perspectivas presentes y futuras en las enfermedades de … · 2013-05-19 · mpsiv (morquio)...
TRANSCRIPT

Perspectivas presentes y futuras en las enfermedades de acúmulo lisosomial.
Dr. Antonio Baldellou Vázquez.

PATOLOGÍA DE ORGANELASCELULARES

BIOGÉNESIS DE LOS LISOSOMAS (I)
Formados por el retículo endoplasmático
rugoso y luego empaquetados por el
complejo de Golgi, contienen enzimas
hidrolíticas y proteolíticas que sirven para
digerir los materiales de origen externo o
interno que llegan a ellos.
Rodeados de una membrana, compuesta
por lípidos y proteínas glicosiladas, que
protege al resto de la célula de la acción de
sus enzimas .
Estas, para más seguridad actúan
fundamentalmente en un medio ácido y
son capaces de digerir polisacáridos,
proteínas, sulfatos, fosfatos y lípidos
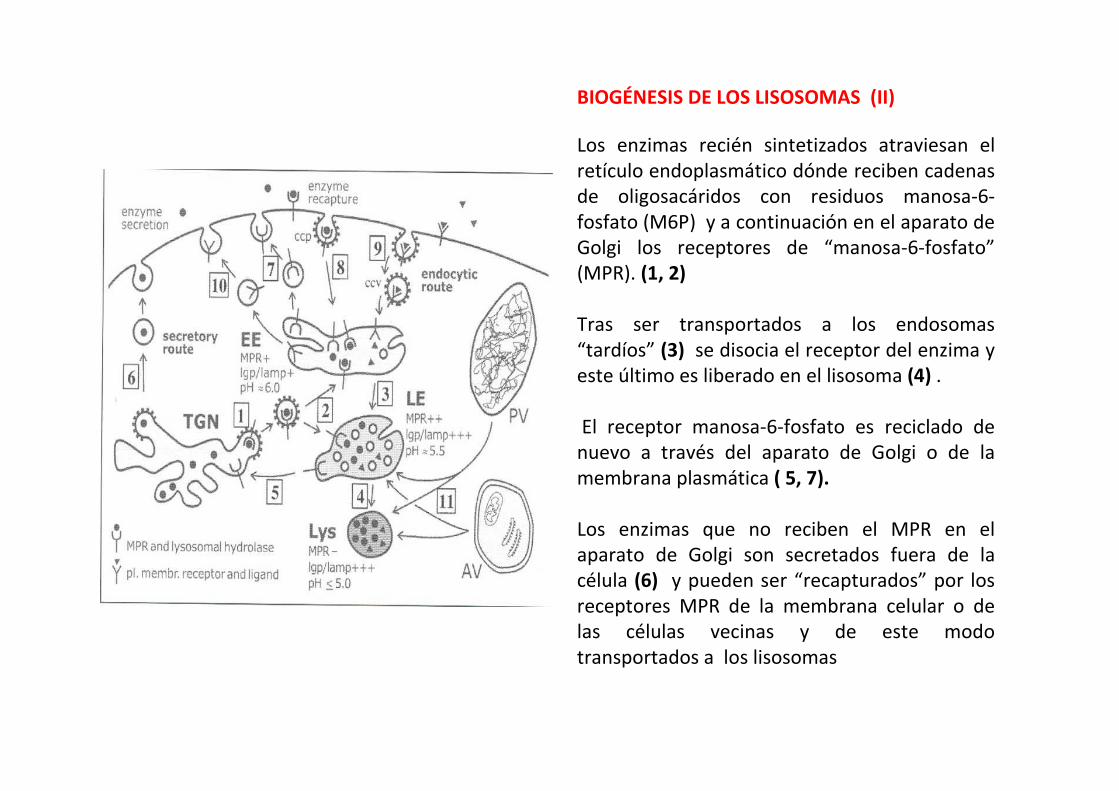
BIOGÉNESIS DE LOS LISOSOMAS (II)
Los enzimas recién sintetizados atraviesan el
retículo endoplasmático dónde reciben cadenas
de oligosacáridos con residuos manosa-6-
fosfato (M6P) y a continuación en el aparato de
Golgi los receptores de “manosa-6-fosfato”
(MPR). (1, 2)
Tras ser transportados a los endosomas
“tardíos” (3) se disocia el receptor del enzima y
este último es liberado en el lisosoma (4) .
El receptor manosa-6-fosfato es reciclado de
nuevo a través del aparato de Golgi o de la
membrana plasmática ( 5, 7).
Los enzimas que no reciben el MPR en el
aparato de Golgi son secretados fuera de la
célula (6) y pueden ser “recapturados” por los
receptores MPR de la membrana celular o de
las células vecinas y de este modo
transportados a los lisosomas

Christian De Duve. 1955.
FUNCIONES DEL LISOSOMA
I. Desempeñan un papel fundamental en los procesos de
depuración celular de sustratos que tiene lugar a través del
sistema “endosomas-autofagosomas-lisosomas” y en los de
endocitosis mediada por receptores y utilizado por muchas
células para internalizar diversas moléculas procedentes de
células vecinas o segregadas por ellas mismas.
II. Intervienen en otras varias funciones biológicas como son
la homeostasis del colesterol, la regulación de los
receptores celulares, la inactivación de organismos
patógenos y el remodelado óseo.
III. Participan en la patogénesis de algunas enfermedades
neurodegenerativas tales como Parkinson, Alzheimer y
Huntington. (synucleinopathies)

Vias que utiliza el lisosoma para la depuración del material celular . Platt, 2012.

ENFERMEDADES LISOSOMALES
Son un grupo de alrededor de 70 enfermedades hereditarias originadas por la deficiencia de una proteína lisosomal o de una proteína no-lisosomal implicada en la biogénesis de los lisosomas.
Su frecuencia global ha venido siendo estimada hasta ahora de alrededor de 1 por cada 5.000 - 8.000 recién nacidos, pero probablemente estas cifras deberán ser revisadas al alza en un inmediato futuro.Henri G. Hers
1963.

ENFERMEDADES LISOSOMALES. ETIOLOGIA
� Regidas por una herencia de carácter autosómico recesivo � Excepto tres de ellas: Fabry y Hunter (X-LR), Danon (X-LD).� Un “master gen” localizado en 6p21.1, codifica el factor de transcripción TFEB.� Los genes responsables de la biogénesis y función de los lisosomas forman parte de la red CLEAR junto a otros genes implicados en el metabolismo celular y función de organelas, porque tienen un dominio común, GTCACGTGAC, llamado Coordinated Lysosomal Expression and Regulation, que es la diana del TFEB.� El TFEB regula la expresión de los genes CLEAR.� En condiciones anómalas el TFEB pasa del citoplasma al núcleo activando los genes diana.
Locus gen
Proteína
490 aa
Gen

TFEB direct targets with a known role in lysosomal function (Palmieri M, 2011)Category Gene name
Lysosomal hydrolases andaccessory proteins ASAH1, CTSA, CTSB, CTSD, CTSF, GAA,
GALNS, GBA, GLA, GLB1, GNS,
GUSB, HEXA, HEXB, IFI30, NAGLU,
NEU1, PLBD2, PPT1, PSAP, SCPEP1,
SGSH, TPP1
Lysosomal membrane C1orf85, CD63, CLCN7, CLN3, CTNS,
MCOLN1, SLC36A1, LAMP1,
TMEM55B
Lysosomal acidification ATP6AP1, ATP6V0A1, ATP6V0B,
ATP6V0C, ATP6V0D1, ATP6V0D2,
ATP6V0E1, ATP6V1A, ATP6V1B2,
ATP6V1C1, ATP6V1D, ATP6V1E1,
ATP6V1G1, ATP6V1H
Non-lysosomal proteins involvedin lysosomal biogenesis NAGPA, GNPTG, IGF2R, M6PR,
BLOC1S1, BLOC1S3, HPS1, HPS3,
HPS5, SUMF1
Autophagy BECN1, GABARAP, HIF1A, NRBF2,
PRKAG2, RAB7A, RRAGC, SQSTM1,
STK4, UVRAG, VPS8, VPS11, VPS18,
VPS26A, VPS33A, VPS35, WDR45

(A) Lysosomal function.
(B) cellular compartment
(C) biological process
Palmieri M, 2011.
Composition of the CLEAR network.

ENFERMEDADES LISOSOMALESCLASIFICACIÓN TRADICIONAL : MOLÉCULA ACUMULADA
• Lipidosisesfingolipidosis
glicoesfingolipidosis: Fabry, Gaucher, GM1, GM2, LMD
otras esfingolipidosis: Niemann-Pick
otras enfermedades de depósito lipídicoWolman
• Mucopolisacaridosis: MPS
• Glucogenosis: Pompe
• Glucoproteinosis :Aspartilglicosaminuria
• Ceroidolipofusinosis neuronales : Lipofusinosis neuronales
• Mucolipidosis: Mucolipidosis (I-cell disease)
• Otras: Cistinosis

ALTERACIONES CONGÉNITAS LISOSOMALESCLASIFICACIÓN MOLECULAR: FUNCIÓN ALTERADA
Defecto primario de hidrolasa lisosomalGaucher, MPS, Pompe, GM1, Farber, Fabry, etc..
Defecto del procesado post-translacional del enzimaMucosulfatidosis
Defecto del tráfico del enzimaI-cell disease
Defecto de la protección del enzimaGalactosialidosis
Defecto en una proteína soluble no enzimáticaNiemann Pick tipo C
Defecto de una proteína de transmembrana no enzimáticaDanon disease
No bien clasificadosLipofusinosis ceroidea neuronal 4 y 7

Pathogenic cascades in
lysosomal disease—Why
so complex?
S. U. Walkley, 2008
Mutación génica
Alteración función
o estructura proteica
Fisiopatología
Alteraciones orgánicas
Manifestaciones clínicas

Hypothetical cascade of events in LSD pathology.
Endo/autolysosomal
events are confined to
the darker shaded
background,
whereas processes
taking place in the
cytoplasm that affect
autophagosomes, the
ER, Golgi,
peroxisomes,
And mitochondria are
on the lighter
background.
Processes depicted
have been observed
in a number of LSDs
but do not necessarily
apply to all LSDs.
Platt, 2012

Summary of organelles affected in LSDs. Platt, 2012

CARACTERÍSTICAS CLÍNICAS GENERALESDE LAS ENFERMEDADES LISOSOMALES
• Pueden debutar en cualquier edad• Gran heterogeneidad clínica• Habitualmente afectación multisistémica
alta frecuencia del SNC (2/3)• Evolución progresiva• Gran disminución de la calidad de vida
Enfermedad de Fabry

AUMENTO DE LA PREVALENCIA DE LAS E.L. EN EL ADULTO
I. Mejora en la formación sanitariaInborn errors of metabolism in adults. Saudubray JM, Sedel F.
Ann Endocrinol (Paris). 2009 Mar;70(1):14-24
II. Aumento supervivenciacribado neonatal / cribado seleccionado
mejores opciones terapéuticas
centros de referencia para tratamiento integral
mejora en la equidad
III. Diagnóstico mejor en el adultoestrategia diagnóstico por especialidades
Endocrine manifestations related to inherited metabolic diseases in adults.
Vantyghem MC, Dobbelaere D, Mention K, Wemeau JL, Saudubray JM, Douillard C.
Orphanet J Rare Dis. 2012 Jan 28;7:11.
Inborn errors of metabolism in adult neurology].
Sedel F.
Rev Neurol (Paris). 2013 Feb;169 Suppl 1:S63-9

The female Gaucher patient: the impact of enzyme replacement therapy around key reproductive events (menstruation, pregnancy and menopause).Zimran A, Morris E, Mengel E, Kaplan P, Belmatoug N, Hughes DA, Malinova V, Heitner R,
Sobreira E, Mrsić M, Granovsky-Grisaru S, Amato D, vom Dahl S.
Blood Cells Mol Dis. 2009 Nov-Dec;43(3):264-88.
Pregnancy in a patient with mucopolysaccharidosis type IH homozygous for the W402X mutation.
Hendriksz CJ, Moss GM, Wraith JE.
J Inherit Metab Dis. 2004;27(5):685-6
Histologic abnormalities of placental tissues in Fabry disease: a case report and review of the literature.Thurberg BL, Politei JM.
Hum Pathol. 2012 Apr;43(4):610-4
Management of a pregnancy complicated by Pompe disease.Weida J, Hainline BE, Bodkin C, Williams MK.
Case Rep Obstet Gynecol. 2012;2012:137861.
The management of pregnancy in Gaucher disease.
Granovsky-Grisaru S, Belmatoug N, vom Dahl S, Mengel E, Morris E, Zimran A.
Eur J Obstet Gynecol Reprod Biol. 2011 May;156(1):3-8.
EMBARAZO Y ENFERMEDADES LISOSOMALES

ENFERMEDADES LISOSOMALES (LSD). DIAGNÓSTICO
I. Diagnóstico preimplantacionalGheona Altarescu, et al.
Prevention of Lysosomal Storage Diseases and Derivation of. Mutant StemCell Lines by Preimplantation Genetic Diagnosis. Mol Biol International doi:10.1155/2012/797342
(“..20 unaffected children from 17 families….”).
II. Diagnóstico prenatal enzimático/molecular
III. Cribado sistemático neonatalBioquímico: Inmunocuantificación / MS/MS
Fabry, Gaucher, Krabbe, MLD, MPS I, II, IIIA, VI, Pompe, N-Pick A/B, DMS, ML II/IIIInforme ETS , AETSA, 2011/11. Sevilla ¿por el momento no usar?
Molecular. ¿ Secuenciación total ?
IV. Cribado selectivo neurológico. Paneles múltiples (Genroutes: 58 genes)
V. Diagnóstico clínicoSospecha clínicaExámenes complementarios de orientación diagnóstica.
biomarcadores, imagen, electrofisiología, proteoma?
VI. Confirmación diagnósticaEstudio enzimático Estudio molecular

OPCIÓN TERAPÉUTICA E D L PERSPECTIVAS DE FUTURO
Tratamiento sintomático Asistencia global
Aporte Enzimático
TES
Células madre (BMT/CB))
Dosis altas ?
Mejor tolerancia inmunológica
Vida media más larga
Paso BBB
Uso intratecal
Autólogas pluripotenciales
Con hiperexpresión génica (lentivirus,
retrovirus))
Microencapsuladas
Terapia Génica
Genes (AAV)
Modificación expresión génica
Mejora vehículos y seguridad
Uso intratecal
Hiperexpresión CLEAR
Stop codon read-through: gentamicina
Tratamiento Enfermedades Lisosomales I

OPCIÓN TERAPÉUTICA E D L PERSPECTIVAS DE FUTURO
Manipulación enzimática
Chaperones químicos (pirimetamina) Moléculas más eficientes
Reducción de la síntesis de sustrato (RSS)
Miglustat
Genisteina
Bloqueo específico
Terapia combinadaTES + células madre
TES + RSS
RSS + aspirina + antiinflamatorios
Células madre + Terapia génica
Células madre + antioxidantes
Combinar nuevas opciones terapéuticas
OtrasIndometacina
Ibuprofeno
Phyoestrógenos antioxidantes
Estimulo exocitosis regulando homeostasis
del Ca
Regulación homeostasis del Fe
Estímulo autofagia: litio. (Ac. Transretinoico)
Tratamiento Enfermedades Lisosomales II

Apéndice I. TRATAMIENTO EDL
Stem Cell Transplantation in Inherited Metabolic DisordersRobert Wynn Hematology Am Soc Hematol Educ Program. 2011;2011:285-91
Emerging therapies for neurodegenerative lysosomal storage disorders – from concept to realityHemsley KM, Hopwood JJJ Inher Metab Dis, 2011; 34: 1003- 1012
Avances en el tratamiento de las enfermedades lisosomales en la infancia. López Marín L, González LActa Pediatr Esp , 2011; 88 (supl): S43-S50
Neural Stem Cell Transplantation as a Therapeutic. Approach for Treating Lysosomal Storage DiseasesLamya S. Shihabuddin & Seng H. ChengNeurotherapeutics ,2011; 8:659–667
Gene, Stem Cell, and Future Therapies for OrphanDiseasesM Ian PhillipsState of the Art. Nature, 92 (2); 2012
Enzyme-Replacement Therapies for Lysosomal Storage Diseases. Technical BriefAgency for Healthcare Research and Quality U.S. Department of Health and Human ServicesAHRQ Publication No. 12(13)-EHC154-EF, January 2013

Apéndice IITable 2. Current UK Paediatric Bone Marrow Transplant Group indications for HSCT in children with IEM*Robert Wynn, 2012
Standard indication Hurler syndrome
Sly syndrome (MPSVII)
Mannosidosis
X-ALD
Clinical option Aspartylglucosaminuria
Wolman disease
Late infantile metachromatic leukodystrophy
Niemann pick C(2)
Developmental MPSIS, MPSIH/S, MPSII, MPSVI in conjunction with ERT or where alloantibodies
attenuate efficacy of ERT
Pompe where alloantibodies attenuates efficacy of ERT
Juvenile Sandhoff
Juvenile Tay Sachs
Presymptomatic or milder forms of globoid cell leukodystrophy (infantile Krabbe
when newborn and asymptomatic)
Early diagnosis fucosidosis
No evidence for BM transplantation Fabry
Infantile Tay Sachs
Infantile Sandhoff
GM1 gangliosidosis
MPSIV (Morquio)
Niemann pick B and C
* This is to guide commissioners and purchasers of HSCT services on which transplants are
indicated; this is not by any means an exhaustive list and many other publications carry such a list
of indications.

CONCLUSIONES
Las enfermedades lisosomales deben ser consideradas como
enfermedades globales de organelas celulares.
La sospecha clínica justifica el cribado molecular mediante
el uso de paneles múltiples.
El tratamiento aplicado debe estar en consonancia con la
patogenia de la enfermedad.
Es necesario valorar cuidadosamente la eficacia y eficiencia
terapéutica en cada paciente.
La asistencia global y multidisciplinaria mejora siempre
la calidad de vida de los pacientes.