audits af qc lab
TRANSCRIPT

SHARE & LEARN
OM
AUDIT AF QC
LABORATORIE
19. NOVEMBER 2014
VED NUSRET ASANOVSKI

AGENDA Krav til Audit
Aftale Audit
Agenda
Indledende møde
Kvalitetssystem
Personale
Lokaler og Udstyr
Analyse
Kontraktarbejde
Reklamationer & Tilbagekaldelser
Selvinspektion

EudraLex, The Rules Governing Medicinal Products in the European
Union, Volume 4, EU Guidelines to Good Manufacturing Practice,
Medicinal Products for Human and Veterinary Use (hereinafter EU
GMP).
Pharmaceutical Quality Control Labs (7/93) GUIDE TO INSPECTIONS OF PHARMACEUTICAL QUALITY CONTROL
LABORATORIES
Microbiological Pharmaceutical Quality Control Labs (7/93) GUIDE TO INSPECTIONS OF MICORBIOLOGICAL PHARMACEUTICAL
QUALITY CONTROL LABORATORIES
Referencer

QC laboratorium bør GMP evalueres hvert 2. år
Disse inspektioner kan omfatte:
- Den specifikke metode, der skal anvendes til at teste et nyt
produkt
- En fuldstændig vurdering af laboratoriets overensstemmelse
med GMP
- En specifikt del af laboratorie operationer
Krav til Audit af QC laboratorier

Afvigelser
Ansvarsforhold
Arkivering
Audits
Autorisationer
Bemyndigelser
Certifikater
Change Control
Dagbøger
Dokumentation og
dokumentationspraksis
Dokumentstyring
Evaluering af leverandører
Faglige kompetencer
Forskrifter
Forsyninger
Frigivelsesprocedurer
Godkendelsesprocedurer
Hygiejne
Kalibrering
Kontrakter
Rengøring
Risikostyring
Råvarer
Selvinspektioner
SOP’er
Specifikationer
Sporbarhed
Stillingsbeskrivelser
Tilbagekaldelser
Træning og uddannelse
Valideringsmasterplan
Vedligeholdelse af
udstyr
Områder som kan inspiceres
Kvalificering og validering
Kvalitetskontrol
Kvalitetsstyringssystem
Laboratorier, lokaler og udstyr
Laboratoriejournaler
Lagre
Line clearance
Logbøger
Orden
OOS håndtering
Organisation
Pladsforhold
Proceskontrol
Product Quality Review
Produktions batchjournaler
Produktionsfaciliteter; lokaler og udstyr
Prøveudtagning og håndtering
Reagenser og referencesubstanser
Reklamationsbehandling

Auditor skal aftale audittidspunkt med virksomheden
Sørg for, at der foreligger skriftlig bekræftelse af dato, starttidspunkt og sluttids-
punkt.
Auditor skal sikre sig præcise oplysninger om adressen for auditerede
virksomhed
Undersøg hvilke relevante GMP-mæssige aktiviteter der foregår på denne
adresse. (Specielt produktion, pakning, lager, kvalitetskontrol og kvalitetssikring, større virksomheder, har ofte
aktiviteter på flere sites)
Auditor skal sikre sig, at kvalitetschefen for auditerede virksomhed deltager i hele
audit (Andet relevant personale skal som minimum kunne tilkaldes)
For audit af udenlandske virksomheder, hvor audit ikke kan gennemføres på
engelsk, må man arrangere tolkebistand
Planlægning og Gennemførelse af Audit

Undersøg, om virksomheden tidligere har været auditeret og hvornår.
Læs tidligere auditrapporter og undersøg:
Udestående afvigelser, observationer eller afgivet løfter.
Følge op på disse under audit.
Til virksomheder, man kun har begrænset forhåndskendskab til, kan det være
hensigtsmæssigt at fremsende et audit questionnaire, så visse spørgsmål er
afklaret på forhånd
Udbed om
- SMF
- SOP index
Faglige forberedelser af audits

Auditor skal udarbejde agenda og fremsende denne til virksomheden, der skal
auditeres, mindst 1 uge inden audit skal finde sted.
Agendaen kan med fordel være kortfattet og skal betragtes som et tidsskema,
der angiver overordnede emner.
Agendaen skal udformes sådan, at den tillader auditor passende fleksibilitet.
Audit agenda

Følgende skal anføres i agendaen:
Navn og adresse på den virksomhed, der auditeres.
Navn på den virksomhed, der udfører audit.
Dato for audit.
Tidspunkt for indledende møde
Formiddagens program i hovedoverskrifter, med tidspunkter for de enkelte
emner
Tidspunkt for frokost (Kan med fordel begrænses til ½ time på sitet)
Eftermiddagens program i hovedoverskrifter, med tidspunkter for de enkelte
emner.
Tidspunkt for afsluttende møde.
Audit agenda

Agenda for audit of
on: …
Auditors:
DAY 1: …
9.00 Introduction of Participants and Company
9.15 Quality system
12.30 Lunch
13:00 Quality system
14.00 Personnel
16.45 Summary of the day
17.00 Audit End
DAY 2: …
9.00 Follow up from the day before
9.30 Plant tour (Premises, Equipment & Analysis in Chemical, Microbiological lab)
12.30 Lunch
13.00 Contract Manufacturing
14.30 Complaints and Recalls // Stability testing
15.00 Internal Audit
15.30 Follow up
16:45 End Meeting
17:00 Audit End
NB: Changes to the agenda may occur during the audit
Best Regards
Nusret Asanovski
CEO, GMP Specialist, M.Sc.Pharm.
Fruebjergvej 3
DK-2100 Copenhagen, Denmark – Tel.: +45 3091 6335 – E-mail: [email protected] – Web: cgxp.eu

En audit starter med et indledende møde, hvor der gives en introduktion af
deltagerne.
auditor redegør for grunden og forventninger for auditten.
Auditor spørger om der er accept af den fremsendte agenda.
auditor kan nævne den dokumentation, der ønskes præsenteret.
fx specifikke batchjournaler, SOP’er, stillingsbeskrivelser, træningsdokumentation,
Periodiske Quality Review, auditplaner etc.
Indledende møde og audit

Opgaven er at indsamle objektiv vidnesbyrd ud fra observationer baseret på
checklisten eller ud fra andre iagttagelser.
Auditor skal kontrollere, om praktiske forhold og håndteringer svarer til beskrivelser
i SOP’er, således at det kontrolleres om kvalitetsstyringssystemet svarer til
virkeligheden.
Ud fra agendaen har auditor udarbejdet en checkliste til eget brug under audit.
Checklisten indeholder spørgsmål, som er relevante for den pågældende audit.
Audit og Checkliste
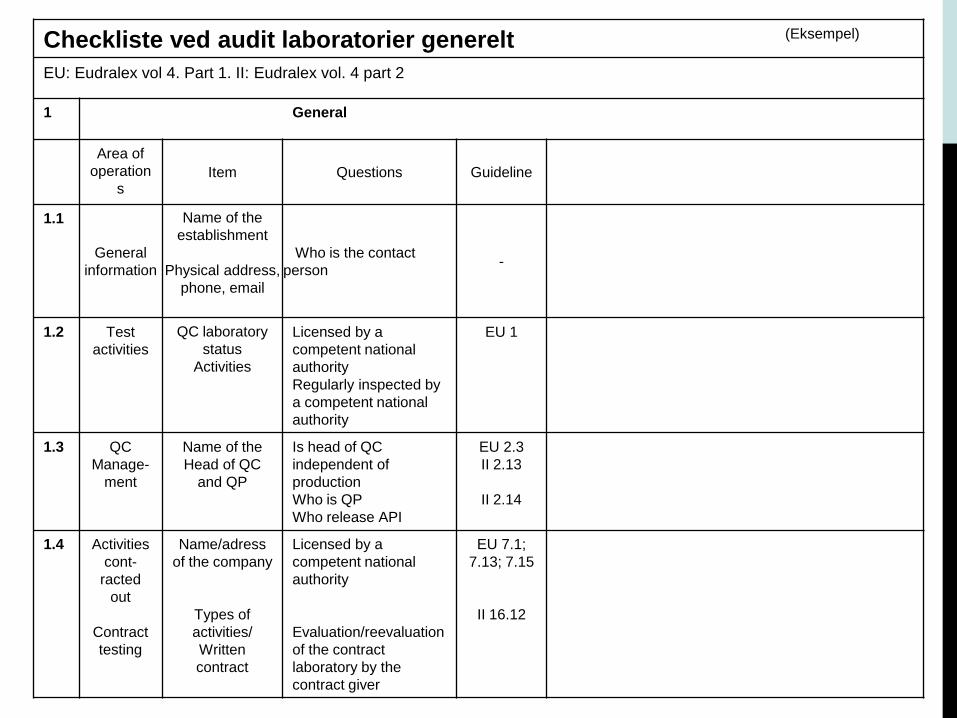
Checkliste ved audit laboratorier generelt (Eksempel)
EU: Eudralex vol 4. Part 1. II: Eudralex vol. 4 part 2
1 General
Area of
operation
s Item Questions Guideline
1.1
General
information
Name of the
establishment
Physical address,
phone, email
Who is the contact
person -
1.2 Test
activities
QC laboratory
status
Activities
Licensed by a
competent national
authority
Regularly inspected by
a competent national
authority
EU 1
1.3 QC
Manage-
ment
Name of the
Head of QC
and QP
Is head of QC
independent of
production
Who is QP
Who release API
EU 2.3
II 2.13
II 2.14
1.4 Activities
cont-
racted
out
Contract
testing
Name/adress
of the company
Types of
activities/
Written
contract
Licensed by a
competent national
authority
Evaluation/reevaluation
of the contract
laboratory by the
contract giver
EU 7.1;
7.13; 7.15
II 16.12

Som afrunding af audit gennemføres et afsluttende møde.
Auditor opsummere de konkret fundne observationer, kritisk, væsentlig, eller
anbefaling.
Auditor giver sit generelle indtryk og en overordnet konklusion af den
gennemførte audit.
Auditor informerer om, hvornår auditrapporten forventes at blive fremsendt.
Virksomheden skal besvare auditrapporten skriftligt indenfor en tidsfrist, der vil
fremgå af rapporten.
Afsluttende auditmøde

Rapporten skal indeholde, men er ikke begrænset til følgende afsnit:
Firmanavn og adresse på auditerede virksomhed.
Dato for audit.
Navne og stillingsbetegnelser for deltagerne fra virksomheden.
Auditor anfører sit navn og stillingsbetegnelse.
Alfabetisk liste over anvendte forkortelser i rapporten.
I indledningen anføres, hvilke ydelser, den auditerede virksomhed leverer eller
påtænkes at skulle levere.
Auditrapport

Der gives en kortfattet beskrivelse af auditerede virksomhed, dens geografiske
placering og dens størrelse.
Virksomhedens aktiviteter beskrives kortfattet.
Beskrivelse af gældende fremstillertilladelse og andre relevante autorisationer.
Det anføres med præcise referencer, hvilke bekendtgørelser og GMP regler, audit er
udført i henhold til.
Observerede afvigelser nævnes i rapporten i den sammenhæng, som de er
konstaterede.
Det bør anføres, hvilken vægt der tillægger de enkelte observationer; kritisk,
væsentlig, eller anbefaling.
Alle afvigelser skal have reference til relevant bekendtgørelse eller GMP Guide.
Auditrapport

Auditrapportens konklusion skal være i overensstemmelse med den mundtlige
konklusion.
Det anføres, om den auditerede virksomhed vurderes som egnet til at levere de
ønskede ydelser.
Den auditerede virksomhed skal give en skriftlig tilbagemelding på observationer og
anbefalinger, og at korrigerende handlinger skal være ledsaget af en tidsplan for
deres gennemførelse.
Den auditerede virksomhed gives 30 dages frist for besvarelse af auditrapporter.
Hvis der er fundet kritiske afvigelser kan hurtigere tilbagemelding være påkrævet.
Udleverede kopier kan vedlægges som bilag til auditrapporten.
Rapporten skal fremsendes til auditerede virksomhed senest 3 uger efter datoen for
audits afholdelse.
Auditrapport
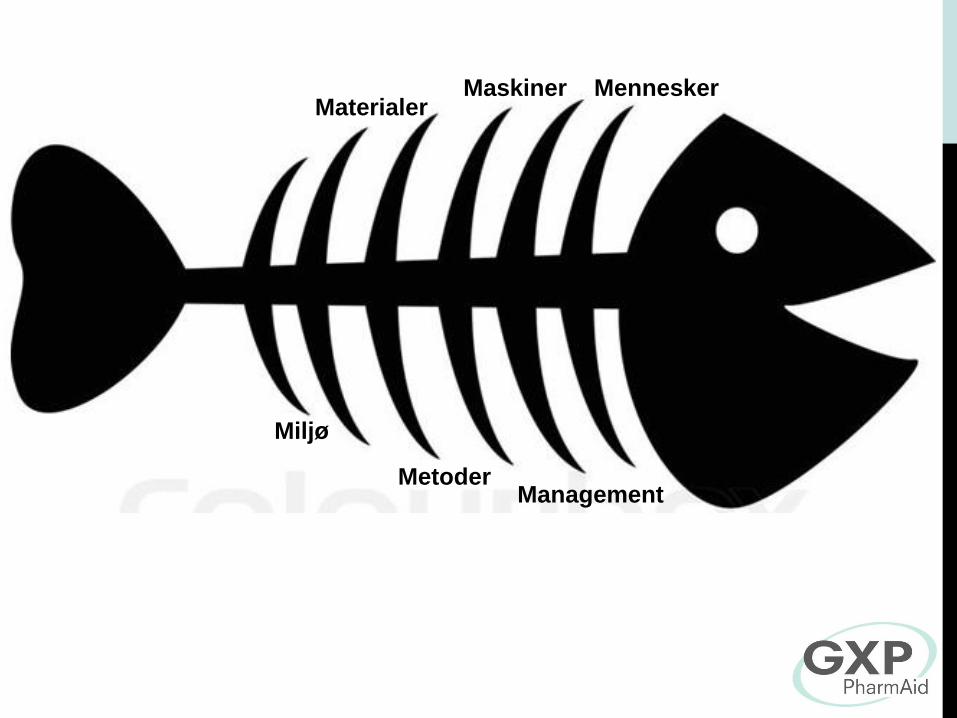
Mennesker Maskiner Materialer
Miljø
Metoder Management

Omfangets af laboratoriets ansvar
Laboratoriet har et dokumenteret kvalitetssystem,
Laboratoriet råder over det fornødne prøvetekniske udstyr og over
lokaler af en tilstrækkelig god standard
Prøvning skal udføres efter egnede og validerede metoder
Quality Management

Overordnet ledelse af laboratoriet
Styring af personalts roller og ansvarsområder
Resurseplanlægning (Er der tilstrækkelig kompetente medarbejdere)
Laborant certificering
Kvalitetssikring og risikovurdering
Senior Management Involvement

Uddannelse (kemiker, laborant, laboratorie tekniker, ingen uddannelse)
- Procenten for forskellige personalegrupper.
Laborant certificering
Efteruddannelser (apparater, analyser)
Kurser (GMP, statistik, ledelse)
Faglig kompetence og praktisk erfaring
Forefindes en ajourført uddannelseslog/træningsjournal for hver
medarbejderne
Ansvarsfordeling defineret
Er der en klar nedskrevet ansvarsfordeling
Er der en stillingsbeskrivelse
Personale

Træning i analyser
Gennemført tre gange med godkendt resultat
Er personerne trænet i den gældende analyseinstruktion
Er personerne trænet i de gældende almene laboratorie SOP’er
Gennemført tre gange med godkendt resultat og/eller trænet i den
gældende laboratorie SOP’er
Personale

Apparaters kvalificeringsstatus
URS: User Requirement Specification
DQ (protokol og rapport)
IQ (protokol og rapport)
OQ (protokol og rapport)
PQ (protokol og rapport)
Udstyr

Apparaters brugsstatus
Service status (Logbøger)
System Suitability Log
Er apparaterne i brug/ikke i brug?
(Apparatlog og QA liste over apparater)
Er analytisk udstyr ID opmærkede, serviceret og kontrolleret? (SOP)
Er apparaterne blevet serviceret (Vedligeholdt/kalibreret) efter reglerne i
SOP
Er serviceteknikeren uddannet? (Uddannelsesjournal)
Sporbar kalibrering af udstyr
Udstyr

Apparaters brugsstatus
Hjælpeapparatur
Er pipetterne kontrolleret, justeret, kalibreret og korrekt opmærkede
med dato og ID?
Er der benyttet de korrekte glasvare/utensilier til fortyndings rækker?
Er logbøger ført ajour?
Udstyr

Laboratoriets tilstand
Er der ryddeligt?
(Ledninger, slanger, notesbøger, instruktioner, reagensglas, støv)
Er der plads til at bevæge sig rundt?
(frysebokse eller papkasser på gulvet)
Er der tilstrækkeligt med lys?
(Kan en normal tekst læses ved laboratoriebordet)
Er ventilationen i orden? (Er den kontrolleret og serviceret?)
Er installationerne i orden? (Vand, laboratorievand, elektricitet, varme)
Er LAF bænke og stinkskabe ryddelige og rene? (Logbøger)
Lokaler

Reagenser
Er de af den rigtige kvalitet? (analyse, teknisk, syntese)
Er hjælpestofferne friske og opmærkede?
Signeret samt dato for anbrud og udløb?
Materialer

Metodeudvikling
Metodeoverførelses procedure, hvordan er det foregået?
Metodeoverførelses rapport?
Analyse Validering
Valideret efter USP, EP, JP, ICH guideline?
Metoder

Analyse Instruktion
Er kalibrering udført med de til opgaven korrekte kalibreringsvæsker?
(For eksempel: pH, Osmometer og Viskositet, passer kalibreringsvæskens
område til måleområdet?)
Er der benyttet de korrekte kemikalier? Teknisk, analyse eller anden kvalitet
Prøvetagning
Er prøverne repræsentativt udtaget?
Hvordan håndteres prøverne?
Er der benyttet de korrekte prøvetagnings remedier?
Er instruktionen fulgt?
Metoder

Gennemgang af analyseresultater
Analyseresultat OK (Hvordan afgives et sådanne resultat?)
Analyseresultat OOS (Hvordan bliver et sådan resultat behandlet?)
Metoder

Normale observationer under audit
Til trods for forskellige typer af analyser, prøver og produkter dukker samme
problemer op:
- Forberedelse af prøver
- At følge en standardforskrift
- Dokumentation,
- HPLC analyser
Osv. Osv.

Normale afvigelser ved audit
GC, HPLC, HPLC-MS, UV, IR:
Kalibreringsparametre ikke helt i overensstemmelse med anerkendte
standarder
HPLC, GC:
Kromatografiske integration parametre er uklart formuleret
IR:
Ingen klare godkendelseskriterier for passerer ID tests
OOS:
Undersøgelser ikke er tilstrækkeligt gennemført
Afvigelser:
Ikke behørigt dokumenterede
Farmakopé metoder:
Ikke efterprøvet

Væsentlige problemområder og kritiske mangler:
Rapportering af data for prøver der ikke er analyseret
Forfalskning af eksisterende data
Selektiv brug af QC data
Overskriver filer
Top formindskelse, Top forstørrelse (Uvidenskabelig Integrationsmetode)
Ikke rapporteret spikning af prøver for at opnå en ”kunstigt” stigning i styrke
Tidsrejse (skiftende tider og datoer)

Eksempler på hvad auditor kigge på for tegn på data manipulation
Data, der er "for gode til at være sandt".
Forskelle mellem elektroniske data og papir rådata.
Datoer, som ikke passer sammen (datoer for kromatogrammer matcher ikke dem, der hævdes at være benyttet til analysen)
Integration parametre, som ikke kan spores eller genfindes. (Laboratoriet bør være i stand til at påvise, at samme resultater kan opnås, når integration
parametre genindtastes)

Eksempler på hvad auditor kigge på for tegn på data manipulation
Elektronisk data, der ikke kan hentes eller er ødelagt.
Integration parametre er forskellige for standarder og prøver i det samme
analyseopsæt, eller mellem prøveinjektioner i samme analyseopsæt.
Log dataopsamling ikke slået til
Injektion sekvenser, der ikke er klart dokumenterede, eksempelvis ved at
gentage analyser og plukke af de bedste resultater
Elektroniske rådata, der ikke er entydig identificerbare og fraværet af klare
retningslinjer for opbevaring og arkivering af analysedata.

Bedste praksis: kromatografi
Dokumentation!
Brug automatisk integration hvis det er muligt eller mest nøjagtig manuel
integration.
Opstil analysesekvenser, der er skitseret i analyseproceduren
Benyt navngivning af filer der tillader hurtig genfinding og sporbarhed af
elektroniske data.
Slå dataopsamling for ændringslog til
Beskriv klare entydige Integration parametre.
Alle vejesedler er underskrevet og dateret og præparatfremstillingen er
dokumenteret og bevaret.

Bedste praksis: IR
Skriv klare og detaljerede standardprocedurer, der angiver:
En detaljeret beskrivende metode for sammenligning af prøvens spekter med
referencen
(For eksempel en programalgoritme i kombination med en visuel undersøgelse og en
peaktabel for sammenligning med reference standardens spektre).
Klare godkendelseskriterier for de forventede bølgelængder.
Klare godkendelseskriterier for mindstekravet til %-transmission i
overensstemmelse med Pharmakopé krav.
Operatør viden og uddannelse.
Deaktivering af muligheden for baseline eller peak-bølgelængde modifikationer.
Anvendelse af ændringslog data / 21CFR kompatible programmer.

Bedste praksis: Andre vigtige faktorer
Papir data versus elektroniske data:
Den bedste praksis er at print sekvens, alle spektre og alle integration parametre.
Hvis ikke dette gøres, bør alle elektroniske data være let tilgængelige,
identificerbare.
De elektroniske data skal kunne konsulteres for at bekræfte, at integration har
været udført efter analyse SOP.
Komplet data revision skal udføres før batchfrigivelse (Standardforskrifter skal klart definere, hvordan korrekturlæser / QA skal gennemgå data
fra analyser).