potent and selective inhibitors of mth1 probe its role in cancer cell
TRANSCRIPT

S1
SUPPORTING INFORMATION
Potent and selective inhibitors of MTH1 probe its role in cancer cell
survival
Jason G. Kettle,† Husam Alwan,† Michal Bista,§ Jason Breed,¶ , Nichola L. Davies,§ Kay Eckersley,¥
Shaun Fillery,§ Kevin M. Foote,§ Louise Goodwin,† David R. Jones,† Helena Käck,# Alan Lau,† J.
Willem M. Nissink,§ Jon Read,¶ James S. Scott,§ Ben Taylor,¶ Graeme Walker,¥ Lisa Wissler,# Marta
Wylot§
†Oncology Innovative Medicines Unit, AstraZeneca, Mereside, Alderley Park, Macclesfield, Cheshire
SK10 4TG, United Kingdom.
§Oncology Innovative Medicines Unit, AstraZeneca, Unit 310 - Darwin Building, Cambridge Science
Park, Milton Road, Cambridge, CB4 0WG, United Kingdom.
¥Discovery Sciences, AstraZeneca, Mereside, Alderley Park, Macclesfield, Cheshire SK10 4TG,
United Kingdom.
¶Discovery Sciences, AstraZeneca, Unit 310 - Darwin Building, Cambridge Science Park, Milton
Road, Cambridge, CB4 0WG, United Kingdom.
#Discovery Sciences, AstraZeneca, Pepparedsleden 1, 431 83 Mölndal, Sweden
Table of Contents Page
1. Experimental methods S2
2. SPR Characterization S4
3. Synthesis details S4
4. Kinase selectivity S34
5. Secondary pharmacology profiling S46
6. Crystallography S52
7. NMR conformational analysis of macrocycles S57

S2
1. Experimental Methods
Assessment of enzyme activity:
Biochemical Assay Activity of the MTH1 enzyme was assesed by detecting the inorganic
pyrophosphate generated when the nucleoside triphosphate substrate, 8-oxo-dGTP, is hydrolysed.
Detection of the inorganic pyrophosphate is achieved using the Ppi Light Inorganic pyrophosphate kit
(Lonza, Basel, Switzerland). Compounds were acoustically dispensed into White 384-well medium
bind plates (784075 Greiner) using an Echo 555 (Labcyte, Sunnyvale CA). A 12 point half-log
concentration response curve, with a 100 µM top concentration were constructed with a consistent
final volume of compound and DMSO of 40 nl per well to give a final DMSO concentration of 1%. A
2 µl addition of MTH1 enzyme was added to all wells of the assay plate this was left for 15 minutes at
room temperature before the reaction was started by the addition of 2 µl 8-oxo-dGTP substrate, the
reaction was allowed to progress for 20 minutes at room temperature before being quenched with 1 µl
of an inhibitor, TH287. All reagents were prepared in assay buffer (25 mM HEPES pH 7.5, 10 mM
MgCl2, 1 mM DTT, 0.005% Tween 20) with final assay concentrations of 0.2 nM MTH1, 10 µM 8-
oxo-dGTP and 0.2 µM TH287, additions were all completed on the BioRaptr dispenser (Beckman
Coulter, Pasadena CA). Detection was completed by the addition of 2.5 µl PPi light detection reagent
by multi-drop combi to all wells, this was left to equilibrate for 60 minutes at room temperature, and
the luminescence signal was measure on an Envision plate reader (Perkin Elmer, Waltham MA). Data
analysis was completed in Genedata screener (Genedata, Basel Switzerland).
Assessment of cellular activity:
Cell Viability Assay A549, NCI-H358 and MCF7 cells were cultured in cell media composed of
RPMI, 10% (v/v) Foetal Calf Serum and 1% (v/v) L-Glutamine. After harvesting, cells were
dispensed into black, 384-well Costar plates (#3712, Corning) to give 150, 400, and 200 cells per
well, respectively, in a total volume of 40µl cell media, and were incubated overnight at 37°C, 90%
relative humidity and 5% CO2. Test compounds were dosed directly into the cell plates, into the inner
308 wells, using a Labcyte Echo 555 acoustic dispenser. The cells were dosed over a 6 point range
from 30µM down to 0.03µM in order to calculate compound IC50’s,with a total DMSO concentration
in the assay of 0.3%. The cell plates were then incubated for 5 days at 37°C. Cells were fixed by the
addition of 20µl 12% formaldehyde in PBS/A (4% final concentration), with a 20 minute room
temperature incubation and stained with 1:5000 dilution of Hoechst 33342, with a 60 minute room
temperature incubation, and then washed with PBS/A. A cell count was performed on the stained cell
plates using a Cellomics ArrayScanTM VTI imaging platform (Thermo Scientific).

S3
CETSA K562 cells were cultured at 37 degrees, 5% CO2 in RPMI-1640 medium supplemented with
10% fetal bovine serum (FBS, Gibco/Life Technologies), 100 units/ml penicillin and 100 units/ml
streptomycin (Gibco/Life Technologies). The cells were fed with half the volume additional fresh
media a day prior to measurements. Cells of passage 7-12 that reached a density of 1.2 to 1.4
million/ml were used in the experiments. Compounds at a concentration of 10 mM in 100% DMSO
were used to create dilution series in DMSO used in the concentration response experiments were
prepared fresh before the experiments. All compounds were added at 2X final test concentration in
Hank’s Balanced Salt solution (HBSS- Gibco/Life Technologies) prior to addition to the cells.
Western blots were performed using an iBlot2 device (Life Technologies) on nitrocellulose
membranes. Transfer was optimized at 6 minutes at 20 V. Blocking and dilution of antibodies was in
5% non-fat milk in Tris Buffered Saline-TWEEN (TBST). The commercially available primary
antibody sc-271082 (Santa Cruz Biotechnology) was used for detection of MTH1. The primary
antibody was diluted at 1:500 and incubated at 4°C over-night. Chemiluminescent western blot
detection was applied using the horseradish peroxidase (HRP) conjugated secondary antibody sc-2055
(Santa Cruz Biotechnology) together with Clarity Western ECL substrate (BioRad).
Melt and shift curves for MTH1 were previously established in intact K562 cells. Intact K562 cells
were pelleted by centrifugation and washed and re-suspended in HBSS at a cell density of 40 million
cells/ml. The cell suspension was divided into 25 µl aliquots and an equal volume of HBSS
containing 2X the intended compound concentration was added, resulting in a final cell concentration
of 20 million cells/ml. 7 step dilution concentration response series of the ligands in 1% DMSO were
applied together with 1% DMSO only as control. The log10 dilution series ranged from 100 pM to
100 µM. The cells were incubated with ligand at 37°C for 30 min, with gentle mixing every 10 min.
The aliquots were heated to a single specific temperature, 52°C, as determined from the previously
established MTH1 melt and shift curves, for 3 minutes, and lysed by 2 cycles of freeze-thawing.
Precipitated protein and cell debris were pelleted by centrifugation at 20000g for 20 minutes. 30 µl of
the supernatant was mixed with 15 µl LDS sample buffer. Protein amounts were detected after
loading 13 µl of the supernatant/LDS mixture per lane on the gel using standard western blot
techniques.
All experiments were repeated on different days and with different passages of cells to reduce risk of
day-to-day variations (N individual incubations in data figures). The 7 step dilution concentration
response series generated in the IsoT C-R experiments were run in triplicate on gels (n individual gel
determinations in data figures). The western blot intensities were obtained by measuring the
chemiluminescence counts per square mm (I= count/mm2). The obtained intensities were plotted in
Excel for melt curves, either as the luminescence count or normalized to a relevant control count. The

S4
IsoT C-R data was analyzed and normalized to control addition (DMSO only). The normalized
intensities were plotted using GraphPad Prism1 analysis and software. Data points are shown as mean
values with error bars indicating ± SEM. No error bars are shown if SEM is smaller than the symbol.
Concentration response curves were fitted using the modified logistic Hill equation algorithm
included in the GraphPad Prism software. The obtained CETSA™ EC50 concentration response
values represent the half maximal concentration the ligands for stabilizing MTH1 at 52°C. The quoted
EC50 with 95% confidence intervals is therefore a relative measure of Target Engagement of
compound available for interaction with MTH1 in intact K562 cells.
2. SPR Characterization
Surface plasmon resonance (SPR). SPR experiments were performed using a Biacore T200
biosensor (GE Healthcare). Series S NTA sensor chips (GE Healthcare) were used. All experiments
were carried out using Assay buffer containing; 50mM Hepes pH= 7.5, 150mM NaCl, 0.005%
Tween-20 (v/v) and 1% DMSO (v/v) as running buffer. Typically, 10mM DMSO stocks of
compounds were diluted 1:100 (v/v) in 50mM Hepes pH= 7.5, 150 mM NaCl, 0.005% Tween-20
(v/v) to form 100µM intermediate stocks with final DMSO concentration of 1% (v/v). The
intermediate solutions were then subsequently diluted using Assay buffer to achieve final
concentration range. Histidine-tagged MTH1 (isoform p18 (M1-V156)) was immobilized as the
ligand onto NTA sensor chips using a capture coupling method.2 Assay buffer was used as
immobilization buffer. The nitrilotriacetic (NTA) surface was first activated with a 2-min injection of
500 µM NiCl2 in running buffer before the carboxymethyl dextran surface was activated with a 7-min
injection of a 1:1 ratio of 0.4 M EDC and 0.1 M NHS. His-tagged protein was diluted into running
buffer to a concentration of 20 µg/ml and immobilised to the surface with a 7-min injection.
Remaining activated groups were blocked with a 2 consequtive 60-sec pulses of 0.5 M Ethanolamine.
Typical immobilization levels ranged from 500 to 3000 resonance units (RU). Biacore T200
Evaluation software (GE Healthcare) was used for data processing and analysis. Equlibrium binidng
analysis was analysed by ploting steady-state response level vs. compound concentration and fitting
Langmuir equation (describing simple 1:1 binary interaction). To determine the rate constants of
association (kon) or dissociation (koff), kinetic analysis was carried out at a constant flow rate of 30
µL/min in running buffer at 25°C. Zero-buffer blank injections and DMSO calibrations were included
for double referencing. Rate constants were calculated globally from the obtained sensorgram data
(RU) by fitting to a 1:1 interaction model that included a mass transport term. The affinity (KD) was
calculated from the equation KD = koff/kon.
Experimental Methods

S5
Chemistry Unless otherwise stated, commercially available reagents were used as supplied. All
reactions requiring anhydrous conditions were conducted in dried apparatus under an atmosphere of
nitrogen. 1H NMR spectra were recorded using a Bruker AV300 or AV400 NMR. Chemical shifts δ
are reported in ppm and multiplicity of signals are denoted s = singlet, d = doublet, t = triplet and m =
multiplet respectively, with coupling constants (J) reported in hertz (Hz). HRMS were recorded using
a Shimadzu LCMS-2020 instrument (ESI+). Reactions and intermediates were also characterised by
mass spectroscopy following liquid chromatography (LCMS or UPLC). Purity of tested compounds
was assessed to be at least 95% by UV using LCMS or UPLC analysis unless indicated otherwise.;
UPLC was carried out using a Waters UPLC fitted with Waters QDa mass spectrometer (Column
temp 40, UV = 190-400 nm, MS = ESI with pos/neg switching) at a flow rate of 1ml/min using a
solvent system of 95% A + 5% B to 5% A to 95% B over 8 minutes (total runtime with equilibration
back to starting conditions was 10 min), where A = 0.1% Formic acid or 0.05% TFA in water (for
acid work) or 0.04% Ammonia in water (for base work) B = Acetonitrile. For acid or base analysis the
column used was Waters Acquity BEH 1.7um 2.1 x 100mm. LCMS was carried out using a
Shimadzu UFLC fitted with a Shimadzu LCMS-2020 mass spectrometer and a Waters BEH C18
(50x2.1, 1.7 µm) or Shim-pack XR-ODS (50x3.0, 2.2 µm) or Phenomenex Gemini –NX 3u C18 110A
(50x3.0, 3 µm) column at a flow rate of 1.2ml/min 95% A to 95% B over 2.0 min with a 0.6 min hold.
Ion exchange purification was generally performed using a SCX-2 (Biotage, Propylsulfonic acid
functionalized silica. Manufactured using a trifunctional silane. Non end-capped) cartridge. Column
chromatography was performed on a Biotage system using a SiO2 column with UV detection.
Individual purification methods referred to here are detailed in the Supplementary section.

S6
Ethyl (6-chloro-2-(methylthio)-5-nitropyrimidin-4-yl)(3-methoxybenzyl)carbamate. 1-
(Bromomethyl)-3-methoxybenzene (0.84 g, 4.2 mmol) was added to ethyl (6-chloro-2-(methylthio)-5-
nitropyrimidin-4-yl)carbamate 8 (1.22 g, 4.2 mmol), sodium iodide (0.63 g, 4.2 mmol) and K2CO3
(0.86 g, 6.3 mmol) in acetone (30 ml) at 20°C. The resulting mixture was stirred at room temperature
for 16 hours. The solid was filtered and the solvent was removed under reduced pressure. The crude
product was purified by flash silica chromatography, elution gradient 0 to 8% ethyl acetate in
petroleum ether. Pure fractions were evaporated to dryness to afford ethyl (6-chloro-2-(methylthio)-
5-nitropyrimidin-4-yl)(3-methoxybenzyl)carbamate (1.56 g, 91%) as a yellow gum; 1H NMR
(DMSO-d6, 300 MHz) δ 1.10 (3H, t), 2.44 (3H, s), 3.72 (3H, s), 4.09 (2H, q), 5.16 (2H, s), 6.76-6.97
(3H, m), 7.24 (1H, td); m/z (ES+), [M+H]+ = 413; acidic, HPLC tR = 1.35 min.
Ethyl 3-methoxybenzyl(6-(methylamino)-2-(methylthio)-5-nitropyrimidin-4-yl)carbamate.
Methylamine (2.82 ml, 5.63 mmol) was added to ethyl (6-chloro-2-(methylthio)-5-nitropyrimidin-4-
yl)(3-methoxybenzyl)carbamate (1.55 g, 3.75 mmol) and triethylamine (1.05 ml, 7.51 mmol) in THF
(30 ml) at 20°C . The resulting mixture was stirred at room temperature for 3 hours. The reaction
mixture was quenched with water (50 ml), extracted with DCM (2 x 100 ml), the organic layer was
dried over Na2SO4, filtered and evaporated to afford ethyl 3-methoxybenzyl(6-(methylamino)-2-
(methylthio)-5-nitropyrimidin-4-yl)carbamate (1.53 g, 100%) as a yellow gum which was used
without further purification; 1H NMR (DMSO-d6, 300 MHz) δ 1.09 (3H, t), 2.44 (3H, s), 2.98 (3H,
d), 3.71 (3H, s), 4.05 (2H, q), 5.07 (2H, s), 6.80 (1H, dd), 6.87-7.01 (2H, m), 7.22 (1H, t), 8.58 (1H,
q); m/z (ES+), [M+H]+ = 408; acidic, HPLC tR = 1.32 min.
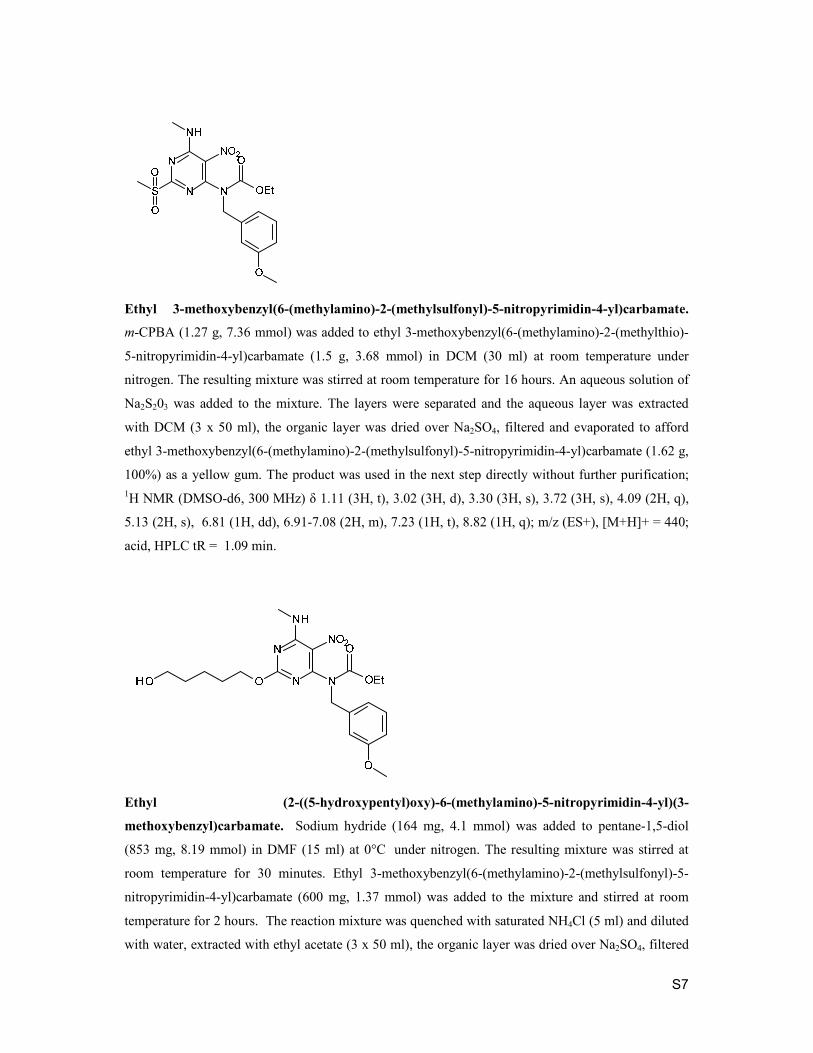
S7
Ethyl 3-methoxybenzyl(6-(methylamino)-2-(methylsulfonyl)-5-nitropyrimidin-4-yl)carbamate.
m-CPBA (1.27 g, 7.36 mmol) was added to ethyl 3-methoxybenzyl(6-(methylamino)-2-(methylthio)-
5-nitropyrimidin-4-yl)carbamate (1.5 g, 3.68 mmol) in DCM (30 ml) at room temperature under
nitrogen. The resulting mixture was stirred at room temperature for 16 hours. An aqueous solution of
Na2S203 was added to the mixture. The layers were separated and the aqueous layer was extracted
with DCM (3 x 50 ml), the organic layer was dried over Na2SO4, filtered and evaporated to afford
ethyl 3-methoxybenzyl(6-(methylamino)-2-(methylsulfonyl)-5-nitropyrimidin-4-yl)carbamate (1.62 g,
100%) as a yellow gum. The product was used in the next step directly without further purification; 1H NMR (DMSO-d6, 300 MHz) δ 1.11 (3H, t), 3.02 (3H, d), 3.30 (3H, s), 3.72 (3H, s), 4.09 (2H, q),
5.13 (2H, s), 6.81 (1H, dd), 6.91-7.08 (2H, m), 7.23 (1H, t), 8.82 (1H, q); m/z (ES+), [M+H]+ = 440;
acid, HPLC tR = 1.09 min.
Ethyl (2-((5-hydroxypentyl)oxy)-6-(methylamino)-5-nitropyrimidin-4-yl)(3-
methoxybenzyl)carbamate. Sodium hydride (164 mg, 4.1 mmol) was added to pentane-1,5-diol
(853 mg, 8.19 mmol) in DMF (15 ml) at 0°C under nitrogen. The resulting mixture was stirred at
room temperature for 30 minutes. Ethyl 3-methoxybenzyl(6-(methylamino)-2-(methylsulfonyl)-5-
nitropyrimidin-4-yl)carbamate (600 mg, 1.37 mmol) was added to the mixture and stirred at room
temperature for 2 hours. The reaction mixture was quenched with saturated NH4Cl (5 ml) and diluted
with water, extracted with ethyl acetate (3 x 50 ml), the organic layer was dried over Na2SO4, filtered

S8
and evaporated to afford a yellow residue. The crude product was purified by flash silica
chromatography, elution gradient 0 to 2% methanol in DCM. Pure fractions were evaporated to
dryness to afford ethyl (2-((5-hydroxypentyl)oxy)-6-(methylamino)-5-nitropyrimidin-4-yl)(3-
methoxybenzyl)carbamate (490 mg, 77%) as a pale yellow oil; ms detection: m/z (ES+), [M+H]+ =
464; acidic, HPLC tR = 1.54 min.
2-((5-Hydroxypentyl)oxy)-9-(3-methoxybenzyl)-6-(methylamino)-7H-purin-8(9H)-one. Iron (578
mg, 10.36 mmol) was added to ethyl (2-((5-hydroxypentyl)oxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(3-methoxybenzyl)carbamate (480 mg, 1.04 mmol) in acetic acid (10 ml) and water (2 ml) at room
temperature. The resulting mixture was stirred at 50°C for 4 hours. The reaction mixture was made
basic with saturated Na2CO3. The reaction mixture was diluted with DCM (50 ml) and the resulting
mixture was filtered through a pad of celite. The celite was washed with DCM/methanol (80/20). The
organic layer was dried over MgSO4, filtered and the solvent was evaporated to afford 2-((5-
hydroxypentyl)oxy)-9-(3-methoxybenzyl)-6-(methylamino)-7H-purin-8(9H)-one (400 mg, 100%) as a
green oil which solidified on standing. The product was used in the next step directly without further
purification; 1H NMR (Chloroform-d, 300 MHz) δ 1.40-1.70 (4H, m), 1.70-1.85 (2H, m), 2.95 (3H,
d), 3.66 (2H, t), 3.69 (3H, s), 3.71 (2H, m), 4.32 (2H, m), 5.30 (2H, s), 6.74-6.93 (3H, m), 7.12-7.19
(1H, m), 10.27 (1H, s); m/z (ES+), [M+H]+ = 388; acidic, HPLC tR = 0.95 min.
2-((5-Bromopentyl)oxy)-9-(3-hydroxybenzyl)-6-(methylamino)-7H-purin-8(9H)-one and 9-(3-

S9
hydroxybenzyl)-2-((5-hydroxypentyl)oxy)-6-(methylamino)-7H-purin-8(9H)-one. BBr3 in DCM
(5.16 ml, 5.16 mmol) was added dropwise to 2-((5-hydroxypentyl)oxy)-9-(3-methoxybenzyl)-6-
(methylamino)-7H-purin-8(9H)-one (400 mg, 1.03 mmol) in DCM (30 ml) at -60°C under nitrogen.
The resulting mixture was stirred below -10°C for 2 hours. The reaction mixture was quenched with
methanol (1 ml) at -60°C. The solvent was removed under reduced pressure. Crude material (70 mg)
from a previous batch was mixed with this crude product and the combined amount was purified
together by flash silica chromatography, elution gradient 0 to 5% methanol (containing 0.1% NH3) in
DCM. The pure fractions were evaporated to dryness to afford firstly 2-((5-bromopentyl)oxy)-9-(3-
hydroxybenzyl)-6-(methylamino)-7H-purin-8(9H)-one (80 mg, 18%) as a yellow solid; 1H NMR
(DMSO-d6, 400 MHz) δ 1.35-1.60 (2H, m), 1.65-1.73 (2H, m), 1.73-1.92 (2H, m), 2.93 (3H, d), 3.54
(2H, t), 4.21 (2H, t), 4.77 (2H, s), 6.48-6.75 (4H, m), 7.10 (1H, t), 9.36 (1H, s), 9.92 (1H, s); m/z
(ES+), [M+H]+ = 436, 438; acidic, HPLC tR = 1.03 min. This was followed by 9-(3-
hydroxybenzyl)-2-((5-hydroxypentyl)oxy)-6-(methylamino)-7H-purin-8(9H)-one (170 mg, 44%) as a
yellow solid; 1H NMR (DMSO-d6, 400 MHz) δ 1.35-1.50 (4H, m), 1.60-1.70 (2H, m), 2.92 (3H, d),
3.35-3.45 (2H, m), 4.19 (2H, t), 4.77 (2H, s), 6.51 (1H, q), 6.54-6.75 (3H, m), 7.10 (1H, t), 9.36 (1H,
s), 9.91 (1H, s); m/z (ES+), [M+H]+ = 374; acidic, HPLC tR = 0.84 min.
2-((5-Bromopentyl)oxy)-9-(3-hydroxybenzyl)-6-(methylamino)-7H-purin-8(9H)-one. PBr3 (0.17
ml, 1.87 mmol) was added to 9-(3-hydroxybenzyl)-2-((5-hydroxypentyl)oxy)-6-(methylamino)-7H-
purin-8(9H)-one (140 mg, 0.37 mmol) in DCM (5 ml) and THF (2 ml) at room temperature. The
resulting mixture was stirred at room temperature for 4 hours. The reaction mixture was quenched
with saturated NaHCO3 (20 ml), extracted with DCM (2 x 50 ml), the organic layer was dried over
Na2SO4, filtered and evaporated to afford pale yellow residue. The crude product was purified by
flash silica chromatography, elution gradient 0 to 5% methanol in DCM. Pure fractions were
evaporated to dryness to afford 2-((5-bromopentyl)oxy)-9-(3-hydroxybenzyl)-6-(methylamino)-7H-
purin-8(9H)-one (93 mg, 57%) as a pale yellow solid; 1H NMR (DMSO-d6, 400 MHz) δ 1.35-1.60
(2H, m), 1.65-1.73 (2H, m), 1.73-1.92 (2H, m), 2.93 (3H, d), 3.54 (2H, t), 4.21 (2H, t), 4.77 (2H, s),
6.48-6.75 (4H, m), 7.10 (1H, t), 9.36 (1H, s), 9.92 (1H, s); m/z (ES+), [M+H]+ = 436, 438; acidic,
HPLC tR = 1.03 min.

S10
20-(Methylamino)-5,6,7,8-tetrahydro-4H,15H-2,19-(azenometheno)-14,10-
(metheno)imidazo[5,1-d][1,12,3,5]dioxadiazacycloheptadecin-17(18H)-one (28). Potassium
carbonate (57 mg, 0.41 mmol) was added to 2-((5-bromopentyl)oxy)-9-(3-hydroxybenzyl)-6-
(methylamino)-7H-purin-8(9H)-one (90 mg, 0.21 mmol) in DMF (3 ml) at room temperature. The
resulting mixture was stirred at 30°C for 20 hours. The solid was filtered and the mixture was purified
by preparative HPLC (XSelect CSH Prep C18 OBD column, 5µ silica, 19 mm diameter, 150 mm
length), using decreasingly polar mixtures of water (containing 0.1% NH4HCO3) and acetonitrile as
eluents. Fractions containing the desired compound were evaporated to dryness to afford 20-
(methylamino)-5,6,7,8-tetrahydro-4H,15H-2,19-(azenometheno)-14,10-(metheno)imidazo[5,1-
d][1,12,3,5]dioxadiazacycloheptadecin-17(18H)-one (20 mg, 27%) as a white solid; 1H NMR
(DMSO-d6, 300 MHz) δ 1.40 (2H, q, J = 6.7, 7.3 Hz), 1.5 – 1.75 (4H, m), 2.86 (3H, d, J = 4.7 Hz),
3.98 (2H, t, J = 7.6 Hz), 4.51 (2H, t, J = 6.9 Hz), 4.81 (2H, s), 6.58 (1H, s), 6.80 (1H, dd, J = 1.9, 7.8
Hz), 6.91 (1H, d, J = 7.4 Hz), 7.20 (1H, t, J = 7.8 Hz), 7.40 (1H, s), 9.97 (1H, s); m/z (ES+), [M+H]+
= 356; acidic, HPLC tR = 2.70 min; HRMS ESI+ m/z observed 356.1765, C18H22N5O3 requires
356.1723
6-Chloro-N4-(3-methoxybenzyl)-2-(methylthio)pyrimidine-4,5-diamine. 3-Methoxybenzylamine
(2.74 g, 20 mmol) was added dropwise to 4,6-dichloro-2-(methylthio)-5-nitropyrimidine 6 (4.8 g, 20

S11
mmol) and triethylamine (4.18 ml, 29.99 mmol) in THF (50 ml) at -20°C under nitrogen. The
resulting mixture was stirred below 0°C for 2 hours. The reaction was concentrated and the residue
partitioned between DCM and water, dried and concentrated to give a solid (6.8 g). Iron (2.22 g, 39.8
mmol) was added to a portion of this solid (1.5 g) in methanol (20 ml) and acetic acid (10 ml) at
20°C. The resulting mixture was stirred at 35°C for 3 hours. The reaction mixture was filtered
through celite. The solvent was removed under reduced pressure. The reaction mixture was diluted
with DCM (100 ml), and washed sequentially with saturated NaHCO3 (50 ml x 2) and brine (50 ml x
2). The organic layer was dried over Na2SO4, filtered and evaporated to afford 6-chloro-N4-(3-
methoxybenzyl)-2-(methylthio)pyrimidine-4,5-diamine (1.1 g) as a grey solid, and used without
further purification; 1H NMR (DMSO-d6, 300 MHz) δ 2.33 (3H, s), 3.73 (3H, s), 4.56 (2H, d), 4.82
(2H, s), 6.76 - 6.97 (3H, m), 7.25 (1H, t), 7.48 (1H, t); m/z (ES+), [M+H]+ = 311; acidic, HPLC tR =
1.09 min.
6-Chloro-9-(3-methoxybenzyl)-2-(methylthio)-9H-purine. Triethoxymethane (0.95 g, 6.43 mmol)
was added to 6-chloro-N4-(3-methoxybenzyl)-2-(methylthio)pyrimidine-4,5-diamine (1 g, 3.22
mmol) in acetic acid (20 ml) at 20°C. The resulting solution was stirred at 60°C for 8 hours. The
solvent was removed under reduced pressure. The solid was dried under vacuum to afford 6-chloro-
9-(3-methoxybenzyl)-2-(methylthio)-9H-purine (1.4 g, 99%) as a brown solid, containing 2 eqivalents
of acetic acid. The product was used in the next step directly without further purification; 1H NMR
(DMSO-d6, 400 MHz) δ 1.92 (6H, s), 2.60 (3H, s), 3.73 (3H, d), 5.42 (2H, s), 6.81-6.97 (3H, m),
7.02 (1H, t), 7.28 (1H, t), 11.98 (2H, s); m/z (ES+), [M+H]+ = 321; acidic, HPLC tR = 1.12 min.
9-(3-Methoxybenzyl)-N-methyl-2-(methylthio)-9H-purin-6-amine. Methylamine (4.91 ml, 9.82
mmol) was added to 6-chloro-9-(3-methoxybenzyl)-2-(methylthio)-9H-purine X (1.05 g, 3.27 mmol)

S12
in THF (20 ml) at room temperature. The resulting mixture was stirred at room temperature for 3
hours. The solvent was removed under reduced pressure. The solid was dried under vacuum to afford
9-(3-methoxybenzyl)-N-methyl-2-(methylthio)-9H-purin-6-amine (1.2 g) as a yellow solid. The
product was used in the next step directly without further purification; 1H NMR (DMSO-d6, 300
MHz) δ 2.91 (3H, d), 3.74 (3H, s), 5.27 (2H, s), 6.72-6.98 (4H, m), 7.28 (1H, t), 7.78 (1H, s), 8.10
(1H, s); m/z (ES+), [M+H]+ = 316; acidic, HPLC tR = 1.39 min.
9-(3-Methoxybenzyl)-N-methyl-2-(methylsulfonyl)-9H-purin-6-amine. m-CPBA (1.37 g, 7.93
mmol) was added to 9-(3-methoxybenzyl)-N-methyl-2-(methylthio)-9H-purin-6-amine (1 g, 3.17
mmol) in DCM (20 ml) at room temperature. The resulting mixture was stirred at room temperature
for 2 hours. The reaction mixture was diluted with DCM (50 ml), and washed sequentially with
saturated NaHCO3 (50 ml x 3) and saturated brine (50 ml x 2). The organic layer was dried over
MgSO4, filtered and evaporated to afford 9-(3-methoxybenzyl)-N-methyl-2-(methylsulfonyl)-9H-
purin-6-amine (1.1 g, 100%) as a yellow solid. The product was used in the next step directly without
further purification; m/z (ES+), [M+H]+ = 348; acidic, HPLC tR = 1.29 min.
4-((9-(3-Methoxybenzyl)-6-(methylamino)-9H-purin-2-yl)oxy)butan-1-ol. Sodium hydride (0.35
g, 8.6 mmol) was added to butane-1,4-diol (1.56 g, 17.3 mmol) in THF (20 ml) at 20°C under
nitrogen. The resulting mixture was stirred at room temperature for 45 minutes. A solution of 9-(3-
methoxybenzyl)-N-methyl-2-(methylsulfonyl)-9H-purin-6-amine (1 g, 2.9 mmol) in THF (10 ml) was
added dropwise to the mixture. The resulting mixture was stirred at room temperature for 2 hours. The
reaction mixture was quenched with saturated NH4Cl (1 ml). The solvent was removed under reduced
pressure. The crude product was purified by flash silica chromatography, elution gradient 0 to 70%
methanol in water. Pure fractions were evaporated to dryness to afford 4-((9-(3-methoxybenzyl)-6-

S13
(methylamino)-9H-purin-2-yl)oxy)butan-1-ol (0.56 g, 54%) as a pale yellow oil which solidified on
standing; 1H NMR (DMSO-d6, 300 MHz) δ 1.51-1.58 (2H, m), 1.63-1.80 (2H, m), 2.85-2.95 (3H,
broad s), 3.43 (2H, td), 3.71 (3H, s), 4.24 (2H, t), 4.41 (1H, t), 5.22 (2H, s), 6.87 (2H, d), 6.92 (1H, d),
7.24 (1H, t), 7.68 (1H, broad s), 8.01 (1H, s); m/z (ES+), [M+H]+ = 358; acidic, HPLC tR = 0.92
min.
4-((8-Bromo-9-(3-methoxybenzyl)-6-(methylamino)-9H-purin-2-yl)oxy)butan-1-ol. NBS (274
mg, 1.54 mmol) was added to 4-((9-(3-methoxybenzyl)-6-(methylamino)-9H-purin-2-yl)oxy)butan-1-
ol (550 mg, 1.54 mmol) in acetonitrile (10 ml) at room temperature. The resulting solution was stirred
at room temperature for 1 hour. The solvent was removed under reduced pressure. The solid was dried
under vacuum to afford 4-((8-bromo-9-(3-methoxybenzyl)-6-(methylamino)-9H-purin-2-
yl)oxy)butan-1-ol (810 mg) as a yellow residue. The product was used in the next step directly
without further purification; m/z (ES+), [M+H]+ = 436,438; acidic, HPLC tR = 1.03 min.
4-((8-Methoxy-9-(3-methoxybenzyl)-6-(methylamino)-9H-purin-2-yl)oxy)butan-1-ol. Sodium
methanolate (30% in methanol) (2 ml) was added to 4-((8-bromo-9-(3-methoxybenzyl)-6-
(methylamino)-9H-purin-2-yl)oxy)butan-1-ol (670 mg, 1.54 mmol) in methanol (5 ml) at room
temperature. The resulting solution was stirred at 60°C for 16 hours. The reaction mixture was
quenched with water (10 ml), neutralized with 2M HCl, extracted with DCM (3 x 50 ml), the organic
layer was dried over Na2SO4, filtered and evaporated to afford 4-((8-methoxy-9-(3-methoxybenzyl)-6-
(methylamino)-9H-purin-2-yl)oxy)butan-1-ol (580 mg, 97%) as a pale yellow gum. The product was
used in the next step directly without further purification; 1H NMR (DMSO-d6, 300 MHz) δ 1.48-1.58
(2H, m), 1.62-1.78 (2H, m), 3.31 (3H, s), 3.43 (2H, q), 3.71 (3H, s), 4.03 (3H, s), 4.10-4.26 (2H, m),

S14
4.40 (1H, t), 5.02 (2H, s), 6.70-6.91 (3H, m), 7.17-7.32 (2H, m); m/z (ES+), [M+H]+ = 388; acidic,
HPLC tR = 1.22 min.
2-(4-Hydroxybutoxy)-9-(3-methoxybenzyl)-6-(methylamino)-9H-purin-8-ol. 4-((8-Methoxy-9-(3-
methoxybenzyl)-6-(methylamino)-9H-purin-2-yl)oxy)butan-1-ol (550 mg, 1.42 mmol) was added to
4N HCl (20 ml) at room temperature. The resulting mixture was stirred at 80°C for 2 hours. The
solvent was removed under reduced pressure. The residue was dissolved in methanol (20 ml), filtered
and the mixture were evaporated to dryness to afford 2-(4-hydroxybutoxy)-9-(3-methoxybenzyl)-6-
(methylamino)-9H-purin-8-ol (530 mg, 100%) as a white solid. The product was used in the next step
directly without further purification; 1H NMR (DMSO-d6, 300 MHz) δ 1.39-1.59 (2H, m), 1.66-1.75
(2H, m), 2.92 (3H, d), 3.36-3.50 (2H, m), 3.68 (3H, s), 4.19 (2H, t), 4.84 (2H, d), 6.78-6.98 (3H, m),
7.22 (1H, t), 10.41 (1H, s); m/z (ES+), [M+H]+ = 374; acidic, HPLC tR = 0.88 min.
2-(4-Bromobutoxy)-9-(3-hydroxybenzyl)-6-(methylamino)-9H-purin-8-ol. BBr3 (1M in DCM)
(7.10 ml, 7.10 mmol) was added to 2-(4-hydroxybutoxy)-9-(3-methoxybenzyl)-6-(methylamino)-9H-
purin-8-ol (530 mg, 1.42 mmol) in DCM (20 ml) at -20°C under nitrogen. The resulting mixture was
stirred at room temperature for 17 hours. The reaction mixture was quenched with methanol (5 ml) at
-60°C and made basic with saturated NaHCO3. The mixture was extracted with DCM/methanol
(10:1) (3 x 50 ml), the organic layer was dried over Na2SO4, filtered and evaporated to afford pale
yellow solid. The crude product was purified by flash silica chromatography, elution gradient 0 to 3%
methanol in DCM. Pure fractions were evaporated to dryness to afford 2-(4-bromobutoxy)-9-(3-
hydroxybenzyl)-6-(methylamino)-9H-purin-8-ol (380 mg, 63%) as a off-white solid; 1H NMR
(DMSO-d6, 300 MHz) δ 1.64-1.99 (4H, m), 2.92 (3H, d), 3.55 (2H, t), 4.22 (2H, t), 4.78 (2H, d),

S15
6.46-6.75 (3H, m), 7.09 (1H, t), 9.36 (1H, s), 9.91 (1H, s), 1x OH not observed; m/z (ES+), [M+H]+ =
422, 424; acidic, HPLC tR = 1.04 min.
19-(Methylamino)-4,5,6,7-tetrahydro-14H-2,18-(azenometheno)-13,9-(metheno)imidazo[5,1-
d][1,12,3,5]dioxadiazacyclohexadecin-16(17H)-one (29). K2CO3 (236 mg, 1.71 mmol) was added
to 2-(4-bromobutoxy)-9-(3-hydroxybenzyl)-6-(methylamino)-9H-purin-8-ol (360 mg, 0.85 mmol) in
DMF (8 ml) at room temperature. The resulting mixture was stirred at 60°C for 16 hours. The solid
was filtered off and the reaction mixture concentrated. The crude product was purified by preparative
HPLC (XSelect CSH Prep C18 OBD column, 5µ silica, 19 mm diameter, 150 mm length), using
decreasingly polar mixtures of water (containing 0.1% NH4HCO3) and acetonitrile as eluents.
Fractions containing the desired compound were evaporated to dryness to afford 19-(Methylamino)-
4,5,6,7-tetrahydro-14H-2,18-(azenometheno)-13,9-(metheno)imidazo[5,1-
d][1,12,3,5]dioxadiazacyclohexadecin-16(17H)-one (43 mg, 15%) as a white solid; 1H NMR
(DMSO-d6, 300 MHz) δ 1.47 – 1.7 (4H, m), 2.83 (3H, d, J = 4.7 Hz), 4.27 (2H, t, J = 6.5 Hz), 4.58
(2H, t, J = 6.3 Hz), 4.82 (2H, s), 6.54 (1H, s), 6.73 (1H, dd, J = 2.4, 8.0 Hz), 6.81 (1H, d, J = 7.4 Hz),
7.15 (1H, t, J = 7.8 Hz), 7.93 (1H, s), 9.16 (1H, s); m/z (ES+), [M+H]+ = 342; acidic, HPLC tR =
2.35 min; HRMS ESI+ m/z observed 342.1594, C17H20N5O3 requires 342.1566
Ethyl 2-((4-((3-((tert-butyldimethylsilyl)oxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-
nitropyrimidin-2-yl)oxy)acetate and ethyl 2-((4-((ethoxycarbonyl)(3-hydroxybenzyl)amino)-6-
(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate. Triethylamine (3.95 ml, 28.4 mmol) was added
to a mixture of ethyl 3-((tert-butyldimethylsilyl)oxy)benzyl(6-(methylamino)-2-(methylsulfonyl)-5-

S16
nitropyrimidin-4-yl)carbamate (5.1 g, 9.5 mmol) and ethyl 2-hydroxyacetate (20 ml) . The resulting
mixture was stirred at 100°C for 1 hour. The solvent was removed under reduced pressure. The crude
product was purified by flash silica chromatography, elution gradient 0 to 40% ethyl acetate in
petroleum ether to give firstly ethyl 2-((4-((3-((tert-
butyldimethylsilyl)oxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetate (1.7 g, 32%) as a yellow gum; 1H NMR (DMSO-d6, 300 MHz) δ 0.11 (6H, s), 0.90
(9H, s), 1.01-1.23 (6H, m), 2.92 (3H, d), 3.96-4.15 (4H, m), 4.84 (2H, s), 5.01 (2H, s), 6.66-6.76 (1H,
m), 6.83 (1H, t), 6.88-6.97 (1H, m), 7.18 (1H, t), 8.63 (1H, broad d); m/z (ES+), [M+H]+ = 564;
acidic, HPLC tR = 1.50 min. This was followed by ethyl 2-((4-((ethoxycarbonyl)(3-
hydroxybenzyl)amino)-6-(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate (1.66 g, 39%) as a yellow
gum; 1H NMR (DMSO-d6, 300 MHz) δ 1.01-1.26 (6H, m), 2.93 (3H, d), 3.93-4.21 (4H, m), 4.84 (2H,
d), 4.98 (2H, s), 6.55-6.69 (1H, m), 6.69-6.78 (2H, m), 7.08 (1H, t), 8.63 (1H, broad q), 9.31 (1H, s);
m/z (ES+), [M+H]+ = 450; acidic, HPLC tR = 1.09 min.
Ethyl 2-((4-((3-(2-((tert-butoxycarbonyl)amino)ethoxy)benzyl)(ethoxycarbonyl)amino)-6-
(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate. DTBAD (0.87 g, 3.8 mmol) was added to ethyl
2-((4-((ethoxycarbonyl)(3-hydroxybenzyl)amino)-6-(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate
(0.85 g, 1.9 mmol), tert-butyl (2-hydroxyethyl)carbamate (0.61 g, 3.8 mmol) and triphenylphosphine
(0.99 g, 3.8 mmol) in THF (30 ml) at room temperature under nitrogen. The resulting mixture was
stirred at 40°C for 17 hours. The solvent was removed under reduced pressure. The crude product was
purified by flash silica chromatography, elution gradient 10 to 20% ethyl acetate in petroleum ether.
Pure fractions were evaporated to dryness to afford ethyl 2-((4-((3-(2-((tert-
butoxycarbonyl)amino)ethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetate (0.7 g, 63%) as a yellow gum; 1H NMR (DMSO-d6, 300 MHz) δ 1.07 (3H, t), 1.13
(3H, t), 1.37 (9H, s), 2.92 (3H, d), 3.20 – 3.41 (2H, m), 3.90 (2H, t), 4.03 (2H, q), 4.07 (2H, q), 4.86
(2H, s), 5.03 (2H, s), 6.80 (1H, dd), 6.90 (1H, d), 6.92 (1H, s), 6.97 (1H, t), 7.21 (1H, t), 8.63 (1H, q);
m/z (ES+), [M+H]+ = 593; acidic, HPLC tR = 1.26 min.

S17
Ethyl 2-((4-((3-(2-aminoethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-
nitropyrimidin-2-yl)oxy)acetate. TFA (0.91 ml, 11.8 mmol) was added to ethyl 2-((4-((3-(2-((tert-
butoxycarbonyl)amino)ethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetate (0.7 g, 1.2 mmol) in DCM (20 ml) at 0°C. The resulting solution was stirred at room
temperature for 3 hours. The solvent was removed under reduced pressure to dryness to afford ethyl
2-((4-((3-(2-aminoethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetate (0.8 g) as a yellow gum. The product was used in the next step directly without further
purification; m/z (ES+), [M+H]+ = 493; acidic, HPLC tR = 1.24 min.
2-((4-((3-(2-Aminoethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetic acid. LiOH (0.14 g, 5.8 mmol) was added to ethyl 2-((4-((3-(2-
aminoethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate
(0.7 g, 1.2 mmol) in THF (15 ml) / water (15 ml) at 0°C. The resulting mixture was stirred at room
temperature for 3 hours. The reaction mixture was adjusted to pH = 2~3 with 3M HCl at 0°C. The
reaction mixture was extracted with ethyl acetate (3 x 50 ml), the organic layer was dried over
Na2SO4, filtered and evaporated to afford 2-((4-((3-(2-aminoethoxy)benzyl)(ethoxycarbonyl)amino)-
6-(methylamino)-5-nitropyrimidin-2-yl)oxy)acetic acid (0.4 g, 75%) as a yellow solid. The product
was used in the next step directly without further purification; m/z (ES+), [M+H]+ = 465; acidic,
HPLC tR = 0.85 min.

S18
Ethyl 18-(methylamino)-19-nitro-13-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1~4,8~]henicosa-1(20),4(21),5,7,16,18-hexaene-2-carboxylate. EDC (495
mg, 2.6 mmol) was added to 2-((4-((3-(2-aminoethoxy)benzyl)(ethoxycarbonyl)amino)-6-
(methylamino)-5-nitropyrimidin-2-yl)oxy)acetic acid (400 mg, 0.86 mmol), diisopropylethylamine
(0.75 ml, 4.3 mmol) and HOBT (396 mg, 2.6 mmol) in DMF (10 ml) at room temperature. The
resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was poured into
water (100 ml), extracted with ethyl acetate (3 x 75 ml), the organic layer was dried over Na2SO4,
filtered and evaporated to afford ethyl 18-(methylamino)-19-nitro-13-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1~4,8~]henicosa-1(20),4(21),5,7,16,18-hexaene-2-carboxylate (300 mg, 78%)
as a yellow gum. The product was used in the next step directly without further purification; m/z
(ES+), [M+H]+ = 447; acidic, HPLC tR = 1.02 min.
20-(Methylamino)-7,8-dihydro-4H,15H-2,19-(azenometheno)-14,10-(metheno)imidazo[5,1-
d][1,12,3,5,15]dioxatriazacycloheptadecine-5,17(6H,18H)-dione (30). Iron (375 mg, 6.7 mmol)
was added to ethyl 18-(methylamino)-19-nitro-13-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1~4,8~]henicosa-1(20),4(21),5,7,16,18-hexaene-2-carboxylate (300 mg, 0.7 mmol)
in acetic acid (15 ml) and water (4 ml) at room temperature. The resulting mixture was stirred at 60°C
for 3 hours. The solid was filtered out and washed with methanol, the filtrate was concentrated to
dryness under reduced pressure. The crude product was purified by flash silica chromatography,
elution gradient 0 to 5% methanol in DCM. The fractions containing desired product were evaported
to dryness and the crude product was purified by preparative HPLC (XSelect CSH Prep C18 OBD
column, 5µ silica, 19 mm diameter, 150 mm length), using decreasingly polar mixtures of water
(containing 0.5% NH3) and acetonitrile as eluents. Fractions containing the desired compound were

S19
evaporated to dryness to afford 20-(methylamino)-7,8-dihydro-4H,15H-2,19-(azenometheno)-14,10-
(metheno)imidazo[5,1-d][1,12,3,5,15]dioxatriazacycloheptadecine-5,17(6H,18H)-dione (12 mg, 5%)
as a white solid; 1H NMR (DMSO-d6, 300 MHz) δ 2.88 (3H, d, J = 4.6 Hz), 3.38 (2H, s), 4.02 – 4.26
(2H, m), 4.73 (2H, s), 4.74 (2H, s), 6.69 (1H, s), 6.80 (1H, dd, J = 2.4, 7.8 Hz), 6.97 (1H, d, J = 7.4
Hz), 7.18 (1H, t, J = 7.8 Hz), 7.37 (1H, t, J = 1.9 Hz), 8.50 (1H, t, J = 5.5 Hz), 10.05 (1H, s); m/z
(ES+), [M+H]+ = 371; acidic, HPLC tR = 1.30 min; HRMS ESI+ m/z observed 371.1492,
C17H19N6O4 requires 371.1468
Ethyl 2-((4-((3-(2-((tert-butoxycarbonyl)(methyl)amino)ethoxy)benzyl)(ethoxycarbonyl)amino)-
6-(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate. DTBAD (1.03 g, 4.5 mmol) was added to
ethyl 2-((4-((ethoxycarbonyl)(3-hydroxybenzyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetate (1 g, 2.2 mmol), tert-butyl (2-hydroxyethyl)(methyl)carbamate (0.78 g, 4.5 mmol) and
triphenylphosphine (1.17 g, 4.5 mmol) in THF (30 ml) at room temperature under nitrogen. The
resulting mixture was stirred at 40°C for 17 hours. The solvent was removed under reduced pressure.
The crude product was purified by flash silica chromatography, elution gradient 10 to 20% ethyl
acetate in petroleum ether. Pure fractions were evaporated to dryness to afford ethyl 2-((4-((3-(2-
((tert-butoxycarbonyl)(methyl)amino)ethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-
nitropyrimidin-2-yl)oxy)acetate (1.1 g, 81%) as a yellow gum; 1H NMR (DMSO-d6, 300 MHz) δ 1.08
(3H, t), 1.16 (3H, t), 1.38 (9H, s), 2.80 (3H, s), 3.18 (2H, t), 3.46 (3H, d), 4.00-4.11 (6H, m), 4.85 (2H,
s), 5.03 (2H, s), 6.81 (1H, dd), 6.90 (1H, d), 6.92 (1H, s), 7.21 (1H, t), 8.61 – 8.66 (1H, m); m/z
(ES+), [M+H]+ = 607; acidic, HPLC tR = 1.24 min.

S20
Ethyl 2-((4-((ethoxycarbonyl)(3-(2-(methylamino)ethoxy)benzyl)amino)-6-(methylamino)-5-
nitropyrimidin-2-yl)oxy)acetate. TFA (1.27 ml, 16.5 mmol) was added to ethyl 2-((4-((3-(2-((tert-
butoxycarbonyl)(methyl)amino)ethoxy)benzyl)(ethoxycarbonyl)amino)-6-(methylamino)-5-
nitropyrimidin-2-yl)oxy)acetat (1 g, 1.7 mmol) in DCM (20 ml) at room temperature. The resulting
solution was stirred at room temperature for 3 hours. The solvent was removed under reduced
pressure to afford ethyl 2-((4-((ethoxycarbonyl)(3-(2-(methylamino)ethoxy)benzyl)amino)-6-
(methylamino)-5-nitropyrimidin-2-yl)oxy)acetate (1.5 g) as a yellow gum. The product was used in
the next step directly without further purification; m/z (ES+), [M+H]+ = 507; acidic, HPLC tR = 0.95
min
2-((4-((Ethoxycarbonyl)(3-(2-(methylamino)ethoxy)benzyl)amino)-6-(methylamino)-5-
nitropyrimidin-2-yl)oxy)acetic acid. LiOH (0.19 g, 8.1 mmol) was added to ethyl 2-((4-
((ethoxycarbonyl)(3-(2-(methylamino)ethoxy)benzyl)amino)-6-(methylamino)-5-nitropyrimidin-2-
yl)oxy)acetate (1 g, 1.6 mmol) in THF (15 ml) / water (15 ml) at 0°C. The resulting mixture was
stirred at room temperature for 3 hours. The reaction mixture was adjusted to pH=2~3 with 3M HCl at
0°C. The reaction mixture was extracted with ethyl acetate (3 x 50 ml), the organic layer was dried
over Na2SO4, filtered and evaporated to afford 2-((4-((ethoxycarbonyl)(3-(2-
(methylamino)ethoxy)benzyl)amino)-6-(methylamino)-5-nitropyrimidin-2-yl)oxy)acetic acid (0.48 g,
62%) as a yellow gum. The product was used in the next step directly without further purification;
m/z (ES+), [M+H]+ = 479; acidic, HPLC tR = 0.86 min.

S21
Ethyl 12-methyl-18-(methylamino)-19-nitro-13-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1~4,8~]henicosa-1(20),4(21),5,7,16,18-hexaene-2-carboxylate. EDC (553
mg, 2.9 mmol) was added to 2-((4-((ethoxycarbonyl)(3-(2-(methylamino)ethoxy)benzyl)amino)-6-
(methylamino)-5-nitropyrimidin-2-yl)oxy)acetic acid (460 mg, 0.96 mmol), diisopropylethylamine
(0.84 ml, 4.8 mmol) and HOBT (442 mg, 2.9 mmol) in DMF (10 ml) at room temperature. The
resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was poured into
water (100 ml), extracted with ethyl acetate (3 x 75 ml), the organic layer was dried over Na2SO4,
filtered and evaporated to afford ethyl 12-methyl-18-(methylamino)-19-nitro-13-oxo-9,15-dioxa-
2,12,17,20-tetraazatricyclo[14.3.1.1~4,8~]henicosa-1(20),4(21),5,7,16,18-hexaene-2-carboxylate (420
mg, 95%) as a yellow gum. The product was used in the next step directly without further
purification; m/z (ES+), [M+H]+ = 461; acidic, HPLC tR = 1.45 min.
6-Methyl-20-(methylamino)-7,8-dihydro-4H,15H-2,19-(azenometheno)-14,10-
(metheno)imidazo[5,1-d][1,12,3,5,15]dioxatriazacycloheptadecine-5,17(6H,18H)-dione (31).
Iron (485 mg, 8.7 mmol) was added to ethyl 12-methyl-18-(methylamino)-19-nitro-13-oxo-9,15-
dioxa-2,12,17,20-tetraazatricyclo[14.3.1.1~4,8~]henicosa-1(20),4(21),5,7,16,18-hexaene-2-
carboxylate (400 mg, 0.9 mmol) in acetic acid (15 ml) and water (4 ml) at room temperature. The
resulting mixture was stirred at 60°C for 4 hours. The solid was filtered and washed with methanol,
the filtrate was concentrated to dryness under reduced pressure. The crude product was purified by
flash silica chromatography, elution gradient 0 to 5% methanol in DCM. The fractions containing
desired product were evaported to dryness to afford 150 mg of impure desired product. The crude
product was purified by preparative HPLC (XSelect CSH Prep C18 OBD column, 5µ silica, 19 mm

S22
diameter, 150 mm length), using decreasingly polar mixtures of water (containing 0.5% NH3) and
acetonitrile as eluents. Fractions containing the desired compound were evaporated to dryness to
afford 6-methyl-20-(methylamino)-7,8-dihydro-4H,15H-2,19-(azenometheno)-14,10-
(metheno)imidazo[5,1-d][1,12,3,5,15]dioxatriazacycloheptadecine-5,17(6H,18H)-dione (30 mg, 9%)
as a white solid; 1H NMR (500 MHz, DMSO) 2.89 (3H, d, J = 4.7 Hz), 2.91 – 2.96 (1H, m), 3.09 (3H,
s), 4.03 – 4.13 (2H, m), 4.14 – 4.24 (1H, m), 4.59 (1H, d, J = 15.1 Hz), 4.67 (1H, d, J = 14.6 Hz), 4.83
(1H, d, J = 14.6 Hz), 5.48 (1H, d, J = 15.1 Hz), 6.53 (1H, d, J = 5.2 Hz), 6.85 (1H, dd, J = 1.5, 8.2
Hz), 6.97 (1H, d, J = 7.6 Hz), 7.16 (1H, d, J = 3.9 Hz), 7.20 (1H, t, J = 7.8 Hz), 9.90 (1H, s); m/z
(ES+), [M+H]+ = 385; base, HPLC tR = 3.28 min; HRMS ESI+ m/z observed 385.1634, C18H21N6O4
requires 385.1624.
Methyl 2-(3-(((6-chloro-2-(methylthio)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate. Sodium idoide (2.05 g, 13.7 mmol) was added
to ethyl (6-chloro-2-(methylthio)-5-nitropyrimidin-4-yl)carbamate (4 g, 13.7 mmol), methyl 2-(3-
(chloromethyl)phenoxy)acetate (3.23 g, 15 mmol) and K2CO3 (2.83 g, 20.5 mmol) in acetone (50 ml).
The resulting mixture was stirred at 40°C for 2 days. The reaction mixture was filtered through celite.
The solvent was removed under reduced pressure to afford methyl 2-(3-(((6-chloro-2-(methylthio)-5-
nitropyrimidin-4-yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (8 g) as a yellow oil. The
product was used in the next step directly without further purification; m/z (ES+), [M+H]+ = 471;
acid, HPLC tR = 1.67 min.
Methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(methylthio)-5-nitropyrimidin-4-

S23
yl)amino)methyl)phenoxy)acetate. Methylamine (20 ml, 17 mmol, 2N in THF) was added to
methyl 2-(3-(((6-chloro-2-(methylthio)-5-nitropyrimidin-4-yl)(ethoxycarbonyl)amino)methyl)
phenoxy)acetate (8 g, 17 mmol) in THF (20 ml). The resulting mixture was stirred at room
temperature for 2 hours. The crude product was purified by flash silica chromatography, elution
gradient 0 to 20% ethyl acetate in petroleum ether. Pure fractions were evaporated to dryness to afford
methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(methylthio)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate (3.6 g, 46%) as a yellow gum; m/z (ES+), [M+H]+ = 466; acid,
HPLC tR = 1.65 min.
Methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(methylsulfonyl)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate. m-CPBA (3.34 g, 19.3 mmol) was added to methyl 2-(3-
(((ethoxycarbonyl)(6-(methylamino)-2-(methylthio)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate (3.6 g, 7.7 mmol) in DCM (200 ml). The resulting mixture was
stirred at room temperature for 2 hours. The reaction mixture was diluted with DCM (150 ml), and
washed sequentially with saturated NaHCO3 (150 ml x 2), water (125 ml x 2), and saturated brine
(150 ml x 2). The organic layer was dried over Na2SO4, filtered and evaporated to afford desired
product methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(methylsulfonyl)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate (3.9 g) as a yellow solid which was used without further
purification; m/z (ES+), [M+H]+ = 498; acid, HPLC tR = 1.47 min.
Methyl 2-(3-(((2-(2-((tert-butoxycarbonyl)amino)ethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate. Triethylamine (1.51 ml, 10.9 mmol) was

S24
added to methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(methylsulfonyl)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate (1.8 g, 3.6 mmol) in tert-butyl (2-hydroxyethyl)carbamate (15 ml) .
The resulting mixture was stirred at 60°C for 2 hours. The reaction mixture was diluted with DCM
(150 ml), and washed sequentially with water (100 ml x 5) and brine (100 ml x 2). The organic layer
was dried over Na2SO4, filtered and evaporated to afford crude product. The crude product was
purified by flash silica chromatography, elution gradient 0 to 20% ethyl acetate in petroleum ether.
Pure fractions were evaporated to dryness to afford methyl 2-(3-(((2-(2-((tert-
butoxycarbonyl)amino)ethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (3.3 g) as a colourless oil which was used without
further purification; m/z (ES+), [M+H]+ = 579; acid, HPLC tR = 1.24 min.
Methyl 2-(3-(((2-(2-aminoethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate. TFA (8 ml, 103.8 mmol) was added to methyl
2-(3-(((2-(2-((tert-butoxycarbonyl)amino)ethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (3.3 g, 5.7 mmol) in DCM (60 ml) at 0°C over a
period of 2 minutes under nitrogen. The resulting mixture was stirred at room temperature for 20
hours. The reaction mixture was poured into ice (150 ml), and made basic with saturated aqueous
Na2CO3. The mixture was then extracted with DCM / methanol (10:1) (5 x 125 ml), the organic layer
was dried over Na2SO4, filtered and evaporated to afford methyl 2-(3-(((2-(2-aminoethoxy)-6-
(methylamino)-5-nitropyrimidin-4-yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (1.1 g, 40%) as
a yellow gum which was used without further purification; m/z (ES+), [M+H]+ = 479; acid, HPLC tR
= 1.13 min.

S25
2-(3-(((2-(2-Aminoethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetic acid. Lithium hydroxide (0.06 g, 2.3 mmol) was
added to methyl 2-(3-(((2-(2-aminoethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (1.1 g, 2.3 mmol) in THF / water (30 ml) at 0°C.
The resulting mixture was stirred at room temperature for 16 hours. The reaction mixture was poured
into ice (125 ml) and the mixture was adjusted to pH=3 with 2M HCl, then extracted with ethyl
acetate (3 x 100 ml) and the organic layer dried over Na2SO4, filtered and evaporated to afford 2-(3-
(((2-(2-aminoethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetic acid (0.96 g, 90%) as a yellow gum which was
used without further purification; m/z (ES+), [M+H]+ = 465; acid, HPLC tR = 0.95 min.
Ethyl 18-(methylamino)-19-nitro-11-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1^{4,8}]henicosa-1(19),4,6,8(21),16(20),17-hexaene-2-carboxylate. EDC
(371 mg, 1.9 mmol) was added to 2-(3-(((2-(2-aminoethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetic acid (900 mg, 1.9 mmol), HOBT (297 mg, 1.9
mmol) and diisopropylethylamine (0.34 ml, 1.9 mmol) in DMF (2 ml). The resulting mixture was
stirred at room temperature for 18 hours. The crude product was purified by flash silica
chromatography, elution gradient 0 to 5% methanol in DCM. Pure fractions were evaporated to
dryness to afford ethyl 18-(methylamino)-19-nitro-11-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1^{4,8}]henicosa-1(19),4,6,8(21),16(20),17-hexaene-2-carboxylate (300 mg,
35%) as a yellow solid; m/z (ES+), [M+H]+ = 447; acid, HPLC tR = 1.06 min.

S26
17-(Methylamino)-8,14-dioxa-1,11,16,19,22-pentaazatetracyclo[13.5.2.1^{3,7}.0^{18,21}]tricosa-
3,5,7(23),15(22),16,18(21)-hexaene-10,20-dione (32). Iron (38 mg, 0.7 mmol) was added to ethyl
18-(methylamino)-19-nitro-11-oxo-9,15-dioxa-2,12,17,20-tetraazatricyclo[14.3.1.1^{4,8}]henicosa-
1(19),4,6,8(21),16(20),17-hexaene-2-carboxylate (300 mg, 0.7 mmol) in acetic acid / water (2 ml).
The resulting mixture was stirred at 60°C for 2 hours. The crude product was purified by preparative
HPLC (Waters XBridge Prep C18 OBD column, 5µ silica, 30 mm diameter, 100 mm length), using
decreasingly polar mixtures of water (containing 0.1% NH4HCO3) and acetonitrile as eluents.
Fractions containing the desired compound were evaporated to dryness to afford 17-(methylamino)-
8,14-dioxa-1,11,16,19,22-pentaazatetracyclo[13.5.2.1^{3,7}.0^{18,21}]tricosa-
3,5,7(23),15(22),16,18(21)-hexaene-10,20-dione (13 mg, 5%) as a white solid; 1H NMR (300 MHz,
DMSO) 2.89 (3H, d, J = 4.6 Hz), 3.56 – 3.79 (2H, m), 4.17 – 4.38 (2H, m), 4.52 (2H, s), 4.72 (2H, s),
6.1 – 6.28 (1H, m), 6.41 – 6.59 (1H, m), 6.85 (1H, d, J = 7.5 Hz), 6.96 (1H, d, J = 7.4 Hz), 7.06 – 7.31
(2H, m), 9.56 (1H, s); m/z (ES+), [M+H]+ = 371; acid, HPLC tR = 1.35 min; HRMS ESI+ m/z
observed 371.1453, C17H19N6O4 requires 371.1468.
Methyl 2-(3-(((2-(2-((tert-butoxycarbonyl)(methyl)amino)ethoxy)-6-(methylamino)-5-
nitropyrimidin-4-yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate. Triethylamine (1.6 ml, 11.5
mmol) was added to methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(methylsulfonyl)-5-
nitropyrimidin-4-yl)amino)methyl)phenoxy)acetate (1.9 g, 3.8 mmol) in tert-butyl (2-
hydroxyethyl)(methyl)carbamate (12 ml, 3.8 mmol). The resulting mixture was stirred at 100°C for 2
hours. The reaction mixture was diluted with DCM (200 ml), and washed sequentially with water
(125 ml x 5) and brine (125 ml x 2). The organic layer was dried over Na2SO4, filtered and evaporated

S27
to afford crude product which was purified by flash silica chromatography, elution gradient 0 to 20%
ethyl acetate in petroleum ether to give methyl 2-(3-(((2-(2-((tert-
butoxycarbonyl)(methyl)amino)ethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (5 g) as a yellow oil which was used without
further purification; m/z (ES+), [M+H]+ = 593; acid, HPLC tR = 1.6 min.
Methyl 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(2-(methylamino)ethoxy)-5-nitropyrimidin-
4-yl)amino)methyl)phenoxy)acetate. TFA (6.5 ml, 84.4 mmol) was added to methyl 2-(3-(((2-(2-
((tert-butoxycarbonyl)(methyl)amino)ethoxy)-6-(methylamino)-5-nitropyrimidin-4-
yl)(ethoxycarbonyl)amino)methyl)phenoxy)acetate (5 g, 8.4 mmol) in DCM (100 ml) at 0°C over a
period of 3 minutes under nitrogen. The resulting mixture was stirred at room temperature for 15
hours. The reaction mixture was poured into ice (100 ml), extracted with DCM / methanol (10:1) (5 x
100 ml), the organic layer was dried over Na2SO4, filtered and evaporated to to afford methyl 2-(3-
(((ethoxycarbonyl)(6-(methylamino)-2-(2-(methylamino)ethoxy)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate (1.5 g, 36%) as a yellow oil; m/z (ES+), [M+H]+ = 493; ACID,
HPLC tR = 1.07 min.
2-(3-(((Ethoxycarbonyl)(6-(methylamino)-2-(2-(methylamino)ethoxy)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetic acid. Lithium hydroxide (0.37 g, 15.2 mmol) was added to methyl
2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(2-(methylamino)ethoxy)-5-nitropyrimidin-4-
yl)amino)methyl)phenoxy)acetate (1.5 g, 3.1 mmol) in THF / water (30 ml) at 0°C . The resulting
mixture was stirred at room temperature for 16 hours. The reaction mixture was poured into ice (150
ml) then made basic with saturated Na2CO3. The reaction was extracted with DCM (3 x 150 ml), the

S28
organic layer dried over Na2SO4, filtered and evaporated to afford 2-(3-(((ethoxycarbonyl)(6-
(methylamino)-2-(2-(methylamino)ethoxy)-5-nitropyrimidin-4-yl)amino)methyl)phenoxy)acetic acid
(1.2 g, 82%) as a orange gum which was used without further purification; m/z (ES+), [M+H]+ = 479;
acid, HPLC tR = 0.99 min.
Ethyl 12-methyl-18-(methylamino)-19-nitro-11-oxo-9,15-dioxa-2,12,17,20-
tetraazatricyclo[14.3.1.1^{4,8}]henicosa-1(19),4,6,8(21),16(20),17-hexaene-2-carboxylate. EDC
(0.42 g, 2.2 mmol) was added to 2-(3-(((ethoxycarbonyl)(6-(methylamino)-2-(2-
(methylamino)ethoxy)-5-nitropyrimidin-4-yl)amino)methyl)phenoxy)acetic acid (1.1 g, 2.2 mmol),
HOBT (0.3 g, 2.2 mmol) and diisopropylethylamine (0.38 ml, 2.2 mmol) in DMF (2 ml). The
resulting mixture was stirred at room temperature for 16 hours. The crude product was purified by
flash silica chromatography, elution gradient 0 to 5% methanol in DCM. Pure fractions were
evaporated to dryness to afford ethyl 12-methyl-18-(methylamino)-19-nitro-11-oxo-9,15-dioxa-
2,12,17,20-tetraazatricyclo[14.3.1.1^{4,8}]henicosa-1(19),4,6,8(21),16(20),17-hexaene-2-carboxylate
(0.35 g, 35%) as a pale yellow oil which was used without further purification; m/z (ES+), [M+H]+ =
461; acid, HPLC tR = 1.17 min.
11-Methyl-17-(methylamino)-8,14-dioxa-1,11,16,19,22-
pentaazatetracyclo[13.5.2.1^{3,7}.0^{18,21}]tricosa-3,5,7(23),15(22),16,18(21)-hexaene-10,20-
dione (33). Iron (36 mg, 0.7 mmol) was added to ethyl 12-methyl-18-(methylamino)-19-nitro-11-
oxo-9,15-dioxa-2,12,17,20-tetraazatricyclo[14.3.1.1^{4,8}]henicosa-1(19),4,6,8(21),16(20),17-
hexaene-2-carboxylate (300 mg, 0.7 mmol) in acetic acid / water (2 ml). The resulting mixture was
stirred at 60°C for 3 hours. The crude product was purified by preparative HPLC (Waters XBridge

S29
Prep C18 OBD column, 5µ silica, 30 mm diameter, 100 mm length), using decreasingly polar
mixtures of water (containing 0.1% NH4HCO3) and acetonitrile as eluents. Fractions containing the
desired compound were evaporated to dryness to afford 11-methyl-17-(methylamino)-8,14-dioxa-
1,11,16,19,22-pentaazatetracyclo[13.5.2.1^{3,7}.0^{18,21}]tricosa-3,5,7(23),15(22),16,18(21)-
hexaene-10,20-dione (7 mg, 3%) as a white solid.; 1H NMR (DMSO-d6, 300 MHz) δ 2.94 (3H, d, J =
4.7 Hz), 3.12 (3H, s), 3.9 – 4.08 (2H, s), 4.37 – 4.45 (2H, s), 4.49 (2H, s), 4.71 (2H, s), 6.22 (1H, q, J
= 4.9 Hz), 6.85 (1H, dd, J = 2.4, 8.3 Hz), 6.96 (1H, d, J = 7.4 Hz), 7.10 (1H, s), 7.22 (1H, dd, J = 7.4,
8.2 Hz), 9.57 (1H, s); m/z (ES+), [M+H]+ = 385; alkali, HPLC tR = 5.35 min; HRMS ESI+ m/z
observed 385.1634, C18H21N6O4 requires 385.1624
4-((4-Chloro-2-fluorophenyl)amino)-6,7-dimethoxyquinoline-3-carboxylic acid. 2N/NaOH(aq) (6
ml, 11.12 mmol) was added to ethyl 4-((4-chloro-2-fluorophenyl)amino)-6,7-dimethoxyquinoline-3-
carboxylate (1.50 g, 3.71 mmol) in methanol (30 ml). The resulting mixture was stirred at 60°C for 2
hours. The solvent was removed under reduced pressure and water (100 ml) was added. The reaction
mixture was adjusted to pH=6 with 2M HCl. The precipitate was collected by filtration, washed with
water (50 ml) and dried under vacuum to afford the title compound (1.35 g, 97%) as a grey solid,
which was used without further purification. 1H NMR (DMSO-d6, 300 MHz) δ 3.49 (3H, s), 3.91 (3H,
s), 6.98 (1H, s), 7.23 (1H, d), 7.29 (1H, dd), 7.37 (1H, s), 7.60 (1H, dd), 8.94 (1H, s); m/z (ES+),
[M+H]+ = 377; acid, HPLC tR = 0.917 min.
4-((4-Chloro-2-fluorophenyl)amino)-6,7-dimethoxyquinoline-3-carboxamide (34). HATU (563
mg, 2.34 mmol) was added to 4-((4-chloro-2-fluorophenyl)amino)-6,7-dimethoxyquinoline-3-
carboxylic acid (800 mg, 2.12 mmol), NH4Cl (341 mg, 6.37 mmol) and diisopropylethylamine (1.854
ml, 10.62 mmol) in DMF (10 ml). The resulting mixture was stirred at room temperature for 24 hours.
The reaction mixture was diluted with ethyl acetate (200 ml), and washed sequentially with water (2 x

S30
150 ml) and brine (2 x 150 ml). The organic layer was dried over Na2SO4, filtered and evaporated to
afford crude product that was purified by flash silica chromatography, elution gradient 0 to 5%
methanol in DCM. Pure fractions were evaporated to dryness to afford the title compound (750 mg,
94%) as a grey solid. 1H NMR (DMSO-d6, 500 MHz) δ 3.52 (3H, s), 3.93 (3H, s), 6.81 (1H, t, J = 8.9
Hz), 6.90 (1H, s), 7.12 (1H, ddd, J = 1.1, 2.4, 8.6 Hz), 7.34 (1H, s), 7.48 (1H, dd, J = 2.4, 10.9 Hz),
7.66 (1H, s), 8.25 (1H, s), 8.87 (1H, s), 10.41 (1H, s); m/z (ES+), [M+H]+ = 376; acid, HPLC tR =
0.910 min; HRMS ESI+ m/z observed 376.0842, C18H16N3O3ClF requires 376.0864
Ethyl 4-((4-chloro-2-fluorophenyl)amino)quinoline-3-carboxylate. Triethylamine (0.118 ml, 0.85
mmol) was added to ethyl 4-chloroquinoline-3-carboxylate (200 mg, 0.85 mmol) and 4-chloro-2-
fluoroaniline (124 mg, 0.85 mmol) in dioxane (5 ml). The resulting mixture was stirred at 100°C for 1
hour. The crude product was purified by flash silica chromatography, elution gradient 0 to 10% ethyl
acetate in petroleum ether. Pure fractions were evaporated to dryness to afford the title compound
(150 mg, 51%) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 1.21 (3H, t), 4.00 (2H, q), 7.31-
7.38 (2H, m), 7.60-7.73 (2H, m), 7.89 (1H, dd), 8.01 (1H, t), 8.09 (1H, d), 8.38 (1H, d), 9.04 (1H, s);
m/z (ES+), [M+H]+ = 345; acid, HPLC tR = 0.988 min.
4-((4-Chloro-2-fluorophenyl)amino)-N-methylquinoline-3-carboxamide (36).
Trimethylaluminium (2M in Hexane, 1 ml, 1.0 mmol) was added to ethyl 4-((4-chloro-2-
fluorophenyl)amino)quinoline-3-carboxylate (240 mg, 0.70 mmol) and methylamine (2M in THF, 2
ml, 2.0 mmol) in THF (5 ml) . The resulting mixture was stirred at room temperature for 1 hour. The
reaction mixture was poured into ice water (150 ml), extracted with ethyl acetate (2 x 100 ml) and the
organic layer was dried over Na2SO4, filtered and evaporated to afford crude product that was purified
by preparative HPLC (Waters XBridge Prep C18 OBD column, 5µ silica, 30 mm diameter, 100 mm
length), using decreasingly polar mixtures of water (containing % NH4HCO3) and acetonitrile as
eluents. Fractions containing the desired compound were evaporated to dryness to afford the title
compound (100 mg, 44%) as a yellow solid. 1H NMR (methanol-d4, 300 MHz) δ 2.76 (3H, s), 7.01

S31
(1H, t, J = 8.7 Hz), 7.11 (1H, ddd, J = 1.2, 2.4, 8.6 Hz), 7.29 (1H, dd, J = 2.3, 10.6 Hz), 7.49 (1H, t, J
= 7.7 Hz), 7.78 (1H, ddd, J = 1.4, 6.9, 8.4 Hz), 7.98 (1H, d, J = 8.5 Hz), 8.02 (1H, d, J = 8.6 Hz), 8.77
(1H, s), 2x NH not observed; m/z (ES+), [M+H]+ = 330; acid, HPLC tR = 1.468 min; HRMS ESI+
m/z observed 330.0806, C17H14N3OFCl requires 330.0809.
Ethyl 6-(benzyloxy)-4-chloro-7-methoxyquinoline-3-carboxylate. Ethyl 6-(benzyloxy)-4-hydroxy-
7-methoxyquinoline-3-carboxylate (30 g, 84.90 mmol) was added to POCl3 (150 ml) at 20°C under
nitrogen. The resulting mixture was stirred at 100°C for 3 hours. The solvent was removed under
reduced pressure then diluted with DCM (750 ml) and washed sequentially with saturated NaHCO3 (3
x 300 ml), water (2 x 300 ml) and saturated brine (3 x 400 ml). The organic layer was dried over
Na2SO4, filtered and evaporated to afford crude product that was purified by flash silica
chromatography, elution gradient 0 to 30% ethyl acetate in petroleum ether. Pure fractions were
evaporated to dryness to afford the title compound (5 g, 16%) as a yellow solid. 1H NMR
(Chloroform-d, 300 MHz) δ 1.46 (3H, t), 4.08 (3H, s), 4.49 (2H, q), 5.35 (2H, s), 7.30-7.59 (6H, m),
7.68 (1H, s), 9.06 (1H, s) ; m/z (ES+), [M+H]+ = 372; ACID, HPLC tR = 1.297 min.
Ethyl 6-(benzyloxy)-7-methoxy-4-(phenylamino)quinoline-3-carboxylate. Acetic acid (1.0 ml,
17.47 mmol) was added to ethyl 6-(benzyloxy)-4-chloro-7-methoxyquinoline-3-carboxylate (4.8 g,
12.91 mmol) and aniline (1.202 g, 12.91 mmol) in i-PrOH (3 ml). The resulting mixture was stirred at
100°C for 2 hours. The solvent was removed under reduced pressure and the crude product was
purified by flash silica chromatography, elution gradient 0 to 50% ethyl acetate in petroleum ether.
Pure fractions were evaporated to dryness to afford the title compound (4.2 g, 76%) as a pale yellow
solid. 1H NMR (DMSO-d6, 300 MHz) δ 1.17 (3H, t), 3.96 (3H, s), 3.97 (2H, q), 4.88 (2H, s), 7.17
(2H, d), 7.31-7.45 (9H, m), 7.58 (1H, s), 8.86 (1H, s), 10.24 (1H, s) ; m/z (ES+), [M+H]+ = 429; acid,
HPLC tR = 1.074 min.

S32
6-(Benzyloxy)-7-methoxy-N-methyl-4-(phenylamino)quinoline-3-carboxamide.
Trimethylaluminum (2M in hexane, 15 ml, 30 mmol) was added to ethyl 6-(benzyloxy)-7-methoxy-4-
(phenylamino)quinoline-3-carboxylate (4.2 g, 9.80 mmol) and methylamine (2M in THF, 20 ml, 40
mmol) in THF (10 ml). The resulting mixture was stirred at room temperature for 16 hours then at
40°C for a further 3 hours. The reaction mixture was poured into ice (150 ml), extracted with DCM (5
x 100 ml) and the organic layer was dried over Na2SO4, filtered and evaporated to afford the title
compound (2.7 g, 67%) as a yellow solid. 1H NMR (DMSO-d6, 300 MHz) δ 2.66 (3H, d), 3.95 (3H,
s), 4.75 (2H, s), 6.93 (2H, d), 6.99-7.05 (1H, m), 7.17 (1H, s), 7.21-7.31 (5H, m), 7.32-7.36 (3H, m),
8.55 (1H, q), 8.72 (1H, s), 9.98 (1H, s) ; m/z (ES+), [M+H]+ = 414; acid, HPLC tR = 1.399 min.
6-Hydroxy-7-methoxy-N-methyl-4-(phenylamino)quinoline-3-carboxamide. Palladium on carbon
(0.695 g, 6.53 mmol) was added to 6-(benzyloxy)-7-methoxy-N-methyl-4-(phenylamino)quinoline-3-
carboxamide (2.7 g, 6.53 mmol) in methanol (110 ml) under hydrogen. The resulting mixture was
stirred at room temperature for 18 hours. The reaction mixture was filtered through celite and the
solvent was removed under reduced pressure to afford the title compound (1.95 g, 92 %) as a yellow
solid. 1H NMR (DMSO-d6, 300 MHz) δ 2.64 (3H, d), 3.95 (3H, s), 6.91 (2H, d), 7.02 (1H, t), 7.15
(1H, s), 7.25 (2H, dd), 7.34 (1H, s), 8.64 (1H, q), 8.73 (1H, s), 9.86 (1H, s), 10.19 (1H, s); m/z (ES+),
[M+H]+ = 324; acid, HPLC tR = 0.741 min.

S33
tert-Butyl 4-(((7-methoxy-3-(methylcarbamoyl)-4-(phenylamino)quinolin-6-
yl)oxy)methyl)piperidine-1-carboxylate. Potassium iodide (51.3 mg, 0.31 mmol) was added to 6-
hydroxy-7-methoxy-N-methyl-4-(phenylamino)quinoline-3-carboxamide (100 mg, 0.31 mmol), tert-
butyl 4-(bromomethyl)piperidine-1-carboxylate (86 mg, 0.31 mmol) and potassium carbonate (42.7
mg, 0.31 mmol) in DMF (2 ml). The resulting mixture was stirred at 60°C for 16 hours. The crude
product was purified by flash C18-flash chromatography, elution gradient 0 to 50% methanol in
0.1%TFA (in water). Pure fractions were evaporated to dryness to afford the title compound (130 mg,
81 %) as a yellow solid. m/z (ES+), [M+H]+ = 521; acid, HPLC tR = 1.027 min.
7-Methoxy-N-methyl-4-(phenylamino)-6-(piperidin-4-ylmethoxy)quinoline-3-carboxamide
hydrochloride salt. tert-Butyl 4-(((7-methoxy-3-(methylcarbamoyl)-4-(phenylamino)quinolin-6-
yl)oxy)methyl)piperidine-1-carboxylate (130 mg, 0.25 mmol) in HCl/ethanol (3 ml). The resulting
mixture was stirred at rt for 2 hours. The solvent was removed under reduced pressure to afford 7-
methoxy-N-methyl-4-(phenylamino)-6-(piperidin-4-ylmethoxy)quinoline-3-carboxamide
hydrochloride salt (110 mg, 96%) as a yellow solid. m/z (ES+), [M+H]+ = 421; acid, HPLC tR =
0.639 min.

S34
7-Methoxy-N-methyl-6-((1-methylpiperidin-4-yl)methoxy)-4-(phenylamino)quinoline-3-
carboxamide (39). Sodium cyanoborohydride (166 mg, 0.78 mmol) was added to 7-methoxy-N-
methyl-4-(phenylamino)-6-(piperidin-4-ylmethoxy)quinoline-3-carboxamide (110 mg, 0.26 mmol),
formaldehyde solution (1 ml, 0.26 mmol) and sodium acetate (21.45 mg, 0.26 mmol) in methanol (3
ml). The resulting mixture was stirred at room temperature for 3 hours. The crude product was
purified by preparative HPLC (Waters XBridge Prep C18 OBD column, 5µ silica, 30 mm diameter,
100 mm length), using decreasingly polar mixtures of water (containing 0.1% Formic acid) and
acetonitrile as eluents. Fractions containing the desired compound were evaporated to dryness to
afford the title compound (40 mg, 35%) as a yellow solid. 1H NMR (methanol-d4, 300 MHz) δ 1.63
(2H, q, J = 12.3, 12.9 Hz), 1.97 – 2.22 (3H, m), 2.65 (3H, s), 2.89 (3H, s), 3.06 (2H, t, J = 12.7 Hz),
3.58 (2H, d, J = 12.5 Hz), 3.68 (2H, d, J = 5.6 Hz), 4.04 (3H, s), 7.28 (1H, s), 7.3 – 7.43 (3H, m), 7.49
(3H, dd, J = 7.6, 15.4 Hz), 8.62 (1H, s), 2x NH not observed; m/z (ES+), [M+H]+ = 435; alkali,
HPLC tR = 1.635 min; HRMS ESI+ m/z observed 435.2409, C25H31N4O3 requires 435.2396
N-Methyl-4-phenylquinazolin-2-amine (40). A suspension of 2-chloro-4-phenylquinazoline (200
mg, 0.83 mmol) in 2M Ammonia in THF (2 ml, 14.00 mmol) was sealed into a microwave tube. The
reaction was heated to 100°C for 1 hour in the microwave reactor and cooled to room temperature.
The reaction mixture was evaporated and the crude product was purified by flash silica
chromatography, elution gradient 0 to 50% ethyl acetate in DCM. Pure fractions were evaporated to
dryness to afford the title compound (157 mg, 80%) as a pale yellow solid. 1H NMR (400 MHz,
DMSO) 2.93 (3H, d, J = 4.8 Hz), 7.18 (1H, ddd, J = 1.2, 6.9, 8.1 Hz), 7.32 (1H, s), 7.54 – 7.61 (4H,
m), 7.65 – 7.72 (4H, m); m/z (ES+), [M+H]+ = 236; HRMS ESI+ m/z observed 236.1208, C15H14N3
requires 236.1188
4-Phenylquinazolin-2-amine (42). 2-Chloro-4-phenylquinazoline (200 mg, 0.83 mmol) was
suspended in THF (1 ml) and 7M Ammonia in methanol (1 ml, 7.00 mmol) and sealed into a
S35
microwave tube. The reaction was heated to 150°C for 3 hours in the microwave reactor and cooled to
room temperature. The reaction mixture was evaporated to dryness and then purified by flash silica
chromatography, elution gradient 0 to 50% ethyl acetate in DCM. Pure fractions were evaporated to
dryness to afford the title compound (175 mg, 95%) as a yellow foam. 1H NMR (DMSO-d6, 400
MHz) δ 6.83 (2H, s), 7.18 (1H, ddd, J = 1.2, 7.0, 8.3 Hz), 7.51 (1H, dd, J = 1.2, 9.0 Hz), 7.55 – 7.61
(3H, m), 7.64 – 7.73 (4H, m); m/z (ES+), [M+H]+ = 222; HRMS ESI+ m/z observed 222.1053,
C14H12N3 requires 222.1053
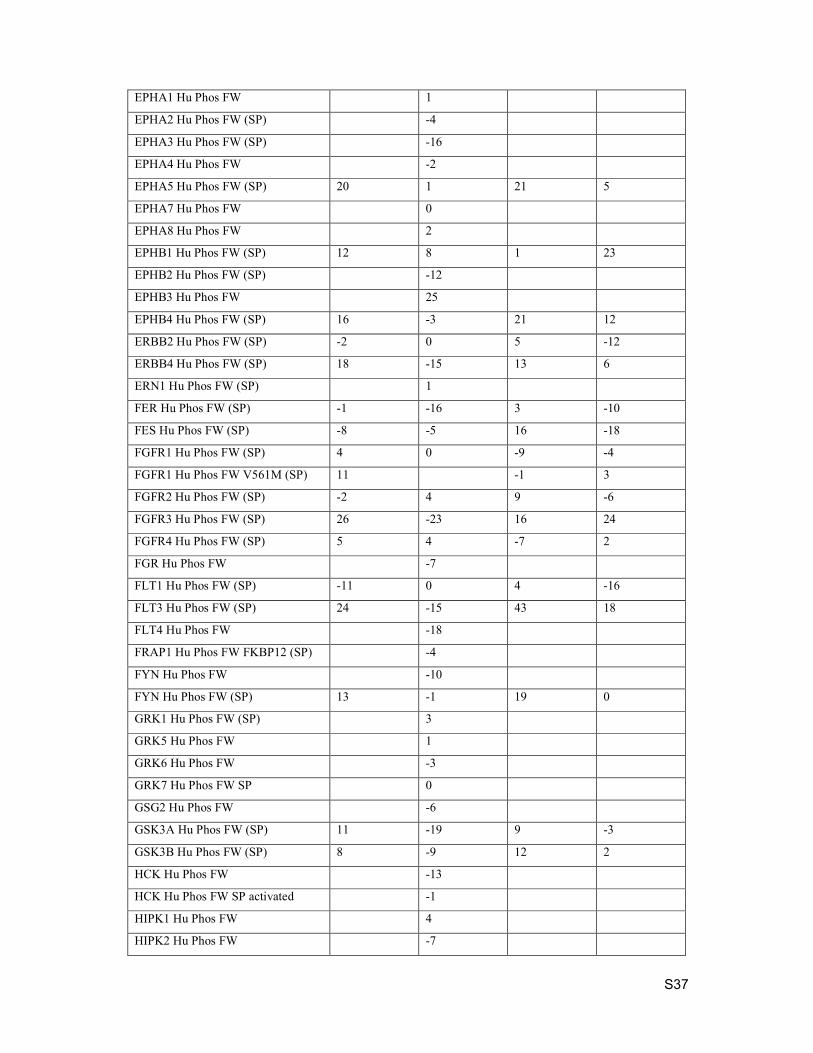
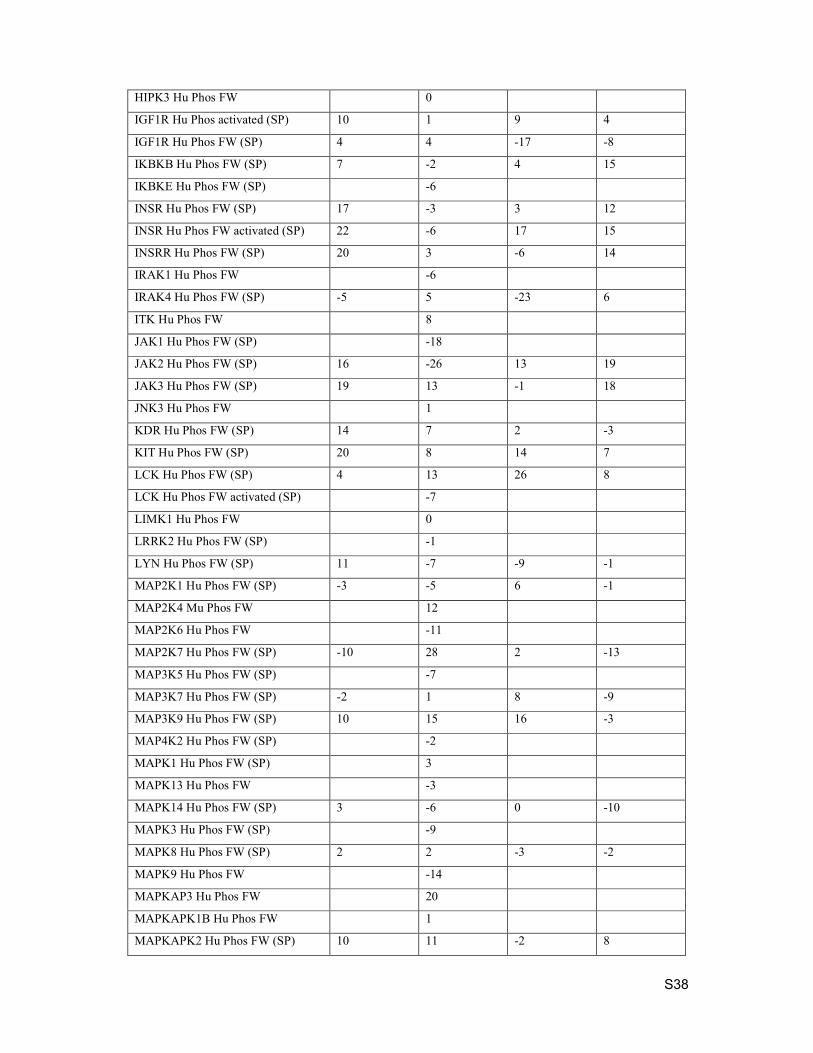
4. Kinase Selectivity
Selectivity testing was performed by Eurofins. Data below is percent inhibition at 1 µM compound
concentration.
Kinase Assay Description Compound 15 Compound 19 Compound 21 Compound 24
ABL1 Hu Phos FW (SP) 19 3 -1 4
ABL2 Hu Phos FW (SP) 16 0 3 17
ACVR1 Hu Phos FW (SP) -6
ACVR1B Hu Phos FW (SP) -6 4 -3 -1
AKT1 Hu Phos FW (SP) 10 -5 -2 2
AKT2 Hu Phos FW (SP) 16 8 23 13
AKT3 Hu Phos FW -17
ALK Hu Phos FW (SP) -2 -8 12 -4
AURKA Hu Phos FW (SP) 2
AURKB Hu Phos FW (SP) 3 -4 4 -6
AURKC Hu Phos FW (SP) 37 3 1 12
AXL Hu Phos FW (SP) -16 -10 7 -10
BLK Hu Phos FW (SP) -12 -4 3 5
BMX Hu Phos FW (SP) 8 -5 16 2
BRAF Hu Phos FW (SP) -15
BRSK1 Hu Phos FW (SP) -7
BRSK2 Hu Phos FW (SP) -15
BTK Hu Phos FW (SP) -2 -8 -5 -6
CaMK1 Hu Phos FW (SP) 14 -1 6 7
CAMK1d Hu Phos FW -2
CAMK2B Hu Phos FW (SP) 13 -10 6 15
CAMK2d Hu Phos FW 0
CAMK2g Hu Phos FW (SP) -2
CAMK4 Hu Phos FW -20

S36
CAMKK2 Hu Phos FW (SP) 2
CDC42BPa Hu Phos FW 7
CDC42BPb Hu Phos FW 8
CDK1/CCNB1 Hu Phos FW (SP) 7 6 12 4
CDK2/CCNA2 Hu Phos FW (SP) 6 0 8 -8
CDK2:CE Hu Phos FW (SP) -7
CDK3 Hu Phos FW 3
CDK5:p25 Hu Phos FW (SP) -39
CDK5:p35 Hu Phos FW (SP) 1
CDK6/CCND3 Hu Phos FW (SP) 22 0 -7 19
CDK7/CCNH/MNAT1 Hu Phos (SP) -1 -4 -8 -2
CDK9/CCNT1 Hu Phos FW (SP) 11 -6 15 1
CHEK1 Hu Phos FW (SP) 16 -18 -5 7
CHK2 Hu Phos FW (SP) -17
CHUK Hu Phos FW (SP) -6
CK1 Yeast Phos FW (SP) -9
CK2 Hu Phos FW 1
CLK1 Hu Phos FW (SP) -3
CLK2 Hu Phos FW SP -3
CLK3 Hu Phos FW 4
CLK4 Hu Phos FW (SP) -8
CSF1R Hu Phos FW (SP) -40 -2 32 -32
CSK Hu Phos FW (SP) -5
CSNK1d Hu Phos FW 2
CSNK1G1 Hu Phos FW (SP) 4 -8 14 -8
CSNK1g2 Hu Phos FW 1
CSNK1g3 Hu Phos FW 5
CSNK2A2 Hu Phos FW (SP) 1 5 3 8
DAPK1 Hu Phos FW 9
DAPK2 Hu Phos FW 19
DCAMKL2 Hu Phos FW 27
DDR2 Hu Phos FW (SP) 3 0 -8 -11
DMPK Hu Phos FW (SP) 1 0 -5 -3
DYRK2 Hu Phos FW (SP) 4 -4 12 3
EEF2K Hu Phos FW (SP) -11 -13 16 -21
EGFR Hu Phos FW (SP) 4 4 3 2
EGFR Hu Phos T790M L858R (SP) -13 17 -12
EIF2AK3 Hu Phos FW (SP) -16
EIF2AK4 Hu Phos FW (SP) -2
S37
EPHA1 Hu Phos FW 1
EPHA2 Hu Phos FW (SP) -4
EPHA3 Hu Phos FW (SP) -16
EPHA4 Hu Phos FW -2
EPHA5 Hu Phos FW (SP) 20 1 21 5
EPHA7 Hu Phos FW 0
EPHA8 Hu Phos FW 2
EPHB1 Hu Phos FW (SP) 12 8 1 23
EPHB2 Hu Phos FW (SP) -12
EPHB3 Hu Phos FW 25
EPHB4 Hu Phos FW (SP) 16 -3 21 12
ERBB2 Hu Phos FW (SP) -2 0 5 -12
ERBB4 Hu Phos FW (SP) 18 -15 13 6
ERN1 Hu Phos FW (SP) 1
FER Hu Phos FW (SP) -1 -16 3 -10
FES Hu Phos FW (SP) -8 -5 16 -18
FGFR1 Hu Phos FW (SP) 4 0 -9 -4
FGFR1 Hu Phos FW V561M (SP) 11 -1 3
FGFR2 Hu Phos FW (SP) -2 4 9 -6
FGFR3 Hu Phos FW (SP) 26 -23 16 24
FGFR4 Hu Phos FW (SP) 5 4 -7 2
FGR Hu Phos FW -7
FLT1 Hu Phos FW (SP) -11 0 4 -16
FLT3 Hu Phos FW (SP) 24 -15 43 18
FLT4 Hu Phos FW -18
FRAP1 Hu Phos FW FKBP12 (SP) -4
FYN Hu Phos FW -10
FYN Hu Phos FW (SP) 13 -1 19 0
GRK1 Hu Phos FW (SP) 3
GRK5 Hu Phos FW 1
GRK6 Hu Phos FW -3
GRK7 Hu Phos FW SP 0
GSG2 Hu Phos FW -6
GSK3A Hu Phos FW (SP) 11 -19 9 -3
GSK3B Hu Phos FW (SP) 8 -9 12 2
HCK Hu Phos FW -13
HCK Hu Phos FW SP activated -1
HIPK1 Hu Phos FW 4
HIPK2 Hu Phos FW -7
S38
HIPK3 Hu Phos FW 0
IGF1R Hu Phos activated (SP) 10 1 9 4
IGF1R Hu Phos FW (SP) 4 4 -17 -8
IKBKB Hu Phos FW (SP) 7 -2 4 15
IKBKE Hu Phos FW (SP) -6
INSR Hu Phos FW (SP) 17 -3 3 12
INSR Hu Phos FW activated (SP) 22 -6 17 15
INSRR Hu Phos FW (SP) 20 3 -6 14
IRAK1 Hu Phos FW -6
IRAK4 Hu Phos FW (SP) -5 5 -23 6
ITK Hu Phos FW 8
JAK1 Hu Phos FW (SP) -18
JAK2 Hu Phos FW (SP) 16 -26 13 19
JAK3 Hu Phos FW (SP) 19 13 -1 18
JNK3 Hu Phos FW 1
KDR Hu Phos FW (SP) 14 7 2 -3
KIT Hu Phos FW (SP) 20 8 14 7
LCK Hu Phos FW (SP) 4 13 26 8
LCK Hu Phos FW activated (SP) -7
LIMK1 Hu Phos FW 0
LRRK2 Hu Phos FW (SP) -1
LYN Hu Phos FW (SP) 11 -7 -9 -1
MAP2K1 Hu Phos FW (SP) -3 -5 6 -1
MAP2K4 Mu Phos FW 12
MAP2K6 Hu Phos FW -11
MAP2K7 Hu Phos FW (SP) -10 28 2 -13
MAP3K5 Hu Phos FW (SP) -7
MAP3K7 Hu Phos FW (SP) -2 1 8 -9
MAP3K9 Hu Phos FW (SP) 10 15 16 -3
MAP4K2 Hu Phos FW (SP) -2
MAPK1 Hu Phos FW (SP) 3
MAPK13 Hu Phos FW -3
MAPK14 Hu Phos FW (SP) 3 -6 0 -10
MAPK3 Hu Phos FW (SP) -9
MAPK8 Hu Phos FW (SP) 2 2 -3 -2
MAPK9 Hu Phos FW -14
MAPKAP3 Hu Phos FW 20
MAPKAPK1B Hu Phos FW 1
MAPKAPK2 Hu Phos FW (SP) 10 11 -2 8

S39
MARK1 Hu Phos FW (SP) 9 9 0 1
MARK2 Hu Phos FW (SP) -2 7 6 -6
MELK Hu Phos FW -10
MERTK Hu Phos FW 13
MET Hu Phos FW (SP) -14 -13 -19 -29
MINK1 Hu Phos FW (SP) 6 2 -2 -2
MKNK2 Hu Phos FW (SP) 1 -1 5 -8
MST1R Hu Phos FW (SP) 7 6 7 -13
MST4 Hu Phos FW (SP) -10
MTOR Hu Phos FW (SP) 6 -1 -2 3
MuSK Hu Phos FW (SP) 1
MYLK Hu Phos FW (SP) 11 -13 6 5
NEK11 Hu Phos FW (SP) -1
NEK2 Hu Phos FW (SP) 30 0 -6 31
NEK3 Hu Phos FW 7
NEK6 Hu Phos FW -10
NEK7 Hu Phos FW 5
NEK9 Hu Phos FW (SP) 1
NLK Hu Phos FW 2
NTRK1 Hu Phos FW (SP) 23 -1 9 -16
NTRK2 Hu Phos FW (SP) -21
NTRK3 Hu Phos FW (SP) -5
NUAK1 Hu Phos FW (SP) 32 0 12 14
p38b Hu Phos FW 1
p38g Hu Phos FW 4
PAK1 Hu Phos FW (SP) 9 -7 15 1
PAK2 Hu Phos FW (SP) 6 11 17 7
PAK4 Hu Phos FW (SP) 1 -2 1 -3
PAK6 Hu Phos FW -11
PAK7 Hu Phos FW (SP) 10 -7 6 -1
PASK Hu Phos FW -12
PDGFRa Hu Phos FW (SP) 4
PDGFRB Hu Phos FW (SP) 13 -7 -12 4
PDPK1 Hu Phos FW (SP) -1 5 -5 -3
PHKG2 Hu Phos FW -3
PIK3C2A Hu Phos TRF (SP) 2
PIK3C2G Hu Phos TRF (SP) 2 5 -3 9
PIK3CA p85a Hu Phos TRF (SP) 2 2 2 2
PIK3CB p85a Hu Phos TRF (SP) 4 4 3 7
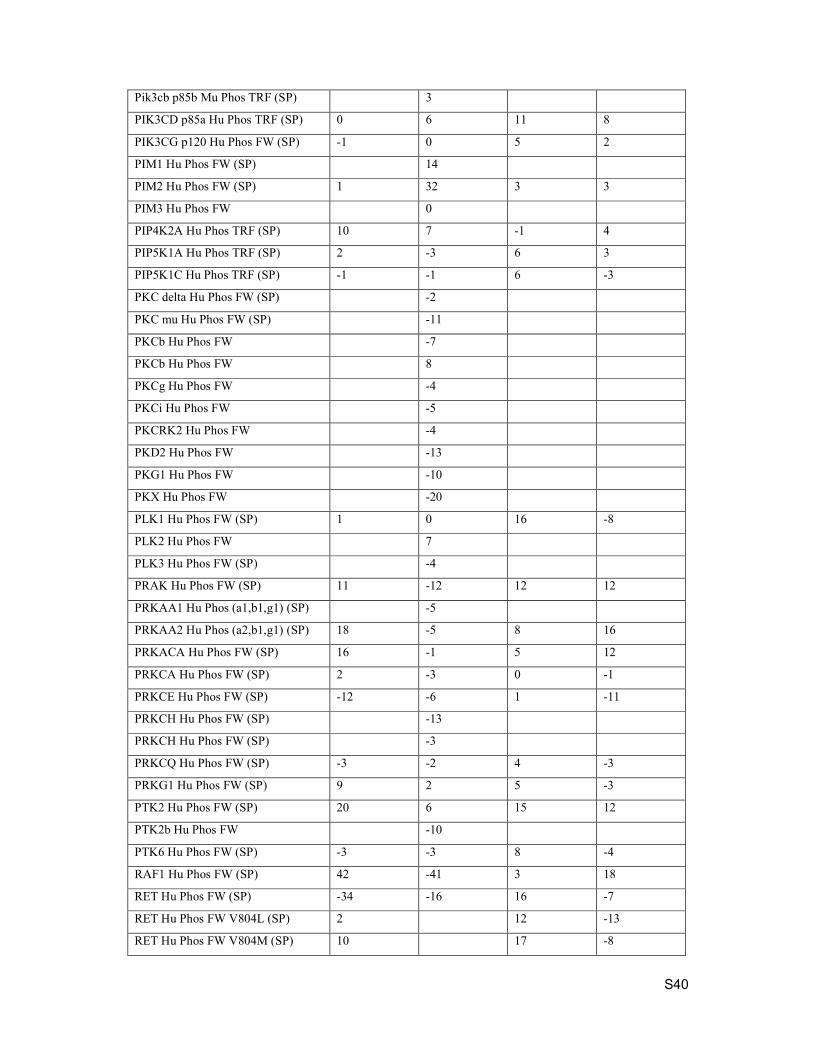
S40
Pik3cb p85b Mu Phos TRF (SP) 3
PIK3CD p85a Hu Phos TRF (SP) 0 6 11 8
PIK3CG p120 Hu Phos FW (SP) -1 0 5 2
PIM1 Hu Phos FW (SP) 14
PIM2 Hu Phos FW (SP) 1 32 3 3
PIM3 Hu Phos FW 0
PIP4K2A Hu Phos TRF (SP) 10 7 -1 4
PIP5K1A Hu Phos TRF (SP) 2 -3 6 3
PIP5K1C Hu Phos TRF (SP) -1 -1 6 -3
PKC delta Hu Phos FW (SP) -2
PKC mu Hu Phos FW (SP) -11
PKCb Hu Phos FW -7
PKCb Hu Phos FW 8
PKCg Hu Phos FW -4
PKCi Hu Phos FW -5
PKCRK2 Hu Phos FW -4
PKD2 Hu Phos FW -13
PKG1 Hu Phos FW -10
PKX Hu Phos FW -20
PLK1 Hu Phos FW (SP) 1 0 16 -8
PLK2 Hu Phos FW 7
PLK3 Hu Phos FW (SP) -4
PRAK Hu Phos FW (SP) 11 -12 12 12
PRKAA1 Hu Phos (a1,b1,g1) (SP) -5
PRKAA2 Hu Phos (a2,b1,g1) (SP) 18 -5 8 16
PRKACA Hu Phos FW (SP) 16 -1 5 12
PRKCA Hu Phos FW (SP) 2 -3 0 -1
PRKCE Hu Phos FW (SP) -12 -6 1 -11
PRKCH Hu Phos FW (SP) -13
PRKCH Hu Phos FW (SP) -3
PRKCQ Hu Phos FW (SP) -3 -2 4 -3
PRKG1 Hu Phos FW (SP) 9 2 5 -3
PTK2 Hu Phos FW (SP) 20 6 15 12
PTK2b Hu Phos FW -10
PTK6 Hu Phos FW (SP) -3 -3 8 -4
RAF1 Hu Phos FW (SP) 42 -41 3 18
RET Hu Phos FW (SP) -34 -16 16 -7
RET Hu Phos FW V804L (SP) 2 12 -13
RET Hu Phos FW V804M (SP) 10 17 -8

S41
RIPK2 Hu Phos FW 0
ROCK1 Hu Phos FW (SP) -2 10 5 -12
ROCK2 Hu Phos FW (SP) 24 -1 17 8
ROS1 Hu Phos FW (SP) 10 3 3 5
RPS6KA1 Hu Phos FW (SP) 22 -12 -2 11
RPS6KA2 Hu Phos FW -4
RPS6KA4 Hu Phos FW (SP) -3
RPS6KA5 Hu Phos FW (SP) 7 -11 -5 6
RPS6KA6 Hu Phos FW -3
RPS6KB1 Hu Phos FW (SP) 9 4 11 -2
SGK1 Hu Phos FW (SP) -7 0 14 -21
SGK2 Hu Phos FW 5
SGK3 Hu Phos FW -8
SIK1 Hu Phos FW (SP) -10
SNRK Hu Phos FW (SP) -11
SRC Hu Phos FW (SP) 7 -2 4 3
SRC Hu Phos FW 1-530 (SP) 27 38 12
SRPK1 Hu Phos FW (SP) -2 4 -9 -13
SRPK2 Hu Phos FW 0
STE20 Hu Phos FW 6
STK10 Hu Phos FW SP 15 1 7 10
STK11 Hu Phos FW (SP) 11 -13 11 10
STK17A Hu Phos FW (SP) 2 4 -10 3
STK23 Hu Phos FW 9
STK24 Hu Phos FW -3
STK25 Hu Phos FW (SP) -7
STK33 Hu Phos FW 0
STK4 Hu Phos FW (SP) 6 -6 10 4
SYK Hu Phos FW -6
TAO1 Hu Phos FW (SP) -4
TAO2 Hu Phos FW -3
TAO3 Hu Phos FW -2
TBK1 Hu Phos FW (SP) 5 -6 2 2
TEC Hu Enz FW (SP) 7
TGFBR1 Hu Phos FW (SP) 4 10 -15 -5
TIE2 Hu Phos FW -15
TLK1 Hu Phos FW (SP) 5
TLK2 Hu Phos FW -1
TNK2 Hu Phos FW (SP) -2
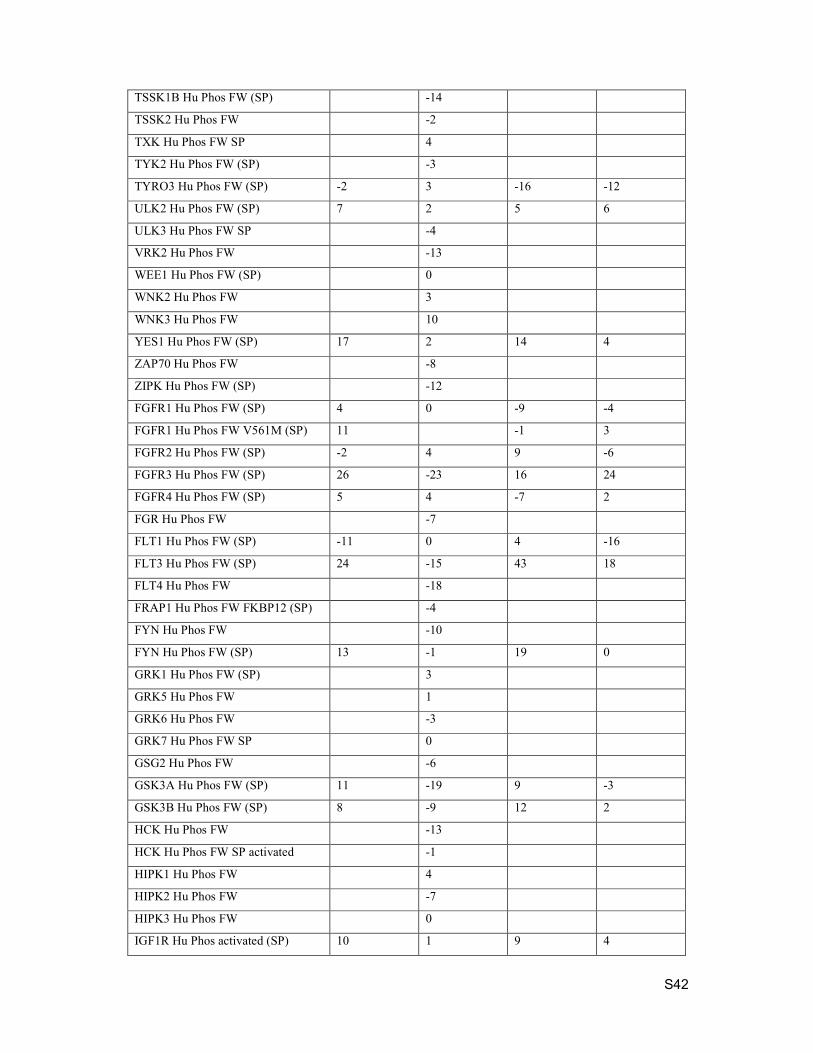
S42
TSSK1B Hu Phos FW (SP) -14
TSSK2 Hu Phos FW -2
TXK Hu Phos FW SP 4
TYK2 Hu Phos FW (SP) -3
TYRO3 Hu Phos FW (SP) -2 3 -16 -12
ULK2 Hu Phos FW (SP) 7 2 5 6
ULK3 Hu Phos FW SP -4
VRK2 Hu Phos FW -13
WEE1 Hu Phos FW (SP) 0
WNK2 Hu Phos FW 3
WNK3 Hu Phos FW 10
YES1 Hu Phos FW (SP) 17 2 14 4
ZAP70 Hu Phos FW -8
ZIPK Hu Phos FW (SP) -12
FGFR1 Hu Phos FW (SP) 4 0 -9 -4
FGFR1 Hu Phos FW V561M (SP) 11 -1 3
FGFR2 Hu Phos FW (SP) -2 4 9 -6
FGFR3 Hu Phos FW (SP) 26 -23 16 24
FGFR4 Hu Phos FW (SP) 5 4 -7 2
FGR Hu Phos FW -7
FLT1 Hu Phos FW (SP) -11 0 4 -16
FLT3 Hu Phos FW (SP) 24 -15 43 18
FLT4 Hu Phos FW -18
FRAP1 Hu Phos FW FKBP12 (SP) -4
FYN Hu Phos FW -10
FYN Hu Phos FW (SP) 13 -1 19 0
GRK1 Hu Phos FW (SP) 3
GRK5 Hu Phos FW 1
GRK6 Hu Phos FW -3
GRK7 Hu Phos FW SP 0
GSG2 Hu Phos FW -6
GSK3A Hu Phos FW (SP) 11 -19 9 -3
GSK3B Hu Phos FW (SP) 8 -9 12 2
HCK Hu Phos FW -13
HCK Hu Phos FW SP activated -1
HIPK1 Hu Phos FW 4
HIPK2 Hu Phos FW -7
HIPK3 Hu Phos FW 0
IGF1R Hu Phos activated (SP) 10 1 9 4

S43
IGF1R Hu Phos FW (SP) 4 4 -17 -8
IKBKB Hu Phos FW (SP) 7 -2 4 15
IKBKE Hu Phos FW (SP) -6
INSR Hu Phos FW (SP) 17 -3 3 12
INSR Hu Phos FW activated (SP) 22 -6 17 15
INSRR Hu Phos FW (SP) 20 3 -6 14
IRAK1 Hu Phos FW -6
IRAK4 Hu Phos FW (SP) -5 5 -23 6
ITK Hu Phos FW 8
JAK1 Hu Phos FW (SP) -18
JAK2 Hu Phos FW (SP) 16 -26 13 19
JAK3 Hu Phos FW (SP) 19 13 -1 18
JNK3 Hu Phos FW 1
KDR Hu Phos FW (SP) 14 7 2 -3
KIT Hu Phos FW (SP) 20 8 14 7
LCK Hu Phos FW (SP) 4 13 26 8
LCK Hu Phos FW activated (SP) -7
LIMK1 Hu Phos FW 0
LRRK2 Hu Phos FW (SP) -1
LYN Hu Phos FW (SP) 11 -7 -9 -1
MAP2K1 Hu Phos FW (SP) -3 -5 6 -1
MAP2K4 Mu Phos FW 12
MAP2K6 Hu Phos FW -11
MAP2K7 Hu Phos FW (SP) -10 28 2 -13
MAP3K5 Hu Phos FW (SP) -7
MAP3K7 Hu Phos FW (SP) -2 1 8 -9
MAP3K9 Hu Phos FW (SP) 10 15 16 -3
MAP4K2 Hu Phos FW (SP) -2
MAPK1 Hu Phos FW (SP) 3
MAPK13 Hu Phos FW -3
MAPK14 Hu Phos FW (SP) 3 -6 0 -10
MAPK3 Hu Phos FW (SP) -9
MAPK8 Hu Phos FW (SP) 2 2 -3 -2
MAPK9 Hu Phos FW -14
MAPKAP3 Hu Phos FW 20
MAPKAPK1B Hu Phos FW 1
MAPKAPK2 Hu Phos FW (SP) 10 11 -2 8
MARK1 Hu Phos FW (SP) 9 9 0 1
MARK2 Hu Phos FW (SP) -2 7 6 -6

S44
MELK Hu Phos FW -10
MERTK Hu Phos FW 13
MET Hu Phos FW (SP) -14 -13 -19 -29
MINK1 Hu Phos FW (SP) 6 2 -2 -2
MKNK2 Hu Phos FW (SP) 1 -1 5 -8
MST1R Hu Phos FW (SP) 7 6 7 -13
MST4 Hu Phos FW (SP) -10
MTOR Hu Phos FW (SP) 6 -1 -2 3
MuSK Hu Phos FW (SP) 1
MYLK Hu Phos FW (SP) 11 -13 6 5
NEK11 Hu Phos FW (SP) -1
NEK2 Hu Phos FW (SP) 30 0 -6 31
NEK3 Hu Phos FW 7
NEK6 Hu Phos FW -10
NEK7 Hu Phos FW 5
NEK9 Hu Phos FW (SP) 1
NLK Hu Phos FW 2
NTRK1 Hu Phos FW (SP) 23 -1 9 -16
NTRK2 Hu Phos FW (SP) -21
NTRK3 Hu Phos FW (SP) -5
NUAK1 Hu Phos FW (SP) 32 0 12 14
p38b Hu Phos FW 1
p38g Hu Phos FW 4
PAK1 Hu Phos FW (SP) 9 -7 15 1
PAK2 Hu Phos FW (SP) 6 11 17 7
PAK4 Hu Phos FW (SP) 1 -2 1 -3
PAK6 Hu Phos FW -11
PAK7 Hu Phos FW (SP) 10 -7 6 -1
PASK Hu Phos FW -12
PDGFRa Hu Phos FW (SP) 4
PDGFRB Hu Phos FW (SP) 13 -7 -12 4
PDPK1 Hu Phos FW (SP) -1 5 -5 -3
PHKG2 Hu Phos FW -3
PIK3C2A Hu Phos TRF (SP) 2
PIK3C2G Hu Phos TRF (SP) 2 5 -3 9
PIK3CA p85a Hu Phos TRF (SP) 2 2 2 2
PIK3CB p85a Hu Phos TRF (SP) 4 4 3 7
Pik3cb p85b Mu Phos TRF (SP) 3
PIK3CD p85a Hu Phos TRF (SP) 0 6 11 8
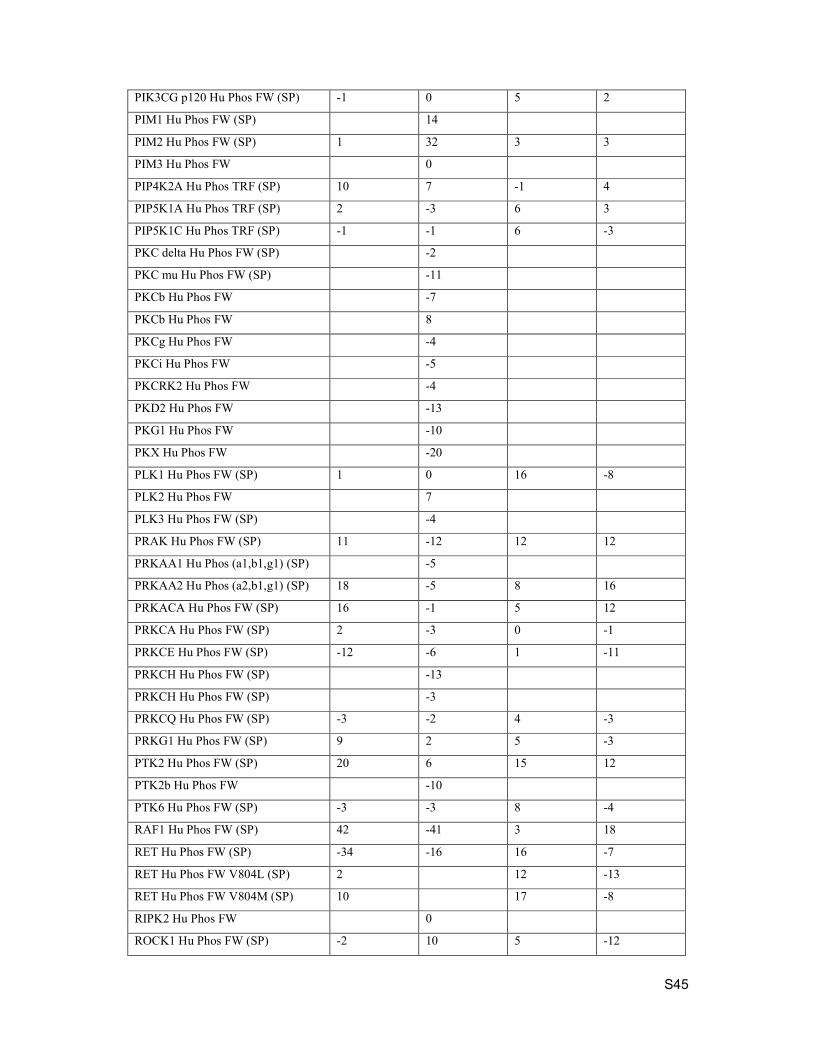
S45
PIK3CG p120 Hu Phos FW (SP) -1 0 5 2
PIM1 Hu Phos FW (SP) 14
PIM2 Hu Phos FW (SP) 1 32 3 3
PIM3 Hu Phos FW 0
PIP4K2A Hu Phos TRF (SP) 10 7 -1 4
PIP5K1A Hu Phos TRF (SP) 2 -3 6 3
PIP5K1C Hu Phos TRF (SP) -1 -1 6 -3
PKC delta Hu Phos FW (SP) -2
PKC mu Hu Phos FW (SP) -11
PKCb Hu Phos FW -7
PKCb Hu Phos FW 8
PKCg Hu Phos FW -4
PKCi Hu Phos FW -5
PKCRK2 Hu Phos FW -4
PKD2 Hu Phos FW -13
PKG1 Hu Phos FW -10
PKX Hu Phos FW -20
PLK1 Hu Phos FW (SP) 1 0 16 -8
PLK2 Hu Phos FW 7
PLK3 Hu Phos FW (SP) -4
PRAK Hu Phos FW (SP) 11 -12 12 12
PRKAA1 Hu Phos (a1,b1,g1) (SP) -5
PRKAA2 Hu Phos (a2,b1,g1) (SP) 18 -5 8 16
PRKACA Hu Phos FW (SP) 16 -1 5 12
PRKCA Hu Phos FW (SP) 2 -3 0 -1
PRKCE Hu Phos FW (SP) -12 -6 1 -11
PRKCH Hu Phos FW (SP) -13
PRKCH Hu Phos FW (SP) -3
PRKCQ Hu Phos FW (SP) -3 -2 4 -3
PRKG1 Hu Phos FW (SP) 9 2 5 -3
PTK2 Hu Phos FW (SP) 20 6 15 12
PTK2b Hu Phos FW -10
PTK6 Hu Phos FW (SP) -3 -3 8 -4
RAF1 Hu Phos FW (SP) 42 -41 3 18
RET Hu Phos FW (SP) -34 -16 16 -7
RET Hu Phos FW V804L (SP) 2 12 -13
RET Hu Phos FW V804M (SP) 10 17 -8
RIPK2 Hu Phos FW 0
ROCK1 Hu Phos FW (SP) -2 10 5 -12
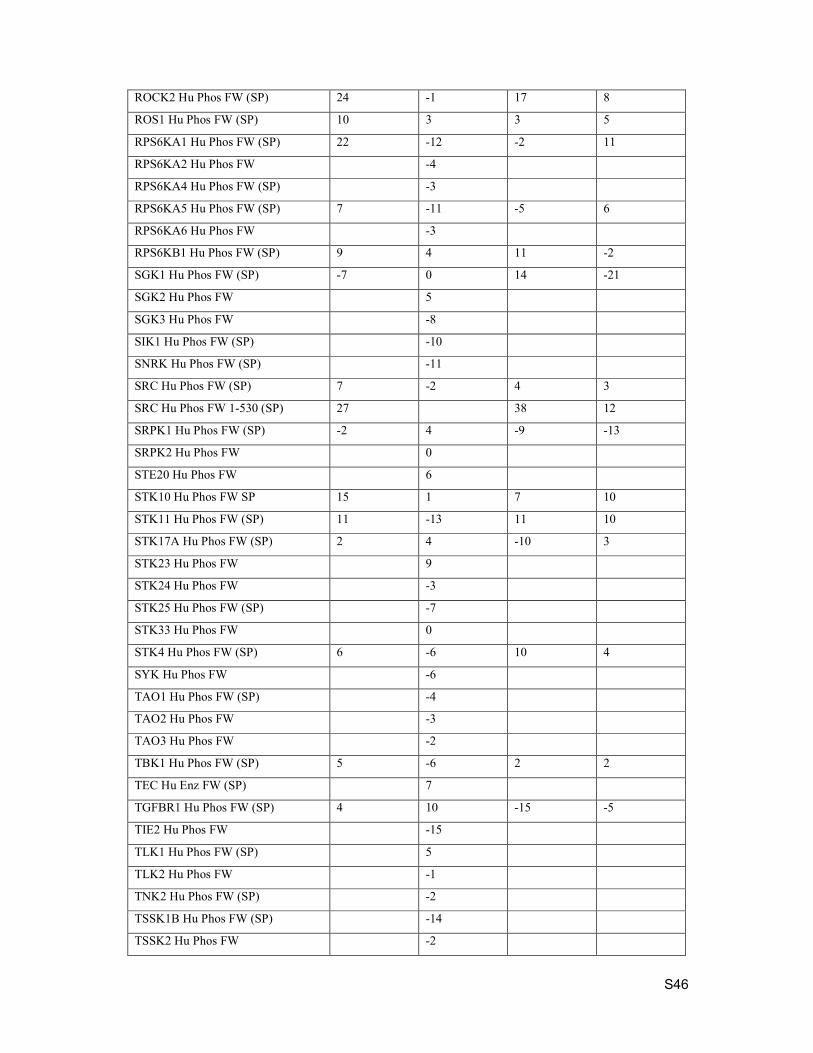
S46
ROCK2 Hu Phos FW (SP) 24 -1 17 8
ROS1 Hu Phos FW (SP) 10 3 3 5
RPS6KA1 Hu Phos FW (SP) 22 -12 -2 11
RPS6KA2 Hu Phos FW -4
RPS6KA4 Hu Phos FW (SP) -3
RPS6KA5 Hu Phos FW (SP) 7 -11 -5 6
RPS6KA6 Hu Phos FW -3
RPS6KB1 Hu Phos FW (SP) 9 4 11 -2
SGK1 Hu Phos FW (SP) -7 0 14 -21
SGK2 Hu Phos FW 5
SGK3 Hu Phos FW -8
SIK1 Hu Phos FW (SP) -10
SNRK Hu Phos FW (SP) -11
SRC Hu Phos FW (SP) 7 -2 4 3
SRC Hu Phos FW 1-530 (SP) 27 38 12
SRPK1 Hu Phos FW (SP) -2 4 -9 -13
SRPK2 Hu Phos FW 0
STE20 Hu Phos FW 6
STK10 Hu Phos FW SP 15 1 7 10
STK11 Hu Phos FW (SP) 11 -13 11 10
STK17A Hu Phos FW (SP) 2 4 -10 3
STK23 Hu Phos FW 9
STK24 Hu Phos FW -3
STK25 Hu Phos FW (SP) -7
STK33 Hu Phos FW 0
STK4 Hu Phos FW (SP) 6 -6 10 4
SYK Hu Phos FW -6
TAO1 Hu Phos FW (SP) -4
TAO2 Hu Phos FW -3
TAO3 Hu Phos FW -2
TBK1 Hu Phos FW (SP) 5 -6 2 2
TEC Hu Enz FW (SP) 7
TGFBR1 Hu Phos FW (SP) 4 10 -15 -5
TIE2 Hu Phos FW -15
TLK1 Hu Phos FW (SP) 5
TLK2 Hu Phos FW -1
TNK2 Hu Phos FW (SP) -2
TSSK1B Hu Phos FW (SP) -14
TSSK2 Hu Phos FW -2

S47
TXK Hu Phos FW SP 4
TYK2 Hu Phos FW (SP) -3
TYRO3 Hu Phos FW (SP) -2 3 -16 -12
ULK2 Hu Phos FW (SP) 7 2 5 6
ULK3 Hu Phos FW SP -4
VRK2 Hu Phos FW -13
WEE1 Hu Phos FW (SP) 0
WNK2 Hu Phos FW 3
WNK3 Hu Phos FW 10
YES1 Hu Phos FW (SP) 17 2 14 4
ZAP70 Hu Phos FW -8
ZIPK Hu Phos FW (SP) -12
5. Secondary Pharmacology Profiling
Data shown below is IC50 data in Cerep Secondary pharmacology screening panel.
Assay Compound 15 Compound 19 Compound 21 Compound 24
D2 Hu Bind FW CR >100 >100 >100 >100
a1B Hu Bind FW CR >100 >100 0.3935 >100
a2C Hu Bind FW CR >100 >100 >100 >100
Ang2 AT1 Hu Bind FW CR >100 >100 >100 >100
EtA Hu Bind FW CR >100 >100 >100 >100
GABAa1b2g2 Hu Bind FW CR >100 >100 >100 >100
H1 Hu Bind FW CR >100 5.991 >100 >100
H2 Hu Bind FW CR >100 >100 >100 >100
GR Hu Cyto Bind FW CR >100 >100 >100 >100
DOP2 Hu Bind FW CR >100 >100 >100 >100
b1 Hu HEK cAMP TRF CR >100 >100 >100 >100
D1 Hu CHO cAMP TRF CR >100 >100 >100 >100
Ang2 AT1 Hu HEK [Ca2+] Ag CR >100 >100 >100 11.79
Ang2 AT1 Hu [Ca2+] Antag CR >100 27.32 >100
DAT Hu Bind FW CR >100 >100 >100 >100
D3 Hu Bind FW CR >100 >100 >100 >100
A1 Hu CHO Imped CDS Ag CR >30 >30 >30
A1 Hu CHO Imped CDS Antag CR >30 >30 28.55 >30
a1A Hu CHO [Ca2+] Ag CR >100 85.73 >100 >100
a1A Hu CHO[Ca2+] Antag CR >100 7.459 71.88 >30
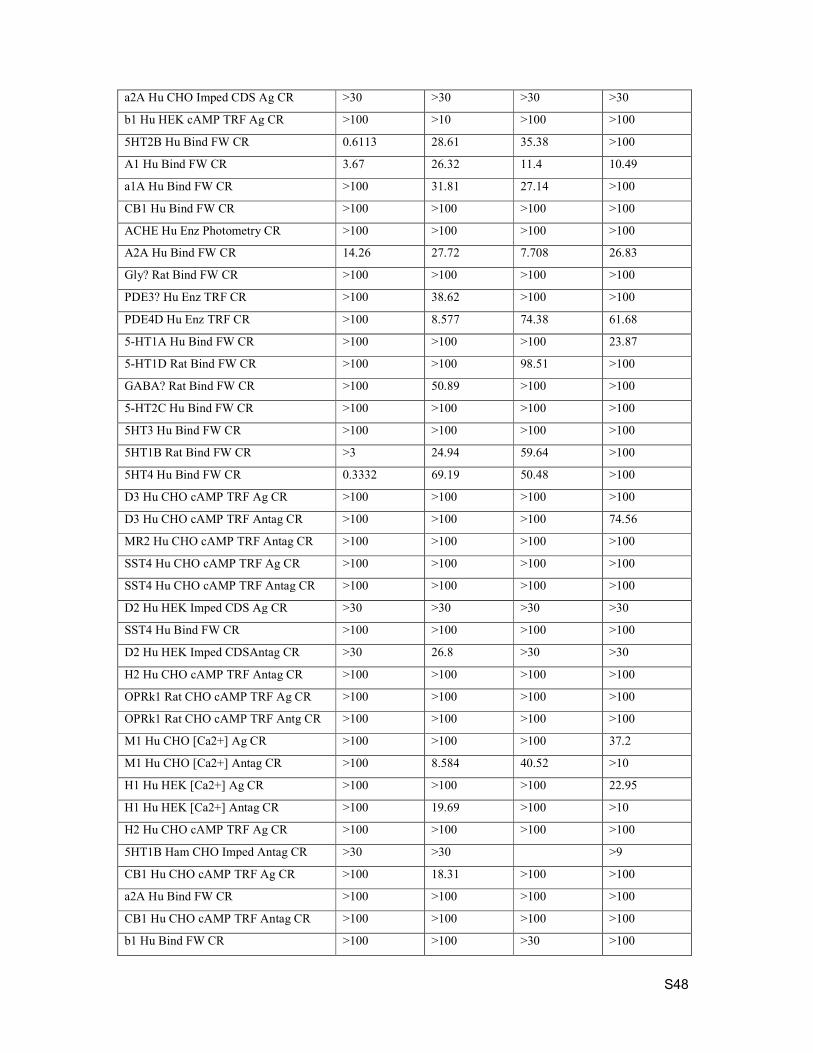
S48
a2A Hu CHO Imped CDS Ag CR >30 >30 >30 >30
b1 Hu HEK cAMP TRF Ag CR >100 >10 >100 >100
5HT2B Hu Bind FW CR 0.6113 28.61 35.38 >100
A1 Hu Bind FW CR 3.67 26.32 11.4 10.49
a1A Hu Bind FW CR >100 31.81 27.14 >100
CB1 Hu Bind FW CR >100 >100 >100 >100
ACHE Hu Enz Photometry CR >100 >100 >100 >100
A2A Hu Bind FW CR 14.26 27.72 7.708 26.83
Gly? Rat Bind FW CR >100 >100 >100 >100
PDE3? Hu Enz TRF CR >100 38.62 >100 >100
PDE4D Hu Enz TRF CR >100 8.577 74.38 61.68
5-HT1A Hu Bind FW CR >100 >100 >100 23.87
5-HT1D Rat Bind FW CR >100 >100 98.51 >100
GABA? Rat Bind FW CR >100 50.89 >100 >100
5-HT2C Hu Bind FW CR >100 >100 >100 >100
5HT3 Hu Bind FW CR >100 >100 >100 >100
5HT1B Rat Bind FW CR >3 24.94 59.64 >100
5HT4 Hu Bind FW CR 0.3332 69.19 50.48 >100
D3 Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
D3 Hu CHO cAMP TRF Antag CR >100 >100 >100 74.56
MR2 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
SST4 Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
SST4 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
D2 Hu HEK Imped CDS Ag CR >30 >30 >30 >30
SST4 Hu Bind FW CR >100 >100 >100 >100
D2 Hu HEK Imped CDSAntag CR >30 26.8 >30 >30
H2 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
OPRk1 Rat CHO cAMP TRF Ag CR >100 >100 >100 >100
OPRk1 Rat CHO cAMP TRF Antg CR >100 >100 >100 >100
M1 Hu CHO [Ca2+] Ag CR >100 >100 >100 37.2
M1 Hu CHO [Ca2+] Antag CR >100 8.584 40.52 >10
H1 Hu HEK [Ca2+] Ag CR >100 >100 >100 22.95
H1 Hu HEK [Ca2+] Antag CR >100 19.69 >100 >10
H2 Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
5HT1B Ham CHO Imped Antag CR >30 >30 >9
CB1 Hu CHO cAMP TRF Ag CR >100 18.31 >100 >100
a2A Hu Bind FW CR >100 >100 >100 >100
CB1 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
b1 Hu Bind FW CR >100 >100 >30 >100

S49
D1 Hu Bind FW CR >100 >100 70.37 >100
5HT2B Hu CHO IP1 TRF Antag CR 2.514 5.211 34.24 82.95
D1 Hu CHO cAMP TRF Ag CR >100 >10 >100 >100
M2 Hu CHO cAMP TRF Ag CR >100 >10 >100 >100
EtA Hu SKN [Ca2+] Antag CR >100 28.76 >100
M2 Hu Bind FW CR >100 >100 68.13 >100
OPRm1 Hu Bind FW CR >100 94.84 59.26 97.01
NAchA1 Hu Bind FW CR >100 >100 >100 >100
NAchA4 Hu Bind FW CR >100 >100 >100 >100
NMDA? Rat Bind FW CR >100 >100 >100 >100
NET Hu Bind FW CR >100 85.11 >100 >100
5HT1B Ham CHO Imped Ag CR >30 >30 >30 >30
EtA Hu [Ca2+] Ag CR >100 74.11 >100 30.66
a2C Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
a2C Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
OPRk1 Rat Bind FW CR >100 61.63 79.92 14.76
a1B Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
M1 Hu Bind FW CR >100 46.98 60.44 >100
M5 Hu Bind FW CR >100 >100 >100 >100
MR2 Hu Bind FW CR 53.84 48.39 >100 >100
NK1 Hu Bind FW CR >100 52.87 17.03 >100
a1B Hu CHO cAMP TRF Antag CR >100 97.64 >100 >100
M2 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
OPRm1 Hu CHO cAMP TRF Ag CR >100 99.51 >100 >100
OPRm1 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
5HT2B Hu CHO IP1 TRF Ag CR >100 >10 >100 >100
5-HT1D Rat CHO Imped Antag CR >30 >30 >30 >30
5HT4 Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
GABAA? Rat Bind FW CR 1.171 >100 14.24 >100
ARG1 Hu Enz Photometry CR >100
5HT4 Hu CHO cAMP TRF Antag CR 0.193 >100 >100 >100
5-HT7 Hu CHO cAMP TRF Ag CR >100 >100 >100
5-HT7 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
BK2 Hu CHO [Ca2+] Ag CR >100 81.36 >100 41.82
BK2 Hu CHO [Ca2+] Antag CR 0.7226 >30 54.72
b2 Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
b2 Hu CHO cAMP TRF Antag CR >100 >100 >100 >100
NAchA7 Hu Bind FW CR >100 >100 >100 >100
NMDA? Rat Bind FW CR >100 >100 >100 >100

S50
Sigma1 Hu Bind FW CR >100 >100 >100 >100
5-HT1A Hu HEK Imped CDS Ag CR >30 21.26 >30 >30
5-HT1D Rat CHO Imped Ag CR >30 >30 >30 >30
CatS Hu Enz Fluorimetry CR >100 >100 >100 >10
PPARgamma Hu Bind FW CR >100 >100 >100 >100
5-HT2C Hu HEK IP1 TRF Ag CR >100 >100 >100 >100
5-HT1A Hu HEK Imped Antag CR >30 >30 >30
ROCK2 Hu Enz TRF CR >100 >100 >100 >100
5HT7 Hu Bind FW CR >100 >100 25.34 >100
TSPO Rat Bind FW CR 20.97
GABAB Hu Bind FW CR >100 >100 >100 >100
Ghre Hu Binding CR >100 >100 >100 >100
COX1 Hu Enz TRF CR >100 >100 >100 30.02
cKIT Hu Enz TRF CR >100 >100 >100 >100
GSK3b Hu Enz TRF CR >100 >100 >100 >100
PDE6 Bov Enz CR >100 8.364 12.58 >100
PDK1 Hu Enz TRF CR >100 >100 >100 >100
ROCK1 Hu Enz TRF CR >100 >100 >100 >100
MAO-B Hu Enz Lumin CR >100 >100 >100 >100
MMP2 Hu Enz Fluorimetry CR >100 >100 >100 >10
PDE10A1 Hu Enz TRF CR 37.29 4.53 3.456 15.71
ALK4 Hu Enz TRF CR >100 >100 >100 >100
FGFR1 Hu Enz FRET CR >100 >100 >100 29.66
a2A Hu CHO Imped Antag CR >30 >30 >30 >30
A2A Rat PC12 cAMP TRF Ag CR >100 >100 >100 >100
A2A Ra PC12 cAMP TRF Antag CR >100 >100 28.96 >100
DOP2 Rat PC12 CDS Ag CR >30 >30 >30 >30
DOP2 Rat PC12 CDS Antag CR >30 >30 >30 >30
MR2 Hu CHO cAMP TRF Ag CR >100 >100 >100 >100
Na+/K+? ATPase Pig Photomet CR >100 >100 >100 >100
EGFR kinase Hu Enz TRF CR >100 51.18 31.01 >100
src Hu Enz TRF CR >100 >100 >100 >100
MAP3K7 Hu Enz TRF CR >100 16.97 40.31 >100
TRKA Hu Enz TRF CR >100 >100 >100 >100
eNOS Hu HUVEC Enz CR >100 >100 >100 >100
NK1 Hu U-373 MG [Ca2+] Ag CR >100 >100 >100 41.67
NK1 Hu U-373 [Ca2+] Antag CR >100 >30 >100
TXA2 sy Hu Platelet Enz EIA CR >100 25.67 89.63 >100
5-HT2C Hu HEK IP1 TRF Antag CR >100 >100 70.7 >100

S51
AT Gpig Bind FW CR 69.1 15.7 31.98 26.17
BK2 Hu Bind FW CR >100 >100 >100 >100
ECE1 Hu Enz Fluorimetry CR >100 >100 >100 >1
b2 Hu Bind FW CR >100 >100 >100 >100
KDR Hu Enz TRF CR >100 >100 >100 31.89
SET Hu Bind FW CR >100 >100 >100 >100
INSR Hu Enz TRF CR >100 >100 >100 >100
COX2 Hu Enz TRF CR >100 >100 >100 >100
RARa Hu Binding SPA CR >100 >100 >100 >100
Ghre Hu CHO [Ca2+] Ag CR >100 94.68 >100 31.12
Ghre Hu CHO [Ca2+] Antag CR >100 16.65 46.88 >10
AurKA Hu Enz TRF CR >100, >100, >100, >100,
GABAB Hu CHOImped CDS Ag CR >30 >30 >30
GABAB Hu CHO ImpedCDS Antag CR >30 >30 >30
CaV-L? Rat extract Bind CR >100, 74.76,37.38 80.3,40.15 >100,
CaV-L? Rat extract Bind CR >100, 19.63,15.24 38.74,30.07 >100,
TSPO Hu Bind FW CR >100, 48.69,46.92 >100,
PI3Ka Hu Enz FRET CR >100, >100, >100, >100,

S52
6. Crystallography
Protein expression and purification. Full length p18 isoform MTH1 (M1-156) with an N-terminal
6-His TEV protease cleavable tag was obtained from the Structural Genomics Consortium (SGC)
(PDBID: SGC:3Q93) and expressed in E. coli Gold BL21(DE3) cells. Cultures were performed in
Terrific Broth (TB) media in the presence of Kanamycin (100 µg/ml) and Tetracyclin (12.5 µg/ml).
Cells were initially grown at 37 oC and when OD600 0.6 was reached; cells were cooled to 18 oC for 20
hours and then harvested by centrigugation at 12,000 x g. Cells were re-suspended in Lysis buffer (40
mM HEPES pH8.0, 300 mM NaCl, 20 mM Imidazole, 1 mM TCEP, 1 mg/ml lysozyme, 1/10,000
dilution benzonase) supplemented with EDTA free Complete Protease inhibitors (Roche). Cells were
lysed using a cell disruptor (Constant Systems TS Series). The lysate was centrifuged at 35,000 x g
for 60 mins using a JLA-16.250 Beckman rotor. The supernatant was loaded onto 10 mls of NiNTA
resin which had been washed with 100ml of Equilibration buffer (40 mM HEPES pH8.0, 300 mM
NaCl, 20 mM Imidazole, 1 mM TCEP). The resin was then washed with with 100 ml of Equilibration
buffer and 3 x 10 ml of Wash buffer (40 mM HEPES pH 8.0, 300 mM NaCl, 40 mM Imidazole, 1
mM TCEP). MTH1 was eluted using 3 x 10ml of Elution buffer (40 mM HEPES pH 8.0, 300 mM
NaCl, 500 mM Imidazole, 1 mM TCEP). Recombinant TEV (Tobacco Etch Virus) protease was then
used to remove the N-terminal 6-His tag during an overnight dialysis step at 4 °C vs. 40 mM HEPES
pH 8.0, 300 mM NaCl, 10 mM Imidazole, 1 mM TCEP. The dialysed protein was re-passed over a
3.5 ml gravity-flow NiNTA column at 4 °C. The unbound fraction was concentrated to 5 ml using a
10 K MWCO centrifugal concentrator. This 5 ml sample was loaded onto a 120 ml S75 column which
had been equilibrated in 20 mM Tris pH 7.4, 150 mM NaCl, 5% Glycerol, 2 mM TCEP. The protein
was concentrated using a 10 K MWCO centrifugal concentrator at 4 °C to ~41 mg/ml and snap frozen
using liquid nitrogen in 50 µl aliquots.
Protein Crystallization, data collection and structure solution. For compound 25, MTH1 protein
was crystallized with the sitting drop method at 4 °C using 0.2 µL well solution containing 1.5 M
Ammonium Sulphate and 0.1M Sodium Chloride and 0.2 µL protein solution containing 3 mM of
inhibitor. Protein crystals were transferred to a cryo-protection solution of 1.5 M Ammonium
Sulphate, 0.1M Sodium Chloride and 20% Glycerol and frozen directly in liquid nitrogen.
For compound 27, MTH1 protein was crystallized with the sitting drop method at 4 °C using 0.2 µL
well solution containing 20% PEG6000, 0.2 M Lithium Chloride, 0.1 M Hepes pH 7.5 and 0.2 µL
protein solution containing 3 mM of inhibitor. Protein crystals were transferred to a cryo-protection
solution of 20% PEG6000, 0.2 M Lithium Chloride, 0.1 M Hepes pH 7.5, 20% Glycerol and frozen
directly in liquid nitrogen.
For compounds 15, 19 and 24, MTH1 protein was crystallized using the hanging drop method at 20
°C using 2 µL well solution containing 25% (w/v) PEG3350, 200 mM lithium sulphate and 100 mM

S53
sodium acetate pH 4.5 mixed with 2 µL protein solution. Protein crystals were transferred to a
solution containing 27% (w/v) PEG3350 200 mM lithium sulphate 100 mM sodium acetate pH 4.5
20% (v/v) DMSO and 2 mM compound. After 20 hours, crystals were removed from the soaking
solution and frozen directly in a stream of nitrogen at a temperature of 100 K.
Data Collection. Diffraction data for MTH1 in complexes with 25 and 27 were collected at ESRF, at
beamline ID29 equipped with a Si111 monochromator, toroidal mirrors and an ADSC Q210 detector
set to wavelength 0.98 Å. Diffraction data were for MTH1complex with 15 were collected at Soleil
beamline Proxima1 with an Si111 monochromator with the beam focused by a pair of Kirkpatrick-
Baez mirrors on a Dectris Pilatus6M using a wavelength of 0.97857 Å. Diffraction data were for
MTH1 complex with 19 were collected at the Diamond beamline I03 with an Si111 monochromator
with the beam focused by a pair of Kirkpatrick-Baez mirrors on a Dectris Pilatus 6M using a
wavelength of 0.97625 Å. Diffraction data were for MTH1complex with 24 were collected on a
Rigaku FRe+ equipped with a Saturn 944 CCD X-ray detector, using Nickel filtered CuKα
wavelength of 1.5418 Å.
Structure Solution and Refinement. For the MTH1 complex with compounds 25 and 27, data were
processed using MOSFLM and SCALA and reduced using CCP4 software3. The structure was solved
by molecular replacement using coordinates of apo-MTH1, previously solved in-house (data not
shown) as a starting model using CCP4 software. X-ray refinement restraints for 27 were generated
with CORINA4. Protein and inhibitor were modelled into the electron density using the program O5.
The model was refined using REFMAC56.
For MTH1 complexes with compounds 15, 19 and 24, data were processed using XDS7 or MOSFLM
and AIMLESS and reduced using CCP4. The structures were solved by molecular replacement using
pdb entry 5ans as a trial model using CCP4 software. X-ray refinement restraints were generated with
Grade8 and refined with MOGUL9. Protein and inhibitor were modelled into the electron density
using, COOT10. The models were initially refined using REFMAC5 and completed with BUSTER11.
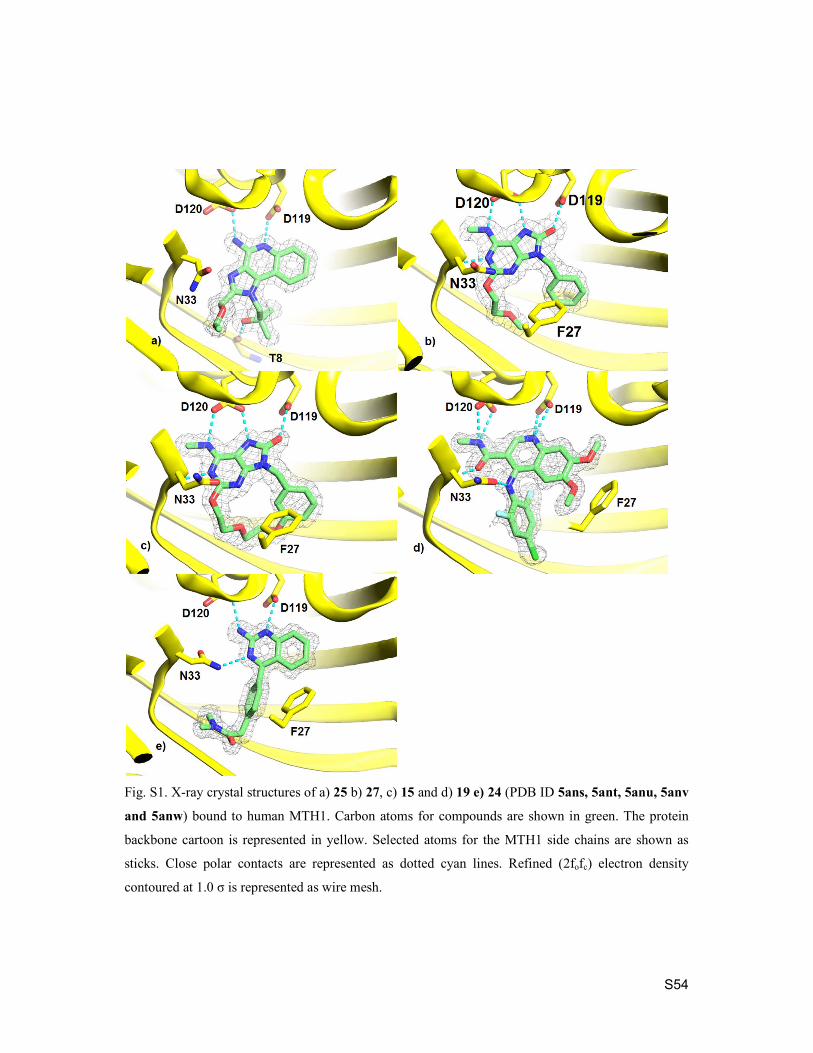
S54
Fig. S1. X-ray crystal structures of a) 25 b) 27, c) 15 and d) 19 e) 24 (PDB ID 5ans, 5ant, 5anu, 5anv
and 5anw) bound to human MTH1. Carbon atoms for compounds are shown in green. The protein
backbone cartoon is represented in yellow. Selected atoms for the MTH1 side chains are shown as
sticks. Close polar contacts are represented as dotted cyan lines. Refined (2fofc) electron density
contoured at 1.0 σ is represented as wire mesh.

S55
Data collection and protein structure refinement statistics for MTH1 complexes with compounds 25,
27, 15, 19 and 24 are shown in table S1. Structure figures were prepared with PyMOL.12
Table S1: Data collection and structure refinement statistics for MTH1 complexes. Numbers in
brackets indicate statistics for the highest resolution shells.
PDB Code 5ANS 5ANT 5ANU 5ANV 5ANW Compound 25 27 15 19 24
Space group: P41212 P21212 P21212 P21212 P21212 Cell constants: a b c (Å),
45.33, 45.33, 151.7
105.1, 120.11,53.0
60.2, 66.8, 36.1
60.4, 66.1, 36.2
61.13 66.07 35.98
Resolution range (Å)
33.7 – 1.6 29.2 – 2.0 44.7 - 1.80 44.6 - 1.16 61.1 - 1.37
Highest resolution shell (Å)
1.69 – 1.60 2.11 – 2.0 1.91-1.80 1.19 - 1.16 1.41 - 1.37
Completeness overall (%)
99.3(99.5) 99.7(99.9) 93.6(92.4) 99.9(100.0) 80.0 (17.0)
Total number of observations
149042 162581 56659 313840 126095
Reflections, unique
21613 44330 13086 50884 25048
Redundancy 6.9(7.0) 3.6(3.6) 4.1(3.4) 6.2(5.0) 5.0 (1.1) Rmerge overall
1 0.070(0.300) 0.089(0.323) 0.081(0.407) 0.041(0.577) 0.052 (0.405)
I/SigI2 17.0 (5.6) 5.6(2.2) 10.3(2.2) 19.8(2.6) 24.6 (4.8) CC1/2 n/a6 n/a6 0.996(0.856) 0.999(0.734) 0.997(0.517) Rvalue overall (%)3
20.5 21.4 20.7 20.4 19.9
Rvalue free (%)4 24.2 25.1 26.0 21.1 22.5
Non hydrogen protein atoms
1262 3759 1257 1292 1256
Non hydrogen ligand atoms
23 72 26 32 22
Solvent molecules 128 227 64 140 73 R.m.s. deviations from ideal values Bond lengths (Å) 0.006 0.009 0.012 0.010 0.008 Bond angles (º) 1.1 1.2 1.5 1.0 1.0 Average B values (Å2) Protein main chain atoms
13.8 28.9 25.4 17.0 12.4
Protein all atoms 14.5 29.5 27.7 19.9 14.5 Ligand 11.5 29.4 21.9 19.4 9.1 Solvent 28.9 34.3 40.1 35.4 24.4 Φ, Ψ angle distribution for residues 5 In most favoured regions (%)
93.9 95.7 94.6 93.7 93.1
In additional allowed regions (%)
6.1 4.1 5.4 6.3 6.9
In generously regions (%)
0.0 0.3 0.0 0.0 0.0
In disallowed regions (%)
0.0 0.0 0.0 0.0 0.0
1 Rmerge = Σhkl [( Σi |Ii - ‹I›| )/ Σi Ii]
2 I/sigI avg is the mean I/sig for the unique reflections in the output file

S56
3 Rvalue = Σhkl ||Fobs| - |Fcalc|| / Σhkl |Fobs|
4 Rfree is the cross-validation R factor computed for the test set of 5 % of unique reflections
5 Ramachandran statistics as defined by PROCHECK13
6 Data collected prior to CC1/2 value generation

S57
NMR Conformational analysis.
NMR spectra were recorded on a Bruker 500 MHz instrument equipped with a 5mm QNP probe.
NMR data was acquired in DMSO-d6, or mixtures of DMSO-d6 and D2O or D2O buffered to pH 7 (50
mM phosphate). All spectra were acquired at 27°C. For the structural assignment of 27 and 29 1D 1H, 13C and 2D HSQC and HMBC spectra were acquired using the standard pulse sequences available in
Top Spin 3.2 (Bruker GmbH).
Data for conformational analysis for 27 and 29 was gathered using 2D ROESY and selective 1D
NOESY experiments. ROESY data were acquired using the EASY ROESY sequence14 with 2048 and
256 data points in F2 and F1 respectively, a spectral width of 12 ppm, mixing times of 300 and 500
ms, and a relaxation delay of 1.5 s and 5 s. 1D NOESY data were acquired using the DPFGSENOE
sequence15 with 65536 data points, a spectral width of 12 ppm, a mixing time of 300 ms and a
relaxation delay of 2 s. Both 1D NOESY and 2D ROESY experiments employed a zero-quantum
filter element.16
Conformational analysis was carried out in DMSO-d6 due to the low aqueous solubility of the
compounds. To check that similar solution conformations were present in more physiologically
relevant systems, D2O or buffered D2O was slowly titrated into a concentrated solution in DMSO-d6.
A 1H NMR was acquired just before precipitation. The 1H NMR chemical shifts are consistent
between solvents, indicating limited dependence of conformational ensembles on the solvent used.
Further experimental details and assignments are located below, Tables NMR-1 and NMR-2.
Conformational ensembles of 27 and 29 were generated using Omega2 (version 2.5.1.5, OpenEye
Scientific Software)17 using default settings, and the resulting energy-minimized conformer sets were
used as input to the MSPIN v. 2.0.2 nOe fitter algorithm.18 This algorithm fits NMR data to ensembles
of conformers rather than a priori assuming the presence of a single, low energy conformation in
solution.
Chemical shifts (δ values) are given in parts per million (ppm) and referenced to the DMSO-d6
residual solvent signal (2.50 ppm). The coupling constants (J) are given in hertz (Hz), and peak
multiplicities are reported as s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), m
(multiplet), br (broad).
Three different methods to determine the relative nOe intensities for pairs of spins were used;
selective 1D nOe, direct integration of cross-peak volume in 2D ROESY, and integration of peak area
in F2-slices at the F1-chemical shift of each resonance. To improve accuracy, the PANIC method
(Peak Amplitude Normalisation for improved cross-relaxation) was applied,19,20 where the NOE
intensities were normalised relative to the diagonal. Correction factors were then applied to
compensate for the number of spins in each environment (corrected integral). The distance between

S58
H20 and H21 (ortho-aromatic protons) was used as reference to calibrate the other interproton
distances in the molecule. Each integration method was used to determine inter-proton distances for
27 using the equation below2 and all gave results within 4% error.
����
����=����
����
where ��� is the intensity of the NOE between I and S (S being the inverted spin) and ��� is the
internuclear distance between I and S.

S59
Table NMR-1 : Assignment and conformational analysis of 29:
1H NMR (500 MHz, DMSO-d6) 1H NMR (500 MHz, DMSO-d6+ 40% 50 mM phosphate in D2O)
Atom # δ multiplicity integral δ multiplicity integral 3 1.56 m 2H 1.52 br s 2H 2 1.63 m 2H 1.55 br s 2H 17 2.82 d, J = 4.7 Hz 3H 2.74 s 3H 1 4.26 t, J = 6.7 Hz 2H 4.09 –
4.31 br m 2H
4 4.58 t, J = 6.4 Hz 2H Not seen*
18 4.81 s 2H 4.81 s 2H 16 6.48 br s 1H Not seen 22 6.72 dd, J = 2.2, 8.1
Hz 1H 6.69 d, J = 8.4
Hz 1H
20 6.80 d, J = 7.2 Hz 1H 6.84 d, J = 7.3 Hz
1H
21 7.14 t, J = 7.8 Hz 1H 7.12 t, J = 7.8 Hz
1H
24 7.92 s 1H 7.80 s 1H 9 9.91 br s 1H Not seen
* due to close proximity to the suppressed water peak NOE correlations and inter-proton distances calculated from analysing F2-slices of 2D ROESY
Atom 1 Atom 2 Normalized integral
Corrected integral
Calculated distance
1 CH2 2 CH2 36.02 9.01 3.00 1 CH2 3 CH2 22.73 5.68 3.24 1 CH2 4 CH2 14.02 3.51 3.52 1 CH2 24 CH 76.58 38.29 2.36 2 CH2 4 CH2 31.41 7.85 3.07 2 CH2 24 CH 16.18 8.09 3.06 3 CH2 4 CH2 40.07 10.02 2.95 3 CH2 24 CH 12.62 6.31 3.19 4 CH2 24 CH 24.42 12.21 2.86 16 NH 17 CH3 7.75 2.58 3.70 18 CH2 20 CH 22.12 11.06 2.90 18 CH2 24 CH 23.46 11.73 2.88 20 CH 21 CH 31.86 31.86 2.43 21 CH 22 CH 33.71 33.71 2.41
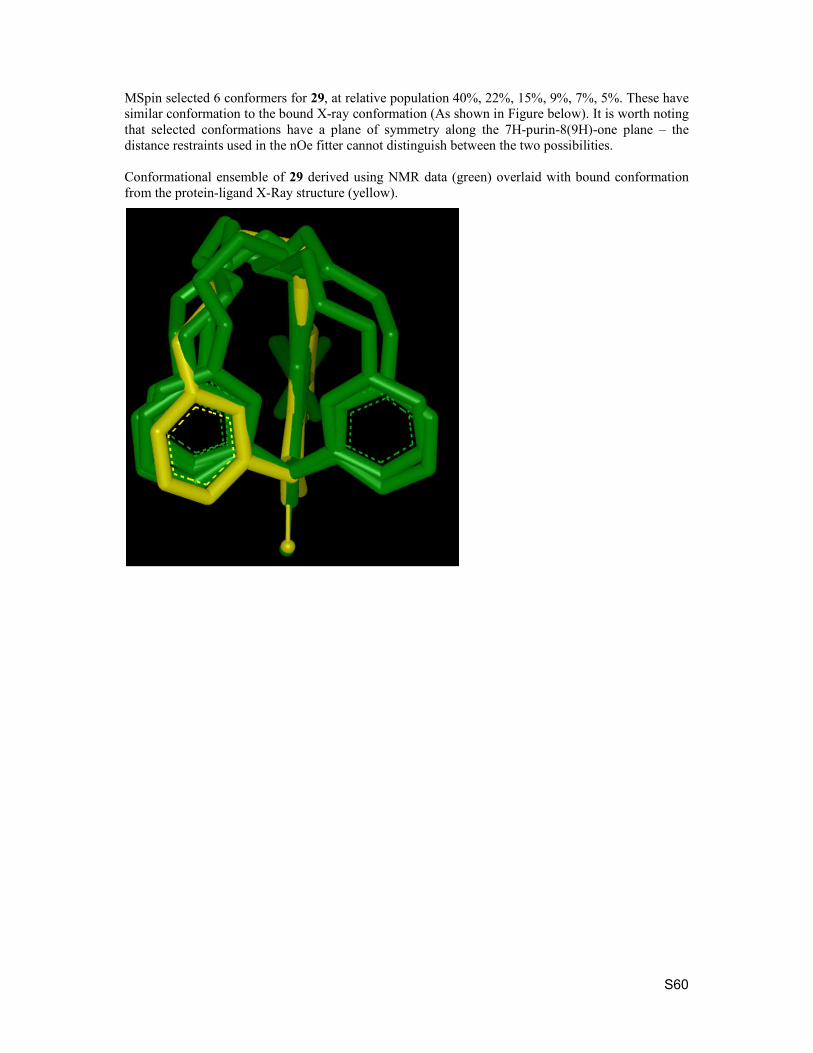
S60
MSpin selected 6 conformers for 29, at relative population 40%, 22%, 15%, 9%, 7%, 5%. These have similar conformation to the bound X-ray conformation (As shown in Figure below). It is worth noting that selected conformations have a plane of symmetry along the 7H-purin-8(9H)-one plane – the distance restraints used in the nOe fitter cannot distinguish between the two possibilities. Conformational ensemble of 29 derived using NMR data (green) overlaid with bound conformation from the protein-ligand X-Ray structure (yellow).
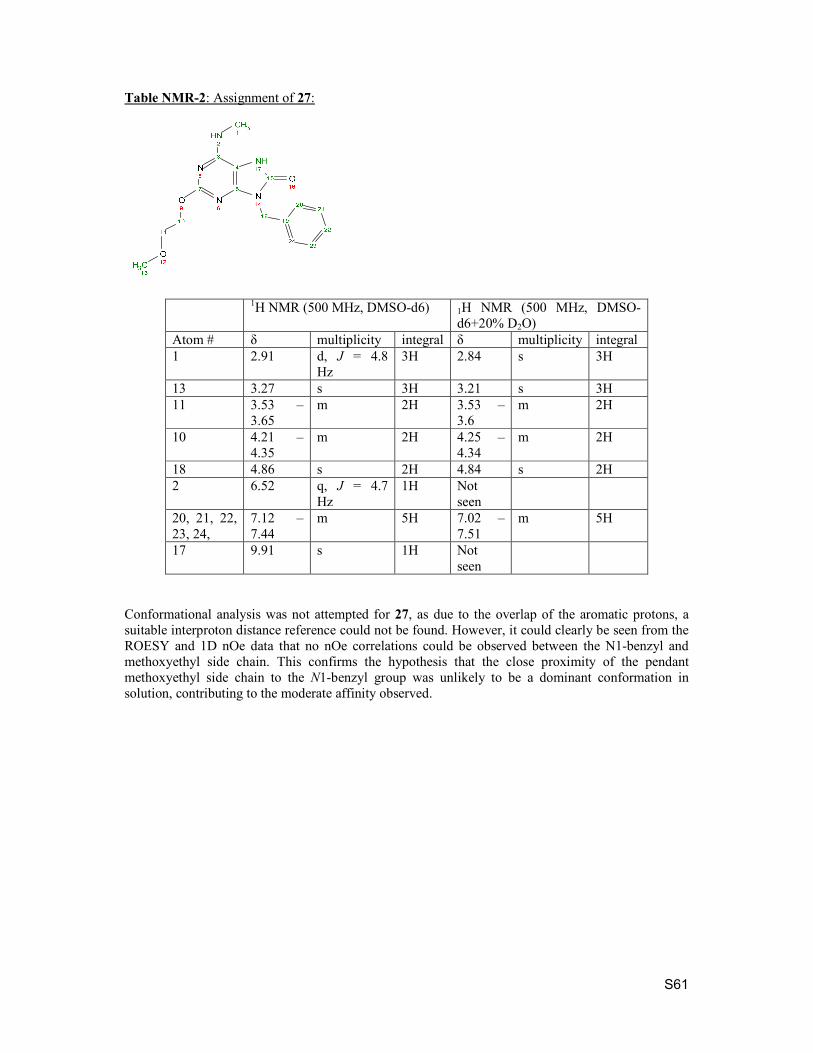
S61
Table NMR-2: Assignment of 27:
1H NMR (500 MHz, DMSO-d6) 1H NMR (500 MHz, DMSO-d6+20% D2O)
Atom # δ multiplicity integral δ multiplicity integral 1 2.91 d, J = 4.8
Hz 3H 2.84 s 3H
13 3.27 s 3H 3.21 s 3H 11 3.53 –
3.65 m 2H 3.53 –
3.6 m 2H
10 4.21 – 4.35
m 2H 4.25 – 4.34
m 2H
18 4.86 s 2H 4.84 s 2H 2 6.52 q, J = 4.7
Hz 1H Not
seen
20, 21, 22, 23, 24,
7.12 – 7.44
m 5H 7.02 – 7.51
m 5H
17 9.91 s 1H Not seen
Conformational analysis was not attempted for 27, as due to the overlap of the aromatic protons, a suitable interproton distance reference could not be found. However, it could clearly be seen from the ROESY and 1D nOe data that no nOe correlations could be observed between the N1-benzyl and methoxyethyl side chain. This confirms the hypothesis that the close proximity of the pendant methoxyethyl side chain to the N1-benzyl group was unlikely to be a dominant conformation in solution, contributing to the moderate affinity observed.

S62
NMR Spectra for 29:

S63
S64

S65
NMR Spectra for 27
S66

S67
S68

S69
1 GraphPad Software, San Diego California USA, www.graphpad.com
2 Kimple, A. J., Muller, R. E.; Siderovski, D. P.; Willard, F. S. A capture coupling method for the
covalent immobilization of hexahistidine tagged proteins for surface plasmon resonance. Methods
Mol Biol 2010, 627, 91-100. 3 CCP4 Acta Cryst. D 1994, D50, 760–763. 4 CORINA: Gasteiger, J.; Rudolph, C.; Sadowski, J. Automatic Generation of 3D Atomic Coordinates
for Organic Molecules. Tetrahedron Comp. Method. 1990, 3, 537-547. 5 Jones T.A., Zou, J.Y. ; Cowan S.W. Improved methods for building protein models in electron
density maps and the location of errors in these models. Acta Cryst. A 1991, 47, 110-119. 6 Refmac version 5.8.73: Murshudov, G. N.; Vagin, A. A.; Dodson, E. J. Acta Cryst. D 1997, 53, 240. 7 XDS : Kabsch, W. Acta Cryst. D Biol Cryst. 2010, 66, 125-132. 8 O. S. Smart, T. O. Womack, A. Sharff, C. Flensburg, P. Keller, W. Paciorek, C. Vonrhein, and G.
Bricogne.(2011) grade, version 1.2.9. Cambridge, United Kingdom, Global Phasing Ltd.,
http://www.globalphasing.com. 9 Mogul: Bruno, I.J. et al. J. Chem. Inf. Comp. Sci. 2004, 44, 2133-2144. 10 Coot: Emsley, P.; Cowtan, K. Acta Cryst.., Sect. D 2004, 60, 2126. 11 Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P, Sharff A., Smart
O.S., Vonrhein C., Womack T.O. (2011). BUSTER version 2.11.6. Cambridge, United Kingdom:
Global Phasing Ltd. 12 PYMOL: Molecular Graphics System, Version 1.5.0.1 Schrödinger, LLC. 13 Procheck: Laskowski, R.A., Rullmannn, J.A., MacArthur, M.W., Kaptein, R.; Thornton, J.M. J.
Biomolecular NMR 1996, 8, 477-486. 14 Thiele, C. M.; Petzold, K.; Schleucher, J. EASY ROESY: Reliable Cross-Peak Integration in
Adiabatic Symmetrized ROESY. Chem. Eur. J. 2009, 15, 585–588; Stott, K.; Keeler, J.; Van, Q. N.;
Shaka, A. J. One-dimensional NOE experiments using pulsed field gradients. J. Magn. Reson. 1997,
125, 302–324. 15 Butts, C. P.; Jones, C. R.; Towers, E. C.; Flynn, J. L.; Appleby, L.; Barron, N. J. Interproton
distance determinations by NOE – surprising accuracy and precision in a rigid organic molecule. Org.
Biomol. Chem. 2011, 9, 177-184. 16 Thrippleton, M. J.; Keeler, J. Elimination of Zero-Quantum Interference in Two-Dimensional
NMR spectra. Angew. Chem., Int. Ed. 2003, 42, 3938–3941. 17 Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer
Generation with OMEGA: Algorithm and validation using high quality structures from the Protein
Databank and the Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572-584. 18 A. Navarro-Vázquez, MSpin 1.3, MestReLab Research S. L.

S70
19 Macura, S.; Farmer, B.T.; Brown, II, L.R. An improved method for the determination of cross-
relaxation rates from NOE data. J. Magn. Reson. 1986, 70, 493–499. 20 Krishnamurthy, K. Revisiting the initial rate approximation in kinetic NOE measurements. J. Mag.
Reson. 2006, 182, 173–177.