faculteit farmaceutische wetenschappen · 2010-06-07 · faculteit farmaceutische wetenschappen...
TRANSCRIPT

FACULTEIT FARMACEUTISCHE WETENSCHAPPEN
Vakgroep Geneesmiddelenleer
Laboratorium voor Algemene Biochemie en Fysische Farmacie
Academiejaar 2008 – 2009
EVALUATIE VAN CHROMATINE‐BINDENDE PEPTIDEN VOOR pDNA‐TRANSFECTIES IN DELENDE CELLEN
Tieme LEYSEELE Eerste Master in de Geneesmiddelenontwikkeling
Promotor
Prof. dr. apr. S. De Smedt
Commissarissen
Dr. lic. K. Tilleman
Dr. apr. C. Stove

De auteur en de promotor geven de toelating deze scriptie voor consultatie beschikbaar te
stellen en delen ervan te kopiëren voor persoonlijk gebruik. Elk ander gebruik valt onder de
beperkingen van het auteursrecht, in het bijzonder met betrekking tot de verplichting
uitdrukkelijk de bron te vermelden bij het aanhalen van de resultaten uit deze scriptie.
26/05/2009
Tieme Leyseele Prof. dr. apr. S. De Smet

DANKWOORD
In de eerste plaats wil ik graag prof. dr. apr. S. De Smedt bedanken om mij
de kans te geven rond dit boeiend onderwerp mijn scriptie te schrijven.
In het bijzonder wil ik ook Nathalie Symens bedanken voor de begeleiding
tijdens het onderzoek en het schrijven van deze scriptie. Zij was altijd en overal
bereikbaar voor vragen en hulp bij alle experimenten.
Ook mogen alle andere personeelsleden van het laboratorium niet vergeten
worden. Ze zorgden voor een aangename werksfeer en stonden steeds
klaar voor het oplossen van allerhande kleine en grote problemen.
Ten slotte wil ik mijn ouders en zus bedanken voor alle steun, niet alleen
tijdens deze onderzoekstage, maar gedurende mijn volledige studie.

LIJST MET GEBRUIKTE AFKORTINGEN
AZ aminozuur
bp baseparen
DAPI 4’,6‐diamidino‐2‐fenylindool
DLS dynamic light scattering
GFP groen fluorescent proteïne
H2A‐H2B histon2A‐histon2B
LAF laminar air flow
mRNA messenger ribonucleïnezuur
NPC nucleaire poriecomplexen
NLS nucleaire lokalisatie signalen
NES nucleaire export signalen
ORI origin of replication site
PBS phosphate buffered saline
pDNA plasmide desoxyribonucleïnezuur
PEI poly‐ethyleen‐imine
siRNA small interfering ribonucleïnezuur

INHOUDSOPGAVE
Dankwoord Inhoudsopgave Lijst met gebruikte afkortingen
1. INLEIDING ............................................................................................................................................ 1
1.1. ALGEMEEN ..................................................................................................................................... 1
1.2. BARRIÈRES BUITEN EN BINNEN DE CEL ......................................................................................... 2
1.3. VECTOREN VOOR PDNA ................................................................................................................ 4
1.3.1. Virale vectoren ...................................................................................................................... 5
1.3.2. Fysische methoden en niet‐virale vectoren ......................................................................... 5
2. DOELSTELLING ....................................................................................................................................... 9
3. MATERIALEN EN METHODEN ............................................................................................................. 11
3.1. AANMAAK VAN COMPLEXEN ...................................................................................................... 11
3.1.1. Algemeen ............................................................................................................................ 11
3.1.2. pDNA ................................................................................................................................... 11
3.1.3. Peptiden .............................................................................................................................. 12
3.1.4. Complexen .......................................................................................................................... 13
3.1.5. Kwaliteitscontrole ............................................................................................................... 14
3.2. CELKWEEK .................................................................................................................................... 17
3.2.1. Algemeen ............................................................................................................................ 17
3.2.2. Uitsplitsen van cellen .......................................................................................................... 18
3.2.3. Cellen tellen ........................................................................................................................ 18
3.2.4. Celsynchronisatie en –arrestatie ........................................................................................ 19
3.3. TRANSFECTIES ............................................................................................................................. 20
3.3.1. Soorten transfecties ............................................................................................................ 20
3.3.2. pDNA ................................................................................................................................... 20

3.3.3. Transfectie door vrije opname van pDNA/peptide‐complexen ........................................ 22
3.3.4. Transfectie met tertiaire complexen .................................................................................. 23
3.3.5. Transfectie d.m.v. elektroporatie ...................................................................................... 23
3.4. XENOPUS SPERMA CHROMATINE BINDING ................................................................................ 24
3.4.1. Miruslabeling ...................................................................................................................... 24
3.4.2. DAPI ..................................................................................................................................... 25
3.5. FLOW CYTOMETRIE ..................................................................................................................... 25
3.6. CONFOCALE LASER SCANNING MICROSCOPIE ............................................................................ 26
3.6.1. Tokaï Hit stage top incubator ............................................................................................. 27
4. RESULTATEN EN DISCUSSIE ................................................................................................................ 28
4.1. KARAKTERISATIE VAN PDNA/PEPTIDE COMPLEXEN ................................................................... 28
4.1.1. Aanmaak complexen .......................................................................................................... 28
4.1.2. Maat van complexatie a.d.h.v. gelelektroforese ............................................................... 28
4.1.3. Deeltjesgrootte a.d.h.v. DLS ............................................................................................... 29
4.1.4. ζ‐potentiaal ......................................................................................................................... 31
4.2. XENOPUS CHROMATINE BINDING ............................................................................................... 34
4.3. NAGAAN SYNCHRONISATIE VAN HELA‐CELLEN ........................................................................... 36
4.4. OPNAME VAN COMPLEXEN DOOR HELA‐CELLEN ....................................................................... 36
4.4.1. Opname van de complexen ................................................................................................ 36
4.4.2. Controle van complexatie in verschillende media ............................................................ 37
4.5. TRANSFECTIES MET TERTIAIRE PDNA‐COMPLEXEN .................................................................... 38
4.6. GEBRUIK VAN ELEKTROPORATIE VOOR TRANSFECTIES MET PDNA/PEPTIDE‐COMPLEXEN ....... 41
4.6.1. Elektroporatie met gWIZ‐GFP/peptide‐complexen ........................................................... 41
4.6.2. Elektroporatie met EGFP‐N1/peptide‐complexen............................................................. 45
5. BESLUIT ............................................................................................................................................... 47
6. BIBLIOGRAFIE ...................................................................................................................................... 48

1. INLEIDING
1.1. ALGEMEEN
De laatste decennia werd het menselijk genoom volledig gesequeneerd. Tal van
genen die betrokken zijn in cellulaire processen en pathogenese werden geïdentificeerd
en gezien als eventuele nieuwe targets voor de genezing van genetische ziektes. Het
grote verschil tussen klassieke geneesmiddelen en gentherapie is dat gentherapie de
oorzaak van de ziekte aanpakt in plaats van de symptomen. Door het on‐target principe,
zou gentherapie een veel specifiekere en meer doeltreffende therapie zijn dan de
klassieke methodes.
Genetische ziektes uiten zich enerzijds door de aanmaak van bepaalde
(messenger) RNA (mRNA)‐moleculen die worden getransleerd tot pathologische
proteïnen die aan de basis liggen van de symptomen van een genetische aandoening.
Anderzijds kunnen essentiële proteïnen volledig ontbreken. De productie van een fout
proteïne kan worden tegengegaan met behulp van oligonucleotiden of small interfering
RNA (siRNA). Deze uit 20 tot 25 tal nucleotiden bestaande RNA‐moleculen beïnvloeden
zowel de mRNA degradatie en translatie, als het chromatine zelf. Hierdoor hebben ze
een effect op de translatiegraad (Valencia‐Sanchez et al., 2006). Een nieuw proteïne
daarentegen kan tot expressie gebracht worden nadat het overeenkomstige
therapeutisch plasmide DNA (pDNA) of mRNA aan de doelcel wordt toegediend.
Gentherapie kan zowel in vivo als ex vivo worden toegepast. Bij ex vivo therapie
worden cellen van de patiënt gecollecteerd om deze in vitro te transfecteren. Het
voordeel hier is dat er een meer efficiënte gentransfer kan plaatsvinden en dat er een
groter aantal getransfecteerde cellen kan aangemaakt worden. Een nadeel is dat het
een heel patiëntspecifieke methode is als gevolg van celimmunogeniciteit. Bij in vivo
therapie worden de therapeutische nucleïnezuren met behulp van vectoren direct bij de
1

patiënt toegediend. Hierdoor is het niet patiëntspecifiek en zal het dus een minder
kostelijke methode zijn. Het is hier wel moeilijker om de specifieke doelcellen te
bereiken (A. Mountain et al., 2000).
1.2. BARRIÈRES BUITEN EN BINNEN DE CEL
Om de celinhoud te beschermen tegen negatieve externe factoren bestaat de cel
uit een aantal verschillende barrières die uiteindelijk zullen moeten overwonnen
worden om siRNA, mRNA en oligonucleotiden in het cytoplasma of pDNA in de celkern
te krijgen. De cel bevat gereguleerde transportmechanismen zodat lichaamseigen
moleculen uitgewisseld kunnen worden met de externe omgeving. Deze barrières en
transportmechanismen zijn echter niet onfeilbaar: tal van virussen en bacteriën hebben
zich zodanig aangepast dat zij gebruik kunnen maken van dezelfde
transportmechanismen om cellen te infiltreren. Heel wat onderzoek heeft
therapeutische strategieën blootgelegd om intracellulaire targets te bereiken.
Op de eerste plaats kunnen de externe pH, nucleasen, scavenger systemen en
het immuunsysteem de vrij toegediende DNA‐ of RNA‐moleculen afbreken. Het is dus
belangrijk dat deze beschermd worden tegen dit soort degradatie door het vormen van
eventuele complexen of vesikels met andere moleculen (M. Belting et al., 2005). Ook
wanneer het pDNA terecht komt in het bloed, bijvoorbeeld na intraveneuze injectie, zal
er een snelle afbraak optreden. Doordat bij in vivo testen een grotere afbraak
voorkwam dan bij in vitro, concludeerde men dat een groot deel van de afbraak het
gevolg moet zijn van enzymen bij passage door de lever (Hashida et al., 1996). Ook de
carrier‐systemen van pDNA en siRNA kunnen verstoord worden bij transport door het
bloed. Zo kunnen bepaalde negatief geladen serumproteïnen, zoals albumine, de
oppervlaktelading van de carrier‐complexen wijzigen waardoor aggregatie zal optreden.
Daarnaast kan door een gewone binding van geladen serumproteïnen aan de carrier‐
complexen, de carrier uitgeschakeld worden (Buyens K et al, 2008).
2

Het plasmamembraan is een dubbele fosfolipidelaag die zich aan het oppervlak van de
cel bevindt. De graad van diffusie door dit membraan hangt af van de grootte, de
hydrofobiciteit en de lading van de moleculen. DNA‐ en RNA‐moleculen zijn bij een
fysiologische pH negatief geladen. Dit bemoeilijkt de passage doorheen het
celmembraan doordat het celoppervlak ook negatief geladen is en dit
afstotingsverschijnselen met zich meebrengt. DNA‐ en RNA‐moleculen, die sterk
geladen zijn, worden matig opgenomen via endocytotische pathways (M. Belting et al.,
2005). Met behulp van carriers voor de DNA‐ of RNA‐moleculen wordt de
endocytotische opname verbeterd.
Endosomen die de opgenomen DNA‐ of RNA‐moleculen bevatten, gaan na een
zekere tijd versmelten met lysosomen. Die laatste zullen de enzymatische afbraak van
de DNA‐ of RNA‐moleculen initiëren. Door de lage pH op dat moment in de endosomen
zal het DNA denatureren en zullen eventuele complexen gevormd met het DNA
uiteenvallen. Lysosomen zijn aanwezig in de cel om vreemde moleculen, die toch door
de cel worden opgenomen, af te breken. Het is dus van belang dat de DNA‐ of RNA‐
moleculen kunnen ontsnappen uit de endosomen vooraleer ze versmelten met
lysosomen. Eenmaal uit de endosomen ontsnapt, moeten de DNA‐ of RNA‐moleculen
stabiel genoeg zijn om hun doelwit te bereiken doorheen het cytoplasma dat ook
nucleasen bevat. Deze stabiliteit kan eventueel verbeterd worden door de moleculen
vooraf met een carrier te binden. pDNA heeft nog een extra barrière te overwinnen
namelijk het kernmembraan. Dit blijkt de grootste barrière te zijn bij gentherapie met
pDNA.
Het kernmembraan bestaat uit een dubbel fosfolipidemembraan en nucleaire
poriecomplexen (NPC’s). Een NPC is een doorgang voor het wederzijds transport van
RNA’s en proteïnen doorheen het kernmembraan. De partikels die passief doorheen
een NPC kunnen migreren zijn maximum 9 nm groot. Via actief transport is dit 30 tot 40
nm (Van der Aa et al., 2006). Actief transport wordt geregeld door nucleaire lokalisatie
3

Fig 1.1: Nucleaire poriecomplexen (Cook et al., 2007)
signalen (NLS’s) en nucleaire
export signalen (NES’s). Wanneer
het importin α/β dimeer een NLS‐
bevattend proteïne herkent wordt
dit proteïne aangeboden aan een
NPC voor opname in de kern (fig
1.1). Tal van onderzoeksgroepen
evalueren het gebruik van NLS’s om
de nucleaire import van pDNA te
verhogen. Tot op heden blijkt deze strategie weinig effectief te zijn (Mesika A. et al.,
2005). Er werd echter vastgesteld dat celdeling een positieve invloed heeft op de
transfectie‐efficiëntie met pDNA. Tijdens de celdeling wordt het kernmembraan
ontmanteld en valt de barrière tijdelijk weg (Lénart P. et al., 2006). Er wordt
verondersteld dat pDNA dat zich in de buurt van het chromatine bevindt per toeval kan
worden ingesloten in de kern bij de heropbouw van het kernmembraan na mitose
(Wilke M. et al., 1996). Hier wordt verder op ingegaan onder 1.3.2.
1.3. VECTOREN VOOR PDNA
Er zijn verschillende methoden om het pDNA tot in de celkern te krijgen. Omwille
van de eerder genoemde barrières dient het pDNA te worden toegediend met behulp
van carriers, ook wel vectoren genoemd. Virale vectoren zijn de meest gebruikte. Het
betreft vooral de virusafgeleiden van retrovirussen, adenovirussen en adeno‐
geassocieerde virussen. Niet‐virale vectoren zijn voornamelijk kationische lipiden en
kationische polymeren. Vaak wordt ook beroep gedaan op een fysische methode zoals
elektroporatie en gene‐gun.
4

1.3.1. Virale vectoren
Adenovirussen kunnen zowel prolifererende als rustende cellen transfecteren.
Ze integreren het transgen niet in het chromosoom van de gastcel, waardoor de
transfectie van voorbijgaande aard is. Uit veiligheidsmaatregelen werden ze
aangepast zodat ze niet meer kunnen repliceren. Er wordt dan gesproken van
adeno‐geassocieerde virussen. De adenovirusvectoren worden gebruikt voor
celdoding, immunotherapeutische doeleinden en acute ziektes. Ze zijn niet
bruikbaar als therapie voor chronische aandoeningen (Chen et al., 1994). Een groot
nadeel aan adenovirussen is dat zij sterk immunogene en inflammatoire reacties
uitlokken. Dit kan een probleem vormen bij het binden aan de doelwitcellen en de
gentransfectie na herhaalde toediening. Sommige adenovirusvectoren leveren
naast het transgen ook nog andere virale gensequenties af die, als ze tot expressie
komen, zorgen voor een cytotoxische T‐cel respons tegen de getransfecteerde
cellen. Een combinatie met een immunosuppresief middel zou deze problemen
eventueel kunnen verhelpen (A. Mountain et al., 2000).
Retrovirussen daarentegen integreren het transgen wel in het chromosoom
van de gastcel. Dit leidt tot een langere expressie in de getransfecteerde cellen. De
retrovirusvectoren kunnen alleen maar binnen in de kern tijdens de mitose. Er kan
dus alleen transfectie plaatsvinden bij prolifererende cellen, wat in de meeste
gevallen een beperking is. Ze zijn ook veel minder immunogeen dan
adenovirusvectoren, omdat ze geen virale gensequenties overbrengen naar de cel
(Miller et al., 1990).
1.3.2. Fysische methoden en niet‐virale vectoren
Na lokale injectie van vrij pDNA in vivo, bijvoorbeeld in de spier of de
levervenen, kan er transfectie voorkomen. Bovendien is deze transfectie vaak
groter dan bij andere niet‐virale vectoren (Wolff et al., 1992). Een groot voordeel
van deze techniek is dat vrij pDNA gemakkelijk en goedkoop kan aangemaakt
worden in bacteriën. (zie 3.3.2)
5

Enkele fysische methoden zoals elektroporatie: een techniek waarbij door het
aanleggen van een elektrisch veld poriën in het celmembraan ontstaan die na
enige tijd spontaan herstellen, kunnen grotere partikels zoals pDNA binnen in de
cel loodsen (C. Favard et al., 2007). Een andere fysische techniek is gene‐gun. In
deze techniek zal men, onder hoge helium druk, gouden micropartikels die
gesuspendeerd zijn in een vloeistof en beladen zijn met het transgen in subcutane
weefsels zoals spieren en tumoren katapulteren (J. Dileo et al., 2003).
Kationische lipiden of polymeren zijn de meest gebruikte niet‐virale vectoren.
DNA is een negatief geladen molecule bij fysiologische pH en zal dus spontaan
complexeren met kationische moleculen als gevolg van elektrostatische interacties.
Als deze kationische molecule een lipide is, spreekt men van lipoplexen. Er kan ook
een vorming van liposomen optreden, hier zullen de kationische lipiden een
membraan vormen rondom het pDNA. Dat membraan zal dan samensmelten met
het celmembraan en zo het pDNA in de cel loodsen (Mönkkönen et al, 1998). De
gevormde pDNA‐complexen beschermen het pDNA tegen enzymatische afbraak en
verbeteren de opname via endocytose. De niet‐virale vectoren moeten in staat zijn
het pDNA te helpen ontsnappen uit de endosomen. Een gekend polymeer dat met
behulp van het proton sponge effect het pDNA in het cytosol vrijstelt, is poly‐
ethyleen‐imine (PEI) (Bieber T. et al., 2002). Het pDNA moet ook tijdens het
transport doorheen het cytosol zo lang mogelijk beschermd worden tegen
degradatie. Tot op vandaag wordt echter aangenomen dat de niet‐virale vectoren
het pDNA in het cytosol moeten vrijstellen. Dit omdat pDNA‐complexen veel te
groot zijn om door de poriën van een (intact) nucleair membraan te diffunderen.
Het transport blijkt heel inefficiënt aangezien de transfectie van rustende cellen
heel laag is (Fasbender A. et al, 1997). Het blijft dus een hele uitdaging voor de
niet‐virale gentherapie wereld om niet‐delende cellen te transfecteren.
6

Delende cellen daarentegen vertonen een veel hogere transfectie‐efficiëntie.
Dit werd bewezen aan de hand van een experiment waar men zowel gearresteerde
cellen als asynchroon delende cellen probeerde te transfecteren met lipoplexen.
Bij de gearresteerde cellen was er zo goed als geen transfectie, wat wijst op de
nood aan mitose om de complexen binnenin de celkern te krijgen (Mortimer et al.,
1999).
In een andere experiment werd gebruik gemaakt van liposomen bestaande uit
1,2‐dioleoyl‐3‐trimethylammonium‐propaan, dioleoyl‐fosfatidylethanolamine en
pDNA. HeLa‐cellen werden hiermee getransfecteerd en gearresteerd in de G1‐fase.
Het percentage cellen dat het transgen tot expressie bracht, steeg immens als de
cellen doorheen de M‐fase gingen. Wat opnieuw wijst op het belang van mitose
om een hogere transfectie‐efficiëntie te bekomen (Tseng et al., 1999).
De transfectie met behulp van fysische methoden of niet‐virale vectoren blijft
echter veel minder efficiënt en doelgericht in vergelijking met die met virale
vectoren. Daarom zou het interessant zijn om niet‐virale vectoren te ontwikkelen
met bepaalde ‘slimme’ eigenschappen. De kennis hiervoor kan worden opgedaan
door het bestuderen van celeigen pathways en transportmechanismen. Een tot nu
toe weinig effectief voorbeeld hiervan is het gebruik van NLS’s om de opname van
pDNA in de intacte kernen te vergroten. Anderzijds kan de studie van het
mechanisme dat virussen gebruiken om cellen en kernen binnen te dringen voor
ideeën zorgen.
Een voorbeeld hiervan is het Kaposi sarcoma herpesvirus. Dit virus maakt
gebruik van het eiwit LANA (latency‐associated nucleair antigen), voor de
vasthechting van het virale genoom aan mitotische chromosomen (Roussel et al.,
2008). Een ander voorbeeld van een virus die mitose nodig geeft om zijn genoom
in de celkern te krijgen is het Moloney murine leukemia virus. Dit werd bewezen
7

door een experiment waarin gesynchroniseerde rat‐ en muiscellen werden
geïnfecteerd met viraal DNA. De integratie van het viraal DNA en de productie van
de virale proteïnen kwam pas op gang toen de cellen door de M‐fase van de
celcyclus gingen (Roe et al., 1993).
Het is intressant de mechanismen van dit soort virussen te achterhalen en na
te bootsen om op eenzelfde manier de cellen te transfecteren met het
therapeutisch pDNA.
8

2. DOELSTELLING
Gentherapie met pDNA wordt voor een groot deel geremd door verschillende barrières en
afbraakmechanismen voor pDNA in de cel. Zoals eerder besproken is het kernmembraan de
grootste barrière die overwonnen moet worden. Uit de literatuurstudie kan besloten worden
dat pDNA mitose nodig heeft voor een effectieve transfectie van de cel. Bij celdeling valt het
kernmembraan tijdelijk weg. Er wordt verondersteld dat pDNA dat zich op dat moment in de
buurt van het chromatine bevindt at random wordt ingesloten in de dochterkernen (Mortimer
et al., 1999).
Aangezien het pDNA in afwachting tot mitose in het cytoplasma verblijft wordt het
blootgesteld aan allerlei nucleasen en scavenger‐systemen die de afbraak van pDNA zullen
initiëren. Men zal dus op zoek moeten gaan naar moleculen die complexen of vesikels vormen
met pDNA die het beschermen tegen dit soort degradatie (M. Belting et al., 2005). Ideaal zou
zijn dat de pDNA‐partikels volledig worden ingesloten in de dochterkernen bij celdeling en dat
ze pas daarna het pDNA gaan vrijstellen. Er moet onderzocht worden of het gebruik van
chromatine‐bindende peptiden in de complexen, met als doel een betere targeting naar het
chromatine, binding op het chromatine en dus een efficiëntere insluiting in de dochterkernen,
een positieve invloed heeft op de transfectie‐efficiëntie.
In de experimenten zal gebruik gemaakt worden van twee soorten chromatine‐bindende
peptiden. Een histonbindend peptide dat bepaalde motieven draagt die binden op histon2A‐
histon2B (H2A‐H2B) dimeren. En een DNA‐bindend peptide dat bestaat uit 2 AT‐Hooks. Deze
motieven binden op de minorgroeve van AT‐rijke sequenties in het DNA. Als pDNA wordt
gebruik gemaakt van gWIZ‐GFP en pEGFP‐N1, twee pDNA’s die coderen voor het groen
fluorescent proteïne (GFP).
In dit onderzoeksproject wordt nagegaan of deze chromatine‐bindende peptiden in staat
zijn om een grotere transfectie‐efficiëntie te bekomen bij HeLa‐cellen in vergelijking met twee
controlepeptiden, nl. Oligo‐D‐lysine en Oligo‐L‐lysine. Er zal gebruik gemaakt worden van
elektroporatie om de pDNA‐complexen in het cytosol te loodsen. Aangezien er vanuit gegaan
9

wordt dat het pDNA door mitose in de kern terecht komt zullen de experimenten zowel op
onbehandelde, als gearresteerde en gesynchroniseerde HeLa‐cellen uitgevoerd worden. Dit om
de transfectie‐efficiëntie in delende en niet‐delende cellen te kunnen vergelijken. Deze
transfectie‐efficiëntie zal bepaald worden door het meten van het percentage GFP‐positieve
cellen met de flow cytometer.
10

3. MATERIALEN EN METHODEN
3.1. AANMAAK VAN COMPLEXEN
3.1.1. Algemeen
De binding tussen de kationische peptiden en het pDNA is een ionische interactie.
pDNA is bij een fysiologische pH negatief geladen door de aanwezigheid van de
fosfaatgroepen, terwijl de peptiden bij deze pH positief geladen moleculen zijn. Er wordt
verondersteld dat door gewoon mengen van pDNA met de kationische peptiden er
spontaan complexen zullen gevormd worden door elektrostatische interacties. Door een
binding met een kationisch peptide wordt de negatieve lading van het pDNA teniet
gedaan, waardoor passage doorheen het celmembraan gemakkelijker gemaakt wordt.
Terzelfder tijd zal dit complex een extra stabiliteit geven aan het pDNA zowel
extracellulair als in het cytosol door het te beschermen tegen degradatie gekatalyseerd
door allerlei enzymen.
3.1.2. pDNA
Er wordt gebruik gemaakt van de plasmiden gWIZ‐GFP (5757 bp) en pEGFP‐N1
(4700 bp) (fig. 3.1). Deze coderen beide voor het groen fluorescent proteïne (GFP). pDNA
bestaat uit een circulaire DNA‐streng die oneindig kan gerepliceerd worden in bacteriën.
Hiervoor bezitten pDNA’s een origin of replication site (ORI). pDNA bevat daarnaast ook
een gensequentie die codeert voor een bepaalde antibioticumresistentie. In dit geval
dragen de gebruikte gWIZ‐GFP en pEGFP‐N1 plasmiden een kanamycine‐resistentiegen.
Dit antibioticum wordt toegevoegd aan het milieu waarin de bacteriën worden
opgekweekt, zodat alleen de bacteriën die goed getransformeerd zijn met het pDNA in
leven blijven en verder opgekweekt worden. Beide plasmiden bevatten ook een virale
promotor noodzakelijk voor de expressie van het GFP‐gen. Deze cytomegalovirus (CMV)‐
promotor is actief in een groot aantal celtypen. De expressie van dit GFP‐gen kan
11

gedetecteerd worden met onder andere flow cytometrie, door de hoeveelheid groen
fluorescent proteïne in de celsuspensie te meten (GFP). Dit soort pDNA wordt gebruikt
omdat hiermee snel en gemakkelijk kan gecontroleerd worden of de toegepaste
methode het pDNA tot in de kern kan loodsen.
Fig 3.1: Schematische voorstelling van gWIZ‐GFP en pEGFP‐N1 plasmiden.
3.1.3. Peptiden
Er wordt gebruik gemaakt van twee chromatinebindende peptiden: een DNA‐
bindend en een histonbindend. De geconserveerde regio’s worden onderlijnd.
Het histonbindend peptide, MYFMWLRSGMIKK, bevat een bepaald motief dat bindt op
Histon2A‐Histon2B (H2A‐H2B) dimeren. Dit motief wordt teruggevonden in IL‐33
(interleukine‐33) en LANA (Kaposi sarcoma herpesvirus latency‐associated nuclear
antigen). Er werd aangetoond dat de geconserveerde regio (MxLRSG) over sterke
chromatine‐targeting eigenschappen beschikt (Roussel et al, 2008). Het gebruikte
peptide werd besteld onder de vorm (KKKKKKMYFMWLRSGMIKKKKKK). Het
histonbindende motief werd aan beide zijden verlengd met lysines om bij fysiologische
pH een positief geladen peptide te bekomen zodat deze elektrostatische interacties kan
aangaan met het negatief geladen pDNA. Het DNA‐bindend peptide werd ook uit de
literatuur gehaald. Het motief RPRGRPR is de meest geconserveerde sequentie in AT‐
Hooks (Aravind et al., 1998). Deze binden op de minor‐groeve van AT‐rijke regio’s in
12

DNA. Het gebruikte peptide bestaat uit twee van deze sterk geconserveerde motieven
omringd door lysines (KKKKRPRGRPRKKKKRPRGRPRKKKK) om het peptide een positieve
lading te bezorgen bij een fysiologische pH.
Verder wordt ook nog gebruik gemaakt van twee niet‐chromatinebindende
controlepeptides, nl. Oligo‐D‐lysine en Oligo‐L‐lysine, die respectievelijk 10 en 20
aminozuren lang zijn.
3.1.4. Complexen
Er wordt gewerkt bij een fysiologische pH, wat zorgt voor een negatieve lading bij
pDNA en een positieve lading bij de peptiden. Door het eenvoudig mengen van deze
twee zal er spontaan complexatie optreden op basis van elektrostatische interacties. De
complexen worden zowel in 20 mM Hepesbuffer bij een pH van 7,4 , als in water
gemaakt. pDNA‐oplossingen met een concentratie van 1 µg/µl bestaan altijd uit 3 nmol
negatieve ladingen per µl. Het histonbindend eiwit bestaat per molecule uit 12 positieve
ladingen, de AT‐hook uit 19 positieve ladingen, het oligo‐D‐lysine uit 10 positieve
ladingen en het oligo‐L‐lysine uit 20 positieve ladingen. Aan de hand van de moleculaire
gewichten van de bestelde peptiden en het aantal positieve ladingen die zij per molecule
dragen worden bepaalde concentraties peptide‐oplossing berekend en aangemaakt om
gemakkelijk pDNA/peptide‐complexen te kunnen maken met de juiste +/‐ ladingsratio’s.
Door een bepaalde hoeveelheid pDNA te mengen met verschillende concentraties van
het positief geladen peptide, worden verschillende +/‐ ladingsratio’s verkregen. Dit is de
verhouding van het aantal positieve ladingen van de peptiden tegenover het aantal
negatieve ladingen van het pDNA. Bijvoorbeeld: om een +/‐ ladingsratio van 40 te
bekomen wordt 10 µl van een 20 ng/µl pDNA‐oplossing bij 10 µL 1/50
peptideverdunning gebracht (Zie tabel 3.1 voor de andere ladingsratio’s). Deze oplossing
wordt gevortext en 30 minuten geïncubeerd bij kamertemperatuur.
13

Ratio 5.0 7.5 10.0 12.5 15.0 17.5 20.0 22.5 25.0 27.5 30.0 32.5 35.0 37.5 40.0
peptide (1/50) 5,625 6.35 6,875 7.50 8,725 8.75 9,375 10.00 µl
peptide (1/100) 2.50 3.75 5.00 6.25 7.50 8.75 10.00 µl
Hepes of water 7.50 6.25 5.00 3.75 2.50 1.25 0.00 4,375 3.75 3,125 2.50 1,875 1.25 0,625 0.00 µl
3.1.5. Kwaliteitscontrole
3.1.5.1. Bepaling van deeltjesgrootte met Dynamic Light Scattering (DLS)
Deze techniek wordt gehanteerd ter bepaling van de grootte van partikels, in dit
geval de grootte van de pDNA/peptide‐complexen, met behulp van de Malvern
Autosizer 4700. De techniek is gebaseerd op het meten van de Brownse beweging,
die voortkomt uit botsingen met de naburige solventmoleculen die hen omringen.
Hoe groter het partikel, hoe trager de Brownse beweging zal zijn. De partikels
worden bestraald met een laser en verstrooien dit licht. Een detector die in een hoek
van 90° tegenover de lichtbron staat zal hierdoor een bepaald
lichtverstrooiingspatroon waarnemen. Nu de laserstraal invalt op een dispersie van
bewegende deeltjes, zal de detector een fluctuatie van dit lichtverstrooiingspatroon
waarnemen in functie van de tijd. De snelheid waarmee dit patroon verandert is dan
een maat voor de snelheid waarmee de deeltjes bewegen en dus ook een maat voor
de grootte van de deeltjes. Zo kan de grootte van de verschillende complexen
bepaald worden. De metingen zijn ook gebaseerd op de viscositeit van de oplossing.
Het is dus belangrijk dat de temperatuur constant gehouden wordt, want bij hogere
temperaturen zal de viscositeit dalen en zullen de deeltjes sneller bewegen, wat een
verkeerd beeld zou kunnen geven over de grootte van de partikels.
De grootte van de partikels wordt berekend met volgende formule:
Tabel 3.1: Aanmaak peptideverdunningen voor verschillende ladingsratio’s
d(H)= kT/3πŋD
d(H):Hydrodynamische diameter D: Diffusie coëfficiënt
k: Bolttzmann’s constante T: Absolute temperatuur
ŋ: Viscositeit
14

Voor de experimenten in deze master thesis worden de complexen stofvrij
aangemaakt zoals beschreven in onderstaande tabel 3.2. De aangemaakte 150 µl
complexen worden onmiddellijk gevortext en 30 minuten geïncubeerd. Nadien
wordt 850 µl water of 20mM Hepes‐buffer toegevoegd afhankelijk van het medium
waarin ook het pDNA en de peptiden werden verdund. De stalen zijn nu klaar voor
meting m.b.v. de Malvern Autosizer 4700. Alle complexen worden zowel in water als
in 20mM Hepes‐buffer gemeten.
+/‐ ladingsratio 5 10 15 20 30 40
Peptide (1/100) (µl) 18,75 37,50 56,25 75 / /
Peptide (1/50) (µl) 56,25 75
Water of 20mM Hepes (µl) 56,25 37,50 18,75 0 18,75 0
gWIZ‐GFP (1µg/µl) (µl) 75 75 75 75 75 75
Tabel 3.2 Complexen aangemaakt voor grootte‐ en Zeta‐potentiaal‐meting.
3.1.5.2. Bepaling van de oppervlaktelading door ζ‐potentiaalmeting
De gevormde pDNA/peptide‐complexen zijn positief geladen. Hierdoor trekken
zij negatief geladen ionen aan, die elektrostatisch worden gebonden aan het
oppervlak van het geladen deeltje. Deze eerste laag wordt de Sternlaag genoemd.
Daarrond zal er zich een laag ionen bevinden die kunnen uitgewisseld worden met
andere ionen uit de elektroneutrale oplossing. De rand van deze laag wordt de
afschuiflaag genoemd en het is deze lading die zal gemeten worden. De potentiaal
van deze laag is de ζ‐potentiaal. Er wordt gebruik gemaakt van ζ‐cellen (cuvetten met
twee elektroden) om een stroom te kunnen aanleggen door de oplossing. De
complexen zullen dan bewegen naar de pool die de tegenovergestelde lading draagt
als hun eigen lading. De snelheid van beweging van de complexen is dan een maat
voor de lading. Deze snelheid wordt bepaald op dezelfde manier als de
groottebepaling van de complexen met de Malvern Autosizer 4700.
15

3.1.5.3. Agarosegelelektroforese
Elektroforese is een techniek waarbij geladen deeltjes gescheiden worden op
basis van hun mobiliteit doorheen een medium waarover een elektrisch veld werd
aangelegd, in dit geval een agarosegel. De gel vormt een macromoleculair netwerk
waarin verschillende moleculen zich op basis van grootte en lading sneller of trager
kunnen bewegen. Een gel is vrij stabiel en zal in normale opstandigheden niet van
vorm of viscositeit veranderen als er een elektrisch veld wordt aangelegd. Hierdoor
blijft de scheiding van de moleculen ongestoord. Er wordt hier opnieuw gewerkt bij
een fysiologische pH waardoor de pDNA‐moleculen negatief geladen zullen zijn, en
de peptiden positief geladen. De mate van migratie naar de positieve pool zal dus
afhangen van de +/‐ ladingsratio’s van de complexen, maar ook van de grootte van
de complexen omdat deze moeten migreren doorheen de poriën van de agarosegel.
Op deze manier worden fragmenten van verschillende groottes en lading
gescheiden.
Voor de experimenten in deze master thesis werd 10 µl van de aangemaakte
complex‐oplossingen volgens een bepaald patroon op de gel geladen.
De 1% agarosegel wordt aangemaakt door 1 g agarose op te lossen in 100 ml 1x TBE
buffer en dit mengsel nadien op te warmen tot kooktemperatuur. Een 10x TBE buffer
wordt aangemaakt door 107,8 g Tris‐base, 55,8 g boorzuur en 7,4 g EDTA op te
lossen in 1 l gedestilleerd water. Na afkoelen tot <65°C wordt de oplossing in de
gelhouder gegoten die voorzien is van een kam die de slots zullen vormen. Eenmaal
de gel gestold is, wordt de kam verwijderd en wordt de gel in een elektroforesebak
gebracht. Nadien wordt de gel overgoten met 1x TBE buffer totdat deze volledig
ondergedompeld is. Vooraf wordt een ladingsbuffer (50% sucrose) met een kleurstof
(broomfenolblauw) toegevoegd aan de stalen. De sucrose dient tot het niet vooraf
uit de slots diffunderen van de stalen. Het broomfenolblauw dient enkel om te
visualiseren hoever het staal al gemigreerd is. Na afsluiten van de elektroforesebak,
16

legt men een elektrisch veld aan tussen de elektroden van 100V gedurende 30
minuten. Na de elektroforese volgt de kleuring van de gel. Hierbij wordt de gel in een
ethidiumbromide (EtBr)‐bad gelegd gedurende 30 minuten. Nadien wordt de
overmaat EtBr weggewassen door de gel te spoelen in een gedestilleerd waterbad.
EtBr is een planaire molecule die gaat intercaleren tussen de basen van het DNA en
wordt daardoor veel fluorescenter. Het DNA wordt nu gevisualiseerd door de gel te
bekijken op een UV‐lichtbak. Als controle wordt meestal ook een basenparenladder
mee op de gel geladen.
3.2. CELKWEEK
3.2.1. Algemeen
Cellen worden gebruikt om de transfectie‐efficiëntie met de complexen na te gaan.
Deze cellen worden in vitro in leven gehouden door ze in een vloeibaar cultuurmedium
te laten groeien en vermenigvuldigen. Om overgroei, met als gevolg celmutaties en
celdood, van de cellen te voorkomen moeten deze tijdig gesplitst en in een vers
cultuurmedium gebracht worden. In deze master thesis wordt gebruik gemaakt van
HeLa‐cellen (humane cervixcarcinoomcellen), dit is een cellijn die in theorie oneindig
blijft delen. De cellen groeien op de bodem van polystyreen cultuurflessen bij een
temperatuur van 37°C, in een milieu van 5% CO2 en luchtvochtigheid van 95%. Het
gebruikte cultuurmedium bestaat uit Dulbecco’s Modified Eagle Medium F12 (DMEM‐
F12) (Gibco Life Technologies), 10% Fetal Bovine Serum (FBS) (Gibco Life Technologies),
2% Penicilline/Streptomycine oplossing (100 IU/ml penicilline en 100 µg/ml
streptomycine) en 1% 2 mM L‐glutamineoplossing. Het is van groot belang dat alles wat
in aanraking komt met de cellen steriel wordt bereid en uitgevoerd in een Laminaire Air
Flow (LAF)‐kast.
17

3.2.2. Uitsplitsen van cellen
Confluentie is een maat voor de hoeveelheid van begroeiing van een cultuurfles.
Wanneer een cultuurfles voldoende confluent is, moet deze worden uitgesplitst over
een aantal nieuwe cultuurflessen waar de cellen opnieuw vrij kunnen delen.
Eerst wordt het cultuurmedium van een confluente fles verwijderd. De cellen zitten
vastgehecht aan de bodem dus deze zullen bijgevolg ook in de fles blijven. Nu worden
deze drie maal gewassen met steriele Phosphate Buffered Saline ‐Ca/‐Mg (PBS) (Gibco
Life Technologies) om resten van dode cellen te verwijderen. De cellen van een 75 cm2
cultuurfles worden van de bodem losgemaakt door toevoegen van 3 ml 0,25%
trypsine/EDTA oplossing (T/E) (Sigma), die vooraf werd gefilterd om onzuiverheden te
verwijderen. De fles wordt nu 5 minuten geïncubeerd in de incubator bij dezelfde
condities waarin de cellen worden gekweekt. Nadien wordt de trypsine geïnactiveerd
door toevoegen van 7 ml vers cultuurmedium (37°C). Nu kan de gewenste hoeveelheid
celoplossing overgebracht worden in de nieuwe cultuurflessen en aangelengd worden
met het juiste volume vers medium. De cultuurflessen worden nu opnieuw in de
incubator gebracht voor verdere deling van de cellen, om na 3 à 4 dagen (afhankelijk van
de confluentie) opnieuw uitgesplitst te worden of de cellen te gebruiken voor
experimenten.
3.2.3. Cellen tellen
Doordat eenzelfde experiment vaak op verschillende stalen wordt uitgevoerd
moeten deze resultaten met elkaar te vergelijken zijn. Hiervoor moeten de cellen
uitgezaaid worden met eenzelfde dichtheid over de verschillende stalen. Bijvoorbeeld:
voor de experimenten in deze master‐thesis worden de cellen uitgezaaid in 6‐well
platen. Hierbij worden in elke well 2,5 x 105 cellen uitgezaaid voor de onbehandelde
cellen, 1,0 x 105 cellen voor de gesynchroniseerde cellen en 4,0 x 105 cellen voor de
gearresteerde cellen. Bij het gebruik van elektroporatie werden 1,2x106 cellen vanuit
één elektroporatie‐cuvet in 1 well overgebracht (zie 3.3.5). Na trypsinisatie van de
kweekfles moeten de cellen in de suspensie geteld worden. Er wordt 50 µl celsuspensie
18

gemengd met 100 µl tryptaanblauw. Dit kleurt de dode cellen blauw zodat deze kunnen
onderscheiden worden van de levende cellen, om op die manier enkel de levende cellen
te tellen. Een deel van deze suspensie wordt dan onder het dekglas van een telkamer
van Bürker gezogen. Een telkamer van Bürker telt 18 grote vierkanten die elk bestaan uit
16 kleine hokjes. 1 groot vierkant komt overeen met 1 µl celsuspensie. Door de cellen te
tellen in een aantal vierkanten onder een lichtmicroscoop kan men berekenen hoeveel
cellen de suspensie bevat per ml.
3.2.4. Celsynchronisatie en –arrestatie
Cellen delen in normale omstandigheden asynchroon ten opzichte van elkaar. Dit
wil zeggen dat alle cellen onafhankelijk van elkaar in een andere fase van de celcyclus
kunnen verblijven. Aan de hand van celsynchronisatie en –arrestatie zal men alle cellen
op dezelfde plaats in de celcyclus trachten te krijgen. Zo kunnen deze cellen onder
andere gebruikt worden in experimenten waar men onderzoekt of celdeling een invloed
heeft op de resultaten, dus om niet‐delende cellen met delende cellen te vergelijken.
Voor de arrestatie van de HeLa‐cellen wordt hetzelfde medium zoals eerder
besproken gebruikt, maar aangevuld met 2mM thymidine. Het thymidine geeft een
negatieve feedback aan de nucleotide‐biosynthese, waardoor een tekort aan nieuwe
nucleotides ontstaat. Bij de cellen die in de S‐fase van de celcyclus verblijven, of deze die
in de S‐fase willen overgaan stopt dus de celcyclus. Na 19h wordt een grote populatie
gearresteerde cellen bekomen die in de S‐fase verblijven of op het einde van de G1‐fase.
Voor een gesynchroniseerde populatie zal men nu terug thymidine‐vrij medium op de
cellen brengen, waardoor de celcyclus hervat wordt. Na 9h zijn alle cellen uit de S‐fase
en wordt opnieuw een medium met thymidine op de cellen gebracht. Aangezien er nu
nog geen cellen in de S‐fase verblijven, zullen alle cellen zich na 17h uniform in de vroege
S‐fase bevinden. Dit noemt men de dubbele thymidine blok. Voor celsynchronisatie
moeten de cellen enkele uren voor het experiment opnieuw vrijgelaten worden zodat ze
19

de celcyclus hervatten, door het op de cellen brengen van thymidine‐vrij medium.
3.3. TRANSFECTIES
3.3.1. Soorten transfecties
Er worden drie technieken gebruikt voor de transfecties: door de complexen vrij op
de cellen te brengen, door gebruik te maken van lipofectamine en door gebruik te
maken van elektroporatie. Eerst worden de complexen aangemaakt in een LAF‐kast,
volgens dezelfde bereidingswijze beschreven onder 3.1.4. Per staal wordt 150 µl
complex‐oplossing aangemaakt. De gebruikte complexen zijn terug te vinden in tabel
3.3. De transfecties met lipofectamine werden uitgevoerd met gWIZ‐GFP. De transfecties
d.m.v. elektroporatie werden zowel met gWIZ‐GFP als met pEGFP‐N1 gedaan.
3.3.2. pDNA
3.1.2.1. Soorten pDNA
Zie 3.2.1
3.1.2.2. Kweek
Het labo beschikt over bacteriën die getransformeerd werden met de gebruikte
pDNA’s. Dit pDNA wordt in grote hoeveelheden gerepliceerd in deze bacteriën. De
bacteriën worden opgekweekt in LB‐medium bestaande uit 25g NaCl, 12,5g Yeast
extract en 25g trypton opgelost in 2500ml gedestilleerd water. Deze oplossing wordt
nadien geautoclaveerd. Na sterilisatie en vóór het gebruik van het medium wordt het
antibioticum kanamycine toegevoegd. Hierdoor zullen alleen de gewenste bacteriën
in leven blijven in het medium.
3.1.2.3. Qiagen Plasmid Giga Kit pDNA opzuivering
De bacteriënculturen die gWIZ‐GFP of pEGFP‐N1 bevatten worden in grote
centrifugepotten gebracht en afgecentrifugeerd zodat een bacteriële pellet bekomen
wordt. Het supernatans wordt verwijderd en de pellet wordt geresuspendeerd in P1‐
20
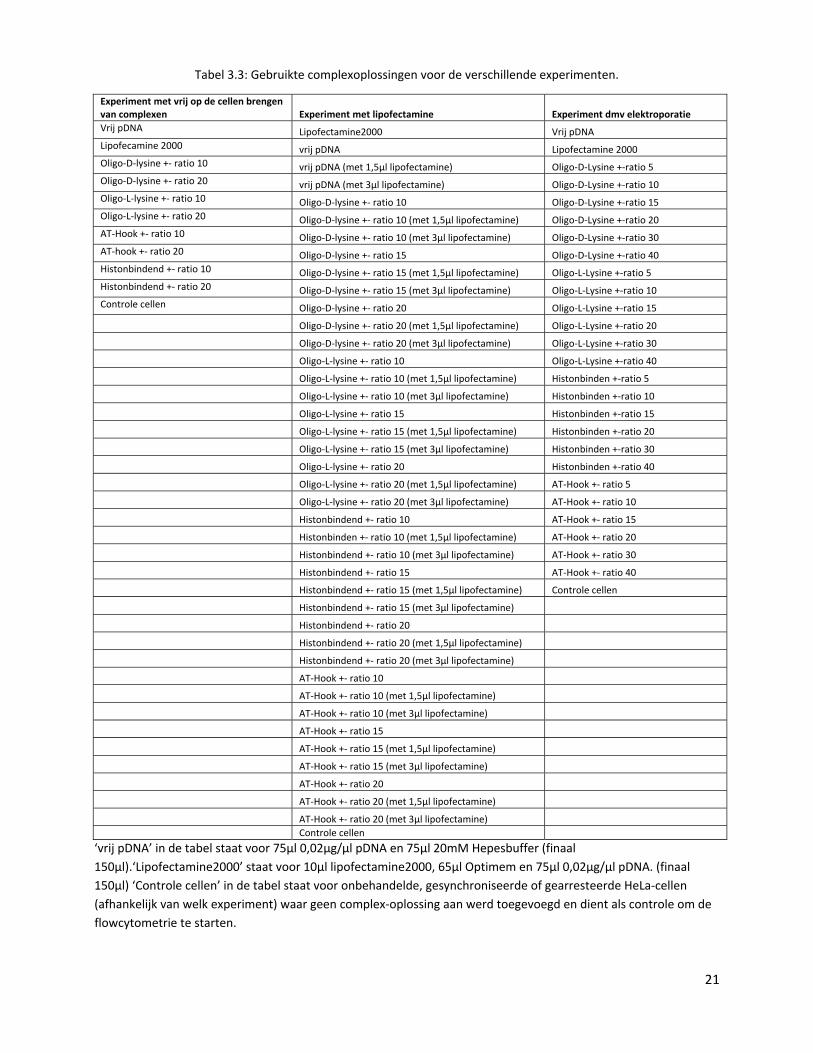
Tabel 3.3: Gebruikte complexoplossingen voor de verschillende experimenten.
Experiment met vrij op de cellen brengen van complexen Experiment met lipofectamine Experiment dmv elektroporatie Vrij pDNA Lipofectamine2000 Vrij pDNA Lipofecamine 2000 vrij pDNA Lipofectamine 2000 Oligo‐D‐lysine +‐ ratio 10 vrij pDNA (met 1,5µl lipofectamine) Oligo‐D‐Lysine +‐ratio 5 Oligo‐D‐lysine +‐ ratio 20 vrij pDNA (met 3µl lipofectamine) Oligo‐D‐Lysine +‐ratio 10 Oligo‐L‐lysine +‐ ratio 10 Oligo‐D‐lysine +‐ ratio 10 Oligo‐D‐Lysine +‐ratio 15 Oligo‐L‐lysine +‐ ratio 20 Oligo‐D‐lysine +‐ ratio 10 (met 1,5µl lipofectamine) Oligo‐D‐Lysine +‐ratio 20 AT‐Hook +‐ ratio 10 Oligo‐D‐lysine +‐ ratio 10 (met 3µl lipofectamine) Oligo‐D‐Lysine +‐ratio 30 AT‐hook +‐ ratio 20 Oligo‐D‐lysine +‐ ratio 15 Oligo‐D‐Lysine +‐ratio 40 Histonbindend +‐ ratio 10 Oligo‐D‐lysine +‐ ratio 15 (met 1,5µl lipofectamine) Oligo‐L‐Lysine +‐ratio 5 Histonbindend +‐ ratio 20 Oligo‐D‐lysine +‐ ratio 15 (met 3µl lipofectamine) Oligo‐L‐Lysine +‐ratio 10 Controle cellen Oligo‐D‐lysine +‐ ratio 20 Oligo‐L‐Lysine +‐ratio 15 Oligo‐D‐lysine +‐ ratio 20 (met 1,5µl lipofectamine) Oligo‐L‐Lysine +‐ratio 20 Oligo‐D‐lysine +‐ ratio 20 (met 3µl lipofectamine) Oligo‐L‐Lysine +‐ratio 30 Oligo‐L‐lysine +‐ ratio 10 Oligo‐L‐Lysine +‐ratio 40 Oligo‐L‐lysine +‐ ratio 10 (met 1,5µl lipofectamine) Histonbinden +‐ratio 5 Oligo‐L‐lysine +‐ ratio 10 (met 3µl lipofectamine) Histonbinden +‐ratio 10 Oligo‐L‐lysine +‐ ratio 15 Histonbinden +‐ratio 15 Oligo‐L‐lysine +‐ ratio 15 (met 1,5µl lipofectamine) Histonbinden +‐ratio 20 Oligo‐L‐lysine +‐ ratio 15 (met 3µl lipofectamine) Histonbinden +‐ratio 30 Oligo‐L‐lysine +‐ ratio 20 Histonbinden +‐ratio 40 Oligo‐L‐lysine +‐ ratio 20 (met 1,5µl lipofectamine) AT‐Hook +‐ ratio 5 Oligo‐L‐lysine +‐ ratio 20 (met 3µl lipofectamine) AT‐Hook +‐ ratio 10 Histonbindend +‐ ratio 10 AT‐Hook +‐ ratio 15 Histonbinden +‐ ratio 10 (met 1,5µl lipofectamine) AT‐Hook +‐ ratio 20 Histonbindend +‐ ratio 10 (met 3µl lipofectamine) AT‐Hook +‐ ratio 30 Histonbindend +‐ ratio 15 AT‐Hook +‐ ratio 40 Histonbindend +‐ ratio 15 (met 1,5µl lipofectamine) Controle cellen Histonbindend +‐ ratio 15 (met 3µl lipofectamine) Histonbindend +‐ ratio 20 Histonbindend +‐ ratio 20 (met 1,5µl lipofectamine) Histonbindend +‐ ratio 20 (met 3µl lipofectamine) AT‐Hook +‐ ratio 10 AT‐Hook +‐ ratio 10 (met 1,5µl lipofectamine) AT‐Hook +‐ ratio 10 (met 3µl lipofectamine) AT‐Hook +‐ ratio 15 AT‐Hook +‐ ratio 15 (met 1,5µl lipofectamine) AT‐Hook +‐ ratio 15 (met 3µl lipofectamine) AT‐Hook +‐ ratio 20 AT‐Hook +‐ ratio 20 (met 1,5µl lipofectamine) AT‐Hook +‐ ratio 20 (met 3µl lipofectamine) Controle cellen
‘vrij pDNA’ in de tabel staat voor 75µl 0,02µg/µl pDNA en 75µl 20mM Hepesbuffer (finaal 150µl).‘Lipofectamine2000’ staat voor 10µl lipofectamine2000, 65µl Optimem en 75µl 0,02µg/µl pDNA. (finaal 150µl) ‘Controle cellen’ in de tabel staat voor onbehandelde, gesynchroniseerde of gearresteerde HeLa‐cellen (afhankelijk van welk experiment) waar geen complex‐oplossing aan werd toegevoegd en dient als controle om de flowcytometrie te starten.
21

buffer. Na toevoegen van achtereenvolgens de P2‐ en P3‐buffer, wordt de
bacteriële pellet geprecipiteerd. Het pDNA bevindt zich in het supernatans, alle
andere celcomponenten bevinden zich in het precipitaat. Na het affiltreren van het
precipitaat wordt het supernatans op een anionuitwisselingskolom gebracht die
vooraf werd geëquilibreerd met QBT‐buffer. Het pDNA wordt weerhouden op de
kolom, maar ook negatief geladen peptiden, RNA en metabolieten blijven achter op
de kolom. Deze worden weggewassen met de QC‐buffer. Eenmaal al deze storende
componenten zijn weggewassen wordt het pDNA van de kolom geëlueerd met QF‐
buffer. Er wordt isopropanol toegevoegd aan het eluaat. Dit zorgt voor het neerslaan
van het pDNA, de zouten blijven echter in oplossing. Deze oplossing wordt
gecentrifugeerd en na centrifugatie wordt de pellet nogmaals gewassen met een
70% ethanoloplossing om resten van overtollige zouten te verwijderen. Na
centrifugatie en weghalen van het supernatans wordt het pDNA 10 tot 20 minuten
gedroogd aan de lucht om nadien opgelost te worden in een geschikte hoeveelheid
Hepesbuffer.
3.1.2.4. Concentratiebepaling
De concentratie van de bekomen pDNA‐oplossing wordt spectrofotometrisch
bepaald. De meting wordt uitgevoerd ten opzichte van een blanco (Hepesbuffer) in
UV‐cuvetten, dit bij een golflengte van 260 nm, omdat een densiteit van 1,000 bij
260 nm overeenkomt met 50 µg/ml dubbelstrengig DNA. Op die manier kan de
concentratie van het pDNA berekend worden. De absorptie wordt ook bekeken bij
een golflengte van 280 nm om de zuiverheid te bepalen. Het staal kan als voldoende
zuiver beschouwd worden als de verhouding van de absorptie bij 260 nm ten
opzichte van de absorptie bij 280 nm tussen 1,8 en 2,0 ligt.
3.3.3. Transfectie door vrije opname van pDNA/peptide‐complexen
De HeLa‐cellen worden vooraf uitgezaaid op 6‐well platen (Zie 3.1.3 uitzaaiing‐
concentratie). De 150 µl gWIZ‐GFP/peptide‐complexen (zie tabel 3.3) gemengd met 1350
µl Optimem worden bovenop de cellen gebracht en 24h geïncubeerd bij 37°C, 5% CO2 en
22

95% luchtvochtigheid. Na 24h en 48h worden de cellen gewassen met PBS (‐Ca/‐Mg) en
voorbereid voor flow cytometrie (zie 3.6).
3.3.4. Transfectie met tertiaire complexen
Het gebruikte lipofectamine 2000 is een transfectie‐reagent dat gebruikt wordt om
cellen in vitro te transfecteren met siRNA of pDNA door middel van lipofectie. Lipofectie
is een methode om genetische materiaal in de cel te brengen gebruik makende van
liposomen. Het lipofectamine vormt vesikels met de complexen waarvan de buitenzijde
qua samenstelling gelijkaardig is met die van het celmembraan. Als gevolg hiervan zullen
de vesikels samensmelten met het celmembraan en zijn inhoud in de cel loodsen.
De gebruikte complexen worden weergegeven in tabel 3.3. De pre‐complexen
worden in drievoud (3x150 µl) aangemaakt, waarna tertiaire complexen met
verschillende hoeveelheden lipofectamine worden gevormd: een complex‐oplossing
waaraan geen lipofectamine2000 wordt toegevoegd en 2 complex‐oplossingen waaraan
respectievelijk 1,5 µl en 3 µl lipofectamine2000 wordt toegevoegd. Deze oplossingen
worden in 1350 µl Optimem bovenop de cellen gebracht, die vooraf uitgezaaid werden
in 6‐well platen (Uitzaaiingsconcentratie zie 3.1.3). De 6‐well platen worden 24h en 48h
geïncubeerd bij 37°C, 5% CO2 en 95% luchtvochtigheid. Na 24h en na 48h worden de
stalen voorbereid voor flow cytometrie (zie 3.6). De transfectie‐efficiëntie wordt bepaald
bij onbehandelde, gearresteerde en gesynchroniseerde HeLa‐cellen.
3.3.5. Transfectie d.m.v. elektroporatie
Elektroporatie is een techniek die door het aanleggen van een extern elektrische
veld een verhoging van de elektrische geleiding en de permeabiliteit van het
celmembraan als gevolg heeft. Het is een methode om extracellulaire partikels in de cel
te brengen. De elektroporatiecondities moeten zodanig gekozen worden dat na
introductie van de extracellulaire partikels het celmembraan zich spontaan hersteld en
dat de schade aan de cellen zoveel mogelijk beperkt blijft.
23

De elektroporatie bij HeLa‐cellen wordt uitgevoerd bij 260 V en 850 µF gedurende ±
30 msec. De gevormde complexen (150 µl) (zie tabel 3.3) worden gemengd met 250 µl
Optimem (Optimem met 2mM thymidine bij gearresteerde cellen) waarin 1,2x106 cellen
gesuspendeerd zitten. Deze suspensie wordt nadien in elektroporatie‐cuvetten gebracht
en geëlektroporeerd. Na elektroporatie wordt de suspensie overgebracht in 15ml tubes,
gewassen met PBS (+Ca/+Mg) en gecentrifugeerd. De bekomen pellet wordt
geresuspendeerd in het juiste medium en uitgezaaid in een 6‐well plaat. Waarna het 24h
en 48h zal incuberen bij 37°C, 5% CO2 en 95% luchtvochtigheid om dan geanalyseerd te
worden m.b.v. flow cytometrie.
3.4. XENOPUS SPERMA CHROMATINE BINDING
Sperma chromatine afkomstig van Xenopus laevis of de klauwkikker wordt in het
onderzoek van deze master thesis gebruikt om de bindingscapaciteit van de gebruikte
peptiden te bestuderen. Alle pDNA/peptide‐complexen in dit experiment werden
aangemaakt met een ladingsratio 10 in water. Het pDNA werd gelabeld met fluoresceïne (zie
3.6.1). Vooreerst werd 20 µl cytosolisch extract van Xenopus‐oöcyten gemengd met 0,6 µl
Xenopus sperma chromatine. Deze oplossing wordt 10 minuten geïncubeerd bij 20°C.
Nadien wordt 1 µl van een complexverdunning van 1:10 in water toegevoegd aan de
oplossing en 20 minuten geïncubeerd bij 20°C. Hierna voegt men nog 1µl DAPI toe die het
chromatine fluorescent maakt. Hiervan wordt dan 5 µl bestudeerd onder een
fluorescentiemicroscoop en worden het aantal complexen gebonden op het chromatine
geteld.
3.4.1. Miruslabeling
Voor deze labeling wordt gebruik gemaakt van de Mirus label IT Nucleic Acid
labeling kit. Het is een niet‐enzymatische, niet‐destructieve methode die zorgt voor de
covalente hechting van labels op nucleïnezuren waarmee elk type DNA of RNA kan
24

gelabeld worden. In de eerste stap wordt 120 µl RNAse vrij water gemengd met 20 µl
10x labeling buffer A, 40 µl 1 µg/µl pDNA en 20 µl Label IT reagent. Deze oplossing wordt
overnacht geïncubeerd en beschermd van het licht. De dag nadien volgt de opzuivering
m.b.v. microspinkollometjes. De concentratie is nu maximaal 0,2 µg/µl.
3.4.2. DAPI
4’,6‐diamidino‐2‐fenylindool (DAPI) is een fluorescente merker die sterk bindt aan
DNA‐moleculen. DAPI wordt onder de fluorescentiemicroscoop geëxciteerd met
ultraviolet licht. Het heeft een emissie van blauw licht, waardoor het in één staal kan
gecombineerd worden met andere fluorescente moleculen zoals fluoresceïne in dit
experiment. DAPI kan zowel bij dode als levende cellen aan het DNA binden en wordt
vandaar als toxisch en mutageen beschouwd.
3.5. FLOW CYTOMETRIE
Flow cytometrie is een techniek waarmee men de eigenschappen van elk partikel in
een staal individueel kan onderzoeken. Omdat de flow cytometer niet zomaar in een
driedimensionale ruimte alle partikels kan detecteren moeten zij apart bij de detector
gebracht worden. Dit gebeurt via het fluidics systeem. Het staal bevindt zich in een centrale
ruimte waarlangs de sheathbuffer vloeit. Doordat de sheathbuffer een veel grotere snelheid
heeft dan het staal ontstaat er een trekkracht van de buffer op het staal waardoor er een
enkelvoudige rij van partikels gevormd wordt, die zo apart kunnen worden gedetecteerd. Dit
effect wordt hydrodynamisch focussen genoemd. Het staal wordt bestraald met een laser
waardoor de partikels op hun beurt het licht zullen verstrooien. Als de partikels fluoroforen
bevatten komt emissie voor na excitatie. Op deze manier kunnen eigenschappen van de
partikels bepaald worden. Licht dat voorwaarts verstrooid en gedetecteerd wordt zal een
maat zijn voor de grootte van de partikels en kan ook gebruikt worden om onderscheid te
maken tussen cellulair afval en levende cellen. Licht dat gedetecteerd wordt over een hoek
25

van 90° ten opzichte van de lichtbron noemt men de side scatter. Deze geeft informatie over
de granulariteit van de partikels. Zowel de forward scatter als de side scatter zijn uniek voor
elke cel, de combinatie van de twee kan gebruikt worden om verschillende celtypes van
elkaar te onderscheiden. Fluorecentiemetingen geven kwantitatieve en kwalitatieve
informatie over de cellen. Zo kan de mate van transfectie en het aantal getransfecteerde
cellen bepaald worden met deze techniek. Het emissie‐licht dat wordt doorgelaten naar de
detector wordt geselecteerd door een aantal optische filters. De fotodetector werkt door het
opgewekt worden van een kleine stroom als deze bestraald wordt met fotonen. Hoe meer
licht op de detector valt, hoe groter de stroom en dus ook hoe groter het detectiesignaal zal
zijn.
In de experimenten van deze master thesis worden de stalen voorbereid uit de 6‐well
platen zoals besproken onder 3.3. Deze platen worden getrypsiniseerd na 24h en 48h met
500 µl 0,25% trypsine/EDTA, nadien worden de platen 5 minuten geïncubeerd in de
incubator waarin ook de cellen bewaard worden. Nu de cellen los zijn van de bodem wordt
2000 µl van het gepaste medium (met of zonder thymidine, afhankelijk van wel of niet
gearresteerde cellen) aan elke well toegevoegd. 1500 µl vanuit elke well wordt nu
overgebracht in 1500 µl‐epjes en de 6‐well plaat met de resterende cellen wordt na
toevoegen van 1 ml extra medium terug in de incubator gebracht. Hierna wordt de oplossing
gecentrifugeerd met een vaste pellet van cellen als gevolg. Het medium wordt nu
weggenomen en vervangen door 300 µL flowbuffer. M.b.v. flow cytometrie wordt per staal
het percentage GFP‐positieve cellen, alsook de gemiddelde expressie van GFP per cel
verkregen.
3.6. CONFOCALE LASER SCANNING MICROSCOPIE
Coherent licht van de laser valt in op een dichroïsche spiegel en wordt gereflecteerd
naar een objectieflens. Op die manier scant men het staal met de laserpunt in verschillende
focale vlakken. Fluorescent licht geëmiteerd door het staal, vanuit dezelfde focale vlakken
26

waar de laser het staal exciteert, worden terug door de dichroïsche spiegel gestuurd en
worden gefocust als een confocaal punt aan het diafragma bij de detector. De dichroïsche
spiegel laat dit emissielicht door, omdat de emissiegolflengte groter is dan de cut‐off van de
spiegel. Dit fenomeen treedt enkel op als er fluoroforen in het staal aanwezig zijn die
kunnen geëxciteerd worden door die specifieke golflengte van de laser. Het licht dat niet in
focus is, dus het emissielicht uit hogere of lagere focale vlakken, wordt tegengehouden door
het diafragma.
3.6.1. Tokaï Hit stage top incubator
De tokaï hit is een kleine incubator die op de microscoop geplaatst kan worden. In de
tokaï hit kunnen de cellen in leven gehouden worden in een milieu van 5% CO2, bij 37°C
en een luchtvochtigheid van 95%. Dankzij de Tokaï hit kan aan live cell imaging gedaan
worden. Men kan gedurende een aantal uur de verandering in levende cellen filmen en
bestuderen onder een microscoop.
27

4. RESULTATEN EN DISCUSSIE
4.1. KARAKTERISATIE VAN PDNA/PEPTIDE COMPLEXEN
4.1.1. Aanmaak complexen
Oligo‐D‐lysine, Oligo‐L‐lysine, het DNA‐bindend en het histonbindend peptide
worden elk apart gecomplexeerd met het pDNA gWIZ‐GFP volgens verschillende +/‐
ladingsratio’s zoals beschreven in 3.1.4. Voorafgaand aan de transfectie‐experimenten
worden deze complexen gecontroleerd op grootte, lading en maat van complexatie en
decomplexatie in verschillende media.
4.1.2. Maat van complexatie a.d.h.v. gelelektroforese
Om de maat van complexatie van de verschillende peptiden met het pDNA na te
gaan worden de complexen onderworpen aan gelelektroforese. De migratielengte van
het mengsel van de peptiden en het pDNA in de gel is dan een maat voor de
complexatie‐sterkte. De gel werd als volgt geladen van links naar recht: 1kb ladder;
pDNA/peptidecomplex met ladingsratio 0 (vrij pDNA); 0,25; 0,5; 1; 2,5; 5; 7,5; 10; 12,5;
15; 17,5; 20; 25; 30; 1kb ladder. De pDNA/oligo‐D‐lysine complexen blijven vanaf
ladingsratio 5 in de slots zitten en pDNA/oligo‐L‐lysine complexen vanaf ladingsratio 7,5.
We kunnen dus stellen dat er vanaf ladingsratio’s 5 en 7,5 van respectievelijk de pDNA/
oligo‐D‐lysine en pDNA/oligo‐L‐lysine stabiele complexen worden gevormd. De
pDNA/chromatine‐bindende peptide‐complexen migreren niet meer uit de slots vanaf
ladingsratio 2,5 (foto 5.1). Ze kunnen bij deze ladingsratio dan ook als stabiel beschouwd
worden.
Foto 5.1 Agarosegel van pDNA/histonbindend peptide‐mengsels met verschillende +/‐ ladingsratio’s
28

4.1.3. Deeltjesgrootte a.d.h.v. DLS
Voor dit experiment worden de complexen stofvrij aangemaakt met ladingsratio’s
5, 10, 15, 20, 30 en 40 zoals beschreven onder 3.1.4.1.
Vanuit de grafieken 5.1, 5.3 en 5.4 kan gesteld worden dat de complexen met oligo‐D‐
lysine, het DNA‐bindend en het histonbindend peptide kleiner zijn als deze in water
aangemaakt worden. Wat bij oligo‐L‐lysine niet voor alle ladingsratio’s geldt (grafiek 5.2).
Er zal voor verdere experimenten in deze master thesis enkel nog gebruik gemaakt
worden van complexen aangemaakt in water.
Er kan gezegd worden dat complexen met het DNA‐bindend en het histonbindend
peptide gemiddeld veel kleiner zijn dan die met oligo‐D‐ en oligo‐L‐lysine. Een oorzaak
van de hoge waarden bij oligo‐D‐lysine zou kunnen zijn dat dit peptide 10 AZ’en lang is
met slechts 10 positieve ladingen en bijgevolg te klein is om bij lage ladingsratio’s pDNA
te complexeren.
De grafieken tonen zoals verwacht bij een hogere ladingsratio een lagere
complexgrootte. Dit komt doordat het aantal peptiden en dus het aantal positieve
ladingen stijgt, waardoor het negatief geladen pDNA compacter kan gecomplexeerd
worden.
0.00
200.00
400.00
600.00
800.00
1000.00
1200.00
1400.00
1600.00
5 10 15 20 30 40
Grootte (n
m)
+/‐ ladingsratio
Grootte Oligo‐D‐lysine
Grootte Oligo‐D‐Lysine Hepes
Grootte Oligo‐D‐lysine Water
.
Grafiek 5.1 Grootte van pDNA/peptide‐complexen met oligo‐D‐lysine aangemaakt in water of hepesbuffer
29

0.00200.00400.00
600.00800.001000.00
1200.001400.001600.00
1800.00
5 10 15 20 30 40
Grootte (n
m)
+/‐ ladingsratio
Grootte Oligo‐L‐lysine
Grootte Oligo‐L‐Lysine Hepes
Grootte Oligo‐L‐lysine Water
Grafiek 5.2 Grootte van pDNA/peptide‐complexen met oligo‐L‐lysine aangemaakt in water of hepesbuffer.
0.00
50.00
100.00
150.00
200.00
250.00
5 10 15 20 30 40
Grootte (n
m)
+/‐ ladingsratio
Grootte histonbindend peptide
Grootte Histondbindend peptide Hepes
Grootte Histonbindend peptide Water
Grafiek 5.3 Grootte van pDNA/peptide‐complexen met het histonbindend peptide aangemaakt in water of hepesbuffer.
30
0.00
50.00
100.00
150.00
200.00
250.00
300.00
350.00
5 10 15 20 30 40
Grootte (n
m)
+/‐ ladingsratio
Grootte DNA‐bindend peptide
Grootte DNA‐bindend peptide Hepes
Grootte DNA‐bindend peptide Water
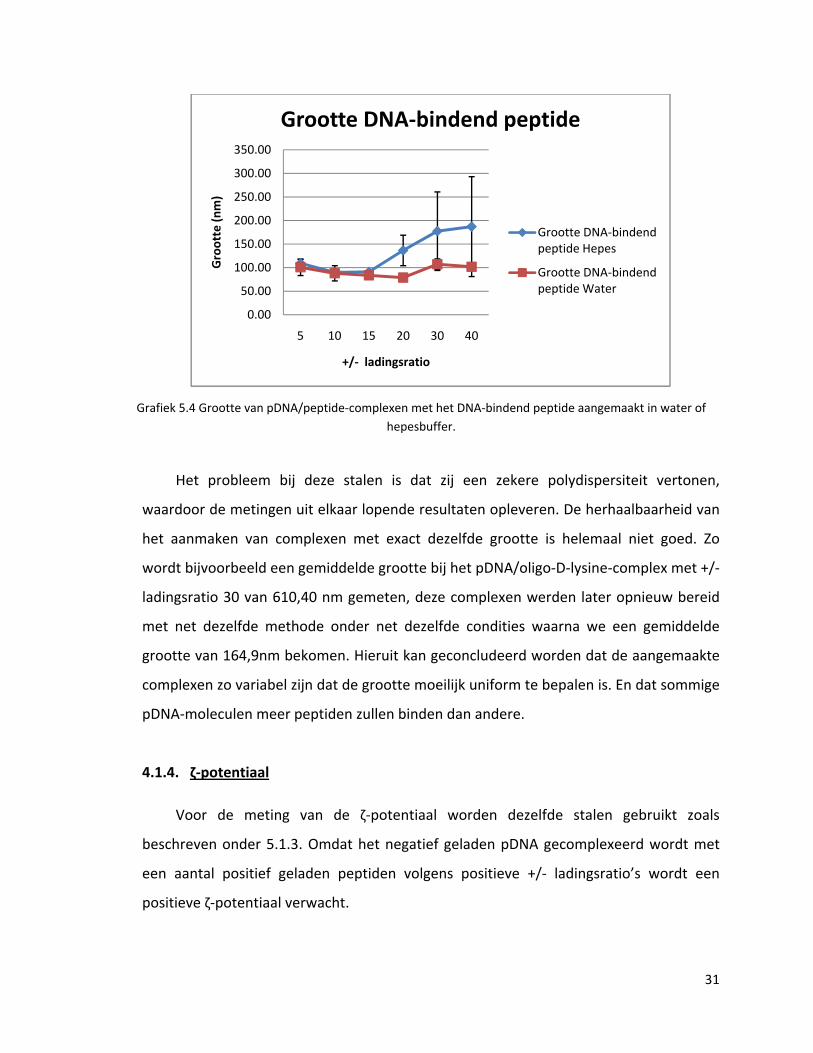
Het probleem bij deze stalen is dat zij een zekere polydispersiteit vertonen waardoor de
Grafiek 5.4 Grootte van pDNA/peptide‐complexen met het DNA‐bindend peptide aangemaakt in water of hepesbuffer.
Het probleem bij deze stalen is dat zij een zekere polydispersiteit vertonen,
waardoor de metingen uit elkaar lopende resultaten opleveren. De herhaalbaarheid van
het aanmaken van complexen met exact dezelfde grootte is helemaal niet goed. Zo
wordt bijvoorbeeld een gemiddelde grootte bij het pDNA/oligo‐D‐lysine‐complex met +/‐
ladingsratio 30 van 610,40 nm gemeten, deze complexen werden later opnieuw bereid
met net dezelfde methode onder net dezelfde condities waarna we een gemiddelde
grootte van 164,9nm bekomen. Hieruit kan geconcludeerd worden dat de aangemaakte
complexen zo variabel zijn dat de grootte moeilijk uniform te bepalen is. En dat sommige
pDNA‐moleculen meer peptiden zullen binden dan andere.
4.1.4. ζ‐potentiaal
Voor de meting van de ζ‐potentiaal worden dezelfde stalen gebruikt zoals
beschreven onder 5.1.3. Omdat het negatief geladen pDNA gecomplexeerd wordt met
een aantal positief geladen peptiden volgens positieve +/‐ ladingsratio’s wordt een
positieve ζ‐potentiaal verwacht.
31

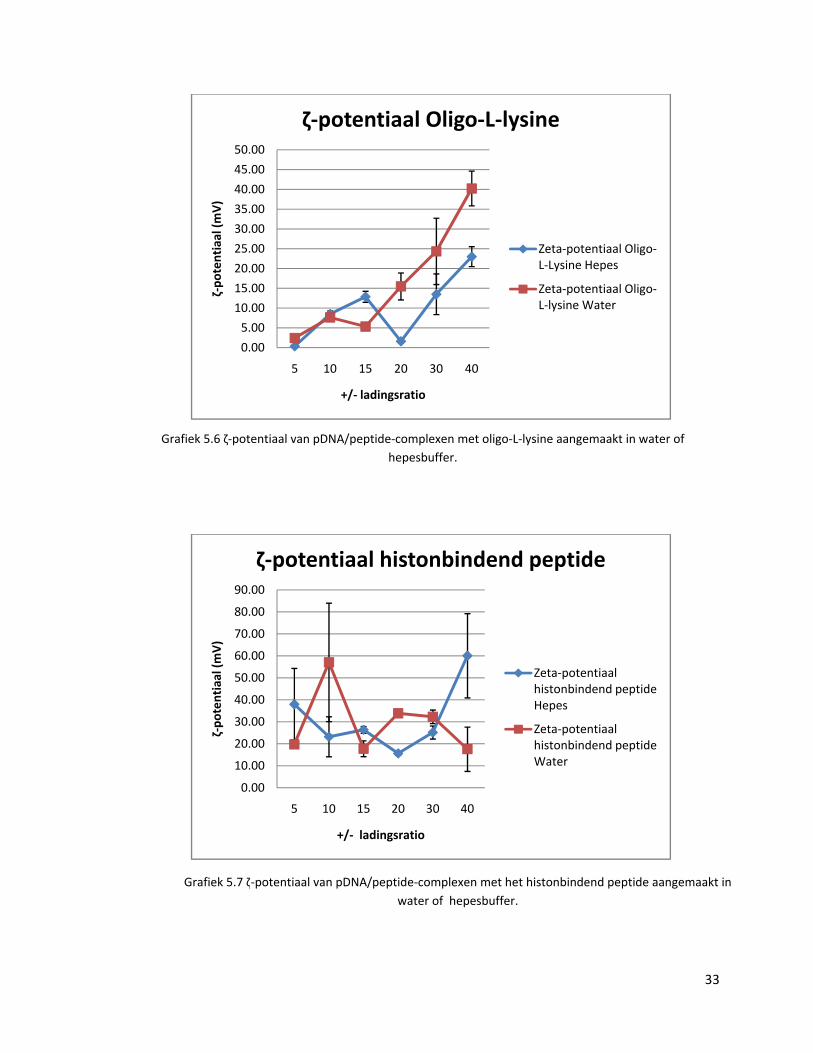
De resultaten worden hieronder samengevat in grafieken 5.5, 5.6, 5.7, 5.8. Er kan
gesteld worden dat alle stalen positief geladen zijn. De verwachte stijgende trend van de
potentiaal volgens stijgende +/‐ ladingsratio’s is echter alleen in grafieken 5.5 en 5.6 te
zien. Ook worden grote foutenbalken bekomen, wat opnieuw duidt op een slechte
herhaalbaarheid van de aanmaak van complexen. Hieruit kan opnieuw afgeleid worden
dat sommige pDNA‐moleculen meer peptiden zullen complexeren dan andere.
De ζ‐potentiaal ligt bij de pDNA/peptide‐complexen in hepesbuffer ook duidelijk lager
dan in water. Dit komt doordat de hepesbuffer de ladingen van de pDNA/peptide‐
complexen afschermt.
0.00
5.00
10.00
15.00
20.00
25.00
30.00
5 10 15 20 30 40
ζ‐po
tentiaal (m
V)
+/‐ ladingsratio
ζ‐potentiaal oligo‐D‐lysine
Zeta‐potentiaal oligo‐D‐lysine Hepes
Zeta‐potentiaal oligo‐D‐lysine Water
Grafiek 5.5 ζ‐potentiaal van pDNA/peptide‐complexen met oligo‐D‐lysine aangemaakt in water of hepesbuffer.
32
0.00
5.00
10.00
15.00
20.00
25.00
30.00
35.00
40.00
45.00
50.00
5 10 15 20 30 40
ζ‐po
tentiaal (m
V)
+/‐ ladingsratio
ζ‐potentiaal Oligo‐L‐lysine
Zeta‐potentiaal Oligo‐L‐Lysine Hepes
Zeta‐potentiaal Oligo‐L‐lysine Water
Grafiek 5.6 ζ‐potentiaal van pDNA/peptide‐complexen met oligo‐L‐lysine aangemaakt in water of hepesbuffer.
0.00
10.00
20.00
30.00
40.00
50.00
60.00
70.00
80.00
90.00
5 10 15 20 30 40
ζ‐po
tentiaal (m
V)
+/‐ ladingsratio
ζ‐potentiaal histonbindend peptide
Zeta‐potentiaal histonbindend peptide Hepes
Zeta‐potentiaal histonbindend peptide Water
Grafiek 5.7 ζ‐potentiaal van pDNA/peptide‐complexen met het histonbindend peptide aangemaakt in water of hepesbuffer.
33

0.00
10.00
20.00
30.00
40.00
50.00
60.00
5 10 15 20 30 40
ζ‐po
tentiaal (m
V)
+/‐ ladingsratio
ζ‐potentiaal DNA‐bindend peptide
Zeta‐potentiaal AT‐Hook Hepes
Zeta‐potentiaal AT‐Hook Water
Grafiek 5.8 ζ‐potentiaal van pDNA/peptide‐complexen met het DNA‐bindend peptide aangemaakt in water of hepesbuffer.
4.2. XENOPUS CHROMATINE BINDING
Dit experiment (zie 3.5) wordt uitgevoerd om te achterhalen welk van de vier gebruikte
peptiden, gecomplexeerd met pDNA, het best aan het chromatine zal binden.
Uit de resultaten (grafiek 5.9) kan afgeleid worden dat de pDNA/oligo‐L‐lysine‐complexen de
grootste affiniteit hebben voor Xenopus chromatine, gevolgd door de complexen op basis
van het DNA‐bindend peptide, oligo‐D‐lysine en tenslotte het histonbindend peptide dat het
minst affiniteit vertoont. Dat ze allemaal kunnen binden is logisch, omdat de complexen
positief geladen zijn en het chromatine netto negatief geladen is (foto 5.2).
De resultaten van de verschillende pDNA/peptide‐complexen mogen echter niet met
elkaar vergelen worden, omdat de complexen niet dezelfde grootte en lading hebben. Het
beter binden van de pDNA/oligo‐L‐lysine‐complexen kan te maken hebben met de
polydispersiteit van deze oplossing. De grotere en meer positief geladen complexen zullen
34
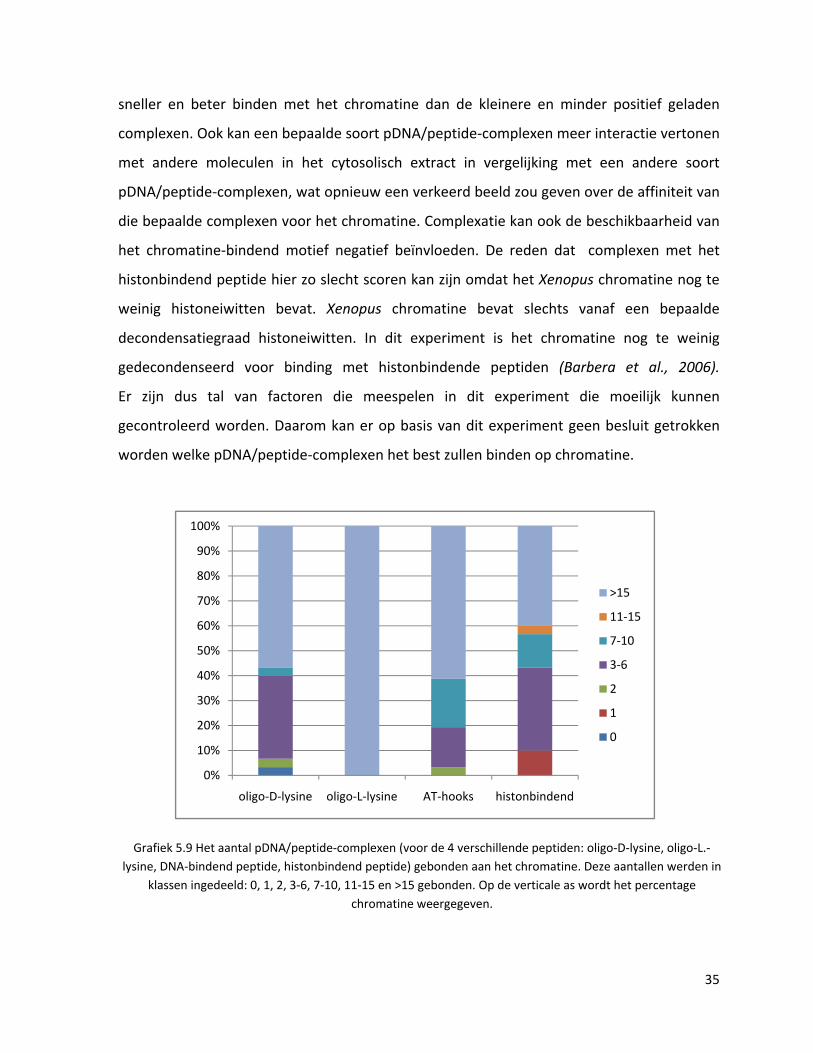
sneller en beter binden met het chromatine dan de kleinere en minder positief geladen
complexen. Ook kan een bepaalde soort pDNA/peptide‐complexen meer interactie vertonen
met andere moleculen in het cytosolisch extract in vergelijking met een andere soort
pDNA/peptide‐complexen, wat opnieuw een verkeerd beeld zou geven over de affiniteit van
die bepaalde complexen voor het chromatine. Complexatie kan ook de beschikbaarheid van
het chromatine‐bindend motief negatief beïnvloeden. De reden dat complexen met het
histonbindend peptide hier zo slecht scoren kan zijn omdat het Xenopus chromatine nog te
weinig histoneiwitten bevat. Xenopus chromatine bevat slechts vanaf een bepaalde
decondensatiegraad histoneiwitten. In dit experiment is het chromatine nog te weinig
gedecondenseerd voor binding met histonbindende peptiden (Barbera et al., 2006).
Er zijn dus tal van factoren die meespelen in dit experiment die moeilijk kunnen
gecontroleerd worden. Daarom kan er op basis van dit experiment geen besluit getrokken
worden welke pDNA/peptide‐complexen het best zullen binden op chromatine.
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
oligo‐D‐lysine oligo‐L‐lysine AT‐hooks histonbindend
>15
11‐15
7‐10
3‐6
2
1
0
Grafiek 5.9 Het aantal pDNA/peptide‐complexen (voor de 4 verschillende peptiden: oligo‐D‐lysine, oligo‐L.‐
lysine, DNA‐bindend peptide, histonbindend peptide) gebonden aan het chromatine. Deze aantallen werden in klassen ingedeeld: 0, 1, 2, 3‐6, 7‐10, 11‐15 en >15 gebonden. Op de verticale as wordt het percentage
chromatine weergegeven.
35

Foto 5.2: Links: blauw DAPI‐gelabeld Xenopus chromatine met groene fluoresceïne‐gelabelde
pDNA/histonbindend peptide complexen. Rechts: blauw DAPI‐gelabeld Xenopus chromatine met groene
fluoresceïne‐gelabelde pDNA/oligo‐D‐lysine complexen.
4.3. NAGAAN SYNCHRONISATIE VAN HELA‐CELLEN
De celsynchronisatie werd uitgevoerd zoals beschreven onder 3.1.4. Nadien werden de
cellen opgevolgd a.d.h.v. live cell imaging met de confocale microscoop. Op die manier werd
waargenomen dat alle cellen delen tussen de 20 en 25h nadat de dubbele thymidine blok
werd opgeheven.
4.4. OPNAME VAN COMPLEXEN DOOR HELA‐CELLEN
4.4.1. Opname van de complexen
De stalen werden aangemaakt zoals beschreven onder 3.3.3 en daarna
onderworpen aan flow cytometrie.
Uit de flow resultaten kunnen we afleiden dat de pDNA/peptide‐complexen niet tot
in de kern geraken. Enkel de controle stalen met lipofectamine2000 die lipoplexen
gevormd hebben met het vrij pDNA geven zoals verwacht een positief resultaat.
36

Het feit dat de cellen niet worden getransfecteerd door de pDNA/peptide‐complexen
gewoon op de cellen te brengen kan allerlei verschillende verklaringen hebben. Het kan
zijn dat er helemaal geen endocytose optreedt, of dat de pDNA/peptide‐complexen niet
kunnen onstnappen uit de gevormde endosomen. Er kan eventueel d.m.v. microscopie
gecontroleerd worden of de pDNA/peptide‐complexen in de cel terecht komen. Het is
ook mogelijk dat de pDNA/peptide‐complexen in het cytoplasma uiteen vallen en dat het
pDNA hierna wordt afgebroken.
20
25
0
5
10
15
cellen
Transfectie Hela‐cellen
4.4.2. Controle van complexatie in verschillende media
Nog een andere hypothese voor de negatieve transfectie‐resulaten uit 5.4.1, is dat de
pDNA/peptide‐complexen terug uit elkaar vallen in het medium waarmee ze aan de
cellen worden toegediend, dus vóór ze door de cellen zouden kunnen opgenomen
worden.
Deze mogelijkheid wordt getest door dezelfde complexen zoals in 5.1.2 éénmaal te
bereiden in Optimem (geladen in de bovenste rij van gel 5.4), en éénmaal in serumvrij
% getransfecteerde
Transfectie na 24h
Transfectie na 48h
Grafiek 5.10 Percentage GFP‐positieve HeLa‐cellen 24h en 48h na transfectie met vrij gWIZ‐GFP, gWIZ‐GFP‐lipoplexen en gWIZ‐GFP/peptide‐complexen.
37

DMEM‐F12 (geladen in de onderste rij van gel 5.4) en deze op dezelfde manier op een
1% agarosegel te laden. Dit werd gedaan voor de complexen op basis van het
histonbindend peptide.
Als de resultaten van deze gel nu vergeleken wordt met gel 5.3 uit 5.1.2., zien we
geen verschil (gel 5.4). Hieruit kan besloten worden dat de pDNA/peptide‐complexen
niet uiteenvallen in de media gebruikt voor celtransfecties.
Foto 5.4 Agarosegel van pDNA/histonbindend peptide‐mengsels met dezelfde +/‐ ladingsratio’s als bij 5.1.2, op dezelfde manier geladen als 5.1.2. Bovenste rij toont de complexen gesuspendeerd in
Optimem, onderste rij zijn complexen gesuspendeerd in DMEM‐F12.
4.5. TRANSFECTIES MET TERTIAIRE PDNA‐COMPLEXEN
De stalen worden aangemaakt zoals besproken onder 3.3. Elk staal wordt echter in
drievoud aangemaakt. Een complex‐oplossing waar geen lipofectamine2000 wordt aan
toegevoegd en 2 complex‐oplossingen waaraan respectievelijk 1,5 µl en 3 µl
lipofectamine2000 wordt toegevoegd. De transfectie‐effeciëntie wordt bepaald bij
onbehandelde, gearresteerde en gesynchroniseerde HeLa‐cellen m.b.v. flow cytometrie.
De percentages GFP‐positieve cellen van alle stalen worden samengevat in grafieken 5.11,
5.12 en 5.13.
38
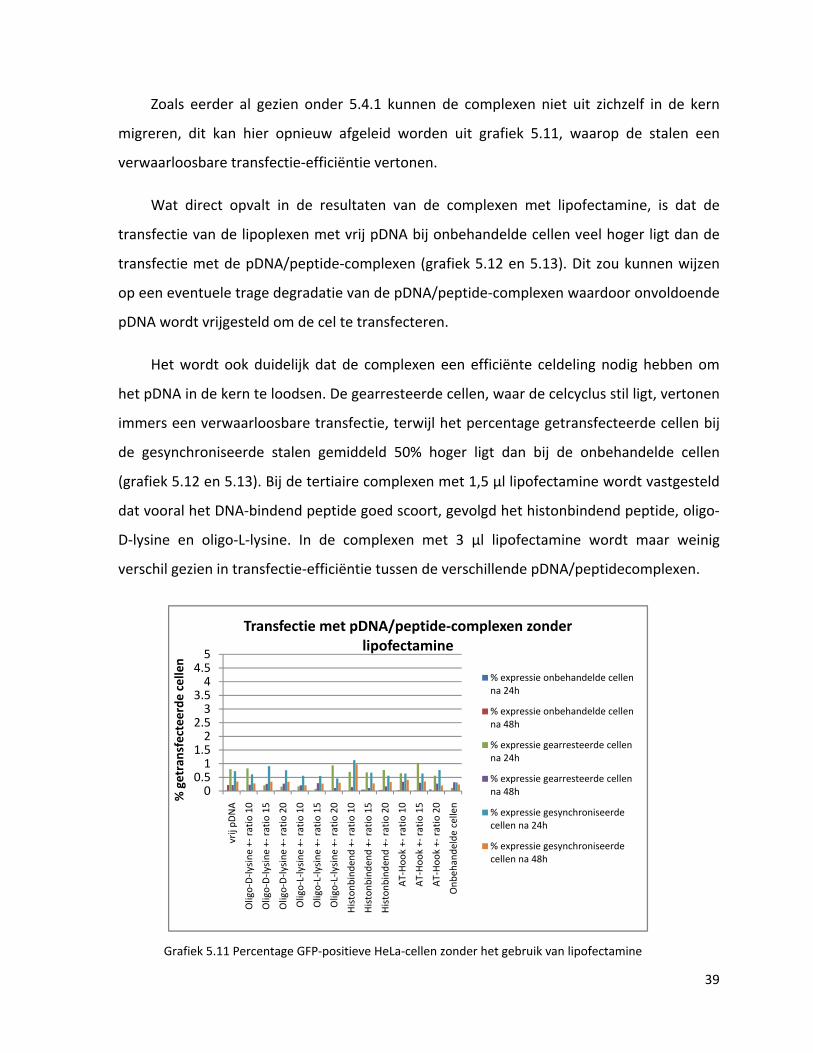
Zoals eerder al gezien onder 5.4.1 kunnen de complexen niet uit zichzelf in de kern
migreren, dit kan hier opnieuw afgeleid worden uit grafiek 5.11, waarop de stalen een
verwaarloosbare transfectie‐efficiëntie vertonen.
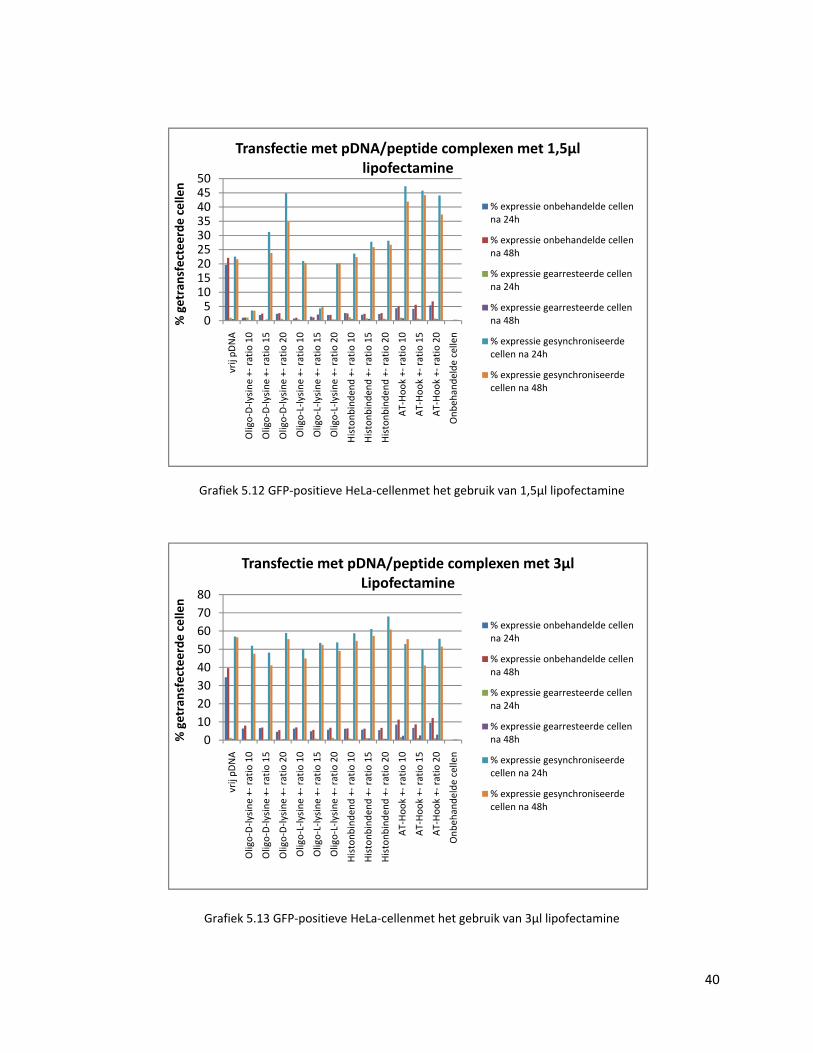
Wat direct opvalt in de resultaten van de complexen met lipofectamine, is dat de
transfectie van de lipoplexen met vrij pDNA bij onbehandelde cellen veel hoger ligt dan de
transfectie met de pDNA/peptide‐complexen (grafiek 5.12 en 5.13). Dit zou kunnen wijzen
op een eventuele trage degradatie van de pDNA/peptide‐complexen waardoor onvoldoende
pDNA wordt vrijgesteld om de cel te transfecteren.
Het wordt ook duidelijk dat de complexen een efficiënte celdeling nodig hebben om
het pDNA in de kern te loodsen. De gearresteerde cellen, waar de celcyclus stil ligt, vertonen
immers een verwaarloosbare transfectie, terwijl het percentage getransfecteerde cellen bij
de gesynchroniseerde stalen gemiddeld 50% hoger ligt dan bij de onbehandelde cellen
(grafiek 5.12 en 5.13). Bij de tertiaire complexen met 1,5 µl lipofectamine wordt vastgesteld
dat vooral het DNA‐bindend peptide goed scoort, gevolgd het histonbindend peptide, oligo‐
D‐lysine en oligo‐L‐lysine. In de complexen met 3 µl lipofectamine wordt maar weinig
verschil gezien in transfectie‐efficiëntie tussen de verschillende pDNA/peptidecomplexen.
00.51
1.52
2.53
3.54
4.55
vrij pD
NA
Oligo‐D‐lysine
+‐ratio 10
Oligo‐D‐lysine
+‐ratio 15
Oligo‐D‐lysine
+‐ratio 20
Oligo‐L‐lysine
+‐ratio 10
Oligo‐L‐lysine
+‐ratio 15
Oligo‐L‐lysine
+‐ratio 20
Histonb
inde
nd +‐ratio 10
Histonb
inde
nd +‐ratio 15
Histonb
inde
nd +‐ratio 20
AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
Onb
ehande
lde cellen
% getransfecteerde cellen
Transfectie met pDNA/peptide‐complexen zonder lipofectamine
% expressie onbehandelde cellen na 24h
% expressie onbehandelde cellen na 48h
% expressie gearresteerde cellen na 24h
% expressie gearresteerde cellen na 48h
% expressie gesynchroniseerde cellen na 24h
% expressie gesynchroniseerde cellen na 48h
39
Grafiek 5.11 Percentage GFP‐positieve HeLa‐cellen zonder het gebruik van lipofectamine
05101520253035404550
vrij pD
NA
Oligo‐D‐lysine
+‐ratio 10
Oligo‐D‐lysine
+‐ratio 15
Oligo‐D‐lysine
+‐ratio 20
Oligo‐L‐lysine
+‐ratio 10
Oligo‐L‐lysine
+‐ratio 15
Oligo‐L‐lysine
+‐ratio 20
Histonb
inde
nd +‐ratio 10
Histonb
inde
nd +‐ratio 15
Histonb
inde
nd +‐ratio 20
AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
Onb
ehande
lde cellen
% getransfecteerde cellen
Transfectie met pDNA/peptide complexen met 1,5µllipofectamine
% expressie onbehandeldena 24h
cellen
% expressie onbehandeldena 48h
cellen
% expressie gearrestna 24h
eerde cellen
% expressie gearrestna 48h
eerde cellen
% expressie cellen na 24h
gesynchroniseerde
% expressie cellen na 48h
gesynchroniseerde
01020304050607080
vrij pD
NA
Oligo‐D‐lysine
+‐ratio 10
Oligo‐D‐lysine
+‐ratio 15
Oligo‐D‐lysine
+‐ratio 20
Oligo‐L‐lysine
+‐ratio 10
Oligo‐L‐lysine
+‐ratio 15
Oligo‐L‐lysine
+‐ratio 20
Histonb
inde
nd +‐ratio 10
Histonb
inde
nd +‐ratio 15
Histonb
inde
nd +‐ratio 20
AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
Onb
ehande
lde cellen
% getransfecteerde cellen
Transfectie met pDNA/peptide complexen met 3µl Lipofectamine
% expressie onbehandelde cellen na 24h
% expressie onbehandelde cellen na 48h
% expressie gearresteerde cellen na 24h
% expressie gearresteerde cellen na 48h
% expressie gesynchroniseerde cellen na 24h
% expressie gesynchroniseerde cellen na 48h
Grafiek 5.12 GFP‐positieve HeLa‐cellenmet het gebruik van 1,5µl lipofectamine
Grafiek 5.13 GFP‐positieve HeLa‐cellenmet het gebruik van 3µl lipofectamine
40

4.6. GEBRUIK VAN ELEKTROPORATIE VOOR TRANSFECTIES MET PDNA/PEPTIDE‐COMPLEXEN
Vooreerst worden cellen, die samen
met 150 µl complex‐oplossing werden
geëlektroporeerd, bekeken onder de
fluorescentiemicroscoop waarbij te zien
was dat sommige cellen getransfecteerd
waren en GFP tot expressie brachten.
Hieruit kan afgeleid worden dat
elektroporatie een bruikbare techniek is
om de pDNA/peptide‐complexen in de
cel te loodsen. Figuur 5.2 HeLa‐cellen geëlektroporeerd met
pDNA/oligo‐L‐lysine complexen. Groene cellen zijn getransfecteerd en vertonen GFP‐expressie.
De stalen werden aangemaakt en geëlektroporeerd zoals beschreven onder 3.3 en na
24h en 48h m.b.v. flow cytometrie bestudeerd. De experimenten werden tweemaal
uitgevoerd op zowel onbehandelde, gearresteerde als gesynchroniseerde cellen, éénmaal
met het pDNA gWIZ‐GFP en éénmaal met het pDNA pEGFP‐N1.
4.6.1. Elektroporatie met gWIZ‐GFP/peptide‐complexen
De flow‐resultaten worden samengevat in grafieken 5.13, 5.14 en 5.15.
De algemene trend bij de drie grafieken is dat de transfectie‐percentages bij de
complexen met het histonbindend peptide telkens lager liggen dan de rest van de stalen.
Eén van de mogelijke verklaringen hiervoor zou kunnen zijn dat de degradatie van deze
complexen veel minder vlot verloopt dan die van de complexen met de overige
peptiden, waardoor het pDNA onvoldoende vrij komt in de kern voor transfectie.
41

Bij deze experimenten komt een groot transfectie‐percentage voor bij de
gearresteerde cellen, die soms zelfs hoger ligt dan het percentage bij de onbehandelde
cellen (grafiek 5.13 en 5.14). Dit wordt normaal niet verwacht omdat de onbehandelde
cellen nog asynchroon delen en de gearresteerde cellen helemaal niet meer delen. Een
mogelijke verklaring hiervoor is dat het kernmembraan ook aangetast wordt door de
elektroporatie waardoor de complexen rechtstreeks in de kern terecht kunnen komen
zonder dat hiervoor celdeling nodig is. Dit kan ook de reden zijn waarom de transfectie‐
efficiëntie van de stalen met het vrij pDNA zo hoog ligt (grafiek 5.13; 5.14; 5.15). De
resultaten tonen ook aan dat het percentage GFP‐positieve cellen bij de gearresteerde
cellen bijna verdubbeld na 48h (grafiek 5.14). Dit kan wijzen op het heropstarten van de
celcyclus doordat het thymidine aanwezig in het medium van de gearresteerde cellen
waarschijnlijk uitgewerkt is. Hierdoor gaan de cellen terug delen en kunnen de
pDNA/peptide‐complexen die nog aanwezig zijn in het cytosol, en dus nog niet in de
kern geloodst waren door elektroporatie, in de kern migreren dankzij de celdeling.
De stalen met de lipoplexen op basis van pDNA en lipofectamine scoren ook zowel
bij de onbehandelde, de gearresteerde als de gesynchroniseerde cellen slecht (grafiek
5.13; 5.14; 5.15). Dit komt doordat de gevormde lipoplexen zijn lipiden waarschijnlijk
nergens kwijt kan, waardoor het pDNA niet kan worden vrijgesteld. In normale
omstandigheden zouden de lipiden van de lipoplexen samensmelten met het
celmembraan of endosoommembraan en zo het pDNA in het cytosol loodsen. Maar door
de elektroporatie wordt een deel van de lipoplexen in zijn geheel in het cytosol en de
celkern geloodst. De transfectie die hier voorkomt is dan waarschijnlijk afkomstig van vrij
pDNA dat niet werd ingesloten in lipoplexen en pDNA dat uit de lipoplexen kon
ontsnappen door samensmelting met membraanstructuren in de cel.
De resultaten bij de gesynchroniseerde cellen tonen een bijna dubbel zo groot
transfectie‐percentage in vergelijking met dat van de onbehandelde en gearresteerde
cellen (grafiek 5.13; 5.14; 5.15). Dit onderstreept nogmaals het belang van de celdeling
om de pDNA/peptide‐complexen in de kern te kunnen loodsen. De resultaten na 48h zijn
42

ongeveer gelijk aan die na 24h. Een extra celdeling zorgt hier dus niet voor een groter
percentage getransfecteerde cellen.
De controle‐peptiden scoren in grafieken 5.14 en 5.15 minstens even goed, en
soms beter dan de chromatine‐bindende peptiden. Er kan echter geen besluit genomen
worden over het nut van het gebruik van chromatine‐bindende peptiden voor het
verhogen van celtransfecties in delende cellen. Elektroporatie is namelijk geen goede
methode om het verschil tussen chromatine‐bindende peptiden en controle‐peptiden
voor transfectie aan te tonen, aangezien ook het kernmembraan wordt aangetast. Dit
kan gesteld worden doordat de stalen met het vrij pDNA bijna even goede resultaten
hebben als de andere stalen. De peptiden hebben in deze experimenten dus geen
toegevoegde waarde.
05
1015202530354045
Vrij pD
NA
Lipo
fectam
ine 2000
Oligo‐D‐Lysine +‐ratio
5Oligo‐D‐Lysine +‐ratio
10
Oligo‐D‐Lysine +‐ratio
15
Oligo‐D‐Lysine +‐ratio
20
Oligo‐D‐Lysine +‐ratio
30
Oligo‐D‐Lysine +‐ratio
40
Oligo‐L‐Lysine
+‐ratio 5
Oligo‐L‐Lysine
+‐ratio 10
Oligo‐L‐Lysine
+‐ratio 15
Oligo‐L‐Lysine
+‐ratio 20
Oligo‐L‐Lysine
+‐ratio 30
Oligo‐L‐Lysine
+‐ratio 40
Histonb
inde
n +‐ratio
5Histonb
inde
n +‐ratio
10
Histonb
inde
n +‐ratio
15
Histonb
inde
n +‐ratio
20
Histonb
inde
n +‐ratio
30
Histonb
inde
n +‐ratio
40
AT‐Hoo
k +‐
ratio
5AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
AT‐Hoo
k +‐
ratio
30
AT‐Hoo
k +‐
ratio
40
Controle cellen% getransfecteerde cellen
Percentage getransfecteerde ONBEHANDELDE cellen met gWIZ‐GFP/peptide‐complexen
transfectie na 24h
transfectie na 48h
Grafiek 5.13 Percentage GFP‐positieve onbehandelde cellen na elektroporatie met vrij gWIZ‐GFP, gWIZ‐GFP‐
lipoplexen of gWIZ‐GFP/peptide‐complexen.
43
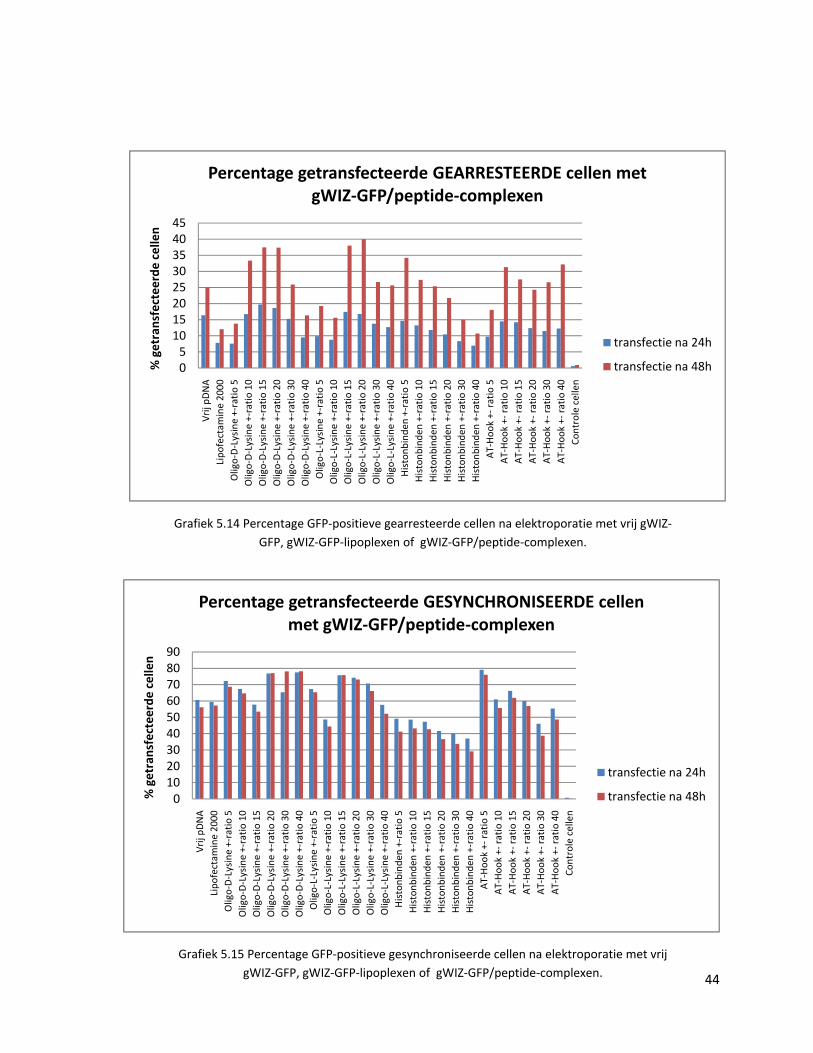
44
05
1015202530354045
Vrij pD
NA
Lipo
fectam
ine 2000
Oligo‐D‐Lysine +‐ratio
5Oligo‐D‐Lysine +‐ratio
10
Oligo‐D‐Lysine +‐ratio
15
Oligo‐D‐Lysine +‐ratio
20
Oligo‐D‐Lysine +‐ratio
30
Oligo‐D‐Lysine +‐ratio
40
Oligo‐L‐Lysine
+‐ratio 5
Oligo‐L‐Lysine
+‐ratio 10
Oligo‐L‐Lysine
+‐ratio 15
Oligo‐L‐Lysine
+‐ratio 20
Oligo‐L‐Lysine
+‐ratio 30
Oligo‐L‐Lysine
+‐ratio 40
Histonb
inde
n +‐ratio
5Histonb
inde
n +‐ratio
10
Histonb
inde
n +‐ratio
15
Histonb
inde
n +‐ratio
20
Histonb
inde
n +‐ratio
30
Histonb
inde
n +‐ratio
40
AT‐Hoo
k +‐
ratio
5AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
AT‐Hoo
k +‐
ratio
30
AT‐Hoo
k +‐
ratio
40
Controle cellen
% getransfecteerde cellen
Percentage getransfecteerde GEARRESTEERDE cellen met gWIZ‐GFP/peptide‐complexen
transfectie na 24h
transfectie na 48h
0102030405060708090
Vrij pD
NA
Lipo
fectam
ine 2000
Oligo‐D‐Lysine +‐ratio
5Oligo‐D‐Lysine +‐ratio
10
Oligo‐D‐Lysine +‐ratio
15
Oligo‐D‐Lysine +‐ratio
20
Oligo‐D‐Lysine +‐ratio
30
Oligo‐D‐Lysine +‐ratio
40
Oligo‐L‐Lysine
+‐ratio 5
Oligo‐L‐Lysine
+‐ratio 10
Oligo‐L‐Lysine
+‐ratio 15
Oligo‐L‐Lysine
+‐ratio 20
Oligo‐L‐Lysine
+‐ratio 30
Oligo‐L‐Lysine
+‐ratio 40
Histonb
inde
n +‐ratio
5Histonb
inde
n +‐ratio
10
Histonb
inde
n +‐ratio
15
Histonb
inde
n +‐ratio
20
Histonb
inde
n +‐ratio
30
Histonb
inde
n +‐ratio
40
AT‐Hoo
k +‐
ratio
5AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
AT‐Hoo
k +‐
ratio
30
AT‐Hoo
k +‐
ratio
40
Controle cellen
% getransfecteerde cellen
Percentage getransfecteerde GESYNCHRONISEERDE cellen met gWIZ‐GFP/peptide‐complexen
transfectie na 24h
transfectie na 48h
Grafiek 5.14 Percentage GFP‐positieve gearresteerde cellen na elektroporatie met vrij gWIZ‐GFP, gWIZ‐GFP‐lipoplexen of gWIZ‐GFP/peptide‐complexen.
Grafiek 5.15 Percentage GFP‐positieve gesynchroniseerde cellen na elektroporatie met vrij gWIZ‐GFP, gWIZ‐GFP‐lipoplexen of gWIZ‐GFP/peptide‐complexen.

4.6.2. Elektroporatie met EGFP‐N1/peptide‐complexen
De flowresultaten werden samengevat in grafieken 5.16, 5.17 en 5.18.
De besluiten uit de resultaten zijn gelijkaardig met deze van 5.6.1. Hier ligt het
transfectie‐percentage van de complexen met het histonbindend peptide en van de
complexen met het DNA‐bindend peptide lager dan bij de complexen met de controle‐
peptiden (grafieken 5.16; 5.17; 5.18). Een mogelijke verklaring hiervoor is dat deze twee
peptiden sterkere complexen vormen met pEGFP‐N1 in vergelijking met de twee
controle‐peptiden. Hierdoor zal het uiteenvallen van de complexen minder vlot
verlopen, met een lagere vrijstelling van pDNA en dus een kleiner aantal
getransfecteerde cellen als gevolg.
De resultaten met pEGFP‐N1 zijn duidelijk beter dan die met gWIZ‐GFP. Dit kan
afgeleid worden uit het aantal GFP‐positieve cellen in de stalen met vrij pDNA. (Grafiek
5.13 tot 5.18).
05
101520253035404550
Vrij pD
NA
Lipo
fectam
ine 2000
Oligo‐D‐Lysine +‐ratio
5Oligo‐D‐Lysine +‐ratio
10
Oligo‐D‐Lysine +‐ratio
15
Oligo‐D‐Lysine +‐ratio
20
Oligo‐D‐Lysine +‐ratio
30
Oligo‐D‐Lysine +‐ratio
40
Oligo‐L‐Lysine
+‐ratio 5
Oligo‐L‐Lysine
+‐ratio 10
Oligo‐L‐Lysine
+‐ratio 15
Oligo‐L‐Lysine
+‐ratio 20
Oligo‐L‐Lysine
+‐ratio 30
Oligo‐L‐Lysine
+‐ratio 40
Histonb
inde
n +‐ratio
5Histonb
inde
n +‐ratio
10
Histonb
inde
n +‐ratio
15
Histonb
inde
n +‐ratio
20
Histonb
inde
n +‐ratio
30
Histonb
inde
n +‐ratio
40
AT‐Hoo
k +‐
ratio
5AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
AT‐Hoo
k +‐
ratio
30
AT‐Hoo
k +‐
ratio
40
Controle cellen
% getransfecteerde cellen
Percentage getransfecteerde onbehandelde cellen met EGFP‐N1/peptide‐complexen
transfectie na 24h
transfectie na 48h
Grafiek 5.16 Percentage GFP‐positieve onbehandelde cellen na elektroporatie met vrij pEGFP‐N1, pEGFP‐N1‐lipoplexen of pEGFP‐N1/peptide‐complexen.
45

05
1015202530354045
Vrij pD
NA
Lipo
fectam
ine 2000
Oligo‐D‐Lysine +‐ratio
5Oligo‐D‐Lysine +‐ratio
10
Oligo‐D‐Lysine +‐ratio
15
Oligo‐D‐Lysine +‐ratio
20
Oligo‐D‐Lysine +‐ratio
30
Oligo‐D‐Lysine +‐ratio
40
Oligo‐L‐Lysine
+‐ratio 5
Oligo‐L‐Lysine
+‐ratio 10
Oligo‐L‐Lysine
+‐ratio 15
Oligo‐L‐Lysine
+‐ratio 20
Oligo‐L‐Lysine
+‐ratio 30
Oligo‐L‐Lysine
+‐ratio 40
Histonb
inde
n +‐ratio
5Histonb
inde
n +‐ratio
10
Histonb
inde
n +‐ratio
15
Histonb
inde
n +‐ratio
20
Histonb
inde
n +‐ratio
30
Histonb
inde
n +‐ratio
40
AT‐Hoo
k +‐
ratio
5AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
AT‐Hoo
k +‐
ratio
30
AT‐Hoo
k +‐
ratio
40
Controle cellen
% getransfecteerde cellen
Percentage getransfecteerde gearresteerde cellen met EGFP‐N1/peptide‐complexen
transfectie na 24h
transfectie na 48h
Grafiek 5.17 Percentage GFP‐positieve gearresteerde cellen na elektroporatie met vrij pEGFP‐N1, pEGFP‐N1‐lipoplexen of pEGFP‐N1/peptide‐complexen.
010
405060708090
Vrij pD
NA
Lipo
fectam
ine 2000
Oligo‐D‐Lysine +‐ratio
5Oligo‐D‐Lysine +‐ratio
10
Oligo‐D‐Lysine +‐ratio
15
Oligo‐D‐Lysine +‐ratio
20
Oligo‐D‐Lysine +‐ratio
30
Oligo‐D‐Lysine +‐ratio
40
Oligo‐L‐Lysine
+‐ratio 5
Oligo‐L‐Lysine
+‐ratio 10
Oligo‐L‐Lysine
+‐ratio 15
Oligo‐L‐Lysine
+‐ratio 20
Oligo‐L‐Lysine
+‐ratio 30
Oligo‐L‐Lysine
+‐ratio 40
Histonb
inde
n +‐ratio
5Histonb
inde
n +‐ratio
10
Histonb
inde
n +‐ratio
15
Histonb
inde
n +‐ratio
20
Histonb
inde
n +‐ratio
30
Histonb
inde
n +‐ratio
40
AT‐Hoo
k +‐
ratio
5AT‐Hoo
k +‐
ratio
10
AT‐Hoo
k +‐
ratio
15
AT‐Hoo
k +‐
ratio
20
AT‐Hoo
k +‐
ratio
30
AT‐Hoo
k +‐
ratio
40
Controle cellen
% getransfecteerde cellen
Percentage getransfecteerde gesynchroniseerde cellen met EGFP‐N1/peptide‐complexen
2030
transfectie na 24h
Grafiek 5.17 Percentage GFP‐positieve gesynchroniseerde cellen na elektroporatie met vrij pEGFP‐N1, pEGFP‐N1‐lipoplexen of pEGFP‐N1/peptide‐complexen.
46

5. BESLUIT
De gevormde pDNA/peptide‐complexen zijn stabiel in water, hepes, Optimem en DMEM‐
F12 en kunnen dus gebruikt worden in de transfectie‐experimenten van HeLa‐cellen (4.1.2 en
4.4.2).
Uit de resultaten van de Xenopus chromatine binding kan besloten worden dat alle
gebruikte pDNA/peptide‐complexen in staat zijn tot binden met het Xenopus chromatine. Er kan
echter niet gezegd worden dat de chromatine‐bindende peptiden beter binden dan de controle‐
peptiden door verschillende factoren zoals besproken onder 4.2.
Bij de experimenten besproken onder 4.4., kan besloten worden dat de pDNA/peptide‐
complexen niet voor transfectie zorgen als zij gewoon op de cellen gebracht worden, tenzij er
lipoplexen worden gevormd met lipofectamine.
De transfecties met de tertiaire complexen tonen aan dat de lipoplexen met vrij pDNA
minstens voor een even goede transfectie zorgen als die met pDNA/peptide‐complexen. Dit kan
het gevolg zijn van een trage decomplexatie van de pDNA/peptide‐complexen zoals besproken
onder 4.5.
De resultaten uit de elektroporatie‐experimenten tonen geen betere transfectie‐efficiëntie
met de pDNA/chromatineindende peptide‐complexen dan met de pDNA/controle peptide‐
complexen. Er kan echter niet besloten worden dat de chromatine‐bindende peptiden minder
effectief zijn dan de controle‐peptiden omdat elektroporatie niet de geschikte methode blijkt
om deze met elkaar te vergelijken. Elektroporatie gaat de kern mee elektroporeren waardoor
het vrij pDNA en pDNA‐complexen niet alleen door celdeling (zoals gewild) in de kern kunnen
belanden . Er is echter wel opnieuw aangetoond dat celdeling een héél positieve invloed heeft
op celtransfectie (4.6). Er zal dus op zoek moeten gegaan worden naar een geschikte methode
om de pDNA‐complexen in het cytosol te loodsen, voordat een besluit kan genomen worden
over de effectiviteit van de chromatine‐bindende peptiden als transfectiemiddel voor pDNA in
delende cellen.
47

6. BIBLIOGRAFIE
Aravind, L.; Landsman, D. (1998). AT‐hook motifs identified in a wide variety of DNA‐binding
proteins. Nucleic Acids Research. Vol. 26, 19, 4413‐4421.
Barbera, A.J. et al. (2006). The nucleosomal surface as a docking station for Kaposi’s sarcoma
herpesvirus LANA. Science. 311, 856‐861.
Belting, M.; Sandgren, S.; Wittrup, A. (2004) Nuclear delivery of macromolecules: barriers and
carriers. Advanced Drug Delivery Reviews. 57, 505– 527.
Bieber, T.; Meissner, W.; Kostin, S., et al. (2002). Intracellular route and transcriptional
competence of polyethyleneimine‐DNA complexes. Journal of controlled release. Vol.82, 2‐3,
441‐454.
Buyens, K.; Lucas, B.; Raemdonck, K.; Braeckmans, K., et al. (2008). A fast and sensitive method
for measuring the integrity of siRNA‐carrier complexes in full human serum. Journal of
Controlled Release. 126, 67–76.
Chen, S‐H.; Shine, H.D.; Goodman J.C., et al. (1993). Gene therapy for brain tumors: Regression
of experimental gliomas by adenovirus‐mediated gene transfer in vivo. Medical Sciences. 91,
3054‐3057.
Dileo, J.; Miller, T.E.; Chesnoy, S.; Huang, L. (2003). Gene transfer to subdermal tissues via a new
gene gun design. Human Gene Therapy. 14, 79–87.
48

Fasbender, A.; Zabner, J.; Zeiher, B.G.; Welsh, M.J. (1997). A low rate of cell proliferation and
reduced DNA uptake limit cationic lipid‐mediated gene transfer to primary cultures of ciliated
human airway epithelia. Gene Therapy. 4, 1173‐1180.
Favard, C.; Dean, S.D.; Rols M‐P. (2007). Electrotransfer as a Non Viral Method of Gene Delivery.
Current Gene Therapy. 7, 67‐77.
Hashida, M.; Mahato, R.I.; Kawabata, K.; Miyao, T.; Nishikawa, M.; Takakura, Y. (1996).
Pharmacokinetics and targeted delivery of proteins and genes. Journal of Controlled Release. 41,
91‐97.
Lénárt, P.; Ellenberg, J. (2006). Monitoring the permeability of the nuclear envelope during the
cell cycle. Methods. 38, 17–24.
Mesika, A.; Kiss, V.; Brumfeld, V.; Ghosh, G.; Reich, Z. (2005). Enhanced intracellular mobility
and nuclear accumulation of dna plasmids associated with a karyophilic protein. Human gene
therapy. 16, 200–208.
Miller, D.G.; Adam M.A.; Miller, A.D. (1990). Gene transfer by retrovirus vectors occurs only in
cells that are actively replicating at the time of infection. Molecular and Cellular Biology. Vol.10,
8, 4239‐4242.
Mönkkönen, J.; Urtti, A. (1998). Lipid fusion in oligonucleotide and gene delivery with cationic
lipids. Advanced Drug Delivery Reviews. 34, 37–49.
49

50
Mortimer, I.; Tam, P.; Maclachlan, I.; Graham, R.W., et al. (1999). Cationic lipid‐mediated
transfection of cells in culture requires mitotic activity. Gene Therapy. 6, 403‐411.
Mountain, A. (2000). Gene therapy: The first decade. Trends in Biotechnology. Vol.18, 3, 119‐
128.
Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. (1993). Integration of murine leukemia virus DNA
depends on mitosis. The EMBO Journal. Vol. 12, 5, 2099‐2108.
Roussel, L.; Erard, M.; Cayrol, C., et al. (2008). Molecular mimicry between IL‐33 and KSHV for
attachment to chromatin through the H2A‐H2B acidic pocket. EMBO reports. Vol.9, 10, 1006‐
1012.
Tseng, W‐C.; Haselton, F.R.; Giorgio T.D. (1999). Mitosis enhances transgene expression of
plasmid delivered by cationic liposomes. Biochimica et Biophysica Acta. 1445, 53‐64.
Valencia‐Sanchez, M.A.; Liu, J.; Hannon G.J. (2006). Control of translation and mRNA
degradation by miRNA’s and siRNA’s. Genes Dev. 20, 515‐524.
Van der Aa, M.A.E.M.; Mastrobattista, E.; Oosting R.S., et al. (2006). The Nuclear Pore Complex:
The Gateway to Successful Nonviral Gene Delivery. Pharmaceutical Research. Vol.23, 3, 447‐
459.
Wilke, M.; Fortunati E.; van den Broeck, M., et al. (1996). Efficacy of a peptide‐based gene
delivery system depends on mitotic activity. Gene Therapy. 3, 1133‐1142.
Wolff, J.A.; Ludtke, J.; Acsadi, G., et al. (1992). Very long‐term persistence of plasmid DNA and
foreign gene‐expression in mouse muscle. Clinical Research. Vol.40, 2, A339‐A339.