katalytisch-spektralphotometrische eisenspurenbestimmung in gemischt-wäßrigen systemen
TRANSCRIPT

Tabelle 1 Kinetisch-katalytische Bestimmung von Kupfer unter Verwendung verschiedener Indikationstechniken
Indikation Regressionsgleichung*) Arbeitsbereich Nachweis- relative Verstiirkungs. [Cu] 107/mol/l grenze **) Standard- faktor ***)
[Cu] 107/mol/l abweichung ~~ ~
Spektralphotometrie AE = 3,6 * lo5 [Cu] 1-10 1,7 0,080 38 Amperometrie A[O,] = 409 [CU] 1-10 1,5 0,066 409 (Clark-Elektrode) Kalorimetrie AQ = 3,45.104 [cu] 0,l-0,7 0,19 0,098 ca. 300
*) Als Signal wurden die Differenzen der Extinktion (AE) oder der Sauerstoffkonzentration (A[O,] in mol/l) nach 10 Minuten bzw. die Wirmeaustauschgeschwindigkeit ( A d in J s-l) zwischen katalysierter und Blindreaktion vermessen, mit [Cu] in mol/l.
***) fur At = 10 min **) nach dem 3s-Kriterium
thoden wiedergegeben. Nur bei Vermessen der Anfangsreaktions- geschwindigkeit erhiilt man eine iihnliche pH-Abhingigkeit wie bei amperometrischer Indikation [2]. Wertet man nach der ge- briiuchlicheren Methode der fixierten Zeit nach 10 min Reaktions- verlauf aus, ergibt sich ein Maximum bei einem pH-Wert von 7,6, da bei hoheren pH-Werten der Indikatorstoff, ein o-Benzo- chinon [6], instabil ist. Da die Stabilitiit des Indikatorstoffes auch durch hohe H,O,-Konzentrationen beeintriichtigt wird, wahlten wir fur die spektralphotometrische Bestimmung als KompromiD folgende Bedingungen: 7,5 - 10-9 M Sulfanilsilure; 0,2 M H,O,; pH 7,6; 2,5 - lo-, M Pyridin, 45°C und 10 min Reaktionszeit. Die unter diesen Bedingungen erhaltene Eichkurve zur katalytisch- spektralphotometrischen Bestimmung von CuII sowie die unter optimalen Bedingungen (pH 8,8; 2,5 - lo-, M Pyridin; 0,39 M H,O,; 25°C) ermittelten Ergebnisse bei amperometrischer [3] und kalorimetrischer Indikation sind in Tab. 1 zusammen rnit den Arbeitsbereichen und den Nachweisgrenzen zusammengestellt. Zur Unterscheidung von Verstiirkungsfaktoren, die von der ka- talytischen Reaktion bzw. von der Indikationsart herruhren, wurde der sogenannte Verstiirkungsfaktor (At * Nk) herangezogen, der den molaren Umsatz an Indikatorstoff (AX) pro mol Kata- lysator ([Cu],) beschreibt [l]:
N k Zyklenzahl. Die Konzentration dee Indikatorstoffes wird dam aus dem Eich- faktor (p) berechnet, der den Zusammenhang zwischen dem Signal (8) und der Konzentration des Indikatorstoffes wider- s p i e g e 1 t
I m Falle der spektralphotometrischen Indikation entspricht der Faktor p - entsprechend dem Lambed-Beerschen Gesetz - dem Produkt aus molarem Extinktionskoeffizient (E = 1,9 * los mol-l om-1 [6]) und der Schichtdicke der Kuvette (d = 5 om). Bei Ver- wendung der Clark-Elektrode entfiillt die Berechnung des Eich- faktors, da das MeBgeriit gegen den Indikatorstoff Sauerstoff ge- eicht wurde ( p = l [Z], [3]). Fur die kalorimetrische Bestimmung ergibt sich der Eichfaktor aus dem AH-Wert der katalytischen Reaktion, der aus der Korre- lation kalorimetrischer und amperometrischer Messungen [2] grob zu 70,5 k J mol-1 abgeschitzt wurde. DieVerstiirkungsfaktoren fur die drei Indikationsmethoden sind in Tab. 1 angegeben. Der fur kalorimetrische Messungen gefundene hohe Verstiirkungsfaktor fiir die katalysierte Reaktion verbunden mit einer empfindlichen Erfassung des Reaktionsumsatzes erkliirt die niedrige Nachweisgrenze kalorimetrischer Bestimmungen. Der geringe Verstiirkungsfaktor spektralphotometrischer Indi- kationen (Tab. 1) ist darauf zuriickzufiihren, daB Nebenreaktionen der Sulfanilsiiure und ihrer Oxydationsprodukte auftreten und damit nur ein Teil der katalytisch oxydierten Sulfanilsiiure als Indikatorstoff bei 370 nm vermessen werden kann. Interessant ist die vergleichbare Empfindlichkeit amperometri- scher und spektralphotometrischer Indikationen, die neue Mog- lichkeiten bei der Untersuchung optisch undurchliissiger Proben eroffnet . Wie die Ergebnisse belegen, sollte die Wahl der Indikations- technik bei der Optimierung einer katalytischen Bestimmung mit einbezogen werden. Leider stellt die kalorimetrische Indikation,
S = p X bzw. X (2)
mit deren Hilfe eine Verbesserung des Nachweisvermogens ca. um den Faktor 10 erreicht wurde, noch keine echte Alternative zu spektralphotometrischen und elektrochemischen Techniken dar, da ihre Anwendung mit liingeren Analysenzeiten und hoheren Kosten verbunden ist.
E x p e r i m e n t e l l e s Die Herstellung der Reagenzlosungen und die Durchfuhrung der kalorimetrischen [2], amperometrischen [3] und spektralphoto- metrischen [5] Messungen wurde bereits beschriehen.
L i t e r a t u r [l] Yatsimirskii, K. B.: Kinetic Methods of Analysis, Oxford,
Pergamon Press, 1965 [2] Otto, M.; Lerchner, J.; Pdp, T.; Zwanziger, H.; Hoyer, E.;
Inczt?dy, J.; Werner, G.: J. inorg. nuclear Chem. 43 (1981) 1101
[3] Otto, M.; Werner, G.: Analyt. chim. Acta 129 (1981) 177 [4] Alexiev, A.; Bontchev, P. R.; Gantcheoa, 8.: Mikrochim. Acta
[5] Otto, M.; Bontchev, P. R.; Mueller, H.: Mikrochim. Acta
[6] Otto, M.; Werner, C.: Z. Chem. 20 (1980) 379
Matthias Otto und Johanne8 Lerchner, Sektion Chemie der Karl- Marx-Universitiit Leipzig, DDR-7010 Leipzig, Liebigstr. 18
eingegangen am 21. Mai 1982
[Wien] 1976 11, 487
[Wien] 1977 I, 193
ZCM 7355
Hetalytisoh-spektralphotometrisohe Eisenspurenbestimmung in gemischt-wanrigen Systemen
Herrn Prof. Dr. Gerhard Ackemnann zum 60. Cteburtstag gewidmet Spurenanalytische Aufgabenstellungen nehmen insbesondere mit der Entwicklung mikroelektronischer Bauelemente weiter an Bedeutung zu. Dabei ist es notwendig, schon die Grund- und Hilfs- chemikalien in entsprechender Reinheit zur Verfugung zu stellen. Die vorliegende Arbeit befaI3t sich mit der Ausarbeitung eines Ver- fahrens zur Bestimmung von Eisenspuren in Methanol, Ethanol und Aceton der Qualitiit ,,spezialrein fur die Mikroelektronik".*) Eisenspuren lassen sich mit katalytisch-spektralphotometrischen Methoden [l] sehr nachweisstark bestimmen. In dieser Arbeit wurde die eisenkatalysierte und durch 1,lO-Phenanthrolin akti- vierte Oxydation von p-Phenetidin (zu cinem Oxydationsprodukt vom Chinonimintyp) durch H,O, in gemischt-wiiBrigen Systemen untersncht [2], [3]. Der Grundgedanke der Arbeit besteht drtrin, die zu untersuchenden Lijsungsmittel aIs Reaktionsmedien fur die katalysierte Reaktion einzusetzen und alle notwendigen Rea- genzien sehr konzentriert zuzugeben, damit ihr Anteil am Ge- samtvolumen gering bleibt. Unterscheidet sich die katalytische Aktivitiit des Eisens in den verschiedenen Reaktionsmedien (methanolisch-, ethanolisch- und acetonisoh-wiil3rige Losungen) wenig, kann uber nur eine Eichfunktion der Eisengehalt in den
*) Dem VEB Laborchemie Apolda gilt Dank f i i r die zur Verfu- gungstellung von entsprechenden Proben.
390 2. Chem., 22. Jg. (1982) Heft 10
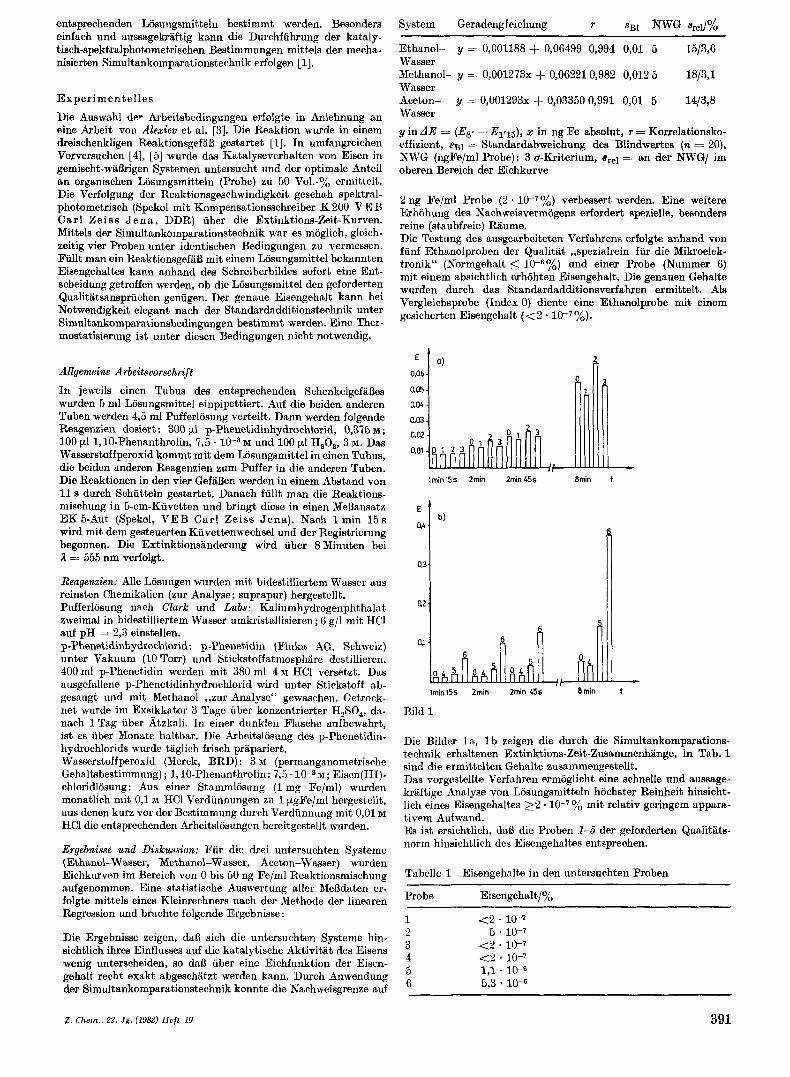
entsprechenden Losungsmitteln bestimmt werden. Besonders einfach und aussagekriiftig kann die Durchfuhrung der kataly- tisch-spektralphotometrischen Bestimmungen mittels der mecha- nisierten Simult ankomparationstechnik erfolgen [l].
Exper imente l les
Die Auswahl der Arbeitsbedingungen erfolgte in Anlehnung an eine Arbeit von Aleziev et al. [3]. Die Reaktion wurde in einem dreischenkligen ReaktionsgefLB gestartet [l]. I n umfangreichen Vorversuchen [4], [S] wurde das Katalyseverhalten von Eisen in gemischt-wiiBrigen Systemen untersucht und der optimale Anteil an organischen Losungsmitteln (Probe) zu 50 Vol.-yo ermittelt. Die Verfolgung der Reaktionsgeschwindigkeit geschah spektral- photometrisch (Spekol mit Kompensationsschreiber K 200 V E B C a r l Ze iss J e n a , DDR) uber die Extinktions-Zeit-Kurven. Mittels der Simultankomparationstechnik war es moglich, gleich- zeitig vier Proben unter identischen Bedingungen zu vermessen. Fiillt man ein ReaktionsgefLB mit einem Losungsmittel bekannten Eisengehaltes kann anhand des Schreiberbildes sofort eine Ent- scheidung getroffen werden, ob die Losungsmittel den geforderten Qualitiitsanspriichen genugen. Dcr genaue Eisengehalt kann bei Notwendigkeit elegant nach der Standardadditionstechnik unter Simultankomparationsbedingungen bestimmt werden. Eine Ther- mostatisierung ist unter diesen Bedingungen nicht notwendig.
Allgemeine Arbeitsvorschrift
In jeweils einen Tubus des entsprechenden SchenkelgefiiBes wurden 5 ml Losungsmittel einpipettiert. Auf die beiden anderen Tuben werden 4,5 ml Pufferlosung verteilt. Dann werden folgende Reagenzien dosiert : 300 pl p-Phenetidinhydrochlorid, 0,375 M ; 100 p1 1,lO-Phenanthrolin, 7,5 * M und 100 pl H,O,, 3 M. Das Wasserstoffperoxid kommt mit dem Losungsmittel in einen Tubus, die beiden anderen Reagenzien zum Puffer in die anderen Tuben. Die Reaktionen in den vier GefiiBen werden in einem Abstand von 11 s durch Schiitteln gestartet. Danach fullt man die Reaktions- mischung in 5-cm-Kuvetten und bringt diese in einen MeBansatz E K 5-Aut (Spekol, V E B Car l Zeiss Jena). Nach 1 min 15 s wird mit dem gesteuerten Kuvett,enwechsel und der Registrierung begonnen. Die Extinktionsgnderung wird iiber 8 Minuten bei L = 555 nm verfolgt.
Reagenzien: Alle Losungen wurden rnit bidestilliertem Wasser aus reinsten Chemikalien (zur Analyse ; suprapur) hergestellt. Pufferlosung nach Clark und Lubs: Kaliumhydrogenphthalat zweimal in bidestilliertem Wasser umkristallisieren; 6 g/l mit HCI auf pH = 2,3 einstellen. p-Phenetidinhydrochlorid: p-Phenetidin (Flnka AG, Schweiz) unter Vakuum (10 Torr) und Stickstoffatmosphare destillieren. 400 ml pPhenetidin werden mit 380 ml 4 w HC1 versetzt. Das ausgefallene p-Phenetidinhydrochlorid wird unter Stickstoff ab- gesaugt und rnit Methanol ,,zur Analyse" gewaschen. Getrock- net wurde im Exsikliator 3 Tage uber konzentrierter H,SO,, da- nach 1 Tag uber Atzkali. In einer dunklen Flasche aufbewahrt, ist es uber Monate haltbar. Die Arbeitslosung des p-Phenetidin- hydrochlorids wurde tLglich frisch prapariert. Wasserstoffperoxid (Merck, BRD) : 3 M (permanganometrische Gehaltsbestimmung) ; 1,lO-Phenanthrolin: 7,5. 1 0 - 3 ~ ; Eisen(II1)- chloridlosung : Aus einer Stammlosung (1 mg Fe/ml) wurden monatlich mit 0,l M HCl Verdunnungen zu 1 ygFe/ml hergestellt, aus denen kurz vor der Bestimmung durch Verdunnung mit 0,Ol M HCl die entsprechenden Arbeitslosungen bereitgestellt wurden.
Ergebnisse und Diskussion: Fur die drei untersuchten Systeme (Ethanol-Wasser, Methanol-Wasser, Aceton-Wasser) wurden Eichkurven im Bereich von 0 bis SO ng Fe/ml Reaktionsmischung aufgenommen. Eine statistische Auswertung aller MeBdaten er- folgte mittels eines Kleinrechners nach der Methode der linearen Regression und hrachte folgende Ergebnisse :
Die Ergebnisse zeigen, daB sich die untersuchten Systeme hin- sichtlich ihres Einflusses auf die katalytische Aktivitkt des Eisens wenig unterscheiden, so daB iiber eino Eichfunktion der Eisen- gehalt recht exakt abgeschiitzt werden kann. Durch Anwendung der Simultankomparationstechnik konnte die Nachweisgrenze auf
System Geradengf eichung ?' sB1 m G aced% ~~
Ethanol- y = 0,001188 + 0,06499 0,994 0,Ol 5 15/3,6 Wasser Methanol- y = 0,001273~ + 0,06221 0,982 0,012 5 18/3,1 Wasser Aceton- y = 0,001293~ + 0,03350 0,991 0,Ol 5 14/3,8 Wasser y in AE = (Eg - E1f15), z in ng Fe absolut, r = Korrelationsko- effizient, P B ~ = Standardabweichung des Blindwertes (n = 20), NWG (ngFe/ml Probe): 3 a-Kriterium, 8,,1 = an der NWG/ im oberen Bereich der Eichkurve
2 ng Fe/ml Probe ( 2 . 10-7Y0) verbessert werden. Eine weitere Erhohung des Nachweisvermogens erfordert spezielle, besonders reine (staubfreie) Riiume. Die Testung des ausgearbeiteten Verfahrens erfolgte anhand von funf Ethanolproben der Qualitiit ,,spezialrein fiir die Mikroelek- tronik" (Normgehalt 5 und einer Probe (Nummer 6) mit einem absichtlich erhohten Eisengehalt. Die genauen Gehalte wurden durch das Standardadditionsverfahren ermittelt. Als Vergleichsprobe (Index 0) diente eine Ethanolprobe mit einem gesicherten Eisengehalt ( < 2 lO-'yO).
0.06
0.05
0.04
0.m
0.02
0.01
lmin15s Zmin Zrnin45s 8rnin t
IrninlSs Zmin lrnin 45s 8 min
Bild 1
L t
Die Bilder l a , l b zeigen die durch die Simultankomparations- technik erhaltenen Extinktions-Zeit-Zusammenhiinge, in Tab. 1 sind die ermittelten Gehialte zusiammengestellt. Das vorgestellte Verfahren ermoglicht eine schnelle und aussage- kriiftige Analyse von Losungsmitteln hochster Reinheit hinsicht- lich eines Eisengehaltes 2 2 * 10-7y0 mit relativ geringem appara- tivem Aufwand. Es ist ersichtlich, daB die Proben 1-5 der geforderten Qualitiits- norm hinsichtlich des Eisengehaltes entsprechen.
Tabelle 1
Probe Eisongehalt/%
1 (2 - lo-'
Eisengehalte in den untersuchten Proben
2 5 . 10-7 3 <2.10-7 4 ( 2 . 10-7 5 1,l . 10-6 6 5,3 ' 10-6
Z. Chein., 22. Jg. (J882) Ilcft 10 391

Li tera tur [l] Muller, H.; Otto, M.; Werner, G.: Katalytische Methoden in der
Spurenanalyse, Leipzig, Akademische Verlagsgesellschaft 1980 [2] Kriaa, E. E.; Savichenko, Y . 8.; Jacimiraktj, K . B.: 2. analit.
Chimii 24 (1969) 875 [3] Alexiev, A.; Bontchev, P . R.; Raykowa, D.: Mikrochim. Acta
[Wien] 1974, 751 [4] Miiller, H.: Leipzig, Univ., Dissertation zur Promotion B 1978 [5] Brauer, H.: Leipzig, Univ., Diplomarbeit 1980
Helmut MiUer, Technische Hochschule ,,Carl Schorlemmer" Leuna-Merseburg, Sektion Chemie, 4200 Merseburg, Geusaer Str., und Hagen Brauer, Karl-Marx-Universitiit Leipzig, Sektion Chemie, DDR-7010 Leipzig, Liebigstr. 18
eingegangen am 26. Mai 1982 ZCM 7361
Stabilitlten von Komplexen zweiwertiger Metalle rnit Tiron und 1,lO-Phenanthrolin in Dioxen-Wasser-NIischungen Hewn Prof. Dr. aerlucrd Ackermann zum GO. Oeburtstag gewidmet
Im Zusammenhang mit der Erarbeitung einer nachweisstarken Be- stimmung von Mangan(I1) auf Grund seines katalytischen Effek- tes auf die Oxydation von Tiron durch Wasserstoffperoxid [l] wurde es notwendig, die Komplexgleichgewichte von MnII und von eventuell storenden Metallionen mit dem Aktivator 1,lO-Phe- nanthrolin und dem Reagens Tiron zu untersuchen. Bei derartigen analytisch genutzten Reaktionen sind neben biniiren Spezies auch terniire Komplexe zu berucksichtigen wie dies von Ackermann und seiner Schule ausfuhrlich gezeigt worden ist [2]. Ternare Komplexe rnit Phenanthrolin und Tiron sind bisher nur fur CuII bekannt [3]. Leider reicht die Loslichkeit des katalytisch inter- essanten terniiren MnII-Komplexes in Wasser nicht &us, um die Komplexgleichgewichte auf pH-potentiometrischem Wege be- stimmen zu konnen. Die Untersuchungen wurden daher in 60%igem wiiBrigen Dioxan durchgefuhrt. Die Komplexgleich- gewichte protonierter bzw. biniirer und terniirer Komplexe lassen sich mit Hilfe der Bruttostabilitatskonstanten&,qra folgendermanen beschreiben:
wobei M, T, P und H entsprechend fur Metallion, deprotoniertes Tiron, Phenanthrolin und Wasserstoffion stehen. Biniire Komplexe: Die ermittelten Stabilitiitskonstanten fiir Me- tall-Tiron-Komplexe in Dioxan-Wasser-Mischungen sind in Tab. 1 zusammengestellt. Eine Untersuchung der Gleichgewichte von Cd-Komplexen war nicht moglich, da die Loslichkeit von CdII- Tiron-Komplexen in diesem Losungsmittelgemisch ungenugend ist. Verglichen mit den verfugbaren Daten fur die 1:l-Komplexe zwischen Metallen und Tiron (Tab. 1) ergeben sich etwas hohere Werte, was auf eine Stabilisierung der Metall-Sauerstoff-Bindung in Losungen geringerer Polaritiit hindeutet. Die pH-potentiometrische Bestimmung von Komplexgleichgewich- ten im Falle der Phenanthrolinkomplexe bereitet Schwierigkeiten. Obwohl die Komplexbildungstendenz von Phenanthrolin mit Me- tallen grol3 ist, besteht eine relativ geringe Neigung gegenuber dem Proton. Dies hat zur Folge, dal3 das Proton rnit den Metallionen nur wenig konkurrieren kann und die Stabilitiitskonstanten fur beson- ders stabile 1: l-Komplexe wie mit &I1, NiIIund CuII zu klein aus- fallen [a]. Generell werdenfur Konstanten von Phenanthrolinkom- plexen bei Zugabe von organischenLosungsmitteln zu Wasser gerin- gere Stabilitiiten gemessen [9]. Trotz dieses Sachverhaltes ergaben sichfiir die Mehrzahl der Metallkomplexe weitaus zu kleine Gleich- gewichtskonstanten und es konnen als verliifiliche Werte nur die- jenigen fur die in dieser Reihe instabilsten &fnI1-Phenanthrolin- komplexe angegeben werden: Ig &p = 3,72 0,03; lg BMp, =
Termirer Xnll-lCompZez: Die Stabilitat des katalytisch aktiven MnII-Komplexes mit Phenanthrolin und Tiron wurde zu 13,24 f 0,lO bestimmt. Damit ergibt sich die Moglichkeit, die Verteilung der Komplexe des MnII mit den in der katalytischen Reaktion ver-
6,94 j, 0,15; lg &p, = 9,51 & 0,07.
TabelIe 1 in 50%igen Dioxan-Wasser-Mischugen
Binke Komplexe fur zweiwertige Metalle mit Tiron
MnII 9,69 & 0,04 15,37 j, 0,07 18,90 & 0,lO (836) b,
(9,49)9
(W.3 dl
COII 10,51 & 0,04 16,90 & 0,08 -
NiII 9,93 & 0,03 17,30 0,06 -
CUII 14,58 & 0,06 27,13 j, 0,08 - (14,23)e)
ZnII 10,93 f 0,06 19,14 & 0,lO - ( W 4 ) dl
a) Standardabweichung b),e) Stabilitiitskonstanten bei 25°C und 0,l M KNOB in Wasser entsprechend [6] und [5] O), d, Werte bei 0,l M KCl und 20°C in Wasser entsprechend [7] und 151
wendeten Liganden Phenanthrolin als Aktivator und Tiron als Reagens bei verschiedenen Bedingungen zu simulieren. Bild 1 gibt als Beispiel die Abhiingigkeit vom pH-Wert fur ein Metall- Ligand-Verhiiltnis von 1 : 2 : 2 wieder. Auf Grund der notwendigen Zugabe von Dioxan als Losungsvermittler ist die Stabilitiitskon- stante des terniiren MnII-Komplexes nur in einem qualitatiwn Modell fiir das komplexchemische Verhalten von MnII in der kata- lytischen Reaktion in rein wiiBriger Losung zu venvenden. Ver- gleiche mit einem iihnlichen System, in dem 2,2'-Bipyridyl als Aktivator verwendet wird und in dem siimtliche Komplexe wasaer- loslich sind, zeigen allerdings eine gute obereinstimmung [lo], [14].
t M nn
2 4 6 8 10 PH -
Bild 1 pH-Abhiingigkeit der Komplexverteilung im System MnII-Phenanthrolin (P)-Tiron (T) bei einem Metall-Ligand- Ligand-Verhiiltnis von 1:2:2
Experimentelles Die verwendeten Reagenzien sowie die Menanordnungen werden an anderer Stelle beschrieben [lo]. Besonderer Wert wurde iLuf die Reinigung des Dioxans gelegt [13], da ansonsten eine kilta- lytische Oxydation von Tiron durch vorhandene Peroxide unver- meidbar ist. Die Auswertung der Titrationskurven erfolgte unter Einbezieliung verschiedener Komplexmuster rnit Hilfe des Computerprogramms SCOGS [ll]. Die Korrektur der gemessenen pH-Werte fur 50%ige Dioxan-Wasser-Mischungen fuBte auf Titrationen verdunnter HNOa mit 0,l M BOH entsprechend [12] ($' = 0,504, vgl. [ll]). Das Ionenprodukt; des Wassers (pK,) wurde in diesem Medium bei 0 , l ~ KN03 zu 16,60 bestimmt.
392