳糖胺
麦芽凝集素 36000 2 N乙酰葡糖胺 N乙酰葡糖胺 Ca2+,Mn2+,Zn2+
大豆凝集素 约110000 4 αDN乙酰半乳糖胺 αDN乙 酰 半 乳 糖
胺,半乳糖
植物血球凝集素 128000 4 αDN乙酰半乳糖胺 αDN乙酰半乳糖胺 Ca2+,Mn2+
糖蛋白上的寡糖可分为几种类型,每种类型都有其特点,这些特点影响着它
们与凝集素的结合,从而可作为选择亲和色谱凝集素的一个重要参数。在不清楚
待纯化糖蛋白的结构和性质时,应从一系列凝集素中筛选合适的。现在有许多凝集素可以直接购得,甚至可以直接购买已偶联好的凝集素亲和
吸附剂。如果没有商品化凝集素亲和吸附剂供应,可以自行将纯的目的凝集素固
定到合适的活化载体上。(2)凝集素与载体的偶联 凝集素可以偶联到不同的活化载体上,常见的有
以下几种类型:①固定到CNBr活化的Sepharose4B上;②固定到Affigel10或
Affigel15上;③固定到CDI活化的琼脂糖上。Affigel10和Affigel15是交联琼
脂糖的N羟基琥珀酰亚胺酯,分别带10个或15个原子长的连接臂,蛋白质通
过其自由氨基与 它 们 进 行 偶 联 (图58);这 里 仅 介 绍 将 配 体 偶 联 到 Affigel及
CDI活化的琼脂糖上的过程,而将配体偶联到CNBr活化的琼脂糖上的方法将在
免疫亲和色谱中进行介绍。
帿帿帿帿帿OAffigel10:10个原子
Affigel15:15个原子
帿帿帿帿帿 O N
O
O
+NH2— →配体 O NH—帿帿帿帿帿帿帿 配体 + NHO
O
O
图58 Affigel与配体的偶联
在将凝集素偶联到活化载体上时,对于不同的凝集素,应在偶联缓冲液中加

135
入相应的特异糖类和所需的二价离子。因为糖可以保护凝集素上的糖蛋白结合位
点,防止这些位点被载体上的活化基团所交联,从而最大程度地保留糖蛋白结合
位点。对于需要二价金属离子的凝集素,二价金属离子能引起凝集素三维结构的
转化,从而导致糖结合位点的形成,因此在偶联之前应加入所需的二价离子。偶
联时凝集素的浓度应低于10mg/ml,因为当载体上固定过多 的 凝 集 素 后,会 产
生空间位阻效应,干扰糖蛋白的结合。将凝集素偶联到Affigel载体上的过程:(1)清 洗 Affigel 将 Affigel置 于 漏 斗 内 抽 滤 除 去 异 丙 醇,并 用 含100
μmol/L所需 二 价 金 属 离 子 和5% (50g/L)特 异 糖 的 pH=84、01mol/LNaHCO3 (偶联缓冲液)清洗,勿使载体干燥。
(2)配制凝集素溶液 用偶联缓冲液配制5~7mg/ml凝集素溶液。(3)偶联 在每克清洗过的湿Affigel中加入1ml凝集素溶液,4℃轻微混合
过夜。(4)封阻未反应位点 加入1mol/L甘氨酸、甘氨酸乙酯或01mol/L乙醇
胺,室温下放置2h。(5)储存 用偶联缓冲液洗凝集素Affigel,将其储存于含005%叠氮化钠
的pH=72PBS中。将凝集素偶联到CDI活化的琼脂糖上的过程如下:(1)清洗CDI活化的琼脂糖载体 将CDI活化的琼脂糖置于漏斗内,抽滤
除去丙酮,并用冰蒸馏水彻底清洗,切勿使凝胶块干燥。(2)配 制 凝 集 素 溶 液 用 含05mol/L 特 异 糖 的 pH=95、01mol/L
NaHCO3 溶液 (偶联缓冲液)配制20mg/ml凝集素溶液。(3)偶联 将1体积湿凝胶加到1体积凝集素溶液中,4℃轻微混合48h。(4)封阻 用pH=95、01mol/LTris清洗,以封阻活化载体上未反应的
位点。(5)储存 依次用3体积pH=40、01mol/L醋酸钠缓冲液,3体积pH=
80、01mol/LNaHCO3 洗两 次,再 用 含002%叠 氮 化 钠 的PBS洗 并 储 存 于
其中。
5312 凝集素亲和色谱的色谱方法
(1)亲和柱的平衡 制备好固定化凝集素后,就可以将其装柱,然后用5倍
体积的平衡缓冲液 (如含01mol/LNaCl和1mmol/L所需二价金属离子的pH=72,20mmol/L磷酸缓冲液)清洗、平衡。亲和柱的大小决定于样品量及凝集素
对糖蛋白 的 吸 附 容 量。为 了 提 高 凝 集 素 结 合 效 率,可 以 在 平 衡 缓 冲 液 中 加 入
01mmol/L二价金属离子,为了改善样品稳定性可加入05%洗涤剂。

136
(2)样品的制备 用凝集素亲和色谱分离的糖蛋白主要分为两类:水溶性糖
蛋白和膜结合糖蛋白。前者包括 细 胞 分 泌 蛋 白 和 结 构 蛋 白,如 激 素、胶 原 蛋 白
等;后者有转运蛋白和细胞表面识别标记,如受体等。在制备水溶性糖蛋 白 样 品 时,应 将 样 品 溶 解 于 生 理pH 的 磷 酸 或TrisHCl
缓冲液中,缓冲液可以采用比较高的离子强度。样品缓冲液中应含有1mmol/L所需的二价金属离子氯化物,且不能含有游离糖,以防止它们与糖蛋白竞争结合
凝集素。样 品 缓 冲 液 中 应 加 适 量 抑 菌 剂,如005%叠 氮 化 钠,以 防 止 微 生 物
污染。在制备膜结合糖蛋白样品时,除了上面对水溶性糖蛋白的要求外,还必须设
法将糖蛋白从细胞或膜上溶解下来,这时就需要在样品缓冲液中加入一些试剂,以破坏膜上蛋白质脂、蛋白质蛋白质间的相互作用。这些试剂包括:强变性剂
(如尿素、盐酸胍)、破膜试剂 (如硫氰酸盐、碘化物)、有机溶剂 (如乙醇、异
丙醇)和洗涤剂。前三组试剂会导致蛋白质变性,因此多使用洗涤剂,但是对不
同的膜结合糖蛋白,对其有效的洗涤剂也不同,且洗涤剂有可能影响糖蛋白与凝
集素的结合效 率,因 此 洗 涤 剂 的 浓 度 最 好 低 于1%。一 般 来 说,非 离 子 洗 涤 剂
TritonX100,NonidetP40的效果比较好。用洗涤剂破坏细胞后,溶酶体蛋白酶会释放出来,因此最好加入蛋白酶抑制
剂,常用的有二硫苏糖醇DTT (抑制巯基蛋白酶)、EDTA或EGTA (抑制金属
蛋白酶)、PMSF (抑制丝氨酸蛋白酶)、抑胃酶肽 (抑制酸性蛋白酶)等。(3)加样及吸附 样品按上述方式制备好后,离心除去沉淀就可直接上样,
若要更好地将目的糖蛋白吸附在凝集素柱上,可降低上样时的流速。然后用平衡
缓冲液洗2~5个床体积,以除去非结合蛋白质或同液洗脱蛋白质。(4)目的糖蛋白的洗脱 在洗脱结合的目的糖蛋白时,通常在平衡缓冲液中
加入对凝集素 有 亲 和 性 的 糖 或 其 类 似 物 (表55),以 将 目 的 糖 蛋 白 置 换 下 来,洗脱糖类的浓度一般为0~05mol/L。
由于疏水相互作用在糖蛋白与凝集素柱的结合中起重要作用,为了有效地洗
脱,需要时可在加了特异性糖的缓冲液中再加入50%1,2乙二醇,以减少疏水
作用。由于硼酸根离子能与一些多糖形成复合物,因此也可以尝试采用硼酸缓冲
液洗脱糖蛋白。有时改变pH至酸性 (但不低于30)或碱性 (但不高于100),也能有效地将糖蛋白从固定化凝集素上洗脱下来,但必须保证目的糖蛋白的稳定
性。将结合了糖蛋白的亲和柱于洗脱缓冲液中放置过夜,延长其解离时间,也可
以提高产量。总之,对不同的凝集素和目的糖蛋白,应采用不同的洗脱缓冲液,甚至将几
种洗脱液组合起来,进行分步洗脱或梯度洗脱。

137
(5)固定化凝集素的再生和储存 色谱后,先用含1mol/LNaCl的平衡 缓
冲液洗色谱柱,再用含抑菌剂 [如005% (05g/L)叠氮化钠]的平衡缓冲液
再生亲和柱,然后就可将固定化凝集素在4℃储存于此缓冲液中。
532 免疫亲和色谱
免疫亲和色谱是利用抗原和抗体之间的高特异性亲和力进行色谱分离的,它
能区分非常相近的抗原,并且只需一步就能从大量的复杂蛋白质溶液中分离出极
少量的目的抗原,因此是目前最具选择性和最有效的纯化方法之一。在进行免疫
纯化时,必须使用单克隆抗体或多克隆抗体,最好是单抗,由于要先制备纯的针
对性的单克隆抗体,因此免疫亲和色谱是最昂贵的亲和方法之一。不过随着单克
隆抗体技术的发展,以及大规模生产方法的应用,能有效降低其成本。一般亲和色谱很少用作蛋白质纯化的前期步骤,主要原因可能在于防止蛋白
质粗溶液中的有效成分被蛋白酶水解以及被杂质污染,特别是对于以蛋白质作为
配体的亲和色谱。但是免疫亲和色谱有时可用于一系列纯化步骤的前期阶段,因
为抗体对蛋白酶具有特别的抗性,而且只能选择性地被几种酶水解。当然免疫亲
和色谱只能用于纯化具有合适的单克隆抗体的蛋白质。在进行免疫亲和色谱时,首先要针对目的抗原,制备出纯的单克隆抗体,然
后将此单抗固定到合适的载体上,得到免疫亲和吸附剂,再将其装柱,接着于合
适的吸附、洗脱条件下,将含目的蛋白的粗溶液上样、吸附、解吸,就可以得到
纯的目的蛋白。如果有了单克隆抗体,免疫亲和色谱就变得非常简单,并能得到很好的纯化
结果。由于现在有许多预活化载体供应,因此可以不必为了活化载体而接触高毒
性的化学试剂。这种预活化载体通常可以简单地与抗体混合、偶联,然后洗去并
封阻。在寻找合适的洗脱剂时应注意抗原和抗体的稳定性。幸运的是,大多数抗
体都能承受极端pH和离子强度,而且抗原能自动识别洗脱液成分的变化。
5321 抗体的选择和纯化
成功的免疫亲和色谱依赖于特定性质的单克隆抗体的选择,而且单抗对抗原
要具有一定强度的亲和力,以便于有效吸附和洗脱。生产单克隆抗体时可以用相
对不纯的抗原免疫获得,这时经常会产生很多抗体,因此必须区分出合适性质的
抗体,并将其纯化以用于制备免疫亲和试剂。纯化的抗体不仅可用于亲和色谱,还可用于许多其他医药及生化分析等领域。
生产免疫纯化试剂主要分为四步:(1)杂交瘤的选择 在选择抗体时第一个重要的步骤 就 是 分 辨 出 对 抗 原 有
一定亲和性和特异性的抗体。应选择高亲和性的抗体,以便于从复杂的 混 合 液
中有效地回收稀释的抗原。但是,抗体的亲和性又不可太高,以防止吸 附 的 抗

138
原难以解吸,最好使其结合 常 数 达107mol/L左 右。一 般 培 养50~200种 杂 交
瘤细胞,但 是 将 所 有 细 胞 系 都 生 长 出 足 够 的 量 并 纯 化 各 抗 体 是 不 现 实 的。因此,必须采用在杂交瘤成长 阶 段 就 能 应 用 的 扫 描 分 析 方 法,最 好 用 在96孔
微滴定板中生长的细胞系上清。以固定在微滴定板上的抗原来滴定抗 体,从 而
计算出抗体 的 相 对 滴 定 值。此 步 骤 的 目 的 是 分 辨 出10~15种 抗 体 以 用 于 下
一步。(2)杂交瘤的培养 可以用腹水、血清或细胞培养液来生产选择出的10~
15种抗体,每种抗体需提取10~100mg。在这一步中可去除那些生长速度低或
抗体产量低或不稳定的细胞系,只保留其中的5~8种细胞系就可以了。接着将
各细胞系中的单克隆抗体用多种技术进行纯化。(3)单克隆抗体的纯化 抗体的纯化方法可以采用传统的生化分离方法,如
硫酸铵沉淀、离子交换色谱等,也可以采用前面介绍的蛋白质A或蛋白质G亲
和色谱法。在纯化过程中可获得有关抗体产量、纯度及其等电点、溶解性、稳定
性的信息。那些难以纯化或不稳定的单克隆抗体在这一步中可以去除,筛选出其
中4~6种优良的细胞系。(4)免疫纯化试剂的制备 更进一步选择是基于抗体偶联于合适载体上之后
的性质。这一步主要考虑的是固相化抗体的产量,回收抗原的洗脱条件,抗原的
吸附容量,抗原的产量,抗原的纯度,免疫纯化试剂的重复使用性能,IgG的渗
漏等。最终选择出一种最好单抗。
5322 抗体的偶联
选择好合适的单克隆抗体并纯化好之后,就可以将它偶联到合适的载体上。抗体偶联时最普遍使用的活化载体是CNBr活化的琼脂糖和Affigel10、Affigel15。在这里仅介绍将抗体偶联于CNBr活化的琼脂糖上的方法,而配体与 Affigel的偶联方法同凝集素亲和色谱法。
CNBr活化的琼脂糖适合于大多数的研究目的,但有时这种材料可能不太有
用,这时可选择其他合适的替代物。CNBr活化的琼脂糖有三个方面不足:作为
一种相对的 软 胶,它 无 法 承 受 高 流 速 或 是 用 于 大 色 谱 柱;其 排 阻 极 限 为20×106,因此高分子量抗原无法进入胶内;配体与载体间的共价键相对不稳定,固
定化配体可能渗漏于抗原内。(1)抗体的制备 在4℃用新鲜的偶联缓冲液 (含05mol/LNaCl的pH=
83、01mol/L硼酸钠 缓 冲 液)透 析 抗 体。所 需 的 抗 体 量 大 约 为:对 单 抗1~10mg/ml胶,对多抗10~20mg/ml胶。由于空间位阻效应,抗体量过多会导致
效率降低。一般将抗体浓度调整为5~10mg/ml。(2)抗体 的 偶 联 用 溴 化 氰 法 将 抗 体 偶 联 到Sepharose4B上 分 为 两 步:

139
Sepharose4B的活化和抗 体 的 偶 联。由 于 溴 化 氰 剧 毒,目 前 多 购 买 预 活 化 好 的
Sepharose4B粉末,临用前将其溶胀平衡于偶联缓冲液中,然后再与抗体进行偶
联,偶联成功后即可封阻载体上未偶联的活化基团。活化的Sepharose4B粉 末
一旦溶胀,整个偶联、封阻过程中都不可使凝胶干燥。
① 预活化Sepharose4B的平衡 称取所需量的CNBr活化Sepharose4B粉
末 (03g粉末约等于1ml溶胀胶),注意勿使其吸潮而破坏CNBr活化位点,然
后将其缓慢倒入10倍体积4℃的1mmol/LHCl溶液中,轻微搅拌、分散凝胶颗
粒。接着在室温下溶胀凝胶约15min,再用4℃的1mmol/LHCl溶液流洗凝胶,最后用10倍凝胶体积的4℃偶联缓冲液洗凝胶。
② 抗体的偶联 4℃下将5体积抗体溶液加到1体积平衡好 的 活 化 琼 脂 糖
中,低速搅拌偶联16~20h。
③ 测试 取1ml反应上清液,测定OD280值,若其值接近0则 表 示 偶 联 成
功;然后用新鲜的偶联缓冲液洗胶;(3)封阻未反应的活化基团 载体上剩余的CNBr活化基团可用过量伯胺化
合物封阻,如乙醇胺、甘氨酸、1氨基丙二醇等。最常用的封阻试剂是乙醇胺。
4℃下加入pH=80、1mol/L乙醇胺或1氨基丙二醇封闭载体上未偶联的活化
基团,然 后 依 次 用 高 和 低 pH 的 缓 冲 液 (含05mol/L NaCl的 pH=40、
01mol/L醋酸钠缓冲 液;含05mol/LNaCl的pH=83、01mol/L碳 酸 钠 缓
冲液)交替清洗凝胶三次,再用平衡缓冲液和洗脱缓冲液交替清洗,最后将其浸
泡在含01% (1g/L)叠氮钠的平衡缓冲液中,4℃保存。
5323 免疫亲和色谱的色谱过程
免疫亲和色谱同样可以采用分批式或连续柱式。免疫亲和色谱分为正向色谱
和负向色谱。正向亲和色谱是从粗溶液中纯化抗原,负向色谱则是从部分纯化的
溶液中除去特殊的抗原污染物。在进行纯化时需要重点考虑如何将抗原有效吸附
到免疫亲和柱上,以及如何有效解吸吸附的抗原,所有操作条件都应保证免疫亲
和吸附剂和抗原的稳定性。这里仅介绍正向免疫纯化,负向纯化与其基本相似,但不需过分在意吸附抗原的稳定性。
(1)平 衡 平 衡 缓 冲 液 通 常 为 中 性、低 离 子 强 度 缓 冲 液,如pH=72、
40mmol/L磷酸钠缓冲液或磷酸盐缓冲液。在临用前应将免疫亲和吸附剂放置至
室温,并用新鲜的无叠氮化钠的平衡缓冲液置换,然后将其装柱,用10倍以上
床体积的平衡缓冲液平衡。(2)上样 上样前粗抗原应溶于室温下的平衡缓冲液中,并通过离心、过滤
等方法使其澄清,以防止免疫亲和柱被其堵塞。上样时的流速应尽量低,不可超
过载体凝胶的推荐流速,否则可能抗原来不及吸附就被洗出;必要时样品可循环

140
上样多次,以使亲和柱内的所有可结合位点都被抗原饱和。理论上讲,每个固定
化的单克隆抗体能结合两个抗原分子,但实际上抗体共价固定到载体上后,一般
只有10%左右抗原结合位点还可以被结合。(3)洗去 杂 蛋 白 用 约10倍 凝 胶 体 积 的 平 衡 缓 冲 液 流 洗 色 谱 柱,直 至
280nm的吸收值达到0,这样可洗去非特异性结合的杂质分 子。如 果 抗 原抗 体
复合物的亲和力很强,可以在平衡 缓 冲 液 中 加 入05mol/LNaCl或 KCl,充 分
洗去杂蛋白,这样色谱产物会更纯。(4)目的抗原的洗脱 破坏了抗原抗体复合物之间的结合键,就能把抗原
洗脱下来。这些结合键是一些弱相互作用,如离子键、氢键、范德华力等。洗脱
时可以通过改变离子强度、pH、温度、表面张力和介电常数等方法而达到解吸
抗原的目的。为了保留抗原和免疫吸附剂的活力,应先试用温和的洗脱条件,即
先试一下增加离子强度,再试极端pH、螯合剂、蛋白质变性剂和有机溶剂 (见
5213节)。洗脱时一般只要求2倍凝胶体积的洗脱液。如果洗脱条件的变化
过于激烈,为了减少对纯化抗原的损害,可以将洗脱液收集到含中和缓冲液的收
集管中或是立即进行凝胶过滤。(5)亲和柱的再生 为了能重复使用免疫吸附剂,每次用后都应小心再生。
先用2~5倍凝胶体积的洗脱缓冲液,再用10倍以上凝胶体积的平衡缓冲液流洗
免疫亲和柱,最后将其储存于含01%叠氮化钠的平衡缓冲液中。
533 环氧乙烷丙烯酰胺珠亲和色谱
传统的溴化氰活化琼脂糖常用于亲和色谱,它们对蛋白质具有较好的吸附容
量,能结合小分子配体,流动性好,且载体相对惰性。但最近其他载体在蛋白质
纯化方面显示出新的优势。这里介绍载体EupergitC的应用,这种载体 除 了 偶
联方法简单外,还具有很好的化学和物理稳定性,因此应用范围广泛。EupergitC是一种含环氧乙烷的聚丙烯酰胺载体,可从RohmPharmaGmbh购得。
5331 EupergitC的化学性质
(1)化学结构 EupergitC是 由 甲 基 丙 烯 酰 胺、N,N′亚 甲 基双 甲 基 丙 烯
酰胺和烯丙基缩水甘油醚等聚合而成的 (图59),它先聚合成微珠,再组装成
较大的珠状结构。这种载体为电 中 性,且 相 对 亲 水,因 此 具 有 既 非 离 子 交 换 色
谱,又非疏 水 色 谱 用 载 体 的 性 质。它 的 一 些 疏 水 性 质 可 能 来 源 于 其 分 子 上 的
甲基。(2)物理性 质 EupergitC的 物 理 稳 定 性 很 好,不 易 变 形,当 水 化 时,在
pH=2~12,离子强度001~10范围内,它不会显著地膨胀、收缩。它可以抵
抗3×107Pa的压力,因此装柱后不会发生填充柱的压缩情况,而且即使在低压
下也具有良好的流速。EupergitC能耐受搅拌,但是磁力搅拌或贴壁剪切搅拌会

141
破坏珠。有四种EupergitC供应 (表56),它们在排阻极限、表面积、结合容量方
面有差别。最小的珠可装入 HPLC柱,由于这种珠不是多孔性物质,因此比其
他形式的EupergitC表面积低。未衍生的珠在-20℃稳定,干珠 的 吸 水 量 大 约
是25ml/gEupergitC。
CH3C
CH
2
C
O
N
H
CH 2N
H
C
O
C CH 3
CH
2
CH CH 2O CH 2C
O
CH 2
CH
2
CCH 3
CH
2
C
O
NH 2
图59 EupergitC的化学结构
表56 EupergitC的性质
EupergitC颗粒大小
/μm
排阻极限
/Da
表面积
/(m2/g干重)结合效率
/(mg白蛋白/g干重)
EupergitC250L 200~250 1000000 15EupergitC 140~180 200000 180 48EupergitC30N 30~50 200000 40 52EupergitC1Z 06~14 6 5
(3)配体结合性质 EupergitC上缩水甘油部分的环氧乙烷基团是配体固定
化的位点,其含量约为1mmol/g干珠,它们可与一些不同的基团发生相互作用
(见图54)。偶联 反 应 的 最 佳 条 件 取 决 于 功 能 基 团 的 性 质。如 色 氨 酸 残 基 在 低
pH时结合程度最大,而伯胺则在高pH时更易结合。由于环氧乙烷基团能与许多生物缓冲系统发生反应,因此偶联缓冲液的选择
非常重要,切记未偶联的载体不能暴露于高反应性的 铵 盐、Tris或GOODs缓
冲液中,偶联后进行亲和色谱纯化操作时,缓冲液的选择就不必这么严格了。
5332 EupergitC的配体偶联过程
(1)环氧乙烷聚丙烯酰胺珠的小批量配体偶联方法 在对EupergitC载体
进行配体偶联之前,应先进行小型试验以获得最佳结合条件。方法如下所示:
① 将配体以尽可 能 高 的 浓 度 溶 解 在 适 当 的 缓 冲 液 中,最 好 是 无 机 缓 冲 液,如磷酸缓冲 液,应 根 据 配 体 和 环 氧 乙 烷 基 团 的 稳 定 性 慎 重 考 虑 缓 冲 液 的pH,

142
pH>10时,环氧乙烷非常易转变成二醇衍生物。
② 在15ml微离心管内将10ml配体溶液加到100mg干EupergitC珠中,并称重。
③ 在室温或配体稳定的温度下以翻转或旋转方式混合珠悬浮液16~48h。
④ 在水浴过程的不同时间内,以10000g离心珠悬浮液10s,使珠沉降,取
少量上清液测定残留配体量。自由的和结合的配体量很大程度上决定于要偶联的配体的量。用这种方法应
该能区分出与载体共价偶联的配体和非共价偶联的配体,因为非共价偶联的配体
与载体能快速结合。进行小型试验时,加到EupergitC珠里的配体溶液 体 积 可
以很大,珠分散液可以离心,并可用上清液测定非共价结合的配体量。简单地将
此方法扩大就能进行大批量偶联。(2)环氧乙烷聚丙烯酰胺珠的制备型配体偶联方法 此方法可用于更大批量
的配体偶联过程。与前述方法相比,加到干EupergitC珠里的配体溶液 的 体 积
大幅度减少,这样就增加了配体的局部浓度,并会使偶联更加有效。其偶联过程
如下:
① 配体溶液的配制 将配体溶解在合适的缓冲液中,对于大多数蛋白质或
小分子量配体而言,pH=75、01mol/L磷酸缓冲液都是合适的。
② 活化载体与配体溶液的混合 于每克干环氧乙烷丙烯酰胺珠内加入4ml配体溶液,珠将占满所有缓冲液空间,从而可提高偶联效率。
③ 偶联 将潮湿珠于室温下放置16~48h,如果配体不稳定则于4℃放置。
④ 洗涤 将珠 分 散 于 合 适 的 缓 冲 液 中,如pH=75、01mol/L磷 酸 缓 冲
液,然后用至少10倍体积的相同缓冲液抽滤清洗偶联好的珠。
⑤ 封阻珠上未反应的环氧乙烷基团 对于每克珠,加入4mlpH=78、5%(体积分数)巯基乙醇或1mol/L甘氨酸或1mol/L乙醇胺溶液,室温下反应16h。
⑥ 清洗 在布氏漏斗上先用重蒸水,再用10倍体积色谱缓冲液彻底清洗衍
生的封阻珠。
⑦ 储存 在4℃储 存 衍 生 珠,需 要 的 话 加 入 合 适 的 抑 菌 剂,如1滴 甲 苯、
002% (02g/L)叠氮化钠或01% (体积分数)甲醛。目前环氧乙烷聚丙烯酰胺珠的应用还相对较少,需摸索新的方法才能使这种
偶联方式得到广泛应用。环氧乙烷聚丙烯酰胺珠的机械和物理稳定性是它们作为
合适载体的优势。其 偶 联 化 学 温 和、安 全,且 偶 联 后 的 材 料 容 易 用 于FPLC和
HPLC,这预示着它们具有作为高分辨率蛋白质纯化方法的潜力。
534 金属螯合亲和色谱
金属螯合亲和色谱又称固定化金属离子亲和色谱,金属离子相互作用色谱以
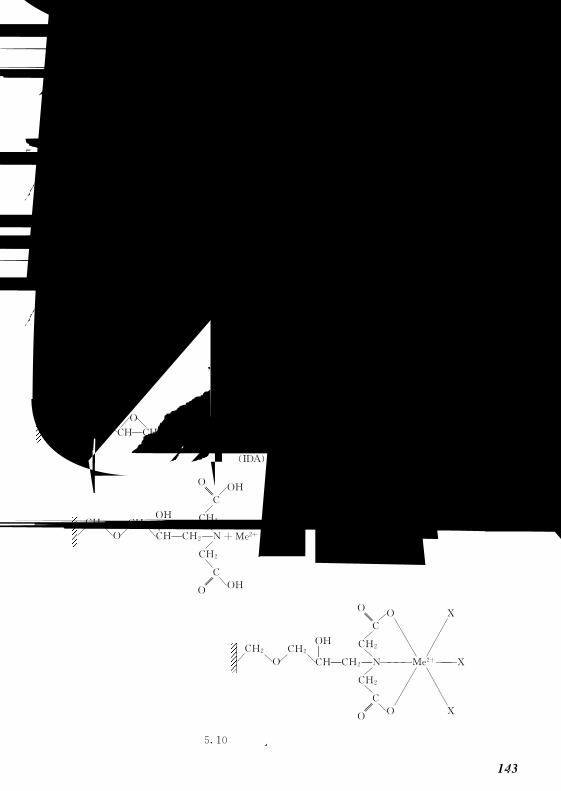
143
及配体交换色谱。这种方法介于高特异性的生物亲和分离方法和低特异性的吸附
方法 (如离子交换色谱)之间。这种色谱对肽或蛋白质表面的特殊基团如组氨酸
残基有特异性。它是以普通凝胶作为载体,连接上合适的螯合配体和金属离子,从而制成亲和吸附剂,固定相中金属离子的数目没有限制,但必须有足够的暴露
出来,以便于与蛋白质发生相互作用。
5341 金属螯合亲和色谱的色谱材料
金属螯合亲和 色 谱 是 基 于 蛋 白 质 的 组 氨 酸 咪 唑 基 和 半 胱 氨 酸 巯 基 能 与 铜、锌、镍离子间形成稳定的螯合物而进行分离的。载体、螯合配体和金属离子的选
择并不 复 杂。常 用 的 螯 合 配 体 有 亚 氨 基 二 醋 酸 (IDA)、三 (羧 甲 基)乙 二 胺
(TED)等。如果刚开始没有某一特定蛋白质的信息,可先选亚氨基二醋酸与较
强的亲和转化离子Cu2+进行固定化。如果其相互作用太强,可换用别的金属离
子,如镍离子或锌离子;或选用别的对蛋白质低亲和性的螯合配体。图510给
出了以环氧乙烷活化的琼脂糖作为载体,以亚氨基二醋酸作为配体,以铜离子或
锌离子作为螯合离子的金属螯合亲和吸附剂的作用示意图。此亲和色谱材料已有
商品出售,其合成过程可查阅有关文献。对于金属螯合亲和色谱,通过改变pH能够有效地吸附和解吸蛋白质,当然也可以用别的可与金属发生竞争性螯合作用
的物质进行洗脱。
CH2OCH2
CHOCH 2
环氧乙烷活化的琼脂糖
+NHCH2COOH
CH2COOH
亚氨基二醋酸 (IDA
→
)
CH2OCH2
CHOH
CH 2NCH2COOH
CH2COOH
CH2
OCH2
CHOH
CH 2N
CH2
CO OH
CH2
C
O OH
+Me2++
金属离子
X蛋白质X
X
→
蛋白质
CH2OCH2
CHOH
CH 2N
CH2
CO O X
CH2
C
O O X
Me2+X
图510 金属螯合亲和吸附剂

144
5342 金属螯合亲和色谱的色谱过程
(1)金属离子于亲和吸附剂上的装载 将亚氨基二醋酸配体偶联到环氧乙烷
活化的琼脂糖载体上得到的色谱材料可以直接购买到。将此IDA凝胶分散后倒
入布氏漏斗,用10床体积蒸馏水清洗以除去储存液中的乙醇。然后加入2~3倍
床体积的50mmol/L金属离子溶液进行混合,再用3倍体积适当缓冲液洗去过量
金属离子。对于铜离子,可以用含05mol/LNaCl的pH=38、01mol/L醋酸
钠缓冲液。接着用5倍体积的含05mol/LNaCl的pH=70、20mmol/L磷酸缓
冲液平衡凝胶,就得到了金属螯合色谱材料。(2)平衡 将以上金属螯合凝胶脱气后装柱,以30cm/h的速度平衡。(3)加样 将样品溶于上述pH=70的平衡缓冲液中,经离心或过滤澄清
后即可上样。然后用平衡缓冲液洗去杂蛋白,直至OD280回到基线。(4)洗脱 结合蛋白质的洗脱有多种方法,可采用连续缓冲系统或不连续缓
冲系统降低pH进行pH梯度洗脱,或用咪唑进行亲和梯度洗脱。进行pH梯 度 洗 脱 时,可 先 用5倍 床 体 积 含05mol/LNaCl的pH=58、
01mol/L醋酸钠缓冲液洗 脱;再 用10~20倍 床 体 积pH=58~38的 缓 冲 液
(含05mol/LNaCl的01mol/L醋 酸 钠 缓 冲 液)进 行 线 性 梯 度 洗 脱;最 后 用
pH=38的缓冲液洗至pH 稳 定,并 使 所 有 蛋 白 质 都 洗 脱 下 来。或 是 直 接 用 约
15倍床体积pH=70~40的缓冲液 (含05mol/LNaCl的20~50mmol/L磷
酸钠缓冲液)进行线性梯度洗脱。(5)柱的再生 先用含50mmol/LEDTA的平衡 缓 冲 液 洗5个 床 体 积,再
用10倍床体积蒸馏水清洗,此时柱子就可以重新用金属离子装载了。如果采用亲和梯度洗脱,则在制备好金属螯合色谱材料后,先用5倍床体积
含20mmol/L咪唑和05mol/LNaCl的pH=70、20mmol/L磷 酸 钠 缓 冲 液 平
衡,再用5~10倍床体积含2mmol/L咪唑的以上缓冲液平衡IDA凝胶柱。上样
后,先用含2mmol/L咪唑的pH=70缓冲液洗去杂蛋白,再转变成含20mmol/
L咪唑的pH=70缓冲液进行线性梯度洗脱,将目的蛋白解吸下来。
535 疏水作用色谱
疏水作用色谱是以亲和吸附剂疏水基团与蛋白质疏水区域间的相互作用为基
础的,相对而言其亲和力不太高。疏水作用色谱中亲和吸附剂上的疏水基团主要是苯基或辛基。碳氢链越长则
疏水性越强,只能用于纯化疏水性很强的 少 量 蛋 白 质,洗 脱 时 需 采 取 极 端 的 方
式,这可能使蛋白质变性。苯基的疏水性相对较弱。由于高盐浓度会加强疏水作
用,因此常采用高盐上样、低盐浓度洗脱。其他改变疏水作用强度的方法也可以
用于吸附和洗脱目的蛋白,如:改变pH使其远离或接近目的蛋白的等电点,改

145
变温度,或是采用特异性洗脱方 法。这 里 不 作 具 体 介 绍 了,需 要 时 可 参 考 有 关
文献。
54 应用
541 抗体和抗原的纯化
抗原和抗体之间具有高度特异性和亲和性,非常适合于采用亲和色谱法进行
分离纯化。如可以采用前面所介绍的免疫亲和色谱法,将对应的纯抗体或抗原偶
联到适当的载体上制成亲和吸附剂,通过其免疫亲和作用进行分离纯化,这种方
法一步就能达到很高的纯化效果,并获得很纯的样品。抗体的纯化也可以用蛋白
质A或蛋白质G亲和色谱柱,但是这种方法是针对IgG的Fc区,因此特异性
不高。
542 酶的纯化
酶纯化时,可以将其底物、辅助因子或抑制剂偶联到载体上,通过酶与其底
物或辅助因子或抑制剂的特异性相互作用进行分离纯化,这方面的例子很多,如
尿激酶的纯化、乳酸脱氢酶的纯化等。如果目的酶是糖蛋白,也可以采用凝集素
亲和色谱法;如果酶蛋白上含有特定的辅助因子,如金属离子、NAD+等,也可
以采用金属螯合亲和色谱、固定化染料亲和色谱等方法。总之,应根据待纯化酶
蛋白的性质选择合适的配体和亲和色谱类型。
543 糖蛋白的纯化
糖蛋白的纯化可采用凝集素亲和色谱法,方法见531节,但凝集素色谱的
特异性不高。或者以苯基硼酸盐作为配体,通过苯基硼酸盐与顺二醇之间共价复
合物的形成来分离纯化某些糖蛋白。如果要提高分离的专一性,也可以将某一特
定糖蛋白的特异性受体制备成亲和吸附剂,通过它们之间的专一性相互作用进行
分离纯化。
544 脂蛋白的纯化
脂蛋白的纯化可针对其脂基团的疏水性能采用疏水色谱进行分离,或是制备
特异性更高亲和吸附剂进行分离纯化。另外,某些特定化合物的结合蛋白或受体蛋白等的纯化也可以采用亲和色谱
法。将这些化合物偶联到合适的活化载体上制备成亲和吸附剂,通过它们与结合
蛋白或受体蛋白之间的亲和力进行分离纯化。

146
6 共 价 色 谱
用来分离蛋白质和其他生物大分子的色谱技术,如离子交换色谱和疏水相互
作用色谱都是基于待分离的分子和吸附剂之间的非共价相互作用而实现的,而共
价色谱则是应用固定相与溶质之间共价键的形成或断裂来实现分离。在共价色谱中,含有Y基团的蛋白质与含有活性基团X的固定相反应,洗
去不结合的蛋白 质,再 利 用 低 分 子 量 的 化 合 物 RY,将 结 合 的 蛋 白 质 释 放 出 来
(图61)。
图61 共价色谱示意
通常用于生物活性蛋白的色谱技术,要求在温和的条件下,既能形成稳定的
键,又能在释放固定蛋白时不破坏其结构。当蛋白质中的氨基和羧基被用于与不
溶性基质固定时,能发生化学反应的潜在位点主要 是 具 有 各 种 功 能 的 氨 基 酸 侧
链。在此条件下,能有效地形成稳定的共价键,并且在温和的条件被分离的功能
团只有巯基基团。现今的共价色谱通常是利用巯基的二硫相互作用来分离含巯基蛋白质,这一
方法是在1973年由Bocklehurst等提出的。只有极少数的共价色谱是基于非巯基
化合物的共价化学反应而进行分离的,如Patchornik等设计 开 发 的 使 蛋 氨 酰 和
色氨酰侧链与活化聚丙烯酰胺基质可逆共价相连,用来分离含有蛋氨酸或色氨酸
的多肽。共价结合具有特异性、牢固并且在适当的条件下可获得较高收率和纯度等特
点,在一些领域,特别是在蛋白质序列分析过程和结构研究中有特殊作用。主要
应用在分离半胱氨酸、含半胱氨酸的肽段 (蛋白质 测 序 中 蛋 白 酶 的 降 解 产 物),含巯基的蛋白分离或进一步分离该蛋白质中巯基附近的肽段等方面。
蛋白质中常常含有巯基基团,并且往往涉及酶、激素和受体等的功能。蛋白
质中巯基反应性的可变,以及用化学法引入巯基等状况使共价色谱可成为一种广
泛应用的技术。

147
本章是针对固定相带有活性二硫化物或二硫氧化物基团、能 与 巯 基 反 应 的
共价色谱,简要描述巯基及其衍生物发生的主要反应类型,详细讨论了 共 价 色
谱中所应用的巯基与活性二硫基团、二硫氧化物之间的反应以及蛋白质 的 可 逆
固定,并介绍了有关共价色 谱 中 凝 胶 衍 生 物 的 性 质、共 价 色 谱 的 原 理 和 应 用
实例。
61 巯基的化学性质
611 离子化、氧化、金属联结、烷基化
6111 离子化
巯基能被解离成质子和巯基阴离子,由于巯基阴离子在中性到弱碱性的条件
下,具有高亲核性,因而巯基基团可参与许多反应,是蛋白质中最活跃的基团。
R— →SH R—S-+H+ (61)式中 R—SH———硫醇;
—SH———巯基。不同巯基化合物的解离常数 (pKa)在8~105的范围内,表61中列出了
一些代表性巯基化合物的pKa 值。
表61 低分子量巯基化合物的pKa 值
化 合 物 pKa 化 合 物 pKa
巯基N,N′二乙胺 78巯基乙胺 83谷胱甘肽 92
巯基乙醇 95巯基丙酸 103
6112 氧化
巯基很容易氧化成相对不活跃的二硫化物,氧化形成的二硫键对于蛋白质三
级结构的稳定性非常重要。氧化反应的速率随着巯基离子浓度以及pH的增大而
增大,在低pH (pH<5)、无强氧化剂的条件下,巯基基团是稳定的。
2R— →SH R—S—S—R(二硫化物)+2H++2e- (62)
巯基的氧化反应有两条途径,沿哪条 路 径 发 生 反 应,取 决 于 所 使 用 的 氧 化
剂。两条氧化途径的终产物是相同的。(1)单氧化途 径 经 过 两 步 脱 电 子 反 应,将 巯 基 转 化 为 次 磺 酸 (R—S—
OH)、亚磺酸 (R—SO2H)和磺酸 (R—SO3H)。(2)双氧化途径 将巯基转化为二硫化物 (R—S—S—R),再进一步氧化形
成二硫氧化物———巯基亚磺酸 (R—SO—S—R)和巯基磺酸 (R—SO2—S—R),最终分别形成亚磺酸和磺酸。

148
6113 金属联结
巯基极易与重金属离子形成配位体,例如形成稳定的锌、铜、汞复合物。最
初的共价色谱就是采用凝胶中的有机汞制剂来分离含有巯基的蛋白质。
6114 烷基化
巯基基团烷基化后形成稳定的硫醚,可永久阻断巯基基团。
I—CH2—COO-+R— →SH R—S—CH2—COO-+HI (63)
最常用的烷基化试剂是碘乙酸或碘乙酰胺,和 其 他 的 巯 基 亲 核 置 换 反 应 一
样,该反应的反应速率随着pH的增大而加快,形成羧甲基衍生物。
612 巯基二硫化物互换反应
应用于共价色谱中的巯基二硫化物互换反应是一种特殊形式的烷基化 (S烷基化)反应,这一反应为两步亲核置换 反 应,形 成 的 中 间 产 物 为 混 合 型 二 硫
化物。
R1—S—S—R1+R— →SH R1—SH+R1—S—S—R (64a)
R1—S—S—R+R— →SH R1—SH+R—S—S—R (64b)
两步反应的总反应式为:
R1—S—S—R1+2R— →SH 2R1—SH+R—S—S—R (64)
在生理状态下 (pH=7,25℃),脂肪族巯基化合物与二硫化物反应的速率
常数为600mol/(L·min)。通过这一互换反 应,利 用 巯 基 化 合 物 还 原 蛋 白 质 分
子内或分子间的二硫键。最常用的巯基化合物是β巯基乙醇 (pKa 为95),注
意必须过量使用。这是因为此互换反应极易可逆发生,且正逆反应的平衡常数接
近一致,因此必须用过量的巯基来还原二硫化物,反之用过量的二硫化物才能氧
化巯基。
R—S—S—R+2HO—CH2CH2—SH(过量 →) 2R—SH+HO(CH2)2—S—S—(CH2)2OH
(65)如果反 应 中 使 用 巯 基 二 元 醇 [二 硫 赤 藓 糖 醇 (DTE)或 二 硫 苏 糖 醇
(DTT)],则无需过量使用。如图62,由于此时形成的混合二硫化物和脂肪族
二硫化物都不稳定,会发生内部巯基二硫化物互换,而形成稳定的六元环内二
硫化物,促使正反应的完全进行,因此可用等物质的量的DTE或DTT还原脂
肪族二硫化物。巯基二硫化物互换反应在许多生化过程,如蛋白质的生物合成、某些蛋白
质的聚集聚合作用、部分胞内酶的活性调节、效应物受体相互作用、跨膜输送

149
等过程中起非常重要的作用。
RSSR+CHOH
H2C
SH
CHOH
CH2
→
HS
RSH+CHOH
H2C
S S R
CHOH
CH2HS
(DTT)
CHOH
H2C
S S R
CHOH
CH2
→
HS
RSH+CHOH
H2C
S S
CHOH
CH2
图62 DTT还原脂肪族二硫化物
613 与活性二硫化物的反应
在巯基二硫化物互换反应中,脂肪族巯基化合物和活性二硫化物间的反应
尤其受到关注。所谓活性二硫化物是指这类二硫化物对应的巯基形式,通过共振
或硫醇硫酮互变异构而稳定,亲核性降低,因而反应活性降低。活性二硫化物分为同质 (图63)和混合型,分子两半部分完全一致的为同
质活性二硫化物,而在 混 合 型 中 二 硫 化 物 的 一 半 (R—S—S—)由 脂 肪 族 巯 基
(RSH,R表示脂肪族基团)衍生出来的
師師師師
師師
。
N
師師師師
師師S S N
師師師師
S N
H
(Ⅰa) (Ⅰb)
-師師師師
師師OOC N
師師師師
師師S S N
COO -師師師師
S COO -
N
H
(Ⅱa) (Ⅱb
師師
師師
帩帩
)
O2 N
-OOC
師師
師師
帩帩S S
COO-
NO 2 師師
師師
帩帩 HS
COO-
NO 2
(Ⅲa) (Ⅲb)
图63 同质活性二硫化物及其相应的还原态
Ⅰa—2,2′二硫双吡啶;Ⅰb—硫代吡啶酮;Ⅱa—5羧基2吡啶二硫;Ⅱb—5羧基2硫代吡啶酮;
Ⅲa—5,5′二硫二 (2硝基苯甲酸酯);Ⅲb—2硝基5巯基苯甲酸酯
脂肪族巯基化合物与同质活性二硫化物2,2′二 硫 双 吡 啶 (2PDS)的 反 应
见图64,反应通过硫醇硫酮互变而促使反应完全,1分子脂肪族巯基可被定量

150
转变为1分子混合活性二硫化物,伴随形成等量的硫酮。
師師師師
師師RSH+ N
師師師師
師師S S
→師師師師
師師NRSS
N
師師師師
師師+ HS
師師師師
師師
N
HS
→師師師師
N
S N
H
硫醇式 硫酮式
图64 巯基化合物与同质活性二硫化物的反应
在酸性条件下,脂肪族巯基化合物与混合活性二硫化物 (2吡啶二硫化物)反应 (图65),由于脂肪族一侧的硫原子亲电子性强,和巯基硫原子形成脂肪
族二硫化物,而2硫代吡啶结构脱开形成硫酮。巯基化合 物 与 活 性 二 硫 化 物 的
反应中释放出的硫酮很容易用分光光度计进行定量分析。
RSH+ R1 師師師師
師師S S
→N
RSSR1師師師師
師師+ HS
師師師師
師師
N
HS
→師師師師
N
S N
H
图65 巯基化合物与混合活性二硫化物的反应
活性二硫化物的特性被大量开发应用,同质活性二硫化物可用于蛋白质中巯
基的滴定,而混合活性二硫化物在共价色谱中有重要应用。
614 巯基与二硫氧化物的反应
在中性或微碱性pH 条 件 下,巯 基 可 特 异 性 地 与 脂 肪 族 二 硫 氧 化 物 定 量 反
应,如巯基亚磺酸 盐 和 巯 基 磺 酸 盐。而 且 如 果 反 应pH 接 近 或 大 于 巯 基 基 团 的
pKa,反应还会加速。巯基磺酸盐与巯基化合物的反应,类似巯基二硫化物互换反应,生成混合
二硫化物和亚磺酸盐 (式66a);巯基亚磺酸盐与2分子巯基化物反应,形成2分子混合二硫化物 (式66b)。
R—SH+R1—SO2—S—R →2 R—S—S—R2+R1—SO2H (66a)
2R—SH+R1—SO—S—R →2 R—S—S—R2+R1—S—S—R+H2O (66b)
这些反应具 有 实 际 应 用,脂 肪 族 巯 基 磺 酸 盐 (如 甲 基 甲 烷 巯 基 磺 酸 盐,
MMTS)广泛用于可逆地封闭巯基依赖型酶中的巯基基团,而芳 香 族 巯 基 磺 酸
酯则用于蛋白质中巯基的滴定。

151
62 含巯基的蛋白质
下面介绍各种不同生物来源蛋白质中巯基的特性以及应用条件下巯基基团活
性的变化。
621 生物体中的氧化还原态
在生物体中,细胞内的氧化还原状态完全不同于胞外基质或溶液,胞内的环
境必须维持在还原态,这是由于细胞内的许多反应都严格依赖于含游离巯基的巯
基化合物,如辅酶A、硫辛酸等。细胞通过含半胱氨酸的三肽———谷胱甘肽 (GSH,图66)来维系胞内的还原
态,GSH能清除自由基、氧化物等与巯基反应的分子。生物体通过GSH合成酶的
合成以及谷胱甘肽还原酶还原谷胱甘肽二硫化物 (GSSG),来维持胞内高浓度的谷
胱甘肽,通常胞内GSH的含量约占所有巯基的90%,其总浓度在毫摩尔范围。
- OOC CH
NH
3+
CH 2 CH 2
C
O
NH CH
CH
2
SH
C
O
NH C
H
H
COO -
图66 谷胱甘肽的结构
622 胞内与胞外的蛋白质
绝大多数胞内蛋白质含有暴露的巯基,极少数含有二硫桥或巯基不暴露。在
许多情况下,巯基参与催化反应。如胞内的组织蛋白酶活性依赖于巯基基团,在
氧化还原酶和转移酶中巯基的烷基化则会导致酶活的完全丧失。巯基基团还是金
属离子的配体,如乙醇脱氢酶中。胞外蛋白质则通常含 二 硫 桥,极 少 数 因 特 殊 目 的 才 需 要 巯 基。胞 外 水 解 酶
(丝氨酸蛋白酶、胰蛋白酶、胰凝乳蛋白酶)、一些血液凝结因子都含有数个二硫
桥,而没有巯基。小分子的胞外蛋白,如神经毒素和蛋白酶抑制剂,尤其富含二
硫桥,这对蛋白结构的稳定性很重要。胞外的植物蛋白酶 (无花果蛋白酶、菠萝
蛋白酶和木瓜蛋白酶)属于例外,它们是巯基依赖型,巯基基团位于这些酶活性
部位口袋中,可避免氧化。
623 蛋白质巯基基团的解蔽
蛋白质中巯基的可反应性取决于环境。许多蛋白质所含的巯基,能和亲水试剂反应,而在疏水环境和变性剂条件下
被掩埋。例如,在天然人血红蛋白中,每分子蛋白中2个巯基有活性而另外4个
无活性,从蛋白的三维结构显示,发现这4个巯基被部分掩埋。

152
在许多情况下,蛋白质的完全展开是允许所有巯基基团具有可反应性的必需
条件。例如,在含有EDTA的8mol/L尿素或6mol/L胍中,血浆铜离子蛋白的
所有巯基具有可反应性,能捕获释放的铜形成配体。
624 蛋白质二硫键的还原
使用变性剂和过量还原剂会使得蛋白质中所有二硫键都被还原成巯基。在一定条件下,对于链间的二硫键,可被选择性地还原一至数个,而不破坏
蛋白质的三级结构。如DTT选择性还原免疫球蛋白链间二硫键。链内二硫桥的还原通常是以拉链状协作进行的,在某些情况下,链内二硫键
的还原不会破坏蛋白质的总构象,甚至不影响蛋白质的活性。
625 巯基的引入
通过巯基化反应将巯基引入蛋白质,从而可以使用共价色谱。最常用的巯基
化反应试剂是N乙酰同型半胱巯基内酯,它能与蛋白质中的氨基反应。有的巯基化反应试剂是用来引入保护性巯基基团 (图67),如用琥珀酰亚
胺吡啶二硫代丙酸 (SPDP)引入的2吡啶二硫化物为混合型活性二硫化物,当
用等物质的量的DTT在温和条件下还原时,可保护蛋白质中的二硫键 不 裂 开,而是被吡啶二硫取代固定在巯基凝胶中。
蛋白质NH2師師師師
師師+ N
S S CH 2 CH 2
C
O
O N
→
O
O(SPDP)
蛋白质 NH C
O
CH 2 CH 2 師師師師
師師S S N
+HO N
O
O
图67 保护性巯基基团的引入
626 巯基基团的衍生化作用
用同质活性二硫化物处理巯基,既可使其烷基化封闭,也可使其活化。在共价色谱的巯 基 基 质 上,用 过 量 的2,2′二 吡 啶 二 硫 化 物 活 化 巯 基 蛋 白,
以免蛋白质中新形成的活性巯基与相邻的巯基形成分子内或分子间的二硫键,而
使蛋白形成二聚体或多聚体。反应可在缓冲液或强变性剂条件下进行,此时所有
的巯基均被活化。
63 共价色谱的凝胶
631 原理
在共价色谱中,利用含巯基的分 子 与 固 定 相 所 含 的 二 硫 基 团 之 间 的 相 互 作

153
用,使其附着在固定相上,从而实现巯基蛋白的分离。
632 凝胶的基质
与其他类型的色谱基质一样,共价色谱要求凝胶基质具有优良的填充性和流
动性、低非特异性吸附等特性。交联葡聚糖、琼脂糖、纤维素、聚丙烯酰胺、多
孔玻璃和各种二氧化硅的衍生物都被用作共价色谱的基质,其中最常用的是颗粒
琼脂糖凝胶,该基质还具有衍生化时不改变其优良性质以及在非极性溶剂中不收
缩的特性。
633 凝胶的固定相
混合活性二硫化物最重要的应用就是作为共价色谱的活性基团,通常多采用
2吡啶二硫取代物的固定相,色谱原理见图68。
師師師師
師師S S N
+P— →SH 師師
師師 S S P + S N
H
固定
S S P +R—SH (过量 →) SH+ P SH
+ R S S R
还原洗脱
图68 活性二硫化物共价色谱原理
P—SH—含巯基蛋白质
近来发展起来的二硫氧化物固定相 (巯基磺酸盐和巯基亚磺酸盐),作为选
择性吸附剂,可用于巯基的可逆固定。由于两个硫原子周围的电子的置换,这些
基团具有高的硫反应性。含巯基的蛋白质分子与二硫氧化物中非氧化的硫原子反
应,形成二硫键从而固定蛋白质分子。如图69所示,与巯基分子反应后,二硫氧化物固定相的离去基团 (亚磺酸
或次磺酸)仍附着在基质上,而活性二硫化物固定相则相反。因而固定在二硫氧
化物固定相上的巯基分子用脂肪族小分子巯基化物还原回收时,更容易被释放。
634 固定相活性基团的比较
与琼脂糖结合的固定相活性基团,具有与巯基反应的很高特异性 (表62)。常用的2吡啶二硫基团 (混合活性二硫化物)、巯基亚磺酸基团 (二硫化物的单
氧化物)和巯基磺酸基团 (二硫化物的二氧化物),对于暴露的巯基基团,它们

154
图69 巯基磺酸共价色谱原理
表62 固定相活性基团的比较
性 质
混合活性二硫化物 二硫氧化物
2吡啶二硫基团巯基亚磺酸基团
(二硫化物的单氧化物)巯基磺酸基团
(二硫化物的二氧化物)
与巯基
的结合
暴露的巯基 反应性均很高
掩埋的巯基 结合所有 根本不结合 有一定程度的结合
亲硫吸附特性 具备 不具备 不具备
固定巯基时反应特性 释放生色离去基团,用分光光度法示踪
离去基团仍结合在基质上,不能用分光光度法示踪
凝胶的再生能力 理论上无数次再生 理论上无数次再生 仅能够再生数次
凝胶的稳定性 pH稳定 pH稳定,可采用叠氮钠抑菌
的反应性都很高,但对于掩埋巯基的结合特性则不同。2吡啶二硫固定相的琼脂
糖凝胶能结合所有的巯基化合物,而巯基磺酸琼脂糖根本不与含有掩埋巯基的
巯基白蛋白结合,但巯基亚磺酸琼脂糖有一定程度的结合。因此,可以利用巯
基磺酸琼脂糖的这一特性来分离具有暴露巯基的蛋白质与掩埋巯基的蛋白质。在高离子强度下,2吡啶二硫凝胶通过亲硫吸附相互作用结合一些缺少巯基
的蛋白质,尤其是免疫球蛋白。这一非共价吸附作用已被应用于色谱技术中,而
二硫氧化物并不具备这样的亲硫吸附特性。作为活性基团,活性二硫化物和二硫氧化物最重要区别在于固定巯基时的反
应特性。2吡啶二硫结构作为固定相的活性基团,与巯基化合物反应时,会导致
2巯基吡啶酮的释放,因而固定过程可以用分光光度法来示踪。而二硫氧化物结

155
构作为固定相的活性基团,在固定巯基时,不会释放出任何低分子量化合物,因
为所形成的次磺酸和亚磺酸仍然结合在基质上,所以二硫氧化物凝胶的固定过程
无法用分光光度法来示踪。显然,当不希望溶液中出现硫醇时,应当选用二硫氧
化物凝胶。理论上,巯基亚 磺 酸 琼 脂 糖 和2吡 啶 二 硫 琼 脂 糖 都 可 以 无 数 次 再 生 使 用,
而巯基磺酸琼脂糖则不能。由于形成了结合在凝胶上的无还原性的磺酸基团,使
巯基磺酸琼脂糖凝胶每使用再生一个循环,其巯基结合容量丧失50%,因而事
实上它只能使用数次。在稳定性上,二硫氧化物固定相和2吡啶二硫衍生物一样,对pH具有相同
的稳定性,并且在叠氮化钠存在下,巯基亚磺酸和巯基磺酸凝胶也是稳定的,而
2吡啶二硫凝胶不宜使用叠氮化钠为抑菌剂。
635 凝胶固定相的引入
通常是先将巯基基团引入琼脂糖凝胶,然后用不同的方法将结合在凝胶上的
巯基活化为2吡啶二硫化物或巯基亚磺酸基团或巯基磺酸基团。
6351 引入巯基
将巯基引入琼脂糖凝胶,较早的方法是将谷胱甘肽偶联到溴化氰活化的琼脂
糖凝胶上 (图610)。谷胱甘肽三肽中的两个羧基官能团使得连接巯基与基质之
帒帒O C N +H2N—谷胱甘肽— →SH
O C
NH+
2
NH谷胱甘肽SH
图610 巯基引入凝胶的谷胱甘肽法
间的间隔臂带负电。
另一种获得巯基取代琼脂糖的 方 法 是
先用表氯醇 (图611)或双环氧化物将环
氧乙烷结构引入凝胶,环氧 乙 烷 结 构 (三
元环)与 硫 代 硫 酸 盐 反 应 形 成 的 衍 生 物
(R—S—SO-3 )被过量的低分子量硫醇进一步还原成巯基。相比于谷胱甘肽法,羟丙基间隔臂没有带电结构。
将巯基引入其他各种含羟基的基质中也可采用上述两种方法。
OH+ClCH 2CHOCH 2→ O CH 2CH
OCH 2
O CH 2CHOCH 2 +Na2S2O →3 O CH 2CH
OH
CH 2 SSO 3
O CH 2CHOH
CH 2 SSO 3 →+RSH O CH 2CHOH
CH 2SH
图611 巯基引入凝胶的表氯醇法
6352 凝胶的活化
(1)将结合在凝胶上的巯基基团转化为混合活性二硫化物 如图612所示,

156
用过量的2,2′二吡啶二硫化物与巯基反应。
O CH 2CHOH
CH 2 SH師師師師
師師+ N
師師師師
師師S S
→N
O CH 2CHOH
CH 2 師師師師
師師S S N
師師師師
+ S N
H
图612 活性二硫化物凝胶的制备
无论 是 含 谷 胱 甘 肽 或2羟 丙 基 间 隔 臂 的2吡 啶 二 硫 化 物 凝 胶,在4℃、
pH=6~7下可稳定贮存几个月。琼脂糖衍生物在适当的添加剂下,比如葡聚糖
和半乳糖,可被冷冻干燥。应该避免使用叠氮化钠作为抑菌剂,因为叠氮化物是优良的亲核体,能和吡
啶二硫基团反应,形成固定相的不稳定亚氧硫基叠氮化物和巯基吡啶酮,从而消
耗活性结构。(2)将结合在凝胶上的巯基基团转化成二硫氧化物 在酸性pH条件下,用
过氧化氢氧化硫丙基琼脂20~30h,将基质上的巯基经二硫化物和巯基亚磺酸转
化为巯基磺酸 (图613)。
图613 巯基磺酸琼脂糖的制备
对于要活 化 成 巯 基 亚 磺 酸 凝 胶 的 硫 丙 基 琼 脂 需 要 进 行 两 步 氧 化 反 应 (图
614)。首先在中性pH 条 件 下,凝 胶 与 氰 铁 酸 钾 反 应,然 后 在pH=5~7下,再用等化学当量的单过氧邻苯二酸盐 (MPP)控制氧化作用,将第一步中所形
成的二硫化物,转化成与凝胶结合的巯基亚磺酸,完成硫丙基琼脂的活化。在中性和弱酸性pH下,巯基亚磺酸基团和巯基磺酸基团对水解的稳定性很
高。事实上,含有这些基团的固定相在pH=5、4℃下可贮存至少6个月,并且
其巯基结合能力不会降低。相比于活性二硫化物,凝胶固定相的二硫氧化物不会与01%的叠氮化钠反
应,因而可用于防止凝胶中细菌的生长。

157
图614 巯基亚磺酸琼脂糖的制备
636 凝胶的取代程度和实际容量
不同的凝胶衍生物之间,固定相的取代程度即获得的可反应的巯基基团含量
差别非常大,而且在很大程度上取决于最初固定相引入的巯基含量。采用基于溴化氰的引入法,只能获得相对较低的巯基取代。而引入环氧乙烷
结构制备的固定相,根据其过量使用的环氧乙烷类化合物是什么,获得的固定相
巯基含量或低或高。由于固定相中结合的巯基基团或多或少地会定量转化成活性二硫化物或二硫
氧化物基团,因而最终可反应的巯基含量取决于起始固定相中巯基含量 (表63)。
表63 活性巯基凝胶的容量
凝 胶 类 型 取代程度/(μmol/ml凝胶) 实 际 容 量
2吡啶二硫谷胱甘肽琼脂糖 1 005μmol人血清白蛋白/ml凝胶
2吡啶二硫羟丙基琼脂糖 20 010μmol血浆铜蓝蛋白/ml凝胶
巯基磺酸琼脂糖 10 017μmol牛血清白蛋白/ml凝胶
巯基亚磺酸琼脂糖 17 018μmol巯基化人血清白蛋白/ml凝胶
064μmol还原态本斯琼斯蛋白链/ml凝胶
凝胶的取代程度是指凝胶结合的2吡啶二硫基团或二 硫 氧 化 物 结 构 与 低 分
子量巯基化合物的反应容量,而凝胶的实际容量是指能结合到凝胶中的蛋白质的
数量。将凝胶和过量的低分子巯基化合物 (如GSH)一起反应,然后洗涤,通
过确定所固定的巯基含量,来获知这一特 定 衍 生 物 的 理 论 巯 基 结 合 容 量。对 于
2吡啶二硫凝胶,用分光光度法直接测定释放出来的2硫代吡啶酮,从而确定结
合的低分子巯基化合物的量。对于固定相含二硫氧化物的凝胶,用二硫代吡啶和
未反应的巯基化合物反滴定来确定所固定的量。一般来说,凝胶所具有的实际容量要比活性巯基基团的取代程度低得多,尤
其是高取代的凝胶。这是由于高分子量的底物 (如蛋白质)在结合到凝胶上时,其空间效性应远大于活性基团浓度的影响。因此,在具体应用中应通过预实验计
算出实际所需的凝胶量。

158
64 色谱技术
本节主要介绍2吡啶二硫琼脂糖衍生物的色 谱 技 术,对 于 二 硫 氧 化 物 琼 脂
糖,应根据色谱基质特性进行适当改变。
641 预备
6411 样品的预处理
在使用共价色谱前,建议先分析样品中的巯基含量,以确保不超过凝胶的容
量。用少量样品 (1~5mg/ml)与2,2′二吡啶二硫化物 (01~02mmol/L)反
应,分光光度法测定释放出的2巯基吡啶酮,从而滴定出样品中的巯基含量。对于生物样品常含有的低分子量巯基化合物 (如GSH),应先采用透析或凝
胶过滤方法去除,再使用共价色谱。根据样品来选择 适 当 的 条 件,可 使 用pH=3~8的 缓 冲 液 (甲 酸 盐、醋 酸
盐、磷酸盐、Tris),浓 度 在005~04mol/L的 范 围 中,含 有 或 不 含 强 变 性 剂
(如8mol/L尿素和6mol/L盐酸胍)。建议加入EDTA (5mmol/L)去捕获过渡
态金属离子,因为它们有可能催化巯基的氧化作用。
6412 预备实验
03g膨胀的平衡过的活性硫丙基琼脂糖凝胶在干燥后,与2~3ml预处理的
蛋白质样品在小试管中混合孵育,密封试管,在1h内往复旋转后进行离心,分
析上清液在343nm下的吸光度 (注意要扣除背景的吸光度),确定结合在凝胶上
的蛋白质的巯基量。在与蛋白质孵育时,活性凝胶有 时 会 因 为 机 械 作 用 而 释 放 出 少 量 巯 基 吡 啶
酮。在不含变性剂的缓 冲 液 中,这 种 活 性 结 构 的 “泄 露”每 小 时 达 到002%,而当有高浓度的脲或胍盐存在时,则要高得多。在pH=40下对应的343nm吸
光度释放量达到0004/h。在许多情况下,尤其是具有高分子 量 的 巯 基 蛋 白,在 高 度 取 代 的2吡 啶 二
硫琼脂糖凝胶中存在的活性结构不会超过1%。因此需要用预备实验的结果来确
定适宜的色谱条件和凝胶的实际结合容量。
642 蛋白样品的结合
对于大蛋白的分离,建议采用谷胱甘肽活化的巯基琼脂糖4B,巯基琼脂糖
中交联度的降低以及间隔臂的延长可以允许较大蛋白质的吸附。而在环氧乙烷活
化的巯基琼脂糖6B凝 胶 中,琼 脂 糖 基 质 通 过 羟 丙 基 间 隔 臂 与 吡 啶 二 硫 化 物 相
连,不利于大蛋白的吸附,但是该凝胶的容量大于巯基琼脂糖4B。样品与凝胶的吸附操作可采用柱式或者分批技术。采用分批式操作时,将凝

159
胶悬浮在样品溶液中孵育,完成样品的结合。采用柱式操作时,让样品经过装有
凝胶并预先平衡过的柱子。对柱子的装填和容积没有严格限定,通常使用长而细
的柱子要比短而粗的柱子好,吸附时注意 调 节 好 流 速,以 确 保 样 品 与 凝 胶 的 接
触,在标准操作中至少1h。用流出液在343nm下 的 吸 光 度 来 估 算 与 凝 胶 反 应 的 样 品 巯 基 量。如 果 用
280nm下的吸光度来估算未结合的蛋白量时,注意扣除所释放出的巯基吡啶酮,因为2巯基吡啶酮在280nm和343nm下的吸光度大致相等 (图615)。
图615 2巯基吡啶酮的吸收光谱
643 洗涤
分批式结合中,用玻璃滤器进行洗涤,对于少量凝胶也可以在柱子中进行洗
涤操作。根据样品与柱子的结合情况来选择洗涤缓冲液,必须将未结合的蛋白质和非
特异性吸附的蛋白质从柱子上洗涤下来,建议使用高离子强度缓冲液 (含01~03mol/LNaCl)来抵消电荷作用。
最简单的方法是上样、洗涤和洗脱使用同样的缓冲液,必要时使用含去垢剂
(如Triton和吐温)的洗涤液,洗涤时注意监测流出液的吸光度,通常采用1~2个柱体积洗涤后,再用还原性洗脱缓冲液平衡柱子,完成洗涤操作。
644 还原洗脱
凝胶中固定相内如果存在未使用的活性基团,则会在还原洗脱中释放出硫代
吡啶酮,为避免污染洗脱下的蛋白质,需采用两步进行还原洗脱。(1)由于硫代吡啶基更容易被还原,先用等物质的量的还原试剂去除掉固定相
中未反应的硫代吡啶基团,这步洗脱操作可以在碱性pH也可在酸性pH下进行。(2)适当洗涤后,再用过量的巯基化合物释放出结合在凝胶上的蛋白样品。

160
通常在pH=8下,用低分子量的硫醇或硫代吡啶进行还原性洗脱,最常用10~25mmol/LDTT或25~50mmol/L2巯基乙醇,在343nm下测定洗脱液吸光度。需要注意的是,如果洗脱时使用的是半胱氨酸,由于氧化态 (胱氨酸)的溶解性
不如还原态 (半胱氨酸),一段时间后,很容易在柱子中沉淀。在还原洗脱步骤中,Hillson提出的顺序洗脱法能提高洗脱的选择性。用一
系列不同还原能力的巯基化合物来增加还原强度或者增加 同 一 巯 基 化 合 物 的 浓
度,获得不同结合蛋白的特异性释放。
645 巯基蛋白的回收
在2吡啶二硫琼脂糖色谱中,如果不 采 用 两 步 还 原 洗 脱,未 与 蛋 白 质 反 应
的凝胶活性基团会在洗脱液中产生相当数量的2硫代吡啶酮。在 进 一 步 处 理 和
活性测定前,用凝胶过滤法去除洗脱液中的小分子化合物,并尽可能使用低pH缓冲液,以减小巯基的氧化风险。
如图616所示,用疏水凝胶SephadexLH20在酸性pH条件下进行低分子
量的多肽样品的脱盐,用排空体积洗脱下多肽,而硫代吡啶酮、还原剂巯基乙醇
的二硫化物和盐被滞留。
图616 人血浆铜蓝蛋白巯基多肽的还原洗脱液的色谱图
●—230nm吸光度;○—343nm吸光度;×—电导率
活性硫代吡啶琼脂糖SephadexLH20柱 (32cm×27cm),Tris缓冲液洗脱。洗脱体
积100ml的第一个峰含有巯基多肽,收集,冷冻干燥。洗脱体积约为200ml、240ml、
290ml下的化合物分别对应于巯基乙醇、巯基乙醇二硫化物和硫代吡啶酮
还原洗脱回收巯基蛋白时,可选用适当的挥发性缓冲液,使洗脱物可被冷冻
干燥。通常洗脱回收的样品体积大,使用冷冻 干 燥 很 方 便,如 用2巯 基 乙 醇 和

161
挥发性缓冲盐 (如醋酸铵),所有低分子化合物在冷冻干燥过程中被蒸发去除掉。使用二硫氧化物琼脂糖作为色谱材料时,用过量的低分子巯基化合物还原洗
脱时,从凝胶中惟一释放的化合物为结合的巯基蛋白,同时形成的次磺酸和亚磺
酸结合在凝胶上,释放下来的蛋白质和过量的还原剂 (可能有少量的相应二硫化
物)一起被洗脱下来。
646 巯基凝胶的再生
完成色谱操作后,凝胶为巯基形式,必须再生后才能再使用。凝胶再生时,必须首先进行洗涤,再根据凝胶的活性基团结构是2吡啶二硫化 物 还 是 二 硫 氧
化物进行不同的再生处理。通常以分批式 在 玻 璃 过 滤 漏 斗 中 洗 涤 凝 胶。将 凝 胶 和 含5mmol/LDTT的
01mol/L磷酸钠缓冲液 (pH=75)混合孵育45min,使得洗脱步骤中可能形成的
所有脂肪族二硫化物还原为硫醇,然后用缓冲液洗涤去除过量的DTT及其氧化态。为确保彻底去除过量试剂DTT,可以用活性二硫化物试剂 (2,2′二吡啶二硫化物
或5,5′二硫二硝基苯甲酸)进行巯基滴定,来监测洗涤液中的DTT的含量。洗涤后的凝胶分别进行不同的再生处理,再生后凝胶的贮存方式和新制备的
凝胶一样。(1)2吡啶二 硫 化 物 凝 胶 再 生 处 理 凝 胶 和 含 饱 和2,2′二 吡 啶 二 硫 化 物
(15mmol/L)的01mol/L磷酸缓冲液 (pH=80)混合,孵育45min。对于巯
基含量大于100μmol/g干衍生物的大容量凝胶,必须用20mmol/L的2,2′二吡
啶二硫化物才能获得完全再生,此时再生反应和随后的洗涤都要在含有20%~30%乙醇的缓冲液中进行,以确保试剂的溶解。
最后,通过大量洗涤来去除过量的2,2′二吡啶二硫化物,为确保彻底洗去
过量试剂,可通过加入少量的巯基化合物,在343nm下监测吸光度来确定试剂
含量。推荐使用的硫丙基琼脂糖凝胶的再生步骤 将1倍体积的2,2′二吡啶二硫
化物 (30~40mg/ml)的乙醇液 (或异丙醇)与4倍体积的01mol/L硼酸缓冲
液 (pH=8,1mmol/LEDTA)中的凝胶混合,80℃回流3h,用乙醇洗涤凝胶,并用初始缓冲液预平衡。
(2)二硫氧化物凝胶的再生处理 对于巯基磺酸凝胶仅能再生数次。根据起
始硫丙基琼脂糖的巯基含量不同,巯基磺酸凝胶的再生次数从两次到数次。而巯
基亚磺酸琼脂糖,理论上可以无数次再生使用。凝胶的再生和初始合成一样,都是从巯基凝胶开始的,可以采用如下步骤:
① 巯基磺酸凝胶 将15g真空干燥的巯基琼脂糖悬浮在45ml的02mol/L醋酸钠缓冲 液 (pH=50)中,在 连 续 振 荡 条 件 下,加 入 过 氧 化 氢,开 始 时 加

162
18ml,然后在30min、90min、150min时各加入22ml,保温反应30h。氧化后
的凝胶转移到玻璃滤器中,用01mol/L醋酸钠洗涤,洗尽过氧化氢,活化后的
凝胶用02mol/L醋酸钠缓冲液 (pH=50)平衡并贮存。
② 巯基亚磺酸凝胶 将15g真空干燥的巯基琼脂糖悬浮在30ml的01mol/
L磷酸钠缓冲液 (pH=70)中,加入01mol/L氰铁酸钾,边加边振荡,直到
黄色留存至少30min,然后用含1mol/LNaCl的02mol/L醋酸钠缓冲液 (pH=50),在玻璃滤器中彻底洗涤凝胶。再将凝胶悬浮在30ml含化学当量的单过氧
邻苯二酸 镁 缓 冲 液 (05mol/molS—S基 团,05mol镁 盐/mol单 过 氧 邻 苯 二
酸,pH=50)中,室 温 振 荡2h,凝 胶 衍 生 物 依 次 用50mmol/L、01mol/L、
02mol/L醋酸钠缓冲液 (pH=50)彻 底 洗 涤,最 终 悬 浮 在02mol/L醋 酸 钠
缓冲液 (pH=50)中储存。
647 反向共价色谱
活性蛋白或多肽的色谱,也称反向共价色谱。在色谱中,将蛋白样品用同型
活性二硫化物处理,与凝胶中含有的固定巯基基团相互作用来实现分离。在这种分离方法中,样品的活化步骤后必须去除过量的试剂 (比如采用凝胶
过滤法),这一点非常重要。另一方面,低容量凝胶必须偶联,以减小凝胶中不
希望发生的巯基二硫互换反应,因为这一反应会导致刚刚结合的蛋白质立刻被
释放出来。在弱酸性pH下进行偶联,脂肪族巯基二硫互换反应会减少,从而
降低这种副反应。随后的操作步骤和共价色谱一样。反向共价色谱在分离大蛋白的水解物多肽时特别有效。
65 应用实例
共价色谱的早期应用主要是从复杂的蛋白质混合物中分离出含巯基分子。在
特定的条件下,甚至可以分离出不同的含巯基分子,具有很高的特异性。此外,共价色谱还可以用于浓缩溶液中浓度极低的巯基化合物。
另外一个重要应用是巯基活性吸附剂通过二硫键反向固定酶,具有暴露巯基
基团的酶可被直接固定,通过温和的巯基化过程可固定巯基基团被掩埋的酶,对
于缺少巯基基团的酶可使用SPDP引入巯基。现在,共价色谱作为蛋白质分离技术的有效补充,在选择性地分离含巯基蛋
白上已有广泛的应用。通过调整吸附pH以及采用顺序洗脱法可以进一步提高该
技术的专一性,因而可应用于纯度相对较低的样品,但是大规模应用还是受到高
成本和再生步骤的限制。
651 从刀豆中分离脲酶
脲酶是催化尿素分解成氨和CO2 的酶,含有6个相同的亚基,非共价联结

163
成分子量为500000的聚集体。它富含巯基,其中许多巯基基团对酶的活性是非
必需的,可以被修饰而不损失活性。以刀豆 粉 为 起 始 原 料,60g粗 粉 和300ml含36%乙 醇、01mol/LKCl和
1mmol/LEDTA的005mol/LTrisHCl缓 冲 液 (pH=72)混 合。混 合 物 在
28℃下搅拌5min,过滤,滤液离心 (500g,20min),上清液用005mol/LTrisHCl缓冲液 (pH=72)稀释至300ml,并用05mol/LNaOH调节pH至72。
用活性硫丙 基 琼 脂 糖 (高 容 量 凝 胶)制 备 总 体 积 为63ml的 柱 (1cm×8cm),并用含有01mol/LKCl和1mmol/LEDTA的005mol/LTrisHCl缓冲
液 (pH=72)平衡柱子。250ml刀豆粉抽提液以20ml/h的流速过柱,绝大部
分紫外吸收的成分通过柱子未滞留。上样后,初 始 的150ml洗 脱 液 中 没 有 脲 酶
的活性,在随后的100ml洗脱液中活性逐步增加。用TrisHCl缓冲液洗涤柱子,直到流出液在280nm处的吸光度低于004。用20ml含20mmol/L二硫苏糖醇、
01mol/LKCl和1mmol/LEDTA的005mol/LTrisHCl缓冲液 (pH=80),洗脱释放出脲酶。
用SephadexG25去除了低分子量物质后,洗脱物中脲酶的比活力 (U/mg干物质)是初始的167倍。用Sepharose6B进一步凝胶过滤,去除一些 高 分 子
量低比活的物质后,比活达到初始的280倍。活性硫丙基琼脂糖结合脲酶活性物
质的容量为51mg/ml凝胶,纯化后脲酶于4℃可稳定贮存数周。
652 木瓜蛋白酶的纯化
木瓜蛋 白 酶 是 一 种 存 在 于 热 带 水 果 番 木 瓜 乳 汁 中 的 蛋 白 酶,分 子 量 约
23500,该酶只含有一个对酶活必不可少的巯基基团。以市售木瓜蛋白酶或干木瓜汁溶液的硫酸铵沉淀物为起始材料,适当处理后
用作共价色谱的样品。
①200mg市 售 木 瓜 蛋 白 酶 溶 解 在 含5mmol/LDTT的01mol/LTrisHCl缓冲液 (pH=80)中,将封闭 的 活 性 位 点 半 胱 氨 酸 转 化 为 巯 基 形 式 还 原 活 性
酶。过量的 DTT用SephadexG25去 除,该 柱 用01mol/LTrisHCl缓 冲 液
(pH=80)或者01mol/L醋 酸 钠 缓 冲 液 (pH=40)预 平 衡,缓 冲 液 都 含 有
03mol/LNaCl和1mmol/LEDTA。100mlTris或醋酸缓冲液排空体积中的物
质 (通常含有04~06mol巯基/mol蛋白质)用作共价色谱的样品。
② 含01~02mol巯基/mol蛋白质的干木瓜汁硫酸铵沉淀物100g溶解在
200ml任一种上述两种缓冲液中。处理后的样品上样至活性巯基琼脂糖柱 (低容量凝胶,18cm×30cm)。用
上样缓冲液洗涤柱子,直至洗脱液在280nm和343nm下的吸光度都低于003。然后 用 含03mol/LNaCl和1mmol/LEDTA 的01mol/LTrisHCl缓 冲 液

164
(pH=80)平衡凝胶,再用含有50mmol/L的L半胱氨酸的同一缓冲液洗脱下
木瓜蛋白酶,读取组分在280nm和343nm下的吸光度,并测定其对N苯甲酰精
氨酸乙酯 (BAEE)的水解活力。收集有活力的组 分,并 加 入30g/100ml溶 液 的 硫 酸 铵 沉 淀 蛋 白。用 最 少 体
积的pH=80的 缓 冲 液 重 新 溶 解 沉 淀,在 SephadexG25色 谱 柱 上,用 含
1mmol/LEDTA的01mol/LKCl溶 液,将 蛋 白 质 和 低 分 子 量 物 质 分 离 开 来。为防止在凝胶过滤 中 形 成 木 瓜 蛋 白 酶半 胱 氨 酸 混 合 二 硫 物,上 样 前 加DTT于
SephadexG25色 谱 柱 中,至 终 浓 度 为5mmol/L。采 用 上 述 条 件,可 以 获 得
100mg纯的木瓜蛋白酶 (1mol巯基/1mol蛋白质)。在pH=80下的色谱分离是基于存在暴露巯基来分离蛋白质的。而在pH=
40下,由于微环境效应,天然木瓜蛋白酶中的巯基基团和2吡啶二硫基团的反
应速度要比变性木瓜蛋白酶的巯基或低分子量化合物与活性基团的反应速度快得
多,特异性更 加 显 著。01mmol/L完 全 有 活 性 的 木 瓜 蛋 白 酶 与 高 达5mmol/LL半胱氨酸混合物,在pH=40下采用共价色谱,所有的木瓜蛋白酶与混合二
硫物凝胶反应,而几乎所有的L半胱氨酸通过色谱柱。
653 共价色谱的顺序洗脱
Hillson利用共价色谱的顺序洗脱成功分离了蛋白质二硫化物异构酶 (PDI)和蛋白质二硫化物氧化还原酶 (也称谷胱甘肽胰岛素转氢酶,GIT)。
以来源于牛肝脏的部分纯化的PDI为起始材料,其中还含有几种巯 基 氧 化
还原酶活性,包括GIT。采用两种方法来制 备 样 品:一 种 是 离 子 交 换 色 谱 部 分
纯化制备;另一种是硫酸铵沉淀获得粗制品。用含 有01mmol/LDTT的 TKM/EDTA/NaCl缓 冲 液 (50mmol/LTris
HCl,pH =75,25mmol/L KCl,5mmol/L MgCl2,125mmol/L EDTA,
01mol/LNaCl)对蛋白样品 (25~125mg,浓度10mg/ml)30℃预处理30min,这种温和还原使得存在于混合二硫化物中任何一个掩埋的巯基暴露。
还原后的样品经过离心,在SephadexG25柱 (2cm×25cm)用TKM/EDTA/NaCl缓冲液去 除DTT。然 后,样 品 可 直 接 上 样 到 活 性 硫 丙 基 琼 脂 糖 柱 上
(湿重30~45g),30℃保持30~60min,使其发生结合,随后进行洗脱。也可以
将样品与凝胶分批混合16h,轻微振荡,然后将胶灌入柱子中,而且分批上样的
结合能力更高。使用后的柱子4℃冷却,用TKM/EDTA/NaCl缓冲液洗涤去除未结合的蛋
白质和非特 异 性 吸 附 的 蛋 白 质。用 不 同 的 低 分 子 量 巯 基 化 合 物 (分 别 含 有
20mmol/LL半 胱 氨 酸、50mmol/L谷 胱 甘 肽、20mmol/LDTT 的 TKM/EDTA/NaCl缓冲液,pH=75),连续洗脱以提高还原能力,从凝胶上置换下结合

165
蛋白。每一步还原洗脱时,用1倍排空体积的还原缓冲液过柱,30℃保持30min。
用TKM/EDTA/NaCl缓冲液洗柱,然 后 在4℃,继 续 用2~10ml/h流 速 洗 脱,收集组分,280nm 下 测 定 蛋 白 含 量,343nm 下 测 定 置 换 下 的2硫 代 吡 啶 酮 的
含量。向每一步收集的含蛋白组分中加入固体DTT,至终浓度5mmol/L,25℃保
温30min,以还原任何蛋白质和洗脱液之间形成的混合二硫化物,再用pH=75的TKM缓冲液4℃充分透析。
在这一方案中获得四种蛋白质组分:从柱子上洗涤下来的未结合的蛋白,分
别被L半胱氨酸、谷胱甘肽、DTT置换下的组分。仅仅在半胱氨酸置换组分中
发现PDI活性,而DTT置换组分中只含有GIT活力,从而实现了以往无法做到
的蛋白质二硫异构酶和蛋白质二硫氧化还原酶的分离。
654 巯基多肽的纯化
通常大分子蛋白在降解过程中会产生非常复杂的多肽混合物,要从中快速分
离巯基多肽,采用共价色谱是最便捷的方法。利用反向共价色谱从还原型核糖核酸酶和巯基清蛋白的木瓜蛋白酶降解物中
分离巯基多肽。具 体 方 法 是:水 解 前,让 蛋 白 质 和2,2′二 吡 啶 二 硫 化 物 反 应,然后将水解得到的活性肽混合物上样到还原型硫丙基琼脂糖柱中,在02mol/L醋酸铵缓冲液 (pH=80)中与柱进行结合。用含有50mmol/L2巯基乙醇的上
述缓冲液进行还原性洗脱,冷冻干燥洗脱下的多肽混合物。因为仅使用了挥发性
缓冲物质和巯基乙醇,可以直接获得清蛋白的纯化巯基多肽。人血浆铜蓝蛋白是含有1046个残基的大分子蛋白,先通过所含的巯基基团
将蛋白固定到柱子上,再使用大剂量的蛋白酶,可以顺次使用两种类型的蛋白酶
(木瓜蛋白酶和胰蛋白酶)来获得初始多肽的亚片段。共价色谱中,在pH=40的醋酸钠缓冲液中进行与柱子的结合反应,用含有50mmol/L巯基乙醇的TrisHCl缓冲液 (pH=80)进行洗脱。洗脱下的巯基多肽可用凝胶过滤和其他色谱
技术进一步纯化。
655 大肠杆菌β半乳糖苷酶的可逆固定
β半乳糖苷酶催化乳糖水解成葡萄糖和半乳糖,在乳品工业中可用于乳清中
的乳糖水解,解决废物处理问题,产生的葡萄糖还可用于发酵。大肠杆菌的β半乳糖苷酶是一个寡聚酶,具有高含量的半胱氨酸 (每个四聚
体中含64个半胱氨酸残基),其中大约1/4的巯基可与巯基试剂反应而不影响酶
活。在温和的条件下,通过暴露的、非必需的巯基基团,可以将β半乳糖苷酶固
定在巯基磺酸琼脂糖上。

166
采用巯基磺 酸 琼 脂 糖 (300μmol/g干 凝 胶 的 取 代 程 度)和β半 乳 糖 苷 酶
(Sigma公 司)进 行 实 验。2g真 空 干 燥 的 巯 基 磺 酸 琼 脂 糖 若 干 份,分 别 与 含
09~46mg蛋白的β半乳糖苷酶溶液75ml(pH=70,01mol/L磷 酸 钾 缓 冲
液配置)混合,轻微搅拌,4℃下保温16h,形成的不溶性衍生物用缓冲液彻底
洗涤,4℃储存。用O硝基苯βD吡喃半乳糖 (溶解在含3mmol/LMgCl2 的01mol/L磷酸
钾缓冲液中,pH=75)分析偶联物的酶活性,获得的稳定偶联物其有效酶含量
为8~114mg蛋白 质/g干 衍 生 物。巯 基 磺 酸 琼 脂 糖 凝 胶 选 择 性 地 结 合 活 性 酶,固定的β半乳糖苷酶的比活,高出初始商品酶50%。
残留的固定相巯基磺酸基团和各种低分子量巯基化合物反应,形成各种不同
的衍生物。由于每种化合物会引发固定化酶的特定微环境,从而使得各衍生物具
有不同的特性,例如,用谷胱甘肽封闭可导致固定化β半乳糖苷酶的衍生物热稳
定性提高。各种 衍 生 物 都 具 有 很 好 的 长 期 稳 定 性,在01mol/L磷 酸 钾 缓 冲 液
(pH=70)中4℃贮存18个月,酶活保持不变。固定化酶能在含有50mmol/LDTT的01mol/L磷酸钾 (pH=85)缓冲液
中被定量释放,可以确定酶与柱子的结合是因为形成了二硫化物。
656 蛋白质亚基的鉴定
在天然状态下,胸苷酸合成酶为含有两个相同亚基的二聚体,每个亚基的活
性位点都含有一个半胱氨酸残基。然而纯的胸苷酸合成酶在贮存过程中,甚至在
含有2巯基乙醇的贮存条件下,会发生酶活下降的情况。通过共价色谱操作,可以将纯酶 蛋 白 分 成 两 个 生 理 生 化 特 性 截 然 不 同 的 组
分,一个具有较高的胸苷酸合成酶活性,而另一个胸苷酸合成酶活性较低。操作
时,在固定相的可反应巯基和酶的催化巯基基团之间引入一可裂开的混合二硫键
来实现固定。通过这一固定步骤可以从纯胸苷酸合成酶收集物中,分离获得高比
活的酶组分。在共价色谱的固定操作中,通过控制固定反应保温时间的长短,来限制天然
酶通过不止一个混合二硫键结合到巯基活性吸附剂的比例。根据这一策略,可将
天然的胸苷酸合成酶仅通过一个亚基被固定,这样在另一个亚基上保留的活性位
点巯基基团可以和N乙基马来酰亚胺反应。用50mmol/L2巯基乙醇从固定相
上洗脱下来的这一异源二聚体,可用于亚基相互作用的研究。这种利用共价色谱和选择性化学修饰的新方法,在特定寡聚蛋白的催化和调
节机理研究中,可用作研究亚基之间相互作用的有效工具。

167
7 电 泳 技 术
71 概述
电泳 (electrophoresis)就是在电场作用下,带电胶体颗粒向着与其电性相
反的电极移动,其分离原理是基于生物大分子的电荷密度。电泳作为一种生化分离手段已广泛用于生物大分子的分离、纯化、分析、制
备,以及用于测定它们的分子量、等电点等。现在,电泳已成为生命科学、医学
等领域以及制药业、农林业、食品工业、生物工程、环境工程和工业分析等行业
中经常使用的实验手段。相对于其他蛋白质分离与纯化手段,电泳具有操作温和、特异性高、分辨率
高等优点。现在被广泛使用的电泳都是以凝胶作为支持介质的。
711 原理
7111 蛋白质的电荷来源
从电泳角度看,蛋白质分子最主要的特性是它的带电行为。由于蛋白质是由氨基酸组成的,而氨基酸带有可解离的氨基 (质子受体)和
羧基 (质子供体),因此是两性电解质。当氨基酸之间通过肽键形成蛋白质后,参
与形成肽键的氨基和羧基就不是可解离基团了,但是蛋白质侧链上仍然有一些酸
碱基团,如Asp的βCOOH、Glu的γCOOH、Lys的εNH2、His的咪唑基、
Arg的胍基等以及蛋白质的氨基末端和羧基末端,使得蛋白质成为两性电解质。在某一pH条件下,蛋白质分子所带的正负电荷数相等,即净电荷为零,此
时为蛋白质的 等 电 点 (pI),蛋 白 质 质 点 在 电 场 中 不 移 动;当 体 系pH>pI时,蛋白质分子会解离出 H+而带负电,从而在电场中向阳极移动;当体系pH<pI时,蛋白质分子会结合 H+而带正电,从而在电场中向阴极移动。
NH+3 —CHR—COOH NH+3 —CHR—COO- NH2—CHR—COO-
A+ Ka1 A± Ka2 A
-
pH<pI pH=pI pH>pI
特定蛋白质的等电点取决于它的氨基酸组成,是一个物理化学常数。对于不
同蛋白质,其等电点范围很宽,在一定pH时,不同蛋白质分子所带电荷的电性

168
和电量不同,由此可进行蛋白质的分离和分析。
7112 迁移率
在一定pH条件下,蛋白质分子会解离而带电,带电的性质和电荷密度取决
于蛋白质分子的性质 (可解离侧链基团)和溶液的pH。不同的带电颗粒在同一
电场的运动速度不同,可用迁移率来表示。迁移率 (m,mobility)即带电颗粒在单位电场强度下的泳动速度:
m=vE=
dtUl
式中 m———迁移率,cm2/(s·V);
v———迁移速度,cm/s;
E———电场强度,V/cm;
d———迁移距离,cm;
t———电泳时间,s;
U———电压,V;
l———凝胶长度,cm。由于球形分子在电场中所受的动力和阻力平衡,即
EQ=6πrηv式中 Q———被分离分子所带净电荷;
η———介质黏度;
r———分子半径。于是有:
m= Q6πrη
即迁移率与球形分子的半径、介质黏度、颗粒所带电荷有关。带电颗粒在电场中的迁移速度与本身所带的净电荷的数量、颗粒大小和形状
有关,一般来说,所带静电荷越多,颗粒越小,越接近球形,则在电场中迁移速
度越快,反之越慢。
7113 影响电泳速度的内在因素
蛋白质分子的氨基酸组成决定其在某个pH条件下的电性和电量,进而决定
它的电泳速度;蛋白质分子的形状与大小也影响它的电泳速度。
7114 影响电泳速度的外在因素
(1)溶液pH 决定带电颗粒的电性和电量,因此需选择合适的pH,使欲分
离的各种蛋白质所带电荷的电性相同但是电量有较大的差异,以利于彼此的分离。

169
(2)溶液离子强度 离子强度越低,则欲分离组分的迁移速度越快;但离子
强度太低则缓冲能力太小,因此合适的离子强度范围在001~02mol/L之间。(3)电场强度 越大则带电颗粒迁移越快,电泳时间越短;反之迁移慢,电
泳时间长。(4)电渗现象 流动相相对于固体支持介质的移动即为电渗,电渗是由于电
泳支持介质上含有带电基团,从而吸附流动相向正极或负极移动。电泳时,带电
颗粒的迁移速度是颗粒本身的迁移速度与由于电渗携带颗 粒 的 移 动 速 度 之 矢 量
和。为消除电渗现象,可选择无电渗的电泳介质。(5)电泳支持介质 要求介质均匀,对样品吸附力小。(6)温度 电泳过程中会产热,造成样品扩散及烧胶,因此需控制电压或电
流,或安装冷却系统。
7115 影响电泳分辨率的因素
电泳中的介质是影响电泳分辨率的一个很重要的因素。由于样品的扩散和对
流会干扰样品的分离,可使用支持介质以防止电泳过程中的对流和扩散。支持介
质必须是化学惰性的、均匀、化学稳定性好,为了减少电内渗,需选择无电渗的
凝胶介质。电压及冷却系统也会影响电泳的分辨率。高电压 (电场强度)可以提高电泳
的分辨率,但高电压会导致凝胶过热甚至烧胶,因此凝胶的良好散热性是非常重
要的,为此可以使用薄的平板胶及采用冷却装置。缓冲系统由于影响样品的电荷密度以及溶解性、稳定性等,会对电泳的分辨
率造成影响。
712 分类
(1)按电泳的分离原理来分
① 移界电泳 样品分子在自由溶液中迁移,从而引起界面的移动。这是最
早的电泳方式,现已被取代。
② 区带电泳 样品分子在介质上或介质中迁移,最终被分离成独立的区带,这是现在最常用的电泳方式。包括PAGE,SDSPAGE等。
③ 稳态电泳 样品分子的电泳迁移在一定时间后达到一个稳态。包括等电
聚焦和等速电泳。(2)按有无固体支持物来分
① 自由电泳 样品在自由溶液中迁移。如移界电泳,某些等速电泳,柱电
泳等。
② 支持物电泳 电泳在固体支持物中进行,这是应用最多的电泳方式。如
PAGE,免疫电泳,等电聚焦,等速电泳等。

170
(3)按用途及介质、电泳装置分类 本章主要介绍以下几种电泳:
① 常规聚丙烯酰胺凝胶电泳 (PAGE);
②SDS聚丙烯酰胺凝胶电泳 (SDSPAGE);
③ 等电聚焦 (载体两性电解质及固相pH梯度);
④ 双向电泳;
⑤ 免疫电泳;
⑥ 蛋白质印迹;
⑦ 毛细管电泳。
713 介质
在自由溶液中进行的移界电泳 已 成 为 历 史,而 采 用 支 持 介 质 的 电 泳 应 用 广
泛。采用支持介质的目的是防止电泳过程中的对流和扩散,使被分离组分得到最
大分辨率的分离。电泳用支持介质分为:(1)无阻滞支持物 化学惰性,能将对流减到最小,其对蛋白质的分离是基
于一定pH条件下蛋白质的电荷密度。介质包括滤纸、醋酸纤维薄膜、纤维素、硅胶等。近年来其使用渐少。
(2)高密度凝胶 不仅能防止对流,降低样品扩散,还可以起到分子筛的作
用,其对蛋白质的分离不仅取决于大分子 的 电 荷 密 度,还 取 决 于 分 子 尺 寸 和 形
状。介质包括淀粉凝胶 (由于质量不稳定且分离效果不好,已很少使用)、聚丙
烯酰胺凝胶 (孔径大小与蛋白质分子同数量级,因此具有分子筛作用)和琼脂糖
凝胶 (孔径较大,对大多数蛋白质仅有很小的分子筛作用)。
7131 聚丙烯酰胺凝胶
聚丙烯 酰 胺 凝 胶 (polyacrylamidegel)是 由 单 体 丙 烯 酰 胺 (acrylamide,
Acr)和交联剂N,N′亚甲基双丙烯酰胺 (N,N′methylenebisacrylamide,Bis)在增速剂和催化剂的作用下聚合而成的三维网状结构的凝胶。聚丙烯酰胺凝胶是
目前最常用的电泳支持介质,它 不 仅 可 用 于 常 规PAGE,也 可 用 作SDSPAGE的凝胶介质,或用作等电聚焦等的介质。
(1)聚合反应 分化学聚合和光化学聚合。化学聚合一般用过硫酸铵 (ammoniumpersulfate,AP)作 催 化 剂,N,N,
N′,N′四甲基乙二胺 (TEMED)作增速剂。碱性条件下TEMED催化AP生成
硫酸自由基,接着硫酸自由基的氧原子激活Acr单体并形成单体长链,Bis将单
体长链连成网状结构。增加AP和TEMED的浓度可加速聚合。碱性条件下胶易
聚合,而酸性条件下由于缺少TEMED的游离碱,难以聚合,这时可用AgNO3作增速剂。低温、氧分子及杂质会阻碍凝胶的聚合。采用此法制备的胶孔径小且

171
重复性好。光化学聚合一般用核黄素作催化剂,TEMED作增速剂,在光照及少量氧的
条件下,黄素被氧化成有自由基的黄素环而引发聚合。此法制备的胶孔径较大且
不稳定,但用此法进行酸性凝胶的聚合效果比较好。(2)聚丙烯酰胺凝胶的孔径及分子筛效应 由 于 聚 丙 烯 酰 胺 凝 胶 是 多 孔 介
质,且孔径大小与蛋白质有相似的数量级,从而能主动参与生物分子的分离,因
此具有分子筛效应。凝胶的孔径大小、机械性能、弹性、透明度、黏着度及聚合
程度取决于T和C,T为Acr和Bis的总浓度 (%),T=Acr%+Bis%;C为交
联浓度 (%),C=[Bis/(Acr+Bis)]×100%。有 效 孔 径 随 着T 的 增 加 而 减 少;
T恒定,C为4%~5%时,孔径最小,高于或低于此 值 则 孔 径 变 大,一 般 不 使
用C>5%的凝胶。由 于 聚 丙 烯 酰 胺 凝 胶 的 孔 径 大 小 与 蛋 白 质 分 子 为 同 数 量 级,因此可利用孔径不同的凝胶来分离不同大小的蛋白质分子。
(3)试剂的性能 Acr和Bis都是神经毒素,应该注意操作过程中采取预防
措施。所有试剂应为分析纯,避光保存,新鲜配制,试剂不纯将会干扰凝胶的聚
合,配试剂应使用重蒸水。(4)聚丙烯酰胺凝胶的优点 在一定浓度时,凝胶透明,有弹性,机械性能
好;化学性能稳定,与被分离物不起化学反应;对pH和温度变化较稳定;几乎
无电渗作用,样 品 分 离 重 复 性 好;样 品 不 易 扩 散,且 用 量 少,其 灵 敏 度 可 达
10-6g;凝胶孔径可调节;分辨率高。(5)聚丙烯酰胺凝胶的应用范围 可用于蛋白质、酶、核酸等生物大分子的
分离、定性、定量及制备,并 可 测 定 分 子 量 和 等 电 点,研 究 蛋 白 质 的 构 象 变 化
等。聚丙烯酰胺凝 胶 可 用 于 常 规 及SDS聚 丙 烯 酰 胺 凝 胶 电 泳,等 电 聚 焦 电 泳,双向电泳,聚丙烯酰胺梯度凝胶电泳及蛋白质印迹等。
7132 琼脂糖凝胶
天然琼脂由琼脂糖和琼脂胶组成,琼脂糖是由半乳糖及其衍生物构成的中性
物质,不带电荷;而琼脂胶是一种含羧基和硫酸根的酸性多糖。琼脂糖也是电泳
最常用的一种支持介质。一般常用1%琼脂糖作为电泳支持物。琼脂糖凝胶应用
于DNA分离、免疫电泳、蛋白质分离、蛋白质印迹、交变脉冲凝胶电泳及等电
聚焦。琼脂糖凝胶作为重要的电泳介质有如下特点:易于制备凝胶;其孔径较大,
因此可用于免疫固定、免疫电泳和微量制备以及分离分子量比较大的物质;其机
械强度较高;琼脂糖无毒;易于染色、脱色及储存电泳结果。琼脂糖的胶凝温度
一般在34~43℃,融化温度在75~90℃。但是琼脂糖凝胶具有不同程度的电内
渗,凝胶容易脱水收缩。

172
7133 醋酸纤维薄膜
醋酸纤维是纤维素的醋酸酯,用醋酸纤维薄膜作支持介质的电泳操作简单、快速、价廉,目前多用于临床检验和医学上的常规分析。
7134 新介质
聚丙烯酰胺和琼脂糖是目前最常用的两种电泳介质。聚丙烯酰胺凝胶的分辨
率高,但孔径相对较小;而琼脂糖凝胶孔径大,因此琼脂糖聚丙烯酰胺混合凝
胶 (如琼脂糖的烯丙基缩水甘油衍生物)可有效地分离大分子。另外,电泳海绵
也是很有前途的新型电泳介质。
714 电泳仪器
现在常规使用的电泳仪器是凝胶电泳仪。凝胶电泳系统一般由电泳槽、电源
和冷却装置组成,还有一些配套装置,如灌胶模具、染色用具、电泳转移仪、凝
胶干燥器、凝胶扫描仪等。其中电泳槽是电泳系统的核心部分。电泳的电压则依
据不同的电泳类型而不同,PAGE、SDSPAGE一般为200~600V;载体两性电
解质等电聚焦可达1000~2000V;固相pH梯度等电聚焦则高达3000~8000V;电泳转移宜采用低电压,大电流。有效的凝胶冷却系统可避免凝胶过热、烧胶,从而可提高电压,加大电场强度,进而加快电泳速度,提高电泳的分辨率。
图71 圆盘
电泳
凝胶电泳按电泳仪形状分为管状凝胶电泳 (圆 盘 电 泳)和 板
状电泳 (包括垂直板状电泳、水平板状电泳)。
7141 圆盘电泳 (discelectrophoresis)圆盘电泳 (图71)有上下两个电泳槽,上电泳槽有若干孔用
于插电泳管。电泳管尺寸早期为长约7cm,内径约5~7mm,现在
则越来越长,越来越细,以提高分辨率和微量化。圆盘电泳的各电
泳管分别制胶,因此不够简便、均一、准确。另外,由于电泳凝胶
与电泳液直接液体接触,易导致液体泄漏,造成短路、断路等。
7142 垂直板状电泳
垂直板状电泳 (图72)有上下两个电泳槽,中间经垂直平板
相连,制胶和电泳在两块垂直放置的平行玻璃板 之 间 进 行,凝 胶
厚度一般为075~3mm。由于同一块板上可同时电泳多个样品,因此均一、可
靠,且凝胶板薄,表面积大,易于冷却,从而分辨率高。垂直电泳也采用直接液
体接触方式。
7143 水平板状电泳
水平板状电泳 (图73)由分置于两侧的缓冲液槽以及中间的凝胶板组成,
缓冲液与凝胶之间通过滤纸桥或凝胶条搭接,即采用半干技术电泳。凝胶下由于

173
有冷却板,因此可采用高压,从而提高了分辨率,缩短了电泳时间。水平电泳的
电极也由固定型转变成可移动型。
图72 垂直板状电泳 图73 水平板状电泳
管状电泳虽然使用最早,但目前已经很少使用,而垂直电泳和水平电泳已成
为主要的电泳方式,其中水平电泳的优势更多些 (表71)。
表71 不同凝胶电泳的特点及应用
电泳 管状电泳(圆盘电泳)板 状 电 泳
垂直板状电泳 水平板状电泳
特点
一管一样;
直接液体接触;
电压 低,分 辨 率 低,速 度 慢,准确度低;
染色效果不好,不利于保存
一板多样;
直接液体接触;
电压较高,分 辨 率 较 高,速 度
较快,准确度高,灵敏度高;
染色效果好,便于保存
一板多样;
半干技术;
电 压 高,分 辨 率 高,速 度 快,准
确度高,灵敏度高;
染色效果好,便于保存;
凝胶 尺 寸 可 调 节,加 样 数 目 和
位置可调节
应用 PAGE,SDSPAGE PAGE,SDSPAGE,蛋 白 质
印迹
PAGE,SDSPAGE,蛋白质印迹,等电聚焦,双向电泳,免疫电泳
715 染色方法
经聚丙烯酰胺、琼脂糖及醋酸纤维薄膜电泳分离的蛋白质需用染色法使其在
支持介质的相应位置上显示出谱带或谱点,从而检测样品的分离情况。现在蛋白
质的染色方法主要有以下几种:(1)考马斯亮蓝R250、G250 染 色 灵 敏 度 达02~05μg/带,此 染 料 是
通过范德华力结合到蛋白质的碱性基团,适用于对蛋白质和肽染色。(2)银染色法 灵敏度比考马斯亮蓝R250高100倍。(3)氨基黑10B 是酸性 染 料,常 用 于 蛋 白 质 染 色,但 对SDS蛋 白 质 染 色
效果不好。

174
(4)荧光染料 包括丹磺酰氯、荧光胺等。
72 常规聚丙烯酰胺凝胶电泳
聚丙烯酰胺凝胶电泳是蛋白质分离与分析中最常用的电泳方法,包括常规聚
丙烯酰胺凝胶电泳 (PAGE)和SDS聚丙烯酰胺凝胶电泳 (SDSPAGE)。
721 原理
7211 分离原理
对于常规PAGE,在电泳过程中蛋白质仍保持其天然构象及生物活性。
PAGE对蛋白质的分离一方面是基于蛋白质的电荷密度,即在恒定缓冲系统
中不同蛋白质间同性净电荷的差异;另一方面是基于分子筛效应,即与蛋白质分
子的大小和形状有关。因此,用PAGE不仅能分离含各种大分子的混合物,还
可以研究生物大分子的特性,如电荷、分子量、等电点乃至构象,并可用于蛋白
质的纯度鉴定。
7212 电泳分离的物理效应
对于不连续聚丙烯酰胺凝胶电泳,它对蛋白质的分离不仅仅基于上面所介绍
的分子筛效应和电荷效应,还基于其对样品的浓缩效应。在不连续聚丙烯酰胺凝胶电泳中,样品首先经过大孔径的浓缩胶被浓缩,再
经过小孔径的分离胶进行分离。浓缩胶与分离胶之间不仅存在凝胶孔径的不连续
性,还存在缓冲系统的不连续性。先导离子 (如Cl-)存在于样品缓冲液及凝胶
缓冲液中,先导离子是与待分离蛋白质具有同性电荷的迁移率很高的离子;尾随
离子 (如Gly-)加在电极缓冲液中,它是随着pH的不同而有不同解离度的蛋
白质的同性离子。在浓缩胶中,对于阳极电泳,由于pH较低 (pH=7左右),尾随离子仅有
01%~1%解离 (Gly-的pKa 为96),因此有效迁移率很低,而带负电的待分
离的蛋白质的迁移率介于先导离子和尾随离子之间。加压后,先导离子由于泳动
快而与尾随离子分开,从而在其后留下一个导电率很低的区带 (电位梯度的不连
续性),这一区带电压梯度高,于是加速蛋白质与尾随离子的迁移,使其赶上但
无法超越先导离子,这样蛋白质就被压缩在先导离子与尾随离子之间一个狭窄的
区带上,得以浓缩,而浓缩胶的孔径较大,所有蛋白质都能顺利通过,同时被浓
缩在一条窄带上。样品进入分离胶后,由于pH升高 (pH=85左右),尾随离子的解离度增
加,其有效迁移率超过蛋白质,于 是 不 存 在 浓 缩 效 应;又 由 于 分 离 胶 的 孔 径 较
小,而蛋白质分子量越大,其迁移 率 越 小,因 此 可 通 过 分 子 筛 效 应 (及 电 荷 效
应)得以分成多个区带。

175
722 常规PAGE的类型
(1)按被分离蛋白质的电性分类 常规PAGE可分为阳极电泳和阴极电泳。
① 阳极电泳 为通常使用的电泳方式,pH=80~95,多数蛋白质带负电,向阳极迁移。
② 阴极电泳 仅用于分离碱性蛋白质,pH=40左右,蛋白质带正电,向
阴极迁移。(2)按电泳槽的类型分类 可分为圆 盘 电 泳,垂 直 平 板 电 泳 和 水 平 平 板 电
泳。其中平板电泳使用较多,因为其分辨率高,电泳结果易保存。(3)按电泳系统的连续性分类 聚丙烯酰胺凝胶电泳可分为连续系统与不连
续系统两大类,前者由于电泳体系中缓冲液pH和凝胶浓度保持不变,因此无浓
缩效应;后者由于电泳体系中缓冲液离子成分、pH 以及凝胶浓度的不 连 续 性,从而有浓缩效应,因此分辨率高。
① 连续电泳 蛋白质的分离主要基于电荷密度,连续电泳只有分离胶,pH恒定,样品缓冲体系、凝胶缓冲体系、电极缓冲体系相同,只是离子强度不同。
② 不连续电泳 蛋白质的分离主要取决于分子大小和形状,电荷密度也有
影响。不连续电泳系统中蛋白质先经浓缩胶浓缩,再经分离胶分离,其中分离胶
可以是均一胶,也可以是梯度胶。均一胶中整块凝胶为同一浓度,而梯度胶是由
一定的浓 度 梯 度 组 成 的,沿 蛋 白 质 迁 移 方 向,浓 度 梯 度 逐 渐 增 大,凝 胶 孔 径
变小。
723 影响分离结果的外在因素
待分离蛋白质的电性决定了电泳时应选择阳极电泳还是阴极电泳,其电量多
少则影响着缓冲系统的选择,而蛋白质分子的大小和形状则决定了凝胶浓度的选
择。反之,电泳缓冲系统和凝胶浓度影响电泳的分离效果。
7231 缓冲系统
电泳时首先要选择合适的缓冲系统,包括缓冲系统的pH和离子组成。其原
则是保证样品的溶解度、稳定性、生物活性以及电泳的速度和分辨率。(1)缓冲系统中pH的选择 对于常规PAGE,由于蛋白质分离时需保持溶
解状态,且蛋白质的分离要依据其电荷密度,因此pH的选择很重要,一方面要
使蛋白质的电荷密度差别大以利于分离,另一方面要使蛋白质荷电多且带同性电
荷以加 速 分 离。由 于 有 近 半 数 蛋 白 质 的 等 电 点 在pH=40~65,因 此 常 使 用
pH=80~95缓 冲 体 系 的 阳 极 电 泳,比 如 TrisHCl(pH=71~89)、TrisGly (pH=83~95)、Tris硼 酸 (pH=83~93)、Tris醋 酸 (pH=72~85)缓冲系统。对于碱性蛋白质可采用pH=30的KOH醋酸缓冲系统。
(2)缓冲系统中离子强度的选择 离子强度低,蛋白质迁移速度快,但电泳

176
带较宽;离子强度高,蛋白质迁移速度慢,电泳带窄细,但由于高导电性而产生
大量热,易导致蛋白质变性及烧胶。另外,还要使缓冲系统有一定的pH缓冲能
力。一般离子强度应为001~01mol/L,常用的为005mol/L。阳极电泳和阴极电泳的缓冲系统列于表72。
表72 常规PAGE的缓冲系统
项 目 阳 极 电 泳 阴 极 电 泳
样品缓冲液 同浓缩胶缓冲液,离子强度适当调整 同浓缩胶
电极缓冲液 pH=70~85(如0025mol/LTris02mol/LGly) pH=40~45(如醋酸Ala)浓缩胶缓冲液 pH=70左右(如001mol/LTrisHCl),样品带负电 pH=50~60(如KOH醋酸)分离胶缓冲液 pH=80~95(如003mol/LTrisHCl),样品带负电 pH=20~40(如KOH醋酸)先导离子 在样品及凝胶缓冲液中,多为Cl- 多为K+
尾随离子 在电极缓冲液中,多为Gly- 多为Ala+或Gly+
反向离子 在样品、电极和凝胶缓冲液中,带正电 带负电
7232 凝胶浓度
对于常规PAGE,凝胶的分子筛效应 (凝胶孔径)与电泳分辨率、电泳速度
密切相关,而凝胶的孔径大小决定于凝胶浓度,因此凝胶浓度的选择很重要。一般可根据样品 中 蛋 白 质 的 分 子 量 范 围 选 择 合 适 的 凝 胶 浓 度 (见 表73)。
对于未知样品,可先选择T为75%的标准均匀胶进行试验,然后根据电泳分离
效果的好坏及区带的迁移距离再选择合适的凝胶浓度,必要时可选择一定浓度范
围的梯度胶分离复杂组分。
表73 蛋白质分子量范围与聚丙烯酰胺凝胶浓度的关系
C=26% C=5%
凝胶浓度(T)/% 分子量范围/kDa 凝胶浓度(T)/% 分子量范围/kDa
5 30~200 5 60~70010 15~100 10 22~28015 10~50 15 10~20020 2~15 20 5~150
724 蛋白质分子量的测定
用常规PAGE测定蛋白质的分子量需排除电荷的影响,因此需测量蛋白质
分子在不同浓度凝胶中的相对迁移率从而得到蛋白质的分子量。具体来说就是先
用不同浓度的凝胶同时测量未知和已知分子量的蛋白质的相对迁移率;然后对不
同蛋白质分子以其相对迁移率的对数对凝胶浓度作图,得出一条直线,直线的斜
率为阻滞系数,它与蛋白质分子量成正比 (确切地说,阻滞系数与凝胶系统的交
联度、分子的形状和分子量有关);最后用已知分子量的蛋白质的阻滞系数对它

177
们的分子量作图,得到一条直线,从此直线上根据未知蛋白质的阻滞系数可查得
它的分子量。
725 常规PAGE的实验方法
任何一种凝 胶 电 泳 都 要 经 历 四 个 主 要 步 骤:制 胶、加 样、电 泳 和 检 测。另
外,还有一些辅助步骤,如样品准备等。天然状态下的PAGE是各种电泳的基础。
7251 制胶
(1)凝胶模具的准备 在制胶之前,首先要将制胶模具准备好。对于管状 (圆盘)电泳,将玻璃管下口封住,以防止胶液泄漏,胶液从顶部
灌注。对于垂直平板电泳,凝胶通常是灌注在中间有隔片的两块玻璃板之间,将模
具垂直放置,左右两边密封住,并用夹子夹紧,底部固定在弹性橡胶条上,以防
止胶液漏出,凝胶溶液从顶部灌注。对于水平平板电泳,也采用垂直灌注方式。垂直平板电泳先灌注分离胶,再
灌注浓缩胶,而水平平板电泳则无顺序要求。在常规PAGE中可进行连续电泳和不连续电泳。连续电泳只有分离胶,因
此灌胶非常方便,而不连续电泳则需分别灌注分离胶和浓缩胶。这里就不连续电
泳中的阳极电泳的制胶方法作一介绍。(2)凝胶贮液的配制
① 丙烯酰胺储备液 (A) T=30%,C固定,26%~3%;
②4×分离胶缓冲液 (B) 如pH=89,015mol/LTrisHCl;
③4×浓缩胶缓冲液 (C) 如pH=68,005mol/LTrisHCl;
④10%过硫酸铵溶液 (AP)。需要注意的是,丙烯酰胺是神经毒剂,应戴手套操作。电泳时的试剂应达到
电泳纯,并使用重蒸水配制溶液。(3)制备分离胶 根据丙烯酰胺的浓度和凝胶的用量配制分离胶溶液,如总
量为10ml,T为x%时,则按照表74的配方操作。
表74 常规PAGE分离胶配方
A液/ml B液/ml 重蒸水/ml 10%AP/μl TEMED/μl
x30×10=x
/3 10/4=25 10-x3-25约50 5~10
其中A液、B液、重蒸水先混合好,再依次缓慢搅拌加入10%AP及TEMED,并立即混合好。溶液中应避免产生气泡,否则凝胶聚合不好。
分离胶溶液混合后,迅速将胶液通过注射器或滴管注入凝胶模具中,达到所

178
需高度后,在胶面上轻轻注入一层1~5mm的蒸馏水,然后静置20~60min,等
待凝胶聚合。刚加入水层时,在分离胶溶液与水层之间有一界面,随后界面迅速
消失,凝胶聚合后,在分离胶和水层之间又会出现一个明显的界面。这时倾斜凝
胶,可判断凝胶是否聚合。凝胶应在20~60min内聚合,若聚合过快或过慢,可通过减少或增加AP及
TEMED的量来调节。若仍然无法达到满意的聚合,应 检 查 各 种 试 剂,如 A液
是否过期,试剂中是否有杂质等。(4)制备浓缩胶 待分离胶聚合后,先除去分离胶上的水层 (用注射器或吸
管吸出,也可以用滤纸吸出),然后根据浓 缩 胶 的 浓 度 和 用 量 配 制 浓 缩 胶 溶 液,配制方法同分离胶,此时B液应换为C液。
将浓缩胶溶液迅速、稳定地 注 入 分 离 胶 之 上,直 至 顶 部。对 于 垂 直 平 板 电
泳,小心地在顶端倾斜插入梳子,待其齿底到达玻璃板顶部时,压平梳子,注意
应避免齿端留有气泡。静置30min等待凝胶 聚 合,然 后 小 心 取 出 梳 子,注 意 不
要撕裂加样孔。对于圆盘电泳,由于样品是直接加在浓缩胶胶面上的,因此无需
用加样梳产生加样孔。对于水平平板电泳,由于可在胶面上直接加样,因此可以
先灌注浓缩胶,再灌注分离胶。将凝胶梳拔出后,用吸管吸取少量电泳缓冲液淋
洗加样孔,以赶去凝胶表面的气泡。将凝胶放入电泳槽内,上下 (或左右)电泳槽中加入电泳缓冲液,使凝胶两
端浸没在缓冲液中,排除凝胶两端的气泡,然后接好电极,准备上样。
7252 加样
(1)制备样品 在加样之前,应首先制备样品溶液,使样品以合适的浓度溶
解在样品缓冲液中。这是电泳成功与否的关键。
5×样品缓冲液:如pH=68、03125mol/LTrisHCl,含甘油25%、溴酚
蓝005%。样品溶液应达到适当 的 离 子 强 度,并 保 持 很 好 的 溶 解 状 态。若 离 子 强 度 过
高,可采用透 析 法、SephadexG25凝 胶 过 滤 法 或 超 滤 法 脱 盐;若 样 品 溶 解 不
佳,可用助溶剂助溶 (如TritonX100),并在10000r/min下离心10min,取上
清加样,以免电泳时脱尾。特别是在冰箱中放置较长时间的样品,上样前一定要
离心除去沉淀物。样品浓度应根据样品组分的复杂程度及目的蛋白的含量来确定。若用考马斯
亮蓝染色,以目的带的浓度为05~2mg蛋白/ml为佳。对未知样品,可作一个
02~20mg蛋白/ml的稀释系列,以寻找最佳浓度。(2)蛋白质标样的准备 将合适分子量范围的5~7种标准蛋白质 (市售或
自行配制)混合溶解在样品缓冲液中,制备成蛋白质标样。

179
(3)加样 对于垂直平板电泳,用微量注射器吸取定量溶液,小心地伸入加
样孔底部,稳定地将样品加成一 窄 带,空 孔 中 应 加 入 样 品 缓 冲 液,以 防 止 边 缘
效应。对于水平电泳,可在胶面上直接加样,或在加样孔中加样。阳极电泳的样品
加在阴极侧;阴极电泳的样品加在阳极侧。对于圆盘电泳,样品直接加在浓缩胶胶面上。
7253 电泳
加样后,接好电极,打开电源,将电压稳定在200V进行电泳。待溴酚蓝前
沿到达电泳槽底部时,关闭电源,取出凝胶,准备染色。在电泳过程中,要确保 电 泳 缓 冲 液 和 凝 胶 保 持 良 好 的 接 触,防 止 短 路 和 断
路,保证电流的稳定。电泳时切勿使凝胶过热或产生温度梯度,否则将会导致边缘
效应,即指示剂前沿呈现两边向上的曲线形,这时可以通过降低电压来改善电泳状
况。电泳时如果出现指示剂前沿呈现两边向下的曲线形,则说明电泳装置不合适。对于水平电泳,应先预电泳20min,以除去凝胶中的不纯物,然后再加样。
加样后,先进行5~10min低电压低电流电泳,使蛋白 质 分 子 顺 利 进 入 凝 胶 后,再调高到合适的电压、电流下进行电泳。
7254 检测
常规PAGE不破坏蛋白质的生物活性,可采用的检测方法很多,其中考马
斯亮蓝染色法最常用,其检测灵敏度为02~05μg/带。若要提高灵敏度可采用
银染色法,检测灵敏度可达纳克 (ng)级。另外还有荧光染色等方法。(1)考马斯亮蓝染 色 考 马 斯 亮 蓝 主 要 有R250和G250两 种。R为 红 蓝
色,G为蓝绿色。R250是三苯基甲烷的衍生物,而G250是R250的甲基取代
物。R250常用于蛋白质的染色,其染色方式多为三步法 (先固定,再染色,最
后脱色),也可固定和染色同时进行;G250多用于对小肽染色,其染色方式多
为两步法 (先同 时 固 定 和 染 色,再 脱 色)。考 马 斯 亮 蓝 含 有 两 个—SO3H 基 团,偏酸性,它结合在蛋白质的碱性基团上,并且与不同的蛋白质结合呈现相同的颜
色,颜色的深浅与蛋白质的含量呈线性关系。在一定pH时,蛋白质染料复合
物又可解聚。考马斯亮蓝的染色方法很多,不尽相同,仅举几例如表75所示。一般固定半小时,固定的时 间 可 以 延 长,固 定 可 使 蛋 白 质 凝 聚,防 止 其 扩
散。而染色时间取决于凝胶厚度等,时间不宜过长,否则增加脱色难度,搅拌和
升温可加速染色和脱色过程。脱色时可多次更换脱色液,直至背景脱干净为止。在染色过程中应避免手指印留在电泳凝胶上,且染色用容器要用盖子或封口
膜密封。

180
表75 考马斯亮蓝染色方法
项 目 固 定 液 染 色 液 脱 色 液
考 马 斯 亮
蓝R250
方法一
57g三氯 醋 酸、17g磺 基
水 杨 酸 溶 于150ml甲 醇、
350ml蒸馏水中
125g考 马 斯 亮 蓝 R250 溶 于 230ml甲 醇 和
230ml蒸 馏 水 中,搅 拌1h后加40ml冰醋酸
500ml冰 醋 酸、1500ml95% 乙 醇,加 蒸 馏 水 至
5000ml
方法二 10g考 马 斯 亮 蓝R250溶 于450ml甲 醇、450ml蒸
馏水和100ml冰醋酸中
100ml甲 醇、100ml冰 醋
酸与800ml蒸馏水混合
考 马 斯 亮
蓝G250
方法一 004%考马斯亮蓝G250溶 于35%过 氯 酸 和20%甲醇溶液中
5%醋酸20%甲醇溶液
方法二
125%三氯醋酸 10g考 马 斯 亮 蓝 G250溶 于1000ml的125%三
氯醋酸中
甲醇∶水∶浓氨水=64∶36∶1
(2)银染色 银染色的灵敏度达2ng/带,银染时蛋白带上的AgNO3被还原成
金属Ag而沉积在蛋白带上。银染色过程中蛋白质先固定、再显色。常用固定剂有
戊二醛、乙醇、甲醇、醋酸、三氯醋酸,固定能阻滞蛋白质在凝胶中的扩散。显色有化学显色和光显色。化学显色包括双胺银染法 (将凝胶加到氢氧化铵
和AgNO3中染色,酸化显像,终止显色)和非双胺化学显色银染法 (凝胶固定,加酸性AgNO3,碱性 条 件 下 用 甲 醛 还 原 使 Ag+ 转 变 成 Ag显 像,酸 化 终 止 显
色)。光显色是通过光使Ag+ 转变成Ag。在这些显色方法中,非双胺化学银染
法应用较多。具体的银染色方法可参考有关资料,目前已经有几家试剂公司提供
银染色试剂盒。(3)荧光染料 荧光染料可以是染料本身发荧光,或是染料与蛋白质的反应
产物发荧光。荧光染料法分为电泳前预标记和电泳后再标记两种方法。电泳前预标记蛋白质主要是采用丹磺酰氯、荧光胺、邻苯二甲酸二醛 (OPA)
等,特别是荧光胺近年来用得较多。由于游离荧光胺及其水解产物不发荧光,只有
其蛋白标记物发荧光,因此其显色无背景干扰,检测灵敏度可达5ng。但是荧光胺
和蛋白质发生反应后,会改变蛋白质在常规PAGE和SDSPAGE中的迁移率。电泳后蛋白带的迅速定位可以用OPA、荧光胺、1苯胺8萘磺酸盐 (ANS)
进行荧光标记。OPA法:将电泳凝胶浸泡在含007~008%巯基乙醇的pH=85,01mol/L巴比妥钠或磷酸缓冲液中10~25min,再于10mg/mlOPA甲醇
溶液中暗处理15min,即可用紫外灯在365nm观察荧光带,可检测到025μg蛋
白质。ANS法:将 电 泳 凝 胶 浸 泡 在 含0003% (003g/L)ANS的pH=68,
01mol/L磷酸缓冲液中5~10min,即可用紫外灯观察荧光带,但不够灵敏,可
检测到20μg以上蛋白质。

181
除了上面这些染色方 法 外,还 有 一 些 其 他 方 法 可 用 于 蛋 白 质 染 色,如 苯 胺
黑、氨基黑、丽春红S、快绿FCF、2,4DNFB与蛋白质的反应等。对于特殊蛋
白质,如糖蛋白、脂蛋白、含铁蛋白等,既可以用一般蛋白质的染色方法,也可
以针对性地利用其功能基团的特殊反应性质而采用更为专一性的染色方法。相对而言,考马斯亮蓝、银染法应用广泛,因为电泳结果可被扫描、保存,
而荧光法虽然灵敏、方便,但难于保存电泳结果,其应用因此而受到限制。银染
法相对于考马斯亮蓝法灵敏度高,但实验繁杂,对试剂要求高,费用昂贵,且会
使某些蛋白质漏检,使别的生物大分子显色,因此在对灵敏度无很高要求的情况
下,多使用考马斯亮蓝染色法。染色后,在加样处染色带过深,则说明样品分子量过大或溶解不好,浓度过
高,应调节电泳的缓冲系统及凝胶浓度,并对样品进行适当处理。电泳凝胶中陷入
气泡,也会导致蛋白带畸变。若出现脱尾现象,则说明样品溶解不佳,可对样品进
行助溶、离心处理。样品溶解不好,也会导致电泳后凝胶表面出现纹理现象。
7255 电泳结果的保存
凝胶经染色后,应马上拍照或扫描,以记录电泳结果。需要的话,可将凝胶
干燥保存。对于平板凝胶也可以浸泡在7%醋酸溶液中低温下密封保存,但保存
时间仅为几个月。厚度大于10mm的厚胶必须用凝胶干燥仪进行干燥,否则会
导致凝胶龟裂,而厚度小于10mm的薄胶既可用凝胶干燥仪干燥,也可采用自
然干燥法。
726 PAGE的应用范围
可测定蛋白质的分子量,对蛋白质组分进行分离、分析、纯化、定性、定量
以及少量的制备。对于常规PAGE,由于电泳后蛋白质还保持其生物活性,因此
尤其适合于分离各种欲保持天然活性的蛋白质,如酶、抗原、抗体、激素等。
73 SDS聚丙烯酰胺凝胶电泳
731 原理
对于SDSPAGE,由于在电泳体系中加入了十二烷基硫酸钠 (SDS)和强还
原剂 (如巯基乙醇,二硫苏糖醇等),其中SDS作为阴离子去污剂会破坏蛋白质
的氢键和疏水相互作用,导致蛋白质构象改变,而巯基乙醇等可以打开蛋白质分
子内的二硫键,使其分解为亚基,因此电泳 过 程 中 蛋 白 质 的 生 物 活 性 丧 失 或 减
弱,但电泳后可恢复或部分恢复其活性。低离子强度时,SDS主要以单 体 形 式 存 在,此 时 它 可 以 与 蛋 白 质 结 合 生 成
蛋白质SDS复合物。当SDS单体浓度大于1mmol/L时,它 与 多 数 蛋 白 质 的 结
合比为14gSDS/g蛋白质。由于SDS带 有 大 量 负 电 荷,当 它 与 蛋 白 质 结 合 后,

182
所带的负电荷大大超过了蛋白质原有的负电荷,从而掩盖了不同蛋白质间电荷的
差异,使所有蛋白质都带有相同密度的负电荷,于是蛋白质间不存在电荷密度的
差别。而SDS蛋白质复合物都是椭圆棒形,无形状差别,棒的长度与蛋白质亚
基分子量有关,所以在SDSPAGE中蛋白质仅存在分子大小的差别,于是可利
用分子量差异将各种蛋白质分开 (分子筛效应),因此SDSPAGE常用于测定蛋
白质亚基的分子量及鉴定纯度。与常规PAGE相似,不连续SDSPAGE对蛋白质的分离除了基于分子筛效
应外,还基于其对样品的浓缩效应。
732 SDSPAGE的类型
在SDSPAGE中,由于蛋白质与SDS结合后都带负电,因此是阳极电泳。与常规PAGE相似,SDSPAGE也可分为圆盘电泳、垂直平板电泳和水平
平板电泳。其中平板电泳较常使用。
SDSPAGE一般不采用连续电泳方式,因为在SDSPAGE中蛋白质间不存
在电荷密度差异,多采用不连续电泳方式。不连续电泳中蛋白质的分离主要取决于
其分子大小和形状。不连续电泳的分离胶可以是均一胶,也可以是梯度胶。
733 影响分离结果的外在因素
与常规PAGE相似,电泳缓冲系统和凝胶浓度是影响SDSPAGE分离效果
的主要因素。
7331 缓冲系统
对于SDSPAGE,由于SDS对 蛋 白 质 的 助 溶 作 用 以 及 负 电 荷 的 包 裹 作 用,蛋白质的分离仅依据于亚基的分子大小,因此缓冲系统的选择较简单,只要利于
分离并保持蛋白质的稳定以及不与SDS发生相互作用即可。最常用的缓冲系统
为TrisGly缓冲液,其余如 磷 酸 缓 冲 液 (多 用 于 连 续 电 泳)、Tris醋 酸 盐 缓 冲
液、Tris硼酸盐缓冲液、咪唑缓冲液以及SDS尿素系统 (用于分离低分子量蛋
白样品)。离子强度一般要求比较低,为001~02mol/L。
SDSPAGE为阳极电泳,其缓冲系统类似于表72中的常规PAGE阳极电
泳。但在凝胶 缓 冲 液 中 加 入01% SDS,在 样 品 缓 冲 液 中 加1%~2% SDS和
1%~6%巯基乙醇 (或3%二硫苏糖醇),并加适量溴酚蓝,在电极缓冲液中加
01%SDS,不连续电泳的凝胶缓冲液和样品缓冲液中还需加适量甘油。
7332 凝胶浓度
由于凝胶浓度决定凝胶孔径大小,而凝胶孔径影响电泳效果 (即凝胶的分子
筛效应)。特别是对于SDSPAGE,由于电泳分离只取决于SDS蛋白质亚基胶束
的大小,因此凝胶浓度的选择尤为重要。应根据样品中蛋白质的分子量范围选择

183
合适的凝胶浓度 (见表76)。
表76 蛋白质分子量范围与SDS聚丙烯酰胺凝胶浓度的关系
C=26% C=5%
凝胶浓度(T)/% 分子量范围/kDa 凝胶浓度(T)/% 分子量范围/kDa
5 25~200 5 60~17010 10~70 10 20~10015 <50 15 10~5020 <40 20 5~40
734 蛋白质分子量的测定
对于SDSPAGE,蛋白质的电泳迁 移 率 仅 取 决 于 亚 基 分 子 量 的 大 小,且 与
其成正比,因此在任何凝胶浓度时,相对迁移率与分子量对数之间都存在线性关
系,选择合适的凝胶浓度只是为了得到最佳分辨率。选择合适的标准蛋白和合适
的凝胶浓度,经过一次电泳 即 可 得 到 蛋 白 质 亚 基 的 分 子 量,而 不 像 常 规PAGE法需作多次 电 泳,因 此SDSPAGE是 测 定 蛋 白 质 分 子 量 的 合 适 方 法。用SDSPAGE测定蛋白质分子量具有设备简单、操作方便、快速、样品用量少且不需纯
样、分辨率高、重复性好等优点,因而优于光散射、超离心、渗透压及凝胶过滤
色谱等 方 法。但 是 对 于 一 些 异 常 蛋 白 质 以 及 有 较 大 辅 基 的 蛋 白 质,用SDSPAGE测得的分子量不太可靠。
735 SDS聚丙烯酰胺凝胶电泳的实验方法
SDSPAGE中,样品缓冲液中必须含有3~4倍于蛋白质的SDS和足以断裂
二硫键的β巯基乙醇或二硫苏糖醇,且蛋白质必须完全变性展开。由于每克伸
展的蛋白质结合14g单 体SDS,因 此 通 常 在 样 品 缓 冲 液 中 加1%~2% (10~20g/L)SDS,在凝胶及电泳缓冲液中加01%SDS。
7351 制胶
SDSPAGE多采用不连续电泳方式,其制胶方法与常规PAGE的制胶方法
基本相同,但由于凝胶中含有SDS,易起泡,低温下又易结晶析出,因此单体储
备液不需 抽 气,聚 合 时 的 凝 胶 也 不 宜 在4℃放 置。制 胶 模 具 的 准 备 也 同 常 规
PAGE一样。下文介绍不连续电泳中的阳极电泳的制胶方法。(1)凝胶储备液的配制
① 丙烯酰胺储备液 T=30%,C固定,26%~3%。
②4×分离胶缓冲液 如pH=88、15mol/LTrisHCl缓冲液,含04%SDS。
③4×浓缩胶缓冲液 如pH=68、05mol/LTrisHCl缓冲液,含04%SDS。
④10%过硫酸铵溶液 (AP)。

184
同样需要注意的是,丙烯酰胺是神经毒剂,应戴手套操作。电泳时的试剂应
达到电泳纯,并使用重蒸水配制溶液。(2)制备分离胶 同常规PAGE一样,也是根据丙烯酰胺的浓度和凝胶的
用量配制分离胶溶液,如体积为10ml,T为x%时,则按照表77配方操作。
表77 SDSPAGE分离胶配方
A液/ml B液/ml 重蒸水/ml 10%AP/μl TEMED/μl
x30×10=x
/3 10/4=25 10-x3-25约50 3~5
根据聚合的快慢来调节AP和TEMED的用量,使凝胶在20~60min内聚合。(3)制备浓缩胶 同常规PAGE。
7352 制备样品和加样
5×样品缓冲液:pH=68、006mol/L (04mol/L)TrisHCl缓冲液,含
25% (50%)甘 油、2% (10%)SDS、5% (体 积 分 数)巯 基 乙 醇 和01%溴
酚蓝。将蛋白质溶液与5×样品缓冲液以4∶1 (体积比)混合 (如果蛋白质溶液浓
度过高,需预先脱盐处理)后,在100℃水浴中加热3~5min,使蛋白质充分变
性,再于10000r/min离心5min,取上清加样。其他同常规PAGE。
7353 电泳
电泳缓冲液:pH=83,0025mol/LTris0192mol/LGly缓 冲 液,含01%SDS。其余同常规PAGE。
7354 染色和检测
多用考马斯亮蓝法和银染法。对于考马斯亮蓝法,在SDSPAGE后,凝胶
的固定和染色时间 应 比 常 规PAGE的 长1倍 左 右,或 用 多 倍 体 积 染 色 液 染 色,以排除SDS的影 响。对 于 银 染 法,在 电 泳 后 染 色 之 前,必 须 将 凝 胶 多 次 漂 洗,以除去SDS。
7355 电泳结果的保存
同常规PAGE。
736 SDSPAGE的应用范围
可测定蛋白质的分子量,对蛋白质组分进行分离,分析、纯化,定性、定量
以及少量的制备。相对 于 光 散 射、色 谱、超 离 心、渗 透 法 等,SDSPAGE是 测
定蛋白质亚基分子量的一种简单、经济、快捷、高分辨的好方法。
74 等电聚焦
等电聚焦 (isoelectrofocusing,IEF)是依据蛋白质分子等电点的不同,在一个

185
连续的、稳定的线性pH梯度中电泳,从而对蛋白质进行分离和分析的技术。由于
IEF具有聚焦效应 (浓缩效应),因此它是目前一向电泳中分辨率最高的技术。等电聚焦的关键 是 在 凝 胶 中 形 成 稳 定 的、连 续 的 线 性pH 梯 度。根 据 建 立
pH梯度原理的不同,IEF可分为载体两性电解质pH梯度IEF和固相pH梯度
IEF。前者是将载体两性电解质 (如脂肪族多氨基多羧酸)溶解在电泳介质溶液
中制胶,形成聚丙烯酰胺或琼脂糖凝胶,然后将凝胶引入电场中等电聚焦。于是
载体两性电解质在凝胶中迁移形成pH梯度,而样品则聚焦在其等电点处,其分
辨率为001pH单位,上样量也不可过高,否则引起载体两性电解质线性pH梯
度的局部改变。后者是将弱酸、弱碱两性基团直接引入丙烯酰胺中,在凝胶聚合
时就形成pH梯度,因此其pH梯度固定,不随环境电场等条件变化,于是其分
辨率更高,达0001pH单位,上样量也可更大。
741 原理
7411 等电聚焦的聚焦效应
常规PAGE的缓冲系统是恒定的,因此样品易扩散,分辨率不高。而对 于
等电聚焦,其分离是在稳定的、连续的线性pH 梯度中进行的。在pH 梯度中,当某一蛋白质处于其非等电点位置时,由于它带有净电荷,势必向异性电极方向
迁移,即向其等电点方向迁移,一旦抵达其等电点位置,因净电荷为0而停止迁
移。即使蛋白质因扩散而进入邻近非等电点区,由于其带电,会被吸引回等电点
处。因此蛋白质只能在其等电点位置被聚焦成一条窄带 (图74),所以等电聚
焦的分辨率大大高于常规PAGE。这种聚焦效应是等电聚焦高分辨率的保证。
图74 等电聚焦的聚焦效应
IEF对蛋白质的分离仅仅取决于蛋白质的等电点,因此可以在凝胶的任何位
置加样,且无需加成窄带。

186
7412 等电聚焦的分辨率
等电聚焦的分辨率决定于pH梯度和场强。pH梯度范围越窄,分辨率越高;场强越高,即电压越大,分辨率越高,但电压过大会在凝胶中产生过多的热而导
致烧胶。为了维持高电压,就必须提供有效的冷却系统,因此等电聚焦电泳多采
用水平平板凝胶。由于等电聚焦的关键是要在凝胶中形成稳定的、连续的线性pH梯度,那么
如何形成这种pH 梯 度 呢?载 体 两 性 电 解 质IEF和 固 相pH 梯 度IEF,在 建 立
pH梯度时的原理和方法不同,对此分别进行介绍。
742 载体两性电解质pH梯度的形成
载体两性电解质pH梯度等电聚焦的凝胶支持介质为聚丙烯酰胺或琼脂糖,其作用仅仅为形成电泳凝胶。而协助pH梯度形成的则为游离的小分子载体两性
电解质,它们通过在凝胶中迁移到固定位置而形成pH梯度。
7421 载体两性电解质
就载体两性电解质IEF而言,建立pH梯度的关键就在于载体两性电解质的
合成。载体两性电解质是一种缓冲能力强、导电性能好的两性电解质。它的分子
量比较小,一般接近1000,易与蛋白质分离。载体两性电解质必须具有良好的
溶解性能,以保证等电聚焦过程中pH梯度的形成和蛋白质的迁移;它必须具有
很强的缓冲能力,以对抗样品对pH梯度的干扰,保证pH梯度的稳定;它必须
具有良好的均匀的导电性能,以保证电场的均匀、高电压和pH梯度的稳定。目前有几种载体两性电解质产品。瑞典LKB公司的Ampholine是脂肪族多
氨基多羧酸;瑞典Pharmacia公司的Pharmalyte是脂肪族多羟基多氨基多羧酸;德国Serva公司的Servalyte是脂肪族多氨基磺酸或多氨基磷酸。电解质的pI在
大多数羧基的pK 和大多数氨基的pK 之间。调节酸和胺的比例,就可得到不同
pH范围的载体两性电解质。脂肪链越长,则载体两性电解质的pH越连续,得
到的pH梯度越平滑。商用载体两性电解质的pH范围有宽pH范围和窄pH范围两种。宽pH范
围一般是pH=3~10,窄pH范围则包括pH从低到高的各种型号,每种型号的
下限和上限之间一般相差2个pH单位左右。
7422 pH梯度的形成
在无电场时,载体两性电解质分子有的带正电,有的带负电,但总的净电荷
为0,此时的pH是其pH范围的平均值。当引入电场时,带不同电荷的载体两
性电解质分子将向其异性电极方向迁移,直至到达其净电荷为0的位置。由于载
体两性电解质具 有 很 强 的 缓 冲 能 力,使 得 它 所 处 环 境 的pH 等 于 它 本 身 的pI。

187
一定时间以后,所有不同pI的载体两性电解质分子都迁移到其对应的pH位置,于是形成从阳极至阴极pH递增的pH梯度。
pH梯度的线性决定于载体两性电解质的质量,将两种或两种以上不同pH范围的载体两性电解质预混合,可产生均 匀 的 缓 冲 能 力、导 电 性 和 更 为 线 性 的
pH梯度。
pH梯度范围的稳定性,决定于载体两性电解质的质量,电泳时的电参数及
凝胶系统的组成。在凝胶中添加甘油、蔗糖或山梨糖醇等 (10%~15%),可增
加黏度,减少电内渗,提高pH梯度的稳定性。
743 载体两性电解质等电聚焦方法
IEF一般采用高电压,以提高分辨率。维持高电压必须具有良好的凝胶冷却
系统,以防止凝胶过热而烧胶,因此一般采用水平平板胶进行等电聚焦电泳。根
据电泳介质的不同,IEF又可分为聚丙烯酰胺凝胶IEF和琼脂糖凝胶IEF。一般
聚丙烯酰胺凝胶只可分析分子量小于300000的蛋白质,但用琼脂糖凝胶可分析
分子量大至2000000的大分子。用于等电聚焦的聚丙烯酰胺凝胶浓度约为5%~8%,交联度约为3%;而用于等电聚焦的琼脂糖浓度为1%;用于等电聚焦的载
体两性电解质常用 的 浓 度 是2%~25%,2%用 于2mm胶,25%用 于05mm胶。载体两性电解质的导电性能越好,缓冲能力越大,就可以加高电压,从而缩
短电泳时间,提高分辨率,但电压过高易引起烧胶。
7431 聚丙烯酰胺凝胶等电聚焦
加有载体两性电解质的丙烯酰胺凝胶的聚合比加有SDS的及常规丙烯酰胺
凝胶的聚合要困难,可提高温度至30~40℃以加快聚合。对于中性和碱性范围
的凝胶聚合可采用APTEMED化学聚合 法,一 般 调 整 催 化 剂 量 使 凝 胶 在40~60min内聚合最好。过多催化剂会导致等电聚焦时烧胶。酸性范围的凝胶聚合比
较困难,可采用亚甲基蓝二苯氯化碘甲苯亚磺酸或核黄素二苯氯化碘甲基亚
磺酸光聚合法。(1)制胶
① 首先组装好灌胶模具,注意在上下两块玻璃板之间的密封垫圈上涂抹硅
油,以获得良好的密封性。
② 配制T为30%,C为3%的丙烯酰胺单体贮液 (注意有毒)。
③ 选取并配制合适pH范围的40%载体两性电解质溶液。
④ 配制凝胶溶液xml(如10ml),见表78。
⑤ 将凝胶溶液沿垫片缓慢稳定地注入灌胶模具内,避免产生气泡。
⑥ 待凝胶聚合后,从模具边缘可以看到光折射,此时即可取胶。
⑦ 将凝胶连同冷却装置放入电泳池中,检查凝胶是否有渗漏。

188
表78 载体两性电解质IEF凝胶配方
试 剂 体积(总xml)/ml 体积(总10ml)/ml 终 浓 度
丙烯酰胺单体贮液 (x×6/30) 20 T=6%,C=3%40%载体两性电解质溶液 (x×24/40) 06 24%
重蒸水 (x-x×630-x×2440) 74
小心混合均匀,避免陷入气泡,必要时可置于抽滤瓶中抽气5~10min10%AP 5xμl 50μl 005%TEMED 05xμl 5μl 005%
轻轻混匀,即刻小心灌胶
(2)等电聚焦
① 采用与PAGE一样的样品处理方法,然后上样。样品可加在凝胶的任意
位置处 (或加样孔内),浓度一般为05~2μg/μl,体积为5~20μl。等电聚焦时
最好将样品溶解在重蒸水中,以避免盐离子对pH梯度的破坏及烧胶。为了促进
样品的溶解,可加入两性离子,如1%甘氨酸或2%载体两性电解质助溶,这些
试剂不会干扰pH梯度;或用一些解聚试剂,如50%二甲基亚砜、50%二甲基亚
酰胺、90%亚酰胺、33%乙烯甘油或四亚基尿素等助溶;对于疏水蛋白,可用尿
素、无离子 去 污 剂,如 TritonX100、NP40、吐 温20、吐 温40、吐 温80等,或两性离子去污剂如Chaps助溶。IEF的加样位置虽说可任意,但将样品加在靠
近等电点处,电泳效果会更好。
② 凝胶两侧分别与阳极电极溶液、阴极电极溶液相连。阳、阴极电极溶液
的作用是为了避免样品或载体两性电解质在阳极氧化或在阴极还原。电极液通常
由不挥发的酸或碱配制而成。阳、阴极电极溶液的pH应比阳、阴极端的pH分
别略低和略高,通常强酸 (10mmol/L或1mol/L磷 酸)或 强 碱 (20mmol/L或
1mol/LNaOH)用作宽pH范围等电聚焦的电极液,弱酸 (1mol/L醋酸、酸性
载体两性电解质溶液)或弱碱 (碱性载体两性电解质溶液)用作窄pH范围的电
极液。阳极电极溶液:10mmol/L或1mol/L磷酸或醋酸溶液,或低2个pH 单
位的2%载体两性电解质溶液。阴极电极溶液:20mmol/L或1mol/LNaOH或
高2个pH单位的载体两性电解质溶液。
③ 将阳极、阴极分别与电源的正、负极相连,接通电源,进行等电聚焦电
泳。不同的电泳装置有不同的电参数,IEF上限电压可达2000V,上限电流可达
50mA。一般采用恒功率方式。
④ 当凝胶的电流达到最小值不再下降时,聚焦基本完成,可终止电泳。(3)等电聚焦后的处理
① 测定pH梯度 聚焦后,可用表面电极从阴极至阳极每隔1cm测定pH,

189
或使用等电点标准来获得pH梯度曲线。
② 固定、染色、脱色 凝胶先在10%三氯醋酸/35%磺基水杨酸固定液中
固定30min,再用脱色液 (25%乙醇8%醋酸溶液)浸洗,以除去凝胶中的载体
两性电解质,防止其与一些蛋白质形成不溶性化合物而导致染色后无法脱尽。之
后用考马斯亮蓝R250染色液染色10min,染色液中加适量硫酸铜,因为它可被
载体两性电解质螯合,以便于除去载体两性电解质,从而易于脱色干净。最后用
脱色液多次脱色直至背景无蓝色为止。
③ 凝胶的保存及IEF结果记录 同PAGE。
7432 琼脂糖凝胶等电聚焦
琼脂糖凝胶的孔径比较大,因此琼脂糖凝胶等电聚焦多用于分离分析大分子
量化合物。(1)制胶
① 组装好灌胶模具,并在70℃烘箱中保温。
② 加热煮沸配制琼脂糖溶液,待温度降至75℃时加入特定pH范围的40%载体两性电解质溶液,至终浓度24% (体积分数),溶解混匀,并使琼脂糖的
终浓度达到1%。
③ 取出灌胶模 具,倾 斜 着 将 上 述 溶 液 灌 注 到 两 块 玻 璃 板 之 间,之 后 放 平、冷却,使凝胶溶液凝固。
④ 取胶,拭去凝胶表面的水以防止电泳时短路。(2)等电聚焦 同聚丙烯酰胺凝胶等电聚焦法。但电极溶液有所不同。阳极
液多为05mol/L醋酸,阴极液多为05mol/LNaOH。(3)等电聚焦后的处理
①pH梯度的测定 采用表面电极测定法
② 固定、染色、脱色 凝胶先在10%三氯醋酸固定液中固定10min,再用
95%乙醇洗10min,吹干;之后用05%考马斯亮蓝R250溶液染色10min,最
后用脱色液 (35%乙醇10%醋酸溶液)脱色直至背景无蓝色为止。相对而言,聚丙烯酰胺凝胶等电聚焦的分辨率比琼脂糖凝胶等电聚焦的高。
由于聚丙烯酰胺电内渗小,可直接用于等电聚焦,而琼脂糖由于有带电基团,其
产生的电内渗将影 响pH 梯 度 及 蛋 白 质 的 分 离,因 此 须 纯 化 后 才 能 用 于 等 电 聚
焦。等电聚焦时,可先 用 宽pH 范 围 等 电 聚 焦 电 泳 找 出 欲 测 蛋 白 质 的pI位 置,再用合适的窄pH范围IEF精确定位并测定其等电点。
744 固相pH梯度的介质及其pH梯度的形成
7441 固相pH梯度的介质
固相pH梯度所用的介质是一些具有弱酸或弱碱性质的丙烯酰胺衍生物,它

190
们与丙烯酰胺和亚甲基双丙烯酰胺具有相似的聚合行为,因此它们除了可以在滴
定终点附近形成pH梯度外,还参与丙烯酰胺的共价交联,形成凝胶,从而形成
固定的pH梯度。固相pH梯度介质包括酸性丙烯酰胺衍生物和碱性丙烯酰胺衍生物。酸性丙
烯酰胺衍生物含有一个弱的羧基,碱性丙烯酰胺衍生物含有一个叔胺基。在聚合
过程中,这些弱酸、弱碱基团就共价结合到聚丙烯酰胺凝胶介质中,参与聚丙烯
酰胺凝胶的形成。现在主要的商品固相pH梯度介质是瑞典LKB公司的Immobiline。Immobi
line的pK 值随实验条件,如温度、离子强度、溶剂的介电常数等而改变,因此
电泳时要严格控制温度为10℃。另外,尿素等添加剂会降低水溶液的介电常数,并进而改变Immobiline的pK值,因此需形成凝胶后再用尿素液泡胀。Immobiline最大的问题就在于它 的 不 稳 定 性。它 易 降 解,脱 去 弱 酸、弱 碱 基 团,从 而 干 扰
pH梯度的形成和准确性;碱性Immobiline尤其易发生自聚合,进而使被分离的
蛋白质分子发生沉淀;另外,碱性Immobiline还会形成N氧化物,从而影响凝
胶聚合。
7442 固相pH梯度的建立
目前主要有六种不同pK 的固相pH梯度介质:pK=36,46,62,70,
85,93。利用不同的组合可配制不同pH范围的凝胶。首先,根据欲建立的pH梯度范围选择合适的缓冲组分和滴定组分,并由此
配制酸性重液和碱性 轻 液。对 于1个pH 范 围 梯 度 的 建 立,可 采 用pHmid≈pK的方式,即设计范围的中点pHmid与缓冲组分的pK 接近,而滴定组分的pK 远
离设计pH范围的两端,将缓冲组分与滴定组分以一定比例分别配制成酸性重液
和碱性轻液 (表79)。对于窄范围pH 梯度的建立,可在此基础上采用 内 插 法
得到。对于宽范围pH 梯度的建立,其配方见表710。现 在 可 利 用 商 业 软 件 从
10种不同pK 的固相pH梯度介质中选择合适的缓冲组分和滴定组分,并以一定
的比例分别配制成酸性重液和碱性轻液。然后将酸性重液置于梯度混合器的混合腔内,其pH为欲建立的pH范围的
酸性起点;将碱性轻液置于储液腔内,其pH 为欲建立的pH 范围的碱性终点。通过酸性重液和碱性轻液的梯度混合,就可得到从酸性到碱性的线性pH梯度。
7443 固相pH梯度等电聚焦的分辨率
固相pH梯度等电聚焦是一向电泳中分辨率最高的。原因在于电泳的分辨率
与电场强度成正比,与pH梯度范围成反比。由于固相pH梯度等电聚焦可以使
用窄范围pH梯度 (<1pH单位),并且凝胶的离子浓度很低,导电性低,从而
可使用高电压,因此电场强度高,所以其分辨率可高达0001pH单位。

191
表79 1个pH范围的固相pH梯度配方
酸性重液的体积/μl
02mol/LImmobilinepK
36 46 62 70 85 93
pH范围
碱性轻液的体积/μl
02mol/LImmobilinepK
36 46 62 70 85 93
— 904 — — — 129 38~48 — 686 — — — 477— 713 — — — 177 41~51 — 803 — — — 659— 682 — — — 235 43~53 — 992 — — — 871— 716 — — — 325 45~55 — 1314 — — — 1208562 600 863 — — — 46~56 — 863 863 — — 105218 863 863 — — — 49~59 — 863 863 — — 248113 863 863 — — — 51~61 — 863 713 — — 4431251 — 1355 — — — 52~62 337 — 724 — — —775 — 903 — — — 55~65 209 — 686 — — —536 — 713 — — — 58~68 144 — 803 — — —416 — 691 — — — 61~71 112 — 1133 — — —972 — — 1086 — — 62~72 262 — — 686 — —635 — — 783 — — 65~75 171 — — 724 — —465 — — 683 — — 68~78 125 — — 934 — —381 — — 736 — — 71~81 103 — — 1422 — —1028 — — 750 750 — 72~82 548 — — 750 750 —938 — — 750 750 — 74~84 458 — — 750 750 —1230 — — — 1334 — 75~85 331 — — — 720 —591 — — — 750 — 80~90 159 — — — 750 —482 — — — 687 — 82~92 130 — — — 893 —389 — — — 720 — 85~95 105 — — — 1334 —1208 — — — — 1314 86~96 325 — — — — 716871 — — — — 992 88~98 235 — — — — 682525 — — — — 707 92~102 141 — — — — 817410 — — — — 694 95~105 111 — — — — 1165
表710 宽范围固相pH梯度配方
酸性重液的体积/μl
02mol/LImmobilinepK
36 46 62 70 85 93
pH范围
碱性轻液的体积/μl
02mol/LImmobilinepK
36 46 62 70 85 93
299 223 157 — — — 35~50 212 310 465 — — —569 99 439 — — — 40~60 390 521 276 — — 722415 240 499 — — — 45~65 — 570 244 235 — 29769 428 414 — — — 50~70 — 474 270 219 — 320— 450 354 113 — — 55~75 347 — 236 287 284 —435 — 323 208 44 — 60~80 286 — 174 325 329 —771 — 276 185 538 — 65~85 192 — 153 278 362 —1349 — — 272 372 845 70~90 484 — — 232 189 546668 — — 445 226 348 75~95 207 — — 925 139 346399 — — 364 355 94 80~100 91 — — 329 366 289

192
续表
酸性重液的体积/μl
02mol/LImmobilinepK
36 46 62 70 85 93
pH范围
碱性轻液的体积/μl
02mol/LImmobilinepK
36 46 62 70 85 93
578 110 450 — — — 40~70 302 738 151 269 — 876702 254 416 133 346 — 50~80 175 123 131 345 346 —
779 — 402 93 364 90 60~90 241 — 161 449 237 225542 — — 378 351 — 70~100 90 — 324 350 280 —
588 254 235 117 170 — 40~80 — 554 360 142 334 288830 582 218 138 795 122 50~90 — 249 263 212 292 230941 — 273 243 260 282 60~100 100 — 333 361 239 326829 235 232 22 250 221 40~90 147 424 360 296 71 663563 463 298 273 227 127 50~100 21 59 34 420 310 2731102 — 455 89 334 — 40~100 — 114 50 488 157 357
745 固相pH梯度等电聚焦方法
固相pH梯度等电聚焦由于使用高电压,因此都采用水平电泳方式。
图75 梯度灌胶器示意
7451 制胶
固相pH梯度凝胶的制胶过程比载体两性电解质凝胶复杂,而与聚丙烯酰胺
梯度凝胶相似。固相pH梯度等电聚焦时,一般T=5%、C=3%。
① 根据实验选择合适的pH范围,并计算缓冲组分和滴定组分的体积。
② 配制T为30%,C为3%的丙
烯酰胺单体贮液 (注意有毒)。
③ 如 图75所 示 组 装 灌 胶 模 具,注意梯度 混 合 器 的 腔 间 阀 和 流 出 管 的
夹子都要关闭。
④ 配制凝胶溶液。
⑤ 灌胶。
a将75ml碱 性 轻 液 (视 凝 胶 大
小而定)注 入 梯 度 混 合 器 的 储 液 腔,小心 打 开 腔 间 阀,赶 去 腔 间 气 泡,再
关上腔 间 阀,并 将 流 入 混 合 腔 的 碱 性
轻液吸回储液腔;
b将同样体积的酸性重液注入 梯
度混合 器 的 混 合 腔,并 通 过 补 偿 棒 使

193
两腔液面相平;
c将流出管插 入 灌 胶 模 具 中 央,其 末 端 距 梯 度 混 合 器 出 口 高 度 相 差5cm,开启磁力搅拌器;
d于两腔中各自先加入TEMED,再加入AP,打开流出管的夹子,当酸液
流到流出管的一半时,打开腔间 阀,则 两 腔 液 面 同 时 下 降,在 混 合 腔 梯 度 混 合
后,缓慢注入垂直平板间;
e灌胶完毕后,室温静置5~20min,于50℃烘箱中聚合。灌胶后立即洗净
梯度混合器。
⑥ 凝胶聚合后,取出凝胶,标上正负极;将凝胶称重后放入重蒸水中,室
温下60次/min振摇6×10min或3×1h,以洗去催化剂和未聚合的单体;吸去
胶面水分,用冷风吹胶至原重。
7452 等电聚焦
① 同载体两性电解质等电聚焦一样处理样品。
② 开启电泳槽循环水浴,设置冷却温度为10℃。
③ 将制好的固相pH梯度凝胶按正负极方向铺在冷却板上,注意在凝胶和
冷却板之间涂以液体石蜡或煤油,以防止气泡产生,否则接触不好。
④ 将合适的电极液 (表711)润湿滤纸电极条,分别放置于凝胶的酸、碱侧。
表711 固相pH梯度等电聚焦电极溶液
阳 极 液 阴 极 液
重蒸水① 重蒸水①
03%~1%载体两性电解质② 03%~1%载体两性电解质②
10mmol/L谷氨酸 10mmol/L赖氨酸
① 适合宽pH范围凝胶和高盐浓度的样品。
② 使用相同或窄于固相pH梯度pH范围的载体两性电解质。
⑤ 上样,样品可加在凝胶的任意位置,但应避免加在紧靠两极或靠近等电
点的位置。
⑥ 将白金电极分别放在滤纸电极条的中心,再将阳极和阴极分别与电源的
正、负极相连,接通电源,进行等电聚焦电泳 (表712)。
表712 固相pH梯度等电聚焦电参数
参 数 第一相预聚焦 第二相聚焦 第三相聚焦
电压/V 500 3500 3500电流/mA 1 5 2功率/W 1 10 5时间/h 1~4 12~16 2

194
7453 等电聚焦后的处理
(1)测定pH梯度 由于固相pH梯度凝胶导电性很低,所以不能用表面电
极测定pH。对于宽范围pH 梯度,可利用等电点标准来测定;对 于 窄 范 围pH梯度,可依据其pH梯度范围根据线性关系推算pH及等电点。
(2)检测 凝胶的染色、干燥、保存、扫描等与载 体 两 性 电 解 质IEF及 常
规PAGE法相同。
746 等电聚焦的应用范围
可测定蛋白质的等电点,对蛋白质组分进行分离,分析、纯化,定性、定量
以及制备。
75 双向电泳
双向电泳就是将样品电泳后,在它的垂直方向再进行第二向电泳。目前的双
向电泳大都是第一向为等电聚焦,第二向为 梯 度SDSPAGE。这 样 经 过 电 荷 和
质量的两次分离后,就可以得到蛋白质等 电 点 和 分 子 量 的 信 息,电 泳 结 果 不 是
带,而是点。这是目前电泳技术中分辨率最高,信息最多的技术。
751 原理
双向电泳的原理与等电聚焦、梯度SDSPAGE完全相同,但是由于是两种
不同电泳体系的组合,为了达到很好的衔接,操作方法上与单向电泳不同,主要
是两向电泳之间的平衡。
752 双向电泳实验方法
双向电泳的第一向可以是载体两性电解质pH梯度IEF,也可以是固相pH梯度IEF。一般其第一向等电聚焦电泳采用水平板式,第二向SDSPAGE采用
垂直或水平板式。
7521 第一向为载体两性电解质pH梯度IEF的双向电泳实验方法
在双向电泳中,需在第一向载体两性电解质pH梯度IEF中加入高浓度尿素
和非离子去污剂 (如Triton、NP40),在样品溶解液中还需再加入二硫苏糖醇。这些试剂不带 电 荷,因 此 不 会 影 响IEF电 泳,但 却 能 使 蛋 白 质 分 子 充 分 伸 展,从而有利于 以 后 与SDS充 分 结 合 形 成SDS蛋 白 质 复 合 物,便 于 第 二 向SDSPAGE的顺利进行。
由于两种电泳体系组成成分及pH不同,在进行第二向电泳之前,应把第一
向电泳后的胶条放在第二向电泳缓冲液中震荡平 衡,以 驱 除 第 一 向 凝 胶 中 的 尿
素、非离子去污剂及载体两性电解质,并使第二向缓冲体系中的β巯基乙醇和
SDS进入凝胶,以利于形成SDS蛋白质复合物。然后将平衡好的胶条封闭固定

195
在第二向凝胶上部进行第二向电泳。(1)等电聚焦电泳的制胶 同载体两性电解质pH梯度IEF中聚丙烯酰胺凝
胶的制胶方法 (表78),但需加8mol/L尿素和05%~2%非离子去污剂。(2)样品准备及加样 可溶蛋白质样品稀释到合适浓度后可直接加样,固体
样品需用含9mol/L尿素和2%非离子去污剂的溶液溶解后才可加样,加样位置
同IEF电泳。(3)等电聚焦电泳 等电聚焦方法同7432节和7452节所述,但电泳
温度应提高至15~20℃,以防止尿素结晶,另外还需适当降低电压和延长聚焦
时间。等电聚焦后依据743节所述方法测定pH梯度。(4)平衡 在第二向SDSPAGE前,需将等电聚焦凝胶剪成胶条,用平衡
液 (表713)平衡5~30min (视胶条厚薄而定)。
表713 双向电泳的平衡液
2巯基乙醇 20%SDS 甘油 pH=68,1mol/LTrisHCl 定容至
5ml 115ml 10ml 625ml 100ml
(5)梯度SDSPAGE的制胶 与SDSPAGE平板胶的制胶过程近似,但由
于分离胶是梯度胶,因此需使用梯度混合器制备分离胶。分离胶配方见表714,分离胶的灌胶过程与固相pH梯度IEF制胶法 (7451节)相同。然后将平衡
好的等电聚焦胶条放置于SDS聚丙烯酰胺凝胶上以进行两向的转移,注意避免
陷入气泡。需要的话可在SDS凝胶的一端加蛋白质分子量标准。
表714 梯度SDSPAGE分离胶的配方
储 液
梯度分离胶(C=3%)终浓度T
4%~225% 8%~18% 10%~225%
重液 轻液 重液 轻液 重液 轻液
30%丙烯酰胺单体储液/ml 13 75 27 6 33 75分离胶缓冲液储液/ml 25 25 25 25 25 2587%甘油/ml 28 28 28重蒸水/ml 34 20 15 1440%过硫酸铵/μl 9 9 9 9 9 9TEMED/μl 5 5 5 5 5 5
(6)SDSPAGE 同735节中SDSPAGE电泳方法。(7)检测 同前。
7522 第一向为固相pH梯度IEF的双向电泳实验方法
作为双向电泳第一向的固相pH梯度凝胶中必须含尿素、非离子去污剂、载

196
体两性电解质、二硫苏糖醇等,以便于第二向SDSPAGE的顺利进行,这些试
剂是在将制备好的干胶水化时加入的,在样品液中同样需含有这些试剂。在进行
第二向电泳 之 前,应 将 第 一 向 电 泳 后 的 胶 条 平 衡,以 适 应SDSPAGE的 电 泳
条件。(1)固相pH梯度凝胶的制备 同7451节。(2)固相pH梯度凝胶的重新水化 用新鲜配制的泡胀液 (表715)将 干
凝胶条水化过夜。
表715 泡胀液
尿素 Triton 载体两性电解质pH=3~10 二硫苏糖醇 重蒸水定容至 OrangeG或溴酚蓝
96g 01ml 01ml 40mg 20ml 少许
(3)样品处理和加样 可溶性样品可直接用于双向电泳。必要时可用裂解液
(表716)裂解样品,之后再用多 倍 体 积 的 样 品 液 (表717)溶 解 裂 解 后 的 样
品。当样品中含有高盐或SDS时,需要用更多的样品液溶解样品。
表716 裂解液
尿素 TritonX100 β巯基乙醇/二硫苏糖醇 载体两性电解质pH=3~10 PMSF 重蒸水定容至
27g 01ml 01ml/100mg 01ml 7g 5ml
表717 样品液
尿素 TritonX100 β巯基乙醇/二硫苏糖醇 载体两性电解质pH=3~10 重蒸水定容至 溴酚蓝
27g 0025ml 01ml/100mg 01ml 5ml 少许
(4)等电聚焦 同前。(5)平衡 在进行第二向电泳之前,将第一向电泳后的凝胶先用第一步平衡
液平衡10min,再用第二步平衡液平衡10min,吸去多余平衡液 (表718)。
表718 平衡液
平 衡 液 pH=68,1mol/LTrisHCl 尿素 甘油 SDS 二硫苏糖醇 碘乙酰胺 重蒸水定容至
第一步平衡液 20ml 72g 6ml 200mg 025~10g — 100ml第二步平衡液 20ml 72g 6ml 200mg — 45g 100ml
(6)梯度SDSPAGE的制胶 同7351节。(7)SDSPAGE 同735节SDSPAGE电泳方法。(8)检测 同前。
753 应用
双向电泳不仅能同时测定蛋白质的等电点、分子量,更重要的是它还可用于

197
了解蛋白质混合物的组成及变化,如不同细胞、亚细胞中的蛋白质组成及含量差
异,从而在病理研究、组织培养以及生物进化等领域中的应用越来越多。
76 免疫电泳
免疫电泳技术是基于抗原的电泳迁移以 及 抗 原 与 抗 体 的 专 一 性 免 疫 沉 淀 反
应。在大部分电泳中,抗体平均分布在pH=86的琼脂糖薄层凝胶中,此时它
不带电,不能迁移;而样品中的抗 原 此 时 带 强 的 负 电,当 它 在 凝 胶 中 电 泳 迁 移
时,就会与抗体发生免疫反应。最初抗原过量,于是产生可溶性的免疫混合物而
向阳极迁移;直至抗原与抗体的浓度达到 当 量 点,此 时 就 形 成 不 溶 性 的 免 疫 沉
淀,沉淀点的高度或面积与抗原的量成正比。
761 原理
抗原和抗体的专一性反应是各 种 免 疫 方 法 的 基 础。目 前 有 许 多 免 疫 分 析 技
术,简述如下:
7611 单向免疫扩散技术
单向扩散技术是最简单的免疫技术,可用于单抗原的定量测定,但这不是电
泳技术。抗原被加在含有单特异性抗体的琼脂糖薄层凝胶的孔中,并充分扩散;当扩
散的抗原浓度与凝胶中的抗体浓度达到当量点时,就 在 孔 的 周 围 形 成 一 个 沉 淀
圈,圈的面积与抗原的量成正比,从而可测定特定抗原的浓度 (图76)。
图76 单向免疫扩散技术 图77 双向免疫扩散技术
7612 双向免疫扩散技术
双向扩散技术可用于测定产生免疫沉淀的正确的抗原抗体浓度。它也不是
电泳技术。在琼脂糖凝胶板中打孔,然后在中央孔中加入特定浓度的抗原/抗体,在周
围的孔中加入连续稀释的抗体/抗原,于是抗原和抗体在凝胶中扩散,当扩散至
当量点时就形成沉淀线。如果沉淀线位于两孔的中间则表明抗原和抗体的浓度适
宜;如果沉淀线偏向一方,则表明远离方过量 (图77)。
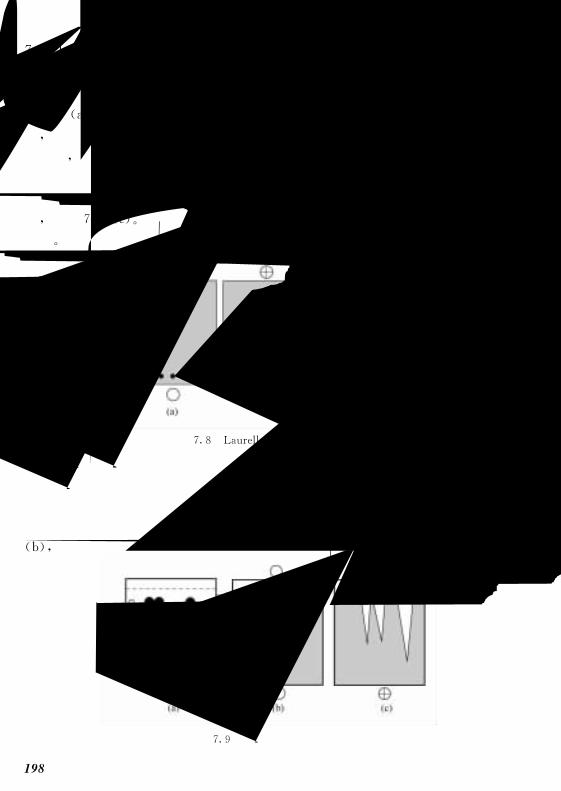
198
7613 “火箭”免疫电泳
Laurell火箭电泳又称单向定量免疫电泳,可用于快速测定单个抗原的浓度。抗原样品被加入到含有单专一性抗体的pH=86的琼脂糖凝胶的孔中,如
图78 (a)。在电场下,抗原 向 正 极 迁 移,走 在 前 面 的 抗 原 与 凝 胶 中 的 抗 体 相
遇,从而形成免疫沉淀而停止迁移;走在后面的抗原继续迁移,当与免疫沉淀物
相遇时,由于抗原过量而使免疫沉淀溶解并再次迁移;可溶性免疫混合物进入新
的凝胶内后再度与抗体相遇而产生新的免疫沉淀物,如图78 (b)。如此不断地
沉淀溶解再沉淀,直至全部抗原都与抗体结合而不再迁移,并形成火箭形沉淀
线,如图78 (c)。火箭峰 的 高 度 与 所 加 抗 原 的 量 成 正 比,由 此 可 测 定 抗 原 的
浓度。
图78 Laurell火箭免疫电泳示意
图79 交叉免疫电泳示意
7614 交叉免疫电泳
交叉免疫电泳是常规琼脂糖凝胶电泳和火箭免疫电泳的两向组合。首先抗原
混合样在常规琼脂糖凝胶中进行第一向电泳分离,如图79 (a),然后此第一向
凝胶条在含有抗 体 的 琼 脂 糖 凝 胶 中 以 垂 直 方 向 进 行 第 二 向 火 箭 电 泳,如 图79(b),从而使每个经第一向分离的抗原组分形成火箭峰,如图79 (c),每个峰

199
的面积与抗原的量成正比,因此交叉免疫电泳可用于对抗原样品进行定性定量。
7615 GrabarWilliams免疫电泳
GrabarWilliams免疫电泳用于在复杂混合物中检测一种或几种抗原。首 先
抗原混合样在常规琼脂糖凝胶中被电泳分离 [图710 (a)],然后抗血清加在与
迁移方向平行的凹槽中 [图710 (b)],凝胶中的抗原与凹槽中的抗体分子双扩
散,形成弧形沉淀线,从而检测出样品中的抗原 [图710 (c)]。
图710 GrabarWilliams免疫电泳
762 电泳方法
7621 溶液的配制
pH=86Tris巴比妥缓冲液:224g二乙基巴比妥酸,443gTris,0053g乳酸钙,0065g叠氮钠,用重蒸水溶解定容至100ml。
1%琼脂糖溶液:称取1g琼 脂 糖,用 上 述 适 当 稀 释 后 的 缓 冲 液 加 热 煮 沸 溶
解,定容至100ml。若 要 在 凝 胶 中 加 抗 体,则 将 融 化 的1%琼 脂 糖 溶 液 冷 却 至
55℃后 (温度不可太高,以防抗体变性、失活;也不可太低,以防琼脂过冷无法
铺板),再加适量抗体,小心搅拌混匀,并即刻倒胶。凝胶中所加抗体的量要合
适,以保证抗原、抗体的比例适当,从而形成完整清晰的沉淀峰,峰高度宜控制
在2~5cm。
7622 制胶
将配好的 (抗体)琼脂糖溶液倒入水平放置的玻璃板上,待其冷却凝固后即
可打孔。
7623 打孔和加样
用合适直径的打孔器打孔,然后用微量注射器在孔内加入抗原样品,注意勿
使样品溢出凝胶表面。
7624 电泳
在电泳槽内加入电极缓冲液 (pH=86Tris巴比妥缓冲液四倍稀释),将抗
原孔端接电泳仪负极,另一端接正极,以电场强度10~20V/cm进行水平电泳。

200
7625 检测
电泳结束后,可直接观 察 沉 淀 线 以 了 解 实 验 结 果,若 要 提 高 沉 淀 线 的 可 见
度,可用考马斯亮蓝染色。将凝胶用09%生理盐水分数次浸泡2天,以洗去未反应的抗体、抗原等蛋
白质;然后在琼脂板上不断压干滤纸,使凝胶干燥,并自然吹干,将琼脂板浸入
考马斯亮蓝染色液中染色;最后用脱色液脱色直至背景无色。
763 应用
由于免疫电泳是基于抗原、抗体间的免疫反应,因此主要用于抗原、抗体的
分离、分析、定性、定量研究。
77 蛋白质印迹
蛋白质印迹就是把从电泳或色谱分离得到的蛋白质转移到固定基质上,以便
用专一性免疫技术探测固定膜上的蛋白质。前文所述的各种电泳方法虽然分辨率
很好,但其进一步检测分析却受到限制,因为生物大分子被嵌在凝胶结构中,难
以让探针分子接近。而将电泳结果转移到固定膜上,就能容易地让探针分子接近
蛋白质,且蛋白带在膜上不会因扩散而降低分辨率。另外,固定膜的操作也相对
更方便、省时,因此能更有效地分析电泳结果。蛋白质印迹法 (Westernblotting)实 际 上 是DNA印 迹 法 (Southernblot
ting)和RNA印迹法 (Northernblotting)的发展。
771 原理
7711 蛋白质印迹的过程
蛋白质印迹一般分为两个步骤:转移和检测。转移就是将蛋白质从凝胶转移
到印迹膜上;检测又分为三步:先用封阻试 剂 封 阻 固 定 膜 上 未 吸 附 蛋 白 质 的 区
域;再用探针检出固定膜上感兴趣的蛋白质;最后用探针上的标记物显示感兴趣
的蛋白质分子。
7712 蛋白质印迹的主要类型
根据转移方法的不同,蛋 白 质 印 迹 主 要 分 为 扩 散 印 迹、毛 细 管 印 迹 和 电 泳
印迹。扩散印迹时,凝胶放在两张固定膜之间,浸在缓冲液中自由扩散2~3天后,
就得到两张互为镜像对称的转移膜,每张膜得到一半蛋白质 (图711)。这种方
法适合于大孔凝胶,虽然简便但费时,而且分辨率降低。毛细管印迹来源于DNA印迹法,在凝胶上放一张固定膜及一叠干滤纸,其
上压以1~2kg重物,借助毛细管作用,当缓冲液流从储液腔流经凝胶和固定膜

201
到滤纸上时,凝胶中的蛋白带随缓冲液流而转移到固定膜上 (如图712)。毛细
管印迹适合于低分子量蛋白质和大孔凝胶。电泳印迹是用电场将凝胶中的 蛋 白 带 转 移 到 印 迹 膜 上,它 需 要 特 定 的 装 置
(图713),转移效率高,转移条件 易 控 制,且 能 保 持 原 有 的 分 辨 率。这 是 目 前
最广泛使用的印迹方法。
图711 大孔径凝胶的双向扩散印迹
图712 毛细管印迹 图713 印迹转移电泳装置
772 试验材料的选择
蛋白质印迹就是把电泳后的蛋白带转移到印迹膜上,以便于进一步分析,因
此蛋白带的转移过程非常重要,影响转移的因素主要有固定膜和转移缓冲液。转
移后,可用不同的方法对印迹膜进行封阻、标记和检测。
7721 固定膜的选择
固定膜应该易与蛋白质分子结合,且不影响随后的检测。目前常用的固定膜
有硝酸纤维素膜、尼龙膜和重氮化纸。硝酸纤维素膜是目前应用最多的膜,其价格便宜、使用方便、无需活化,对
蛋白质的结合能力较高,平均为80μg蛋白质/cm2;而且背景浅,可用各种染色
方法检测。它有045μm、02μm和01μm几种规格,小于02μm的小孔膜适
于转移分子量小于20000的蛋白质。

202
尼龙膜对蛋白质的结合能力更高,达480μg蛋白质/cm2,机械强度也更高;但它的背景很高,封阻过程烦琐,不能用阴离子染料如考马斯亮蓝等染色,而且
价格比较昂贵,因此使用较少。尼龙膜适合于小分子量蛋白质、酸性蛋白质、糖
蛋白的印迹转移。重氮化纸主要有重 氮 苯 氧 甲 基 (DBM)和 重 氮 苯 硫 醚 (DPT)纤 维 素 纸,
这类纸在临用前需进行重氮化处理,其优点是蛋白质共价结合到纸上,因此可作
多次探测,但其重氮化基团不稳定,且与蛋白质的结合能力较低。
7722 转移缓冲液的选择
转移缓冲液的离子强度、pH及其稳定性影响着印迹转移的效果。为了减少
产热,缓冲液的离子强度应适当 降 低,以 便 能 提 高 电 压,加 快 蛋 白 质 的 转 移 速
度。其pH应远离蛋白质pI,以保证蛋白质有最大的可溶性。蛋 白 质 印 迹 转 移
时常用Tris缓冲体系,如Tris甘氨酸缓冲液等,因为它比较稳定。
773 蛋白质印迹试验方法
这里仅介绍电泳印迹实验方法,即电转移过程。
7731 转移单元的组成
转移缓冲液:156mmol/LTris,120mmol/L甘氨酸,用重蒸水配制。有时
图714 转移单元的组成
转移缓 冲 液 中 加 入20%甲 醇,以 增 强
蛋白 质 与 硝 酸 纤 维 素 膜 的 结 合 能 力,并防 止 凝 胶 变 形,但 甲 醇 会 降 低 某 些
蛋白 质 的 转 移 效 率。转 移 缓 冲 液 中 可
加入 低 浓 度SDS (≤01%),以 增 加
转移效率。将电泳后的凝胶剥离下来,固定膜
和凝胶在转移缓冲液中浸泡15~20min,如图714所示,组成转移单元。注意固
定膜应放在凝胶的正确侧,操作时一定要戴手套,每层之间都要避免气泡。
7732 电转移
将转移单元置于转移装置内,以08mA/cm2电流转移05~1h,转 移 时 间
与凝胶孔径、蛋白质分子大小、固定膜及转移缓冲液等有关。
7733 检测
转移后,小心地从固定膜上剥离凝胶,就可以对膜进行染色或用其他方法检
测膜。大部分电泳用染色法可用于膜的染色,如氨基黑法、印度墨水法等。检测
膜时应首先将膜上未吸附蛋白质的区域封闭,常用的封阻试剂有2%~5%牛血

203
清白蛋白、1%白明胶、005%~03%去污剂等;然后用专一性探针标记膜上的
印迹蛋白,最常用的专一探针为抗体;接着再用二抗标记一抗,而二抗上标记有
酶或其他显色系统,如辣根过氧化物酶、碱性磷酸酶、胶体金、125I等;最后通
过二抗上的标记物的显色反应检测出印迹膜上的印迹蛋白质。
774 应用
蛋白质印迹主要用于对极微量蛋白质进行分析、纯化、鉴定、复性等,如氨
基酸的组成分析、序列分析、特异性结合分析等。
78 毛细管电泳
前文所介绍的凝胶电泳技术虽然应用广泛,但是分离时间长、效率比较低而
且难以实现自动化。采用毛细管进行电泳分离,由于毛细管自身的抗对流作用,使得凝胶介质不再是必需的,特别是可以对样品进行快速、高效分离,能够实现
在线检测和自动化操作,因此它已成为一种高效分离技术,并常与质谱等分析仪
器联用,实 现 对 样 品 的 快 速、高 效 分 离 分 析。毛 细 管 电 泳 的 分 离 原 理 同 常 规
电泳。毛细管电泳的主要特点是柱效更高,分析更快,操作灵活、多样。在毛细管
电泳中,扩散对分辨率的影响可以忽略,而毛细管内的电渗、样品同管壁的吸附
作用则是影响分离效率的两个主要因素。由于毛细管内壁表面带有离子化基团,使得管壁不同程度地吸引样品分子,造成非均一性电渗流而影响分离。为此,可
以通过使蛋白质与管壁的硅羟基都不带电或都带负电荷来改善这种吸附和电渗情
况,但更有效的解决方法是对毛细管内壁进行改性。因此在毛细管电泳中,毛细
管电泳柱的制备是一个重要环节,其制备主要包括键合涂层和吸附涂层。通过对
石英毛细管内壁进行涂层处理,能有效地减少毛细管内壁对蛋白质的吸附作用,从而抑制电渗,提高柱效,改善分离效果,如常见的硅烷化处理。
毛细管电泳的另一主要特点是它能实现在线检测,其检测器主要有紫外可
见光检测器、荧光检测器和激光诱导荧光检测器,三者的检测灵敏度不断提高,但后两者需对样品进行衍生化处理。
毛细管电泳的类型主要有三种:毛细管区带电泳、毛细管等电聚焦电泳、毛
细管凝胶电泳。毛细管电泳一般包括三个步骤:电泳柱的制备,平衡;进样;电
泳分析。其电泳柱的制备方式不同于前面所介绍的任何电泳;其进样方式也不同
于常规电泳,而采用流体力学方式,与 HPLC的相似;其电泳分析也不同于常
规电泳的染色后分析,而采用在线分析。具体的实验方法这里不介绍了,可以参
考有关文献。

204
8 特殊用途蛋白质的纯化
81 测序用蛋白质的纯化
蛋白质纯化的目的除了研究蛋白质特性外,在分子生物学中最初主要用于蛋
白质测序,以确定蛋白质的一级结构。随着重组DNA技术的应用,蛋白质测序
的作用已经有所变化。由于DNA测序比较简单快速,大分子或者稀有蛋白完整
的一级结构通常可以通过cDNA序列的翻译而比较容易确定。但在许多应用中,仍然需要测定一些蛋白质的序列,如用于基因克隆的寡核苷酸探针的合成,拓扑
结构的分析,蛋白质加工以及同工酶的确定等。而且近来蛋白质结构和功能区的
重要性也越来越受到关注,这些区域经常是共价修饰的位点。所有这些应用中,蛋白质测序是测定一级结构氨基酸修饰位点的惟一方法。
目前的蛋白质序列分析方法都是定量的,因此可以分析多肽 或 蛋 白 质 样 品
的纯度,也可以确定 多 亚 基 复 合 体 的 亚 基 数。蛋 白 质 测 序 一 般 有 两 类 方 法:
Edman降解法和应用 高 速 原 子 轰 击 (fastatombombardment,FAB)的 质 谱 分
析法。两种方法各 有 优 缺 点,传 统 的Edman降 解 法 可 以 手 工 操 作,利 用 苯 异
硫氰酸酯或者与二甲氨基偶氮苯异硫氰酸酯或荧光异硫氰酸酯合用测 序。一 般
含量分别低于1nmol或20pmol的蛋白质或多肽都能被测序,得到约20个 氨 基
酸残基的 可 信 序 列 长 度。也 可 利 用Edman降 解 法 自 动 测 序,其 灵 敏 度 很 高,可低于10pmol,1nmol的初始样 品 可 以 测 得50~80氨 基 酸 残 基。此 外 利 用 固
相测序法,多肽或蛋白质共价连接到固相载体上,可以降低测序反应中 的 损 失
和化学污染。“气相”或液体 脉 冲 测 序 仪 由 于 其 灵 敏 度 的 提 高,代 替 了 液 体 旋
杯测序仪。这些仪器 都 是 将 多 肽 或 蛋 白 质 非 共 价 吸 附 到 惰 性 载 体 上 而 进 行 工
作的。利用FAB的质谱分析法相对于Edman降解法具有某些优点。如 N末端阻
断的多肽也可以测序。因为可以根据碳水化合物结构得到序列信息,糖蛋白也可
通过一步反应按照蛋白质进行处理。而且含有多至5种多肽的混合物也可以准确
的测序。但是,FAB质谱分析法不能直接用于完整蛋白的测序,必须首先产生
较短的片段。且灵敏度稍低于Edman降解法。本节主要讨论用于测定氨基酸序列的蛋白质纯化过程中一些注意事项以及经
常在测序前所采用的一些纯化技术。

205
811 测序蛋白纯化技术
8111 一般注意事项
有关文献所描述的蛋白质和多肽的纯化 方 法 一 般 都 涉 及 手 工 或 自 动 序 列 分
析。当制备测序用蛋白样品时,需要注意以下一些问题,这些限制因素更多的可
能与Edman降解测序法有关,而FAB质谱法也有其自身的污染等问题。污染物主要来源于试剂中的多种小分子有机和无机化合物。为避免杂质对N
末端的阻断,整个纯化过程中高品质试剂 (分析纯以上)以及采用温和反应条件
是非常重要的。例如,如果尿素溶液中含有氰酸根离子,这些离子会与氨基反应
而影响测序,而在高品质的尿素溶液中其含量较低。为了更谨慎起见,尿素溶液
应在使用前配制,并经过离子交换树脂纯化。这些树脂均有商品,可由厂商 (如
BDH或BioRad)提供。此外,加入20mmol/L的甲胺可能有助于测序,应避免
使用一些会与氨基产生反应的试剂。有一些可能会引起污染而应避免的小分子物质,其中含有伯胺的化合物通常
是最麻烦的,经常会破坏整个测序分析。在一些利用载体吸附的固相测序中,这
类物质会非常有效地与蛋白质和多肽竞争。而在其他自动和手动测序中,它们也
会与Edman降解试剂苯异硫氰酸酯反应,从而导致产率的降低,严重时会产生
明显的重叠问题。一般序列重叠是由一次测序反应循环期间的部分反应引起的,这样从此次循环之后会产生交错的顺序,至少在随后的循环中会观察到两个相同
的氨基酸。最常见的氨基杂质可能是铵盐,特别是碳酸氢铵,该物质通常认为可通过冻
干法完全除去,但是许多研究人员发现该方法不能达到完全去除的目的。其他一
些应避免使用的常用化合物 包 括Tris、嘧 啶、甘 氨 酸、二 羟 乙 基 甘 氨 酸、氨 基
糖、多缓冲体系、两性电解质以及大多数的去污剂。虽然测序 (特别是利用固相
法测序)仍然要在这些物质存在的条件下进行,但污染的存在严重降低了测序程
序的有效性。如果用FAB质谱分析样品,应该除去大多数盐,因为它们可能会
与甘油以及常用于该技术的载体形成所不希望的 加 合 物。大 分 子 污 染 物 (如 磷
脂、碳水化合物和核酸)一般较少遇到,但是它们也会带来特定的问题,必须将
其去除。
8112 最终纯化步骤
除去可能会干扰各类测序程序杂质的方法有很多,以下作一简要介绍。方法
的选择很大程度上取决于测序材料的性质和数量,一 般 应 特 别 注 意SDSPAGE的简便有效性。
(1)透析 水透析在许多情况下是很有效的。有些固相测序过程中,01%~025%的SDS与蛋白质一起保留在溶液中不会对测序产生不利影响 (而实际上可

206
能有助于蛋白质与树脂的结合)。如果要求除去SDS,通常可以通过20%~30%乙醇或2丙醇透析来完成。对于含量少于1nmol的样品,除非采用一些微量 透
析系统,一般不用透析。气相或液相脉冲仪也应避免使用SDS。(2)反相色谱 SepPak试剂盒对此方法比较有效,而且操作简便。蛋白质
吸附到树脂上,而盐和杂质可水洗除去,再用乙腈或甲醇回收蛋白。反相 HPLC对于得到纯样品是一种非常有效的方法。有机溶剂 (特别是高
纯度乙腈)加入001%~01%三 氯 醋 酸 效 果 很 好,样 品 可 通 过 冻 干 除 去 溶 剂
(注意再溶解时溶液可能呈酸性)。如果蛋白质可被低离子强度溶液洗脱,疏水色
谱对于除去两性电解质、多缓冲体系和盐类也是非常有用的。(3)有机溶剂沉淀 对于清洗样品,用10~20倍体积冰丙酮,或5~10倍
体积冰乙醚,或醚醇混合液 [体积比 (1∶1)~(3∶1)]沉淀和洗涤也是一种快
速有效的方法,特别是要求除去去污剂时,不建议使用三氯醋酸。(4)凝胶过滤色谱 利用SephadexG10和G25或者BiogelP2和P4以
1%~5%的 醋 酸 或 甲 酸 溶 液 脱 盐 也 是 一 种 有 效 的 方 法。当 体 系 中 含 有 膜 蛋 白
和/或去污剂,而通常又需要将这些物质作为单体保留在溶液中时,可以将甲酸醋酸氯仿甲醇 (体积比1∶1∶2∶1)混合液加入到SephadexLH60和LH20树脂中。一般不允许加热干燥这些溶液中的样品,否则会导致过多的 N末端甲
酰化。此外,HPLC凝胶渗透柱,比如TSK系列 (Anachem,Beckman,Pharma
ciaLKB公司)或者GF系列 (DuPont公 司)也 可 以 采 用。一 般 最 好 避 免 采 用
凝胶过滤,除非蛋白质或多肽数量为纳摩尔级。(5)聚丙烯酰胺凝胶电泳 (PAGE) 除去少量污染物,特别是大分子物质
的一种快速有效的方法是进行十二烷基硫酸钠 (SDS)聚丙烯酰胺凝胶电泳,以
及电洗脱或电渗析等。为避免阻断N末端,使用高品质的试剂配制凝胶尤为重
要。一般应使用电泳级 试 剂 (如BDH),理 想 的SDS应 在 温 水 溶 液 中 重 结 晶。电泳缓冲液中加入02mg/ml的硫乙醇酸也有作用。
简单的电洗脱操作程序不需要复杂的设备,方法如下:
① 考马斯亮蓝凝胶染 色 (5~10min)后,于 脱 色 液 中 漂 洗10~15min,直
至出现蛋白质条带,为保证收率,染色和脱色时间应该尽可能短。完整切下所需
条带。
② 将 缓 冲 液 [25mmol/LTris甘 氨 酸,pH=85,01% (1g/L)SDS;
50mmol/LTris醋酸,pH=78,01%SDS;01mol/L碳 酸 氢 钠,pH=78,
01%SDS或01mol/L磷酸钠,pH=78,01%SDS]加入到平板电泳槽中,液面高出凝胶架大约1cm。如需要,缓冲液中可加入01% (体积分数)的2巯

207
基乙醇或2~5mmol/L的二硫苏糖醇。对于膜蛋白,缓冲液的SDS浓度可以提
高到1% (10g/L)以确保溶解。
③ 取长度足以装入凝胶切片的透析袋,一端留出约2cm,用夹子夹住,将
电泳槽中的缓冲液加入袋中。凝胶切片放 入 袋 中 后,封 口 前 轻 轻 挤 出 大 部 分 液
体。将凝胶切片置于透析袋一端。注意避免带入气泡。
④ 将透析袋置于电泳槽凝胶架上。调节缓冲液液面,使其恰好淹没透析袋。
25~100V直流 (20~150mA)电泳3~20h,然后反向电泳大约30s(以便使聚
于透析膜表面的蛋白质脱离透析膜)。
⑤ 将凝胶切片从透析袋中除去。重新染色检查蛋白质是否被洗脱。所有残
留的小凝胶片段也必须除去。通常将清液转移,离心或者过滤以去除所有残片。
⑥ 透析袋重新封口,将蛋白质溶液在蒸馏水中透析数遍,透析液可含最多
025% (25g/L)的SDS,以保持蛋白质溶解,但仅限于最后一次蒸馏水透析。考马斯亮蓝染料应与蛋白质充分接触后才可作为电洗脱的指示剂。
对于固相蛋白测序仪,蛋白质可以从凝胶上直接电泳转移到衍生化玻璃纤维
纸或聚二氟乙二烯 (PVDF)上。未预先活化的PVDF膜可用于气相仪。转移后
的蛋白质经考马斯亮蓝或荧光染料显影,将显色斑或显色条带切下后可直接放入
测序仪中。
812 多肽的制备和纯化
由于多种原因,经常要将蛋白质转变为多肽用于测序。比如需要关于制备基
因克隆DNA探针的 序 列 信 息 时,含 有 甲 硫 氨 酸 或 色 氨 酸 的 多 肽 可 能 就 非 常 有
用,因为这 些 氨 基 酸 只 由 一 个 三 联 体 碱 基 密 码 所 编 码。含 色 氨 酸 的 多 肽 可 在
280nm吸光 度 下 检 测。理 想 情 况 下,色 谱 洗 脱 液 可 用 双 通 道 检 测 器 (设 定 为
220nm和280nm)或二极管检测器 (测定各峰的吸收光谱)检 测。含 甲 硫 氨 酸
的多肽可用 氨 基 酸 分 析 或 特 定 标 记 测 定。30个 氨 基 酸 残 基 以 内 的 多 肽 需 要 用
FAB质谱以得到较好的测序结果。制备和纯化这类多肽的常用方法如下文所述,也可参见有关文献。
对于制备分子量较小的多肽有许多方法,首先要使蛋白还原并且不可逆修饰
所有的半胱氨酸残基。这样可防止二硫键的形成或交换,有利于随后测序中半胱
氨酸的确定 (未修饰的半胱氨酸会转变为难以检测的产物)。通常使用的试剂包
括碘乙酸和2乙烯 吡 啶。还 原 和 巯 基 修 饰 的 方 法:将 蛋 白 质 溶 解 于 不 超 过1ml的6mol/L氯化胍或含8mol/L去离子尿素的05mol/L的TrisHCl,pH=80~85,1mmol/LEDTA的溶液中,也可加入不超过1%的SDS以保证蛋白质的溶
解。加入10μl的2巯基乙醇或DTT,浓度达到10mmol/L,N2 环 境 暗 室20~35℃保温1~3h。加入无色碘乙酸或碘乙酰胺 (250mg/ml溶于01mol/LNaOH

208
溶液,体积不超过01ml)或4乙烯吡啶 (25mmol/L乙腈溶液),N2 环境暗室
室温分别保持20min。丙酮沉淀,透析,凝胶过滤回收蛋白。用于制备小分子多肽最常用的 方 法 是:①在 甲 硫 氨 酸 残 基 后 溴 化 氰 化 学 裂
解;②在赖氨酸或精氨酸残基后利用胰蛋白酶,或谷氨酸残基后利用StaphylococcusaureusV8内切蛋白酶GluC酶法降解。还有一些其他的化学方法和酶法
降解。利用各种酶,如内切蛋白酶ArgC、AspN、GluC以及LysC进行有限的
蛋白质降解,可以得到N末端阻断的蛋白质序列数据 (表81)。含有最多1%SDS的电洗脱蛋白液可以进行蛋白质降解,该方法可与SDSPAGE联用。降解
后得到的多肽通过SDSPAGE分离回收。
表81 降解方法
试 剂 条 件
内切蛋白酶ArgC 100mmol/LNH4HCO3 溶液,20~37℃保持8h。冷冻,煮沸或加入10mmol/L的
PMSF终止降解反应
内切蛋白酶AspN 50mmol/L磷酸钠,pH80溶液,20~37℃保 持18h。可 含 有 最 多1mol/L尿 素,
1mol/L氯化胍或001%SDS内切蛋白酶GluC 同ArgC,最多可含05%SDS内切蛋白酶LysC 同ArgC,最多可含01%SDS胰蛋白酶 同ArgC溴化氰(CNBr) 70% HCOOH溶液,最多可比 Met过量1000倍,N2 环境室温暗室保持24h。旋
转蒸发终止反应(注意:CNBr可形成有毒物质)
多肽混合物可借助反相或离子交换 HPLC或FPLC得到充分纯化。由于混
合物一般比较复杂,通常需要利用几种色 谱 方 法。首 先 可 利 用 的 是 离 子 交 换 色
谱,然后是反相色谱。此外,也可尝试在两个不同pH条件下的反相色谱。最终
的色谱分离 (反相体系)最好是在可挥发的缓冲液中进行,如01%的三氟醋酸
和乙腈溶液。这样多肽样品就可以利用离心浓缩器进行充分的真空干燥。以少量
体积溶液溶解多肽,一般1%的三氟醋酸乙腈 (50∶50,体 积 比)溶 液 对 大 多
数多肽都较适合,样品可转移到气相测序仪进行测序。对于固相测序,含有SDS的缓冲液可适用于联用方法。
82 用于晶体学研究的蛋白质纯化
在蛋白质生物化学研究早期中,结晶经常是作为一种纯化技术,结晶度也被
认为是纯度的一项指标。结晶学研究主要集中在一些经过选择的蛋白质,这些蛋
白质来源丰富,易于结晶。蛋白质结晶技术发展至今,完整的蛋白质三维结构分
析已不是一项很费时的工作。而实际上许多蛋白质研究的限制因素通常正是如何

209
制备适宜的晶体。用于结晶的蛋白质样品所要求的高纯度对于制备衍射级晶体常
常是最重要的因素之一,许多结晶试验由于蛋白质样品不如最初设想的那样具有
较高的均一性而陷入困境。通过结晶,蛋白质可以从含有许多杂质的混合物中纯
化出来,随着重结晶纯度的提高,晶体品质也会改善。而对于细菌、酵母、真菌
以及真核细胞表达系统得到的非常微量的蛋白质,已建立了一些晶体生长模式和
纯化方法,使得自然界中不会出现的或者含量非常低的分子或片段的结构研究也
成为可能。虽然晶体生长成功的报道很多,但 遇 到 的 许 多 问 题 却 没 有 完 整 的 记
录。无法得到持续生长的高品质晶体正是困扰结构分析的许多问题之一,同一批
次蛋白制备的结晶显然应该是相同的,但实际上可能常常得到品质不同的晶体,甚至根本得不到晶体。
蛋白质结晶的基本原理在许多方面与小 分 子 (盐 类 或 氨 基 酸)结 晶 原 理 相
同。缓慢获得低度过饱和蛋白质溶液是必需的,也就是说,溶剂中可能含有比平
衡状态下更多的溶解蛋白。这种亚稳态的蛋白质溶液通常可通过沉淀剂的逐步改
变,蛋白质的浓缩,或者pH或温度的变化而得到。当然,同时也必须考虑这些
变化可能会对蛋白质结构和活性产生的影响;对于膜蛋白,还必须注意这些变化
对去污剂状态产生的影响。最终的结晶条件应该是热力学最稳定的结晶状态。利
用透析、蒸汽扩散以及缓慢冷却或加热技术可以达到所要求的条件。在低溶解度
条件下,分子适当的聚集,缓慢地接近一个临界点,形成一个核,晶体从该核上
自发地持续生长。开始形成均匀晶核所要求的过饱和度 (也就是蛋白质聚集形成
的晶核)一般要比继续生长所要求的过饱和度高得多。这就解释了为什么过度的
成核过程可能经常会形成大量的小晶体,而形成少量的大晶体却非常困难。在低
过饱和度下,晶核的形成经常表现出非均一性,晶体在灰尘颗粒、纤维或者毛发
上开始生长,不管如何操作,似乎总可以得到结晶。了解检验均一性的重要性,对于考察蛋白质晶体特性及其形成过程是非常有
益的。蛋白质晶体是一种相当开放的三维立体晶格,每一重复的晶形就是单一的
或一组蛋白质分子。晶体体积很大一部分被水分子所占据 (30%~90%),只有
很小一部分的蛋白质表面与其他蛋白质分子相接触。与蛋白质表面紧密接触的水
分子与蛋白质之间,或者相互间以氢键网 络 的 形 式 排 列 在 一 起,而 在 晶 体 的 外
部,溶解槽中,其性质更像普通的水。蛋白质分子相互接触的作用形式非常类似
于可稳定蛋白质结构的形式,也就是多种弱作用力,包括氢键、离子键、疏水作
用,有时还有金属配位作用。晶体内相互接触的蛋白质表面区域氨基酸成分分析
表明,这些氨基酸与蛋白质表面氨基酸残 基 的 分 布 没 有 明 显 的 差 异。对 于 膜 蛋
白,晶格中也有蛋白质去污剂和胶束样去污剂去污剂相互作 用 的 形 式。因 此,所有蛋白质晶体都是柔软的,而且对湿度非常敏感。近来原子显微得到的蛋白质

210
结晶影像显示,在存在或多或少的晶体球形序列的同时,也可以看到许多局部的
变形 (错位,晶格改变),这说明了许多蛋白质晶体衍射相当弱以及衍射斑摇摆
度较广的原因。在蛋白质纯化期间,可以得到有关蛋白质沉淀和蛋白质稳定性的信息,可以
测定蛋白质随pH变化的溶解度范围并估测等电点。大多数蛋白质在其等电点表
现出溶解度最低,此时净电荷为零,排斥力也最小。但是蛋白质是非常复杂的多
离子体,其表面有特殊的电荷分布模式,因而可能产生多个最小溶解度值。许多
蛋白质晶体在远离等电点几个pH单位也能生长的现象就反映了这一问题。确定和优化形成优质晶体所需要的条件仍是一项比较困难的工作。假设在由
pH、离子强度、相反离子、温度等条件确定的多维参数空间存在某个最优条件,而且没有理由认为这些因素可以独立优化,那么就需要大量的实验考察所有的组
合。利用统计学方法已经能改变研究结晶条件的方式,该方法包括设计数量相对
较少的实验、不完整的因素分析、选择覆盖整个参数空间范围的实验条件的随机
组合等。
Kim及其同事提出了另一种研究方法,采用了一个有98种条件的 “稀疏矩
阵”,有关信息可见于互联网上的结晶数据库 (http:/wwwbmcdnistgov:8080/
bmcd/bmcdhtml)。适当的结 晶 溶 液 已 有 商 品 销 售。该 方 法 利 用 蒸 汽 扩 散 或 微
量法建立筛选条件,然后通过显微镜仔细观察以发现一些非常细小的微晶体,当
筛选条件接近参数空间某一区域时,晶体可能会生长。虽然有时该筛选方法可以
得到比较理想的结果,确实得到晶体,但以pH、离子组成、蛋白质浓度等作为
优化的途径更加常见。表82表明了一个简单的实验过程,以一次 “稀疏矩阵”得到的成功条件进行四变量二水平实验。在远离成功条件时,仔细研究商品溶液
如何制备是非常重要的。具有相同pH的不同缓冲液都可包括在优化过程中,其
表82 适用于四因素二水平筛选的结晶盘设计
项 目pH
低 高蛋白质浓度
沉
淀
剂
低 A2B2
A3B3
A4B4
A5B5
低
高
高 C2D2
C3D3
C4D4
C5D5
低
高
添加剂 低 高 低 高
注:各实验槽垂直方向记为A~D,水平方向记为2~5,对应于Linbro平板的中心区域,记录各变量
及设定的水平。如考察左上角的四个槽,具有相同 的pH和 沉 淀 剂 浓 度,但 添 加 剂 用 量 和 蛋 白 浓 度 不 同。
通过比较这四个实验槽,可以清楚地得到添加 剂 和 蛋 白 浓 度 的 作 用 效 果。比 较 A行 的 实 验 槽,可 以 清 楚
地得到添加剂的作用是否与pH有关……

211
浓度通常比蛋白质高几个数量级,而且相互间很少发生作用也很重要。在建立筛
选方法时,应注意一些基本的无机化学反应,比如磷酸根离子与镁离子在许多筛
选溶液中都起到相当重要的作用,但如果样品中存在这两种离子,就会形成非常
细小的磷酸镁晶体,从而导致结晶成功的假象。对膜蛋白、核酸、DNA结合蛋
白,该结晶筛选方法也衍生出了许多特定的筛选方法。如果仍然得不到晶体,则
需重新考察蛋白质及其特性的问题。
821 蛋白质结晶方法
晶体生长技术有许多种,此处介绍一些已成功的方法,比如分批法、蒸汽扩
散法和透析法。蛋白质结晶所需设备并不昂贵,其中比较贵的是性能良好的显微镜 (最好带
有摄影装置)。带有偏光器/分析器的立体变 焦 显 微 镜 对 于 检 查 晶 体 生 长 相 当 有
用,在透过各种厚度的玻璃、塑料和溶液对晶体聚焦,以及在X射线研究前操
作较大结晶时可能都是必要的。传统或反向显微镜用来确定较小的晶体,而偏振
光用于在无定形沉淀物中定位微晶体,在X射线研究之前定位晶体的光学各向
异性也是非常有用的。分配少量液体需要移液枪和注射器,高速微量离心机对于
除去蛋白溶液中的较大颗粒比较理想,超滤设备用于少量蛋白的浓缩和缓冲液的
快速更换。大部分的结晶生长设备都不是专一性的,而是从其他技术借鉴过来或者仅仅
是实验室标准玻璃和塑料仪器的组合。设备的选用取决于蛋白质的丰富程度以及
所采用的晶体生长条件。许多晶体可通过 改 变 蛋 白 溶 液 的 离 子 强 度、醇 含 量 或
pH而生长,采用下文介绍的透析或蒸汽扩散方法,其过程是相当缓慢的。
8211 分批法
传统的结晶方法就是 “分批法”,通过加入沉淀剂或者改 变pH,直 到 蛋 白
质达到其溶解极限,形成少量的晶核,溶液 静 置 一 段 时 间 后 会 生 长 出 较 大 的 晶
体。分批实验的主要限制因素是沉淀条件要求保持静置,实验过程中惟一的变化
是蛋白质浓度的降低。将系统脱离晶核形成区,防止晶核过度形成,有助于结晶
化,但每次实验仅考察一套沉淀条件,所以设计实验之前需要先了解结晶条件。克服该限制因素的传统方法是缓慢降低蛋白质的溶解度,通过加热样品,置于绝
热的真空瓶中,于冷房中缓慢冷却来完成。传统分批法所采用的大量样品 (100μl以上)适合于大晶体生长,但不适用
于结晶条件的筛选。微量分批法最新的操作为,将一小滴蛋白质溶液浸入到石蜡
油中 (如图81),这样可避免溶液的蒸发,极微量的样品 (小于1μl)也可以应
用,因而该方法对于最初的筛选比较理想。该技术的进一步改进是采用石蜡油与
聚硅氧烷油混合。聚硅氧烷油允许水分缓慢地扩散,随着水分的蒸发,液滴中蛋

212
白质和沉淀剂的浓度会缓慢上升,这样,蛋白质和沉淀剂的浓度范围可在各小液
滴中自动实现。该方法的缺点是液滴会在几星期内干透,除非保存在4~10℃范
围内,所以必须定时检查结晶。
图81 蛋白质结晶方法
(a)挂滴实验;(b)微量分批实验;(c)微量透析扣实验
微量分批法的操作过程如下。将足量的石蜡油 注 入Terasaki平 板 (Hampton法,该平板含72个 锥 形 槽,每 个 槽 可 容 纳 最 多10μl液 体),覆 盖 所 有 样 品
槽;将1~5μl沉淀剂分别注入各样品槽,沉淀剂液滴注于油面下,可下沉到槽
底;将1~5μl蛋白质溶液以同样方式分别注入各样品槽,蛋白质溶液与沉淀剂
可通过扩散相混合;静置平板,控制好温度;1~2周后检查平板;之后6个月

213
中每月检查平板,液滴中的水分透过石蜡油缓慢蒸发 (蒸发速度可通过混合聚硅
氧烷油与石蜡油加以调节,增加聚硅氧烷油量可使液滴蒸发速度加快),使液滴
中蛋白质浓度增加;将过量的油 从 平 板 上 排 掉,只 在 样 品 槽 中 留 有 油,收 集 结
晶,在含有结晶的样品槽中注满15μl适当的沉淀剂以代替油,之后就可按常规
操作处理结晶了。
8212 蒸汽扩散法
蒸汽扩散法是目前广泛应用于衍射级晶体制备的一种方法。该方法将蛋白质
液滴与沉淀剂混合,在密封的槽中通过与槽内高浓度沉淀剂相平衡而缓慢脱水。蛋白质液滴 也 可 挂 在 密 封 槽 的 盖 片 上 (即 挂 滴),或 置 于 槽 内 的 载 体 上 (即 点
滴)。通常将等体积的蛋白质溶液和槽中溶液混合。蒸汽扩散法有利于自动筛选
沉淀剂浓度范围,但与微量分批脱水实验不同,它有一个明确的终点。理论上该
方法可以优化结晶,当沉淀剂浓度缓慢达到溶解度相图的晶核形成区域后,应快
速增加蛋白质浓度以弥补结晶所造成的蛋白质浓度降低。这不仅需要实验终点的
优化,也需要平衡速度的优化。改变平衡速度最简单的方法是改变液滴比表面。一般较大的液滴比小液滴平衡速度慢得多,比如32μl的挂滴比8μl的挂滴平衡
时间要长两倍多。加快实验时间进程的另一种简便方法为移取200μl石蜡/聚硅
氧烷油层置于槽内溶液的顶部。选用的沉淀剂在液滴平衡动力学中也具有重要作
用。含有盐类沉淀剂,如硫酸铵的样品槽可在一两天内达到平衡,而含有PEG类沉淀剂的样品槽则需要1个多月。因此在只含有PEG的结晶过程中,经过几
天生长得 到 的 晶 体 可 以 认 为 类 似 于 微 量 批 次 试 验。加 入 适 量 的 NaCl(如
200mmol/L)可以将PEG结晶过程的平衡时间缩短至大约10d,但一般情况下,较长的平衡时间得到的晶体质量较好。
挂滴蒸汽扩散法的操作方法:用移液枪将聚硅氧烷脂涂布Linbro组织培养
平板各样品槽边框,可使盖片与样品槽密封;将1ml沉淀剂 溶 液 分 配 至 各 样 品
槽中;将2~8μl蛋白质溶液置于盖片中心;从样品槽中小心移取2~8μl沉淀剂
溶液,置于蛋白质液滴顶部;将盖片翻转置于同一样品槽上,并将盖片压入聚硅
氧烷脂中形成密封;静置平板,控制温度。
8213 透析法
透析法也已成功应用,沉 淀 条 件 范 围 也 可 自 动 筛 选,但 与 前 面 两 种 方 法 不
同,蛋白质浓度保持恒定。一般用作预处理的各类半透膜都可用来包裹置于厚壁
毛细管或塑胶玻璃微量透析扣中的蛋白质溶液 (见图81)。透析膜用 “O”形环
或从适当直径的聚硅氧烷橡胶管切割下来的橡圈所固定。将该装置悬浮或浸于可
透析的沉淀剂,低离子强度缓冲液或水中。分步改变外部条件,每次维持几天的
平衡时间。在蛋白质溶液的毛细管柱中,沉淀剂强度梯度的形成可以在特定高度

214
使晶体生长,并有助于选择确定理想的沉淀剂强度。透析法对于蛋白质一般并不
经济。塑胶扣需要 容 纳5μl液 体,此 外 通 常 在 注 入 液 体 时 还 要 求 膜 内 不 产 生 气
泡。但透析法有更多的灵活性,因为在晶体制备失败时得到的无定形沉淀,可以
用适当溶液再次溶解,利用同一样品来考察其他条件。
8214 晶体的保存
一旦晶体生长,通常在其生长的母液中可长期保存一段时间,也可转移到相
同组分的无蛋白溶液中,作为结晶混合物。在缺少沉淀剂的高浓度蛋白溶液中生
长的结晶经常会在这样的条件下重新溶解,而转移到含有沉淀剂的与初始生长环
境相同的溶液中可使其稳定,将晶体短时间 (15min)置于05%戊二醛溶液中,可在晶体表面形成交联外壳,极大地提高了晶体对于不同条件的耐受性。有观察
表明,在硫酸铵或聚乙二醇沉淀剂中生长的晶体长期在盐溶液中更为稳定,可能
是由于光诱导乙二醇的降解和产生损害蛋白的活性成分。沉淀剂经常以高摩尔浓
度存在,因而一般实验级试剂中的痕量杂质就会变得明显,所以与蛋白质接触的
试剂采用最高质量级别为好。用于晶体生长或保存的缓冲溶液类型并没有严格的
限制,但应注意,类似磷酸盐的物质以及随后结构分析中的重金属可能会产生严
重的不溶性问题,而且在如甲基戊二醇、乙 醇 或 高 浓 度 的 低 分 子 量 乙 二 醇 存 在
时,特别是在4℃下,甚至在相当稀的溶液中磷酸盐结晶也容易形成,因而采用
磷酸缓冲液时经常会过早地认为晶体生长。
822 不均一性对蛋白质结晶的影响
8221 杂质的影响
蛋白质样品纯度不足可能会导致:结晶的彻底失败,或只能得到小晶体,或
不能得到良好衍射X射线的大晶体。如果蛋白质没有预结晶,那么蛋白质的纯度就成为需要额外考虑的变量。目
的蛋白的微观不均一性可能比一些完全不相关的分子所造成的污染更为麻烦。在
有些纯化情况下,利用晶格堆积来选择具 有 适 当 结 构 的 分 子 时,重 结 晶 比 较 有
效。例如,在治疗糖尿病的高品质药剂的生产中,胰岛素结晶是非常重要的。但
胰岛素的重复结晶不能除去残留的污染物,其中包括大约5%的胰岛素原、转化
反应的各种中间体以及提取过程中的其他化学成 分,这 些 物 质 可 以 通 过 凝 胶 过
滤、离子交换和反相色谱除去。由于蛋白性质的变化可能相当小,微观不均一性
提出了更为重要的纯化问题,而且大多数蛋白质制品都有可能遇到,甚至是比较
容易制备高品质晶体的商品鸡蛋溶菌酶,也发现其中含有少量的卵清蛋白和溶菌
酶二聚体。X射线拓扑测定表明,晶体中的这些杂质引起晶体晶格产生一定的张
力,降低了衍射的质量。为模拟微观不均一性的影响,在火鸡蛋溶菌酶结晶过程
中加入鸡蛋溶菌酶作为污染物,这两种蛋白的序列95%相同,而所有不同的残

215
基都位于分子表面。由于人工引入的微观不均一性,其效果是通过抑制晶体某一
面的生长而改变了晶体的形态。一般这样的影响可能会形成针状或薄片状晶体,不适合衍射实验。
8222 微观不均一性的来源
微观不均一性可能来自于天冬酰胺或谷氨酰胺的脱酰胺作用,半胱氨酸的氧
化,蛋白质水解,序列多晶型现象,转译后修饰的不确定性,如丝氨酸的磷酸化
作用或者N末端甲基化或乙酰化等,举例如下:(1)脱酰胺作用 牛胰岛素的六个侧链酰胺中,有一个酰胺的脱酰胺作用在
极端pH条件下比其他酰胺要快得多,而脱酰胺后的产品也具有完全的活性,与
未降解的胰岛素同形结晶。但由于结晶面的生长速度相对不稳定,与完整胰岛素
的典型菱形形态相比,该晶体形态几乎没有类似之处。如果胰岛素制剂被脱酰胺
产物严重污染,则会在结晶中形成严重的 “雪花状”多晶聚合体。(2)氧化作用 游离硫醇的反应能力随溶剂的变化而变化。酶活性位点的几
何形状会增强此类基团的反应,除非受到保护,这些基团是试剂缓冲液中痕量金
属非常有效的清除剂,或者很容易被氧化,从而导致酶失活。比如Lactobacilluscasei的胸苷酸合成酶晶体衍射X射线的能力就与硫醇氧化状态有关。
Ecoli胆色素原脱氨酶在pH=55时用PEG沉淀很容易结晶,但该酶的共
价结合辅因子二吡咯甲烷在此pH下容易氧化,产生黄色的失活蛋白。部分氧化
的蛋白可形成大晶体,产生X射线值达到017nm的解析度。虽然在DTT过量
存在时晶体可以有效生长,但要得到非氧化形式的大蛋白质单晶体比较困难。对
此的解释是氧化的辅因子具有刚性的平面结构,似乎对周围的蛋白质区域具有组
织作用,而未氧化的辅因子则更具柔性。关键是氧化蛋白对于光非常敏感,似乎
氧化型辅因子对于光吸收可以进行异构化反应,类似于视网膜视色素的反应,从
而干扰了与周围蛋白的相互反应,显然使整个结构不稳定。因此氧化型蛋白质的
晶体只能生长在暗处,一旦晶体长成,暴露于实验室通常光照下几天就足以完全
打乱这些晶体而得到一种韧性结构。(3)磷酸化作用 糖原磷酸化酶活性部分受靠近 N末端的一个丝氨酸残基
磷酸化状态所控制,磷酸化的a型更为活跃。在安静的肌肉中a型占优,负责转
化为b型的酶可以迅速从提取物中除去。另外,b型也可以积累。这样化学相似
的物质在分离时遇到的困难就小多了。晶 体 研 究 已 经 显 示 葡 萄 糖 抑 制 的a型 和
IMP激活的b型构象上的差别,特别是在磷酸化位点区域。(4)蛋白质水解过程 鼠颌下腺神经生长因子的制备表明,经蛋白质水解可
从一个大分子得到链长不同的分子,N末端的八个残基和C末端的一个残基可
能丢失。由于蛋白质可能作为多种不同链长分子组合而成的稳定二聚体而进行分

216
离,所以情况更为复杂。不同分子的等电点非常接近,而X射线衍射结构在 N末端也没有获得什么信息。
(5)同工酶 真菌华根霉 (Rizopuschinensis)产生两种形式的胞外天冬氨
酸蛋白酶,pI分别为56和60。在共纯化时,可以生长成比较好的晶体。等电
聚焦分离后,pI为56的 蛋 白 酶 形 成 大 晶 体,对X射 线 衍 射 有 较 高 的 解 析 度,而且对于辐射损坏较不敏感;而pI=60的蛋白酶则没有结晶。
(6)碳水化合物组分 人白细胞弹性蛋白酶在凝胶电泳上显示5个条带,主
要是由碳水化合物所造成,但蛋白质结晶情况良好。其他许多糖基化血清蛋白也
已成功结晶。但是与蛋白质结合的碳水化合物还是经常被认为可能是一种杂质。有时在电子密度图上可以清楚看到这些链的存在,比如免疫球蛋白、流感病毒壳
血凝素以及唾液酸酶等。现在有一类酶可以除去所有碳水化合物链,但处理成本
比较高,而且裂解物的除去也有困难。糖裂解酶已与谷胱甘肽硫转移酶作为融合
蛋白得到表达,在谷胱甘肽柱上通过亲和色谱可从去糖基蛋白中快速分离。对于
人绒膜促性腺素 (HCG),利用 HF部分去除碳水化合物,可以进行结晶和结构
分析。通过基因操作在微生物中表达的蛋白可提供无碳水化合物的有效蛋白源,而从现存的糖蛋白上完全去除糖原并不容易。
8223 微观不均一性的检测
等电聚焦分析与高于和低于等电点条件下聚丙烯酰胺凝胶电泳联用,能够检
测大多数潜在的微观不均一性问题。链内断裂可以通过还原条件下的SDS电泳
检测到。在蛋白质制剂结晶前,可以利用这些方法进行分析。所有色谱分离的纯度标准应根据形成的对称洗脱峰而定。对于离子交换色谱
可利用相反离子的平缓洗脱梯度实现,而对于凝胶过滤色谱则需确保使用正确的
分级分离范围。复杂的混合物如果用较快的盐梯度洗脱或者在凝胶过滤柱中以较
大的空隙率洗脱,也可能表现为相当纯。银染色法分析电泳已经足够灵敏,但在
进样量较大的凝胶上很少能得到单一的银染色条带。日益广泛应用的蛋白质定性
技术是电喷质谱技术,只需要50μg完全脱盐的蛋白,就可能测定精确的蛋白质
分子量 (±001%),可以确定蛋白质共价结构中可能存在的有害于结晶的问题,如Ecoli表达蛋白N末端蛋氨酸的缺失等。图82为电喷质谱技术的一个例子,过量表达的Ecoli酶电喷质谱 结 果 表 明,由 于 不 完 全 的 N末 端 脱 蛋 氨 酸 作 用,导致蛋白的不均一性。成熟酶应为31820Da。而该样品离子交换色谱和凝胶过滤
色谱分析均为单一的,在SDS电泳上也是单一条带。对于不涉及电荷改变的序列变化,反相色谱处理改变较小的蛋白质是非常有
效的。在柔性更强的蛋白质中,不均一性也可能与一些不太容易被检测的其他因
素有关,如蛋白质的部分伸展或促进明显的异构物形成等。当蛋白质高效表达,

217
图82 过量表达的Ecoli酶电喷质谱结果
而胞内折叠机制不适应时可能会导致包含体的形成,同时也可能得到部分折叠的
可溶性蛋白质。亲和色谱可以除去这些物质。利用亲和标记,Ni螯合色谱可从
Ecoli破碎液中非常有效地 “钓出”重组产物的组氨酸尾,但如果不进一步纯化
的话,也不一定能得到理想的结晶材料。组氨酸尾本身就能随pH改变而增加蛋
白质的实际电荷,影响其溶解性。不过许多在适当位置带有标记的蛋白质已经得
到结晶。大多数构建的商业化产品中都包含一个蛋白质裂解位点以便除去标记。
8224 添加剂与蛋白质水解
除了沉淀剂,非肽类物质,包括酶活性所要求的辅因子以及金属离子,在结
晶过程中也是很重要的,比如鼠肝苹果酸酶结 晶 时 就 需 要 NADP。但 蛋 白 质 与
这些物质的部分结合可能也是微观不均一性的重要原因。纯化可能也会除去一些
结晶所必需的成分,如螯合剂或解离条件在粗组织提取液中经常用于协助抗蛋白
质水解,但同时会除去晶体生长所需要的金属。胰岛素结晶需要锌离子,氧化型
硫氧化还原蛋白结晶需要铜离子,氯霉素乙酰转移酶结晶需要钴离子,在这些例
子中,金属离子似乎仅仅是连接晶格中的分子。控制蛋白质水解特别适合于结晶,相关例子很多 (如免疫球蛋白,刀豆球蛋
白,延伸因子TU,核糖核酸酶S,细胞色素b5)。有时,似乎是蛋白质水解除
去了蛋白质柔软或突出的部分,使得结构更为紧凑,易于压缩到一起。另外,多
肽的连接链也可能断裂,释放出完整的结构域。通过晶体研究,流感病毒血凝素
和唾液酸酶这两种天然蛋白都是通过一个疏水域结合到病毒外壳膜上,该疏水域
在制备过程中被菠萝蛋白酶或链霉蛋白酶所裂解,其强烈的糖基化作用可能防止

218
了更广泛的蛋白质水解。
83 融合蛋白的纯化
基因工程提供了人们改变蛋白质性质的机会,因而可以借此改善蛋白质的纯
化特性。通过在目的DNA的3′端或5′端插入DNA序列,可以改变蛋白质两端
的氨基酸序列,进而作为可纯化的融合蛋白。这些融合子可帮助蛋白质形成包含
体,用于稳定蛋白质,免受蛋白酶的攻击,还可以赋予蛋白质特定的纯化性质,使其适用于免疫亲和、金 属 螯 合、离 子 交 换、疏 水 色 谱 以 及 其 他 分 离 操 作。此
外,对于已知特性的蛋白质,改变其中特定氨基酸可引入具有特定基质吸附亲和
力的片段。纯化融合蛋白的原理简单 (图83),但每种蛋白都有其特性,蛋白质融合
设计需要引入基因工程和蛋白质工程的观点。在考虑采用何种纯化融合时,需要
考虑蛋白质的最终用途。如果蛋白质是作为生物分析试剂或生物催化剂,融合片
段只要不干扰蛋白质的活性,将其留在蛋白质上也没有关系。但如果蛋白质是作
为长效的药用产品,那么就需要去除融合蛋白,以防止产生不利的免疫反应。
图83 融合蛋白的纯化
在实际操作中,需要考虑三个问题:采用何种融合来解决微生物系统表达哺
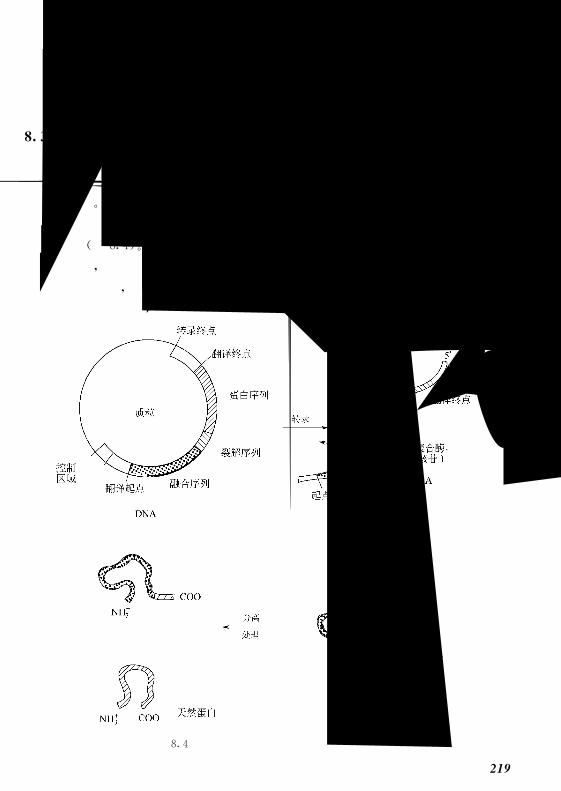
219
乳动物蛋白所遇到的技术问题;是否有助于满足制备大量蛋白的要求,而成本又
是主要因素;在研究中采用何种融合可以发现基因序列已知而生理功能未知的蛋
白质的生物功能。本节除了介绍融合蛋白纯化的基本原理和过程之外,还将介绍
融合蛋白纯化技术的一些新进展以及相关实例。
831 融合蛋白的纯化过程
DNA合成和基因拼接技术已用于培养工程细胞生产具有新特性的蛋白质,其
中一项应用就是使蛋白融合。融合蛋白可以改善其稳定性和纯化特性,有助于从培
养物中分离。虽然纯化的融合蛋白由于具有免疫或酶活性可以加以应用,但一般只
是作为天然蛋白的前体,利用蛋白质化学技术水解特定肽键,可以从融合子上释放
天然蛋白 (图84)。融合技术既可在实验室中用于蛋白质的快速分离,进行结构
和功能研究,也可用于大量生产酶催化剂和高纯度的药物。设计稳定性和纯化特性
提高的融合蛋白,使天然蛋白从融合蛋白上释放是很关键的。此外,纯化过程也包
括一些融合蛋白以及由融合蛋白前体产生的天然蛋白的提取、纯化和分析。
图84 细菌生产的融合蛋白的合成和分离

220
8311 融合蛋白的设计
这里主要介绍提高蛋白稳定性和纯化特性的融合蛋白的设计,并提出一些实
践原则。(1)融合蛋白的水解 融合蛋白设计主要考虑的是最终除去融合子生产天然
蛋白,并为此做好相应的准备。如果融合蛋白缺乏所需的特异性或生物活性就更
需要这样。一般通过在融合子和蛋白的结合处引入可化学或酶水解的裂解肽来实
现水解。有许多蛋白酶和化学方法可以采用,如裂解多肽链中的肽键的内切肽酶
或从蛋白C末端或N末端顺序水解氨基酸的外切肽酶等 (表83)。
表83 特定裂解序列
肽 链 方 法 产 物
NMetC 溴化氰 NMet和CNAspProC 酸解 NAsp和ProCNLys/ArgC 胰蛋白酶 NLys/Arg和CNGlu/AspC V8蛋白酶 NGlu/Asp和CNLysArgC Clostropain NLysArg和CN(Lys)n/(Arg)m 羧肽酶B N和nLys与mArgNAspAspLysC 肠激酶 NAspAspLys和CGluAlaGluC 氨肽酶 GluAlaGlu和CNIleGluGlyArgC Ⅹa因子 NIleGluGlyArg和CNProXGlyProC 胶原酶 NProX和GlyProC
注:以上氨基酸序列 (X为任意氨基酸)已被用作融合蛋白连接子,产生特定的裂解点以便将大部分
或全部融合多肽从目的蛋白上除去。与融合多肽连接的肽链 既 可 连 接 到 N末 端 也 可 连 接 到C末 端,甚 至
两端都可连接,分别表示为N多肽和C多肽。
在设计裂解肽时,应考虑以下因素:
① 应确定天然蛋白对各种裂解反应的抗性 由于化学和酶的作用可能会受
到蛋白构象的影响,因此即使存在敏感的氨基酸,蛋白质也可能具有抗性。将天
然蛋白中敏感氨基酸加以替代或缺失也可提高其抗性,但得到的变异蛋白活性可
能改变。
② 必须考虑反应条件对天然蛋白的影响 一般化学水解在变性条件下进行,需要蛋白质的重新折叠,而蛋白酶可在非变性的生理条件下作用。
③ 应确定反应的专一性 极端pH 可能会修饰氨基酸,蛋白酶中的杂质也
可能引起非专一性水解。(2)用于稳定性的融合 蛋白质的稳定可通过形成细胞内的包含体或将蛋白
质排放到细胞周质空间或培养基中而实现 (图85)。Ecoli细胞内可溶性肽的水解
是造成蛋白质提取产率较低的主要因素。多肽融合可以促进包含体的形成,虽然蛋
白质会变性并与其他细胞成分聚集,但却是稳定的 (表84)。为了分泌蛋白,可

221
与分泌引导序列进行融合,使其分泌到细胞周质和培养基中,而分泌引导序列则被
细胞膜上的酶所裂解。表85列举了一些通过形成融合蛋白稳定蛋白质的例子。
图85 工程蛋白在Ecoli中的定位
表84 利用基本的小肽融合形成包含体稳定人表皮生长因子
融 合 多 肽 溶解部分/% 半衰期/min 不溶部分/% 稳定性
无 >90 6 — —N末端TrpE 67 25 33 稳定
C末端聚精氨酸 61 19 39 稳定
TrpE和聚精氨酸 83 36 17 稳定
注:利用脉冲追加实验研究 基 本 小 肽 融 合 子 的 加 入 对Ecoli表 达 的 人 表 皮 生 长 因 子 稳 定 性 的 影 响。
表达的人表皮生长因子可溶与不溶部分的百分数以及相应的半衰期列于表中。结果表明包含体中的不溶性
蛋白是稳定的。
表85 用于稳定的蛋白融合子
多 肽 机 制 蛋 白
β半乳糖苷酶 包含体 生长激素抑制素
β半乳糖苷酶 包含体 胰岛素A和B链
β半乳糖苷酶 包含体 β内啡肽
TrpE 包含体 尿抑胃素
聚精氨酸 包含体 尿抑胃素
分泌信号和蛋白A 胞外 类胰岛素生长因子(IGF1)分泌信号 细胞周质 尿抑胃素

222
制备形成包含体的融合蛋白,以稳定蛋白质,需考虑以下因素:
① 包含体可能与部分表达良好的Ecoli蛋白结合而形成,得到的蛋白不能
折叠,形成聚合体,富含融合蛋白;
② 为了提高培养产率,减少天然蛋白释放过程中的副反应,应采用小分子
融合肽来稳定蛋白;
③ 应考虑将分离得到的包含体作为富含融合蛋白的原料,并增强融合蛋白
自身的纯化特性来提高纯化;
④ 包含体中的蛋白质可能是变性的,需要重折叠,这对于小分子蛋白通常
不是问题,如果确有问题,应考虑利用细菌或酵母表达系统将蛋白分泌到细胞周
质或培养基中。(3)用于纯化的融合 蛋白质纯化可利用蛋白质在电荷,疏水性和分子大小
等方面的物理化学差异,或者利用基于专一结构或功能特性的生物亲和方法来实
现。通过设计具有独特纯化特性的融合蛋白,可以实现快速而简便的纯化。此外
也应考虑利用纯化融合检测或分析目的蛋白的潜力,光谱、色谱、酶学和抗原特
性都可用于开发简便的分析方法,此类融合蛋白纯化的例子列于表86中。
表86 改善纯化的蛋白融合子
纯化方法 纯化融合子 纯化方法 纯化融合子
离子交换 多 聚(精 氨 酸)/(赖 氨 酸)/(赖 氨 酸精
氨酸)金属螯合 富含组氨酸的多聚物
底物亲和 β半乳糖苷酶,氯霉素转氨酶
免疫亲和 蛋白A,Flag肽,受体A
设计纯化融合蛋白时,首先应考虑工作的规模和目的。如果要大规模分离蛋
白,则纯化基质的成本比较关键,离子交换适合于此类大规模操作;如果用于实
验目的,所需数量较少,或者蛋白 质 价 格 很 高,那 么 较 昂 贵 的 亲 和 法 也 可 以 接
受。其次应考虑蛋白纯化过程中由裂解反应产生的残基,除非天然蛋白可以很容
易地从纯化裂解产物中分离,否则会丧失纯化融合的许多优点。最后,如果蛋白
质作为包含体产生,应避免可能会干扰重折叠过程的融合子。
8312 融合蛋白的提取
工程蛋白可以在细胞培养不同部位,不同数量表达。优化表达时必须明确目
的蛋白的数量和位置。提取方法的选择取决于重组蛋白的位置。酵母或细菌培养
分泌的蛋白可通过离心或过滤除去细胞后进行纯化。在细胞质、细胞周质或包含
体中积累的蛋白,则在纯化前需要破碎细胞,除去细胞碎片。对于某些蛋白,蛋
白质水解可能是个问题,应采用低温或蛋白酶抑制剂。膜结合蛋白可用非离子去污剂溶解,但提取包含体中的蛋白可能需要适当的

223
蛋白质变性剂。一般操作是在溶解包含体前,先除去可溶性的胞质蛋白。由于采
用变性剂溶解,所以在纯化之前或之后需要蛋白质重折叠步骤。
8313 融合蛋白的重折叠
蛋白质的重折叠很复杂,而且很少能够100%有效。操作条件一般由经验确
定,蛋白质浓度、氧化还原电位、离子强度、pH、时间和温度都很重要。吐温20,β辛基葡 糖 苷 以 及 甘 油 等 添 加 剂 也 有 成 功 应 用。以 下 条 件 可 作 为 试 验 的
起点:(1)变性 将蛋白质以1mg/ml溶解于6mol/L的尿素或6mol/L盐酸胍溶
液中,与25mmol/L二硫苏糖醇、50mmol/LTrisHCl(pH=80)溶液一起沸
水浴加热5min,冷却,离心澄清。(2)复性 将复性缓冲液于37℃迅速稀释样品10~100倍,浓度达到01~
10mg/ml。复性缓冲液含10%甘油,5mmol/L谷胱甘肽 (还原型),05mmol/L谷胱甘肽 (氧化型)。缓冲液 (a)50mmol/LTrisHCl,pH=8;(b)50mmol/L
β丙氨酸,pH=38;(c)磷酸盐缓冲液;(d)50mmol/L磷酸钠,pH=6。去
塞保存24h,使空气氧化硫醇。测定生物活性,并选择最优缓冲液。重复优化过
程确定蛋白浓度和还原剂。
8314 融合蛋白的纯化
纯化方法取决于设计阶段所引入的纯化特性。注意以下问题,有助于纯化的
进行:(1)离子交换色谱 聚阳离子融合子可与核酸结合,特别是纯化可溶性胞内
蛋白时。相反,HPLC离子交换基质,如 MonoS(Pharmacia公司),通常在核
酸过量下结合蛋白,并能实现一步纯化。为避免降低融合蛋白对离子交换基质的
亲和力,样品的预处理如下所述:
① 沉淀 聚乙烯胺是一种聚阳离子,可与核酸强烈结合,形成沉淀,应避
免过量添加,防止干扰随后的离子交换步骤。含有10mg/ml蛋白质的01mol/LTrisHCl(pH=80)溶液的澄清溶菌液中,加入008% (08g/L)polyminP(BDH)。于4℃放置5min,离心澄清。
② 消化 在高盐浓度下,核酸会与蛋白分离,可用核酸酶降解。含有2mg/
ml蛋白澄清溶菌液中,加入等体积的4mol/LNaCl溶液和100μgDNaseⅠ/mg蛋白质。4℃下透析16h,离心澄清。
(2)亲和色谱 有时亲和色谱中观察到的亲和力低于预期值,这可能有几种
原因,如色谱柱上配基浓度较低、空间位阻或融合蛋白上的结合位点不足等。通
过小心制备色谱柱以及利用高盐缓冲液降低非特异性反应可以减少这些问题。采
用免疫亲和时,应尽量避免抗体,因为抗体要求比较苛刻的洗脱条件。

224
(3)变性蛋白的纯化 有时融合蛋白不得不用变性剂提取,如尿素或胍。在此
情况下,采取亲和纯化前几乎都需要复性。如果纯化融合子未有效重折叠,产率就
会降低。因此,小的简单融合,如聚精氨酸和Flag肽可能具有优势。利用离子交
换也可进行纯化,尽管蛋白质在尿素中是变性的。胍提取物也可用尿素稀释以便结
合。吸附后,可进行蛋白质重折叠,此时蛋白质仍然结合在离子交换基质上。
8315 天然蛋白的回收
从融合蛋白上释放天然蛋白的水解方法在设计阶段就已确定。此处介绍肽键
特异性水解方法的选择及其局限性,所列条件可作为进一步优化的起点。(1)化学裂解 用于裂解肽键的化学方法比较有限。反应通常在变性条件下
效果最好。利用低pH和较高的温度可以很好地进行天冬酰胺和谷氨酰胺残基的
脱酰胺作用。
① 低pH AspPro肽键在低pH条件下通常不易水解。在缺乏变性剂的条
件下,水解反应缓慢,而位阻因素可能引起在其他天冬氨酸残基处的裂解。根据
融合子是处于羧基 还 是 氨 基 末 端,Asp或Pro会 留 在 蛋 白 上。该 方 法 也 具 有 优
点,AspPro键在蛋白质中相当少,因而选择性很高。具体操作时,将蛋白质以
05mg/ml置于7mol/L盐 酸 胍,10% (体 积 分 数)醋 酸 溶 液 中,以 吡 啶 调 节
pH=25,于56℃保持24h。以10mol/L氢氧化铵于冰水浴中中和样品。
② 溴化氰 (CNBr) 该反应在甲硫氨酸的羧基侧水解肽键。将蛋白以2mg/
ml溶解于6mol/L盐酸胍、01mol/LHCl(pH=10)溶液中,加入等量的 溴
化氰。维持4h,以5mol/L氢氧化铵中和。(2)蛋白质酶法水解 有许多酶能够 裂 解 肽 键,必 须 使 用 高 度 纯 化 的 蛋 白
酶,或者杂质蛋白酶必须经过选择性抑制剂失活。为提高稳定性,推荐使用固定
化蛋白酶,并可重复使用,减少蛋 白 酶 对 产 物 的 污 染。这 些 酶 可 从 许 多 公 司 购
得,如Sigma公司、BoehringeMannheim公司、CooperBiomedical&ICNImmunobiologicals公司等。
将蛋白酶固定化于琼脂糖的方法:将20mg蛋白酶溶解于10ml01mol/L碳
酸氢钠 (pH=83)缓 冲 液 中。加 入10mlCNBr活 化 的 琼 脂 糖,于4℃反 应
16h。以磷酸缓冲盐 (PBS)洗涤,并保存于4℃PBS和01%叠氮化钠溶液中。蛋白酶包括外肽酶和内肽酶。外肽酶从羧末端或氨末端顺序降解氨基酸 (比
如羧肽酶B),而内肽酶裂解蛋白质内易作用的氨基酸 (比如胰蛋白酶)。由于水
解过程中释放酸,为保持最佳酶活性需要控制pH。
8316 分析
许多纯化融合都可以提供灵敏的对融合蛋白定量分析。采用亲和融合 (如氯
霉素转氨酶和β半乳糖苷酶融合)时,可通过适当底物的酶法测定来完成分析。

225
对于离子交换融合,如聚精氨酸,可采用 HPLC,或者可 直 接 对 羧 肽 酶B消 化
后所释放的精氨酸进行分析。抗原性融合可通过标准免疫学方法分析。在需要去
除融合时,也可采用这样的分析来确定裂解效果。(1)HPLC分析 HPLC分 析 取 决 于 融 合 蛋 白 的 特 性,下 面 的 操 作 方 法 适
合于电荷纯化融合。对于离子交换,尿素溶解的蛋白可直接分析。以09ml适当的低盐缓冲液稀释01ml提 取 物,离 心 或 过 滤 澄 清;对 于 碱
性蛋白,采用20℃下5mm×50mm 的 TSK5PW (BioRad公 司)HPLC色 谱
柱。以pH=60、20mmol/L的 MesNaOH 缓 冲 液 平 衡,30ml线 性 梯 度 (0~15mol/LNaCl)洗脱,流 速1ml/min;对 于 酸 性 蛋 白,采 用DEAE5PW 色 谱
柱,20mmol/LTrisHCl(pH=80)溶 液;对 于 尿 素 溶 解 蛋 白,于6mol/L超
纯或去离子尿素溶液中配制各缓冲液,并于48h内使用。(2)采用免疫分析 典型的ELISA分析适合于抗原性肽类。将样品与标准
溶液用于血细胞凝集 (HA)平 板 (Millipore公 司),各 样 品 槽 的 浓 度 为40ng,室温下保持30min;与Tris缓冲盐 (pH=70)溶液,3%牛血清白蛋白 (BSA)共同培养1h以阻断非特异性蛋白结合位点;加入确定量的小鼠单克隆抗体,培
养1h;以PBS洗涤,加入碱性磷酸酶标记的山羊抗小鼠抗体 (Sigma公司);培
养1h后,以PBS洗 涤 平 板 数 次,然 后 加 入 比 色 试 剂 (底 物 片,Sigma公 司);将各 HA平板所含物质转移至96孔的聚苯乙烯平板,于450nm测定吸光度。
832 蛋白质的水解保护
在融合蛋白的纯化过程中,蛋白质的水解具有重要影响。一方面,天然蛋白
从融合蛋白上释放需要利用蛋白质水解;另一方面,融合蛋白的异种表达又需要
防止蛋白质的水解。面临的实际问题是如何得到高水平的表达以及如何保护蛋白质免受微生物蛋
白酶的作用。由于内源细菌蛋白不会遇到这些问题,将目的蛋白融合到某一高表
达细菌蛋白的N末 端 就 可 避 免 此 类 问 题。一 旦 表 达,融 合 蛋 白 会 高 水 平 积 累。蛋白积累的原因并不是因为蛋白酶不再降解外源蛋白,而是由于融合蛋白表达、沉淀、变性,成为包含体。这样,高密度的蛋白质团保护了蛋白质免受水解。
蛋白质若成功表达形成包含体,就可以直接通过细胞破碎和离心来完成与细
菌其他蛋白的分离,得到显著的纯化。为表达一个小 肽 (<10000Da),利 用 具
有适当的裂解位点并能在Ecoli中表达的多肽融合仍是一种特殊而简便的方法。下面介绍将蛋白质连接到表达良好的细菌蛋白 N末端从而在Ecoli中成功表达
的小蛋白,再利用商品蛋白酶选择性裂解得到目的产物的例子。
8321 融合APⅢ蛋白的纯化及利用V8蛋白酶释放蛋白
心房肽Ⅲ(APⅢ)会引起血管舒张,也可作为利尿剂,这两种作用都能帮助

226
治疗高血压。该蛋白仅由24个氨基酸构成,在一般的Ecoli表达系统中由于蛋
白质快速降解而不稳定。当与顶端切除的细菌蛋白RecA融合表达时,得到的蛋
白是稳定的。已有大量有关融合连接子的研究,包括Ⅹa因子、凝血酶和V8等。
V8蛋白酶并不适合于这一蛋白,因为它在谷氨酸或天冬氨酸残基后裂解。虽然
APⅢ不含谷氨酸,却含有一个天冬氨酸。不过,相关基因已经构建,该基因表
达的融合蛋白在RecA蛋白与APⅢ连接处有一个谷氨酸,意外地得到了所需的
裂解 (见图86)。该方法表明蛋白质折叠时可能会产生保护作用,显示出利用
多种可能性寻找适合的裂解位点时所具有的优势。密码子经优化在Ecoli中表
达后,得 到 的 蛋 白 质 以 稳 定 的 形 式 存 在,经 离 心 和 纯 化,于4℃ 50mmol/LNH4HCO3 中透析6h,APⅢ半胱氨酸发生氧化。该氧化作用对于减慢内部天冬
氨酸的裂解速度是非常必要的。应注意防止蛋白质 过 度 降 解 而 产 生 APⅢ1~9和
APⅢ10~24。
RecA蛋白末端↓
裂解连接子
1 2 3 4 5 6 7 8 9 10↓
内部裂解位点
帿帿 GluPheGluSerSerCysPheGlyGlyArgIleAspArg
20 19 18 17 16 15 14 13 12 11
AsnCysGlyLeuGlySerGlnAlaGlyIle
SerPheArgTyr21 22 23 24
图86 利用一个谷氨酸连接子作为V8蛋白酶裂解位点,
APⅢ融合到RecAC末端的一级序列
氧化型 RecAGluAPⅢ的 裂 解 与 分 析:将 APⅢ(100ml)和 V8蛋 白 酶
(1mg/ml)存储液于37℃放置;加入01mlV8蛋白酶,温和振荡,保温,移取
05ml样品与05mlHPLC (C18反 相 柱)平 衡 缓 冲 液 [10% (体 积 分 数)乙
腈/水和005%三氟醋酸]混合;进样10μl,利用100%乙腈,005%三氟醋酸
以0~100%线性梯度洗脱;计算APⅢ/RecAGluAPⅢ峰面积比,当峰面积比
达到95%时,反应混合物冷却至4℃,并加入10ml01mol/LHCl终止反应。
8322 利用胰蛋白酶从trypE融合子中分离EGF人表皮生长因子 (EGF)是由53个氨基酸组成的多肽激素,具有治疗溃疡和
愈合伤口的作用。该蛋白以前曾从人尿中分离出微量样品,随着基因工程的出现,该蛋白有可能大量制备。如果该天然蛋白在Ecoli中单独表达,很快会被降解。但若作为融合蛋白表达,则形成包含体而比较稳定。该蛋白本身抗胰蛋白酶分解,所以可作为N末端和C末端融合子进行表达,精氨酸作为胰蛋白酶的裂解肽。
胰蛋白酶水解蛋白质释放出酸,因此,蛋白质分解后在稀释缓冲液中加入一
定量的碱,以维持pH在适当的范围。pH=80是水解的最适pH。利用自动滴

227
定仪可以使反应连续进行。利用胰蛋白酶除去N融合物和C融合物:将EGF融合蛋白溶液 (90mg/
ml)置于1L的玻璃烧杯中,于37℃水浴保温,温和搅拌;插入装有100mmol/LNaOH的 自 动 滴 定 仪 探 头,设 置 pH 为80,打 开 记 录 仪,加 入 胰 蛋 白 酶
(100mg/ml),连续水 解 直 至 无 酸 产 生,关 闭 滴 定 仪;加 入500ml终 止 缓 冲 液
(50mmol/L醋酸钠,50mmol/L氯化钠),醋酸调节pH至36。从水解产物中纯化EGF:20L缓冲液 A (500mmol/L醋酸钠,50mmol/L
氯化钠,pH=36)平 衡 色 谱 柱 (TSK5PW),500ml蛋 白 水 解 物 (2mg/ml)进样分离;用20L缓冲液 A洗 柱,再 用20L缓 冲 液B (500mmol/L醋 酸 钠,
500mmol/L氯 化 钠,pH=36)进 行 洗 脱,收 集 50ml洗 脱 液;以 500ml01mol/L的氢氧化钠清洗色谱柱。重复色谱分离,可以得到纯化的蛋白质。
833 常用融合技术
为研究蛋白质及其功能,需要进行蛋白质的大量表达和纯化。但问题是如何
最大限度地增加获得表达蛋白的机会,得到的不仅仅是经过纯化的蛋白,而且是以
其天然生物活性形式存在的蛋白。比较好的解决方法是利用不会干扰蛋白质活性的
纯化融合子,既可以将其很容易地去除,又不会影响蛋白质的折叠。采用的表达系
统也必须尽可能在遗传上接近材料的来源,以便尽量增大蛋白质正确折叠的机会。适用于纯化蛋白的 “尾巴”类型相当广泛 (表87),任何较小的Tag都可
能是最有效的。生产厂商已根据特定的融合技术提供了多种试剂盒及药品,使得
蛋白质表达和纯化技术达到了非常高的水平。如果目的蛋白适用融合技术,这些
试剂盒就能帮助使用者克隆蛋白质,在合适的宿主中表达并通过某种亲和方法进
行纯化。但在分离由重组DNA技术生产的蛋白质时,没有一种单一的方法可以解
决所有可能遇到的问题,因此最好尝试多种方法,找到对于该体系最有效的一种。
8331 聚组氨酸
Sigma公司提供了一种试剂盒用于生产和纯化带有聚组氨酸标记 (六个组氨
酸的一段序列)的蛋白。该标记对于二价金属离子具有很高的亲和性,可以形成
螯合物。由于六聚组氨酸并不占据所结合金属的全部配位点,金属离子可以同时
与聚组氨酸标记的融合蛋白复合,复合物可利用固定化金属亲和色谱 (IMAC)进行色谱纯化。将次氮基三醋酸 (NTA)固定化于琼脂糖上,NTA琼脂糖一般
以镍 (Ni2+)预负载,然后可用于结合聚组氨酸标记的融合蛋白。聚组氨 酸 对
于镍NTA琼脂 糖 有 很 好 的 亲 和 性,而 且 可 用 许 多 方 法 洗 脱,如 浓 度01~02mol/L的咪唑缓冲液。Sigma公 司 还 提 供 了 另 外 五 种 金 属NTA琼 脂 糖,为
不同链长和大小 的 目 的 融 合 分 子 进 行 优 化 分 离 提 供 了 更 大 的 选 择 余 地。AmershamPharmacia公司和Novagen公司也提供聚组氨酸融合子和IMAC试剂盒。

228
表87 蛋白融合选项
普 通 类 型 大小 C末端或N末端 分 离 方 法
1酶
βgalGSTCATtrypE
116kDa26kDa24kDa27kDa
N,CNNN
IS,IAIS(GSH)
ISHIC
2多肽结合蛋白
SPASPG
14~31kDa28kDa
NC
IIgGI清蛋白
3碳水化合物结合域
MBPSBDCBD(CenA)
CBD(Cex)
40kDa119aa111aa128aa
NCNC
ISISISIS
4生物素结合域 8kDa N I抗生物素蛋白
5抗原决定基
RecAFlag
144aa8aa
CN
IAIA
6带电氨基酸
聚Arg聚Asp谷氨酸盐
5~15aa5~16aa1aa
CCN
IEC,pptnIEC,pptn,RMEIEC
7聚 His尾 1~9aa N,C IMAC
8其他聚氨基酸尾
聚Phe聚Cys
11aa4aa
NN
苯基琼脂糖凝胶
硫代丙基琼脂糖凝胶
注:aa—氨基酸;βgal—β半乳糖苷酶;CAT—氯霉素乙酰基转移酶;CBD—纤维素结 合 域;GSH—
还原型谷胱甘肽;GST—谷胱甘肽转移酶;HIC—疏水作用色谱;IA—固定化抗体;IEC—离子交换色谱;
IIgG—固定化免疫球蛋白G;IMAC—固 定 化 金 属 亲 和 色 谱;IS—固 定 化 底 物;MBP—麦 芽 糖 结 合 蛋 白;
pptn—选择性沉淀;RME—反相微胶囊萃取;SBD—淀粉结合域;SPA—葡萄球菌蛋 白 A;SPG—链 球 菌
蛋白G。
8332 Flag系统
这是一种促进重组 蛋 白 在Ecoli和 酵 母 中 表 达 和 纯 化 的 专 利 系 统,由Immunex和Kodak公司开发,现已由Sigma公司以试剂盒形式销售。完整的系统
包括载体和强启动子,以促进重组蛋白质 的 合 成,Flag肽 编 码 在 目 的 蛋 白 的C末端 或 N 末 端。Flag是 一 段 八 肽,序 列 如 下:NAspTyrLysAspAspAspAspLysC。Flag肽被固定化的抗Flag抗体所识别,是蛋白质纯化中抗Flag亲

229
和凝胶上的结合配基,抗体/抗原结合为钙依赖型。该N末端肽也含有一个高度
特异性的肠激酶裂解位点,因此利用此酶也可在纯化后选择性地除去融合子。用
于Ecoli表达的试剂盒有两种形式,分别允许Flag肽在N末端或C末端表达。酵母表达试剂盒只设置为N末端表达。EcoliN末端Flag表达试剂盒包含表达
载体,测序引物,抗Flag单克隆抗体,亲和凝胶Flag肽,对照质粒和蛋白,肠
激酶。
8333 重组噬菌体抗体系统
重组噬菌体抗体系统 (RPAS)纯 化 模 块 的 试 剂 盒 由 AmershamPharmacia公司提供。该系统用于可溶性单链片段易变 (ScFv)抗体的 一 步 亲 和 纯 化,该
抗体携带一个C末端13个氨基酸的多肽标记 (ETag),基于Pharmacia公司的
抗ETagSepharose高效介质进行纯化。重链和轻链基因从鼠杂交瘤或脾细胞中
分离,并利用RPAS鼠ScFv模块拼接入一个ScFv基因。一旦构建成功,ScFv基因插入到表达载体中,产生噬菌体标记物或含有ETag的可溶性ScFv抗体。
ETag被共价结合到Sepharose上NHS活化的抗ETag单克隆抗体所识别,进
行ScFv抗体的亲和纯化。该试剂盒包括预装填的抗ETag亲和柱,饱和结合容
量约07mgScFv,10次纯化用缓冲液 (抗Tag柱可使用至少20次)和柱接头。
8334 融合标记的检测和纯化:STag系统
该蛋白标记系统是基于15个氨基酸的STag肽与胰腺核糖核酸酶A的一个
104个氨基酸的S蛋白的相互作用,形成具有酶活性的复合物。由Novagen公司
提供。检测STag融合蛋白也可利用试剂盒,包括STag快 速 定 量 测 定 试 剂 盒
和根据印迹检测融合蛋白的STagWestern印迹试剂盒。
8335 T7Tag亲和纯化试剂盒
Novagen公司还提供了T7Tag亲和纯化试剂盒,用于快速免疫亲和纯化带
有11个氨基酸的T7Tag序列 (也就是T7基因10蛋白的最初11个氨基酸)的
目的蛋白。利用目的蛋白结合到抗T7Tag单克隆抗体上,而该单克隆抗体又与
交联的琼脂糖珠共价结合进行纯化。结合蛋白通常在pH=22洗脱,但需要中
和缓冲液,防止蛋白质暴露于低pH环境。处理能力随目的蛋白不同而不同。试
剂盒包括T7Tag抗体琼脂糖,缓冲液和纯化柱。
8336 基于麦芽糖结合蛋白的融合
NewEnglandBiolads公 司 提 供 了 一 种 基 于 目 的 蛋 白 与 麦 芽 糖 结 合 蛋 白
(maltosebindingprotein,MBP,由malE基因编码)融合的试剂盒。该融合子
利用一个强启动子和 MBP翻译起始信号进行大量表达,对 MBP的一步亲和纯
化就可以纯化得到融合蛋白。据称该系统融合蛋白产率每升培养液可达100mg。

230
该试剂盒包括pMAl载体,其中含有目的基因克隆位点,也包括编码特定蛋白酶
识别位点的序列,如Ⅹa因子,可经直链淀粉树脂亲和柱纯化后裂解目的蛋白。
8337 亲和几丁质结合标记的Intein介质纯化
NewEnglandBiolads公 司 还 提 供 了 基 于 蛋 白 质 拼 接 机 制 研 究 的 试 剂 盒。
IMPACTI系统利用了酿酒酵母 (Saccharomycescerevisiae)VMA1基因的蛋白
拼接部分—Intein。Intein经 改 造,可 在 低 温,2巯 基 乙 醇 或 半 胱 氨 酸 等 硫 醇 存
在的条件下在其N末端自裂解。编码目的蛋白的基因插入到pCYB载体的多克
隆位点 (MCS),在目的蛋白的C末 端 和Intein基 因 N末 端 之 间 形 成 融 合 (图
87)。编码一小段 来 自Bacilluscirculans的5kDa大 小 几 丁 质 结 合 域 (ChBD)的DNA接到Intein的C末端,进行这三部分融合蛋白的亲和纯化。诱导Ecoli表达系统,得到的粗细胞提取液经过几丁质柱时,融合蛋白结合到几丁质柱上,而其他所有杂质则通过柱子被洗掉。然后融合子在DDT或2巯基乙醇存在条件
下,于4℃放置过夜,在柱上进行Intein介质的自裂解,释放目的蛋白而Intein几丁质结合域融合部分仍结合在柱上 (图87)。
图87 IMPACTT7亲和融合原理

231
834 融合蛋白纯化实例
此处介绍几个利用融合蛋白 纯 化 的 实 例:①利 用 多 肽 的C末 端 和 N末 端
融合来 稳 定 和 提 高 小 肽 激 素———Ecoli生 产 的 包 含 体 (尿 抑 胃 素)的 纯 化 特
性,可以得 到 适 合 于 临 床 研 究 的 高 纯 度 的 人 表 皮 生 长 因 子;②利 用 C末 端
ArgLys融合,有助于利用离子交换色谱 纯 化Ecoli可 溶 性 胞 内 酶,该 技 术 适
合于生产大量的高度纯化的蛋白;③利用小肽融合,通过免疫亲和色谱 从 培 养
酵母的胞外基 质 中 纯 化 菌 落 刺 激 因 子 (CSF1),该 方 法 能 够 生 产 用 于 研 究 的
高纯度蛋白。
8341 人表皮生长因子 (EGF,尿抑胃素)的纯化
典型的利用融合蛋白前体纯化EGF的过程:(1)提 取 8L发 酵 罐 培 养 Ecoli细 胞,培 养 基 为 改 良 的 M9,培 养 至
670nm吸光度 约 为30时;离 心 收 集 细 胞,于1L01mol/LTrisHCl(pH=80)溶液中均质化破碎,离心回收包含体;将包含体悬浮于1L5mol/L尿 素,
40mmol/LTrisHAcNaOH (pH=90)溶 菌 缓 冲 液 中,超 声 波 处 理,离 心
澄清。(2)重折叠和氧化 EGF于5mol/L尿素中进行重折叠和氧化,将溶解的包
含体于溶菌缓冲液中4℃保持24h,以 HCl调节pH=55,离心澄清。(3)预处理和色谱分离 以polyminP沉淀核酸;100mlSPSepharose色谱
柱 以 40mmol/L Tris醋 酸,5mol/L 尿 素 溶 液 (pH=55)平 衡,并 用 含
400mmol/LNaCl的上述缓冲液洗脱EGF;超滤浓缩至8mg/ml,并 于40mmol/LTris醋酸缓冲液 (pH=55)中透析。
(4)酶消化及再次色谱分离 10mgEGF用胰 蛋 白 酶 (用 量1/1000)进 行
消化,并利用阳离子交换 HPLC分析产生的EGF,然后通过制备 HPLC再次纯
化EGF。
8342 转氨酶的纯化
C末端聚ArgLys融合结合 羧 肽 酶B (CPB)消 化 可 使 天 然 蛋 白 通 过 阳 离
子交换 色 谱 高 纯 度 分 离,该 融 合 方 法 已 被 用 于 纯 化 细 菌 天 冬 氨 酸 转 氨 酶
(EC2611)。转氨酶经克隆并与另外7个带正电的氨基酸残基在C末 端 融
合,于Ecoli中表达。CPB消 化 的 优 点 在 于 可 以 特 异 性 地 降 解C末 端 的 碱 性
氨基酸 (Arg或Lys),然后中止反 应,因 此 融 合 蛋 白CPB反 复 消 化 并 不 会 引
起酶活性 的 明 显 变 化。得 到 的 融 合 蛋 白 也 具 有 与 天 冬 氨 酸 转 氨 酶 同 样 的 专
一性。酶的纯化过程如下:(1)提取 直接以10μl重组Ecoli甘油保存液接种于含 M9复合盐和酪蛋

232
白氨基酸的摇瓶 (250ml)中振荡培养过夜,接种到10L发酵罐中,在同一培养
图88 天冬氨酸转氨酶纯化SDSPAGECrude—粗溶菌液;NA—经过第一次离子交换柱后
未结合的蛋白;04mol/LNaCl—第一次离子交换
柱洗脱液;CPB—羧肽酶B消化的洗脱液;
NA′—消化后经过第二次离子交换柱未结
合的转氨酶;04mol/LNaCl—第二
次离子交换柱洗脱液
基中生长至670nm吸光度为25。离心
收 集 细 胞,于 500ml 的 50mmol/LMesNaOH溶 菌 缓 冲 液 中 均 质 化 破 碎
细胞,离心澄清。上清液中含有可溶性
的胞内酶,用于离子交换色谱。(2)离子 交 换 色 谱 将 澄 清 的2ml
溶菌液 通 过SPSephadex阳 离 子 交 换 柱
(50mm×200mm)分 离,流 速 为10ml/
min,以1L50mmol/LMesNaOH 缓 冲
液 (pH=65)洗涤色谱柱,再以1L含
有075mol/LNaCl的该缓冲液洗脱。(3)多聚精氨酸的消化及再次色谱
分 离 20℃下 以50mlCPBSepharose消化 已 部 分 纯 化 的 天 冬 氨 酸 转 氨 酶
45min,10L50mmol/L MesNaOH 缓
冲液 透 析3h,再 次 色 谱 分 离 (同 前)。未吸附部分中含有纯化后的转氨酶。纯
化过程SDSPAGE见图88。天冬氨酸转氨酶的纯化结果列于表
88中。
表88 天冬氨酸转氨酶的纯化
样 品 体积/ml 蛋白/mg 酶活/U 比活性/(U/mg) 回收率/%
粗酶 400 2460 70100 28 100色谱柱1次分离 300 662 54300 82 775消化 270 575 46000 80 656色谱柱2次分离 500 275 39800 145 568
8343 Flag肽菌落刺激因子1的纯化
Flag肽用于免疫亲和纯化,序列为AspTyrLysAspAspAspAspLys,包含
肠激酶裂解位点。由于含有酵母分 泌 序 列,Flag肽 和 菌 落 刺 激 因 子1 (CSF1)融合蛋白可由酵母细胞分泌。分泌序列在排出细胞 时 于 细 胞 膜 上 被 蛋 白 酶 所 裂
解,而FlagCSF1融合蛋白则在培养基中积累。纯化过程如下:(1)提取 重 组 酵 母 (Saccharomycescerevisiae)于 复 合 培 养 基 中30℃培

233
养,直至达到生长稳定期,离心,保留上清液,将上清液经045μm膜过滤。(2)免疫亲和色谱 过滤后的1L上清液加入50mmol/LHepesNaOH (pH=
70,含01mmol/LCaCl2 和015mol/LNaCl)缓冲液;以1ml免疫亲和色谱
柱分离过夜,流速为1ml/min;以50ml含01mg/mlCaCl2 的PBS缓冲液洗涤
色谱柱,再以01mol/L的甘氨酸HCl(pH=30)缓冲液10ml洗脱FlagCSF1。(3)Flag肽 的 裂 解 将 收 集 的FlagCSF1配 制 在10mmol/L的 TrisHCl
(pH=80)溶液中,以1mol/L的NaOH调节pH为80,进行肠激酶消化。
84 包含体的初步纯化
重组DNA技术促进了许多重要的药用蛋白制品以及具有商业价值的蛋白制
品的生产。同源或非同源蛋白质的高水平表达经常会引起宿主细胞质中高浓度无
定形不溶性聚合物 (即包含体)的形成。本节讨论包含体的一些性质,并介绍制
备包含体最常用的一些方法。由于大部分数据都是在Ecoli中得到的,本节将
着重介绍该宿主细胞包含体的制备和纯化。
841 影响包含体形成的因素
重组蛋白在Ecoli中高水平表达可能会得到占细胞总蛋白40%~50%的表
达蛋白质。蛋白质处于这样高的表达水平,经常会在细胞质中形成包含体,有时
每个细胞中可能存在不止一个包含体。非同源蛋白表达时包含体的形成最先报道
于Ecoli中胰岛素原的制备。大多数关于包含体形成的研究是利用Ecoli作为宿主细胞,而其他宿主在
高水平表达时也能形成包含体,包括酵母菌、芽孢杆菌以及昆虫细胞等。蛋白质
的性质、表达速度、表达水平、生长温度以及培养基组成等对于不溶性蛋白包含
体的形成有时都有影响,但各种因素对蛋白质的影响可能不会相同。
842 细胞匀浆中包含体的分离
通常通过离心或过滤从细胞匀浆中回收包含体。最普通的回收方法是利用实
验室规模的离心机,10000~20000g离心10~30min。对 于 各 种 不 同 的 蛋 白 质,离心条件有所不同,因为包含体的大小可能随蛋白质和表达水平的不同而不同。对于大规模回收包含体,也可利用其他离心设备,比如连续碟式离心机。
843 包含体的洗涤
洗涤包含体的目的是为了使目的蛋白随后更方便地重折叠和纯化,洗涤也可
能会除去蛋白酶,从而防止在后续纯化过程中蛋白质的不稳定,洗涤还可在包含
体溶解之前除去DNA、脂质以及可溶性的细胞蛋白和其他组分。是否进行洗涤
取决于随后采用的纯化步骤以及包含体的初始纯度。由于未破碎的细 胞 会 与 包 含 体 共 沉 淀,通 常 将 细 胞 于 高 压 下 通 过 Manton

234
Gaulin匀浆器作用2~3次,以有效破碎95%以上的细胞。为得到比较纯的包含
体,一般推荐用水或碱性缓冲液洗涤包含体数次。除去包含体杂质有许多不同的
洗涤方法,如牛促生长激素包含体的细胞匀浆 液 中 加 入 溶 菌 酶,EDTA和 脱 氧
胆酸盐,可除去大部分 核 酸,脂 多 糖 和 磷 脂,但 不 溶 解 牛 促 生 长 激 素。TritonX100、蔗糖、尿素、脱氧胆酸盐和辛基葡糖苷都已成功用于除去包含体中的杂
质。洗涤的pH也很关键,因为过高pH下包含体一般更容易溶解,而对Ecoli包含体,低pH洗涤液 (pH=3~6)通常是无效的,因为Ecoli主要的污染蛋
白在该pH范围也是 不 溶 的。由 于 包 含 体 中 的 各 蛋 白 都 有 其 各 自 独 特 的 溶 解 特
性,没有一种洗涤缓冲液适用于所有的包 含 体。洗 涤 缓 冲 液 中 含 有 蛋 白 酶 抑 制
剂,对于防止不希望发生的蛋白质水解也是必要的。抑制剂的选择取决于目的蛋
白酶,但是蛋白酶可能是未知的。一般丝氨酸蛋白酶抑制剂如PMSF经常加入
到洗涤缓冲液及细胞匀浆缓冲液中。经验表明,进行有效洗涤通常有益于抑制未
知来源的蛋白酶。影响包含体洗涤效率的主要因素包括:包含体蛋白的溶解性,pH,离子强
度,温度,暴露时间,均质化 效 果,缓 冲 液 与 蛋 白 质 的 比 例,添 加 剂 浓 度 (尿
素,去污剂)等。一般而言,较高的pH对除去污染物更为有效,只要不溶解包含体,不会对
目的蛋白进行化学修饰,利用高pH缓冲液经常能够制备高质量的包含体。这里
介绍碱性pH条件下洗涤包含体的方法。碱性pH条件下洗涤包含体:4℃10000g离心20min收集包含体,弃去上清
液,注意不要破坏包含体沉淀;在包含体上部如果有明显的细胞碎片,可用刮刀
刮去表层,包含 体 通 常 为 浅 褐 色 至 微 红 色;每 克 包 含 体 加 入20ml洗 涤 缓 冲 液
(10mmol/LNaHCO3,1mmol/LEDTA,pH=100),将 含 有 包 含 体 的 试 管 置
于冰上,20s超 声 波 脉 冲 处 理,重 新 悬 浮 包 含 体,注 意 超 声 处 理 时 保 持 低 温;
10000g离心20min,重新悬浮洗涤,直至上清液澄清无色;通过SDS凝胶电泳
或其他适合于该蛋白的分析方法,确保包含体中的目的蛋白不会由于这样的处理
而溶解。
85 膜蛋白的纯化
膜蛋白与磷脂双分子层的缔合模式决定了所采用的纯化方法。如果蛋白嵌入
在疏水相中,将内在膜蛋白以可溶形式释放则需要破坏磷脂双分子层或者裂解膜
定位多肽链,而且所有后续的纯化操作中,都会要求膜内疏水界面与水介质之间
屏蔽,通常可利用去污剂来实现。而对于外周膜蛋白,可以通过改变缓冲液离子
强度或者在更宽松的条件下引起一定程度的变性,将蛋白从膜上分离。

235
由于膜蛋白一般只占细胞总蛋白的很小一部分,任何污染都会产生很大的影
响,因此,最有效的初始步骤就是得到尽可能纯的适当的膜制剂。一般从全细胞
或组织开始。有些细胞 (如红细胞和肿瘤细胞)通常不经过普通的胞溶作用,就
可以得到原生质膜或含有分散的小蛋白集合的典型微泡。脊椎动物和无脊椎动物
的视网膜也是较好的材料,膜上含有主要的光反应结构,通过温和搅拌,几乎可
以完整无缺地从细胞上释放出来。膜片段或亚细胞器的分离可通过不同的方法实
现,一般使用离心技术,其他的方法包括多聚物双相分离、高压自由流动电泳以
及 (免疫)亲和色谱和凝胶过滤色谱。
851 蛋白质的溶解性
有许多方法可以快速确定目的蛋白是内在蛋白还是外周蛋白。大多数涉及膜
的操作都包括以下的洗涤或剥离过程。现在电荷转换电泳也已成功应用,但更快
速的方法是利用 去 污 剂TritonX114中 的 相 分 配,其 优 点 是 随 着 温 度 从0℃升
高,微胶囊发生聚合,当温度高于20~25℃,最终分离出来形成第二相,只有
比例非常小的TritonX114残留在水相中。因此,溶解的内在膜蛋白在0~4℃优先分配到富含去污剂的区域中,可通过离心回收,而外周蛋白几乎总是在水相
中。在应用此方法前,确保目的蛋白在采用去污剂处理时能保持其生物活性。利用TritonX114的相分配方法:(1)去污剂的纯化 加入10gTritonX114 (Sigma公司或Calbiochem公司)
和8mg丁基羟基甲苯至pH=75、015mol/LKCl的200ml20mmol/L磷酸钾溶
液中;冷却混合物至0℃左右,离心除去不溶性物质 (3000g离心10min),然后
将可溶性去污剂于35℃保持18~20h;缓慢离心 (100g离心10min),保留去污
剂相 (下相),纯化的去污剂可于室温下保存3个月以上。(2)膜的 溶 解 与 分 配 于0~4℃加 入 纯 化 的 TritonX114至10mmol/L
TrisHCl或磷酸缓冲 盐 的 膜 溶 液 中,pH 为74,其 中 含 有 最 多150mmol/L的
NaCl或KCl,最 终 去 污 剂 浓 度 可 达2%~3% (20~30g/L),膜 蛋 白 浓 度1~3mg/ml;0~4℃保持1h,100000g离心1h,除去不溶物质;于合适的离心管中
以6%蔗糖溶液缓冲层覆盖可溶性部分,加热至25~30℃,在温度高于20℃下
100g离心10min;去 污 剂 富 集 相 出 现 在 试 管 底,类 似 油 层。水 相 可 用05%(5g/L)TritonX114重新平衡,重复该过程。利用水相缓冲液同样可再次得到
去污剂相。
8511 外周膜蛋白
有许多除去膜外周成分的方法,比较温和的处理方法包括使用低离子强度缓
冲液、01~1mmol/LEDTA。为 防 止 蛋 白 质 不 可 逆 变 性,pH 应 在6~8范 围
内,在低温下操作,应避免使用碘 离 子 和 二 碘 水 杨 酸 等 阴 离 子,因 为 作 为 破 膜

236
剂,它们具有类似去污剂的溶解 特 性。处 理 过 程 中,有 时 应 考 虑 加 入 蛋 白 酶 抑
制剂。但是不少外周膜蛋白并不会在这些较温和的条件下释放,需要更为强烈的处
理方式,这常常会导致蛋白质变 性,而 且 还 是 不 可 逆 的。这 样 的 处 理 条 件 包 括
6mol/L盐酸胍,8mol/L尿素,1mmol/L对氯汞苯甲酸,pH=20~30的酸和
pH=95~110的碱等。碱处理好于酸处理,因为酸有时会导致 蛋 白 质 沉 淀 而
干扰随后的膜回 收。为 防 止 肽 键 的 断 裂,极 端pH 条 件 下 膜 的 剥 离 通 常 在0~4℃下进行,并尽快恢复至中性。
经过各种处理,50%以 上 的 总 蛋 白 可 从 膜 上 释 放,但 经 常 会 引 起 膜 的 囊 泡
化、破碎或转化,因此,回收条件、离心、过滤等操作都应优化调整以保证膜的
最大得率。
8512 内在膜蛋白
内在膜蛋白根据与双分子层疏水相结合的结构比例可以分为四类。第一类通
过1~2个跨膜蛋白片段固定在膜中,第二类与脂肪酸或脂质共价结合,这两类
中大部分蛋白都是在水相中的,一般其生物活性并不涉及脂质双分子层。双分子
层只是作为结构载体或者蛋白定位和结构组成的方式,例如大量的酶类,包括肽
酶、酯酶和磷酸酶等。此类蛋白经常可利用蛋白酶 (2%,pH=70,25~37℃,
3h)或磷酸酯酶 (比如Sigma公司磷酸酯酶C,2%,pH=74,25~30℃,3h)从膜上释放而不会破坏其天然活性。第三类蛋白是目前相对较少但却非常重要的
一类,具有惟一的跨膜片段,胰岛素、生长因子是此类蛋白的代表。它们与第一
类蛋白的区别在于比较重要的分子部分在膜的内外表面,其完整的生物活性不仅
取决于这两个亲水域,也取决于跨膜片段。但在水相的球形部分可通过蛋白质水
解作用从分子其他部分游离出来,此时,胞外域仍然保持一定的活性,但由于已
经无法与胞内域联系,其完整的生物活性受到破坏。第四类内在膜蛋白分子多次
横贯双分子层,分子结构中的主要部分在疏水相中。转运蛋白和视紫红质类受体
属于此类。虽然蛋白质水解也可释放此类蛋白片段进入水相,但常常需要有机溶
剂或去污剂将其从双分子层上溶解。有机溶剂脱脂是基于大部分磷脂很容易溶解在多数纯有机溶剂中,而大多数
内在膜蛋白 却 不 溶 解 的 原 理。采 用 有 机 溶 剂 处 理 冻 干 膜 (1ml/mg),溶 剂 为 氯
仿,氯仿与甲醇、乙醇或丁醇的混合物 (比例为2∶1、1∶1或1∶2,体积比)等,在此条件下,大多数膜蛋白迅速沉淀。
去污剂加入到膜制剂中,则迅速分配进双分子层中,直至达到某临界点膜不
再稳定,开始破裂。如果加入足量的适当去污剂,膜最终溶解为脂质和蛋白质成
分,蛋白质存在于去污剂单分子层所包围的溶液中,去污剂单分子层介于蛋白质

237
疏水表面与大量的水介质之间。但是需要注意,不同的膜和蛋白对不同的去污剂
反应方式有所不同。去污剂的选择应该尽可能纯,成本低,保存时间不宜长。去
污剂对膜的处理通常会导致结构的破裂,但这并不一定意味着目的蛋白作为单分
子已经释放。有时,可能会产生微泡或者维持一种寡聚状态。例如细胞骨架微绒
毛核在双分子层已经完全溶解的条件下仍然主要以未离解的形式回收得到。
852 膜蛋白的纯化
蛋白质一般可以根据分子大小、电荷性或疏水性及专一性进行分离。对于内
在膜蛋白虽然同样适用,但其疏水特性和溶解所要求的特殊条件经常成为关键的
限制因素,而对于外周膜蛋白,采用水溶性多肽的处理方法比较合适。在此主要
介绍内在膜蛋白处理方法的应用。
8521 根据分子量的分离
(1)酸化有机溶剂 大部分糖基 “软性”树脂 (Sepharose,Sephadex,Superose)并不适用于纯的或酸化的有机溶剂,此时,其溶胀效果并不满意,分离
能力也大大降低,而且可能会严重吸附蛋白。相反,合成的聚合树脂 (如羟化聚
醚)和 羟 丙 醇 葡 聚 糖 (SephadexLH20和 LH60)在 该 条 件 下 却 可 用 于 常 规
HPLC或者FPLC系统。此外,硅胶载体在有机混合物中作用充分,但在酸性溶
液中缓慢降解。聚丙烯酰胺在酸性水溶液中表现良好 (即使甲酸达到70%),但
在非水相条件下效果不太满意。接触有机溶剂的所有器件都应由玻璃或聚四氟乙
烯制成。在酸化条件下,蛋白质会有部分甚至完全变性。采用强酸条件时,尽可
能保持较短的操作时间和较低的温度,并在使用后以较温和的溶剂清洗树脂。(2)去污剂和变性剂 使用水溶性去污剂的凝胶过滤经常是一种非常有效的
初步分离步骤。实际上所有树脂都可成功应用各种去污剂和变性剂 (尿素、盐酸
胍),但需要注意以下几点:
① 柱缓冲液应含有接近或高于其CMC的去污剂,有助于在色谱过程中确保
蛋白质不会聚集,沉淀或丧失活性;
② 去污剂与蛋白质的结合会产生比蛋白质本身分子量更大的分子,高孔隙
率树脂 (如Sepharose)易实现有效分离;
③ 膜中的脂质与去污剂会形成混合胶束;
④ 少数情况下,蛋白质可能与树脂不可逆吸附,特别是葡聚糖基载体,改
用惰性较强的材料如聚丙烯酰胺可以弥补这一点。
8522 根据电荷分离
(1)电泳 聚丙烯酰胺凝胶电泳和电荷转换电 泳 可 以 应 用 于 内 在 膜 蛋 白 分
离。聚丙烯酰胺凝胶电泳在处理内在膜蛋白时,电泳可在非变性去污剂如TritonX100或脱氧胆 酸 中 进 行,但 有 时 溶 解 情 况 不 好,还 会 出 现 聚 合 和 “拖 尾”现

238
象。电荷转换电泳利用了内在膜蛋白在其暴露的疏水区域与去污剂结合的特性。由于使用的非变性去污剂可能为中性,或带有电荷,结合到蛋白质上就可能改变
其带电状态,因而改变电泳迁移率。因此,不同的去污剂可以引起不同的电泳行
为,可以作为一种天然蛋白是否含有重要的暴露疏水区域 (因而是否属于内在膜
蛋白类)的指示剂。对于回收少量非常纯的蛋白,特别是在应用电洗脱时,等电
聚焦是一种很有效的实用工具。膜蛋白具有很宽的等电点范围,假如能得到非聚
合状态的蛋白,去污剂中的等电聚焦是非常成功的,因此同一蛋白的不同磷酸化
形式也可被分离。内在膜蛋白的处理方法与一般水溶性蛋白处理方法略有不同,只是包含了去污剂,通常是非离子型去污剂 (两性离子去污剂也可采用),以防
止其掩盖蛋白质的原有电荷。(2)离子交换色谱 常用于蛋白质纯化的所有离子交换系统也可用于纯化内
在膜蛋白。该方法与等电聚焦一样,也需要去污剂,而且去污剂的选择受到保持
蛋白质天然结构和带电特性要求的影响。非离子型去污剂通常比较适合,而两性
离子类也可采用。非离子型的CHAP系列去污剂,DEAE类载体和CM类载体
以及羟基磷灰石已经成功用于膜蛋白分离。(3)色谱聚焦 该技术所用的载体在pH=3~12范围内抗降解,最有效的
范围是pH=5~9,它可与中性有机溶剂,变性剂如8mol/L尿素或6mol/L盐酸
胍以及中性或两性离子去污剂一起使用。如果蛋白质保持溶解而且不聚合,该方
法快速而有效。去污剂如毛地黄皂苷和单月桂酸钠在高于其CMC浓度条件下使
用处理视紫红质时,该方法很有效。
8523 亲和色谱
在膜蛋白领域,亲和色谱已成为一项特别有价值的技术。由于许多膜蛋白都
是糖基化蛋白,与碳水化合物结合的凝集素是很有效的。它们很容易与多种载体
结合,而且在非变性去污剂中十分稳定。由于其具有不同的糖特异性,因而可用
于区分糖蛋白。含有糖基的去污剂应进行检验以确保不会与所用凝集素的活性位
点相结合。当目的糖蛋白含有一个末端唾液酸残基时,大部分其他膜糖蛋白可利
用不同特异性凝集素组合 (例如Lensculinaris植物血球凝集素和小麦胚凝集素)从混合物中除去。未结合的含唾液酸部分可用唾液酸苷酶处理除去这一残基,从
而产生一个新的末端糖基而被凝集素亲和载体所识别。
86 治疗用蛋白质的纯化
治疗用蛋白可以分为四类:调节因子 (包括激素,细胞分裂素,淋巴细胞活
素以及其他细胞生长和代谢的调节因子),血制品 (包括血清中的血液因子和酶
类血纤维蛋白原激活因子),疫苗和单克隆抗体。

239
无论何种污染物,在随着治疗用蛋白一起使用甚至进入人体时,即使极低的
剂量都会带来很大的风险,甚至是致命的。因此,对于治疗用蛋白,污染问题显
得尤为重要,对此类蛋白的纯化要求是最 高 的,其 产 品 纯 度 的 要 求 也 是 最 严 格
的。治疗用蛋白质的纯化必须考虑:(1)蛋白质来源 因为特定的杂质存在与否取决于制备路线。传统来源是动
物组织或人血清,现在也包括利用微生物培养、各种病毒感染的哺乳动物细胞、所有原核和真核重组宿主载体表达系统以及鼠杂交瘤等多种系统,因此杂质来
源更为广泛。除了通 常 的 污 染 蛋 白、DNA以 及 目 的 蛋 白 的 聚 合 或 降 解 产 物 外,还包括细菌内 毒 素 等 热 原 物 质 和 动 物 组 织 或 细 胞 中 的 病 毒 以 及 培 养 基 的 部 分
成分。(2)纯化过程可能带来的污染 比如一些色谱基质,特别是亲和基质由于长
时间使用可能引起的渗漏,也会污染蛋白产品。本节主要介绍治疗用蛋白质的纯化,重点是在治疗用蛋白质中可能会出现的
各种污染物类型。在实际应用中,应充分分析评估特定污染物含量,并设计和确
定治疗用蛋白质的纯化工艺。
861 治疗用蛋白纯化的关键问题
治疗用蛋白的纯化方法与前面提到的其他蛋白的纯化方法没有很大区别,使
用的技术很多是相同的。由于此类蛋白的特殊性,纯化最关键的问题是蛋白质纯
度和污染物的检测。常用蛋白质纯度检测技术包括各种凝胶电泳检测和 HPLC检测等,治疗用
蛋白在必须满足传统色谱和电泳纯度标准的同时,还必须有一套完全独立的分析
技术,用于测定临床上的特定污染物。
8611 免疫法
免疫法根据可能污染物特定抗血清的产生原理,既可用于定量也可用于定性
测定相应污染物。适用于此类分析的技术比较多,读者可参考有关免疫化学应用
的书籍,这里介绍一些实例。(1)抗血清的生产 对于重组DNA系统,如Ecoli中生产的蛋白质,制备
能够识别多种宿主细胞蛋白的抗血清,可 通 过 抗 宿 主 细 胞 的 溶 解 液 而 得 到。抗
EcoliK12广谱抗血清的制备 方 法:培 养EcoliK12至 对 数 生 长 期,离 心 收 集
菌体;将032gEcoli菌体悬浮于1ml溶解缓冲液 (80mol/L尿素,2%诺乃
洗涤剂P40,5%2巯基乙醇)中,冻融3~5次溶解细胞,于200μl溶菌液中加
入300μl水和15mlFreund完全佐剂,混合乳化;将2ml乳化液注射到白兔背
部20~40处,各注射点都应在皮层内;8周后,每隔3周于单一位点肌内注射
相似剂量的乳化于Freund不完全佐剂中的Ecoli溶菌液;第五次注射后10天,

240
放出兔血制备血清。这里,抗原制剂是通过去污剂溶解从而造成宿主细胞蛋白的变性而制备的。
当产物通过同样的方法从重组Ecoli中释放时,这样的操作是可取的。对于重
组哺乳动物细胞体系,释放产物需要的条件较温和,抗原的制备也应该以相似的
方式进行。另一种策略,特别是对于重组DNA体系生产的蛋白质,就是构建一
个与生产体系完全相同的宿主载体系统,但缺失编码产物的插入基因,然后经
过同样的纯化过程,得到一系列与产物生化性质相似的蛋白,用以产生抗体。(2)免疫化学技术 通过多种技术,如凝胶扩散、琼脂糖凝胶电泳和SDS
聚丙烯酰胺凝胶电泳分离之后,可采用利用抗体显示污染物的技术。其中最有效
的可能就是多种免疫印迹技术 (Western印迹)。
Ecoli蛋白SDS凝胶免疫印迹测定:将纯化蛋白质样品分成几部分 (200~500μg)进行SDSPAGE检 测,样 品 中 也 应 包 括Ecoli本 身 (01~10μg总 蛋
白);将蛋白质从电泳胶上电转移至硝酸纤维素纸上;将硝酸纤维素纸置于含适
当封 阻 蛋 白 (例 如 3% 牛 血 红 蛋 白 或 5% 干 脱 脂 乳)的 磷 酸 缓 冲 盐 溶 液
(8mmol/LNa2HPO4,15mmol/LKH2PO4,014mol/LNaCl)中保持1h;室
温下于50ml含025ml兔抗Ecoli血清的封阻缓冲液中温和振荡过夜;洗涤硝
酸纤维素纸,换 洗 数 次 封 阻 缓 冲 液,每 次 洗 涤10min;以 放 射 性 碘 标 记 的 抗 兔
IgG (107Bq/50ml)培养4h;用PBS洗涤数遍;干燥硝酸纤维素纸,并在装有
增光屏的暗箱中通过放射自显影1~3天在KodakXS1胶片上显示条带。该技术已经有许多改进,以提高灵敏度和操作的方便性。最常用的两种方法
是抗血清的亲和纯化和标记酶的使用 (碱性磷酸酶或过氧化物酶),而不用放射
性标记的抗体显示抗体抗原复合物。(3)免疫分析技术 部分纯化的宿主细胞蛋白制品一般认为可能有污染物,
由此引起的抗体已被用于放射免疫分析 (RIA),放射性标记的宿主细胞蛋白被
冷 (未标记)的宿主细胞蛋白所代替,用于进行宿主细胞蛋白含量的定量测定。该方法在许多方面可以改进,在建立此类 操 作 时,读 者 可 参 考 有 关 免 疫 分 析 的
书籍。
Ecoli蛋白的放射性免疫分析方法:(1)抗血清滴定曲线 各试管中装有100μl测定缓冲液,100μl示踪物 (放
射性碘标记蛋白 制 剂)以 及 抗Ecoli抗 血 清 各100μl,稀 释 范 围 从1/500~1/
500000,或100μl测 定 缓 冲 液 (非 特 异 性 结 合,NSB);4℃培 养 过 夜;加 入
300μlPEG/丙 种 球 蛋 白 试 剂 (5g聚 乙 二 醇6000,30mg牛 丙 种 球 蛋 白,20ml005mol/LTrisHCl,pH=85)沉淀抗体结合部分;4℃培养1h,2500g离心
30min,吸去上清液,计量沉淀。

241
(2)测定 选择适当浓度的抗血清,在0抗原量时有15%~40%的特异性
结合 [特异性结合=结合的比例 (%)-NSB],记为B0;各试管中装有100μl测定缓冲液,100μl示踪物,100μl稀释的抗血清以及100μl未标记的Ecoli蛋
白制剂,每次稀释2倍,每个试管稀释范围从1pg到1μg;进行培养和分离,测
定工作范围为可以得到抗体结合示踪物完全置换的Ecoli蛋白制剂的浓度范围,从Ecoli蛋白浓度零水平下降到非特异性结合水平;测定试管内产物的污染水
平,从置换曲线读出结果。此外,还有一些超灵敏染色技术 (比如银染色法)可以用于蛋白质污染物的
检测,一般免疫分析灵敏极限接近5mg/L,免疫印迹接近25~50mg/L,银染色
接近200~400mg/L。不过各种技术都有其各自的优势,免疫分析具有极高的灵
敏性,免疫印迹法可以提供关于抗原的定性和定量信息,而银染色法不需要依靠
实验动物体内产生抗体来检测污染物。
8612 核酸杂交技术
皮克数量级DNA的检测技术在很大程度上依靠宿主细胞或者用于生产的重
组载体与放射性标记DNA探针的杂交。
EcoliDNA的斑点印迹杂交方法:将硝酸纤维素膜浸于2倍浓度的标准柠
檬酸盐缓冲液 (SSC,以20倍浓缩制备,1753gNaCl和882g柠檬酸钠溶解于
800ml水中,氢氧化 钠 调 节pH 至70,制 备1L溶 液)中,将 参 考 (未 标 记)
EcoliDNA制剂以01pg/ml~100ng/ml浓度范围作为系列斑点进样硝酸纤维
素滤纸,使用商品斑点印迹设备 (如BioRad公司)进样体积可多达150μl,另
一斑点区域,待测蛋白浓度范围为1μg/ml~10mg/ml;晾干滤纸,并于80℃烘
烤1h固定斑点;将滤纸于装有最小体积 (对5cm×5cm滤纸,最少7ml)的预
杂交溶液 (1%Ficoll,1%聚乙烯吡咯烷酮,1%牛血清白蛋白,5倍浓度SSC,
05ng/ml鲑鱼精液DNA)的 密 封 塑 料 袋 中,50℃缓 慢 振 荡 培 养 过 夜 进 行 预 杂
交;除去袋中的预杂交溶液,装入同样体积的含107 脉冲/min放射性标记EcoliDNA的预杂交溶液,同上培养过夜;从袋中取出滤纸,于50℃2倍浓度SSC,
01%SDS溶液中洗涤3次;晾干滤纸,利用放射自显影测定结合物的放射性,样品中的DNA含量可通过与稀释系列测试比较进行分析。
8613 病毒检测
(1)人类病毒 对于人类组织和血清的纯化蛋白,最为关注的病毒是AIDS病毒 (HIV)和乙肝病毒。利用免疫测试检测病毒抗原的存在,可以检测到蛋白
制剂中的这两 种 病 毒。相 关 试 剂 盒 已 有 许 多 厂 家 生 产,典 型 的 如 HIV抗 体 的
Wellcozyme检验、肝炎抗原的 Hepatest,都已由 Wellcome公司销售。(2)鼠类病毒 在鼠类杂交瘤单克隆抗体中,超过20种鼠类起源的病毒被

242
确定为可能污染物。其中 Hanta病毒 (出血热)和淋巴细胞脉络丛脑膜炎病毒
可能会引起严重的人类疾病,还有许多病毒也可能与人类免疫缺陷有关。检测制
剂中的特定鼠类病毒最有效的方法是鼠抗体形成测试 (mouseantibodyproduction,MAP)。将待测制剂注射入健康鼠体内,经过适当时间后检测鼠血清是否
有目的病毒抗体的存在。由于此类检测方法只能在设施完备的病毒学实验室中进
行,此处不 作 详 细 介 绍。此 类 检 验 是 验 证 治 疗 用 单 克 隆 抗 体 纯 度 的 一 个 重 要
方面。
862 工艺确认
既然许多潜在的污染物已经得到确证,下一步就是设计纯化工艺将这些污染
物从产品中特异性地除去。
8621 DNA一般说来,纯化过程不需要包括特异性除去DNA的步骤,因为通常通过离
子交换步骤就可以有效地去除。其他色谱或大量分离步骤通常也可降低DNA含
量1~2个数量级。
8622 蛋白质
蛋白质也许是最难以从产品中分离的一类污染物,因为在纯化后仍然留在产
物中的蛋白可能具有与目的蛋白相似的分子量、等电点和疏水性。除了在纯化过
程后期再次使用亲和纯化色谱,还没有其他特异性的方法可以去除与目的产物性
质相似的蛋白。也可以采用一些额外的纯化步骤,如阳离子和阴离子交换色谱来
降低这些物质含量,以达到所期望的水平。
8623 聚合或降解产物
受到某些降解的目的蛋白,由于 形 式 不 同 也 看 作 一 种 特 殊 类 型 的 蛋 白 污 染
物。在纯化过程中有时会产生大量的蛋白质变化形式,包括:共价或非共价的二
聚作用或聚合作用;氧化作用,通常在甲硫氨酸残基上;天冬酰胺和谷氨酰胺残
基的脱酰胺作用;不正确折叠;不正确的二硫键形成;β天冬氨酰转移,通过天
冬氨酸β羧基形成β肽键;内切或外切肽酶的裂解等。蛋白的氧化和脱酰胺产物
是其带电荷的衍生物,可以通过离子交换色谱除去。聚合形式可通过凝胶过滤色
谱去除。错误的二硫键形式与原蛋白性质通常差别相当大,可以很容易地去除。其他形式如错误折叠,β天冬氨酰转移以及末端部分降解的产物去除可能要困难
些。如果产物适用,通常制备 HPLC比较有效。
8624 热原
细菌内毒素为脂多糖,其物理化学性质与大多数蛋白质差别相当大。但是尽
管蛋白质纯化技术可以降低热原的水平,但要将其完全去除可能需要特异性的去

243
热原步骤。关于内毒素的性质已有较多介 绍,一 些 特 性 已 被 用 于 设 计 去 热 原 操
作。单体脂多糖 (LPS)分 子 量 为10000~20000,但 在 水 中 容 易 聚 集,形 成 的
聚集物分子量达1000000。因而通过100000分子量截留膜进行超滤是很有效的。相反,对于高分子量的产品,内毒素可以利用螯合剂解聚,通过50000分子量截
留膜超滤,保留产物。内毒素带负电荷,因而也可以使用正电荷膜如聚酰胺或结
合胺膜将其去除。脂肪族聚合物如聚丙烯可结合内毒素,也已用于去热原药物的
制备。应该注意,为避免纯化过程中重新引入热原物质,操作必须十分谨慎。所
有使用的试剂必须是无菌无热原的,最简单的处理方法就是高压灭菌 (120psi?,
20min,循环3次)。玻璃仪器也必须240℃干热灭菌1h。实验室塑料器具如管子
等可以用酸或碱处理去热原。所有操作必须在无菌条件下进行。也有许多试剂如
抗体亲和柱可以不用这种方法处理,对其最好的处理方法就是用无热原缓冲液大
量洗涤,直至洗出液经测试无热原。细菌内毒素可通过鲎测试进行测定。
8625 病毒
类似热原和DNA,病毒颗粒与大多数具有药用价值的蛋白质生物化学性质
不同,通常也不需要特异性步骤将其除去。但是蛋白质产品也可能会受到人体致
病病毒的污染,特别是从人组织或细胞中纯化的蛋白质,因此可能有必要采取一
些特殊步骤使病毒休止。热处理 (60℃,10h)已广泛用于休止血清蛋白中的病
毒。其他方法包括UV照射、致电离辐射以及甲醛或β丙酰内酯化学修饰等。但
具体采用的条件对于特定病毒需要重新确定,因为病毒对于各类休止方法的敏感
性差别非常大。虽然这些方法都可以成功地休止病毒,但它们也可能会引起蛋白
质的变性或化学变化,应谨慎使用。病毒的去除效果或污染水平可通过清除率研
究进行预测。
8626 基质的渗漏产物
亲和基质 (如抗体Sepharose的组合)经过较长时间使用可能不够稳定,结
合的配位体可能会从柱上渗漏下来从而污染产物,或者由于重复使用,亲和柱可
能由于杂质污染而中毒,最终在洗脱液中开始出现杂质。由于这些原因,通常认
为亲和纯化不应是 纯 化 过 程 的 最 后 步 骤,其 后 需 要 进 行 凝 胶 过 滤 或 离 子 交 换
色谱。
? 1psi=689476Pa。

244
9 不同来源蛋白质的纯化
91 微生物细胞培养蛋白质的纯化
微生物来源的蛋白质种类繁多,性质各异,相关的纯化研究较多,在工业上
也有广泛的应用,有不少技术已经比较成熟。随着蛋白质工程、材料领域等研究
的不断发展,又开发出了许多新的纯化技术。本节介绍一些与微生物来源的蛋白
质及相关生物分子纯化有关的问题。蛋白纯化步骤可以分为上游和下游操作,为
方便起见,将下游操作表述为利用澄清后的原料进行色谱分离操作,而上游操作
则为前面的澄清步骤,本节内容只限于下游操作。
911 色谱纯化方法
多种色谱技术可用于微生物来源的蛋白质的纯化,包括盐析、离子交换、疏
水作用、亲和、凝胶过滤、手性、亲硫作用等。每种技术都利用了各种蛋白独特
的物理化学特性。在这些技术中,一般可放大到生产规模的是离子交换、疏水作
用、凝胶过滤以及少量的亲和分离。离子交换、疏水作用和亲和色谱都是利用吸
附技术,可选择性地吸附/解吸原料中一种或多种成分从而实现纯化。实际操作
中,有些方法利用了吸附色谱,比如离子交换的策略,如图91所示。根据原料
的特性和工艺要求,阴离子或阳离子交换色谱可以按正向或反向步骤进行,疏水
作用色谱或亲和色谱也可以按相似的方式以正向 或 反 向 步 骤 进 行。各 种 吸 附 步
骤,无论是正向或反向,都可以在柱或分批处理体系中进行。不管怎样,操作中
都应该考虑到色谱技术的选择和操作模式等方面的问题。
目标为阴离子?如pH>pI ↓
↓
是
是否保留目标,不保留杂质? ↓
是
采用阴离子交换↓
即正向步骤
↓
否
采用阳离子交换↓
即反向步骤
↑
↑否
目标为阳离子?如pH<pI ↓
↓
是
是否保留目标,不保留杂质? ↓
是
采用阳离子交换↓
即正向步骤
↓
否
采用阴离子交换↓
即反向步骤
图91 确定采用离子交换色谱操作的方法
9111 正向或反向步骤
正向吸附步骤不仅可用于选择性吸附、纯化,如果洗脱体积少于原料体积,

245
还可用于浓缩。如果需要选择性吸附,分步或连续梯度洗脱可能较好。应该注意
的是,洗脱缓冲液中的产物可作为下一步处理的原料,因此应预先考虑其流动相
是否适合于下一步操作,否则可能需要大量附加操作 (如透滤,脱盐或溶剂萃取
等),从而增加成本。反向步骤没有浓缩作用,经常起到明显的纯化效果,而且在整个色谱阶段流
动相保持一致,不需要为洗脱而进行流动相调整。
9112 分批或柱处理器
对于正向步骤,建议采用梯度洗脱的柱系统。而对于反向步骤,分批系统可
能在时间和成本上更为有利,因为不需要高解析率的产物解吸附作用。微生物来
源的蛋白质纯化中,对于上述问题应加以考虑,以优化工艺路线。在确定工艺时
也应考虑这些问题,特别是在产物可能用于医药等情况下。
9113 梯度洗脱
大多数吸附操作中,目的蛋白吸附到固定相上,然后经选择性解吸附,从基
质上洗脱下来。高选择性分离时,如亲和分离,吸附作用一般选择性很高,只需
一步洗脱就能达到满意的效果;但对于疏水作用色谱或离子交换色谱,吸附阶段
可能有多种蛋白质结合到吸附剂上。为了达到选择性解吸附,必须仔细控制洗脱
缓冲条件。一种简便的方法就是在柱处理系统中进行梯度洗脱,通常采用线性梯
度洗脱,虽然可能产生凸形或凹形梯度。线性梯度作为时间或体积的函数,在流
动相中会发生缓冲液A对B组成的线性变化。但线性梯度容易制备,已有报道
应用25L纤维素和琼脂糖离子交换色谱柱,其线性梯度有效体积可达400L。制
备和运行线性梯度的操作参考前文 (4542节)。
912 蛋白质离子交换纯化
以下是一些微生物来源蛋白质通过离子交换进行纯化的实例。
9121 酵母酶的分离
酵母富含蛋白质,特别是一些酶类。为了实现有效分离,细胞碎片需预先去
除。以下介绍一种3磷酸甘油醛脱氢酶 (GAPDH),3磷酸甘油酸激酶 (PGK)和磷酸丙糖异构酶 (TIM)从酵母中释放的方法。
将100g干酵母分散于1000ml去离子水中,加入氢氧化钠至pH=10,溶解
细胞3h,在此期间,加入氢氧化钠以维持pH为10。以醋酸调节pH至52,过
滤澄清,滤液中加入硫酸铵至浓度为20% (200g/L),4℃保 持12h。过 滤 除 去
沉淀,滤液中 加 入 硫 酸 铵 至 浓 度 为47% (470g/L),4℃保 持12h以 上,利 用
10μm膜 过 滤,收 集 沉 淀,溶 解 于200ml50mmol/L磷 酸 钠 (pH=70,含
2mmol/LEDTA)缓冲液中,于-20℃保存备用。上述 三 种 酶 从 酵 母 细 胞 释 放

246
过程的活力变化如表91所示。
表91 酶从酵母细胞释放过程中的变化
实 验 阶 段总活力/U
GAPDH PGK TIM
细胞溶解 463050 284850 202500020%(NH4)2SO4 上清液 302460 285420 86025047%(NH4)2SO4 沉淀 189255 208310 752950
注:来源于100g面包酵母细胞。
酵母提取物可利用阴离子交换色谱进行分离,得到部分分离的各种酶。根据
目的蛋白及其所要求的纯度,利用洗脱缓冲液优化色谱分离,以得到所希望的分
离结果。以TIM为例,可通过阴离子交换色谱实现分离。将酵母提取物在10倍体积
的10mmol/L三乙醇胺HCl缓冲液 (pH=90,含2mmol/LEDTA)中低温透
析12h,重复进行2~3次,直 到 透 析 后 的 缓 冲 液 电 导 率 和pH 与 起 始 值 相 同。收集透析后的酵母提取物用于随后的色谱分离。将DE52、QA52或ExpressIonQ (Whatman公 司)于 上 述 相 同 缓 冲 液 中 平 衡 至 pH 和 电 导 率 恒 定,调 为
20%~30% (200~300g/L)的浆状,填充于15cm内径的色谱注中,床高度为
10cm,将40ml透 析 后 的 酵 母 提 取 物 上 柱 分 离,流 速 为2~4ml/min,然 后 以
100ml相同 缓 冲 液 洗 涤,流 速 也 为2~4ml/min。再 采 用200ml上 述 缓 冲 液 与
200ml含 有500mmol/LNaCl的 上 述 相 同 缓 冲 液 以 线 性 梯 度 进 行 洗 脱,流 速 为
2~4ml/min,检测洗脱液280nm处的吸光度,收集样品用于随后的分析。利用QA52按照上述方法,可分离得到基本无PGK活性的TIM。操作中,
PGK只微弱地与QA52结合,洗涤时释放出来。TIM 与QA52结合,盐梯度洗
脱,得到约90%的回收率。实验结果见表92。
表92 利用QA52对面包酵母提取物中TIM的纯化
实 验 阶 段 PGK活力/U TIM活力/U TIM回收率/%
上柱前 7080 37120 —洗脱后 550 33360 899
9122 枯草杆菌 (Bacillussubtilis)淀粉酶的分离
α淀粉酶水解淀粉分子中的α1,4键,产生麦芽糖、麦芽三糖和α糊精,淀
粉酶的水解作用在食品工业中非常重要,应用广泛,包括生产甜味剂,调节渗透
压,增加热稳定性、糖化作用等。商品淀粉酶来源于一些微生物宿主,包括枯草
杆菌 (Bacillussubtilis)的 变 种amyloliquefaciens、地 衣 芽 孢 杆 菌 (Bacillus

247
lichenoformis)、黑曲霉 (Aspergillusniger)以 及 根 霉 (Rhizopusspp)等。各种类型淀粉酶的特性,特别是热稳定性,都由产酶微生物所决定。
纳瓦内斯 (退 浆 酶)是 一 种Bsubtilis的 变 种amyloliquefaciens的 商 品 提
取物,对其中的淀粉酶进行离子交换色谱分离,该过程可分为两步:样品澄清和
阴离子交换色谱。样品澄清可以采用硫酸铵沉淀或超滤两种方法。采用硫酸铵沉淀法,首先添加硫酸铵至60% (600g/L),4℃静置过夜,离
心收集沉淀,然后溶解于20mmol/LTrisHCl缓冲液 (pH=87)中,于4℃透
析,用于随后的色谱分离。该操作几乎可以完全回收原料中的淀粉酶,也适用于
进行柱色 谱 分 离。但 用 于 工 业 规 模 时,由 于 大 量 使 用 硫 酸 铵 而 缺 乏 实 际 可 操
作性。采用超滤操作也可以几乎完全回收原料中的淀粉酶,适用于大规模柱色谱分
离。利用 MWCO10000的超滤膜,可得到290L含有1148kg总蛋白和574000单位淀粉酶的样品,进行下一步离子交换。
阴离子交换色谱 的 操 作:于20mmol/LTrisHCl缓 冲 液 (pH=87)中 平
衡25kg的ExpressIonQ直至pH和电导率恒定,调为30% (300g/L)的浆液,将ExpressIonQ 填 充 于45cm 内 径 的 色 谱 柱 中,得 到25L的 色 谱 床 (床 高
16cm)。将290L酶样品上柱分离,流速为150cm/h,以上述缓冲液洗涤色谱柱,再以100L含250mmol/LNaCl的20mmol/LTrisHCl缓 冲 液 洗 脱 结 合 的 淀 粉
酶,流速为150cm/h。纯化结果见表93,可分离得到总量为369g的 蛋 白,含
有408320U的淀粉酶,回收率71%,纯化了22倍。纯化至此的淀粉酶如有必
要可通过其他色谱步骤进一步纯化。色谱柱可以用上述洗脱缓冲液再生。
表93 利用ExpressIonQ从纳瓦内斯 (退浆酶)中纯化淀粉酶
项 目 淀粉酶活力/U 蛋白量/g 淀粉酶回收率/%
样品 574000 1148 —离子交换 408320 369 71
9123 DNA修饰酶的分离
进行重组DNA操作时,常常用到许多DNA修饰酶,包括在特定核苷序列
处裂解DNA的限制性酶 (内切核酸酶),连接核酸的连接酶以及体外复制DNA分子的聚合酶。对于所有重组DNA实验,Ⅱ型内切核酸酶已成为必要的工具。这些酶在整个细菌界均可发现,需要从不同的细菌中纯化各种酶。一般以该菌种
属名的第一个字母和种名的前两个字母命名该酶。例如,EcoRⅠ来自于Ecoli菌株RY13,罗马数字Ⅰ表示该酶是从该菌种中分 离 的 第 一 个 酶。PstⅠ,来 自
Providenciastuartii;KpnⅠ,来 自Klebsiellapneumoniae;AluⅠ,来 自Ar

248
throbacterluteus。
DNA连接酶在重组DNA实验中也是必需的,因为其催化连接由Ⅱ型 限 制
性酶裂解得到的DNA片段。常用的连接酶是T4DNA连接酶,由Ecoli噬菌体
T4 编码。该酶通常由带有该基因克隆的Ecoli制备。T4DNA连接酶能够连接
带有黏性和平头末端的DNA片段,使得该酶在克隆应用中具有很好的通用性。
DNA聚合酶在分子生物学中也广泛的应用,如体外标记DNA、DNA测序,特别是通过聚合酶链反应 (PCR)对少量DNA的体外扩增等。除了大量来自嗜
热微生物的酶,也有应用嗜温噬菌体和细菌的酶。分子生物学家对大量DNA修
饰酶的需求使得有必要建立一种通用的纯化方法,既对DNA结合蛋白具有相对
的专一性,又 能 快 速 处 理 不 同 细 菌 的 提 取 物。Whatman公 司 的 磷 酸 纤 维 素
(P11)能够满足上述要求,该磷酸纤维素可作为阳离子交换树脂,也可 以 作 为
拟亲和基质纯化结合核酸的酶,如内切核酸酶、激酶和聚合酶等。从细 菌 细 胞 分 离 纯 化 DNA 修 饰 酶 的 方 法:将1L 培 养 液12000g 离 心
10min,收集细胞,于4℃下 重 新 悬 浮 于50ml10mmol/L磷 酸 钾 缓 冲 液 (pH=75,含7mmol/L2巯基乙醇)中,12000g离心10min,再次收集细胞,将细胞
泥再悬浮于20ml上述相同缓冲液中,加入20mg溶 菌 酶,37℃保 温10min,将
悬浮液冷却,超 声 破 碎30s,冷 却30s,重 复9次。4℃下50000g离 心40min,将上清液进行P11色 谱 柱 分 离,色 谱 柱 洗 涤 后,以100ml上 述 缓 冲 液 和100ml含1mol/L盐的10mmol/L磷酸钾缓 冲 液,线 性 梯 度 洗 脱,流 速 为30cm/h,测
定酶活性。该方法对于分离纯化22种不同的DNA修饰酶 (列于表94)都是有效的。
表94 利用P11色谱制备DNA修饰酶
酶 来 源 酶 名 称 酶 来 源 酶 名 称
EcoliRY13 EcoRⅠHaemophilusinfluenzaeRd HindⅡ,HindⅢProvidenciastuartii PstⅠSerratiamarcescens SmaⅠBacillusamyloliquefaciensH BamHⅠBglobigii BglⅠ,BglⅡKlebsiellapneumoniae KpnⅠArthrobacterluteus AluⅠStreptomycesalbusG SalGⅠSachromogenes SacⅠ,SacⅡProteusvulgaris PvuⅠ,PvuⅡ
Bacilluscaldolyticus BclⅠHaemophilusaegyptius HaeⅡ,HaeⅢHhaemolyticus HhaⅡHaemophilusparainfluenzae HpaⅠ,HpaⅡStaphylococcusaureus3A Sau3AStreptomycesphaechromogenes SphⅠThermusaquaticus TaqⅠXanthomasbadrii XbaⅠXanthomonasholicola XhoⅠ,XhoⅡ带有T4DNA连接酶基因的Ecoli T4DNA连接酶
带有DNA聚合酶基因的Ecoli DNA聚合酶Ⅰ
9124 微生物培养物中核酸的纯化
核酸提供了编码蛋白质的模板。纯化的DNA可用于各种目的,包括PCR、

249
测序以及基因治疗等,而在微生物宿主中表达的质粒需要大量分离。由于DNA与RNA在中性pH下均为阴离子,因而可通过阴离子交换色谱分离。DEAE纤
维素纸,比如 Whatman公司的DE81可结合DNA 片段和tRNA,质粒DNA可
利用DEAE纤维素吸附色谱从细胞溶解液中分离。质粒DNA通常可利用含有顺磁性成分的阴离子交换琼脂糖颗粒进行分离,
其应用的质粒为pBluescript,该方法具有比较广泛的适用性。细菌细胞中分离质粒DNA:表达质粒pBluescript的EcoliJM109细 胞 于
含有100μg/ml氨 苄 青 霉 素 (氨 苄 西 林)的LB培 养 基 中 生 长 至 对 数 后 期,取
15ml细胞培养液10000g离心1min收集菌体细胞。将细胞重新悬浮于100μl悬
浮缓冲液 (50mmol/LTrisHCl,pH=80,10mmol/LEDTA,400μg/ml核糖
核酸酶A)中,加 入200μl溶 解 缓 冲 液 [200mmol/LNaOH,1% (10g/L)十
二烷基 硫 酸 钠],混 合,溶 解 细 胞,冰 冷5min,再 加 入150μl4℃沉 淀 缓 冲 液
(3mol/L醋酸钾,pH=55)沉淀基因组DNA和其他杂质,冰冷5min,10000g离心5min,上 清 液 中 加 入 4% (40g/L)DEAEMagarose的 STET 缓 冲 液
[10mmol/L TrisHCl,pH=80,100mmol/L NaCl,1mmol/L EDTA,1%(10g/L)TritonX100]150μl,混合5min,于磁力分离器中固定化琼脂糖颗粒,并弃去上清液,将胶粒重新悬浮于400μl的10mmol/LTrisHCl缓冲液 (pH=80,400mmol/LNaCl,1mmol/LEDTA)中 洗 涤,重 复 上 述 固 定 化 和 洗 涤 步
骤。以200μl10mmol/LTrisHCl缓冲液 (pH=80,1mol/LNaCl,1mmol/LEDTA)洗脱 结 合 的 DNA,固 定 化 胶 粒,收 集 上 清 液,再 加 入500μl预 冷 至
-20℃的无水乙 醇 和20μl75mol/L醋 酸 铵 缓 冲 液 沉 淀 DNA,于-20℃保 持
10min,4℃下15000g离心30min,收集沉淀,弃去上清液,以50μl70% (体积
分数)冷乙醇洗涤沉淀,4℃15000g再离心10min,弃去上清液,沉淀于室温下
空 气 干 燥30min,将 沉 淀 重 新 溶 解 于50μl10mmol/LTrisHCl(pH=80,
1mmol/LEDTA)缓冲液中,制得pBluescriptDNA。由于微生物细胞溶解液相当黏稠,在柱操作中可能会出现问题。而色谱载体
中的顺磁性成分有助于在分批色谱分离中快速固定化,所以一般以分批操作为主。
Magarose胶粒对于磁场反应非常迅速,一般不到10s内就可从悬浮液中固定化。
DEAEMagarose对于pBluescriptDNA的结合特性列于表95中。在质粒
DNA分离过程中,按照碱性溶解法,DEAEMagarose[4% (40g/L)悬 浮 液,
150μl]可 从15mlEcoliJM109细 胞 培 养 液 中 分 离 约82μg的pBluescriptDNA。A260/A280的结果表明,DNA纯度很高。琼脂糖凝胶电泳也证实了这一结
果。从DEAEMagarose上洗脱的DNA很容易用冷乙醇沉淀收集。其他一些基
于离子交换的质粒DNA分离试剂盒要求利用异丙醇沉淀。

250
表95 pBluescriptDNA与DEAEMagarose的结合
所用胶粒体积/μl DNA产率/μl A260/A280
150 82(n=5) 194
92 哺乳动物细胞培养蛋白质的纯化
哺乳动物细胞培养作为生物技术工业中一项主要的成就,已经从19世纪60年代开始迅速发展成为一项充满活力的生物制药生产工艺。如今的生物制药工业
中,大约一半是来自于哺乳动物细胞培养的产品。一般动物细胞生产的重组蛋白主要性质可归纳如下:以天然的正确折叠形式
分泌到培养基中;正确进行翻译后修饰;重组蛋白分子量没有限制;哺乳动物细
胞不产内毒素;产物浓度低;培养基中的蛋白含量可能较高;生产流程可能含有
病毒、DNA或细胞蛋白,必须通过下游工艺去除。对哺乳动物细胞培养蛋白质的纯化,需要考虑两方面的问题。首先,重组哺
乳动物细胞之所以能够正确生产复制人类蛋白,是 因 为 它 们 控 制 基 因 表 达 的 方
式、所用的合成和加工酶以及蛋白质的折叠和分泌机制都与人类细胞一致。这种
机制的类似性有利于保持重组蛋白产品的纯正,同时也提出了特定的纯化和特性
问题。生产细胞得到的蛋白可能与人体同种蛋白发生免疫交叉反应。某些生产类
型细胞可能大量产生如生长因子、细胞分裂素等蛋白,而这些蛋白在很小剂量下
就具有生物活性。消除这些潜在的风险要强化对哺乳动物细胞重组产品纯化方法
特异性的要求。其次,细胞生长和繁殖所需的培养基也可能在相当程度上增加纯
化蛋白质产品的难度。与微生物发酵所用的简单培养基不同,哺乳动物细胞培养
基通常需要一些不确定的动物来源的营养添加物,如血清。这会明显增加原料蛋
白的处理量,引入由病毒或其他危险因素带来的额外的污染风险。因此,哺乳动物细胞来源的蛋白纯化策略不仅要能够可重复生产,满足一定
纯度要求和特定形式,而且应消除可能来源于制备过程中的潜在有害污染物,包
括细胞或培养物中的动物蛋白、病毒以及DNA,或者用于细胞培养的抗生素等。以下事项需要注意:
① 卫生要求,所有分离步骤必须能在平静的条件下操作,而且不应引入生
物负荷或内毒素,纯化原料应是无菌的,而且内毒素含量很低,不使用抗生素。
② 不能蒸汽灭菌的工艺材料 (如色谱载体)必须采用其他有效方式清洗和
脱热原。典 型 的 方 法 包 括 用 消 毒 溶 液 如02~05mol/LNaOH 或 酸 性 乙 醇
(60%乙 醇/05mol/L 醋 酸)处 理,氢 氧 化 钠/乙 醇 混 合 物 (如50%乙 醇/
02mol/LNaOH)也可采用,处理时间一般为室温下4~24h。

251
③ 工艺的完整性,确 保 产 物 能 够 从 一 个 操 作 步 骤 直 接 进 入 到 下 一 个 步 骤,中间不需要额外的操作,是比较理想的。
④ 无论是对于机械力还是不利的培养条件,哺乳动物细胞比微生物细胞要
脆弱得多,所以必须注意保持细胞的完整。需要细心控制培养和细胞分离条件,避免由于细胞溶解而释放的胞内蛋白对产物的污染。
哺乳动物细胞生产蛋白质的另一种方式就是转基因动物。转基因动物乳汁进
行重组人蛋白的生产是最近发展起来的,对于需求量大的蛋白 (如某些治疗用单
克隆抗体可能年需求>100kg),利用转基因动物生产可能比 利 用 哺 乳 动 物 细 胞
培养在商业上要可行得多。适用于哺乳动物细胞培养或转基因动物生产的重组蛋白或单克隆抗体的典型
纯化方案一般包括初步分离 (过滤或离心、超滤浓缩)、离子交换 (或亲和)色
谱、凝胶过滤等,最终得到纯度99%~100%的产品。
921 工艺设计
工艺设计主要考虑采用一些最有效的措施,确保所处理的产品尽可能富集和
浓缩,又不含有较难处理的污染物,对于动物细胞产物,仔细选择细胞、细胞生
长方式以及生长所用的培养基,对后续的纯化会有很大的帮助。选择生产细胞时,除了要求具有较好的生长能力和较高产率外,还需要了解
何种病毒可由细胞释放,以确定纯化策略。细胞中不能含有潜在污染病毒或其他
传染物质。生产细胞需低温贮藏,保存在液氮中作为 “主细胞库”,其中一些细
胞制成 “工作细胞库”。每个生产批次都从工作细 胞 库 中 新 鲜 的 细 胞 试 管 开 始。除了生物安全性方面的考虑,该方法也非常有助于生产的可重复性。
选用的培养基对于重组蛋白产品的纯化策略也是极其重要的。哺乳动物细胞
的培养基非常复杂,含有盐、维生素、氨基酸、糖类以及各种添加剂,常用物质
包括血清,使用浓度通常为10% (体积分数),有时也利用其他一些蛋白质混合
物来代替血清。含血清培养基在生物医药工业中的应用十分普遍,但也存在一些
下游处理的实际问题,比 如:污 染 蛋 白 含 量 较 高 (10% FCS含 有 约4g/L的 蛋
白),血清性能和成分会产生批次间的变化,血清中的病毒和其他物质如BSE可
能污染产品,成本较高 (一般为其他培养基成本的10~20倍)等。通常在使用血清前需分批筛选,检查是否存在病毒,但未知传染物质也可能
会产生严重的后果。通过纯化过程清除潜在传染物仍然是主要的安全措施。培养
基蛋白组成和目的蛋白间物理化学的相似性也可能会引出问题。标准组织培养基
通常含有作为pH指示剂的酚红,可能会与纯化过程所采用的某些色谱基质发生
不可逆结合,从而产生问题。这些在选择培养基时都应有所考虑。不同类型的哺乳动物细胞要求不同的培养条件。有些细胞可在搅拌发酵罐中

252
生长,较容易控制和放大,但需要额外的步 骤 来 分 离 细 胞 和 含 有 分 泌 产 物 的 培
养基。
922 细胞分离
哺乳动物细胞非常容易被机械剪切力所损伤,需要采用温和的分离方法。实
际上哺 乳 动 物 细 胞 很 容 易 沉 淀 (甚 至 可 以 利 用 原 始 的 重 力 沉 降),可 以 在
(1000~1500)g下有效地离心分离而无损伤。近来开发的专用于动物细胞分离的
完全封闭的离心分离机,具有低离心力、低剪切蠕动泵、无旋转密封的特点。近来,一些色谱基质已经被开发应用,使得可以从细胞培养物中直接有效地
捕获目的产物而不需要预先进行细胞分离。比如Bioprocessing公司基于刚性玻
璃珠载体系统的PROSEP类产品,和Pharmacia公司利用石英核周围交叉连接
琼脂糖的大孔珠 的STREAMLINE类 产 品,这 两 类 载 体 都 是 非 常 稳 定 的 吸 附
剂,可耐受常用的大部分清洁消毒剂。需要注意的是,产物的最优结合条件不一
定与细胞存活所要求的最优条件相一致,这可能导致细胞的溶解和细胞内容物的
释放,从而增加下游处理的负荷。
923 产品的初步回收和分离
由于只有很少量细胞溶解,哺乳动物细胞培养上清液所含核酸很少,一般不
需要利用核酸酶处理以降低原料黏度。澄清后的培养液中目的蛋白浓度相对较低,含有高浓度的来自培养基的小分
子物质,初步分离纯化可减少主要的污染物。具体方法包括蛋白质沉淀 (盐或有
机溶剂),目的蛋白的吸附和液液萃取等。超滤法现在已被广泛使用,因为超滤
易于操作,不会使蛋白质处于极端或变性条件,易于放大规模,不会引入潜在的
污染物。但超滤没有很好的分离能力,因为可透过膜的颗粒大小范围很宽,而且
也会浓缩可能存在的病毒颗粒。超滤的另一个问题是由于微粒或者某些蛋白在膜
上形成胶层,可能会产生膜阻塞的问题,用于哺乳动物细胞培养的某些防沫剂也
可能堵塞超滤膜。将样品适当澄清能够对此问题的解决有所帮助。乳汁是一种复杂的胶状悬浮物,含有高浓度的酪蛋白胶粒和其他乳汁蛋白、
脂肪液滴、宿主细胞以及宿主细胞溶解产生的蛋白和核酸。乳汁中重组蛋白质产
品的初步回收和分离采用乳品加工业所 建 立 的 技 术,包 括:撇 沫 和 离 心 除 去 脂
肪,酸法或盐法沉淀酪蛋白,过滤去除颗粒。这些步骤增加了从乳品中制备纯蛋
白的成本,比通常从细胞培养上清液中得到的最终产品百分收率还要低很多。含
有目的蛋白的澄清乳清部分可作为浓缩原料,用于后续纯化步骤。
924 主要纯化方法
哺乳动物细胞生产蛋白质主要是用于生物医药。对于这些高附加值产品纯化
步骤的主要任务是制备安全、纯净、可重复的蛋白。为得到较高的产率,增加处
253
理强度以及优化过程的经济性,以尽可能少的处理步骤实现这一目的非常重要。这就需要选择对动物细胞产物具有最高解析能力,可提供必要的蛋白纯度,特别
是对可能存在的病毒和DNA污染物具有满意去除率的一系列互补的特异性单元
操作。商业上大部分实际处理方法可经3~5步纯化达到这一要求,还有许多其
他技术也可达到这一要求。实际上,柱色谱法是涉及哺乳动物细胞产物纯化最常
考虑的方法,比如离子交换色谱、疏水作用色谱、亲和色谱等。利用两种不同类
型色谱分离柱,将其串联使用,由于操作条件要求不同,有时也许更具优势,比
如经离子交换柱得到的高离子强度洗脱液,也许可直接适用于疏水作用色谱,反
之亦然。亲和色谱是高选择性的蛋白质纯化方法,在某些情况下可以获得绝对专
一的产物,例如固定化单克隆抗体可通过 “免疫亲和”纯化相应抗原,固定化激
素可纯化其受体等。但固定化抗体本身据认为具有 被 病 毒 或 其 他 物 质 污 染 的 可
能,因此所用抗体本身应是医药级的。在 抗 体 纯 化 领 域,蛋 白 A (来 源 链 球 菌
Streptococcusaureuse)已广泛应用并获得 成 功。该 蛋 白 对 大 部 分 免 疫 球 蛋 白 亚
型的Fc区具有高亲和力,可专一性结合。主要来自于植物的凝集素可选择性结
合糖蛋白中的不同糖部分,虽然对于特定蛋白并不是绝对专一,但凝集素亲和色
谱可在一步内完成非常有效的纯化。凝集素通常是有毒性的,比如强效的血球凝
集素或促分裂素,这一点应引起注意。近来,染料亲和分离方法得到了快速发展,该方法蛋白质结合容量非常高,
合适的染料既便宜又易于大量获得,与非毒性试剂配合使用简单,利用适当的基
质,配基渗漏可尽量降低,可得到较高的解析率。新型染料亲和柱可经受包括氢
氧化钠等消毒溶液的处理而不渗漏。当蛋白质已经达到必要的纯度和浓缩形式后,最后步骤通 常 是 精 制,一 般
采用凝胶过滤色谱,对于从可能已经形成的凝聚体中分离蛋白质,去除 残 留 的
添加剂等都特别有效。哺乳动物细胞生产生物医药产品时,去除内源或 同 源 蛋
白质的可能来源是一个问题,包括细胞培养中的宿主细胞和用于培养基 的 动 物
蛋白,或者转基因动物的宿主细胞和血清蛋白。仔细适当的分离条件对 于 除 去
污染物是很有帮助的。动物来源的痕量蛋白,包括同源蛋白的检测经常 通 过 灵
敏的免疫 学 测 试 而 完 成,比 如 MolecularDevices公 司 的 ThresholdTM系 统 测 试
方法。
925 样品纯化举例———单克隆抗体的纯化
单克隆抗体作为一类最具有工业生产经验的特定蛋白代表,已经积累和阐明
了哺乳动物细胞生产纯净蛋白产品中所遇到的问题。单克隆抗体在诊断领域的广
泛应用以及不断增长的产品需求量,对蛋白质纯化的技术和经济性提出了更高的
要求。抗体分子的整体相似性使建立通用纯化系统成为可能,相同的柱系统可用

254
于几种不同的产品。
9251 利用蛋白A分离
蛋白A亲和色谱已广泛用于抗体纯化,在某些场合,几乎是特定抗体纯度
超过95%的理想的高得率单步纯化方法。蛋白A色谱去除内毒素、DNA和病毒
非常有效,但由于某些免疫球蛋白异构物的无效结合,有时还需要苛刻的条件进
行洗脱,从而可能破坏抗体或导致蛋白 A从柱上脱落。该方法也存在有一些实
际问题。通过改善固定化技术和优化蛋白A在柱上的定位来拓宽结合Ig种类的
范围,可处理未澄清培养基的蛋白A柱床材料也已开发出来。利用蛋白A吸附剂 (PROSEPA)捕获组织培养上 清 液 中 单 克 隆 抗 体 的
方法:根据操作说明准备填充柱。以5~10倍柱体积的25mmol/LTrisHCl缓
冲液 (pH=74,含150mmol/LNaCl)在适当流速下洗涤,通过045μm过滤
器过滤澄清培养上清液,将样品上柱,以5~10倍柱体积的上述缓冲液洗涤,用
01mmol/L柠檬酸盐 (pH=30)缓冲液每小时50倍柱体 积 的 流 速 洗 脱 产 物,直接将样品收集到中和缓冲液 (1mol/LTrisHCl,pH=90)中,避免产物暴
露于酸性pH而受到破坏。色谱柱可用乙醇/醋酸混合液处理,不建议用氢氧化
钠处理。
9252 单克隆抗体离子交换纯化
阳离子交换 法 纯 化 单 克 隆 抗 体,具 有 高 容 量、良 好 的 化 学 和 机 械 稳 定 性、
pH和离子强度与原料差别不大的条件下捕获抗体的能力。已经开发的高效载体
有 Pharmacia 公 司 的 STREAMLINE 载 体,BioRad 公 司 的 BioScaleS2,
JTBaker公司的BakerbondABx以及Sterogene生物分离公司的CELLTHRUBIGBEAD离子交换树脂等。
利用BakerbondTM ABx捕获培养上清液中的单克隆抗体:准备适当尺寸的
色谱 柱,并 以 约 15 倍 柱 体 积 的 10mmol/L2 (N吗 啉)乙 烷 磺 酸 (MES)(pH=56)缓冲液平衡,对于5cm直径的色谱柱流速约为90ml/min。当色谱柱
充分平衡后,洗脱液pH应为56。以4倍体积上述缓冲液稀释细胞培养上清液,然后以60ml/min流速上柱,所有同型免疫球蛋白都可有效结合,色谱柱经洗涤
后,利用500mmol/L醋 酸 钠 缓 冲 液 (pH=70)按0~100%的 线 性 梯 度,以
90ml/min流速进行洗脱。以20倍柱体积的高离子强度盐溶液 (如2mol/L醋酸
钠或1mol/L磷酸钾缓冲液,pH=7~8)流经色谱 柱 进 行 再 生。如 有 必 要,可
利用蛋白溶解剂或去污剂清洗色谱柱。
926 消除污染
在哺乳动物细胞生产的生物医药蛋白制品中,可能存在的污染物主要包括病
毒、杂质DNA以及其他一些生物活性物质,这些污染物的存在,会给生物蛋白

255
制品带来很大的风险,必须加以清除。
9261 消除病毒污染风险
病毒污染风险可对细胞库中的生产细胞进行病毒传染性测定,通过仔细筛选
而部分消除。细胞的培养过程应避免可能激活细胞内潜伏感染病毒的条件。纯化
过程中要求包括至少两步作用机制不同的强烈的病毒失活或去除步骤,而且还要
通过严格的病毒清除研究验证,以证明该纯化步骤能够有效地清除病毒。在下游
处理操作中一般没有必要包括特异性的病毒去除步骤,因为所采用的许多纯化方
法能有效降低病毒效价。为便于测定准确的病毒清除率,通常利用添加 “同类指示剂”实验,即在某
一纯化步骤前故意加入大量的感染病毒。出于安全和方便的原因,这些测试都利
用与可能污染的致病病毒紧密相关的 “模型病毒”。实际上有必要使病毒具有很
高的效价,以便进行清除率的测定 (通常数量级为104~105 倍)。表96列出了
用于清除率研究的重组模型病毒。
表96 用于清除率研究的重组模型病毒
病 毒 类 型 大小/nm RNA/DNA 被 膜 稳定性①
犬细小病毒 细小病毒 22 DNA 无 高
脊髓灰质炎病毒 细小核糖核酸病毒 25 RNA 无 中等
呼肠孤病毒 呼肠孤病毒 60 RNA 无 高
SV40 乳多空病毒 45 DNA 无 高
鼠白血病毒 反转录病毒 80 RNA 有 低
腺病毒 腺病毒 70 DNA 无 中等
HIV 反转录病毒 90 RNA 有 低
VSV 棒状病毒 80 RNA 有 低
副流感病毒 副黏病毒 150 RNA 有 低
IBR 疱疹病毒 100 DNA 有 中等
假狂犬病毒 疱疹病毒 120 DNA 有 中等
痘苗病毒 痘病毒 600 DNA 有 高
① 蛋白纯化过程中在可能遇到的各种条件下以活性形式存在的能力。
病毒清除率应作为 “折算系数”,即对于某一特定步骤起始材料中同类指示
剂病毒负荷 (效价×体积)的对数 (lg)与所得产物中病毒负荷对数之比。如果
各步骤是独立的,而且根据不同的物理化学参数实现分离,则每步得到的折算系
数可用于计算总折算系数。重复应用相同的分离步骤得到的折算系数不一定是加
成的。表97列举了各种操作步骤中不同病毒预计的折算系数,但这只是一个普
通范围,对于特定系统必须验证其清除率。
9262 消除DNA污染
用于生产重组蛋白和单克隆抗体的多种细胞含有活性致癌基因,因而可能存

256
在对患者具有 致 癌 作 用 的 风 险。根 据 WHO在1986年 公 布 的 原 则,每 次 剂 量
100pg水平认为对患 者 构 成 的 风 险 可 以 忽 略,因 此 可 作 为 比 较 合 适 的 纯 化 操 作
目标。
表97 经过重组蛋白和单克隆抗体的各种纯化步骤,可预计的一些病毒① 典型的折算系数②
操 作 步 骤 脊髓灰质炎病毒 异型鼠白血病毒 风疹病毒
阴离子交换 (3~4)lg >4lg >5lg蛋白A亲和分离 (3~4)lg (3~4)lg >6lg凝胶过滤 >3lg >3lg >3lg酸处理(pH=35或更低) <1lg >4lg >3lg过滤(35nm孔径) <1lg (5~7)lg (6~8)lg
① 异型鼠白血病毒是一种逆转录病毒,经常用于 模 拟 可 能 存 在 于 鼠 杂 交 瘤 中 的 逆 转 录 病 毒。风 疹 病
毒是特别与人或灵长类动物生产细胞有关的病毒类型之一。
②lg体积×输入材料的病毒效价
体积×( )产物的病毒效价
清除DNA的方法类似于对病毒的处理方法。大多数蛋白质纯化方法很容易
分离蛋白质和DNA,而离子交换对此特别有效。与病毒一样,DNA清除验证也
包括用已知数量DNA同类指示剂材料进行纯化,在该纯化系统的缩小模型中测
定清除率,得到DNA折算系数。与病毒清除率研究一样,必须注意同类指示剂
中的DNA负荷不会干扰纯化步骤的表现特性验证。皮克级灵敏度的DNA分析方法有 MolecularDevices公司极限总DNA分析
法和直接荧光DNA定量方法等。
93 动物组织蛋白质的纯化
本节介绍从动物组织中分离蛋白质所涉及的一些原则,包括组织的选择、组
织破碎方法以及适当的亚细胞成分的分离过程等。所介绍的大部分方法已经应用
于实际。同时介绍三个动物蛋白的纯化实例,包括:从猪肾脏皮质膜中分离血管
紧张肽转化酶和氨肽酶P,从猪肝胞液中分离果糖1,6二磷酸酶。
931 组织的选择
对目的蛋白起始组织材料的选择由一些因素决定。首先,理想的原料组织应
含丰富蛋白质,如肾脏皮质中许多细胞表面肽酶含量很高,而且这一器官相对较
大,可以经常分离相当数量的蛋白 质,如 从200g的 肾 脏 皮 质 可 纯 化10~15mg的血管紧张肽转化酶。但在初次分离蛋白质时,可能并不考虑来源是否丰富。其
次要考虑的是动物组织是否易获得。当地的屠宰场可以是猪和牛等家畜的大部分
组织的有效来源,而小型动物 (鼠类,兔子 等)的 组 织 通 常 从 公 共 动 物 机 构 获
得。因此,鼠类的组织适合于纯化少量蛋白,而猪内脏可能更适用于大量制备。

257
一般而言,人组织很难获得,虽然也能够利用当地妇产医院得到胎盘。在处理动
物组织时,应遵循严格的实验室原则,随着近来关于传染性海绵体脑病如 “疯牛
病”等的病毒来源和传播途径的争论,在处理牛、羊组织时,采用一些预防措施
也是必要的。其他还需要考虑的因素包括组织的新鲜程度和破碎的方便性等,对
于某些蛋白质的纯化,可能需要采用新鲜的而非冷冻的组织,这也会影响原料的
来源和纯化策略。
932 组织破碎
组织的破碎方法取决于蛋白质是胞外还是胞内的,可溶性的还是膜结合的,或者位于亚细胞器中等。对于大型器官,如猪肾脏或人胎盘,首先就是用剪刀或
解剖刀将其切成约1cm2 的碎片。对于大量组织的均质化处理,可采用搅切器快
速破碎。对于少量组织,可采用其他均质器。对于软组织,可采用玻璃玻璃或
玻璃聚四氟乙烯均质器。根据蛋白质的不同,均质缓冲液中可能需要含有蛋白
酶抑制剂。均质化后,残留的大块组织可通过细棉布或细纱布过滤或者低速离心
除去。均质及以后的亚细胞分离应在4℃进行,均质化时间应尽量短,以减少不
必要的蛋白质水解和变性。
933 防止蛋白质的有害水解
组织均质化后,通常在不同亚细胞结构内的蛋白酶将释放到溶液中,并与目
的蛋白相接触,很可能将其降解。根据一般规律,存在于细胞表面或以可溶形式
分泌的蛋白以及糖基化蛋白对于降解有较强的抗性。在4℃预冷的缓冲液中进行
均质化操作及后续的分离操作,对于降低蛋白质有害水解会有帮助。通过在各种
缓冲液中添加蛋白酶抑制剂可进一步控制蛋白质水解。表98列出了一些商品蛋
白酶抑制剂。采用以下两种混合抑制剂可防止哺乳动物蛋白分离过程中的有害蛋
白 质 水 解:①EDTA (5mmol/L),DipF (01mmol/L),E64 (10μmol/L),抗痛 素 (10μg/ml),亮 肽 素 (10μg/ml),抑 肽 素 A (1μmol/L);②EDTA(1mmol/L),1,10二 氮 杂 菲 (1mmol/L),DipF (01mmol/L),PMSF(1mmol/L),E64 (10μmol/L),抑肽素A (1μmol/L)。
在使用此类抑制剂时需要考虑的关键因素之一是成本,特别是在大规模重复
分离一种蛋白时。另一个因素是在分离一种蛋白酶并通过分析酶活性了解其纯化
过程时,应避免使用此类蛋白酶的抑制剂。
934 亚细胞分级分离
分离目的蛋白所在的亚细胞成分,最普通的方法是通过离心进行。如果纯化
一种存在于亚细胞器中的蛋白,如线粒体中的丙酮酸脱氢酶,则需要另外的步骤
打开细胞器。根据亚细胞器和蛋白质特性,采用的方法包括超声波处理、渗透溶
解或匀浆器均质作用等。

258
表98 类别专一的蛋白酶抑制剂特性
抑制蛋白
酶类别抑 制 剂 方式① 储备浓度 有效浓度 备 注②
金属酶 EDTA R 50mmol/L 1~5mmol/L
1,10二氮杂菲 R 100mmol/L 1mmol/L 溶于甲醇
丝氨酸 牛胰蛋白酶抑制剂 R 1~10mg/ml 2~10μg/ml
二异 丙 基 氟 磷 酸 盐
(DipF)I 1mol/L 01mmol/L 极 毒。溶 于 2丙 醇。
pH=75水溶液中半衰期
为1h
苯基甲磺酰氟(PMSF) I 100mmol/L 1mmol/L 溶 于2丙 醇,于4℃保
存。pH=75水溶液中半
衰期为1h
3,4二氯 异 香 豆 素(3,4DCI)
I 10mmol/L 5~100μmol/L 溶于DMSO。pH=75水溶液中半衰期为20min
半胱氨酸 E64[反环氧琥珀酰L亮
氨酰氨基(4胍基)丁烷]I 1mmol/L 10μmol/L
碘乙酰胺 I 100mmol/L 1mmol/L 按需制备新鲜试剂
p羟基酚汞磺酸 I 100mmol/L 1mmol/L 丝 氨 酸/半胱氨酸③
抗痛素
抑凝乳蛋白酶素
RR
1mmol/L10mmol/L
1~10μmol/L10~100μmol/L
溶于甲醇
溶于DMSO
亮肽素 R 10mmol/L 10~100μmol/L
酸性酶 抑胃肽 R 1mmol/L 1μmol/L 溶于甲醇
① 抑制模式分为可逆的 (R)和不可逆的 (I)。
② 除非另外注明,a抑制剂应以注明的储备浓度溶于水中,并于-20℃保存;b储备溶液于-20℃保存可稳定6个月;c抑制剂可从Sigma公司购得。
③ 类胰蛋白酶丝氨酸蛋白酶及一些半胱氨酸蛋白酶。
猪肾脏亚细胞分级分离的方法 (所有操作均在4℃进行):20g猪肾脏皮质于
200ml的均质 缓 冲 液 (50mmol/LHepes/NaOH,pH=75,033mol/L蔗 糖)中均质化,600g离心匀浆25min。从细胞核和细胞碎片沉淀上 面 移 取 上 清 液,于6000g离心8min,从 溶 酶 体 和 线 粒 体 沉 淀 上 面 移 取 上 清 液,于40000g离 心
30min,再从微粒体沉淀上面移取上清液,并于100000g离心90min,最后从轻
微粒体和核糖体沉淀上面移取上清液部分。所有沉淀部分应按要求重新悬浮于适
当缓冲液中。对于大规 模 纯 化 原 生 质 膜 结 合 蛋 白,可 利 用 简 化 的 差 速 离 心 方 法,使 用
GSA/SLA1500Sorvall转子可进行较大体积 (15L)的 匀 浆。虽 然 只 得 到 粗 微
粒体膜成分,该方法也适用于进一步分离膜结合蛋白。从猪肾脏中分离粗微粒体膜成分的方法:将100g猪肾脏皮质放入搅切器中,
置于1L的4℃均 质 缓 冲 液 (50mmol/LHepes/NaOH,pH=74,033mol/L蔗糖)中均质化,8000g离心匀浆液15min,小心取出上清液,26000g离心2h,

259
得到的 微 粒 体 沉 淀 重 新 悬 浮 于 缓 冲 液 (10mmol/L Hepes/NaOH,pH=74)中,最终浓度达到约10mg蛋白/ml。该重新悬浮的微粒体沉淀可作为膜结合蛋
白的原料。肾脏的顶端微绒毛或刷状缘膜以及肠上皮细胞,富含大量的胞外酶,包括碱
性磷酸酶、5′核苷酸酶、血管紧张肽转化酶和许多其他肽酶。其他亚细 胞 结 构
与 Mg2+聚合,使得需 要 分 离 的 膜 成 分 高 度 富 集 在 微 绒 毛 膜 中,基 本 上 不 会 污
染膜。从猪肾脏中分离微绒毛膜成分的方法:将50g猪肾脏皮质放入搅切器中,于
4℃均质缓冲液 (10mmol/L甘露醇,2mmol/LTrisHCl,pH=71)中均质化,得到10% (100g/L)的匀浆液。将匀浆液放入破碎机中,置于冰上,边搅拌边
加入10gMgCl2·6H2O,将悬浮液置于冰 上 冷 却15min,间 歇 搅 拌,1500g离
心12min,沉淀其他亚细胞结构。将上清液15000g离心15min,沉淀 微 绒 毛 膜
成分,用移液管从松散的浅粉红色沉淀上面小心移取上清液,将沉淀重新悬浮于
50ml上述均质缓冲液中,再搅拌加入05gMgCl2·6H2O,冰冷却15min,间歇
搅拌,2200g 离 心12min,沉 淀 其 他 亚 细 胞 结 构,移 取 上 清 液,15000g 离 心
15min,小心 移 取 上 清 液,将 膜 沉 淀 重 新 悬 浮 于25ml的10mmol/L Hepes/
NaOH (pH=74)中。对于可溶性胞外蛋白,通过离心沉淀细胞后很容易得到。这里介绍分级分离
新鲜猪血的方法。于1L新鲜血液中尽快加入167ml柠檬酸葡萄糖溶液 (251g柠檬酸,418g柠檬酸钠,334g葡 萄 糖 溶 于167ml水 中),混 合,以 防 止 血 液
凝固。所有后续步骤均在4℃下进行。血液1000g离心10min,将上清液移入干
净离心管中,30000g离心20min,沉 淀 血 小 板,移 取 上 清 液,于10mmol/L醋
酸钠溶液 (pH=54)中透析,将透析液10000g离心30min。得到的上清液于
适当缓冲液中透析,以备目的蛋白进一步纯化。
935 膜蛋白的溶解
膜蛋白溶解的方法取决于蛋白质与脂质双分子层是如何联结的。对于具有多
个跨膜区域或与脂质部分 (如肉豆蔻酸酯、棕榈酸酯或异戊二烯基团)共价结合
的蛋白,必须用去污剂破坏膜结构。去污剂的选择取决于溶解目的蛋白以及维持
蛋白质活性构象的有效性、实用性和成本。虽 然TritonX100是 一 种 常 用 于 溶
解膜蛋白的去 污 剂,但 其 主 要 的 缺 陷 是 具 有 较 高 的 UV吸 收。使 用 去 污 剂 后,在随后纯化过程中所有的缓冲液都必须含有该去污剂,否则由于疏水部分的聚集
及其与色谱柱和器皿表面的非特异性结合,几乎肯定会引起蛋白质的损失。对于
具有单一跨膜区域的膜蛋白,可用去污剂将其从膜上溶解。而对于多肽链大部分
位于膜某一侧的蛋白质,可以用胰蛋白酶或木瓜蛋白酶裂解。许多哺乳动物的糖

260
基化磷脂酰肌醇 (GPI)定 位 蛋 白 容 易 被 细 菌 磷 脂 酰 肌 醇 特 异 性 磷 脂 酶C (PIPLC)所释放,细菌PIPLC可选择性裂解GPI。由于有限的蛋白裂解,只有部
分膜蛋白被细菌PIPLC释放,因而获得了有效的初始纯化,而且释放的蛋白形
式也是亲水的。膜蛋白的溶解方法:将重新悬浮的微粒体膜沉淀与以下溶液之一混合处理。
①20% TritonX100:蛋 白 为7∶1 (体 积/质 量)比 例 的 TritonX100溶 液,
4℃下保持1h;②最 终 浓 度60mmol/L的n辛 基βD吡 喃 葡 糖 苷 于4℃下 保 持
1h;③胰蛋白酶:蛋 白 为1∶10 (质 量 比)比 例 的 胰 蛋 白 酶 溶 液,37℃下 保 持
1h;④细菌PIPLC (01U/mg蛋白)于37℃下保持2h。保温一段时间后,样
品于31000g离心90min,移出含有溶解蛋白的上清液。
936 动物蛋白质纯化举例
通过以上方法一般可以得到含有目的蛋白的可溶性部分,而蛋白质的进一步
纯化则取决于蛋白质特性以及是否可以采用亲和色谱或其他选择性色谱。以下介
绍三种蛋白质 的 纯 化,首 先 是 以 TritonX100或 胰 蛋 白 酶 将 蛋 白 从 膜 上 溶 解,利用抑制剂亲和色谱纯化血管紧张肽转化酶,其次介绍如何利用常规色谱组合操
作纯化GPI定位氨肽酶P,最后是细胞质果糖1,6二磷酸酶的纯化。
9361 血管紧张肽转化酶的纯化
血管紧张肽转化酶 (EC34151)是一种类型Ⅰ内在膜蛋白,也有以可
溶形式存在于原生质中,对控制血压具有关键作用。该酶为锌金属酶,其抑制剂
如 卡 普 多 普 瑞 尔 (巯 甲 丙 脯 氨 酸)、依 拉 普 利 (enalapril)和 雷 米 普 利
(ramipril)目前临床上用作抗高血压剂。依拉普利的衍生物———赖诺普利 (lisinopril)已证明对该酶的亲和纯化是一种良好配基。这里介绍从猪肾脏皮质中分
离微粒体膜成分,并以有限胰蛋白酶处理从膜上溶解该酶后,亲和纯化血管紧张
肽转化酶的过程。该方法从200g猪肾脏皮质中基本上可一步纯化血管紧张肽转
化酶达15mg。
lisinopril28nmSepharose亲和纯化血管紧张肽转化酶:200ml工作缓冲液
(10mmol/L Hepes,03mol/L KCl,01mmol/LZnCl2,pH=75)以 流 速
10~20ml/h预 平 衡1cm×6cmlisinopril28nmSepharose色 谱 柱 和15cm×10cm未处理的SepharoseCL4B预处理柱,将胰蛋白酶溶解的蛋白质样品以流
速10~20ml/h上柱分离,收集所有流出液,确定未结合蛋白。一旦所用样品流
经色谱柱,除去预处理柱,用400ml上述工 作 缓 冲 液 洗 涤 亲 和 色 谱 柱,以 含 有
10μmol/Llisinopril的上述工作缓冲液或50mmol/LpH=90硼酸钠缓冲液从柱
上洗脱 结 合 蛋 白。集 中280nm 具 有 高 吸 光 度 的 样 品,于5mmol/LTrisHCl(pH=80)缓冲液中透析,然后浓缩蛋白质。
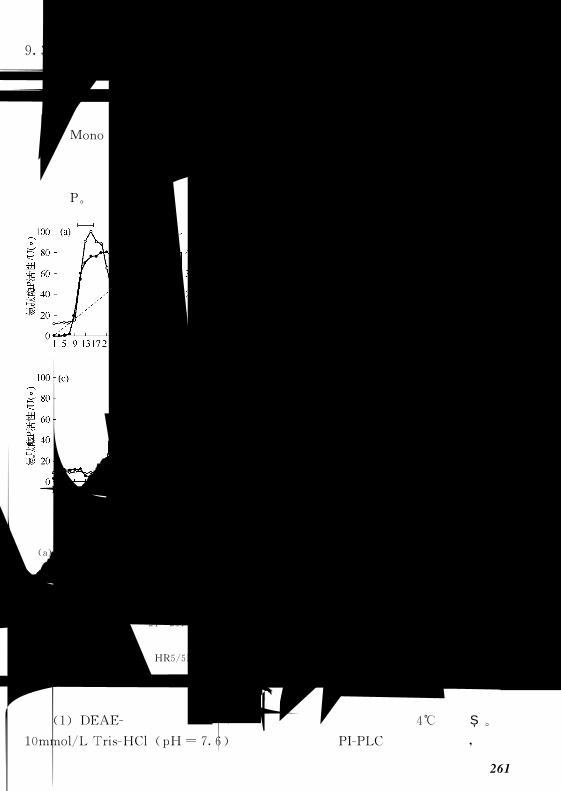
261
9362 氨肽酶P的纯化
氨肽酶P(EC34119)是一种广泛分布的哺乳动物细胞表面肽酶,包含
在体内血管舒缓激肽的代谢过程中。该GPI定位蛋白被细菌PIPLC裂解而从膜
上释放,该蛋白的亲水形式通过阴离子交换和疏水作用色谱联用进一步纯化,最
后利用 MonoQ的阴离子交换色谱浓缩氨肽酶P,经SDS聚丙烯酰胺凝胶分析,可纯化为单一蛋白。图92为纯化氨肽酶P所得到的结果。按照各个阶段酶纯度
最高而不是回收率最大的原则收集有效部分,可从400g猪肾脏皮质纯化12mg氨肽酶P。
图92 从猪肾脏皮质中纯化氨肽酶P(a)经DEAE纤维素色谱柱分离,以0~05mol/LNaCl线性梯度洗脱结合蛋白,合并馏分10~16,
于10mmol/LTrisHCl(pH76)溶液中透析;(b)再以 HR5/5MonoQ色谱柱分离,
0~05mol/LNaCl非线性洗脱结合蛋白,合并馏分29~35,于20mmol/L磷酸钠 (pH75)
溶液中透析;(c)HR5/5烷基Superose色谱柱分离,20~0mol/L(NH4)2SO4 非线性洗脱
结合蛋白,合并馏分24~26,以cilastatinSepharose和lisinoprilSepharose混合亲和色谱分离,
该柱的全部流出液于10mmol/LTrisHCl(pH=80)溶液中透析;
(d)以平衡的 HR5/5MonoQ色谱柱分离,0~05mol/LNaCl非线性梯度洗脱
结合蛋白,合并馏分32和33作为纯化的氨肽酶P
氨肽酶P活性; 蛋白质
(1)DEAE纤维素 阴 离 子 交 换 色 谱 所 有 纯 化 操 作 均 在4℃下 进 行。在
10mmol/LTrisHCl(pH=76)缓 冲 液 中 透 析 PIPLC 溶 解 的 样 品,样 品

262
31000g离心90min,除去沉淀物质,得到的上清液流经已预平衡的DEAE纤维
素色谱柱 (30ml床体 积),流 速 为20~30ml/h,洗 涤 色 谱 柱 后 以200ml含0~05mol/LNaCl的上 述 TrisHCl缓 冲 液 非 线 性 洗 脱 结 合 蛋 白,收 集 样 品。于
280nm测定吸光度,并分析酶活性。(2)烷基Superose疏水作用色谱 将上述活性部分于20mmol/L磷酸钠缓
冲液 (pH=75)中透析,透析后的样品与等体积的 含4mol/L(NH4)2SO4 的
相同磷酸钠缓冲液混合,采用预平衡的HR5/5烷基Superose色谱柱分离,用相
同磷酸 钠 缓 冲 液 以20~0mol/L (NH4)2SO4 线 性 梯 度 洗 脱,收 集 样 品,于
280nm测定吸光度,并分析酶活性。(3)MonoQ阴离子交换色谱 在10mmol/LTrisHCl(pH=80)缓冲液
中透析上 述 样 品,以 HR5/5MonoQFPLC色 谱 柱 分 离,10mmol/LTrisHCl(pH=80)缓冲 液 以0~05mol/LNaCl梯 度 洗 脱 结 合 蛋 白,收 集 活 性 部 分,于280nm测定吸光度,并分析酶活性,得到纯化后的氨肽酶P。
9363 果糖1,6二磷酸酶的纯化
果糖1,6二磷酸酶存在于肝细胞的胞液中。这里介绍利用硫 酸 铵 沉 淀 和 两
步不同洗脱方式的CM纤维素阳离子交换色谱从猪肝中纯化该酶的方法,其 中
第二步色谱利用底物洗脱。整个纯化过程所有缓冲液都含有二硫苏糖醇,以保持
胞液的还原环境,而在初始的均质缓冲液中含有PMSF,尽量减少有害的蛋白水
解。该方法可从50g猪肝中纯化得到2mg果糖1,6二磷酸酶。所有纯化 操 作 均 在4℃下 进 行,用200ml含015mol/LKCl、01mmol/L
DTT、1mmol/LPMSF的缓冲液均质化50g猪肝脏,48000g离心匀浆1h,移取
上清液,并于100000g离心1h。再次移取上清液,加入5mol/L冷NaOH溶液,调节pH 至 65±01,于 100000g 再 次 离 心 1h,向 上 清 液 中 缓 慢 加 入
(NH4)2SO4 (327g/100ml),搅拌30min,48000g离 心20min,再 向 上 清 液 中
缓慢加入(NH4)2SO4 (66g/100ml),搅拌30min,48000g离心20min,弃去上
清液,将 沉 淀 溶 解 于30ml缓 冲 液 A (5mmol/L丙 二 酸/NaOH,01mmol/LDTT,pH=60)中,并于相同缓冲液中透析。以缓冲液A预平衡的25cm×10cmCM纤 维 素 色 谱 柱,分 离 透 析 液,色 谱 柱 洗 涤 后,以 100ml的 5~30mmol/L丙二酸/NaOH、01mmol/LDTT (pH=60)溶液线性梯度洗脱结
合蛋白,合 并 具 有 高 的 果 糖1,6二 磷 酸 酶 活 性 的 部 分,浓 缩 至 pH=65、
5mmol/L丙二酸/NaOH (含01mmol/LDTT)缓冲液中,并 以 此 缓 冲 液 预 平
衡CM纤维素色谱柱 (1cm×5cm),分离浓缩后的样品,以含01mmol/L果糖1,6二磷酸的上述缓冲液洗脱结合蛋白,收集、合并、浓缩酶活高的部分,得到
纯酶。

263
94 植物组织蛋白质的纯化
植物在蛋白质纯化方面存在一些特殊的问题,使得单位体积蛋白得率比动物
或原核生物要低,而且组织的破坏、干扰纯化和具有氧化作用的天然物质的存在
会影响酶的活性。谨慎选择材料来源以及恰当的组织破碎方法就显得很重要。一
旦蛋白质成为可分离的形式,植物蛋白就可像其他蛋白一样,采用一般的纯化方
法进行处理。本节介绍一些关于植物来源蛋白质纯化的情况,同时对于比较重要
的植物系统所特有的酶,介绍一些成功的纯化方法。
941 影响植物组织蛋白质纯化的因素
9411 蛋白质浓度低
与大部分其他生物不 同,植 物 的 大 量 固 体 物 质 并 不 是 蛋 白 质,而 是 包 括 淀
粉、纤维素、果胶和半纤维素以及多酚等在内的其他大分子物质。最初的均质化
必须在大量的缓冲液中进行,以便初步过滤。大量的不溶性物质由于凝胶作用或
者简单的吸附作用会影响过滤,因此最初只能得到低浓度的蛋白质,需要经过沉
淀或浓缩。均质化后硫酸铵沉淀、PEG沉淀对于植物蛋白质很常用。如果采用
超滤浓缩,应考虑植物蛋白与膜结合的能力,以免产生负面影响。
9412 蛋白质水解
植物蛋白质纯化有时会出现蛋白质水解的问题,常伴有初始蛋白质浓度的降
低,这可能由于初始步骤操作时间过长,或者存在蛋白酶的作用。植物含有的主
要蛋白水解酶类包括丝氨酸蛋白酶、半胱氨酸蛋白酶和金属蛋白酶,可选用相应
的抑制剂。1mmol/L的PMSF适用于丝氨酸蛋白酶的抑制 (表99)。
9413 抑制和褐变
许多植物组织富含氧化酶,它们作用于内源底物,产生活性产物,与酶反应
后使其钝化,这可能是植物天然防御机制的一部分。多酚氧化酶存在于许多植物
中,如马铃薯、苹果、香蕉和甜菜,从而产生褐变反应,特别是当游离酪氨酸存
在时。其他植物如豆类,组织破碎时所大量 释 放 的 过 氧 化 物 酶 也 会 引 起 褐 变 反
应。通过除去底物或抑制氧化酶可以减少褐变反应。阳离子树脂 (如Dowex1×2)或疏水作用树脂 (如AmberliteXAD2)可用于去除反应底物。反应抑制剂
可以使用硫醇、2巯基乙醇或二硫苏糖醇,也可选择抗坏血酸 (但应注意对均质
缓冲液pH的影响)或偏亚硫酸氢钠 (表99)。
942 材料来源
植物很少整体用于蛋白质纯化,通常是将一些器官如叶或茎作为主要原料,也可能利用一些特殊植物结构,制备相对均一的细胞类型作为起始材料,这种情

264
况相对比较少。
表99 植物组织提取缓冲液常用的保护剂
保 护 剂 最终浓度 保 护 剂 最终浓度
渗压剂
蔗糖 033~05mol/L
山梨醇 033mol/L
甘油 10%(体积分数)抗氧化剂
2巯基乙醇 1~10mmol/L
二硫苏糖醇 1~10mmol/L
抗坏血酸(维生素C) 1~20mmol/L酚氧化抑制剂
PVP 1%~5%(10~50g/L)
苯酚甲醛离子交换树脂XAD2 1%~5%(10~50g/L)
Dowex1×2树脂 1%~5%(10~50g/L)
偏亚硫酸氢钠 1~5mmol/L蛋白酶抑制剂
牛血清白蛋白 1%~5%(10~50g/L)
PMSF 1mmol/L
对氯汞苯甲酸 1mmol/L
1,10二氮杂菲 1mmol/L
EDTA 1mmol/L
亮肽素 10~100μmol/L
抑胃肽 1μmol/L
抑凝乳蛋白酶素 10~100μmol/L
9421 叶
叶来源比较丰富,叶细胞主要是初级细胞壁,比较脆弱,因此容易破碎。配
备双筒显微镜可检查细胞的破裂,搅切破碎率通常应在90%左右。过度均质化
会导致质体的破裂,在纯化可溶性蛋白质时会遇到困难。叶是很好的原料,一般
不会有很多问题。但如果目的产物是抗体,由于最终产物受到二磷酸核酮糖羧化
酶/加氧酶的污染,纯化会困难一些。而要完全提取所有光合作用酶,则需要利
用研磨法。叶上的腺毛和毛状体结构经常富含特定的蛋白,从其他原料纯化困难
的酶可利用这些结构纯化。马铃薯叶毛状体的多酚氧化酶和薄荷腺毛中单萜生物
合成的酶就是两例。后者的研究取得了成功,完成了许多酶的纯化,并克隆了编
码这些酶的cDNA,如果没有初始的细胞富集过程,这些结果很可能难以实现。一般采用玻璃珠温和研磨叶子表面进行富集,利用该方法约90%的总叶毛状体
可以完整的形式高纯度分离。从植物腺毛毛状体中分离分泌细胞的过程如下:收集顶芽和叶子,浸泡在冷
的去离子水中1h,加入20g植物材料和150g玻璃珠 (直径05mm),XAD4树
脂于破 碎 器 中,将 破 碎 缓 冲 液 [200mmol/LHepes,pH=73,含 有200mmol/L山梨醇、10mmol/L蔗 糖、5mmol/L二 硫 苏 糖 醇、10mmol/LKCl、5mmol/LMgCl2、05mmol/L磷酸钾、01mmol/LPPi(焦磷酸)、06% (6g/L)甲基纤维
素和1% (10g/L)分子量40000的聚乙烯基吡咯烷酮]注满容器,研磨叶和芽
操作4次,每次1min,中间置于冰水冷却30s,以350μm的尼龙滤网除去植物
材料、玻璃珠和XAD4树脂,以不含PVP或甲基纤维素的破碎缓冲液洗涤截留
物,将滤液通过105μm的尼龙滤网,除去小组织片段后,再 通 过20μm过 滤 收

265
集分泌细胞。
9422 茎
茎材料蛋白质回收率低,其纤维性质使得组织破碎很困难。对于草本植物,茎可切成小段,于研钵中与研磨剂 (如酸洗的粗沙)一起研磨。如果酶在极端温
度下稳定,可以先在液氮中冷冻,干磨,再加入缓冲液形成浆液,但是短时间内
难以破碎很多细胞。
9423 根
根通常比茎更为脆弱,含有丰富的可溶性酚类物质,一般取决于生长条件。有时根也含有丰富的氧化酶,如过氧化物酶 (如玉米),主根和块茎含有多酚氧
化酶 (如甜菜和马铃薯)。常规抗氧化褐变的预防措施 (PVP,Dowex,抗氧化
剂,硫醇等)可以改善以此为材料的纯化工作。
9424 种子和储藏器官
种子和储藏器官有相似的问题,它们都有高含量的储藏蛋白,这些蛋白会与
目的蛋白一起纯化,因而将其分离非常困难。马铃薯块茎就是典型的例子,含有
丰富的储藏蛋白———马铃薯糖蛋白。对这些原料进行蛋白质纯化之前,预先了解
其中的蛋白质分子量范围是有益的。得到任何与目的蛋白分子量相似的纯化结果
都应谨慎 作 出 结 论。如 果 某 一 步 骤 可 以 有 效 吸 附 主 要 储 藏 蛋 白,也 能 有 助 于
纯化。
9425 果实
果实是经历明显发育变化的器官,应该对目的蛋白的出现时间有所了解。过
熟的果实可能会含有过量的蛋白酶和糖苷酶,造成可溶性多糖增加,纯化初期的
澄清过滤和离心阶段可能会促使蛋白质沉淀。绿色果实在提取蛋白质前可于黑暗
中存放数天,以降低由于破损所释放的质体蛋白的污染。有色果实一般不会出现
特殊问题。
9426 组织培养物
组织培养物作为一种丰富的原料来源,可提供相对单一的细胞类型,提供了
纯化次级代谢酶的有效原料。虽然自养细胞培养是可行的,但大多数培养物却是
异养的,因而不需要叶绿体,以避免绿色组织可能带来的一些问题,通常也可以
避免整个植物原料中会引起问题的一些酶和化合物。由于组织培养物蛋白质含量
丰富而没有活性,酶的纯化一般不从完整的植物组织开始。激发是一种非常有用
的操纵方法。在实际操作中,将真菌材料加入到培养物中,从而打开其特有的次
级代谢途径。此方法特别有助于多酚、单萜和生物碱代谢酶的纯化。有些如来自
面包酵母的激发物应用相当普遍,而有些则具有物种专一性。

266
9427 细胞器
有些主要的细胞结构为植物细胞所特有,如叶绿体、液泡和细胞壁等。其他
一些结构如内膜系统,在内质网ER和高尔基体的增殖水平上表现出差异,使其
与其他细胞类型区分开来。这些结构本身 可 以 作 为 原 料,而 且 可 以 加 快 纯 化 进
程。细胞壁是分泌系统的产物,由于细胞壁成分的独特性,ER、高尔基体和泡
囊都含有植物所特有的酶。(1)叶绿体和有色体 制备完整的、具有活性的叶绿体已经有许多方法,但
其中有些方法并不包括叶绿体分离步骤,而是直接对过滤的匀浆液进行硫酸铵沉
淀。对于一些位于叶绿体中的涉及其他代谢途径的酶,叶绿体的富集也是一个必
要的步骤。叶绿体可在各种渗压剂中分离,最常用的是033mol/L山 梨 醇,也
可使用蔗糖和甘露醇。缓冲液并不关键,Tris、MES和 Hepes都广泛使用。一
般添加剂为 MgCl2、MnCl2、无机磷酸盐和EDTA,异抗坏血酸和BSA为常用
保护剂。叶片经短时间混合,过滤,低速离心 (如1000g),完整的叶绿体可通
过温和搅拌或用软笔重新悬浮。叶绿体可经糖 (如蔗糖)、多糖 (Ficoll)或无机
物 (如Percoll)的密度梯度 分 离 进 一 步 纯 化。果 汁 有 色 体 也 可 密 度 梯 度 分 离,但对 于 辣 椒 中 的 有 色 体,蔗 糖 是 惟 一 适 合 的 渗 压 剂,细 胞 器 集 合 在084/
145mol/L的界面上。粗的或纯化的质体制剂可在适当的缓冲液中破碎,一般使
用100mmol/LTrisHCl(pH78),含有1mmol/LEDTA、10mmol/LMgCl2、
15mmol/L2巯基乙醇和005% TritonX100缓冲液。用于酶纯化的叶绿体和有色体的分离 方 法:将 去 茎 叶 片、绿 色 果 实 (2kg)
或有 色 果 实 (2kg)切 碎,置 于 2L 预 冷 的 分 离 介 质 [50mmol/L TrisHCl(pH=80)溶液,含1mmol/LEDTA、04mol/L蔗糖、1mmol/L2巯基乙醇]中,组织破碎 数 次,细 棉 布 过 滤,150g离 心5min,对 于 叶 绿 体,将 上 清 液 于
2200g离心30s(对于有色体,将上清液于2200g离心10min),弃去上清液,以
上述分离介质洗涤粗细胞器,并在所需离心条件下沉淀,采用蔗糖梯度离心纯化
质体。对于叶绿体,先悬浮于10ml分离介质中,在不连续蔗糖梯度 [075mol/
L,1mol/L,15mol/L蔗 糖 (各10ml)溶 于50mmol/LTrisHCl(pH=76)溶液中,含1mmol/L2巯基乙醇]上分层,1000g离心15min,完整的叶绿体条
带于1/15mol/L界面处,破碎的膜于075/1mol/L界面处 (对于有色体,先悬
浮于2ml分离介质中,在不连续蔗糖梯度 [045mol/L,084mol/L,145mol/
L蔗糖 (各9ml)溶于50mmol/LTrisHCl(pH=76)溶液中,含1mmol/L2巯基乙醇]上分层,62000g离心1h,完整的有色体条带于084/145mol/L界
面处,破碎的膜于045/084mol/L界面处。(2)微粒体 植物微粒体蛋白的纯化由于含量低以及可操作体积的限制显得

267
比较困难,可利用复杂的梯度离心浮选法进行富集。沉淀微粒体所要求的高离心
速度通常意味着可操作的最大体积约为200ml,但利用 Mg2+ 沉淀可有效地从大
量样品体积中得到大量的微粒体,采用该方法,经1000g离 心20min除 去 细 胞
壁、叶绿体和胞核,得到大量的匀浆液,加入适当体积的2mol/LMgCl2 使其浓
度达到25mmol/L。上清液于4℃保持30min,使微粒体聚集,然后在所用转子
最大允许转速下离心收集,可沉淀大部分ER和高尔基体。以该方法制备的大量
微粒体可进一步分离为富含ER或高尔基体膜的制剂。微粒体易于溶解,溶解后
有比较高的活力回收率。适合的溶解缓冲液应为50mmol/LTrisHCl溶液,含
有10mmol/LMgCl2、1mmol/L二硫苏糖醇和1%还原的TritonX100。大量制备和分离用于酶纯化的植物微粒体的方法 按1∶1将植物材料或组
织培养细 胞 于 均 质 介 质 [50mmol/LTrisHCl(pH=76)溶 液,含1mmol/LMgCl2、1mmol/L二硫苏糖醇、033mol/L蔗糖、1%PVP]中均质化,细棉布
过滤,于1000g 离 心20min,上 清 液 于10000g 再 离 心10min,加 入2mol/LMgCl2 至最终浓度为25mmol/L,微粒体于4℃下絮凝30min,高速离心1h,收
集微 粒 体。弃 去 上 清 液,将 沉 淀 重 新 悬 浮 于 悬 浮 介 质 [50mmol/LTrisHCl(pH=76)溶液,含10mmol/LMgCl2、1mmol/L二硫苏糖醇、033mol/L蔗
糖]中,粗 微 粒 体 部 分 不 连 续 蔗 糖 梯 度 [05mol/L、1mol/L、16mol/L蔗 糖
(各10ml)溶于50mmol/LTrisHClpH=76溶 液,含1mmol/L2巯 基 乙 醇、
10mmol/LMgCl2]100000g离心1h,富集高尔基体膜,高尔基体条带位于05/
10mol/L界面处,10/16mol/L界面处富含ER。
943 确定植物蛋白种类的直接和间接策略
蛋白质通过色谱进行分离,并直接分析确定,可认为是直接策略。对于植物
主要考虑的是组织富集和破碎技术。至于色谱材料以及运行条件等因素根据经验
与采用其他原料时相似。前面已经提到,植物的最终产率较低,初始蛋白质浓度
低,需要浓缩,因而必须考虑浓缩步骤和最终收率。有许多纯化在最后回收阶段
失败,主要是由于蛋白质黏附在浓缩膜上,而数量又不足以形成稳定的沉淀,因
此经常导致相当数量的蛋白质损失。有的实验室倾向于最后采用氯仿/甲醇沉淀,而不 是 硫 酸 铵 或 三 氯 乙 酸 沉 淀。如 果 需 要 用 于 蛋 白 质 测 序,可 以 使 用 反 相
HPLC,最终产物通过SDSPAGE测 定。如 果 银 染 色 显 示 单 一 条 带,可 认 为 是
纯的。氯仿/甲醇沉淀法和反相 HPLC法是蛋白质测序前常用的最后分离方法。氯
仿/甲醇沉淀法的具体操 作 如 下。于1体 积 的 样 品 中 加 入4体 积 的 甲 醇,混 合,加入1体积的高纯度氯仿混合,再加入1体积的水,混合,离心形成两相,弃去
上层,注意不要破坏两相界面上的白色蛋白质层。再加入3体积的甲醇,混合,

268
离心沉淀蛋白,除去上 清 液,溶 解 蛋 白 于 适 当 溶 剂 中。反 相 HPLC法 的 操 作:将蛋白质样品于01% (体积分数)的三氟乙酸 (TFA)中透析,浓缩以减少体
积,利用AquaporeRP300C8 色 谱 柱,以 含01% (体 积 分 数)TFA的90%(体积分数)乙腈溶液0~70%的梯度进行 HPLC,收集蛋白,洗脱的蛋白完全
适合于N末端蛋白测序。在许多情况下,考虑到活力损失,不允许进行多步柱分离,或者最终产品显
示出多条蛋白质条带。此时可以采用间接分析技术,如凝胶电泳法可用于标示目
的酶,此方法可在自然凝胶中进行,或者以活性底物类似物,通常为叠氮衍生物
标记蛋白。含血红素的蛋白可通过280nm和405nm两种波长检测进行纯化,此方法可用
于富含血红素的蛋白如过氧化物酶,以及其他一些纯化较为困难的酶,如细胞色素
P450的纯化,该酶由于需要与NADPH细胞色素P450还原酶重组,还没有直接的分
析方法。有时也可利用氢过氧化物作为供体进行测定,或通过与底物和抑制剂的结
合谱进行检测。利用这些方法已经纯化了一些细胞色素P450 (表910)。
表910 血红素和铜氧化酶纯化显示分析系统及色谱步骤
酶 分析 1 2 3 4 5 6
CYP71 HP SM DE GFCYP73肉桂酸羟化酶 BS SM 2P DE HACYP74丙二烯氧化物合酶 HP AP AS HI MQ MPCYP79NTyr羟化酶 BS SM DE/GF RACYP80黄芦木碱合酶 R SM HI HA MQ MP木质素过氧化物酶 S WW LC GF漆酶 OE WW AS LC NG多酚氧化酶 S AS DE GF LC HI GF
注:HP—氢 过 氧 化 物 分 析;BS—结 合 谱 分 析;R—辐 射 分 析;S—光 谱 分 析;OE—氧 电 极 分 析;
SM—溶解微粒体;AP—丙酮粉;WW—细 胞 壁 盐 洗;AS—硫 酸 铵 沉 淀;DE—DEAE离 子 交 换;2P—双
相TritonX114分离;GF—凝胶过滤;HI—疏水作用色谱;RA—红琼脂糖;HA—羟磷灰石色谱;LC—
ConASepharose;MP—MonoP色谱聚焦 (Pharmacia公司);MQ—MonoQ (Pharmacia公 司);NG—非
变性凝胶电泳。
944 样品纯化举例
9441 糖代谢酶类
植物是糖代谢最典型的代表,除 了 在 所 有 真 核 生 物 中 发 现 的 分 解 代 谢 途 径
外,它们还能固碳,而且可大量生产结构和储藏多糖。尽管所有卡尔文循环的酶
基因都已被克隆,但并不是所有的酶都已纯化得到纯酶 (表911),其中有些酶
具有胞液和质体形式,需要区别和分离。目前大多数纯化方法是直接从全细胞匀

269
浆液开始。
表911 成功纯化卡尔文循环和C4 光合作用酶的方法
酶 分析 1 2 3 4 5
核酮糖1,5二磷酸羟化/加氧酶 R DG MQ果糖二磷酸酶 S AS IE GF景天庚酮糖二磷酸酶 S CS DE GF BS核酮糖5磷酸激酶 S AS HI BSPEP羧化酶 S PG IE GF BS GF丙酮酸二激酶 S PG IE GF BSPEP羧激酶 S AS DE DE
注:R—辐射 分 析;S—分 光 光 度 分 析;DG—密 度 梯 度 离 心;AS—硫 酸 铵 沉 淀;CS—叶 绿 体 基 质;
PG—PEG沉淀;DE—DEAE离子交换;GF—凝胶过滤;HI—疏水作用色谱;IE—离子交换色谱;MQ—
MonoQ (Pharmacia公司);BS—BlueSepharose。
淀粉合成酶、ADP葡萄糖焦磷酸化酶、淀粉合酶和分支酶构成了一条相对
较短的代谢途径,但却是重要的商业酶。出于纯化目的,这些酶一般作为可溶性
酶处理,但淀粉合酶却是从分离的淀粉颗粒中纯化出来的。负责合成细胞壁多糖
的酶难以纯化,部分是由于这些是膜结合酶,任何溶解过程立即会改变其作用的
环境而导致酶的变化 (表912)。
表912 纯化淀粉和细胞壁多糖生物合成酶的方法
酶 分析 1 2 3 4 5
UDP葡萄糖焦磷酸化酶 R PG DE HI HI淀粉合酶 R SG DE HI淀粉分支酶 R SG DE HA GF蔗糖磷酸合酶 R PG DE HI MQ HAUDP葡萄糖脱氢酶 S AS HI GF BS阿拉伯聚糖合酶 R SM DE SE RP愈创葡聚糖合酶 R SM DE SE木聚糖合酶 R SM DE SE岩藻糖基转移酶 R SM GF NG IF
注:R—辐 射 分 析;S—分 光 光 度 分 析;PG—PEG 沉 淀;SG—淀 粉 颗 粒 分 离;AS—硫 酸 铵 沉 淀;
SM—溶解微粒体;BS—BlueSepharose;DE—DEAE离 子 交 换;HI—疏 水 作 用 色 谱;HA—羟 磷 灰 石 色
谱;IF—等电聚焦;GF—凝胶过滤;MQ—MonoQ (Pharmacia公 司);NG—非 变 性 凝 胶 电 泳;RP—反
相 HPLC。
9442 次级代谢酶类
植物具有广泛的次级 代 谢 特 性,具 有 很 大 的 商 业 利 益,也 是 基 因 操 作 的 目
标。虽然通过突变和互补已经分离了基因,但通过蛋白质纯化途径进行基因克隆
仍有一定空间。

270
次级代谢产物的形成和相互转变很复杂,类黄酮的合成从p香豆酰CoA和
丙二酰CoA开始,在生物化学、分子生物学和遗传学方面已有广泛介绍。类黄
酮合成途径中,四羟基苯基苯乙烯酮一般不会积累,由苯基苯乙烯酮异构酶转化
为一般的黄酮柑橘黄素,后由二氢黄酮3羟化酶羟化得 到 二 氢 山 柰 酚,该 物 质
在此途径中处于中心位置,从它开始产生分支,是种间特异性积累黄酮醇和花色
苷的基础。类黄酮生物合成中许多酶的纯化方案列于表913中。
表913 类黄酮生物合成酶的纯化
酶 分析 1 2 3 4 5 6
苯基苯乙烯酮异构酶 S AS IE DL二氢黄酮3羟化酶 R AS IE GF HA MP HI黄酮合酶 R AS QS GF MP HI二氢黄酮4还原酶 R AS QS DL MQ异黄酮氧化还原酶 R AS QS DL GF紫檀烷合酶 TC AS QS DL GF糖基转移酶 R AS AD AC MP GF
注:S—分光光度分析;R—辐射分析;TC—TLC分析;AS—硫酸铵沉淀;QS—QSepharose;AD—
ADPSepharose;DL—染料配基离心;GF—凝 胶 过 滤;AC—亲 和 色 谱;HA—羟 磷 灰 石 色 谱;HI—疏 水
作用色谱;IE—离子交换色谱;MP—MonoP (Pharmacia公司);MQ—MonoQ (Pharmacia公司)。
生物碱的生物合成酶主要来源于组织培养。涉及的生物合成酶的纯化方案列
于表914。
表914 涉及生物碱生物合成酶的纯化
酶 分析 1 2 3 4 5
长春花朵灵酰基转移酶 S AS GF QS HA AC色氨酸脱羧酶 R AS DE HA GF MQ颠茄酮还原酶 S AS DE HA HI MP
注:R—辐射 分 析;S—分 光 光 度 分 析;AS—硫 酸 铵 沉 淀;GF—凝 胶 过 滤;DE—DEAE离 子 交 换;
HA—羟磷灰石色谱;HI—疏 水 作 用 色 谱;QS—QSepharose;MP—MonoP (Pharmacia公 司);MQ—
MonoQ (Pharmacia公司);AC—亲和色谱 (辅酶A)。

271
10 蛋白质的常用指标分析
在蛋白质纯化过程中,涉及多种理化指标的分析,常用的包括蛋白质含量的
分析测定、纯度的检测、分子量的测定、等电点的测定以及N末端和C末端测
定。这些指标的分析对于认识某一蛋白质,指导纯化过程都有很重要的作用。
101 蛋白质的含量分析
1011 测定蛋白质浓度的方法
蛋白质的含量测定是生物化学研究中最常用、最基本的分析方法之一。常用
的经典方法有定氮法、双缩脲法 (Biuret法)、Folin酚试剂法 (Lowry法)、紫
外吸收法。近10年来普遍使用的新测定法有Bradford法 (考马斯亮蓝法)和银
染色法。需要注意的是,这些方法并非在任何条件下适用于任何蛋白质,同一蛋
白质溶液采用不同的测定方法,可能获得不同的结果。在蛋白质的分离纯化过程中,测定溶液中蛋白质的总量或某一蛋白质的含量
是常规要求,在选择测定方法时应考虑:测定方法的灵敏度及精确度能否满足要
求,所需蛋白质样品的量以及测定后能否回收,蛋白质的性质,可能存在的干扰
物质,所需的仪器和花费时间以及操作是否简单容易,测定结果是绝对值 (近似
值)还是相对值 (标准的比值)等。每种测定方法都有优缺点。定氮法操作复杂但结果较准确,其他方法中使用
的标准蛋白质多以定氮法来测定蛋白质含量。双缩脲法虽然灵敏度差,但是干扰
物质少,可用于无需十分精确的蛋白质快速测定。Bradford法和Lowry法的灵
敏度比紫外吸收法高10~20倍,比双缩脲法灵敏100倍以上。Bradford法具有
快速、高灵敏度的突出优点,应用越来越广泛。利用微波改良的Lowry法和二
喹啉甲酸 (BCA)法可在极短的时间内精确测定蛋 白 质 浓 度,非 常 有 利 于 常 规
检测。以下简要介绍各种测定方法的基本原理、测定方法、要点以及改良操作。
1012 紫外分光光度法
蛋白质分子中的酪氨酸、苯丙氨酸和色氨酸残基的苯环含有共轭双键,使蛋
白质具有吸收紫外光的性质。最大吸收峰 (λmax)280nm处的吸光度与蛋白质含
量成正比。此外,蛋白质溶液在238nm的光吸收值与肽键含量成正比。利用一
定波长下,蛋白质溶液的光吸收值与蛋白质浓度的正比关系,通过简单的分光光

272
度计可以定量测定溶液中的蛋白质含量。紫外吸收法简便、灵敏、快速,不消耗样品,测定后仍能回收使用。大多数
缓冲液和低浓度的盐不干扰测定,因而特别适用于 柱 色 谱 洗 脱 液 的 快 速 连 续 检
测,此时只需测定蛋白质浓度的变化,而非绝对值。此法的特点是测定蛋白质含量的准确度较差,干扰物质多。在用标准曲线法
测定蛋白质含量时,对那些与标准蛋白质中酪氨酸和色氨酸含量差异大的蛋白质
有一定的误差。故该法适于用测定与标准蛋白质氨基酸组成相似的蛋白质。当样品中含有嘌呤、嘧啶及核酸等吸收紫外光的物质时,干扰较大。虽然可
通过校正计算适当消除核酸的干扰,但测定结果仍存在一定误差,因为不同的蛋
白质和核酸的紫外吸收是不相同的。溶液的pH会改变 蛋 白 质 吸 收 高 峰,样 品 测 定 要 与 测 定 标 准 曲 线 在 相 同 的
pH下进行。吸光测量值最好在005~10,吸光度在03附近的精度最高。浑
浊溶液应采取过滤 (02μm过滤)或 离 心 使 溶 液 澄 清,或 者 用A280-A310进 行
修正。
10121 近UV (280nm)光吸收法
测定蛋白质溶液在280nm的吸光度值是最常用的紫外吸收法。在近UV区,蛋白质的吸光度主要取决于其 中 的 酪 氨 酸 和 色 氨 酸 残 基 的 含
量,此外,在很小程度上,还取决于苯丙氨酸和二硫键的含量。因此,不同蛋白
质的消光系数 变 化 较 大,对 于 大 多 数1mg/ml的 蛋 白 质 溶 液 的A1mg/ml280 为05~15 (表101),而某些富含酪氨酸的绵状蛋白达到4。因此,对于某一特定蛋白
质测定比吸光值,可以比较准确计算出蛋白质浓度。
表101 标准蛋白质溶液A1mg/ml280
蛋 白 质 A280 蛋 白 质 A280
牛血清白蛋白 070 γ球蛋白 138核糖核酸酶A 077 胰蛋白酶 160卵白蛋白 079 胰凝乳蛋白酶 202肠毒素 133 α淀粉酶 242
这种测定方法操作简单、快速、灵敏,且样品可回收,低浓度的盐不干扰测
定,因而被广泛应用于蛋白质制备中,特别适用于柱色谱分离中的样品测定,便
于连续自动装置的使用。但是测定准确度较差,不是严格的定量方法,适用于粗
提蛋白的测定。许多在紫外区引起光吸收的物质会干扰测定,若存在非蛋白类的
发色基团 (如血红素、吡哆醛)则A280会增大。在280nm下核酸的消光大约是
蛋白质的10倍,有强干扰作用。测定时根据需要,对待测蛋白质溶液用合适的缓冲液 (无 UV吸收)稀释,

273
确保测量精度。以水或缓冲液为空白对照,用石英比色皿在紫外分光度计上直接
读取待测蛋白质溶液在280nm的吸光度值,控制蛋白质浓度在01~10mg/ml,检测的灵敏度为02~2mg/ml,比色杯的最小测量体积为01ml。
10122 肽键测定法
肽键在远UV区有强吸收,最大吸收峰λmax在190nm,但由于氧的吸收和分
光光度计的输出困难,一般选用238nm测定。蛋白质溶液在238nm处的光吸收
强弱与肽键的含量成 正 比。用 标 准 蛋 白 质 溶 液 配 制 系 列 浓 度 (50~500μg/ml)的蛋白质溶液50ml,测定光吸收值A238,绘制标准曲线。常用09%NaCl溶液
适当稀释蛋白溶液进行测定,再由标准曲线求得未知样品的浓度。这种测定方法简单、灵敏,样 品 可 回 收,对 于 不 同 蛋 白 质 的 光 吸 收 变 化 很
小。蛋白质溶 液 进 行 柱 色 谱 分 离 时,用238nm 检 测 洗 脱 液 的 蛋 白 质 峰 位,比
280nm吸收法灵敏。但是测定时需要在远 UV区校正分光光 度 计 的 精 度,许 多
缓冲液和多种有机化合物 (如醇、酮、醛、醚、有机酸、酰胺类和过氧化物、血
红素等)在此区域有强吸收,干扰因素较多。最好用无机盐、无机碱和水溶液进
行测定。含有有机溶剂的样品,经蒸干或其他方法去除干扰物质后,用水、稀酸
或稀碱溶解测定。
10123 吸光度差异法
当蛋白质样品中存在具有紫外吸收的干扰物时,利用在不同紫外光波下的光
吸收值的差异,来测定蛋白质的含量。(1)含有核酸的蛋白质浓度测定 核酸 对 紫 外 光 有 很 强 的 吸 收,在280nm
处的吸收比蛋白质强10倍,但吸收高峰在260nm附近,核酸260nm处的消光系数
是280nm处的2倍,纯核酸的光吸收比值A280/A260≈05。而蛋白质在280nm紫
外吸收值大于260nm的吸收值,纯蛋白质的光吸收比值A280/A260≈18。通常对含有核酸的蛋白质溶液,分别测定A280和A260,根 据 吸 收 差 值,用
经验公式计算出精确的蛋白质浓度:蛋白质浓度(mg/ml)=145×A280-074×A260
采用相似的吸光度差异的原理,还可采用下面的公式来测定含有核酸的蛋白
质样品:
蛋白质浓度(mg/ml)=A235-A280251
蛋白质浓度(mg/ml)=0183×A230-00758×A260(2)低浓度的蛋白质溶液测定 低浓度的蛋白质会由于比色杯的吸收,而从
溶液中损失。采用高离子强度,或在含有非离子表面活性剂的缓冲液中测定,有
助于防止这种损失。

274
通常不用280nm的 光 吸 收 测 定 蛋 白 质 的 稀 溶 液 浓 度,而 是 利 用215nm与
225nm吸收值之差,通过标准曲线来测定。蛋白质浓度在20~100μg/ml,蛋白
质浓度与吸光度差成正比。当pH在4~8之间,蛋白质在215~225nm范围内
的吸收值与pH无关。配制系列 低 浓 度 的 标 准 蛋 白 质 (20~100μg/ml)50ml,分 别 测 定215nm
和225nm的 吸 光 度 值,并 计 算 出 吸 收 差:Δ=A215-A225。以 吸 收 差Δ为 纵 坐
标,蛋白质浓度为横坐标,绘出标准曲线。再测出未知样品的吸收差,由标准曲
线查出未知样品的蛋白质浓度。在特定条件下,可以精确测定蛋白浓度在5μg/L的溶液。
NaCl、(NH4)2SO4 以及01mol/L磷酸、硼酸和三羟甲基氨基甲烷 (Tris)等缓冲液,都无显著干扰作用,但是01mol/L醋酸、NaOH、琥珀酸、邻苯二
甲酸、巴比妥等缓冲液在215nm光吸收值较大,必须将其浓度降到5mmol/L以
下才无显著影响。蛋白质浓度(μg/ml)=144(A215-A225)
1013 Lowry法
10131 原理
Lowry法又称为Folin酚 试 剂 法,最 早 是 由Lowry确 定 蛋 白 质 浓 度 测 定 的
基本步骤,以往在生物化学领域中广 泛 应 用。Lowry法 的 显 色 原 理 是 根 据 双 缩
脲反应,蛋白质中的肽键在碱性条件下与Cu2+ 反应,生成紫红色Cu+ 复合物。
Folin酚试剂在Cu+的催化下,磷钼酸磷 钨 酸 盐 被 蛋 白 质 中 的 芳 香 族 氨 基 酸 残
基还原,产生深蓝色的钼蓝和钨蓝混合物,其最大吸收峰在745~750nm处。在
一定的浓度范围内,所形成蓝色的深浅与蛋白质含量之间有线性关系,可用于蛋
白质含量的测定。由于产生的混合物的蓝色强度部分取决于酪氨酸和色氨酸含量,而它们在各
种蛋白质中含量不同,因此会随不同的蛋白质而出现显色深浅的变化。通常用于
酪氨酸和色氨酸的定量测定和蛋白质的相对浓度测定。由于Folin试剂的主要成分为磷钼酸和磷钨酸,试剂仅在酸性pH条件下稳
定,而还原反应又只在pH=10~105范围内发生,因此在进行测定时,Folin酚试剂加到碱性的铜蛋白质溶液中,必须立即混匀。此外,Folin试剂的配制较
为困难,近年来逐渐被考马斯亮蓝法取代。在Lowry法中,由于加入Folin酚试剂强化了双缩脲反应,使得显色量增
加,检测灵敏度提高且比较恒定,因而被广泛应用于不同环境中的蛋白质混合物
或粗提物的测定。通常测定范围是20~250μg,最低可检测5μg的蛋白含量。检测灵敏度范围

275
在5~100μg/ml。
10132 材料
(1)复合物形成试剂 试剂:a.2% Na2CO3 溶液;b.1% CuSO4·5H2O溶液;c.2%酒石酸钠钾溶液。使用前将三种试剂a∶b∶c以100∶1∶1 (体积
比)混合配制而成。(2)1mol/L的Folin试剂 配制较为困难,可购买商品化试剂。(3)标准蛋白 4mg/ml的牛血清蛋白储液,-20℃保存。用蒸馏水稀释制
备系 列 浓 度 的 标 准 蛋 白 稀 释 液 (0μg/ml,10μg/ml,20μg/ml,50μg/ml,
100μg/ml,200μg/ml,500μg/ml,1000μg/ml,2000g/ml)。
10133 方法
① 向01ml的样品 (或标样)中加入01ml的2mol/LNaOH,沸水浴水解
10min。对于沉淀物样品,采用2mol/LNaOH溶解沉淀,沸水浴水解10min后,取02ml水解液。对于整个细胞或其他复杂样品需要经过适 当 的 预 处 理 后 进 行
Lowry分析。
② 水解 液 冷 却 到 室 温,加1ml新 鲜 配 置 的 复 合 物 形 成 试 剂,室 温 放 置
10min。时间无需严格控制,在几小时内,不会影响最后的吸光度。由于Lowry反应的pH必须维持在10~105,所以在处理非此pH范围内的待测样品时要很
小心。
③ 加01mlFolin试 剂,用 旋 涡 混 合 仪 混 合,室 温 放 置30~60min。Folin试剂在碱中不稳定,必须充分混 合,这 对 结 果 的 重 复 性 也 非 常 重 要。加 入 试 剂
20~30min后呈色达到饱和,此后每小时颜色信号减弱1%,因而放置不要超过
60min。
④ 若蛋白质浓度低于500μg/ml时,在750nm读取吸光度;若蛋白质浓度
在100~2000μg/ml时,550nm测定吸光度。
⑤ 根据标准蛋白质稀释液的浓度和相应的吸光度绘制标准曲线,测定样品
的吸光度,建议测定2~3份平行样,来计算样品蛋白质的浓度。注意,高浓度时标准曲线非线性,应控制待测样品的浓度在标准曲线的线性
范围内。
10134 要点
(1)蛋白质的Lowry测定法专一性较差,干扰物质较多而且影响大。
① 表102列出了Lowry测定法中部分干扰性物质的效应,干扰性物质主要
包括缓冲液、药 物、核 酸 和 糖 类、EDTA 等。低 浓 度 尿 素 (05%)、硫 酸 钠
(1%)、硝 酸 钠 (1%)、三 氯 醋 酸 (05%)、乙 醇 (5%)、乙 醚 (5%)、丙 酮

276
(05%)等溶液对显色无影响,但这些物质浓度高时,均需作校正曲线。
表102 Lowry测定法中部分干扰性物质的影响
含50μgBSA和下列
物质的样品
Lowry法检测
的BSA/μg
水空白
校正
干扰物空白
校正
含50μgBSA和下列
物质的样品
Lowry法检测
的BSA/μg
水空白
校正
干扰物空白
校正
对照:50μgBSA水溶液 5000 —
01mol/LHCl 4420 438001mol/LNaOH 5060 506010mol/LNaCl 5020 501010%SDS 沉淀 沉淀
02%叠氮化钠 4920 4900002%叠氮化钠 4950 4960100mmol/LEDTA 13850 51050mmol/LEDTA 9670 68010mmol/LEDTA 3360 127050mmol/LEDTA,pH=1125 7230 50010%辛基葡萄糖苷 沉淀 沉淀
40%蔗糖 490 289010%蔗糖 4290 41101%蔗糖 4840 481002mol/L山梨糖醇 6370 310002mol/L山梨糖醇,pH=1125 6860 266020%硫酸铵 沉淀 沉淀
10%硫酸铵 沉淀 沉淀
3%硫酸铵 2120 214010%硫酸铵,pH=11 3260 3280
40mol/L胍基盐酸 沉淀 沉淀
30mol/L脲 5320 450010%TritonX100 沉淀 沉淀
10%Brij35 沉淀 沉淀
10%Lubrol 沉淀 沉淀
10%Chaps 沉淀 沉淀
10%Chapso 沉淀 沉淀
05mol/LTris 1020 880025mol/LTris 2790 281001mol/LTris 3890 3890025mol/LTris,pH=1125 4080 408001mol/L碳酸氢铯 沉淀 沉淀
100mmol/L葡萄糖 6810 617050mmol/L葡萄糖 6270 584010mmol/L葡萄糖 5260 512010mol/L甘氨酸 730 77010mol/L甘氨酸,pH=11 3250 279020mol/L醋酸钠,pH=55 540 33002mol/L醋酸钠,pH=55 4750 476010mol/L磷酸钠 730 53001mol/L磷酸钠 4660 4660
② 在蛋白质浓度高的情况下,通过稀释到有效检测浓度可以将干扰效应降
到最小。如果无法用稀释减少干扰时,应选择恰当的空白。
③ 当测定的蛋白质溶液浓度低于10μg/ml,可在检测前通过沉淀将样品从
干扰物质中分离出来,去除干扰物的影 响。沉 淀 操 作:加01ml015%脱 氧 胆
酸盐到10ml蛋白样品,混合在室温下放置10min,加01ml72%的TCA,混
匀,(1000~3000)g离心30min,倾去上清,沉淀用2mol/LNaOH溶解。除去
干扰的同时蛋白样品被浓缩,再测定稀释液中的蛋白质。
④ 由去垢剂、蔗糖 和EDTA引 起 的 干 扰 可 通 过 在Lowry试 剂 中 添 加SDS来消除。
(2)用改良的Lowry测定法能提高反应的灵敏度。Folin试剂加到两个组分
中,分别混合,灵敏度可提高20%;加入Folin试剂后3min,再加入DTT,灵

277
敏度可以提高50%。(3)Lowry检测法反应速度 慢,所 需 时 间 较 长。采 用 微 波 炉 改 良 操 作 可 进
行快速分析,而且形成线性的标准曲线。微波Lowry检测方法如下:在聚苯乙烯管中,混合样品与Lowry检测试剂;
将样品置于全塑料试管架上,放于微波炉中央,旁边放置一烧杯,杯中装有一定
体积的室温水,使微波室内的液体总体积为100ml;在微波最高设置下辐射10s;在750nm下测定样品的吸光度,并根据标准曲线确定蛋白质的浓度。
1014 二喹啉甲酸检测法
10141 原理
由Smith等首先提出的 二 喹 啉 甲 酸 (BCA)检 测 法,反 应 简 单 而 且 没 有 太
多的干扰物影响。反应原理和Lowry法类似,在碱性条件下,蛋白质分子中的
肽键能与Cu2+络合生成络合物,同时将Cu2+还原成Cu+。Cu+ 与BCA试剂特
异性结合,形成稳定的深紫红色复 合 物,其 最 大 吸 收 在562nm。因 为 反 应 中 产
生的Cu+是蛋白浓度和保温时间的函数,颜色的深浅与蛋白质浓度成正比,所
以可根据吸光度值与已知浓度的蛋白质标准进行 比 较,来 测 定 未 知 蛋 白 样 品 的
浓度。加入BCA强化双缩脲反应的BCA法和加入Folin酚试剂强化双缩脲反应的
Lowry法相比,由于BCA在碱性条件下稳定,操作可一步完成,而Lowry检测
法需进行两步 操 作。BCA法 的 灵 敏 度 和Lowry法 相 似,还 能 抵 抗 干 扰 物 的 存
在。该方法不受去垢剂、变性剂 (脲 和 胍 基 盐 酸)影 响,但 对 存 在 还 原 糖 更 加
敏感。有标准检测和微量检测两种类型,检测灵敏度分别为10~1200μg/ml和05~
10μg/ml。
10142 材料和方法
(1)标准检测 适用于样品蛋白含量为100μg/ml~1000μg/ml的检测
① 试剂
a二喹啉甲酸钠01g,Na2CO3·H2O20g,酒石酸钠 (二水物)016g,
NaOH04g,NaHCO3095g,配 成100ml,NaHCO3 或 NaOH 调 节 pH 至
1125。
b04gCuSO4·5H2O溶于10ml水。室温下试剂很稳定。
② 标准工作试剂 (SWR) 以体积比100∶2将试剂A和试剂B混合。溶液
呈绿色,室温下1周内稳定。
③ 检测方法 取100μl样品,加入2ml标准工作试剂,60℃保温30min。冷

278
却至室温,测定在562nm下的吸光度。用1mg/ml的牛血清蛋白储液 进 行 不 同
浓度的稀释,作标准曲线,来确定样品的相对浓度。(2)微量检测 适用于样品蛋白含量为05~10μg/ml的检测。
① 试剂
aNa2CO3·H2O08g,酒石酸钠 (二 水 物)16g,NaOH16g,用 水 配
成100ml,用10mol/LNaOH调节pH至1125。
b二喹啉甲酸钠4g溶于100ml水。
cCuSO4·5H2O04g溶于10ml水。室温下试剂都很稳定。
② 浓缩标准工作试剂 将1倍体积试剂C与25倍体积试剂B混合,再加入
26倍体积的试剂A。
③ 检测方法 取10ml样品,加入1ml浓缩标准工作试剂,60℃保温1h。冷却至室温,测定562nm下的吸光度。用1mg/ml的牛血清蛋白储液 进 行 不 同
浓度的稀释,作标准曲线,来确定样品的相对浓度。
10143 要点
(1)若延长样品的保温时间可提高检测的灵敏度。反之,如果颜色太深,可
提早停止保温。注意标准样品的处理时间必须相同。加入工作试剂保温形成的复
合物颜色可稳定1h。(2)表103列出了一些试剂对BCA检测的干扰影响。对于任何试剂的干扰,采用稀释法可以克服,此时须采用合适的对照样品,
允许形成残留色。例如,影响Cu2+利用的螯合剂EDTA会严重干扰检测,样品
中若存在脂类物质则会导致吸光值过高,都可以通过将样品稀释至有效检测浓度
来克服干扰。对于巯基和还原糖类化合物的干扰可采用以下方法去除:将蛋白质
结合到尼 龙 膜 上,彻 底 洗 涤 去 除 干 扰 物 质,然 后 对 尼 龙 膜 结 合 的 蛋 白 质 进 行
BCA检测。(3)BCA法反应时间较长,采用快速测定的微波炉改良法可促进反应进程,
改良的BCA操作形成线性的标准曲线。微波BCA检测方法如下:在聚苯乙烯管中,混合样品与BCA检测试剂;将
样品置于全塑料试管架上,放于微波炉中央,旁边放置一烧杯,杯中装有一定体
积的室温水,使微波 室 内 的 液 体 总 体 积 为100ml;在 微 波 最 高 设 置 下 辐 射20s,在562nm下测定样品的吸光度,并根据标准曲线确定蛋白质的浓度。
1015 考马斯亮蓝染色法
10151 原理
1976年,Bradford根据蛋白质与染料相结合的原理建立了考马斯亮蓝染色

279
表103 BCA检测法中干扰性物质的影响
含50μgBSA和下列
物质的样品
BCA法检测的BSA/μg
水空白
校正
干扰物空白
校正
含50μgBSA和下列
物质的样品
BCA法检测的BSA/μg
水空白
校正
干扰物空白
校正
对照:50μgBSA水溶液 5000 —
01mol/LHCl 5070 508001mol/LNaOH 4900 494010mol/LNaCl 5130 511010%SDS 4920 489002%叠氮化钠 5110 5090002%叠氮化钠 5110 5100100mmol/LEDTA 无色 无色
50mmol/LEDTA 2800 294010mmol/LEDTA 4880 491050mmol/LEDTA,pH=1125 3150 328010%辛基葡萄糖苷 5090 508040%蔗糖 5540 487010%蔗糖 5250 50501%蔗糖 5130 512002mol/L山梨糖醇 4290 378002mol/L山梨糖醇,pH=1125 4070 362020%硫酸铵 560 12010%硫酸铵 1600 12003%硫酸铵 4490 420010%硫酸铵,pH=11 4810 4520
40mol/L胍基盐酸 4830 469030mol/L脲 5130 501010%TritonX100 5020 498010%Brij35 5100 509010%Lubrol 5070 507010%Chaps 4990 495010%Chapso 5180 510005mol/LTris 3620 3290025mol/LTris 4660 440001mol/LTris 5080 4960025mol/LTris,pH=1125 5200 503001mol/L碳酸氢铯 4950 4970100mmol/L葡萄糖 24500 571050mmol/L葡萄糖 14400 477010mmol/L葡萄糖 7000 491010mol/L甘氨酸 无色 无色
10mol/L甘氨酸,pH=11 5070 489020mol/L醋酸钠,pH=55 3550 345002mol/L醋酸钠,pH=55 5080 504010mol/L磷酸钠 3710 362001mol/L磷酸钠 5080 5040
法,也称Bradford检测法,这是目前灵敏度最高的蛋白质测定方法。其检测原
理为:在酸性分析试剂溶液中,染料考马 斯 亮 蓝G250与 蛋 白 质 结 合 的 主 要 是
阴离子形式的蓝色,其最大吸收峰 (λmax)位置在590nm,形成的复合物颜色的
深浅与蛋白浓度的高低成正比关系。因此,通过测定染料的蓝色离子态可以定量
测定蛋白质,通常测定595nm下的吸光度。研究认为染料最容易与蛋白质中的精氨酰和赖氨酰残基结合,由于各种蛋白
质中的含量不同,与染料的相互作用有强 有 弱,导 致 测 定 不 同 蛋 白 质 时 偏 差 较
大。进行了一些改良后,可以克服波动问题,但通常会导致检测更容易受其他化
合物的干扰。考马斯亮蓝法目前已成为应用最广泛的蛋白质含量测定方法,BioRad公司
的蛋白质定量检测试剂盒就是应用该检测法。与Lowry法相比,Bradford法操
作更方便简单,而且快速,灵敏度更高,受生物样品中的非蛋白质组分和常用试
剂的干扰更少。

280
10152 材料和方法
(1)染料试 剂 100mg考 马 斯 亮 蓝 G250溶 解 在50ml95%乙 醇,然 后 与
100ml85%磷酸混合,加入蒸馏水配成1L。棕色瓶中室温下可储存数周,使用
前必须用滤纸过滤除去储存试剂中沉淀的染料。在常规蛋白测定中,对染料的品
质无严格要求,考马斯亮蓝G250中可溶性染料的含 量 因 来 源 和 供 应 商 不 同 变
动很大,通常认为Serva蓝G含量最高,往往用于改良测定法中。(2)蛋白质标准 在考马斯亮蓝法中,由于最常用的蛋白质标准牛血清蛋白
与染料的反应偏大,导致测定的样品蛋白含量偏低。因此,选择标准蛋白质建立
标准曲线时,尽量采用与待测样品蛋白相同的蛋白质,或者选用与染料结合能力
接近于蛋白质平均值的牛γ球蛋白为蛋白质标准,以减小偏差。在 具 体 报 告 测
定量时,必须说明所用的蛋白质标准。蒸馏水 配 置 牛γ球 蛋 白 储 液,-20℃存
放。使用前用280nm 下 的 吸 光 度 测 定 标 准 溶 液 中 精 确 的 蛋 白 浓 度,γ球 蛋 白
A1mg/ml280 (1cm光程)为135。(3)检测器皿要求 使用洁净无洗涤剂的塑料器皿或玻璃器皿,用甲醇或去
污剂漂洗去除器皿上的染料痕迹。请勿使用石英、硅胶比色杯,因为染料会结合
在此类材料上。(4)检测方 法 有 标 准 检 测 和 微 量 检 测 两 种 分 析 类 型,分 别 适 用 于10~
100μg蛋白质和1~10μg蛋白质的测定。由于检测试剂因存储时间会导致吸光度
的变化,所以每次检测必须作标准曲线。
① 样品稀释 吸取100μl蛋白样品稀释液 (100,10-1,10-2,10-3)双份。
② 制备标准曲线 吸取γ球蛋白储液 (标准检测中储液浓度1mg/ml,微量
检测中储液浓度100μg/ml),分别取10μl、20μl、40μl、60μl、80μl、100μl各双
份,用蒸馏水配成100μl,以蒸馏水为试剂空白。
③ 测定 加入染料试剂 (加入量:标准检测5ml,微量检测1ml),混合均
匀 (倒置翻转或轻轻振荡,避免气泡)。染料与蛋白质结合的过程只需要2min,复合物的颜色在1h内保持稳定 (5~20min内稳定性最好)。因此加入染料试剂
2~60min内,测定各管的吸光度,不需要严格控制时间。
④ 计算 标准曲线轻微非线性,在标准检 测 中,100μgγ球 蛋 白 的A595约
为04;在微量检测中,10μgγ球蛋白的A595为045。根据待测样品的吸光度,从标准曲线上计算出样品蛋白质浓度。
10153 要点
(1)相对来说,考马斯亮蓝法不受绝大多数常用生物化学试剂的干扰。不过
有一些 化 合 物 会 明 显 改 变 空 白 的 吸 光 度 或 是 减 缓 蛋 白 质 与 染 料 的 反 应 (表
104)。

281
表104 考马斯亮蓝法中各种试剂对检测的影响①
化 合 物OD595的
变化
相当于的
BSA/μg化 合 物
OD595的
变化
相当于的
BSA/μg
1mol/LKCl 0000 000 1mol/L巯基乙醇 0004 0365mol/LNaCl 0000 000 01mol/LEDTA 0004 0361mol/LMgCl2 0000 000 1mol/L蔗糖 0013 1171mol/L硫酸铵 0000 000 5%苯酚 0046 41495%乙醇 0000 000 丙酮 0069 62101%SDS 0011 099 01% TritonX100 0013 1171%SDS 0495 4455 1% TritonX100 0590 53102mol/LTris 0026 234 99%甘油 0012 108
① 表中的数值为标准检测中含01ml化合物测定的结果。
在碱性条件下,由于游离染料的解离平衡向阴离子形式移动,从而使碱性缓
冲液中的蛋白质吸光度增大。此外,盐酸胍、抗坏血酸 (维生素C)能和染料竞
争与蛋白质的反应,导致测得的蛋白质含量降低。通常认为测 定 中 的 主 要 干 扰 物 为 去 污 剂、TritonX100、十 二 烷 基 硫 酸 钠
(SDS)以及01mol/L的 NaOH。因 此,测 定 前 用 凝 胶 过 滤 或 透 析 去 除 样 品 溶
液中的洗涤剂和两性电解质。在测定含有膜成分的蛋白质时,往 往 采 用 膜 裂 解 剂 (NaOH 或 洗 涤 剂)预
处理样品,此时对测定结果的处理要谨慎。(2)在改良检测法中,提高染料的浓度或溶液pH,增大游离染料中能与蛋
白质反应的蓝色离子态的比例,可提高检测的灵敏度,大大降低不同蛋白质引起
的波动,但是样品对表面活性剂的干扰更敏感。(3)考马斯亮蓝法测定的蛋白质溶液浓度不能太高,因为蛋白质浓度高时,
游离染料的含量大为减少,会导致标准曲线的非线性。根据染料试剂在未与蛋白
质结合之前本身为棕褐色,在465nm下有吸收,可采用595nm和465nm下吸光
度的比值作图,来修正游离染料的耗尽,提高标准曲线的线性和测定的精确度。(4)由于蛋白质与染料结合后形成的复合物有更高的消光系数,因此随着蛋
白质浓度 的 变 化,光 吸 收 值 变 化 也 大,灵 敏 度 很 高,蛋 白 质 的 最 低 检 测 量 达
25~1μg。
102 蛋白质的分子量测定
蛋白质的分子量一般在10000~1000000之间。测定蛋白质分子及亚单位的
分子量,是为了进一步开展一级结构的研究。蛋白质分子量的测定方法很多,通常是根据蛋白质的物理化学性质来测定或

282
者根据化学分析的结果来计算分子量。先后用于测定蛋白质分子量的方法有:渗
透压、黏度、光散射、超离心 (沉降速度、沉降平衡)、凝胶过滤色谱 (柱色谱、薄板色谱)、聚丙烯酰胺凝胶电泳 (PAGE)及十二烷基硫酸钠聚丙烯酰胺凝胶
电泳 (SDSPAGE)、电喷射电离 (ESI)质谱和基质辅助激光解吸电离 (MALDI)质谱等方法。这些根据蛋白质分子的物理化学性质方法 测 得 的 分 子 量 称 为
物理分子量。常使用的方法有超离心法、凝胶过滤和SDS凝胶电泳法,其中超
离心法可直接测定蛋白质的分子量,而在凝胶过滤和SDS凝胶电泳法中的测定
结果为与分子量标准相比较的相对值 (相对分子质量Mr),一般认为超离心法测
得的结果更接近真实值。而根据蛋白质的化学分析,如氨基酸定量分析结果计算
出的蛋白质最小分子量称为化学分子量。不同方法测定的结果准确度相差很大。采用质谱法测定时,利用不同的离子
化技术形成气态蛋白质离子,根据电磁场中离子的折射程度依赖于质量来测定分
子量。测定的准确 度 达 到0001%,可 用 于 检 测 翻 译 后 的 修 饰 以 及 进 行 序 列 分
析,但是需要昂贵的特殊仪器。在大多数情况下,仅仅需要粗略估算分子大小,最常采用SDSPAGE凝胶电泳和凝胶过滤色谱法来测定蛋白质的分子量,这两
种方法快速简单,但测量值的误差在5%~10%左右。无论采用哪种方法测定分
子量,都要求蛋白样品均一,否则测定的结果不可靠。
1021 SDSPAGE凝胶电泳法测定蛋白质的分子量
10211 原理
SDSPAGE是蛋白质分析中最常用的凝胶 电 泳 系 统,广 泛 用 于 蛋 白 质 混 合
物的定性分析、纯化监测。聚丙烯酰胺凝胶由丙烯酰胺单体、亚甲基双丙烯酰胺交联剂在引发剂四甲基
乙二胺和催化剂的存在下室温聚合而成。调整丙烯酰胺浓度和交联度可以有效地
改变凝胶的孔径,从而对大多数大分子颗粒起分子筛效应。在外加电场进行电泳
时,蛋白质分子在电场中的泳动速度取决于它所带的净电荷的多少、颗粒的大小
及形状。由于蛋白质的氨基酸组成不同,等电点不同,在同一pH条件下所带的
净电荷不同,利用在同一电场中的迁移速度不同使各蛋白质分子相互分离。在SDSPAGE中,向聚丙烯酰胺凝胶电泳体系中加入一定量的十二烷基硫
酸钠 (SDS),一般加入量为01%。SDS是 能 与 蛋 白 质 强 结 合 的 变 性 剂,能 使
蛋白质的氢键、疏水键打开,并定量结合到蛋白质分子上,形成SDS蛋白质复
合物。平均1分子SDS结合有2分子的氨基酸残基,SDS与大多数蛋白质的结
合比为14gSDS/g蛋白质。由于十二烷基硫酸根带负电,电荷量远远超过蛋白
质分子的原有天然电荷量,因而掩盖了不同蛋白质之间的原有电荷差异,使荷电
互不相同的蛋白质具有相同密度的负电荷。

283
另外,蛋白质样品在 含 有β巯 基 乙 醇 和SDS的 样 品 缓 冲 液 中 煮 沸 处 理 后,所有的蛋白质都完全变性,维系二、三级结构的二硫桥被还原,肽链伸展,使空
间结构彼此不同的蛋白质具有相同的构象。由于分子筛效应,在聚丙烯酰胺凝胶
中电泳,肽链越长,迁移速度越慢,分子量的对数与蛋白质肽链的迁移率之间有
线性关系。因而在SDSPAGE中,蛋白质分子在电场中的迁移率主要取决于它的分子
大小,而与原有的净电荷及分子形状无关,所以可以用来测定蛋白质的分子量。选择聚丙烯酰胺的孔径,在凝胶中将未知蛋白质和一系列分子量标准蛋白质进行
电泳,测量每种标准蛋白在凝胶中的迁移距离,根据标准蛋白质的分子量的对数
与迁移率 之 间 的 关 系 曲 线,测 量 未 知 蛋 白 质 的 迁 移 率,从 而 计 算 出 相 应 的 分
子量。对于任何给定的凝胶浓度,分子量的对数和相对迁移率只在有限的分离范围
内呈线性关系。即15%聚丙烯酰胺凝胶中分子量的线性范围为10000~50000;
10%聚丙烯酰胺凝胶中分子量的线性范围为15000~70000;5%聚丙烯酰胺凝胶
中分子量的线性范围为60000~200000。必须注意,在SDSPAGE中分子量的对数和相对迁移率的线性关系仅仅在
蛋白质结合有恒定量的SDS比率时才是正确的。对于一些电荷异常或构象异常
的蛋白质,如非常碱性的蛋白质、糖蛋白、胶原蛋白,用SDSPAGE凝胶电泳
法测定的分子量结果不可靠。在组蛋白中,由于带正电的氨基酸比例高,减小了
整个蛋白质所带的负电荷,迁移比预想的要慢很多。分析电泳结果时要注意,一
般至少用两种方法测定,相互验证。
蛋白质的相对迁移率(Rf)=样品迁移距离
指示染料迁移的距离
在实际应用上,SDSPAGE凝胶电泳法是测 定 蛋 白 质 分 子 量 的 最 常 用 的 方
法。测定时需要至少3~4种标准蛋白质,各种标准分子量混合物都可使用。应
选用分子量分布在未知蛋白质两侧的标准蛋白质。注意,该方法测定的是亚单位
肽链的分子量,适用的蛋 白 质 分 子 量 跨 度 较 大 (10000~300000),测 定 方 法 快
速、易于操作,样品用量很少,一次操作可同时测定多个样品,但花费的时间较
长。此外,由于测量前沿和样品迁移距离的测量误差,误差较大,对操作的要求
较高。
10212 材料
(1)丙烯酰胺 储 液 30%丙 烯 酰 胺,08%亚 甲 基 双 丙 烯 酰 胺,滤 纸 过 滤,
4℃密封存放。注意:丙烯酰胺单体是一种神经毒素,操作时要非常小心,在通
风橱中称量,戴口罩和手套。

284
(2)缓冲液
a1875mol/LTrisHCl,pH88。
b06mol/LTrisHCl,pH68。(3)10%过硫酸铵 (APS) 新鲜配置。(4)10%SDS 室温储存。低温下SDS从 溶 液 中 析 出,可 将 试 剂 瓶 在 水 浴
中加热溶解。(5)四甲基乙二胺 (TEMED)(6)电泳缓冲液 12gTris,576g甘氨酸,20gSDS,用水配制成2L,无
需调pH。(7)样品缓冲液 50ml06mol/LTrisHCl(pH=68),05gSDS,50g
蔗糖,025mlβ巯基乙醇,05%溴酚蓝储液50ml,用蒸馏水配制成50ml。缓
冲液中的β巯基乙醇的还原力随储存时间下降,通常2周后须重新配制或补加巯
基乙醇。(8)蛋白质染色液 溶解在50%甲醇中的01%考马斯亮蓝R250,10%冰
醋酸。先将染料溶解在甲醇中,再加水,然后加冰醋酸。必要时用滤纸过滤。(9)脱色液 10%甲醇,7%冰醋酸。(10)微量加样注射器或微量移液器枪头。
10213 方法
(1)电泳 样 品 的 准 备 蛋 白 质 样 品 与 样 品 缓 冲 液 混 合,95~100℃加 热
5min。
① 固体样品直接溶解在样品缓冲液中,纯蛋白或简单 混 合 物 的 浓 度 为1~05mg/ml。复杂样品的合适浓度则需要测定。
② 溶液样品用等体积的两倍样品缓冲液稀释。加入酸性样品后缓冲液由蓝
色变为黄色。电泳样品的正确pH很重要,因此对处于pH不接近65的强缓冲
液中的蛋白质样品,必须先透析。
③ 对已经加入样品缓冲液的蛋白质样品,分装冷冻储存一段时间后,在使
用前也必须补加巯基乙醇。(2)用去垢剂清洁凝胶板,干燥后组装,并用铁夹固定垂直。具体的组装方
式根据所用的凝胶槽类型而定。(3)分离胶的制备
① 混合下列溶液,真空脱气30s。
a15%凝胶:80mlpH=88、1875mol/LTrisHCl,114ml去离子蒸馏
水,200ml丙烯酰胺储液,04ml10%SDS,02ml10%APS。
b10%凝胶:80mlpH=88、1875mol/LTrisHCl,181ml去离子蒸馏

285
水,133ml丙烯酰胺储液,04ml10%SDS,02ml10%APS。
② 加入14ml的TEMED,并迅速轻轻旋转,充分混匀。TEMED引发聚合
反应的开始,因此操作应相当迅速。
③ 立即将分离胶 混 合 物 转 移 到 凝 胶 框 中,小 心 地 加 入 到 两 块 玻 璃 板 之 间。加入量控制在距离加样孔底部1cm处,多余的凝胶不能倒入水池。
④ 为确保分离胶的表面平滑,用移液枪头在胶面上非常小心地加入蒸馏水,加入量为超过胶层2mm。因为水和凝胶的密度差异很大,水溶液不会与凝胶混
合,而是在表面伸展。或者在通风橱中加入水不溶性的有机溶剂,如异丁醇。
⑤ 凝胶放置待聚合。触摸凝胶板可察觉凝胶形成过程的放热,当清晰看到
凝胶和覆盖的水层之间的光折射变化时,表明聚合完成。(4)浓缩胶的制备
① 在分离胶聚合 时 准 备 浓 缩 胶 溶 液 (4℃),混 合 以 下 溶 液:10mlpH=68、06mol/LTrisHCl,75ml去离子蒸馏水,135ml丙烯酰胺储液,01ml10%SDS,005ml10%APS,真空脱气。
② 吸去分离胶面覆盖的水层。
③ 加14mlTEMED到浓缩胶溶液中,迅速混匀。用2ml洗涤分离胶表面,吸去洗涤液,加入浓缩胶溶液,插入梳子,放置待聚合。当梳子周围产生折射变
化时,表明凝胶已形成,大约需要20min。
④ 聚合完成后,小心地拔去梳子,用电泳缓冲液冲洗净未聚合的丙烯酰胺
溶液。(5)加样和电泳
① 凝胶框放入电泳装置中,向上槽中注入电泳缓冲液,检查有无渗漏。再
在下槽中加入电泳缓冲液,并倾斜装置赶掉凝胶底部的气泡。
② 用微量加样 枪 头 吸 取 制 备 好 的 样 品,穿 过 缓 冲 液,慢 慢 释 放 到 加 样 孔,加样缓冲液的密度会使样品沉到加样孔的底部。通常加样量为5~10μl,记录加
样的位置。
③ 电泳仪接通电源,正确连接电极。恒流30mA进行电泳,直至溴酚蓝到
达凝胶的底部,大约需要25~30h。(6)凝胶的染色和脱色
① 拆除凝胶装置,撬开凝胶板,取出凝胶,去掉浓缩胶,将分离胶放入染
色液。
② 振 荡 染 色 至 少2h。通 常 采 用 的 考 马 斯 亮 蓝 染 色 可 检 测01μg的 蛋 白 条
带,也可采用更高灵敏度的银染色。
③ 染色后的凝胶用脱色液进行振荡脱色,间隔一段时间更换脱色液,或者

286
在脱色液中放置固体材料 (如聚苯乙烯泡沫)吸附洗脱下的染料可加速脱色。(7)测量标准蛋白质的迁移距离,计算Rf,并和分子量的对数作图,建立
标准曲线。(8)测量未知蛋白质的迁移距离,计算Rf,从标准曲线上求出未知蛋白质
的分子量。
10214 要点
(1)根据所研究的蛋白质的大小选择 适 当 的 凝 胶。典 型 的 分 离 胶 大 多 使 用
15%聚丙烯酰胺凝胶,蛋白质有效分离范围为10000~100000 (即凝胶孔径允许
分子量10000的蛋白质不受阻碍通过,而分子量100000的蛋白质只能刚刚进入
凝胶孔隙)。对于分子量为150000的蛋白质必须采用低浓度大孔径的凝胶。分离
分子量范围宽广的蛋白质时,适合采用梯度凝胶。(2)凝胶溶液混合后进行脱气,有助于防止溶液中的氧对聚合反应的抑制,
建议将脱气作为常规方式。另一种防止氧对聚合抑 制 的 方 法 是 提 高 催 化 剂 的 浓
度,但由于聚合为放热反应,释放 出 的 热 量 会 导 致 凝 胶 中 形 成 小 气 泡,尤 其 在
15%的凝胶中比较常见。(3)将浓缩胶溶液直接小心地加到分离胶的顶部,省略覆盖这一步,可节省
时间。(4)制备浓缩胶时,用记号笔标出梳孔的底部位置,或在浓缩胶混合液中加
入少量的溴酚蓝,使浓缩胶呈淡蓝色,有利于加样孔的辨别。(5)通过指示剂溴酚蓝可以看到,样品通过浓缩胶时被压缩成一条细带。首
次分析未知样品时,应在溴酚蓝到达凝胶底部时停止电泳,因为低分子量的蛋白
质和染料的泳动距离接近,继续电泳可能会丢失。如果发现蛋白质位于距分离胶
顶部2/3的情况,可以继续电泳30~60min,增大条带的分离,此时染料将跑出
凝胶。
1022 凝胶过滤色谱法测定天然蛋白质的分子量
10221 原理
凝胶过滤色谱是蛋白质纯化的一个标准色谱模式,广泛应用于生物大分子的
分离、纯化、浓缩、脱盐等操作中,也可用于测定蛋白质的分子量。方法简单,样品用量少,而且可用于粗制品的测定。有关色谱基质、排阻效应及分离原理等
内容已经在前文中介绍。在理想的凝胶过滤色谱中,支持介质不与溶质分子相互作用,形状相同的多
聚物的分子量的对数与分配系数Kd 之间呈直线关系。根据分子量和洗脱体积之间
的关系,用几种已知分子量的蛋白质标准进行色谱分析,绘制出标准曲线。然后在
同样的条件下对未知蛋白质进行色谱分析,根据洗脱体积,从标准曲线上求出此未

287
知蛋白质对应的分子量。这就是凝胶过滤色谱法测定蛋白质的分子量的原理。要注意用凝胶过滤色谱法测定蛋白质分子量时,决定蛋白质洗脱体积的是溶
质分子的大小,而非分子量。所以用球蛋白质标准所作的校准曲线不能用于其他
形状的蛋白质的分子量测定。下面所述的天然蛋白质分子量的凝胶过滤色谱测定
条件不适用于碱性、酸性和疏水的蛋白质。
10222 材料
(1)装置 高效液相色谱系统,包括泵、注射器、凝胶过滤柱、保护柱、紫
外线检测器、数据记录仪。以下使用的凝胶过滤柱为ZorbaxBioseriesGF250柱
(094cm×25cm,杜邦公司),硅胶粒度4μm,最大流速2ml/min,球蛋白的排
阻极限为几十万。(2)化学药品
①02mol/L磷酸钠缓冲液 (pH=70) 610ml的02mol/LNa2HPO4 和
390ml的02mol/LNaH2PO4 混合,022μm膜过滤。
② 蓝色葡聚 糖 溶 液 将 蓝 色 葡 聚 糖 (Sigma,平 均 分 子 量2000000)溶 于
02mol/L磷酸钠缓冲液 (pH=70)配成1mg/ml,045μm膜过滤。
③ 甘氨酸溶液 将 甘 氨 酸 溶 于02mol/L磷 酸 钠 缓 冲 液 (pH=70)配 成
10mg/ml,045μm膜过滤。
④ 标准蛋白溶液 用02mol/L磷酸钠缓冲液 (pH=70)配制05mg/ml标准蛋白溶液,045μm膜过滤。
常用的蛋白质分子量标准见表38。根据凝胶过滤柱的排阻范围选择购买商
品化的蛋白质分子量标准。注意:一些不具备理想排阻行为的蛋白质 (如碱性蛋
白和非常酸性的蛋白质)不适合用作标准。因为即 便 是 在 高 离 子 强 度 的 缓 冲 液
中,这些蛋白质的排阻行为仍受到离子作用的影响。如细胞色素C (等电点10)和溶菌酶 (等电点11),由于离子交换作用不能完全被抑制,使Kd>10,而胃
蛋白酶 (等电点1)会被过早洗脱。
10223 方法
(1)用02mol/L磷 酸 钠 缓 冲 液 (pH=70)以1ml/min的 流 速 来 平 衡 柱
子,直到214nm的吸光度恒定。(2)进20μl蓝色葡聚糖溶液来测定Vo,再以20μl甘氨酸溶液重新进样来测
定Vt。(3)进20μl标准蛋白质混合溶液,测定各种蛋白质的洗脱体积Ve。计算每
种蛋白质的Kd,绘制分子量的对数与Kd的曲线。(4)进20μl待测蛋白质溶液,根据吸光度图测定洗脱体积Ve,样品中含有
多种蛋白质时,收集各组分进行分析,以确定吸收峰。

288
(5)计算未知蛋白的Kd,根据校准曲线测算出分子量。
10224 要点
(1)用凝胶过滤色 谱 法 测 定 蛋 白 质 的 分 子 量 时,通 常 在pH=6~8的 范 围
内,分子量的对数与分配系数Kd 之间的线性关系较好。而在极端pH下,可能
因蛋白质变性使测 定 结 果 发 生 偏 离。糖 含 量 超 过5%的 糖 蛋 白 测 得 的 分 子 量 偏
大,而含铁蛋白的测定值偏小。(2)在非理想的凝胶过滤色谱中,荷电蛋白质 (蛋白质所带的电荷取决于等
电点和流动相pH)的排阻行为受离子相互作用的影响。如连接有疏水相的凝胶仍
含有未衍生的硅烷醇基团,导致非理想的凝胶过滤色谱。当流动相pH>3时,会
有大量的阴离子与溶质相互作用。使得带正电的蛋白质进行离子交换,导致排出延
迟;而带负电的蛋白质因为离子排阻被提早排出。在低流动相pH (pH=2)时柱子
带有净正电荷,则色谱中带正电的蛋白质被提早洗脱,而带负电的蛋白质发生滞后。在这类非理想的凝胶过滤色谱中,必须使用高离子强度的流动相来减少离子
相互作用,但是增加离子强度会促进疏水作用的形成,因此控制流动相的离子强
度在02~05mol/L范围内,可将离子相互作用和疏水作用的影响减到最小。(3)在变性条件下,用凝胶过滤色谱测定分子量最可靠。通常采用在缓冲液
中加入二硫苏糖醇来还原二硫键,破坏蛋白质的二、三级结构,也可以采用在缓
冲液中加入胍基盐酸或SDS。不过,变性条件下的凝胶过滤色谱也存在一些问题:
① 在变性条件,蛋白质分解成亚基和多肽链,无法获得整蛋白质的分子量。
② 不纯制备物的生物活性被破坏,无法进行酶活检测。
③ 在某些柱子中,由于蛋白分子从球形变为无规线圈,回转半径增加,造
成 可 分 离 的 分 子 量 范 围 减 小,但 这 有 利 于 小 蛋 白 或 多 肽 的 分 离。例 如,
TSKG3000SW柱对天然蛋白的分离范围是2000~70000,对变性蛋 白 的 分 离 范
围为10000~500000。
④ 变性 剂 常 在 远 紫 外 区 有 光 吸 收,不 能 选 择 蛋 白 质 最 灵 敏 区 域 (200~220nm)监测吸光度。
⑤ 含有变性剂的流动相黏度升高,必须降低流速以避免形成高柱压,而且
使用后很难完全去除变性剂 (尤其是SDS),柱子以后只能使用于这类流动相。
⑥ 高浓度的盐,尤其含卤化物离子的盐,不利于泵和不锈钢材质的维护。
103 蛋白质的等电点测定
1031 蛋白质的等电点
等电点 (pI)是蛋白质的一个物理化学常数,取决于蛋白质分子的氨基酸的

289
组成和构象。作为两性分子的蛋白质,在不同pH环境中氨基酸侧链所带的电荷
不同,当处于某一pH时,蛋白质分子的净电荷为零,这一pH称为该蛋白质的
等电点。不同蛋白质的等电点范围很宽,根据等电点的不同可以进行蛋白质的分
离和分析。
1032 等电聚焦技术
等电聚焦 (isoelectrofocusing,IEF)是20世纪60年代建立的一种高分辨率的
蛋白质分离分析技术。它是利用蛋白质的pI不同,在一个稳定、连续、线性的pH梯度中进行蛋白质的分离,并根据蛋白质的位置来测定蛋白质的pI。在等电聚焦
电泳中,分离仅仅取决于蛋白质的等电点,而与分子大小和形状无关。根据建立pH梯度的原理不同,分为载体两性电解质pH梯度和固相pH梯
度。前者利用两性缓冲液离子在电场中建立pH梯度;后者中缓冲基团成为凝胶
介质中的一部分,在凝胶聚合时便形成pH梯度,所用的介质是一些具有弱酸性
或弱碱性的丙烯酰胺衍生物。固相pH梯度等电聚焦是一种新型的聚焦技术,是
目前分辨率最高的电泳方法。将IEF和SDS凝胶电泳结合使用的双向电泳会获
得极高的分辨率。
1033 超薄聚丙烯酰胺凝胶等电聚焦法测定蛋白质的等电点
10331 原理
在载体两性电解质pH梯度等电聚焦中,pH梯度的建立是利 用 载 体 两 性 电
表105 一些蛋白质的等电点
蛋 白 质 等 电 点 蛋 白 质 等 电 点
鱼精蛋白(鲑鱼精) 121 血清乙种球蛋白 56溶菌酶(鸡) 110 纤维蛋白原(人) 58组蛋白(牛) 108 胰岛素(牛) 54细胞色素C(马) 106 β乳球蛋白 52糜蛋白酶原 95 牛乳清蛋白 512核糖核酸酶A(牛) 78 脲酶 50血红蛋白(人) 71 原肌球蛋白 51肌红蛋白(马) 70 血清甲种球蛋白 51生长激素(人) 69 白明胶(动物皮) 480~485胶原蛋白 66 血清白蛋白(人) 49血清丙种球蛋白 63~72 甲状腺素球蛋白 46
γ球蛋白(人) 66 酪蛋白(牛乳) 46肌凝蛋白 62~66 牛乳球蛋白 45~55小麦麦胶蛋白 65 卵清蛋白(鸡) 46羧肽酶 60 丝蛋白(蚕丝) 20~24麻仁蛋白 55~60 胃蛋白酶 <10过氧化氢酶 56

290
解质 (carrierampholyte)在电场的作用下自然形成的。载体两性电解质不同于
两性电解质,为人工合成的低分子量多氨基多羧酸,由许多脂肪族的多氨基多羧
基异构体和同系物组成,有连续变化的氨基与羧基比。载体两性电解质具有良好
的导电性,能运载电流并且有足够的缓冲能力。外加电场时,从正级到负级,载
体两性电解质以pI逐步增加的方向自我排列,每个载体两性电解质会维持局部
的pH等于它的pI,这样就形成了一个均衡的pH梯度。载体两性电解质的pH范围有许多种,通常使用的是广泛pH 范围 (35~
95),可分离样品中各种蛋白质,但是非常碱性的蛋白质会跑出凝胶。根据待分
离蛋白质的pI范围 (表105),选择载体两性电解质的pH范围 (如4~6,5~7,
4~8等)。使用窄pH范围的载体两性电解质,可以分开pI非常接近 (例如只
相差001单位)的蛋白质。在大部分情况下,载体两性电解质的功能基团与蛋
白质可逆结合,且分子量小,通过透析、凝胶过滤、离子交换等方法可从聚焦的
蛋白质条带中除去。在这一电泳方法中使用的支撑介质有聚丙烯酰胺凝胶和琼脂糖凝胶。常用低
浓度 (4%)的聚丙烯酰胺凝胶,所具有的大孔径可以使蛋白质在外加电场中能
够无障碍自由迁移。蛋白质样品加到凝胶表面通过扩散进入凝胶,在外加电场的作用下,根据其
荷电情况发生迁移,直到净电荷为零的pH梯度位置固定下来。如果蛋白质从这
一位置稍微向正级扩散,由于所带的微弱正电荷使其向负级回迁,直到与其pI相应的pH梯度位置;同样,如果向负级扩散,带有微弱的负电荷,也会使蛋白
质迁移回相同的位置。在等电聚焦中,蛋白质只能在它的等电点位置稳定,聚焦成一条窄带,这是
一个稳态过程,其分辨率大大高于常规的凝胶电泳技术。在实际电泳中,外加电
场后,蛋白质的迁移和pH梯度的形成同时进行。等电聚焦电泳法具有分辨率高、重复性好等优点,但是由于载体两性电解质
的价格昂贵,导致等电聚焦的成本很高,限制了它的应用。现今多使用超薄板等
电聚焦,凝胶厚度仅为015mm,以电绝缘带作为凝胶板之间的间隔。相比于传
统凝胶 (1~2mm厚)的 等 电 聚 焦,大 大 降 低 了 制 备 成 本,分 析 材 料 的 用 量 更
少,染色和脱色更快捷,干胶更容易。
10332 材料
(1)丙 烯 酰 胺 储 液 丙 烯 酰 胺388g,亚 甲 基 双 丙 烯 酰 胺012g,蔗 糖
100g,溶解在80ml水中。使用前数天制备,4℃储存。添加蔗糖可提高凝胶的
机械稳定性,还会大大降低pH的梯度漂移。(2)核黄素溶液 新鲜配制,10mg核黄素溶解在100ml水中,搅拌20min,

291
放置使不溶物沉淀或短时间离心。利用核黄素在有光时降解释放出游离基来引发
聚合反应,光聚合形成聚丙烯酰胺凝胶。也可以使用 过 硫 酸 铵 和TEMED来 聚
合凝胶,但是存在两性电解质被过硫酸氧化的风险,而且凝胶中高浓度的过硫酸
还会导致蛋白条带的变形。(3)两性电解质 广泛pH范围35~95。(4)滤纸电极条 22cm×06cm的 Whatman17号滤纸,与凝胶玻板等长。(5)加样滤纸 05cm2的 Whatman1号滤纸。(6)电解液 10mol/LH3PO4 为正级电解液,10mol/LNaOH为负级电
解液。(7)固定液 150ml的甲醇和350ml的蒸馏水混合,加入175g磺基水杨酸
和575g三氯醋酸。(8)蛋白质染色液 溶解在50%甲醇中的01%考马斯亮蓝R250,10%冰
醋酸。先将染料溶解在甲醇中,再加水、冰醋酸。(9)玻璃板 22cm×12cm,厚度为1mm的玻璃,有利于冷却,2mm厚的
玻璃也可以。(10)PVC电绝缘带 厚度约为015mm,可以用测微尺测量。(11)明亮的光源 选用不会散发太多的热量的光源。
10333 方法
(1)制胶准备
① 清洗玻璃板 用去污剂及甲基乙醇彻底清洗两块玻璃板的表面,洗净晾
干。玻璃板的洁净是基本要求。
② 胶模的准备 将05cm宽的PVC电绝缘带沿着一块玻璃板的四边粘贴,注意不能有任何重叠,否则将会影响凝胶板之间的间隔厚度。
③ 上层玻璃板的处理 取另一块玻璃板用三甲基硅烷进行硅烷化处理,目
的是确保剥胶时,凝胶不黏附于上层玻璃板,而黏附在具有间隔边带的下层玻璃
板上。(2)凝胶制备
① 配 制 凝 胶 溶 液 将90ml的 丙 烯 酰 胺,04ml的 两 性 电 解 质 溶 液 和
006ml的核黄素溶液混合。注意,丙烯酰胺单体是一种神经毒素,操作时应戴
防护手套。
② 将胶模放在盘中,用移液器将全部的凝胶溶液沿着胶模的短边转移到胶
模中。将处理后的上层玻璃板置于胶模的一侧短边的间隔带,逐步放低,慢慢接
近凝胶溶液,凝胶溶液在板间扩散,小心不要产生气泡。若产生气泡,小心地抬
高上板赶去气泡后再慢慢放低。

292
③ 两板合拢后,压边并用夹子夹紧玻璃板的边,并清除板上及盘中多余的
丙烯酰胺溶液。
④ 凝胶模放 置 在 光 源 下,聚 合 至 少3h。使 用 热 光 源 时,应 将 胶 模 邻 近 放
置,以免凝胶蒸发干,产生小缝隙。聚合前后变化不明显,可以观察胶模间隔带
边的光折射或是在制备时边缘引入小气泡来判定聚合情况。
⑤ 聚合好的凝胶可在4℃下至少储 存2个 月,也 可 以 立 即 使 用。若 立 即 使
用,可将凝胶板在4℃先放置15min,以便使玻璃板容易分开。(3)取胶、铺板
① 凝胶模置于工作台,在两块板之间插入解剖刀片,慢慢扭转,并移去上
层玻璃板。通常凝胶黏着在带有绝缘间隔带的下层玻璃板上,不要去除绝缘带,以防止在染色和脱色步骤中凝胶脱落。清洁凝胶下层板。
② 打开冷却水装置,10℃冷水流经冷却板。
③ 打开电泳槽的上盖,在冷却板上铺上液体石蜡,将带有凝胶的下层板轻
轻放置到冷却板上,不能有气泡,确保接触良好。(4)加样
① 浸润滤纸条 分别用10mol/L磷酸和10mol/LNaOH均一浸润与凝胶
等长的正级滤纸条和负级滤纸条。注意,滤纸电极条必须完全浸润,但不能有液
体滴下。分别放置在凝胶的长边,平行放置。
② 样品的处理 样品最好用重蒸水溶解,对于盐含量过多的样品必须用凝
胶过滤或者透析脱盐。因为即使是低浓度的盐离子也会产生pH梯度的扭曲,形
成波形条带。而高浓度的盐离子还会产生高电流,导致烧胶。
③ 加样量 正方 形 的 加 样 滤 纸 用 蛋 白 质 样 品 完 全 浸 润 后 放 置 在 凝 胶 表 面,每个样品之间间隔05cm。加样滤纸上过多的液体会导致凝胶表面有液体,扭曲
该区域的电泳。根据滤纸的吸收情况决定适合的加样体积,通常约为5μl。
④ 加样位置 理论上样品可以加在正级和负级之间的任何位置,但是要尽
量避免将样品加在聚焦位置,这会导致迁移的缓慢和条带扭曲。另一方面,还必
须考虑蛋白质的稳定性,例如酸性pH下容易变性的蛋白质在负级加样比较好,加样位置宜离开负级滤纸电极条1cm。
(5)电泳
① 所有的样品加好后,在滤纸电极条上放置铂电极。注意保持铂电极和滤
纸电极条之间的良好接触 (如利用有机玻璃板沿铂电极的伸展和拉紧)。
② 在板间加500V的 电 压,此 时 电 流 约 为4~6mA,10min后 电 压 增 大 到
1000V,再过10min后增大到1500V。
③ 电泳1h后,关 闭 电 源,用 镊 子 小 心 取 出 加 样 滤 纸。这 步 操 作 不 是 必 需

293
的,但除去加样滤纸可避免聚焦在该区域的条带发生扭曲。
④ 继续用1500V电压再电泳1h,电流值随着凝胶的聚焦而慢慢降低。
⑤ 理论上完全聚焦时,凝胶中没有荷电粒子,电流为0。但事实上在完全聚
焦时,由于凝胶中缓冲液的电渗 现 象,仍 有 很 小 的 电 流。为 确 认 电 泳 聚 焦 的 完
成,可以以血样为有色标记,将2~3个稀释到10-2的血样加在凝胶的两端进行
电泳,当它们在凝胶中的泳动位置相同时,可以肯定聚焦发生,结束电泳。血样
的聚焦位置在凝胶 (pH=35~10)的中央。
⑥ 电泳结束时,检测到的电流大约为05mA。(6)蛋白质的染色脱色
① 固定 从电泳装置上移开凝胶板,轻轻地在200ml固定液中洗涤20min,过度剧烈的洗涤会使凝胶从玻璃板上脱落。在这一步操作中,可看到一些沉淀蛋
白质条带。电泳后并不能对等电聚焦凝胶立即进行蛋白质染色,因为两性电解质
也会被染成均一的蓝色。通过固定操作使蛋白质在凝胶中沉淀,而可溶性的两性
电解质可通过洗涤去除。
② 洗涤 倒掉固定液,用100ml脱色液洗涤凝胶2min,丢弃洗涤液。短暂
的洗涤非常重要,否则染液加到仍含有固定液的湿凝胶中,会有一定量的染色蛋
白质沉淀析出。
③ 染色 加入蛋白质染色液,轻轻搅动凝胶约10min。
④ 脱色 倾弃蛋白质染色液,用脱色液洗涤凝胶,观察凝胶中被染色的蛋
白条带。
⑤ 干胶 凝胶板吹干过夜,永久保存。(7)pI的测定
①pH梯度校准曲线的绘制 一种方法是,将等电点标准 (已知pI的蛋白
质混合物)加在待测的未知样品邻近的泳道中。应根据所用的载体两性电解质的
pH范围来选择等电点标准,商品化标准中所含的有色组分可用于监测聚焦的发
生。染色后以标准蛋白质的pI为纵坐标,离开电极的距离为 横 坐 标,绘 制pH梯度校准曲线。另一种方法是,加样时在样品的邻近留一空白泳道。在凝胶染色
前切割下来,并切成1mm的薄片,每一个薄片用1ml的水均质,用微电极测量
形成的溶液pH。同样绘制pH和距离的校准曲线。
② 测定计算pI 测定未知蛋白质的迁移距离,从曲线中求出它的pI。
10334 要点
在等电聚焦中最常见的问题是pH梯度的扭曲,在聚焦模式下会产生波形条
带。引起的原因各种各样,除了最常见的样品含盐量过多外,还包括:铂电极和
滤纸电极条之间的电接触不良,凝胶板的厚度变化,滤纸电极条的不均匀浸润,

294
冷却不够导致的局部发热点等。
104 蛋白质的纯度分析
1041 纯度标准
要确定一个蛋白质样品的纯度不是一件简单的事,往往受很多因素的限制。通常,污染蛋白的含量极低,当它低于所用分析方法的检测极限时,就很难从蛋
白质样品中检测出。单独使用某种检测方法时,污染蛋白与蛋白质样品的表现行
为类似,同样会被误认为是均一的。因此,只用一种方法作为纯度检验的标准是
不可靠的,必须选择多种检验纯度的方法。理 论 上 只 有 当 样 品 通 过 了 所 有 的 检
验,才能确定这个样品是纯的。最好的纯度标准是建立多种分析方法,从不同的角度来测定蛋白质样品的均
一性。任何单独一种方法的鉴定结果只能作为蛋白质均一性的必要条件,而非充
分条件。样品的纯度最终取决于所用检测方法的类型和分辨能力。
1042 常用的纯度检测方法
通常用于蛋白质样品纯度检测的方法有物理化学法,如电泳、沉降、HPLC和溶解度分析等。此外,化学分析法以及利用生物活性、免疫学特性的方法也在
纯度检测中使用。对于不同用途的蛋白质样品,根据所要求的不同纯度,选用相
宜的检测方法。一般来说,对样品的纯度要求越高,应当采用越多的检验方法。
10421 分光光度法
纯蛋白质的A280/A260应为175,此法可用来快速检测蛋白质样品中有无核
酸污染。
10422 超速离心沉降法
纯的蛋白质在离心场中以单一的沉降速度移动,在离心管中均一蛋白质溶液
只形成一个界面。分步取出离心管中的样品,作图得到的样品浓度分布对称,表
明样品是均一的。用此法检测纯度所需样品用量少、时间短,但由于沉降系数主
要是由分子大小和形状决定的,而与化学组成无关,因此灵敏度较差,难以检测
出微量杂质。
10423 溶解度分析
纯的蛋白质在一定的溶剂系统中具有恒定的溶解度,而不依赖于溶液中未溶
解固体的数量。用恒浓度法鉴定蛋白质纯度在理论上是严格的,在实验方法上简
单易行。在严格规定的条件下,以加入的固体蛋白质对溶解的蛋白质作图。纯蛋
白质的溶解度曲线只呈现一个折点,折点前的直线斜率为1,折点后的斜率为0。而不纯的蛋白质的溶解度曲线常常呈现2个或2个以上的折点。

295
10424 电泳法
目前采用的 电 泳 分 析 有PAGE和SDSPAGE、等 电 聚 焦、毛 细 管 电 泳 等。在聚丙烯酰胺凝胶电泳上显现出一条带,只说明样品在荷质比 (电荷/质量)方
面均一。如果一系列 不 同pH 条 件 下 的 电 泳 都 为 一 条 带,则 结 果 更 可 靠。SDSPAGE电泳法只适用于含有相同亚基的蛋白质,等电聚焦法是基于蛋白质等电点
的差异来 分 离 的,也 可 用 于 纯 度 检 测 中,纯 蛋 白 质 的 等 电 聚 焦 电 泳 呈 现 单 一
条带。
10425 色谱法
用线性梯度的离子交换色谱或凝胶过滤色谱分析样品时,纯蛋白质样品的洗
脱图谱上呈现出单一的对称峰,具有恒定 的 比 活。如 果 洗 脱 的 各 部 分 比 活 都 相
同,则表明样品在色谱性质上均一。HPLC常用于多肽、蛋白质纯度的鉴定。
10426 生物活性法
在酶蛋白的纯度检测中,酶活力是一个很好的评价指标。随着杂质的除去,比活增加,当样品不能进一步纯化时,比活达到恒定的最大值。如果蛋白质含有
容易检测到的金属或结合紧密的辅助因子,其含量会随着纯化过程而增加,当样
品达到均一状态时,含量也恒定。此外,污染蛋白质生物活性的消失也是一个间
接的纯度评价标准。对于激素、氧载体等具有特殊功能的蛋白质,通过其生理作用、催化性能进
行纯度检验,可检出浓度低到几千分之一或百万分之一的杂质,灵敏度远远高于
许多物理或化学方法。
10427 蛋白质化学结构分析法
氨基酸组成的定量分析可用来鉴定蛋白质的纯度,纯蛋白质的所有氨基酸都
成整数比。常 用 精 确、灵 敏、快 速 的 氨 基 酸 定 量 分 析 仪 来 检 测 样 品 的 氨 基 酸
组成。此外,蛋白质的末端分析也用于纯度鉴定。均一的单链蛋白质样品中,N末
端残基只可能有一种氨基酸。定量分析纯蛋白质的 N末端,可以发现每摩尔蛋
白质含有整摩尔数的N末端氨基酸,少量其他末端基的存在,常常表示存在着
杂质。
10428 免疫化学法
免疫技术是鉴定纯度的有用方法,很多蛋白质和某些多糖、脂类都有抗原性,给适当动物注射时,刺激特异性抗体的形成。注射均一性的物质只形成一种特异性
抗体,而注射几种具有抗原性物质时则产生几种抗体,此法只适用于杂质为抗原性
物质。放射免疫分析和酶联免疫分析广泛用于各种检测中,灵敏度很高。

296
105 糖蛋白质中的糖含量分析
由于糖基取代的非均一化,在凝胶电泳中糖基化蛋白通常出现典型的扩散条
带或迁移形成明显的拖尾,蛋白质染色时出现这类现象表明存在聚糖。一些高度
糖基化的蛋白质,如黏蛋白和蛋白聚糖并不进入一般的凝胶,要用琼脂糖凝胶或
丙烯酰胺琼脂糖凝胶进行电泳。对样品的性质进行鉴定之前以及在去糖基化之后时,分析是否含糖很重要。
例如在测定分子量时,由于SDS仅仅结合到糖蛋白的部分多肽链上,导致其在
SDSPAGE中的电泳行为非理想化,不能测 定 到 准 确 的 分 子 量。同 样,用 凝 胶
过滤色谱法测定的糖蛋白分子量偏大。根据样品类型和量来选择糖检测方法,对于蛋白样品证明阳性糖反应的方法
较多 (图101)。蛋白质样品
↓
→
化学分析
内切糖苷酶糖酰胺酶外切糖苷酶化学处理→
SDSPAGE
→
↓
放射自显影
→
蛋白染色糖染色
→用单抗或凝集素层叠
图101 蛋白样品的糖检测
用内切糖苷酶 (如肽N糖苷酶F,内切糖苷酶F2,内切糖苷酶 H)处理蛋
白,如果导致凝胶上一个或多个条带的迁移率改变,表明存在N聚糖。采用专
一降解的O唾液酸糖蛋白酶可以确定含有成簇的唾液酸O聚糖的糖蛋白 (黏蛋
白),经过唾液酸酶或糖胺聚糖裂合酶处理,同样引起迁移率的改变。用化学方法 (如水解、β消除或氟化氢处理)也能实现N聚糖和O聚糖的
完全去除,但通常肽的破坏会妨碍采用凝胶电泳进行进一步的分析。对于特殊检
测目的可使用高灵敏度的检测系统,将蛋白质样品与膜杂交后,用凝集素或专一
性抗体进行检测。
1051 糖蛋白的化学分析
10511 原理
对于凝胶中的糖蛋白和溶液中的寡糖,通过高碘酸盐在两个浓度下的氧化,先检测唾液酸,然后检测任何具有两个游离的邻近羟基基团的单糖。高碘酸在两
个羟基基团之间切割,形成活性乙醛,它能被硼氢化钠 (NaB[3H4])还原或者
与商品化试剂盒中的高灵敏度的探针偶合而被检测,然后用苯酚硫酸试剂快速
显色分析溶液中存在的单糖或含有C2羟基基团的寡糖,这一方法对单糖和寡糖

297
有相对的特异性。
10512 材料
(1)高 碘 酸 氧 化 01mol/L醋 酸 缓 冲 液 (pH=55),含 有8mmol/L或
15mmol/L的高碘酸钠,高碘酸试剂暴露在光下会降解,必须新鲜配制;1,2亚乙
基二醇;01mol/L氢氧化钠;还原剂:硼氢化钠,氚化的硼氢化钠 (NaB[3H4])或者氟硼酸钠;冰醋酸。
(2)苯酚硫酸分析 H2O (HPLC色谱级),4%苯酚溶液,浓硫酸,1mg/
ml半乳糖,1mg/ml甘露糖。
10513 方法
(1)高碘酸氧化
① 将01~10mg的糖蛋白溶解在2ml含高碘酸钠的醋酸缓冲液中,高碘
酸钠 的 浓 度 为:单 糖 用15mmol/L,糖 醇 用8mmol/L,专 门 氧 化 唾 液 酸 用
1mmol/L。
② 寡糖在室温下暗室中用高碘酸氧化1h,糖醇在4℃氧化48h,唾液酸在0℃氧化1h。高碘酸氧化必须在暗处进行,以避免非特异性氧化,这一点非常重要。
③ 加入25μl的1,2亚乙基二醇分解过量的高碘酸,4℃放置过夜。
④ 加入01mol/L氢氧化钠使pH达到70,大约需加15ml。
⑤ 用25mg的NaB[3H4]还原氧化物,4℃放置过夜。活性乙醛也可与含胺
化合物如地高新配基偶联而被检测。
⑥ 加入醋酸调pH为40,样品浓缩干燥。
⑦ 每次用100μl的甲醇蒸发3次,由于在酸性环境中加入甲醇后导致形成
挥发性的甲基硼酸,可去除硼酸。(2)苯酚硫酸分析
① 将含量约1μg/10μl的未知样品溶液,连同系列浓度范围的己糖标准 (半
乳糖或甘露糖,通常用量1~10μg)一起加到微孔滴定板上。
② 每孔中加入25μl的4%苯酚溶液,彻底混合,放置5min。
③ 每孔中加入200μl的浓硫酸,混合,在492nm下测定吸光度。有盐存在
时,浓硫酸加入到苯酚/糖醇中时可能有喷溅,应小心操作。每孔中不能装得太
多,以防浓硫酸溢出,损害微孔滴定板的检测器。
1052 SDS凝胶中的糖蛋白染色法
10521 原理
SDSPAGE凝胶电泳是常用蛋白质分离技 术,凝 胶 中 的 蛋 白 质 通 常 用 考 马
斯亮蓝或银染色。对于高度糖基化的糖蛋白 (蛋白质低聚糖)或蛋白聚糖 (蛋

298
白质黏多糖),由于糖链干扰银离子的结合,导致染色微弱,灵敏度很低,甚至
无法检测。传统的染色方法用改良的Schiff碱反应检测糖蛋白,先用高碘酸氧化
糖,随后用Schiff试剂 (PAS)、爱茜蓝或者肼衍生物进行染色,但是检测灵敏
度低,非糖基化的蛋白质不被染色,有糖特异性。在爱茜蓝银染色中,以爱茜蓝作为第一步的染色试剂,与氧化的糖蛋白结
合,随后用中性银来增强染色效果。由于爱茜蓝染料对非糖基化蛋白的银染色没
有妨碍,因此糖基化和非糖基化的蛋白质均被染上色。比传统方法灵敏度提高2倍,可以检测SDS凝胶中纳克浓度的高度糖基化蛋白,非常适合于很稀的混合
样品及少量纯样品中的糖蛋白检测。一些糖蛋白单独使用爱茜蓝银染色时,获得的染色条带很微弱,可能是由
于与爱茜蓝结合的带负电的基团含量低。采用高碘酸氧化先进行糖的氧化可以克
服这一问题。下面介绍糖蛋白的高碘酸氧化和随后的爱茜蓝银染色法。
10522 试剂和材料
所有的化合物均为分析纯,必须使用高纯度的水。(1)固定液 10% (体积分数)三氯醋酸水溶液。(2)洗涤液 洗涤液Ⅰ:25% (体积分数)甲醇、10% (体积分数)的醋酸
的水溶液。洗涤液Ⅱ:10% (体积分数)甲醇、5% (体积分数)的醋酸的水溶
液。洗涤液Ⅲ:5% (体积分数)醋酸水溶液。(3)氧化液 1% (10g/L)高碘酸水溶液。(4)还原液 05% (5g/L)焦亚硫酸钾水溶液。(5)染色液 0125% (125g/L)爱 茜 蓝 溶 于 洗 涤 液Ⅰ,大 量 搅 拌,使 用
前过滤。(6)光敏处理液 5%的戊二醛水溶液,使用时新鲜配制。(7)显影储液 25% (25g/L)的碳酸钠水溶液。(8)银液 04% (4g/L)硝酸银水溶液,使用时新鲜配制。(9)显影液 向显影储液中加入甲醛至终浓度为0013% (体积分数) (如
加入35μl的37%甲醛溶液到100ml显影储液中,搅拌数秒),使用时新鲜配制。(10)终止液 10% (体积分数)的醋酸、10% (体积分数)甘油的水溶液。
10523 分析方法
(1)电泳结束后,将凝胶转移到有盖的培养皿中,加入固定液在30℃固定
10min。也可以用醋酸乙醇溶液固定凝胶中的蛋白质,但效果不如三氯醋酸好。(2)洗涤液Ⅲ洗 涤 凝 胶2次,每 次2min。凝 胶 浸 没 在 氧 化 液 中30℃氧 化
20min。再用洗涤液Ⅲ洗涤凝胶2次,每次2min,然后水洗涤2次,每次2min。再加入还原液30℃还原12min。用水洗涤凝胶2次,每次2min。然后在50℃用

299
洗涤液Ⅰ洗涤凝胶2次,每次2min。(3)加入染色液,凝胶在50℃下染色15min。(4)用洗涤液Ⅰ洗涤凝胶3次,洗涤时间分别为1min、4min、5min,然后
用洗涤液Ⅱ在50℃洗涤2次,洗涤时间分别为2min、4min。爱茜蓝在随后的银
染色中将不可逆地固定在凝胶中,形成墨绿色的背景。用稀醋酸冲洗至背景仅保
留微弱的蓝色。(5)加入光敏处理液在50℃处理6min。注意,戊二醛有害健康,必须在密
封箱或通风橱中使用。(6)用洗涤液Ⅱ洗涤凝胶2次,洗涤时间分别为3min、5min,随后在50℃
用水洗涤2次,每次2min。(7)凝胶浸没在银溶液中,40℃浸没65min。(8)30℃下用水洗涤凝胶2次,每次30s。这步操作可冲掉多 余 的 银 离 子,
不损失结合到爱茜蓝糖蛋白的银离子。注意,过少的洗涤会形成背景的金属银,而太强烈的洗涤则会降低灵敏度。
(9)用新鲜配制的显影液在室温下显影30s,产生黑色的沉淀,将凝胶转移
到新鲜的显影液中,再显影4~8min,显影时间应根据所要求的灵敏度和背景染
色情况 以 及 特 异 性 进 行 变 动。1~3min后 看 到 糖 蛋 白,而 非 糖 基 化 蛋 白 在4~8min后显现,对凝胶顺序拍照。
(10)加入终止液保持5min,终止显影。凝胶中的蛋白质用普通的银染色法
进行染色,作为对照,与爱茜蓝银染色法比较来确定糖蛋白。
10524 要点
(1)方案最适用于PhastSystem (Pharmacia公司)的带有支撑介质的小凝
胶,可以在自动的显影冲洗箱中设定程序进行自动染色,迅速获得结果,而且重
复性好。如果使用其他系统或更大的凝胶,通常每一步都需要延长时间。无支撑
的1mm厚的凝胶用洗涤液Ⅰ时,增加洗涤时间至15h,更换4次洗涤液,其他
每一步的保持时间加倍,也能获得很好的结果。(2)操作时,触摸凝胶应戴手套或使用镊子,因为指印也会被染色。由于染
色方法高度灵敏,设备 (显影冲洗箱或染色盘)完全洁净以及使用高质量的水是
基本要求,所有的染色和洗涤操作中应搅动凝胶。(3)从凝胶中去除SDS必须进行大量的洗涤操作,否则会沉淀爱茜蓝,导
致背景过高。如果凝胶是非变性的常规PAGE,可以缩短洗涤时间。
106 蛋白质和肽的N末端氨基酸测定
测定肽链N末端残基的化学方法主要有:2,4二硝基氟苯法 (FDNB),二

300
甲基氨基萘 磺 酰 氯 法 (DNSCl)和 苯 异 硫 氰 酸 酯 法 (PTH 法,即 基 本Edman降解法)。另外还有酶解法 (氨肽酶法)。一般来说,在N末端氨基酸的测定中,以化学方法为主,很少用酶解法。
1061 FDNB法
经典的FDNB法 (2,4二硝基氟苯法)是FSanger在1945年提出的 N末
端氨基酸的分析方法,是蛋白质化学中最重要的方法之一,可用于测定蛋白质或
肽的N末端基团、蛋白质中肽链的数目、蛋白质的最小分子量及蛋白质水解产
物的氨基酸组成。
FDNB法的反应原理如图102所示。2,4二硝基氟苯 (FDNB)在很温和的
条件下 (室温,pH=8~9),能与蛋白质的游离氨基反应,生成二硝基氟苯衍生
物———DNP蛋白。DNP蛋白完全水解释放出N末端的DNP氨基酸,通过色谱
分离后与标准DNP氨基酸对照进行鉴定。蛋白质 FDNB DNP蛋白质
R—NH2 師師
師師
帩帩+ F NO 2
NO2
pH=8~9→ 師師
師師
帩帩室温 NHR NO2+ HF
NO2
H+水解↓105℃
氨基酸+DNP氨基酸 (黄色)
图102 FDNB法的反应原理
FDNB的反应性很强,能在不引起肽链断裂的温和条件下,定量与蛋白质反
应,因而在测定过程中不会因肽链断裂而人为形成 “新”末端。除了某些链内氨
基酸衍生物外,DNP氨基酸都呈黄色,非常有利于检测。虽然FDNB也能与α氨基外的其他少数基团反应,但生成物并不影响N端
氨基酸的分析,如oDNP酪 氨 酸 (无 色)、imDNP组 氨 酸 (无 色)、εDNP赖
氨酸 (黄色,醚不溶),因而FDNB是一个理想的端基分析试剂。
FDNB法的具体分析过程如下:
① 蛋白质的二硝基苯化 反应在微碱性介质中进行,在NaHCO3 存在下,暗室中反应,反应时间主要取决于蛋白质样品的溶解度和反应性。
② 除去过量试剂 反应结束后酸化处理,离心,并用醇、醚洗涤,DNP蛋
白质避光储存。
③ DNP蛋 白 质 的 水 解 用 盐 酸 水 解 时,大 部 分 DNP氨 基 酸 不 会 被 破 坏
(表106)。
④ 分 离 DNP氨 基 酸 根 据 各 自 的 溶 解 度 以 有 机 溶 剂 乙 醚 从 水 解 产 物 中
提取。

301
表106 酸水解时的DNP氨基酸的回收率
DNP氨基酸57mol/L盐酸水解
水解时间/h 回收率/%
12mol/L盐酸105℃水解16h回收率/%
DNPAla 12 80 75DNPArg 12 90 75DNPAsp 24 60 75双DNPCys 12 25 0DNPGlu 12 75 75DNPGly 8 40 50DNPIle 12 80 75DNPLeu 12 80 75双DNPLys 8 95 75
εDNPLys 12 95 75DNPMet 12 75 75DNPPhe 12 70 50DNPPro 2 10 50DNPSer 12 90 75DNPThe 24 90 75DNPTrp 12 90 0双DNPTyr 12 75 50DNPVal 12 80 75
⑤ DNP氨基酸的 分 析 采 用 单 相 或 双 相 纸 色 谱、硅 胶 薄 板 色 谱,与 标 准
DNP氨基酸对照进行定量分析。用FDNB法鉴定出αNDNP氨基酸,并确定N末端氨基酸的数目,从而计
算出多肽链的数目。但在有些情况下,会检测不到末端基团:末端封闭,无游离
α氨基的 环 状 化 合 物;末 端 乙 酰 化;DNP氨 基 酸 被 分 解,如 DNPPro、DNPGly;酸解未形成DNP氨基酸,如末端为Val、Ile时,水解生成DNP小肽。
在FDNB法中,由于DNP氨基酸对光敏感,实验必须避光在暗处进行。此
外,酸水解DNP蛋白质获得氨基酸和DNP氨基酸的混合物,必须反复抽提来
分离。因此,作为端基分析的经典方法,FDNB法近年来逐步被操作更简单、灵
敏度更高的DNS法代替。
1062 DNS分析法
10621 原理
DNS法 (二甲基氨基萘磺酰氯法)是1956年Gray和Hartley提出的测定N末端的方法,最初用于肽的末端氨基酸测定。用DNS法分析蛋白质时,由于不
溶性限制了与DNSCl反应的蛋白数量,使DNS衍生物的得率低,可以用改良
DNS法。

302
DNS法的测定原理 (图103)与FDNB法类似,在碱性介质中,荧光性的
二甲基氨基萘磺酰氯 (简称DNSCl)与肽或蛋白质的自由氨基反应,由于丹酰
基团和N末端氨基酸之间的键能抗酸水解,因而在DNS肽或蛋白质的总水解物
中,包含了游离氨 基 酸 和 N末 端 氨 基 酸 的 丹 酰 化 衍 生 物———DNS氨 基 酸,在
UV灯下DNS氨基酸生成荧光,可以用聚酰胺薄板色谱鉴定
師師師師帪帪
。
SO O
師師師師
Cl
N
H3C CH3
+ H2 N CHR1
CO NH CHR2
幆COpH90~
→ 師師師師帪帪
95
SO ONH CHR1
CO NH CHR2
幆
師師師師
CO
N
H3C CH3
+HCl
DNSCl 肽或蛋白质 DNS肽或DNS蛋白质
H+→ 師師
師師帪帪105℃18h
SO O NH CH COOHR
師師師師1
N
H3C CH3
DNS氨基酸
+ H2 N CHR(2→n)COOH
氨基酸的混合物
图103 DNS法的测定原理
与FDNB法相比,在DNS法中水解后无需提取水解物,可直接用电泳或色
谱法鉴定氨基酸,操作简单,而且分辨率和准确度高。此外,DNS氨基酸的荧
光十分强烈,很容易检测,灵敏度达到1~5ng的DNS氨基酸,比FDNB法灵
敏度高100倍。因此,近年来DNS法广泛用于肽或蛋白质的氨基酸组成和N末
端分析。需要注 意 的 是,酪 氨 酸 和 赖 氨 酸 残 基 的 侧 链 也 会 和 DNSCl反 应,形 成
οDNSTyr和εDNSLys,同时还产生双DNSTyr和双DNSLys,不过在色谱
中的位置都有别于DNSα氨基酸。His的侧链也会形成DNS衍生物,但它对酸
不稳定,因而也不会出现双DNS衍生物。而当肽或蛋白质末端是两个疏水残基
时,由于它们之间的肽键能在很大程度上抗酸水解,因此会形成DNSN末端氨
基酸和DNSN末端二 肽 的 混 合 物。此 外,疏 水 的 N末 端 氨 基 酸 与 脯 氨 酸 相 邻
时,也会产生一些二肽衍生物,不过这些都不影响N末端氨基酸的鉴定。绝大多数DNS衍生物具有高回收率 (>90%),但也有一些在酸水解时被破

303
坏,如色氨酸的DNS衍生物被全部分解,脯氨酸、丝氨酸、苏氨酸的DNS衍生
物收率分别 为25%、65%、70%。用 聚 酰 胺 薄 层 色 谱 可 鉴 定 所 有 的DNS氨 基
酸,板两面都涂有聚酰胺薄层,一面点上待测的DNS氨基酸样品,另一面点上
标准DNS氨基酸和待测样品的混合物,利用色谱分析后板的透明性鉴定未知的
DNS氨基酸。
DNS法最大的缺点是:天冬酰胺和谷氨酰胺的DNS衍生物水解时会生成相
应的DNS天冬氨酸和DNS谷氨酸,因此鉴定为DNSAsp或DNSGlu的残基通
常称为Asx或Glx,即该残基是酸或酰胺还是未知的。
10622 主要试剂和材料
(1)25mg/ml的DNS丙酮溶液,4℃避光存放,可稳定数月。(2)02mol/L碳酸氢钠溶液,应定期检查有无微生物生长。(3)6mol/LHCl溶液。(4)测定管50mm×6mm。(5)双面涂层的聚酰胺薄板。(6)色谱溶剂系统
① 溶剂1:甲酸∶水=15∶100 (体积比)
② 溶剂2:甲苯∶醋酸=9∶1 (体积比)
③ 溶剂3:乙酸乙酯∶甲醇∶醋酸=2∶1∶1 (体积比)(7)标准DNS氨基酸的 丙 酮 溶 液:脯 氨 酸、亮 氨 酸、苯 丙 氨 酸、苏 氨 酸、
谷氨酸、精氨酸,浓度均为约50μg/ml。(8)具有265nm长波或者254nm短波的UV光源。
10623 具体分析方法
(1)与DNSCl的反应
①1~5nmol样 品 用 适 当 体 积 的 水 溶 解 后 转 移 至 测 定 管 中,真 空 干 燥 成
膜片。
② 用10μl02mol/L碳酸氢钠溶液溶解膜片,加入10μl的DNS丙酮溶液,混合。
注意:pH对DNSCl和肽的偶合反应很重要,pH过高DNSCl水解产生副
产物DNSOH,用含50%丙酮的碳酸氢钠缓冲液保证pH=95~105的环境要
求。在DNSCl过量时产生DNSNH2。
③ 管口用封口膜密封,37℃保温1h,或者室温下3h。(2)DNS肽或蛋白的水解
① 反应后样品真空干燥,加入6mol/LHCl,用氧火焰密封,105℃过夜水
解18h。

304
② 反应管变凉后用玻璃刀切开管子的上部,样品真 空 干 燥30min,以 五 氧
化二磷为干燥剂,干燥器置于50~60℃的水浴中。(3)DNS氨基酸的分析鉴定
① 点样 用50%吡啶10μl溶解干燥后的样品,在通风橱中点1μl样品于聚
酰胺薄板一侧,并在薄板的另一侧点05μl标准DNS氨基酸的混合样。干燥后
进行色谱分析。
② 色谱分析 放入色谱溶剂1中展开,溶剂前沿距薄板顶端1cm取出吹干,在UV灯下能看到从原点铺展开的蓝色荧光痕迹和其中的一些绿色的荧光斑点。沿溶剂1展开的直角方向,将聚酰胺薄板在溶剂2中展开,溶剂前沿距薄板顶端
1cm取出在通风橱中吹干 (溶剂中含甲苯),在 UV灯下观察薄板的样品面,有
三个主要的 荧 光 区 域:薄 板 的 底 部 的 蓝 色 荧 光 区 域 为 DNS水 解 形 成 的 DNSOH,位于薄板顶端约1/3处的蓝绿色荧光为DNSCl的副反应产物,这两点在
所有的DNS薄板上都可以看见,可以用作内标。肽或蛋白质的N末端氨基酸的
DNS衍生物呈绿色荧光。如果肽不纯,会看到其他衍生物。溶剂2能将疏水的
DNS衍生物与一些中性氨基酸分离,而荷电衍生物与其他中性氨基酸仍然位于
色谱膜的下端。经过溶剂2后的所有快速运动的DNS衍生物都可以通过与内标
的位置比较而得出合理的鉴定。将薄板翻过来,比较另一面的标准衍生物的位置
可获得明确的鉴定结果。记录溶剂2后的衍生物的鉴定结果。需要注意的是:薄
板两面完全是独立色谱。不提倡透过薄板从一侧看另一侧的荧光点。再将薄板放在溶剂3中,与 溶 剂2的 展 层 方 向 一 致,展 层 至 溶 剂 距 离 顶 边
1cm处,取出吹干。在UV灯下观察,在溶剂2中移动缓慢的衍生物经过溶剂3后已经分开,可以被鉴定。而在溶剂2中快速运动的衍生物此时运动至薄板的顶
部,通常已无法辨别,因此必须在溶剂2后记录是何衍生物。溶剂3后获得的分
离结果见图104 (b)。(4)经过溶剂2和溶剂3后的结果之和可以确定初始样品中 N末端氨基酸
的数目和相对强度。(5)三种溶剂 系 统 难 以 鉴 定DNSArg、DNSHis、DNSCys,须 采 用 第 四
种溶剂。对于 Arg和 His,采 用005mol/L的 磷 酸 三 钠∶乙 醇 (3∶1,体 积
比),对于Cys采用1mol/L氨水∶乙醇 (1∶1,体积比)。展层方向和溶剂2、3相同,与另一面的标准衍生物比较来鉴定。
1063 Edman降解法
1950年PehrEdman首先提出了用异硫氰酸苯酯测定肽链的 N末端氨基酸
的PTH法,又称为Edman降解法 (异硫氰酸苯酯法)。Edman降解法的基本原
理是异硫氰酸苯酯 (PITC)能在温和条件下与含有自由氨基的多肽或蛋白质发

305
图104 聚酰胺薄板上DNS氨基酸的色谱图
(a)两系统溶剂色谱分析后图谱;(b)三系统溶剂色谱分析后图谱,交叉点为点样位置;
黑色斑点表示所用的标准DNS氨基酸;a—DNSOH;b—DNSNH2;
1—οDNSTyr;2—εDNSLys;3—bisDNSHis
生偶联反应,生成的苯氨基硫甲酰衍生物经过环化,从肽链上断裂下来,然后转
变为PTH氨基 酸。利 用PTH氨 基 酸 在 紫 外 光 下 有 强 吸 收,可 以 用 色 谱 进 行
鉴定。
Edman降解法包括以下步骤 (图105):
① 偶联反应 碱性条件下、在氮气中,异硫氰酸苯酯与N末端的氨基反应
形成苯氨基硫甲酰肽 (PTC肽)的衍生物。
② 洗涤 除去过量的PITC和缓冲液后的样品被彻底干燥。
③ 裂解反应 在无水酸的作用下,PTC肽发生特异性裂解,产生游离的初
始N末端氨基酸的噻唑啉酮和丢失了原N末端残基的变短的多肽。
④ 萃取 将噻唑啉酮萃取至疏水有机溶剂中,与变短的多肽分离。
⑤ 转化反应 在稀酸作用下,使不稳定的噻唑啉酮转换为稳定的乙内酰苯
硫脲氨基酸 (PTH氨基酸)。
⑥ 鉴定 用TLC或 HPLC分析,与标准图谱比较鉴定PTH氨基酸。在Edman降解 法 中 将 偶 联、洗 涤、裂 解、萃 取 和 转 化 这 个 过 程 称 为 一 个
“循环”,在手工测定中大约需要2h。基本Edman降解法最大的优点是:在切下
N末端氨基酸残基时,不会发生整个肽链的全水解,切短的肽再经过下一个循环
的Edman降解步骤,产生下一个残基的PTH衍生物。因此,Edman降解法后
来用于测定从N端开始的氨基酸排列顺序,并在此基础上于1967年设计出第一

306
图105 Edman降解反应和转化反应
台自动测序仪。目前所有测定N端序列的方法都是以Edman降解反应为基础发
展起来的改良方法。
107 蛋白质N末端序列分析
随着重组DNA技术的应用,蛋白质测序的作用有所改变。由于DNA测序
简单快速,现今大多数蛋白质序列都是通过基因或cDNA的核苷酸序列推导 出
来的,但是蛋白质直接测序技术仍是基因表达产物的鉴定及研究蛋白质结构与功
能不可缺的工具。它用于蛋白质或多肽的纯度分析,确定多亚基复合体系的亚基
数,只有获得惟一的N末端的序列或N末端的氨基酸才能很好地证实某一蛋白
质或多肽是纯的;蛋 白 识 别,将 一 个 短 的 N末 端 序 列 与 已 经 发 表 的 序 列 比 较,有助于识别某一蛋白;从蛋白质肽链间的短序列获得构建DNA探针的信息,用
于基因克隆的寡核苷酸探针的合成;拓扑结构的分析、蛋白质加工、同工酶的确
定;蛋白质结构和功能区经常是共价修饰的位点,测序是测定一级结构氨基酸修
饰位点的惟一方法。目前用于N末端序列测定的方法除了基本Edman降解法,还有在提高循环操
作效率和检测灵敏度方面发展出的DNSEdman法和DABITC/PITC双偶合法。肽或蛋白质序列测定的一般流程如下:(1)对肽或蛋白质的纯样进行一定的样品处理 (化学裂解、酶催化、侧链修
饰),制备测序的小段肽,并分离纯化。
① 特殊的蛋白水解酶催化 胃蛋白酶只切割赖氨酸 (K)或精氨酸 (R)之
后的肽键,V8蛋白酶只在谷氨酸 (E)后切割。

307
② 化学裂解 化学试剂溴化氰在甲硫氨酸残基后切割肽键。(2)序列测定 采用一定的手段和方法对系列小段肽进行序列测定。测序方法分为两大类:Edman降解法和高速原子轰击 (FAB)质谱分析法。
Edman降解法及发展出的DNSEdman法和DABITC/PITC双偶合法应用普遍,
FAB质谱法可以 测 定 N末 端 阻 断 的 多 肽 序 列,并 且 能 够 准 确 测 定 多 肽 混 合 物
(多至5种)的肽序列,但是灵敏度不高,不能直接用于完整蛋白的测序。(3)序列确定 根据重叠片段的序列 重 排 法,再 现 原 始 样 品 蛋 白 质 的 肽 链
序列 (图106)。HLMGSHLVDALELVMGDRGFEYTPKAWLV… 序列
(a)→
T1:HLMGSHLVDALELVMGDRT2:GFEYTPKT3:AWLV
(c)
(b) → V1:HLMGSHLVDALE
V2:LVMGDRGFEV3:YTPKAWLV
(c
)
(d)
T1:HLMGSHLVDALELVMGDR V1:HLMGSHLVDALE V2: LVMGDRGFE T2: GFEYTPK V3: YTPKAWLV T3: AWLV
HLMGSHLVDALELVMGDRGFEYTPKAWLV…
↓
序列
图106 序列测定图解
(a)胰蛋白酶裂解多肽链;(b)V8蛋白酶的裂解;(c)Edman降解法测定胰蛋白
裂解的肽片段 (T1~T3)和V8蛋白酶裂解的肽片段 (V1~V3)的氨基酸序列;
(d)根据重叠肽段组装完整的全长序列肽链
测定肽和蛋白质的序列从手段上分,有自动测序和手工测序两种,两者各有
优缺点 (表107)。表107 两种序列测定手段的比较
项 目 手工序列测定 自动序列测定
实验设备和要求 常规生物化学实验设备,低成本 昂贵的自动设备,需要专业
操作技术和高额维持费用
测 定 的 N 末 端 序 列 长 度
(残基数) 10~20个残基 的 范 围,50个 残 基 需 要 专 家
级经验 高达100个
重复率 不超过95% 98%
一个操 作 者 每 个 工 作 日 处
理和识别的量 不超过3~5个循环,实验密集型,分批同时
对多个肽进行测序 达到30个循环或更多
需要的材料 05~5nmol(初 始 材 料),约5~50nmol粗
原料(最灵敏的手工法) 1~20pmol(自 动 序 列 仪),20~200pmol(常规测定)
特点 广泛使用,实用性强 复杂而高效

308
1071 DNSEdman法
10711 原理
对基 本 Edman降 解 法 的 一 个 重 要 改 进 是 用 二 甲 氨 基 磺 酰 氯 标 记 蛋 白 质,
1970年 Hartley提出了将Edman降解和丹酰化过程结合起来的DNSEdman法。利用DNS法高灵敏度地鉴定 N末端的氨基酸,以及Edman降解能对多肽或蛋
白质按顺序去除N末端氨基酸,结合了两种方法的优点,实现N端的序列测定。少量样品用DNS法测定 N末端 氨 基 酸,剩 余 样 品 进 行Edman降 解———偶
合、洗涤、裂解和抽提,丢 弃 抽 提 出 的 衍 生 物 噻 唑 啉 酮。取 少 量 的 切 短 的 残 留
肽,用DNS法测定样品中新暴露的N末端氨基酸,其余的再进行另一次Edman降解。在每个周期 中,用TLC鉴 别DNS氨 基 酸,由 于DNS法 的 灵 敏 度 很 高,因而可以补偿样品肽用于丹酰化反应的少量损失。
10712 材料
(1)具有磨砂玻璃塞的试管。(2)50%吡啶水溶液 (AR级),4℃避光在氮气中存储。(3)5%的异硫氰酸苯酯 (高纯度或测序级)吡啶溶液,4℃避光在氮气中存
储,新鲜配置,1个月内使用。(4)水饱和的醋酸丁酯,室温存储。(5)无水三氟醋酸 (TFA),室温氮气存储。
10713 方法
(1)用 适 当 体 积 的 水 将 待 测 序 的 肽 溶 解,转 移 到 测 序 管 中,真 空 干 燥 成
膜片。(2)肽膜溶解在200μl的50%吡啶溶液中,取出5μl用DNS法分析N末端
的氨基酸。(3)Edman降解循环:
① 偶联 加5%的异硫氰酸苯酯溶液100μl于测序管中,轻轻混合,冲入氮
气,在具塞试管中50℃保温45min。
② 真空干燥 结束后,以五氧化二磷为干燥剂,干燥器放在50~60℃的水
浴中真空干燥。需要注意的是:偶联反应后样品必须彻底干燥,因为任何痕量的
水会导致下步反应中肽链内的裂解,从而增加 N末端氨基酸。对偶联反应后很
难彻底干燥的肽,可加入100μl乙醇,混匀后再真空干燥。干燥时须避免有机化
合物和酸进入泵中。
③ 裂解 加入200μl的TFA至测定管中,冲入氮气,在具塞试管中50℃保
温15min,保温 结 束 后,真 空 干 燥,完 成 裂 解 反 应。挥 发 性 的 TFA 会 很 快 蒸

309
发掉。
④ 萃取 用200μl水溶解管中的物质,加入醋酸丁酯15ml,剧烈振荡10s,然后离心3min。小心移出上层有机相丢弃,注意不要碰到下层水相,否则会大
大减少测序的肽量。醋酸丁酯反 复 抽 提 数 次 后,试 管 中 的 水 相 真 空 干 燥,直 至
干透。(4)用50%的吡啶200μl重新溶解干燥后的物质,取出5μl用DNS法测定
新释放形成的N末端氨基酸 (即肽链序列上的第二个残基)。每次循环开始时,可适当增加用于DNS法的样品量。
(5)返回第 (3)步再进行下一次的Edman降解循环,直至肽链被完全测序。
10714 要点
(1)DNSEdman法完 成 一 次 循 环 大 约 耗 时25h,重 复 率 通 常 为90%~95%,允许测序长度可达15个残基或更长。一个工作日内可同时对8~12肽进
行3~4次循环,在保温和干燥步骤中,对 前 一 日 测 序 的 丹 酰 化 样 品 进 行 鉴 定,可提高效率。
(2)DNSEdman法中利用Edman降解去除 N末端氨基酸,而丢 弃 了 抽 提
出N末端氨基酸的噻唑啉酮衍生物,对于N末端氨基酸的鉴定是利用了高灵敏
度的DNS法。因 此,和 基 本Edman法 相 比,DNSEdman法 最 主 要 的 缺 点 是:丹酰化无法区分残基是酸还是酰胺,在序列测定结果中常常包含鉴定为 Asx和
Glx的残基。不过可以从肽在电泳中的迁移推导出残基的酸或酰胺特性。(3)在DNS法中,色氨酸在酸水解时被破坏而无法鉴定。不过在DNSEd
man法中,如果末端为色氨酸,在TFA裂解中可以看到深紫色,能明确鉴定为
色氨酸残基。(4)在Edman降解中,当谷氨酸暴露为N末端氨基酸时,通常会看到一个
微弱的DNSGlu。由于谷氨酸会环化形成没有自由氨基的焦谷 氨 酰 衍 生 物,从
而锁定Edman降解,在后续的循环中检测不到其他残基。(5)由于Edman降解无法形成100%的裂解,随着循环次数的增 加 会 导 致
背景N末端氨基酸的累积。在较长的过程 (10~20个循环)中,可将连续循环
的DNS薄板邻近放置,同时观察,来鉴定新形成的N末端氨基酸。
1072 DABITC/PITC双偶合法
10721 原理
为了克服DNSEdman法的缺点,1976年张瑞跃合成了一种有色的异硫氰酸酯
衍生物 (4N,N对二甲氨基偶氮苯4′异硫氰酸酯,简称DABITC)替代PITC。在偶 联 反 应 中,DABITC与 N 末 端 氨 基 酸 残 基 生 成 的 DABTH氨 基 酸
(4N,N对二甲氨基偶氮苯4′乙内酰硫脲氨基酸)有明显的颜色,在聚酰胺薄

310
膜上色谱分析后,经盐酸蒸气熏即能显出红色斑点,而杂质与试剂反应物显示蓝
色或紫色斑点,能 方 便 而 准 确 测 定 氨 基 酸,灵 敏 度 达 到1nmol。因 为DABITC降解N末端氨基酸的原理类似于Edman降解,又称为有色Edman降解。
用此试剂测定多肽和蛋白质序列比经典的Edman方法灵敏25倍,样品量仅
需2~8nmol。惟 一 的 不 足 是,DABITC与 氨 基 偶 合 反 应 产 率 较 低,一 般 不 到
50%。因此,如果仅仅用DABITC进行偶联反应,则在第二个周期时为初始肽
和被切去一个残基的肽的混合物,所以在第二个周期之后会发现两个DABTH衍生物。其后 问 题 迅 速 扩 大,几 个 周 期 后 就 无 法 鉴 定 序 列。为 解 决 这 一 问 题,
1978年张瑞跃又设计了在DABITC后再用异硫氰酸苯酯 (PITC)试剂将余下没
有与DABITC偶合的氨基进行第二次偶合,保证100%的衍生化,即双偶合法。由于PITC与多肽的偶合反应近于定量,从而可以定量去除N末端,并暴露出第
二个氨基。暴露的N末端氨基又能与DABITC/PITC双偶合降解,这样可逐个
测定出蛋白质或多肽的N末端氨基酸序列。
DABITC/PITC双偶合法灵敏度高、操作简便,不需要特殊设备,鉴定方法
可靠,无色的PTH衍生物和其他的紫外吸收副产物不会干扰有色的DABTH衍
生物的鉴别。Gln、Asn、Trp不被破坏,可直接检出。
DABITC/PITC双偶合法是多肽和蛋白质手工测序中最令人满意的方法,现
在大多数实验室选择此法。作为液相序列测定法发展起来的双偶合法同样适用于
手工或自动固相测序方法,在固相测序法中多肽或 蛋 白 质 共 价 连 接 在 固 相 载 体
上,可以降低测序反应中的损失和化学污染。
10722 材料和试剂
(1)吡啶、异硫氰酸苯酯 (PITC)和三氟醋酸 (TFA) 测序级,重蒸水配
制,PITC必须装在可密封的管中,-20℃,氮气中储存。(2)DABITC 可 直 接 使 用 或 将 1gDABITC 溶 于 70ml丙 酮 重 结 晶,
DABITC溶解在无水吡啶中,配成28mg/ml使用。(3)庚烷、醋酸乙酯、丁基醋酸酯、甲苯和醋酸 分析纯。(4)TEM缓冲液 50ml重蒸水,50ml丙酮,05ml三乙胺,02mol/L醋
酸5ml,pH=1065。(5)反应管 厚 壁 的 硼 硅 酸 盐 玻 璃 管,带 有 磨 口 塞 (不 可 用15ml的Ep
pendorf管)。(6)恒温装置 采用水浴控温装置,偶合和裂解温度50~55℃,转化温度
55~80℃。(7)真空泵 最好采用两个泵,一个用于去除偶合和萃取后的挥发性碱和溶
剂,另一个留作去 除 裂 解 和 转 化 后 的TFA。真 空 泵 应 设 有 低 温 恒 温 保 护 装 置,

311
而且在用于TFA的泵和低温恒温装置之间应放置KOH阱保护。(8)TLC薄板和色谱分析设备 聚酰胺薄板 (30mm×30mm)
10723 液相手工序列测定方法
(1)偶合
① 用50%的吡啶80μl溶解5~10nmol样品,加DABITC (28mg/ml吡啶
溶液)40μl(400nmol),通入氮气,密封管,52℃保温50min。
② 加10μl的PITC,通入氮气,密封管,彻底混匀,52℃保温20min。(2)洗涤
① 加入500μl的庚烷醋酸乙酯 (2∶1),旋涡混匀,短暂离心 (1min)。
② 吸去上层有机相,丢弃。再用庚烷醋酸乙酯抽提下层水相两次,每次都
弃去上层有机相。洗涤步骤中,离心后必须非常小心,不要搅动有机相和水相之
间的界面,因为有些肽在洗涤中变为不溶性,会在这一界面上沉淀。每次抽提须
留1~2mm有机相。
③ 真空干燥下层水相,样品必须完全干燥后,再进行裂解。(3)裂解 加50μl无水TFA,通入氮气,密封管,52℃保温15min,真空
干燥除去TFA。如果肽链中包含特殊的疏水残基 (Pro、Val、Ile),尤其是位于
长肽的起始,建议裂解步骤重复2~3次。(4)抽提
① 加200μl醋酸丁酯和50μl水,充分混合,短暂离心,将上层有机相转移
到转化管中。
② 真空干燥萃取物和水相。
③ 干燥后的水相可进行下一循环,干燥有机相将噻唑啉酮转化为乙内酰脲。(5)转化
① 向转化管中加入50μl的50%TFA,密封试管,80℃保温10min。
② 真空干燥,再用2~5μl的乙醇溶解,取适量用聚酰胺薄板进行TLC。(6)标准物的制备
① 色谱参考标记物的制备 吸50μl吡啶置于反应管中,加入30μl二乙胺、
30μl乙醇胺,250μl浓度为28mg/ml的DABITC吡啶溶液。通入氮气,盖上塞
子充分混合,52℃反应1h。离心真空干燥,用1ml无水乙醇溶解,存储在带螺
旋盖的试管中备用。
② 标准DABTH氨基 酸 的 制 备 称1mg (约5μmol)的 氨 基 酸,加100μlTEM缓冲液 溶 解,加 入50μl(200nmol)DABITC。密 封 管 子,52℃反 应1h。离心真空干燥。用50μl的50%TFA重新溶解,52℃反应45min,或者80℃反应
10min。加入无水乙醇使标样的浓度为5~10pmol/μl。

312
(7)聚酰胺薄板色谱
① 在左下角距离边界4mm处点样 (色谱标记物和样品),样品的点样量取
决于初始肽的浓度,并且随着循环数的增大而增加,以补偿抽提损失的累积。
图107 DABTH氨基酸衍生物的双向薄板色谱图
1,2分别表示第Ⅰ向和第Ⅱ向的展层方向
用氨基酸编码的单字母表示 衍 生 物:d,e—参 考 标 记 物 二 乙 胺 和 乙 醇 胺 的 衍 生 物,均 为
紫色;T2,T3,S2,S3,S4—苏氨 酸 和 丝 氨 酸 在 测 序 的 裂 解 反 应 中 无 水 TFA的 降 解 产 物,
在标准DABTH氨基酸制备中不会形成;T2 和S2—测序中最普遍出现的形式,T2为蓝
紫色,其他衍生物均为红色;K—αDABTHεDABTCLys,通常出现在标样中;
K2,K3—双偶合法中DABITC/PITC衍生物的混合物,仅仅在侧序中与K一起出现
羧甲基半胱氨酸 (CMCys)位于乙醇胺的衍生物的下面,显示不出来
② 在加盖的展开槽中进行色谱分析,第Ⅰ向的展层溶剂为冰醋酸水(1∶2,体积比),展层 方 向 为 样 品 位 于 右 角 底 部,允 许 溶 剂 跑 到 薄 板 的 顶 边,大 约 要
2~3min。然后立刻取出,彻底吹干,要求闻不到醋酸味。
③ 将薄板转90°进行第Ⅱ向色谱分析,溶剂为甲苯正己烷醋酸 (2∶1∶1,体积比)。展层至溶剂距离薄板顶部2~3mm,大约要1~2min。取出聚酰胺薄
板吹干。(8)DABTH衍生物的显色鉴定
① 用浓盐酸蒸气熏板,DABTH氨基酸显出红色斑点,参考标记物为蓝色。通常随着循环进行的次数,形成的副产物强度增加。注意勿使浓盐酸直接接触聚
酰胺薄板,因为这会使其溶解。
② 对照标准色谱图和参考标记物的相对位置进行鉴定 (图107)。

313
10724 要点
(1)样品准备 测定一个未知蛋白的全序列,至少要获得05~1μmol的纯
化材料。要求至少用两种标准证明纯度,最好是测定 N末端氨基酸是惟一的或
者测定一小段N末端序列。(2)选择合适的蛋白裂解方法,产生最少量的测序适用的最大长度的肽。选
择裂解方法前应测定蛋白质的分子量和精确的氨基酸组成,避免某些残基受裂解
影响。例如蛋白质中出现半胱氨 酸,通 常 应 先 还 原 蛋 白 质,并 且 在 降 解 前 修 饰
巯基。(3)盐污染会导致重复率降低和干燥时间延长,必须选择合适的纯化方法以
减少盐浓度,比如采用挥发性的缓冲液、避免采用硫酸铵沉淀和使用SDS。如果
存在盐,仅仅用透析还无法完全脱盐,必须至少采用两步独立的脱盐步骤后,再
进行序列测定。在肽的纯化中,使用反向 HPLC进行有机溶剂 (如乙腈)的梯
度洗脱再冻干,可以很好脱盐。在测序过程中,必须确保洗涤后下层水相彻底干
燥,再加入TFA进行裂解。避免含TFA样品的干燥器与含吡啶样品的干燥器
共用同一个真空线路,防止盐的累积。(4)要求采用良好的真空系统,以保证迅速干燥。(5)需要中断循环时,最好在裂解反应去除了TFA后再中断,并且第二天
必须完成该循环。(6)准备进行偶合反应的样品放在50%吡啶中比干燥存储好。(7)聚酰胺薄层色谱中点样大小控制在1mm以内。转化后的干燥DABTH
衍生物为红色至紫色,溶于乙醇的溶液为淡黄色,在TLC薄板上吹干后为棕褐
色。颜色 的 亮 度 与 浓 度 相 对 应。通 常 DABTH衍 生 物 的 点 样 量 达 到 50~100pmol,可以获得明确的结果。
(8)如果只要测定N末端氨基酸,则可省略用PITC的第二步偶合。
108 蛋白质和肽的C末端分析
1081 概述
肽链的羧基末端称为C末端。C末端序列信息在分子生物学中很有用,尤其
是检测已知DNA序列的基因表达产物的翻译后修饰。例如,真核细胞内80%的
可溶蛋白由于翻译后修饰的结果,N末端氨基都被封闭,难以进行Edman降解。此外,可用于确证初始密码子和阅读框的正确位置,设计用于筛 选cDNA文 库
的寡核苷酸探针,确定蛋白质切割位置等。另一方面,当蛋白质和肽的 N末端
乙酰化、甲酰化,或者N末端的谷氨酸残基环化形成焦谷氨酸以及丝氨酸或苏
氨酸残基的ο酰基转移作用锁定了Edman降解反应,则无法从N末端测定序列

314
信息。测定蛋白质和肽的C末端氨基酸序列的方法有化学分析法 (如肼解法和重
氢标记法)、酶解法 (羧肽酶法)以及质谱法。相比于N末端序列测定的Edman降解法,至今还未能设计出可连续切除C末端残基的有效方便的化学序列分析
方法。而质谱法需要分辨率极高的昂贵光谱分析仪,并要求极高的专门技术知识
和丰富的操作经验。利 用 羧 肽 酶 可 以 很 容 易 快 速 获 得 一 定 的C末 端 序 列 信 息,因而羧肽酶法是测定C末端氨基酸序列的主要方法。
1082 C末端序列分析的羧肽酶Y法
10821 原理
羧肽酶是一类外切蛋白水解酶,从肽链的羧基端开始每次切除一个L氨基
酸或残基。有许多来源于植物或动物的羧肽酶,它们在理化特性和特定氨基酸的
释放速率上各不相同。有四种羧肽酶被广泛应用于蛋白质和肽的顺序测定中,它
们是来 源 于 牛 胰 腺 的 羧 肽 酶 A (EC34171)、来 源 于 猪 胰 腺 的 羧 肽 酶 B(EC34172)、来源于橘叶的羧肽酶C (EC34121)和来源于面包酵母
的羧肽酶Y (EC34161)。在测定C末端的氨基酸序列时,用羧肽酶消化蛋白质或肽,在 不 同 的 时 间
间隔中,取出一份,用氨基酸自动分析仪定量分析释放出的游离氨基酸。被释放
的氨基酸数目和种类随时间发生变化,以时间为横坐标,释放出的氨基酸的量为
纵坐标作图,得到氨基酸释放的动力学曲线。从每个氨基酸的相对释放速率可以
推导出C末端序列。在C末端的酶法测定中,羧肽酶A和B最先被应用。羧肽酶A特异性较广
泛,能释放绝大多数的C末端氨基酸,对非极性氨基酸残基的切割最佳,但 是
不能释放精 氨 酸、脯 氨 酸、羟 脯 氨 酸 或 赖 氨 酸 残 基。羧 肽 酶B的 特 异 性 有 限,只能切割释放精氨酸和赖氨酸残基。由于羧肽酶A和羧肽酶B具有相似的最适
作用pH,并且在很大程度上特异性互补,因此羧肽酶A和B可以被一起使用,但是遇到脯氨酸残基时酶解仍然被锁定。羧肽酶C具有广泛的特异性,可以释
放所有的C末端氨基酸,包括脯氨酸。羧肽酶Y (CPY)和羧肽酶C同样具有
广泛的特异性,并且它对蛋白质底物有强作用,能在存在尿素和去污剂的条件下
发挥作用,它是目前在蛋白质和肽的C末端序列分析中常用的工具酶。羧肽酶Y从肽链的C末端开始每次切下一个氨基酸残基,不过各个氨基酸
的释放速率不同。释放速度取决于所切割的氨基酸侧链性质,还在一定程度上取
决于相邻氨基酸残基的性质。当倒数第二个或末端残基为芳香族或脂肪族侧链氨
基酸时释放最快,而当C末端为甘氨酸或天冬氨酸,以及倒数第二个位置为赖
氨酸或精氨酸时,释放速率很慢。羧肽酶Y对三肽的切割困难,二肽完全抗水

315
解。C末端为脯氨酸时可以很好切除,但是若脯氨酸在甘氨酸的羧端一侧,则很
可能无法释放。羧肽酶Y水解酸性氨基酸的最适pH为55,而对于中性和碱性
氨基酸,最适pH为70。
10822 材料
(1)降解缓冲液 01mol/L吡啶醋酸缓冲液 (pH=56),01mol/L亮氨
酸,1%SDS。(2)羧肽酶Y 酶的冻干物可在4℃存储6~12个月。悬浮在饱和硫酸铵中
的酶 液-20℃存 储。溶 解 在 水 中 并 用 水 透 析 配 制 的1%酶 溶 液,等 份 分 装,
-20℃可至少存储2年。稀释液 (<01mg/ml)很快失活,必须于使用前新鲜
配制。
10823 方法
(1)用200μl降解缓冲液溶解20nmol的蛋白质样品,60℃保温20min,使
蛋白质变性。(2)冷却后,取出25μl作为初始取样 (即时间t=0)。(3)2nmol羧肽酶Y溶于5~10μl的01mol/L吡啶醋酸缓冲液 (pH=56),
加入到样品溶液,彻底混匀,室温 (25℃)保温,分别在时间t为1min、2min、
5min、10min、20min、30min和60min时各取出25μl样品。分别向每份取样中
加入5μl冰醋酸以终止反应。(4)将样品冷冻干燥,除去挥发性的吡啶和醋酸,进行氨基酸分析。(5)作出各个氨基酸的释放量与时间的关系图,从每个氨基酸的相对释放速
率推导C末端的氨基酸顺序。
10824 要点
(1)降解缓冲液的pH超过6时,羧肽酶会发生不可逆失活。其中加入1%SDS或6mol/L的尿素有助于蛋白质样品的溶解。降解缓冲液中的亮氨酸是作为
氨基酸分析时的内标,用以补偿任何操作损失或者样品误差。使用柱后衍生的离
子交换色谱分离氨基酸时,亮氨酸是合适的内标,根据氨基酸分析仪的灵敏度调
整亮氨酸的浓度。但对于其他系统,例如采用柱前衍生的色谱,可以选用其他内
标 (ο邻苯二醛、β丙氨酸、氨基乙醇、缬氨酸)。在氨基酸的分析中,每份取样
中的少量SDS不会干扰洗脱曲线或者影响仪器的安全性。(2)商品化的羧肽酶含有一定量的游离氨基酸,此外酶溶液反复冷冻、解冻
或者在室温下存储时间延长,也会导致自溶释放出游离氨基酸,必须在使用前除
去。酶的硫酸铵悬液离心,并用饱和硫酸铵洗涤后再溶解于缓冲液中。溶解的酶
可以用吡啶醋酸缓冲液透析。(3)本方法中的取样时间对 大 多 数 蛋 白 质 是 合 适 的,但 也 存 在 许 多C末 端

316
序列为释放缓慢的氨基酸的情况,此时应进行重复实验,延长保温时间或提高酶
与底物的比率。待分析的样品为肽时,在缓冲液中可以省去SDS,因为C末端
已可被接近,同样加热步骤也可以被省略。此外,可以缩短取样的时间间隔,因
为游离氨基酸的释放速率比蛋白质样品快,采用的取样时间t为0min、05min、
1min、2min、5min、10min和20min。(4)理论上讲,遵循释放出的氨 基 酸 动 力 学 曲 线 可 以 推 导 出C末 端 的 氨 基
酸顺序,但是由于酶对不同侧链基团的亲和性不相同,各个氨基酸的释放速度变
化相当大,因而对测定结果的解释要小心。对于多条肽链的蛋白质用该法测定C末端的结果比较复杂,比较难以解释清楚。

317
11 实 验 部 分
111 用于蛋白质纯化的仪器 (工作站)
在实验室中,一套自己组装的蛋白质色谱系统通常由恒 流 泵、色 谱 柱、检
测器和分部收集器等几部分组成。这样的一套装置成本很低,色谱柱一 般 是 自
装柱,可以根据需要选择柱的尺寸和色谱介质的类型,还可根据待分离 物 质 的
性质选择合适的检测器,如果操作条件和技术控制得好,能够获得比较 理 想 的
分离效果。正因为如此,这种自装色谱系统目前仍然被广泛采用。但 是,随 着
技术的进步,成套的蛋白质 纯 化 系 统 开 始 出 现,使 得 自 装 系 统 暴 露 出 很 多 缺
点:自装系统中常用的蠕动泵能够提供的压力不大,当使用了细颗粒的 高 效 色
谱介质后,色谱柱的背景压力往往很大,用蠕动泵无法驱动液流按一定 的 流 速
进入色谱柱;自装柱在色谱柱的设计、填充技术上远远比不上预装柱,柱 效 率
通常比预装柱要低很多;加样过程是手工完成,此过程很容易因加样不 均 匀 及
破坏色谱床面而导致洗脱峰 峰 形 变 差;洗 脱 过 程 中 洗 脱 剂 的 更 换 要 手 工 完 成,进行梯度洗脱要使用梯 度 混 合 器,很 难 形 成 复 杂 的 混 合 型 梯 度;操 作 时 间 长,分离速度慢……因此,人们在进行生物大分子的分离纯化时越来越多地 依 赖 于
设计良好的蛋白质纯化系统,这种工作站形式的纯化系统为人们提供了 一 个 纯
化平台,配合不同类型和规格的色谱柱,通过对过程控制软件的设定,可 以 方
便地实现特定物质纯化方法的探索和优化,在不同分离规模上都能获得 比 自 装
系统更好的效果。以下简要介绍一下蛋白质纯化工作站的主要部件及几 种 常 见
的产品。
1111 蛋白质纯化系统的主要部件
目前市售蛋白质纯化工作站的结构主要由进样系统、泵系统、检测器、收集
系统、循环系统、控制和输入输出系统等组成。
11111 进样系统
蛋白质液相色谱系统中进样方式主要有隔膜进样、停流进样、阀进样、进样
器进样等,目前又以阀进样和进样器进样最为常用。阀进样是目前绝大多数液相色谱系统都提供的一种进样方式,其缺点是阀接
头和连接管存在死体积,导致柱效率稍低于隔膜进样,但该方式耐压高、再现性
好、操作方便,因此成为常用的进 样 方 式,其 中 又 以 六 通 进 样 阀 最 为 常 用 (图

318
111)。该阀能在采样位置和进样位置之间切换,首先将阀切换至采样位置,用
注射器将样品在常压下注入量管内 (用黑色部分表示),量管的容量控制了最大
加样量,若注入的样品体积超过了最大加样量,多出部分的样品会从废液排出口
4流出。将阀转过60°后切换至进样位置,启动泵将溶剂从入口1注入,可将样
品从管口3顶入色谱柱。
图111 六通进样阀的结构
1—溶剂入口;2—量管;3—连接色谱柱管口;4—废液排出口
将阀与特定的进样装置结合产生了进样器进样方式,进样器的种类很多。自
动进样器是一种适合对多份不同样品在相同条件下进行色谱分析的进样方式,通
过计算机控制,可以在无人看管情况下进行操作。超级进样器适合于对同一样品
进行多次不同条件的色谱,或对大体积样品进行进样。
11112 泵系统
泵系统的作用是提供各种流速稳定的液流,液相色谱系统中常用的有往复泵
和气动泵,往复泵的缺点是有脉冲,而气动泵的缺点是会将气体带入流动相中,对色谱柱产生不良影响。泵系统的主要参数是输液能力和能提供的压力。色谱系
统所需的输液能力由分离的规模决定,分析型分离和实验室小规模制备分离对输
液能力要求低,通常不超过10ml/min,但是对泵的精密度要求高;而制备型分
离对输液能力要求高,需达到100ml/min,但是对泵的精密度要求低,小的波动
不会对色谱结果造成很大影响。
11113 检测器
检测器是根据所分离物质的性质来进行选择的,最常用的是紫外检测器和示

319
差折 光 检 测 器。对 于 蛋 白 质,通 常 在280nm 有 最 大 吸 收,而 核 酸 类 物 质 在
254nm附近有最大吸收,所以这两类物质的检测 一 般 都 采 用 紫 外 吸 收 法。在 远
紫外区能产生吸收的物质种类很多,因此选用波长小于220nm的紫外线对样品
进行检测时,杂质和流动相成分很可能对检测产生干扰。很多紫外检测器都提供
了双波长、三波长甚至更多波长的同时检测,从而有助于对样品中不同成分进行
对照比较,生成较全面的色谱图。对于在紫外区无吸收或虽然有吸收但受其他物
质干扰较大的样品,采用示差折光检测器进行检测是比较合适的,例如在检测糖
类或糖蛋白等物质的分离时常用到该检测器。有时将不同的检测器串联在一起使
用可以获得更好的效果。液相色谱系统中除了对目的组分进行检测外,还需要检测色谱柱中洗脱液的
pH、电导等参数,这些通过配置特定的电极来实现。
11114 收集系统
对于最简单的样品分离,可以采用手工收集的方法,但液相色谱系统一般都
配置了全自动分部收集器来对样品进行收集。收集的模式可以有多种,常用的是
按规定的时间或洗脱液体积进行分部收集,也可根据检测器检测到的洗脱情况,按洗脱峰进行收集。由于在检测器和分部收集器之间存在一定的死体积,收集时
应注意有迟后现象。
11115 循环系统
对于复杂的混合物一步色谱很难实现完全分离,通常需要通过若干步纯化才
能获得纯品,具体操作时可以将色谱后分部收集到的目的蛋白作为样品再次进行
色谱分离,这种色谱多数情况下与上一次色谱采用的是不同的色谱技术,有时也
会采用同一种介质再次色谱。很多液相色谱系统提供循环回路,色谱柱下端流出
的洗脱液经检测器后可以不用收集而直接作为样品加样到同一根或不同的色谱柱
上,进行再次色谱。如果在检测器后面配置一个多路阀,可以控制杂质组分形成
的洗脱峰直接排出而不进入再分离步骤。循环系统的一个关键指标就是死体积要
小,否则第一次色谱后已分离的组分在系统中会因 为 谱 带 扩 张 而 重 新 混 合 在 一
起,并可能造成二次色谱时洗脱峰峰宽过大。
11116 控制和输入输出系统
目前市售的液相色谱系统都是 全 电 脑 控 制 的,生 产 商 开 发 出 相 应 的 控 制 软
件,人们在选定色谱柱进行色谱时,在软件 界 面 中 可 以 对 各 种 色 谱 参 数 进 行 设
定,还可以规定检测器检测的范围、分部收集器收集的方式以及色谱柱的再生和
清洗方案等。待各种所需溶剂、样品准备完毕,各种参数设定好以后,在软件界
面上点击运行,色谱系统就会根据预设方案进行色谱过程,而色谱图会输出在电
脑的显示器上,并保存在电脑内。人们还可以根据需要选择在色谱图中显示的参

320
数,以免曲线过多造成图谱过于烦琐。
1112 几种常见的蛋白质色谱系统
Pharmacia公司的AKTAdesign系列色谱系统是一类适合于从分析型到中小规
模制备 使 用 的 中 低 压 液 相 色 谱 系 统,其 中 包 括 了 AKTAprime、AKTA FPLC、
AKTApurifier和AKTAexplorer四 种 由 简 单 到 复 杂 的 系 统。AKTAexplorer被称为快速纯 化 工 艺 开 拓 系 统,是 AKTA系 列 中 配 置 最 完 整 的 液 相 色 谱 系 统
(图112),具有自动缓冲液配置、自 动 加 样 和 在 线 缓 冲 液 监 控、多 柱 转 换、多
波长连续检测等特点,配合各种类型的预装柱,可以进行包括凝胶过滤、离子交
换、亲和色谱、疏水色谱、反相色谱等多种色谱。与系统配套的Unicorn控制软
件可以对色谱过程进行各类参数设定和全方位的输入输出控制。对于大规模分离
纯化过程,Pharmacia公司还提供了BioProcess系统。
图112 AKTAexplorer快速纯化工艺开拓系统
美国AppliedBiosystems公司的BioCAD700E快速纯化工作站是同时适用
于高、中、低压纯化和分析的液相色谱系统 (图113),具有压力和流量应用范
围广、提供多种模板可快速分离、柱串联功能、全部参数在线检测、系统开放性
强、适用于各种不同规格和类型的预装柱和自装柱等特点,配合专门开发的软件
系统,可以方便地进行纯化工艺的探索及样品的分析和制备。表111列出了该
系统的部分性能规格。

321
图113 BioCAD700E快速纯化工作站
表111 BioCAD700E快速纯化工作站的部分性能规格
泵系统 双 柱 塞 泵 组分/废料阀 手工和自动单组分收集
梯度能力 低压四元百分比混合
洗脱或pH增量为0.1% 组分混合准确度±1.0%
流速范围 0.2~100ml/min,精确度±0.3%最大压力 3000psi进样方式 多通道自动进样,手动进样器,通过
泵系统大体积进样检测
紫外/可见 190~800nm 连 续 双 波 长,自 动 调零,流动池(分 析 型6mm,9μl;制 备 型
3mm,4.5μl),0.0005~3.0AUFS pH 范围2~12,自动校正
电导率 范围0~200mS/cm,自动校正
外部收集器 可选,支持多种组分收集器压缩气体 空气或氮气(气动阀)
5psi氦气(用于在线脱气)材料 100% 生 物 相 容:PEEK,Teflon,
Kelf,Kynar,Sapphire柱 ①单 柱,旁 路、反 相;②双 柱,旁 路、
串联、并联;③三 柱;④低 分 散;⑤直 接控制;⑥ 可 选 SCOUT 柱 切 换 装 置(8柱),支持各种柱子和填料
压力范围 0~3000psi辅助输入 12bitA/D连接其他检测器
旁路检测器 pH/电导率旁路自动切换
图114 WatersDelta3000蛋白质快速制备色谱仪

322
Waters公司也是全球重要的液相色谱系统生产商,其产品较多涉及高效液
相色谱 (HPLC)及液质联用 (LCMS)领域,Delta系列是其应用于自动快速
提纯蛋白质的制备型色谱仪 (图114)。此外,还有BioRad公 司 的ECONOSystem 系 列 产 品、Labomatic公 司 的
HPMPLC高效中压液相制备色谱系统等。
112 蛋白质分离纯化实例
1121 蔗糖酶的分离纯化
蔗糖酶是一种水解酶,它能使蔗糖水解为葡萄糖和果糖。选用蔗糖酶含量丰
富的活性干酵母为原料,先通过自溶法使酶游离出来,采用有机溶剂分级沉淀法
进行粗分离,用SephadexG25进行缓冲液交换后,采用DEAE纤维素离子交
换色谱进行纯化,可以得到较纯的蔗糖酶,用SDS聚丙烯酰胺凝胶电泳法进行
纯度鉴定。酵母蔗糖酶属胞内酶,因此必须先将细胞破碎,然后进行抽提。破碎抽提的
方法很多,对于活性干酵母可采取氨解法,其原理与自溶法相同,通过加入甲苯
并保温搅拌让酵母自身酶系发挥作用,使菌体自溶破碎。在加入甲苯的同时,加
入了一定量的氨水,为活性干酵母的自溶提供适宜的pH条件,便于蔗糖酶的有
效抽提。具 体 操 作 为:称 取50g活 性 干 酵 母 粉 于250ml烧 杯 中,加 入100ml05mol/L的氨水与之混合,再加入该混合液总体积5%~10%的甲 苯,混 合 均
匀,用磁力搅拌器搅拌16~20h,然后向溶液中加入15倍体积的水,用4mol/L的醋酸调节pH至58,低温下于4000r/min离心10min,所得上清液即为粗酶
E1 (不清时则需提高离心转速再次离心或过滤)。通过细胞自溶,从酵母中抽提出来含有大量杂质的粗酶,还需进一步分离纯
化。首先采用一些初步分离手段,比如盐析法、有机溶剂沉淀法等。有机溶剂分
级沉淀是一种常用的有效沉淀和纯化酶蛋白的方法,它利用有机溶剂的脱水作用
破坏了蛋白质分子的水化膜,使蛋白质分子易凝集沉淀。实验中具体操作为:粗
酶E1用4mol/L醋 酸 调 节pH 至45,添 加 经 预 冷 却 的 乙 醇 至 乙 醇 终 浓 度 为
32%,注意 乙 醇 要 缓 慢 加 入,同 时 搅 拌 均 匀。添 加 完 毕,4000r/min 离 心
10min,此时蔗糖酶未被沉淀,将上清液倒入另一烧杯中,弃去沉淀。往上清液
中继续添加乙醇使其终浓度达到475%,于4000r/min离心10min,此时蔗糖酶发
生沉淀,弃去上清液得少量沉淀,用30mlpH=58、005mol/L的醋酸醋酸钠缓
冲液溶解沉淀,于10000r/min离心10min除去不溶物,得上清液为蔗糖酶E2。乙醇分级沉淀后的蔗糖酶E2仍含有较多的杂质,采用DEAE纤维素阴离子
交换法进一步纯化,在离子交换之前采用SephadexG25凝胶过滤色谱来进行缓

323
冲液交换,使蔗糖酶溶解于离子交换的起始缓冲液中。凝胶过滤色谱和离子交换
色谱均采用自装色谱系统。具体操作为:填充一根SephadexG25凝胶柱,尺寸
为16cm×50cm,用2~3倍柱体积的pH=58、005mol/L的醋酸醋酸钠缓冲
液平衡 凝 胶 柱。将 上 一 步 收 集 得 到 的 蔗 糖 酶E2加 样 至SephadexG25凝 胶 柱,用同样的缓冲液洗脱,流速为2ml/min。得到一个平台状的很大的洗脱峰,用小
烧杯收集 整 个 洗 脱 峰。填 充 一 根 DEAE纤 维 素 DE52阴 离 子 交 换 柱,尺 寸 为
26cm×20cm,填充前应注意用醋酸将交换剂的pH调节至起始pH=58,用5倍柱体积的起始缓冲液pH=58、005mol/L的醋酸醋酸钠平衡柱床,然后将
缓冲液交换后收集的洗脱峰加样至离子交换柱,用起始缓冲液洗涤色谱柱,流速
为2ml/min,将此条件下不吸附的杂质洗去,紫外检测器检测到一个很大的穿透
峰。采用pH阶段洗脱法,用pH=35、005mol/L的醋酸醋酸钠作为洗脱缓
冲液,流速仍为2ml/min,用分部收集器收集,出峰完全后通过测定蔗糖酶活力
的方法确定活性峰,得到较纯的蔗糖酶。
1122 转谷氨酰胺酶的分离纯化
转谷氨酰胺酶分子量约38000,是一种催化酰基转移反应的转移酶,它以肽
链中谷氨酰胺残基的γ羧酰胺基作为酰基供体,而酰基受体可以是多肽链中赖
氨酸残基的ε氨 基、伯 胺 基 或 者 水,从 而 可 以 形 成 蛋 白 质 分 子 内 和 分 子 间 的
ε(γ谷氨酰)赖氨酸异肽键,蛋白质分子和小分子 伯 胺 之 间 的 连 接 或 者 水 解 蛋
白质中谷氨酰胺残基脱去氨基生成谷氨酸残基。由于该酶具有催化蛋白质交联及
修饰蛋白质的作用,在食品、医药等领域具有广泛的应用。由Streptoverticilliummobaraense菌株经25L发酵罐发酵产生的含转谷氨酰
胺酶的发酵液作为原料。该酶属于胞外酶,由于菌株在发酵后期形成菌丝球,用
小型板框压滤机压滤或离心机3000r/min离心10min可以除去菌体,得到104L粗谷氨酰胺酶滤液。对上述滤液进行超滤浓缩,选用截留分子量为10000的超滤
膜,控制浓缩倍数在3~35倍之间,得到3108ml浓缩液。粗分级方法选择乙醇
沉淀,按浓缩液∶乙醇(体积比)=1∶15的比例缓慢加入预先冷却的乙醇,沉淀
目的蛋白,4℃、3000r/min离心10min,弃去上清液,将沉淀冻干即得到转谷氨酰
胺酶粗酶。测定各步骤所得样品酶活力,得到酶活性回收率,列于表112。
表112 粗酶提取各步骤收率
步 骤 酶 活 酶 量 总酶活单位/U 回收率/%除去菌体后 2870U/ml 104L 29848 100超滤浓缩后 8607U/ml 3108ml 26751 896乙醇沉淀、冻干后 71765U/g 2948g 21158 709
粗酶进一步 采 用 凝 胶 过 滤 和 离 子 交 换 进 行 纯 化,操 作 平 台 为Pharmacia的

324
AKTAexplorer100快速纯化工艺开拓系统,通过专业软件 Unicorn实现自动化
操作。凝胶过滤色谱选用的色谱柱为 HiLoadTM26/60Superdex75pg,其分级分
子量范围为3000~70000。洗脱剂为含有02mol/LNaCl的pH=65、002mol/L磷酸钠缓冲液,其中02mol/LNaCl的作用是提高离子强度,减小蛋 白 质 和 凝
胶介质之间的非特异性吸附作用。实验显示用该洗脱剂比用不含 NaCl的pH=65、002mol/L磷酸钠缓冲液作为洗脱剂进行色谱分辨率高。将粗酶溶于缓冲
液,过滤或10000r/min离 心 除 去 不 溶 物,取20ml上 样,流 速 为18ml/min,图谱如图115所 示。测 定 纯 化 前 后 的 蛋 白 质 浓 度 和 酶 活,此 过 程 纯 化 倍 数 为
206倍,活性回收率为802%。
图115 凝胶过滤色谱图
注:带 “”的为活性峰
将分部收集得到 的 活 性 峰 在 离 子 交 换 色 谱 所 使 用 的pH=65、002mol/L磷酸钠缓冲液中进行透析。由于转谷氨酰胺酶的等电点为89,而最适作用pH在6左右,在 此pH 条 件 下 酶 蛋 白 带 正 电 荷,实 验 中 选 用 了 强 阳 离 子 交 换 柱
HiPrepTM16/10Source30S进行色谱。所用的离子交换剂以聚苯乙烯二乙烯苯
颗粒为基质,颗 粒 的 直 径 为30μm,功 能 基 团 为 磺 酸 基 (—SO-3 )。采 用 提 高
NaCl浓度的方 法 进 行 梯 度 洗 脱,极 限 缓 冲 液 为 含 有10mol/LNaCl的pH=65、002mol/L磷酸钠缓冲液。先初步试用了以下洗脱方案:洗脱流速30ml/
min,上样后先用2个柱体积 (40ml)的起始缓冲液洗去不吸附的杂质,然后用
复合线性梯度进行洗脱。先在12个柱体积 (240ml)内将NaCl的浓度从0mol/
L提高到03mol/L,再在4个柱体积 (80ml)内将NaCl的浓度从03mol/L提
高到05mol/L,最后用含有10mol/LNaCl的pH=65、002mol/L磷酸钠缓

325
冲液洗去吸附牢固的杂质。实验结果如图116所示,从图中可以看出,转谷氨
酰胺酶和离子交换剂之间吸附作用并不强,在较低的盐浓度下就被洗脱下来。因
此将洗脱方案优化为:洗脱流速30ml/min,上样后先用2个柱体积 (40ml)的
初始缓冲液洗去不吸附的杂质,然后用NaCl浓度线性梯度进行洗脱,在10个
柱体积 (200ml)内将NaCl的浓度从0提高到015mol/L。最后用含有10mol/LNaCl的pH=65、002mol/L磷 酸 钠 缓 冲 液 洗 去 吸 附 牢 固 的 杂 质,结 果 如 图
117所示。通过测定纯化前后蛋白质浓度和酶活力可知,此阶段的纯化倍数为
328倍,活性回收率为705%。纯化后所得活性分部进行SDSPAGE,经考马
斯亮蓝染色后呈单一条带,证明已获得良好的纯化效果。
图116 初次离子交换图谱
注:带 “”的为活性峰
1123 纯化重组蛋白Pseudomonasaeruginosa外毒素A重组Ecoli周质中表达的Pseudomonasaeruginosa外毒素A (Mw=55000)在
纯化后经修饰连接上糖链可用作疫苗,本实验讨论了一种可用于大规模纯化此重组
蛋白的方法。此方法从原料到最终得到高纯度的外毒素A,只经过四步色谱。在提取阶段,Ecoli匀浆后不进行澄清操作而直接利用膨胀床吸附技术从悬
浊液中提取目的蛋白。采用STREAMLINE系列吸附剂,选择合适 的 条 件 可 以
使目的蛋白连接在吸附剂上,而颗粒物质和其他杂质不受阻碍,通过色谱柱,然
后再选择另一条 件 将 目 的 蛋 白 洗 脱。实 验 中 选 用STREAMLINE200柱 (内 径

326
图117 优化后离子交换图谱
注:带 “”的为活性峰
200mm),柱中填充了47L的STREAMLINEDEAE吸附剂,色谱系统为BioProcessModular。将47kgEcoli细胞渗透休克后悬浮于pH=74、50mmol/L
图118 STREAMLINEDEAE膨胀床吸附色谱图
的Tris缓 冲 液 中,最 终 得 到180L样 品。加 样 至STREAMLINE200柱,用
pH=74、50mmol/L 的 Tris缓 冲 液 完 成 洗 涤 过 程,加 样 和 洗 涤 流 速 均 为
400cm/h。然后用含05mol/LNaCl的pH=74、50mmol/L的 Tris缓 冲 液 作
洗脱剂进行洗脱,流速为100cm/h。色谱图见图118,出现两个峰,一 个 大 而

327
宽的是不吸附的穿透峰,洗脱阶段出现一个主峰。在中间纯化阶段采用了疏水色谱和离子交换色谱。首先进行疏水色谱,选用
图119 PhenylSepharose6FastFlow疏水色谱图
BPG200柱 (200mm×150mm),其 中 填
充 了 47L 的 PhenylSepharose6 FastFlow介 质。往 前 一 步 骤 中 收 集 到 的 样 品
中添加 硫 酸 铵 至 终 浓 度 为06mol/L,将
此样 品 上 柱,用 含 07mol/L 硫 酸 铵 的
pH=74、50mmol/L的 磷 酸 盐 缓 冲 液 洗
涤柱子,等 穿 透 峰 出 完 后 换 用pH=74、
20mmol/L的磷 酸 盐 缓 冲 液 进 行 洗 脱,得
到一个含目的蛋白的洗脱峰,最后用蒸馏
水清洗色谱柱,各阶段流速均为120cm/h,图谱见图119。经过疏水色谱步骤,除去
了包括核酸在内的大部分具有紫外吸收的杂质。由于疏水色谱是在高的离子强度
下加样,低的离子强度下 洗 脱,而 离 子 交 换 色 谱 则 相 反,在 低 的 离 子 强 度 下 加
样,高的离子强度下洗脱,因此在蛋白质纯化时常常将这两种技术连续使用,可以
简化操作。离子交换选用了FineLINE100柱 (100mm×50mm),其中填充了375ml的SOURCE30Q离子交换介质。将上步收集到的样品用蒸馏水稀释1~3倍可直接
加样,每轮色谱加样量为15L。加样后用pH=74、20mmol/L的磷酸盐缓冲液洗
涤除去不吸附组分,然后用线性NaCl浓度梯度洗脱,极限缓冲液为含10mol/LNaCl的pH=74、20mmol/L的磷酸盐缓冲液,梯度为20个柱体积内NaCl浓度从
0mol/L增至05mol/L,流速为600cm/h,得到色谱曲线见图1110。
图1110 SOURCE30Q离子交换色谱图

328
图1111 SOURCE15PHE疏水色谱图
经上述色谱过程后,所得样品已经比
较纯,最后 的 精 制 阶 段 再 次 使 用 疏 水 色
谱,选用内径为35mm的SOURCE15PHE柱。往上步收集得到的样品中添加硫酸铵
至10mol/L,每 轮 色 谱 加 样 量 为05L。加样后用含10mol/L硫酸铵的pH=74、
50mmol/L的 磷 酸 盐 缓 冲 液 洗 涤 色 谱 柱。然后用线性下降的硫酸铵浓度梯度洗脱,极限缓冲液为pH=74、50mmol/L的磷酸
盐缓冲液,梯度为15个柱体积内硫酸铵浓
度从10mol/L降至055mol/L,流速为200cm/h,得到色谱曲线见图1111。收集得到的最终样品经聚丙烯酰胺凝胶电泳得到单一条带 (图1112),证
明为纯品,各纯化步骤的回收率见表113,最终总回收率为51%。
图1112 纯化样品的电泳分析
(a)SDSPAGE结果:1,5—分子量标准;2—SOURCE15PHE色谱后样品;
3—SOURCE30Q色谱后样品;4—PhenylSepharose6FastFlow色谱后样品
(b)NativePAGE结果:1—PhenylSepharose6FastFlow色谱后样品;
2—SOURCE30Q色谱后样品;3—SOURCE15PHE色谱后样品
表113 外毒素A各纯化步骤所得结果
纯 化 阶 段 体积/L 总蛋白/g 外毒素A/g 回收率/%抽提液 180 351 108 100STREAMLINEDEAE 135 140 854 79PhenylSepharose6FastFlow 114 41 660 61SOURCE30Q 302 126 604 56SOURCE15PHE 122 — 55 51

329
1124 Ecoli中RNA聚合酶的提取和纯化
对 于RNA聚 合 酶 以 及 其 他 众 多 的DNA结 合 蛋 白,常 使 用 亲 和 色 谱 技 术
来 分 离。利 用 此 类 蛋 白 质 与 DNA的 特 异 性 结 合 能 力,可 以 将 小 段 含 有 特 定
序 列 的DNA作 为 配 基 连 接 到 支 持 基 质 上 构 成 亲 和 色 谱 的 介 质。但 是 这 样 的
配 基 在 细 胞 外 是 很 容 易 受 到 核 酸 酶 的 攻 击 而 发 生 降 解 的,因 此 人 们 多 数 情 况
下 并 不 选 用 核 酸 本 身 作 为 配 基,而 是 选 择 一 些 具 有 类 似 生 物 学 特 性 的 其 他 亲
和 配 基,例 如 肝 素,由 于 该 分 子 既 有 亲 和 力,又 带 电 荷,能 与 蛋 白 质 发 生 离
子 作 用,因 此 是 用 亲 和 色 谱 法 分 离 DNA结 合 蛋 白 的 理 想 配 基。这 里 介 绍 采
用 亲 和、凝 胶 过 滤 和 离 子 交 换 三 步 色 谱 的 方 法 从 大 肠 杆 菌 培 养 液 中 分 离
RNA聚 合 酶。将Ecoli培 养 液 离 心 后 收 获 细 胞,向 其 中 加 入 两 倍 体 积 的 裂 解 缓 冲 液
[20mmol/LTrisHCl,pH=8,含5% (体 积 分 数)甘 油、1mmol/LEDTA、
025mol/LNaCl、20mmol/L2巯基乙醇、1mmol/LPefabloc]使之悬浮,在冰
浴中进行超声破碎,然 后 于5℃、12100g离 心1h,弃 去 沉 淀 得 到 上 清 液 作 为
原料。向上清液 中 添 加 硫 酸 铵 至 终 浓 度 为50%饱 和 度,于 冰 浴 中 搅 拌30min使RNA聚合酶充分 沉 淀,于5℃、12100g离 心90min,弃 上 清,将 沉 淀 溶 解
于起 始 缓 冲 液 [20mmol/LTrisHCl,pH=8,含5% (体 积 分 数)甘 油、
01mmol/LEDTA、10mmol/L2巯 基 乙 醇、01mmol/LPefabloc、02mol/L
图1113 HiTrapHeparin5ml亲和色谱图
NaCl],并利用PD10凝 胶 柱 进 行 缓
冲液交换。样品用045μm滤 膜 过 滤
后进 行 亲 和 色 谱,色 谱 柱 为 HiTrapHeparin5ml,取35ml样 品 上 样,并
用起 始 缓 冲 液 洗 涤 色 谱 柱,流 速 为
1ml/min (30cm/h),洗 去 穿 透 峰 后
进行 洗 脱,极 限 缓 冲 液 为20mmol/LTrisHCl,pH=8,含5% (体积分数)甘 油、01mmol/L EDTA、10mmol/L2巯 基 乙 醇、01mmol/L Pefabloc、
10mol/LNaCl。采 用 NaCl浓 度 线
性 梯 度 洗 脱,斜 率 为20个 柱 体 积 内 NaCl浓 度 从02mol/L增 至10mol/L。分 部 收 集,结 果 见 图1113。
上步收集 得 到 的 活 性 分 部 经 浓 缩 后 进 行 凝 胶 过 滤 色 谱,采 用 的 色 谱 柱 是
Superdex200HR10/30。取05ml样 品 上 样,洗 脱 缓 冲 液 为20mmol/LTrisHCl,pH=8,含5% (体积分数)甘油、01mmol/LEDTA、10mmol/L2巯基

330
乙醇、01mmol/LPefabloc、02mol/LNaCl,流 速 为025ml/min (19cm/h),得到结果如图1114所示。
图1114 Superdex200HR10/30凝胶过滤色谱图
图1115 HiTrapQ1ml离子交换色谱图
凝胶过滤分离的同时也实现了缓冲液交换,将上步收集得到的样品直接进行
离子交换色谱。色谱柱选用 HiTrapQ1ml,加样量为1ml,加样后用起始缓冲
液洗涤,流速为1ml/min (158cm/h)。起始缓冲液和极限缓冲 液 的 成 分 均 与 亲
和色谱时相同。采用NaCl浓度线性梯度洗脱,斜率为30个柱体积内NaCl浓度
从02mol/L增至05mol/L,分部收集,每分部为05ml,所得结果如图1115所示。纯化后的样品经SDSPAGE分析纯度为90%。
1125 从Ecoli中分离制备高纯度的去乙酰头孢菌素C合成酶
去乙酰头孢菌素C合成酶 (DAOCS)是对氧敏感的不稳定的酶,在Streptomyces中参与头孢菌素/头霉素的生物合成。从DAOCS过量表达 的Ecoli中
分离纯化出高纯度的酶,需尽可能缩短操 作 时 间。根 据 该 酶 的 一 部 分 已 知 的 特
性,采取离子交换、疏水和凝胶过滤色谱 (表114),最终得到10mg有活性的
纯酶。
表114 DAOCS的部分特性和设计分离的方案
参 数 性 质 设 计
等电点 48 用阴离子交换剂在中性pH下分离
分子量 34500 选用分级范围在3000~70000的Superdex75prepgrade进行
凝胶过滤分离
稳定性 对氧敏感,与空气接触易失活 尽可能缩短纯化时间,添加DTT以减少氧化
DAOCS存在于Ecoli的细胞质中,将细胞匀浆后,首先迅速除去抽提液中

331
对目的蛋白有害的杂质 (如蛋白酶等),由于该酶等电点为48,在中 性pH 条
件下进行阴离子交换色谱是合适的,选用 HiPrep16/10QXL色 谱 柱,柱 体 积
为20ml,先采用NaCl浓度线性梯度洗脱 [图1116 (a)],根 据 色 谱 结 果,以
目的蛋白洗脱峰的纯度、峰体积和目的蛋白的降解程度 (活性回收率)作为优化
的指标,采用 了 NaCl浓 度 阶 段 洗 脱 的 方 法 [图1116 (b)],洗 脱 结 果 仍 不 理
想,对阶段洗脱的条件进一步 进 行 了 优 化,得 到 图1116 (c)所 示 的 比 较 理 想
的纯化结果。色 谱 时 每 次 加 样 量 为40ml,起 始 缓 冲 液 为pH=75、50mmol/L的TrisHCl,含2mmol/LDTT、02mmol/L盐酸苯甲脒、02mmol/LPMSF,极限缓冲液为含10mol/LNaCl的起始缓冲液,流速为该色谱柱的最大建议流
速10ml/min。
图1116 通过调节洗脱方式对离子交换色谱效果进行优化
离子交换后常使用疏水色谱,这两者之间的样品处理过程最简化,向离子交
换收集得到组分中添加硫酸铵至2mol/L后即可。该环节中选择三种不同的色谱
柱进行色谱操作,以确定最优条件,三根色谱柱分别为RESOURCEISO、RESOURCEETH和RESOURCEPHE,它们都以15μm的SOURCE填料为基质,分别连接 上 异 丙 基 或 苯 基 而 成。加 样 量 为10ml,起 始 缓 冲 液 为pH=75,含
20mol/L硫酸铵、1mmol/LEDTA、1mmol/LDTT的50mmol/LTrisHCl缓
冲液,极限缓冲液比起始缓冲液少了20mol/L硫酸铵,其余组分完全相同。采
用下降 的 硫 酸 铵 浓 度 线 性 梯 度 洗 脱,梯 度 为20个 柱 体 积,硫 酸 铵 浓 度 从
20mol/L降至0mol/L,流 速 为3ml/min,结 果 见 图1117,决 定 选 用 尺 寸 为
16mm×50mm的RESOURCEISO色谱柱。实验还发现缓冲液中添加甘油可以
提高酶活性的收率。最后的精制采用凝胶过滤,疏水色谱收集得到的样品浓缩后可直接进行凝胶
过滤,采用的色谱柱是 HiLoad16/60Superdex75prepgrade,该酶的分子量处
于此介质的分级范围内。

332
图1117 分别采用三种不同的疏水色谱柱分离结果
经过多次的色谱条件优化,最后确定DAOCS的分离纯化步骤为:
①Ecoli抽提液用HiPrep16/10QXL离子交换色谱,加样量40ml,起始
缓冲液为pH=75,含2mmol/LDTT、02mmol/L盐 酸 苯 甲 脒、02mmol/LPMSF的50mmol/LTrisHCl缓冲液,极 限 缓 冲 液 为 含10mol/LNaCl的 起 始
缓冲液。采用阶段洗脱,先用5个柱体积 的 起 始 缓 冲 液 洗 涤 色 谱 柱,然 后 用5个柱体积的含03mol/LNaCl的 缓 冲 液 进 行 洗 脱,DAOCS在 此 步 被 洗 脱,再
用极限缓冲液洗5个柱体积,流速均为10ml/min(300cm/h)。
② 收集所得活性分部添加硫酸铵至16mol/L用来上样,选用尺寸为16mm×50mm,填充SOURCE15ISO离子交换剂 的 色 谱 柱 进 行 离 子 交 换 色 谱,加 样 量
40ml,起始缓冲液为pH=75,含16mol/L硫酸铵、10%甘油、1mmol/LEDTA、
2mmol/LDTT、02mmol/L 盐 酸 苯 甲 脒、02mmol/LPMSF 的 50mmol/LTrisHCl缓冲液,极限缓冲液比起始 缓 冲 液 少 了16mol/L硫 酸 铵,其 余 组 分
完全相同。采用下降的硫酸铵浓度复合梯度进行洗脱,在4个柱体积内 硫 酸 铵
浓 度 从16mol/L降 至134mol/L,再 在8个 柱 体 积 内 降 至122mol/L,然
后4个 柱 体 积 内 降 至104mol/L,最 后 用 极 限 缓 冲 液 洗4个 柱 体 积,流 速 为
5ml/min。
③ 收集活性分部经浓缩后进行 凝 胶 过 滤,采 用 HiLoad16/60Superdex75
prepgrade色谱柱,加样量3ml,洗脱缓冲液为pH=75,含1mmol/LEDTA、
2mmol/LDTT、02mmol/L 盐 酸 苯 甲 脒、02mmol/LPMSF 的100mmol/LTrisHCl缓冲液,流速为1ml/min,分部收集。
整个色谱过程所 得 色 谱 曲 线 见 图1118,将 纯 化 后 样 品 进 行SDSPAGE分

333
图1118 三步色谱法纯化乙酰头孢菌素C合成酶 (DAOCS)(a)离子交换;(b)疏水色谱;(c)凝胶过滤
图1119 DAOCS纯化样品的SDSPAGE分析1,6—分子量标准;2—细胞匀浆抽提物;3—离子
交换后;4—疏水色谱后样品;5—凝胶过滤后纯酶
析,银染色后结果见图1119,证明得
到了纯酶。从以上蛋白质分离纯化的实例中可
以看出,不同的蛋白质分离纯化时虽然
并不存在一套普遍适用的方法,但还是
有一些共同的原则、一般的操作程序可
以遵 循,也 有 一 些 共 同 的 方 法 可 以 采
用。蛋白质的分离纯化实际上是根据目
的蛋白的理化性质和生物学性质,将各
种分离纯化方法按一定的顺序组合运用
的过程。对于不同的蛋白质,按其特性
对纯化方案进行设计和不断优化,最终
可以得到满足纯化要求的方法。

334
参 考 文 献
1 “Acrylamidegelcastinghandbook”.PharmaciaBiotechABUppsala,Sweden.1994.21~28
2 “HorizontalSDSelectrophoresisin05mmthinporegradientgel”ApplicationNote348.PharmaciaLKB
BiotechnologyABUppsala.Sweden.1991
3 “ImmobilineRDryStripKitfor2DElectrophoresiswithImmobilineDryStripandExcelGelTMSDS”In
structions.PharmaciaLKBBiotechnologyAB.1993.36~37
4 Acrylamidegelcastinghandbook.PharmaciaBiotechAB.UppsalaSweden,1994.20
5 AndrewsAT.Electrophoresis:Theory,TechniquesandBiochemicalandClinicalApplications.Oxford:
Clarendonpress,1986.29
6 AngalSandDeanPDG.BiochemJ,1977,167:301
7 AngalS,DeanPDG,etal.PurificationbyExploitingofActivity.In:ScopesRK,ed,ProteinPurifi
cation.SpringerVerlagPress,1998.245~291
8 AvrameasS,TernynckT,etal.ScandJImmunol,1978,8:7
9 BarondesSH.AnnuResBiochem,1981,50:207
10 BatasB,JonesHR,etal.JChromatogrA,1997,766:109~119
11 BatasB,ChaudhuriJB.BiotechnolBioeng,1996,50:16~23
12 BethellGS,AyersJS,etal.JBiolChem,1979,254:2572
13 BeuveryEC,GriffithsJB,etal.Animalcelltechnology:developmentstowardsthe21stCentury.
Dordrecht:KluwerAcademicPublishers,1995.597
14 BeynonRJandBondJS.Proteolyticenzymes:apracticalapproach.Oxford:OxfordUniversityPress,
1989.259
15 BhikhabhaiR,JohanssonG,etal.JApplBiochem,1984,6:336~345
16 BittnerM M,GoldbergS,HeerenBandGalluppiG.FedProc,1986,45:3049
17 BradshawTM,DunlapRB.Biochemistry,1993,32:12774
18 BrocklehurstK,CarlssonJ,KierstanMPJ,etal.BiochemJ,1973,133:573
19 BollagDM,RozyckiMD.ProteinMethods.NewYork:WileyLiss,Inc.,1996
20 BoardmanNK,PartridgeSM.BiochemJ,1955,59:543~552
21 BonnerjeaJ,OhS,etal.Biotechnology,1986,4:954~958
22 BoschettiE.JChromatogr,1994,658:203~235
23 BouvierF,HuguenyP,etal.PlantJ,1994,6:45
24 BreewerSJandSassenfeldH M.TrednsinBiotechnology,1985,3:119
25 BrewerSJ,DickersonCH,etal.JChromatogr,1986,362:443
26 BurnettWN.AnalBiochem,1981,112:195~203
27 CarlssonJ,BatistavieraF,RydénL.CovalentChromatography.In:JansonJC,RydenL,ed,Protein
Purification:Principles,HighResolution Methods,and Applications.2nded.WileyVCH,Inc.,
1998
28 CheyenNE.JCrystGrowth.1992,122:176
29 CheyenNE.JApplCrystallogr,1997,30:198
30 ChiariM,RighettiPG.Electrophoresis,1995,16:1815~1829

335
31 ChongS.JBiolChem,1996,271:22159
32 ClarkeM HG,FreemanT.ClinSci,1968,35:403
33 ClarlssonJ,OlssonI,AxenR,etal.ActaChemScand,1976,B30:180
34 CopelandRA.Methodsforproteinanalysis:apracticalguidetolaboratory.NewYork:Chapmanand
Hall,1994
35 CutlerP.AffinityChromatography,In:DoonanSed,ProteinPurificationProtocols.Totowa,New
Jersey:HumanaPress,1996.157~168
36 DArcyA,ElmoreC,etal.JCrystGrowth,1996,168:175
37 DavitzMA,HereldD,etal.Science,1987,238:81
38 DeanPDG,JohnsonWSandMiddleFA.AffinityChromatography:APracticalApproach.Oxford:
OxfordUniversityPress,1985
39 DentonKA,TateSA.JChromatogr,1997,697:111~121
40 DriouichA,VianB,etal.PlantJ,1992,2:13
41 EjimaD,WatanabeM,etal.BiotechnolBioeng,1999,62:301
42 FaheyEM,ChaudhuriJB,BindingP.SepSciTechnol,2000,35:1743
43 FindlayJBCandEvansW H.Biologicalmembranes:apracticalapproach.Oxford:OxfordUniversity
Press,1987.1
44 FindlayJBCandGeisow M.ProteinSequencing:APracticalApproach.Oxford:OxfordUniversity
Press,1989
45 FrederikssonG,NilssonS,OlssonH,etal.ImmunolMethods,1987,97:65
46 GabriacB,wreckReichhartD,TeutschH,etal.ArchBiochemBiophys,1991,288:302
47 GengXD,ChangXQ.JChromatogr,1992,599:185~194
48 GerminoJandBastiaD.ProcNatlAcadSciUSA,1984,81:4692
49 GershenzonJ,McCaskillD,etal.AnalBiochem,1992,200:130
50 GershoniJM,PaladeGE.AnalBiochem,1982,124:396~405
51 GershoniJM,PaladeGE.AnalBiochem,1982,131:1~15
52 GiaffredsE,TonaniC,RighettiPG.JChromatogr,1993,630:313~327
53 GillilandGL,TungM,BlakesleeDM,etal.ActaCrystallogr,1994,50:408
54 GlenneyJ.AnalBiochem,1986,156:315~319
55 GoeddelDV,KleidDG,BolivarF,etal.ProcNatlAcadSciUSA,1979,76:106
56 GooderhamK.TransferTechniquesinProteinBlotting,in:Wailer,MethodsinMolecularBiology.
Vol1.Clifton:HumanaPress,1984.165~178
57 HamesBD,RickwoodD.GelElectrophoresisofProteins.2nded.Oxford:OxfordUniversityPress,
1990.9
58 HamesBD,HigginsSJ.NucleicAcidHybridization:APracticalApproach.OxfordUniversityPress,
1985
59 HarringtonMG,LeeKH,BailyJetal.Eletrophoresis,1994,15:187~194
60 HarrisELVandAngalS.ProteinPurificationMethods:APracticalApproach.Oxford:OxfordUni
versityPress,1989
61 HeigerD N.HighPerformanceCapillaryElectrophoresis:anIntroduction.US:HewlettPackard

336
Corp,1992
62 HeywoodSP.[PhDthesis].UniversityofLeeds,UK:London,1996
63 HigginsIJ,BestDJandJonesJ.Biotechnology:Principlesandapplications.Oxford:BlackwellScien
tificPublications,1985.143
64 HillsonDA,JBiochemBiophysMeth,1981,4:101
65 HoltonTAandCornishEC.PlantCell,1995,7:1071
66 HooperNMandTurnerAJ.Lipidmodificationofproteins:apracticalapproach.Oxford:OxfordUni
versityPress,1992.89
67 HoppTP,PrickettKS,PriceVL,etal.Bio/Technology,1988,6:1204
68 HopperNM,HryszkoJandTurnerAJ.BiochemJ,1990,267:509
69 HuguenyP,RomerS,KuntzM,etal.EurJBiochem,1992,209:399
70 HutchensTWandYipTT.JChromatogr,1992,604:133~141
71 HutchensTWandYipTT.JChromatogr,1991,536:1~15
72 HutchensTWandYipTT.JChromatogr,1990,500:531~542
73 HutchensTW,NelsonRW,etal.JChromatogr,1992,604:125~132
74 IonExchangeChromatography:PrinciplesandMethods.AmershamPharmaciaBiotechAB,1999
75 ItakuraK,HiroseT,CreaR,etal.Science,1997,198:1056
76 ItoK,IkemaseT,IshikawaT.BiotechBiochem,1992,56:906~912
77 JackGWandBeerDJ.ImmunoaffinityChromatography,In:DoonanSed,ProteinPurificationProto
cols.Totowa,NewJersey:HumanaPress,1996.187~196
78 JancarikJandKimSHJ.ApplCrystallogr,1991,24:409
79 JansonJCandRydenL.ProteinPurification:Principles,HighResolutionMethods,andApplications.
2nded.NewYork:JohnWiley&SonsInc,Publication,1998
80 JohnstoneA,ThorpeR.ImmunochemistryinPractice.Oxford:BlackwellScientific,1982
81 KarlssonE,EakerD,PonteriusG.BiochimBiophysActa,1972,57:235~248
82 KennedyRM.HydrophobicChromatography.MethEnzymol,1990,182:339~343
83 KenneyAandFowellS.MolBiol,1992,11:125~133
84 KhanRH,AppaRaoKBC,EshwariANS,etal.BiotechnolProg,1998,14:722
85 KohnJandWilchekM.TIBTECH,1987,5:220
86 KrugerNJandHammondJBW.In:WalkerJMed,MethodsinMolecularBiology.VolⅢ.1988.363
87 LapercheY,BulleF,AissaniT,etal.ProcNatlAcadSciUSA,1986,83:937
88 LaurellCB,ScandJ.ClinLabInvest,1972,124 (29Suppl):21~37
89 LevisonPR,JonesRM H,ToomeDV,etal.JChromatogr:A,1996,734:137
90 LevisonPR,BadgerSE,DennisJ,etal.JChromatogr:A,1998,816:107
91 MarshallT.AnalBiochem,1984,136:340
92 MeagherM M,BarlettRT,etal.Biotechnol,Bioeng,1994,43:969
93 NiederauerMQandGlatzCE.AdvBiochemEngBiotechnol,1992,47:159
94 OppongSYandHopperNM.BiochemJ,1993,292:597
95 OubridgeC,ItoN,TeoCH,etal.JMolBiol,1995,249:409
96 OvsejeviK,BrenaB,BatistaVieraF,etal.EnzymeMicrobTechnol,1995,17:151

337
97 PackerL.Methodsinenzymology.Vol214.SanDiego:AcademicPress,1993.274
98 PartingtonJCandBolwellGP.Phytochemistry,1996,42:1499
99 PetersonEAandTorresAR.MethEnzymol,1984,104:113~133
100 PetersonGL.MethodsEnzymol,1983,91:95
101 Pharmacia.AffinityChromatography:PrinciplesandMethods,1993
102 PorathJandOlinB.Biochemistry,1983,22:1621~1630
103 ProteinPurification.AmershamPharmaciaBiotechAB,1999
104 RabilloudT.Electrophoresis,1996,17:813~829
105 RabilloudT,VinconM,GarinJ.Electrophoresis,1995,16:1414
106 RighettiPG.ImmobilizedpHgradients:TheoryandMethodology.Elsevier,Amsterdam,1990.69~79
107 RighettiPG.ImmobilizedpHgradients:TheoryandMethodology,Elsevier,Amsterdam,1990.
132~139
108 RighettiPG,TudorG,EkK.JChromatogr,1981,220:115~194
109 RydénL,EakerD.EurJBiochem,1983,132:241
110 SammonsDW,AdamsLD,NishizawaEE.Electrophoresis,1981,2:135
111 SaravisCA.Electrophoresis,1984,5:54~55
112 SassenfeldH M.TrendsBiotechnol,1990,8:88
113 SchwerC.Electrophoresis,1995,16:2121~2126
114 ScopesRK.ProteinPurification:PrinciplesandPractice.Berlin:Springer,1994
115 SeetharamRandSharmaSK.Purificationandanalysisofrecombinantproteins.NewYork,USA:
MarcelDekkerInc.,1991.239~266
116 ShineJ,FettesI,LanNCY,etal.Nature,1980,285:456
117 SmithMC,FurmanTC,etal.JBiolChem,1988,263:7211
118 SoferGandHagelL.HandbookofProcessChromatography.optimization,scaleupandvalidation.
California,SanDiego:AcademicPress,1997
119 StadlerRandZenkM H.JBiolChem,1993,268:823
120 SzokoE.Electrophoresis,1997,18:74~81
121 ToverERandBaldoBA.Electrophoresis,1987,8:452~463
122 UsersManualofMultiphorⅡ ElectrophoresisSystem.PharmaciaLKBBiotechnologyAB.Uppsala,
Sweden,1992
123 WalkerJM.TheProteinProtocolsHandbook.Totowa,NewJersey:HumanaPress,1996
124 WardJM,WallaceLJ,CowanD,etal.AnalChimActa,1991,249:195
125 WernerM H,CloreGM,GronenbornAM,etal.FEBSLett,1994,345:125~130
126 WestIandGoldringO.LectinAffinityChromatography,In:DoonanSed,ProteinPurificationProto
cols.Totowa,NewJersey:HumanaPress,1996.177~185
127 WestermeierR.ElectrophoresisinPractice.Germany:VCHPress,1993.53~63
128 WestonRDandAvrameasS.BiochemBiophysResCommun,1971,45:1574
129 WilsonKandWalkerJM.Principlesandtechniquesofpracticalbiochemistry.5thed.NewYork:
CambridgeUniversityPress,1999
130 WongHH,ONeill,MiddlebergAP.Bioseparations,1996,6:185

338
131 WorrallDM.DyeLigandAffinityChromatography,In:DoonanSed,ProteinPurificationProtocols.
Totowa,NewJersey:HumanaPress,1996.169~176
132 WrayW,BonlikasT,WrayVP,etal.AnalBiochem,1981,118:197
133 YipTTandHutchensTW.ImmobilizedMetalIonAffinityChromatography,In:DoonanSed,Pro
teinPurificationProtocols.Totowa,NewJersey:HumanaPress,1996.197~210
134 YipTTandHutchensTW.ProteinExpressPurification,1991,2:355~362
135 ZimmerlinA.,WojtaszekP,BolwellGP.BiochemJ,1994,299:747
136 CR洛著.亲和色谱导论.刘毓秀译.北京:科学出版社,1983
137 蔡武成,袁厚积主编.生物物质常用化学分析法.北京:科学出版社,1982.93~99
138 陈盛编著.生物高分子化学.厦门:厦门大学出版社,2003.85~94
139 大连轻工业学院主编.生物化学.北京:轻工业出版社,1980.22,45
140 达世禄.色谱学导论.武汉:武汉大学出版社,1999.384
141 傅若农编.色谱分析概论.北京:化学工业出版社,2000.180
142 郭尧君,郭强,黄力力等.生物化学与生物物理进展,1994,21:356~359
143 郭尧君,郭强.生物化学与生物物理进展,1994,21:143~146
144 郭尧君,李辛辉,朱海珍等.生物化学与生物物理进展,1997,24:540~542
145 郭尧君.蛋白质电泳实验技术.北京:科学出版社,1999
146 何忠效.生物化学实验技术.北京:化学工业出版社,2004
147 金冬雁,金奇,侯云德编著.核酸和蛋白质的化学合成与序列分析.北京:科学出版社,1996
148 李建武等.生物化学实验原理和方法.北京:北京大学出版社,1994
149 师治贤,王俊德编著.生物大分子的液相色谱分离和制备.北京:科学出版社,1999
150 彭秀玲,袁汉英,谢毅等.基因工程实验技术.第2版.长沙:湖南科学技术出版社,1998.246~279
151 荣国斌,苏克曼.大学有机化学基础.上海:华东化工学院出版社,2000
152 陶慰孙等编著.蛋白质分子基础.北京:人民教育出版社,1981
153 王镜岩,朱圣庚,徐长法主编.生物化学.第3版.北京:高等教育出版社,2002
154 汪家政,范明主编.蛋白质技术手册.北京:科学出版社,2000
155 王立等编.色谱分析样品处理.北京:化学工业出版社,2000
156 (英)罗伯·里 德 (RobReed),大 卫·荷 尔 姆 斯 (DavidHolmes),琼 纳 坦·威 也 斯 (Jonathan
Weyers),阿兰·琼斯 (AllanJones)著.生 物 分 子 科 学 实 验 技 术.王 晓,李 德 红,孟 祥 春 译.长
沙:湖南科学技术出版社,2001.147~149
157 吴冠芸,潘华珍等主编.生物化学与分子生物学实验常用数据手册.北京:科学出版社,1999
158 伍越寰.有机化学.合肥:中国科技大学出版社,2002
159 夏其昌主编.蛋白质化学研究技术与进展.北京:科学出版社,1997
160 徐秀璋编著.蛋白质顺序分析技术.北京:科学出版社,1988
161 叶勤主编.现代生物技术原理及其应用.北京:中国轻工业出版社,2003
162 于世林编.高效液相色谱方法及应用.北京:化学工业出版社,2000
163 于自然,黄熙泰主编.现代生物化学.北京:化学工业出版社,2001.53
164 张承圭,王传怀等合编.生物化学仪器分析及技术.北京:高等教育出版社,1990
165 赵永芳主编.生物化学技术原理及应用.第3版.北京:科学出版社,2002
166 张龙翔.生化实验方法和技术.第2版.北京:高等教育出版社,1997