mechanism of oncogenic signal activation by the novel ... · with cancer with oncogenic fusion...
TRANSCRIPT

Cancer Biology and Signal Transduction
Mechanism of Oncogenic Signal Activation by theNovel Fusion Kinase FGFR3–BAIAP2L1Yoshito Nakanishi, Nukinori Akiyama,Toshiyuki Tsukaguchi,Toshihiko Fujii,Yasuko Satoh,Nobuya Ishii, and Masahiro Aoki
Abstract
Recent cancer genome profiling studies have identified manynovel genetic alterations, including rearrangements of genesencoding FGFR family members. However, most fusion genesare not functionally characterized, and their potentials intargeted therapy are unclear. We investigated a recently discov-ered gene fusion between FGFR3 and BAI1-associated protein2-like 1 (BAIAP2L1). We identified 4 patients with bladdercancer and 2 patients with lung cancer harboring the FGFR3–BAIAP2L1 fusion through PCR and FISH assay screens. Toinvestigate the oncogenic potential of the fusion gene, weestablished an FGFR3–BAIAP2L1 transfectant with Rat-2 fibro-blast cells (Rat-2_F3-B). The FGFR3–BAIAP2L1 fusion hadtransforming activity in Rat2 cells, and Rat-2_F3-B cells werehighly tumorigenic in mice. Rat-2_F3-B cells showed in vitro
and in vivo sensitivity in the selective FGFR inhibitorCH5183284/Debio 1347, indicating that FGFR3 kinase activityis critical for tumorigenesis. Gene signature analysis revealedthat FGFR3–BAIAP2L1 activates growth signals, such as theMAPK pathway, and inhibits tumor-suppressive signals, suchas the p53, RB1, and CDKN2A pathways. We also establishedRat-2_F3-B-DBAR cells expressing an FGFR3–BAIAP2L1 variantlacking the Bin–Amphiphysin–Rvs (BAR) dimerization domainof BAIAP2L1, which exhibited decreased tumorigenic activity,FGFR3phosphorylation, and F3-B-DBARdimerization, comparedwith Rat-2_F3-B cells. Collectively, these data suggest that con-stitutive dimerization through the BAR domain promotes consti-tutive FGFR3 kinase activation and is essential for its potentoncogenic activity. Mol Cancer Ther; 14(3); 704–12. �2015 AACR.
IntroductionChromosomal translocations/rearrangements are major dri-
vers of tumorigenesis. Since the discovery of the BCR–ABL genefusion in chronic myelogenous leukemia, several fusion kinaseshave been identified in hematologic and epithelial malignancies,including anaplastic lymphoma kinase (ALK) fusions, RETfusions, and ROS1 fusions (1–3). Generally, the partner proteinsof fusion kinases possess dimerization domains, which promotekinase domain dimerization and constitutive activation. Severalsmall-molecule inhibitors have been developed to treat patientswith cancer with oncogenic fusion kinases. For instance, the ALKinhibitors crizotinib or alectinib have demonstrated excellenttargeted activity against cancers harboring EML4–ALK genefusions (4, 5).
Normally, the FGFR receptor tyrosine kinase is transientlyactivated FGF ligand–mediated homo/heterodimerization. TheFGFR signaling pathway is constitutively activated by geneticalterations, such as gene amplifications, point mutations, or
chromosomal translocations/rearrangements, which promotecell growth, angiogenesis, cell migration, invasion, and metas-tasis (6). FGFR1 amplification is a key genetic alteration insquamous cell lung carcinoma and hormone receptor–positivebreast cancer (7, 8), whereas FGFR2 is amplified in gastriccancer (9). FGFR2 and FGFR3 point mutations are mainlyobserved in endometrial cancer and bladder cancer, respective-ly (10, 11). Since the first reports of FGFR1 and FGFR3 fusiongenes in hematologic malignancies (12, 13), several novelchromosomal translocations/rearrangements of FGFRs havebeen discovered in patients with glioblastoma, bladder cancer,breast cancer, and cholangiocarcinoma by next-generationsequencing (NGS) technology (14–17).
Awell-characterized gene fusionoccurs between FGFR3 and thetransforming acidic coiled-coil containing protein 3 (TACC3)gene. TACC3 contains a coiled-coil domain and exerts ligand-independent activation upon dimerization (17). The constitu-tively activated FGFR3–TACC3 protein can promote ERK andSTAT3 signaling (16, 18). miR-99a targets the 30-untranslatedregion (UTR) of FGFR3 to suppress FGFR3 expression in normaltissues. However, because the FGFR3 fusion loses its 30-UTR, thefusion protein is highly expressed (19).
The FGFR3–BAI1-associated protein 2-like 1 (BAIAP2L1)fusion gene has not been identified in clinical tumor samples.Although FGFR3–BAIAP2L1 dimerization and increased ERK andSTAT1 phosphorylation in FGFR3–BAIAP2L1-positive cells havebeen reported (18), themechanismsof constitutive activation andassociated signaling pathways are unclear. Therefore, we assessedthe prevalence of the FGFR3–BAIAP2L1 fusion gene in clinicalsamples, studied its tumorigenic activity in vitro and in vivo, andinvestigated its signaling pathway. BAIAP2L1 has a Bin–Amphi-physin–Rvs (BAR) domain, which is themost conserved feature in
Research Division, Chugai Pharmaceutical Co., Ltd., Kamakura, Kana-gawa, Japan.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Y. Nakanishi and N. Akiyama are co–first authors who contributed equally to thisarticle.
CorrespondingAuthor:YoshitoNakanishi, Chugai Pharmaceutical Co., Ltd., 200Kajiwara, Kamakura, Kanagawa 247-8530, Japan. Phone: 81-467-47-6262;Fax: 81-467-46-5320; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-14-0927-T
�2015 American Association for Cancer Research.
MolecularCancerTherapeutics
Mol Cancer Ther; 14(3) March 2015704
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T

amphiphysins. The BAR domain forms a crescent-shaped dimerthat preferentially binds highly curved, negatively charged mem-branes (20). In the FGFR3–BAIAP2L1 fusion, the BAIAP2L1fragment, including the BAR domain, is fusedwith the C-terminaldomain of FGFR3, and therefore FGFR3–BAIAP2L1 retains theentire kinase domain of FGFR3. To clarify the role of BAR domainin the FGFR3–BAIAP2L1 fusion kinase, we compared the tumor-igenic and dimerization activities of FGFR3–BAIAP2L1 andFGFR3–BAIAP2L1 lacking the BAR domain.
Materials and MethodsReagents and cell lines
A 1-(1H-benzimidazol-5-yl)-5-aminopyrazole derivative,CH5183284/Debio 1347, was synthesized at Chugai Pharmaceu-tical Co. Ltd., as described previously (21). SW780, RT4, 3T3, 293,HT1376, and HCT116 were purchased from the American TypeCulture Collection, Rat-2 was purchased from the Health ScienceResearchResources Bank, andRT112/84 is purchased fromHealthProtection Agency Culture Collections. All cell lines wereobtained more than one year ago from each experiment and werepropagated for less than 6 months after thawing and culturedaccording to suppliers' instructions.
PCR, Sanger sequencing of FGFR3–BAIAP2L1 gene fusions, andbreak-apart FISH assays
Sequencing of clinical samples was conducted under Institu-tional Review Board–approved protocols conducted at ChugaiPharmaceutical Co., Ltd. cDNAs from clinical samples derivedfrom patients with bladder cancer (n¼ 46), lung cancer (n¼ 83),head and neck cancer (n¼ 17), and gastroesophageal cancer (n¼18) were obtained from OriGene Technologies, Inc. or TriStarTechnology Group, LLC. The information of primers of PCR isavailable in Supplementary Materials and Methods. For FGFR3break-apart FISH experiments, 4-mm-thick formalin-fixed, paraf-fin-embedded sections were deparaffinized and treated withPretreatment Reagent (Abbott) for 10 minutes at 95�C. Pepsinsolution (ZytoVision) was added and samples were incubated at37�C for 10 minutes. FGFR3 gene rearrangements were detectedwith the FGFR3 Split Dual Color FISH probe (GSP Lab., Inc.).Slides and probes were denatured simultaneously at 75�C for 10minutes, followed by hybridization at 37�C overnight with Ther-moBrite (Abbott Molecular, Inc.). Slides were washed with 0.3%NP-40/2x saline-sodium citrate at 72�C for 2minutes and stainedwith DAPI (Life Technologies).
Colony formation assays, spheroid formation assays, andxenograft transplantation studies
Rat-2 cells were infected with lentiviruses, and stable transduc-tants were selected in puromycin (1 mg/mL medium). Colonyformation activities of stably transduced Rat-2 cells were mea-sured with a CytoSelect 96-Well Cell Transormation assaykit (CELL BIOLABS, INC.). Stably transduced Rat-2 cells wereseeded in 6-well or spheroid plates (Sumilon Celltight Spheroid96U; Sumitomo Bakelite, Inc.) and incubated for 3 to 4 days at37�C. Cell morphologies were observed microscopically, and cellviabilities were measured using the CellTiter-Glo LuminescentCell Viability Assay Kit (Promega). Female BALBnu/nu mice(CAnN.Cg-Foxn1<nu>/CrlCrlj nu/nu) were obtained fromCharles River Laboratories Japan and housed under specificpathogen-free conditions. Stably transduced Rat-2 cells were
suspended in serum-free medium, and 5� 106 cells were injectedsubcutaneously into the right flanks of 7- to 9-week-old mice.Tumors were measured using a gauge twice weekly, and tumorvolumes (TV) were calculated as: TV ¼ a � b2/2, where a is thetumor length, and b is the width (22). All in vivo studies wereapproved by the Chugai Institutional Animal Care and UseCommittee.
In vitro and in vivo efficacy studiesCell lines were seeded in 96-well plates (Sumilon Celltight
Spheroid 96U) in medium containing final CH5183284/Debio1347 concentrations of 0.003 to 20,000 nmol/L and incubatedat 37�C for 4 days. Subsequently, cell viabilities were measuredusing the CellTiter-Glo Luminescent Cell Viability Assay Kit.For in vivo efficacy studies, cells were implanted into mice asdescribed above. After tumors reached approximately 200 to300 mm3, animals were randomized into groups (4–5/group)and received oral CH5183284/Debio 1347 administrationonce per day.
Western blot analysisCells were treated with CH5183284/Debio 1347 or a solvent
control (0.1% dimethyl sulfoxide; DMSO) for 2 hours andlysed in Cell Lysis Buffer (Cell Signaling Technology) contain-ing protease and phosphatase inhibitors. For animal studies,xenograft tumors were homogenized using a BioMasher (K.K.Ashisuto) before lysis. Cell lysates were denatured with SampleBuffer Solution with Reducing Reagent for SDS-PAGE (LifeTechnologies) and resolved on precast 10% or 5% to 20%SDS-PAGE gels (Wako Pure Chemical Industries, Ltd.). Afterelectroblotting, Western blot analysis was performed as describ-ed (23). Antibody references are available in SupplementaryMaterials and Methods.
RNA-Seq and expression analysisCellular RNAwas extracted using the RNeasyMini Kit (Qiagen,
Inc.). Quality assessment, poly-A selection, and sequencingwith aHiSeq 2000 Sequencing System (Illumina) were performed byMacrogen, Inc. Cellular RNA samples were prepared for sequenc-ing using a TruSeq RNA Sample Preparation kit (Illumina) togenerate an mRNA library, and 100 bases were sequenced fromboth ends of the library. RSEM software was used to align readsagainst RefSeq transcripts and calculate expression values for eachgene (24). Fold changes in expression levels were calculated toidentify downregulated genes (<80% expression) and upregu-lated genes (>120% expression), relative to Rat-2_mock cells andother cell lines. We also purified and sequenced total RNA fromRat-2_F3-B cells treated for 24 hours with either 0.1%DMSO or 1mmol/L CH5183284/Debio 1347. Fold changes were calculatedby normalizing gene expression levels in CH5183284/Debio1347–treated cells to DMSO control cells, identifying suppressed(<50%expression) or induced genes (>200%expression), relativeto DMSO controls.
Phosphorylation levels and dimerization activities of FGFR3constructs
cDNAs encoding wild-type (WT) FGFR3, F3-B, F3-B-DBAR,BAIAP2L1, F3-B lacking the SH domain (aa 342–401; F3-B-DSH),F3-B lacking the SH and BAR domains (F3-B-DBAR/DSH),and kinase dead (K508M) F3-B (F3-B-KD) were inserted into
Oncogenic Mechanisms and Prevalence of FGFR3–BAIAP2L1
www.aacrjournals.org Mol Cancer Ther; 14(3) March 2015 705
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T
the pCXND3 vector (Kaketsuken) and used to transfect 293 cells.At 72 hours after transfection, cells were lysed in Cell Lysis Buffer(Cell Signaling Technology). Lysates were then studied by West-ern blot analysis. In separate experiments, 293 cells were trans-fected with the FLAG-tagged or Myc-tagged expression constructalone or in combination, using the FuGene HD reagent (Pro-mega). At 3 days after transfection, cells were lysed and immu-noprecipitation was performed with Anti-FLAG M2 Affinity Gel(Sigma-Aldrich). Precipitates were washed 10 times with CellLysis Buffer and eluted at 95�C for 5 minutes with ReducingReagent for SDS-PAGE (Life Technologies).
ResultsIdentification of patients harboring FGFR3 rearrangements
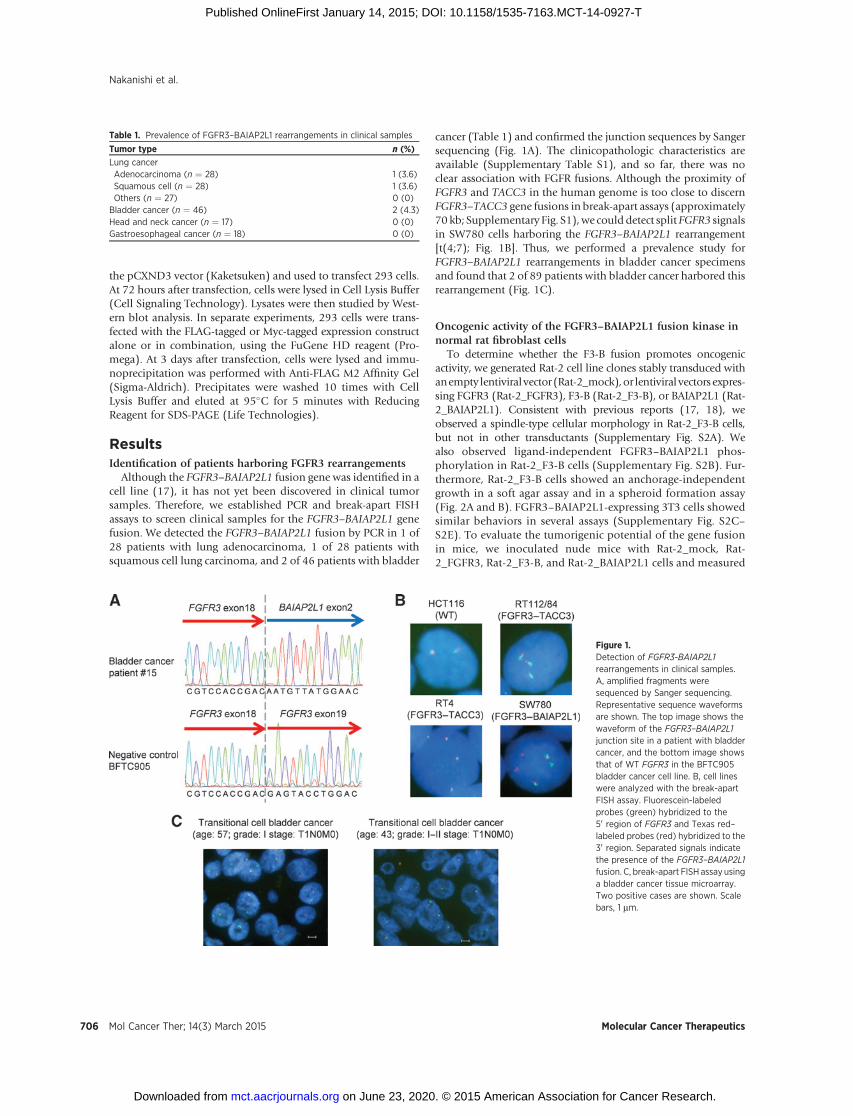
Although the FGFR3–BAIAP2L1 fusion gene was identified in acell line (17), it has not yet been discovered in clinical tumorsamples. Therefore, we established PCR and break-apart FISHassays to screen clinical samples for the FGFR3–BAIAP2L1 genefusion. We detected the FGFR3–BAIAP2L1 fusion by PCR in 1 of28 patients with lung adenocarcinoma, 1 of 28 patients withsquamous cell lung carcinoma, and 2 of 46 patients with bladder
cancer (Table 1) and confirmed the junction sequences by Sangersequencing (Fig. 1A). The clinicopathologic characteristics areavailable (Supplementary Table S1), and so far, there was noclear association with FGFR fusions. Although the proximity ofFGFR3 and TACC3 in the human genome is too close to discernFGFR3–TACC3 gene fusions in break-apart assays (approximately70 kb; Supplementary Fig. S1),we could detect split FGFR3 signalsin SW780 cells harboring the FGFR3–BAIAP2L1 rearrangement[t(4;7); Fig. 1B]. Thus, we performed a prevalence study forFGFR3–BAIAP2L1 rearrangements in bladder cancer specimensand found that 2 of 89 patients with bladder cancer harbored thisrearrangement (Fig. 1C).
Oncogenic activity of the FGFR3–BAIAP2L1 fusion kinase innormal rat fibroblast cells
To determine whether the F3-B fusion promotes oncogenicactivity, we generated Rat-2 cell line clones stably transduced withanempty lentiviralvector (Rat-2_mock),or lentiviral vectors expres-sing FGFR3 (Rat-2_FGFR3), F3-B (Rat-2_F3-B), or BAIAP2L1 (Rat-2_BAIAP2L1). Consistent with previous reports (17, 18), weobserved a spindle-type cellular morphology in Rat-2_F3-B cells,but not in other transductants (Supplementary Fig. S2A). Wealso observed ligand-independent FGFR3–BAIAP2L1 phos-phorylation in Rat-2_F3-B cells (Supplementary Fig. S2B). Fur-thermore, Rat-2_F3-B cells showed an anchorage-independentgrowth in a soft agar assay and in a spheroid formation assay(Fig. 2A and B). FGFR3–BAIAP2L1-expressing 3T3 cells showedsimilar behaviors in several assays (Supplementary Fig. S2C–S2E). To evaluate the tumorigenic potential of the gene fusionin mice, we inoculated nude mice with Rat-2_mock, Rat-2_FGFR3, Rat-2_F3-B, and Rat-2_BAIAP2L1 cells and measured
Table 1. Prevalence of FGFR3–BAIAP2L1 rearrangements in clinical samples
Tumor type n (%)
Lung cancerAdenocarcinoma (n ¼ 28) 1 (3.6)Squamous cell (n ¼ 28) 1 (3.6)Others (n ¼ 27) 0 (0)
Bladder cancer (n ¼ 46) 2 (4.3)Head and neck cancer (n ¼ 17) 0 (0)Gastroesophageal cancer (n ¼ 18) 0 (0)
Figure 1.Detection of FGFR3-BAIAP2L1rearrangements in clinical samples.A, amplified fragments weresequenced by Sanger sequencing.Representative sequence waveformsare shown. The top image shows thewaveform of the FGFR3–BAIAP2L1junction site in a patient with bladdercancer, and the bottom image showsthat of WT FGFR3 in the BFTC905bladder cancer cell line. B, cell lineswere analyzed with the break-apartFISH assay. Fluorescein-labeledprobes (green) hybridized to the50 region of FGFR3 and Texas red–labeled probes (red) hybridized to the30 region. Separated signals indicatethe presence of the FGFR3–BAIAP2L1fusion. C, break-apart FISH assay usinga bladder cancer tissue microarray.Two positive cases are shown. Scalebars, 1 mm.
Nakanishi et al.
Mol Cancer Ther; 14(3) March 2015 Molecular Cancer Therapeutics706
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T
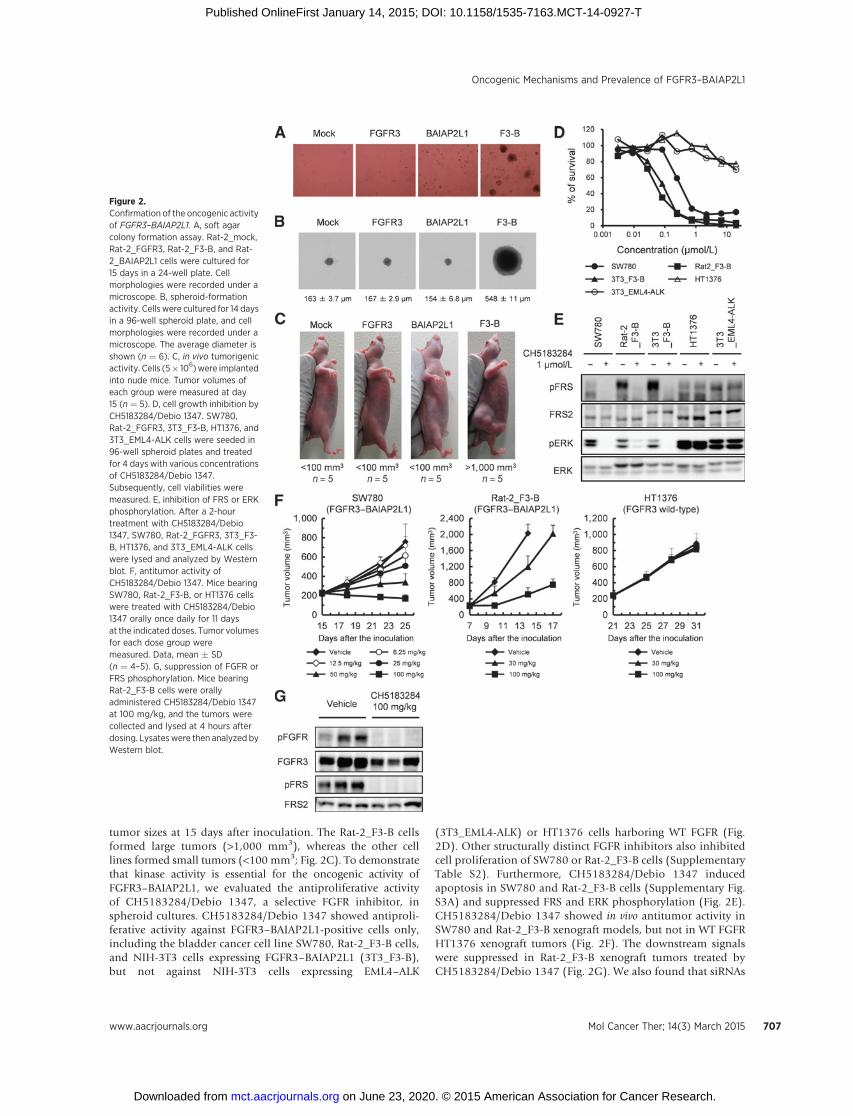
tumor sizes at 15 days after inoculation. The Rat-2_F3-B cellsformed large tumors (>1,000 mm3), whereas the other celllines formed small tumors (<100 mm3; Fig. 2C). To demonstratethat kinase activity is essential for the oncogenic activity ofFGFR3–BAIAP2L1, we evaluated the antiproliferative activityof CH5183284/Debio 1347, a selective FGFR inhibitor, inspheroid cultures. CH5183284/Debio 1347 showed antiproli-ferative activity against FGFR3–BAIAP2L1-positive cells only,including the bladder cancer cell line SW780, Rat-2_F3-B cells,and NIH-3T3 cells expressing FGFR3–BAIAP2L1 (3T3_F3-B),but not against NIH-3T3 cells expressing EML4–ALK
(3T3_EML4-ALK) or HT1376 cells harboring WT FGFR (Fig.2D). Other structurally distinct FGFR inhibitors also inhibitedcell proliferation of SW780 or Rat-2_F3-B cells (SupplementaryTable S2). Furthermore, CH5183284/Debio 1347 inducedapoptosis in SW780 and Rat-2_F3-B cells (Supplementary Fig.S3A) and suppressed FRS and ERK phosphorylation (Fig. 2E).CH5183284/Debio 1347 showed in vivo antitumor activity inSW780 and Rat-2_F3-B xenograft models, but not in WT FGFRHT1376 xenograft tumors (Fig. 2F). The downstream signalswere suppressed in Rat-2_F3-B xenograft tumors treated byCH5183284/Debio 1347 (Fig. 2G). We also found that siRNAs
Figure 2.Confirmation of the oncogenic activityof FGFR3–BAIAP2L1. A, soft agarcolony formation assay. Rat-2_mock,Rat-2_FGFR3, Rat-2_F3-B, and Rat-2_BAIAP2L1 cells were cultured for15 days in a 24-well plate. Cellmorphologies were recorded under amicroscope. B, spheroid-formationactivity. Cells were cultured for 14 daysin a 96-well spheroid plate, and cellmorphologies were recorded under amicroscope. The average diameter isshown (n ¼ 6). C, in vivo tumorigenicactivity. Cells (5� 106)were implantedinto nude mice. Tumor volumes ofeach group were measured at day15 (n ¼ 5). D, cell growth inhibition byCH5183284/Debio 1347. SW780,Rat-2_FGFR3, 3T3_F3-B, HT1376, and3T3_EML4-ALK cells were seeded in96-well spheroid plates and treatedfor 4 days with various concentrationsof CH5183284/Debio 1347.Subsequently, cell viabilities weremeasured. E, inhibition of FRS or ERKphosphorylation. After a 2-hourtreatment with CH5183284/Debio1347, SW780, Rat-2_FGFR3, 3T3_F3-B, HT1376, and 3T3_EML4-ALK cellswere lysed and analyzed by Westernblot. F, antitumor activity ofCH5183284/Debio 1347. Mice bearingSW780, Rat-2_F3-B, or HT1376 cellswere treated with CH5183284/Debio1347 orally once daily for 11 daysat the indicated doses. Tumor volumesfor each dose group weremeasured. Data, mean � SD(n ¼ 4–5). G, suppression of FGFR orFRS phosphorylation. Mice bearingRat-2_F3-B cells were orallyadministered CH5183284/Debio 1347at 100 mg/kg, and the tumors werecollected and lysed at 4 hours afterdosing. Lysateswere then analyzed byWestern blot.
Oncogenic Mechanisms and Prevalence of FGFR3–BAIAP2L1
www.aacrjournals.org Mol Cancer Ther; 14(3) March 2015 707
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T

targeting the FGFR3, BAIAP2L1, and FGFR3–BAIAP2L1 fusiontranscripts showed antiproliferative activities against SW780cells (Supplementary Fig. S3B and S3C). Collectively, these datademonstrated that the FGFR3–BAIAP2L1 fusion kinase pro-moted tumor growth in vivo and that its kinase activity wasimportant for oncogenic activity.
Downregulation of tumor-suppressive pathways by the FGFR3–BAIAP2L1 fusion kinase
A previous study reported that the FGFR3–BAIAP2L1 fusionkinase induced ERK and STAT1 phosphorylation (18); howev-er, the mechanisms of tumor growth promotion in vivo byFGFR3–BAIAP2L1 have not been demonstrated. Therefore,we conducted a comprehensive gene expression analysis byNGS using 4 cell lines (Rat-2_mock, Rat-2_FGFR3, Rat-2_F3-B,
and Rat-2_BAIAP2L1). These cell lines were cultured undernormal growth conditions, and total RNAs were preparedfor RNA-Seq with an Illumina HiSeq2000. We identified1,566 upregulated genes and 1,916 downregulated genes inRat-2_F3-B cells, compared with expression levels observed inRat-2_mock cells or Rat-2_FGFR3 cells (Fig. 3A). To selectgenes that are regulated by FGFR3 kinase, we identified asubset of genes whose differential expression levels werereversed by CH5183284/Debio 1347 treatment (Supplemen-tary Fig. S4). Finally, we selected 210 genes comprising anFGFR3–BAIAP2L1-regulated gene signature (SupplementaryTable S3), which was analyzed by Ingenuity Pathway Analysis(IPA) software (Ingenuity Systems) to identify upstream reg-ulators. The results indicated that the RB1, p53, and p16pathways were suppressed in Rat-2 cells expressing FGFR3–BAIAP2L1, whereas the E2F2 and E2F1 pathways were activat-ed (Table 2). To confirm that these pathways are similarlyregulated in vivo, xenograft tumors from mice implanted withRat-2_mock, Rat-2_FGFR3, Rat-2_F3-B, and Rat-2_BAIAP2L1cells were analyzed by Western blot (Fig. 3B). As previouslyreported (18), phospho-ERK and phospho-MEK were upregu-lated only in Rat-2_F3-B tumors. Cyclin D1 protein levelswere elevated because of MAPK pathway activation, whereasphospho-AKT was downregulated. Consistent with the IPAresults, p53 and p21 expressions were markedly decreased.Similarly, decreased phosphorylation of RB1 and p27 wasobserved in Rat-2_F3-B–derived xenograft tumors. Collective-ly, these results suggested that FGFR3–BAIAP2L1 activated theMAPK pathway and downregulated tumor-suppressive path-ways involving RB, p53, and CDKN2A.
A
215
284
1,566
193 540
2,151220
273
119
1,916
328 135
33697
FGFR3
F3-B BAIAP2L1
FGFR3
F3-B BAIAP2L1
Upregulated genes Downregulated genes
B Mock FGFR3 F3-B BAIAP2L1
pFGFR
FGFR3
pERK
BAIAP2L1
Cyclin D1
ERK
pAKT
AKT
p21
p27
pMEK
MEK
pRB1
RB1
p53
Mock FGFR3 F3-B BAIAP2L1
Figure 3.Analysis of FGFR3–BAIAP2L1pathways. A, Venn diagrams showingthe number of commonly anddifferentially upregulated genes (left)and downregulated genes (right) inRat-2_F3-B, Rat-2_FGFR3, and Rat-2BAIAP2L1 cells. B, xenograft tumorsderived from implantation of Rat-2_mock, Rat-2_FGFR3, Rat-2_F3-B,and Rat-2_BAIAP2L1 cells into micewere lysed and analyzed by Westernblot (n ¼ 3).
Table 2. List of pathways activated or inhibited by FGFR3–BAIAP2L1, assuggested by analysis with IPA software
Upstreamregulator
Predicted state inRat-2_F3-B cells
Activationz-score
P valueof overlap
RB1 Inhibited �2.95 9.5E�17TP53 Inhibited �2.05 1.4E�12RBL1 Inhibited �2.75 1.1E�11CDKN2A Inhibited �2.31 4.9E�10NUPR1 Inhibited �3.21 2.2E�06E2F2 Activated 2.00 1.4E�13E2F1 Activated 3.52 1.3E�12TBX2 Activated 2.83 1.9E�08CEBPB Activated 2.36 1.9E�05STAT3 Activated 2.40 2.7E�04
Nakanishi et al.
Mol Cancer Ther; 14(3) March 2015 Molecular Cancer Therapeutics708
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T

The FGFR3–BAIAP2L1 fusion kinase BAR domain is essentialfor oncogenic activity
Most partner genes of fusion kinases have dimerizationmotifs such as coiled-coil domains, and kinase domain dimer-ization can lead to constitutive kinase domain activation (25).The BAR domain of BAIAP2L1 contains a known dimerizationmotif, but it is uncertain if the BAR domain facilitates dimer-ization or promotes oncogenic activity through constitutivekinase activation. To study BAR domain function in FGFR3–BAIAP2L1, we established Rat-2 cells expressing FGFR3–BAIAP2L1 without the BAR domain (Rat-2_F3-B-DBAR, Fig.4A) and compared its oncogenic potential with that observedwith Rat-2_F3-B cells. We first measured the spheroid forma-tion capacity of Rat-2_F3-B-DBAR cells relative to those of Rat-2, Rat-2_mock, Rat-2_FGFR3, and Rat-2_F3-B cells. Rat-2_F3-B-DBAR formed 1.4-fold more spheroids than Rat-2_mock cells,whereas Rat-2_F3-B formed 26-fold higher spheroids (Fig. 4B).Next, we evaluated tumorigenesis of Rat-2_F3-B cells innude mice. Xenograft tumors derived from Rat-2_F3-B cellsformed large tumors (average size: 1,600 mm3) by 14 daysafter tumor implantation (Fig. 4C). However, Rat-2_F3-B-DBAR cells formed tumors that were >10-fold smaller (averagesize: 89 mm3). We confirmed that the FGFR3 constructs wereexpressed approximately equally in each cell line (Supplemen-tary Fig. S5). The observations that the spheroid formingactivities of Rat-2_F3-B and Rat-2_F3-B-DBAR cells were essen-tially the same in the presence of FGF1 ligand (Fig. 4D) and thatFGF1 induced F3-B-DBAR phosphorylation (SupplementaryFig. S6) both suggested that differences in tumorigenic activity
without FGF1 were not due to conformational alterations of thefusion proteins that affected kinase activity, but were solelydependent on BAR domain function. These data demonstratedthat the BAR domain of FGFR3–BAIAP2L1 was essential for theobserved oncogenic activity.
Contribution of the FGFR3–BAIAP2L1 fusion kinase BARdomain to dimerization activity
To study the requirement of the FGFR3–BAIAP2L1 BARdomain for oncogenic activity, we examined activation statuseswith an FGFR3 deletion series in Rat-2 cells. 293 cells weretransfected with the empty pCXND3 vector, or pCXND3 con-structs encoding FGFR3, F3-B, F3-B-DBAR, F3-B with the SHdomain deleted (F3-B-DSH), F3-B with the BAR and SH3domains deleted (F3-B-DBAR/DSH), and kinase dead F3-B(F3-B-KD; Fig. 5A). Next, FGFR3 phosphorylation in transfec-tants was analyzed in Western blots (Fig. 5B). Compared withFGFR3, F3-B phosphorylation was markedly increased, and nophosphorylation of the kinase dead mutant (K508M) wasdetected, suggesting that FGFR3 phosphorylation results fromautophosphorylation. Among the F3-B deletion mutants stud-ied, FGFR3 phosphorylation in cells expressing the F3-B-DBARand F3-B-DBAR/DSH variants was remarkably diminished.Then, we validated FGFR3 dimerization in 293 cells transfectedwith FLAG-tagged and/or Myc-tagged FGFR3 fusion variants byimmunoprecipitation of FLAG-tagged proteins (Fig. 5C).Although the Myc-tagged F3-B was coimmunoprecipitated withFLAG-tagged F3-B, we only detected a minor portion of coim-munoprecipitated Myc-tagged F3-B-DBAR with FLAG-tagged
B
C
Rat
io to
moc
k
Par
ent
FG
FR
3
F3-
B
F3-
B-Δ
BA
R
BA
IAP
2L1
Moc
k
0123456789
Rat
io to
moc
k
D
Par
ent
FG
FR
3
F3-
B
F3-
B-Δ
BA
R
BA
IAP
2L1
Moc
kFGF1
0
500
1,000
1,500
2,000
2,500
0
10
20
30
Tum
or v
olum
e (m
m3 )
FG
FR
3
F3-
B
F3-
B-Δ
BA
R
BA
IAP
2L1
Moc
k
ATM
FGFR3–BAIAP2L1 (F3-B)
FGFR3
FGFR3–BAIAP2L1 ΔBAR (F3-B-ΔBAR)
TK1 TK2
BARTK1 SH3TK2
TK1 SH3TK2
Coiled-coil
BAR SH3
BAIAP2L1
Figure 4.Essentiality of the BAR domain in theFGFR3–BAIAP2L1 fusion kinase. A,schematic diagram of full-lengthFGFR3, FGFR3–BAIAP2L1 (F3-B), andthe BAR deletion mutant of FGFR3–BAIAP2L1 (F3-B-DBAR). B, spheroidformation activity. Rat-2, Rat-2_mock,Rat-2_FGFR3, Rat-2_F3-B, Rat-2_F3-B-DBAR, and Rat-2_BAIAP2L1 cellswere seeded into 96-well spheroidplates and cultured for 4 days.Subsequently, cell viabilities weremeasured (n ¼ 3). C, in vivotumorigenic activity. Cells (5 � 106)were implanted subcutaneously intonude mice. Tumor volumes of eachgroup were measured at day 14(n ¼ 5). D, ligand dependency. Cellswere cultured in the presence of FGF1(0, 0.1, 1, 10, and 100 ng/mL) andheparin (10 mg/mL) for 96 hours, afterwhich cell viabilities were measured(n ¼ 3).
Oncogenic Mechanisms and Prevalence of FGFR3–BAIAP2L1
www.aacrjournals.org Mol Cancer Ther; 14(3) March 2015 709
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T
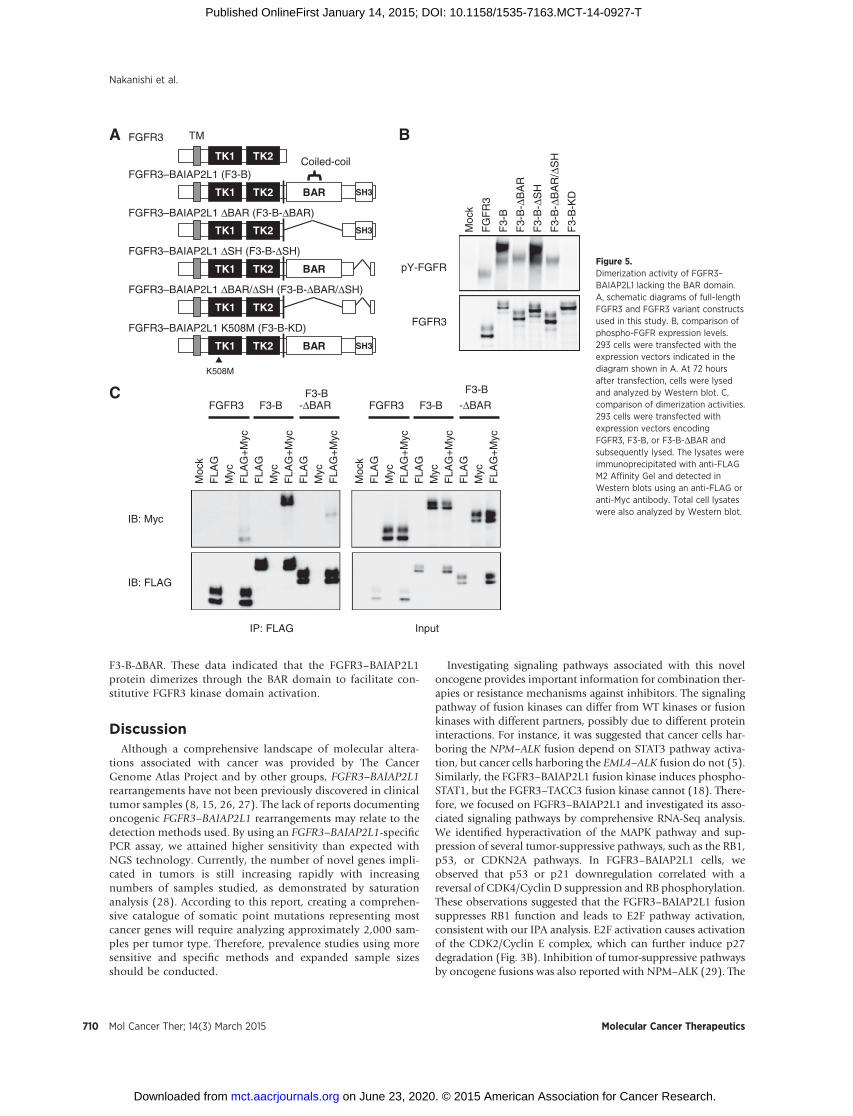
F3-B-DBAR. These data indicated that the FGFR3–BAIAP2L1protein dimerizes through the BAR domain to facilitate con-stitutive FGFR3 kinase domain activation.
DiscussionAlthough a comprehensive landscape of molecular altera-
tions associated with cancer was provided by The CancerGenome Atlas Project and by other groups, FGFR3–BAIAP2L1rearrangements have not been previously discovered in clinicaltumor samples (8, 15, 26, 27). The lack of reports documentingoncogenic FGFR3–BAIAP2L1 rearrangements may relate to thedetection methods used. By using an FGFR3–BAIAP2L1-specificPCR assay, we attained higher sensitivity than expected withNGS technology. Currently, the number of novel genes impli-cated in tumors is still increasing rapidly with increasingnumbers of samples studied, as demonstrated by saturationanalysis (28). According to this report, creating a comprehen-sive catalogue of somatic point mutations representing mostcancer genes will require analyzing approximately 2,000 sam-ples per tumor type. Therefore, prevalence studies using moresensitive and specific methods and expanded sample sizesshould be conducted.
Investigating signaling pathways associated with this noveloncogene provides important information for combination ther-apies or resistance mechanisms against inhibitors. The signalingpathway of fusion kinases can differ from WT kinases or fusionkinases with different partners, possibly due to different proteininteractions. For instance, it was suggested that cancer cells har-boring the NPM–ALK fusion depend on STAT3 pathway activa-tion, but cancer cells harboring the EML4–ALK fusion do not (5).Similarly, the FGFR3–BAIAP2L1 fusion kinase induces phospho-STAT1, but the FGFR3–TACC3 fusion kinase cannot (18). There-fore, we focused on FGFR3–BAIAP2L1 and investigated its asso-ciated signaling pathways by comprehensive RNA-Seq analysis.We identified hyperactivation of the MAPK pathway and sup-pression of several tumor-suppressive pathways, such as the RB1,p53, or CDKN2A pathways. In FGFR3–BAIAP2L1 cells, weobserved that p53 or p21 downregulation correlated with areversal of CDK4/Cyclin D suppression and RB phosphorylation.These observations suggested that the FGFR3–BAIAP2L1 fusionsuppresses RB1 function and leads to E2F pathway activation,consistent with our IPA analysis. E2F activation causes activationof the CDK2/Cyclin E complex, which can further induce p27degradation (Fig. 3B). Inhibition of tumor-suppressive pathwaysby oncogene fusions was also reported with NPM–ALK (29). The
B
C
Moc
k
FG
FR
3
F3-
B
F3-
B- Δ
BA
R
F3-
B- Δ
SH
F3-
B-Δ
BA
R/Δ
SH
F3-
B-K
D
FGFR3
pY-FGFR
FGFR3
FLA
GM
ycF
LAG
+M
ycF
LAG
Myc
FLA
G+
Myc
FLA
GM
ycF
LAG
+M
yc
Moc
k
IB: Myc
IB: FLAG
F3-BF3-B
-ΔBAR FGFR3F
LAG
Myc
FLA
G+
Myc
FLA
GM
ycF
LAG
+M
yc
FLA
GM
ycF
LAG
+M
yc
Moc
k
F3-B
F3-B
-ΔBAR
IP: FLAG Input
A TMFGFR3
TK1 TK2
FGFR3–BAIAP2L1 (F3-B)
BARTK1 SH3TK2
FGFR3–BAIAP2L1 ΔBAR (F3-B-ΔBAR)
TK1 SH3TK2
Coiled-coil
FGFR3–BAIAP2L1 ΔSH (F3-B-ΔSH)
BARTK1 TK2
FGFR3–BAIAP2L1 ΔBAR/ΔSH (F3-B-ΔBAR/ΔSH)
TK1 TK2
FGFR3–BAIAP2L1 K508M (F3-B-KD)
BARTK1 SH3TK2
K508M
Figure 5.Dimerization activity of FGFR3–BAIAP2L1 lacking the BAR domain.A, schematic diagrams of full-lengthFGFR3 and FGFR3 variant constructsused in this study. B, comparison ofphospho-FGFR expression levels.293 cells were transfected with theexpression vectors indicated in thediagram shown in A. At 72 hoursafter transfection, cells were lysedand analyzed by Western blot. C,comparison of dimerization activities.293 cells were transfected withexpression vectors encodingFGFR3, F3-B, or F3-B-DBAR andsubsequently lysed. The lysates wereimmunoprecipitated with anti-FLAGM2 Affinity Gel and detected inWestern blots using an anti-FLAG oranti-Myc antibody. Total cell lysateswere also analyzed by Western blot.
Nakanishi et al.
Mol Cancer Ther; 14(3) March 2015 Molecular Cancer Therapeutics710
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T

theory of "oncogene-induced senescence" supports the suppres-sive effect of the FGFR3–BAIAP2L1 fusion kinase (30).Oncogene-induced senescence is a mechanism used by cells to stay in apremalignant stage, wherein cells do not become malignantwithout an additional genetic alteration(s). This phenomenonhas been documented in cells harboring variants of BRAF (31),KRAS (32), and EGFR (33), and involves p53 or RB pathwayactivation (34). Therefore, bothMAPKpathway activation and theescape from senescence through p53 or RB suppression maypromote FGFR3–BAIAP2L1-expressing cells to undergo transfor-mation. This suggests that the combined inhibition of suppressiveactivity and FGFR may result in synergistic antitumor activityagainst cancers harboring FGFR3–BAIAP2L1.
The FGFR3–BAIAP2L1 fusion kinase is the first fusion kinaseutilizing a BARdomain as a dimerizationmotif.We demonstratedthat the BAR domain is essential for aberrant FGFR3 kinaseactivation and oncogenic activity (Fig. 4). Thus, small-moleculeFGFR inhibitors may facilitate targeted therapy for patients har-boring the FGFR3–BAIAP2L1 rearrangement (Fig. 2F). An emerg-ing clinical issue is the development of drug resistance, and next-generation inhibitors against the same target are under clinicalinvestigation because these tumors may depend upon the sametargets, even after resistance development. Similarly, point muta-tions in the FGFR kinase domain are implicated in resistanceagainst FGFR kinase inhibitors (35, 36). Therefore, the develop-ment of alternative therapeutic antagonists of the same targets isimportant. Deletion of the BAR domain dramatically impairsdimerization activity (Fig. 5), suggesting that targeting theBAIP2L1 BAR domain may be a viable therapeutic approach.Interestingly, BAIAP2L1 siRNA did not cause cytotoxicity inRT112/84 or HT1376 cells, which have WT BAIAP2L1 (Supple-mentary Fig. S3). Therefore, the inhibition of BAIAP2L1 isexpected to be safe, although further investigation is necessary.
In summary, we demonstrated that FGFR3–BAIAP2L1 exertspotent tumorigenic activity through ligand-independent andconstitutive dimerization via the BAR domain, and a cell lineharboring this gene fusion was sensitive to the FGFR inhibitor
CH5183284/Debio 1347. We also detected this rearrangement inhuman clinical bladder and lung cancer specimens. Therefore,treating patients harboring FGFR gene fusions such as FGFR3–BAIAP2L1 with CH5183284/Debio 1347 or other FGFR inhibi-tors may be a promising approach in the future. CH5183284/Debio 1347 is currently under phase I clinical investigation byDebiopharm International S.A. in patients harboring FGFR genet-ic alterations (NCT01948297).
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: Y. NakanishiDevelopment of methodology: Y. NakanishiAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Nakanishi, N. Akiyama, T. Tsukaguchi, Y. SatohAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): Y. Nakanishi, T. FujiiWriting, review, and/or revision of the manuscript: Y. Nakanishi, N. IshiiAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): Y. Nakanishi, N. AkiyamaStudy supervision: Y. Nakanishi, N. Ishii, M. Aoki
AcknowledgmentsThe authors thank Toshikazu Yamazaki and Yuko Aoki for helpful discus-
sions. They also thank Kiyoaki Sakata, Yasue Nagata, Yukari Nishitoh, YuhsukeIde, and Tsutomu Takahashi for performing pharmacologic assays and AnneVaslin, H�el�ene Maby-El Hajjami, and Corinne Moulon from DebiopharmInternational S.A. for their helpful discussions and technical support.
Grant SupportThis study was funded by the Chugai Pharmaceutical Co., Ltd.The costs of publication of this articlewere defrayed inpart by the payment of
page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received October 24, 2014; revised December 26, 2014; accepted December29, 2014; published OnlineFirst January 14, 2015.
References1. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al.
Identification of the transforming EML4-ALK fusion gene in non-small-celllung cancer. Nature 2007;448:561–6.
2. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global surveyof phosphotyrosine signaling identifies oncogenic kinases in lung cancer.Cell 2007;131:1190–203.
3. Takahashi M, Cooper GM. ret transforming gene encodes a fusion proteinhomologous to tyrosine kinases. Mol Cell Biol 1987;7:1378–85.
4. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al.Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer.N Engl J Med 2010;363:1693–703.
5. Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, FukamiTA, et al. CH5424802, a selective ALK inhibitor capable of blocking theresistant gatekeeper mutant. Cancer Cell 2011;19:679–90.
6. Turner N, Grose R. Fibroblast growth factor signalling: from developmentto cancer. Nat Rev Cancer 2010;10:116–29.
7. TurnerN, PearsonA, Sharpe R, LambrosM,Geyer F, Lopez-GarciaMA, et al.FGFR1 amplification drives endocrine therapy resistance and is a thera-peutic target in breast cancer. Cancer Res 2010;70:2085–94.
8. Cancer Genome Atlas Research N. Comprehensive genomic characteriza-tion of squamous cell lung cancers. Nature 2012;489:519–25.
9. Hara T, Ooi A, Kobayashi M, Mai M, Yanagihara K, Nakanishi I. Ampli-fication of c-myc, K-sam, and c-met in gastric cancers: detection by fluo-rescence in situ hybridization. Lab Invest 1998;78:1143–53.
10. Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-GarauX, et al. Frequent activating mutations of FGFR3 in human bladder andcervix carcinomas. Nat Genet 1999;23:18–20.
11. Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H,et al. Frequent activating FGFR2 mutations in endometrial carcinomasparallel germline mutations associated with craniosynostosis and skeletaldysplasia syndromes. Oncogene 2007;26:7158–62.
12. Xiao S, Nalabolu SR, Aster JC,Ma J, Abruzzo L, Jaffe ES, et al. FGFR1 is fusedwith a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lympho-ma syndrome. Nat Genet 1998;18:84–7.
13. Yagasaki F,WakaoD, Yokoyama Y, Uchida Y,Murohashi I, KayanoH, et al.Fusion of ETV6 to fibroblast growth factor receptor 3 in peripheral T-celllymphoma with a t(4;12)(p16;p13) chromosomal translocation. CancerRes 2001;61:8371–4.
14. Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, et al.Fibroblast growth factor receptor 2 tyrosine kinase fusions define a uniquemolecular subtype of cholangiocarcinoma.Hepatology 2014;59:1427–34.
15. Majewski IJ, Mittempergher L, Davidson NM, Bosma A, Willems SM,Horlings HM, et al. Identification of recurrent FGFR3 fusion genes in lungcancer through kinome-centred RNA sequencing. J Pathol 2013;230:270–6.
16. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al.Transforming fusions of FGFR and TACC genes in human glioblastoma.Science 2012;337:1231–5.
www.aacrjournals.org Mol Cancer Ther; 14(3) March 2015 711
Oncogenic Mechanisms and Prevalence of FGFR3–BAIAP2L1
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T

17. Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions inbladder cancer. Hum Mol Genet 2013;22:795–803.
18. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al.Identification of targetable FGFR gene fusions in diverse cancers. CancerDiscov 2013;3:636–47.
19. Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, et al. Thetumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation inglioblastoma. J Clin Invest 2013;123:855–65.
20. Peter BJ, KentHM,Mills IG, Vallis Y, Butler PJ, Evans PR, et al. BARdomainsas sensors ofmembrane curvature: the amphiphysin BAR structure. Science2004;303:495–9.
21. Nakanishi Y, Akiyama N, Tsukaguchi T, Fujii T, Sakata K, Sase H, et al. Thefibroblast growth factor receptor genetic status as a potential predictor ofthe sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhib-itor. Mol Cancer Ther 2014;13:2547–58.
22. Ishii N, Harada N, Joseph EW, Ohara K, Miura T, Sakamoto H, et al.Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor,CH5126766, that suppresses feedback reactivation of RAF activity. CancerRes 2013;73:4050–60.
23. Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, et al.The selective class I PI3K inhibitor CH5132799 targets human cancersharboring oncogenic PIK3CA mutations. Clin Cancer Res 2011;17:3272–81.
24. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seqdata with or without a reference genome. BMC Bioinforma 2011;12:323.
25. Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase generearrangements in epithelial malignancies. Nat Rev Cancer 2013;13:772–87.
26. Cancer Genome Atlas Research N. Comprehensive molecular characteri-zation of urothelial bladder carcinoma. Nature 2014;507:315–22.
27. Omberg L, Ellrott K, YuanY,KandothC,WongC, KellenMR, et al. Enablingtransparent and collaborative computational analysis of 12 tumor typeswithin The Cancer Genome Atlas. Nat Genet 2013;45:1121–6.
28. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, GolubTR, et al. Discovery and saturation analysis of cancer genes across 21tumour types. Nature 2014;505:495–501.
29. Cui YX, Kerby A, McDuff FK, Ye H, Turner SD. NPM-ALK inhibits the p53tumor suppressor pathway in an MDM2 and JNK-dependent manner.Blood 2009;113:5217–27.
30. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic rasprovokes premature cell senescence associated with accumulation of p53and p16INK4a. Cell 1997;88:593–602.
31. Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, vanderHorst CM, et al. BRAFE600-associated senescence-like cell cycle arrest ofhuman naevi. Nature 2005;436:720–4.
32. Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al.Tumour biology: senescence in premalignant tumours. Nature 2005;436:642.
33. SunC,Wang L,Huang S,HeynenGJ, Prahallad A, Robert C, et al. Reversibleand adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature2014;508:118–22.
34. Kuilman T,MichaloglouC,MooiWJ, PeeperDS. The essence of senescence.Genes Dev 2010;24:2463–79.
35. Byron SA, Chen H, Wortmann A, Loch D, Gartside MG, Dehkhoda F, et al.The N550K/H mutations in FGFR2 confer differential resistance toPD173074, dovitinib, and ponatinib ATP-competitive inhibitors. Neopla-sia 2013;15:975–88.
36. Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, et al.Tumour cell responses to new fibroblast growth factor receptor tyrosinekinase inhibitors and identification of a gatekeepermutation in FGFR3 as amechanism of acquired resistance. Oncogene 2013;32:3059–70.
Mol Cancer Ther; 14(3) March 2015 Molecular Cancer Therapeutics712
Nakanishi et al.
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T

2015;14:704-712. Published OnlineFirst January 14, 2015.Mol Cancer Ther Yoshito Nakanishi, Nukinori Akiyama, Toshiyuki Tsukaguchi, et al.
BAIAP2L1−Kinase FGFR3 Mechanism of Oncogenic Signal Activation by the Novel Fusion
Updated version
10.1158/1535-7163.MCT-14-0927-Tdoi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2015/01/14/1535-7163.MCT-14-0927-T.DC1
Access the most recent supplemental material at:
Cited articles
http://mct.aacrjournals.org/content/14/3/704.full#ref-list-1
This article cites 36 articles, 11 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/14/3/704.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/14/3/704To request permission to re-use all or part of this article, use this link
on June 23, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst January 14, 2015; DOI: 10.1158/1535-7163.MCT-14-0927-T