genética de las displasias óseas - sccalp.org · tipo de herencia, tal como los recogidos en las...
TRANSCRIPT

Bol Pediatr 1989, 30 365 - 373
Genética de las displasias Óseas
Las Displasias Oseas son anomalías del crecimiento, desarrollo y modelación del cartílago y10 hueso, debidas probable- mente a errores bioquímicos innatos e in- trínsecos al esqueleto y hasta donde se sa- be, todas ellas de base génica monogéni- ca: Dominantes y Recesivas, más Autosó- micas que Gonosómicas (&) (1-5).
Su manifestación clínica, en forma de enanismo disarmónico, fragilidad ósea, bultos y deformidades, hizo de estos seres humanos objeto de burlas, miedo ances- tral, moda de la corte española de los Austria (las sabandijas), reflejados por Velázquez: Mari-Bárbola en «Las Meninaso y «Don Sebastián de Morra,>, ambos pro- bablemente acondroplásicos, don Diego de Acedo en «El Filósofou, verosímilmente pseudoacondroplásico; y tema literaric desde eEl licenciado Vidrierau de Cervan- res a los múltiples cuentos infantiles en donde el ser minúsculo (Los 7 enanitos, El enano saltarín ...) es el protagonista de la bondad o la maldad. Todo este mundo de admiración y respeto sigue vigente en al- guna parte de nuestra sociedad y de ahí la importancia de explicar a sus padres, cuando accidentalmente nace un niño así, las razones cientzcas del ¿por qué? Ver- daderamente su estudio es complejo pues
integran este capítulo múltiples enferme- dades, raras en su mayoría, aunque en conjunto significan un 3 11.000 de recién nacidos vivos y muertos y 5,6 nuevos casoslaño en nuestra consulta de Genética Pediátric$(Bbla 1) (85 c-os en 15 años), complicándose aún más la situación, por los cambios evolutivos de la propia enfer- medad y la terminología frecuentemente no concordante.
Su conocimiento. diferenciación y pa- trón hereditario interesan al genetista cií- nico, dado que estas enfermedades y ano- malías se pueden prevenir en parte, a tra- vés del hecho más práctico que hoy día la genética puede ofrecer: el Consejo Genéti- co (6). Este, como información de la exis- tencia o no de un riesgo de enfermar, de lo que puede ocurrir y no de lo que va a wceder, es un acto médico científico y hu- &no, no limitado a dar una cifra de apa- rición o de recurrencia, sino extensivo a explicar el alcance evolutivo de la afec- ción, las perspectivas vitales, las posibili- dades terapéuticas y las preventivas. Por ello, es necesario que el médico, dé todo el apoyo humano en esos momentos de frustración biológica en que unos padres descubren que los genes dados al hijo han fallado. Se ha de ser claro y objetivo, eli-
Rerumen de ia Participación en /a Mera Redondo: rDiipiaiai óiearn en el 11 Memona Pmf rGui(iermo Arceu. Sontmder 6-20-1989.
Sección de Genética Pediátnba. Horpifai Oñiveriitan'o nNtra. Sra. de Cou~dongau. Depart~mento de Medi- cina de ia Uniuerridsd de Ouiedo.

366 J. FERNANDEZ TORAL Y COLS.
TABLA 1. CASUISTICA PERSONAL (SECCION DE GENETICA PEDIATRICA-HTAL. UNIVER- SITARIO ~Nt r a . Sra. de Covadonga~. OVIEDO)
OSTEOCONDRODISPLASIAS (Clasifinrión dc Paris. 1983)
1." Dcfcrros dcl rrcrimicnro dc los hucsos largos yio vérrcbras
a) Idcntificablcs al natimicnto: Parrón Gcnériro
- Letales:
Acondrogénerir . . . . . . . . . . . . . . . . . . . 2 (ambos tipo 11) . . . . . . . . . . . . . . . . . . . . . AR Displasia Tanarófora . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD y AR Síndromcs Cosrillas Coiras . . . 1 (forma lcral dc la displasia rorácira) . AR
- Habirualmcnrc no lcralcs (o no can prccozmcnrc):
Condrodisplasia punrtata . . . . . . . . . . 2 (1 gravc y aria lcvc) . . . . . . . . . . . . . . . . . AR y ADIFenocopias Displasia cmpomélVa . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR Acondioplaria . . . . . . . . . . . . . . . . . . . . 7 (todas erpoiádicas) . . . . . . . . . . . . . . . . . AD (Muración por
anosidad par.) Dirplasia Diasrrótica . . . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR Displasia Metatrópica . . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Aü y A8 D. Condraccradéimica (Ellis V.C.) . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR Displasia Toraco.Arfuiante Ueunc) . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR Dirplasia de Knicrr . . . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD Displasia mcsomélira de Langcr . 2 (hermanar) (padicr ron Dirrondrosrcosis) AR (Discondrosrroiis
Homzig) Displasia Acromeromélica . . . . . . . . . . 2 (no em~arentadai) . . . . . . . . . . . . . . . . . . AR Displasia Clcidoirancal . . . . . . . . . . . . 4 (2 son hermanos. 2 esporádicos) . . . . AD y AR ? Sindromc dc Larrcn . . . . . . . . . . . . . . . 1 (2 hcmanos. su madrc y 2 rrporádicos) . AD y AR
b) Idcnrificablci cn cl curso dcl ciccimicnco:
Hipocondroplasia . . . . . . . . . . . . . . . . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD Dircondrorrcorir de Lcri . . . . . . . . . . . . 2 (padres de 2 Langer) . . . . . . . . . . . . . . . . . AD Condrodisplasia Mciafisaria . . . . . . . . . 1 (Schmid) . . . . . . . . . . . . . . . . . . . . . . . . . . AD Displasia EspondilomcnfUaria . . . 3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD Displasia pciférica dc Braislford . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD y (AR? Seudoacondroplasia . . . . . . . . . . . . . . . 3 (padre y dos hijos) . . . . . . . . . . . . . . . . . . AD y AR Triro Rino Rlangia (Gicdion) . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD
2 . - Desarrollo Anárquico del Cartílago y Componentes Fibraros del kqucieto
Exorroris carrilaginosa Múlriplc . . . . . . 12 (8 familias. 4 casos son crporádicos) . . . Aü Eniondromatoris con Hemagiomas . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ¿NO? ¿AD?
3." Anomdias de la Densidad dc la Corrical Diafisaria y10 dcl Modclado Mcraiirario
Osrcogéncris Impcifecra . . . . 18
CLASIFICACION DE SIILENCE: Tipo IA. . . . 6 ............ AD IB. . . . . 4 ............ AD 11.. . . . 3 ............ AD y AR 111. . . . . 4 ............ AD y AR IVA. . . . 1 ............ AD
Pirnodisástosis . . . . . . . . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR Osrcopcrrosis . . . . . . . . . . . . . . . . . . . . . 2 (1 grave prccoz. 1 iardia rbcnigna,) . AR y AD Dispiasia Crancomctafiraria . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD Dirorreorclerorir . . . . . . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . .. ... . AR y EX Hipcrosrorir Corticd Infantil . . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AD
- TOTAL . . . . . . . . . . . . . . . . . 81 raros
AD: auraiámica dominante. AR: autorómira rcccriva. DJX: dominante ligada a X. RLX: rcccsiva ligada a X

GENETICA DE LAS DISPLASIAS OSEAS 367
tivo, eliminando erróneos sentimientos de culpabilidad o castigo en los progenitores, y comprobando que las explicaciones han sido bien comprendidas siendo preciso evi- tar caer en la tentación de «dirigir» la con- ducta de los afectados respecto a actitudes futuras en relación a su desdencencia. Así, la pareja puede tomar una decisión libre y responsable sobre los hijos nos vemos, va- lorando argumentos de riesgo numérico, gravedad, supervivencia. . . y otros sociales, religiosos y privados.
Para que el asesoramiento genético sea eficaz, es preciso que se apoye en el hecho fundamental del Diagnóstico exacto, ex- cluyendo causas no genéticas como origen de la situación a estudiar y así, la presen- cia de epífisis punteadas en un recién na-
cido, puede correqonder a Fetopatía Al- cohólica, Embriopatia por Warfarina, Tri- somías 18 y 21, S. de Zellweger o bien a las dos formas de Condrodisplasia Puncta- ta: la AD de Conradi-Humermann y la Ri- zomélica, grave y frecuentemente mortal, de patrón AR (fig. l), expresiones de la heterogeneidad de muchas displasias óseas. ~e:;e?tos ejemplos se deduce la ne- cesaria apo6tura en el estudio multidisci- plinario: clínico, radiológico, analítico, histológico.. . en ocasiones con la presión del tiempo vital, pues la gravedad del pa- ciente puede llevarlo a la muerte sin haber podido hacer un diagnóstico preciso. En .la Tabla 11 hemos recogido las Osteocondio- displasias letales pre o connatalmente y las que tienen alto riesgo de serlo precozmen-
FIG. 1. a y b: Epífisis punteadas en un recién nacido, afecto de forma grave autosómica rece- siva
368 J. FERNANDEZ TORAL Y COLS
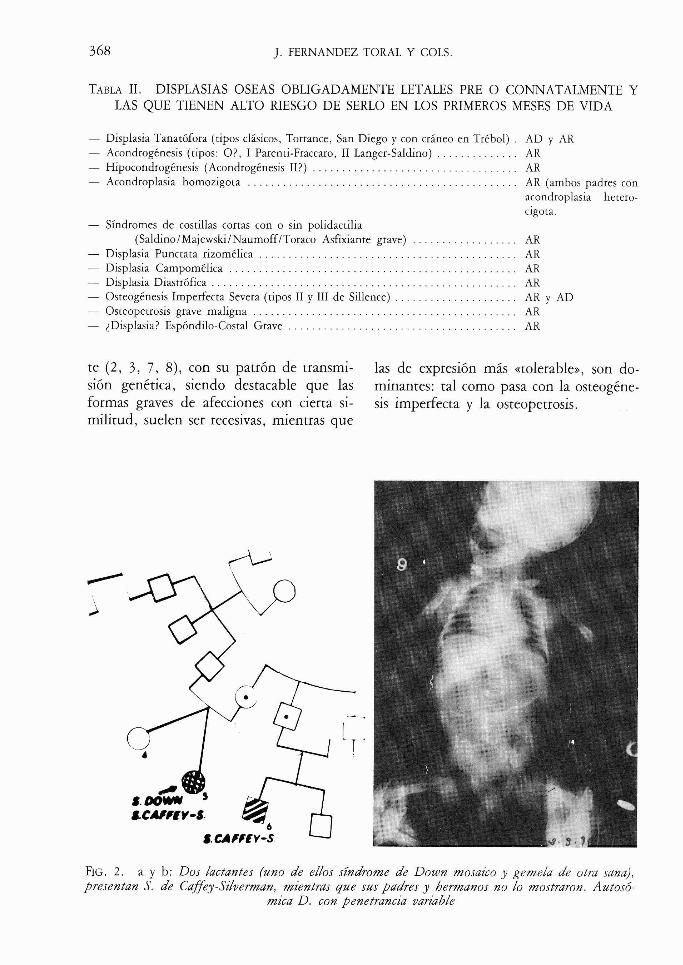
TABLA 11. DISPLASIAS OSEAS OBLIGADAMENTE LETALES PRE O CONNATALMENTE Y LAS QUE TIENEN ALTO RIESGO DE SERLO EN LOS PRIMEROS MESES DE VIDA
- Displasia Tanatófora (tipos clásicos, Torrance, San Diego y con cráneo en Trébol) . AD y AR . . . . . . . . . . . . . . - Acondrogénesis (tipos: O? , 1 Parenti-Fraccaro, 11 Langer-Saldino) AR
- Hipocondrogénesis (Acondrogénesis II?) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR - Acondroplasia homozigota . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR (ambos padres con
acondroplasia hetero- cigota.
- Síndromes de costillas cortas con o sin polidactilia . . . . . . . . . . . . . . . . . . (Saldino/Majewski/NaumoffíToraco Asfixiante grave) AR
- Displasia Punctata rizomélica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR - Displasia Campomélica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR - Displasia Diastrófica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR - Osteogénesis Imperfecta Severa (tipos 11 y 111 de Sillence) . . . . . . . . . . . . . . . . . . . . . AR y AD - Osteopetrosis grave maligna . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR - iDisplasia? Espóndilo-Costal Grave . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . AR
te (2, 3, 7 , 8), con su patrón de transmi- las de expresión más «tolerable», son do- sión genética, siendo destacable que las minantes: tal como pasa con la osteogéne- formas graves de afecciones con cierta si- sis imperfecta y la osteopetrosis. militud, suelen ser recesivas, mientras que
FIG. 2. a y b: Dos lactantes (uno de ellos sindrome de Down mosaico y gemela de otra sana), presentan S. de Caffey-Szlverman, mzentras que sus padres y hermanos no lo mostraron. Autosó-
mica D. con penetrancza variable

GENETICA DE LAS DISPLASIAS OSEAS 369
Otra consideración a tomar es la Pene- trancia o capacidad de manifestarse el gen. Así, en la Hiperóstosis Cortical de Caffey, de patrón AD, se dan fenómenos de gene- ración saltada, o sea, que algunas personas que llevan el gen de la afección no la ma- nifiestan, como apreciamos en la fig. 2 en donde 2 niños de la misma familia presen- taron la enfermedad y los familiares que los unen no la tuvieron (9). Cuando el gen está presente pero la manifestación clínica es variable, de más o menos gravedad o le- vedad, se habla de Expresividad y así ve- mos formas de Exóstosis Múltiples familia- res (10) AD muy floridas (habitualmente varones) frente a ouas atenuadas (fig. 3) o la familia en la que el hijo con Osteogéne- sis Imperfecta Tarda AD tuvo sólo 2 fractu- ras mientras su madre hizo 14 en el mismo plzzo de tiempo.
Lo visto hasta ahora ha de reflejarse en el Arbol Genealógico ( l l ) , elaborado ex- haustivamente, con detalle, registrando consanguinidad, tallas, deformidades.. . a través de documentos lo más objetivos po- sibles como en este caso, las fotografías o la radiología. Vean por ejemplo cómo a Toulouse Lautrec (fig. 4), debido a su ba- : ja talla, la hipoplasia mandibular oculta bajo la barba, el acortamiento de miem- bros, las fracturas de fémur y el ser hijo de pareja consanguínea, se le tipificó (Ma- roteaux) de Picnodisóstn~i~ diagnóstico
FIG. 3 . Exostosis ósea como muestra de afec- FIG. 4. Henri de Toulouse Lautrec, probable- ción generalizada que se transmite bajo patrón mente padeció Picnodisóstosis autosómica rece-
autosómico dominante siva

370 J. FERNANDEZ TORAL Y COLS
imposible si no existieran autorretratos. fotografías y literatura sobre su vida.
A pesar de todo, la duda surge como en cualquier otro aspecto de la medicina. Una afección rara, de diagnóstico no segu- ro, de genética dudosa, que surge esporá- dicamente en una familia «limpia», plan- tea serios problemas al genetista a la hora del asesoramiento. Incluso en entidades bien conocidas, lo excepcional puede dar- se, como en la familia de la fig. 5 en don- de 2 de 3 hijos, varón y hembra, mues- tran la forma completa de la Osteocondro- displasia Cleido Pubo Dento Craneal ( i 2 ) , siendo sus padres clínica y radiológicamen- te normales. La chica ha tenido un hijo no afecto. No hay consanguinidad parental, pero la primera sugerencia sería la de estar ante un patrón AR. Sin embargo, en una familia previa publicada por Hawkins (13), uno de los 3 hermanos afectos hijos de padres indemnes, tuvo 1 hijo con la displasia. Se plantea la duda entre un gen no penetrante en uno de los dos abuelos, la repetición de mutaciones germinales en los gametos de uno de éstos, el mosaico gonadal para esa genopatía específica o in-
FIG. 5 . a) Paciente 111-3 afecto de Dirplasia Cleidocraneal @ente amplia, barbilla pequena, cla- vícuhs hipoplásicas). b) Arbolgenealógico: se plantea la duda de un patrón recesivo

GENETICA DE LAS DISPLASIAS OSEAS 371
FIG. 6 . Deformidad y acortamiento mesoméli- co en un ni60 con S. de Langer: talla baja, ti- bias cortas, piernas incurvadas. . . y padres con
Discondrosteosis
cluso el divorcio entre la paternidad bioló- las situaciones recesivas a fin de evitar sen- gica y la legal. timientos de culpabilidad. Otra considera-
ción es que el consejo genético pierde efi- Estas situaciones excepcionales, de es-
@asa o nula manifestación de un gen do-
Si hemos llegado a una conclusión res- pecto al caso planteado, viene la hora de la información en la que se explica a los padres el resultado del estudio, acompa- ñándonos de esquemas sencillos para cada tipo de herencia, tal como los recogidos en las figuras 7 y 8, que son las situacio- nes más frecuentes. Es preciso ser claros y concretos en la explicación, pues se suele comprender bien la causa de un trauma- tismo o la consecuencia de una infección, pero la relación entre gen y enfermedad es
minante, se dan asimismo en Osteogénesis Imperfecta Tarda (14-15) y de ahí la nece- sidad de objetivar la osteoporosis, la altura de los cuerpos vertebrales.. . estudiar la composición del colágeno en algunas for- mas así como plantear el diagnóstico por DNA; o detectar la deformidad de Made- lung en los padres de niños con Discon- drosteosis de Leri o ésta en los de la Dis-
7. Modelo teórico de probabilidades de .misión AD en la que e lgen mutado es A
y el recesivo <sano», es «a))
MEREUUA A~!iü)oUiU W l m ; f
QT; RP
- . más etérea para la generalidad, salvo si es
FIG. 8. Esquema en el que se ejemplariza la una forma Es preciso no descendencia & 2 portadores ~ n n o . ~ de un gen el hecho justificativo de la endogamia en recesivo «a»
plasia Mesomélica de Langer (fig. 6).

372 J. FERNANDEZ TORAL Y COLS.
cacia si se efectúa cuando el siguiente em- barazo está en curso, y de ahí la impor- tancia de su realización precoz.
En la transmisión AD, a partir del sur- gimiento del primer caso, por neomuta- ción -caso por ejemplo de la Acondro- plasia que tiende a aparecer con la edad añosa del padre (16, 17) aunque con más mortalidad si el progenitor es joven-, el 50 % de los hijosias de los afectos es de esperar lo sean igualmente, mientras que los hijos de los sanos así lo serán (a excep- ción de alguna familia extraordinaria) (18). De la unión de dos personas afectas las probabilidades teóricas son las recogi- das en la fig. 9, siendo destacable que por ejemplo el enfermo de Acondrosplasia Homozigota lo es en grado tan severo que suele fallecer precozmente.
FIG. 9. De la unión de dos afectos de enfer- medad autosónica dominante, surgen formas homoz2gota (AA), graves o nortaLes, como en
/a Acondroplasla
En las formas AR (fig. 8), se halla con frecuencia, consanguinidad entre los pa- dres, sanos por lo demás, pero portadores
del gen recesivo y por definición heterozigo- tos. La probabilidad de hijos enfermos, des- conocido en general aa priori», es del 25 % en cada embarazo, con la consideración de que dependiendo de la entidad, la sobrevi- vencia postnatal de los afectos tiende a ser menor o incluso nula: Osteogénesis Imper- fecta Letal (tipo 11), Acondrogénesis y Dis- plasia Tanatófora, Osteopetrosis Precoz. .;
Queda por último el comentar algunos aspectos del Diagnóstico Prenatal de las Displasias óseas. Se realiza fundamental- mente a través de la ecografía de tiempo real y alta resolución a partir de la semana 16 -un caso de Ellis Van Creveld a las 13 semanas (19)-, con medición exhaustiva de diámetros y longitudes, así como valora- ción de ecogenicidad de cuerpos vertebrales y osificación craneal, relación per.cef.iper. abd., polidactilia, incurvaciones.. . Su reali- zación exige alta experiencia y ha de hacerse fundamentalmente en casos de hijos previa- mente afectados; así se han diagnosticado Acondrogénesis, Acondroplasias, Displasia Tanatófora, Campomélica, Osteogénesis Imperfectas.. . (20-21). Se han ensayado otros métodos tales como la medición del pirofosfato en líquido amniótico en casos de Osteogénesis Imperfecta (22), con resulta- dos aleatorios; la radiación X que ha de ser tardía y a modo confirmatorio si acaso del hallazgo ecográfico; la resonancia magnéti- ca (23), que precisa de la inmovilización fe-
\tal con curare para su realización y que jun- to a la inseguridad aún de sus efectos secun- darios en el feto, la hacen no práctica; y el estudio del DNA en vellosidades coriales pa- ra algunas formas dominantes en familias informativas de Osteogénesis Imperfecta (24-26), única displasia con localización ge- nética confirmada en cromosoma 7q (21-22) y 17q (21-22) codificando la cadena alfa 2 y alfa 1 del cológeno 1, respectivamente que abre las puertas a un futuro ilusionante que permita descubrir las causas íntimas de estas afecciones invalidantes y su curación.

GENETICA DE LAS DISPLASIAS OSEAS 373
MAROTEAUX, P.: O~téochondmdyrp/a~iei. En MaiBdiar Orreurei de /'enfani. de MAnorrAux, P. Flamarion Méd-Sc. 1974. Paiis. BUENO, M.: Enfimedader óreai conititz~ciona- /m. En TrBtado de P e d i d , de CRUZ. M . Es- paxs. 1989, Barcelona. SAWIOUYA, J . M. y DELGADO. A.: Dirp/ariai órear. Salvat. 1968. Barcelona. , . . S ~ N C H E Z VILLARES. E. y FERNANDEZ CALVO. J . L.: Anomoiíor órea conrtituciona/ar en e/ niño. Bol. Soc. Cast. Asr. Leon. de'pediarr. 1971. . ~
XIV: 159-181. CRESPO HERNANDEZ, M. y cok.: Oiteocondm~ diipia~ia~. Bol. Soc. Cart. Ast. Leon. de Pe- diatr. 1973, XIV: 293-313. PLASENCIA AMELA, A. y COIS.: Coniejo Genéti~ co. An. Esp. Pediatr. 1987. 27: 130-134. Intern~tiono/ nomenciatgre ofconrtitutionai di- reerer ofbone. Ann. Radiology. 1983; 26: 417- 462. LnNOrn. L. O . y S c o n . CH. 1.: Birth Dflectr: At/m and Compendium. Bergsma D. 1979; N. York. ALVAREZ BERCIANO. F. y COIS.: Hipemitorii c o r ti<=/ 172fBntiL (Síndrome de Caffev-SihemonJ. .. Bol. s i c . ~ a ; . Ast. Leon. de Pediair. 1980; XXI: 47-16. ~ A R E Z BERCIANO, F. y cok.: f i o ~ t ~ l i i c#rt</&- ginora mú/ti&: A propóiito de doi obretvacio- nei. Bol. Soc. Cast. Ast. Leon. de Pediatr. 1977; XVIII: 379-386. FERN~NDEZ TORAL. J . y COIS.: Conrejo Genético y Pediatno. Bol. Soc. Cast. Asc. Leon. de Pe- diatr. 1979: XX: 19-36. FERNANDEZ TORAL. J . y COIS.: Orteoondrodi~~ plmu? cleido pub" dentm craneai (Síndrome de ~ ~ n e 4 a i t o n ) . Bol. Soc. Casr. Asr. Leon. de Pediatr. 1976; XVII: 421-438. HAWKINS, F. C. y COIS.: DO, CBJOI de diió~torir c/eidacrdnea/ en una familia. Bol. Soc. Ped. Madrid 1976; XXII: 82-86. SYKES. B. y COIS.: Orteogeneiir Imppe+ect# ir linRed to both type 1 coiiagen rtructurBigener. Lancet ii. 86: 69-72.
WALLIS. G. y col.: Mztutions kninded to the pro d fa 2 (1) coliagen gene ore rerpon~ibie for re ve^
rd carei of orteogenerir impe+ecta type 1. J . Med. Genet. 1986; 23: 411-416. T~OMPSON, J. N. y ~01s.: Achondmpiari~ ond p~rental age. The New England J . of Med. 1986: 314: 521-122. LENZ, W. : AnomalíBi de/ crecimiento y j oma corporal. En Genética Humana 11, de BECKER, P. E. Pág.: 65.117 Toray. 1966; Barcelona. FRYNS. J . P. y COIS.: Geninal moraicirm in achondropiuis: a fomil)l ui th 3 ~ffeected iib- iingr of nomo/p~rent i . Clin. Genet. 1983. 24: 116-158. WELDNER. B. M.; PERSSON. P. H. e IvARSSON. S. A,: Prennto( diizgagnorir of dwi1g61m ¿y ~ i t r u ~ roundrcreening. Arch. of Dis. in Child. 1985; 60: 1.070-1.072. SILLENCE, D. W.; RIMOIN. D. L. y LA~HMAN. R.: Neonatal Dworfum. Ped. Clin. Nort. Am. 1987; 21: 453-411. BRONS. J. T. y cok.: Prenatal u/traronographic diagnorir of orteqgenerir imperfecta. Am. J . Obsret. Gynecol. 1988; 159: 176-181. GARNER, K. L. y COIS.: Evaiutionr of inorganic pyrophoiph~te in ~ m n * ~ t i c f i i d ai a mode of prenataL dzignorir of osteogenerir imperfecta. Pren. Diagn. 1984: 4: 109-112. SARDA. P. y COIS.: Strategie actue/ie dei dépis- tage et diagnortic prénatd. La Médic. Infanti- le. 1989: 5: 311-357. . , ~ ~
TSIPOURAS. P. Y cols.: Prenatalprediction o fo i - teo~en& im#e+cto (O1 ñpe IV): exc/urion of . . . inheritance uring n coilagen gene probe. J . Med. Genet. 1987; 24: 406-409. SARDA. P. y CLAUSTES, M.: Diagnoitic préna- tal der M&& monogéniquer par génie géné~ tique. Pn>c@er génér~ux. La Médic. Infantile. 1989; 1: 371-377. McKusic~, V. A : Mandeiicn Inheritnnce in Man. Johns Hopkins H. Med. P. 1988; Balri- morc.
Petición de Separatar:
JOAQU~N ERNANDEZ TORAL Avda. de Galicia, 8-2 33005 OVlEDO