neue therapieoptionen des bullösen pemphigoids und der ... · zwischen einer akuten und einer...
TRANSCRIPT

Aus der Klinik für Dermatologie, Allergologie und Venerologie
der Universität zu Lübeck
Direktor: Prof. Dr. med. D. Zillikens
Neue Therapieoptionen des bullösen Pemphigoids
und der Epidermolysis bullosa acquisita:
Untersuchungen am Gefrierschnittmodell
Inauguraldissertation
zur
Erlangung der Doktorwürde
der Universität zu Lübeck
-Aus der Sektion Medizin-
vorgelegt von
Nicole Möckel
aus Schlema
Lübeck 2013

1. Berichterstatter: Prof. Dr. med. Ralf Ludwig
2. Berichterstatter: Priv.-Doz. Dr. med. Ingeborg Bos
Tag der mündlichen Prüfung: 17.01.2014
Zum Druck genehmigt. Lübeck, den 17.01.2014
-Promotionskommission der Sektion Medizin-

Inhaltsverzeichnis I
Inhaltsverzeichnis
1. Einleitung ................................................................................................................. 1
1.1 Blasenbildende Autoimmundermatosen .............................................................. 1
1.2 Das bullöse Pemphigoid ..................................................................................... 2
1.2.1 Epidemiologie .............................................................................................. 2
1.2.2 Risikofaktoren .............................................................................................. 2
1.2.3 Klinik ............................................................................................................ 3
1.2.4 Verlauf und Prognose .................................................................................. 5
1.2.5 Immunpathologie ......................................................................................... 5
1.2.6 Diagnostik ...................................................................................................12
1.3 Epidermolysis bullosa acquisita .........................................................................16
1.3.1 Epidemiologie .............................................................................................16
1.3.2 Klinik ...........................................................................................................16
1.3.3 Assoziierte Erkrankungen ...........................................................................17
1.3.4 Immunpathologie ........................................................................................18
1.3.5 Diagnostik ...................................................................................................20
1.4 Therapie ............................................................................................................23
1.4.1 Therapie des BP .........................................................................................23
1.4.2 Therapie der EBA .......................................................................................30
1.5 Ziele dieser Arbeit ..............................................................................................32
2. Material und Methoden ...........................................................................................33
2.1 Das Gefrierschnittmodell....................................................................................33
2.1.1 Verwendete Seren ......................................................................................34
2.1.2 Herstellung der Gefrierschnitte ...................................................................35
2.1.3 Isolierung von Leukozyten, Zellvitalität und Zellzählung ..............................35
2.1.4 Vorbereitung der Wirkstoffe ........................................................................37
2.1.5 Vorbereitung der Gefrierschnitte .................................................................40
2.1.6 Fixierung und Färbung der Schnitte ............................................................41
2.1.7 Mikroskopische Auswertung .......................................................................41
2.1.8 Statistik .......................................................................................................41
2.2 Das NBT-Gefrierschnittmodell ...........................................................................42
2.2.1 Isolierung von Leukozyten ..........................................................................42

Inhaltsverzeichnis II
2.2.2 Vorbereitung der NBT-Lösung ....................................................................42
2.2.3 Vorbereitung von Methylprednisolon ...........................................................43
2.2.4 Vorbereitung der Gefrierschnitte .................................................................43
2.2.5 Fixierung der Gefrierschnitte .......................................................................44
2.3 Der ROS-Release ..............................................................................................44
2.3.1 Isolierung von neutrophilen Granulozyten mit Polymorphprep ....................44
2.3.2 Vorbereitung von Methylprednisolon ...........................................................45
2.3.3 Durchführung des ROS-Release ................................................................46
2.3.4 Auswertung ................................................................................................47
3. Ergebnisse ..............................................................................................................48
3.1 Verwendete Seren .............................................................................................48
3.1.1 Testung der EBA-Seren im Gefrierschnittmodell .........................................48
3.1.2 Testung der BP180-Seren im Gefrierschnittmodell .....................................49
3.2 Vergleich der DES auf humaner Haut und Maushaut .........................................51
3.3 Effekte von Methylprednisolon auf die Bildung von ROS
im Gefrierschnittmodell mit Nitroblautetrazolium (NBT) ......................................51
3.4 Effekte von Methylprednisolon auf die Bildung von ROS im ROS-Release ........54
3.5 Effekte der PI3-Kinase-Inhibitoren PI-103 und TGX-221 auf die
dermo-epidermale Spaltbildung im Gefrierschnittmodell ....................................56
3.5.1 Effekte von PI-103 auf die dermo-epidermalen Spaltbildung .......................56
3.5.2 Effekte von TGX-221 auf die dermo-epidermalen Spaltbildung ...................60
3.6 Effekte des löslichen Fcγ – Rezeptors IIb SM101 auf die
dermo-epidermale Spaltbildung im Gefrierschnittmodell ....................................65
3.6.1 SM101 im Gefrierschnittmodell mit Maushaut und EBA-Serum
immunisierter Kaninchen ............................................................................66
3.6.2 SM101 im Gefrierschnittmodell mit humaner Haut und humanem
BP-Serum .................................................................................................. 67
4. Diskussion ..............................................................................................................70
4.1 Das Gefrierschnittmodell....................................................................................70
4.1.1 Antikörper ...................................................................................................72
4.1.2 Komplement ...............................................................................................72
4.1.3 Neutrophile Granulozyten ...........................................................................72
4.1.4 Gefrierschnitte ............................................................................................73
4.1.5 Vergleich der Gefrierschnittmodelle auf humaner Haut und Maushaut........73
4.2 Effekte von Methylprednisolon auf die Bildung von ROS ...................................74

Inhaltsverzeichnis III
4.2.1 Allgemeines zu Methylprednisolon ..............................................................74
4.2.2 Methylprednisolon im NBT-Gefrierschnittmodell und im ROS-Release .......76
4.3 Effekte von PI3-Kinase-Inhibitoren auf die dermo-epidermale Spaltbildung .......77
4.3.1 Allgemeines zu PI3-Kinasen und deren Funktion ........................................77
4.4 Effekte des löslichen humanen Fcγ – Rezeptors IIb auf die
dermo-epidermale Spaltbildung ex vivo .............................................................85
4.4.1 Allgemeines zu Fcγ-Rezeptoren .................................................................85
4.4.2 SM101 ........................................................................................................86
4.5 Ausblick .............................................................................................................89
5. Zusammenfassung .................................................................................................91
6. Literaturverzeichnis ................................................................................................93
7. Anhang .................................................................................................................. 123
7.1 Voten der Ethikkommission ............................................................................. 123
7.2 Abbildungsverzeichnis ..................................................................................... 124
7.3 Tabellenverzeichnis ......................................................................................... 125
8. Danksagungen ...................................................................................................... 126
9. Lebenslauf ............................................................................................................. 127
10. Publikationen ..................................................................................................... 128

Abkürzungsverzeichnis VI
Abkürzungsverzeichnis
BP – bullöses Pemphigoid
BSA – bovines Serumalbumin
CL – Chemilumineszenz
ddH2O – double-distilled H2O (zweifach destilliertes H2O)
DES – dermo-epidermale Spaltbildung
DEJ – dermo-epidermale Junktionszone
DIF – direkte Immunfluoreszenz
DMSO – Dimethylsulfoxid
EBA – Epidermolysis bullosa acquisita
EDTA – Ethylendiamintetraacetic acid (Ethylendiamintetraessigsäure)
ELISA – Enzyme-linked immunosorbent assay
FcR – Fc-Rezeptor
IIF – indirekte Immunfluoreszenz
LPS – Lipopolysaccharid
MP – Methylprednisolon
NADPH – Nicotinsäureamid-adenin-dinukleotid-phosphat
NAOH – Natriumhydroxid
NBT – Nitroblautetrazolium
NC – non-collagen (nicht-kollagen)
NK – Negativkontrolle
PBS – Phosphate-buffered saline
PK – Positivkontrolle
PI3K – Phosphatidylinositid-3-Kinase
ROS – reactive oxygen species (reaktive Sauerstoffspezies)
RPMI – Roswell Park Memorial Institute medium
SEM – standard error of the mean (Standardfehler)
z0,25 – 25 %-Perzentil
z0,75 – 75 %-Perzentil

1. Einleitung 1
1. Einleitung
1.1 Blasenbildende Autoimmundermatosen
Bei blasenbildenden Autoimmundermatosen handelt es sich um eine Gruppe
heterogener Erkrankungen, die sich klinisch durch Blasen und Erosionen an Haut
und hautnahen Schleimhäuten auszeichnen (Schmidt and Zillikens, 2011).
Gemeinsame pathologische Grundlage aller Formen ist die Schädigung
spezifischer Adhäsions- und Strukturmoleküle der Haut durch pathogene
Autoantikörper. Je nach Zielstruktur unterscheidet man die
Pemphiguserkrankungen, die Pemphigoiderkrankungen, die Epidermolysis bullosa
acquisita und die Dermatitis herpetiformis. (siehe Tab.1).
Im Folgenden soll das Augenmerk auf zwei Autoimmundermatosen gerichtet sein,
dem bullösen Pemphigoid (BP) und der Epidermolysis bullosa acquisita (EBA).
Erkrankungsgruppe Unterform Hauptzielantigene
Pemphigus Pemphigus vulgaris
Pemphigus foliaceus
paraneoplastischer Pemphigus
IgA-Pemphigus
Dsg 3, Dsg 1
Dsg 1
Plakine
Dsc 1, Dsg 3
Pemphigoid bullöses Pemphigoid
Pemphigoid gestationis
Schleimhautpemphigoid
a
lineare IgA-Dermatose
Lichen planus pemphigoides
Anti-p200/Laminin γ1 Pemphigoid
BP180, BP230
BP180
BP180, Laminin 332,
α6β4-Integrin
LAD-1
BP180
Laminin γ1
Epidermolysis bullosa
acquisita
Typ-VII-Kollagen
Dermatitis herpetiformis
Duhring
Gewebstransglutaminase
Tabelle 1: Blasenbildende Autoimmundermatosen und deren Zielantigene (nach (Schmidt
and Zillikens, 2011; Sitaru et al., 2004)). Anhand der Zielantigene werden vier Hauptgruppen
unterschieden. Je nach Lage der Zielstruktur kommt es zur intraepidermalen (Pemphigus-
Erkrankungen) oder subepidermalen (Pemphigoid-Gruppe, Epidermolysis bullosa acquisita,
Dermatitis herpetiformis) Spaltbildung. Dsg: Desmoglein, Dsc: Desmocollin

1. Einleitung 2
1.2 Das bullöse Pemphigoid
1.2.1 Epidemiologie
Das bullöse Pemphigoid (BP) ist eine blasenbildende Autoimmundermatose,
welche sich klinisch durch die Bildung von Blasen an der Haut auszeichnet. Es
handelt sich dabei um eine Erkrankung des älteren Menschen, bei
Diagnosestellung sind die Patienten im Mittel 75 Jahre alt (Kippes et al., 1999).
Darüber hinaus steigt die Inzidenz nach dem 70. Lebensjahr signifikant an und
erreicht ihren Höhepunkt im Alter von 90 Jahren (Marazza et al., 2009). Die
mittlere Inzidenz der Erkrankung liegt zwischen 7 und 14 Fällen pro eine Million
Einwohner (Bernard et al., 1995; Bertram et al., 2009; Gudi et al., 2005; Marazza
et al., 2009), wobei in einer Studie in Unterfranken eine Inzidenz von 13,4
Neuerkrankungen pro eine Million Einwohner pro Jahr angegeben wurde (Bertram
et al., 2009). Bei über 90-jährigen Männern wird derzeit sogar eine Inzidenz von
knapp 400 Neuerkrankungen pro eine Million Einwohner pro Jahr angenommen
(Gudi et al., 2005; Jung et al., 1999). In einer retrospektiven Studie aus
Großbritannien konnte außerdem ein Anstieg der Inzidenz von 17 % pro Jahr
gezeigt werden, wobei hier aktuell eine Inzidenz von 43 Neuerkrankungen pro
eine Million Einwohner pro Jahr beschrieben wird (Langan et al., 2008). Es wird
davon ausgegangen, dass die steigende Inzidenz unter anderem an den
verbesserten diagnostischen Möglichkeiten zur Erkennung des BP und der sich
ändernden Altersstruktur der Bevölkerung liegt (Bertram et al., 2009). Es wurde
beschrieben, dass Männer fast zweimal so häufig wie Frauen betroffen sind (Jung
et al., 1999).
1.2.2 Risikofaktoren
Für das Auftreten des BP wurden mehrere mögliche Auslöser beschrieben, wie
zum Beispiel Trauma, Verbrennungen, Radiotherapie oder ultraviolettes Licht
(Schmidt et al., 2011). Auch die Einnahme bestimmter Medikamente über einen
längeren Zeitraum stellt einen Risikofaktor dar. So konnte die chronische
Einnahme von Aldosteronantagonisten und Neuroleptika in Zusammenhang mit
dem Auftreten des BP gebracht werden (Bastuji-Garin et al., 1996), als auch von

1. Einleitung 3
Spironolaktonen und Phenothiazinen mit aliphatischen Seitenketten (Bastuji-Garin
et al., 2011).
Des Weiteren wurde eine Assoziation zwischen dem Vorkommen eines
bestimmten Allels des humanen Leukozyten-Antigens (HLA), HLA-DQβ1*0301,
und dem Auftreten des BP beschrieben (Delgado et al., 1996). HLA-Proteine
dienen der Präsentation von Antigenfragmenten auf Antigen-präsentierenden
Zellen (APC) und der Aktivierung von T-Zellen. In vitro konnte so bei der Mehrzahl
von BP-Patienten, die das HLA-DQβ1*0301-Allel exprimierten, eine T-Zell-Antwort
auf BP180 beobachtet werden. Gleiches traf auf gesunde, HLA-DQβ1*0301-
positive Spender zu. Mononukleäre Zellen gesunder Spender, die andere HLA-
DQ-Allele exprimierten, zeigten hingegen keine T-Zell-Antwort gegenüber BP180
(Budinger et al., 1998).
Weiterhin konnte ein Polymorphismus des Fcγ-Rezeptors IIIa im Bereich des
Nukleotids 559 mit dem Auftreten von BP in Verbindung gebracht werden. Die
Unterschiede der Allele äußern sich dabei in der Affinität des Rezeptors durch
Kodierung und Einbau der Aminosäuren Phenylalanin (niedrigaffiner Rezeptor)
oder Valin (hochaffiner Rezeptor). Es konnte gezeigt werden, dass etwa 60 % der
untersuchten BP-Patienten homozygot für den niedrigaffinen FcγRIIIa-Allotyp sind,
bei den gesunden Kontrollen waren es nur halb so viele. Das Vorkommen des
niedrigaffinen FcγRIIIa könnte somit die Ausbildung eines BP begünstigen
(Weisenseel et al., 2007).
1.2.3 Klinik
Das BP wurde erstmals 1953 in einem von Lever veröffentlichten Artikel über
Pemphigus erwähnt (Lever, 1953). Schon vorher wurde eine Unterscheidung
zwischen einer akuten und einer chronischen Form des Pemphigus getroffen
(Talbott et al., 1940), wobei die als akut bezeichnete Form mikroskopisch eine
Akantholyse der Epidermis zeigte, die chronische Form jedoch nicht. Auch konnte
Lever histologisch beim sogenannten Pemphigus vulgaris acutus eine
intraepidermale Blasenbildung nachweisen, bei der chronischen Form lag die
Blasenbildung subepidermal. So bezeichnete Lever den Pemphigus vulgaris
chronicus, welcher histologisch eigentlich kein Pemphigus war, als bullöses
Pemphigoid (Ghohestani et al., 2001).

1. Einleitung 4
Das klinische Bild des BP ist jedoch vielgestaltig. Das zunächst auftretende
prämonitorische Stadium, welches mehrere Wochen bis Monate oder sogar Jahre
andauern kann, zeichnet sich durch Juckreiz aus. Es können aber auch
ekzemartige Reaktionen, lokal begrenzte Erytheme oder papulovesikulöse
beziehungsweise urtikarielle Plaques auftreten, welche exkoriiert werden
(Ghohestani et al., 2001; Hertl and Schuler, 2002a; Zillikens, 2005). Diese
unspezifischen Symptome können die einzigen Zeichen der Erkrankung bleiben
(Schmidt et al., 2011). Das darauffolgende blasenbildende Stadium zeichnet sich
durch Bläschen und Blasen auf normaler oder erythematös veränderter Haut aus,
wobei sich auch hier weiterhin urtikarielle oder infiltrierte Plaques finden. Diese
können gelegentlich annulär angeordnet sein (Hertl and Schuler, 2002a; Schmidt
et al., 2011). Die Blasen sind prall, wenige Millimeter bis Zentimeter groß, weisen
einen klaren oder gelegentlich hämorrhagischen Inhalt auf (Zillikens, 2005) und
heilen nach mehreren Tagen als krustöse Erosionen ab (Hertl and Schuler,
2002a). Typischerweise sind die verschiedenen Entwicklungsstadien von Blasen,
Erosionen und Krusten erkennbar (siehe Abb.1). Prädilektionsstellen sind die
Beugen von Armen und Beinen, das untere Abdomen, die Innenseiten der
Oberschenkel, sowie die intertriginösen Bereiche, wobei die Läsionen meist
symmetrisch auftreten (Zillikens, 2005). Ein Befall der Schleimhäute ist auch
möglich, allerdings kommt dies nur in 10 bis 30 % der Fälle vor (Hertl and Schuler,
2002a). Es findet sich ein negatives Nikolski-Phänomen I, das Nikolski-Phänomen
II kann jedoch positiv ausfallen, die intakten Blasen lassen sich also durch Druck
seitlich verschieben (Hertl and Schuler, 2002a). Auch reißen die Blasen nicht so
leicht wie beim Pemphigus. Es kommen Spontanremissionen und Exazerbationen
vor, wobei erneute Schübe meist weniger stark ausgeprägt sind (Ghohestani et
al., 2001).

1. Einleitung 5
Abbildung 1: Klinisches Erscheinungsbild des bullösen Pemphigoids. Charakteristisch für
das Erscheinungsbild des bullösen Pemphigoids ist das Nebeneinander von prallen Blasen,
Erosionen und Krusten auf normal erscheinender oder erythematöser Haut. Quelle: Klinik für
Dermatologie, Universität zu Lübeck
1.2.4 Verlauf und Prognose
Das BP verläuft chronisch rezidivierend und ist im Verlauf von mehreren Jahren
oft selbstlimitierend, wobei eine Remission durchschnittlich innerhalb von fünf
Jahren erfolgt (Wojnarowska et al., 2001). Rezidive verlaufen meist weniger
schwer (Ghohestani et al., 2001; Korman, 1987). Als prognostisch ungünstig
wurden ein hohes Lebensalter, ein niedriger Karnofsky-Index, eine hochdosierte
Therapie mit Glukokortikoiden, ein niedriges Serumalbumin und eine hohe
Blutsenkungsgeschwindigkeit beschrieben (Joly et al., 2005; Joly et al., 2009;
Rzany et al., 2002). Die Mortalität innerhalb des ersten Jahres der Erkrankung
liegt in Europa zwischen 19 und 41 % (Bystryn and Rudolph, 2005), was vor allem
mit dem Alter der Patienten und der Polymorbidität zu begründen ist (Parker et al.,
2008). Dabei wurden vor allem Sepsis, kardiovaskuläre Erkrankungen (Roujeau et
al., 1998) oder Erkrankungen der Atemwege (Gudi et al., 2005) als Todesursache
beschrieben. Insgesamt ist die Mortalität der an BP Erkrankten im Vergleich zur
Kontrollpopulation zweifach erhöht (Langan et al., 2008).
1.2.5 Immunpathologie
Für die Pathogenese des BP sind Autoantikörper gegen zwei Strukturproteine der
Haut von Bedeutung, BP180 und BP230. Dabei handelt es sich um Bestandteile
der Desmosomen, die für die dermo-epidermale Adhärenz von essentieller

1. Einleitung 6
Bedeutung sind (siehe Abb.2). Durch Bindung der Antikörper an diese
Strukturproteine entstehen Immunkomplexe, die Komplement aktivieren.
Aktiviertes Komplement führt zur Degranulation von Mastzellen und der
Rekrutierung neutrophiler Granulozyten, die an die Immunkomplexe binden und
dadurch aktiviert werden. Aktivierte neutrophile Granulozyten setzen
proteolytische Enzyme und reaktive Sauerstoffspezies (ROS) frei, was zur
Schädigung der dermo-epidermalen Junktionszone (DEJ) führt (Jordon et al.,
1985).
Abbildung 2: Autoantigene der Haut (nach (Bieber et al., 2010)). Die Strukturproteine der
Desmosomen (blau-lila) umfassen unter anderem BP180 und BP230, Zielantigene des BP. Die
hemidesmosomalen Strukturproteine (grün) sind typische Zielstrukturen für
Pemphiguserkrankungen. Bei der EBA sind Autoantikörper gegen das Typ-VII-Kollagen gerichtet,
das sich in der Sublamina densa der Basalmembran befindet.
1.2.5.1 BP 180
a. Struktur
BP180, auch als Typ-XVII-Kollagen bekannt, ist ein 180 kD schweres
transmembranes Glykoprotein, welches eine Typ-II-Orientierung aufweist und
einen intrazellulären N-Terminus, eine transmembranen Domäne sowie einen
Desmoglein 1 und 3
Desmocollin1 und 3
DesmoplakinKeratin
BP230
BP180
Plectin
α6β4 Integrin
Laminin 332
Typ VII Kollagen
p200
Lamina lucida
Lamina densa
Sublamina densa
Ep
iderm
isB
asalm
em
bra
n
Pemphigus
Pemphigoid
Epidermolysis
bullosa acquisita

1. Einleitung 7
extrazellulär liegenden C-Terminus besitzt (Giudice et al., 1992; Giudice et al.,
1993; Hopkinson et al., 1992). Der carboxyterminale, extrazelluläre Bereich
umfasst circa 1.000 Aminosäuren und besteht aus 15 Kollagendomänen
unterschiedlicher Länge (15 bis 242 Aminosäuren), welche voneinander durch
kurze Abschnitte nicht-kollagener (NC)-Sequenzen getrennt sind (Giudice et al.,
1992) (siehe Abb.3). BP180 ist über den intrazellulären N-terminalen Anteil mit der
hemidesmosomalen Plaque der basalen Keratinozyten assoziiert, überspannt die
Lamina lucida und reicht mit dem C-terminalen Anteil bis in die Lamina densa der
epidermalen Basalmembran (Bedane et al., 1997; Masunaga et al., 1997). Es
wurde gezeigt, dass die extrazelluläre Domäne in vivo mindestens eine Loop-
Struktur in der Lamina densa aufweist und wahrscheinlich durch Interaktion mit
anderen Komponenten der Basalmembran wichtig für die dermo-epidermalen
Adhäsion ist (Nonaka et al., 2000). Die NC16A-Domäne, welche sich extrazellulär
proximal der Zytoplasmamembran befindet, interagiert unter anderem mit dem α6-
Integrin (Hirako et al., 1998). Über die NC1-Domäne im extrazellulären Anteil
interagiert BP180 außerdem mit Laminin 5 (Rousselle et al., 1997). Über die
intrazellulären Anteile des BP180 besteht eine Verbindung zu BP230, β4-Integrin
und zu Keratin 18 (Aho and Uitto, 1999; Borradori and Sonnenberg, 1999;
Hopkinson and Jones, 2000).
Abbildung 3: Schematische Darstellung von BP180 (nach (Ujiie et al., 2010)). Der
extrazelluläre Anteil des Strukturproteins besteht aus Kollagendomänen, die durch nicht-kollagene
(NC-) Domänen unterbrochen sind. Hauptantigen des BP stellt das plasmamembrannahe NC16A
dar. TM: transmembrane Domäne
Plasma-membran
TM
NC16A
NH2 COOH
Kollagendomänen
IntrazelluläreDomäne
ExtrazelluläreDomäne

1. Einleitung 8
b. Pathogenität
Die extrazelluläre NC16A-Domäne wurde als immundominante Region von BP180
beschrieben. Es konnte mittels Epitop-Mapping gezeigt werden, dass die meisten
Seren von Patienten mit BP mit dem Bereich der NC16A-Domäne korrelieren
(Giudice et al., 1993). Auch mithilfe eines ELISA konnte nachgewiesen werden,
dass über 90 % der Patientenseren mit der NC16A-Domäne reagieren (Nakatani
et al., 1998; Zillikens et al., 1997a). Insgesamt konnten vier verschiedene
immundominante Loki innerhalb des 45 Aminosäuren langen N-terminalen
Abschnitts von NC16A bestimmt werden, wohingegen der restliche, 28
Aminosäuren lange Abschnitt keine für BP relevanten Epitope aufweist (Zillikens
et al., 1997b). Die pathogenetische Bedeutung von Antikörpern gegen NC16A
konnte auch mithilfe des passiven Mausmodells gezeigt werden. Durch die
strukturellen Unterschiede des BP180 der Maus, war eine Blasenbildung durch
Injektion von aufgereinigtem IgG aus BP-Seren nicht erfolgreich (Anhalt and Diaz,
1987). Daher wurden Kaninchen mit einem rekombinanten Fragment des murinen
BP180, das NC16A beinhaltete, immunisiert. Das gebildete Kaninchen-IgG wurde
danach in neonatale Mäuse injiziert, die daraufhin eine Blasenbildung entwickelten
(Liu et al., 1993). Weiterhin konnte in vitro die Bedeutung von humanen
Antikörpern gegen NC16A mithilfe des Gefrierschnittmodells nachgewiesen
werden. Dafür wurden Gefrierschnitte humaner Haut mit humanen Leukozyten
und mit gegen rekombinantes NC16A aufgereinigte IgG aus BP-Patientenseren
inkubiert, was zur Spaltbildung zwischen Epidermis und Dermis führte. Auch
konnte gezeigt werden, dass IgG, welches gegen NC16A nicht reaktiv war, im
Gefrierschnittmodell auch keine Spaltbildung erzeugte (Sitaru et al., 2002b). Des
Weiteren wurde gezeigt, dass Antikörper nicht nur an die extrazelluläre, sondern
auch an die intrazelluläre Domäne binden, was Auswirkungen auf die Schwere der
Erkrankung haben könnte (Dresow et al., 2009; Perriard et al., 1999). Die
Immunantwort gegen BP180 wird vor allem über IgG1, IgG4 und IgE vermittelt,
seltener durch IgG2 und IgG3 (Dopp et al., 2000; Laffitte et al., 2001). Dabei
wurde ein Vorkommen von IgG1 vor allem in der akuten Phase der Erkrankung
beschrieben, wogegen IgG4 vor allem bei Patienten in Remission gefunden wurde
(Hofmann et al., 2002). Weiterhin konnte gezeigt werden, dass die Serumlevel von
anti-BP180 Antikörpern mit der Krankheitsaktivität korrelieren (Dopp et al., 2000;
Mutasim, 2000; Schmidt et al., 2000).

1. Einleitung 9
1.2.5.2 BP 230
a. Struktur
Das 230 kD schwere Protein BP230 wurde erstmals 1981 beschrieben (Stanley et
al., 1981). BP230 gehört zur Familie der Plakine und ist ein zytoplasmatischer
Bestandteil des Hemidesmosoms (Ruhrberg and Watt, 1997). Das Polypeptid
besteht aus einer zentralen coiled-coil Domäne, welche von zwei globulären
Domänen flankiert wird (Borradori and Sonnenberg, 1999). Mit der C-terminalen
Domäne agiert BP230 mit den Intermediärfilamenten, vor allem mit dem Keratin
5/Keratin14-Heterodimer, was eine wichtige Rolle bei der Bindung von
Intermediärfilamenten an den hemidesmosomalen Plaque spielt (Fontao et al.,
2003). N-terminal interagiert BP230 mit der zytoplasmatischen Domäne von
BP180 und wahrscheinlich auch mit β4-Integrin (Hopkinson and Jones, 2000;
Schaapveld et al., 1998).
b. Pathogenität
Als immundominante Regionen wurden die C-terminale B- und C-Subdomäne
beschrieben, an welche vor allem Antikörper vom Typ IgG4 und IgG1 binden. Aber
auch N-terminal konnten Epitope nachgewiesen werden (Skaria et al., 2000). Etwa
80 % der BP-Seren binden im ELISA an rekombinantes BP230 (Thoma-Uszynski
et al., 2004). Im Moment ist jedoch noch unklar, ob Antikörper gegen BP230 direkt
zur Spaltbildung führen. In einem Mausmodell konnten bei Injektion von
polyklonalen Kaninchen-Antikörpern gegen humanes BP230 klinische und
immunpathologische Zeichen des BP beobachtet werden (Kiss et al., 2005). In
einer anderen Studie war dies erst verzögert nach Bestrahlung mit UVB auslösbar
(Hall et al., 1993). Letztendlich ist die pathogenetische Relevanz von Antikörpern
gegen BP230 noch nicht eindeutig nachgewiesen.
1.2.5.3 Komplement
Die Autoantikörper binden an die oben beschriebenen Strukturproteine der DEJ.
Beim BP wird jedoch nur eine Spaltbildung ausgelöst, wenn Komplement aktiviert
wird (Gammon, 1989). Dabei erfolgt die Aktivierung des Komplements durch den
Fc-Teil des pathogenen IgG (Liu et al., 1995). Die Notwendigkeit von Komplement
(C) wurde im Mausmodell gezeigt, indem bei C5-defizienten Mäusen durch
Injektion von murinem IgG gegen BP180 keine Spaltbildung ausgelöst werden

1. Einleitung 10
konnte, wohl aber bei Mäusen mit C5 (Liu et al., 1995). Aktiviertes C5 (C5a) ist ein
potenter inflammatorischer Mediator, bindet über einen C5a-Rezeptor an
Mastzellen und führt damit zu deren Degranulation und der Freisetzung
proinflammatorischer Zytokine wie IL-1, IL-6, TNF-α oder GM-CSF und somit zur
Rekrutierung von neutrophilen und eosinophilen Granulozyten und Monozyten
(Heimbach et al., 2011; Liu et al., 1993; Marshall, 2004). In einem anderen
Mausmodell konnte gezeigt werden, dass nicht nur der alternative sondern auch
der klassische Weg der Komplementaktivierung von Bedeutung zu sein scheint
(Nelson et al., 2006). Im Gefrierschnittmodell ist zur Induktion der Spaltbildung
jedoch kein Komplement nötig. Dies liegt darin begründet, dass bei Inkubation der
Gewebsschnitte mit Leukozyten diese sich bereits im Bereich der DEJ befinden
und so keine Rekrutierung über Entzündungsmediatoren nötig ist (Sitaru et al.,
2002b).
1.2.5.4 Mastzellen
Es konnte im Mausmodell gezeigt werden, dass Mastzellen eine grundlegende
Rolle bei der Chemotaxis von neutrophilen Granulozyten spielen (Chen et al.,
2001b; Heimbach et al., 2011). Mastzellen können viele verschiedene Mediatoren,
wie Leukotriene, Plättchen-aktivierender Faktor oder Zytokine, produzieren (Galli,
1993; Galli et al., 1991). In der Blasenflüssigkeit von Patienten mit BP konnten
hohe Werte von Histamin, Leukotrien B4, IL-1, -2, -4, -5, -6, -8 und -10 sowie von
TNF-α und IFN-γ nachgewiesen werden (Ameglio et al., 1998; Katayama et al.,
1984; Kawana et al., 1990; Schmidt et al., 1996). Unabhängig davon wurde ein
Mastzell-unabhängiger, direkter Einfluss auf die Aktivierung und Rekrutierung
neutrophiler Granulozyten durch Komplement C3a und C5a beschrieben (Chen et
al., 2001b). Im Mausmodell wurde jedoch gezeigt, dass Mastzell-defiziente Mäuse
nach Injektion von murinem anti-BP180 keine Zeichen des BP entwickelten, bei
zusätzlicher intradermaler Injektion von IL-8 aber sowohl klinische als auch
histologische Zeichen der Infiltration neutrophiler Granulozyten und Blasenbildung
zeigten. Dies gibt Hinweise darauf, dass Mastzellen durch die Freisetzung
bestimmter Chemokine eine essentielle Rolle bei der Rekrutierung und Aktivierung
von neutrophilen Granulozyten spielen (Chen et al., 2001b). Es wurde weiterhin
beschrieben, dass die Level von IL-1, IL-8, TNF-α und besonders von IL-5 in den

1. Einleitung 11
Blasen und die Spiegel von IL-6 und IL-8 im Serum mit der Krankheitsschwere
korrelieren (Ameglio et al., 1998; Inaoki and Takehara, 1998).
1.2.5.5 Neutrophile Granulozyten
Neutrophile Granulozyten spielen eine wesentliche Rolle in der Pathogenese des
BP. Im Mausmodell mit neutropenen Mäusen konnten mit murinem anti-BP180
IgG keine histologischen oder klinischen Zeichen des BP ausgelöst werden (Liu et
al., 1997). Auch im Gefrierschnittmodell konnte eine Spaltbildung nur in
Anwesenheit von neutrophilen Granulozyten erzeugt werden (Gammon et al.,
1982b; Sitaru et al., 2002b). Die Aktivierung der neutrophilen Granulozyten erfolgt
durch Bindung an Immunkomplexe über den Fc-Rezeptor der Zellen (Zhao et al.,
2006). Dabei sind beim Menschen vor allem der Fcγ-Rezeptor IIa und der Fcγ-
Rezeptor IIIb von Bedeutung (Yu et al., 2010). Auch spielen für die Pathogenese
des BP beim Menschen weitere Entzündungszellen eine Rolle. So sind in frühen
Stadien in der betroffenen Haut vor allem eosinophile Granulozyten zu finden, was
einen Hinweis darauf gibt, dass diese möglicherweise einen Beitrag zur
Pathogenese leisten (Zone et al., 2007). In Hautbiopsaten von erkrankten
Patienten sind meist mehr eosinophile als neutrophile Granulozyten nachweisbar
(Borrego et al., 1996). Im ex-vivo-Modell kommt hingegen den neutrophilen
Granulozyten eine entscheidende Rolle zu (Gammon et al., 1982b; Sitaru et al.,
2002b). Weiterhin sind in den betroffen Hautarealen Lymphozyten, Monozyten und
Mastzellen nachweisbar (Liu et al., 1997).
1.2.5.6 Proteolytische Enzyme und reaktive Sauerstoffspezies
Die aktivierten neutrophilen Granulozyten setzen ROS und Enzyme frei (Jordon et
al., 1985). Neutrophile Granulozyten enthalten in ihren Granula verschiedene
proteolytische Enzyme wie Kollagenase, Elastase, Cathepsin G oder Gelatinase
B, welche direkt die Basalmembranzone schädigen und zur DES führen (Weiss,
1989). Im Mausmodell kommt besonders der Gelatinase B (Matrix-Metallo-
Proteinase 9) und der Neutrophilen-Elastase eine Bedeutung zu (Liu et al., 1998;
Liu et al., 2000). Dabei werden der Gelatinase B vor allem regulierende Effekte auf
die Neutrophilen-Elastase durch Inaktivierung von Proteinaseinhibitoren
zugeschrieben. Die Neutrophilen-Elastase ist hauptsächlich für die Spaltbildung
verantwortlich (Liu et al., 2000). Bei Patienten mit BP konnten sowohl in der

1. Einleitung 12
Hautbiopsie als auch in der Blasenflüssigkeit Neutrophilen-Elastase und
Gelatinase B nachgewiesen werden (Verraes et al., 2001).
1.2.6 Diagnostik
Das BP zeichnet sich durch ein weites Spektrum an klinischen
Manifestationsformen und atypischen Varianten aus. Oftmals geht der
Blasenbildung zudem ein prämonitorisches Stadium mit starkem, chronischen
Juckreiz voraus (Schmidt and Zillikens, 2011). Die Diagnose wird in der direkten
Immunfluoreszenz (DIF) durch Ablagerung von IgG an der DEJ und über
Nachweis von pathogenem IgG im Serum mithilfe des ELISA gesichert.
1.2.6.1 Histopatholgie
Die für die histologische Beurteilung nötige Biopsie sollte sich über eine Stelle mit
einer makroskopisch sichtbaren Blase erstrecken, aber auch einen Anteil
gesunder Haut beinhalten (Sitaru et al., 2004). Typisch im lichtmikroskopischen
Bild sind eine subepidermale Blasenbildung mit einem Infiltrat von eosinophilen
und neutrophilen Granulozyten in der oberen Dermis und in der Blase selbst
(siehe Abb.4), in frühen Formen kann auch eine eosinophile Spongiose
vorkommen (Schmidt et al., 2011). Aufgrund des histologischen Bildes allein kann
ein BP jedoch nicht eindeutig diagnostiziert werden (Hertl and Schuler, 2002b).
Auch sind typische histologische Veränderungen nur für etwa 60 % der BP-
Patienten beschrieben (Chan et al., 2003; Courville et al., 2000).
Abbildung 4: Histopathologischer Befund eines bullösen Pemphigoids. Typisch für die
Erkrankung sind die subepidermale Blasenbildung und das Vorkommen eosinophiler und
neutrophiler Infiltrate. Quelle: Klinik für Dermatologie, Universität zu Lübeck

1. Einleitung 13
1.2.6.2 Direkte Immunfluoreszenz
Die direkte Immunfluoreszenz (DIF) stellt den diagnostischen Goldstandard für
das BP dar (Schmidt and Zillikens, 2011). In der DIF können Ablagerungen vor
allem von IgG und C3 entlang der DEJ nachgewiesen werden, gelegentlich finden
sich auch Ablagerungen von IgM, IgE und IgA (Hertl and Schuler, 2002b; Schmidt
et al., 2011). Es sollte periläsionale, also unmittelbar neben der Blase befindliche
Haut, oder erythematöse, noch nicht blasenbildende Haut biopsiert werden. Wird
die Haut direkt aus der Blase verwendet, kommt es häufig zu falsch-negativen
Ergebnissen, da durch die vorliegende Entzündung gebundene Autoantikörper
und Komplement abgebaut werden können (Sitaru et al., 2004). Die
Autoantikörper und Komplement lagern sich in bestimmten Mustern entlang der
Basalmembranzone ab, was zur Differenzierung der unterschiedlichen
blasenbildenden Erkrankungen hilfreich sein kann. Beim BP lagern sich Antikörper
und Komplement in Form von n-Zacken ab (Vodegel et al., 2004).
1.2.6.3 Indirekte Immunfluoreszenz
Zum Nachweis von im Serum zirkulierenden Antikörpern dient die indirekte
Immunfluoreszenz (IIF) auf Organschnitten (Sitaru et al., 2004). Dabei hat sich
beim BP als geeignetes Substrat eine mit 1 M NaCl-Lösung gespaltene humane
Hautprobe (Salt-Split-Skin) bewährt (Schmidt and Zillikens, 2011). Es binden
zirkulierendes IgG, aber auch IgM, IgE und IgA an die epidermalen Bereiche, also
an das Blasendach des vorher erzeugten Spaltes (Schmidt et al., 2011) (siehe
Abb.5). Die Sensitivität dieser Methode wurde auf etwa 80 % beziffert, die
Spezifität auf 60 % (Roussel et al., 2011).

1. Einleitung 14
Abbildung 5: Indirekte Immunfluoreszenz beim bullösem Pemphigoid. Die für das bullöse
Pemphigoid verantwortlichen Antikörper gegen BP180 und BP230 binden an den epidermalen
Bereichen des mit 1 M NaCl künstlich erzeugten Spalts. Quelle: Klinik für Dermatologie, Universität
zu Lübeck
1.2.6.4 Immunoblot
Für den Immunoblot werden humane Epidermis-, Dermis- oder
Keratinozytenextrakte oder rekombinante Formen von BP180 und BP230
verwendet, die auf eine Trägermembran übertragen und mit Patientenseren
inkubiert werden (Chan et al., 2003; Schmidt et al., 2011). Die Autoantikörper
binden an die jeweiligen fixierten Antigene und werden angefärbt (Chan et al.,
2003). So kann eine Reaktion vor allem bei den 240 kD- und 180 kD-Banden
gezeigt werden (Labib et al., 1986).
1.2.6.5 ELISA
Eine genauere Diagnose kann sich mit dem Enzyme-linked immunosorbent Assay
(ELISA) stellen lassen, für welchen rekombinante Proteine für bestimmte
Abschnitte von BP180 oder BP230 verwendet werden (Schmidt et al., 2011). Beim
BP reagieren die Autoantikörper vor allem mit der immundominanten NC16A-
Domäne des BP180, im ELISA lag bei etwa 90 % der eingesetzten BP-
Patientenseren eine positive Reaktion vor (Sitaru et al., 2007; Zillikens et al.,
1997a). Die Sensitivität für den Nachweis von Antikörpern gegen BP180 wird mit
etwa 85 % beschrieben, die Spezifität mit circa 95 % (Roussel et al., 2011;
Tampoia et al., 2009). Autoantikörper gegen BP230 sind im ELISA mit einer
Sensitivität von 60 - 70 % nachweisbar (Kromminga et al., 2004; Yoshida et al.,
2006). Die Kombination eines BP180- und BP230-ELISA bringt eine nur

1. Einleitung 15
unwesentliche Verbesserung der Sensitivität, der BP230-ELISA ist deshalb vor
allem bei Schleimhautbeteiligung oder einem fehlenden Nachweis von BP180-
Antikörpern im ELISA bei positivem Befund in der Immunfluoreszenz von
Bedeutung (Roussel et al., 2011; Schmidt et al., 2011). Es wurde mehrfach eine
Korrelation der Antikörpertiter von BP180-Antikörpern mit der Krankheitsaktivität
beschrieben (Dopp et al., 2000; Mutasim, 2000; Schmidt et al., 2000). Dies scheint
bei den Titern der BP230-Antikörper nicht der Fall zu sein (Yoshida et al., 2006).
Bei der Mehrheit der BP-Patienten konnte neben den IgG-Antikörpern auch noch
IgE und IgA nachgewiesen werden (Iwata et al., 2008; Kromminga et al., 2000;
Messingham et al., 2009).

1. Einleitung 16
1.3 Epidermolysis bullosa acquisita
1.3.1 Epidemiologie
Die Epidermolysis bullosa acquisita (EBA) ist eine erworbene, chronische
Erkrankung der Haut und Schleimhäute und zeichnet sich durch subepidermale
Blasenbildung aus (Hertl and Schuler, 2002a). Es handelt sich um eine seltene
Erkrankung, die Inzidenz in Westeuropa liegt zwischen 0,17 und 0,26
Neuerkrankungen pro eine Million Einwohner pro Jahr (Bernard et al., 1995;
Zillikens et al., 1995), wobei in einer aktuellen Studie in Unterfranken eine Inzidenz
von 0,5 Neuerkrankungen pro eine Million Einwohner angegeben wurde (Bertram
et al., 2009). Die EBA kommt weltweit und ohne ethnische oder
Geschlechtsprädisposition vor (Gammon and Briggaman, 1993; Nanda et al.,
2004; Wong and Chua, 2002) und kann in jedem Lebensalter auftreten, meist
jedoch zwischen dem 40. und 50. Lebensjahr (Gammon, 1988).
1.3.2 Klinik
Die EBA wurde erstmals vor über hundert Jahren als eine im Erwachsenenalter
auftretende, der Epidermolysis bullosa hereditaria dystrophica ähnlichen
Erkrankung von Elliot beschrieben (Elliott, 1895). Die mechanobullöse, nicht-
inflammatorische Form der EBA wurde erstmals 1971 von Roenigk definiert durch
eine erhöhte Hautfragilität und durch Trauma induzierte Blasenbildung und
Erosionen, die typischerweise an den Streckseiten der Extremitäten lokalisiert sind
und unter Narben- und Milienbildung abheilen (Roenigk et al., 1971). Schon durch
minimale Traumen kommt es zur Bildung praller Blasen mit akraler Betonung an
Händen und Füßen, sowie an Ellenbogen, Knien und im Sakralbereich .Häufig
kommt es auch zu einer Beteiligung der Mundschleimhaut. (Hertl and Schuler,
2002a) (siehe Abb.6 a, d). Schwere Formen ähneln der hereditären Epidermolysis
bullosa und können zu einer Fibrosierung an Händen und Fingern,
Ösophagusstenosen, Haar- und Nagelverlust und bei okulärer Beteiligung zur
Erblindung führen. Leichte Verlaufsformen ähneln der Porphyria cutanea tarda
(Hertl and Schuler, 2002a; Ishii et al., 2010). Neben dieser mechanobullösen Form
wurden später verschiedene inflammatorische Subtypen der EBA beschrieben, die

1. Einleitung 17
klinisch dem BP, der linearen IgA-Dermatose oder dem Schleimhautpemphigoid
ähneln können (Dahl, 1979; Gammon et al., 1982a; Gammon et al., 1984a;
Hashimoto et al., 1996; Kurzhals et al., 1991). Bei diesen inflammatorischen
Unterformen findet sich keine erhöhte Hautfragilität, kaum Narben- und
Milienbildung und ein abweichendes Verteilungsmuster der Blasen und Erosionen
zum Beispiel an Rumpf, Extremitäten und Intertrigines (Hertl and Schuler, 2002a)
(siehe Abb.6 b, c). Oftmals können EBA-Patienten, die zu Beginn der Erkrankung
einen inflammatorischen Phänotyp aufweisen, gleichzeitig oder im späteren
Verlauf zusätzlich mechanobullöse Merkmale entwickeln (Gammon et al., 1984a;
Stewart et al., 1991).
Abbildung 6: Klinisches Erscheinungsbild der Epidermolysis bullosa acquisita (EBA). Das
Erscheinungsbild der EBA kann erheblich variieren. Beim klassischen Typ wird unter anderem eine
akrale Betonung der Blasen und Erosionen beschrieben (a), wohingegen sich bei der
inflammatorischen Form der Erkrankung Läsionen besonders an Rumpf und Extremitäten finden
(b) und die mitunter dem klinischen Bild des bullösen Pemphigoids ähneln kann (c). Auch die
Mundschleimhaut kann betroffen sein (d). Quelle: Klinik für Dermatologie, Universität zu Lübeck
1.3.3 Assoziierte Erkrankungen
Die für die EBA verantwortlichen Autoantikörper gegen Typ-VII-Kollagen sind auch
für die bullöse Form des systemischen Lupus erythematodes (SLE) verantwortlich
(Chan et al., 1999; Gammon et al., 1988; Shirahama et al., 1998). Es konnte
außerdem gezeigt werden, dass das Auftreten der EBA und des bullösen SLE mit
der Expression eines bestimmten MHC-II-Proteins der Zellmembran, dem HLA-
DR2, assoziiert ist (Gammon et al., 1988). Des Weiteren wurde das Auftreten der
EBA mit anderen Systemerkrankungen, wie rheumatoider Arthritis, Psoriasis und
Kryoglobulinämie beschrieben (Endo et al., 1997; Hertl and Schuler, 2002a; Krivo
and Miller, 1978).
ba dc

1. Einleitung 18
Auch eine Assoziation mit chronisch entzündlichen Darmerkrankungen wurde
beobachtet (Ray et al., 1982), wobei insbesondere der Morbus Crohn von
Bedeutung zu sein scheint (Chen et al., 2002). Im Zusammenhang damit konnte
gezeigt werden, dass Typ-VII-Kollagen im Magen-Darm-Trakt exprimiert wird und
Autoantikörper an die gastrointestinale Basalmembran binden, was zur Aktivierung
von Komplement und der Rekrutierung von Leukozyten führte (Ishii et al., 2011).
1.3.4 Immunpathologie
1.3.4.1 Typ-VII-Kollagen
a. Struktur
Typ-VII-Kollagen, Autoantigen der EBA, ist der Hauptbestandteil der Ankerfibrillen,
welche die Epidermis und die Basalmembranzone mit der papillären Dermis
verbinden (Briggaman and Wheeler, 1975; Sakai et al., 1986; Woodley et al.,
1984). Typ-VII-Kollagen setzt sich aus drei identischen α-Ketten zusammen. Jede
dieser Ketten beinhaltet eine 145 kDa schwere zentrale kollagenen Tripelhelix,
welche von einer 145 kDa aminoterminalen nichtkollagenen NC1-Domäne und
einer carboxyterminalen 34 kDa nichtkollagenen NC2-Domäne flankiert wird
(Parente et al., 1991). Typ-VII-Kollagen bildet extrazellulär antiparallele End-zu-
End Dimere, die über Disulfidbrücken überlappender Moleküle carboxyterminal
miteinander verbunden sind, wobei ein Fragment der NC2-Domäne proteolytisch
abgespalten wird (Bruckner-Tuderman et al., 1995; Chen et al., 2001a). Die
Dimere lagern sich dann seitlich aneinander und bilden so die Ankerfibrillen, die
an beiden Enden die NC1-Domäne aufweisen (Burgeson, 1993). Die NC1-
Domänen befinden sich in der Lamina densa und führen so zu einer
halbkreisförmigen Anordnung der Fibrillen (Shimizu et al., 1997). Des Weiteren
interagiert die NC1-Domäne mit verschiedenen Strukturproteinen, wie Fibronektin,
Typ I Kollagen und der β3-Kette von Laminin 5 (Chen et al., 1999; Chen et al.,
1997b). Fibronektin interagiert auch mit der kollagenen Domäne von Typ-VII-
Kollagen (Lapiere et al., 1994). Die NC1-Domäne wird als immundominanter
Abschnitt des Typ-VII-Kollagens beschrieben (Gammon et al., 1993; Jones et al.,
1995; Lapiere et al., 1993; Tanaka et al., 1994; Woodley et al., 1988). Seltener
werden auch die NC2-Domäne oder die kollagene Domäne erkannt (Fukumoto et

1. Einleitung 19
al., 2004; Ishii et al., 2004; Mayuzumi et al., 2006; Schmidt et al., 2002; Tanaka et
al., 1997).
b. Pathogenität
Für die Blasenbildung werden verschiedene Mechanismen diskutiert. Es wird
angenommen, dass EBA-Autoantikörper die Interaktion von Typ-VII-Kollagen mit
den assoziierten Strukturproteinen wie Laminin 5 oder Fibronektin beeinträchtigen
(Chen et al., 1999; Gammon et al., 1993). Außerdem können Autoantikörper auch
direkt mit der Bildung der antiparallelen Dimere interferieren (Chen et al., 2001a).
Über diese Mechanismen lassen sich die Fragilität der Haut und eine nicht-
inflammatorische Reaktion bei der klassischen Form der EBA erklären (Remington
et al., 2008). Weiterhin ist eine durch Immunkomplexe vermittelte Aktivierung von
Komplement und Leukozyten beschrieben, was zu einer inflammatorischen
Reaktion führt und klinisch ein dem BP ähnliches Bild zeigt (Gammon et al.,
1984a).
1.3.4.2 Antikörper
Die für die EBA verantwortlichen Antikörper gegen Typ-VII-Kollagen gehören vor
allem der IgG-Klasse an, seltener finden sich Antikörper vom IgA-Typ (Sitaru et
al., 2004). Es konnten alle vier IgG-Subklassen in den Seren von EBA-Patienten
nachgewiesen werden, vor allem aber IgG1 und IgG4 (Bernard et al., 1991; Cho et
al., 1998). Bezüglich der Fähigkeit zur Bindung von Komplement weisen die
zirkulierenden und gewebsständigen Antikörper Unterschiede auf, so hat IgG4
eine niedrige Affinität zu Komplement, IgG1 und IgG3 eine hohe (Cho et al.,
1998). Es wurde gezeigt, dass die Verteilung der Antikörper-Subklassen und ihre
komplementbindenden Eigenschaften mit dem klinischen Phänotyp nicht
korrelieren (Cho et al., 1998; Gandhi et al., 2000; Mooney and Gammon, 1990).
Im Mausmodell konnte jedoch eine Korrelation zwischen den IgG-Titern und der
Krankheitsschwere beobachtet werden (Sitaru et al., 2005).
1.3.4.3 Komplement
Die Bedeutung von Komplement für die Pathogenese der EBA konnte im
Mausmodell gezeigt werden, indem bei immunisierten C5-defizienten Mäusen
durch Antikörper-Transfer keine Blasenbildung auslösbar war (Sitaru et al., 2005).

1. Einleitung 20
Es konnte außerdem gezeigt werden, dass EBA-Antikörper effektiver Komplement
aktivieren als BP-Antikörper (Gammon et al., 1984b; Mooney et al., 1992).
1.3.4.4 Neutrophile Granulozyten
Die Aktivierung von Komplement führt zur Chemotaxis von neutrophilen
Granulozyten, welche ROS und Proteasen wie Elastase und Gelatinase B
freisetzen und somit eine Blasenbildung induzieren (Chiriac et al., 2007;
Shimanovich et al., 2004). Im Gefrierschnittmodell konnte unter Einsatz von EBA-
Patientenserum die Bedeutung von neutrophilen Granulozyten für die DES
nachgewiesen werden (Sitaru et al., 2002a).
1.3.5 Diagnostik
1.3.5.1 Histopathologie
Histologisch zeigt sich zunächst ein papilläres Ödem, später kommt es zur
Ausbildung subepidermaler Blasen. Bei der klassischen, mechanobullösen
Variante findet sich nur wenig Entzündungsinfiltrat, wohingegen bei der
entzündlichen Variante dermale Infiltrate aus neutrophilen und eosinophilen
Granulozyten, Monozyten und Lymphozyten nachweisbar sind (siehe Abb.7).
Aufgrund der unterschiedlichen klinischen und histologischen Präsentationsformen
ist es nicht möglich, anhand dieser allein die Diagnose einer EBA zu stellen
(Hallel-Halevy et al., 2001; Sitaru et al., 2004).
Abbildung 7: Histopathologischer Befund bei der Epidermolysis bullosa acquisita (EBA).
Typisch für die EBA ist eine subepidermale Blasenbildung. Vorwiegend bei der entzündlichen Form
der Erkrankung sind dermale Leukozyteninfiltrate nachweisbar. Quelle: Klinik für Dermatologie,
Universität zu Lübeck

1. Einleitung 21
1.3.5.2 Direkte Immunfluoreszenz
Die DIF periläsionaler Hautbiopsate von an EBA erkrankten Patienten kann lineare
Ablagerungen vor allem von IgG, sowie von C3, IgA, IgM, Properdin (Faktor P)
und C4 entlang der DEJ zeigen (Nieboer et al., 1980; Smoller and Woodley, 1992;
Vodegel et al., 2002). Ein ähnliches Bild liegt bei Hautbiopsaten von Patienten mit
BP vor (Yaoita et al., 1981). Es konnte gezeigt werden, dass die Ablagerungen
von Antikörpern und Komplement nicht streng linear sind, sondern bei den
verschiedenen blasenbildenden Erkrankungen unterschiedliche Muster zeigen. So
konnte bei der EBA eine Ablagerung entlang der Basalmembranzone in Form von
u-Zacken beobachtet werden (Vodegel et al., 2004).
1.3.5.3 Indirekte Immunfluoreszenz
Mithilfe der IIF lassen sich zirkulierende Antikörper in EBA-Seren nachweisen.
Dafür werden Organschnitte verwendet, meist humane Hautproben, die vorher mit
1 M NaCl behandelt wurden (Ishii et al., 2010). Die zirkulierenden Antikörper
binden wie bei der DIF an den dermalen Bereichen des artifiziellen Spaltes (siehe
Abb.8). Allerdings ist eine dermale Bindung von Antikörpern nicht
pathognomonisch für EBA, sondern lässt sich auch bei dem bullösen SLE, dem
vernarbenden Pemphigoid oder dem anti-p200 Pemphigoid zeigen (Ghohestani et
al., 1997; Hallel-Halevy et al., 2001). Des Weiteren lassen sich nur bei etwa der
Hälfte der EBA-Patienten zirkulierende Antikörper nachweisen (Sitaru et al., 2004).
Abbildung 8: Indirekte Immunfluoreszenz bei der Epidermolysis bullosa acquisita (EBA). Die
für die EBA verantwortlichen Antikörper gegen Typ-VII-Kollagen binden an den dermalen Bereich
des durch 1 M NaCl künstlich erzeugten Spalts. Quelle: Klinik für Dermatologie, Universität zu
Lübeck

1. Einleitung 22
1.3.5.4 Immunelektronenmikroskopie
Elektronenmikroskopisch konnte gezeigt werden, dass die Blasenbildung im
Bereich unterhalb der Lamina densa auftritt (Nieboer et al., 1980; Yaoita et al.,
1981). Bei manchen Patienten lässt sich jedoch auch eine Spaltbildung in der
Lamina lucida zeigen (Fine et al., 1989). Mithilfe der direkten
Immunelektronenmikroskopie lassen sich in Biopsaten gebundene EBA-Antikörper
und Komplement in der Lamina densa und Sublamina densa nachweisen (Nieboer
et al., 1980). In der indirekten Immuneletronenmikroskopie konnte gezeigt werden,
dass zirkulierende Antikörper vor allem in der Lamina densa binden, in der die
NC1-Domäne lokalisiert ist. In manchen Fällen ist allerdings auch eine Bindung in
der Dermis unterhalb der Lamina densa möglich, bei einer Reaktion mit der NC2-
Domäne oder der tripelhelikalen kollagenen Domäne (Ishii et al., 2004; Ishii et al.,
2009; McMillan et al., 2003).
1.3.5.5 Immunoblot
Im Immunoblot mit Extrakten normaler humaner Dermis erkennen die Seren vom
EBA-Patienten das 290 kDa schwere Typ-VII-Kollagen oder seine
immundominante 145 kDa schwere NC1-Domäne (Woodley et al., 1984). Auch
rekombinantes NC1 oder NC2 können im Immunoblot erkannt werden (Ishii et al.,
2004)..
1.3.5.6 ELISA
Mithilfe des ELISA können Antikörper gegen Typ-VII-Kollagen über rekombinantes
NC1 und NC2 mit einer hohen Sensitivität bestimmt werden. Dabei wurde gezeigt,
dass bei der Kombination von rekombinantem NC1 und NC2 mit einer Sensitivität
von etwa 94 % und eine Spezifität von 98 % Antikörper nachgewiesen werden
(Chen et al., 1997a; Saleh et al., 2011).

1. Einleitung 23
1.4 Therapie
Für die Therapie des BP und der EBA gibt es aufgrund der geringen Zahl
prospektiver Studien noch keine festen Leitlinien. Meist setzt sich die Therapie
beider Erkrankungen jedoch aus einem Steroid mit einem Adjuvanz zusammen.
Die Kombination kann dabei beispielsweise mit Dapson oder mit
Immunsuppressiva wie Azathioprin, Methotrexat, Cyclophosphamid, Cyclosporin
oder Mycophenolat-Mofetil erfolgen (Hertl and Schuler, 2002b; Schmidt, 2009). In
schweren und therapierefraktären Erkrankungsfällen steht außerdem der Einsatz
von intravenösen Immunglobulinen (IVIG), Rituximab oder einer Immunadsorption
zur Verfügung (Schmidt and Zillikens, 2011).
1.4.1 Therapie des BP
Derzeit gibt es noch keine feste Standardtherapie des BP (Wojnarowska et al.,
2002). Eine Umfrage in 42 deutschen Hautkliniken ergab jedoch, dass in 27 % der
Kliniken eine topische Behandlung mit Clobetasol-Propionat als Therapie der
ersten Wahl gilt und in mehr als 50 % systemische Steroide in einer Dosierung
von ≤ 1 mg/kg Prednisolonäquivalent angewendet wurden. In knapp 70 % wurde
Azathioprin adjuvant als erste Wahl verwendet (Hofmann et al., 2009).
1.4.1.1 Steroide
In den meisten Fällen erfolgt die Therapie des BP zunächst mit topischen oder
systemischen Steroiden. Diese wirken immunsuppressiv über genomische und
nicht-genomische Wege, wodurch es zur Bildung antiinflammatorischer Proteine
oder zur Hemmung proinflammatorischer Zytokine kommt (Buttgereit et al., 2005;
Buttgereit et al., 1998). Vor allem eine längerfristige Gabe von Steroiden ist jedoch
mit einer erhöhten Morbidität und Mortalität verbunden, was sich besonders bei
den meist älteren, polymorbiden Patienten problematisch gestalten kann (Claudy,
2001; Rzany et al., 2002).
Bei lokalisierten Formen des BP kann zunächst eine topische Therapie
beispielsweise mit 0,05 % Clobetasol-Propionat (10 - 30 g/d) begonnen werden,
die häufig bereits zu einer Besserung führt und aufgrund der geringeren
Nebenwirkungen der systemischen Therapie vorgezogen wird (Schmidt and
Zillikens, 2011). In einer Studie konnte außerdem gezeigt werden, dass topische

1. Einleitung 24
Steroide, wie Clobetasol-Propionat, die gleiche Wirksamkeit zeigen wie
systemische Steroide (Prednisolon) und in schweren Fällen einer systemischen
Therapie sogar überlegen sind. Dabei lag die Einjahresüberlebensrate bei einer
topischen Therapie um 20 % höher und die Nebenwirkungsrate um 25 % niedriger
als bei systemischer Therapie mit Steroiden (Joly et al., 2002). In einer weiteren
Studie wurde das Standardregime einer lokalen Therapie (Clobetasol-Propionat
40 g pro Tag für 12 Monate) mit einem milderen Regime (10 - 30 g pro Tag)
verglichen, wobei in leichteren Erkrankungsfällen das Risiko für schwerwiegende
Nebenwirkungen zweifach niedriger lag als bei Einsatz des milden Regimes. Mit
einer Einsparung von 70 % der kumulativen Steroiddosis konnte so die Prognose
der Patienten verbessert werden (Joly et al., 2009). Systemisch werden Steroide
in einer Dosierung von 0,5 - 1 mg Prednisolon pro kg Körpergewicht pro Tag
eingesetzt, wobei diese Initialdosis logarhythmisch über Wochen bis Monate
hinweg reduziert wird. Als Nebenwirkungen beim Einsatz von Steroiden spielen
besonders eine erhöhte Infektneigung, eine diabetische Stoffwechsellage,
Osteoporose, gastrointestinale Ulzera oder Thrombosen eine Rolle (Goebeler et
al., 2004; Rosenberg et al., 1976).
Häufig erfolgt deshalb eine Kombination mit Dapson, Tetrazyklinen oder mit
Immunsuppressiva wie Azathioprin, Methotrexat, Chlorambucil oder
Mycophenolat-Mofetil (Hertl and Schuler, 2002b; Schmidt, 2009). Die Verwendung
von Adjuvanzien ermöglicht eine Einsparung von Steroiden, ein adjuvanter Einsatz
von Azathioprin konnte beispielsweise die erforderliche Dosis an Prednisolon fast
halbieren (Khumalo et al., 2005). Allerdings führen auch die oben aufgeführten
Immunsuppressiva zu verschiedenen, mitunter schweren Nebenwirkungen. Im
Folgenden sind die einzelnen Therapeutika, die aktuell Einsatz finden, dargestellt.
1.4.1.2 Azathioprin
Azathioprin gehört zur Gruppe der Antimetabolite und ist ein Purinanalogon, das
mit der DNA-Synthese und der Aktivierung von T-Zellen interferiert (Patel et al.,
2006; Tirado-Sanchez et al., 2011). Es ist das am häufigsten beim BP eingesetzte
Adjuvanz und wird in einer Dosis von 1 bis 3 mg/kg verwendet, wobei Steroide
eingespart werden können (Beissert et al., 2007; Kazlow Stern et al., 2005; Patel
et al., 2006). In einer Studie mit 38 Patienten konnte bei 92 % eine komplette
Remission erreicht werden (Beissert et al., 2007). Vor Einleitung einer Therapie

1. Einleitung 25
sollte jedoch die Aktivität der Thiopurinmethyltransferase (TPMT) bestimmt
werden. Die TPMT ist eines von drei Enzymen, welches am Metabolismus von
Azathioprin beteiligt ist (Snow and Gibson, 1995). Bei Patienten mit geringer
Aktivität des Enzyms kann es bei Einsatz von Azathioprin zu einer ausgeprägten
Myelosuppression mit Panzytopenie kommen (Jackson et al., 1997).
1.4.1.3 Methotrexat
Methotrexat gehört ebenfalls zu den Antimetaboliten. Es handelt sich dabei um ein
Folsäureanalogon, das kompetitiv verschiedene Enzyme, wie die
Dihydrofolsäurereduktase (DHFR), welche zur DNA-Synthese benötigt wird,
hemmt. Auch die Methionin-Synthetase, die zur Bildung des proinflammatorischen
S-adenyl-Methionins nötig ist, wird durch Methotrexat inhibiert. Der Wirkstoff hat
somit sowohl antiproliferative als auch antiinflammatorische Effekte (Olsen, 1991).
In einer retrospektiven Studie mit 62 BP-Patienten, welche mit Methotrexat
therapiert wurden, konnte eine Verbesserung der klinischen Beschwerden bei
95 % beobachtet werden. Dabei wurden wöchentlich 5 - 25 mg Methotrexat
entweder als Monotherapie oder zusammen mit Steroiden eingesetzt (Gurcan and
Ahmed, 2009a). In einer weiteren Studie konnte eine Monotherapie mit 5 mg
Methotrexat wöchentlich eine Remission bei 43 % der Patienten mit BP bewirken.
In der Vergleichsgruppe wurde Methotrexat mit Prednisolon kombiniert, hier
konnte eine Remission bei 35 % beobachtet werden (Kjellman et al., 2008).
Nebenwirkungen wie Anämie, Neutropenie oder gastrointestinale Beschwerden
wurden beobachtet, wobei bei letzterem die zusätzliche Gabe von 5 mg Folsäure
pro Woche als protektiv beschrieben wird (Ortiz et al., 2000). Auch regelmäßige
Blutbildkontrollen und Leberfunktionstests sollten während einer Therapie
durchgeführt werden (Gurcan and Ahmed, 2009a).
1.4.1.4 Mycophenolat-Mofetil
Eine weitere Möglichkeit besteht in der Therapie mit Mycophenolat-Mofetil. Es
handelt sich dabei um einen nichtkompetitiven Inhibitor der Iosin-Monophosphat-
Dehydrogenase, welche zur de-novo Synthese von Guanin-Nukleotiden bei der
DNA- und RNA-Synthese während der T- und B-Zell Proliferation nötig ist
(Grundmann-Kollmann et al., 1999). Mycophenolat-Mofetil wurde beim BP
eingesetzt in Kombination mit Steroiden oder als Monotherapie (Bohm et al., 1997;
Grundmann-Kollmann et al., 1999). In einer Studie mit 35 BP-Patienten, die mit

1. Einleitung 26
Mycophenolat-Mofetil in Kombination mit Methylprednisolon behandelt wurden,
konnte bei allen eine Remission erreicht werden. Dabei wurden 0,5 mg/kg
Prednisolon mit 2 g Mycophenolat-Mofetil pro Tag verabreicht (Beissert et al.,
2007). Als Nebenwirkungen werden gastrointestinale Beschwerden, Infektionen
und dosisabhängige hämatologische Effekte beschrieben (Beissert et al., 2007;
Grundmann-Kollmann et al., 1999).
1.4.1.5 Chlorambucil
Auch die Therapie mit Chlorambucil wurde mehrfach beschrieben. Es handelt sich
dabei um ein Alkylanz, das die DNA-Synthese der B- und T-Zellen stört (Shah et
al., 2000). Beim BP wurde Chlorambucil erfolgreich in einer Dosis von 0,1 mg/kg
pro Tag in Kombination mit 20 - 60 mg Prednisolon eingesetzt. Darunter kam es
bei allen Patienten zu einer Remission und zu einer Steroideinsparung von 34 %
(Chave et al., 2004). Als Nebenwirkung konnte eine vorübergehende
Thrombozytopenie beobachet werden (Milligan and Hutchinson, 1990). Weitere
Nebenwirkungen sind unspezifisch, abhängig von der Dauer der Therapie und der
Dosierung wurde jedoch das Auftreten von malignen Tumoren, wie der akuten
myeloischen Leukämie beschrieben (Palmer and Denman, 1984).
1.4.1.6 Cyclophosphamid
Cyclophosphamid ist ein weiteres Alkylanz, das vor allem bei schweren
generalisierten und therapieresistenten Fällen des BP verwendet wird (Dawe et
al., 1997; Itoh et al., 1996; Ogawa et al., 2004). Dabei wird Cyclophosphamid
entweder als Dauertherapie mit 100 - 200 mg pro Tag oder als Stoßtherapie mit
500 mg intravenös eingesetzt (Hertl and Schuler, 2002b). Auch eine Kombination
mit Steroiden wird als effektiv beschrieben (Appelhans et al., 1993; Dawe et al.,
1997). Als Nebenwirkungen können gastrointestinale Beschwerden,
Myelosuppression mit Panzytopenie, Infektionen oder eine Hepatopathie
auftreten. Außerdem wurde ein erhöhte Inzidenz von Tumorerkrankungen
beschrieben (Appelhans et al., 1993; Hertl and Schuler, 2002b).
1.4.1.7 Cyclosporin A
Cyclosporin A ist ein Calcineurin-Inhibitor, der die Calcineurin-Phosophatase
hemmt und somit die Bildung von IL-2 in den T-Zellen stört. Durch fehlendes IL-2
kommt es zu einer Hinderung der Aktivierung von T-Zellen, außerdem wird die

1. Einleitung 27
Bildung von INF-γ und GM-CSF beeinflusst (Amor et al., 2010). Der Einsatz von
Cyclosporin A sollte beim BP niedrigdosiert mit 3 - 5 mg/kg pro Tag in
Kombination mit einem Steroid erfolgen (Capella et al., 2001). Als
Nebenwirkungen bei Therapie mit Cyclosporin A werden gastrointestinale
Beschwerden, Nephrotoxizität und Hypertension beschrieben (Capella et al.,
2001; Mrowietz et al., 2009).
1.4.1.8 Dapson
Dapson kann adjuvant zu einem topischen oder systemischen Steroid eingesetzt
werden (Khumalo et al., 2005). Der genaue Wirkmechanismus von Dapson ist
noch nicht vollständig geklärt, die antiinflammatorischen Effekte von Dapson
werden jedoch vor allem auf die Hemmung der Chemotaxis und der Funktion von
neutrophilen Granulozyten zurückgeführt (Harvath et al., 1986; Schmidt and
Zillikens, 2011) zum Beispiel durch Hemmung der Freisetzung des für die
Chemotaxis wichtigen IL-8 (Schmidt et al., 2001) oder durch Beeinflussung des für
die Adhärenz von neutrophilen Granulozyten bedeutenden β2-Integrins (Booth et
al., 1992). Die Ansprechrate beim adjuvanten Einsatz von Dapson zur Behandlung
des BP wurde in einer retrospektiven Studie mit 81 % angegeben (Gurcan and
Ahmed, 2009b). Außerdem wurde bei adjuvantem Einsatz von Dapson eine
schnellere Reduktion der eingesetzten Steroiddosen beschrieben (Jeffes and
Ahmed, 1989; Schmidt et al., 2005). Dabei werden 1 - 2 mg/kg Dapson pro Tag
eingesetzt (Hertl and Schuler, 2002b). Es wurde eine genauso gute Wirksamkeit
wie bei Einsatz von Azathioprin als Adjuvanz beschrieben (Tirado-Sanchez et al.,
2011). Generell sind die Nebenwirkungen bei der Gabe von Dapson bis zu einer
Plasmakonzentration von 5 mg/l relativ mild (Zuidema et al., 1986). Wichtig ist
jedoch, vor Therapiebeginn die Aktivität der Glucose-6-Phosphat-Dehydrogenase
zu bestimmen, um das Risiko für das Auftreten einer hämolytischen Anämie zu
verringern (Coleman, 1993; Zhu and Stiller, 2001). Als weitere Nebenwirkungen
einer Therapie mit Dapson können eine Methämoglobinämie oder eine
Agranulozytose auftreten (Goebeler et al., 2004). Um den hämatologischen
Effekten entgegenzuwirken, wurde der Einsatz von Antioxidanzien wie Vitamin E
oder von Cimetidin, welches die Metabolisierung von Dapson zu Hydroxylamin in
der Leber hemmt, empfohlen (Coleman et al., 1992; Prussick et al., 1992).

1. Einleitung 28
1.4.1.9 Tetrazykline
Tetrazykline, wie Minozyklin oder Doxycyclin, werden allein oder in Kombination
mit Niktotinamid oder Steroiden eingesetzt (Miida et al., 2011; Thomas et al.,
1993). Es konnte gezeigt werden, dass Tetrazykline ähnlich effektiv wirken wie
Steroide (Fivenson et al., 1994). Der Wirkmechanismus von Tetrazyklinen beim
BP richtet sich gegen die Rekrutierung von Neutrophilen und Eosinophilen und die
Hemmung von proteolytischen Enzymen und der Antikörperbildung durch
Blockade der Protein- und DNA-Synthese (Berk and Lorincz, 1986; Schneir et al.,
1990; Thong and Ferrante, 1979). Tetrazykline wirken somit sowohl
antiinflammatorisch als auch immunsuppressiv (Loo et al., 2001). Als
unerwünschte Wirkungen sind Hyperpigmentierung, gastrointestinale Symptome
und Lichtempfindlichkeit beschrieben (Fivenson et al., 1994; Loo et al., 2001).
Meist werden 2 - 4 mal 500 mg am Tag eingesetzt, allein oder in Kombination mit
0,5 - 2 g/d Nikotinamid (Hertl and Schuler, 2002b). Der zusätzliche positive Effekt
von Nikotinamid beruht wahrscheinlich auf der Hemmung der Freisetzung von
Proteasen aus den Granulozyten und auf der Hemmung der Degranulation von
Mastzellen (Hornschuh et al., 1997).
1.4.1.10 Intravenöse Immunglobuline
Bei schweren Fällen des BP steht als weitere Behandlungsmöglichkeit der Einsatz
von intravenösen Immunglobulinen (IVIG) zur Verfügung. IVIG werden aus
gepooltem Plasma von gesunden Spendern gewonnen und enthalten intaktes IgG,
wobei die einzelnen IgG-Subklassen entsprechend normalem Serum verteilt sind.
Der Wirkmechanismus ist jedoch noch nicht ausreichend geklärt. Generell dient
IgG als Mediator für antiinflammatorische aber auch proinflammatorische
Reaktionen durch Bindung des Fc-Fragments an den jeweiligen Fcγ-Rezeptor.
Therapeutisch verabreichtes IgG und dessen Fc-Fragmente haben einen
antiinflammatorischen Effekt, was durch eine N-terminal lokalisierte Seitenkette
des Fc-Fragments zustande kommt (Anthony et al., 2008; Kaneko et al., 2006). Im
Mausmodell konnte außerdem gezeigt werden, dass IVIG besonders an neonatale
Fc-Rezeptoren (FcRn) bindet, zirkulierendes pathogenes IgG verdrängt und
Blasenbildung durch Injektion reduziert wird. Es konnte auch gezeigt werden, dass
FcRn-defiziente Mäuse unempfindlich gegen Autoantikörper blasenbildender
Erkrankungen sind und keine Blasen entwickeln (Li et al., 2005; Sesarman et al.,

1. Einleitung 29
2008). Im therapeutischen Einsatz eignen sich IVIG nur in Kombination mit einem
Immunsuppressivum, mithilfe dessen durch sinkende Antikörperlevel ein Rebound
verhindert werden soll (Czernik and Bystryn, 2008). Durch den steroidsparenden
Effekt können Nebenwirkungen reduziert werden (Daoud and Amin, 2006). Auch
die Nebenwirkungen von IVIG selbst sind mild (Katz et al., 2007).
1.4.1.11 Immunadsorption
Außerdem stellt die Immunadsorption eine effektive Methode zum Senken der
Antikörperspiegel und somit zum schnellen Erreichen einer klinischen Remission
dar (Schmidt and Zillikens, 2010). Mithilfe der Immunadsorption können durch
verschiedener Absorbersysteme selektiv Immunglobuline aus dem Blut entfernt
werden (Schmidt, 2009). Die pathogenetische Rolle von Antikörpern gegen
NC16A für das BP konnte klinisch und experimentell nachgewiesen werden, zum
Beispiel im Mausmodell oder im Gefrierschnittmodell (Liu et al., 1993; Sitaru et al.,
2002b). Auch konnte gezeigt werden, dass die Serumkonzentrationen von
Antikörpern mit der Erkrankungsaktivität korrelieren (Schmidt et al., 2000). In
schweren, therapieresistenten Fällen des BP wurde die Immunadsorption bereits
mit Erfolg eingesetzt (Herrero-Gonzalez et al., 2005; Ino et al., 1997) .
1.4.1.12 Rituximab
Rituximab ist ein monoklonaler chimerer Antikörper, der an das CD20-Antigen von
B-Lymphozyten (prä-B Zellen und prä-Plasmazellen) bindet, wodurch eine Lyse
der B-Lymphozyten über direkte Apoptose oder über komplementinduzierte oder
antikörperinduzierte Zytotoxizität ausgelöst wird (Arin and Hunzelmann, 2005;
Hertl et al., 2008). Eine Therapie mit Rituximab führt zu einer Senkung pathogener
Autoantikörper produzierender B-Zellen (Hertl et al., 2008). Rituximab wird
erfolgreich in der Behandlung therapieresistenter bullöser Autoimmundermatosen
eingesetzt (Hertl et al., 2008; Nagel et al., 2009). Als Nebenwirkungen können
Kopfschmerzen, Fieber, Urtikaria, Pruritus oder Hypotension auftreten, welche
meist aber mild ausfallen (Schmidt et al., 2006b). Rituximab wurde beim BP mit
Steroiden und Immunsuppressiva kombiniert (Schmidt et al., 2007). Die
Wirksamkeit der Therapie des BP mit Rituximab ist durch die geringe Anzahl
bisher behandelter Patienten noch nicht ausreichend beurteilbar (Hertl et al.,
2008).

1. Einleitung 30
1.4.2 Therapie der EBA
Die EBA ist eine chronische Erkrankung, deren Therapie sich oft schwierig
gestaltet. Aufgrund der geringen Zahl prospektiver Studien ist keine allgemeine
Standardtherapie definiert (Kirtschig et al., 2002). Die initiale Therapie der EBA
setzt sich meist aus hochdosierten systemischen Steroiden (1 - 2 mg/kg/Tag) in
Kombination mit anderen Immunsuppressiva wie Methotrexat, Azathioprin,
Cyclophosphamid oder Mycophenolat-Mofetil zusammen (Schmidt, 2009). Bei der
Mehrzahl an Patienten mit der klassischen, nicht-inflammatorischen Form der EBA
kann damit jedoch keine oder nur eine vorübergehende Remission errreicht
werden (Engineer and Ahmed, 2001). Die inflammatorische Form scheint besser
auf eine Therapie anzusprechen.
1.4.2.1 Dapson
Zusätzlich zu einer immunsuppressiven Therapie kann durch den Einsatz von
Dapson eine Besserung erreicht werden. Dabei wird eine Ausgangsdosis von
50 mg/Tag schrittweise erhöht, bis eine klinische Remission eintritt. Dies erfolgt
meist bei einer Dosis von 100 - 250 mg, welche dann über mehrere Monate
beibehalten wird (Mutasim, 2004). Dapson wirkt über eine Hemmung der
Chemotaxis und Funktion neutrophiler Granulozyten (Harvath et al., 1986).
1.4.2.2 Colchizin
Des Weiteren wurde bei therapierefraktärer EBA in einigen Fällen der erfolgreiche
adjuvante oder alleinige Einsatz von Colchizin bei der klassischen und
inflammatorischen Form der EBA beschrieben, wobei die Tagesdosis zwischen
1 - 2 mg lag (Berbis and Privat, 1989; Cunningham et al., 1996; Megahed and
Scharffetter-Kochanek, 1994). Colchizin hemmt die Polymerisation von Mirkotubuli
und interferiert so mit verschiedenen zellulären Funktionen, wie zum Beispiel der
Mitose und der Chemotaxis (Ehrenfeld et al., 1980). Weiterhin wurden Einflüsse
auf die Freisetzung lysosomaler Enzyme aus neutrophilen Granulozyten, die
Sekretion von Antikörpern aus den Plasmazellen und die Produktion von
Kollagenasen beschrieben (Antoine et al., 1980; Bauer and Valle, 1982; Rudolph
et al., 1977). Es wurden gastrointestinale Nebenwirkungen bei hohen Dosierungen
von Colchizin beobachtet, was den Einsatz bei assoziierter chronisch
entzündlicher Darmerkrankung limitiert (Engineer and Ahmed, 2001).

1. Einleitung 31
1.4.2.3 Cyclosporin A
Bei schweren Fällen wurde auch der Einsatz von Cyclosporin A beschrieben,
welches durch die Hemmung von T-Zellen in die Stimulation
antikörperproduzierender B-Zellen eingreift (Amor et al., 2010). In schweren,
therapieresistenten Fällen konnte eine Remission mit Cyclosporin, als
Monotherapie oder zusammen mit Steroiden, in einer Dosierung von 5 - 9 mg/kg
pro Tag erreicht werden. Therapielimitierend können mögliche Nebenwirkungen
wie Pankreatitis, Urtikaria, Nephrotoxizität oder Diarrhoe sein (Clement et al.,
1993; Connolly and Sander, 1987; Crow et al., 1988; Gupta et al., 1990; Layton
and Cunliffe, 1990; Merle et al., 1990; Zachariae, 1987).
1.4.2.4 Intravenöse Immunglobuline
Eine weitere vielversprechende Therapiemöglichkeit bei refraktärem Verlauf ist der
Einsatz von intravenösen Immunglobulinen (IVIG). Dabei wurde in den meisten
Fällen IVIG hochdosiert eingesetzt, mit 400 mg/kg pro Tag über 5 Tage und in
Intervallen von 4 bis 6 Wochen, entweder als Monotherapie oder in Kombination
mit einem Steroid oder einem weiteren Adjuvanz (Caldwell et al., 1994; Campos et
al., 2006; Gourgiotou et al., 2002; Harman and Black, 1999; Kofler et al., 1997;
Meier et al., 1993; Mohr et al., 1995; Segura et al., 2007).
1.4.2.5 Rituximab
Auch Rituximab, ein chimerer monoklonaler anti-CD20 Antikörper, stellt eine
wirksame Alternative bei schweren und therapierefraktären Verläufen dar (Sadler
et al., 2007; Schmidt et al., 2006a). Rituximab ist gegen das B-Zell-spezifische
CD-20 gerichtet und interagiert vor allem mit reifen B-Zellen, die dadurch nicht
mehr zu Plasmazellen umgewandelt werden und Antikörper produzieren können.
Die Zerstörung der B-Zellen erfolgt durch komplement- oder antikörpervermittelte
Zytotoxizität oder über direkte Apoptose (Hertl et al., 2008). Die geringen
Fallzahlen lassen bisher jedoch nur eine begrenzte Aussagekraft zur Wirksamkeit
bei der EBA zu.

1. Einleitung 32
1.5 Ziele dieser Arbeit
Die Therapiemöglichkeiten des BP und der EBA sind momentan größtenteils auf
eine generelle immunsuppressive Therapie begrenzt. Diese ist vor allem bei der
EBA oft nicht ausreichend, um die Symptome zu kontrollieren (Engineer and
Ahmed, 2001). Beim BP kommt es nach Absetzen der Therapie regelhaft zu
Rezidiven (Bernard et al., 2009). Eine weitere Problematik stellen die teils
schweren Nebenwirkungen der bekannten Therapien und die erhöhte Mortalität
bei Einsatz von hochdosierten Steroiden dar (Rzany et al., 2002).
Ziel dieser Arbeit war es deshalb, neue mögliche Therapieansätze zu
untersuchen. Dies erfolgte im Gefrierschnittmodell, mithilfe dessen die Wirkungen
zweier PI3-Kinase-Inhibitoren und eines löslichen Fcγ-Rezeptors betrachtet
wurden.
Mit dem Gefrierschnittmodell lassen sich die Vorgänge bei der Blasenbildung
simulieren, indem Hautschnitte mit Antikörperseren und Leukozyten inkubiert
werden, was zur Spaltbildung im Hautschnitt führt (Gammon et al., 1982b). Im
Gefrierschnittmodell konnte bereits gezeigt werden, dass für die Spaltbildung die
NADPH-Oxidase, welche für die Bildung von ROS zuständig ist, eine besondere
Rolle spielt (Chiriac et al., 2007). Die NADPH-Oxidase wird unter anderem durch
Phosphoinositide reguliert, die durch PI3-Kinasen gebildet werden
(Vanhaesebroeck et al., 2001).
In weiteren Untersuchungen konnte zudem gezeigt werden, dass es nur dann zur
Spaltbildung kommt, wenn neutrophile Granulozyten über einen Fcγ-Rezeptor an
die Immunkomplexe binden, was zur Aktivierung der Zellen führt (Sitaru et al.,
2002a). Auch wurde bereits ein löslicher Fcγ-Rezeptor beschrieben, der an
Immunkomplexe bindet und so die Reaktion mit zellgebundenen Fcγ-Rezeptoren
kompetitiv hemmen kann (Gavin et al., 1995).
Die in dieser Arbeit verwendeten PI3-Kinase-Inhibitoren und der lösliche Fcγ-
Rezeptor wurden deshalb hinsichtlich ihrer Wirkung auf die Spaltbildung beim BP
und der EBA im Gefrierschnittmodell untersucht.
Auch wenn Steroide Mittel der ersten Wahl in der Therapie des BP und der EBA
sind, ist ihr Wirkmechanismus noch nicht vollständig geklärt. Zusätzlich wurde
deshalb mithilfe des NBT-Gefrierschnittmodells und des ROS-Release die
Wirkung von Methylprednisolon weiter charakterisiert.

2. Material und Methoden 33
2. Material und Methoden
2.1 Das Gefrierschnittmodell
Mit dem Gefrierschnittmodell wurden die Effekte der PI-3-Kinase-Hemmer TGX-
221 und PI-103, sowie des Fcγ-Rezeptors IIb (SM101) auf die DES in vitro
untersucht. Im Versuch wurden dazu Gefrierschnitte von Maushaut oder humaner
Haut verwendet, die mit Serum immunisierter Kaninchen (EBA) oder mit Serum
von Patienten mit BP inkubierten, wobei die Seren in vorgeschalteten
Gefrierschnittmodellen auf ihre Fähigkeit zur Spaltbildung untersucht wurden.
Danach wurden die zu testenden Wirkstoffe zusammen mit vorher isolierten
humanen Leukozyten auf die Gefrierschnitte gegeben. Die Präparate wurden
gewaschen, H-E gefärbt und am Mikroskop ausgewertet (siehe Abb.9).
Entsprechende Stellungnahmen der Ethikkommission wurden eingeholt und sind
im Anhang aufgelistet.
Abbildung 9: Das Prinzip des Gefrierschnittmodells. Gefrierschnitte werden zunächst mit
Antikörper-haltigem Serum inkubiert (1). Danach werden zu den Gefrierschnitten isolierte humane
Leukozyten gegeben und erneut inkubiert (2). Die Beurteilung der Schnitte erfolgt am Mikroskop
(3), hier wird die dermo-epidermale Spaltbildung prozentual bestimmt. Die Pfeile kennzeichnen
Bereiche mit Spaltbildung.

2. Material und Methoden 34
2.1.1 Verwendete Seren
2.1.1.1 Positivseren EBA
Es wurden Positivseren von 5 verschiedenen Kaninchen verwendet. Diese wurden
vorher mit GST-mCol7c immunisiert, einem Abschnitt des murinen Typ-VII-
Kollagens (Col7), der sich innerhalb der NC1-Domäne befindet. Dabei lagen die in
der IIF ermittelten Titer zwischen 5.000 und 45.000, im Durchschnitt bei 15.000.
Die Seren wurden in gesonderten Versuchsdurchläufen in den Verdünnungen 1:2,
1:4, 1:8, 1:16 (verdünnt mit PBS) und pur getestet und am Mikroskop die
Spaltbildung als Anteil der beschädigten DEJ an der gesamten DEJ des
Hautschnittes prozentual bewertet. Mithilfe der Ergebnisse dieser Versuche
wurden dann die Seren und die Verdünnungen mit der höchsten prozentualen
Spaltbildung für die Testung der Versuchsdurchläufe mit Wirkstoff ausgewählt. Es
wurden Seren in den Verdünnungen 1:2, 1:4 und 1:8 und pur ohne Verdünnung
verwendet, meist wurde 1:2 verdünnt. Dabei wurde eine Spaltbildung von 10 bis
93 % erreicht.
2.1.1.2 Positivseren BP
Humane Seren wurden von 22 Spendern mit BP gewonnen. Die Titer im ELISA
mit rekombinantem BP180 NC16A lagen zwischen 465 und 12.990 U/ml,
durchschnittlich bei 3.589 U/ml. Die Seren wurden im Verhältnis 1:2, 1:4, 1:8 oder
1:16 mit PBS verdünnt, wobei die Verdünnung den im ELISA ermittelten Titern
und der Spaltbildung in der Positivkontrolle (ohne Zugabe von Wirkstoffen)
angepasst wurde. Des Weiteren wurden alle Seren in gesonderten
Versuchsdurchläufen in den Verhältnissen 1:2, 1:4, 1:8 und 1:16 im
Gefrierschnittmodell getestet und die DES prozentual bewertet. Aus den
Ergebnissen dieser Versuche wurden dann die Seren und die Verdünnung mit der
höchsten prozentualen Spaltbildung für die Testung der Wirkstoffe ausgewählt.
Dabei wurde in den Versuchsdurchläufen mit Wirkstoff eine Spaltbildung in der
Positivkontrolle von 10 bis 100 % erzielt.
2.1.1.3 Negativseren
Für die Versuche auf humaner Haut wurden humane Negativseren gesunder
Spender verwendet, welche keine Hinweise auf eine bullöse Dermatose zeigten.

2. Material und Methoden 35
Als Negativkontrolle bei den Versuchen mit Maushaut dienten Seren gesunder
Kaninchen.
2.1.2 Herstellung der Gefrierschnitte
2.1.2.1 Maushaut
Für die Herstellung der Gefrierschnitte wurde Maushaut gesunden C57Bl/6-
Mäusen entnommen. Die Herstellung der Hautschnitte erfolgte wie im Folgenden
für humane Haut beschrieben.
2.1.2.2 Humane Haut
Für die Herstellung der humanen Gefrierschnitte wurde neonatale Vorhaut
verwendet, welche in RPMI gelagert wurde und zur Vorbereitung zunächst in
kaltem PBS gewaschen und anschließend in jeweils 5 - 15 mm große Stücke
geteilt wurde. Diese Stücke wurden in Cryomolds (Tissue-Tek Cryomold, Sakura
Finetec Inc., USA) mit Tissue-Tek O.C.T. Compound-Einbettmedium (Sakura
Finetec Inc., USA) überschichtet und bei -80°C gehärtet und aufbewahrt. Die
Hautschnitte wurden dann mit einem Kryostaten (Leica CM 3050 S, Leica
Mikrosysteme Vertrieb GmbH, Wetzlar) bei -20°C in 6 µm dicke Schnitte geteilt
und auf chemisch gereinigte Objektträger (SuperFrost/Plus, Gerhard Menzel,
Glasbearbeitungswerk GmbH & Co. KG, Braunschweig) gezogen, wobei jeweils
zwei Hautschnitte in der Größe von etwa 10x10 mm platziert wurden. Die fertigen
Objektträger wurden bis zur Verwendung bei -20°C gelagert
.
2.1.3 Isolierung von Leukozyten, Zellvitalität und Zellzählung
Zur Gewinnung von Leukozyten wurde gesunden, freiwilligen Spendern 80 ml Blut
abgenommen, das mit Heparin antikoaguliert wurde. Die Blutentnahme erfolgte
nach mündlicher und schriftlicher Aufklärung und Unterzeichnen einer
Einverständniserklärung durch die Freiwilligen.
Die Spender befanden sich in einem Alter zwischen 19 und 43 Jahren, das
durchschnittliche Alter betrug 27 Jahre. Es nahmen 18 Frauen und 14 Männer teil.
Die Blutentnahme wurde mit einer Butterflykanüle (BD Valu Set, Becton Dickinson
GmbH, Heidelberg) und vier 20 ml Spritzen (BD Discardit II, Becton Dickinson

2. Material und Methoden 36
GmbH, Heidelberg) durchgeführt. Dabei wurde vor der Blutentnahme jeder Spritze
1 ml Heparin (Heparin-Natrium 25.000, ratiopharm GmbH, Ulm) zur
Antikoagulation zugefügt. Außerdem wurden vier 50 ml-Tubes (Sarstedt AG & Co.,
Nümbrecht) mit jeweils 20 ml einer 3 %-Dextranlösung (Dextran 500, Carl Roth
GmbH + Co. KG, Karlsruhe), die mit 0,9 % NaCl versetzt war (Natriumchlorid, J.T.
Baker, Deventer, Holland), vorbereitet. Das entnommene Blut wurde vorsichtig in
die Tubes überführt, wobei jeweils eine 20 ml Spritze mit Vollblut in ein
vorbereitetes Tube gegeben wurde. Anschließend wurde die Suspension für
20 - 30 min bei Raumtemperatur inkubiert, bis sich zwei Phasen bildeten. Dabei
enthielt die untere Phase die Mehrheit der Erythrozyten und die obere Phase die
benötigten Leukozyten. Die obere Phase wurde im nächsten Schritt vorsichtig mit
einer automatischen Pipette (Pipetus-Akku, Hirschmann Laborgeräte GmbH & Co.
KG, Eberstadt) mit einem 10 ml-Aufsatz (Sarstedt AG & Co., Nümbrecht) in drei
neue 50 ml-Tubes überführt. Diese wurden für 12 min bei 1.200 rpm und 19°C
zentrifugiert (Varifuge 3.0 R, Heraeus Instruments GmbH, Hanau). Der Überstand
wurde danach mit einer Wasserstrahlpumpe abgesaugt (Brand GmbH & Co. KG,
Wertheim) und verworfen. Den Pellets der drei Tubes wurden dann jeweils mit der
automatischen Pipette und einem 25 ml-Aufsatz 10 ml RPMI (Lonza RPMI 1640
with L-Glutamin, BioWhittaker, Verviers, Belgien) zugegeben, die Lösung
resuspendiert und in einem Tube vereinigt. Dieses Tube mit 30 ml Suspension
wurde nochmals für 12 min bei 1.200 rpm und 19°C zentrifugiert und im
Nachfolgenden wurde der Überstand wieder abgesaugt. Das Pellet wurde dann in
20 ml eiskalter 0,2 %-NaCl-Lösung resuspendiert und nach 20 Sekunden erneut
mit 20 ml eiskalter 1,6 %-NaCl-Lösung resuspendiert und danach erneut für
12 min bei 1.200 rpm und 4°C zentrifugiert. Der Überstand wurde mit einer
Wasserstrahlpumpe abgesaugt und verworfen. Das Pellet wurde dann in 30 ml
RPMI resuspendiert und 7 min bei 1.200 rpm und 4°C zentrifugiert. Der Überstand
wurde abgesaugt und die isolierten Leukozyten wurden mit 7 - 9 ml RPMI
resuspendiert und auf Eis gestellt. Je nach ermittelter Zellzahl wurden die
Leukozytensuspensionen weiter mit RPMI verdünnt.
Die Zellvitalität der gewonnenen Leukozyten wurde mittels Trypanblau (Trypan-
Blue-Solution 0,4 %, Sigma-Aldrich Chemie GmbH, Deisenhofen) getestet. Dafür
wurden in einem 1,5 ml Eppendorf-Reaktionsgefäß (Eppendorf AG, Hamburg)
10 µl der Zellsuspension mit 440 µl PBS und 50 µl Trypanblau gemischt. Aus

2. Material und Methoden 37
dieser Suspension wurden 10 µl in eine Neubauer Zählkammer (Assistent,
Sondheim) injiziert und unter dem Mikroskop (Wilovert S, Helmut Hund GmbH,
Wetzlar) in 20-facher Vergrößerung ausgewertet. Dabei konnten nicht mehr vitale
Zellen durch die defekte Zellwand den blauen Farbstoff aufnehmen, vitale Zellen
erschienen weiterhin als durchscheinend farblos. Es wurden nur Suspensionen mit
einer Vitalität von mindestens 95 % weiterverwendet. Zur Ermittlung der Zellzahl
erfolgte außerdem eine Auszählung zweier in der Zählkammer vorgegebener
Quadrate. Die Zellzahl je Milliliter ließ sich dann wie folgt berechnen: Summe der
zwei Quadrate x ½ x 104 Zellen/ml x 50. Die ermittelte Zellzahl lag zwischen
2,5 und 7 x 107 Zellen/ml, im Durchschnitt bei 4,87 x 107 Zellen/ml.
2.1.4 Vorbereitung der Wirkstoffe
2.1.4.1 PI-103
PI-103 ist ein ATP-kompetitiver PI3-Kinase Hemmer, welcher die DNA-PK, PI3K
p110α und mTOR hemmt (Knight et al., 2006). Der Wirkstoff PI-103 mit der
Summenformel C19H16N4O3 hat eine molekulare Masse von 348,4 g/mol.
Von PI-103 (Cayman Chemical Company, Ann Arbor, USA) wurde 1 mg mit einer
elektrischen Feinwaage (Elektrische Feinwaage PT 150, Sartorius AG, Göttingen)
abgewogen und in 1 ml DMSO (Sigma-Aldrich Chemie GmbH, Deisenhofen)
gelöst, wodurch man eine Lösung mit einer Konzentration von 2.870 µM an
Wirkstoff erhielt. Von dieser wurden jeweils 15 µl in 1,5 ml Eppendorfgefäße
aliquotiert und bei -20°C gelagert. Durch Verdünnen von 10 µl der Stocklösung mit
190 µl RPMI im Verhältnis 1:20 wurde eine Ausgangslösung mit einer
Konzentration von 100 µM bereitet, welche nunmehr 5 % DMSO enthielt. Zum
Verdünnen wurde ein Verdünnungsmedium aus 1 ml RPMI und 50 µl DMSO
vorbereitet, welches somit ebenfalls 5 % DMSO aufwies. Die Ausgangslösung von
100 µM wurde dann über eine Verdünnungsreihe durch verdünnen von jeweils
100 µl der vorangegangenen Lösung mit 200 µl Verdünnungsmedium im
Verhältnis 1:3 in die Konzentrationen von 30 µM, 10 µM, 3 µM und 1 µM gebracht.
Durch einen weiteren Verdünnungsschritt im Verhältnis 1:100 mit jeweils 990 µl
der vorbereiteten Zellsuspension und 10 µl der Pharmakonzentrationen von
100 µM, 10 µM und 1 µM erhielt man die zu testenden Wirkstoffkonzentrationen

2. Material und Methoden 38
von 1 µM, 0,1 µM und 0,01 µM. Um die Effekte des Lösungsmittels DMSO auf die
Spaltbildung beurteilen zu können, wurde 990 µl der Zellsuspensionen der
Positivkontrollen und Negativkontrollen jeweils die gleiche Menge an DMSO
beigemengt, also pro Probe 10 µl des Verdünnungsmediums. Von den
hergestellten Positivkontrollen, Negativkontrollen und den Wirkstoffkontrollen
1 µM, 0,1 µM und 0,01 µM wurden dann 500 µl auf die Gefrierschnitte gegeben,
wobei alle Kontrollen doppelt angelegt wurden.
2.1.4.2 TGX-221
TGX-221 hemmt selektiv die PI3-Kinase p110β (Jackson et al., 2005). Der
Wirkstoff TGX-221 mit der Summenformel C21H24N4O2 besitzt eine molekulare
Masse von 364,4 g/mol. Von dem verwendeten Wirkstoff TGX-221 (Cayman
Chemical Company, Ann Arbor, USA) lag 1 mg gelöst in 1 ml Ethanol (70%) vor,
was als Stocklösung verwendet wurde und eine Konzentration von 2.740 µM
aufwies. Es wurden jeweils 15 µl des gelösten Wirkstoffs in mehrere 1,5 ml
Eppendorfgefäße aliquotiert und bei -20°C gelagert. Die Lösung war so für 1 Jahr
haltbar. Ausgehend von dieser Stocklösung wurde eine Verdünnungsreihe
hergestellt. Durch verdünnen von 10 µl der Stocklösung mit 190 µl RPMI im
Verhältnis von 1:20 erhielt man eine 100 µM Ausgangslösung für die
Verdünnungsreihe. Zum Verdünnen wurde ein Verdünnungsmedium aus 1 ml
RPMI und 50 µl Ethanol vorbereitet, das somit 5 % Ethanol enthielt. In der
Verdünnungsreihe wurde jeweils im Verhältnis 1:3 mit Verdünnungsmedium
verdünnt. Dazu wurden jeweils 100 µl der Lösung des vorangegangenen
Verdünnungsschritts mit 200 µl Verdünnungsmedium verdünnt, wobei Lösungen
mit Konzentrationen von 30 µM, 10 µM, 3 µM, 1 µM und 0,3 µM entstanden.
Danach wurden 10 µl der Lösungen mit den Konzentrationen von 30 µM, 3 µM
und 0,3 µM jeweils im Verhältnis 1:100 mit 990 µl der vorbereiteten Zellsuspension
verdünnt um die gewünschten Lösungen von 0,3 µM, 0,03 µM und 0,003 µM zu
erhalten. Um die Wirkung des Lösungsmittels Ethanol beurteilen zu können,
wurde den Zellsuspensionen der Positivkontrolle (990 µl), sowie der
Negativkontrolle (990 µl) jeweils 10 µl des Verdünnungsmediums hinzugefügt. Von
den hergestellten Positivkontrollen (hier gleichzeitig die Lösungsmittelkontrollen),
Negativkontrollen und den Wirkstoffkontrollen in den Konzentrationen 0,3 µM,

2. Material und Methoden 39
0,03 µM und 0,003 µM wurden jeweils 500 µl auf die Gefrierschnitte gegeben,
wobei alle Kontrollen doppelt angelegt wurden.
2.1.4.3 SM101
SM101 ist ein löslicher humaner Fcγ-Rezeptor IIb (Sondermann and Jacob, 1999).
Der Wirkstoff SM101 (Suppremol, Martinsried, Germany) lag als Stocklösung von
20 mg/ml vor, wurde bei -80°C gelagert und nach dem Auftauen zu je 55 µl in
1,5 ml Eppendorfgefäße aliquotiert und bei -20°C gelagert. 50 µl der Stocklösung
wurden mit 150 µl RPMI zunächst 1:4 verdünnt auf eine Konzentration von
5 mg/ml. Diese Ausgangslösung wurde dann in einer Verdünnungsreihe mit RPMI
auf die übrigen benötigten Konzentrationen verdünnt. Dabei wurden zunächst
40 µl der 5 mg/ml-Lösung mit 160 µl RPMI im Verhältnis 1:5 verdünnt, wobei man
eine Lösung von 1 mg/ml erhielt. 20 µl dieser Lösung wurden dann mit 180 µl
RPMI verdünnt um 1:10 auf eine Konzentration von 0,1 mg/ml. Man erhielt somit
Wirkstofflösungen in den Konzentrationen 5 mg/ml, 1 mg/ml und 0,1 mg/ml. Der
Wirkstoff SM101 wurde in zwei Schritten mit den Hautschnitten inkubiert. Im
ersten Schritt wurden die drei vorbereiteten Wirkstoffkonzentrationen im Verhältnis
1:10 mit den gleichen Mengen an Positivseren verdünnt. Dabei gab man zu 45 µl
der vorbereiteten Seren jeweils 5 µl der Wirkstofflösungen, womit man die
endgültigen Wirkstoffkonzentrationen von 0,5 mg/ml, 0,1 mg/ml und 0,01 mg/ml
erhielt. Die Negativseren und Proben für die Positivkontrollen wurden im gleichen
Verhältnis von 1:10 mit PBS verdünnt. Das hergestellte Wirkstoff-Serum Gemisch
wurde auf die entsprechenden Gefrierschnitte gegeben und im
Begasungsbrutschrank bei 37°C für 1 Stunde inkubiert. Im zweiten Schritt wurden
die Ausgangskonzentrationen 5 mg/ml, 1 mg/ml und 0,1 mg/ml wiederum 1:10
verdünnt, diesmal mit der vorbereiteten Zellsuspension. Dabei wurde zu 900 µl
Zellsuspension 100 µl der jeweiligen Wirkstofflösung gegeben. Man erhielt
Lösungen mit den gewünschten Endkonzentrationen von 0,5 mg/ml, 0,1 mg/ml
und 0,01 mg/ml. Die Zellsuspensionen für die Positivkontrollen und
Negativkontrollen wurden im gleichen Verhältnis 1:10 mit 100 µl RPMI verdünnt,
um eine vergleichbare Zahl an Zellen zu gewährleisten. Von den hergestellten
Proben der Positivkontrollen, Negativkontrollen und der Wirkstoffkontrollen
0,5 mg/ml, 0,1 mg/ml und 0,01 mg/ml wurden jeweils 500 µl auf die Gefrierschnitte

2. Material und Methoden 40
gegeben und diese bei 37°C für 3 Stunden inkubiert. Jede Kontrolle wurde in
Doppelbestimmung angelegt.
2.1.5 Vorbereitung der Gefrierschnitte
Die Gefrierschnitte wurden aufgetaut und die Qualität der Hautschnitte unter dem
Mikroskop bei 200facher Vergrößerung beurteilt. Danach wurde zwischen die
einzelnen Hautschnitte eine Trennlinie mit einem Ölstift (DAKO-Pen, DAKO
Denmark A/S, Glostrup, Dänemark) gezogen, um eine Vermischung der
verschiedenen Seren zu vermeiden. Dann wurden auf die Hautschnitte die
entsprechenden Positiv- und Negativseren aufgetragen, wobei ein Schnitt mit
jeweils 50 µl humanem Serum beziehungsweise 20 µl Mausserum vollständig
bedeckt wurde. Pro Versuchsreihe wurden jeweils 4 verschiedene Positivseren
verwendet, die in entsprechendem Verhältnis mit PBS verdünnt wurden. Die
verwendeten Negativseren wurden in der Verdünnung an die Positivseren
angepasst. Die Positivkontrollen als auch die Kontrollen der einzelnen
Wirkstoffkonzentrationen wurden doppelt angelegt. In einer schwarzen
Feuchtigkeitskammer (Werner Hassa GmbH, Lübeck) wurden die vorbereiteten
Hautschnitte eine Stunde bei 37°C und 5 % CO2 im Begasungsbrutschrank
(Binder GmbH, Tuttlingen) inkubiert und danach 5 min in PBS gewaschen.
Danach wurden Injektionskammern für die Zellsuspension hergestellt, indem der
Objektträger mit den Gefrierschnitten mit einem weiteren Objektträger (Heinz
Herenz Medizinalbedarf GmbH, Hamburg) abgedeckt wurde, welcher an beiden
Seiten mit Tesafilm umwickelt war. Somit entstand zwischen den beiden
Objektträgern eine Kammer von etwa 1 mm Höhe. Mithilfe von Parafilm (Parafilm
M Laboratory Film, Pechiney Plastic Packaging, Menasha, Wisconsin, USA)
wurden die beiden Objektträger aneinander fixiert. In die entstandene Kammer
wurde dann jeweils 500 µl der vorbereiteten Zellsuspension injiziert. Diese enthielt
entweder den jeweiligen Wirkstoff in entsprechender Konzentration oder wurde für
die Positiv- und Negativkontrollen entsprechend der Menge an zugefügtem
Wirkstoff mit RPMI verdünnt. Danach wurden die Schnitte bei 37°C und 5 % CO2
für 3 Stunden im Begasungsbrutschrank inkubiert und dann vorsichtig in PBS
5 - 10 Minuten gewaschen bis mikroskopisch bei 200facher Vergrößerung

2. Material und Methoden 41
(Mikroskop Wilovert S, Helmut Hund GmbH, Wetzlar) die DEJ gut einsehbar und
frei von Zellen war.
2.1.6 Fixierung und Färbung der Schnitte
Die Schnitte wurden zunächst 5 Minuten in Formalin fixiert und unter
Leitungswasser gewaschen. Die Färbung erfolgte dann mit Hämatoxylin und
Eosin. Dafür wurden die Schnitte erst 4 Minuten mit der Kernfärbung nach
Papanicolaou gefärbt und danach unter Leitungswasser abgewaschen. Dann
wurden die Schnitte 20 Sekunden in Essigsäure-Alkohol gestellt und danach
nochmals abgespült. Danach wurden die Schnitte 10 Sekunden in
Ammoniakwasser gestellt, gewässert und 1 - 2 Minuten mit Eosin gegengefärbt.
Schließlich wurden die Schnitte in einer aufsteigenden Alkoholreihe von 96 %-,
96 %- und 100 %- Alkohollösung sowie Xylol dehydriert und mittels eines
Glaseindeckautomaten (Leica CV 5030, Leica Mikrosysteme Vertrieb GmbH,
Wetzlar) eingedeckt. Die fertig gefärbten und eingedeckten Schnitte wurden dann
am Mikroskop (Olympus BX40, Olympus Deutschland GmbH, Hamburg) in
200facher und 400facher Vergrößerung ausgewertet.
2.1.7 Mikroskopische Auswertung
Die mikroskopische Auswertung (Mikroskop Olympus BX40, Olympus
Deutschland GmbH, Hamburg) wurde geblindet in 200facher und 400facher
Vergrößerung durchgeführt. Dabei wurde die Spaltbildung prozentual als Anteil
der zerstörten DEJ an der gesamten dermo-epidermalen Übergangszone
angegeben. Die Fotografien der mikroskopischen Schnitte wurden mit dem
Programm ProgRes Capture 2.1 angefertigt.
2.1.8 Statistik
Die statistische Auswertung erfolgte mithilfe von Microsoft Excel und SigmaPlot
11.1.

2. Material und Methoden 42
2.2 Das NBT-Gefrierschnittmodell
Mithilfe des NBT-Gefrierschnittmodells kann die Bildung von ROS in aktivierten
neutrophilen Granulozyten dargestellt werden. NBT (Nitroblautetrazoliumchlorid)
ist ein löslicher Redoxfarbstoff, der durch ROS zu einem unlöslichen blauen
Farbstoff (Formazan) reduziert wird (Roth and Kaeberle, 1981). Mithilfe des NBT-
Gefrierschnittmodells wurde so der Effekt von Methylprednisolon (MP) auf die
ROS-Produktion und somit der Aktivierung neutrophiler Granulozyten untersucht.
Im Versuch wurden dazu Gefrierschnitte von humaner Haut mit BP-
Patientenseren inkubiert. Danach wurde MP mit den vorher isolierten humanen
Leukozyten und der NBT-Lösung auf die Gefrierschnitte gegeben und inkubiert.
Die Präparate wurden dann gewaschen und am Mikroskop ausgewertet.
2.2.1 Isolierung von Leukozyten
Die Isolierung neutrophiler Granuloyzten erfolgte entsprechend dem
Gefrierschnittmodell ohne NBT.
2.2.2 Vorbereitung der NBT-Lösung
Der hier verwendete Stoff NBT (Sigma-Aldrich Chemie GmbH, Deisenhofen) lag
als fester Stoff in einer Konzentration von 0,05 % (0,5 mg/ml) vor. Davon wurden
mithilfe einer elektrischen Feinwaage (Ohaus Analytical Plus, Nänikon, Schweiz)
5 mg abgewogen und unter schütteln (Schütteltisch Thermostat 5320, Eppendorf
AG, Hamburg) titrierend in 10 µl-Schritten DMF (Sigma-Aldrich Chemie GmbH,
Deisenhofen) zugefügt, bis der Stoff komplett gelöst vorlag. Dies war nach Zugabe
von insgesamt 50 µl DMF der Fall. Die Lösung wurde dann mit RPMI auf 100 µl
aufgefüllt. Man erhielt so eine NBT-Lösung mit einer Konzentration von 5 %
(50 mg/ml). Die NBT-Lösung wurde dann durch Zugabe der vorbereiteten
Leukozytensuspension im Verhältnis 1:100 wieder auf 0,05 % verdünnt. Dazu
wurden zu 7 ml Zellsuspension 70 µl NBT-Lösung hinzugefügt.

2. Material und Methoden 43
2.2.3 Vorbereitung von Methylprednisolon
Für den Versuch wurde MP (Urbason solubile 16 mg, Sanofi-Aventis GmbH,
Frankfurt/M.) verwendet. MP (C22H30O5) ist ein synthetisches Glukokortikoid mit
einer molekularen Masse von 374,47 g/mol. Für den vorliegenden Versuch wurde
eine Stocklösung hergestellt, indem eine Ampulle mit 16 mg festem Wirkstoff in
1 ml Wasser gelöst wurde. Diese wies somit eine Konzentration von 16 mg/ml,
umgerechnet 42,47 mM, auf. Durch Verdünnung der Stocklösung wurden dann die
einzelnen gewünschten Konzentrationen von 3 mM, 1 mM, 0,3 mM, 0,1 mM und
0,03 mM hergestellt. Dazu wurde eine Verdünnungsreihe angelegt. Durch
verdünnen von 300 µl der Stocklösung mit 100 µl RPMI erhielt man eine 30 mM
Ausgangslösung für die Verdünnungsreihe. In allen anderen Schritten der
Verdünnungsreihe wurde dann jeweils im Verhältnis 1:3 mit RPMI weiter verdünnt.
Dazu wurden jeweils 100 µl der Lösung des vorangegangenen
Verdünnungsschritts mit 200 µl RPMI verdünnt, wodurch die Verdünnungen von
10 mM, 3 mM, 1 mM und 0,3 mM entstanden. Danach wurden die entstandenen
Lösungen jeweils im Verhältnis 1:10 mit der vorbereiteten Zellsuspension, der
bereits NBT zugefügt wurde, verdünnt. Dazu wurden je 100 µl der
Pharmakonlösungen mit den Konzentrationen 30 mM, 10 mM, 3 mM, 1 mM und
0,3 mM mit jeweils 900 µl Zellsuspension verdünnt. Dadurch erhielt man die
gewünschten Konzentrationen an MP von 3 mM, 1 mM, 0,3 mM, 0,1 mM und
0,03 mM. Für die Kontrolle ohne Wirkstoff wurden 900 µl der Zellsuspension mit
100 µl RPMI verdünnt. Nach Zugabe von Zellsuspension zu den vorbereiteten
Wirkstoffkonzentrationen wurden jeweils 500 µl auf die Gefrierschnitte
aufgetragen, wobei alle Kontrollen in Doppelbestimmung getestet wurden.
2.2.4 Vorbereitung der Gefrierschnitte
Der Ablauf entsprach dem des Gefrierschnittmodells ohne NBT. Allerdings erfolgte
in diesem Versuchsaufbau eine Inkubation der Schnitte im Begasungsbrutschrank
für nur 1,5 Stunden.

2. Material und Methoden 44
2.2.5 Fixierung der Gefrierschnitte
Die Schnitte wurden nach vorsichtigem Abwaschen überflüssiger Zellen in PBS
mit Aquatex (Merck KGaA, Darmstadt) und Deckgläsern (Cover glasses lab
solute, Th. Geyer Gruppe, Renningen) eingedeckelt. Die Auswertung erfolgte
mikroskopisch (Olympus BX40, Olympus Deutschland GmbH) in 200facher und
400facher Vergrößerung.
2.3 Der ROS-Release
Mithilfe des ROS-Release wurden die Effekte von MP auf die Bildung von ROS in
neutrophilen Granulozyten untersucht.
2.3.1 Isolierung von neutrophilen Granulozyten mit Polymorphprep
Zur Gewinnung von neutrophilen Granulozyten wurde gesunden, freiwilligen
Spendern 18 ml Blut abgenommen, das mit EDTA antikoaguliert wurde. Dafür
wurden Spender, die zwischen 22 und 31 Jahren alt waren, verwendet. Das
durchschnittliche Alter betrug 26 Jahre, es nahmen zwei Frauen und ein Mann teil.
Die Abnahme erfolgte mit einer Butterflykanüle (BD Valu Set, Beckton Dickinson
GmbH, Heidelberg) und zwei 9 ml-Röhrchen, denen EDTA zur Antikoagulation
zugesetzt war (Monovette EDTA, Sarstedt AG & Co., Nümbrecht). Es wurden
zusätzlich vier 15 ml-Tubes (Sarstedt AG & Co., Nümbrecht) vorbereitet, in die
jeweils 4,5 ml Polymorphprep (Fresenius Kabi Norge AS, Oslo, Norwegen)
pipettiert wurde. Das entnommene Blut wurde anschließend vorsichtig in die
Tubes überführt, wobei in jedem Tube das Polymorphprep mit jeweils 4,5 ml Blut
vorsichtig überlagert wurde. Anschließend wurden die Röhrchen 30 - 35 min bei
1.600 rpm und Raumtemperatur unter Ausschalten der Bremse zentrifugiert
(Varifuge 1.0 R, Heraeus Instruments GmbH, Hanau). Nach dem Zentrifugieren
bildeten sich in jedem Tube Zellbanden aus, wobei sich in der obersten Bande
mononukleäre Zellen, wie Lymphozyten und Monozyten sammelten und in der
zweiten Bande die benötigten Granulozyten. Im nächsten Schritt wurde deshalb in
jedem Tube zunächst die oberste Bande vorsichtig mit einer automatischen
Pipette (Pipetus-Akku, Hirschmann Laborgeräte GmbH & Co. KG, Eberstadt) mit
einem 5 ml-Aufsatz (Sarstedt AG & Co., Nümbrecht) abgenommen und verworfen.

2. Material und Methoden 45
Danach wurde vorsichtig die zweite Zellbande abgenommen und in ein 50 ml-
Tube (Sarstedt AG & Co., Nümbrecht) überführt. Dadurch erhielt man etwa 10 ml
Granulozytensuspension. Dieser wurde danach die gleiche Menge an
Halbmedium (RPMI 1640 verdünnt mit LPS-freiem ddH2O im Verhältnis 1:1)
zugefügt und mit RPMI (Lonza RPMI 1640 mit L-Glutamin, BioWhittaker, Verviers,
Belgien) auf 50 ml aufgefüllt. Danach wurde das Tube nochmals für 10 min bei
1.400 rpm und Raumtemperatur zentrifugiert. Der Überstand wurde danach mit
einer Wasserstrahlpumpe abgesaugt. Dem Pellet wurde mit der automatischen
Pipette mit 10 ml-Aufsatz (Sarstedt & Co., Nümbrecht) 6 ml eiskalter Lysepuffer
(LPS-freies D-PBS verdünnt mit LPS-freiem ddH2O im Verhältnis 1:5) zugefügt
und die Lösung resuspendiert. Die Lösung wurde 30 Sekunden inkubiert und
danach mit RPMI auf 50 ml aufgefüllt. Das Tube wurde dann erneut 10 min bei
1.400 rpm und Raumtemperatur zentrifugiert und der Überstand danach mit der
Wasserstrahlpumpe abgesaugt. Das Pellet wurde dann in 20 ml
Chemilumineszenzmedium (CL-Medium, RPMI-1640 ohne Phenolrot mit 25 mM
HEPES, Biochrom AG, Berlin) resuspendiert und wiederholt 10 min bei 1.400 rpm
und Raumtemperatur zentrifugiert. Danach wurde der Überstand mit der
Wasserstrahlpumpe abgesaugt und das Pellet mit 4 ml CL-Medium resuspendiert.
Die Suspension mit isolierten Granulozyten wurde dann bis zur Weiterverwendung
auf Eis gestellt. Je nach ermittelter Zellzahl erfolgte eine weitere Verdünnung der
Granulozytensuspension mit CL-Medium.
2.3.2 Vorbereitung von Methylprednisolon
Für den Versuch wurde MP (Urbason solubile 16 mg, Sanofi-Aventis GmbH,
Frankfurt/M.) verwendet. Für den vorliegenden Versuch wurde eine Stocklösung
hergestellt, indem eine Ampulle mit 16 mg festem Wirkstoff in 1 ml Wasser gelöst
wurde. Diese wies somit eine Konzentration von 42,47 mM auf. Durch
Verdünnung der Stocklösung wurden dann die einzelnen gewünschten
Konzentrationen von 3 mM, 1 mM, 0,3 mM, 0,1 mM und 0,03 mM hergestellt.
Dazu wurde eine Verdünnungsreihe angelegt. Durch verdünnen von 150 µl der
Stocklösung mit 850 µl CL-Medium erhielt man eine 6 mM Ausgangslösung für die
Verdünnungsreihe. In allen anderen Schritten der Verdünnungsreihe wurde dann
jeweils im Verhältnis 1:3 mit CL-Medium weiter verdünnt. Dazu wurden jeweils

2. Material und Methoden 46
300 µl der Lösung des vorangegangenen Verdünnungsschritts mit 600 µl CL-
Medium verdünnt, wodurch die Verdünnungen von 2 mM, 0,6 mM, 0,2 mM und
0,06 mM entstanden. Danach wurden die entstandenen Lösungen jeweils im
Verhältnis 1:2 mit der vorbereiteten Zellsuspension verdünnt. Dazu wurden je
600 µl der Pharmakonlösungen mit den Konzentrationen 6 mM, 2 mM, 0,6 mM,
0,2 mM und 0,06 mM mit jeweils 600 µl Zellsuspension verdünnt. Dadurch erhielt
man die gewünschten Konzentrationen an MP 3 mM, 1 mM, 0,3 mM, 0,1 mM und
0,03 mM. Für die Kontrolle ohne Wirkstoff wurden 600 µl der Zellsuspension mit
600 µl CL-Medium verdünnt. Nach Zugabe von Zellsuspension zu den
vorbereiteten Wirkstoffkonzentrationen erfolgt eine dreistündige Inkubation bei
37°C und 5 % CO2 im Begasungsbrutschrank (Heraeus Instruments GmbH,
Hanau). Danach wurden je 100 µl der Lösungen in die jeweiligen Wells pipettiert.
2.3.3 Durchführung des ROS-Release
Zunächst erfolgte die Vorbereitung einer Mikrotiterplatte (Greiner Bio-One GmbH,
Frickenhausen) durch Coating mit NC16A-Antigen (Euroimmun Medizinische
Labordiagnostika AG, Lübeck). Dafür wurde die vorliegende Antigenlösung mit der
Konzentration von 1 mg/ml auf 20 µg/ml mit PBS verdünnt. Danach wurden jeweils
50 µl der verdünnten Lösung in jedes Well der Platte pipettiert und bei 4°C über
Nacht inkubiert. Dann wurde die Platte mit Waschpuffer (PBS mit 0,05 % Tween)
zur Entfernung überschüssiger, nicht gebundener Antigene im ELISA-Washer
(Columbus Pro, Tecan Group Ltd., Männedorf, Schweiz) gewaschen und mit
300 µl Blockingpuffer (PBS mit 1 % biotinfreiem BSA und 0,05 % Tween) je Well
geblockt. Die Platte wurde dann für 1 Stunde bei Raumtemperatur auf dem
Schütteltisch (TPM 4, Sarstedt AG & Co., Nümbrecht) inkubiert. Danach wurde die
Platte erneut im ELISA-Washer gewaschen. Dann wurden jeweils 50 µl der
vorbereiteten BP-Antikörperseren und das humane Negativserum in die jeweiligen
Wells pipettiert und für 2 Stunden bei Raumtemperatur auf dem Schütteltisch
inkubiert. Die Antikörperseren wurden vorher mit biotinfreiem Blockingpuffer im
Verhältnis 1:300 verdünnt. Nach der Inkubation wurde die Platte erneut im ELISA-
Washer gewaschen und auf Eis gestellt. Den vorbereiteten Wirkstofflösungen mit
unterschiedlichen Lösungen an MP wurde dann die vorbereitete
Granulozytensuspension hinzugefügt und für 3 Stunden bei 37°C und 5 % CO2 im

2. Material und Methoden 47
Begasungsbrutschrank inkubiert. Die jeweiligen Lösungen aus Zellsuspension
ohne MP oder mit Pharmakon in unterschiedlicher Konzentration wurden danach
mit jeweils 60 µl Luminollösung versetzt. Diese wurde vorher vorbereitet aus 2 mg
Luminol (5-amino-2,3-dihydro-1,4-phthalazindion, Roche Diagnostics, Mannheim),
das in 8 µl NaOH gelöst und mit 1 ml ddH2O aufgefüllt wurde. Danach wurden je
100 µl der Suspension mit MP in der Konzentration von 3 mM, 1 mM, 0,3 mM,
0,1 mM, 0,03 mM oder 0 mM je Well pipettiert und die Platte sofort in den ELISA-
Reader (Perkin-Elmer LAS, Rodgau, Germany) zur photometrischen Auswertung
gestellt. Die optische Dichte (OD) wurde bei 425 nm und 37°C in 60 Minutenzyklen
gemessen. Die Daten wurden als relative Lichteinheiten (RU) dargestellt. Als
Leerwertkontrolle diente die OD von Hexa-Lösung (5 % Hexadecyltimethyl-
ammoniumbromid in ddH2O). Diese wurde nach Bestimmung der OD aller Proben
subtrahiert. Die gemessene OD korreliert dabei mit der ROS-Produktion der
neutrophilen Granulozyten.
2.3.4 Auswertung
Die Auswertung der gemessenen Werte erfolgte mit dem Wallac 1420 Manager.
Die statistische Auswertung erfolgte mit Sigma 11.1.

3. Ergebnisse 48
3. Ergebnisse
3.1 Verwendete Seren
Es wurden insgesamt 47 Seren von Patienten mit BP verwendet. Als
Negativkontrollen dienten nach Testung auf eine mögliche Spaltbildung im
Gefrierschnittmodell Seren gesunder Spender.
Für die Versuchsreihen auf Maushaut wurden insgesamt 11 verschiedene Seren
von Kaninchen, die mit mCol7c, einem immunogenen Protein des murinen Typ-
VII-Kollagens, immunisiert wurden, verwendet. Als Negativkontrollen dienten
Kontrollseren gesunder Kaninchen.
3.1.1 Testung der EBA-Seren im Gefrierschnittmodell
Die 11 Seren wurden hinsichtlich ihrer Fähigkeit zur DES auf Maushaut getestet.
Dabei konnte eine Spaltbildung mit 5 der 11 Seren erzeugt werden (siehe Abb.10).
Die Spaltbildung lag bei den jeweiligen Proben zwischen 30 und 80 %, der
Mittelwert ± SEM lag bei 70 ± 10,49 %. Die in der IIF ermittelten Titer dieser Seren
lagen zwischen 5.000 und 15.000, der Mittelwert ± SEM betrug 9.000 ± 2449,49
(siehe Tab.2). Die jeweiligen Verdünnungen der Seren wurden experimentell im
Gefrierschnittmodell ermittelt unter Verwendung der unverdünnten Seren sowie
der Verdünnungen 1:2, 1:4, 1:8 und 1:16. Die Verdünnung, die im
Gefrierschnittmodell die höchste Spaltbildung zeigte, wurde ausgewählt. Die meist
verwendete Verdünnung war 1:2.
Serumnummer IIF-Titer Verdünnung DES (%)
SA 6333 15000 01:02 80
SA 6334 15000 01:02 80
SA 6303 5000 pur 30
SA 6304 5000 pur 90
SA 6305 5000 01:02 70
Tabelle 2: Ergebnisse der Serumtestung von EBA-Seren im Gefrierschnittmodell. Dargestellt
sind die Verdünnungen, in denen die höchste Spaltbildung ausgelöst werden konnte. IIF: indirekte
Immunfloureszenz, DES: dermo-epidermale Spaltbildung

3. Ergebnisse 49
Abbildung 10: Testung der EBA-Seren auf Gefrierschnitten von Maushaut. 5 der getesteten
Seren führten zur eine Spaltbildung (b,c), unter Verwendung von Seren nicht-immunisierter
Kaninchen ließ sich keine Spaltbildung auslösen (a). In der hier angewendeten H-E-Färbung stellt
sich die Epidermis lila, die Dermis rosa dar. Die Pfeile kennzeichnen Bereiche mit dermo-
epidermaler Spaltbildung. (Vergrößerung a,b: 200fach, c: 400fach)
3.1.2 Testung der BP180-Seren im Gefrierschnittmodell
Die Patientenseren wurden hinsichtlich der DES auf Proben humaner Haut im
Gefrierschnittmodell getestet (siehe Abb.11). Dabei konnte mit 21 der
verwendeten Seren eine Spaltbildung erzeugt werden. Diese lag zwischen 20 und
100 %, wobei der Mittelwert ± SEM 66,75 ± 5,84 % betrug. Die ELISA-Werte mit
rekombinantem BP180 NC16A befanden sich für diese Seren zwischen 77 U/ml
und 12.990 U/ml, der Mittelwert ± SEM lag bei 3.421 ± 794,52 U/ml. Die einzelnen
Seren wurden in den Verdünnungen 1:2, 1:4, 1:8 und 1:16 im Gefrierschnittmodell
verwendet und die Verdünnung mit der höchsten Spaltbildung ausgewählt (siehe
Tab.3). Die meist verwendete Verdünnung betrug 1:12.
a b c
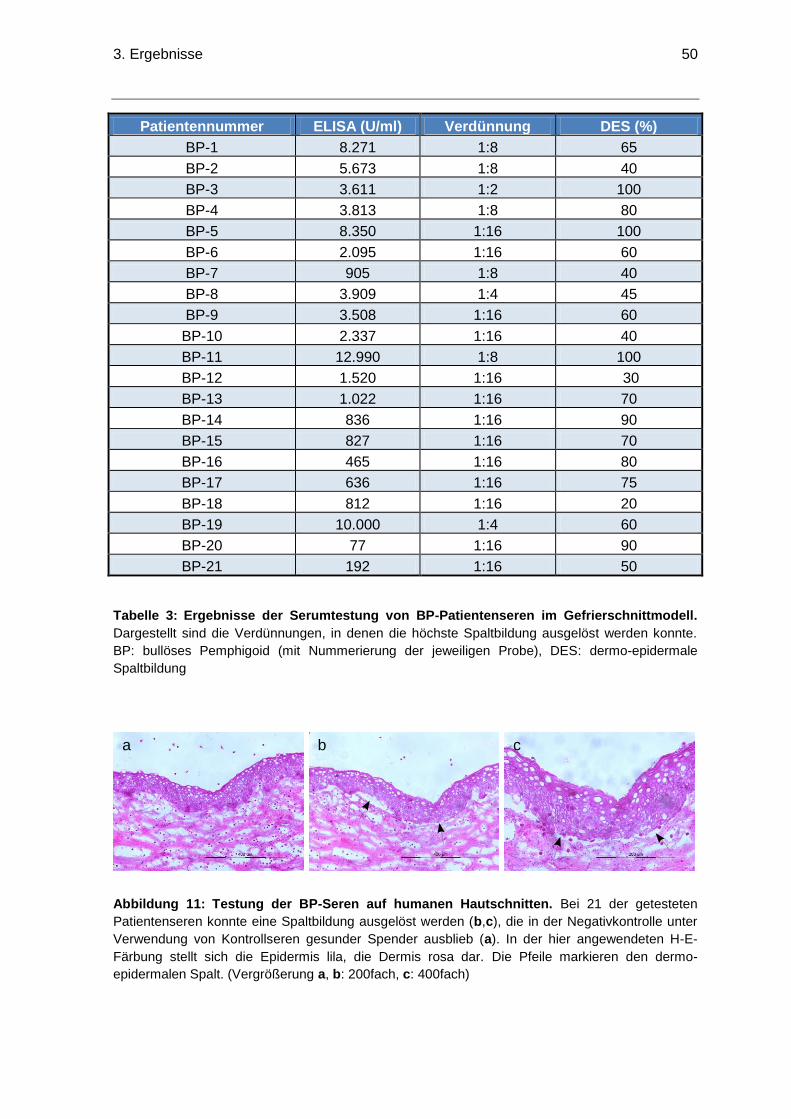
3. Ergebnisse 50
Patientennummer ELISA (U/ml) Verdünnung DES (%)
BP-1 8.271 1:8 65
BP-2 5.673 1:8 40
BP-3 3.611 1:2 100
BP-4 3.813 1:8 80
BP-5 8.350 1:16 100
BP-6 2.095 1:16 60
BP-7 905 1:8 40
BP-8 3.909 1:4 45
BP-9 3.508 1:16 60
BP-10 2.337 1:16 40
BP-11 12.990 1:8 100
BP-12 1.520 1:16 30
BP-13 1.022 1:16 70
BP-14 836 1:16 90
BP-15 827 1:16 70
BP-16 465 1:16 80
BP-17 636 1:16 75
BP-18 812 1:16 20
BP-19 10.000 1:4 60
BP-20 77 1:16 90
BP-21 192 1:16 50
Tabelle 3: Ergebnisse der Serumtestung von BP-Patientenseren im Gefrierschnittmodell.
Dargestellt sind die Verdünnungen, in denen die höchste Spaltbildung ausgelöst werden konnte.
BP: bullöses Pemphigoid (mit Nummerierung der jeweiligen Probe), DES: dermo-epidermale
Spaltbildung
Abbildung 11: Testung der BP-Seren auf humanen Hautschnitten. Bei 21 der getesteten
Patientenseren konnte eine Spaltbildung ausgelöst werden (b,c), die in der Negativkontrolle unter
Verwendung von Kontrollseren gesunder Spender ausblieb (a). In der hier angewendeten H-E-
Färbung stellt sich die Epidermis lila, die Dermis rosa dar. Die Pfeile markieren den dermo-
epidermalen Spalt. (Vergrößerung a, b: 200fach, c: 400fach)
a b c

3. Ergebnisse 51
3.2 Vergleich der DES auf humaner Haut und Maushaut
Es wurde außerdem das Versuchsmodells mit humaner Haut, humanem Serum
und humanen Leukozyten mit dem Modell unter Verwendung von Maushaut,
Kaninchenserum und humanen Leukozyten verglichen. Dafür wurden die
Positivkontrollen (PK/DMSO) der Versuchsreihen von TGX-221 (n = 31) und
PI-103 (n = 22) betrachtet. Dabei ergab sich in den Versuchen mit humaner Haut
eine mittlere DES ± SEM von 65 ± 4,04 %, während die mittlere DES ± SEM auf
Maushaut 30 ± 5,21 % betrug. Im t-Test zeigte sich somit ein signifikanter
Unterschied mit P ≤ 0,001.
Das Lösungsmittel DMSO scheint zudem auf die DES einen Effekt zu haben. Im
Vergleich der PK in den Versuchen mit PI-103 und TGX-221 (PK/DMSO) mit den
PK der Versuchsreihen mit SM101 ergab sich eine signifikant geringere DES
(P ≤ 0,001).
3.3 Effekte von Methylprednisolon auf die Bildung von ROS im
Gefrierschnittmodell mit Nitroblautetrazolium (NBT)
Aufgrund langjähriger klinischer Erfahrung sind Steroide Therapie der Wahl beim
BP und der EBA (Joly et al., 2002; Schmidt and Zillikens, 2011). Der genaue
Wirkmechanismus der Glukokortikoide ist jedoch nach wie vor nicht vollständig
geklärt. Es wurde beispielsweise eine hemmende Wirkung von Glukokortikoiden
auf die Enzyme ERK-1 und -2 beschrieben, die an der Regulierung der NADPH-
Oxidase und der Bildung von ROS beteiligt sind (Kassel et al., 2001). Daher wurde
in dieser Arbeit untersucht, ob die Behandlung mit Steroiden die Generierung von
ROS im Gefrierschnittmodell inhibieren kann.
Die Rolle von ROS bei der durch BP-Antikörper induzierten Spaltbildung wurde
dabei im Gefrierschnittmodell mit NBT untersucht. NBT ist ein Redoxfarbstoff, der
bei der Aktivierung neutrophiler Granulozyten durch die gebildeten ROS zu einem
blauen Farbstoff (Formazan) reduziert wird. Formazan lagert sich in den
aktivierten neutrophilen Granulozyten im Gefrierschnitt entlang der DEJ ab.
Die hemmenden Effekte von Methylprednisolon (MP) auf die Bildung von ROS
(Aktivierung der neutrophilen Granulozyten) konnten so ebenfalls untersucht
werden.
Für den Versuch wurde MP in den Konzentrationen 3x10-2 mM, 1x10-1 mM,

3. Ergebnisse 52
3x10-1 mM und 1 mM getestet. Diese wurden aus einer 42,5 mM Stocklösung
durch Verdünnen hergestellt und mit einer 5 % NBT-Lösung versetzt. Es ergaben
sich so die Versuchsgruppen: Versuchsgruppe I als Positivkontrolle (PK) mit dem
Lösungsmittel RPMI, als Kontrollen mit MP in unterschiedlichen Konzentrationen
Versuchsgruppe II (3x10-2 mM), III (1x10-1 mM), IV (3x10-1 mM) und V (1 mM) und
Versuchsgruppe VI als Negativkontrolle (NK). Es wurden 3 Versuchsreihen (n = 3)
durchgeführt.
Dabei wurde die Ablagerung von Formazan entlang der DEJ mikroskopisch nach
folgenden Kriterien ausgewertet: keine Ablagerung von Formazan entlang der DEJ
wurde mit 0 bewertet, vereinzelte Ablagerung mit 1, vereinzelt
zusammenhängende Ablagerung entlang der DEJ mit 2 und eine durchgehende
Ablagerung von Formazan entlang der DEJ mit 3.
Dabei ergab sich in der Versuchsgruppe I (PK) eine mediane Bewertung von 1
(25%-Perzentil (z0,25): 1, 75 %-Perzentil (z0,75): 2), in Versuchsgruppe II von 1
(z0,25: 1, z0,75: 2), in Versuchsgruppe III ergab sich eine mediane Präzipitation von
1 (z0,25: 0,25, z0,75: 2,75), in Versuchsgruppe IV von 1 (z0,25: 0, z0,75: 2) und in
Versuchsgruppe V (höchste Konzentration von MP) ergab sich eine mediane
Bewertung von 0 (z0,25: 0, z0,75: 0). Versuchsgruppe VI (NK) zeigte keine
Ablagerungen. Beim Vergleich der Versuchsgruppen I bis V untereinander zeigte
sich im Kruskal-Wallis-Test ein signifikanter Unterschied mit P ≤ 0,001. Der
Vergleich der Versuchsgruppen II - V mit der Versuchsgruppe I wurde mithilfe von
Dunn´s Methode durchgeführt. Dabei ergab sich lediglich zwischen
Versuchsgruppe I und Versuchsgruppe V (höchste Konzentration an MP, 1 mM)
ein signifikanter Unterschied (P < 0,05). Der Vergleich von Versuchsgruppe I mit
Versuchsgruppe II bis IV zeigte keinen signifikanten Unterschied.
Im Gefrierschnittmodell mit NBT konnte somit nur für die höchste Konzentration an
MP (1 mM) eine hemmende Wirkung auf die Bildung von ROS durch die
verminderte Reduktion und Präzipitation von NBT entlang der DEJ beobachtet
werden (siehe Abb.12 und 13).

3. Ergebnisse 53
3
2
1
0
Abbildung 12: Die Ablagerung von Formazan ist abhängig von der Methylprednisolon (MP)-
Dosis (nach (Hellberg et al., 2013)). Die Ablagerung von Formazan entlang der DEJ wurde auf
einer Skala von 0 – 3 bewertet. Dies entsprach keiner (0), vereinzelter (1), vereinzelt
zusammenhängender (2) und durchgehender (3) Präzipitation, im Diagramm zusätzlich dargestellt
als Blauabstufung. Hinsichtlich der Ablagerung von Formazan ergab sich so für die Positivkontrolle
(PK) ein Median von 1 (25 %-Perzentil (z0,25): 1, 75 %-Perzentil (z0,75): 2). Die Präzipitation von
Formazan war abhängig von der Konzentration an MP, in der höchsten Dosis von MP (1 mM)
zeigten sich signifikant geringere Ablagerungen als in der PK (P < 0,05).
Abbildung 13: Ablagerung von Formazan entlang der dermo-epidermalen Junktionszone
(DEJ) (nach (Hellberg et al., 2013)). Die Inkubation von Gefrierschnitten mit neutrophilen
Granulozyten und Kontrollserum führte zu minimalen Ablagerungen (a), wohingegen die
Positivkontrolle mit BP-Patientenserum eine fast durchgehende Formazan-Präzipitation entlang
der DEJ zeigte (b). Methylprednisolon in der höchsten Dosierung von 1 mM hemmte die
Ablagerung (c). *: DEJ. (Vergrößerung 200fach)
Konzentration Methylprednisolon (mM)
Fo
rma
zan
-Prä
zip
itatio
n
32
10
0 3x10-2 1x10-1 3x10-1 1
**
a
***
*****
**
*** * *
* * * ** * *
** *
*
b
* **
c
****
**
**
**

3. Ergebnisse 54
3.4 Effekte von Methylprednisolon auf die Bildung von ROS im ROS-
Release
Die Bildung von ROS kann auch mittels Chemilumineszenz im ROS-Release
gemessen werden. Dazu wurde eine Mikrotiterplatte mit NC16A-Antigen gecoatet
und mit Antikörperseren von BP-Patienten und isolierten humanen neutrophilen
Granulozyten inkubiert.
Mithilfe eines ROS-Release unter Einsatz von MP konnten die Ergebnisse aus
dem Gefrierschnittmodell mit NBT bestätigt werden.
Dafür wurde MP in den Konzentrationen 3x10-2 mM, 1x10-1 mM, 3x10-1 mM, 1 mM
und 3 mM verwendet. Damit ergaben sich die folgenden Versuchsgruppen:
Versuchsgruppe I als PK, für die jeweiligen Konzentrationen von MP die
Versuchsgruppen II (3x10-2 mM), III (1x10-1 mM), IV (3x10-1 mM), V (1 mM) und
VI (3 mM) und Versuchsgruppe VII als NK. Es wurden 4 Versuchsreihen
durchgeführt.
Dabei konnte bei normalisierten Werten ausgehend von einem medianen Signal
von 1 RU (Relative Unit)/ml in Versuchsgruppe I (PK) für die Versuchsgruppe II
ein medianes Signal von 0,87 RU/ml (z0,25: 0,65 RU/ml, z0,75: 0,98 RU/ml)
bestimmt werden. Für Versuchsgruppe III ergaben sich 1,00 RU/ml
(z0,25: 0,87 RU/ml, z0,75: 1,13 RU/ml). Für die Versuchsgruppen IV wurde ein
medianer Wert von 0,84 RU/ml (z0,25: 0,63 RU/ml, z0,75: 0,94 RU/ml) bestimmt, bei
Versuchsgruppe V lag der mediane Wert bei 0,84 RU/ml (z0,25: 0,68 RU/ml,
z0,75: 0,90 RU/ml) und bei Versuchsgruppe VI lag ein medianes Signal von
0,57 RU/ml (z0,25: 0,51 RU/ml, z0,75: 0,69 RU/ml) vor. Zum Vergleich der einzelnen
Versuchsgruppen wurde ein Kruskal-Wallis-Test durchgeführt. Dieser zeigte einen
signifikanten Unterschied zwischen den Signalen der einzelnen Gruppen
(P ≤ 0,001). Mithilfe von Dunn´s Methode konnte die Versuchsgruppe I (PK) mit
den Versuchsgruppen II bis VI (Wirkstoffgruppen) verglichen werden. Dabei ergab
sich ein signifikanter Unterschied zwischen Versuchsgruppe I und den
Versuchsgruppen IV bis VI (P < 0,05). Zwischen Versuchsgruppe I und den
Versuchsgruppen II und III zeigte sich kein signifikanter Unterschied (siehe
Abb.14).
Damit konnte im ROS-Release gezeigt werden, dass MP in den Konzentrationen
3x10-1 mM, 1 mM und 3 mM die durch BP180-Antikörper induzierte Bildung von

3. Ergebnisse 55
ROS in neutrophilen Granulozyten hemmt. Geringere Konzentrationen von MP
haben keinen Einfluss auf den ROS-Release.
Abbildung 14: Die Freisetzung von reaktiven Sauerstoffspezies (ROS) ist abhängig von der
Methylprednisolon (MP)-Dosis (nach (Hellberg et al., 2013)). Gezeigt ist die relative Freisetzung
von ROS ohne und mit MP in verschiedener Dosierung. Die Daten sind dargestellt als Median
(schwarze Linie), 25 %-Perzentil (unterer Rand der Box), 75 %-Perzentil (oberer Rand der Box),
obere und untere Antennen (Fehlerbalken, 5%- und 95%-Perzentil) und Ausreißer (Punkte). Die
Sterne kennzeichnen die Konzentrationen, in denen ein signifikant niedrigeres Signal bezüglich der
PK gemessen wurde (P < 0,05). Der Median für die Positivkontrolle (PK, entspricht 0 mM MP)
wurde unter Verwendung von normalisierten Werten auf 1,0 RU/ml festgelegt. In den MP-
Konzentrationen 3x10-1
mM, 1 mM und 3 mM zeigte sich eine signifikant niedrigere Freisetzung
von ROS verglichen mit der PK (P < 0,05) .
Die Ergebnisse dieses Versuches wurden bereits im Journal of Investigative
Dermatology veröffentlicht (Hellberg et al., 2013).
Konzentration Methylprednisolon (mM)
RO
S-F
reis
etz
un
g (
rela
tive
un
its)
3x10-20 1x10-1 3x10-1 1 3

3. Ergebnisse 56
3.5 Effekte der PI3-Kinase-Inhibitoren PI-103 und TGX-221 auf die dermo-
epidermale Spaltbildung im Gefrierschnittmodell
Phosphatidylinosit-3-Kinasen (PI3-Kinasen) spielen in der Regulation der NADPH-
Oxidase und der Produktion von ROS eine wichtige Rolle. Dabei sind für die
Produktion von ROS in neutrophilen Granulozyten vor allem die PI3Kβ, die PI3Kγ
und die PI3Kδ von Bedeutung (Condliffe et al., 2005; Kulkarni et al., 2011).
Inhibitoren dieses Enzyms könnten somit einen möglichen Therapieansatz in der
Behandlung des BP und der EBA darstellen. In dieser Arbeit wurden deshalb die
beiden PI3-Kinase-Inhibitoren PI-103 und TGX-221 hinsichtlich ihrer Effekte auf
die DES im Gefrierschnittmodell getestet.
3.5.1 Effekte von PI-103 auf die dermo-epidermalen Spaltbildung
Der PI3-Kinase-Inhibitor PI-103 hemmt die Klasse-IA-PI3-Kinasen α, β und δ ab
einer Konzentration von 2x10-3 µM. Ab einer Konzentration von 15x10-3 µM wird
zusätzlich die PI3Kγ sowie mTOR und die DNA-Proteinkinase (DNA-PK) inhibiert
(Raynaud et al., 2007; Workman et al., 2010).
Im Gefrierschnittmodell wurde PI-103 in den Dosierungen 1x10-2 µM, 1x10-1 µM
und 1 µM getestet. Diese wurden aus einer 2870 µM Stocklösung (PI-103 gelöst in
DMSO) über eine Verdünnungsreihe hergestellt.
Es ergaben sich folgende Versuchsgruppen: Versuchsgruppe I als PK mit dem
Lösungsmittel DMSO, als Kontrollen mit dem Wirkstoff PI-103 in unterschiedlicher
Dosis die Versuchsgruppen II (1x10-2 µM), III (1x10-1 µM) und IV (1 µM) und
Versuchsgruppe V als NK.
3.5.1.1 PI-103 im Gefrierschnittmodell mit Maushaut und EBA-Serum
immunisierter Kaninchen
In allen Versuchsreihen (n = 9) konnte eine DES erzeugt werden. Dabei ergab
sich für die Versuchsgruppe I (PK/DMSO) eine mediane Spaltbildung von 30 %
(z0,25: 15 %, z0,75: 47,5 %), für die Versuchsgruppe II eine mediane DES von 20 %
(z0,25: 10 %, z0,75: 95 %), für Versuchsgruppe III ergab sich eine DES von 0 %
(z0,25: 0 %, z0,75: 95 %) und in Versuchsgruppe IV betrug die mediane DES 15 %
(z0,25: 0 %, z0,75: 65 %). In der Versuchsgruppe V (NK) konnte keine Spaltbildung
erzeugt werden (0 %). Im Vergleich der Versuchsgruppen I bis IV zeigte sich in

3. Ergebnisse 57
der Varianzanalyse mithilfe des Kruskal-Wallis-Tests kein signifikanter
Unterschied mit P = 0,590 (siehe Abb.15 und 16).
Im Gefrierschnittmodell mit EBA-Seren hatte PI-103 somit keinen wesentlichen
Effekt auf die DES, was möglicherweise durch das unspezifische Wirkspektrum
des Inhibitors erklärt werden könnte.
Abbildung 15: Effekte von PI-103 auf die Spaltbildung in Maushaut. Die Daten sind dargestellt
als Median (schwarze Linie), 25 %-Perzentil (z0,25, unterer Rand der Box), 75 %-Perzentil (z0,75,
oberer Rand der Box), untere und obere Fehlerbalken (5 %- und 95 %-Perzentil). In der
Positivkontrolle mit DMSO (PK/DMSO) ergab sich eine mediane dermo-epidermale Spaltbildung
(DES) von 30 % (z0,25: 15 %, z0,75: 47,5 %). Zwischen der PK und den Wirkstoffkontrollen bestand
kein signifikanter Unterschied hinsichtlich der DES, P = 0,590. Der Median der
Wirkstoffkonzentration 1x10-1
µM lag bei 0 % (z0,25: 0 %, z0,75: 95 %).
Konzentration PI-103 ( M)
0 (PK/DMSO) 0,01 0,1 1
DE
S (
%)
0
20
40
60
80
100
0 (PK/DMSO) 1x10-2 1x10-1 001

3. Ergebnisse 58
Abbildung 16: Repräsentative Bilder des Effekts von PI-103 auf die Spaltbildung in
Maushaut. Auf die durch EBA-Antikörper induzierte Spaltbildung hatte PI-103 in den
Konzentrationen 1x10-2
µM, 1x10-1
µM und 1 µM (c-e) verglichen mit der Positivkontrolle (a,b)
keinen Effekt. Vgl. Negativkontrolle (f). Die Pfeile markieren Bereiche mit dermo-epidermaler
Spaltbildung. (Vergrößerungen a: 400fach, b-f: 200fach)
3.5.1.2 PI-103 im Gefrierschnittmodell mit humaner Haut und humanem BP-
Serum
In allen Versuchsreihen (n = 13) konnte eine DES induziert werden. Dabei zeigte
sich in der Versuchsgruppe I (PK/DMSO) eine mediane Spaltbildung von 75 %
(z0,25: 42,5 %, z0,75: 87,5 %), in der Versuchsgruppe II von 70 % (z0,25: 40 %,
z0,75: 80 %), in Versuchsgruppe III von 60 % (z0,25: 50 %, z0,75: 80 %), in
Versuchsgruppe IV von 60 % (z0,25: 35 %, z0,75: 80 %) und in Versuchsgruppe V
(NK) lag keine Spaltbildung vor (0 %). Beim Vergleich der einzelnen
Versuchsgruppen I bis IV zeigte sich in der Varianzanalyse mithilfe des Kruskal-
Wallis Tests kein signifikanter Unterschied (P = 0,792).
PI-103 wies somit in keiner der getesteten Konzentrationen signifikante Effekte auf
die DES auf, was mit dem unspezifischen Wirkspektrum des Inhibitors in
Zusammenhang stehen könnte (siehe Abb.17 und 18).
a b c
d e f

3. Ergebnisse 59
Abbildung 17: Effekte von PI-103 auf die Spaltbildung in humaner Haut. Die Daten sind
dargestellt als Median (schwarze Linie), 25 %-Perzentil (z0,25, unterer Rand der Box),
75 %-Perzentil (z0,75, oberer Rand der Box), obere und untere Fehlerbalken (5 %- und
95%-Perzentil) und Ausreißer (Punkte). Die mediane Spaltbildung (DES) in der Positivkontrolle
(PK/DMSO) lag bei 75 % (z0,25: 42,5 %, z0,75: 87,5 %). Es zeigte sich kein signifikanter Unterschied
zwischen den Versuchsgruppen hinsichtlich der DES, P = 0,792.
Konzentration PI-103 ( M)
0 (PK/DMSO) 0,01 0,1 1
DE
S (
%)
0
20
40
60
80
100
0 (PK/DMSO) 1x10-2 1x10-1 001
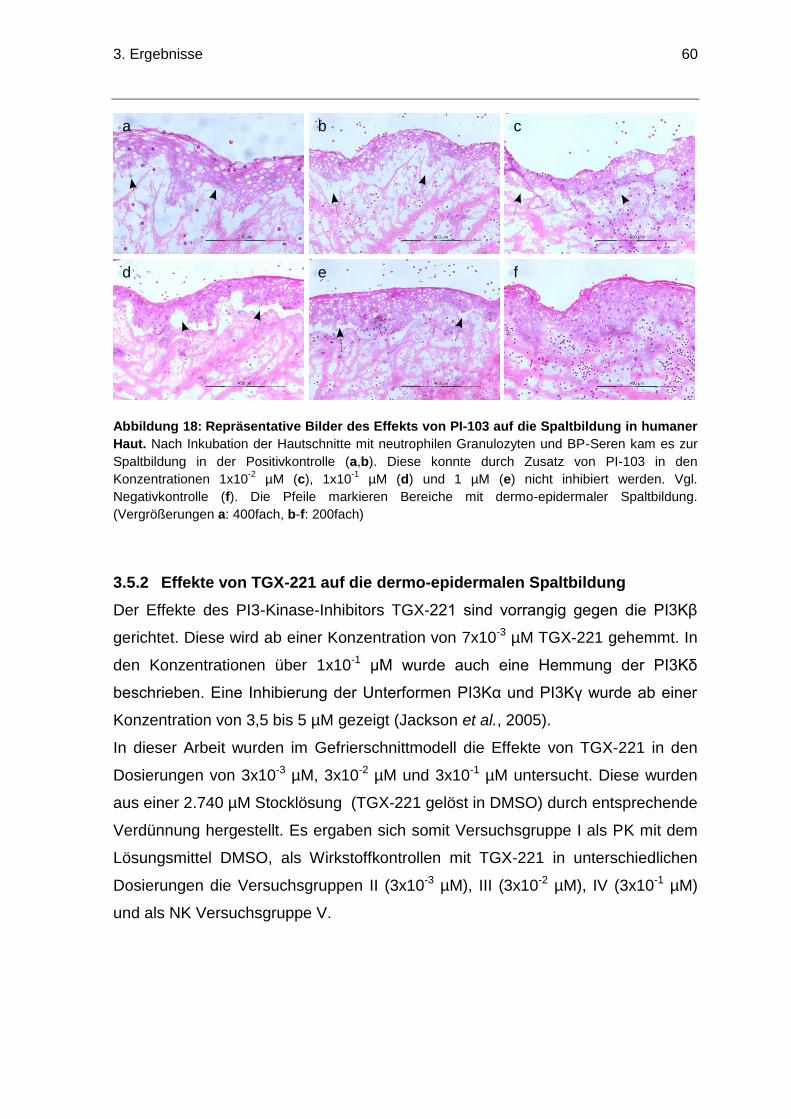
3. Ergebnisse 60
Abbildung 18: Repräsentative Bilder des Effekts von PI-103 auf die Spaltbildung in humaner
Haut. Nach Inkubation der Hautschnitte mit neutrophilen Granulozyten und BP-Seren kam es zur
Spaltbildung in der Positivkontrolle (a,b). Diese konnte durch Zusatz von PI-103 in den
Konzentrationen 1x10-2
µM (c), 1x10-1
µM (d) und 1 µM (e) nicht inhibiert werden. Vgl.
Negativkontrolle (f). Die Pfeile markieren Bereiche mit dermo-epidermaler Spaltbildung.
(Vergrößerungen a: 400fach, b-f: 200fach)
3.5.2 Effekte von TGX-221 auf die dermo-epidermalen Spaltbildung
Der Effekte des PI3-Kinase-Inhibitors TGX-221 sind vorrangig gegen die PI3Kβ
gerichtet. Diese wird ab einer Konzentration von 7x10-3 µM TGX-221 gehemmt. In
den Konzentrationen über 1x10-1 µM wurde auch eine Hemmung der PI3Kδ
beschrieben. Eine Inhibierung der Unterformen PI3Kα und PI3Kγ wurde ab einer
Konzentration von 3,5 bis 5 µM gezeigt (Jackson et al., 2005).
In dieser Arbeit wurden im Gefrierschnittmodell die Effekte von TGX-221 in den
Dosierungen von 3x10-3 µM, 3x10-2 µM und 3x10-1 µM untersucht. Diese wurden
aus einer 2.740 µM Stocklösung (TGX-221 gelöst in DMSO) durch entsprechende
Verdünnung hergestellt. Es ergaben sich somit Versuchsgruppe I als PK mit dem
Lösungsmittel DMSO, als Wirkstoffkontrollen mit TGX-221 in unterschiedlichen
Dosierungen die Versuchsgruppen II (3x10-3 µM), III (3x10-2 µM), IV (3x10-1 µM)
und als NK Versuchsgruppe V.
a b c
d e f

3. Ergebnisse 61
3.5.2.1 TGX-221 im Gefrierschnittmodell mit Maushaut und EBA-Serum
immunisierter Kaninchen
Es konnte in allen Versuchsreihen (n = 9) eine DES erzeugt werden. Dabei zeigte
Versuchsgruppe I (PK) eine mediane Spaltbildung von 25 % (z0,25: 12,5 %,
z0,75: 30 %), Versuchsgruppe II von 65 % (z0,25: 1,3 %, z0,75: 90 %),
Versuchsgruppe III von 30 % (z0,25: 20 %, z0,75: 70 %) und Versuchsgruppe IV von
0 % (z0,25: 0 %, z0,75: 7,5 %). In Versuchsgruppe V (NK) konnte keine Spaltbildung
erzeugt werden (0 %). Beim Vergleich der Versuchsgruppen I bis IV zeigte sich in
der Varianzanalyse mithilfe des Kruskal-Wallis-Tests ein signifikanter Unterschied
mit P = 0,011. Der Vergleich der Versuchsgruppen II bis IV mit der
Versuchsgruppe I wurde mit Dunn’s Methode durchgeführt. Dabei ergab sich im
Vergleich von Versuchsgruppe I (PK) mit Versuchsgruppe IV (3x10-1 µM TGX-221)
ein signifikanter Unterschied mit P < 0,05. Der Vergleich von Versuchsgruppe I mit
den Versuchsgruppen II und III (3x10-3 µM, 3x10-2 µM) zeigte keinen signifikanten
Unterschied (siehe Abb.19 und 20).
Die Ergebnisse zeigen, dass TGX-221 in der höchsten verwendeten Konzentration
von 3x10-1 µM im Gefrierschnittmodell unter Verwendung von Seren EBA-
immunisierter Kaninchen und humaner neutrophiler Granulozyten einen Effekt auf
die DES hat.

3. Ergebnisse 62
Abbildung 19: Effekte von TGX-221 auf die Spaltbildung in Maushaut. Die Daten sind
dargestellt als Median (schwarze Linie), 25 %-Perzentil (z0,25, unterer Rand der Box),
75 %-Perzentil (z0,75, oberer Rand der Box). Die mediane dermo-epidermale Spaltbildung (DES)
der Positivkontrolle (PK/DMSO) lag bei 25 % (z0,25: 12,5 %, z0,75: 30 %). In der höchsten
verwendeten Konzentration von 3x10-1
µM TGX-221 wurde die DES signifikant gehemmt (*), der
Median lag bei 0 % (z0,25: 0%, z0,75: 7,5 %), P = 0,011.
Konzentration TGX-221( M)
0 (PK/DMSO) 3x10^-3 3x10^-2 3x10^-1
DE
S (
%)
0
20
40
60
80
100
0 (PK/DMSO) 3x10-3 3x10-2 03x10-1
*
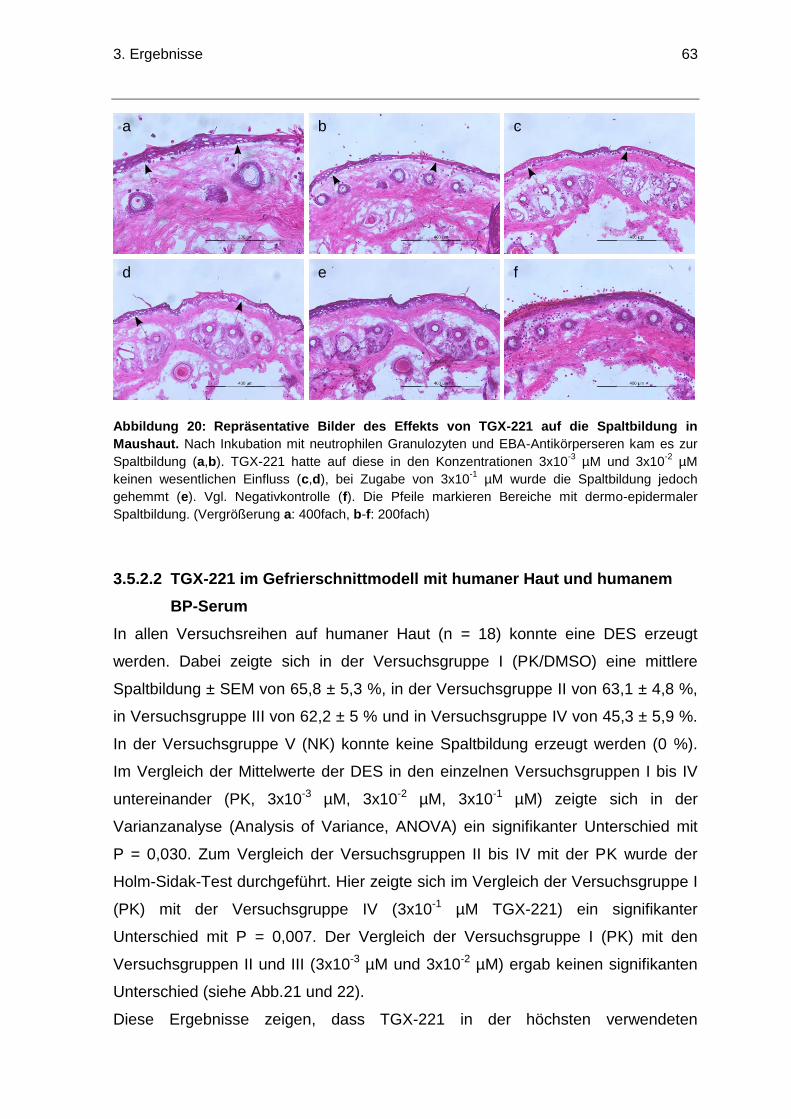
3. Ergebnisse 63
Abbildung 20: Repräsentative Bilder des Effekts von TGX-221 auf die Spaltbildung in
Maushaut. Nach Inkubation mit neutrophilen Granulozyten und EBA-Antikörperseren kam es zur
Spaltbildung (a,b). TGX-221 hatte auf diese in den Konzentrationen 3x10-3
µM und 3x10-2
µM
keinen wesentlichen Einfluss (c,d), bei Zugabe von 3x10-1
µM wurde die Spaltbildung jedoch
gehemmt (e). Vgl. Negativkontrolle (f). Die Pfeile markieren Bereiche mit dermo-epidermaler
Spaltbildung. (Vergrößerung a: 400fach, b-f: 200fach)
3.5.2.2 TGX-221 im Gefrierschnittmodell mit humaner Haut und humanem
BP-Serum
In allen Versuchsreihen auf humaner Haut (n = 18) konnte eine DES erzeugt
werden. Dabei zeigte sich in der Versuchsgruppe I (PK/DMSO) eine mittlere
Spaltbildung ± SEM von 65,8 ± 5,3 %, in der Versuchsgruppe II von 63,1 ± 4,8 %,
in Versuchsgruppe III von 62,2 ± 5 % und in Versuchsgruppe IV von 45,3 ± 5,9 %.
In der Versuchsgruppe V (NK) konnte keine Spaltbildung erzeugt werden (0 %).
Im Vergleich der Mittelwerte der DES in den einzelnen Versuchsgruppen I bis IV
untereinander (PK, 3x10-3 µM, 3x10-2 µM, 3x10-1 µM) zeigte sich in der
Varianzanalyse (Analysis of Variance, ANOVA) ein signifikanter Unterschied mit
P = 0,030. Zum Vergleich der Versuchsgruppen II bis IV mit der PK wurde der
Holm-Sidak-Test durchgeführt. Hier zeigte sich im Vergleich der Versuchsgruppe I
(PK) mit der Versuchsgruppe IV (3x10-1 µM TGX-221) ein signifikanter
Unterschied mit P = 0,007. Der Vergleich der Versuchsgruppe I (PK) mit den
Versuchsgruppen II und III (3x10-3 µM und 3x10-2 µM) ergab keinen signifikanten
Unterschied (siehe Abb.21 und 22).
Diese Ergebnisse zeigen, dass TGX-221 in der höchsten verwendeten
a b c
d e f
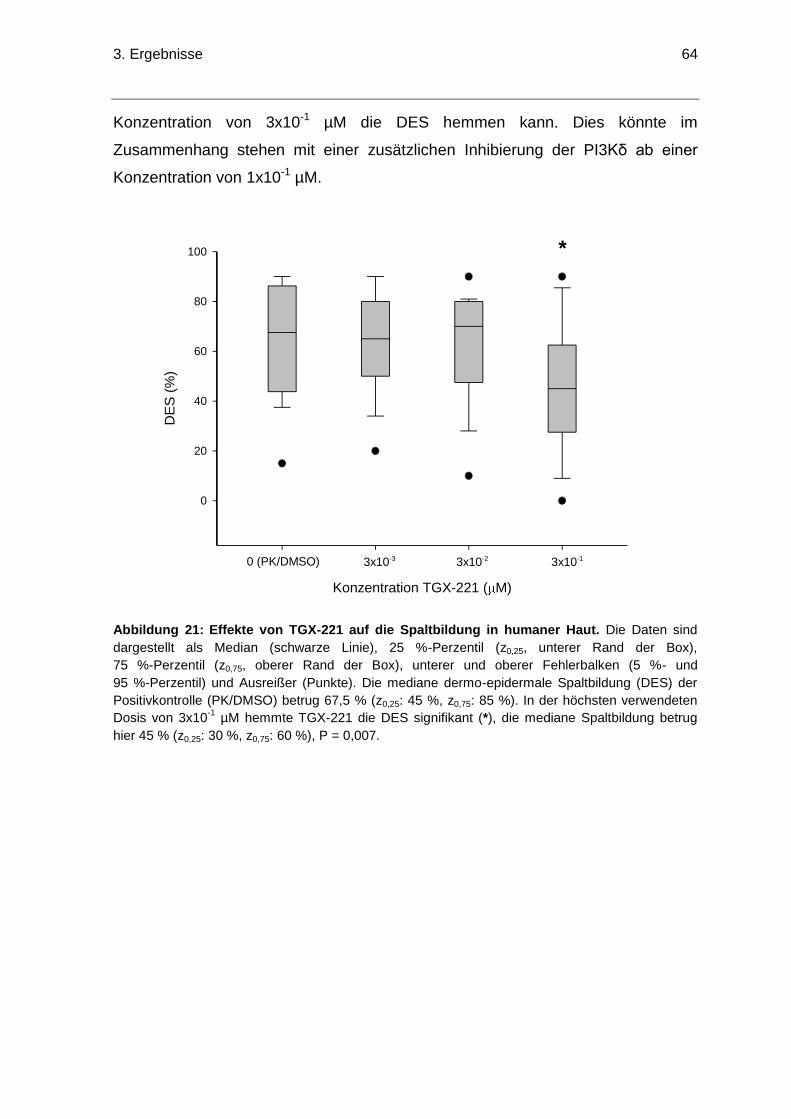
3. Ergebnisse 64
Konzentration von 3x10-1 µM die DES hemmen kann. Dies könnte im
Zusammenhang stehen mit einer zusätzlichen Inhibierung der PI3Kδ ab einer
Konzentration von 1x10-1 µM.
Abbildung 21: Effekte von TGX-221 auf die Spaltbildung in humaner Haut. Die Daten sind
dargestellt als Median (schwarze Linie), 25 %-Perzentil (z0,25, unterer Rand der Box),
75 %-Perzentil (z0,75, oberer Rand der Box), unterer und oberer Fehlerbalken (5 %- und
95 %-Perzentil) und Ausreißer (Punkte). Die mediane dermo-epidermale Spaltbildung (DES) der
Positivkontrolle (PK/DMSO) betrug 67,5 % (z0,25: 45 %, z0,75: 85 %). In der höchsten verwendeten
Dosis von 3x10-1
µM hemmte TGX-221 die DES signifikant (*), die mediane Spaltbildung betrug
hier 45 % (z0,25: 30 %, z0,75: 60 %), P = 0,007.
Konzentration TGX-221 ( M)
0 (PK/DMSO) 3x10^-3 3x10^-2 3x10^-1
DE
S (
%)
0
20
40
60
80
100
0 (PK/DMSO) 3x10-3 3x10-2 03x10-1
*

3. Ergebnisse 65
Abbildung 22: Repräsentative Bilder des Effekts von TGX-221 auf die Spaltbildung in
humaner Haut. Eine deutliche Spaltbildung zwischen Epidermis und Dermis war in der
Positivkontrolle (a,b) und bei den Wirkstoffkonzentrationen 3x10-3
und 3x10-2
µM erkennbar (c,d).
Dies konnte mit 3x10-1
µM TGX-221 gehemmt werden (e). Vgl. Negativkontrolle (f). Die Pfeile
markieren Bereiche mit dermo-epidermaler Spaltbildung. (Vergrößerung a: 400fach, b-f: 200fach)
3.6 Effekte des löslichen Fcγ – Rezeptors IIb SM101 auf die dermo-
epidermale Spaltbildung im Gefrierschnittmodell
Die Aktivierung neutrophiler Granulozyten wird vermittelt über die Bindung von
Immunkomplexen an einen zellgebundenen Fcγ-Rezeptor (Sitaru et al., 2002a;
Zhao et al., 2006). Dabei exprimieren humane neutrophile Granulozyten vor allem
den FcγRIIIb und den FcγRIIa (Li et al., 1996; Veri et al., 2007). Für den in dieser
Arbeit getesteten löslichen rekombinanten FcγRIIb konnte die Bindung von
pathogenem IgG und somit die kompetitive Hemmung der Reaktion mit
zellgebundenen Fcγ-Rezeptoren bereits in vitro unter Verwendung von murinem
IgG gezeigt werden (Magnusson et al., 2008). In dieser Arbeit wurde der FcγRIIb
(SM101) im Gefrierschnittmodell hinsichtlich seiner Effekte auf die DES
untersucht. Dabei wurde SM101 in den Konzentrationen 1x10-2 mg/ml,
1x10-1 mg/ml und 5x10-1 mg/ml verwendet. Diese wurden aus einer Stocklösung
von 20 mg/ml durch entsprechendes Verdünnen hergestellt. Somit ergaben sich
Versuchsgruppe I als PK, die Kontrollgruppen mit SM101 als Versuchsgruppen II
(1x10-2 mg/ml), III (1x10-1 mg/ml) und IV (5x10-1 mg/ml), sowie Versuchsgruppe V
als NK.
a b c
d e f

3. Ergebnisse 66
3.6.1 SM101 im Gefrierschnittmodell mit Maushaut und EBA-Serum
immunisierter Kaninchen
In allen Versuchsreihen (n = 18) konnte eine DES induziert werden. Dabei ergab
sich in Versuchsgruppe I (PK) eine mediane Spaltbildung von 92,5 % (z0,25: 90 %,
z0,75: 96,3 %), in Versuchsgruppe II von 95 % (z0,25: 83,8 %, z0,75: 96,3 %), in
Versuchsgruppe III von 92,5 % (z0,25: 90 %, z0,75: 98,8 %), in Versuchsgruppe IV
von 90 % (z0,25: 85 %, z0,75: 96,3 %) und in Versuchsgruppe V (NK) lag keine
Spaltbildung vor (0 %). Beim Vergleich der einzelnen Versuchsgruppen I bis IV
zeigte sich in der Varianzanalyse mithilfe des Kruskal-Wallis-Tests kein
signifikanter Unterschied (P = 0,892).
Im Gefrierschnittmodell mit EBA-Kaninchenserum konnte somit in keiner der
verwendeten Konzentrationen ein Effekt von SM101 auf die DES nachgewiesen
werden (siehe Abb.23 und 24).
Abbildung 23: Effekte von SM101 auf die Spaltbildung in Maushaut. Die Daten sind dargestellt
als Median (schwarze Linie), 25 %-Perzentil (z0,25, unterer Rand der Box), 75 %-Perzentil (z0,75,
oberer Rand der Box), unterer und oberer Fehlerbalken (5 %- und 95 %-Perzentil), Ausreißer
(Punkte). Die mediane dermo-epidermale Spaltbildung (DES) in der Positivkontrolle (PK) lag bei
92,5 % (z0,25: 90 %, z0,75: 96,3 %). Zwischen den Versuchsgruppen zeigte sich kein signifikanter
Unterschied, P = 0,892.
Konzentration SM101 (mg/ml)
0 (PK) 0,01 0,1 0,5
DE
S (
%)
40
50
60
70
80
90
100
00000 (PK) 1x10-2 1x10-1 05x10-1

3. Ergebnisse 67
Abbildung 24: Repräsentative Bilder des Effekts von SM101 auf die Spaltbildung in
Maushaut. Im Versuch mit Maushaut und EBA-Seren zeigte sich gegenüber der Positivkontrolle
(a,b) nach Zugabe von SM101 in den Konzentrationen 1x10-2
mg/ml (c), 1x10-1
mg/ml (d) und
5x10-1
mg/ml (e) keine Inhibierung der Spaltbildung. Vgl. Negativkontrolle (f). Die Pfeile markieren
Bereiche mit dermo-epidermaler Spaltbildung. (Vergrößerung a: 400fach, b-f: 200fach
3.6.2 SM101 im Gefrierschnittmodell mit humaner Haut und humanem BP-
Serum
In allen Versuchsreihen (n = 21) konnte eine DES erzeugt werden. Dabei zeigte
sich in Versuchsgruppe I (PK) eine mittlere Spaltbildung ± SEM von 58,6 ± 5,7 %,
in der Versuchsgruppe II von 39,5 ± 5,8 %, in Versuchsgruppe III von
38,1 ± 4,8 %, in Versuchsgruppe IV von 34,8 ± 4,8 % und in Versuchsgruppe V
(NK) keine Spaltbildung (0 %). Beim Vergleich der Mittelwerte der DES der
einzelnen Versuchsgruppen I bis IV (PK, 1x10-2 mg/ml, 1x10-1 mg/ml,
5x10-1 mg/ml SM101) ergab sich in der Varianzanalyse (Analysis of variance,
ANOVA) ein signifikanter Unterschied (P = 0,009). Zum Vergleich der
Versuchsgruppen II bis IV mit der Versuchsgruppe I (PK) wurde der Holm-Sidak-
Test angewendet. Dabei ergab sich zwischen Versuchsgruppe I (PK) und
Versuchsgruppe II (1x10-2 mg/ml SM101) ein signifikanter Unterschied mit
P = 0,013. Zwischen Versuchsgruppe I und Versuchsgruppe III (1x10-1 mg/ml
SM101) ergab sich ein signifikanter Unterschied mit P = 0,007 und zwischen
Versuchsgruppe I und Versuchsgruppe IV (5x10-1 mg/ml SM101) lag ein
signifikanter Unterschied mit P = 0,002 vor (siehe Abb.25 und 26).
a b c
d e f

3. Ergebnisse 68
Diese Ergebnisse zeigen, dass der lösliche FcγRIIb SM101 in allen verwendeten
Konzentrationen (1x10-2 – 5x10-1 mg/ml) die DES in-vitro hemmen kann.
Abbildung 25: Effekte von SM101 auf die Spaltbildung in humaner Haut. Die Daten sind
dargestellt als Mittelwert (schwarze Linie), 25 %-Perzentil (z0,25, unterer Rand der Box),
75 %-Perzentil (z0,75, oberer Rand der Box), untere und obere Fehlerbalken (5 %- und
95 %-Perzentil), Ausreißer (Punkte). Die mediane dermo-epidermale Spaltbildung (DES) der
Positivkontrolle (PK) lag bei 70 % (z0,25: 37,5 %, z0,75: 80 %). SM101 konnte in allen verwendeten
Konzentrationen die DES signifikant inhibieren (*). Abgebildet sind die mediane DES unter
Verwendung der niedrigsten Wirkstoffkonzentration (1x10-2
mg/ml) mit 35 % (z0,25: 20 %,
z0,75: 55 %), unter Anwendung von 1x10-1
mg/ml SM101 lag die mediane DES bei 40 %
(z0,25: 20 %, z0,75: 50 %) und in der höchsten verwendeten Konzentration (5x10-1
mg/ml) betrug die
mediane Spaltbildung 30 % (z0,25: 20 %, z0,75: 45 %). Zwischen den Versuchsgruppen bestand ein
signifikanter Unterschied mit P = 0,009.
Konzentration SM101 (mg/ml)
0 (PK) 0,01 0,1 0,5
DE
S (
%)
0
20
40
60
80
100
00000 (PK) 1x10-2 1x10-1 05x10-1
***

3. Ergebnisse 69
Abbildung 26: Repräsentative Bilder des Effekts von SM101 auf die Spaltbildung in humaner
Haut. Im Vergleich zur Spaltbildung in der Positivkontrolle (a,b), konnte in den Kontrollen unter
Zugabe des Wirkstoffs SM101 in allen Konzentrationen 1x10-2
mg/ml (c), 1x10-1
mg/ml (d) und
5x10-1
mg/ml (e) die Spaltbildung gehemmt werden. Vgl. Negativkontrolle (f). Die Pfeile markieren
Bereiche mit dermo-epidermaler Spaltbildung. (Vergrößerungen a: 400fach, b-f: 200fach)
a c
d e f
b

4. Diskussion 70
4. Diskussion
Das BP und die EBA zählen zu den bullösen Autoimmundermatosen, die mit
subepidermaler Blasenbildung einhergehen. Ausschlaggebend für die Entstehung
beider Erkrankungen sind Autoantikörper gegen Strukturproteine der Haut. Beim
BP handelt es sich dabei um Antikörper gegen Bestandteile der
Hemidesmosomen, BP180 und BP230 (Giudice et al., 1992; Stanley et al., 1981).
Die EBA zeichnet sich durch Autoantikörper gegen Typ-VII-Kollagen aus (Woodley
et al., 1988). Durch Bindung der Autoantikörper an die jeweiligen Strukturproteine
kommt es zur Ausbildung von Immunkomplexen und zur Generierung eines pro-
entzündlichen Milieus in der Haut. Dies führt zur Rekrutierung und nachfolgenden
Aktivierung neutrophiler Granulozyten und zur Freisetzung von proteolytischen
Enzyme und ROS, welche die DEJ schädigen. Histologisch ist dies erkennbar an
einer subepidermale Spaltbildung (Chiriac et al., 2007; Shimanovich et al., 2004).
Die Therapiemöglichkeiten des BP und der EBA sind momentan vor allem auf eine
generelle immunsuppressive Therapie begrenzt. Diese führt bei der EBA oft nicht
zur Remission, beim BP kommt es nach dem Absetzen der Therapie regelhaft zu
Rezidiven (Joly et al., 2009). Aus diesem Grund wurden in dieser Arbeit neue
mögliche Therapieansätze untersucht. Dies erfolgte im Gefrierschnittmodell,
mithilfe dessen eine hemmende Wirkung des PI3K-Inhibitors TGX-221 auf die
Spaltbildung im Modell mit BP- und EBA-Seren beobachtet wurde. Der lösliche
Fcγ-Rezeptor SM101 zeigte im Modell mit humanen BP-Seren eine Wirkung.
Zusätzlich wurde mithilfe des NBT-Gefrierschnittmodells und des ROS-Release
gezeigt, dass MP die Bildung von ROS hemmen kann.
4.1 Das Gefrierschnittmodell
Das Gefrierschnittmodell stellt ein Modell dar, das verschiedene Aspekte der
Pathogenese des BP und der EBA in einfacher und schneller Weise untersuchen
kann. Dabei können sowohl die Interaktionen zwischen Antigen und Antikörper,
als auch zwischen Immunkomplexen (Fc-Teil der Antikörper) und neutrophilen
Granulozyten (Fc-Rezeptor) betrachtet werden. Auch die Aktivierung neutrophiler
Granulozyten wird in diesem Modell abgebildet und kann beispielsweise unter

4. Diskussion 71
Einsatz von NBT sichtbar gemacht werden.
Das Gefrierschnittmodell wurde von Gammon et. al entwickelt und ermöglichte es,
erstmals ex vivo auf Gefrierschnitten humaner Haut nach Inkubation mit Seren von
Patienten mit BP und Zugabe humaner Leukozyten eine Spaltbildung zwischen
Epidermis und Dermis zu induzieren (Gammon et al., 1982b). Später konnte auch
mit Seren von Patienten mit EBA im Gefrierschnittmodell eine DES ausgelöst
werden (Sitaru et al., 2002a). Mithilfe des Gefrierschnittmodells konnten bisher so
unter anderem die Bedeutung der Elastase, der Gelatinase B und des
Fc-Fragments der Antikörper für die Spaltbildung nachgewiesen werden
(Shimanovich et al., 2004; Sitaru et al., 2002a). Außerdem wurde die wichtige
Rolle der NADPH-Oxidase und der Bildung von ROS bezüglich der DES mithilfe
des Gefrierschnittmodells beschrieben (Chiriac et al., 2007). Dabei wird die
NADPH-Oxidase unter anderem PI3-Kinase-vermittelt reguliert (Vanhaesebroeck
et al., 2001). Für die Pathogenese des BP konnte außerdem die Bedeutung des
FcγRIIa und FcγRIIIb im Gefrierschnittmodell gezeigt werden (Yu et al., 2010).
In dieser Arbeit wurden mithilfe des Gefrierschnittmodells zwei PI3-Kinase-
Inhibitoren und ein löslicher Fcγ-Rezeptor hinsichtlich einer möglichen
inhibitorischen Wirkung auf die Spaltbildung untersucht. Dies erfolgte unter
Einsatz von humanen peripheren Leukozyten gesunder Spender, humanen
Hautproben und Seren von Patienten mit BP. Dies ermöglichte
Untersuchungsbedingungen, die den Prozessen im menschlichen Organismus
sehr ähnlich sind. Ein weiterer Versuchsaufbau mit humanen Leukozyten, Proben
von Maushaut und Seren von Kaninchen, die gegen EBA immunisiert wurden,
ermöglichte auch für diese Erkrankung, die in der Klinik weitaus seltener als das
BP vorkommt, ein funktionierendes Modell zur Untersuchung der pathogenen
Prozesse. Außerdem können mithilfe des Gefrierschnittmodells durch den
einfachen Versuchsaufbau und die schnelle Durchführbarkeit einzelne Versuche
gut reproduziert werden. Im Vergleich zu klinischen Studien bietet das
Gefrierschnittmodell die Möglichkeit, Wirkstoffe zu testen, deren mögliche toxische
Wirkungen noch unklar sind.
Allerdings können einige Aspekte der Pathogenese des BP und der EBA, wie die
Migration neutrophiler Granulozyten oder der Einfluss anderer Entzündungszellen,
im Gefrierschnittmodell nicht ausreichend abgebildet werden.
Insgesamt stellt das Gefrierschnittmodell eine einfache und effektive Methode dar,

4. Diskussion 72
mit der einzelne pathogenetische Prozesse beleuchtet und mögliche neue
Wirkstoffe, die in diese Prozesse eingreifen, primär getestet werden können. So
konnte in dieser Arbeit ein Effekt des PI3-Kinase-Inhibitors TGX-221 und des
löslichen Fcγ-Rezeptors SM101 gezeigt werden.
4.1.1 Antikörper
Mit den in dieser Arbeit verwendeten BP- und EBA-Seren konnte in den
Versuchen eine DES erzeugt werden. Unter Verwendung von Seren gesunder
Spender (negative Kontrollseren) war hingegen keine Spaltbildung induzierbar.
Damit konnte bestätigt werden, dass die in den Seren enthaltenen Autoantikörper
entscheidend für die Spaltbildung sind, dabei spielt vor allem das Fc-Fragment
eine Rolle (Sitaru et al., 2002a).
4.1.2 Komplement
Die Bedeutung von Komplement in vivo konnte im Mausmodell mit C5-defizienten
Mäusen gezeigt werden, die nach Transfer von Antikörpern gegen BP180 keine
Blasenbildung zeigten (Liu et al., 1995). Im Gefrierschnittmodell kann die Rolle
von Komplement jedoch nicht untersucht werden, da die Leukozyten direkt auf die
Hautschnitte gegeben wurden und sich somit in unmittelbarer Nähe zur DEJ
befanden. Im Gegensatz zur Situation in vivo entfällt damit die Rekrutierung der
Leukozyten entlang eines chemotaktischen Gradienten aus den Blutgefäßen in
das Gewebe und an die DEJ, was unter anderem über aktiviertes Komplement
gesteuert wird (Sitaru et al., 2002a).
4.1.3 Neutrophile Granulozyten
Neutrophile Granulozyten wurden aus Vollblut gesunder Spender isoliert. Durch
die Verwendung von Blutproben unterschiedlicher Versuchspersonen wurden bei
konstanten Versuchsbedingungen unterschiedliche Zellzahlen ermittelt, welche
durch Verdünnen mit RPMI jedoch an einen Zielwert angeglichen wurden. Bei
konstanter Zellzahl konnte unter Verwendung der Zellen der einzelnen
Blutspender im Versuch trotzdem ein Unterschied in der Spaltbildung beobachtet

4. Diskussion 73
werden. Dies könnte auf mögliche genetische Schwankungen wie
Polymorphismen der Fcγ-Rezeptoren der neutrophilen Granulozyten
zurückgeführt werden.
Durch die direkte Zugabe der Antikörper werden im Gefrierschnittmodell
außerdem die Schritte, die zur Antikörperbildung führen, nicht betrachtet. Dabei
handelt es sich um den Toleranzverlust gegen die Strukturproteine, die T- und
B-Zell-Aktivierung und die Antikörperproduktion in der Plasmazelle (Bieber et al.,
2010). Auch die Rolle anderer Entzündungszellen kann nicht ausreichend
betrachtet werden.
4.1.4 Gefrierschnitte
Bei der Herstellung von Gefrierschnitten wurden Hautproben auf Objektträgern
fixiert. Durch die Verbindung der Basallamina mit elastischen Fasern der Dermis
besteht weiterhin eine natürliche Spannung in der DEJ. Die Spaltbildung auf den
Gefrierschnitten kommt dadurch zustande, dass die von den neutrophilen
Granulozyten freigesetzten Proteasen den Verankerungskomplex auflösen und so
zur Retraktion der Dermis von der Epidermis führen. Zu unterscheiden war diese
von der Schädigung der Hautschnitte bei der Herstellung oder während der
einzelnen Versuchsschritte und der daraus resultierenden artifiziellen
Spaltbildung.
4.1.5 Vergleich der Gefrierschnittmodelle auf humaner Haut und Maushaut
Die Testung der einzelnen Wirkstoffe erfolgte in zwei verschiedenen
Versuchsmodellen: unter Verwendung humaner Leukozyten, humaner Haut und
humanen BP-Seren und unter Verwendung humaner Leukozyten, Maushaut und
EBA-Kaninchenseren. Dabei war auf Maushaut in der Positivkontrolle eine
signifikant geringere Spaltbildung auslösbar (P ≤ 0,001). Dies könnte durch eine
geringere Bindungsaffinität der Kaninchenantikörper zu den humanen neutrophilen
Granulozyten aufgrund struktureller Unterschiede erklärt werden (Nimmerjahn and
Ravetch, 2008).

4. Diskussion 74
4.2 Effekte von Methylprednisolon auf die Bildung von ROS
4.2.1 Allgemeines zu Methylprednisolon
Glukokortikoide sind aufgrund ihrer ausgeprägten entzündungshemmenden und
immunmodulatorischen Wirkung Therapie der ersten Wahl beim BP und der EBA
(Goebeler et al., 2004; Joly et al., 2002). Der genaue Wirkmechanismus der
Kortikosteroide ist jedoch noch nicht vollständig geklärt. Es wurden verschiedene
Effekte beschrieben, die in genomische und nicht-genomische Wirkungen
eingeteilt werden können (Buttgereit et al., 1998). Dabei scheinen in niedrigerer
Dosierung die genomischen Wirkungen, bei steigender Dosis die nicht-
genomischen Wirkungen zu überwiegen (Buttgereit et al., 1998).
Die genomischen Effekte werden über einen zytosolischen Glukokortikoid-
Rezeptor (cGCR) vermittelt. Dieser liegt in zwei Isoformen vor, einer α-Isoform, die
das Glukokortikoidmolekül bindet und einer β-Isoform, der vor allem regulierende
Eigenschaften zugeschrieben werden (Bamberger et al., 1995). Der inaktive
cGCR liegt im Zytoplasma im Komplex mit Hitzeschockproteinen (HSP) und
anderen Chaperonen vor. Nach Bindung des Glukokortikoidmoleküls an den
cGCR kommt es unter anderem zur Abspaltung von HSP90 und damit zur
Aktivierung des Komplexes. Dieser wird daraufhin in den Zellkern transloziert, wo
der Komplex an die Glukokortikoid-responsiven Elemente (GRE) der DNA bindet
(Buttgereit et al., 2005). Dies führt zur Transkription von Genen und damit zur
Synthese verschiedener Proteine, wie Annexine, Endonukleasen, neutrale
Endopeptidase, Angiotensin converting enzyme oder von Lipocortin 1, das die
Phospholipase A2 hemmt und damit antiinflammatorisch wirkt (Buttgereit et al.,
1998). Die Wirkung der Glukokortikoide durch Induktion der Proteinsynthese wird
als Transaktivierung bezeichnet und wird für viele der unerwünschten Wirkungen
verantwortlich gemacht (Buttgereit et al., 2005). Glukokortikoide können die
Synthese bestimmter Proteinen außerdem hemmen, was als Transrepression
bezeichnet wird. Diese erfolgt durch Bindung des Kortikosteroid-Rezeptor-
Komplexes an Transkriptionsfaktoren wie das Aktivator-Protein-1 (AP-1) oder NF-
κB, wodurch die Synthese von Zytokinen wie TNF-α, IL-2, IL-6 und COX-2
gehemmt wird (Buttgereit et al., 1998). Die genomischen Wirkungen der
Glukokortikoide sind charakterisiert durch eine Latenz von 30 Minuten bis zu

4. Diskussion 75
mehreren Stunden bis zum Wirkeintritt (Buttgereit et al., 1998).
Zusätzlich zu den genomischen Effekten wurden nicht-genomische Wirkungen
beschrieben, die vor allem ab einer höheren Dosierung und innerhalb von
Sekunden bis wenigen Minuten auftreten. Diese Wirkungen werden
wahrscheinlich über einen membrangebundenen Glukokortikoidrezeptor (mGCR)
oder unspezifisch vermittelt (Buttgereit et al., 1998). Die unspezifischen Wirkungen
beruhen auf den lipophilen Eigenschaften der Glukokortikoide, was deren
Einlagerung in Membranen ermöglicht. Möglichweise wird dadurch der Transport
von Kationen, vor allem von Ca2+ und Na2+, über die Plasmamembran beeinflusst
(Buttgereit et al., 1997; Buttgereit and Scheffold, 2002). Es wird davon
ausgegangen, dass hohe intrazelluläre Ca2+-Konzentrationen für die
Degranulation neutrophiler Granulozyten von Bedeutung sind (Sengelov et al.,
1993). Mit hohen Dosen von Methylprednisolon (10-3 - 10-1 mM) konnte so in
neutrophilen Granulozyten eine Degranulation und damit die Freisetzung
lysosomaler Enzyme gehemmt werden (Liu et al., 2005). Außerdem scheint der
Protonenfluss über die Mitochondrienmembran und die mitochondriale
Atmungskette durch Glukokortikoide beeinflusst zu werden, was die Bildung von
ATP, das als Energiequelle von essentieller Bedeutung für die Zellfunktion ist,
beeinträchtigt (Buttgereit et al., 1994; Buttgereit and Scheffold, 2002). Die über
einen mGCR vermittelten Wirkungen wurden für neutrophile Granulozyten noch
nicht gezeigt, allerdings konnte in Monozyten und B-Lymphozyten ein
membranständiger Rezeptor für Glukokortikoide nachgewiesen werden
(Bartholome et al., 2004). Durch Bindung von Kortikosteroiden an den mGCR
wurde die Aktivierung verschiedener second messenger-Systeme durch Ca2+, IP3,
cAMP oder die Proteinkinase C (PKC) beschrieben (Buttgereit and Scheffold,
2002; Buttgereit et al., 1998).
Weiterhin wurde für Glukokortikoide eine hemmende Wirkung auf die extrazellulär
regulierten Kinasen (ERK)-1 und -2, die zu den Mitogen-aktivierten Proteinkinasen
(MAPK) gehören, nachgewiesen. Dabei konnte in Mastzellen gezeigt werden,
dass Glukokortikoide die Expression des MAP-Kinase-Phosphatase-1 (MKP1)-
Gens induzieren und dessen proteasomalen Abbau verhindern. Die MKP-1 ist ein
Enzym, das die ERK-1 und -2 dephosphoryliert und dadurch inaktiviert (Kassel et
al., 2001). ERK sind über die Regulation der p47-Untereinheit der NADPH-
Oxidase an der Bildung von ROS beteiligt (Dewas et al., 2000).

4. Diskussion 76
Die Bedeutung von ROS für die Pathogenese blasenbildender
Autoimmundermatosen wurde bereits in einer Studie mit Mäusen, die gegen EBA
immunisiert wurden, beschrieben. Diese Mäuse wiesen eine nicht funktionsfähige
NADPH-Oxidase auf und zeigten keine typische Blasenbildung. Entsprechend
konnte ex vivo im Gefrierschnittmodell unter pharmakologischer Hemmung der
NADPH-Oxidase keine DES ausgelöst werden (Chiriac et al., 2007).
4.2.2 Methylprednisolon im NBT-Gefrierschnittmodell und im ROS-Release
In dieser Arbeit wurde das synthetische Glukokortikoid Methylprednisolon (MP)
hinsichtlich seiner Effekte auf die Bildung von ROS in einem Gefrierschnittmodell
mit NBT und im ROS-Release unter Einsatz von Antikörperseren von Patienten
mit BP getestet. Dabei konnte zunächst bestätigt werden, dass ROS bei der durch
BP-Antikörper induzierten Aktivierung von neutrophilen Granulozyten gebildet
werden. Weiterhin war eine dosisabhängige Hemmung der ROS-Bildung durch
MP zu beobachten. Im Gefrierschnittmodell trat so eine Wirkung in der höchsten
Dosierung von 1 mM ein. Im ROS-Release wirkte MP in den Konzentrationen
3x10-1 mM, 1 mM und 3 mM.
Der Wirkmechanismus, der hierbei eine Rolle spielte kann allerdings nicht genau
festgelegt werden. Die beschriebene Latenz von mehreren Stunden bis zum
Wirkeintritt bei der genomischen Wirkung im Vergleich zu Sekunden bis Minuten
bei nicht-genomischen Wirkungen (Buttgereit et al., 1998) gibt leider keinen
Aufschluss über den Wirkmechanismus, da sowohl im Gefrierschnittmodell als
auch im ROS-Release MP zusammen mit den isolierten Leukozyten für 2 bis 3
Stunden inkubiert und erst nachfolgend ausgewertet wurde und damit über den
zeitlichen Eintritt der Wirkung keine Aussage getroffen werden kann.
Für MP wurde allerdings bereits eine unspezifische nicht-genomische Wirkung ab
einer Konzentration von 10-3 - 10-1 mM beschrieben (Buttgereit and Scheffold,
2002). In dieser Arbeit zeigte MP ab einer Konzentration von 3x10-1 mM eine
Wirkung. Dies würde für einen unspezifischen Effekt bei höherer Dosierung
sprechen. Dieser nicht-genomische Effekt von hochdosiertem MP wurde bereits
für die Degranulation von humanen neutrophilen Granulozyten beschrieben (Liu et
al., 2005). Eine schnelle, nicht-genomische Wirkung über mGCR kann ebenfalls
nicht ausgeschlossen werden, auch wenn der mGCR bisher nur in Monozyten und

4. Diskussion 77
Lymphozyten nachgewiesen werden konnte (Bartholome et al., 2004). Durch die
beschriebenen Effekte von MP auf die Expression der MKP-1 und damit der
Inaktivierung von ERK mit Hemmung der NADPH-Oxidase (Dewas et al., 2000),
sind auch genomische Wirkungen denkbar. Der hemmende Effekt von MP speziell
auf die ROS-Produktion könnte somit von Bedeutung für die beobachteten
Wirkungen im NBT-Gefrierschnittmodell und im ROS-Release gewesen sein.
Es konnte so mithilfe des NBT-Gefrierschnittmodells und des ROS-Release belegt
werden, dass ROS bei der Aktivierung von neutrophilen Granulozyten durch BP-
Antikörper gebildet werden. Weiterhin konnte eine hemmende Wirkung von MP
auf die ROS-Produktion gezeigt werden, womit auch die Wirksamkeit von MP
beim BP ex vivo erneut bestätigt wurde.
Aufgrund der unerwünschten klinischen Wirkungen von MP, die auch durch den
nicht vollständig geklärten Wirkmechanismus bedingt sind und vor allem durch
hochdosierte und über einen längeren Zeitraum eingesetzte Glukokortikoide
verursacht werden, wird jedoch eine Optimierung der Therapie des BP und der
EBA angestrebt. Dies könnte unter anderem über selektiv wirkende
Glukokortikoide (Buttgereit et al., 2005) oder durch eine Dosisreduktion mithilfe
einer Kombinationstherapie erreicht werden. In dieser Arbeit wurden deshalb
mögliche neue Wirkstoffe zur Therapie untersucht, die im Folgenden diskutiert
werden.
4.3 Effekte von PI3-Kinase-Inhibitoren auf die dermo-epidermale
Spaltbildung
4.3.1 Allgemeines zu PI3-Kinasen und deren Funktion
Phosphatidylinositid-3-Kinasen (PI3-Kinasen, PI3K) sind Lipidkinasen, welche
Phosphatidylinositol, einen Bestandteil der zellulären Membran, an der 3-OH-
Position des Inositolringes phosphorylieren (Rameh and Cantley, 1999). Die
phosphorylierten Moleküle, auch Phosphoinositide genannt, dienen als second
messenger und sind an zahlreichen intrazellulären Signalwegen beteiligt (Fruman
et al., 1998; Vanhaesebroeck et al., 2001).
In neutrophilen Granulozyten finden sich alle bisher bekannten Unterformen der

4. Diskussion 78
PI3K, die in Klasse I, II und III eingeteilt werden können. Die PI3K der Klasse I
lassen sich weiter unterteilen in Klasse IA und Klasse IB. Zur Klasse IA gehören
die PI3Kα, PI3Kβ und die PI3Kδ, welche nach ihren 110 kDa schweren
katalytischen Untereinheiten p110α, p110β und p110δ benannt sind. Die
katalytischen Untereinheiten bilden Heterodimere mit den regulatorischen
Untereinheiten p85 oder p55, die jeweils zwei Src-Homologie-2 (SH2)-Domänen
besitzen. Zusammen mit anderen proteinbindenden Domänen innerhalb der
regulatorischen und katalytischen Untereinheit interagiert die SH2-Domäne mit
membranassoziierten Signalkomplexen. Klasse IA Kinasen werden typischerweise
Tyrosinkinase-gekoppelt durch Zytokine oder über Fc-Rezeptoren aktiviert
(Fruman and Cantley, 2002; Wymann et al., 2000). Zu den Klasse-IB-Kinasen
gehört die PI3Kγ, welche sich aus einer katalytischen p110γ-Einheit und einer
regulierenden p101-Einheit zusammensetzt und über G-Protein-gekoppelte
Rezeptoren, an die zum Beispiel Komplement oder Chemokine binden, aktiviert
wird (Wymann et al., 2000). Die PI3K der Klasse I sind verantwortlich für die
Bildung von Phosphatidylinositol (3,4,5)-trisphosphat (PIP3) und
Phosphatidylinositol (3,4)-bisphosphat (PI(3,4)P2). Beiden Phosphoinositiden wird
unter anderem eine Bedeutung für die Regulation der NADPH-Oxidase und der
Produktion von ROS, der Chemotaxis und der Degranulation neutrophiler
Granulozyten zugeschrieben (Fruman and Cantley, 2002; Rommel et al., 2007;
Vanhaesebroeck et al., 2001). Beide Phosphoinositide können dabei an
Pleckstrin-Homologie (PH)-Domänen binden. Für die Produktion von ROS binden
PIP3 und PI(3,4)P2 an die PH-Domänen verschiedener Proteinkinasen, wie der
PDK1 (3-Phosphoinositid-dependent proteinkinase), der Akt-Kinase/Proteinkinase
B, der ERK-Kinase und einigen Isoformen der Proteinkinase C (PKC) und
regulieren diese (Chen et al., 2003; Parekh et al., 2000). Diese Kinasen
phosphorylieren die p47phox-Komponente der NADPH-Oxidase, wodurch diese
aktiviert wird (Chen et al., 2003). PIP3 und PI(3,4)P2 können auch direkt an
p47phox binden (Kanai et al., 2001). Des Weiteren reguliert PIP3 die Aktivität von
P-REX1, einem Guanin-Nukleotid-Austauschfaktor, der die GTPase RAC aktiviert,
welche dann die NADPH-Oxidase reguliert (Welch et al., 2002). Die PDK1, Akt
und RAC scheinen durch Regulation der Polymerisierung von Aktin und der damit
verbundenen Zellpolarisierung auch die Chemotaxis zu vermitteln (Fruman and
Cantley, 2002; Wymann et al., 2000). Die Sekretion proteolytischer Enzyme erfolgt

4. Diskussion 79
über die Aktivierung kleiner GTPasen (Wymann et al., 2000).
In aktivierten neutrophilen Granulozyten konnte ein vorübergehender Anstieg von
PIP3 und PI(3,4)P2 beobachtet werden (Traynor-Kaplan et al., 1989). Dabei konnte
in neutrophilen Granulozyten die PI3Kγ durch Chemokine wie IL-8, durch C5a
oder durch das bakterielle Peptid Formyl-Methionyl-Leucyl-Phenylalanin (fMLP)
aktiviert werden (Hirsch et al., 2000; Li et al., 2000; Sasaki et al., 2000). In durch
fMLP aktivierten humanen neutrophilen Granulozyten konnte für diese Isoform
außerdem eine Bedeutung für die Produktion von ROS nachgewiesen werden
(Condliffe et al., 2005). Des Weiteren scheint die PI3Kγ eine Rolle hinsichtlich der
Chemotaxis von neutrophilen Granulozyten zu haben (Hirsch et al., 2000; Sasaki
et al., 2000).
Auch die PI3K der Klasse IA scheinen an der Bildung von ROS in neutrophilen
Granulozyten beteiligt zu sein. Dies konnte für die PI3Kδ in humanen neutrophilen
Granulozyten nachgewiesen werden (Condliffe et al., 2005). Weiterhin spielt auch
die PI3Kδ eine Rolle in der Chemotaxis neutrophiler Granulozyten (Puri et al.,
2004). Durch Inhibierung der PI3Kδ konnte außerdem die Freisetzung
proteolytischer Enzyme aus neutrophilen Granulozyten gehemmt werden (Sadhu
et al., 2003a).
In der Fcγ-Rezeptor-abhängigen Aktivierung neutrophiler Granulozyten, die auch
für die Pathogenese bullöser Autoimmundermatosen eine grundlegende Rolle
spielt, konnte außerdem die Bedeutung der PI3Kβ für die Bildung von ROS
gezeigt werden (Kulkarni et al., 2011). Dabei scheint die Aktivierung der PI3Kβ
zusätzlich über einen G-Protein-gekoppelten Rezeptor (BLT1) zu erfolgen
(Kulkarni et al., 2011). In einem Mausmodell mit EBA-immunisierten Mäusen, die
keine PI3Kβ aufwiesen, konnte außerdem eine verringerte Blasenbildung
nachgewiesen werden (Kulkarni et al., 2011).
Auch die pathogene Bedeutung von ROS für die Blasenbildung konnte bereits in
vivo mit Mäusen ohne funktionierende NADPH-Oxidase gezeigt werden, indem
durch Injektion von Antikörpern gegen Typ-VII-Kollagen keine Blasenbildung
induziert werden konnte (Chiriac et al., 2007). Auch ex vivo mithilfe des
Gefrierschnittmodells konnte bereits durch pharmakologische Inhibierung der
NADPH-Oxidase eine DES unterdrückt werden (Chiriac et al., 2007).
Ausgehend von den Kenntnissen, dass PI3-Kinasen vor allem über die
Regulierung der NADPH-Oxidase eine Rolle in der immunkomplexvermittelten

4. Diskussion 80
Gewebsschädigung spielen, könnte die Inhibierung der PI3K einen Ansatzpunkt
zur Intervention in die Pathogenese des BP und der EBA bieten.
4.3.1.1 PI-103
PI-103 gehört zur Gruppe der Pyridofuropyrimidine und ist ein potenter ATP-
kompetitiver unselektiver Inhibitor, der vor allem die PI3K der Klasse IA PI3Kα,
PI3Kβ und PI3Kδ mit einer mittleren inhibitorischen Konzentration (IC50) von
2x10-3 µM inhibiert. In höherer Konzentration wird auch die PI3Kγ mit einer IC50
von 15x10-3 µM sowie mTOR und die DNA-PK gehemmt (Raynaud et al., 2007;
Workman et al., 2010). In den vorliegenden Versuchen wurde PI-103 in
unterschiedlichen Dosierungen eingesetzt, sodass in der geringsten Konzentration
von 1x10-2 µM lediglich die PI3-Kinasen der Klasse IA und in allen anderen
verwendeten Konzentrationen (1x10-1 µM und 1 µM) zusätzlich die PI3Kγ, mTOR
und die DNA-PK gehemmt wurden. Dabei konnte in dieser Arbeit sowohl im
Versuch mit EBA-Kaninchenseren als auch mit humanen BP-Seren die DES durch
keine der eingesetzten Wirkstoffkonzentrationen gehemmt werden.
In neutrophilen Granulozyten kommen alle Klassen der PI3K vor, wobei allen
Isoformen der PI3K Klasse I eine Beteiligung an der Bildung von PIP3 und
PI(3,4)P2 zugeschrieben wird (Condliffe et al., 2005). In humanen neutrophilen
Granulozyten, die mit fMLP und TNF-α aktiviert wurden, wurde vor allem die PI3Kγ
für die Aktivierung der NADPH-Oxidase verantwortlich gemacht (Condliffe et al.,
2005). In der Pathogenese der EBA und des BP spielt jedoch vor allem die
Aktivierung neutrophiler Granulozyten durch Immunkomplexe eine Rolle. Es
scheinen hier also besonders die PI3K der Klasse IA von Bedeutung zu sein, da
diese Tyrosinkinase-vermittelt über den Fc-Teil der Antikörper aktiviert werden
können (Fruman and Cantley, 2002; Wymann et al., 2000). So konnte
beispielsweise für die PI3Kδ eine Bedeutung für die Bildung von ROS in humanen
neutrophilen Granulozyten nachgewiesen werden (Condliffe et al., 2005). In
humanen neutrophilen Granulozyten, die über Immunkomplexe aktiviert wurden,
konnte außerdem gezeigt werden, dass PI3Kβ für die Bildung von ROS eine
entscheidende Rolle spielt. Auch hier konnte bei höheren
Immunkomplexkonzentrationen eine zusätzliche Aktivierung der PI3Kδ beobachtet
werden (Kulkarni et al., 2011). PI-103 hemmt zusätzlich die PI3Kα. Dieser Isoform
wird zusammen mit der PI3Kβ eine nichtredundante essentielle Rolle für

4. Diskussion 81
verschiedene zelluläre Prozesse zugeschrieben. Im Mausmodell mit p110α- und
p110β-defizienten Mäusen wurde so eine erhöhte Letalität beobachtet (Bi et al.,
2002; Bi et al., 1999).
Im Mausmodell wurde weiterhin gezeigt, dass durch Inhibierung der PI3Kδ die
Freisetzung von Elastase aus neutrophilen Granulozyten gehemmt wird (Sadhu et
al., 2003a). Die PI3Kδ wird durch PI-103 schon in geringen Konzentrationen
gehemmt. Da jedoch neben der Elastase auch der Gelatinase B eine Bedeutung
für die DES zukommt, könnte diese im Versuch trotz des Einsatzes von PI-103 zu
einer Spaltbildung geführt haben (Shimanovich et al., 2004).
Außerdem spielen die PI3Kγ und die PI3Kδ eine Rolle bei der Chemotaxis
neutrophiler Granulozyten. Im Mausmodell mit PI3Kγ- und PI3Kδ-defizienten
Mäusen, sowie unter pharmakologischer selektiver Hemmung der PI3Kδ wurde
eine Störung der Migration der neutrophilen Granulozyten beschrieben (Hirsch et
al., 2000; Puri et al., 2004; Puri et al., 2005; Sasaki et al., 2000). Dabei konnte
auch ein Vorkommen der PI3Kγ und der PI3Kδ in vaskulären Endothelzellen
nachgewiesen werden (Puri et al., 2004; Puri et al., 2005). Dieser Ansatzpunkt
kann jedoch im Gefrierschnittmodell nicht berücksichtigt werden, da die isolierten
Leukozyten direkt auf die Hautschnitte gegeben wurden und sich so in
unmittelbarer Nähe zur DEJ befanden. Für die Situation in vivo könnte dies aber
von weiterer Bedeutung für die Inhibierung der Blasenbildung sein.
Durch die unspezifische Hemmung aller Klasse-IA-Kinasen durch PI-103, sowie in
höheren Dosierungen auch der PI3Kγ, mTOR und der DNA-PK, könnten
unerwünschte Wechselwirkungen, wie zum Beispiel die Interaktion mit anderen
Signaltransduktionswegen oder untereinander, zu einer mangelnden Inhibierung
der NADPH-Oxidase geführt haben. Durch Hemmung aller Isoformen wäre auch
das Auftreten toxischer Wirkungen denkbar.
Die Effekte von PI-103 könnten somit durch die mangelnde Selektivität des
Inhibitors limitiert sein, wodurch eine Inhibierung der Spaltbildung im
Gefrierschnittmodell nicht möglich war. Die vielfältigen Einflüsse der PI3-Kinasen
sind zudem noch nicht vollständig geklärt. Die bisherigen Erkenntnisse
zusammenfassend würde sich daher für den therapeutischen Einsatz bei BP und
EBA möglicherweise ein selektiver Inhibitor der PI3Kβ und der PI3Kδ besser
eignen.

4. Diskussion 82
4.3.1.2 TGX-221
TGX-221 ist ein selektiver Inhibitor der PI3Kβ (IC50 = 7x10-3 µM). In höheren
Konzentrationen werden auch die PI3Kδ (IC50 = 1x10-1 µM) und in sehr hohen
Konzentrationen (IC50 = 3,5 - 5 µM) zusätzlich die PI3Kα und die PI3Kγ gehemmt
(Jackson et al., 2005). Die in dieser Arbeit verwendeten Dosierungen zielten auf
eine Hemmung der PI3Kβ ab, in der höchsten verwendeten Dosierung von 3x10-1
µM wurde auch die PI3Kδ gehemmt. Dabei war im Gefrierschnittmodell sowohl
unter Verwendung humaner BP-Seren als auch mit EBA-Kaninchenseren in dieser
höchsten Dosierung von TGX-221 eine Inhibierung der Spaltbildung zu
beobachten.
Die Rolle der einzelnen PI3K-Isoformen für die ROS-Produktion konnte in
humanen neutrophilen Granulozyten gezeigt werden, die mit TNF-α und fMLP
aktiviert wurden. Eine vorrangige Rolle für die Regulierung der NADPH-Oxidase
wurde hier der PI3Kγ und der PI3Kδ zugeschrieben. Für die beiden weiteren
Isoformen der Klasse IA, PI3Kα und PI3Kβ, wird jedoch nur von einer
untergeordneten Rolle ausgegangen (Condliffe et al., 2005).
Die Bedeutung der PI3Kδ für die ROS-Bildung konnte weiterhin gezeigt werden
durch selektive Hemmung dieser Isoform. Dabei wurde die Aktivität der Akt-Kinase
und der PDK1 in Endothelzellen gehemmt (Puri et al., 2004). Diese Proteinkinasen
spielen eine Rolle in der Aktivierung der NADPH-Oxidase und werden durch die
von PI3K gebildeten Phosphoinositide PI(3,4)P2 und PIP3 reguliert (Chen et al.,
2003; Parekh et al., 2000). Auch in neutrophilen Granulozyten konnte durch
selektive Hemmung der PI3Kδ die Bildung von ROS inhibiert werden (Sadhu et
al., 2003a). Neben den Wirkungen der PI3K auf die Bildung von ROS, konnte bei
neutrophilen Granulozyten außerdem durch selektive Hemmung der PI3Kδ die
Freisetzung der Elastase inhibiert werden (Sadhu et al., 2003a). Die Elastase ist
zusammen mit der Gelatinase B wesentlich an der Zerstörung von
Strukturproteinen und somit an der DES beteiligt (Shimanovich et al., 2004).
Neben der Inhibierung der ROS-Bildung kann dies zusätzlich zu einer
verminderten DES geführt haben. Des Weiteren spielt die PI3Kδ eine wichtige
Rolle in der Chemotaxis neutrophiler Granulozyten. So konnte im Mausmodell mit
PI3Kδ-defizienten Mäusen und unter pharmakologischer selektiver Hemmung der
PI3Kδ eine beeinträchtigte Migration von neutrophilen Granulozyten beobachtet
werden (Puri et al., 2004; Sadhu et al., 2003b). Außerdem wurde gezeigt, dass die

4. Diskussion 83
PI3Kδ auch in vaskulären Endothelzellen vorkommt, deren Funktionsweise
ausschlaggebend für die Neutrophilen-Migration ist (Puri et al., 2004). Im
Gefrierschnittmodell konnte dieser Ansatzpunkt jedoch nicht untersucht werden,
da die isolierten Leukozyten direkt auf die Hautschnitte gegeben wurden und sich
damit in unmittelbarer Nähe zur DEJ befanden.
Die Isoform, die in allen verwendeten Konzentrationen von TGX-221 gehemmt
wurde, ist jedoch die PI3Kβ. Bisher ist wenig bekannt über die PI3Kβ, eine Isoform
die ubiquitär vorkommt und eine essentielle Bedeutung für verschiedene zelluläre
Prozesse einzunehmen scheint. Dies zeigte sich im Mausmodell mit p110β-
defizienten Mäusen, bei denen eine erhöhte Letalität beschrieben wurde (Bi et al.,
2002). Es konnte außerdem gezeigt werden, dass bei der Hemmung der PI3Kβ
durch TGX-221 in humanen neutrophilen Granulozyten, die durch fMLP aktiviert
wurden, sowohl die Bildung von PIP3 als auch die Produktion von ROS sank
(Condliffe et al., 2005). In einer weiteren Studie konnte die Bedeutung der PI3Kβ
für die Regulierung der NADPH-Oxidase gezeigt werden, indem neutrophile
Granulozyten PI3Kβ-defizienter Mäuse keine ROS bildeten. Weiterhin konnten
bereits die hemmenden Effekte des Wirkstoffes TGX-221 auf die ROS-Produktion
in murinen neutrophilen Granulozyten gezeigt werden. In der gleichen Studie war
außerdem im Mausmodell mit EBA-immunisierten, PI3Kβ-defizienten Mäusen
keine Blasenbildung induzierbar. Schließlich konnte bereits die hemmende
Wirkung von TGX-221 in durch EBA-Immunkomplexe aktivierten humanen
neutrophilen Granulozyten nachgewiesen werden (Kulkarni et al., 2011). Die
Ergebnisse der vorliegenden Arbeit decken sich mit diesen Erkenntnissen und
deuten darauf hin, dass die Bildung von ROS bei der immunkomplexvermittelten
Aktivierung von neutrophilen Granulozyten von der PI3Kβ abhängig ist. Auch für
das BP gilt der gleiche Pathomechanismus der Aktivierung über Immunkomplexe.
In der gleichen Studie konnte außerdem gezeigt werden, dass bei höheren
Antikörpertitern die ROS-Produktion in murinen neutrophilen Granulozyten, die
zusätzlich keine Aktivität der PI3Kδ aufwiesen, weiter gesenkt werden konnte
(Kulkarni et al., 2011). Die Hemmung der PI3Kδ könnte hier neben der Hemmung
der ROS-Produktion zusätzliche Effekte auf die DES durch Hemmung der
Elastase und der Chemotaxis haben. Dies steht im Einklang mit den Ergebnissen
dieser Arbeit, da in den vorliegenden Versuchen die Hemmung der DES nur in der
höchsten verwendeten Konzentration von TGX-221 möglich war. In dieser

4. Diskussion 84
Konzentration erfolgte eine zusätzliche Hemmung der PI3Kδ.
In vivo ist die Selektivität von TGX-221 auch im Hinblick auf unerwünschte
Wirkungen von Bedeutung. So wird die PI3Kδ fast ausschließlich in hämatogenen
Zellen exprimiert. Im Modell mit p110δ-defizienten Mäuse zeigten diese
ausschließlich immunologische Beeinträchtigungen (Puri et al., 2004). Des
Weiteren konnte gezeigt werden, dass ein Verlust der PI3Kβ zu keiner
Beeinträchtigung des humoralen Immunsystems führt (Kulkarni et al., 2011).
Generell war möglicherweise durch das selektivere Wirkspektrum von TGX-221,
verglichen mit dem unspezifischen Inhibitor PI-103, eine bessere Wirksamkeit im
Gefrierschnittmodell zu beobachten. Dies könnte durch geringer ausgeprägte
Wechselwirkungen erklärt werden. Dabei war sowohl im Modell mit BP-
Patientenseren als auch im Modell mit EBA-Kaninchenseren unter Verwendung
der höchsten Konzentration von TGX-221 (3x10-1 µM) eine Hemmung der
Spaltbildung möglich. Es ist außerdem davon auszugehen, dass bei Einsatz von
TGX-221 in vivo durch Einbeziehung der Chemotaxis möglicherweise eine noch
ausgeprägter inhibitorische Wirkung zu erwarten ist.
Hinsichtlich der Therapie von Immunkomplex-vermittelten Erkrankungen wie dem
BP und der EBA könnte der Einsatz von TGX-221 somit durchaus eine effektive
neue Therapieoption darstellen. Weiterhin ist jedoch eine weitere Klärung der
einzelnen Signalwege der PI3K und der Effekte der Isoformen nötig. Die
Wirksamkeit von TGX-221 sollte außerdem durch eine Testung in vivo,
beispielsweise im Mausmodell, weiter bestätigt werden. Für die EBA liegen bereits
die oben beschriebenen Ergebnisse aus dem Mausmodell vor, für das BP stehen
diese bisher noch aus. Die bisherigen Ergebnisse sprechen allerdings für einen
aussichtsreichen therapeutischen Ansatz.

4. Diskussion 85
4.4 Effekte des löslichen humanen Fcγ – Rezeptors IIb auf die dermo-
epidermale Spaltbildung ex vivo
4.4.1 Allgemeines zu Fcγ-Rezeptoren
Neutrophile Granulozyten spielen eine wesentliche Rolle in der Pathogenese
blasenbildender Autoimmundermatosen wie dem BP und der EBA (Gammon et
al., 1982b; Liu et al., 1997; Sitaru et al., 2002a; Sitaru et al., 2002b). Von
besonderer Bedeutung ist dabei die Bindung von Immunkomplexen an einen auf
neutrophilen Granulozyten befindlichen Fcγ-Rezeptor, was zur Aktivierung der
Zellen führt (Sitaru et al., 2002a; Sitaru et al., 2002b; Zhao et al., 2006).
Es sind verschiedene Klassen von Fcγ-Rezeptoren bekannt, beim Menschen
finden sich der FcγRI, FcγRIIa, FcγRIIb, FcγRIIc, FcγRIIIa und der FcγRIIIb
(Nimmerjahn and Ravetch, 2008). Der FcγRIIa, FcγRIIb und FcγRIIc bestehen
lediglich aus einer α-Kette, die Antikörper bindet. Alle anderen Fcγ-Rezeptoren
setzen sich aus einer α-Kette und einer dimeren γ-Kette, welche für die
Signaltransduktion zuständig ist und ein Immunrezeptor-Tyrosinbasiertes
aktivierendes Motiv (ITAM) beinhaltet, zusammen. Mit Ausnahme des FcγRIIb und
des FcγRIII, enthalten alle Fcγ-Rezeptoren ein ITAM (Daeron, 1997; Nimmerjahn
and Ravetch, 2008). In Folge des Crosslinkings der Fcγ-Rezeptoren durch die
Immunkomplexe erfolgt eine Phosphorylierung von ITAM, wodurch weitere
Signalwege aktiviert werden, die zur Freisetzung von Zytokinen, Proteasen oder
zur Bildung von ROS führen (Nimmerjahn and Ravetch, 2008). Der FcγRIIb ist der
einzige bekannte Fcγ-Rezeptor mit hemmenden Eigenschaften, was über ein
Immunrezeptor-Tyrosinbasiertes inhibitorisches Motiv (ITIM) vermittelt wird
(Daeron, 1997; Nimmerjahn and Ravetch, 2008). Der FcγRIIIb ist der einzige
Glykosyl-Phoyphatidylinositol (GPI)-gekoppelten Rezeptor (Nimmerjahn and
Ravetch, 2008). Bei allen anderen Fcγ-Rezeptoren handelt es sich um Typ-I-
Transmembran-Glykoproteine (Nimmerjahn and Ravetch, 2008).
Die Bedeutung der Fc-Rezeptoren für die Pathogenese blasenbildender
Autoimmundermatosen konnte anhand von Mäusen mit fehlendem neonatalen Fc-
Rezeptor gezeigt werden, indem diese nach passiver Immunisierung kein BP
entwickelten und im Vergleich zur Kontrollgruppe niedrigere Serumlevel für
zirkulierendes pathogenes IgG aufwiesen (Li et al., 2005). Auch im

4. Diskussion 86
Gefrierschnittmodell konnte die Relevanz des Fc-Fragments der Antikörper und
des Fcγ-Rezeptors für die Rekrutierung und Aktivierung von neutrophilen
Granulozyten und somit für die DES nachgewiesen werden (Sitaru et al., 2002a;
Sitaru et al., 2002b). Humane neutrophile Granulozyten exprimieren vor allem den
FcγRIIIb und den FcγRIIa (Li et al., 1996; Veri et al., 2007). Im Gegensatz dazu
finden sich auf murinen neutrophilen Granulozyten vor allem der FcγRIII, FcγRIV
und der FcγRIIb (Nimmerjahn and Ravetch, 2008). Dabei scheint für die
Pathogenese des BP im Mausmodell vor allem der FcγRIII von Bedeutung zu sein
(Zhao et al., 2006). Für die EBA hingegen konnte im Mausmodell dem FcγRIV
eine besondere Rolle zugeschrieben werden (Kasperkiewicz et al., 2012) Dabei
ähnelt der murine FcγRIII dem humanen FcγRIIa, der murine FcγRIV entspricht in
etwa dem humanen FcγRIIIa (Nimmerjahn and Ravetch, 2008). Die Bedeutung
des FcγRIIa und des FcγRIIIb für die Pathogenese des BP konnte unter
Verwendung humaner neutrophiler Granulozyten im Gefrierschnittmodell bereits
nachgewiesen werden (Yu et al., 2010).
Weiterhin konnte bereits gezeigt werden, dass ein rekombinanter humaner
löslicher FcγRIIb Effekte auf die Pathogenese verschiedener entzündlicher
Erkrankungen, wie dem systemischen Lupus erythematodes, der rheumatoiden
Arthritis oder der Arthus-Reaktion, hat (Ierino et al., 1993; Werwitzke et al., 2008;
Wines et al., 2003). Der lösliche FcγRIIb bindet dabei pathogenes IgG und hemmt
somit die Bindung der Antikörper an membrangebundene Fcγ-Rezeptoren
kompetitiv, wodurch die zelluläre Aktivierung ausbleibt (Magnusson et al., 2008).
Durch die zentrale Rolle der Interaktion zwischen Immunkomplexen und Fcγ-
Rezeptoren für die inflammatorischen Reaktionen beim BP und der EBA, wäre ein
therapeutischer Ansatzpunkt, in diese Bindung pharmakologisch zu interferieren.
4.4.2 SM101
In dieser Arbeit wurde deshalb der lösliche humane FcγRIIb (SM101) im
Gefrierschnittmodell auf seine Wirksamkeit bei diesen beiden
Autoimmundermatosen getestet. Fcγ-Rezeptoren finden sich in löslicher Form
auch im menschlichen Organismus und werden durch Proteolyse der
membrangebundenen Formen und durch alternatives Spleißen gebildet (Fridman
et al., 1993). Sie sind vermehrt nachweisbar beim Vorliegen inflammatorischer

4. Diskussion 87
Prozesse und besitzen wahrscheinlich eine immunregulatorische Funktion
(Huizinga et al., 1990; Masuda et al., 2003). Es wird davon ausgegangen, dass
der lösliche FcγRIIb pathogenes IgG bindet und so die Bindung an
membrangebundene Fcγ-Rezeptoren kompetitiv hemmt (Gavin et al., 1995). In
vitro war eine Bindungsfähigkeit des FcγRIIb an murines IgG nachweisbar
(Magnusson et al., 2008; Werwitzke et al., 2008). Außerdem konnte im
Mausmodell durch die Bindung eines löslichen humanen FcγRIIb an
Immunkomplexe die Arthus-Reaktion inhibiert werden (Ierino et al., 1993).
Weiterhin wurde ein rekombinanter humaner löslicher FcγRIIb bereits erfolgreich
für verschiedene Autoimmunerkrankungen getestet. Im Mausmodell konnten so
die Wirkungen des löslichen FcγRIIb auf die Pathogenese des systemischen
Lupus erythematodes und der rheumatoiden Arthritis gezeigt werden (Magnusson
et al., 2008; Werwitzke et al., 2008). Beide Erkrankungen beruhen, wie das BP
und die EBA, auf der Bildung von Immunkomplexen und deren Bindung an Fcγ-
Rezeptoren von Leukozyten, wodurch eine Entzündungsreaktion ausgelöst wird.
Damit könnte der lösliche humane FcγRIIb auch bei entzündlichen
Autoimmundermatosen eine Wirkung haben.
In dieser Arbeit wurde deshalb der lösliche humane FcγRIIb verwendet, um im
Gefrierschnittmodell mit humaner Haut und Antikörperseren von Patienten mit BP
die Effekte auf diese Erkrankung zu untersuchen. Dabei konnte SM101 in allen
verwendeten Konzentrationen die DES inhibieren. Der Wirkstoff wurde zunächst
mit den Antikörperseren zusammen auf den Hautschnitten inkubiert, wodurch der
FcγRIIb an die Immunkomplexe binden konnte. Erst danach erfolgte die Zugabe
der Leukozytensuspension. Durch die geringere Spaltbildung im Vergleich zur
Positivkontrolle konnte gezeigt werden, dass der FcγRIIb auch an BP-Antikörper
bindet, wodurch eine Reaktion der später hinzugefügten neutrophilen
Granulozyten mit den Immunkomplexen nicht möglich war. Damit konnte
außerdem erneut bestätigt werden, dass neutrophile Granulozyten bei dem BP
über Immunkomplexe aktiviert werden und die DES wesentlich auf dieser
Aktivierung beruht. Dies entspricht den bisher gewonnen Erkenntnissen zur Rolle
der Fcγ-abhängigen Aktivierung neutrophiler Granulozyten im Gefrierschnittmodell
(Sitaru et al., 2002a; Sitaru et al., 2002b). Auch im Mausmodell konnte an FcγRIII-
defizienten Mäusen die Rolle der Fcγ-Rezeptoren für die Aktivierung neutrophiler
Granulozyten und die Pathogenese des BP gezeigt werden (Zhao et al., 2006).

4. Diskussion 88
Der FcγRIIb ist ein niedrigaffiner Rezeptor, der mit den auf humanen neutrophilen
Granulozyten gebundenen, ebenfalls niedrigaffinen FcγRIIa und FcγRIIIb um die
Bindung an den Immunkomplexen konkurrieren muss (Nimmerjahn and Ravetch,
2008). Außerdem wird wahrscheinlich auch der hochaffine FcγRI auf aktivierten
neutrophilen Granulozyten exprimiert (Nimmerjahn and Ravetch, 2008; Zhao et
al., 2006). Möglicherweise ist deshalb eine ausreichende Konzentration des
löslichen FcγRIIb nötig, um alle Immunkomplexe zu besetzen und damit die
Bindung der neutrophilen Granulozyten hemmen zu können.
Im Gegensatz dazu konnten die hemmenden Wirkungen von SM101 im
Gefrierschnittmodell auf Maushaut unter Verwendung der Seren von Kaninchen
mit EBA nicht reproduziert werden. Dies könnte durch Strukturunterschiede des
Kaninchen-IgG begründet werden, das möglicherweise mit einer geringeren
Affinität von humanem FcγRIIb gebunden wird (Nimmerjahn and Ravetch, 2008).
Die möglicherweise hemmenden Effekte von SM101 konnten in diesem Modell
deshalb für die EBA nicht gezeigt werden.
Im Mausmodell für die rheumatoide Arthritis wurde weiterhin unter Einsatz des
FcγRIIb ein Abfall des pathogenen zirkulierenden IgG im Serum sowie der Level
für inflammatorische Zytokine beschrieben (Magnusson et al., 2008). Diese
Effekte des FcγRIIb würden in vivo durchaus auch eine Rolle für das BP spielen,
da gezeigt werden konnte, dass sowohl die Zytokinlevel in der Blasenflüssigkeit
und im Serum (Ameglio et al., 1998; Inaoki and Takehara, 1998) als auch die
Serumtiter der Autoantikörper mit der Krankheitsaktivität korrelieren (Mutasim,
2000; Schmidt et al., 2000).
Auch wenn die genauen Wirkungen der einzelnen Fcγ-Rezeptoren noch nicht
geklärt sind, ist ein hemmender Effekt durch den löslichen FcγRIIb bei
verschiedenen Autoimmunerkrankungen in vitro und in vivo nachgewiesen. Aktuell
befindet sich SM101 für den systemischen Lupus erythematodes und für die
chronisch idiopathische thrombozytopenische Purpura (ITP) bereits in Phase II der
klinischen Testung (SuppreMol, 11.07.2011, 12.04.2010). In der klinischen Studie
ergab sich eine gute Verträglichkeit des Wirkstoffes (SuppreMol, 14.02.2012).
Weitere Untersuchungen mit SM101 bezüglich seiner Wirkungen auf entzündliche
Autoimmundermatosen wie dem BP und der EBA sind nötig, der lösliche FcγRIIb
stellt jedoch möglicherweise einen vielversprechenden neuen Therapieansatz dar.

4. Diskussion 89
4.5 Ausblick
In der vorliegenden Arbeit wurden neue mögliche Therapieansätze des BP und
der EBA untersucht. Dies erfolgte im Gefrierschnittmodell, in dem die PI3-Kinase-
Inhibitoren TGX-221 und PI-103, sowie der lösliche Fcγ-Rezeptor (SM101)
betrachtet wurden.
Der unselektive PI3K-Inhibtor PI-103 zeigte dabei weder im Modell mit humanen
BP-Seren noch im Modell mit EBA-Kaninchenseren eine hemmende Wirkung auf
die Spaltbildung, was möglichweise durch unerwünschte Wechselwirkungen
aufgrund der Inhibierung aller Isoformen der PI3K der Klasse I erklärt werden
kann. Für weitere Versuche würden sich daher selektivere PI3K-Inhibitoren
eignen.
Ein solcher selektiver Inhibitor ist TGX-221, der sowohl im Modell mit humaner
Haut und humanen BP-Seren als auch im Modell mit Maushaut und EBA-
Kaninchenseren eine Hemmung der Spaltbildung in der höchsten verwendeten
Konzentration von 3x10-1 µM zeigte. In dieser Konzentration werden zwei
Isoformen der PI3K gehemmt, die PI3Kβ und die PI3Kδ. Der PI3Kδ wird eine
Bedeutung für die Bildung von ROS, sowie für die Freisetzung der Elastase und
der Chemotaxis neutrophiler Granulozyten zugeschrieben (Condliffe et al., 2005;
Puri et al., 2004; Sadhu et al., 2003a). Auch die PI3Kβ spielt eine Rolle bei der
Regulierung der NADPH-Oxidase (Kulkarni et al., 2011). Die Bedeutung der ROS-
Produktion für die Spaltbildung im Gefrierschnittmodell konnte bereits gezeigt
werden (Chiriac et al., 2007). Kulkarni et al. konnten mit TGX-221 außerdem
bereits in humanen neutrophilen Granulozyten, die durch EBA-Immunkomplexe
aktiviert wurden, die ROS-Bildung inhibieren (Kulkarni et al., 2011). Auch die
Ergebnisse dieser Arbeit deuten darauf hin, dass die PI3Kβ, möglicherweise
zusammen mit der PI3Kδ, eine wichtige Rolle bei der Bildung von ROS in der
immunkomplexvermittelten Aktivierung neutrophiler Granulozyten spielt. Damit
könnte TGX-221 einen interessanten neuen Ansatzpunkt in der Therapie des BP
und der EBA darstellen. Um den exakten Wirkmechanismus von TGX-221
genauer aufzuschlüsseln, sind jedoch weitere Untersuchungen nötig. Die Effekte
von TGX-221 auf die Bildung von ROS könnten zum Beispiel im
Gefrierschnittmodell unter Einsatz von Nitroblautetrazolium (NBT) untersucht
werden. Auch in-vivo-Modelle sind beispielsweise hinsichtlich der Effekte von
TGX-221 auf die Chemotaxis neutrophiler Granulozyten von Bedeutung.

4. Diskussion 90
Des Weiteren zeigte der lösliche humane FcγRIIb (SM101) im Gefrierschnittmodell
mit humaner Haut und BP-Patientenserum einen hemmenden Effekt auf die
Spaltbildung. Durch Bindung des FcγRIIb an die Immunkomplexe wird die
Reaktion mit Fcγ-Rezeptoren auf neutrophilen Granulozyten und damit deren
Aktivierung gehemmt (Gavin et al., 1995). Die Fcγ-abhängige Aktivierung
neutrophiler Granulozyten im Gefrierschnittmodell wurde bereits durch Sitaru et al.
beschrieben (Sitaru et al., 2002a). SM101 zeigte im Modell mit EBA-
Kaninchenserum jedoch keine Reaktion, was möglichweise mit einer geringeren
Affinität des FcγRIIb zu Kaninchen-IgG begründet werden kann. Die Effekte von
SM101 auf die Pathogenese der EBA sollten deshalb in einem Modell unter
Verwendung humaner Antikörperseren nochmals untersucht werden. Weitere
Versuche in vivo, zunächst am Tiermodell, sind in Hinblick auf die Anwendung von
SM101 beim BP von großer Bedeutung. SM101 befindet sich aktuell bereits in der
Phase II der klinischen Testung für den systemischen Lupus erythematodes und
die idiopathische thrombozytopenische Purpura, zwei Erkrankungen, die ebenfalls
über Immunkomplexe vermittelt werden (SuppreMol, 11.07.2011, 19.04.2010).
Die Ergebnisse dieser Arbeit liefern somit erste Anhaltspunkte für mögliche neue
und spezifischere Therapieansätze für das BP und die EBA. Im Folgenden sind
weitere Untersuchungen, wie die Testung der Wirkstoffe am Tiermodell, von
großer Bedeutung. Hier können weitere Aussagen zu der Wirkung in vivo und
möglichen unerwünschten Wirkungen getroffen werden.

5. Zusammenfassung 91
5. Zusammenfassung
Das bullöse Pemphigoid (BP) und die Epidermolysis bullosa acquisita (EBA) sind
blasenbildende Autoimmundermatosen, die sich klinisch durch Ausbildung praller
Blasen an der Haut, bei der EBA zusätzlich durch Milien und Narben,
auszeichnen. Beide Erkrankungen sind mit Autoantikörpern gegen
Strukturproteine der Haut assoziiert. Beim BP ist die Immunantwort gegen BP180
und BP230 gerichtet, wobei die pathogenetische Relevanz bisher nur für BP180-
Antikörper nachgewiesen wurde. Für die Pathogenese der EBA sind
Autoantikörper gegen Typ-VII-Kollagen verantwortlich. Antikörper und
Strukturproteine bilden Immunkomplexe, an die neutrophile Granulozyten über
einen Fcγ-Rezeptor binden, wodurch die Zellen aktiviert werden. Es kommt zur
Entzündung und Blasenbildung.
Die Therapiemöglichkeiten beider Erkrankungen sind momentan noch begrenzt
und beruhen auf dem Einsatz von Steroiden und anderen Immunsuppressiva.
Diese führen meist zur Remission, durch generelle Immunsuppression kommt es
jedoch zu erhöhter Morbidität und Mortalität. Außerdem sind Rezidive nach
Absetzen der Steroide beschrieben. Ziel dieser Arbeit war es daher, neue
Therapieansätze zu untersuchen.
Dafür wurde das Gefrierschnittmodell verwendet. Es handelt sich dabei um eine
Methode, bei der ex vivo Gewebsschnitte mit Antikörper-Seren und Leukozyten
inkubiert werden, was zur Freisetzung proteolytischer Enzyme und reaktiver
Sauerstoffspezies und infolge zur dermo-epidermalen Spaltbildung führt. In dieser
Arbeit wurden Gefrierschnitte humaner Haut mit BP-Patientenserum verwendet.
Im Modell für die EBA wurde Maushaut mit Serum EBA-immunisierter Kaninchen
inkubiert.
Mithilfe dieses Modells wurden die PI3-Kinase-Inhibitoren PI-103 und TGX-221
getestet. Diese hemmen unter anderem die PI3Kβ, die eine zentrale Rolle bei der
Aktivierung neutrophiler Granulozyten spielt. Der lösliche Fcγ-Rezeptor IIb
(SM101) wurde hinsichtlich der Fähigkeit zur Bindung von Immunkomplexen und
der Hemmung einer Interaktion mit Fcγ-Rezeptoren auf Leukozyten untersucht.
Dabei wurde gezeigt, dass TGX-221 in beiden Modellen die Spaltbildung durch

5. Zusammenfassung 92
selektive Inhibierung der PI3Kβ hemmen konnte. PI-103 zeigt weder im Versuch
mit BP noch mit EBA eine Wirkung, wahrscheinlich aufgrund der unspezifischen
Inhibierung der PI3K. SM101 konnte im BP-Modell die Spaltbildung durch
Interaktion mit humanen Autoantikörpern hemmen. Für die EBA war dies nicht
möglich, da Kaninchenantikörper eine geringere Bindungsaffinität zu SM101 zu
haben scheinen.
Somit ergaben sich mithilfe des Gefrierschnittmodells Anhaltspunkte für neue,
möglicherweise effektivere Therapiemöglichkeiten dieser beiden Erkrankungen.
Es wurden außerdem die Wirkungen von Methylprednisolon weiter charakterisiert.
Dies erfolgte im Gefrierschnittmodell unter Verwendung von Nitroblautetrazolium,
das durch die in aktivierten neutrophilen Granulozyten gebildeten reaktiven
Sauerstoffspezies reduziert wird und in den Zellen als blauer Farbstoff präzipitiert.
Auch im ROS-Release wurde Methylprednisolon untersucht. Dabei konnte in
beiden Versuchen die Bildung reaktiver Sauerstoffspezies durch
Methylprednisolon gehemmt werden.

6. Literaturverzeichnis 93
6. Literaturverzeichnis
Aho S, Uitto J (1999) 180-kD bullous pemphigoid antigen/type XVII collagen: tissue-
specific expression and molecular interactions with keratin 18. J Cell Biochem 72:356-67.
Ameglio F, D'Auria L, Bonifati C, Ferraro C, Mastroianni A, Giacalone B (1998) Cytokine
pattern in blister fluid and serum of patients with bullous pemphigoid: relationships with
disease intensity. Br J Dermatol 138:611-4.
Amor KT, Ryan C, Menter A (2010) The use of cyclosporine in dermatology: part I. J Am
Acad Dermatol 63:925-46; quiz 47-8.
Anhalt GJ, Diaz LA (1987) Animal models for bullous pemphigoid. Clin Dermatol 5:117-25.
Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV (2008)
Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science
320:373-6.
Antoine JC, Maurice M, Feldmann G, Avrameas S (1980) In vivo and in vitro effects of
colchicine and vinblastine on the secretory process of antibody-producing cells. J Immunol
125:1939-49.
Appelhans M, Bonsmann G, Orge C, Brocker EB (1993) [Dexamethasone-
cyclophosphamide pulse therapy in bullous autoimmune dermatoses]. Hautarzt 44:143-7.
Arin MJ, Hunzelmann N (2005) Anti-B- cell-directed immunotherapy (rituximab) in the
treatment of refractory pemphigus--an update. Eur J Dermatol 15:224-30.
Bamberger CM, Bamberger AM, de Castro M, Chrousos GP (1995) Glucocorticoid
receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin
Invest 95:2435-41.
Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, Bienert M,
Radbruch A, Burmester GR, Lauster R, Scheffold A, Buttgereit F (2004) Membrane
glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood
mononuclear cells and up-regulated after in vitro stimulation and in patients with
rheumatoid arthritis. FASEB J 18:70-80.

6. Literaturverzeichnis 94
Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E, Roujeau JC,
Bernard P, Guillaume JC, Ingen-Housz-Oro S, Maillard H, Pauwels C, Picard-Dahan C,
Dutronc Y, Richard MA (2011) Risk factors for bullous pemphigoid in the elderly: a
prospective case-control study. J Invest Dermatol 131:637-43.
Bastuji-Garin S, Joly P, Picard-Dahan C, Bernard P, Vaillant L, Pauwels C, Salagnac V,
Lok C, Roujeau JC (1996) Drugs associated with bullous pemphigoid. A case-control
study. Arch Dermatol 132:272-6.
Bauer EA, Valle KJ (1982) Colchicine-induced modulation of collagenase in human skin
fibroblast cultures. I. Stimulation of enzyme synthesis in normal cells. J Invest Dermatol
79:398-402.
Bedane C, McMillan JR, Balding SD, Bernard P, Prost C, Bonnetblanc JM, Diaz LA, Eady
RA, Giudice GJ (1997) Bullous pemphigoid and cicatricial pemphigoid autoantibodies
react with ultrastructurally separable epitopes on the BP180 ectodomain: evidence that
BP180 spans the lamina lucida. J Invest Dermatol 108:901-7.
Beissert S, Werfel T, Frieling U, Bohm M, Sticherling M, Stadler R, Zillikens D, Rzany B,
Hunzelmann N, Meurer M, Gollnick H, Ruzicka T, Pillekamp H, Junghans V, Bonsmann
G, Luger TA (2007) A comparison of oral methylprednisolone plus azathioprine or
mycophenolate mofetil for the treatment of bullous pemphigoid. Arch Dermatol 143:1536-
42.
Berbis P, Privat Y (1989) [Value of colchicine in treating acquired epidermolysis bullosa].
Ann Dermatol Venereol 116:301-7.
Berk MA, Lorincz AL (1986) The treatment of bullous pemphigoid with tetracycline and
niacinamide. A preliminary report. Arch Dermatol 122:670-4.
Bernard P, Prost C, Aucouturier P, Durepaire N, Denis F, Bonnetblanc JM (1991) The
subclass distribution of IgG autoantibodies in cicatricial pemphigoid and epidermolysis
bullosa acquisita. J Invest Dermatol 97:259-63.
Bernard P, Reguiai Z, Tancrede-Bohin E, Cordel N, Plantin P, Pauwels C, Vaillant L,
Grange F, Richard-Lallemand MA, Sassolas B, Roujeau JC, Lok C, Picard-Dahan C,
Chosidow O, Vitry F, Joly P (2009) Risk factors for relapse in patients with bullous
pemphigoid in clinical remission: a multicenter, prospective, cohort study. Arch Dermatol
145:537-42.

6. Literaturverzeichnis 95
Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, Lorette G,
Bonnetblanc JM, Prost C (1995) Incidence and distribution of subepidermal autoimmune
bullous skin diseases in three French regions. Bullous Diseases French Study Group.
Arch Dermatol 131:48-52.
Bertram F, Brocker EB, Zillikens D, Schmidt E (2009) Prospective analysis of the
incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch
Dermatol Ges 7:434-40.
Bi L, Okabe I, Bernard DJ, Nussbaum RL (2002) Early embryonic lethality in mice
deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome 13:169-72.
Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL (1999) Proliferative defect
and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of
phosphoinositide 3-kinase. J Biol Chem 274:10963-8.
Bieber K, Sun S, Ishii N, Kasperkiewicz M, Schmidt E, Hirose M, Westermann J, Yu X,
Zillikens D, Ludwig RJ (2010) Animal models for autoimmune bullous dermatoses. Exp
Dermatol 19:2-11.
Bohm M, Beissert S, Schwarz T, Metze D, Luger T (1997) Bullous pemphigoid treated
with mycophenolate mofetil. Lancet 349:541.
Booth SA, Moody CE, Dahl MV, Herron MJ, Nelson RD (1992) Dapsone suppresses
integrin-mediated neutrophil adherence function. J Invest Dermatol 98:135-40.
Borradori L, Sonnenberg A (1999) Structure and function of hemidesmosomes: more than
simple adhesion complexes. J Invest Dermatol 112:411-8.
Borrego L, Maynard B, Peterson EA, George T, Iglesias L, Peters MS, Newman W, Gleich
GJ, Leiferman KM (1996) Deposition of eosinophil granule proteins precedes blister
formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule
proteins. Am J Pathol 148:897-909.
Briggaman RA, Wheeler CE, Jr. (1975) The epidermal-dermal junction. J Invest Dermatol
65:71-84.
Bruckner-Tuderman L, Nilssen O, Zimmermann DR, Dours-Zimmermann MT, Kalinke DU,
Gedde-Dahl T, Jr., Winberg JO (1995) Immunohistochemical and mutation analyses
demonstrate that procollagen VII is processed to collagen VII through removal of the NC-2
domain. J Cell Biol 131:551-9.

6. Literaturverzeichnis 96
Budinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, Merk HF, Yancey
K, Hertl M (1998) Identification and characterization of autoreactive T cell responses to
bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest 102:2082-9.
Burgeson RE (1993) Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J
Invest Dermatol 101:252-5.
Buttgereit F, Grant A, Muller M, Brand MD (1994) The effects of methylprednisolone on
oxidative phosphorylation in Concanavalin-A-stimulated thymocytes. Top-down elasticity
analysis and control analysis. Eur J Biochem 223:513-9.
Buttgereit F, Krauss S, Brand MD (1997) Methylprednisolone inhibits uptake of Ca2+ and
Na+ ions into concanavalin A-stimulated thymocytes. Biochem J 326 ( Pt 2):329-32.
Buttgereit F, Scheffold A (2002) Rapid glucocorticoid effects on immune cells. Steroids
67:529-34.
Buttgereit F, Song IH, Straub RH, Burmester GR (2005) [Current insights into the
development of new glucocorticoid receptor ligands]. Z Rheumatol 64:170-6.
Buttgereit F, Wehling M, Burmester GR (1998) A new hypothesis of modular
glucocorticoid actions: steroid treatment of rheumatic diseases revisited. Arthritis Rheum
41:761-7.
Bystryn JC, Rudolph JL (2005) Why is the mortality of bullous pemphigoid greater in
Europe than in the US? J Invest Dermatol 124:xx-xxi.
Caldwell JB, Yancey KB, Engler RJ, James WD (1994) Epidermolysis bullosa acquisita:
efficacy of high-dose intravenous immunoglobulins. J Am Acad Dermatol 31:827-8.
Campos M, Silvente C, Lecona M, Suarez R, Lazaro P (2006) Epidermolysis bullosa
acquisita: diagnosis by fluorescence overlay antigen mapping and clinical response to
high-dose intravenous immunoglobulin. Clin Exp Dermatol 31:71-3.
Capella GL, Casa-Alberighi OD, Finzi AF (2001) Therapeutic concepts in clinical
dermatology: cyclosporine A in immunomediated and other dermatoses. Int J Dermatol
40:551-61.

6. Literaturverzeichnis 97
Chan LS, Lapiere JC, Chen M, Traczyk T, Mancini AJ, Paller AS, Woodley DT,
Marinkovich MP (1999) Bullous systemic lupus erythematosus with autoantibodies
recognizing multiple skin basement membrane components, bullous pemphigoid antigen
1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol 135:569-73.
Chan YC, Sun YJ, Ng PP, Tan SH (2003) Comparison of immunofluorescence
microscopy, immunoblotting and enzyme-linked immunosorbent assay methods in the
laboratory diagnosis of bullous pemphigoid. Clin Exp Dermatol 28:651-6.
Chave TA, Mortimer NJ, Shah DS, Hutchinson PE (2004) Chlorambucil as a steroid-
sparing agent in bullous pemphigoid. Br J Dermatol 151:1107-8.
Chen M, Chan LS, Cai X, O'Toole EA, Sample JC, Woodley DT (1997a) Development of
an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis
bullosa acquisita. J Invest Dermatol 108:68-72.
Chen M, Keene DR, Costa FK, Tahk SH, Woodley DT (2001a) The carboxyl terminus of
type VII collagen mediates antiparallel dimer formation and constitutes a new antigenic
epitope for epidermolysis Bullosa acquisita autoantibodies. J Biol Chem 276:21649-55.
Chen M, Marinkovich MP, Jones JC, O'Toole EA, Li YY, Woodley DT (1999) NC1 domain
of type VII collagen binds to the beta3 chain of laminin 5 via a unique subdomain within
the fibronectin-like repeats. J Invest Dermatol 112:177-83.
Chen M, Marinkovich MP, Veis A, Cai X, Rao CN, O'Toole EA, Woodley DT (1997b)
Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with
extracellular matrix components. A potential role in epidermal-dermal adherence in human
skin. J Biol Chem 272:14516-22.
Chen M, O'Toole EA, Sanghavi J, Mahmud N, Kelleher D, Weir D, Fairley JA, Woodley
DT (2002) The epidermolysis bullosa acquisita antigen (type VII collagen) is present in
human colon and patients with crohn's disease have autoantibodies to type VII collagen. J
Invest Dermatol 118:1059-64.
Chen Q, Powell DW, Rane MJ, Singh S, Butt W, Klein JB, McLeish KR (2003) Akt
phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J
Immunol 170:5302-8.
Chen R, Ning G, Zhao ML, Fleming MG, Diaz LA, Werb Z, Liu Z (2001b) Mast cells play a
key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest
108:1151-8.

6. Literaturverzeichnis 98
Chiriac MT, Roesler J, Sindrilaru A, Scharffetter-Kochanek K, Zillikens D, Sitaru C (2007)
NADPH oxidase is required for neutrophil-dependent autoantibody-induced tissue
damage. J Pathol 212:56-65.
Cho HJ, Lee IJ, Kim SC (1998) Complement-fixing abilities and IgG subclasses of
autoantibodies in epidermolysis bullosa acquisita. Yonsei Med J 39:339-44.
Claudy A (2001) Evaluation of the safety and efficacy of a potent topical cortico-steroid in
the treatment of bullous pemphigoid. Clin Dermatol 19:778-80.
Clement M, Ratnesar P, Thirumoorthy T, McGrath J, Black MM (1993) Epidermolysis
bullosa acquisita--a case with upper airways obstruction requiring tracheostomy and
responding to cyclosporin. Clin Exp Dermatol 18:548-51.
Coleman MD (1993) Dapsone: modes of action, toxicity and possible strategies for
increasing patient tolerance. Br J Dermatol 129:507-13.
Coleman MD, Rhodes LE, Scott AK, Verbov JL, Friedmann PS, Breckenridge AM, Park
BK (1992) The use of cimetidine to reduce dapsone-dependent methaemoglobinaemia in
dermatitis herpetiformis patients. Br J Clin Pharmacol 34:244-9.
Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K,
Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M,
Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT (2005) Sequential
activation of class IB and class IA PI3K is important for the primed respiratory burst of
human but not murine neutrophils. Blood 106:1432-40.
Connolly SM, Sander HM (1987) Treatment of epidermolysis bullosa acquisita with
cyclosporine. J Am Acad Dermatol 16:890.
Courville P, Kupfer I, Gilbert D, Thomine E, Metayer J, Joly P (2000) [Evaluation of
histological criteria for bullous pemphigoid. Correlation with antigens recognized by
immunoblotting of anti-epidermal autoantibodies]. Ann Pathol 20:564-9.
Crow LL, Finkle JP, Gammon WR, Woodley DT (1988) Clearing of epidermolysis bullosa
acquisita with cyclosporine. J Am Acad Dermatol 19:937-42.
Cunningham BB, Kirchmann TT, Woodley D (1996) Colchicine for epidermolysis bullosa
acquisita. J Am Acad Dermatol 34:781-4.

6. Literaturverzeichnis 99
Czernik A, Bystryn JC (2008) Improvement of intravenous immunoglobulin therapy for
bullous pemphigoid by adding immunosuppressive agents: marked improvement in
depletion of circulating autoantibodies. Arch Dermatol 144:658-61.
Daeron M (1997) Fc receptor biology. Annu Rev Immunol 15:203-34.
Dahl MG (1979) Epidermolysis bullosa acquisita--a sign of cicatricial pemphigoid? Br J
Dermatol 101:475-84.
Daoud YJ, Amin KG (2006) Comparison of cost of immune globulin intravenous therapy to
conventional immunosuppressive therapy in treating patients with autoimmune
mucocutaneous blistering diseases. Int Immunopharmacol 6:600-6.
Dawe RS, Naidoo DK, Ferguson J (1997) Severe bullous pemphigoid responsive to
pulsed intravenous dexamethasone and oral cyclophosphamide. Br J Dermatol 137:826-
7.
Delgado JC, Turbay D, Yunis EJ, Yunis JJ, Morton ED, Bhol K, Norman R, Alper CA,
Good RA, Ahmed R (1996) A common major histocompatibility complex class II allele
HLA-DQB1* 0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci U S A
93:8569-71.
Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J (2000) The mitogen-activated
protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-
methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J
Immunol 165:5238-44.
Dopp R, Schmidt E, Chimanovitch I, Leverkus M, Brocker EB, Zillikens D (2000) IgG4 and
IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous
pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad
Dermatol 42:577-83.
Dresow SK, Sitaru C, Recke A, Oostingh GJ, Zillikens D, Gibbs BF (2009) IgE
autoantibodies against the intracellular domain of BP180. Br J Dermatol 160:429-32.
Ehrenfeld M, Levy M, Bar Eli M, Gallily R, Eliakim M (1980) Effect of colchicine on
polymorphonuclear leucocyte chemotaxis in human volunteers. Br J Clin Pharmacol
10:297-300.
Elliott G (1895) Two cases of epidermolysis bullosa. J Cutan Genitourin Dis 13:10.

6. Literaturverzeichnis 100
Endo Y, Tamura A, Ishikawa O, Miyachi Y, Hashimoto T (1997) Psoriasis vulgaris
coexistent with epidermolysis bullosa acquisita. Br J Dermatol 137:783-6.
Engineer L, Ahmed AR (2001) Emerging treatment for epidermolysis bullosa acquisita. J
Am Acad Dermatol 44:818-28.
Fine JD, Tyring S, Gammon WR (1989) The presence of intra-lamina lucida blister
formation in epidermolysis bullosa acquisita: possible role of leukocytes. J Invest Dermatol
92:27-32.
Fivenson DP, Breneman DL, Rosen GB, Hersh CS, Cardone S, Mutasim D (1994)
Nicotinamide and tetracycline therapy of bullous pemphigoid. Arch Dermatol 130:753-8.
Fontao L, Favre B, Riou S, Geerts D, Jaunin F, Saurat JH, Green KJ, Sonnenberg A,
Borradori L (2003) Interaction of the bullous pemphigoid antigen 1 (BP230) and
desmoplakin with intermediate filaments is mediated by distinct sequences within their
COOH terminus. Mol Biol Cell 14:1978-92.
Fridman WH, Teillaud JL, Bouchard C, Teillaud C, Astier A, Tartour E, Galon J, Mathiot C,
Sautes C (1993) Soluble Fc gamma receptors. J Leukoc Biol 54:504-12.
Fruman DA, Cantley LC (2002) Phosphoinositide 3-kinase in immunological systems.
Semin Immunol 14:7-18.
Fruman DA, Meyers RE, Cantley LC (1998) Phosphoinositide kinases. Annu Rev
Biochem 67:481-507.
Fukumoto T, Umekawa T, Higuchi M, Hashimoto T, Shumann H, Bruckner-Tuderman L,
Asada H, Miyagawa S (2004) Childhood epidermolysis bullosa acquisita with
autoantibodies against all 3 structural domains of type VII collagen. J Am Acad Dermatol
50:480-2.
Galli SJ (1993) New concepts about the mast cell. N Engl J Med 328:257-65.
Galli SJ, Gordon JR, Wershil BK (1991) Cytokine production by mast cells and basophils.
Curr Opin Immunol 3:865-72.
Gammon WR (1988) Epidermolysis bullosa acquisita. Semin Dermatol 7:218-24.

6. Literaturverzeichnis 101
Gammon WR (1989) Immune complex and complement-mediated leukocyte recruitment
in bullous pemphigoid. Immunol Ser 46:509-25.
Gammon WR, Briggaman RA (1993) Epidermolysis bullosa acquisita and bullous
systemic lupus erythematosus. Diseases of autoimmunity to type VII collagen. Dermatol
Clin 11:535-47.
Gammon WR, Briggaman RA, Wheeler CE, Jr. (1982a) Epidermolysis bullosa acquisita
presenting as an inflammatory bullous disease. J Am Acad Dermatol 7:382-7.
Gammon WR, Briggaman RA, Woodley DT, Heald PW, Wheeler CE, Jr. (1984a)
Epidermolysis bullosa acquisita--a pemphigoid-like disease. J Am Acad Dermatol 11:820-
32.
Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA (1988)
Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa
acquisita antigen: evidence that the expression of autoimmunity to type VII collagen is
HLA class II allele associated. J Invest Dermatol 91:228-32.
Gammon WR, Inman AO, 3rd, Wheeler CE, Jr. (1984b) Differences in complement-
dependent chemotactic activity generated by bullous pemphigoid and epidermolysis
bullosa acquisita immune complexes: demonstration by leukocytic attachment and organ
culture methods. J Invest Dermatol 83:57-61.
Gammon WR, Merritt CC, Lewis DM, Sams WM, Jr., Carlo JR, Wheeler CE, Jr. (1982b)
An in vitro model of immune complex-mediated basement membrane zone separation
caused by pemphigoid antibodies, leukocytes, and complement. J Invest Dermatol
78:285-90.
Gammon WR, Murrell DF, Jenison MW, Padilla KM, Prisayanh PS, Jones DA, Briggaman
RA, Hunt SW, 3rd (1993) Autoantibodies to type VII collagen recognize epitopes in a
fibronectin-like region of the noncollagenous (NC1) domain. J Invest Dermatol 100:618-
22.
Gandhi K, Chen M, Aasi S, Lapiere JC, Woodley DT, Chan LS (2000) Autoantibodies to
type VII collagen have heterogeneous subclass and light chain compositions and their
complement-activating capacities do not correlate with the inflammatory clinical
phenotype. J Clin Immunol 20:416-23.
Gavin AL, Wines BD, Powell MS, Hogarth PM (1995) Recombinant soluble Fc gamma RII
inhibits immune complex precipitation. Clin Exp Immunol 102:620-5.

6. Literaturverzeichnis 102
Ghohestani RF, Nicolas JF, Rousselle P, Claudy AL (1997) Diagnostic value of indirect
immunofluorescence on sodium chloride-split skin in differential diagnosis of subepidermal
autoimmune bullous dermatoses. Arch Dermatol 133:1102-7.
Ghohestani RF, Novotney J, Chaudhary M, Agah RS (2001) Bullous pemphigoid: from the
bedside to the research laboratory. Clin Dermatol 19:690-6.
Giudice GJ, Emery DJ, Diaz LA (1992) Cloning and primary structural analysis of the
bullous pemphigoid autoantigen BP180. J Invest Dermatol 99:243-50.
Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA (1993) Bullous
pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous
site on the BP180 ectodomain. J Immunol 151:5742-50.
Goebeler M, Sitaru C, Zillikens D (2004) [Blistering autoimmune dermatoses (II): therapy].
J Dtsch Dermatol Ges 2:774-91; quiz 92-3.
Gourgiotou K, Exadaktylou D, Aroni K, Rallis E, Nicolaidou E, Paraskevakou H,
Katsambas AD (2002) Epidermolysis bullosa acquisita: treatment with intravenous
immunoglobulins. J Eur Acad Dermatol Venereol 16:77-80.
Grundmann-Kollmann M, Korting HC, Behrens S, Kaskel P, Leiter U, Krahn G, Kerscher
M, Peter RU (1999) Mycophenolate mofetil: a new therapeutic option in the treatment of
blistering autoimmune diseases. J Am Acad Dermatol 40:957-60.
Gudi VS, White MI, Cruickshank N, Herriot R, Edwards SL, Nimmo F, Ormerod AD (2005)
Annual incidence and mortality of bullous pemphigoid in the Grampian Region of North-
east Scotland. Br J Dermatol 153:424-7.
Gupta AK, Ellis CN, Cooper KD, Nickoloff BJ, Ho VC, Chan LS, Hamilton TA, Tellner DC,
Griffiths CE, Voorhees JJ (1990) Oral cyclosporine for the treatment of alopecia areata. A
clinical and immunohistochemical analysis. J Am Acad Dermatol 22:242-50.
Gurcan HM, Ahmed AR (2009a) Analysis of current data on the use of methotrexate in the
treatment of pemphigus and pemphigoid. Br J Dermatol 161:723-31.
Gurcan HM, Ahmed AR (2009b) Efficacy of dapsone in the treatment of pemphigus and
pemphigoid: analysis of current data. Am J Clin Dermatol 10:383-96.

6. Literaturverzeichnis 103
Hall RP, 3rd, Murray JC, McCord MM, Rico MJ, Streilein RD (1993) Rabbits immunized
with a peptide encoded for by the 230-kD bullous pemphigoid antigen cDNA develop an
enhanced inflammatory response to UVB irradiation: a potential animal model for bullous
pemphigoid. J Invest Dermatol 101:9-14.
Hallel-Halevy D, Nadelman C, Chen M, Woodley DT (2001) Epidermolysis bullosa
acquisita: update and review. Clin Dermatol 19:712-8.
Harman KE, Black MM (1999) High-dose intravenous immune globulin for the treatment of
autoimmune blistering diseases: an evaluation of its use in 14 cases. Br J Dermatol
140:865-74.
Harvath L, Yancey KB, Katz SI (1986) Selective inhibition of human neutrophil chemotaxis
to N-formyl-methionyl-leucyl-phenylalanine by sulfones. J Immunol 137:1305-11.
Hashimoto T, Ishiko A, Shimizu H, Tanaka T, Dodd HJ, Bhogal BS, Black MM, Nishikawa
T (1996) A case of linear IgA bullous dermatosis with IgA anti-type VII collagen
autoantibodies. Br J Dermatol 134:336-9.
Heimbach L, Li Z, Berkowitz P, Zhao M, Li N, Rubenstein DS, Diaz LA, Liu Z (2011) The
C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous
pemphigoid. J Biol Chem 286:15003-9.
Hellberg L, Samavedam UK, Holdorf K, Hansel M, Recke A, Beckmann T, Steinhorst K,
Boehncke WH, Kirchner T, Mockel N, Solbach W, Zillikens D, Schmidt E, Ludwig RJ,
Laskay T (2013) Methylprednisolone Blocks Autoantibody-Induced Tissue Damage in
Experimental Models of Bullous Pemphigoid and Epidermolysis Bullosa Acquisita through
Inhibition of Neutrophil Activation. J Invest Dermatol.
Herrero-Gonzalez JE, Sitaru C, Klinker E, Brocker EB, Zillikens D (2005) Successful
adjuvant treatment of severe bullous pemphigoid by tryptophan immunoadsorption. Clin
Exp Dermatol 30:519-22.
Hertl M, Schuler G (2002a) [Bullous autoimmune dermatoses. 1: Classification]. Hautarzt
53:207-19; quiz 20-1.
Hertl M, Schuler G (2002b) [Bullous autoimmune dermatoses. 3: Diagnosis and therapy].
Hautarzt 53:352-65; quiz 66-7.

6. Literaturverzeichnis 104
Hertl M, Zillikens D, Borradori L, Bruckner-Tuderman L, Burckhard H, Eming R, Engert A,
Goebeler M, Hofmann S, Hunzelmann N, Karlhofer F, Kautz O, Lippert U, Niedermeier A,
Nitschke M, Pfutze M, Reiser M, Rose C, Schmidt E, Shimanovich I, Sticherling M, Wolff-
Franke S (2008) Recommendations for the use of rituximab (anti-CD20 antibody) in the
treatment of autoimmune bullous skin diseases. J Dtsch Dermatol Ges 6:366-73.
Hirako Y, Usukura J, Uematsu J, Hashimoto T, Kitajima Y, Owaribe K (1998) Cleavage of
BP180, a 180-kDa bullous pemphigoid antigen, yields a 120-kDa collagenous extracellular
polypeptide. J Biol Chem 273:9711-7.
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S,
Mantovani A, Altruda F, Wymann MP (2000) Central role for G protein-coupled
phosphoinositide 3-kinase gamma in inflammation. Science 287:1049-53.
Hofmann S, Thoma-Uszynski S, Hunziker T, Bernard P, Koebnick C, Stauber A, Schuler
G, Borradori L, Hertl M (2002) Severity and phenotype of bullous pemphigoid relate to
autoantibody profile against the NH2- and COOH-terminal regions of the BP180
ectodomain. J Invest Dermatol 119:1065-73.
Hofmann SC, Kautz O, Hertl M, Sticherling M, Zillikens D, Bruckner-Tuderman L (2009)
Results of a survey of German dermatologists on the therapeutic approaches to
pemphigus and bullous pemphigoid. J Dtsch Dermatol Ges 7:227-33.
Hopkinson SB, Jones JC (2000) The N terminus of the transmembrane protein BP180
interacts with the N-terminal domain of BP230, thereby mediating keratin cytoskeleton
anchorage to the cell surface at the site of the hemidesmosome. Mol Biol Cell 11:277-86.
Hopkinson SB, Riddelle KS, Jones JC (1992) Cytoplasmic domain of the 180-kD bullous
pemphigoid antigen, a hemidesmosomal component: molecular and cell biologic
characterization. J Invest Dermatol 99:264-70.
Hornschuh B, Hamm H, Wever S, Hashimoto T, Schroder U, Brocker EB, Zillikens D
(1997) Treatment of 16 patients with bullous pemphigoid with oral tetracycline and
niacinamide and topical clobetasol. J Am Acad Dermatol 36:101-3.
Huizinga TW, de Haas M, Kleijer M, Nuijens JH, Roos D, von dem Borne AE (1990)
Soluble Fc gamma receptor III in human plasma originates from release by neutrophils. J
Clin Invest 86:416-23.
Ierino FL, Powell MS, McKenzie IF, Hogarth PM (1993) Recombinant soluble human Fc
gamma RII: production, characterization, and inhibition of the Arthus reaction. J Exp Med
178:1617-28.

6. Literaturverzeichnis 105
Inaoki M, Takehara K (1998) Increased serum levels of interleukin (IL)-5, IL-6 and IL-8 in
bullous pemphigoid. J Dermatol Sci 16:152-7.
Ino N, Kamata N, Matsuura C, Shinkai H, Odaka M (1997) Immunoadsorption for the
treatment of bullous pemphigoid. Ther Apher 1:372-6.
Ishii N, Hamada T, Dainichi T, Karashima T, Nakama T, Yasumoto S, Zillikens D,
Hashimoto T (2010) Epidermolysis bullosa acquisita: what's new? J Dermatol 37:220-30.
Ishii N, Recke A, Mihai S, Hirose M, Hashimoto T, Zillikens D, Ludwig RJ (2011)
Autoantibody-induced intestinal inflammation and weight loss in experimental
epidermolysis bullosa acquisita. J Pathol 224:234-44.
Ishii N, Yoshida M, Hisamatsu Y, Ishida-Yamamoto A, Nakane H, Iizuka H, Tanaka T,
Hashimoto T (2004) Epidermolysis bullosa acquisita sera react with distinct epitopes on
the NC1 and NC2 domains of type VII collagen: study using immunoblotting of domain-
specific recombinant proteins and postembedding immunoelectron microscopy. Br J
Dermatol 150:843-51.
Ishii N, Yoshida M, Ishida-Yamamoto A, Fritsch A, Elfert S, Bruckner-Tuderman L,
Hashimoto T (2009) Some epidermolysis bullosa acquisita sera react with epitopes within
the triple-helical collagenous domain as indicated by immunoelectron microscopy. Br J
Dermatol 160:1090-3.
Itoh T, Hosokawa H, Shirai Y, Horio T (1996) Successful treatment of bullous pemphigoid
with pulsed intravenous cyclophosphamide. Br J Dermatol 134:931-3.
Iwata Y, Komura K, Kodera M, Usuda T, Yokoyama Y, Hara T, Muroi E, Ogawa F,
Takenaka M, Sato S (2008) Correlation of IgE autoantibody to BP180 with a severe form
of bullous pemphigoid. Arch Dermatol 144:41-8.
Jackson AP, Hall AG, McLelland J (1997) Thiopurine methyltransferase levels should be
measured before commencing patients on azathioprine. Br J Dermatol 136:133-4.
Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche
V, Anderson KE, Dopheide SM, Yuan Y, Sturgeon SA, Prabaharan H, Thompson PE,
Smith GD, Shepherd PR, Daniele N, Kulkarni S, Abbott B, Saylik D, Jones C, Lu L,
Giuliano S, Hughan SC, Angus JA, Robertson AD, Salem HH (2005) PI 3-kinase
p110beta: a new target for antithrombotic therapy. Nat Med 11:507-14.

6. Literaturverzeichnis 106
Jeffes EW, 3rd, Ahmed AR (1989) Adjuvant therapy of bullous pemphigoid with dapsone.
Clin Exp Dermatol 14:132-6.
Joly P, Benichou J, Lok C, Hellot MF, Saiag P, Tancrede-Bohin E, Sassolas B, Labeille B,
Doutre MS, Gorin I, Pauwels C, Chosidow O, Caux F, Esteve E, Dutronc Y, Sigal M, Prost
C, Maillard H, Guillaume JC, Roujeau JC (2005) Prediction of survival for patients with
bullous pemphigoid: a prospective study. Arch Dermatol 141:691-8.
Joly P, Roujeau JC, Benichou J, Delaporte E, D'Incan M, Dreno B, Bedane C, Sparsa A,
Gorin I, Picard C, Tancrede-Bohin E, Sassolas B, Lok C, Guillaume JC, Doutre MS,
Richard MA, Caux F, Prost C, Plantin P, Chosidow O, Pauwels C, Maillard H, Saiag P,
Descamps V, Chevrant-Breton J, Dereure O, Hellot MF, Esteve E, Bernard P (2009) A
comparison of two regimens of topical corticosteroids in the treatment of patients with
bullous pemphigoid: a multicenter randomized study. J Invest Dermatol 129:1681-7.
Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, Vaillant L, D'Incan M,
Plantin P, Bedane C, Young P, Bernard P (2002) A comparison of oral and topical
corticosteroids in patients with bullous pemphigoid. N Engl J Med 346:321-7.
Jones DA, Hunt SW, 3rd, Prisayanh PS, Briggaman RA, Gammon WR (1995)
Immunodominant autoepitopes of type VII collagen are short, paired peptide sequences
within the fibronectin type III homology region of the noncollagenous (NC1) domain. J
Invest Dermatol 104:231-5.
Jordon RE, Kawana S, Fritz KA (1985) Immunopathologic mechanisms in pemphigus and
bullous pemphigoid. J Invest Dermatol 85:72s-8s.
Jung M, Kippes W, Messer G, Zillikens D, Rzany B (1999) Increased risk of bullous
pemphigoid in male and very old patients: A population-based study on incidence. J Am
Acad Dermatol 41:266-8.
Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB (2001)
The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol
3:675-8.
Kaneko Y, Nimmerjahn F, Ravetch JV (2006) Anti-inflammatory activity of immunoglobulin
G resulting from Fc sialylation. Science 313:670-3.

6. Literaturverzeichnis 107
Kasperkiewicz M, Nimmerjahn F, Wende S, Hirose M, Iwata H, Jonkman MF,
Samavedam U, Gupta Y, Moller S, Rentz E, Hellberg L, Kalies K, Yu X, Schmidt E, Hasler
R, Laskay T, Westermann J, Kohl J, Zillikens D, Ludwig RJ (2012) Genetic identification
and functional validation of FcgammaRIV as key molecule in autoantibody-induced tissue
injury. J Pathol 228:8-19.
Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC (2001)
Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation
of MKP-1. EMBO J 20:7108-16.
Katayama I, Doi T, Nishioka K (1984) High histamine level in the blister fluid of bullous
pemphigoid. Arch Dermatol Res 276:126-7.
Katz U, Achiron A, Sherer Y, Shoenfeld Y (2007) Safety of intravenous immunoglobulin
(IVIG) therapy. Autoimmun Rev 6:257-9.
Kawana S, Ueno A, Nishiyama S (1990) Increased levels of immunoreactive leukotriene
B4 in blister fluids of bullous pemphigoid patients and effects of a selective 5-lipoxygenase
inhibitor on experimental skin lesions. Acta Derm Venereol 70:281-5.
Kazlow Stern D, Tripp JM, Ho VC, Lebwohl M (2005) The use of systemic immune
moderators in dermatology: an update. Dermatol Clin 23:259-300.
Khumalo N, Kirtschig G, Middleton P, Hollis S, Wojnarowska F, Murrell D (2005)
Interventions for bullous pemphigoid. Cochrane Database Syst Rev:CD002292.
Kippes W, Schmidt E, Roth A, Rzany B, Brocker EB, Zillikens D (1999)
[Immunopathologic changes in 115 patients with bullous pemphigoid]. Hautarzt 50:866-72.
Kirtschig G, Murrell D, Wojnarowska F, Khumalo N (2002) Interventions for mucous
membrane pemphigoid/cicatricial pemphigoid and epidermolysis bullosa acquisita: a
systematic literature review. Arch Dermatol 138:380-4.
Kiss M, Husz S, Janossy T, Marczinovits I, Molnar J, Korom I, Dobozy A (2005)
Experimental bullous pemphigoid generated in mice with an antigenic epitope of the
human hemidesmosomal protein BP230. J Autoimmun 24:1-10.
Kjellman P, Eriksson H, Berg P (2008) A retrospective analysis of patients with bullous
pemphigoid treated with methotrexate. Arch Dermatol 144:612-6.

6. Literaturverzeichnis 108
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R,
Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM (2006) A
pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling.
Cell 125:733-47.
Kofler H, Wambacher-Gasser B, Topar G, Weinlich G, Schuler G, Hintner H, Romani N,
Fritsch P (1997) Intravenous immunoglobulin treatment in therapy-resistant epidermolysis
bullosa acquisita. J Am Acad Dermatol 36:331-5.
Korman N (1987) Bullous pemphigoid. J Am Acad Dermatol 16:907-24.
Krivo JM, Miller F (1978) Immunopathology of epidermolysis bullosa acquisita.
Association with mixed cryoglobulinemia. Arch Dermatol 114:1218-20.
Kromminga A, Scheckenbach C, Georgi M, Hagel C, Arndt R, Christophers E, Brocker
EB, Zillikens D (2000) Patients with bullous pemphigoid and linear IgA disease show a
dual IgA and IgG autoimmune response to BP180. J Autoimmun 15:293-300.
Kromminga A, Sitaru C, Hagel C, Herzog S, Zillikens D (2004) Development of an ELISA
for the detection of autoantibodies to BP230. Clin Immunol 111:146-52.
Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, Hirose M, Juss
J, Oxley D, Chessa TA, Ramadani F, Guillou H, Segonds-Pichon A, Fritsch A, Jarvis GE,
Okkenhaug K, Ludwig R, Zillikens D, Mocsai A, Vanhaesebroeck B, Stephens LR,
Hawkins PT (2011) PI3Kbeta plays a critical role in neutrophil activation by immune
complexes. Sci Signal 4:ra23.
Kurzhals G, Stolz W, Meurer M, Kunze J, Braun-Falco O, Krieg T (1991) Acquired
epidermolysis bullosa with the clinical feature of Brunsting-Perry cicatricial bullous
pemphigoid. Arch Dermatol 127:391-5.
Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA (1986) Molecular heterogeneity of
the bullous pemphigoid antigens as detected by immunoblotting. J Immunol 136:1231-5.
Laffitte E, Skaria M, Jaunin F, Tamm K, Saurat JH, Favre B, Borradori L (2001)
Autoantibodies to the extracellular and intracellular domain of bullous pemphigoid 180, the
putative key autoantigen in bullous pemphigoid, belong predominantly to the IgG1 and
IgG4 subclasses. Br J Dermatol 144:760-8.

6. Literaturverzeichnis 109
Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J (2008) Bullous
pemphigoid and pemphigus vulgaris--incidence and mortality in the UK: population based
cohort study. BMJ 337:a180.
Lapiere JC, Chen JD, Iwasaki T, Hu L, Uitto J, Woodley DT (1994) Type VII collagen
specifically binds fibronectin via a unique subdomain within the collagenous triple helix. J
Invest Dermatol 103:637-41.
Lapiere JC, Woodley DT, Parente MG, Iwasaki T, Wynn KC, Christiano AM, Uitto J (1993)
Epitope mapping of type VII collagen. Identification of discrete peptide sequences
recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest
92:1831-9.
Layton AM, Cunliffe WJ (1990) Clearing of epidermolysis bullosa acquisita with
cyclosporine. J Am Acad Dermatol 22:535-6.
Lever WF (1953) Pemphigus. Medicine (Baltimore) 32:1-123.
Li M, Wirthmueller U, Ravetch JV (1996) Reconstitution of human Fc gamma RIII cell type
specificity in transgenic mice. J Exp Med 183:1259-63.
Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, Roopenian DC, Liu Z
(2005) Complete FcRn dependence for intravenous Ig therapy in autoimmune skin
blistering diseases. J Clin Invest 115:3440-50.
Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D (2000) Roles of PLC-beta2 and -beta3
and PI3Kgamma in chemoattractant-mediated signal transduction. Science 287:1046-9.
Liu L, Wang YX, Zhou J, Long F, Sun HW, Liu Y, Chen YZ, Jiang CL (2005) Rapid non-
genomic inhibitory effects of glucocorticoids on human neutrophil degranulation. Inflamm
Res 54:37-41.
Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, Giudice GJ (1993) A passive
transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using
antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest
92:2480-8.
Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, Diaz LA (1995) The role of
complement in experimental bullous pemphigoid. J Clin Invest 95:1539-44.

6. Literaturverzeichnis 110
Liu Z, Giudice GJ, Zhou X, Swartz SJ, Troy JL, Fairley JA, Till GO, Diaz LA (1997) A
major role for neutrophils in experimental bullous pemphigoid. J Clin Invest 100:1256-63.
Liu Z, Shipley JM, Vu TH, Zhou X, Diaz LA, Werb Z, Senior RM (1998) Gelatinase B-
deficient mice are resistant to experimental bullous pemphigoid. J Exp Med 188:475-82.
Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, Senior RM, Werb Z (2000)
The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in
vivo. Cell 102:647-55.
Loo WJ, Kirtschig G, Wojnarowska F (2001) Minocycline as a therapeutic option in bullous
pemphigoid. Clin Exp Dermatol 26:376-9.
Magnusson SE, Andren M, Nilsson KE, Sondermann P, Jacob U, Kleinau S (2008)
Amelioration of collagen-induced arthritis by human recombinant soluble FcgammaRIIb.
Clin Immunol 127:225-33.
Marazza G, Pham HC, Scharer L, Pedrazzetti PP, Hunziker T, Trueb RM, Hohl D, Itin P,
Lautenschlager S, Naldi L, Borradori L (2009) Incidence of bullous pemphigoid and
pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol 161:861-8.
Marshall JS (2004) Mast-cell responses to pathogens. Nat Rev Immunol 4:787-99.
Masuda M, Morimoto T, Kobatake S, Nishimura N, Nakamoto K, Dong XH, Komiyama Y,
Ogawa R, Takahashi H (2003) Measurement of soluble Fcgamma receptor type IIIa
derived from macrophages in plasma: increase in patients with rheumatoid arthritis. Clin
Exp Immunol 132:477-84.
Masunaga T, Shimizu H, Yee C, Borradori L, Lazarova Z, Nishikawa T, Yancey KB (1997)
The extracellular domain of BPAG2 localizes to anchoring filaments and its carboxyl
terminus extends to the lamina densa of normal human epidermal basement membrane. J
Invest Dermatol 109:200-6.
Mayuzumi M, Akiyama M, Nishie W, Ukae S, Abe M, Sawamura D, Hashimoto T, Shimizu
H (2006) Childhood epidermolysis bullosa acquisita with autoantibodies against the
noncollagenous 1 and 2 domains of type VII collagen: case report and review of the
literature. Br J Dermatol 155:1048-52.
McMillan JR, Matsumura T, Hashimoto T, Schumann H, Bruckner-Tuderman L, Shimizu H
(2003) Immunomapping of EBA sera to multiple epitopes on collagen VII: further evidence
that anchoring fibrils originate and terminate in the lamina densa. Exp Dermatol 12:261-7.

6. Literaturverzeichnis 111
Megahed M, Scharffetter-Kochanek K (1994) Epidermolysis bullosa acquisita--successful
treatment with colchicine. Arch Dermatol Res 286:35-46.
Meier F, Sonnichsen K, Schaumburg-Lever G, Dopfer R, Rassner G (1993) Epidermolysis
bullosa acquisita: efficacy of high-dose intravenous immunoglobulins. J Am Acad
Dermatol 29:334-7.
Merle C, Blanc D, Zultak M, van Landuyt H, Drobacheff C, Laurent R (1990) Intractable
epidermolysis bullosa acquisita: efficacy of cyclosporin A. Dermatologica 181:44-7.
Messingham KA, Noe MH, Chapman MA, Giudice GJ, Fairley JA (2009) A novel ELISA
reveals high frequencies of BP180-specific IgE production in bullous pemphigoid. J
Immunol Methods 346:18-25.
Miida H, Fujiwara H, Ito M (2011) Association between effective dose of prednisolone,
alone or in conjunction with other immunosuppressants, and titre of anti-bullous
pemphigoid 180 antibody: a retrospective study of 42 cases. Clin Exp Dermatol 36:485-8.
Milligan A, Hutchinson PE (1990) The use of chlorambucil in the treatment of bullous
pemphigoid. J Am Acad Dermatol 22:796-801.
Mohr C, Sunderkotter C, Hildebrand A, Biel K, Rutter A, Rutter GH, Luger TA, Kolde G
(1995) Successful treatment of epidermolysis bullosa acquisita using intravenous
immunoglobulins. Br J Dermatol 132:824-6.
Mooney E, Falk RJ, Gammon WR (1992) Studies on complement deposits in
epidermolysis bullosa acquisita and bullous pemphigoid. Arch Dermatol 128:58-60.
Mooney E, Gammon WR (1990) Heavy and light chain isotypes of immunoglobulin in
epidermolysis bullosa acquisita. J Invest Dermatol 95:317-9.
Mrowietz U, Klein CE, Reich K, Rosenbach T, Ruzicka T, Sebastian M, Werfel T (2009)
Cyclosporine therapy in dermatology. J Dtsch Dermatol Ges 7:474-9.
Mutasim DF (2000) Levels of antibodies to BP180 correlate with disease activity in bullous
pemphigoid. Arch Dermatol 136:253-4.
Mutasim DF (2004) Management of autoimmune bullous diseases: pharmacology and
therapeutics. J Am Acad Dermatol 51:859-77; quiz 78-80.

6. Literaturverzeichnis 112
Nagel A, Hertl M, Eming R (2009) B-cell-directed therapy for inflammatory skin diseases.
J Invest Dermatol 129:289-301.
Nakatani C, Muramatsu T, Shirai T (1998) Immunoreactivity of bullous pemphigoid (BP)
autoantibodies against the NC16A and C-terminal domains of the 180 kDa BP antigen
(BP180): immunoblot analysis and enzyme-linked immunosorbent assay using BP180
recombinant proteins. Br J Dermatol 139:365-70.
Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA (2004) Spectrum of
autoimmune bullous diseases in Kuwait. Int J Dermatol 43:876-81.
Nelson KC, Zhao M, Schroeder PR, Li N, Wetsel RA, Diaz LA, Liu Z (2006) Role of
different pathways of the complement cascade in experimental bullous pemphigoid. J Clin
Invest 116:2892-900.
Nieboer C, Boorsma DM, Woerdeman MJ, Kalsbeek GL (1980) Epidermolysis bullosa
acquisita. Immunofluorescence, electron microscopic and immunoelectron microscopic
studies in four patients. Br J Dermatol 102:383-92.
Nimmerjahn F, Ravetch JV (2008) Fcgamma receptors as regulators of immune
responses. Nat Rev Immunol 8:34-47.
Nonaka S, Ishiko A, Masunaga T, Akiyama M, Owaribe K, Shimizu H, Nishikawa T (2000)
The extracellular domain of BPAG2 has a loop structure in the carboxy terminal flexible
tail in vivo. J Invest Dermatol 115:889-92.
Ogawa Y, Adachi A, Okamoto M, Hashimoto T, Tomita Y (2004) A case of refractory
bullous pemphigoid with plasmapheresis-associated thrombopenia: efficacy of pulsed
intravenous cyclophosphamide therapy. J Dermatol 31:651-4.
Olsen EA (1991) The pharmacology of methotrexate. J Am Acad Dermatol 25:306-18.
Ortiz Z, Shea B, Suarez Almazor M, Moher D, Wells G, Tugwell P (2000) Folic acid and
folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid
arthritis. Cochrane Database Syst Rev:CD000951.
Palmer RG, Denman AM (1984) Malignancies induced by chlorambucil. Cancer Treat Rev
11:121-9.

6. Literaturverzeichnis 113
Parekh DB, Ziegler W, Parker PJ (2000) Multiple pathways control protein kinase C
phosphorylation. EMBO J 19:496-503.
Parente MG, Chung LC, Ryynanen J, Woodley DT, Wynn KC, Bauer EA, Mattei MG, Chu
ML, Uitto J (1991) Human type VII collagen: cDNA cloning and chromosomal mapping of
the gene. Proc Natl Acad Sci U S A 88:6931-5.
Parker SR, Dyson S, Brisman S, Pennie M, Swerlick RA, Khan R, Manos S, Korman BD,
Xia Z, Korman NJ (2008) Mortality of bullous pemphigoid: an evaluation of 223 patients
and comparison with the mortality in the general population in the United States. J Am
Acad Dermatol 59:582-8.
Patel AA, Swerlick RA, McCall CO (2006) Azathioprine in dermatology: the past, the
present, and the future. J Am Acad Dermatol 55:369-89.
Perriard J, Jaunin F, Favre B, Budinger L, Hertl M, Saurat JH, Borradori L (1999) IgG
autoantibodies from bullous pemphigoid (BP) patients bind antigenic sites on both the
extracellular and the intracellular domains of the BP antigen 180. J Invest Dermatol
112:141-7.
Prussick R, Ali MA, Rosenthal D, Guyatt G (1992) The protective effect of vitamin E on the
hemolysis associated with dapsone treatment in patients with dermatitis herpetiformis.
Arch Dermatol 128:210-3.
Puri KD, Doggett TA, Douangpanya J, Hou Y, Tino WT, Wilson T, Graf T, Clayton E,
Turner M, Hayflick JS, Diacovo TG (2004) Mechanisms and implications of
phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue.
Blood 103:3448-56.
Puri KD, Doggett TA, Huang CY, Douangpanya J, Hayflick JS, Turner M, Penninger J,
Diacovo TG (2005) The role of endothelial PI3Kgamma activity in neutrophil trafficking.
Blood 106:150-7.
Rameh LE, Cantley LC (1999) The role of phosphoinositide 3-kinase lipid products in cell
function. J Biol Chem 274:8347-50.
Ray TL, Levine JB, Weiss W, Ward PA (1982) Epidermolysis bullosa acquisita and
inflammatory bowel disease. J Am Acad Dermatol 6:242-52.

6. Literaturverzeichnis 114
Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, Henley A, Di-Stefano F,
Ahmad Z, Guillard S, Bjerke LM, Kelland L, Valenti M, Patterson L, Gowan S, de Haven
Brandon A, Hayakawa M, Kaizawa H, Koizumi T, Ohishi T, Patel S, Saghir N, Parker P,
Waterfield M, Workman P (2007) Pharmacologic characterization of a potent inhibitor of
class I phosphatidylinositide 3-kinases. Cancer Res 67:5840-50.
Remington J, Chen M, Burnett J, Woodley DT (2008) Autoimmunity to type VII collagen:
epidermolysis bullosa acquisita. Curr Dir Autoimmun 10:195-205.
Roenigk HH, Jr., Ryan JG, Bergfeld WF (1971) Epidermolysis bullosa acquisita. Report of
three cases and review of all published cases. Arch Dermatol 103:1-10.
Rommel C, Camps M, Ji H (2007) PI3K delta and PI3K gamma: partners in crime in
inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol 7:191-201.
Rosenberg FR, Sanders S, Nelson CT (1976) Pemphigus: a 20-year review of 107
patients treated with corticosteroids. Arch Dermatol 112:962-70.
Roth JA, Kaeberle ML (1981) Evaluation of bovine polymorphonuclear leukocyte function.
Vet Immunol Immunopathol 2:157-74.
Roujeau JC, Lok C, Bastuji-Garin S, Mhalla S, Enginger V, Bernard P (1998) High risk of
death in elderly patients with extensive bullous pemphigoid. Arch Dermatol 134:465-9.
Roussel A, Benichou J, Randriamanantany ZA, Gilbert D, Drenovska K, Houivet E, Tron
F, Joly P (2011) Enzyme-linked immunosorbent assay for the combination of bullous
pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol
147:293-8.
Rousselle P, Keene DR, Ruggiero F, Champliaud MF, Rest M, Burgeson RE (1997)
Laminin 5 binds the NC-1 domain of type VII collagen. J Cell Biol 138:719-28.
Rudolph SA, Greengard P, Malawista SE (1977) Effects of colchicine on cyclic AMP levels
in human leukocytes. Proc Natl Acad Sci U S A 74:3404-8.
Ruhrberg C, Watt FM (1997) The plakin family: versatile organizers of cytoskeletal
architecture. Curr Opin Genet Dev 7:392-7.

6. Literaturverzeichnis 115
Rzany B, Partscht K, Jung M, Kippes W, Mecking D, Baima B, Prudlo C, Pawelczyk B,
Messmer EM, Schuhmann M, Sinkgraven R, Buchner L, Budinger L, Pfeiffer C, Sticherling
M, Hertl M, Kaiser HW, Meurer M, Zillikens D, Messer G (2002) Risk factors for lethal
outcome in patients with bullous pemphigoid: low serum albumin level, high dosage of
glucocorticosteroids, and old age. Arch Dermatol 138:903-8.
Sadhu C, Dick K, Tino WT, Staunton DE (2003a) Selective role of PI3K delta in neutrophil
inflammatory responses. Biochem Biophys Res Commun 308:764-9.
Sadhu C, Masinovsky B, Dick K, Sowell CG, Staunton DE (2003b) Essential role of
phosphoinositide 3-kinase delta in neutrophil directional movement. J Immunol 170:2647-
54.
Sadler E, Schafleitner B, Lanschuetzer C, Laimer M, Pohla-Gubo G, Hametner R, Hintner
H, Bauer JW (2007) Treatment-resistant classical epidermolysis bullosa acquisita
responding to rituximab. Br J Dermatol 157:417-9.
Sakai LY, Keene DR, Morris NP, Burgeson RE (1986) Type VII collagen is a major
structural component of anchoring fibrils. J Cell Biol 103:1577-86.
Saleh MA, Ishii K, Kim YJ, Murakami A, Ishii N, Hashimoto T, Schmidt E, Zillikens D,
Shirakata Y, Hashimoto K, Kitajima Y, Amagai M (2011) Development of NC1 and NC2
domains of type VII collagen ELISA for the diagnosis and analysis of the time course of
epidermolysis bullosa acquisita patients. J Dermatol Sci 62:169-75.
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B,
Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A,
Penninger JM (2000) Function of PI3Kgamma in thymocyte development, T cell
activation, and neutrophil migration. Science 287:1040-6.
Schaapveld RQ, Borradori L, Geerts D, van Leusden MR, Kuikman I, Nievers MG,
Niessen CM, Steenbergen RD, Snijders PJ, Sonnenberg A (1998) Hemidesmosome
formation is initiated by the beta4 integrin subunit, requires complex formation of beta4
and HD1/plectin, and involves a direct interaction between beta4 and the bullous
pemphigoid antigen 180. J Cell Biol 142:271-84.
Schmidt E (2009) [Optimizing therapy in patients with severe autoimmune blistering skin
diseases]. Hautarzt 60:633-40.
Schmidt E, Bastian B, Dummer R, Tony HP, Brocker EB, Zillikens D (1996) Detection of
elevated levels of IL-4, IL-6, and IL-10 in blister fluid of bullous pemphigoid. Arch Dermatol
Res 288:353-7.

6. Literaturverzeichnis 116
Schmidt E, Benoit S, Brocker EB, Zillikens D, Goebeler M (2006a) Successful adjuvant
treatment of recalcitrant epidermolysis bullosa acquisita with anti-CD20 antibody
rituximab. Arch Dermatol 142:147-50.
Schmidt E, della Torre R, Borradori L (2011) Clinical features and practical diagnosis of
bullous pemphigoid. Dermatol Clin 29:427-38, viii-ix.
Schmidt E, Hopfner B, Chen M, Kuhn C, Weber L, Brocker EB, Bruckner-Tuderman L,
Zillikens D (2002) Childhood epidermolysis bullosa acquisita: a novel variant with
reactivity to all three structural domains of type VII collagen. Br J Dermatol 147:592-7.
Schmidt E, Hunzelmann N, Zillikens D, Brocker EB, Goebeler M (2006b) Rituximab in
refractory autoimmune bullous diseases. Clin Exp Dermatol 31:503-8.
Schmidt E, Kraensel R, Goebeler M, Sinkgraven R, Brocker EB, Rzany B, Zillikens D
(2005) Treatment of bullous pemphigoid with dapsone, methylprednisolone, and topical
clobetasol propionate: a retrospective study of 62 cases. Cutis 76:205-9.
Schmidt E, Obe K, Brocker EB, Zillikens D (2000) Serum levels of autoantibodies to
BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol
136:174-8.
Schmidt E, Reimer S, Kruse N, Brocker EB, Zillikens D (2001) The IL-8 release from
cultured human keratinocytes, mediated by antibodies to bullous pemphigoid autoantigen
180, is inhibited by dapsone. Clin Exp Immunol 124:157-62.
Schmidt E, Seitz CS, Benoit S, Brocker EB, Goebeler M (2007) Rituximab in autoimmune
bullous diseases: mixed responses and adverse effects. Br J Dermatol 156:352-6.
Schmidt E, Zillikens D (2010) Immunoadsorption in dermatology. Arch Dermatol Res
302:241-53.
Schmidt E, Zillikens D (2011) The diagnosis and treatment of autoimmune blistering skin
diseases. Dtsch Arztebl Int 108:399-405, I-III.
Schneir M, Ramamurthy N, Golub L (1990) Minocycline-treatment of diabetic rats
normalizes skin collagen production and mass: possible causative mechanisms. Matrix
10:112-23.

6. Literaturverzeichnis 117
Segura S, Iranzo P, Martinez-de Pablo I, Mascaro JM, Jr., Alsina M, Herrero J, Herrero C
(2007) High-dose intravenous immunoglobulins for the treatment of autoimmune
mucocutaneous blistering diseases: evaluation of its use in 19 cases. J Am Acad
Dermatol 56:960-7.
Sengelov H, Kjeldsen L, Borregaard N (1993) Control of exocytosis in early neutrophil
activation. J Immunol 150:1535-43.
Sesarman A, Sitaru AG, Olaru F, Zillikens D, Sitaru C (2008) Neonatal Fc receptor
deficiency protects from tissue injury in experimental epidermolysis bullosa acquisita. J
Mol Med (Berl) 86:951-9.
Shah N, Green AR, Elgart GW, Kerdel F (2000) The use of chlorambucil with prednisone
in the treatment of pemphigus. J Am Acad Dermatol 42:85-8.
Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Brocker EB, Opdenakker G, Zillikens
D, Sitaru C (2004) Granulocyte-derived elastase and gelatinase B are required for dermal-
epidermal separation induced by autoantibodies from patients with epidermolysis bullosa
acquisita and bullous pemphigoid. J Pathol 204:519-27.
Shimizu H, Ishiko A, Masunaga T, Kurihara Y, Sato M, Bruckner-Tuderman L, Nishikawa
T (1997) Most anchoring fibrils in human skin originate and terminate in the lamina densa.
Lab Invest 76:753-63.
Shirahama S, Furukawa F, Yagi H, Tanaka T, Hashimoto T, Takigawa M (1998) Bullous
systemic lupus erythematosus: detection of antibodies against noncollagenous domain of
type VII collagen. J Am Acad Dermatol 38:844-8.
Sitaru C, Dahnrich C, Probst C, Komorowski L, Blocker I, Schmidt E, Schlumberger W,
Rose C, Stocker W, Zillikens D (2007) Enzyme-linked immunosorbent assay using
multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and
specific detection of pemphigoid autoantibodies. Exp Dermatol 16:770-7.
Sitaru C, Goebeler M, Zillikens D (2004) [Bullous autoimmune dermatoses (I):
Pathogenesis and diagnosis]. J Dtsch Dermatol Ges 2:123-8; quiz 39-40.
Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D (2002a) Autoantibodies to
type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-
epidermal separation in cryosections of human skin. Am J Pathol 161:301-11.

6. Literaturverzeichnis 118
Sitaru C, Mihai S, Otto C, Chiriac MT, Hausser I, Dotterweich B, Saito H, Rose C, Ishiko
A, Zillikens D (2005) Induction of dermal-epidermal separation in mice by passive transfer
of antibodies specific to type VII collagen. J Clin Invest 115:870-8.
Sitaru C, Schmidt E, Petermann S, Munteanu LS, Brocker EB, Zillikens D (2002b)
Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in
cryosections of human skin. J Invest Dermatol 118:664-71.
Skaria M, Jaunin F, Hunziker T, Riou S, Schumann H, Bruckner-Tuderman L, Hertl M,
Bernard P, Saurat JH, Favre B, Borradori L (2000) IgG autoantibodies from bullous
pemphigoid patients recognize multiple antigenic reactive sites located predominantly
within the B and C subdomains of the COOH-terminus of BP230. J Invest Dermatol
114:998-1004.
Smoller BR, Woodley DT (1992) Differences in direct immunofluorescence staining
patterns in epidermolysis bullosa acquisita and bullous pemphigoid. J Am Acad Dermatol
27:674-8.
Snow JL, Gibson LE (1995) The role of genetic variation in thiopurine methyltransferase
activity and the efficacy and/or side effects of azathioprine therapy in dermatologic
patients. Arch Dermatol 131:193-7.
Sondermann P, Jacob U (1999) Human Fcgamma receptor IIb expressed in Escherichia
coli reveals IgG binding capability. Biol Chem 380:717-21.
Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI (1981) Characterization
of bullous pemphigoid antigen: a unique basement membrane protein of stratified
squamous epithelia. Cell 24:897-903.
Stewart MI, Woodley DT, Briggaman RA (1991) Epidermolysis bullosa acquisita and
associated symptomatic esophageal webs. Arch Dermatol 127:373-7.
SuppreMol (11.07.2011) SuppreMol initiates Phase IIa clinical trial in Systemic Lupus
Erythematosus (SLE) with its lead candidate SM101.
<http://www.suppremol.com/tl_files/pdf/press_releases/2011/2011_07_11_SM101_Ph2_S
tart_EN_final.pdf> Accessed 20.11.2011.
SuppreMol (12.04.2010) SuppreMol initiates Phase Ib/IIa clinical trial with its lead
candidate SM101.
<http://www.suppremol.com/tl_files/pdf/press_releases/2010/2010_04_12_SM101_Ph1b-
2a_EN_final.pdf> Accessed 20.11.2011.

6. Literaturverzeichnis 119
SuppreMol (14.02.2012) SuppreMol releases positive interim Phase Ib/IIa results on
SM101 in Primary Immune Thrombocytopenia (ITP) trials
<http://www.suppremol.com/tl_files/pdf/press_releases/2012/2012-02-
14_SuppreMol_ITP_PhIb_EN_final.pdf> Accessed 05.06.2013.
SuppreMol (19.04.2010) SuppreMol Receives U.S. Orphan Drug Designation for SM101.
<http://www.suppremol.com/tl_files/pdf/press_releases/2010/2010-04-
19_SuppreMol_PR_ODD_US_en_final.pdf> Accessed 20.11.2011.
Talbott JH, Lever WF, Consolazio WV (1940) Metabolic studies on patients with
pemphigus J Invest Dermatol 3:31-68.
Tampoia M, Lattanzi V, Zucano A, Villalta D, Filotico R, Fontana A, Vena GA, Di Serio F
(2009) Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous
pemphigoid. Ann N Y Acad Sci 1173:15-20.
Tanaka H, Ishida-Yamamoto A, Hashimoto T, Hiramoto K, Harada T, Kawachi Y, Shimizu
H, Tanaka T, Kishiyama K, Hopfner B, Takahashi H, Iizuka H, Bruckner-Tuderman L
(1997) A novel variant of acquired epidermolysis bullosa with autoantibodies against the
central triple-helical domain of type VII collagen. Lab Invest 77:623-32.
Tanaka T, Furukawa F, Imamura S (1994) Epitope mapping for epidermolysis bullosa
acquisita autoantibody by molecularly cloned cDNA for type VII collagen. J Invest
Dermatol 102:706-9.
Thoma-Uszynski S, Uter W, Schwietzke S, Hofmann SC, Hunziker T, Bernard P, Treudler
R, Zouboulis CC, Schuler G, Borradori L, Hertl M (2004) BP230- and BP180-specific auto-
antibodies in bullous pemphigoid. J Invest Dermatol 122:1413-22.
Thomas I, Khorenian S, Arbesfeld DM (1993) Treatment of generalized bullous
pemphigoid with oral tetracycline. J Am Acad Dermatol 28:74-7.
Thong YH, Ferrante A (1979) Inhibition of mitogen-induced human lymphocyte
proliferative responses by tetracycline analogues. Clin Exp Immunol 35:443-6.
Tirado-Sanchez A, Diaz-Molina V, Ponce-Olivera RM (2011) Efficacy and safety of
azathioprine and dapsone as an adjuvant in the treatment of bullous pemphigoid. Allergol
Immunopathol (Madr).

6. Literaturverzeichnis 120
Traynor-Kaplan AE, Thompson BL, Harris AL, Taylor P, Omann GM, Sklar LA (1989)
Transient increase in phosphatidylinositol 3,4-bisphosphate and phosphatidylinositol
trisphosphate during activation of human neutrophils. J Biol Chem 264:15668-73.
Ujiie H, Shibaki A, Nishie W, Shimizu H (2010) What's new in bullous pemphigoid. J
Dermatol 37:194-204.
Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R,
Parker PJ, Waterfield MD (2001) Synthesis and function of 3-phosphorylated inositol
lipids. Annu Rev Biochem 70:535-602.
Veri MC, Gorlatov S, Li H, Burke S, Johnson S, Stavenhagen J, Stein KE, Bonvini E,
Koenig S (2007) Monoclonal antibodies capable of discriminating the human inhibitory
Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A):
biochemical, biological and functional characterization. Immunology 121:392-404.
Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P (2001) Respective
contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of
BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol 117:1091-6.
Vodegel RM, de Jong MC, Pas HH, Jonkman MF (2002) IgA-mediated epidermolysis
bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol 47:919-25.
Vodegel RM, Jonkman MF, Pas HH, de Jong MC (2004) U-serrated immunodeposition
pattern differentiates type VII collagen targeting bullous diseases from other subepidermal
bullous autoimmune diseases. Br J Dermatol 151:112-8.
Weisenseel P, Martin S, Partscht K, Messer G, Prinz JC (2007) Relevance of the low-
affinity type of the Fcgamma-receptor IIIa-polymorphism in bullous pemphigoid. Arch
Dermatol Res 299:163-4.
Weiss SJ (1989) Tissue destruction by neutrophils. N Engl J Med 320:365-76.
Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H,
Tempst P, Hawkins PT, Stephens LR (2002) P-Rex1, a PtdIns(3,4,5)P3- and
Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell 108:809-21.
Werwitzke S, Trick D, Sondermann P, Kamino K, Schlegelberger B, Kniesch K, Tiede A,
Jacob U, Schmidt RE, Witte T (2008) Treatment of lupus-prone NZB/NZW F1 mice with
recombinant soluble Fc gamma receptor II (CD32). Ann Rheum Dis 67:154-61.

6. Literaturverzeichnis 121
Wines BD, Gavin A, Powell MS, Steinitz M, Buchanan RR, Mark Hogarth P (2003) Soluble
FcgammaRIIa inhibits rheumatoid factor binding to immune complexes. Immunology
109:246-54.
Wojnarowska F, Kirtschig G, Highet AS, Venning VA, Khumalo NP (2002) Guidelines for
the management of bullous pemphigoid. Br J Dermatol 147:214-21.
Wojnarowska F, Kirtschig G, Khumalo N (2001) Treatment of subepidermal
immunobullous diseases. Clin Dermatol 19:768-77.
Wong SN, Chua SH (2002) Spectrum of subepidermal immunobullous disorders seen at
the National Skin Centre, Singapore: a 2-year review. Br J Dermatol 147:476-80.
Woodley DT, Briggaman RA, O'Keefe EJ, Inman AO, Queen LL, Gammon WR (1984)
Identification of the skin basement-membrane autoantigen in epidermolysis bullosa
acquisita. N Engl J Med 310:1007-13.
Woodley DT, Burgeson RE, Lunstrum G, Bruckner-Tuderman L, Reese MJ, Briggaman
RA (1988) Epidermolysis bullosa acquisita antigen is the globular carboxyl terminus of
type VII procollagen. J Clin Invest 81:683-7.
Workman P, Clarke PA, Raynaud FI, van Montfort RL (2010) Drugging the PI3 kinome:
from chemical tools to drugs in the clinic. Cancer Res 70:2146-57.
Wymann MP, Sozzani S, Altruda F, Mantovani A, Hirsch E (2000) Lipids on the move:
phosphoinositide 3-kinases in leukocyte function. Immunol Today 21:260-4.
Yaoita H, Briggaman RA, Lawley TJ, Provost TT, Katz SI (1981) Epidermolysis bullosa
acquisita: ultrastructural and immunological studies. J Invest Dermatol 76:288-92.
Yoshida M, Hamada T, Amagai M, Hashimoto K, Uehara R, Yamaguchi K, Imamura K,
Okamoto E, Yasumoto S, Hashimoto T (2006) Enzyme-linked immunosorbent assay
using bacterial recombinant proteins of human BP230 as a diagnostic tool for bullous
pemphigoid. J Dermatol Sci 41:21-30.
Yu X, Holdorf K, Kasper B, Zillikens D, Ludwig RJ, Petersen F (2010) FcgammaRIIA and
FcgammaRIIIB are required for autoantibody-induced tissue damage in experimental
human models of bullous pemphigoid. J Invest Dermatol 130:2841-4.

6. Literaturverzeichnis 122
Zachariae H (1987) Cyclosporine A in epidermolysis bullosa acquisita. J Am Acad
Dermatol 17:1058-9.
Zhao M, Trimbeger ME, Li N, Diaz LA, Shapiro SD, Liu Z (2006) Role of FcRs in animal
model of autoimmune bullous pemphigoid. J Immunol 177:3398-405.
Zhu YI, Stiller MJ (2001) Dapsone and sulfones in dermatology: overview and update. J
Am Acad Dermatol 45:420-34.
Zillikens D (2005) Blasenbildende Autoimmundermatosen. In: Dermatolgie und
Venerologie (Braun-Falco O, Wolff HH, eds) Vol. 5, Berlin: Springer Verlag, 607-38.
Zillikens D, Mascaro JM, Rose PA, Liu Z, Ewing SM, Caux F, Hoffmann RG, Diaz LA,
Giudice GJ (1997a) A highly sensitive enzyme-linked immunosorbent assay for the
detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J
Invest Dermatol 109:679-83.
Zillikens D, Rose PA, Balding SD, Liu Z, Olague-Marchan M, Diaz LA, Giudice GJ (1997b)
Tight clustering of extracellular BP180 epitopes recognized by bullous pemphigoid
autoantibodies. J Invest Dermatol 109:573-9.
Zillikens D, Wever S, Roth A, Weidenthaler-Barth B, Hashimoto T, Brocker EB (1995)
Incidence of autoimmune subepidermal blistering dermatoses in a region of central
Germany. Arch Dermatol 131:957-8.
Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L (2007) IgE basement membrane zone
antibodies induce eosinophil infiltration and histological blisters in engrafted human skin
on SCID mice. J Invest Dermatol 127:1167-74.
Zuidema J, Hilbers-Modderman ES, Merkus FW (1986) Clinical pharmacokinetics of
dapsone. Clin Pharmacokinet 11:299-315.

7. Anhang 123
7. Anhang
7.1 Voten der Ethikkommission
Alle durchgeführten Untersuchungen wurden von der zuständigen
Ethikkommission positiv begutachtet und sind im Folgenden aufgeführt.
Aktenzeichen (Datum des Genehmigungsschreibens): 09-140 (13.11.2009), 05-
056 (21.04.2005), 04-144 (28.10.2004), 04-061 (11.05.2004)

7. Anhang 124
7.2 Abbildungsverzeichnis
Abbildung 1: Klinisches Erscheinungsbild des bullösen Pemphigoids ................................. 5
Abbildung 2: Autoantigene der Haut (nach (Bieber et al., 2010)).. ........................................ 6
Abbildung 3: Schematische Darstellung von BP180 (nach (Ujiie et al., 2010)) ................... 7
Abbildung 4: Histopathologischer Befund eines bullösen Pemphigoids. ............................ 12
Abbildung 5: Indirekte Immunfluoreszenz beim bullösem Pemphigoid. ............................. 14
Abbildung 6: Klinisches Erscheinungsbild der Epidermolysis bullosa acquisita (EBA). ... 17
Abbildung 7: Histopathologischer Befund bei der
Epidermolysis bullosa acquisita (EBA) .............................................................. 20
Abbildung 8: Indirekte Immunfluoreszenz bei der
Epidermolysis bullosa acquisita (EBA) .............................................................. 21
Abbildung 9: Das Prinzip des Gefrierschnittmodells. ............................................................. 33
Abbildung 10: Testung der EBA-Seren auf Gefrierschnitten von Maushaut. ..................... 49
Abbildung 11: Testung der BP-Seren auf humanen Hautschnitten ..................................... 50
Abbildung 12: Die Ablagerung von Formazan ist abhängig von der
Methylprednisolon (MP)-Dosis (nach (Hellberg et al., 2013)) ...................... 53
Abbildung 13: Ablagerung von Formazan entlang der dermo-epidermalen
Junktionszone (DEJ) (nach (Hellberg et al., 2013)) ..................................... 53
Abbildung 14: Die Freisetzung von reaktiven Sauerstoffspezies (ROS) ist abhängig
von der Methylprednisolon (MP)-Dosis (nach (Hellberg et al., 2013) ......... 55
Abbildung 15: Effekte von PI-103 auf die Spaltbildung in Maushaut.. ................................ 57
Abbildung 16: Repräsentative Bilder des Effekts von PI-103 auf die Spaltbildung
in Maushaut. ........................................................................................................ 58
Abbildung 17: Effekte von PI-103 auf die Spaltbildung in humaner Haut.. ......................... 59
Abbildung 18: Repräsentative Bilder des Effekts von PI-103 auf die Spaltbildung
in humaner Haut .................................................................................................. 60
Abbildung 19: Effekte von TGX-221 auf die Spaltbildung in Maushaut.. ............................ 62
Abbildung 20: Repräsentative Bilder des Effekts von TGX-221 auf die Spaltbildung
in Maushaut. ........................................................................................................ 63
Abbildung 21: Effekte von TGX-221 auf die Spaltbildung in humaner Haut. ..................... 64
Abbildung 22: Repräsentative Bilder des Effekts von TGX-221 auf die Spaltbildung
in humaner Haut .................................................................................................. 65
Abbildung 23: Effekte von SM101 auf die Spaltbildung in Maushaut.................................. 66
Abbildung 24: Repräsentative Bilder des Effekts von SM101 auf die Spaltbildung
in Maushaut. ........................................................................................................ 67
Abbildung 25: Effekte von SM101 auf die Spaltbildung in humaner Haut .......................... 68
Abbildung 26: Repräsentative Bilder des Effekts von SM101 auf die Spaltbildung
in humaner Haut .................................................................................................. 69

7. Anhang 125
7.3 Tabellenverzeichnis
Tabelle 1: Blasenbildende Autoimmundermatosen und deren Zielantigene
(nach (Schmidt and Zillikens, 2011; Sitaru et al., 2004)) ........................................ 1
Tabelle 2: Ergebnisse der Serumtestung von EBA-Seren im Gefrierschnittmodell ........... 48
Tabelle 3: Ergebnisse der Serumtestung von BP-Patientenseren im
Gefrierschnittmodell ................................................................................................... 50

8. Danksagungen 126
8. Danksagungen
Ich danke Herrn Prof. Dr. med. D. Zillikens, Direktor der Klinik für Dermatologie,
Allergologie und Venerologie der Universität zu Lübeck, für die Möglichkeit der
Durchführung meiner Arbeit, sowie der Bereitstellung des dermatologischen
Forschungslabors.
Mein großer Dank gilt auch Herrn Prof. Dr. med. Ralf Ludwig für die Überlassung
des Themas, die Anregungen während der experimentellen Arbeit und der
Erstellung der Arbeit sowie die Möglichkeit der Veröffentlichung meiner
Ergebnisse.
Mein besonderer Dank gilt Herrn Dr. med. Andreas Recke für die Ratschläge und
Unterstützung während der experimentellen Arbeit.
Ganz herzlich möchte ich mich bei allen Mitarbeitern der Arbeitsgruppe
Autoimmunität bedanken, die mir vor allem während der Durchführung der
Experimente immer hilfreich zur Seite standen. Mein besonderer Dank gilt
Rebecca Cames und Claudia Kauderer.
Auch gilt den Patienten und den gesunden Blutspendern großer Dank, mit deren
Hilfsbereitschaft die Durchführung der Experimente ermöglicht wurde.
Zuletzt danke ich meiner Familie für die Unterstützung während meiner gesamten
Studienzeit und während der Fertigstellung dieser Arbeit.

9. Lebenslauf 127
9. Lebenslauf
Persönliche Daten
Name: Möckel
Vorname: Nicole
Geburtsdatum: 02.06.1988
Geburtsort: Schlema
Hochschulstudium
2006 - 2008 Studium der Humanmedizin, Martin-Luther-Universität
Halle-Wittenberg
08 / 2008 1. Staatsexamen
2008 - 2013 Studium der Humanmedizin, Universität zu Lübeck
04 / 2013 2. Staatsexamen
Famulaturen
2009 Anästhesie: Helios Klinikum Aue
2009 Innere Medizin: Sana Kliniken Lübeck
2010 Chirurgie: Sana Kliniken Lübeck
2011 Ambulanz der Chirurgie: Universität zu Lübeck
Praktisches Jahr
02 - 06 / 2012 Innere Medizin: Sana Kliniken Lübeck
06 - 10 / 2012 Chirurgie: Hospital Regional, Jinotepe, Nicaragua
10 / 2012 - 01 / 2013 Wahlfach Dermatologie: Klinik für Dermatologie,
Allergologie und Venerologie der Universität zu Lübeck
Zeitraum der Dissertation
03 / 2010 - 09 / 2011 Durchführung der Experimente: Forschungslabor der
Klinik für Dermatologie, Allergologie und Venerologie,
Universität zu Lübeck
10 / 2011 - 02 / 2012 Promotionssemester zum Verfassen der Arbeit
06 / 2013 Fertigstellung der Arbeit

10. Publikationen 128
10. Publikationen
(1) Hellberg L, Samavedam UK, Holdorf K, Hansel M, Recke A, Beckmann T,
Steinhorst K, Boehncke WH, Kirchner T, Mockel N, Solbach W, Zillikens D,
Schmidt E, Ludwig RJ, Laskay T (2013) Methylprednisolone Blocks
Autoantibody-Induced Tissue Damage in Experimental Models of Bullous
Pemphigoid and Epidermolysis Bullosa Acquisita through Inhibition of
Neutrophil Activation. J Invest Dermatol. doi:10.1038/jid.2013.91
Eine Veröffentlichung der Daten zur Hemmung der dermo-epidermalen
Spaltbildung durch den löslichen FcγRIIb (SM101) wird in Kürze folgen.