i sn 186 -29 x journal -...
TRANSCRIPT

JOURNALTUMORZENTRUM ERFURT
INHALT Seite 4n Klinische Krebsregister – Zur Halb-
zeit gibt es noch einiges zu tun
Seite 5n Klinik, Genetik und Management von Patienten mit Neurofibroma- tose Typ 2
Seite 20n Primäre Chemohormontherapie des hormonnaiven metastasierten Prostatakarzinoms
Seite 22n 15 Jahre Radiochirurgie in Erfurt
Seite 26n Therapiesysteme für die Radiochirurgie
Seite 32n 28. Onkologische Konferenz des Tumorzentrum Erfurt e.V. fand am 6. und 7. November 2015 in Erfurt statt
Seite 33n Neoadjuvante Therapiekonzepte beim Pankreaskarzinom als neuer Meilenstein – Darstellung des Status quo anhand einer Falldemonstration
Seite 40n Was ist tumor-associated tissue eosinophilia (TATE)
Seite 42n Interdisziplinäre Therapie eines lokal fortgeschrittenen Basal- zellkarzinoms
Seite 45n Ewing-Sarkom der Mandibula – Fallbericht einer seltenen Tumorentität
Seite 46n Bericht von der Mitglieder- versammlung des Tumorzentrum Erfurt e.V. am 15.04.2015
Seite 49n Veranstaltungsverzeichnis
Seite 50n Angebote des Tumorzentrum Erfurt e.V.
01/2015
ISSN 1868-291X
Das klinische Programm Autologe Stammzelltransplantation am HELIOSKlinikum Erfurt ist gestartet. Am 7. Juli dieses Jahres wurde die erste au-tologe Stammzelltransplantation (SZT) bei einem Patienten mit einemMultiplen Myelom durchgeführt. Nach umfangreichen Umbauten konnte eine Stammzelltransplantations-einheit mit hocheffizienter Partikelfiltration der Raumluft und Schleusen-systemen in der 4. Medizinischen Klinik (Hämatologie, internistische Onkologie und Hämostaseologie; Chefarzt: Priv.-Doz. Dr. med. Herbert G. Sayer) eingeweiht werden. Prüfungen und Vorgaben der Kostenträgerund der Landesbehörden mussten beachtet und eingehalten werden. Die Hochdosischemotherapie mit anschließender autologer Blutstamm-zelltransplantation ist heutzutage fester Bestandteil in den Leitlinien beider Behandlung des MultiplenMyeloms und bei fortgeschritte-nen malignen Lymphomen. DerAblauf einer solchen Behandlungbesteht meist zunächst aus einereinleitenden Chemotherapie mitanschließender Wachstumsfaktor-gabe zur Mobilisierung derStammzellen aus dem Knochen-mark ins periphere Blut. Die Ab-sammlung der Stammzellen wirdbeim Erfurter SZT-Programm vomHaema-Blutspendedienst durch-geführt. Die Lagerung und dienach Arzneimittelgesetz notwen-dige Freigabe der Stammzellenübernimmt die Firma Seracell inRostock. Besondere Hygiene-Maßnahmensind bei der Hochdosischemothe-rapie angezeigt, da in der Zeit biszum Anwachsen der Stammzelleneine mehrtägige Aplasiephase mitstarker Einschränkung der Körper-abwehr resultiert. Nach dem Um-bau 2014/2015 verfügt die Stationüber 6 Betten mit jeweils über eineVorschleuse zu betretenden 4 Zim-mern. Damit ist die 4. MedizinischeKlinik nun in der Lage, den Patien-ten in Erfurt auch diese Therapie-option anzubieten. Bis zum No-vember 2015 sind bereits 11 Pa-tienten transplantiert worden.
Stammzelltransplantationseinheit eröffnetneue Möglichkeiten der Tumortherapiein Erfurt
Transplantationsbereich
Einzelzimmer mit Luftfilterung
Schleuse

SUTENT®: So individuell wie Ihr Patient.
POTENZIALE
MAXIMAL AUSSCHÖPFEN –
MIT AKTIVEM
THERAPIEMANAGEMENT1,2
First-line-Therapie beim fortgeschrittenen, metastasierten Nierenzellkarzinom#ersiatsatem
Ther--Thereinl-tsirF
akllezeneri Nentergtro fmie beipaTher
#monizr
,enentitrhcseg
an 107 J0. 2del J Mgn. N Elt aJ er Rezto. M1510i 2nud Jantn Soitmaofnihca F®ttenuS#
.42–511:)2(653;1an 1
0. 2vet Raerr Tecan. Clt ao D eanlletsa. C2
Pfizer Pharma GmbH, Linkstraße 10, 10785 Berlin
. (Dysgeusie, Ageusie); Hypertonie; Dyspnoe, Nasenbluten, Husten; Stomatitis/aphthöse Stomatitis, Abdominalschm. (Bauchschm., Sstörintravasale Koagulopathie, periton. Blut., Nebenniereninsuff., Pneumothorax, Schock u. plötzl. T
ährend Wwird. fortgesetzt Behandl. d. wenn schlossen verminderten Appetit, Beeinträchtig. d. Geschmackssinns, Hypertonie, Erschöpf., gastrointest. Störtödl., sind Nierenversagen, Herzinsuff., Lungenembolie, gastrointest. Perforat. u. Hämorrhagie (z.
umoren (pNET) m. Krankheitsprogression. D. Erfahrung m. Sutent als First-line-Behandlung ist begrenzt. . Tdifferenz. pankreat. neuroendokru./od. metast. maligner gastrointest. Stromatumoren (GIST), wenn e. Behandl. m. Imatinib wg. Resistenz od. Unverträglichk. fehl
.), Gelatine, Eisen(III)-oxid (EEurMagnesiumstearat (Ph.Hartkapsel.mg mg/50mg/2512,5®Sutent
.04–032:)3(93;ya3 M1
Pfizer Pharma GmbH, Linkstraße 10, 10785 Berlin
. (Dysgeusie, Ageusie); Hypertonie; Dyspnoe, Nasenbluten, Husten; Stomatitis/aphthöse Stomatitis, Abdominalschm. (Bauchschm., SSehr häufig:od. intravasale Koagulopathie, periton. Blut., Nebenniereninsuff., Pneumothorax, Schock u. plötzl. Tintravasale Koagulopathie, periton. Blut., Nebenniereninsuff., Pneumothorax, Schock u. plötzl. Tod.
Hämatol. entwickeln. Hypothyreose e. sich kann Behandl. d. ährend schlossen verminderten Appetit, Beeinträchtig. d. Geschmackssinns, Hypertonie, Erschöpf., gastrointest. Stör
B. Atemwegs-, Gastrointestinaltrakt-, Ttödl., sind Nierenversagen, Herzinsuff., Lungenembolie, gastrointest. Perforat. u. Hämorrhagie (z.umoren (pNET) m. Krankheitsprogression. D. Erfahrung m. Sutent als First-line-Behandlung ist begrenzt.
u./od. metast. maligner gastrointest. Stromatumoren (GIST), wenn e. Behandl. m. Imatinib wg. Resistenz od. Unverträglichk. fehl171), Schellack, Propylenglycol, Natriumhydroxid; 25itandioxid (E172), T.), Gelatine, Eisen(III)-oxid (E
Hartkps. enthält Sunitinibmalat, entspr 1Wirkstoff:Zusammensetzung: Wirkstoff: Sunitinib. Hartkapsel.
- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstör Neutropenie, Thrombozytopenie, Anämie, Leukopenie; Hypothyreose; verminderter Appetit/Appetitlosigk.; Schlaflosigk.; Schwindel
chm. im Unter. (Dysgeusie, Ageusie); Hypertonie; Dyspnoe, Nasenbluten, Husten; Stomatitis/aphthöse Stomatitis, Abdominalschm. (Bauchschm., S Neutropenie, Thrombozytopenie, Anämie, Leukopenie; Hypothyreose; verminderter Appetit/Appetitlosigk.; Schlaflosigk.; SchwindelSehr häufig:
gehören Anämie) u. Thrombozytopenie Neutropenie, B. (z.. StörB. Durchfall, Übelk., Stomatitis, Dyspepsie u. Erbrechen), V
B. Atemwegs-, Gastrointestinaltrakt-, T -, Harnwegs- od. Gehirnblutungen). D. häufigsten Nebenwirk. jeden Grades (b. Pat. i. mRCC-, GIST. (z.schlossen verminderten Appetit, Beeinträchtig. d. Geschmackssinns, Hypertonie, Erschöpf., gastrointest. Stör
-, Harnwegs- od. Gehirnblutungen). D. häufigsten Nebenwirk. jeden Grades (b. Pat. i. mRCC-, GISTB. Durchfall, Übelk., Stomatitis, Dyspepsie u. Erbrechen), V
umorB. Atemwegs-, Gastrointestinaltrakt-, TB. Durchfall, Übelk., Stomatitis, Dyspepsie u. Erbrechen), V
Überempfindlichk. gg. d. Wirkstoff od. sonst. Bestandteile. . zur Behandl. fortgeschritt./metast. Nierenzellkarzinome (mRCC). B. Erw
Gegenanzeigen:umoren (pNET) m. Krankheitsprogression. D. Erfahrung m. Sutent als First-line-Behandlung ist begrenzt. Überempfindlichk. gg. d. Wirkstoff od. sonst. Bestandteile. geschlagen ist. B. Erwu./od. metast. maligner gastrointest. Stromatumoren (GIST), wenn e. Behandl. m. Imatinib wg. Resistenz od. Unverträglichk. fehl
mg zusätzl.: Eisen(III)- hydroxid-oxid x H2O (Emg/50171), Schellack, Propylenglycol, Natriumhydroxid; 25mg Sunitinib. mg/50mg/25. 12,5Hartkps. enthält Sunitinibmalat, entspr
Neutropenie, Thrombozytopenie, Anämie, Leukopenie; Hypothyreose; verminderter Appetit/Appetitlosigk.; Schlaflosigk.; Schwindel- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstör- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstör
Neutropenie, Thrombozytopenie, Anämie, Leukopenie; Hypothyreose; verminderter Appetit/Appetitlosigk.; Schlaflosigk.; Schwindelumfassten Ausgang tödl. m. Ereignisse Nebenwirk. häufigsten d. zu gehören
. Erythrodysästhesie-Syndr-plantarerfärb. d. Haut u. palmarB. Durchfall, Übelk., Stomatitis, Dyspepsie u. Erbrechen), V-, Harnwegs- od. Gehirnblutungen). D. häufigsten Nebenwirk. jeden Grades (b. Pat. i. mRCC-, GIST-, Harnwegs- od. Gehirnblutungen). D. häufigsten Nebenwirk. jeden Grades (b. Pat. i. mRCC-, GIST
Nebenwirkungen: Überempfindlichk. gg. d. Wirkstoff od. sonst. Bestandteile. . zur Behandl. fortgeschritt./metast. Nierenzellkarzinome (mRCC). B. Erw. zur Behandl. nicht resezierb. od. metast., gut. zur Behandl. fortgeschritt./metast. Nierenzellkarzinome (mRCC). B. Erw
Anwendungsgebiete:172). 172), Eisen(II,III)-oxid (Emg zusätzl.: Eisen(III)- hydroxid-oxid x H2O (E421), Croscarmellose-Natrium, Povidon (K-25),.) (EEur Mannitol (Ph.Sonst. Bestandteile:mg Sunitinib.
b-5v
24su
-hk-
0
.),- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstör Neutropenie, Thrombozytopenie, Anämie, Leukopenie; Hypothyreose; verminderter Appetit/Appetitlosigk.; Schlaflosigk.; Schwindelgefühl, Kopfschm., Geschmacks-gefühl, Kopfschm., Geschmacks
dissem.Multiorganversagen, a. u. umfassten . ein. Diese Sympt. können abnehmen,. Erythrodysästhesie-Syndr
-Zulassungsstudien)- u. pNET-, Harnwegs- od. Gehirnblutungen). D. häufigsten Nebenwirk. jeden Grades (b. Pat. i. mRCC-, GIST D. schwersten Nebenwirk., einige davonNebenwirkungen:
. zur Behandl. nicht resezierb. od. metast., gut. zur Behandl. nicht resezierb. B. ErwAnwendungsgebiete:
421), Croscarmellose-Natrium, Povidon (K-25),
. Erythrodysästhesie-SyndrAusschlag m. Juckreiz), Änder
ereinigtes Königreich. 9NJ, VSandwich, Kent CT13Rhabdomyolyse, Nekrolyse; tox.-epidermale gangraenosum,
thrombozytopenische Purpura, hämolytisch-urämisches SyndrCholezystitis/Cholezystitis ohne Gallensteine, Leberfkt. anormal; Osteonekrose d. Kiefers, Fistel; Harnwegsblut.; verzögerte Wuinfarkt, stummer Myokardinfarkt), Herzinsuff., Kardiomyopathie, Perikarderguss, VFasziitis, bakt. Inf. (Abdominalabszess, Abdominalsepsis, Divertikulitis, Osteomyelitis); Panzytopenie; Überempfindlichk.; Hypezahl erniedrigt, Lipaseerhöh., vermind. Thrombozytenzahl, Hämoglobin erniedrigt, Amylase/Amylase erhöht, Aspartataminotransferazahl erniedrigt, Lipaseerhöh., vermind. Thrombozytenzahl, Hämoglobin erniedrigt, Amylase/Amylase erhöht, Aspartataminotransfera
erfärb. d. Nägel); Schm. d. Muskel- u. Skelettsystems, Muskelspasmen, Myalgie, Muskelschwäche; Nierenversagen, akutes Nierenvereränd./V(VHämorrhoiden, Glossodynie, Mundschm., Mundtrockenh., Flatulenz, orale Beschw
Nasenverstopf., trockene Nasenschleimhaut; gastroösophageale Oropharynx/Pharyngolaryngealschm., ., Angina pectoris, instabile Angina pectoris, Koronararterienverschluss), Ejektionsfraktion verringert/abnormal; tiefe VIschämie (akutes KoronarsyndrHarnwegsinf., Candidose), orale u. Ösophagus
(Bronchitis, Inf. d. unteren Atemwege, Pneumonie), Abszess (Abszess an Gliedmaßen, Analabszess, Zahnfleischabszess, Leberabszes(Bronchitis, Inf. d. unteren Atemwege, Pneumonie), Abszess (Abszess an Gliedmaßen, Analabszess, Zahnfleischabszess, LeberabszesÖsophagus
. d. Haarfarbe, trockene Haut; Schm. in e. Extremität, Arthralgie, Rückenschm.; Schleimhautentzünd., Erschöpf./Kraftlosigk., Öd., Ausschlag (psoriasiforme Dermatitis, . Erythrodysästhesie-Syndr
Ausschlag m. Juckreiz), Änderpalmar-plantar. Erythrodysästhesie-Syndr-plantarAusschlag m. Juckreiz), ÄnderpalmarAusschlag m. Juckreiz), Änder
. (Dysgeusie, Ageusie); Hypertonie; Dyspnoe, Nasenbluten, Husten; Stomatitis/aphthöse Stomatitis, Abdominalschm. (Bauchschm., Sstör
PFIZER PHARMA GmbH, LinkstrRepräsentant in Deutschland:ereinigtes Königreich. eitere WWarnhinweise:.. SyndrMyopathie; nephrot. Rhabdomyolyse, .; posteriores revers. Enzephalopathie-Syndrumorlyse-Syndr.); Angioödem; Thyroiditis; Tthrombozytopenische Purpura, hämolytisch-urämisches Syndr
Cholezystitis/Cholezystitis ohne Gallensteine, Leberfkt. anormal; Osteonekrose d. Kiefers, Fistel; Harnwegsblut.; verzögerte Wu-Intervalls im EKG; Terläng. d. QTinfarkt, stummer Myokardinfarkt), Herzinsuff., Kardiomyopathie, Perikarderguss, V
Fasziitis, bakt. Inf. (Abdominalabszess, Abdominalsepsis, Divertikulitis, Osteomyelitis); Panzytopenie; Überempfindlichk.; Hypezahl erniedrigt, Lipaseerhöh., vermind. Thrombozytenzahl, Hämoglobin erniedrigt, Amylase/Amylase erhöht, Aspartataminotransferazahl erniedrigt, Lipaseerhöh., vermind. Thrombozytenzahl, Hämoglobin erniedrigt, Amylase/Amylase erhöht, AspartataminotransferaFasziitis, bakt. Inf. (Abdominalabszess, Abdominalsepsis, Divertikulitis, Osteomyelitis); Panzytopenie; Überempfindlichk.; Hype
erfärb. d. Nägel); Schm. d. Muskel- u. Skelettsystems, Muskelspasmen, Myalgie, Muskelschwäche; Nierenversagen, akutes Nierenver., Aufstoßen; Exfoliation d. Haut, Hautreaktionen/Hauterkrank., Ekzem, Blase, Erythem, Alopezie, Akne, Juckreiz, HauthyperpigmeHämorrhoiden, Glossodynie, Mundschm., Mundtrockenh., Flatulenz, orale Beschw
Refluxerkrank., Dysphagie, Gastrointestinalblut., Ösophagitis, aufgetriebener ., Angina pectoris, instabile Angina pectoris, Koronararterienverschluss), Ejektionsfraktion verringert/abnormal; tiefe V
Nasenverstopf., trockene Nasenschleimhaut; gastroösophageale ., Angina pectoris, instabile Angina pectoris, Koronararterienverschluss), Ejektionsfraktion verringert/abnormal; tiefe V
ymphopenie; LLymphopenie; Schock; Sepsis/septischer Hautinf./Cellulitis, Harnwegsinf., (Bronchitis, Inf. d. unteren Atemwege, Pneumonie), Abszess (Abszess an Gliedmaßen, Analabszess, Zahnfleischabszess, Leberabszes
. d. Haarfarbe, trockene Haut; Schm. in e. Extremität, Arthralgie, Rückenschm.; Schleimhautentzünd., Erschöpf./Kraftlosigk., Öd., Ausschlag (psoriasiforme Dermatitis, exfoliativer Hautausschlag, erythematöser ., Ausschlag (psoriasiforme Dermatitis,
. (Dysgeusie, Ageusie); Hypertonie; Dyspnoe, Nasenbluten, Husten; Stomatitis/aphthöse Stomatitis, Abdominalschm. (Bauchschm., S
2015.JuniStand: Berlin. 10, 10785. PFIZER PHARMA GmbH, LinkstrAbgabestatus:Gebrauchsinformation. u. Fach- s. Informationen eitere
.; Linksherzinsuff., T.; posteriores revers. Enzephalopathie-Syndrndheil.; Kreatinphosphokinase im Blut erhöht, Thyreotropin im Blut erhöht. Cholezystitis/Cholezystitis ohne Gallensteine, Leberfkt. anormal; Osteonekrose d. Kiefers, Fistel; Harnwegsblut.; verzögerte Wu
umorblut.; Lungenblut., respiratorische Insuff.; gastrointestinale Perforation/Darmperforat., Pankreatitis, Analfistel; Leberve-Intervalls im EKG; Trthyreose; Hirnblut., apoplekt. Insult, transitorFasziitis, bakt. Inf. (Abdominalabszess, Abdominalsepsis, Divertikulitis, Osteomyelitis); Panzytopenie; Überempfindlichk.; Hype
zahl erniedrigt, Lipaseerhöh., vermind. Thrombozytenzahl, Hämoglobin erniedrigt, Amylase/Amylase erhöht, Aspartataminotransferase erhöht, Alaninaminotransferase erhöht, Kreatinin im Blut erhöht, Blutdruck erhöht, Harnsäure im Blut erhöht. zahl erniedrigt, Lipaseerhöh., vermind. Thrombozytenzahl, Hämoglobin erniedrigt, Amylase/Amylase erhöht, Aspartataminotransferasagen, Chromurie, Proteinurie; Schm. i. Brustkorb, Schm., grippeähnl. Erkrank., Schüttelfrost; vermind. Körpergewicht, Leukozyterfärb. d. Nägel); Schm. d. Muskel- u. Skelettsystems, Muskelspasmen, Myalgie, Muskelschwäche; Nierenversagen, akutes Nierenver
., Aufstoßen; Exfoliation d. Haut, Hautreaktionen/Hauterkrank., Ekzem, Blase, Erythem, Alopezie, Akne, Juckreiz, HauthyperpigmeRefluxerkrank., Dysphagie, Gastrointestinalblut., Ösophagitis, aufgetriebener
., Angina pectoris, instabile Angina pectoris, Koronararterienverschluss), Ejektionsfraktion verringert/abnormal; tiefe Venenthrombose, Hitzewall., Hitzegefühl; Lungenembolie, Pleuraerguss, Hämoptyse, Belastungsdyspnoe, Schm. imRefluxerkrank., Dysphagie, Gastrointestinalblut., Ösophagitis, aufgetriebener
., Angina pectoris, instabile Angina pectoris, Koronararterienverschluss), Ejektionsfraktion verringert/abnormal; tiefe VRefluxerkrank., Dysphagie, Gastrointestinalblut., Ösophagitis, aufgetriebener
Neuropathie, periph. Depression; Hypoglykämie; Dehydratation, s, Pankreasabszess, perinealer Abszess, perirektaler Abszess, rektaler Abszess, subkutaner Abszess, Zahnabszess), Pilzinf. (Can(Bronchitis, Inf. d. unteren Atemwege, Pneumonie), Abszess (Abszess an Gliedmaßen, Analabszess, Zahnfleischabszess, Leberabszes
eme (Gesichtsödem, peripheres Ödem), FieberHautausschlag, follikulärer Ausschlag, generalisierter Ausschlag, makulöser Ausschlag, makulo-papulöser Ausschlag, papulöser Au
. d. Haarfarbe, trockene Haut; Schm. in e. Extremität, Arthralgie, Rückenschm.; Schleimhautentzünd., Erschöpf./Kraftlosigk., Ödexfoliativer Hautausschlag, erythematöser Hautausschlag, follikulärer Ausschlag, generalisierter Ausschlag, makulöser Ausschlag, makulo-papulöser Ausschlag, papulöser AuHautausschlag, follikulärer Ausschlag, generalisierter Ausschlag, makulöser Ausschlag, makulo-papulöser Ausschlag, papulöser Au
. d. Haarfarbe, trockene Haut; Schm. in e. Extremität, Arthralgie, Rückenschm.; Schleimhautentzünd., Erschöpf./Kraftlosigk., Ödexfoliativer Hautausschlag, erythematöser
. d. Haarfarbe, trockene Haut; Schm. in e. Extremität, Arthralgie, Rückenschm.; Schleimhautentzünd., Erschöpf./Kraftlosigk., Öd
- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstörchm. im Unter. (Dysgeusie, Ageusie); Hypertonie; Dyspnoe, Nasenbluten, Husten; Stomatitis/aphthöse Stomatitis, Abdominalschm. (Bauchschm., S
Hautausschlag, follikulärer Ausschlag, generalisierter Ausschlag, makulöser Ausschlag, makulo-papulöser Ausschlag, papulöser Aueme (Gesichtsödem, peripheres Ödem), Fieber
Unternehmer:Pharmazeutischer erschreibungspflichtig. VAbgabestatus:orsade de pointes; Hepatitis; Erythema multif., Stevens-Johnson-Syndr.; Linksherzinsuff., T
thrombot. Mikroangiopathie (thrombotisch-Selten:ndheil.; Kreatinphosphokinase im Blut erhöht, Thyreotropin im Blut erhöht. umorblut.; Lungenblut., respiratorische Insuff.; gastrointestinale Perforation/Darmperforat., Pankreatitis, Analfistel; Leberve
. ischäm. Attacke; kongestive Herzinsuff., Myokardinfarkt (akuter Myokardrthyreose; Hirnblut., apoplekt. Insult, transitorse erhöht, Alaninaminotransferase erhöht, Kreatinin im Blut erhöht, Blutdruck erhöht, Harnsäure im Blut erhöht.
sagen, Chromurie, Proteinurie; Schm. i. Brustkorb, Schm., grippeähnl. Erkrank., Schüttelfrost; vermind. Körpergewicht, Leukozyt., Hautläsion, Hyperkeratose, Dermatitis, Nagelerkrank.ntier., Aufstoßen; Exfoliation d. Haut, Hautreaktionen/Hauterkrank., Ekzem, Blase, Erythem, Alopezie, Akne, Juckreiz, Hauthyperpigme
., Rektalblut., Zahnfleischblut., Mundulzerat., Proktalgie, Cheilitis,Bauch, abdom. BeschwRefluxerkrank., Dysphagie, Gastrointestinalblut., Ösophagitis, aufgetriebener enenthrombose, Hitzewall., Hitzegefühl; Lungenembolie, Pleuraerguss, Hämoptyse, Belastungsdyspnoe, Schm. imenenthrombose, Hitzewall., Hitzegefühl; Lungenembolie, Pleuraerguss, Hämoptyse, Belastungsdyspnoe, Schm. im
Lidödem, Periorbitalödem, Hyperästhesie; Hypästhesie, Parästhesie, Neuropathie, s, Pankreasabszess, perinealer Abszess, perirektaler Abszess, rektaler Abszess, subkutaner Abszess, Zahnabszess), Pilzinf. (Can
Virusinf. (Nasopharyngitis u. oraler Herpes), Atemwegsinf.Häufig:. eme (Gesichtsödem, peripheres Ödem), FieberHautausschlag, follikulärer Ausschlag, generalisierter Ausschlag, makulöser Ausschlag, makulo-papulöser Ausschlag, papulöser Au
- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstör
Limited,Pfizer Unternehmer:., Pyodermaorsade de pointes; Hepatitis; Erythema multif., Stevens-Johnson-Syndr
thrombot. Mikroangiopathie (thrombotisch-rsagen,umorblut.; Lungenblut., respiratorische Insuff.; gastrointestinale Perforation/Darmperforat., Pankreatitis, Analfistel; Leberve
-. ischäm. Attacke; kongestive Herzinsuff., Myokardinfarkt (akuter Myokard nekrotis.Gelegentlich:se erhöht, Alaninaminotransferase erhöht, Kreatinin im Blut erhöht, Blutdruck erhöht, Harnsäure im Blut erhöht.
-ensagen, Chromurie, Proteinurie; Schm. i. Brustkorb, Schm., grippeähnl. Erkrank., Schüttelfrost; vermind. Körpergewicht, Leukozyt., Hautläsion, Hyperkeratose, Dermatitis, Nagelerkrank.
., Rektalblut., Zahnfleischblut., Mundulzerat., Proktalgie, Cheilitis,enenthrombose, Hitzewall., Hitzegefühl; Lungenembolie, Pleuraerguss, Hämoptyse, Belastungsdyspnoe, Schm. im
myokardiale.; ränensekrTverstärkte Lidödem, didose d.s, Pankreasabszess, perinealer Abszess, perirektaler Abszess, rektaler Abszess, subkutaner Abszess, Zahnabszess), Pilzinf. (Can
Virusinf. (Nasopharyngitis u. oraler Herpes), Atemwegsinf.sschlag u.Hautausschlag, follikulärer Ausschlag, generalisierter Ausschlag, makulöser Ausschlag, makulo-papulöser Ausschlag, papulöser Au
.),- u. Oberbauch), Erbrechen, Diarrhö, Dyspepsie, Übelk., Obstipat.; Hautverfärb. (gelbe Hautfarbe, Pigmentierungsstör
.de.pfizerwww

Wir wünschen allen Mitgliedern, Partnern, Freunden und Förderern
des Tumorzentrum Erfurt e.V. ein frohes Weihnachtsfest
und ein gesundes neues Jahr.
Wir danken Ihnen herzlich für Ihr Engagement und hoffen auf eine
weitere gute Zusammenarbeit.
Prof. Dr. Albrecht Stier
Vorsitzender des Vorstandes
Prof. Dr. Hartwig Kosmehl
Vorsitzender des Wissenschaftlichen Beirates
Dr. Hubert Göbel
Geschäftsführer

n Klinische Krebsregister – Zur Halb-zeit gibt es noch einiges zu tun
Mario GrotheLandesvertretung Thüringen, Verband der Ersatz-kassen e.V. (vdek)
Am 9. April 2013 ist das Gesetz zur Weiterentwicklungder Krebsfrüherkennung und zur Qualitätssicherungdurch klinische Krebsregister (Krebsfrüherkennungs- und-registergesetz – KFRG) in Kraft getreten. Danach sollenbis 2017 flächendeckend klinische Krebsregister, welchevergleichbare Daten erheben, aufgebaut werden. Bisherbestehen länderspezifisch unterschiedliche Regelungenund Rahmenbedingungen.
Hinsichtlich der Krebsregister legt das KFRG die Einrich-tung regionaler klinischer Krebsregister, die als fachlichunabhängige Einrichtungen alle wichtigen Behandlungs-schritte im Verlaufe einer Krebserkrankung (ambulant undstationär) und ihrer Behandlung anfallenden Daten erfas-sen, in allen Bundesländern fest. Durch das Gesetz werdenu.a. einheitliche Voraussetzungen für die Erfassung desgesamten Behandlungsverlaufs von Krebspatienten ein-schließlich des Behandlungsergebnisses sowie für die Dar-stellung der Ergebnisqualität geschaffen. Grundlage fürdie Datenerfassung aller klinischen Krebsregister ist dereinheitliche onkologische Datensatz. Dieser Datensatz giltfür alle Krebsarten und wird fortlaufend um tumorspezi-fische Module ergänzt. In diesem Datensatz wird genauvorgegeben, welche Daten der behandelnde Arzt doku-mentieren muss.
Krebsregistergesetz – Aufgabe der einzelnen Bundes-länder
Die notwendigen Bestimmungen für die Einrichtung undden Betrieb der klinischen Krebsregister müssen durch dieeinzelnen Bundesländer in Form landesrechtlich vergleich-barer Regelungen geschaffen werden.
Stand der Umsetzung der klinischen Krebsregister
a) Krebsregistergesetze der einzelnen BundesländerNach knapp zwei Jahren (Stand: Anfang November 2015)sind erst in fünf Bundesländern (Bremen, Hamburg, Hes-sen, Saarland und Schleswig-Holstein) Krebsregisterge-setze (nach KFRG) in Kraft getreten oder beschlossen wor-den. In drei Bundesländern (Baden-Württemberg, Nord-rhein-Westfalen und Rheinland-Pfalz) liegt ein Entwurfzum Krebsregistergesetz vor. In den restlichen acht Bun-desländern (Mecklenburg-Vorpommern, Bayern, Berlin,Brandenburg, Niedersachsen, Sachsen-Anhalt, Sachsenund Thüringen) liegt noch kein Entwurf für ein Krebsregi-stergesetz vor. Die Bundesländer Berlin und Brandenburgwerden ein gemeinsames länderübergreifendes Krebsre-gister aufbauen.
b) Übergangsvereinbarungen zwischen den Bundeslän-dern und den KrankenkassenverbändenIn acht Bundesländern (Baden-Württemberg, Bayern,Brandenburg, Bremen, Hamburg, Rheinland-Pfalz, Saar-
land, Sachsen-Anhalt) wurden bis jetzt zwischen denKrankenkassenverbänden und den einzelnen Bundeslän-dern Übergangsvereinbarungen zur Einrichtung und Wei-terentwicklung der klinischen Krebsregister abgeschlos-sen. Die Finanzierung der Leistungen ab dem 1. Januar2016 ist in den einzelnen Bundesländern sehr heterogengeregelt und vereinbart.
Investitionskosten für die Errichtung bzw. den Aus-und Umbau der klinischen Krebsregister
Um eine rasche Umsetzung der Landesgesetze und denzügigen Auf-, Aus- und Umbau klinischer Krebsregister zufördern, werden durch die Deutsche Krebshilfe die hierfürnotwendigen Investitionskosten zu 90 % (höchstens je-doch 7,2 Mio. Euro) finanziert. Die Länder tragen (minde-stens) die verbleibenden 10 % (0,8 Mio. Euro).
Aufgaben der gesetzlichen Krankenversicherung
Der Betrieb der klinischen Krebsregister wird durch dieKrankenkassen gefördert. Die dem GKV-Spitzenverbandobliegende Verpflichtung für die Förderung Kriterien un-ter Beteiligung der im KFRG aufgeführten Organisationenund Personen (§ 65c Absatz 3 SGB V) bis Ende 2013 zuentwickeln, wurde durch den Beschluss des Kriterienkata-loges im Dezember 2013 durch den GKV-Spitzenverbanderfüllt. Die Vereinbarung hinsichtlich der Meldevergütungfür jede landesrechtlich vorgesehene Meldung wurde imDezember 2014 geschlossen und die Höhe der Meldever-gütungen durch Schiedsperson nach § 65c Abs. 6 Satz 8SGB V am 24. Februar 2015 festgelegt.
Klinisches Krebsregister in Thüringen
Ein Gesetzentwurf für ein klinisches Krebsregister in Thü-ringen liegt bisher nicht vor. Aus Sicht des vdek ist ein kli-nisches Krebsregister pro Bundesland ausreichend, umdie im Gesetz vorgesehenen Aufgaben zu erfüllen. Da-durch werden Informationsverluste und Fehler an denSchnittstellen bei mehreren Krebsregistern vermieden undes wird eine mehrfache Erfassung von Patienten ausge-schlossen.
Finanzierung der Krebsregister in Thüringen
Die bestehenden Krebsregister (Aufgabengebiet der Tu-morzentren) werden bis zum 31. Dezember 2015 durchZentrumszuschläge nach § 2 Abs.2 S.2 Nr.4 KHEntgG fi-nanziert. Das Krebsfrüherkennungs- und Registergesetz(KFRG) regelt durch eine Änderung am § 17b Abs. 1 S. 4KHG, dass Zuschläge nicht mehr für den Betrieb klinischerKrebsregister vereinbart werden dürfen. Diese Regelungsoll zum 1. Januar 2016 in Kraft treten. Danach wäre eineFinanzierung über Zentrumszuschläge ab dem 1. Januar2016 nicht mehr möglich.
Da der Gesetzgeber das hieraus entstehende Finanzie-rungsproblem erkannt hat, soll diese Regelung des KFRGdurch das Krankenhausstrukturgesetz (KHSG) „Artikel 8des Kabinettsentwurfs“ aufgehoben werden. Sollte dieseGesetzesänderung erfolgen, wovon derzeit auszugehenist, da keine gegenteiligen Aussagen bekannt sind, ist
n Seite 4 n JOURNAL 01/2005JOURNAL 01/2015
n Seite 5 nJOURNAL 01/2015
auch eine Finanzierung der Krebsregister über die Zen-trumszuschläge ab dem 1. Januar 2016 weiterhin mög-lich. Um eine Doppelfinanzierung jedoch auszuschließen,wird gleichzeitig durch das KHSG ein Verbot der doppel-ten Finanzierung geregelt (Artikel 2 Nr. 3 § 2 Abs. 2KHEntgG).
Sollte eine Abrechnung der Krebsregisterpauschalendurch eine Übergangsvereinbarung aufgrund von ver-schiedenen vorliegenden Problemen nicht möglich sein,werden die Krankenkassenverbände in Thüringen die Fi-nanzierung der Krebsregister über Zentrumszuschlägefortführen.
Korrespondenzadresse:
Ass. jur. Mario GrotheVerband der Ersatzkassen e.V. (vdek)Landesvertretung ThüringenLucas-Cranach-Platz 299099 ErfurtTelefon: 0361-4425237E-Mail: [email protected]://ww.vdek.com
n Klinik, Genetik und Managementvon Patienten mit NeurofibromatoseTyp 2
Steffen K. Rosahl, Anna Lawson McLean, Marcel AlbrechtNeurofibromatosezentrum, Klinik für Neurochirurgie,HELIOS Klinikum Erfurt
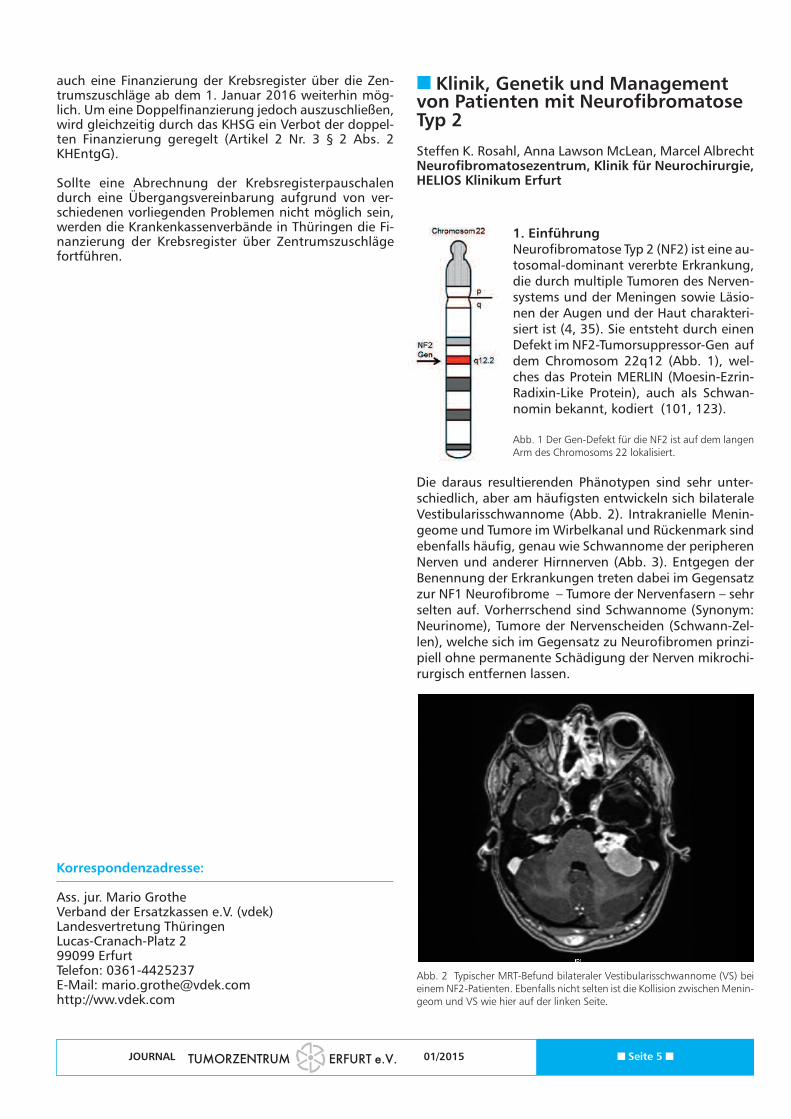
1. EinführungNeurofibromatose Typ 2 (NF2) ist eine au-tosomal-dominant vererbte Erkrankung,die durch multiple Tumoren des Nerven-systems und der Meningen sowie Läsio-nen der Augen und der Haut charakteri-siert ist (4, 35). Sie entsteht durch einenDefekt im NF2-Tumorsuppressor-Gen aufdem Chromosom 22q12 (Abb. 1), wel-ches das Protein MERLIN (Moesin-Ezrin-Radixin-Like Protein), auch als Schwan-nomin bekannt, kodiert (101, 123).
Abb. 1 Der Gen-Defekt für die NF2 ist auf dem langenArm des Chromosoms 22 lokalisiert.
Die daraus resultierenden Phänotypen sind sehr unter-schiedlich, aber am häufigsten entwickeln sich bilateraleVestibularisschwannome (Abb. 2). Intrakranielle Menin-geome und Tumore im Wirbelkanal und Rückenmark sindebenfalls häufig, genau wie Schwannome der peripherenNerven und anderer Hirnnerven (Abb. 3). Entgegen derBenennung der Erkrankungen treten dabei im Gegensatzzur NF1 Neurofibrome – Tumore der Nervenfasern – sehrselten auf. Vorherrschend sind Schwannome (Synonym:Neurinome), Tumore der Nervenscheiden (Schwann-Zel-len), welche sich im Gegensatz zu Neurofibromen prinzi-piell ohne permanente Schädigung der Nerven mikrochi-rurgisch entfernen lassen.
Abb. 2 Typischer MRT-Befund bilateraler Vestibularisschwannome (VS) beieinem NF2-Patienten. Ebenfalls nicht selten ist die Kollision zwischen Menin-geom und VS wie hier auf der linken Seite.

n Seite 6 n JOURNAL 01/2005JOURNAL 01/2015
Abb. 3 In den spinalen MRT von NF2-Patientensieht man oft multiple kleinere Schwannomegehäuft im Bereich der Cauda equina. Bei grö-ßenprogredienten, Rückenmark und Nervenkomprimierenden Schwannomen und Menin-geomen besteht eine Indikation zur chirurgi-schen Entfernung. Intramedulläre Ependymomekönnen meist über sehr lange Zeiträume beob-achtet werden.
2. HistorischesDie erste klinische Beschreibung stammt von Wishart ausdem Jahr 1822 (128). Nachdem von Recklinghausen aus-gangs des 19. Jahrhunderts das klinische Bild der Neuro-fibromatose 1 (NF1) beschrieben hatte und der großeHarvey Cushing 1917 auch noch einen Zusammenhangvon bilateralen Tumoren des 8. Hirnnerven bei Patientenmit Morbus Recklinghausen beschrieb, kam es zu einerjahrzehntelangen Verwirrung der beiden Neurofibroma-tosetypen. Erst als es möglich wurde, die Genloci von NF1und NF2 eindeutig auseinanderzuhalten, konnte die NF2auch formal eindeutig von ihrem Namensvetter abge-grenzt werden (111). Jüngster Zuwachs zur NF-Familie istdie Schwannomatose, genetisch abgrenzbar und klinischvor allem durch multiple schmerzhafte subkutane Tumo-ren auffällig.
3. EpidemiologieDie scheinbare Prävalenz von NF2 ist über die letzten Jahr-zehnte kontinuierlich angestiegen. Dafür sind wahr-scheinlich sowohl die Einführung der Kernspintomografie(MRT) als auch die gestiegene Bekanntheit der Erkrankungverantwortlich.1992 gab die Arbeitsgruppe um Evans in Manchesternoch eine Prävalenz von 1:210.000 an (36). Die gleicheArbeitsgruppe fand in einer neueren Studie allerdingseine Prävalenz von 1:60.000. Die Inzidenz liegt wahr-scheinlich zwischen 1:33.000 bis 1: 87.000 Geburten (3, 34).Das mittlere Alter bei Diagnosestellung liegt bei 25 Jah-ren, allerdings beginnt die Symptomatik im Mittel ca. 7 Jahre vor der Sicherung der Diagnose (47). Unterschiedein der Häufigkeit der Erkrankung bezüglich Geschlechtund Zugehörigkeit zu Bevölkerungsgruppen sind bishernicht berichtet worden.
4. MolekolarbiologieDas NF2-Tumorsuppressorgen wurde 1993 identifiziert.Es enthält 17 Exons die das 69kDa Protein kodieren, wel-ches man MERLIN (Moesin-Ezrin-Radixin-Like-Protein)oder Schwannomin genannt hat (101, 123). In Überein-stimmung mit Knudson’s 2-Hit-Hypothese wird die Tu-morentstehung initiiert, wenn beide Allele des Gens inak-tiviert sind (54). Patienten ererben entweder eine Keim-zellmutation des Allels eines Elternteils oder erkrankendurch eine postzygotische Neumutation eines Allels wäh-rend der Embryogenese. Die Tumoren entstehen vor allemim Nervensystem, aber auch in anderen Zielorganen (z.B.Auge und Haut) aus Zellen, in denen das (normale) Wild-typ-NF2-Allel seine Funktion verliert. Eine somatische In-aktivierung beider Allele wurde auch in sporadischenSchwannomen (>90%), Meningeomen (50%) und Epen-dymomen (5%) nachgewiesen (29, 112). Die beim Menschen am häufigsten vertretenen Isoformenvon MERLIN sind Isoform I und II (14, 94), die nach neu-esten Daten vermutlich beide tumorsupprimierend wirkenkönnen (131). Phosphorylierung ist der entscheidendeMechanismus der Regulierung der Tumorsuppressorakti-vität von MERLIN. Durch Phosphorylierung über cAMPwird das Protein in seine offene Form überführt und inak-tiviert (46, 49, 51, 99, 114, 115).
Abb. 4 Kristall-Struktur der FERM-Domäne des Tumorsuppressor-ProteinsMERLINRCSB Protein Data Bank, An Information Portal to Biological Macromolecu-lar Structures(http://www.rcsb.org/pdb/explore/explore.do?structureId=1h4r)
Entsprechend erfolgt die Umkehrung dieses Prozessesdurch Phosphatasen, welche MERLIN wieder in seine ge-schlossen, aktive Form überführen, die über intramoleku-lare Assoziation mit der FERM-Domäne (ERM bezeichneteine Familie Actin-bindender Proteine, benannt nach denprototypischen Vertretern Ezrin, Radixin und Moesin, wel-che in der finalen, N-terminalen Domäne große Homolo-gie zeigen) wirkt (Abb. 4). MERLIN ist insofern ein unge-wöhnlicher Tumorsuppressor, als es sich an das Membran-Zytoskelett assoziiert und die Plasmamembran damit ver-bindet (110). Es gibt eine ganze Reihe von Mechanismen,welche bei Abwesenheit von MERLIN zur Tumorentste-
n Seite 7 nJOURNAL 01/2015
hung führen. Sie sind bisher nicht vollständig untersuchtund verstanden. Primär wird der Effekt offenbar dadurchvermittelt, dass MERLIN in die Organisation von Mem-branproteinen eingreift (z.B. CD44, EGF-Rezeptor, Laylin),Zell-zu-Zell Adhäsionen beeinflusst (z.B. über β-Catenin,ε-Cadherin, β1-Integrin, Paxillin) und indem es die Archi-tektur des Zytoskeletts verändert (z.B. über βII-Spectrin,F-Actin, Rho guanosine Triphosphatasen oder das neuro-nale Wiskott-Aldrich-Syndrom-Protein). Außerdem inter-agiert MERLIN mit zytosolischen Proteinen. Alle diese Ef-fekte wirken sich downstream auf verschiedene mitogeneSignalwege aus, von denen die wichtigsten der Phosphoi-nositide-3-Kinase-Signalweg (PI3K) und der mitogen-ak-tivierte Proteinkinase (MAPK)-Signalweg sind (4). Dieseonkogenen Signalwege sind entscheidend für die Promo-tion von Zellwachstum, Proteintranslation und zelluläreProliferation. Chemotherapeutika, welche gegen dieseSignalwege gerichtet sind (Sorafenib, Trastuzumab, La-patinib, LY294002, Proteinkinaseinhibitoren, P21-aktivier-te Kinase-Inhibitoren) befinden sich in präklinischer undzum Teil in erster klinischer Erprobung. Relativ etabliert istbereits der Angiogenese-Inhibitor Bevacizumab, der dasWachstum von Vestibularisschwannomen und den damitverbundenen beidseitigen Hörverlust in vielen Fällen ef-fektiv stoppen (73, 87, 89, 92), in Deutschland allerdingsbisher nur off-label eingesetzt werden kann.
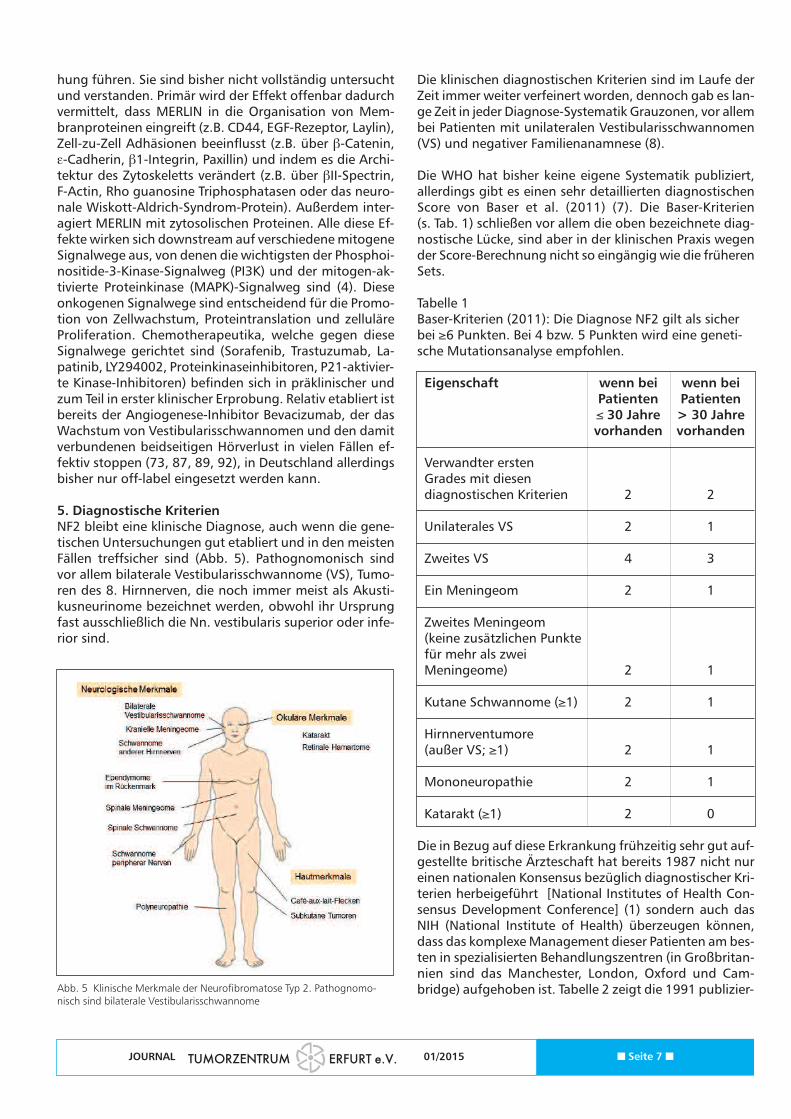
5. Diagnostische KriterienNF2 bleibt eine klinische Diagnose, auch wenn die gene-tischen Untersuchungen gut etabliert und in den meistenFällen treffsicher sind (Abb. 5). Pathognomonisch sindvor allem bilaterale Vestibularisschwannome (VS), Tumo-ren des 8. Hirnnerven, die noch immer meist als Akusti-kusneurinome bezeichnet werden, obwohl ihr Ursprungfast ausschließlich die Nn. vestibularis superior oder infe-rior sind.
Abb. 5 Klinische Merkmale der Neurofibromatose Typ 2. Pathognomo-nisch sind bilaterale Vestibularisschwannome
Die klinischen diagnostischen Kriterien sind im Laufe derZeit immer weiter verfeinert worden, dennoch gab es lan-ge Zeit in jeder Diagnose-Systematik Grauzonen, vor allembei Patienten mit unilateralen Vestibularisschwannomen(VS) und negativer Familienanamnese (8).
Die WHO hat bisher keine eigene Systematik publiziert,allerdings gibt es einen sehr detaillierten diagnostischenScore von Baser et al. (2011) (7). Die Baser-Kriterien (s. Tab. 1) schließen vor allem die oben bezeichnete diag-nostische Lücke, sind aber in der klinischen Praxis wegender Score-Berechnung nicht so eingängig wie die früherenSets.
Tabelle 1 Baser-Kriterien (2011): Die Diagnose NF2 gilt als sicherbei ≥6 Punkten. Bei 4 bzw. 5 Punkten wird eine geneti-sche Mutationsanalyse empfohlen.
Eigenschaft wenn bei wenn beiPatienten Patienten≤ 30 Jahre > 30 Jahrevorhanden vorhanden
Verwandter erstenGrades mit diesendiagnostischen Kriterien 2 2
Unilaterales VS 2 1
Zweites VS 4 3
Ein Meningeom 2 1
Zweites Meningeom(keine zusätzlichen Punktefür mehr als zweiMeningeome) 2 1
Kutane Schwannome (≥1) 2 1
Hirnnerventumore(außer VS; ≥1) 2 1
Mononeuropathie 2 1
Katarakt (≥1) 2 0
Die in Bezug auf diese Erkrankung frühzeitig sehr gut auf-gestellte britische Ärzteschaft hat bereits 1987 nicht nureinen nationalen Konsensus bezüglich diagnostischer Kri-terien herbeigeführt [National Institutes of Health Con-sensus Development Conference] (1) sondern auch dasNIH (National Institute of Health) überzeugen können,dass das komplexe Management dieser Patienten am bes-ten in spezialisierten Behandlungszentren (in Großbritan-nien sind das Manchester, London, Oxford und Cam-bridge) aufgehoben ist. Tabelle 2 zeigt die 1991 publizier-

NIH
Bilaterale VSoderVerwandter ersten Gradesmit NF2+ unilaterales VSoder + einer dieser Tumoren:Neurofibrom, Meningeom,Gliom, Schwannom oderjuvenile Katarakt
Manchester
Bilaterale VSoderVerandter ersten Gradesmit NF2+ unilaterales VSoder + einer dieser Tumoren:Neurofibrom, Meningeom,Gliom, Schwannom oderjuvenile KataraktoderUnilaterales VS+zwei dieser Tumoren:Neurofibrom, Meningeom,Gliom, Schwannomoder + juvenile Kataraktoder>2 Meningeome+ unilaterales VS oder zweidieser Tumoren:Neurofibrom, Gliom, Schwannomoder Katarakt
NNFF
Gesicherte NF2:Bilaterale VSoderVerwandter ersten Grades mit NF2undunilaterales VS mit ED <30 Jahreoder zwei dieser Tumoren:Neurofibrom, Meningeom, Gliom,Schwannomoder juvenile Kataraktwahrscheinliche NF2:Unilaterales VSundmindestens ein Meningeom, Gliomoder Schwannom oder juvenileKatarakt
oder>2 Meningeome+unilaterales VS mit ED <30 Jahreoder einer dieser Tumoren:Meningeom, Gliom, Schwannomoder juvenile Katarakt
n Seite 8 n JOURNAL 01/2005JOURNAL 01/2015
ten diagnostischen Kriterien der Briten im Vergleich zuden so genannten „Manchester Kriterien“ von 1992, wel-che auch Patienten mit negativer Familienanamnese undunilateralem Vestibularisschwannom einbeziehen. Außer-dem sind hier die von der US National NeurofibromatosisFoundation (NNFF) vorgeschlagenen Kriterien zum Ver-gleich dargestellt, welche auf einer Studie von Gutmannet al. aus dem Jahr 1997 basieren (45). Letztere unter-scheiden erstmals „gesicherte“ und „wahrscheinliche“NF2-Fälle.
Die Inzidenz der drei klinischen Hauptkriterien (neurolo-gische, okuläre, dermatologische Manifestationen) vari-iert in der Literatur zum Teil erheblich (Tab. 3).An unserem Neurofibromatosezentrum stützen wir Erst-diagnosen gern auf die Baser-Kriterien, empfehlen aberaktuell eine genetische NF2-Diagnostik bei Patienten ab12 Jahren auch dann, wenn kein Vestibularisschwannomin der MRT-Diagnostik sichtbar ist, aber multiple Menin-geome oder Neurinome im ZNS nachgewiesen wurden.
Andererseits haben wir auch einen inzwischen über 70Jahre alten Patienten mit bilateralen Vestibularisschwan-nomen beraten, dessen genetische Diagnostik unauffälligist. Ein ebensolcher Fall, bei dem erstmals beide Tumorengenetisch sequenziert und als NF2-negativ klassifiziertwurden, ist in diesem Jahr erstmals von der Arbeitsgruppeum Gareth Evans publiziert worden (33).
6. Genetische Untersuchung und genetischer Schwe-regradSeit der Identifikation des NF2-Gens auf dem Chromosom22 ist es möglich, die Diagnose molekulargenetisch zu si-chern. Eine humangenetische Beratung ist schon deshalbwichtig, um mit den Patienten die Vererbbarkeit der Er-
krankung und entsprechende Konsequenzen zu bespre-chen. Die Tests werden am besten an frischem Tumorgewebebei der Entfernung des ersten Tumors durchgeführt. Blut-tests haben eine Treffsicherheit von 70-75% für die Iden-tifizierung des Gendefekts bei bekannten NF2-Patienten.Erst wenn der Defekt nachgewiesen wurde, sind Blutun-tersuchungen auch bei Verwandten sinnvoll. Ein NF2-Screening bei Patienten ohne klinischen Verdacht auf NF2ist nicht zu empfehlen. Neumutationen bei Patienten mitmilderen klinischen Verläufen sind in den meisten FällenMissense-Mutationen, welche oft schwer zu detektierensein können. Wenn bei unauffälligen Bluttests im Tumorbeide Allele betroffen sind, dann handelt es sehr wahr-
Tabelle 2 Diagnostische NF2-Kriterien des National Institute of Health (NIH), Manchester und National NeurofibromatosisFoundation (NNFF)

Tabelle 3 Häufigkeit radiologisch nachweisbarer Tumoren, okulä-rer Läsionen und Hauttumoren bei NeurofibromatoseTyp 2 [modifiziert nach Asthagiri et al. 2009 (3)]
Häufigkeit bei NF2
Neurologische Merkmale
Bilaterale Vestibularisschwannome 90-95 % (114)Schwannome anderer Hirnnerven 24-51 % (9, 41, 70,
80, 81, 109)Intrakranielle Meningeome 45-58 % (35, 70, 85)Spinale Tumoren 63-90 % (28, 70, 74,
81, 85, 98)Extramedullär 45-90 %Intramedullär 18-53 %
Periphere Neuropathie <66 % (70, 81, 120)
Ophthalmologische MerkmaleKatarakte 60-81 % (16, 35, 70,
81, 120)Epiretinale Membranen 12-40 % (16, 95)Retinale Hamartome 6-22 % (35, 70, 96)
HautmerkmaleHauttumoren 59-68 % (35, 63, 71, 82)Café-aux-lait-Flecken 41-48 %Subkutane Tumoren 43-48 %Intradermale Tumoren selten
scheinlich um ein genetisches Mosaik für eines der ge-schädigten Allele (30).Weil Mosaike bei etwa 30% der Patienten auftreten, hatdie genetische Diagnostik im Tumor, die natürlich einerAufklärung und des Einverständnisses des Patienten be-darf, einen entsprechenden Stellenwert. Rein somatischeMosaike sind nicht vererbbar, klinisch sind die Erschei-nungsbilder dabei oft weniger schwer. Trotz aller Heterogenität sind die Erscheinungsbilder undVerläufe der Erkrankung in NF2-Familien meist ähnlich (4,35, 130). Prinzipiell ist es heute aufgrund verschiedener Studienzur Korrelation von Genetik und Klinik möglich, nebender phänotypischen Schwere auch den genetischenSchweregrad zu bestimmen (10, 32, 38, 107, 113, 119).
Ferner et al. haben 2014 anhand von Literaturbefundenund eigenen Beobachtungen die folgende Einstufung ge-netischer Schweregrade der NF2 vorgenommen: Abbruchmutationen (engl.: truncating mutations) alsoNonsense- und Frameshift-Mutationen in den Exons 1-13aller Zellen wurden als „schwer“ klassifiziert. Klinisch wur-de auch früher schon die schwere Verlaufsform vom Wis-hart-Typ mit diesen in Verbindung gebracht (47).
Unter mittelschweren Mutationen wurden drei verschie-dene Mechanismen zusammengefasst:
a) Deletionen außerhalb von Promotorregion und Exon 1
b) Splice Mutationen in den Exons 1-8c) Mosaike von Abbruchmutationen der Exons 1-13
im Bluttest.
Als „milde“ genetische Erkrankungsformen bezeichnetendie Autoren folgende Mutationen:
a) Missense Mutationen oder Deletionen (in frame)b) größere Deletionen, welche die Promotorregion
oder das Exon 1 einbeziehenc) Splice-Mutationen in den Exons 9-15d) Mosaike ohne Abbruchmutationen der Exons 1-13
im Blutteste) fehlender Mutationsnachweis im Blut.
Die Autoren klassifizierten von den 288 in ihrer Serie ein-geschlossenen Patienten 58,4% als mild, 17% als mittel-schwer und 14% als schwer (10,4% der Patienten warennicht genetisch untersucht worden). Obwohl auch in früheren Studien Missense-Mutationenals milde genetische Formen mit dem Gardner-Phänotypkorreliert waren (47), war in dieser Studie die Korrelationzwischen der durch die Patienten angegebenen Lebens-qualität und der genetischen Schwere der Erkrankung nurschwach ausgeprägt. Die Autoren erklären das u.a. damit,dass der genetische Schweregrad den längerfristigen Ver-lauf charakterisiert während die Einschätzung der Lebens-qualität natürlich nur den aktuellen Zustand reflektierenkann. Außerdem könnte es auch sein, dass die Patientenmit einem hohen genetischen Schweregrad bereits früh-zeitig diagnostiziert und behandelt wurden, bevor durchdie Tumoren schwere Symptome und funktionelle Defiziteausgelöst werden konnten. Zudem können auch Patien-ten mit genetisch milder Einstufung der Erkrankung undgeringer Tumorbelastung unter signifikanten Symptomeund neurologischen Defiziten leiden.
7. Klinischer SchweregradDie Bestimmung des Schweregrades der Erkrankung wur-de in den letzten Jahren verfeinert. Während einige Stu-dien zur Lebensqualität bereits vorliegen, fehlen systema-tische Studien zur psychischen Stress-Belastung und zumCoping der Patienten noch. Sie sind aber, federführenddurch die Universität Giessen in Zusammenarbeit mit denZentren in Tübingen, Hamburg und Erfurt in Arbeit. Ge-nerell ist es erstaunlich, wie gut viele dieser Patienten mitzunehmenden funktionellen neurologischen Defiziten,Schmerzen und sozialen Problemen umzugehen wissen.Interessant ist, dass die durch NF2-Patienten empfundeneund in Fragebögen reflektierte Lebensqualität zwar in bis-herigen Studien mit dem radiologischen Phänotyp undschwächer auch mit dem genetischen Schweregrad kor-reliert, entscheidendere Faktoren für die Lebensqualitätaber individuelle Belastung und individueller Leidensdruckzu sein scheinen (40).
Klassisch werden als Verlaufsformen der schwere Wishart-Phänotyp (früher Beginn, rascher Progress, schlechte
n Seite 9 nJOURNAL 01/2015

n Seite 10 n JOURNAL 01/2005JOURNAL 01/2015
Prognose) und der leichtere (Feiling-)Gardner-Phänotyp(Erstdiagnose nach dem 20. Lebensjahr, geringe Tumor-last) unterschieden.
Neuere Untersuchungen unterscheiden klinisch in schwe-re, mittelschwere und leichte Erscheinungsbilder. Ferneret al. (40) zogen die Grenzen 2014 anhand ihrer Untersu-chungen an 288 Patienten so:Eine schwere Erkrankung lag vor, wenn die Patienten beider ersten Symptommanifestation jünger als 20 Jahre wa-ren und zusätzlich zu einem bzw. zwei nachweislichenVestibularisschwannomen mindestens zwei symptomati-sche oder große (>1,5cm) Tumoren hatten, einschließlichder vor Diagnosestellung bereits entfernten Tumoren.Auch Patienten, bei denen die Erstdiagnose vor dem 12.Lebensjahr gestellt wurde und die mindestens einensymptomatischen Tumor hatten, wurden als „schwer“klassifiziert (24,3%).Patienten, die bei der Erstmanifestation älter als 30 Jahrewaren und nicht mehr als zwei symptomatische oder gro-ße (>1,5cm) Tumoren (einschließlich zuvor entfernter Tu-moren) hatten wurden als „mild“ eingestuft (43,1%).Mittelschwer betroffen sind nach dieser Klassifikation allePatienten, die nicht in eine der beiden vorigen Kategorienpassten (32,3%).Da die Ausprägung einzelner Symptome einen erhebli-chen Einfluss sowohl auf die objektivierbare Schwere alsauch auf den subjektiven Leidensdruck hat, erschien unsdiese strikt auf den Zeitpunkt der Erstdiagnose und dieradiologische Tumorbelastung fokussierte Einteilung zukurz zu greifen. Außerdem hatte sich ja gezeigt, dass dieKorrelation zwischen Lebensqualität und klinischemSchweregrad schwächer als erwartet war, wenn man dieklinische Symptomkonstellation aus dem Rating desSchweregrades ausklammert.Wir haben daher die Einteilung der Oxforder Arbeitsgrup-pe als „radiologischen Phänotyp“ übernommen, aber wei-tere klinische Kriterien hinzugefügt, um eine möglichstindividuelle, patientengerechte Einschätzung des klini-schen Schweregrades zu erreichen (Tab. 4). Dabei spielenFunktionseinschränkungen, Defizite im Bereich der Sin-neskanäle und Schmerzsyndrome eine vorrangige Rolle.Aus Untersuchungen bei Patienten mit sporadischen VSist bekannt, dass Schwindel und Gleichgewichtsstörun-gen oft die entscheidenden Faktoren für die vom Patien-ten empfundene Beeinträchtigung der Lebensqualitätsind (18, 19, 21, 76). Wenn man bedenkt, dass NF2-Pa-tienten im Laufe ihres Lebens häufig nicht nur ihre beidenGleichgewichtsnerven einbüßen, sondern auch nochdurch Neuropathien und Tumoren im Bereich des Rücken-marks und des peripheren Nervensystems weitere Rück-meldesysteme der Körperorientierung verlieren, gewinntman eine Vorstellung zumindest von dieser Dimensionder Erkrankung. Dazu kommen der Verlust des Hörens,welcher auch durch Cochlea-Implantate (CI) und audito-rische Hirnstammimplantate (ABI) nicht wirklich zu kom-pensieren ist, und teilweise massive Sehstörungen nichtnur durch beidseitige Katarakte sondern auch durch Tu-more um die Sehnerven oder die Entwicklung von Hirn-
druck (Pseudotumor cerebri, Hydrocephalus). SchwersteVerläufe werden durch Schluckstörungen induziert, wel-che durch Tumoren, aber vor allem auch durch Operatio-nen im Bereich der kaudalen Hirnnerven entstehen. SolcheFaktoren waren für uns entscheidend, um den Patientenbei der klinischen Einschätzung des Schweregrades ihrerErkrankung gerecht zu werden, auch wenn es nach diesenKriterien schwerst betroffene Patienten gibt, die ihre Le-bensqualität höher einschätzen als wesentlich leichter be-troffene.
Tabelle 4 Klinische Schweregrade der Neurofibromatose (ErfurterKlassifikation)
leicht schwersymptomatische Erstmanifes- symptomatische Erstmanifesta-tation >30 Jahre tion >20 Jahre+ +nicht mehr als zwei weitere zumindest 2 weitere TumorenTumoren (symptomatische (symptomatisch oder >1,5 cm)oder >1,5 cm)1) zusätzlich zu den Vestibularis-
schwannomenoderNachweis eines zentralnervö-sen Tumors vor dem 12. Lebens-jahr und mindestens eines wei-teren symptomatischen Tumorsbei genetisch positivem Befundfür NF2
+ eines oder mehrere + eines oder mehrerefolgender Merkmale folgender Merkmale• noch funktionell hörend • vollständige Ertaubung• kein Schwindel • schwerster Schwindel• keine Einschränkungen • Schluckstörungen mitder sprachlichen Expression Ernährungssonde
• keine wesentliche bilaterale • PhonationsstörungSehbehinderung • hochgradige Sehstörung
• keine wesentliche Ein- • Fazialisparese > H&B Grad IIIschränkung des Gehver- • Rollstuhlpflichtigkeit durchmögens (partielle) Querschnittsläh-
• keine wesentliche Schmerzen oder schwere Ataxie• Inkontinenz mit Katheteri-sierungspflicht
• schwere Orientierungsstörung• schwere Gedächtnisstörung• zusätzliche Erkrankungen mitschwerer oder mittelschwererSymptomatik (z.B. Syringo-myelie nach Operationen,Pseudotumor cerebri durchSinusvenenverschluss bei parasagittalen Meningeomen,medikamentös nicht beherrsch-bare Depression)
• schwere chronische Schmerzen
1)einschließlich der Vestibularisschwannome und zuvorchirurgisch entfernter Tumoren

n Seite 11 nJOURNAL 01/2015
Eine mittelschwere Ausprägung liegt bei allen Patientenvor, die nicht unter eine der beiden anderen Kategorienklassifiziert werden. Darunter fallen z.B. auch Patientenmit höchstgradiger Schwerhörigkeit beidseits (mit oderohne CI/ABI), mit starkem Schwindel, mit mittelgradigerFazialisparese (House und Brackman Grad III), mit mode-raten Schluckstörungen, mit schweren bilateralen Seh-störungen, mit mittelgradigen Schmerzen, mit mäßigenGedächtnisstörungen und mit zusätzlichen Erkrankungenmit mittelschwerer Symptomatik (z.B. Epilepsie, Hydroce-phalus, multipler Sklerose).
Hier soll auch erwähnt werden, dass es möglicherweiseregionale Unterschiede in der Beurteilung des klinischenSchweregrades gibt. In der bereits mehrfach zitierten Stu-die der Gruppe um Rosalie Ferner schätzten die Ärzte dieklinische Schwere der Erkrankung bei Patienten in Man-chester geringer ein als in London, Cambridge und Ox-ford. Natürlich kann das auch lediglich Ausdruck klinischheterogener Patientengruppen gewesen sein (40).
8. BildgebungAlle Patienten, bei denen der klinische Verdacht auf dasVorliegen einer NF2 besteht, sollten eine MRT mit i.v.-Kontrastmittelgabe zur Beurteilung beider innerer Gehör-gänge erhalten. Hochauflösende Aufnahmen sind v.a.zum Ausschluss eines Tumors auf der Gegenseite bei ge-sichertem unilateralem Vestibularisschwannom erforder-lich.Wenn die Diagnose sicher ist, dann werden MRT des Kop-fes und der gesamten Neuroachse zum radiologischenStaging der Erkrankung als Ausgangsbefund veranlasst.Einige Zentren empfehlen bei Erstmanifestation einenGanzkörper-Scan als Ausgangsbefund, andere sind hierzurückhaltender und untersuchen Thorax, Abdomen undExtremitäten erst dann, wenn sich hier korrelierte Symp-tome zeigen. In der spinalen Achse findet man meist multiple kleine Tumoren entlang der Cauda equina. Manchmal zeigensich aber auch größere, noch asymptomatische Menin-geome oder Schwannome, welche neurale Strukturen be-reits schwer komprimieren und entfernt werden müssen(Abb. 6).
Die frühzeitige Entfernung dieser Tumoren kann langfris-tig die Morbidität erheblich verringern, zumal eine durchsie ausgelöste Rückenmarks-Symptomatik nicht immer zurehabilitieren ist. Intramedulläre Tumoren (Ependymome,intramedulläre Schwannome) sind oft über viele Jahregrößenstabil. Bei nachweislichem Wachstum gelten aberprinzipiell die oben beschriebenen Vorgehensweisen wiefür rückenmarksnahe Tumoren. Nicht selten findet maneine, meist lokal begrenzte, assoziierte Syringomylie.Bei älteren Patienten ist die Erstdiagnose bilateraler Tu-moren in den inneren Gehörgängen suspekt auf ein me-tastasierendes Leiden. Daher muss sich hier eine Primär-tumorsuche anschließen. Findet sich kein Ausgangstumor,dann sollte eine erneute MRT des Kopfes nach 3 Monatenveranlasst werden.Ansonsten empfehlen wir bei Patienten mit intrakraniellenTumoren eine erneute kranielle MRT-Diagnostik 6 Monatenach dem Erstbefund. Sind die Tumore dann stabil, wirdjährlich ein MRT des Kopfes als Routine empfohlen. Dasgleiche gilt für größere Tumoren im Bereich der Wirbel-säule. Wenn keine spinale Manifestation nachgewiesenwurde, dann sollte eine erneute spinale Bildgebung beiAuftreten einer entsprechenden Symptomatik erfolgen.Bei Nachweis kleiner und mittelgroßer spinaler Tumorenist eine Routine-Nachuntersuchung nach 3 Jahren ausrei-chend. Auch bei sehr stabilen intrakraniellen Befundenkann unter Umständen das Kontrollintervall von einemJahr auf einen längeren Zeitraum ausgedehnt werden.
9. ManagementDie NF2 ist ein relativ anspruchsvolles Krankheitsbild,nicht nur wegen der fast unausweichlichen Ertaubungder meisten Patienten im Verlauf. Ein kompetentes Ma-nagement bedarf eines multidisziplinären Teams unterBeteiligung v.a. der Humangenetik, der Neurochirurgie,der Otolaryngologie, der Augenheilkunde, der Neurolo-gie, der Pädiatrie, der Radiologie, der Pathologie, derStrahlentherapie und der Audiologie (15). Die Lebenser-wartung der Patienten hat sich durch die Betreuung derPatienten in entsprechenden Zentren sicher verbessert.Das prioritäre Ziel des Managements muss die Erhaltungvon Funktionen sein. Damit untrennbar verbunden ist dieLebensqualität der Patienten. Die Erstmanifestation der
NF2 – z.B. eine Mononeuropathie - wirdoft noch verkannt. Insbesondere denNeurologen und den Pädiatern kommtdaher eine besondere Bedeutung imRahmen des Managements und der Zu-ordnung von Frühsymptomen (u.a. Ra-dikulopathien der oberen oder unterenExtremitäten durch extra-axiale spinaleTumoren) zu. Potentiell bietet sich hierdie Chance, durch zeitnahe Veranlassung
Abb. 6 T2-gewichtete MRT mit einem extraaxialenSchwannom mit Rückenmarkskompression in HöheBW1/2. Der Patient ist gehfähig, beklagt aber eine zu-nehmende Gangunsicherheit und Dranginkontinenz.

n Seite 12 n JOURNAL 01/2005JOURNAL 01/2015
einer körperlichen, v.a. kutanen und augenärztlichen Un-tersuchung, den Diagnosezeitpunkt vor zu verlagern, in-dem Hautmerkmale und retinale Hamartome frühzeitigerkannt werden. In Verbindung mit einer genetischenBlutuntersuchung lässt sich so in den meisten Fällen dieDiagnose sichern. Seit der grundlegenden Unterscheidung der Ausprägungin Wishart- und Gardner-Typ haben mehrere Untersuchun-gen inzwischen eine Beziehung zwischen Phänotyp undGenotyp der Erkrankung dokumentiert. Wie oben beschrieben führen alle Abbruch-Mutationen(Nonsense oder Frameshift) zu einem schwereren Verlaufder Erkrankung (9, 38, 52, 83, 106). Deletionen mit kon-sekutivem vollständigem Verlust des ProteinproduktsMERLIN, und Missense-Mutationen sind mit milderen Ver-läufen assoziiert. Splice-Mutationen sind mit unterschiedlicher Krankheits-ausprägung verbunden. Betreffen sie die Exons 1-5 sinddie Verläufe meist schwerer als bei Mutationen in denExons 11-15 (10, 53). Damit verbunden ist offenbar auchdie Lebenserwartung der Patienten (9).Mosaike beeinflussen den Phänotyp ganz entscheidend.Dabei steht der Anteil der von der Mutation betroffenensomatischen Zellen in Zusammenhang mit dem klinischenVerlauf der Erkrankung: Patienten mit einer geringerenAnzahl betroffener Zellen werden voraussichtlich eine mil-dere Ausprägung zu erwarten haben, zum Teil mit asym-metrischer (in Bezug auf die Tumorlast in den Kleinhirn-brückenwinkeln) oder oligolokaler Erkrankung, z.B. reinim Kleinhirnbrückenwinkel gelegener oder zumindest reinintrakranieller Lokalisation (37).
Trotz der o.g. Studien, die eine enge Verknüpfung zwi-schen Geno- und Phänotyp nahelegen, ist dieser Zusam-menhang zwischen Mutationstyp und Mosaikbildungganz sicher nicht linear. Die klinischen Krankheitsverläufevariieren beträchtlich, besonders aber bezüglich desWachstumsverhaltens einzelner Tumoren (56). Besonderseindrucksvoll dokumentiert sich das bei den pathogno-monischen Vestibularisschwannomen (42), deren Wachs-tumsverhalten völlig unabhängig von der Gesamttumor-last sein kann (11).
10. Manifestationen im Nervensystem10.1. VestibularisschwannomeDas Management bei NF2 ruht inzwischen auf vier Säulen:Beobachtung, Mikrochirurgie, stereotaktische Radiothe-rapie (im weiteren Sinn, d.h. unter Einschluss der Radio-chirurgie) und neuerdings Chemotherapie (außerhalb vonStudien mit off-label Bevacizumab in Deutschland). DerEntscheidungsbaum ist dabei außerordentlich komplex,daher sollen hier nur die wesentlichen Kriterien aufge-führt werden. Dies schließt Hörfunktion (Audiometrie,kontralaterales Hörvermögen), Tumorgröße (Hirnstamm-kompression), Tumorausdehnung im inneren Gehörgang(Fundus, Fossa cochlearis), Wachstumsrate, Patientenprä-ferenz, Komorbidität (v.a. bezogen auf die NF2-Erkran-kung selbst) ein. Solange ein konservatives Managementmöglich ist, wird dies zu bevorzugen sein. Eine (seltene)
Ausnahme bildet eine Situation, bei der bilateral kleineTumoren ohne Infiltration der Fossa cochlearis bzw. desFundus des inneren Gehörgangs gefunden wurden undmikrochirurgisch die Möglichkeit zur hörerhaltenden Ent-fernung eines der Tumoren gesehen wird (17, 109, 117).Gelingt dies, dann ändert eine solche Behandlung nichtnur den Verlauf der Erkrankung, sondern auch langfristigdie soziale Stellung des Patienten unter Umständengrundlegend. Das bedeutet nicht, dass nach Erstdiagnosenicht zunächst eine Verlaufskontrolle (6 Monate) sinnvollsein kann, um das biologische Wachstumsverhalten derTumoren zu beobachten, auch wenn ein lineares Wachs-tum nachweislich seltener als ein saltatorisches ist (56,90).Die Indikationsstellung zur Entfernung hirnstammkom-primierender Vestibularisschwannome sollte immer dasNutzen-Risiko-Verhältnis berücksichtigen, welches hierklar als Funktionserhaltung versus chirurgisches Risiko(Vestibulocochlearis, Fazialis, Kaudale Hirnnerven) defi-niert ist. In Großbritannien haben Baser und Kollegen2005 die kumulative Exzisionsrate der VS so beschrieben:1% im Alter von 20 Jahren, 3% im Alter von 25 Jahren,37% im Alter von 50 Jahren (12). Neben den drei klassi-schen chirurgischen Zugängen zur Tumorentfernung (re-trosigmoidal, translabyrinthär, subtemporal) gibt es Kon-zepte zur reinen knöchernen und duralen Dekompressiondes inneren Gehörgangs (insbesondere mit dem Ziel, dieBlutversorgung der Cochlea zu verbessern und dadurchdas Hörvermögen längere Zeit zu erhalten), zur intrakap-sulären Tumorreduktion und zur Kombination mit stereo-taktisch-radiochirurgischen Verfahren.Aus verschiedenen Gründen sind Vestibularisschwanno-me bei NF2 schwieriger zu operieren als sporadische VS.Vor allem sind die Tumoren häufiger gelappt bzw. multi-lokulär und umschließen auch einmal die Hirnnerven an-statt sie rein zu verlagern. Zudem sind sie, ebenfalls ausunterschiedlichen Gründen, oft adhärenter an Nerven undGefäßen als sporadische Tumoren (48). Dadurch ist die Er-haltung z.B. des Fazialis schwieriger und nicht selten sindprimäre Transplantationen erforderlich, um am Ende derRehabilitation zumindest einen kompletten Lidschluss zuerreichen (108). Daher ist die Neigung der Chirurgen zueiner vollständigen Tumorentfernung im Laufe der Zeitgeringer geworden, mehrzeitige und kombinierte Kon-zepte (Radiotherapie, Chemotherapie) haben hier ver-stärkt Einzug gehalten.
Nach anfänglichem Enthusiasmus wird heute auch dieRolle der Radiotherapie als Behandlungsoption für Vesti-bularisschwannome bei NF2-Patienten allerdings auch kri-tischer gesehen. Bereits über kürzere (5-Jahres-) Zeiträu-me betrachtet variieren die „Tumorkontrollraten“ (=Wachstumsstopp und/oder Schrumpfung), wahrschein-lich v.a. abhängig von der applizierten Strahlendosis, be-trächtlich zwischen 66–100% (50, 60, 67, 75, 86, 104,121, 125). Auch die Hörerhaltungsrate wird in diesen Zeit-räumen mit 33–57% recht variabel angegeben (50, 60,67, 75, 86, 104, 121). Es gibt Bemühungen, die Mechanismen der Radioresis-

n Seite 13 nJOURNAL 01/2015
tenz mancher VS näher zu untersuchen, um hier ggf. inZukunft eine Radiosensitisierung zu erreichen (129).Ein Wachstumsstopp durch Radiotherapie ist offenbar ge-genüber sporadischen Tumoren seltener (2) und beson-ders höhere - bezüglich der Tumorkontrolle effektivere -Strahlendosen können zumindest bei sporadischen Tu-moren den Hörverlust beschleunigen (97). Das Risiko signifikanter permanenter, also nicht mehr re-habilitierbarer Fazialisparesen durch die Radiotherapiewird mit 0–10% angegeben, transiente Paresen werdenbei 10-17% der Patienten berichtet (50, 60, 67, 75, 102,104). Ob es ein höheres Risiko der Induktion maligner Tu-moren durch radioaktive Strahlung dieser Tumoren ge-genüber dem Spontanverlauf gibt, ist bislang nicht aus-reichend geklärt, auch wenn etliche NF2-Fälle in der Lite-ratur beschrieben werden, bei denen nach Radiotherapiemaligne Tumoren, darunter maligne periphere Nerven-scheidentumore, maligne Meningeome, Rhabdomyosar-kome und maligne Ependymome auftraten (5, 20, 24, 79,84, 122). Baser et al. berechneten aus diesen Daten ein 7-fach erhöhtes Malignitätsrisiko im Vergleich zu NF2-Pa-tienten, welche keine Radiotherapie erhalten haben (6).Andererseits finden sich in der Literatur bisher nur wenigeBerichte über bösartige de-novo-Tumoren im Bereich desNervus vestibularis. Noch seltener ist die maligne Trans-formation eines histologisch gesichert gutartigen Vesti-bularisschwannoms zu einem anaplastischen Sarkom. Demetriades und Kollegen fanden 2010 neben einem ei-genen Fall 13 weitere maligne Vestibularisschwannomein der Literatur. Sechs dieser Patienten hatten zuvor einekranielle Strahlentherapie erhalten, aber nur bei zwei Pa-tienten war zuvor ein gutartiges VS histologisch gesichertworden (26).Bei drei weiteren Patienten mit histologisch gesichertenmalignen Schwannomen war zuvor eine gezielte radio-chirurgische Behandlung bei radiologischem Verdacht aufdas Vorliegen eines Vestibularisschwannoms durchge-führt worden. Zwei dieser Patienten litten unter Neurofi-bromatose Typ2. In diesem Jahr verglichen Maducdoc und Kollegen aus Ir-vine die in der Literatur berichteten Fälle maligner Trans-formation von VS nach mikrochirurgischer Behandlungund nach stereotaktischer Radiochirurgie (64). Sie schlos-sen in ihre Untersuchungen 8 Fälle ein, bei denen die Ent-artung nach kombinierter Behandlung mit beiden Ver-fahren auftrat. Bei weiteren 4 Fällen war eine maligneTransformation nach Mikrochirurgie aufgetreten, ohnedass eine Bestrahlung erfolgte. Die Autoren fanden je-doch auch 18 Berichte über primär maligne Vestibularis-schwannome ohne vorherige Interventionen bezüglichdes Tumors. Aufgrund der niedrigen berichteten Inzidenzvon malignen Schwannomen de novo lag die Schlussfol-gerung der Studie nahe, dass das Risiko einer maligenTransformation nach Behandlung eines VS zwar nichtgleich Null, aber doch sehr niedrig sei und nicht einmalgesichert ist, ob bei den berichteten Fällen tatsächlich dietherapeutische Intervention ursächlich war. Zu einer ähn-lichen Schlussfolgerung waren auch schon frühere Unter-
suchungen gekommen (103, 105). Die Studie fand aberaußerdem 12 berichtete Fälle einer malignen Entartungbei NF2-Patienten. Wenn man den relativ geringen Anteilvon NF2-Betroffenen an der Weltbevölkerung betrachtet,dann muss man hier wohl einen Zusammenhang mit be-denken.Es wurde vermutet, dass Patienten mit Keimzelldefektenim Tumorsuppressorgen auf dem Chromosom 22 suszep-tibler für sekundäre Malignome nach einer Strahlenbe-handlung sein könnten (20). Histopathologisch wird einvermehrter Pleomorphismus in VS beschrieben, die nacheiner Strahlentherapie wegen erneuten Wachstums ent-fernt wurden (61). Die chirurgische Entfernung mancherbestrahlter VS kann unter Umständen schwieriger sein,(93, 116), das Gleiche trifft aber auch auf voroperierte Tu-moren zu. Aufgrund der intensivierten molekulargenetischen For-schung zu diesem Krankheitsbild in jüngster Zeit mehrensich die Studien zu Versuchen medikamentöser Beeinflus-sung des Verlaufs. Die NF2 stellt dabei eine Art Modell imBereich der benignen Tumorerkrankungen dar. Auch ausklinischer Sicht erfolgreich sind dabei die Untersuchungenzu VEGF (Vascular endothelial growth factor) -Inhibitorenerfolgreich verlaufen. Der monoklonale Antikörper Beva-cizumab ist in der Lage, das VS-Wachstum auch über Zeit-räume von mehreren Jahren zu stoppen und die Tumor-größe insbesondere am Beginn der Behandlung signifi-kant zu reduzieren (72, 91, 92). Damit einher geht in ei-nigen Fällen eine Stabilisierung oder Verbesserung desHörvermögens (87, 92). Die Nebenwirkungen, v.a. Nephrotoxizität sind auf Dauer nicht zu vernachlässigen(118), eine Dosisreduktion kann in einzelnen Fällen er-folgreich sein (39).Andere Substanzen, wie Sorafenib, zielen auf verschiede-ne intrazelluläre Aspekte des Zellteilungszyklus(Integrin/FAK/Src/Ras-Signalweg, Phosphatidylinositol-3-kinase/Protein-Kinase-C/Src/c-Raf-Signalweg, PDGFRbeta-vermittelte ERK1/2-Aktivierung (30, 61, 88)(Ammoun,Flaiz, Ristic, Schuldt, & Hanemann, 2008).Die Rehabilitation des Hörens spielt eine wichtige Rollebei NF2-Patienten. Bei Patienten mit mittelschwerem Hör-verlust kann zunächst ein normales Hörgerät helfen. Istdas nicht mehr ausreichend, kann man je nach der indivi-duellen Dynamik der Vestibularisschwannome die Implan-tation eines CI (Cochlea Implant) oder eines ABI (AuditoryBrainstem Implant) in Betracht ziehen.Cochlea Implantate sind eine Option, solange die bipola-ren Neurone in der Cochlea und ein Hörnerv funktionellnoch intakt sind (62, 77, 124). Die meisten NF2-Patientenprofitieren erheblich auch bezüglich des Sprachverständ-nisses von diesen Implantaten, auch wenn die Ergebnisseschlechter sind als bei nicht von dieser Erkrankung betrof-fenen Patienten. Sind beide Hörnerven zerstört, dann ist eine partielle Wie-derherstellung des Hörvermögens nur durch ein ABI mög-lich (13, 22, 43, 69, 78, 100). Dieses Implantat übernimmtbis auf das Elektrodendesign die Technologie des CI. Ober-flächenelektroden werden dafür im Recessus lateralis des

n Seite 14 n JOURNAL 01/2005JOURNAL 01/2015
4. Ventrikels an den Hirnstamm im Bereich der Nucleicochleares angelegt. Klinische Versuche mit penetrierendeTiefenelektroden (HEI) und Elektroden im Mittelhirn (Au-ditorisches Mittelhirnimplantat) haben die Ergebnisse derOberflächenimplantate bisher nicht übertroffen (23, 57-59). Generell stehen die Ergebnisse hinter den mit CI er-reichbaren Resultaten deutlich zurück. Im Zusammen-hang mit Lippenablesen können einzelne Patienten je-doch ein limitiertes Sprachverständnis erreichen. Die Er-langung eines freien Sprachverständnisses nach einemlängeren audiologischen Anpassungsprozess ist eine Aus-nahme. Zudem empfinden es die Patienten als Gewinn,Geräusche wie Türklingel, auf der Straße vorbeifahrendeAutos oder auch nur das Ansprechen durch andere Men-schen wahrnehmen zu können. Die Rate der Nichtnutzerliegt bei 10-20% (68, 69).Inzwischen implantieren mehrere NF2-Zentren ein ABIauch bei Patienten, die auf der Gegenseite noch hörenkönnen („Sleeper“). Dieses Implantat wird in Intervalleneingeschaltet mit dem Ziel, durch einen zeitweisen audi-torischen Input die neurale Kapazität der Hörbahn zu er-halten und so das Hörvermögen zu optimieren, wennspäter das ABI tatsächlich dauerhaft gebraucht wird.Nach Implantation von CI und ABI ist die Qualität vonMRT-Verlaufsuntersuchungen zumindest im Bereich desipsilateralen Kleinhirnbrückenwinkels und Felsenbeinseingeschränkt. Die für die transkutane Signalübertragungeingesetzten Magnete können im wechselnden Magnet-feld dislozieren (27) und durch Erwärmung Gewebschä-den induzieren. Suszeptibilitätsartefakte durch den Mag-neten im Implantat führen zu Verzerrungen und schrän-ken die Beurteilbarkeit der MRT ein (66). Es wurden auchDemagnetisierungen des Magneten und damit Implan-tatfehlfunktionen beschrieben (65).Dennoch sind die modernen Implantate prinzipiell MRT-sicher in 1,5-Tesla-Geräten (44). Magnetdislokationenkönnen fast immer durch straffe Druckverbände verhin-dert werden (25). Obwohl eine gewisse Verzerrung derMR-Bilder nicht gänzlich zu vermeiden ist, kann man dieseverringern, wenn man den Kopf des Patienten im Scannerso rotiert, dass das Magnetfeld der Implantatspule mitdem Magnetfeld des Scanners gleichgerichtet ist (126).Alternativ muss man den Magneten u.U. in Lokalanästhe-sie oder Kurznarkose temporär zum MRT entfernen. Mit-unter ist es unumgänglich, v.a. zur Beurteilung des ipsila-teralen Kleinhirnbrückenwinkels, ein Computertomo-gramm anzufertigen (Strahlenbelastung und geringereWeichteilauflösung im Vergleich zum MRT).
10.2. MeningeomeKonvexitätsmeningeome sind meist unproblematisch voll-ständig zu entfernen, auch wenn dies bei NF2-Patientennur dann sinnvoll ist, wenn sie durch Wachstum einedeutliche Kompression ausüben oder symptomatisch wer-den. Im Gegensatz dazu sind Meningeome der Schädel-basis oft schwer zu entfernen und das Risiko postoperati-ver Morbidität ist entsprechend höher. Radiochirurgiescheint im Rahmen der NF2 noch weniger effektiv für Me-
ningeome als für Vestibularisschwannome zu sein (55).Wentworth gab 2009 nach Bestrahlung von Meningeo-men bei NF2-Patienten ein progressionsfreies 5-Jahres-Überleben von 86% an (127).Kombinierte mikrochirurgisch-radiochirurgische Strate-gien können vor allem bei hirnstammkomprimierendenpetroklivalen Tumoren mit Ausdehnung in den Sinus ca-vernosus oder parasagittalen Meningeomen mit Einwach-sen in den dorsalen Anteil des Sinus sagittalis superiorsinnvoll sein. Bei ausgeprägter intrakranieller Meninge-omlast kann es sowohl zu einem malresorptiven Hydroce-phalus als auch zu einem Pseudotumor cerebri kommen,insbesondere bei zunehmendem Befall des Sinus sagittaliskommen.
10.3. Spinale TumorenSpinale Neurinome und Meningeome müssen entferntwerden, wenn sie das Rückenmark zunehmend kompri-mieren oder symptomatisch werden. Die meisten dieserTumore sind extraaxial gelegen, Schwannome gehen häu-fig von Nervenfasern aus, deren (sensorische) Funktionbereits über einen längeren Zeitraum und für den Patien-ten unbemerkt verloren gegangen sein kann. Die mikro-chirurgische Exzision der Tumoren ist bei sorgfältiger Prä-paration mit einem geringen Morbiditätsrisiko verbun-den. Intramedulläre Schwannome sind äußerst selten.Ependymome werden bei NF2-Patienten relativ häufig di-agnostiziert, können aber über Jahrzehnte stabil sein undmüssen dann auch nicht reseziert werden. Man sollte al-lerdings bedenken, dass neurologische Defizite, die durchdiese Tumore entstehen, oft auch nach Entfernung desTumors persistieren können. Bei rascherem Wachstumkann daher die mikrochirurgische Exzision durch einenerfahrenen Neurochirurgen für den Patienten die bessereWahl sein, als so lange zu beobachten, bis ein neurologi-sches Defizit eingetreten ist. Sehr selten sind spinale As-trozytome.
10.4. Periphere NervenSchwannome im Bereich des Plexus brachialis und desPlexus lumbosacralis sind nicht selten, bedürfen einer chi-rurgischen Behandlung nur dann, wenn sie symptoma-tisch werden. Oft ist es aber auch dann nicht möglich, diezuweilen ausgedehnten Tumorpakete zu entfernen. Auchhier steht die Funktionserhaltung weit im Vordergrund,zumal die Regeneration der multilokulär geschädigtenund zusätzlich durch Neuropathien betroffenen Nervenoft schlechter ist als bei Patienten ohne NF2. Eine opera-tive Entfernung subkutaner Tumore ist nur dann sinnvoll,wenn sie den Betroffenen Schmerzen, Missempfindun-gen oder mechanische Irritationen bereiten. Sie gehenmeist von sehr zarten sensiblen, bereits vor der Exzisionafunktionellen Nervenästen aus. Bei kachektischen Patien-ten können sie aber auch einmal überraschend von denNervenscheiden größerer gemischter Arm- oder Beinner-ven ausgehen und diese Nerven aufsplitten und ausdün-nen. Deshalb ist immer der Einsatz eines Operationsmi-kroskops sinnvoll. Tumoren in der Zunge, am Gaumen, an

n Seite 15 nJOURNAL 01/2015
der Wangenschleimhaut und im Bereich der Nasenneben-höhlen sind fast immer Schwannome und müssen nurdann operiert werden, wenn sie störend werden.
11. Andere ManifestationenPeriphere Polyneuropathien stellen eine große Herausfor-derung dar, die medikamentöse Behandlung ist sympto-matisch und nicht immer effektiv.Abgesehen von schwereren Kataraktformen bedürfen diemeisten okulären Manifestationen der NF2 keiner Be-handlung. Regelmäßige augenärztliche Kontrollen sindinsbesondere erforderlich, um Stauungspapillen sowie Vi-sus- und Gesichtsfeldstörungen durch Tumore um dieSehnerven frühzeitig zu erkennen. Unbedingt vermiedenwerden müssen Hornhautulzera durch Benetzungsstörun-gen. Vor allem während Operationen an anderen Lokali-sationen sollte man daher darauf achten, durch Augen-verbände eine feuchte Kammer zu schaffen.
12. ScreeningObwohl die Kriterien für die klinische Erstdiagnose derNF2 inzwischen nahezu lückenlos sind, ist die genetischeSicherung durch Bluttests bei Vorliegen eines genetischenMosaiks nicht immer möglich. In diesen Fällen wird Tumorgewebe benötigt, welchesprinzipiell am leichtesten aus kutanen oder subkutanenTumoren gewonnen werden könnte. Dennoch ist der ersteexzidierte Tumor häufig ein Vestibularisschwannom, weildiese Tumoren am häufigsten eine chirurgische Interven-tion erfordern. Wenn die Mutation identifiziert ist, kannman gefährdete Verwandte ebenfalls auf diese spezifischeMutation untersuchen. Verwandte ersten Grades und sol-che Blutsverwandte, bei denen der Verdacht auf das Vor-liegen einer NF2 besteht, haben wegen des autosomal-dominanten Erbgangs ein signifikantes Risiko (Abb.7).
Abb.7 Genetischer Stammbaum einer NF2-Familie. Bei einem autosomol-dominanten Vererbungsgang besteht bei einem betroffenen Elternteil mitheterozygotem Genotyp eine 50%ige Wahrscheinlichkeit der Weiterverer-bung der Erkrankung, unabhängig vom Geschlecht.
Auf der anderen Seite des Spektrums gibt es Patienten,die noch keine klinischen NF2-Zeichen zeigen und erst
später symptomatisch werden. Das betrifft vor allem Pa-tienten mit genetischen Mosaiken.Wenn ein NF2-Risiko identifiziert wurde, dann werden re-gelmäßige genetische Tests empfohlen, um die Diagnosefrühzeitig zu stellen und das entsprechende Managementeinzuleiten (61).Bei Kindern betroffener Eltern kann das klinische Scree-ning bereits bei Geburt mit der Untersuchung auf Kata-rakt beginnen. Das formale Screening mittels kranialerund spinaler MRT sowie audiologischen Tests sollte im Al-ter von 10-12 Jahren beginnen. Einige Autoren empfehleneinen früheren Beginn der bildgebenden Diagnostik abdem 7.Lebensjahr, nämlich sobald das Kind ohne Sedie-rung im MRT-Scanner still liegen kann (47). Eine Tonau-diometrie ist auch schon bei jüngeren Kindern als Scree-ning-Test möglich, wenn ein NF2-Risiko besteht. Ebensowie eine Untersuchung mittels akustisch evozierter Po-tenziale (AEP) ist dieser Test bei kleinen VS allerdings nichtsehr sensitiv. Jedes Kind, bei dem ein typischerweise NF2-assoziiertesSymptom (z.B. Hörverlust oder Fazialisparese) auftritt,sollte sobald wie möglich einer MRT-Bildgebung zuge-führt werden, ggf. unter Sedierung bzw. Narkose.Bei Patienten unter 20 Jahren sollten jährliche Verlaufs-kontrollen erfolgen. Wenn sich das Tumorwachstum da-nach verlangsamt, können die Kontrollintervalle zur Bild-gebung auf 3 Jahre ausgedehnt werden. Bei sehr stabilen,milden Verläufen ist es in einzelnen Fällen auch möglich,ab dem 40. Lebensjahr die radiologischen Kontrollen zubeenden (61).Wenn bei einem Individuum ohne klinische Zeichen eineNF2-Mutation gesichert wurde, dann sollte eine kranielleMRT jährlich und eine spinale MRT alle 3 Jahre durchge-führt werden. Nach Gareth Evans haben Individuen mit klinischen NF2-Merkmalen ein Risiko von mehr als 20% wenn:
· ein unilaterales Vestibularisschwannom vor dem 20. Lebensjahr,
· ein solitäres intrakranielles Meningeom vor dem 20. Lebensjahr,
· ein solitäres Schwannom an einer anderen Lokali-sation im Kindesalter,
· ein retinales Hamartom in der Kindheit oder · die Manchester-Kriterien vor dem 50. Lebensjahr
erfüllt sind und zusätzlich eine Mononeuropathie in der Kindheit bestehen.
Ein Risiko zwischen 1 und 19% besteht, wenn ein unilate-rales VS im Alter von 20-30 Jahren diagnostiziert wurde.
Ein erhöhtes NF2-Risiko besteht, wenn:• bei einem Verwandten ersten Grades die Diagnose NF2 gestellt wurde
• im Alter von <30 Jahre ein unilaterales Vestibularis-schwannom diagnostiziert wurde
• multiple spinale Tumoren gefunden wurden• kutane bzw. subkutane Schwannome diagnosti-ziert wurden

n Seite 16 n JOURNAL 01/2005JOURNAL 01/2015
Bei allen Risiko-Patienten sollten sich eine kranielle undeine spinale MRT sowie audiologische, ophthalmologi-sche und dermatologische Untersuchungen anschließen.Diese Untersuchungen sollten im Alter von 18 und 20Jahren wiederholt werden. Aufgrund der Daten der Arbeitsgruppe aus Manchesterempfiehlt sich eine erneute kranielle MRT in jedem Fall 5,10 und 20 Jahre nach der initialen Risikoeinstufung. Wennbis dahin keine eindeutigen Hinweise für eine NF2 vorlie-gen, sinkt das Risiko, dass sich die Erkrankung doch nochmanifestiert, unter 1%.
Bei gesicherter Diagnose werden jährlich MRT des Kopfesmit Kontrastmittel und alle 1-3 Jahre MRT der Wirbelsäulebei Patienten mit bekannten spinalen Tumoren angefer-tigt. Liegen keine spinalen Tumore vor, dann reichte einespinale MRT im Abstand von 5 Jahren aus, immer unterder Voraussetzung, dass keine neuen Symptome auftre-ten.
Empfohlene Screening-Intervalle für Kinderbetroffener Eltern
• Augenärztliches Screening jährlich ab Geburt (U1)• Audiologie mit akustisch-evozierten Hirnstamm-potenzialen ab Geburt oder früher Kindheit
• Präsymptomatischer genetischer Bluttest ab 10. Lj.*• Kranielle MRT ab 10.-12. Lj.*• Spinale MRT ab 10.-12. Lj.* (alle 2-3 Jahre)
Andere Individuen mit erhöhtem Risiko sollten ein komplettesScreening (exklusive genetisches Screening) zu dem Zeitpunktbekommen, an dem die Risikofaktoren erkannt wurden.*Früher als 10. Lj. bei schwer betroffenen Familien.
Empfehlung nach Asthagiri et al. 2009 (4) aufgrund vonDaten von Evans et al. 2005 (31)
13. LebenserwartungEvans et al. gaben 1992 in einer Kohortenstudie mit 150Patienten die mittlere Lebenserwartung von NF2-Betrof-fenen mit 62 Jahren an (35). Das war schon damals eineerstaunliche Zahl, weil mehr als 40% der Patienten in die-ser Kohorte vor Erreichen des 50. Lebensjahres bereitsverstorben waren. Es ist zu vermuten, dass sich die Le-benserwartung aufgrund der verbesserten diagnosti-schen und therapeutischen Möglichkeiten in den letztenJahren erhöht hat.
14. FazitSchweregrad (Phänotyp) und klinischer Verlauf der Neu-rofibromatose Typ 2 sind tatsächlich zu dem genetischenMuster der zugrundeliegenden Mutationen korreliert,auch wenn die Ausprägung bei einzelnen Familienmit-gliedern erheblich variieren kann und das Wachstumsver-halten individueller Tumoren sehr heterogen ist. Eine frü-he Diagnosestellung, kontinuierliche Verlaufskontrollenund die Anbindung der Patienten an spezialisierte, multi-
disziplinäre Zentren verbessern die Behandlungsergebnis-se. Wesentliche Prinzipien des Managements sind die Er-haltung und Wiederherstellung von neuronalen Funktio-nen, ein Kardinalfehler kann die Behandlung von asymp-tomatischen, stabilen Tumoren sein.
Literatur
Reference list1. Neurofibromatosis. Conference statement. National Institutes of
Health Consensus Development Conference. Arch Neurol 45:575-578, 1988.
2. Anderson BM, Khuntia D, Bentzen SM, Geye HM, Hayes LL, Kuo JS, Baskaya MK, Badie B, Basavatia A, Pyle GM, Tome WA, Mehta MP: Single institution experience treating 104 vestibular schwannomas with fractionated stereotactic radiation therapy or stereotactic ra-diosurgery. J Neurooncol 116:187-193, 2014.
3. Antinheimo J, Sankila R, Carpen O, Pukkala E, Sainio M, Jaaske-lainen J: Population-based analysis of sporadic and type 2 neurofi-bromatosis-associated meningiomas and schwannomas. Neurolo-gy 54:71-76, 2000.
4. Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, Lonser RR: Neurofibromatosis type 2. Lancet 373:1974-1986, 2009.
5. Bari ME, Forster DM, Kemeny AA, Walton L, Hardy D, Anderson JR: Malignancy in a vestibular schwannoma. Report of a case with cen-tral neurofibromatosis, treated by both stereotactic radiosurgery and surgical excision, with a review of the literature. Br J Neurosurg16:284-289, 2002.
6. Baser ME, Evans DG, Jackler RK, Sujansky E, Rubenstein A: Neurofi-bromatosis 2, radiosurgery and malignant nervous system tumours. Br J Cancer 82:998, 2000.
7. Baser ME, Friedman JM, Joe H, Shenton A, Wallace AJ, Ramsden RT, Evans DG: Empirical development of improved diagnostic criteria or neurofibromatosis 2. Genet Med 13:576-581, 2011.
8. Baser ME, Friedman JM, Wallace AJ, Ramsden RT, Joe H, Evans DG: Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology 59:1759-1765, 2002.
9. Baser ME, Kuramoto L, Joe H, Friedman JM, Wallace AJ, Gillespie JE, Ramsden RT, Evans DG: Genotype-phenotype correlations for ner-vous system tumors in neurofibromatosis 2: a population-based study. Am J Hum Genet 75:231-239, 2004.
10. Baser ME, Kuramoto L, Woods R, Joe H, Friedman JM, Wallace AJ, Ramsden RT, Olschwang S, Bijlsma E, Kalamarides M, Papi L, Kato R, Carroll J, Lazaro C, Joncourt F, Parry DM, Rouleau GA, Evans DG: The location of constitutional neurofibromatosis 2 (NF2) splice site mu-tations is associated with the severity of NF2. J Med Genet 42:540-546, 2005.
11. Baser ME, Makariou EV, Parry DM: Predictors of vestibular schwan-noma growth in patients with neurofibromatosis Type 2. J Neuro-surg 96:217-222, 2002.
12. Baser ME, Mautner VF, Parry DM, Evans DG: Methodological issues in longitudinal studies: vestibular schwannoma growth rates in neu-rofibromatosis 2. J Med Genet 42:903-906, 2005.
13. Behr R, Colletti V, Matthies C, Morita A, Nakatomi H, Dominique L, Darrouzet V, Brill S, Shehata-Dieler W, Lorens A, Skarzynski H: New outcomes with auditory brainstem implants in NF2 patients. Otol Neurotol 35:1844-1851, 2014.
14. Bianchi AB, Hara T, Ramesh V, Gao J, Klein-Szanto AJ, Morin F, Menon AG, Trofatter JA, Gusella JF, Seizinger BR, .: Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nat Genet 6:185-192, 1994.
15. Blakeley JO, Evans DG, Adler J, Brackmann D, Chen R, Ferner RE, Hanemann CO, Harris G, Huson SM, Jacob A, Kalamarides M, Kara-jannis MA, Korf BR, Mautner VF, McClatchey AI, Miao H, Plotkin SR, Slattery W, III, Stemmer-Rachamimov AO, Welling DB, Wen PY, Wide-mann B, Hunter-Schaedle K, Giovannini M: Consensus recommen-dations for current treatments and accelerating clinical trials for pa-tients with neurofibromatosis type 2. Am J Med Genet A 158A:24-41, 2012.
16. Bosch MM, Boltshauser E, Harpes P, Landau K: Ophthalmologic fin-

n Seite 17 nJOURNAL 01/2015
dings and long-term course in patients with neurofibromatosis type 2. Am J Ophthalmol 141:1068-1077, 2006.
17. Brackmann DE, Fayad JN, Slattery WH, III, Friedman RA, Day JD, Hit-selberger WE, Owens RM: Early proactive management of vestibular schwannomas in neurofibromatosis type 2. Neurosurgery 49:274-280, 2001.
18. Breivik CN, Nilsen RM, Myrseth E, Finnkirk MK, Lund-Johansen M: Working disability in Norwegian patients with vestibular schwanno-ma: vertigo predicts future dependence. World Neurosurg 80:e301-e305, 2013.
19. Breivik CN, Varughese JK, Wentzel-Larsen T, Vassbotn F, Lund-Johansen M: Conservative management of vestibular schwanno-ma—a prospective cohort study: treatment, symptoms, and quality of life. Neurosurgery 70:1072-1080, 2012.
20. Carlson ML, Babovic-Vuksanovic D, Messiaen L, Scheithauer BW, Neff BA, Link MJ: Radiation-induced rhabdomyosarcoma of the brainstem in a patient with neurofibromatosis type 2. J Neurosurg112:81-87, 2010.
21. Carlson ML, Tveiten OV, Driscoll CL, Goplen FK, Neff BA, Pollock BE, Tombers NM, Castner ML, Finnkirk MK, Myrseth E, Pedersen PH, Lund-Johansen M, Link MJ: Long-term quality of life in patients with vestibular schwannoma: an international multicenter cross-sectional study comparing microsurgery, stereotactic radiosurgery, observa-tion, and nontumor controls. J Neurosurg 122:833-842, 2015.
22. Colletti L, Shannon R, Colletti V: Auditory brainstem implants for neurofibromatosis type 2. Curr Opin Otolaryngol Head Neck Surg20:353-357, 2012.
23. Colletti V, Shannon R, Carner M, Sacchetto L, Turazzi S, Masotto B, Colletti L: The first successful case of hearing produced by electrical stimulation of the human midbrain. Otol Neurotol 28:39-43, 2007.
24. Comey CH, McLaughlin MR, Jho HD, Martinez AJ, Lunsford LD: Death from a malignant cerebellopontine angle triton tumor despite stereotactic radiosurgery. Case report. J Neurosurg 89:653-658, 1998.
25. Crane BT, Gottschalk B, Kraut M, Aygun N, Niparko JK: Magnetic resonance imaging at 1.5 T after cochlear implantation. Otol Neu-rotol 31:1215-1220, 2010.
26. Demetriades AK, Saunders N, Rose P, Fisher C, Rowe J, Tranter R, Hardwidge C: Malignant transformation of acoustic neuroma/vestibular schwannoma 10 years after gamma knife stereotactic radiosurgery. Skull Base 20:381-387, 2010.
27. Deneuve S, Loundon N, Leboulanger N, Rouillon I, Garabedian EN: Cochlear implant magnet displacement during magnetic resonance imaging. Otol Neurotol 29:789-190, 2008.
28. Dow G, Biggs N, Evans G, Gillespie J, Ramsden R, King A: Spinal tu-mors in neurofibromatosis type 2. Is emerging knowledge of geno-type predictive of natural history? J Neurosurg Spine 2:574-579, 2005.
29. Dumanski JP, Carlbom E, Collins VP, Nordenskjold M: Deletion map-ping of a locus on human chromosome 22 involved in the oncoge-nesis of meningioma. Proc Natl Acad Sci U S A 84:9275-9279, 1987.
30. Evans DG: Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis 4:16, 2009.
31. Evans DG, Baser ME, O’Reilly B, Rowe J, Gleeson M, Saeed S, King A, Huson SM, Kerr R, Thomas N, Irving R, Macfarlane R, Ferner R, McLeod R, Moffat D, Ramsden R: Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg 19:5-12, 2005.
32. Evans DG, Bowers N, Huson SM, Wallace A: Mutation type and po-sition varies between mosaic and inherited NF2 and correlates with disease severity. Clin Genet 83:594-595, 2013.
33. Evans DG, Freeman S, Gokhale C, Wallace A, Lloyd SK, Axon P, Ward CL, Rutherford S, King A, Huson SM, Ramsden RT: Bilateral vestibular schwannomas in older patients: NF2 or chance? J Med Genet52:422-424, 2015.
34. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F: Birth incidence and prevalence of tumor-prone syndromes: esti-mates from a UK family genetic register service. Am J Med Genet A152A:327-332, 2010.
35. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Harris R: A clinical study of type 2 neurofibromatosis. Q J Med 84:603-618, 1992.
36. Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, Strachan T, Harris R: A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. J Med Genet29:847-852, 1992.
37. Evans DG, Ingham SL: Reduced life expectancy seen in hereditary diseases which predispose to early-onset tumors. Appl Clin Genet6:53-61, 2013.
38. Evans DG, Trueman L, Wallace A, Collins S, Strachan T: Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): evidence for more severe disease associated with truncating muta-tions. J Med Genet 35:450-455, 1998.
39. Farschtschi S, Kollmann P, Dalchow C, Stein A, Mautner VF: Reduced dosage of bevacizumab in treatment of vestibular schwannomas in patients with neurofibromatosis type 2. Eur Arch Otorhinolaryngol2015.
40. Ferner RE, Shaw A, Evans DG, McAleer D, Halliday D, Parry A, Ray-mond FL, Durie-Gair J, Hanemann CO, Hornigold R, Axon P, Golding JF: Longitudinal evaluation of quality of life in 288 patients with neurofibromatosis 2. J Neurol 261:963-969, 2014.
41. Fisher LM, Doherty JK, Lev MH, Slattery WH, III: Distribution of non-vestibular cranial nerve schwannomas in neurofibromatosis 2. Otol Neurotol 28:1083-1090, 2007.
42. Fisher LM, Doherty JK, Lev MH, Slattery WH: Concordance of bilateral vestibular schwannoma growth and hearing changes in neurofibro-matosis 2: neurofibromatosis 2 natural history consortium. Otol Neurotol 30:835-841, 2009.
43. Grayeli AB, Kalamarides M, Bouccara D, Ambert-Dahan E, Sterkers O: Auditory brainstem implant in neurofibromatosis type 2 and non-neurofibromatosis type 2 patients. Otol Neurotol 29:1140-1146, 2008.
44. Gubbels SP, McMenomey SO: Safety study of the Cochlear Nucleus 24 device with internal magnet in the 1.5 Tesla magnetic resonance imaging scanner. Laryngoscope 116:865-871, 2006.
45. Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D: The diagnostic evaluation and multidis-ciplinary management of neurofibromatosis 1 and neurofibromato-sis 2. JAMA 278:51-57, 1997.
46. Heiska L, Alfthan K, Gronholm M, Vilja P, Vaheri A, Carpen O: Asso-ciation of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5-bisphos-phate. J Biol Chem 273:21893-21900, 1998.
47. Hoa M, Slattery WH, III: Neurofibromatosis 2. Otolaryngol Clin North Am 45:315-32, viii, 2012.
48. Jaaskelainen J, Paetau A, Pyykko I, Blomstedt G, Palva T, Troupp H: Interface between the facial nerve and large acoustic neurinomas. Immunohistochemical study of the cleavage plane in NF2 and non-NF2 cases. J Neurosurg 80:541-547, 1994.
49. Jin H, Sperka T, Herrlich P, Morrison H: Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature442:576-579, 2006.
50. Kida Y, Kobayashi T, Tanaka T, Mori Y: Radiosurgery for bilateral neurinomas associated with neurofibromatosis type 2. Surg Neurol53:383-389, 2000.
51. Kissil JL, Johnson KC, Eckman MS, Jacks T: Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem 277:10394-10399, 2002.
52. Kluwe L, Bayer S, Baser ME, Hazim W, Haase W, Funsterer C, Mautner VF: Identification of NF2 germ-line mutations and comparison with neurofibromatosis 2 phenotypes. Hum Genet 98:534-538, 1996.
53. Kluwe L, MacCollin M, Tatagiba M, Thomas S, Hazim W, Haase W, Mautner VF: Phenotypic variability associated with 14 splice-site mutations in the NF2 gene. Am J Med Genet 77:228-233, 1998.
54. Knudson AG, Jr.: Mutation and cancer: statistical study of retino-blastoma. Proc Natl Acad Sci U S A 68:820-823, 1971.
55. Kondziolka D, Levy EI, Niranjan A, Flickinger JC, Lunsford LD: Long-term outcomes after meningioma radiosurgery: physician and pa-tient perspectives. J Neurosurg 91:44-50, 1999.
56. Lawson McLean A, Rosahl SK. Growth dynamics of intracranial tu-mors in patients with neurofibromatosis type 2. J.Neurosurg. sub-mittted. 2015.
Ref Type: Journal (Full)57. Lenarz T, Lim HH, Reuter G, Patrick JF, Lenarz M: The auditory mid-

n Seite 18 n JOURNAL 01/2005JOURNAL 01/2015
brain implant: a new auditory prosthesis for neural deafness-con-cept and device description. Otol Neurotol 27:838-843, 2006.
58. Lim HH, Lenarz M, Lenarz T: Auditory midbrain implant: a review. Trends Amplif 13:149-180, 2009.
59. Lim HH, Lenarz T: Auditory midbrain implant: research and develop-ment towards a second clinical trial. Hear Res 322:212-223, 2015.
60. Linskey ME, Lunsford LD, Flickinger JC: Tumor control after stereo-tactic radiosurgery in neurofibromatosis patients with bilateral acoustic tumors. Neurosurgery 31:829-838, 1992.
61. Lloyd SK, Evans DG: Neurofibromatosis type 2 (NF2): diagnosis and management. Handb Clin Neurol 115:957-967, 2013.
62. Lustig LR, Yeagle J, Driscoll CL, Blevins N, Francis H, Niparko JK: Cochlear implantation in patients with neurofibromatosis type 2 and bilateral vestibular schwannoma. Otol Neurotol 27:512-518, 2006.
63. MacCollin M, Mautner VF: The diagnosis and management of neu-rofibromatosis 2 in childhood. Semin Pediatr Neurol 5:243-252, 1998.
64. Maducdoc MM, Ghavami Y, Linskey ME, Djalilian HR: Evaluation of Reported Malignant Transformation of Vestibular Schwannoma: De Novo and After Stereotactic Radiosurgery or Surgery. Otol Neurotol36:1301-1308, 2015.
65. Majdani O, Leinung M, Rau T, Akbarian A, Zimmerling M, Lenarz M, Lenarz T, Labadie R: Demagnetization of cochlear implants and tem-perature changes in 3.0T MRI environment. Otolaryngol Head Neck Surg 139:833-839, 2008.
66. Majdani O, Rau TS, Gotz F, Zimmerling M, Lenarz M, Lenarz T, Labadie R, Leinung M: Artifacts caused by cochlear implants with non-removable magnets in 3T MRI: phantom and cadaveric studies. Eur Arch Otorhinolaryngol 266:1885-1890, 2009.
67. Mathieu D, Kondziolka D, Flickinger JC, Niranjan A, Williamson R, Martin JJ, Lunsford LD: Stereotactic radiosurgery for vestibular schwannomas in patients with neurofibromatosis type 2: an analysis of tumor control, complications, and hearing preservation rates. Neurosurgery 60:460-468, 2007.
68. Matthies C, Brill S, Kaga K, Morita A, Kumakawa K, Skarzynski H, Claassen A, Hui Y, Chiong C, Muller J, Behr R: Auditory brainstem im-plantation improves speech recognition in neurofibromatosis type II patients. ORL J Otorhinolaryngol Relat Spec 75:282-295, 2013.
69. Matthies C, Brill S, Varallyay C, Solymosi L, Gelbrich G, Roosen K, Ernestus RI, Helms J, Hagen R, Mlynski R, Shehata-Dieler W, Muller J: Auditory brainstem implants in neurofibromatosis Type 2: is open speech perception feasible? J Neurosurg 120:546-558, 2014.
70. Mautner VF, Lindenau M, Baser ME, Hazim W, Tatagiba M, Haase W, Samii M, Wais R, Pulst SM: The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery 38:880-885, 1996.
71. Mautner VF, Lindenau M, Baser ME, Kluwe L, Gottschalk J: Skin ab-normalities in neurofibromatosis 2. Arch Dermatol 133:1539-1543, 1997.
72. Mautner VF, Nguyen R, Knecht R, Bokemeyer C: Radiographic re-gression of vestibular schwannomas induced by bevacizumab treat-ment: sustain under continuous drug application and rebound after drug discontinuation. Ann Oncol 21:2294-2295, 2010.
73. Mautner VF, Nguyen R, Kutta H, Fuensterer C, Bokemeyer C, Hagel C, Friedrich RE, Panse J: Bevacizumab induces regression of vestibu-lar schwannomas in patients with neurofibromatosis type 2. Neuro Oncol 12:14-18, 2010.
74. Mautner VF, Tatagiba M, Lindenau M, Funsterer C, Pulst SM, Baser ME, Kluwe L, Zanella FE: Spinal tumors in patients with neurofibro-matosis type 2: MR imaging study of frequency, multiplicity, and va-riety. AJR Am J Roentgenol 165:951-955, 1995.
75. Meijer OW, Vandertop WP, Lagerwaard FJ, Slotman BJ: Linear accel-erator-based stereotactic radiosurgery for bilateral vestibular schwannomas in patients with neurofibromatosis type 2. Neuro-surgery 62:A37-A42, 2008.
76. Myrseth E, Moller P, Wentzel-Larsen T, Goplen F, Lund-Johansen M: Untreated vestibular schwannomas: vertigo is a powerful predictor for health-related quality of life. Neurosurgery 59:67-76, 2006.
77. Neff BA, Wiet RM, Lasak JM, Cohen NL, Pillsbury HC, Ramsden RT, Welling DB: Cochlear implantation in the neurofibromatosis type 2 patient: long-term follow-up. Laryngoscope 117:1069-1072, 2007.
78. Nevison B, Laszig R, Sollmann WP, Lenarz T, Sterkers O, Ramsden R, Fraysse B, Manrique M, Rask-Andersen H, Garcia-Ibanez E, Colletti
V, von WE: Results from a European clinical investigation of the Nu-cleus multichannel auditory brainstem implant. Ear Hear 23:170-183, 2002.
79. Noren G: Long-term complications following gamma knife radio-surgery of vestibular schwannomas. Stereotact Funct Neurosurg70 Suppl 1:65-73, 1998.
80. Otsuka G, Saito K, Nagatani T, Yoshida J: Age at symptom onset and long-term survival in patients with neurofibromatosis Type 2. J Neu-rosurg 99:480-483, 2003.
81. Parry DM: NF2 and spinal tumors. J Neurosurg Spine 5:179, 2006.82. Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas
N: Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet52:450-461, 1994.
83. Parry DM, MacCollin MM, Kaiser-Kupfer MI, Pulaski K, Nicholson HS, Bolesta M, Eldridge R, Gusella JF: Germ-line mutations in the neu-rofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet 59:529-539, 1996.
84. Patel TR, Chiang VL: Secondary neoplasms after stereotactic radio-surgery. World Neurosurg 81:594-599, 2014.
85. Patronas NJ, Courcoutsakis N, Bromley CM, Katzman GL, MacCollin M, Parry DM: Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology 218:434-442, 2001.
86. Phi JH, Kim DG, Chung HT, Lee J, Paek SH, Jung HW: Radiosurgical -treatment of vestibular schwannomas in patients with neurofibro-matosis type 2: tumor control and hearing preservation. Cancer115:390-398, 2009.
87. Plotkin SR, Ardern-Holmes SL, Barker FG, Blakeley JO, Evans DG, Ferner RE, Hadlock TA, Halpin C: Hearing and facial function out-comes for neurofibromatosis 2 clinical trials. Neurology 81:S25-S32, 2013.
88. Plotkin SR, Halpin C, McKenna MJ, Loeffler JS, Batchelor TT, Barker FG: Erlotinib for progressive vestibular schwannoma in neurofibro-matosis 2 patients. Otol Neurotol 31:1135-1143, 2010.
89. Plotkin SR, Merker VL, Halpin C, Jennings D, McKenna MJ, Harris GJ, Barker FG: Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: a retrospective review of 31 patients. Otol Neurotol 33:1046-1052, 2012.
90. Plotkin SR, Merker VL, Muzikansky A, Barker FG, Slattery W, III: Na-tural history of vestibular schwannoma growth and hearing decline in newly diagnosed neurofibromatosis type 2 patients. Otol Neuro-tol 35:e50-e56, 2014.
91. Plotkin SR, Singh MA, O’Donnell CC, Harris GJ, McClatchey AI, Halpin C: Audiologic and radiographic response of NF2-related vestibular schwannoma to erlotinib therapy. Nat Clin Pract Oncol 5:487-491, 2008.
92. Plotkin SR, Stemmer-Rachamimov AO, Barker FG, Halpin C, Padera TP, Tyrrell A, Sorensen AG, Jain RK, di TE: Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med 361:358-367, 2009.
93. Pollock BE, Lunsford LD, Kondziolka D, Sekula R, Subach BR, Foote RL, Flickinger JC: Vestibular schwannoma management. Part II. Failed radiosurgery and the role of delayed microsurgery. J Neuro-surg 89:949-955, 1998.
94. Pykett MJ, Murphy M, Harnish PR, George DL: The neurofibromato-sis 2 (NF2) tumor suppressor gene encodes multiple alternatively spliced transcripts. Hum Mol Genet 3:559-564, 1994.
95. Ragge NK, Baser ME, Klein J, Nechiporuk A, Sainz J, Pulst SM, Riccardi VM: Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol120:634-641, 1995.
96. Ragge NK, Baser ME, Riccardi VM, Falk RE: The ocular presentation of neurofibromatosis 2. Eye (Lond) 11 ( Pt 1):12-18, 1997.
97. Rasmussen R, Claesson M, Stangerup SE, Roed H, Christensen IJ, Caye-Thomasen P, Juhler M: Fractionated stereotactic radiotherapy of vestibular schwannomas accelerates hearing loss. Int J Radiat Oncol Biol Phys 83:e607-e611, 2012.
98. Rennie AT, Side L, Kerr RS, Anslow P, Pretorius P: Intramedullary tu-mours in patients with neurofibromatosis type 2: MRI features as-sociated with a favourable prognosis. Clin Radiol 63:193-200, 2008.
99. Rong R, Surace EI, Haipek CA, Gutmann DH, Ye K: Serine 518 phos-phorylation modulates merlin intramolecular association and bin-ding to critical effectors important for NF2 growth suppression. On-

cogene 23:8447-8454, 2004.100. Rosahl SK, Lenarz T, Matthies C, Samii M, Sollmann WP, Laszig R:
Hirnstammimplantate zur Wiederherstellung des Hörvermögens. Dt Aerzteblatt 101:180-188, 2004.
101. Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, .: Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363:515-521, 1993.
102. Rowe J, Grainger A, Walton L, Radatz M, Kemeny A: Safety of radio-surgery applied to conditions with abnormal tumor suppressor genes. Neurosurgery 60:860-864, 2007.
103. Rowe J, Grainger A, Walton L, Radatz M, Kemeny A: Safety of radio-surgery applied to conditions with abnormal tumor suppressor genes. Neurosurgery 60:860-864, 2007.
104. Rowe J, Radatz M, Kemeny A: Radiosurgery for type II neurofibro-matosis. Prog Neurol Surg 21:176-182, 2008.
105. Rowe J, Radatz M, Kemeny A: Radiosurgery for type II neurofibro-matosis. Prog Neurol Surg 21:176-182, 2008.
106. Ruttledge MH, Andermann AA, Phelan CM, Claudio JO, Han FY, Chretien N, Rangaratnam S, MacCollin M, Short P, Parry D, Michels V, Riccardi VM, Weksberg R, Kitamura K, Bradburn JM, Hall BD, Propping P, Rouleau GA: Type of mutation in the neurofibromatosis type 2 gene (NF2) frequently determines severity of disease. Am J Hum Genet 59:331-342, 1996.
107. Ruttledge MH, Rouleau GA: Role of the neurofibromatosis type 2 gene in the development of tumors of the nervous system. Neuro-surg Focus 19:E6, 2005.
108. Samii M, Gerganov V, Samii A: Microsurgery management of vestibular schwannomas in neurofibromatosis type 2: indications and results. Prog Neurol Surg 21:169-175, 2008.
109. Samii M, Matthies C, Tatagiba M: Management of vestibular schwannomas (acoustic neuromas): auditory and facial nerve func-tion after resection of 120 vestibular schwannomas in patients with neurofibromatosis 2. Neurosurgery 40:696-705, 1997.
110. Scherer SS, Gutmann DH: Expression of the neurofibromatosis 2 tumor suppressor gene product, merlin, in Schwann cells. J Neu-rosci Res 46:595-605, 1996.
111. Seizinger BR, Martuza RL, Gusella JF: Loss of genes on chromosome 22 in tumorigenesis of human acoustic neuroma. Nature 322:644-647, 1986.
112. Seizinger BR, Rouleau G, Ozelius LJ, Lane AH, St George-Hyslop P, Huson S, Gusella JF, Martuza RL: Common pathogenetic mechanism for three tumor types in bilateral acoustic neurofibromatosis. Science 236:317-319, 1987.
113. Selvanathan SK, Shenton A, Ferner R, Wallace AJ, Huson SM, Rams-den RT, Evans DG: Further genotype—phenotype correlations in neurofibromatosis 2. Clin Genet 77:163-170, 2010.
114. Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, O’Bryan JP, Gupta V, Ratner N, Der CJ, Jacks T, McClatchey AI: The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell1:63-72, 2001.
115. Sherman L, Xu HM, Geist RT, Saporito-Irwin S, Howells N, Ponta H, Herrlich P, Gutmann DH: Interdomain binding mediates tumor growth suppression by the NF2 gene product. Oncogene 15:2505-2509, 1997.
116. Shuto T, Inomori S, Matsunaga S, Fujino H: Microsurgery for vestibu-lar schwannoma after gamma knife radiosurgery. Acta Neurochir (Wien ) 150:229-234, 2008.
117. Slattery WH, III, Fisher LM, Hitselberger W, Friedman RA, Brackmann DE: Hearing preservation surgery for neurofibromatosis Type 2-re-lated vestibular schwannoma in pediatric patients. J Neurosurg106:255-260, 2007.
118. Slusarz KM, Merker VL, Muzikansky A, Francis SA, Plotkin SR: Long--term toxicity of bevacizumab therapy in neurofibromatosis 2 pa-tients. Cancer Chemother Pharmacol 73:1197-1204, 2014.
119. Smith MJ, Higgs JE, Bowers NL, Halliday D, Paterson J, Gillespie J, Huson SM, Freeman SR, Lloyd S, Rutherford SA, King AT, Wallace AJ, Ramsden RT, Evans DG: Cranial meningiomas in 411 neurofibro-matosis type 2 (NF2) patients with proven gene mutations: clear po-sitional effect of mutations, but absence of female severity effect on age at onset. J Med Genet 48:261-265, 2011.
120. Sperfeld AD, Hein C, Schroder JM, Ludolph AC, Hanemann CO: Oc-currence and characterization of peripheral nerve involvement in
neurofibromatosis type 2. Brain 125:996-1004, 2002.121. Subach BR, Kondziolka D, Lunsford LD, Bissonette DJ, Flickinger JC,
Maitz AH: Stereotactic radiosurgery in the management of acoustic neuromas associated with neurofibromatosis Type 2. J Neurosurg119 Suppl:815-822, 2013.
122. Thomsen J, Mirz F, Wetke R, Astrup J, Bojsen-Moller M, Nielsen E: In-tracranial sarcoma in a patient with neurofibromatosis type 2 treated with gamma knife radiosurgery for vestibular schwannoma. Am J Otol 21:364-370, 2000.
123. Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K, .: A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 72:791-800, 1993.
124. Trotter MI, Briggs RJ: Cochlear implantation in neurofibromatosis type 2 after radiation therapy. Otol Neurotol 31:216-219, 2010.
125. Vachhani JA, Friedman WA: Radiosurgery in patients with bilateral vestibular schwannomas. Stereotact Funct Neurosurg 85:273-278, 2007.
126. Wackym PA, Michel MA, Prost RW, Banks KL, Runge-Samuelson CL, Firszt JB: Effect of magnetic resonance imaging on internal magnet strength in Med-El Combi 40+ cochlear implants. Laryngoscope114:1355-1361, 2004.
127. Wentworth S, Pinn M, Bourland JD, Deguzman AF, Ekstrand K, Ellis TL, Glazier SS, McMullen KP, Munley M, Stieber VW, Tatter SB, Shaw EG: Clinical experience with radiation therapy in the management of neurofibromatosis-associated central nervous system tumors. Int J Radiat Oncol Biol Phys 73:208-213, 2009.
128. Wishart JH: Case of tumours in the skull, dura mater, and the brain. Edinburgh Med Surg J 18:393-397, 1822.
129. Yeung AH, Sughrue ME, Kane AJ, Tihan T, Cheung SW, Parsa AT: Ra-diobiology of vestibular schwannomas: mechanisms of radioresis-tance and potential targets for therapeutic sensitization. Neurosurg Focus 27:E2, 2009.
130. Zhao Y, Kumar RA, Baser ME, Evans DG, Wallace A, Kluwe L, Mautner VF, Parry DM, Rouleau GA, Joe H, Friedman JM: Intrafamilial corre-lation of clinical manifestations in neurofibromatosis 2 (NF2). Genet Epidemiol 23:245-259, 2002.
131. Zoch A, Mayerl S, Schulz A, Greither T, Frappart L, Rubsam J, Heuer H, Giovannini M, Morrison H: Merlin Isoforms 1 and 2 Both Act as Tumour Suppressors and Are Required for Optimal Sperm Matura-tion. PLoS One 10:e0129151, 2015.
132. Ammoun, S., Flaiz, C., Ristic, N., Schuldt, J., & Hanemann, C. O. (2008). Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Research, 68(13), 5236–45. doi:10.1158/0008-5472.CAN-07-5849
133. Zoch, A., Mayerl, S., Schulz, A., Greither, T., Frappart, L., Rübsam, J., … Morrison, H. (2015). Merlin Isoforms 1 and 2 Both Act as Tumour Suppressors and Are Required for Optimal Sperm Maturation. PloS One, 10(8), e0129151. doi:10.1371/journal.pone.0129151
Korrespondenzadresse:
Prof. Dr. med. Steffen RosahlKlinik für NeurochirurgieHELIOS Klinikum ErfurtNordhäuser Str. 7499089 ErfurtTelefon: [email protected]
n Seite 19 nJOURNAL 01/2015

n Seite 20 n JOURNAL 01/2005JOURNAL 01/2015
n Primäre Chemohormontherapie deshormonnaiven metastasiertenProstatakarzinoms
Thomas SteinerKlinik für Urologie, HELIOS ProstatakarzinomzentrumErfurt, HELIOS Klinikum Erfurt
Seit vielen Jahren gilt die Einleitung einer Androgende-privation (ADT) als Standardtherapie beim metastasiertenhormonsensitiven Prostatakarzinom. Diese kann grund-sätzlich als komplette Androgendeprivation (LHRH [Luteinisierungshormon-Releasinghormon]-Analogonbzw. LHRH-Antagonist + Antiandrogen) als auch als Mo-notherapie (LHRH-Analogon bzw. LHRH-Antagonist) er-folgen. Beim alleinigen Einsatz eines LHRH-Analogons istzur Vermeidung eines flare-up Phänomens primär über10-14 Tage ein Antiandrogen einzusetzen.
Die Etablierung der Chemotherapie mit Docetaxel beimkastrationsrefraktären Prostatakarzinom markierte vor 10Jahren einen wichtigen Schritt zur Überlebensverlänge-rung nach Versagen einer hormonablativen Therapie. Inder Primärtherapie konnte sich die Chemotherapie bisherjedoch nicht etablieren, da für die Hinzunahme der Che-motherapie zur Androgendeprivation bis 2014 kein Über-lebensvorteil gegenüber der alleinigen Hormontherapiegezeigt werden konnte. So demonstrierte die GETUG-AFU-15-Studie, in der erstmalig im Rahmen einer pro-spektiven Phase III Studie Docetaxel plus hormonablativeTherapie vs. hormonablative Therapie allein untersuchtwurde, lediglich einen Vorteil beim progressionsfreienÜberleben, jedoch nicht beim Gesamtüberleben. Auch inder aktualisierten Analyse der Überlebensdaten erreichtedie Verlängerung des Gesamtüberlebens zugunsten derChemohormontherapie in dieser Studie keine statistischeSignifikanz (Tab. 1).
Neue Erkenntnisse der Jahre 2014/2015
Auf dem Kongress der „American Society of Clinical On-cology“ (ASCO) 2014 wurden erstmals die Ergebnisse der„Chaarted-Studie“ (ChemoHormonal Therapy versus An-drogen Ablation Randomized Trial for Extensive Diseasein Prostate Cancer) vorgestellt, die aktuell als Vollpubli-kation im New England Journal of Medicine erschienensind. In dieser Studie wurde die Androgendeprivation ran-domisiert gegenüber einer Kombination aus Androgen-deprivation plus maximal 6 Zyklen Chemotherapie mitDocetaxel (75 mg/m2, 3-wöchentlich) verglichen. Dabeiwurden zunächst nur Patienten mit einer hohen Tumorlasteingeschlossen – nach einem Amendment konnten späterauch Patienten mit niedriger Tumorlast in die Studie auf-genommen werden. Analog der auf dem ASCO 2015 erst-mals präsentierten STAMPEDE-Studie war der Großteil derPatienten primär metastasiert, nur 27% der Patienten hat-ten in der Vorgeschichte eine Lokaltherapie der Prostataerhalten.
Beide Studien demonstrieren einen statistisch signifikan-ten Vorteil im Hinblick auf das progressionsfreie Überle-ben und Gesamtüberleben zugunsten der kombiniertenChemohormontherapie (Tab. 1). In der Chaarted Studiefindet sich ein Überlebensvorteil von 17 Monaten für Pa-tienten mit einer hohen Tumorlast, die außer einer visze-ralen Metastasierung auch durch das Vorhandensein von>3 ossären Metastasen inklusive einer Metastasen außer-halb des knöchernen Beckens/Wirbelsäule definiert wur-de. In der Subgruppe der Patienten mit niedriger Tumor-last wurde das mediane Überleben in beiden Therapiear-men noch nicht erreicht. Die STAMPEDE-Studie ist eine Multiarmstudie, in der ver-schiedene Therapieregime gegenüber der Androgende-privation als Standardtherapie verglichen werden. DieTherapie in diesem Kontrollarm bestand aus einer LHRH-Therapie/Orchiektomie ± Addition eines Antiandrogen.Eingeschlossen wurden sowohl Patienten mit einem lokalfortgeschrittenen Prostatakarzinom [cT3/4, prostataspe-zifisches Antigen (PSA) ≥40 ng/ml oder Gleason-Score 8–10], Patienten im Progress nach primärer Lokaltherapie(PSA ≥4 ng/ml + PSADT <6 Monate, PSA ≥20 ng/ml, N+,M+) oder primär metastasierte Prostatakarzinompatien-ten (cTx, N+/M+). In einer ersten Auswertung des Kon-trollarms lag das mediane Überleben dieser Patientennach einem mittleren Follow-up von 20 Monaten bei 42,1Monaten. Als Prognosefaktoren für das Überleben konn-ten der Allgemeinzustand, das Alter, der Gleason-Scoreund die Metastasenlokalisation identifiziert werden. ImVergleich einer ADT+Docetaxel vs. ADT zeigte sich einVorteil im Gesamtüberleben zugunsten der Chemohor-montherapie von 10 Monaten [77 vs. 67 Monate, HazardRatio (HR) 0,76 (0,63–0,91), p=0,003]. In der Subgrup-penanalyse zeigt sich, dass Patienten ohne Fernmetasta-sen (M0) scheinbar nicht durch die Chemohormonthera-pie profitieren. In der Subgruppe der metastasierten Pa-tienten (61% der Patienten) lag der Überlebensvorteil hin-gegen bei 22 Monaten [65 vs. 43 Monate, HR=0,73 (0,59,0,89), p=0,002]. Eine Unterscheidung zwischen Patientenmit niedriger bzw. hoher Metastasenlast bzw. mit/ohnevorherige Lokaltherapie erfolgte in der STAMPEDE Studienicht. Inzwischen wurden die Daten aller drei Studien (Chaarted,GETUG-AFU 15 und STAMPEDE) im Sinne einer Metaana-lyse gepoolt. Die gepoolte Analyse demonstriert eine re-lative Risikoreduktion im Gesamtüberleben von 26% beieiner HR von 0,74. Aufgrund von Unterschieden im Pa-tientenkollektiv (Anzahl eingeschlossener Patienten, me-dianer PSA-Wert zur Baseline, Tumorlast der eingeschlos-senen Patienten, Anteil entdifferenzierter Tumoren) lassensich die Ergebnisse der Chaarted-Studie, der STAMPEDE-Studie und der GETUG-AFU 15-Studie natürlich nicht ab-solut miteinander vergleichen. Die kombinierte Analysedeutet jedoch klar auf einen erheblichen Überlebensvor-teil der mit einer Chemohormontherapie behandelten Pa-tienten hin (Abb. 1).

n Seite 21 nJOURNAL 01/2015
Stellungnahme des AKO (Arbeitskreis Onkologie derDeutschen Gesellschaft für Urologie) und der AUO (Ar-beitsgemeinschaft Urologische Onkologie der Deut-schen Krebsgesellschaft)
Basierend auf den Ergebnissen von insgesamt 3 Pha-se-III-Studien besteht eine ausreichende Evidenz, eineEmpfehlung zum Nutzen der Chemohormontherapie aus-zusprechen. Nach erneuter Abstimmung über die Evi-denzlage durch die Mitglieder des Arbeitskreises Onkolo-gie (AKO) und der Vorstandsmitglieder der Arbeitsge-meinschaft Urologische Onkologie (AUO) sollte Patien-
ten mit einem hormonsensitiven, metastasierten Pros-tatakarzinom eine kombinierte Chemohormonthera-pie angeboten werden. Dabei profitieren v. a. Patientenmit einer hohen Metastasenlast von der Therapie im Sinneeiner Überlebensverlängerung. Gemäß der Patientencha-rakteristika der Chaarted- und der STAMPEDE-Studie soll-ten nur Patienten mit einem guten Allgemeinzustand(ECOG 0/1) die Therapie erhalten. Weitere Empfehlungenzur Charakterisierung, welche Patienten von der Therapieam ehesten profitieren, sind aufgrund der Datenlage bis-lang noch nicht möglich.
Abb. 1 Gepoolte Analyse des Gesamtüberlebens der drei prospektiv randomisierten Studien zur primären Chemohormontherapie des hormon-naiven metastasierten Prostatakarzinoms. A: ausschließlich metastasiertePatienten; B: alle eingeschlossene Patienten (aus Tucci et al. Eur Urol 2015epub).
Korrespondenzadresse:
Prof. Dr. med. Thomas SteinerKlinik für UrologieHELIOS Klinikum ErfurtHELIOS Prostatakarzinomzentrum ErfurtNordhäuser Straße 7499084 ErfurtTelefon: 0361-7812201e-Mail: [email protected]
Tabelle 1 Ergebnisse der drei prospektiv randomisierten Studien zur primären Chemohormontherapie des hormonnai-ven metastasierten Prostatakarzinoms (aus Ohlmann et al. Der Urologe 2015).

n Seite 22 n JOURNAL 01/2005JOURNAL 01/2015
n 15 Jahre Radiochirurgie in Erfurt
Klaus Hamm, Hans-Ulrich Herold, Gunnar SurberCyberKnife Centrum Mitteldeutschland
Ende April 2000 nahm unser interdisziplinäres Team ausder Neurochirurgie, Strahlentherapie und Medizinphysik(nach monatelangen Fortbildungen im In- und Ausland)die Arbeit mit dem damals in Europa ersten Novalis-Ra-diochirurgie-System auf. Nach dem Umbau können wirseit Oktober 2012 mit dem einzigen robotergeführtenRadiochirurgie-System (Cyberknife) unsere Patienten wie-der mit der modernsten Technologie behandeln. In diesenJahren haben wir zunehmend an Erfahrung gewonnenund unsere Ergebnisse auch in Zusammenarbeit mit an-deren Zentren auf Kongressen vorgestellt und publiziert.
Anlässlich „15 Jahre Radiochirurgie in Erfurt“ fand am 8. Mai 2015 wieder eine wissenschaftliche Tagung statt,auf der unsere Erfahrungen und die von namhaften Ex-perten vorgestellt und reflektiert werden konnten. Zu-nächst wurde eine Gemeinschaftsarbeit mit den BonnerKollegen über Langzeit-Ergebnisse nach radiochirurgi-schen Behandlungen von Hypophysenadenomen vorge-stellt, aus der 2 Publikationen hervorgegangen sind: 2014über die hormoninaktiven und 2015 über die hormonak-tiven Adenome mit Akromegalie. Bei letzteren soll nebender Tumorkontrolle vor allem der Hormonexzess beseitigtwerden. Das gelingt aber nicht immer, mitunter durch dieverzögerte Strahlenwirkung erst nach Monaten und kannauch mehrere Jahre dauern. Deshalb ist die mikroneuro-chirurgische Operation die Therapie der Wahl und radio-chirurgische Verfahren kommen erst bei der persistieren-den Akromegalie mit Rest- oder Rezidiv-Tumoren durchdie Beteiligung des Sinus cavernosus zum Einsatz.
Zwei Medizinphysik-Experten haben (für Mediziner ver-ständlich) sehr interessante physikalisch-technische The-men vorgestellt, aus Köln eine jetzt veröffentlichte Ver-gleichsstudie über einen exakten Vergleich ihrer früheren,rahmenbasierten Linearbeschleuniger-Pläne, die sie mitden gleichen Vorgaben am Cyberknife erneut geplant ha-ben und zumindest bei den physikalischen ParameternVerbesserungen feststellten. Aus München wurden eben-falls publizierte Daten zur tatsächlichen Präzision des Cy-berknife bei der spinalen Radiochirurgie vorgetragen,durchaus vergleichbar mit denen der bekannten rahmen-basierten und rahmenlosen Radiochirurgie intrakraniell.Der anschließende Berliner Vortrag über die roboter-ge-führte spinale Radiochirurgie benigner und maligner Lä-sionen in Zusammenarbeit mit den Münchener Kollegendemonstrierte die guten Ergebnisse, die bei 1-3 Befundenim Spinalkanal und in der Wirbelsäule erreichbar sind.
Am Nachmittag wurden dann die radiochirurgischenMöglichkeiten im Abdomen, Thorax und Becken vorge-stellt, die in einem der nächsten Hefte dieses Journals the-matisiert werden sollen:
Zunächst aus Heidelberg die stereotaktische Radiothera-pie von Lebertumoren und Lebermetastasen zusammenmit einer Literaturübersicht respektabler Ergebnisse.
Es folgte die Kölner Gruppe mit einer beachtlichen Fallzahlvon mit dem Cyberknife behandelten nichtkleinzelligenLungentumoren. Überwiegend gelang bei den Lokalisa-tionen sogar das „Tumor-Tracking“ ohne Goldmarker, sodass das Risiko eines Pneumothorax durch die Markerim-plantation entfällt. Die Ergebnisse sind überzeugend,auch bei unserer noch bescheidenen Anzahl von radio-chirurgischen Behandlungen bei Lungenbefunden.Schließlich wurde aus Frankfurt/Güstrow noch das ThemaRadiochirurgie von Metastasen im Bauch- und Beckenbe-reich referiert, das auch durch die Erfahrungen mit derOligometastasierung an Bedeutung zugenommen hat.Ein Vortrag zur Supportivtherapie radiochirurgischer Pa-tienten rundete die Veranstaltung ab. Da diese selten not-wendig ist, liegen dazu nur wenige Erfahrungsberichtevor.
Im Ergebnis sehen wir unsere Argumentation gegenübermanchen restriktiven Kostenträgern bestätigt: Wie beiOperationen ist nicht allein die Verfügbarkeit eines mo-dernen Gerätesystems die Garantie für eine erfolgreicheArbeit mit guten Ergebnissen, noch entscheidender istdas hochspezialisierte, interdisziplinäre und erfahreneTeam, das die entsprechende Technik beherrscht.Besonders interessant ist in den letzten Jahren die Ent-wicklung von der „klassischen“ einmaligen Radiochirurgieintrakranieller Pathologien mit der notwendigen stereo-taktischen Ring-/Rahmen- Fixierung am Kopf (auch als ste-reotaktische Einzeit-/Einzel-Konvergenzbestrahlung be-zeichnet) hin zur rahmenlosen und damit auch extrakra-niell möglichen Präzisionsbestrahlung mit 1-5 Applikatio-nen/Fraktionen, die heute allgemein als Radiochirurgiebezeichnet werden.
Damit hat sich die Radiochirurgie, bei der hohe, ablativeDosen submillimetergenau auf ein kleines Zielvolumenappliziert werden, von der einmaligen (rahmenbasierten)„stereotaktischen“ Bestrahlung zur 1-5maligen „bildge-führten“ (image guided) Bestrahlung weiterentwickelt.Die Radiochirurgie ist eine Behandlungsmethode, die mit3 verschiedenen Gerätesystemen durchgeführt werdenkann (Gammaknife, spezielle Linearbeschleuniger=LI-NAC, Cyberknife). Diese 3 Systeme unterscheiden sichdurch die technische Realisierung der für die Radiochirur-gie notwendigen Strahlen-Konvergenz und –Kollimation.Der Name Strahlenchirurgie wurde gewählt, weil bei die-ser Strahlenbehandlung nicht wie üblich über Wochentäglich kleine Dosen verabreicht werden, sondern ver-gleichbar mit der Chirurgie, zunächst vom Neurochirur-gen ein stereotaktischer Rahmen an den Kopf der Patien-ten geschraubt und die Behandlung damit an dem selbenTag mit einer hohen, sehr exakt platzierten Dosis abge-schlossen wurde (werden musste). Die Bezeichnung „…knife“ resultiert aus dem Umstand, dass die Dosis nahezu

n Seite 23 nJOURNAL 01/2015
„messerscharf“ am Rand des zu behandelnden Zielvolu-mens „abgeschnitten“ wird, also mit einem steilen Dosis-abfall zum gesunden Gewebe, um dieses möglichst ma-ximal schonen zu können.
Historisches
Entwickelt und benannt wurde die Radiochirurgie vondem schwedischen Neurochirurgen Lars Leksell am Karo-linska Institut in Stockholm in den 1950iger und 1960igerJahren zusammen mit dem Physiker Börje Larsson zu-nächst für funktionelle stereotaktische Indikationen. Dieseauf einen Zielpunkt konzentrierte Hochpräzisionsbestrah-lung definierten sie als eine „Methode zur Devitalisierungeines definierten intrakraniellen (Hirn)areals mit einer ho-hen Dosis, stereotaktisch geführt und einmalig appli-ziert“. Dafür konstruierten sie schließlich ein einzigartigesBestrahlungssystem mit den damals für die Strahlenan-wendung verwendeten Kobalt-Quellen, nannten es„Gammaknife“ und brachten es erstmals 1968 am Karo-linska Institut zum klinischen Einsatz.
Die Charakteristika der Radiochirurgie (stereotactic radio-surgery =SRS) sind die perkutane, fokussierte Applikationvon entsprechend kollimierten (eingegrenzten/gebündel-ten) Strahlen aus einer Vielzahl unterschiedlicher Richtun-gen mit der damit verbundenen hohen Konzentration imdefinierten Zielgebiet und einem starkem Dosisabfall amRand zur bestmöglichen Schonung der umgebenden Ge-websstrukturen. Voraussetzungen waren die stereotakti-sche Zielpunkt-Führung durch das Anschrauben eines Ste-reotaxie-Rahmens am Kopf der Patienten (damit be-schränkt auf intrakranielle Pathologien und die einmaligeDosisapplikation) und eine hochauflösende Bildgebungzur exakten Planung der Koordinaten der Zielpunkte (Iso-zentren).
Erst die Verfügbarkeit der Computer-Tomographie (CT)seit 1975 und schließlich die Entwicklung der Magnet-Re-sonanz-Tomographie (MRT) führten schließlich zur Detail-Erkennbarkeit auch bereits kleiner, aber im Verlauf pro-gredienter Tumoren in diesen Schnittbild-Techniken unddamit zu einem rasanten Fortschritt der (intrakraniellen)Radiochirurgie seit den 1980iger Jahren - das Gammakni-fe wurde zum Synonym für die Radiochirurgie und zu-nehmend von Neurochirurgen weltweit eingesetzt. Auchich konnte bereits 1993 während einer Hospitation imGammaKnife Zentrum der Universitätskliniken in Wienerste praktische Erfahrungen mit der Radiochirurgie sam-meln (1. Gammaknife Zentrum in Deutschland 1994 inMünchen, wurde nach der Installation eines Cyberknife2005 stillgelegt).
Ebenfalls in den 1980iger Jahren kamen in der Strahlen-therapie immer mehr Linearbeschleuniger (LINAC)- Syste-me zum klinischen Einsatz und lösten die radioaktivenKobalt- Bestrahlungsgeräte sukzessive ab – die LINAC-Technik wurde ständig weiterentwickelt und auch für die
stereotaktische Einzeit-Konvergenzbestrahlung (die Ra-diochirurgie) nutzbar gemacht (hier nahm auch die Strah-lentherapie mit dem DKFZ in Heidelberg eine führendeRolle ein).
Weltweit wurden nun auch „adaptierte“ oder „dedizierte“LINAC-Geräte für die einmalige (rahmenbasierte) Radio-chirurgie und die fraktionierte (maskenbasierte) stereo-taktische Radiotherapie eingesetzt. Wir haben von 2000bis 2012 mit einem speziellen LINAC (dem Novalis-Sys-tem) gearbeitet und damit mehr als 2000 intrakranielleBehandlungen durchgeführt.
Der Neurochirurg John Adler hat schließlich nach länge-rem Aufenthalt bei Leksell in Stockholm Ende der1980iger Jahre an der Stanford-Univ. in San Francisco dasals Cyberknife benannte, einzige robotergesteuerte Ra-diochirurgie-System entwickelt - ein kompakter LINACwird mit einem Industrieroboter der Fa. KUKA (Augsburg)bildgestützt hochpräzise geführt. Das Cyberknife erhielt1999 die FDA-Zulassung und hat sich seit 2001 auch inEuropa etabliert.
Seit Oktober 2012 können wir im Cyberknife ZentrumMitteldeutschland unseren Patienten die Radiochirurgiemit diesem System nicht nur rahmenlos intrakraniell er-möglichen, sondern auch in ausgewählten Fällen extra-kraniell einschließlich der Atmungs-Synchronisation mit„tracking“ beweglicher Zielvolumina. Mittlerweile sind inDeutschland bereits 10 CyberKnife-Systeme installiert.
Klinische und strahlenbiologische Aspekte
Aufgrund seiner physikalisch-technischen Besonderheitenund dem eingeschränkten Indikationsspektrum ist die Ra-diochirurgie keine Konkurrenz zur sogenannten „konven-tionellen“ Bestrahlung, bei der ein größeres Zielvolumenmit dem tumorspezifisch notwendigen Sicherheitssaumfraktioniert mit einer täglich niedrigen Einzeldosis(z.B.1,8Gy/2Gy) bis zu einer entsprechend viel höherenGesamtdosis (z.B.54-60Gy) aufgesättigt werden muss.Entscheidend dafür sind die strahlenbiologischen Er-kenntnisse, dass Tumorgewebe bei jeder Fraktion mehrgeschädigt wird als gesunde Gewebe, die sich rascher er-holen können. Natürlich versuchen Tumoren, ihre geschä-digten Zellen möglichst rasch zu ersetzen (Repopulation),weshalb eine entsprechend hohe Gesamtdosis mit meh-reren Wochen Behandlungsdauer gewählt werden muss.Maligne Primärtumoren, deren Tumorränder durch dasinfiltrative Wachstum in der Bildgebung nicht klar ab-grenzbar sind, müssen leitliniengerecht mit einem Sicher-heitssaum >1-2 cm bestrahlt werden und bleiben die Do-mäne der konservativen Strahlentherapie. Erst bei einemumschriebenen Rezidiv kann die Radiochirurgie hier einezusätzliche Option sein, wenn eine großvolumige Dosis-erhöhung nicht mehr möglich ist.

n Seite 24 n JOURNAL 01/2005JOURNAL 01/2015
Bei der Radiochirurgie werden dagegen hohe, ablativeDosen (z.B. 12-25Gy einmalig oder 3-5 Tage jeweils 5-9Gy) appliziert, eine Repopulation des Tumors ist dannkaum noch möglich und klonogene Tumorzellen könnendefinitiv vernichtet werden. Voraussetzung ist aber einhoher methodischer Aufwand zur Realisierung der not-wendigen, maximalen Präzision, um gesunde Gewebeweitestgehend zu schonen. Denn durch eine Bestrahlungwerden je nach Gewebe-Empfindlichkeit und Höhe derDosis eine Reihe von physikalischen und biologischen Re-aktionen ausgelöst, in deren Folge vor allem durch ver-schiedene „Radikale“ Tumorzellen devitalisiert und damitan der weiteren Zellteilung gehindert werden, in der Tei-lung befindliche Zellen absterben und schließlich diesegeschädigten Zellen von den körpereigenen Abwehrme-chanismen eliminiert werden. Diese Prozesse können Wo-chen, Monate und sogar Jahre dauern, weshalb Strahlen-wirkungen langfristig beobachtet werden müssen. Bei be-sonders heftigen Umgebungsreaktionen können dannauch temporäre, selten permanente Nebenwirkungenauftreten (nach mehr als 3 Monaten als „Spätschäden“definiert).
Primäres Ziel einer Bestrahlung ist zunächst die Devitali-sierung des Tumors, die sogenannte „Tumorkontrolle“,also den Tumor am weiteren Wachstum zu hindern. Imweiteren Verlauf kann eine Tumorregredienz unterschied-lichen Ausmaßes erreicht werden, weshalb das Anspre-chen mit „no change“(NC), „partial response“(PR) und„complete response“ (CR) bewertet wird.
Insbesondere bei malignen Tumoren incl. deren Metasta-sen ist natürlich eine komplette oder partielle Remissionerwünscht, weshalb eine entsprechend höhere Dosis ge-wählt werden muss. Leider steht dem besseren und ra-scheren Ansprechen auch ein häufigeres Rezidiv-Tumor-wachstum gegenüber, dann als „progressive desease“(PD) bezeichnet. Ursachen sind die rasche Zellteilung undRepopulation der malignen Tumoren. Benigne Tumorenreagieren meist verzögert und mit geringerer Regredienz,jedoch sind Rezidive seltener.
Indikationen
Wie bereits ausgeführt, waren die Indikationen bei derstereotaktischen (rahmenbasierten) Radiochirurgie mitunserem Novalis-System ausschließlich auf die intrakra-niellen Indikationen beschränkt. Allerdings konnten wirmit dem Novalis auch die Möglichkeit der (fraktionierten)stereotaktischen Radiotherapie (SRT) in verschiedenenFraktionierungsschemata über 2-6 Behandlungswochennutzen. Wegen der geringeren Präzision durch das not-wendige Maskensystem für die Einstellung der stereotak-tischen Zielpunktkoordinaten (Isozentrum) von +/- 1-2mm mußte ein entsprechender Sicherheitssaum mit be-strahlt und deshalb die Kombination mit den strahlenbio-logischen Vorteilen der Fraktionierung gewählt werden. Mit dem Cyberknife wird die für die Radiochirurgie not-
wendige Präzision von >1mm durch die permanente Bild-führung und Korrektur des Roboters realisiert. Damit wer-den die intrakraniellen Behandlungen nur mit einer leich-ten Maskenfixierung durchgeführt, die nicht zur Einstel-lung eines Zielpunktes notwendig ist, sondern den Pa-tienten ermöglicht, den Kopf während der befund-abhängig 30-60 minütigen Behandlungszeit entspanntruhig zu halten (denn jede Kopfbewegung würde durchdie permanente Bildkontrolle zur Behandlungsunterbre-chung und damit zur Verlängerung der Behandlungszeitführen). Mit der gleichen (radiochirurgischen) Präzisionkann dadurch die Behandlung auch wiederholt werdenals sogenannte multisession radiosurgery =msRS (Erwei-terung der Radiochirurgie-Definition auf 1-5 Fraktionen).Aufgrund der permanenten Bildführung mit digitalenRöntgenkontrollen während der gesamten Behandlungs-zeit ist eine häufigere Fraktionierung wie bei der SRT mitdem Cyberknife nicht sinnvoll und für die meisten Befun-de auch nicht notwendig. Lediglich bei den Schädelbasis-tumoren, die die besonders strahlenempfindlichen Seh-nerven / Chiasma involvieren (die dann im Dünnschicht-MRT nicht mehr abgrenzbar sind), muss eine SRT mit kon-ventioneller Fraktionierung über 6 Wochen durchgeführtwerden.
Die intrakraniellen Pathologien machen im Indikations-spektrum unserer Patienten immer noch einen Anteil>90% aus, obwohl mit dem Cyberknife auch einzelne Tu-moren, 1-3 Metastasen oder eine Oligometastasierung inWirbelsäule und Spinalkanal, in der Lunge und Leber so-wie im Becken und Abdomen behandelt werden können.Das liegt sicher mit an unseren „traditionellen Überwei-ser-Strukturen“, da andere Zentren teilweise über einenhöheren Anteil extrakranieller Indikationen berichtet ha-ben.
Eine Radiochirurgie in vorbestrahlten Regionen ist ebensomöglich wie eine Kombination mit Operation oder Che-motherapie. Selbstverständlich sollte die individuelle The-rapiestrategie in den interdisziplinären Konferenzen derjeweiligen Organzentren entschieden werden.
Hirnmetastasen
Als eine der onkologisch besonders relevanten Hauptindi-kationen mit einem Anteil von 25-30% an der intrakra-niellen Radiochirurgie abschließend hier noch einige Aus-führungen zu den Hirnmetastasen. Einzelne, in der Regel1-3 kleinere Hirnmetastasen sind eine Domäne der Radio-chirurgie. Vor allem bei wenig strahlensensiblen Primär-tumoren (Melanome, Nierenzellkarzinome), wenn eineGanzhirnbestrahlung (whole brain radiotherapy= WBRT)kaum Aussicht auf Erfolg hat, können auch 6 kleine Me-tastasen in einer Sitzung problemlos mit dem Cyberkniferadiochirurgisch behandelt werden. Überhaupt ist die Fra-ge nach dem richtigen Zeitpunkt für eine WBRT immernoch umstritten und war bereits Gegenstand einiger Stu-dien und vieler Diskussionen. Obwohl die Kombination

n Seite 25 nJOURNAL 01/2015
einer Radiochirurgie und WBRT das progressionsfreie In-tervall durch ein verzögertes Auftreten distanter Metasta-sen verlängert hat, war kein Einfluß auf die Überlebenszeitbei häufigeren kognitiven Einbußen nachzuweisen. Des-halb werden immer mehr namhafte Stimmen laut, die beieiner engmaschigen Kontrolle (ca. 3monatlich MRT) undAusschöpfung der radiochirurgischen Möglichkeiten dieWBRT so lang wie möglich „in Reserve“ halten.
Großvolumige Metastasen sind problematischer und vor-rangig eine OP-Indikation. Sprechen die individuellen Be-dingungen dagegen, haben wir mit der sogenannten„multisession radiosurgery“ (msRS) mit dem Cyberknifebzw. der „hypofraktionierten stereotaktischen Radiothe-rapie“ (hfSRT) mit dem Novalis die besten Erfahrungengemacht. Das wurde jetzt auch durch weitere wissen-schaftliche Studien untermauert.
Durch die Weiterentwicklung der Tumortherapien hin zur„targeting therapy“ mit verbesserten Überlebenszeitensteigt auch die Wahrscheinlichkeit für die Tumorpatien-ten, die häufig durch die Blut-Hirn-Schranke nicht anspre-chenden Hirnmetastasen zu „erleben“. Im Kontext mitdem extrakraniellen Status muss dann die individuell best-mögliche Therapiestrategie unter Berücksichtigung undAbwägung aller Aspekte gesucht werden.
Der Ablauf einer radiochirurgischen Behandlung anhandeines Fallbeispiels: 56jährige Patientin mit einem metastasierten Mamma-karzinom (ED 2008), Z. n. brusterhaltender Operation06/2011, fällt klinisch durch eine Hemiparese rechts undpsychische Veränderungen auf. Im Kopf-MRT zeigen sich2 Hirnmetastasen, eine symptomatische, größere Metas-tase frontoparietal links mit erheblichem Hirnödem sowieeine zweite kleinere, temporoparietal links gelegene Me-tastase ohne auffälliges Begleitödem (Abb. 1).
Abb. 1 Z., Manuela 56 J. – 2 Hirnmetastasen bei Mammakarzinom(ED 01/2011), Kopf-MRT mit einer großen, symptomatischen Metastase li.frontoparietal und einer kleinen Hirnmetastase li. parietal11/2012 Mikrochirurgische Exstirpation der symptomatischen Hirnmetastase
Der extrakranielle Status ist beim „restaging“ stabil. Inder interdisziplinären Tumorkonferenz wird die Indikationzur Operation der symptomatischen Metastase sowie an-schließenden Radiochirurgie des Resektionsbereiches undder kleinen Zweitmetastase gestellt. Die Patientin wirdentsprechend beraten und ausführlich über das jeweiligeProcedere, die Risiken und Erfolgsaussichten aufgeklärtund willigt in die Behandlungsstrategie ein. Die ambulan-te Radiochirurgie ist weiterhin noch keine abrechenbareLeistung der gesetzlichen Krankenkassen, deshalb musszunächst per Eilantrag die Kostenübernahme geklärt wer-den.
Es erfolgt die komplikationslose Operation der sympto-matischen Hirnmetastase und eine dünnschichtigeMRT-Kontrolle am 1.postoperativen Tag (zur operationsbedingtartefaktfreien Beurteilung innerhalb von 24 Stunden er-forderlich), eine der Voraussetzungen für die millimeter-genaue Planung einer Radiochirurgie. Als weitere Vorbe-reitungen muss noch ein ebenfalls dünnschichtiges Pla-nungs-CCT (ca. 1mm lückenlose Schichten) mit der un-mittelbar vorher individuell angepassten Masken-Lagerung durchgeführt werden. Zu der aufwändigen Pla-nung selbst müssen die Patienten nicht anwesend sein –nach dem Datenimport in das Planungssystem erfolgt zu-nächst eine Bildfusion der dünnschichtigen CCT- undMRT-Bilder, dann die „Schicht für Schicht“- Definition derZielvolumina, häufig auch gemeinsam mit den Neurora-diologen (hier der Resektionsbereich und die 2., bereitsprogrediente Metastase parietal links) sowie der zu scho-nenden Strukturen wie z.B. Augen, Tränendrüsen, Seh-nerven, Chiasma, Hypophyse, Hirnstamm (sogenannte„Risikoorgane“, für die es Tabellen mit den einzuhalten-den Dosisgrenzwerten gibt). Die Medizinphysik-Expertenentwickeln nun verschiedene Dosisplan-Vorschläge, ge-meinsam wird der individuell optimierte Plan gewählt undentsprechend für Radiochirurgie mit dem Cyberknife vor-bereitet.
Aufgrund der großvolumigen Resektionshöhle von 21,5cm³ wird dieser Bereich mit einer msRS (3x7Gy Randdosisauf die 70% Isodose) an 3 aufeinanderfolgenden Tagenbehandelt, am 1.Tag zusätzlich die Zweitmetastase mit ei-ner RS (16Gy Randdosis auf die 70% Isodose).
Dünnschichtige MRT-Kontrollen werden bei Hirnmetasta-sen zunächst alle 3 Monate erbeten, um den Behand-lungsverlauf beurteilen und ggf. neue Metastasen früh-zeitig erkennen und wieder behandeln zu können. In die-sem Fall zeigt sich nach 7 Monaten eine neue, noch kleineMetastase temporal links, die ebenfalls mit einer RS (16GyRanddosis auf die 69% Isodose) erfolgreich behandeltwird. 18 Monate später wieder eine neue, noch kleineMetastase frontolateral links, es erfolgt in gleicher Weiseeine Radiochirurgie. Weitere Kontrollen bisher ohne Nach-weis aktiver zerebraler Metastasen (kein Rezidiv im Resek-tionsbereich und allenfalls minimale KM-Aufnahme imBereich der 3 kleinen Metastasen) – Abb. 2

n Seite 26 n JOURNAL 01/2005JOURNAL 01/2015
Abb. 2 Z., Manuela 56 J. – Hirnmetastasen bei Mammakarzinom (ED 01/2011)MRT-Verlauf von 01/2013 bis 04/201511/2012 OP der symptomatischen Hirnmetastase re. frontoparietal01/2013 msRS 3x7Gy Randdosis des Resektionsbereiches und
RS 16Gy Randdosis der progredienten Metastase li. parietal09/2013 RS 16Gy Randdosis der Metastase li. temporal01/2015 RS 16Gy Randdosis der Metastase li. frontolateral04/2015 unauffälliger Resektionsbereich und subtotale Regredienz
der anderen 3 Hirnmetastasen
Mit der Strategie der regelmäßigen MRT-Kontrollen undSalvage-Radiochirurgien im Bedarfsfall kann in diesen ge-eigneten Fällen eine Ganzhirnbestrahlung mit möglichenneurokognitiven Störungen umgangen oder zumindesthinausgeschoben werden.
Die Radiochirurgie bietet eine Erweiterung des Therapie-spektrums in einer komfortablen, ambulanten Form mithoher Effektivität und geringen Nebenwirkungen. Sie istalternativ oder mit anderen Therapieschritten simultanoder additiv einsetzbar sowie im Rezidivfall auch in vor-bestrahlten Regionen.
Korrespondenzadresse:
Priv.-Doz. Dr. med. Klaus Hamm CyberKnife Centrum Mitteldeutschland im HELIOS Klinikum ErfurtNordhäuser Straße 7499089 ErfurtTelefon: 0361-7816718 E-Mail: [email protected]://www.ckcm.de
n Therapiesysteme für die Radio-chirurgie
Gunnar Surber, Hans-Ulrich Herold, Klaus Hamm
Die Radiochirurgie ist charakterisiert als eine Methode, ineiner einmaligen Therapiesitzung eine hohe ablativeStrahlendosis (Energiedosis) im zu behandelnden Zielvo-lumen zu konzentrieren. Dabei soll einerseits dieses Vo-lumen von der therapeutischen Dosis komplett umfasstwerden (Verschreibungsdosis = Randdosis), andererseitssollen aber die die Läsion umgebenden Areale keine odereine hinreichend geringe Dosisbelastung erfahren. Diesengegensätzlichen Ansprüchen perfekt gerecht zu werden,ist wegen der physikalischen Eigenschaften der ange-wendeten Röntgen- oder Gammastrahlung leider nichtmöglich. Es geht also um die Suche nach einem geeigne-ten Kompromiss.
Diese Ausgangssituation hat für jede Strahlenanwen-dung Gültigkeit. Es kommt aber im Detail darauf an, wiegut dieser Kompromiss gestaltet werden kann.
Abb. 1 Typische Dosisverteilung für eine Radiochirurgie des Akustikusneurinoms

n Seite 27 nJOURNAL 01/2015
Im Unterschied zur konventionellen Strahlentherapie, beider mit vielen Fraktionen (ca. 30) und einer geringen Dosispro Fraktion (ca. 2,0 Gy) gearbeitet wird, werden bei derRadiochirurgie in einer einzelnen Sitzung Dosiswerte zwi-schen 12 und 25 Gy, bei der Trigeminusneuralgie sogarbis 90 Gy verabreicht. Würde umliegendes gesundes Ge-webe (z.B. Hirnareale, Myelon) einer solchen Dosisbelas-tung ausgesetzt, wären erhebliche funktionelle Ausfälleals Nebenwirkungen zu erwarten.
Wie gut und nachhaltig kann das vermieden werden?Wie und in welchem Ausmaß kann eine erreichbare Scho-nung während der gesamten Therapiedauer garantiertund kontrolliert werden?
Die Methodik der Radiochirurgie ist auf das Ziel gerichtet,einen außerordentlich steil abfallenden Dosisgradientenvom Rand des zu behandelnden Zielvolumens (z.B. Tumor)in Richtung Normalgewebe zu erzeugen. Das wird durcheine Vielzahl von Einstrahlrichtungen aus jeweils verschie-denem Raumwinkel realisiert (Konvergenzbestrahlung).Je größer deren Anzahl, desto enger „schnüren“ sich dieverschiedenen Dosis-Level (Isodosen) um das zu bestrah-lende Zielvolumen und desto geringer wird das Volumendes Normalgewebes, das unbeabsichtigt mit einer rele-vanten Dosis belastet wird.
Das gilt nicht nur in einer Bestrahlungsebene (2-dimen-sional), sondern muss räumlich, also 3-dimensional, be-trachtet werden. Dabei sind alle einzelnen Strahlen auf ei-nen fixen Zielpunkt in der Läsion gerichtet (Isozentrum,angewendet bei Gammaknife und Linearbeschleuniger)oder die Einstrahlrichtungen können frei im Raum variiertwerden (nicht-isozentrisch, angewendet beim Cyber-knife).
Abb. 2 Dosisgradienten bei fokussierter Bestrahlung mit einem Cyberknife
Darüber hinaus ist die Genauigkeit der Positionierung desPatienten bzw. des zu behandelnden Zielvolumens in Re-lation zum Bestrahlungssystem ein wichtiges Kriteriumfür den Erfolg der Behandlung. Traditionell wird die Radiochirurgie unter stereotaktischenBedingungen durchgeführt. Als Referenz für die räumli-che Zuordnung der CT-Bilddaten und damit für das zu im-plementierende Koordinatensystem dient hier ein invasivan der Schädelkalotte fixierter stereotaktischer Rahmen.Damit kann der Patient unverrückbar und submillimeter-genau in die vorab berechnete Behandlungsposition ge-bracht werden, sodass die Zielpunkt-Koordinaten genaumit dem System-Isozentrum übereinstimmen. Währendder ca. 30-minütigen Behandlung ist die Patientenpositi-on dann nicht mehr verifizierbar.
In den vergangenen 15 Jahren hat sich ein Trend weg vonder klassischen, stereotaktischen Zielpunktlokalisationentwickelt. An deren Stelle tritt die Lokalisation und Posi-tionierung mit Hilfe von Röntgenbildern, die direkt amBestrahlungsgerät mit dem Patienten in Bestrahlungspo-sition angefertigt werden. Von diesen Bilddaten werdendurch Vergleich mit errechneten Bildern aus dem Pla-nungs-CT (digital reconstructed radiography, DRR) dienotwendigen Positionierungs-Vektoren abgeleitet, sodasses möglich wird, die Position des Patiententisches mitdem Patienten adäquat anzupassen. Dieses Verfahrenwird allgemein als bildgeführte Strahlentherapie bezeich-net (image guided radiotherapy, IGRT).
Wie genau diese Positionskorrektur vorgenommen wer-den kann, ist von einer Vielzahl von Details abhängig:
– Zum einen spielt die Auflösung der Tischmechanik eineRolle. Kann der Tisch in x, y und z millimetergenau be-wegt werden oder lassen sich sogar Zehntelmillimetereinstellen?
– Des Weiteren ist die Qualität der für die Konturierungund Planung verwendeten Bildgebung ein wichtigesKriterium. Deren Auflösung sollte in keiner Richtungden Wert von 1 mm überschreiten. Konkret müssen„Field of view“ hinreichend klein (möglichst < 50 cm)und die Matrixgröße hinreichend groß (mind. 512²)gewählt werden, um entsprechend kleine Pixel erzeu-gen zu können. Für die Schichtdicke sollte ca. 1 mmgewählt werden.Das alles ist entscheidend für eine hochaufgelöste Kon-turierung des Zielvolumens in den einzelnen Schich-ten, aber auch für die spätere Positionierung des Pa-tienten mit der IGRT.
– Schließlich sollte die erreichbare Präzision über dengesamten Behandlungszeitraum erfasst und kontrol-liert werden können.
Nachfolgend sollen die drei wichtigsten Gerätekonzeptevorgestellt werden, mit denen radiochirurgische Präzi-sionsbehandlungen möglich sind.
n Seite 28 n JOURNAL 01/2005JOURNAL 01/2015
GammaknifeMit der Entwicklung des Gammaknife-Systems in den spä-ten 1960er Jahren begann die Geschichte der Radiochi-rurgie. Mit diesem System können ausschließlich intra-kranielle Läsionen behandelt werden. In den inzwischenca. 50 Jahren wurde das Gerät zwar weiterentwickelt undteilweise modifiziert, das grundlegende Arbeitsprinzip istjedoch gleich geblieben.
Abb. 3 Gammaknife
Zur Behandlung wird der im stereotaktischen Rahmen fi-xierte Kopf des Patienten mit dem Zielpunkt der zu be-strahlenden Läsion in das Zentrum einer dickwandigen,innen hohlen Halbkugel (Kollimator-Helm) positioniert.Die Wand besteht dabei aus Metallen mit guten Absorp-tionseigenschaften (hohe Ordnungszahl) und hat eine Di-cke von mehreren Zentimetern. In diese Wand sind 201leicht konische Bohrungen eingebracht, die jeweils aufden Kugelmittelpunkt gerichtet sind. Das Innere der Gan-try des Gammaknifes ist ebenfalls als hohle Halbkugel ge-staltet. Auf deren Innenseite sind passend zur Positionender Bohrungen des Helmes 201 relativ stark radioaktiveKobaltquellen angebracht (Energie der Gammastrahlung:1,17 und 1,33 MeV).
Abb. 4 Kollimatorhelm des Gammaknife
Im Ergebnis entstehen ca. 200 Einzelstrahlen aus verschie-denen Raumwinkeln, die jeweils einen kreisförmigenQuerschnitt besitzen und auf den Kugelmittelpunkt ge-richtet sind. Dort laufen alle Einzelstrahlen zusammen,sodass sich die einzelnen Dosisbeiträge addieren und einekugelförmige Dosisverteilung im Patienten erzeugen. Fürdie Anwendung kann zwischen 4 verschiedenen Kollima-tor-Helmen gewählt werden. Diese weisen jeweils einheit-liche Bohrungsdurchmesser auf (4, 8, 14 oder 18 mm).Außerdem können beliebig viele Einzelstrahlen ausge-blockt werden, um die Dosisverteilung zu beeinflussen.
Abb. 5 Isozentrische Überlage-rung von Einzelstrahlen
Die erzeugte Dosisverteilung ist dabei stets annäherndkugelförmig. Da die meisten zu behandelnden Läsionenjedoch irregulär geformt sind, kann die entsprechendeDosisverteilung nur durch Aneinanderreihung einzelnerDosiskugeln erreicht werden. Damit muss der Patient so-wohl für den Helmwechsel als auch für die Ausrichtungauf einen neuen Zielpunkt mehrmals neu positioniert wer-den, was zu relativ langen Behandlungszeiten führt.
Abb. 6 Notwendige Aneinan-derreihung von kugelförmigenDosisverteilungen bei irregulärgeformten Zielvolumina
Die neuen Gammaknife-Systeme (seit 2009) haben des-halb einen modifizierten Aufbau. Es gibt nur noch eineneinzigen, fest eingebauten Kollimator-Helm mit insgesamt192 Kollimatoröffnungen, anteilig mit einem Durchmes-ser von 4, 8 und 16 mm (jeweils 64). Die 192 Kobaltquel-len können sektorweise derart motorisch bewegt werden,dass sie außen jeweils vor gleich großen Kollimatoröff-nungen sitzen. Durch die Aufteilung in Sektoren könnengleichzeitig auch Strahlen mit verschiedenem Durchmes-ser appliziert werden, der Wechsel des Kollimator-Helmeswird dadurch überflüssig.
Außerdem kann der Patiententisch 3-dimensional moto-risch bewegt werden, so dass die Einstellung verschiede-ner Zielpunkte schneller gelingt.

n Seite 29 nJOURNAL 01/2015
Abb. 7 Längsschnitt durch das Gammaknife mit beweglichen Kobalt-Quellen(außen, gelb) vor verschieden großen Kollimatoröffnungen (weiß)
Aktuelle Entwicklungen beinhalten auch ein integriertesBildgebungssystem - eine Kontrolle der Patientenpositionist damit vor, aber nicht während der Behandlung mög-lich. Außerdem versucht man bei dieser zukünftigen Ver-sion „Icon“ auch eine nichtinvasive Maskenfixierung an-zuwenden, um den Patienten auch die unangenehmeRahmenfixierung ersparen und hypofraktionierte Bestrah-lungen durchführen zu können.
Die Behandlungszeit variiert stark in Abhängigkeit vonder Komplexität der Befundausdehnung und der Aktivitätder Kobaltquellen. Durch deren Halbwertszeit von ca. 5,3Jahren ist alle 5-6 Jahre ein Quellenwechsel (mit radioak-tivem Abfall) erforderlich, um zunehmend längere Be-handlungszeiten zu vermeiden.
Dedizierte Linearbeschleuniger (LINAC)Nach der Etablierung von Linearbeschleunigern in derStrahlentherapie wurden diese Geräte auch für die Radio-chirurgie nutzbar gemacht. Daraus ergaben sich hohe An-forderungen an die geometrische Stabilität und Präzisionder verwendeten Systemkomponenten (Gantry, Tisch, Kol-limatorsystem) sowie an die Reproduzierbarkeit der an-gewendeten, ultraharten Röntgenstrahlung. Die Energievon 6 MeV mit einer durchschnittlichen Dosis der erzeug-ten Bremsstrahlung von ca. 2 MeV hat sich etabliert.
Abb. 8 Linearbeschleuniger mitmontierten Rundkollimatoren
Mit den heute verfügbaren, für radiochirurgische Anwen-dungen dedizierten Linearbeschleunigern kann bei sorg-fältiger Justage eine mechanische Stabilität und Zentrie-rung erreicht werden, die die damit einhergehende Unsi-cherheit auf einen Wert von ca. 0,5 mm begrenzt. Daskonnten wir auch durch langjährige Messungen an demdamals ersten dedizierten LINAC „Novalis“ nachweisen,den wir bis 2012 genutzt haben.
Die Formung des Strahlquerschnittes kann beim LINAC(vergleichbar mit dem Gammknife) durch Rundkollimato-ren mit den leicht konischen Bohrungen von 4-50 mm in10-12 Abstufungen erfolgen. Mit diesen runden Strah-
lenfeldern wurden Rotationsbestrahlungen (Arcs) über ei-nen Winkelbereich von 60-140° durchgeführt, für ver-schiedene Tischwinkel wiederholt, so dass im Ergebnisauch eine Vielzahl von Einstrahlrichtungen zustande kam.Aktuell kommt diese Strategie kaum noch zum Einsatz.
Moderne LINACs verfügen alle über einen Multi-Leaf-Kol-limator (MLC), mit dem die Form der Strahlenfelder durchmotorische Bewegungen der Lamellen auf die Kontur derLäsion aus dem jeweils betrachteten Winkel angepasstwerden kann (Konformation). Dabei wird im Regelfallauch bei komplexer geformten Läsionen deren gesamtesVolumen vom Strahl erfasst, d.h. es müssen nicht mehrDosiskugeln aneinandergesetzt werden.
Im Sinne einer statischen Anwendung können mehrereStehfelder (>10) aus verschiedenen Raumwinkeln (Gan-trywinkel und Tischwinkel) sequentiell auf den Zielpunktgelenkt werden. Aber auch Rotationsbestrahlungen mitdynamischer Nachführung der Feldform an die sich än-dernde Projektion der Läsion waren schon mit unserem„Novalis“ möglich (dynamic arcs).
Abb. 9 Linearbeschleuniger No-valis mit MLC für dynamische Ro-tationsbestrahlung
Dabei kann die Feldkontur umso besser an das Zielvolu-men angepasst werden, je dünner die im MLC zur An-wendung kommenden Lamellen sind. Für radiochirurgi-sche Behandlungen sollten diese höchstens 2-3 mm dicksein. Für die Radiochirurgie wurde dem Patienten bis zur Ent-wicklung einer hinreichend genauen IGRT auch ein ste-reotaktischer Rahmen an den Kopf geschraubt, mit demdas Planungs-CT angefertigt und die Bestrahlung durch-geführt wurden (identisches Procedere wie beim Gam-maknife).
Abb. 10 Stereotaktischer Kopf-rahmen mit Schraub-Pins fürdie Befestigung an der Schä-delkalotte
Die bereits beschriebene bildgeführte Patientenpositio-nierung (mit planaren Röntgenprojektionen oder ConeBeam CT) ist heute als Option für alle Linearbeschleuni-gertypen verfügbar. Die Methode wurde für das breite

n Seite 30 n JOURNAL 01/2005JOURNAL 01/2015
Anwendungsspektrum der konventionellen Strahlenthe-rapie entwickelt und wird heute flächendeckend in derStrahlentherapie angewendet.
In den meisten Fällen wird ein Röntgen-Bildgebungssys-tem genutzt, welches direkt am Beschleunigerarm (Gan-try) montiert ist. Sowohl Röntgenröhre als auch Detektorkönnen mit einem Gelenkarm in Position gebracht und ei-ne Aufnahme ausgelöst werden. Das geschieht aus zweizueinander orthogonalen Richtungen vor Beginn der Be-strahlung. Die sich ergebenden 3-dimensionalen Korrek-turwerte können verwendet werden, um die Patienten-position entsprechend zu korrigieren. Hierbei wird in denmeisten Fällen ein Genauigkeitsmaß von ca. 1 mm er-reicht. Nur einzelne Geräte erlauben auch eine Tischposi-tionierung auf +/- 0,1 mm, was einen deutlichen Quali-tätssprung darstellt.
Abb. 11 Linearbeschleunigermit ausfahrbarem Röntgen-Bildgebungssystem
Eine andere, nur in dedizierten Radiochirurgie-Zentrenverfügbare Option besteht in der festen Integration vonRöhre und Detektor in die Raumstruktur. Dabei werdendiese Module an die Decke bzw. versenkt in den Bodenmontiert, womit die mechanischen Unsicherheiten weit-gehend minimiert werden und eine deutlich höhere Prä-zision erreicht werden kann.
Um die erhöhte Genauigkeit der Bildgebung auch in ge-nauere Patientenpositionierung umsetzen zu können,kommen bei derart ausgestatteten Systemen häufig Tisch-aufsätze zur Anwendung, mit denen translatorische Kor-rekturen auf +/- 0,1 mm genau vorgenommen werdenkönnen, aber auch rotatorische Korrekturen um die dreiRaumachsen möglich sind. Ein solches Vorgehen wirdhäufig auch 6D-Positionierung genannt und bietet das
derzeit höchste erreich-bare Genauigkeitsni-veau.
Abb. 12 Dedizierter Linearbe-schleuniger mit fest montier-tem Röntgen- Bildgebungssys-tem
Durch den Übergang zur bildgeführten Patientenpositio-nierung wird das Indikationsspektrum auch auf extrakra-nielle Befunde erweitert.
Die Behandlungszeiten liegen bei ca. 30-60 min, in Einzel-fällen auch darüber.
Cyberknife
Mit der Verfügbarkeit von extrem präzisen Industrierobo-tern entstand die Idee, diese Eigenschaften auch für denradiochirurgischen Einsatz zu nutzen. So wurde in den1990er Jahren in Kalifornien ein robotergestütztes Be-strahlungssystem entwickelt, bei dem der mit 6 Gelenkenausgestattete Roboter (Fa. KUKA, Augsburg) einen kom-pakten 6 MeV LINAC relativ frei im Raum bewegt. Die da-bei erreichbare Genauigkeit liegt bei ca. 0,1 bis 0,2 mmmit sehr hoher Reproduzierbarkeit.
Abb. 13 Cyberknife VSI inErfurt
Die Radiochirurgie erfolgt typischerweise durch 100-300statische Bestrahlungsfelder, wobei deren Zahl prinzipiellnicht nach oben begrenzt ist (abgesehen von der Bestrah-lungszeit).
Im Unterschied zu den beiden vorgenannten Therapie-Systemen werden beim Cyberknife die Einzelstrahlennicht (isozentrisch) auf einen Punkt gerichtet. Vielmehrkönnen die Zielpunkte für jeden Strahl individuell festge-legt werden, so dass man im geometrischen Kontext vonwindschiefen Vektoren spricht. Dies hat den Vorteil, dassdie erzeugten Dosisverteilungen nicht primär kugelförmigsein müssen, sondern der dreidimensionalen Ausdehnungdes Zielvolumens folgen können, dies schließt auch aus-geprägte Konkavitäten ein. Darüber hinaus kann auch dieDosisbelastung in der Läsion selbst gezielt beeinflusstwerden (Dosisgradient im Sinne eines inhärenten Boosts).
Abb. 14 Darstellung der Viel-zahl von Einstrahlrichtungenam Cyberknife

n Seite 31 nJOURNAL 01/2015
Für die Patientenlagerung gibt es zwei Tischvarianten: Als Standardausstattung steht ein 5D-Tisch zur Verfü-gung, mit dem die im Rahmen der IGRT ermittelte Fein-einstellung der Patientenposition vorgenommen werdenkann. Dies umfasst neben den 3 translatorischen Richtun-gen (x, y, z) auch 2 rotatorische Freiheitsgrade (Pitch undRoll). Lediglich die verbleibende Rotation um die Vertikal-achse (Yaw) kann nicht motorisch vorgenommen werden,sondern wird durch sanfte Korrektur der Patientenpositi-on auf dem Tisch realisiert. Die Auflösung der Bewegun-gen wird auf +/- 0,1 mm bzw. 0,1° genau ausgeführt.
Als Option steht die RoboCouch zur Verfügung. Hier wirddie Tischplatte von einem weiteren Roboterarm mit glei-cher Genauigkeit frei im Raum bewegt und schließt alle 6Freiheitsgrade ein.
Abb. 15 Standard-Couch (5D)
Abb. 16 RoboCouch (6D)
Für Behandlungen im Kopfbereich unterstützt eine ana-tomisch vorgeformte Kopfschale sowie eine weiche Mas-ke die „ruhige“ Lagerung.
Der Patient liegt immer auf einer weichen Matte. Für Be-handlungen außerhalb des Kopfes können zusätzlich auchder Körperkontur angepasste Vakuum-Matten verwendetwerden.
Abb. 17 Weiche Kopfmaske Abb. 18 Vakuum-Matte
Das integrierte, digitalisierte Bildgebungssystem bestehtaus 2 fest an der Decke montierten Röntgenröhren und
entsprechenden Detektoren, die im Boden eingelassensind. Die angewandte Methodik der IGRT entspricht zu-nächst der für die Linearbeschleuniger beschriebenen Vor-gehensweise. Ein wesentlicher Unterschied besteht darin,dass die Röntgenaufnahmen nicht nur vor der Bestrah-lung ausgelöst werden, sondern in regelmäßigen, ein-stellbaren Zeitabständen auch während der gesamten Be-handlungszeit. Die dabei ermittelten Abweichungen inallen 3 translatorischen sowie 3 rotatorischen Richtungen(meist wenige Zehntelmillimeter bzw. -grad) werden au-tomatisch an die Robotersteuerung übermittelt, so dassdieser durch gezielte Änderung seiner Position die ermit-telte Abweichung kompensieren kann.
Abb. 19 Orthogonale Rönt-gen-Projektionen
Die Genauigkeit der Patientenpositionierung wird alsopermanent kontrolliert und auftretende Abweichungenkorrigiert, dies wird mit dem Terminus „Tumor-Tracking“beschrieben.
Die Feldformung erfolgte bislang entweder durch Rund-kollimatoren (12 Größen, 5-60 mm) oder durch einenIRIS-Kollimator, durch den von Strahl zu Strahl der Durch-messer motorisch variiert werden kann, aber ebenfallskreisförmig bleibt.
Seit diesem Jahr ist für das Cyberknife ein spezieller, vor-schaltbarer Mini-MLC verfügbar, mit dem der Strahlquer-schnitt auf die Kontur des Zielvolumens oder Teile davonangepasst werden kann. Sehr wahrscheinlich wird sichdadurch die Behandlungszeit, die üblicherweise zwischen30-50 min liegt, um ca. 10-30% reduzieren lassen.
Abb. 20 Rundkollimatoren Abb. 21 Iris-Kollimator

n Seite 32 n JOURNAL 01/2005JOURNAL 01/2015
Abb. 22 Cyberknife M6 mit MLC
Mit dem Cyberknife können neben intrakraniellen Indika-tionen (mit sanfter Maskenfixierung) auch spinale, thora-kale sowie abdominale Befunde bis in den Beckenbereichbehandelt werden.
Insbesondere durch physiologische Atembewegungenunterliegt in diesen Fällen das Zielvolumen einem mehroder weniger rhythmischen Bewegungsmuster. Diese Be-wegung kann durch eine synchrone, räumlich gleichge-richtete Bewegung des Roboters kompensiert werden.Hierzu wird zunächst der Atemzyklus durch exakte Beob-achtung der Brustkorbbewegung mit einem stereoskopi-schen Kamerasystem erfasst. An 8 repräsentativen Punk-ten der Atemkurve werden paarweise Röntgenaufnah-men ausgelöst, auf denen die Position des Tumors mar-kiert wird. Auf diese Weise kann die Software einenBewegungspfad des Tumors lernen und die Bewegungdes Roboters entsprechend anpassen. In diesem Modusführt der Roboter also während der aktiven Strahlerzeu-gung eine zyklische Bewegung aus, die exakt der Tumor-bewegung entspricht. Durch fortgesetzte Aufnahmen infestgelegten Intervallen wird dieses Modell während dergesamten Behandlungszeit fortlaufend aktualisiert.
Korrespondenzadresse:
Dipl.-Ing. Gunnar SurberCyberKnife Centrum Mitteldeutschland im HELIOS Klinikum ErfurtNordhäuser Straße 7499089 ErfurtTelefon: 0361-7816716 E-Mail: [email protected]://www.ckcm.de
n 28. Onkologische Konferenz desTumorzentrum Erfurt e.V. fand am6. und 7. November 2015 inEisenach statt
Oberstes Ziel der Tumorzentren ist es, die Betreuung derTumorpatienten im Einzugsgebiet ständig zu verbessern.Die jährlich vom Tumorzentrum Erfurt e.V. durchgeführ-ten Onkologischen Konferenzen sind dieser Aufgabe inbesonderer Weise verpflichtet. Neben der Vermittlung ak-tuellen Wissens sollen Interdisziplinarität, Kommunikationund Kooperation aller Versorgungsbereiche gefördertwerden. Die Themenauswahl berücksichtigt daher sowohlInnovationen in der Onkologie als auch Probleme derpraktischen Umsetzung und ist gleichermaßen an nieder-gelassene und klinisch tätige Ärzte gerichtet.
Die mit 122 Teilnehmern gut besuchte diesjährige Veran-staltung fand traditionsgemäß wieder im Haus HainsteinEisenach statt.
Schwerpunkte der Tagung waren folgende Themen:- Speicheldrüsentumoren- Moderne Verfahren in der bildgebenden Diagnostik- Gynäkologische Tumoren- Neues aus der Forschung (MALDI-TOF zur Tumor- typisierung)
Auf dem wie bereits im Vorjahr in das Programm aufge-nommenen „Jungen Forum Onkologie“ wurde jüngerenÄrztinnen und Ärzten die Gelegenheit gegeben, eigeneBeiträge zu onkologischen Themen zu präsentieren. Die-ses Angebot fand mit 8 eingereichten Beiträgen eine guteResonanz. Drei Vorträge wurden prämiert. Einige der Bei-träge stellen wir im Folgenden vor.

n Seite 33 nJOURNAL 01/2015
n Neoadjuvante Therapiekonzeptebeim Pankreaskarzinom als neuer Meilenstein – Darstellung des Statusquo anhand einer Falldemonstration
Vortrag zum „Jungen Forum Onkologie“ auf der28. Onkologischen Konferenz des TumorzentrumErfurt e.V. am 6. und 7. November 2015 in Eisenach
Markus Mille1, Michael Glatzel2, Albrecht Stier11Klinik für Allgemein- und Viszeralchirurgie,HELIOS Klinikum Erfurt2Klinik für Strahlentherapie und Radioonkologie,HELIOS Klinikum Erfurt
EinleitungJährlich erkranken in Deutschland etwa 15.000 Menschenan einem duktalen Adenokarzinom des Pankreas. Somitliegt das Pankreaskarzinom in der Statistik der Krebsneu-erkrankungen bei Männern auf dem 9. und bei Frauenauf dem 7. Platz. Trotzdem ist zu beachten, dass in Bezugauf die Krebstodesursache das Pankreaskarzinom bereitsauf Platz 4 rangiert und nach wie vor mit einer durch-schnittlichen 5-Jahresgesamtüberlebensrate von nur 5%einhergeht (1). Dies ist nicht zuletzt der Tatsache geschul-det, dass frühe Symptome fehlen und keine diagnosti-schen Marker existieren, welche eine frühzeitige Diagnoseerlauben würden. Hinzu kommt auch ein aggressives Tu-morwachstum des duktalen Adenokarzinoms mit raschersystemischer Metastasierung. So findet sich bei initialerDiagnosestellung bei der überwiegenden Mehrheit ein lo-kal fortgeschrittenes bzw. metastasiertes Stadium, sodasseine kurative Resektion nur bei 10 – 20% der Patientenmöglich ist (1). Zusätzlich hat sich selbst bei Patienten,welche umgehend einer Resektion zugeführt werden, inden letzten 30 Jahren trotz der Verbesserungen der chi-rurgischen Technik im besten Fall eine mediane Überle-benszeit von nur 24 Monaten sowie eine 5-Jahresüberle-bensrate von 15-20% erreichen lassen (2, 3). Dabei ist dieResektion des Tumors aktuell immer noch die einzigeMöglichkeit, eine definitive Heilung zu erreichen. Es mussjedoch auch beachtet werden, dass die Pankreatikoduo-denektomie trotz der verbesserten chirurgischen Techniknach wie vor mit einer hohen Inzidenz an postoperativenKomplikationen verbunden ist, während die postoperati-ve Letalität aktuell nur noch bei 1 – 4% in entsprechendenZentren liegt. Die überwiegende Mehrheit der operiertenPatienten (ca. 85%) entwickelt hingegen trotz tumorfreierResektionsränder (R0-Resektion) ein Rezidiv oder eineFernmetastasierung, sodass von einer frühen systemi-schen Ausbreitung des Pankreaskarzinoms ausgegangenwerden muss (4, 5). Aufgrund des tumorbiologischen Ver-haltens sowie der Langzeitergebnisse nach kurativer Re-sektion sollte deshalb auch ein möglichst frühzeitiger sys-temischer Therapieansatz in Erwägung gezogen werden.
Prinzipien der neoadjuvanten TherapieWie bereits angeführt ist nur ein kleiner Anteil der Patien-ten mit einem Pankreaskarzinom primär operabel. Hinge-gen liegt bei 50 – 60% der Patienten bereits im Rahmender initialen Diagnosestellung ein metastasiertes Stadiumvor, sodass nur eine palliative Chemotherapie als einzigeOption bleibt. Dagegen findet sich bei 30 – 40% ein soge-nanntes „lokal fortgeschrittenes“ Tumorstadium, sodasseine primäre Resektion fraglich oder eben gar nicht mög-lich ist.
Während neoadjuvante Therapiekonzepte bei anderen so-liden Tumoren mit lokal fortgeschrittenen Tumorstadien,wie z.B. dem Rektumkarzinom oder Ösophaguskarzinom,bereits zu einem unverzichtbaren Bestandteil der onkolo-gischen Therapie gehören, sind derartige Vorgehenswei-sen beim Pankreaskarzinom nur spärlich untersucht undGegenstand ausgeprägter Diskussionen.
Hauptziel einer neoadjuvanten Therapie ist dabei sicher-lich, den Tumor zurückzudrängen und so eine erhöhte Ra-te an negativen Resektionsrändern als auch ein Down-staging zu erreichen, um überhaupt eine Resektion durch-führen zu können. Es existieren aber noch weitere Ratio-nalen, die im Zusammenhang mit einer neoadjuvantenTherapie betrachtet werden sollten. So steht mittlerweileaußer Frage, dass eine multimodale Therapie essentiellfür die Behandlung des Pankreaskarzinoms ist, wobei de-ren vollständige Absolvierung einen entscheidenden Fak-tor für ein verbessertes Überleben darstellt (6). So konntemittlerweile gezeigt werden, dass eine neoadjuvante The-rapie auch mit einer erhöhten Rate an vollständiger Ab-solvierung der multimodalen Therapie einhergeht (7). Zu-sätzlich kommt es nicht selten vor, dass beim initialen Sta-ging des Pankreaskarzinoms kein Hinweis auf eine Metas-tasierung besteht, jedoch intraoperativ eine Filiarisierungfestgestellt werden muss oder innerhalb kurzer Zeit nachder Resektion Fernmetastasen auftreten. Deswegen stellteine weitere Überlegung zur neoadjuvanten Therapie dieDetektion von Patienten dar, welche eine besonders ag-gressive Tumorbiologie bzw. Mikrometastasierung auf-weisen und bereits während der Vorbehandlung eine Pro-gress mit Metastasierung entwickeln. So lassen sich un-nötige „Auf-und-Zu“-Raten vermindern und Patientenmüssen nicht einer Operation mit vermutlich fehlendemNutzen unterzogen werden. Neben der entsprechendenTumortherapie kann ein neoadjuvantes Behandlungskon-zept aber auch dazu dienen, Patienten mit einem grenz-wertig funktionellen Status, welcher mit einer erhöhtenpostoperativen Komplikationsrate und Letalität verbun-den ist, weiter zu evaluieren und ggf. medikamentös zuoptimieren (8). Im schlimmsten Fall kann sich im Rahmender neoadjuvanten Therapie aber auch eine funktionelleInoperabilität offenbaren und den Patienten für eine Pan-kreasresektion als vollkommen ungeeignet identifizieren.Tabelle 1 fasst hierbei noch weitere mögliche Aspekte ei-ner neoadjuvanten Therapie zusammen.

n Seite 34 n JOURNAL 01/2005JOURNAL 01/2015
Tabelle 1 Vorteile und Nachteile einer neoadjuvanten The-rapie beim Pankreaskarzinom; adaptiert nach (4, 8).
Rationale und Möglichkeiten einer neoadjuvanten TherapieErhöhte Rate an R0-ResektionenDownstaging von primär inoperablen Patienten Erhöhte Abschlussrate einer multimodalen TherapieSelektion von Patienten mit aggressiver Tumorbiologie und fehlendem Nutzen einer ResektionVerminderte „Auf-und-Zu“-RatenVerbesserte Einschätzung des funktionellen Status des Patienten durch die neoadjuvante Therapie Erhöhte Effizienz einer Radiotherapie am nativen GewebeAngedeutete, erniedrigte Rate an Pankreasfisteln
Angesichts dieser Aspekte ergeben sich zwei Gruppen,bei denen aktuell eine neoadjuvante Therapie in Abhän-gigkeit von der Befundlage in Frage kommt. So werdenaktuell neoadjuvante Therapiekonzepte bei Patienten mit„resektablen“ Stadien als auch mit „lokal fortgeschritte-nen“ Tumorstadien ohne Hinweis auf eine Fernmetasta-sierung, welche primär jedoch nicht oder nur fraglich re-sektabel sind, evaluiert.
Klassifikation der ResektabilitätDie präoperative Bestimmung der Resektabilität des Pan-kreaskarzinoms hat schon immer eine diagnostische He-rausforderung dargestellt, ist aber gerade für die unter-schiedlichen neoadjuvanten Therapiekonzepte sehr we-sentlich. Aufgrund der komplexen Anatomie im Bereichdes Pankreaskopfes und der engen Lagebeziehung ver-schiedener Gefäß- und Organstrukturen, können auchkleinere Tumore bereits zu einer irresektablen Situationführen. Dabei spielt für eine kurative Resektion vor allemdie Lagebeziehung des Tumors zur Arteria mesentericasuperior (AMS), Arteria hepatica communis (AHC), Venamesenterica superior bzw. Pfortader (VMS-PA) als auchzum Truncus coeliacus (TC) eine wesentliche Rolle. Auf-grund der engen Lagebeziehung der AMS zum Pankreas-kopf bzw. Processus uncinatus innerhalb weniger Milli-meter ist die Beurteilung einer möglichen Infiltration füreine kurative Resektion von herausragender Bedeutung.Zwar ist die Segmentresektion der AMS mit entsprechen-der Rekonstruktion bei lokalisierter Tumorinfiltration inausgewählten Fälle durchaus technisch machbar, diese istjedoch mit einer 5-fach erhöhten perioperativen Letalitätund einer 2-fach erhöhten Letalität im ersten Jahr ver-bunden (9). Im Gegensatz dazu kann eine Infiltration derVMS-PA durchaus chirurgisch durch eine entsprechendeGefäßresektion gelöst werden, ohne die perioperativeMorbidität und Mortalität im Vergleich zur Standardpan-kreasresektion signifikant zu steigern (10). Somit gilt eineInfiltration der AMS aktuell als Kontraindikation für eineResektion des Primärtumors, während venöse Resektio-nen durchaus indiziert sein können. Aus diesem Grundgalt ein Pankreaskarzinom bis dato immer dann als „re-sektabel“, wenn eine freie VMS-PA, kein Hinweis auf eineMetastasierung und keine Infiltration der AMS vorlag.Präoperativ lassen sich jedoch die Tumorausbreitung bzw.
Gefäßinfiltration manchmal schwer einordnen und einedefinitive Resektabilität kann nicht immer klassifiziert wer-den. Aufgrund dieser Tatsache werden Pankreaskarzino-me aktuell in drei Subgruppen in Bezug auf deren Resek-tabilität eingeteilt (siehe Tabelle 2). Die erste Gruppe be-zeichnet dabei „resektable“ Karzinome, welche keine Ge-fäßinfiltration aufweisen und von tumorfreien Resek-tionsrändern ausgegangen werden kann. Eine primäreOperation ist somit bei diesen Patienten indiziert. Diezweite Gruppe beinhaltet die sogenannten „Borderline“-Tumore, welche zwar mit erhöhtem technischem Auf-wand vermutlich reseziert werden können, aber ein er-höhtes Risiko, eine R1-Situation zu erhalten, haben. Dieletzte Gruppe besteht aus den „nicht-resektablen“ Karzi-nomen, welche eine chirurgisch nicht zu versorgende Ge-fäßinfiltration aufweisen, sodass diese Tumore prinzipielleiner Resektion nicht zugänglich sind. Borderline-Tumoresowie nicht resektable Karzinome werden meist auch alslokal fortgeschrittene Karzinome zusammengefasst.
Da die Resektabilität im Rahmen der präoperativen CT-bzw. MRT-Diagnostik beurteilt wird, wurde initial vom M.D. Anderson Cancer Center eine entsprechende Klassifi-kation anhand von CT-Parametern geschaffen (11). Diesewurde in weiterer Folge vom National ComprehensiveCancer Network (NCCN) modifiziert und wird mittlerweileauch von der International Study Group of Pancreatic Sur-gery (ISGPS) zur Beurteilung der Resektabilität empfohlen(12, 13). Tabelle 2 fast diese Klassifikation der NCCN zu-sammen. Es gilt jedoch zu beachten, dass nach wie vorkein einheitlicher Standard zur Definition der Resektabili-tät in der Literatur existiert. Für eine weitere multimodaleBehandlung, gerade im Sinne einer neoadjuvanten Thera-pie, ist eine derartige Einteilung jedoch unerlässlich.
Neoadjuvante Therapie beim resektablen Pankreas-karzinomZur neoadjuvanten Therapie beim resektablen Pankreas-karzinom wurden in den letzten beiden Jahrzehnten eini-ge Phase-II-Studien veröffentlicht. Es gilt jedoch an dieserStelle kritisch anzumerken, dass bis zum jetzigen Zeit-punkt keine Ergebnisse einer randomisierten kontrollier-ten Studie vorliegen, welche eine neoadjuvante Therapiegefolgt von einer Resektion mit der primären Resektionvergleicht (14). Eine Studie, welche aktuell eine neoadju-vante Chemotherapie mit Gemcitabine vs. der primärenOperation untersucht, ist die multizentrische NEOPAC-Studie (NCT 01314027).
In allen anderen bisher veröffentlichten, meist retrospek-tiven Arbeiten finden sich vor allem radiochemotherapeu-tische Therapiemodalitäten mit unterschiedlichen Sche-mata, Strahlungsdosen und Chemotherapeutika. Eine derbisher umfangreichsten Patientenserien wurde vom M. D.Anderson Cancer Center veröffentlicht (15, 16, 17, 18). Sokonnten diese Arbeiten erfolgreich demonstrieren, dassbei Patienten, welche nach Abschluss der neoadjuvantenTherapie keinen Hinweis auf einen Tumorprogress haben,eine höhere R0-Resektionsrate, niedrigere Lokalrezidivrate

n Seite 35 nJOURNAL 01/2015
und verbesserte Überlebensrate erzielt werden konnten(19). Umgekehrt kam es aber auch bei bis zu 26% der Pa-tienten nach abgeschlossener neoadjuvanter Therapie zueinem Progress des Pankreaskarzinoms, sodass eine wei-tere Resektion nicht mehr möglich war (20). Es ist jedochwahrscheinlich, dass gerade diese Patienten auch nichtvon einer primären Resektion ohne neoadjuvante Vorbe-handlung profitiert hätten, da bei diesen offensichtlichvon einer aggressiveren Tumorbiologie auszugehen ist.
Dass die Datenlage zur neoadjuvanten Therapie beim re-sektablen Pankreaskarzinom jedoch nicht unbedingt ho-mogen ist, zeigen zwei rezente Metaanalysen (20, 21). Inbeiden Arbeiten konnte kein signifikanter Vorteil einerneoadjuvanten Vorbehandlung beim primär resektablenPankreaskarzinom nachgewiesen werden. Es zeigten sichsogar ähnliche mediane Überlebensrate von 20.1 – 23.6Monaten bei primärer Resektion, verglichen mit einemmedianen Überleben von 23.4 Monaten nach neoadju-vanter Therapie (21).
Aufgrund der fehlenden Datenlage kann, trotz der ver-meintlichen theoretischen Vorteile, aktuell keine Empfeh-lung zur neoadjuvanten Therapie beim primär resektablenPankreaskarzinom ausgesprochen werden. Die Anwen-dung eines derartigen Therapiekonzeptes bei resektablenTumoren wird deshalb auch von den gültigen S3-Leitliniennicht empfohlen und sollte lediglich auf Studien be-schränkt bleiben.
Neoadjuvante Therapie beim lokal fortgeschrittenenPankreaskarzinom (Borderline- und nicht-resektableTumore)Obwohl, wie oben angeführt, mittlerweile Kriterien zurDefinition von Borderline- und nicht-resektablen Tumorenvorliegen, ist gerade der Vergleich von Studien, welchesich der neoadjuvanten Therapie bei diesem Patientengutwidmen, aufgrund der Heterogenität der Resektabilitäts-definitionen als auch der verwendeten Therapieregimeäußerst schwierig. Es überrascht deswegen nicht, dasssich aktuell noch keine definitive Empfehlung zur neoad-juvanten Therapie beim Borderline- bzw. lokal-fortge-schrittenen Pankreaskarzinom abzeichnet. Die häufigstenTherapieregime, welche bis dato beim lokal fortgeschrit-tenen Pankreaskarzinom zur Anwendung kamen, sindGemcitabine und 5-Fluoruracil (5-FU) als Monotherapieoder eine Induktionschemotherapie mit Gemcitabine ge-folgt von einer Radiochemotherapie mit 5-FU oder Gem-citabine als Radiosensitizer (20, 21). So konnte in einerMetaanalyse unter Einschluss von 111 Studien gezeigtwerden, dass durch derartige Therapieschemata eine Re-sektionsrate von 33.2% bei Patienten mit initialem lokalfortgeschrittenen und teilweise nicht-resektablen Befunderreicht werden kann (21). Zusätzlich zu diesem bereitsbeachtlichen Ergebnis haben diese Patienten auch ein me-dianes Überleben von 20.5 Monaten, welches dem Out-come von primär resezierten Patienten entspricht (Abb. 1).
Abb. 1. Therapeutische Optionen beim Pan-kreaskarzinom mit entsprechenden Überle-bensraten in Monaten nach Gillen et al. (21).
Tabelle 2 Kriterien der NCCN zur Definition der Resektabilität beim Adenokarzinom des Pankreas (13) VMS-PA=Vena mesenterica superior-Pfortader, AHC=Arteria hepatica communis, AMS=Arteria mesenterica superior,TC=Truncus coeliacus.
ResektabelKeine FernmetastasenKein Kontakt zur VMS-PA oder <180° Kontakt ohne Wandunregelmäßigkeit
Kein Kontakt zur AHC, AMS oder TC
Borderline-resektabelKeine FernmetastasenTumorkontakt zur VMS-PA >180° mit Wand-unregelmäßigkeiten oder Thrombose. Venöse Resektion und Rekonstruktion sind aber möglich.Tumorkontakt zur Vena cava inferiorTumorkontakt zur AHC ohne Affektion zum TC mit der Möglichkeit der Resektion undRekonstruktionTumorkontakt zur AMS <180°
Nicht-resektabelFernmetastasierungTumorinfiltration der VMS-PA ohne Möglichkeitder Rekonstruktion
Tumorkontakt zur AMS >180°Tumorkontakt zum TC
Infiltration der Aorta

In den letzten Jahren hat die Kombination von Folinsäure,5-FU, Irinotecan und Oxaliplatin (FOLFIRINOX) einen zu-nehmenden Stellenwert, vor allem beim metastasiertenPankreaskarzinom erhalten, und wenn nicht sogar eineneue Ära eingeleitet. So konnte die ACCORD-11/PRODI-GE-4-Studie, deren Daten 2011 veröffentlicht wurden, ei-ne Verlängerung des medianen Überlebens von 6.8 Mo-nate auf 11.1 Monate durch die Verwendung von FOLFI-RINOX im Vergleich zur Gemcitabinetherapie beim me-tastasierten Pankreaskarzinom nachweisen (22). Eineerhöhte Toxizität dieses Chemotherapieschemas darf da-bei jedoch nicht außer Acht gelassen werden. Nach die-sem überragenden Ergebnis ist es nicht verwunderlich,dass FOLFIRINOX zunehmend zur neoadjuvanten Therapievon Borderline- bzw. nicht-resektablen Tumoren verwen-det wurde, um so ein schnelles und signifikantes Down-staging bei lokal fortgeschrittenen Tumoren zu erreichen.Dabei wird FOLFIRINOX sehr häufig zur Induktionsthera-pie durchgeführt, gefolgt von einer Radiochemotherapiemit 5-FU oder Gemcitabine. Ein derartiger Algorithmusmit Induktionschemotherapie gefolgt von einer kombi-nierten Radiochemotherapie beim lokal fortge- schritte-nen Pankreaskarzinom scheint in einer retrospektivenAnalyse von 4 prospektiven Studien durch die Groupe Co-operatéur Multidisciplinaire en Oncologie (GERCOR) miteinem verbesserten Überleben verbunden zu sein (23). Ei-ne aktuelle Meta-Analyse zur Verwendung von FOLFIRI-NOX konnte so anhand der Analyse von 13 eingeschlos-senen Studien eine R0-Resektionsrate von 40% bei initia-len Borderline- bzw. nicht-resektablen Tumoren nachwei-sen (24). In weiterer Folge soll nun ein Fallbeispiel einesinitial nicht-resektablen Pankreaskarzinoms, welches mit-tels neoadjuvanten Therapieansatz erfolgreich am HELIOSKlinikum Erfurt behandelt wurde, dargestellt werden.
FalldemonstrationEs wird der Fall eines 58-jährigen Patienten dargestellt,welcher sich von November 2014 bis August 2015 in un-serer Behandlung befand und sich erfolgreich einem neo-adjuvanten Konzept unterzogen hat. Initial stellte sich derPatient im Oktober 2014 an einem auswärtige Klinikumwegen seit 8 Wochen bestehender Durchfallsymptomatikmit intermittierenden unspezifischen Oberbauchschmer-zen vor. Zusätzlich beschrieb der Patient einen Gewichts-verlust von ca. 12 kg innerhalb von 8 Wochen. Die Be-stimmung der Tumormarker ergab dabei ein CA 19-9 bei39 E/ml sowie ein CEA bei 6.6 ng/ml. Zur weiteren Abklä-rung wurde eine Computertomographie des Abdomensdurchgeführt. Hier konnte eine Raumforderung im Be-reich des Pankreaskopfes ohne eindeutigen Hinweis aufeine Gefäßinfiltration oder eine Organmetastasierungdargestellt werden (Abb. 2). Ergänzend wurde noch eineMagnetresonanztomographie des Pankreas durchgeführt(Abb. 3). Hier zeigte sich ein möglicher struktureller Kon-takt zum proximalen Bereich der VMS. Der Tumor wurdemit weniger als 90° Kontaktfläche zum Gefäß eingestuft.
Abb. 2. Computertomographie des Abdomens mit Darstellung des Pankre-askopftumors. (A) und (B) zeigen dabei die Raumforderung im Pankreaskopf(weißer Pfeil) mit begleitender Dilatation des Ductus pancreaticus (A). VMSund AMS stellen sich in (A) ohne eindeutigen Hinweis auf eine Tumorinfil-tration dar.
Der Patient wurde in weiterer Folge an unserer Klinik zurResektion des Primärtumors vorgestellt. Da laut Bildge-bung eine resektable Situation vorlag, planten wir im No-vember 2015 die Resektion. Intraoperativ fand sich jedochsowohl eine Infiltration der VMS als auch der AMS, welcheauch histologisch bestätigt wurde. Da zu diesem Zeit-punkt somit eine Resektion weder technisch möglich nochindiziert war, wurde der Eingriff als explorative Laparoto-mie beendet. Der Casus wurde in weiterer Folge in unsererinterdisziplinären Tumorkonferenz besprochen und eineneoadjuvante Chemotherapie mit FOLFIRINOX beschlos-sen. Diese wurde vom Patienten ambulant durchgeführtund nach 4 Zyklen erfolgte ein Re-Staging mittels MRCP.Hier zeigte sich der Lokalbefund im Pankreaskopf größen-konstant ohne Hinweis auf ein Therapieansprechen. Ausdiesem Grund wurde nach nochmaliger Diskussion desCasus in unserer Tumorkonferenz ein Vorgehen analogdes Prüfarmes der CONKO-007-Studie (NCT 01827553)festgelegt. Dabei wurden noch 2 Zyklen FOLFIRINOX zurKomplettierung der Induktionschemotherapie verab-reicht. In Anschluss daran erfolgte eine kombinierte Ra-diochemotherapie mit einer erreichten Gesamtdosis von50,4 Gray (Einzeldosen zu je 1,8 Gy) und der Gabe vonGemcitabine (300 mg/m²/Tag) an Tag 1, 8, 15, 22 und 29.
Abb. 3. Magnetresonanz-Cholangiopankreatikographie (MRCP) im Rahmendes initialen Stagings. (A) Darstellung des Pankreaskopftumors mit mögli-chen Kontakt zur VMS (Pfeil). (B) Zusätzliche Darstellung des „double-duct-Zeichens“ mit einer geringen Dilatation des Ductus hepatocholedochus so-wie erweiterten Ductus pancreaticus als Hinweis für eine Tumorobstruktion.
Diese wurde vom Patienten soweit gut vertragen und the-rapiespezifische Nebenwirkungen traten im Verlauf nichtauf. Nach Abschluss der Radiochemotherapie wurdenochmals eine Computertomographie des Abdomensdurchgeführt. Hier zeigte sich im Wesentlichen nur eingeringfügig kleiner gewordener Pankreaskopftumor. Da
n Seite 36 n JOURNAL 01/2005JOURNAL 01/2015

aber bekannt ist, dass nach neoadjuvanter Radiochemo-therapie ein Therapieansprechen in der radiologischenBildgebung schwer zu beurteilen ist (25), wurde im Au-gust 2015, 7 Wochen nach Abschluss der Radiochemo-therapie, die erneute Operation durchgeführt. Intraope-rativ gestaltete sich die Präparation aufgrund derber Ad-häsionen sowie einer entsprechenden Fibrose etwas er-schwert. Es ließ sich jedoch zu diesem Zeitpunkt keineweitere Gefäßinfiltration nachweisen, sodass nun erfreu-licherweise die geplante pyloruserhaltende Pankreatiko-duodenektomie (Abb. 4) ohne Gefäßresektion komplika-tionslos durchgeführt werden konnte.
Abb. 4. (A) Schematische Darstellung der pyloruserhaltenden Pankreaskopf-resektion (PPPD) mit Pankreatikojejunostomie, biliodigestiver Anastomoseund Duodenojejunostomie. (B) Typisches Präparat nach PPPD bei Pankreas-kopftumor.
Die histologische Aufarbeitung des OP-Präparates er-brachte dabei den Nachweis deutlich regressiv veränder-ter Reste eines duktalen Adenokarzinoms ohne Organ-überschreitung, welche eine maximale Ausdehnung von4 mm aufwiesen. Ein Lymphknotenbefall konnte ebensonicht nachgewiesen werden.
So konnte durch das durchgeführte neoadjuvante Be-handlungskonzept folgende Ausbreitungsklassifikationerzielt werden: ypT1 ypN0 L0 V0 R0. Der Patient wurdeam 12. postoperativen Tag nach unkompliziertem Kost-aufbau und entsprechender Mobilisation in die häuslicheVersorgung entlassen. Entsprechend der S3-Leitlinie undim Sinne des perioperativen Behandlungskonzeptes wur-de in weiterer Folge eine adjuvante Chemotherapie mitGemcitabine beschlossen und ambulant eingeleitet.
DiskussionNeoadjuvante Therapiekonzepte scheinen besonders beilokal fortgeschrittenen Pankreaskarzinomen von beson-derem Nutzen zu sein. Durch ein entsprechendes multi-modales Vorgehen können so bis zu 30% der Patienteneiner Resektion zugeführt werden, bei der immerhin in80% der Fälle eine R0-Situation erreicht werden kann (20,21). Beachtet man die Tatsache, dass diese Patienten bisdato keiner Resektion und somit keiner kurativen Intenti-on zugeführt werden konnten, stellt dies durchaus einbeachtliches Ergebnis dar. Durch die Verwendung vonFOLFIRINOX als Chemotherapeutikum gelingt offenbar
noch eine Erhöhung der Resektionsrate auf 40%, wie ineiner aktuellen Metaanalyse gezeigt werden konnte (24).Da diese Daten jedoch auf retrospektiven Arbeiten beru-hen, wird es an zukünftigen randomisierte Studien liegen,diese Ergebnisse noch besser herauszuarbeiten bzw. zubestätigen. Die multizentrische CONKO-007-Studie, wel-che aktuell Patienten rekrutiert, untersucht dabei die al-leinige neoadjuvante Chemotherapie im Vergleich mit ei-ner Induktionstherapie gefolgt von einer neoadjuvantenRadiochemotherapie (NCT01827553). Dabei kommen alsChemotherapeutika im Rahmen der Indikationstherapiebzw. im Kontrollarm entweder Gemcitabine oder FOLFIRI-NOX zum Einsatz. Die Ergebnisse dieser multizentrischen,randomisierten Studie werden mit großer Spannung er-wartet, um Rückschlüsse auf den definitiven Einfluss vonFOLFIRINOX schließen zu können. Auf der anderen Seitewird auch zum ersten Mal der direkte Vergleich einer al-leinigen Chemotherapie mit einer kombinierten Radio-chemotherapie durchgeführt.
Die Radiotherapie im Rahmen eines multimodalen Ma-nagements wurde vor ca. 3 Jahrzehnten zum ersten Maldurch die Gastrointestinal Tumor Study Group (GITSG)evaluiert. Durch die adjuvante Radiotherapie konnte einmedianes Überleben von 21 Monaten im Vergleich zu 10Monaten in der Kontrollgruppe erzielt werden (26). Wäh-rend der Radiotherapie somit im angloamerikanischenRaum ein gewisser Stellenwert eingeräumt wurde, ist dieadjuvante Radio(chemo-)therapie im europäischen Raumnicht verbreitet und wird kritisch diskutiert. Diese Tatsacheberuht im Wesentlichen auf zwei europäischen Studien.Eine Studie der European Organization for Research andTreatment of Cancer (EORTC) zur adjuvanten Radioche-motherapie erbrachte zwar einen Überlebensvorteil von5.5 Monaten im Vergleich zur Kontrollgruppe (24.5 vs.19.0), konnte jedoch keine statistische Signifikanz errei-chen (p=0.208) (27). Die zweite europäische Studie, dieEuropean Study Group for Pancreatic Cancer-1 (ESPAC-1)Studie, fand sogar eine reduziertes Überleben im Ver-gleich zur Kontrollgruppe (15.5 vs. 16.1) (28, 29). BeideStudien werden zwar heftig in Bezug auf den Studienauf-bau und die mangelnde Durchführung der Strahlenthera-pie kritisiert, aber es fehlen schlussendlich aktuelle rando-misierte Studien zur adjuvanten Radiochemotherapie, umdiese Arbeiten möglicherweise zu widerlegen. Im Gegen-satz zur adjuvanten Therapie konnte sich die Radioche-motherapie als neoadjuvantes Therapieregime bereitssehr wohl etablieren. Während dabei die Chemotherapieeher versucht, der systemischen Ausbreitung bei lokalfortgeschrittenen Pankreaskarzinomen entgegenzuwir-ken, reduziert eine Radiotherapie den Anteil an vitalenZellen im Tumorrandbereich (30). So ist vor allem einekombinierte Radiochemotherapie mit erhöhten Raten vonTumorfibrosen, erhöhten R0-Resektionsraten sowie teil-weise erniedrigten Lokalrezidivraten assoziiert (30, 31).
Auch sollte beachtet werden, dass, ähnlich wie bei derRadiochemotherapie, im Rahmen der FOLFIRINOX-Thera-pie ebenfalls eine gewisse Fibrose, um die mesenterialen
n Seite 37 nJOURNAL 01/2015

Gefäße zurückbleiben kann, ohne jedoch maligne Zellenzu enthalten (32). Dies kann natürlich in der Bildgebungals ein Tumorresiduum interpretiert und so als Inoperabi-litätskriterium fehlgedeutet werden. Eine ähnliche Situa-tion findet sich in dem zuvor beschriebenen Casus, beidem unmittelbar präoperativ kein größerer Befundwan-del festgestellt werden konnte. Erst intraoperativ ließ sicheine fehlende Tumorinfiltration bei bestehender Fibrosebeweisen. Diese wurde ebenso histologisch bestätigt.Umso wichtiger ist also auch die interdisziplinäre Befund-beurteilung, um eine maximalen Response beurteilen zukönnen und das weitere Procedere zu planen. Im Zweifelsollte dann aus unserer Sicht auch immer eine chirurgi-sche Exploration durchgeführt werden, um die lokale Si-tuation makroskopisch verifizieren zu können bzw. umkeinen Patienten seine Chance auf eine mögliche R0-Re-sektion durch eine Fehlinterpretation zu verwehren.
Die aktuelle S3-Leitlinie zum Pankreaskarzinom äußertsich nicht zuletzt aufgrund der eher noch nicht eindeuti-gen Datenlage nur sehr eingeschränkt in Bezug auf eineneoadjuvante Therapie (33). Bei resektablen Befundenwird jedoch außerhalb von Studien aktuell keine neoad-juvante Therapie empfohlen, zudem auch bis dato keineindeutiger Benefit bei dieser Patientengruppe gezeigtwerden konnte (20, 21, 33). Im Gegensatz dazu kann lautS3-Leitlinie bei Patienten mit einem lokal fortgeschritte-nen Tumor eine neoadjuvante Therapie, auch außerhalbvon Studien, durchgeführt werden. Es werden zwar alleOptionen (Chemotherapie, Radiotherapie bzw. Radioche-motherapie) angeführt, aber entsprechende Empfehlun-gen bzw. Angaben zur Durchführung der möglichen The-rapieschemata werden, im Gegensatz zu NCCN-Guideli-nes, nicht erläutert. Aus diesem Grund sollten bei diesenPatienten entsprechende Studienprotokolle, wie z.B. derCONKO-007-Studie, zur Anwendung kommen. Zusätzlichmuss an dieser Stelle auch noch einmal die Notwendigkeitder Behandlung dieser Patienten in einem entsprechen-den Zentrum mit allen multimodalen Therapiemöglich-keiten erwähnt werden, um den korrekten Ablauf bzw.Erfolg einer neoadjuvanten Therapie sicherstellen zu kön-nen. Auch muss gerade bei Patienten mit einem Borderli-ne-Tumor und der Möglichkeit einer intraoperativen ve-nösen Gefäßresektion ein entsprechendes Maß an chirur-gischer Expertise vorhanden sein, um eine kurative Resek-tion sicherstellen zu können.
ZusammenfassungDie neoadjuvante Therapie des Pankreaskarzinom stellteinen neuen Ansatz dar, das Überleben von Patienten, vorallem mit einem lokal fortgeschrittenen Tumor, signifikantzu verbessern. Dabei scheinen aktuell vor allem multimo-dale Konzepte mit FOLFIRINOX und kombinierter Radio-chemotherapie von Vorteil zu sein. Um jedoch endgültigeRückschlüsse ziehen zu können und Empfehlungen in Be-zug auf das optimale Therapieregime aussprechen zu kön-nen, sind dringend randomisiert, kontrollierte Studien er-forderlich. Zusätzlich sollten Patienten aufgrund der In-terdisziplinarität und Komplexität neoadjuvanter Thera-
piekonzepte zur Sicherstellung des Behandlungserfolgesnur an entsprechenden Zentren behandelt werden.
Literatur
(1) Hidalgo M (2010) Pancreatic cancer. The New England journal ofmedicine 362:1605-1617
(2) Winter JM, Brennan MF, Tang LH, D’angelica MI, Dematteo RP,Fong Y, Klimstra DS, Jarnagin WR, Allen PJ (2012) Survival afterresection of pancreatic adenocarcinoma: results from a single in-stitution over three decades. Annals of surgical oncology19:169-175
(3) Sohn TA, Yeo CJ, Cameron JL, Koniaris L, Kaushal S, Abrams RA,Sauter PK, Coleman J, Hruban RH, Lillemoe KD (2000) Resectedadenocarcinoma of the pancreas-616 patients: results, out-comes, and prognostic indicators. Journal of gastrointestinalsurgery 4:567-579
(4) Fathi A, Christians KK, George B, Ritch PS, Erickson BA, Tolat P,Johnston FM, Evans DB, Tsai S (2015) Neoadjuvant therapy forlocalized pancreatic cancer: guiding principles. Journal of gas-trointestinal oncology 6:418-429
(5) Lowy AM (2008) Neoadjuvant therapy for pancreatic cancer.Journal of gastrointestinal surgery 12:1600-1608
(6) Bilimoria KY, Bentrem DJ, Ko CY, Tomlinson JS, Stewart AK, Win-chester DP, Talamonti MS (2007) Multimodality therapy for pan-creatic cancer in the U.S.: utilization, outcomes, and the effectof hospital volume. Cancer 110:1227-1234
(7) Tzeng CW, Tran Cao HS, Lee JE, Pisters PW, Varadhachary GR,Wolff RA, Abbruzzese JL, Crane CH, Evans DB, Wang H, AbbottDE, Vauthey JN, Aloia TA, Fleming JB, Katz MH (2014) Treatmentsequencing for resectable pancreatic cancer: influence of earlymetastases and surgical complications on multimodality therapycompletion and survival. Journal of gastrointestinal surgery18:16-24
(8) Sutton JM, Abbott DE (2014) Neoadjuvant therapy for pancreascancer: past lessons and future therapies. World journal of gas-troenterology 20:15564-15579
(9) Mollberg N, Rahbari NN, Koch M, Hartwig W, Hoeger Y, BüchlerMW, Weitz J (2011) Arterial resection during pancreatectomy forpancreatic cancer: a systematic review and meta-analysis. Annalsof surgery 254:882-893
(10) Zhou Y, Zhang Z, Liu Y, Li B, Xu D (2012) Pancreatectomy com-bined with superior mesenteric vein-portal vein resection forpancreatic cancer: a meta-analysis. World journal of surgery36:884-891
(11) Varadhachary GR, Tamm EP, Abbruzzese JL, Xiong HQ, Crane CH,Wang H, Lee JE, Pisters PW, Evans DB, Wolff RA (2006) Border-line resectable pancreatic cancer: definitions, management, androle of preoperative therapy. Annals of surgical oncology13:1035-1046
(12) Bockhorn M, Uzunoglu FG, Adham M, Imrie C, Milicevic M,Sandberg AA, Asbun HJ, Bassi C, Buchler M, Charnley RM, Con-lon K, Cruz LF, Dervenis C, Fingerhutt A, Friess H, Gouma DJ,Hartwig W, Lillemoe KD, Montorsi M, Neoptolemos JP,Shrikhande SV, Takaori K, Traverso W, Vashist YK, Vollmer C, YeoCJ, Izbicki JR, International Study Group of Pancreatic S (2014)Borderline resectable pancreatic cancer: a consensus statementby the International Study Group of Pancreatic Surgery (ISGPS).Surgery 155:977-988
(13) National Conprehensive Cancer Network. NCCN (2015) ClinicalPractice Guidelines in Oncology. Pancreatic Adenocarcinoma.Version 2. 2015.http://www.nccn.org/professionals/physician_gls/f_guidelines.asp
(14) Puleo F, Marechal R, Demetter P, Bali MA, Calomme A, Closset J,Bachet JB, Deviere J, Van Laethem JL (2015) New challenges inperioperative management of pancreatic cancer. World journalof gastroenterology 21:2281-2293
(15) Evans DB, Rich TA, Byrd DR, Cleary KR, Connelly JH, Levin B,Charnsangavej C, Fenoglio CJ, Ames FC (1992) Preoperativechemoradiation and pancreaticoduodenectomy for adenocarci-noma of the pancreas. Archives of surgery 127:1335-1339
n Seite 38 n JOURNAL 01/2005JOURNAL 01/2015

(16) Pisters PW, Wolff RA, Janjan NA, Cleary KR, Charnsangavej C,Crane CN, Lenzi R, Vauthey JN, Lee JE, Abbruzzese JL, Evans DB(2002) Preoperative paclitaxel and concurrent rapid-fractiona-tion radiation for resectable pancreatic adenocarcinoma: toxici-ties, histologic response rates, and event-free outcome. Journalof clinical oncology 20:2537-2544
(17) Evans DB, Varadhachary GR, Crane CH, Sun CC, Lee JE, PistersPW, Vauthey JN, Wang H, Cleary KR, Staerkel GA, CharnsangavejC, Lano EA, Ho L, Lenzi R, Abbruzzese JL, Wolff RA (2008) Preop-erative gemcitabine-based chemoradiation for patients with re-sectable adenocarcinoma of the pancreatic head. Journal of clin-ical oncology 26:3496-3502
(18) Varadhachary GR, Wolff RA, Crane CH, Sun CC, Lee JE, PistersPW, Vauthey JN, Abdalla E, Wang H, Staerkel GA, Lee JH, RossWA, Tamm EP, Bhosale PR, Krishnan S, Das P, Ho L, Xiong H, Ab-bruzzese JL, Evans DB (2008) Preoperative gemcitabine and cis-platin followed by gemcitabine-based chemoradiation for re-sectable adenocarcinoma of the pancreatic head. Journal of clin-ical oncology 26:3487-3495
(19) Paulson AS, Tran Cao HS, Tempero MA, Lowy AM (2013) Thera-peutic advances in pancreatic cancer. Gastroenterology144:1316-1326
(20) Assifi MM, Lu X, Eibl G, Reber HA, Li G, Hines OJ (2011) Neoad-juvant therapy in pancreatic adenocarcinoma: a meta-analysis ofphase II trials. Surgery 150:466-473
(21) Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H, Kleeff J(2010) Preoperative/neoadjuvant therapy in pancreatic cancer: asystematic review and meta-analysis of response and resectionpercentages. PLoS medicine 7:e1000267
(22) Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Be-couarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, De LaFouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, Groupe Tumeurs Digestives Of U, Inter-group P (2011) FOLFIRINOX versus gemcitabine for metastaticpancreatic cancer. The New England journal of medicine364:1817-1825
(23) Huguet F, Andre T, Hammel P, Artru P, Balosso J, Selle F, Deniaud-Alexandre E, Ruszniewski P, Touboul E, Labianca R, De GramontA, Louvet C (2007) Impact of chemoradiotherapy after diseasecontrol with chemotherapy in locally advanced pancreatic ade-nocarcinoma in GERCOR phase II and III studies. Journal of clini-cal oncology 25:326-331
(24) Petrelli F, Coinu A, Borgonovo K, Cabiddu M, Ghilardi M, LonatiV, Aitini E, Barni S, Gruppo Italiano Per Lo Studio Dei CarcinomiDell’apparato D (2015) FOLFIRINOX-based neoadjuvant therapyin borderline resectable or unresectable pancreatic cancer: ameta-analytical review of published studies. Pancreas 44:515-521
(25) Ferrone CR, Marchegiani G, Hong TS, Ryan DP, Deshpande V, Mc-donnell EI, Sabbatino F, Santos DD, Allen JN, Blaszkowsky LS,Clark JW, Faris JE, Goyal L, Kwak EL, Murphy JE, Ting DT, Wo JY,Zhu AX, Warshaw AL, Lillemoe KD, Fernandez-Del Castillo C(2015) Radiological and surgical implications of neoadjuvanttreatment with FOLFIRINOX for locally advanced and borderlineresectable pancreatic cancer. Annals of surgery 261:12-17
(26) Gastrointestinal Tumor Study Group (1987) Further evidence ofeffective adjuvant combined radiation and chemotherapy fol-lowing curative resection of pancreatic cancer. Cancer 59:2006-2010
(27) Klinkenbijl JH, Jeekel J, Sahmoud T, Van Pel R, Couvreur ML,Veenhof CH, Arnaud JP, Gonzalez DG, De Wit LT, Hennipman A,Wils J (1999) Adjuvant radiotherapy and 5-fluorouracil after cur-ative resection of cancer of the pancreas and periampullary re-gion: phase III trial of the EORTC gastrointestinal tract cancer co-operative group. Annals of surgery 230:776-782
(28) Neoptolemos JP, Dunn JA, Stocken DD, Almond J, Link K, BegerH, Bassi C, Falconi M, Pederzoli P, Dervenis C, Fernandez-Cruz L,Lacaine F, Pap A, Spooner D, Kerr DJ, Friess H, Büchler MW, Euro-pean Study Group for Pancreatic C (2001) Adjuvant chemoradio-therapy and chemotherapy in resectable pancreatic cancer: arandomised controlled trial. Lancet 358:1576-1585
(29) Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, HickeyH, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, Falconi M,Pederzoli P, Pap A, Spooner D, Kerr DJ, Büchler MW, EuropeanStudy Group for Pancreatic C (2004) A randomized trial ofchemoradiotherapy and chemotherapy after resection of pan-creatic cancer. The New England journal of medicine 350:1200-1210
(30) Katz MH, Landry J, Kindler HL (2014) Current controversies in thestage-specific multidisciplinary management of pancreatic can-cer. American Society of Clinical Oncology educational book /ASCO. American Society of Clinical Oncology. Meeting:e157-164
(31) Pingpank JF, Hoffman JP, Ross EA, Cooper HS, Meropol NJ, Freed-man G, Pinover WH, Levoyer TE, Sasson AR, Eisenberg BL (2001)Effect of preoperative chemoradiotherapy on surgical marginstatus of resected adenocarcinoma of the head of the pancreas.Journal of gastrointestinal surgery 5:121-130
(32) Khushman M, Dempsey N, Cudris Maldonado J, Loaiza-Bonilla A,Velez M, Carcas L, Dammrich D, Hurtado-Cordovi J, Parajuli R,Pollack T, Harwood AP, Macintyre J, Tzeng CD, Merchan JR, Re-strepo MH, Akunyili, Ii, Ribeiro A, Narayanan G, Portelance L,Sleeman D, Levi JU, Rocha Lima CM, Hosein PJ (2015) Full doseneoadjuvant FOLFIRINOX is associated with prolonged survival inpatients with locally advanced pancreatic adenocarcinoma. Pan-creatology doi: 10.1016/j.pan.2015.08.010.]
(33) AWMF (2013) S3-Leitlinie zum exokrinen Pankreaskarzinom. Ver-sion 1.0. http://www.awmf.org/leitlinien/detail/ll/032-010OL.html
Korrespondenzadresse:
Dr. Dr. med. univ. Markus MilleKlinik für Allgemein- und ViszeralchirurgieHELIOS Klinikum ErfurtNordhäuser Str. 7499089 ErfurtTelefon: 0361/781 6163 e-Mail: [email protected]
n Seite 39 nJOURNAL 01/2015

n Was ist tumor-associated tissueeosinophilia (TATE)
Abstract des Vortrags zum „Jungen Forum Onkolo-gie“ auf der 28. Onkologischen Konferenz des Tu-morzentrum Erfurt e.V. am 6. und 7. November 2015in Eisenach
Tomáš NácovskýInstitut für Pathologie, HELIOS Klinikum Erfurt
EinleitungTumor-associated tissue eosinophilia (TATE, Tumorassozi-ierte Gewebseosinophilie) wird als „eosinophil granulo-zytäre Infiltration im Tumorstroma ohne Zusammenhangmit Ulzeration oder Nekrose“ definiert. Die Literatur ver-wendet den Begriff jedoch fast ausschließlich nur im Zu-sammenhang mit Karzinomen, wenngleich eine Gewebs-eosinophilie (vermehrte eosinophile Granulozyten in ei-nem Gewebe) auch innerhalb anderer maligner sowie be-nigner Tumoren vorkommen kann (Tabelle 1).
Interessanterweise ist dieses Phänomen bereits seit 1896bekannt, trotzdem wird es aber erst in den letzten Jahrenausführlicher epidemiologisch, immunologisch und histo-logisch studiert. Weil die Definition der TATE keine gene-rell gültige minimale Dichte des eosinophilen Infiltratesangibt, werden in den Studien individuell nummerischeingegrenzte Gruppen mit unterschliedlich starker Ge-webseosinophilie verglichen und abweichende Eigen-schaften der stärker eosinophil durchsetzten Gruppen alsTATE-Eigenschaften interpretiert.
Wo findet sich TATE?Die meisten bisherigen Studien beschäftigen sich mit TATEin Plattenepithelkarzinomen des Kopf-Hals-Bereichs. TATEkann allerdings ebenfalls in zahlreichen anderen Organenauftreten, inklusive distaler Teile vom Verdauungstrakt,des Urogenitalsystems, der Lunge oder der Haut. Auchhistologisch erscheint das Spektrum von TATE-assoziier-ten Karzinomen abwechslungsreich - u.a. Plattenepithel-karzinome, Urothelkarzinome sowie Adenokarzinome(Tabelle 2).
Pathogenese der TATEDie Pathogenese der TATE bleibt unklar. Eine Beteiligungder Gewebsschädigung durch den wachsenden Tumor istmöglich. Initiale chemotaktische Faktoren können entwe-der von Tumorzellen oder von anderen Entzündungszellenim Tumorstroma produziert werden. Sogenannte dama-ge-associated molecular pattern molecules (DAMPs), ein-schließlich Kernprotein high-mobility group box 1 (HMGB-1), stellen einige der chemotaktischen Kandidaten dar.HMGB-1 wird von hypoxischen, oxidativ gestressten oderernährungsbedingt gestressten Zellen sezerniert bzw. ausnekrotischen Zellen ausgeschieden. Sekundär spielt Eota-xin, ein von eosinophilen Granulozyten selbst abgeson-dertes Produkt, eine selbststimulierende Rolle im Sinnevon Anziehen neuer Eosinophiler. Weitere allgemeine che-motaktische Faktoren für eosinophile Granulozyten, miteiner möglichen Rolle bei TATE, sind platelet-activatingfactor (PAF), Histamin, Komplementkomponenten C3a /C5a, Macrophage inflammatory protein (MIP), Monocytechemotactic protein-2 (MCP-2), Eosinophiler chemotak-tischer Faktor A, RANTES und IL-5.
Bakterielle, parasitäre und virale Infektionskrankheiten
Autoimmunerkrankungen
Allergie (Typ I)
nicht nur Eosinophile Ösophagitis /Gastroenteritis / Kolitis
Idiopathischsondern auch z.B. eosinophile Cholezystitis (akut oder chronisch?)
Benigne z.B. Kolorektales Adenom
Maligne Karzinome, T-NHL, HL, Sarkome (bei Sarkomen selten)
EntzündlicheErkrankungen
Tumoren (eo.G. im Stroma)
TATE
Tabelle 1 Gewebseosinophilie-Ätiologie
n Seite 40 n JOURNAL 01/2005JOURNAL 01/2015

Tabelle 2 Wo findet sich TATE?(rot – eigene Beobachtungen)
–> Undifferenziertes Karzinom vom nasopharyngealen Typ–> Sklerosierendes Mukoepidermoidkarzinom–> Plattenepithelkarzinome im Kopf-Hals-Bereich–> Plattenepithelkarzinom des Ösophagus–> Adenokarzinom des Magens–> Kolorektales Adenokarzinom–> Nichtkleinzelliges Lungenkarzinom–> Urothelkarzinom der ableitenden Harnwege–> Plattenepithelkarzinom des Penis–> Adenokarzinom des Corpus Uteri–> Plattenepithelkarzinom der Vagina–> Plattenepithelkarzinom der Haut
Bedeutung der TATE und ihre Assoziation mit anderenTumoreigenschaftenEnzym Gelatinase der eosinophilen Granulozyten degra-diert die Basalmembran sowie die Extrazellularmatrix, waseine Tumorinvasion erleichtert. Zu weiteren tumorunter-stützenden Wirkungen der TATE gehört Immunsuppressi-on sowie Produktion von angiogenetischen Faktoren,möglicherweise einschließlich TGF-alpha. Zugleich weisendie eosinophilen Granulozyten allerdings auch karzinom-hemmende Aktivität auf. Permeabilitätssteigerung für an-titumorale Zytokine ist ein erstes Beispiel dazu. Danebensind mehrere Moleküle wie LFA-1 (CD11a/CD18), TNF,Granzym A, eosinophiles kationisches Protein oder eosi-nophiles Neurotoxin für direkte Apoptose der Karzinom-zellen verantwortlich. Drittens werden die erwähnten im-munsuppressiven Eigenschaften durch Aktivierung ande-rer Leukozyten und Mastzellen ausgeglichen. Die aktivier-ten Mastzellen beteiligen sich anschließend durch einendirekten Kontakt mit Tumorzellen sowie durch Degranu-lation an der karzinomhemmenden Kaskade.
Prognostische Bedeutung des immunologisch doppeldeu-tigen Phänomens erläutern erst Studien von Ausbreitung,Überleben bzw. Rekurrenz der Karzinome mit TATE imVergleich zu den gleichen Karzinomtypen ohne TATE. Diemeisten Studien im Kopf-Hals-Bereich und alle von weni-gen Studien in anderen Organen (Ösophagus, Kolorek-tum, eine Studie der TATE im Penis mit lediglich 17 Fällen)interpretieren TATE als prognostich günstige Erscheinung,die antitumoralen eosinophil granulozytären Wirkungenkönnten also als dominant angesehen werden. Nur eineMinderheit von Studien im Kopf-Hals-Bereich brachte un-terschiedliche Ergebnisse, entweder ohne Einfluss auf diePrognose oder sogar mit prognostisch ungünstiger Be-deutung.
In kleinen Proben ist ein invasives und ein in situ Platten-epithelkarzinom der Mundhöhle manchmal histologischschwierig zu unterscheiden. Pathologen können eineeventuelle TATE in diesen Situationen zur richtigen Diag-nosestellung nutzen. In der Mundhöhle kommt die TATEnämlich, laut einer Studie, ausschließlich nur bei invasivenKarzinomen vor, niemals jedoch bei In-situ-Karzinomen.Bei fehlender TATE kommen allerdings beide Entitäten inFrage.
Eines der zukünftigen Prinzipien von antitumoraler Im-muntherapie könnte auf eosinophile Granulozyten gezieltwerden. Es bleibt aber noch umstritten, in was für einemSinne. Während einige Autoren vorschlagen, diese Zellenaufgrund ihres enzymatischen Anteils an der Tumorinva-sion zu blockieren, scheint eine Stimulation der eosino-philen Granulozyten im Kontext der erwähnten prognos-tischen Studien logischer.
TATE zeigt eine Assoziation mit dem Alter. Patienten miteosinophil reichen laryngealen Plattenepithelkarzinomensind durchschnittlich um 10 Jahre jünger als Patientenmit der gleichen Diagnose ohne TATE (40-60 vs. 50-70Jahre). In der Mundhöhle wird eine signifikante Beziehungder TATE zu Tumorgröße angegeben: Je größer das Karzi-nom, desto dichter ist das eosinophile Infiltrat. Im Gegen-teil dazu korreliert histopathologisches Grading mit TATEnicht, wenigstens in den bislang vorliegenden Studiender Mundhöhle und des Larynx.
Abb. 1 Nierenmetastase eines Adenokarzinoms der Lunge mit TATE (400x)
ZusammenfassungTATE stellt nach den meisten Studien, im Vergleich zu dengleichen Karzinomen ohne TATE, ein prognostisch güns-tiges histologisches Phänomen dar, welches in zahlreichenOrganen sowie mehreren histologischen Karzinomvarian-
n Seite 41 nJOURNAL 01/2015

ten dokumentiert wurde. Daneben kann TATE bei um-strittener Invasivität eines Plattenepithelkarzinoms derMundhöhle für Pathologen diagnostisch hilfreich sein, dasie nicht bei in situ Karzinomen der Mundhöhle vor-kommt. In der Zukunft könnte das Verständnis der TATEauch neue immuntherapeutische Behandlungsmöglich-keiten einbringen, auf eosinophile Granulozyten gezielt.
Abb. 2 Plattenepithelkarzinom der Haut mit TATE (400x)
QuellenFALCONIERI, G., et al. Eosinophil-rich squamous carcinoma of the oral cav-ity: a study of 13 cases and delineation of a possible new microscopic enti-ty.. [online]. [cited 03-09-15]. Available fromhttp://www.ncbi.nlm.nih.gov/pubmed/18774493 .
LEGRAND, F., et al. Human eosinophils exert TNF-� and granzyme A-medi-ated tumoricidal activity toward colon carcinoma cells.. [online]. [cited 05-09-15]. Available from http://www.ncbi.nlm.nih.gov/pubmed/21068403 .
LOTFI, R., et al. Eosinophilic granulocytes and damage-associated molecu-lar pattern molecules (DAMPs): role in the inflammatory response withintumors.. [online]. [cited 24-09-15]. Available fromhttp://www.ncbi.nlm.nih.gov/pubmed/17198080 .
ONO, Y., et al. Tumor-associated tissue eosinophilia of penile cancer.. [on-line]. [cited 05-10-15]. Available fromhttp://www.ncbi.nlm.nih.gov/pubmed/12028296 .
PETER, C. D., et al. Assessment of Tumor Associated Tissue Eosinophilia(TATE) in Oral Squamous Cell Carcinoma Using Carbol Chromotrope Stain.[online]. [cited 05-10-15]. Available from http://www.scielo.cl/pdf/ijodon-tos/v9n1/art14.pdf .
Korrespondenzadresse:
MUDr. Tomáš NácovskýInstitut für PathologieHELIOS Klinikum ErfurtNordhäuser Str. 7499089 ErfurtTelefon: 0361-7812779e-Mail: [email protected]
n Interdisziplinäre Therapie eineslokal fortgeschrittenen Basalzell-karzinoms
Abstract des Vortrags zum „Jungen Forum Onkolo-gie“ auf der 28. Onkologischen Konferenz des Tu-morzentrum Erfurt e.V. am 6. und 7. November 2015in Eisenach
Stephanie Ender, Ivonne Kellner, Rudolf A. HerbstKlinik für Hautkrankheiten und Allergologie, HELIOSHauttumorzentrum Erfurt, HELIOS Klinikum Erfurt
Der 71-jährige Patient wurde notfallmäßig in der Klinikfür Augenheilkunde mit einem seit 3 Jahren bestehendenTumor im Bereich des rechten Auges bei zunehmenderGrößenprogredienz und Verlust des Sehvermögens vor-gestellt. Der Patient gab an, seit über 2 Jahren seine krebs-kranke Ehefrau zu versorgen und stellte sich nun nur we-gen der zunehmenden Schmerzen vor. Bei Aufnahmezeigte sich im Bereich der rechten Orbita und übergrei-fend auf den rechten Nasenrücken ein monströser, scharfabgegrenzter, tief ulzerierter, foetide riechender Tumormit deutlichem teleangiektatischem Randwall. Der rechteAugapfel war klinisch nicht mehr sicher abgrenzbar (Abb. 1).
Abb. 1 Vor Therapie
Die ophtalmologische Untersuchung ergab ein komplettverlorenes Sehvermögen für das rechte Auge; ein kleinerBulbus war sonographisch noch zu erkennen. Ein MRTdes Schädels vom Aufnahmetag zeigte eine mit einer tu-morösen Kontrastmittel aufnehmenden Raumforderungausgefüllte rechte Orbita, vorwiegend von medial, kranialund ventral den Bulbus deutlich komprimierend. Außer-dem stellte sich eine Infiltration der Haut periorbital biszur Nasenwurzel mit Nasenbeinosteolyse sowie V. a. Or-bitadachinfiltration dar (Abb. 2). Bei klinischem Verdachtauf ein ulzeriertes weit fortgeschrittenes Basalzellkarzi-
n Seite 42 n JOURNAL 01/2005JOURNAL 01/2015

Abb. 2 Vor Therapie
nom wurde dermatologischerseits eine Probebiopsie ent-nommen. Die dermatohistologische Untersuchung bestä-tigte diese Verdachtsdiagnose.
In der Interdisziplinären Hauttumorkonferenz wurde un-ter Einbezug von Kollegen der Dermatochirurgie, der Hals-, Nasen- Ohrenklinik, der Augenklinik, der Strahlen-therapie, der Radiologie, der Hämatoonkologie und derNeurochirurgie aufgrund der deutlichen Ausdehnung desBefundes mit fraglicher Durainfiltration und der Nähezum gesunden Auge weder eine operative Therapie nocheine Strahlentherapie empfohlen. Stattdessen wurde einesystemische gezielte Therapie mit Vismodegib (150 mgpro Tag per os) favorisiert und eine erneute Stagingunter-suchung nach 4 Wochen empfohlen. Nach 4-wöchigerVismodegib-Therapie zeigte sich der Tumor sowohl kli-nisch als auch bildgebend deutlich größenregredient: imBereich des Nasenrückens zeigte sich ein deutlicher Rück-gang der Ulzeration bei weiterhin nachweisbaren Telean-giektasien (Abb. 3 und 4). Aufgrund dieses guten Anspre-chens wurde in einer nachfolgenden InterdisziplinärenHauttumorkonferenz eine Fortsetzung der Vismodegib-Therapie über weitere 2 Monate mit dann erneutem Re-Staging zur Klärung einer möglicherweise dann beste-henden Operationsindikation empfohlen.
Abb. 3 Nach 4-wöchiger Vismodegib-Therapie
Abb. 4 Nach 4-wöchiger Vismo-degib-Therapie
Nach 3 Monaten Vismodegib-Therapie zeigten sich dieTumorrandbereiche allesamt narbig konsolidiert und inder Tiefe konnte man nun unter narbigen Veränderungendie Residuen des rechten Bulbus erkennen. (Abb. 5). Diebildgebende (MRT-)Untersuchung ergab einen deutlichenTumorregress extrakonal superior und medial mit regre-dienter Einbeziehung der anliegenden Augenmuskelnund des Orbitadaches sowie der medialen Orbitawand.Es zeigte sich ein teildestruierter Bulbus rechts, der nungering weniger komprimiert war (Abb. 6).
Abb. 5 Nach 3-monatiger Vismodegib-Therapie
Zur dermatohistologischen Einschätzung wurden in meh-reren Arealen der vormaligen Tumorfläche Probebiopsienentnommen, die allesamt Narbengewebe ohne Anhalt fürResiduen eines Basalzellkarzinoms zeigten.
Der Patient wurde daraufhin auf Empfehlung der erneu-ten Interdisziplinären Hauttumorkonferenz in der „Schä-delbasiskonferenz“ unseres Onkologischen Zentrums mitder Frage nach einer nun möglicherweise bestehendenoperativen Sanierungsmöglichkeit vorgestellt. Hier wurdebei deutlicher Tumorreduktion und zumindest in den vor-genommenen Mapping Probebiopsien fehlendem Tumor-nachweis in den aktuellen Mapping-Probeexzisionenempfohlen von einer Operation und auch einer Radiatioweiterhin Abstand zu nehmen und diese erst im Falle ei-nes Rezidivs zu erwägen. Bei dieser Entscheidung spielte
n Seite 43 nJOURNAL 01/2015

auch die Multimorbidität (z.B. kardial) des Patienten unddie damit verbundenen deutlich erhöhten Operationsrisi-ken eines nach wie vor sehr ausgedehnt zu planenden in-terdisziplinären Eingriffes eine Rolle. Auch eine zusätzli-che Probebiopsie aus der Tiefe der Orbita wurde als nichtzielführend – da die Gesamteinschätzung eher nicht ver-ändernd - betrachtet.
Abb. 6 Nach 3-monatiger Vismo-degib-Therapie
Zusammenfassend kann festgestellt werden, dass auchbei lokal ausgedehnten Tumoren die Therapieentschei-dungen immer interdisziplinär erfolgen müssen. Wie indiesem Fall sind hierzu eine ganze Reihe von operativenwie z. B. Hals-, Nasen- Ohren-Klinik, MKG-Chirurgie, Au-genklinik, Dermatochirurgie, Neurochirurgie und konser-vativen Fachdisziplinen wie die Dermatoonkologen, Strah-lentherapeuten und Hämatoonkologen erforderlich. Ge-gebenenfalls kann es auch sinnvoll sein, Patienten in ver-
schieden und unterschiedlich strukturierten Boards (hierInterdisziplinäres Hauttumor-Board und Schädelbasis-gruppe) vorzustellen. Gerade diese Optionenvielfalt dürf-te nur an darauf spezialisierten Onkologischen Zentrenvorgehalten werden können.
Abschließend zeigt sich ein beeindruckendes Ansprechendes Basalzellkarzinoms auf die zielgerichtete Therapie mitVismodegib. Nach bisherigen Erkenntnissen über dieseerst wenige Jahre zur Verfügung stehende Systemtherapiedes metastasierten und/oder nicht operablen bzw. strah-lentherapierbaren Basalzellkarzinoms ist hier jedoch frü-her oder später mit einem Rezidiv, Wieder- oder Weiter-wachsen des Tumors zu rechnen. Deshalb sind zunächstweiterhin engmaschige klinische und bildgebende Kon-trolluntersuchungen indiziert. Auch ist eine engmaschigeBetreuung des Patienten wegen der die Lebensqualitätz.T. deutlich einschränkenden Nebenwirkungen (im kon-kreten Fall insbesondere Muskelkrämpfe mit zeitweiserEinschränkung der Mobilität und Geschmacksverlust)durch ein hierin erfahrenes Behandlungsteam dringenderforderlich.
Korrespondenzadresse:
Stephanie EnderKlinik für Hautkrankheiten und AllergologieHELIOS Hauttumorzentrum ErfurtHELIOS Klinikum ErfurtNordhäuser Straße 7499189 ErfurtTelefon: 0361-781 72706e-Mail: [email protected]
n Seite 44 n JOURNAL 01/2005JOURNAL 01/2015
n Erneute großzügige Spende der Sparda-Bank Berlin an das Erfurter Tumorzentrum
Im Rahmen der Jahresmitgliederversammlung des Tumorzen-trum Erfurt e.V. am 15.4.2015 im Augustinerkloster Erfurtübergab die Leiterin der Erfurter Filiale der Sparda-Bank Ber-lin, Frau Busse, dem Tumorzentrum symbolisch einen Spen-den-Scheck in Höhe von 20.000 €.
Das aus Erträgen einer Gewinnspar-Lotterie stammende Geldsoll für die Krebsforschung und die Verbesserung der Patien-tenbetreuung eingesetzt werden. Der Vorsitzende des Tu-morzentrum Erfurt e.V., Prof. Albrecht Stier, bedankte sichunter dem Beifall der Anwesenden sehr herzlich für die er-neute großzügige Spende, mit deren Hilfe vor allem die Fi-nanzierung einer Study nurse-Stelle für die Studiendokumen-tation in der Hämatologie/Onkologie und Dermatoonkologiefortgeführt werden kann.

n Ewing-Sarkom der Mandibula –Fallbericht einer seltenen Tumorentität
Abstract des Vortrags zum „Jungen Forum Onkolo-gie“ auf der 28. Onkologischen Konferenz des Tu-morzentrum Erfurt e.V. am 6. und 7. November 2015in Eisenach
B. Rödiger1, H. Sayer2, J.-U. Piesold1
1Klinik für Mund-, Kiefer- und Gesichtschirurgie, Plas-tische Operationen2Klinik für Hämatologie und internistische Onkologie(4. Medizinische Klinik) HELIOS Klinikum Erfurt
HintergrundDas Ewing-Sarkom ist der zweithäufigste, primär maligneKnochentumor des Kinder- und jungen Erwachsenenal-ters. Es handelt sich definitionsgemäß immer um einenhochmalignen (G4) Tumor. 20-25% der Patienten weisenzum Zeitpunkt der Erstdiagnose bereits Metastasen auf.Das mittlere Erkrankungsalter liegt bei 15 Jahren, wobeider Anteil an männlichen Patienten mit 1,3:1 überwiegt.Die häufigsten Lokalisationen sind Becken, Femur, Hume-rus sowie die Rippen. Wir berichten hier über einen selte-nen Fall, in dem eine 42-jährige Patientin ein Ewing-Sar-kom der Mandibula präsentierte.
Anamnese und KlinikDie Patientin stellte sich im Januar 2015 in unserer Klinikvor und berichtete initial über eine seit mehreren Wochenbis Monaten progrediente Schwellung der linken Ge-sichtshälfte. An Nebendiagnosen waren eine arterielle Hy-pertonie sowie Adipositas zu eruieren. Die klinische Un-tersuchung zeigte einen derb tastbaren, etwa faustgro-ßen Tumor im Bereich der linken Jochbeinregion. Lymph-knotenschwellungen oder B-Symptomatik waren nichtfestzustellen.
DiagnostikDas sich anschließende MRT offenbarte eine zentral ne-krotische, malignomsuspekte Raumforderung im Bereichder Incisura mandibulae und des Proc. coronoideus linksmit Infiltration des Collum mandibulae sowie der Mm.masseter et temporalis. In den daraufhin angefertigtenStaginguntersuchungen (PET-CT, Thorax- und Abdomen-CT) konnten keine weiteren pathologischen bzw. tumor-verdächtigen Areale festgestellt werden. Das mittelsStanzbiopsie gewonnene, blauzellige Tumorgewebekonnte nach eingehender pathologischer und immunhis-tochemischer Aufarbeitung und in der Zusammenschaumit den klinischen und radiographischen Befunden zurDiagnose eines Ewing-Sarkoms der Mandibula führen.
TherapieNach interdisziplinärer Falldiskussion innerhalb des Kopf-
Hals-Tumorzentrums und des Onkologischen Zentrumsunseres Klinikums wurde der risikoadaptierte Therapieal-gorithmus festgelegt. Dieser bestand analog der Ewing-2008-Studie aus 6 Zyklen Induktions-Chemotherapie (VIDE – Vincristin, Ifosfamid, Doxorubicin, Etoposid) mitanschließender operativer Therapie und 8 Zyklen adju-vanter Polychemotherapie (VAC – Vincristin, ActinomycinD, Cyclophosphamid). Die Dauer der Primärtherapie be-läuft sich hierdurch auf insgesamt 10-12 Monate. Lokalwurden im Juli 2015 eine linksseitige Hemimandibulekto-mie mit Resektion der Kaumuskulatur und anschließenderEinbringung einer temporären Rekonstruktionsplatte mitKiefergelenkendoprothese durchgeführt. Im selben Ein-griff erfolgte eine ipsilaterale Neck-Dissection in 3 Regio-nen. Die histopathologische Aufarbeitung des Unterkie-ferteilresektats ergab eine R0-Situation mit Einschluss ei-ner großen Fibrosezone ausgehend von der Mandibulaals Korrelat eines durch Chemotherapie vorbehandeltenEwing-Sarkoms. Sowohl immunhistochemisch als auchkonventionell histologisch waren keine Tumorreste mehrnachweisbar. Im Neck-Dissection-Präparat konnten 21 tu-morfreie Lymphknoten gefunden werden. Somit ergabsich eine Ausbreitungsklassifikation von ypT0 ypN0 (0/21)cM0. Eine Unterkieferrekonstruktion mittels autologemKnochentransplantat wurde für den 9. - 12. Monat nachAbschluss der Primärbehandlung geplant.
Korrespondenzadresse:
Benjamin RödigerKlinik für Mund-, Kiefer-, Gesichtschirurgie, PlastischeOperationenHELIOS Klinikum ErfurtNordhäuser Straße 7499089 ErfurtTelefon: 0361-781 72672e-Mail: [email protected]
n Seite 45 nJOURNAL 01/2015

n Bericht von der Mitgliederversamm-lung des Tumorzentrum Erfurt e.V.am 15.04.2015
Auszüge aus dem Jahresbericht 2014
Prof. Dr. med. Albrecht Stier, Vorsitzender des Tumorzentrum Erfurt e.V.,berichtete über die Arbeit des vergangenenJahres
Der Bericht orientiert sich an dem im Memorandum derArbeitsgemeinschaft Deutscher Tumorzentren (ADT) ent-haltenen Kriterienkatalog zur regelmäßigen Beurteilungder Leistungsfähigkeit eines Tumorzentrums und den da-raus resultierenden Aufgaben.
1. Krebsregister
Die klinische Tumordokumentation ist eine der Haupt-aufgaben des Tumorzentrums und zugleich ein we-sentliches Element der Qualitätssicherung in der On-kologie. Dies unterstreicht auch der Nationale Krebs-plan für Deutschland und räumt der flächendeckendenEinführung Klinischer Krebsregister mit dem am 9. April 2013 in Kraft getretenen Krebsfrüherken-nungs- und -registergesetz (KFRG) die höchste Prioritätein.
Im Bereich des Erfurter Tumorzentrums bestehen be-reits gute Voraussetzungen für die Umsetzung dieseswichtigen Zieles des Nationalen Krebsplanes.
Die Meldebereitschaft der Krankenhäuser und nieder-gelassenen Ärzte als auch das Interesse an den Datendes Klinischen Krebsregisters nimmt weiterhin stetigzu. Großen Anteil an dieser Entwicklung haben dielaufenden Zertifizierungsverfahren kooperierender Or-gankrebszentren sowie die Screening-Programme fürHaut- und Brustkrebs.
Am 31.12.2014 waren die Krankheitsverläufe von109.069 Patienten mit insgesamt 129.525 Tumorenim klinischen Register des Tumorzentrums gespeichert.6.886 Patienten (9.462 Tumoren) wurden im Be-richtsjahr neu erfasst.
Dafür gilt den meldenden Ärzten ebenso Dank wieden Mitarbeitern des Klinischen Registers, die erneutdie wiederum gestiegene Zahl eingehender Dokumen-te (2014: 35.938 eingegangene Arztbriefe) ohne zu-sätzliches Personal bewältigt haben. Die Grafik zeigt
Abb.1 Neu erfasste Patienten/Tumoren im Klinischen Krebsregister Er-furt
nämlich eines nicht: Die deutliche Steigerung der Do-kumentation von Therapien und Verlaufsdaten wie derTumornachsorge. Das bedeutet: Die Krankheitsverläu-fe werden immer vollständiger dokumentiert.
Wenn ein Register als Instrument zur Messung der Er-gebnisqualität dienen soll, ist es notwendig, zu jedemKrankheitsverlauf mindestens einmal im Jahr den Tu-morstatus zu dokumentieren. Nur so können entitäts-und stadienbezogene Auswertungen der rezidiv- oderprogressionsfreien Überlebenszeit als einem der wich-tigsten Qualitätsindikatoren vorgenommen werden.
Wir wünschen uns dafür eine Fortsetzung der erfreuli-chen Entwicklung und noch mehr aktive Nachsorge-meldungen. Das hilft dem Tumorzentrum und dientder Sache, denn das Nachfragen und Einholen vonNachsorgedaten durch das Register sind sehr mühsamund angesichts fehlender personeller Ressourcen vomRegister kaum zu leisten. Der Aufwand zur Meldungvon Nachsorgeergebnissen (Datum, aktueller Tumor-status) ist vergleichsweise gering. Eine Arztbrief-Kopiean das TZ reicht aus.
In Thüringen gibt es nach wie vor keine Möglichkeit
des Datenabgleichs mit den Einwohnermeldeämternzur Ermittlung des Life-Status der Patienten. Ange-strebt wird ein kostenloser oder zumindest kosten-günstiger jährlicher elektronischer Datenabgleich mitdem Landes-Melderegister. Die Ermittlung des Life-Status über den Totenscheinabgleich mit dem Epide-miologischen Krebsregister in Berlin hat sich zwar inder Vergangenheit bewährt. Die Aktualität und Zuver-lässigkeit der Daten ist jedoch für viele Fragestellungennicht ausreichend.
Das TZ Erfurt arbeitete aktiv im Gesundheitszielepro-zess des Freistaates Thüringen mit. Im Berichtsjahrstand die Entwicklung einer Konzeption zur Umset-zung des Krebsfrüherkennungs- und -registergesetzes(KFRG) im Thüringen im Vordergrund. Verantwortlichfür die Umsetzung des KFRG durch ein eigenes Lan-desgesetz ist das Thüringer Ministerium für Arbeit, So-
1993/94 1995 1996 1997 1998 1999 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013 2014 MAR2015
0
2000
4000
6000
8000
10000
PatientenTumoren
Patienten 836 2039 2384 2768 2752 3527 3744 4069 4782 5361 6575 6487 5813 6490 6510 7696 7709 7668 7516 7457 6886 2147Tumoren 973 2090 2478 2927 3014 3741 3897 4411 5361 6101 6631 7583 7386 7489 7977 9544 9744 9840 9408 9537 9462 2360
n Seite 46 n JOURNAL 01/2005JOURNAL 01/2015

ziales, Gesundheit, Frauen und Familie. Dessen Kon-zept sieht die Gründung einer gGmbH durch die Trä-gereinrichtungen der bestehenden regionalen Registervor, der das Land im Rahmen einer Rechtsbeleihungdie Aufgabe der klinischen Krebsregistrierung über-trägt. Die bisherigen regionalen Registerstellen sollenan ihren Standorten erhalten bleiben und künftig aufeiner gemeinsamen Datenbank arbeiten. Der Serverund die vorgesehene Leitstelle zur Betreuung diesergemeinsamen Datenbank und für die Realisierung dervom KFRG und den Förderkriterien des Bundesverban-des der gesetzlichen Krankenkassen vorgegebenenAufgaben sollen am Uniklinikum Jena eingerichtetwerden.
Das Erfurter Register ist Kooperationspartner des On-kologischen Zentrums HELIOS Klinikum Erfurt mit demModul Kopf-Hals-Tumorzentrum und den EntitätenLeukämien, Lymphome, Plasmozytom, gynäkologischeTumoren und Nierenkarzinom sowie von 11 zertifizier-ten Organtumorzentren (HELIOS Brustzentrum Erfurtund Gotha, Darmkrebszentrum Erfurt [Kath. Kranken-haus Erfurt], Prostatakarzinomzentrum Kath. Kranken-haus Erfurt, Darmkrebszentrum Südthüringen [Klini-kum Meiningen], Prostatakrebszentrum Südthüringen[Klinikum Meiningen], HELIOS Hautkrebszentrum Er-furt, HELIOS Darmzentrum Erfurt mit Modul Pankreas-karzinomzentrum, HELIOS ProstatakarzinomzentrumErfurt, Lungenzentrum Bad Berka) und war auch 2014an der Vorbereitung und Durchführung der aufwendi-gen Zertifizierungsverfahren sowie der jährlichen Au-dits beteiligt. Außerdem ist das Register in die Doku-mentation des Brustzentrums Mittelthüringen (Söm-merda/UH-Kreis) und des Darmkrebszentrums in Mühl-hausen einbezogen. Die Nutzung der vorhandenenInfrastruktur für die Tumordokumentation einschließ-lich der Möglichkeiten statistischer Auswertungen istfür bestehende und künftige Organzentren vor allemwegen der Unterstützung bei der Beschaffung der un-erlässlichen Follow up-Daten vorteilhaft. Außerdem er-füllen die Einrichtungen auf diesem Wege die in Thü-ringen geltende Meldepflicht für Tumorerkrankungen,da die epidemiologischen Daten vom Klinikregister andas Gemeinsame Krebsregister in Berlin weitergeleitetwerden.
Auch von den anderen Ärzten der Region wurden dieServiceleistungen des klinischen Registers regelmäßiggenutzt (täglich Anforderungen von Übersichtsberich-ten zum Krankheitsverlauf, weiterhin Abteilungs- bzw.Praxisstatistiken einschließlich Überlebenszeitanaly-sen).
Die immer bessere Erfassungsrate im Klinischen Regis-ter wirkt sich auch positiv auf die Melderate für dasGemeinsame Krebsregister der neuen Bundeslän-der und Berlins (GKR) in Berlin aus, da fast alle Mel-dungen über die Tumorzentren zum epidemiologi-schen Register gelangen. So wird inzwischen auch die
geforderte 90%-Marke aus Arztmeldungen erfassterKrebserkrankungen für Thüringen erreicht. Dabei leis-tet das Erfurter Register mit mehr als 40 % den größtenBeitrag aller Register in Thüringen.
Abb.2 Eingangsstatistik des Gemeinsamen Krebsregisters der neuenBundesländer (GKR) in Berlin, Anteil der Erstmeldungen aus den Thürin-ger Tumorzentren (Erfassungsstand 12/2014)
2. Interdisziplinäre onkologische Konsile
Seit November 1993 werden vom Tumorzentrum re-gelmäßig interdisziplinäre Konsile organisiert. Auchfür das Jahr 2014 ist eine hohe Zahl beratener Fälle zuverzeichnen. In den 50 durchgeführten Konsilen desBerichtsjahres sind insgesamt 995 Fälle besprochenund protokolliert worden.
Das von einer Arbeitsgruppe unter maßgeblicher Be-teiligung von Prof. Stier und Dr. Göbel entwickelteSAP-gestützte Konzept für die Anmeldung, Organisa-tion und Protokollierung wurde durch die Firma celsi-us37 realisiert und ist seit Februar 2012 im Einsatz.
3. Leitlinien / Projektgruppen
Aktuelle Nachsorgeleitlinien einzelner Tumorentitätenwurden im Journal des Tumorzentrums publiziert.
Nachdem inzwischen anerkannte Leitlinien von derDeutschen Krebsgesellschaft und den medizinischenFachgesellschaften für nahezu alle Tumorentitäten vor-liegen, wurde die Erarbeitung eigener Leitlinien aberweitgehend eingestellt. Das Tumorzentrum sieht seineAufgabe vorrangig darin, die überregionalen Leitlinienstärker in der Region zu propagieren.
Die beiden im Jahre 2012 gegründeten Projektgrup-pen „Magenkarzinom“ (Leiter: PD Dr. Schreiber, BadLangensalza) und „Nierentumoren“ (Leiter: Prof. Dr.Steiner, Erfurt) haben ihre Arbeit fortgesetzt. Ergeb-nisse der Projektgruppe „Nierenkarzinom“ wurden aufder 27. Onkologischen Konferenz im November 2014präsentiert.
n Seite 47 nJOURNAL 01/2015

4. Fort- und Weiterbildungsveranstaltungen
Das Profil der vom Tumorzentrum veranstalteten Fort-und Weiterbildungen wurde beibehalten. Die Veran-staltungen wurden überwiegend als Symposien vonca. 3 Stunden Dauer organisiert. Die OnkologischeKonferenz als zweitägige Hauptveranstaltung des Jah-res fand traditionsgemäß wieder im Haus Hainstein Ei-senach statt.
Insgesamt sind 18 Fort- und Weiterbildungen orga-nisiert worden, bei denen insgesamt 1.462 Teilneh-mer registriert wurden.
5. Psychoonkologie
Seit 1996 führt das TZ Erfurt einen Psychoonkologi-schen Dienst, der inzwischen in das psychologischeBetreuungsangebot des HELIOS Klinikum Erfurt inte-griert ist.
Im Berichtsjahr konnte die Zahl der betreuten onkolo-gischen Patienten weiter gesteigert werden auf 907(zum Vergleich 2013: 835, 2012: 692, 2011: 671).
Die Betreuung wird überwiegend als psychoonkologi-scher Konsiliardienst in den bestehenden Organtumor-zentren und dem Onkologischen Zentrum angebotenund rege in Anspruch genommen.
Ein wichtiger Teil der Arbeit ist die psychologische Un-terstützung des ärztlichen und pflegerischen Personalsbei ihrem belastenden Umgang mit traumatisiertenPatienten. Dazu wurden von den Mitarbeitern des Psy-choonkologischen Dienstes Vorträge im Rahmen vonKlinik-Weiterbildungen gehalten.
6. Patientenberatung, Öffentlichkeitsarbeit, Selbsthilfe
- Die regelmäßig per Telefon und e-Mail eingehendenAnfragen von Patienten und Angehörigen wurdenüberwiegend von den Mitarbeitern der Geschäfts-stelle selbst beantwortet. Schwierige medizinischeAnfragen wurden an entsprechende Fachvertreterweitergegeben, die deren Beantwortung übernah-men. Dafür sei an dieser Stelle nochmals gedankt!
- Im Berichtsjahr wurde eine Informationsveranstal-tungen für Patienten und interessierte Bürger durch-geführt:
01.07.2014 4. Patiententag des OnkologischenZentrums HKE (30 Teilnehmer)
- Ständiger Kontakt besteht zum Erfurter Gesundheits-amt (Geschwulstberatungsstelle, Kontakt- und Infor-mationsstelle für Selbsthilfegruppen) und zur Frau-enselbsthilfe nach Krebs (Veranstaltung des TZE fürdie SHG sowie Teilnahme des Vorstands an derenVeranstaltungen).
- Die Internetseite des TZ wurde ständig aktualisiert. 5Newsletter wurden versandt.
Das Tumorzentrum Erfurt ist eines der ganz wenigenTumorzentren in Deutschland, die ein eigenes Journalherausgeben. Auch im Jahr 2014 wurde wieder einHeft publiziert. Ständiger Bedarf besteht an redaktionellen Beiträ-gen. Alle sind aufgefordert Artikel einzureichen, umdieses ambitionierte Projekt in der bisherigen Quali-tät fortführen zu können.
7. Forschung, Serviceleistungen, Ausbildung
- Unterstützung bei der statistischen Auswertung undbei der Präsentation der Ergebnisse von verschiede-nen Studien und Untersuchungen, die in den Klinikendurchgeführt worden sind.
- 2 Promotionsarbeiten wurden vom Register mitbe-treut.
- Als beauftragter Partner der beteiligten Kliniken im Einzugsgebiet arbeitet das TZE an der MSKK-Studie (Signature Diagnostics, Potsdam) mit.
- 5 Praktikanten wurden im Rahmen ihrer Ausbildung zu Medizinischen Dokumentationsassisten vom Kli- nischen Register betreut.
8. Zusammenarbeit mit anderen Tumorzentren und Fachgesellschaften
Die Zusammenarbeit mit anderen Tumorzentren wirdgepflegt:
In der Interessengemeinschaft der Thüringischen Tu-morzentren spielte das TZ Erfurt eine aktive Rolle.Regelmäßig fanden Treffen der Koordinatoren statt.Dr. Göbel ist Vertreter der Interessengemeinschaftim Vorstand der Thüringischen Krebsgesellschaft(ThKG). Als Vertreter der Thüringer TZ nahm er anden Beratungen des Verwaltungsrates der am Ge-meinsamen Krebsregister beteiligten Bundesländerteil.
- Prof. Stier ist stellvertretender Vorsitzender der Thü-ringischen Krebsgesellschaft.
- Das TZ Erfurt ist Mitglied im Kooperationsverbundklinischer Krebsregister Deutschlands, der sich einebessere Vernetzung lokaler und regionaler Aktivitä-ten zum Ziel gesetzt hat sowie gemeinsame Daten-auswertungen durchführt. Das Erfurter Register warim Berichtsjahr an der überregionalen Datenauswer-tungen zu Prostatakarzinom, Mammakarzinom, ko-lorektalen Karzinomen, Lungenkrebs, Nierentumo-ren und Malignem Melanom für die 4. Qualitätskon-ferenz auf dem 31. Deutschen Krebskongress 2014beteiligt.
9. Vereinsstatistik
Am 31.12.2014 hatte der Verein 294 Mitglieder.3 Mitglieder wurden im Berichtsjahr neu aufgenom-men; 5 Mitglieder sind ausgeschieden.
n Seite 48 n JOURNAL 01/2005JOURNAL 01/2015

n Seite 49 nJOURNAL 01/2015
IMPRESSUM
ISSN 1868-291X (Print-Ausgabe)ISSN 1868-2928 (Internet)
n Herausgeber: Tumorzentrum Erfurt e.V.
n Redaktion: Prof. Dr. med. Hartwig Kosmehl · Dr. rer. nat. Hubert Göbel
n Redaktionsbüro und Versand:Tumorzentrum Erfurt e.V.
Nordhäuser Straße 74 · 99089 ErfurtTelefon: 03 61 / 7 81-48 02 · Telefax: 03 61 / 7 81-48 03
E-Mail: [email protected]
n Layout, Satz und Druck: Handmann Werbung GmbH Erfurt
n Hinweis: Das Tumorzentrum Erfurt erstellt die Artikel nach bestem
Wissen und Gewissen. Die Verantwortung für den Inhalt der medizinischen und wissenschaftlichen Beiträge obliegt den Autoren. Sie stellen keine Handlungsempfehlungen für den
individuellen Fall dar.
n Veranstaltungsverzeichnis
Liebe Kolleginnen und Kollegen,
wir möchten Ihre gezielten und konzentrierten Fortbil-dungsaktivitäten mit diesem Veranstaltungsverzeichnisunterstützen und Ihnen auch 2015 wieder ein breitesSpektrum zertifizierter und hoffentlich für Sie interessan-ter Fort- und Weiterbildungen anbieten. Die nachstehen-de Liste enthält nur die zum gegenwärtigen Zeitpunktterminlich und thematisch feststehenden Veranstaltun-gen und kann daher weder vollständig sein noch umfas-send informieren. Sie soll als Orientierungshilfe dienenund Sie animieren, alle weiteren Informationen und die laufenden Aktualisierungen auf der Internetseitewww.tumorzentrum-erfurt.de nachzulesen und/oder direkt in der Geschäftsstelle zu erfragen.Über eine zahlreiche Teilnahme an den Veranstaltungen,rege Diskussionen sowie die Vertiefung und Ausweitungpersönlicher Kontakte freuen wir uns besonders.
Prof. Dr. med. A. Stier
Vorsitzender | Tumorzentrum Erfurt e. V.
Februar
10.02.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster ErfurtDer alte Tumorpatient
März
16.03.2016, 19.00 bis 20.30 UhrFestsaal, Rathaus ErfurtPatiententag des Onkologischen Zentrums HELIOS Klinikum Erfurt
17.03.2016, 19.00 bis 20.30 UhrHELIOS Klinikum Erfurt, Auditorium47. Erfurter Fortbildung Hämatologie und Onkologie für Kranken-schwestern und -pfleger
23.03.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster ErfurtNeues aus der Pathologie
April
06.04.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster ErfurtAktuelle Diagnostik und Therapie von Lungentumoren
13.04.2016, 16.00 bis 20.00 UhrHELIOS Klinikum Erfurt, Auditorium8. Erfurter Dermatologische Frühjahrstagung
Juni
29.06.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster Erfurt26. Erfurter Fortbildung Hämatologie und Onkologie
September
07.09.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster ErfurtAktuelle Uroonkologie
21.09.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster Erfurt2. Erfurter Symposium Stammzelltherapie
Oktober
05.10.2016, 17.00 bis 20.00 UhrHELIOS Klinikum Erfurt, AuditoriumGynäkologische Onkologie
29.10.2016, 9.00 bis 14.00 UhrEvangelisches Augustinerkloster ErfurtEndokrine Tumoren
November
04. – 05.11.2016Evangelisches Augustinerkloster Erfurt29. Onkologische Konferenz
24.11.2016, 19.00 bis 20.30 UhrHELIOS Klinikum Erfurt, Auditorium48. Erfurter Fortbildung Hämatologie und Onkologie für Kranken-schwestern und -pfleger
Dezember
07.12.2016, 17.00 bis 20.00 UhrEvangelisches Augustinerkloster ErfurtUpdate Nierentumoren

n Seite 50 n JOURNAL 01/2005JOURNAL 01/2015
n ANGEBOTE DES TUMORZENTRUM ERFURT e.V.
KONSILARDIENSTE
• Interdisziplinäres onkologisches KonsilJeden Mittwoch, 7.30 Uhr, Demo-Raum C 1.400 des Insti-tuts für bildgebende Diagnostik, Hauptgebäude 1. OG,HELIOS Klinikum Erfurt, Nordhäuser Straße 74
Anmeldungen über Telefon 03 61 / 7 81-48 02
Leitung: PD Dr. Sayer / Prof. Dr. Scharf
Jeder Arzt kann seine onkologischen Fälle persönlich ei-nem Gremium von Experten aller Fachdisziplinen vorstel-len. Am Ende der (kostenfreien) Beratung erhält er einekonkrete Therapieempfehlung. Zu jeder Fallbesprechungwird ein Protokoll angefertigt, das dem vorstellenden Arztund eventuellen mitbehandelnden Ärzten zugeht.
• Telefonischer KonsilardienstUnkompliziertes Vermitteln von Kontakten zu den speziellen onkologischen Ansprechpartnern aller Fachge-bietef www.tumorzentrum.de
ONKOLOGISCHE LEITLINIEN
Hilfestellung bei der Umsetzung der aktuellen Diagno-se-, Therapie- und Nachsorgeleitlinien der DeutschenKrebsgesellschaft und der medizinischen Fachgesellschaf-ten. In Ergänzung und zur praktischen Durchführung wer-den diese bei Bedarf für die speziellen regionalen Bedin-gungen adaptiert.
KONTAKTE ZU SELBSTHILFEGRUPPEN UNDHOSPIZDIENSTEN IN DER REGION
PSYCHOLOGISCHE BETREUUNGBetreuungsangebote für stationäre Patienten des HELIOSKlinikum Erfurt sowie für Ärzte und Pflegepersonal.
FORT- UND WEITERBILDUNG
• Ärzte• Krankenschwestern und -pfleger• Sozialdienste
DOKUMENTATION• Klinische TumordokumentationIn Erfüllung des Qualitätssicherungsauftrages des Sozial-gesetzbuches (SGB V) wird für jeden Patienten der ge-samte Krankheitsverlauf nach anerkannten Regeln (Tu-morbasisdokumentation) dokumentiert. Die Unterlagenstehen dem Patienten und ihren behandelnden Ärztenzur Verfügung. Im Einzelfall (bei Umzug, Arztwechsel,Verlust von Originalunterlagen) sind sie für den Arzt eineunschätzbare Hilfe.
• Gemeinsames Krebsregister der neuen Bundesländer
Epidemiologisch relevante Daten werden entsprechendgeltender Gesetze an das Gemeinsame Krebsregister derneuen Bundesländer weitergegeben.Mehr als 95 % der Meldungen des Einzugsgebietes kom-men vom Tumorzentrum. Diese Daten werden regelmä-ßig mit den amtlichen Sterbedaten abgeglichen und ste-hen dem meldenden Einrichtungen zur Verfügung.
SERVICE
• Unterstützung der Nachbetreuung, Erinnerungsfunktion
Auf persönlichen Wunsch werden Patienten (und ihre be-treuenden Ärzte) an vereinbarte bzw. vergessene Nach-sorgetermine erinnert.
• Statistiken für Krankenhäuser und PraxenErstellung von Übersichten, Leistungsstatistiken undÜberlebenszeitanalysen für die von der jeweiligen Ein-richtung betreuten Patienten.
n HIER ERREICHEN SIE UNS
HELIOS Klinikum Erfurt GmbHHaus 22, Nordhäuser Straße 74, 99089 Erfurt
Telefon: 03 61 / 7 81-48 02Telefax: 03 61 / 7 81-48 03E-Mail: [email protected]: http://www.tumorzentrum-erfurt.deGeschäftsführer: Dr. rer. nat. Hubert Göbel
Sie können die Arbeit des Tumorzentrum Erfurt e.V.
durch Ihre Spende unterstützen!Sparkasse Mittelthüringen
IBAN: DE6482 0510 0001 3012 3609SWIFT-BIC: HELADEF1DEM
(Spenden sind steuerlich begünstigt!
• InformationenKostenlose Bereitstellung von Tumor-Nachsorgepässenund Informationsmaterialien für Patienten, Ärzte, Pflege-personal und Sozialdienste

n Seite 51 nJOURNAL 01/2015
n WISSENSCHAFTLICHER BEIRAT
Prof. Dr. med. Hartwig Kosmehl (Vorsitzender)
Chefarzt, Institut für Pathologie, HELIOS Klinikum Erfurt
Telefon: 03 61 / 7 81-27 51
Dr. med. Elke Conrad
Chefärztin, Klinik für Nuklearmedizin, HELIOS Klinikum
Erfurt, Telefon: 0361 / 7 81-24 43
Dr. med. Alexander Fichte
Urologe, Geschwister-Scholl-Straße 6, 99085 Erfurt,
Telefon: 0361 / 6 43 73 03
Dr. jur. Arnim Findeklee
Leiter, Landesvertretung Thüringen, Verband der Ersatz-
kassen (vdek), Lucas-Cranach-Platz 2, 99099 Erfurt,
Telefon: 0361 / 4 42 52 11
Dr. med. Michael Glatzel
Chefarzt, Klinik für Strahlentherapie und Radioonkolo-
gie, HELIOS Klinikum Erfurt, Telefon: 0361 / 7 81-24 01
Dr. med. Jörg Kluge
Chefarzt, Klinik für Thoraxchirurgie und thorakale
Endoskopie, HELIOS Klinikum Erfurt,
Telefon: 0361 / 7 81-25 81
Dipl.-Med. Susanne Köhler
Chefärztin, Innere Medizin III, Hämatologie / Onkologie /
Palliativmedizin, HELIOS Kreiskrankenhaus Gotha-Ohr-
druf, Telefon: 03621 / 2 20-1 78
Sebastian Kreutz
Fachbereichsleiter, Bereich Verhandlungsmanagement,
AOK Plus, 98523 Suhl, Telefon: 0800 / 10 59 0 – 81 21 6
Dr. med. Anja Merte
Oberärztin, Klinik für Frauenheilkunde und Geburtshilfe,
HELIOS Klinikum Erfurt, Telefon: 0361 / 7 81-40 01
Priv.-Doz. Dr. med. Jörn-Uwe Piesold
Chefarzt, Klinik für Mund-, Kiefer- und Gesichtschirurgie,
HELIOS Klinikum Erfurt, Telefon: 0361 / 7 81-22 31
Prof. Dr. med. Steffen Rosahl
Chefarzt, Klinik für Neurochirurgie, HELIOS Klinikum
Erfurt, Telefon: 0361 / 7 81-22 61
Dr. med. Claus-Peter Schneider
Leitender Arzt, Abteilung für internistische Onkologie
und Hämatologie, Zentralklinik Bad Berka,
Telefon: 036458 / 5-24 00
Priv.-Doz. Dr. med. Lutz-Dieter Schreiber
Chefarzt, Klinik für Allgemein- und Viszeralchirurgie,
Hufeland Klinikum, Standort Bad Langensalza,
Telefon: 03603 / 8 55-0
n VORSTAND
Prof. Dr. med. Albrecht Stier (Vorsitzender)
Chefarzt, Klinik für Allgemein- und Viszeralchirurgie,
HELIOS Klinikum Erfurt, Telefon: 0361 / 7 81-23 31
Dr. med. Jörg Pertschy (Stellvertr. Vorsitzender)
Chefarzt, Klinik für Allgemein-, Visceral- und
Gefäßchirurgie, Katholisches Krankenhaus St. Nepomuk
Erfurt, Telefon: 0361 / 6 54-12 01
Prof. Dr. med. Rudolf A. Herbst
Chefarzt, Klinik für Hautkrankheiten und Allergologie,
HELIOS Klinikum Erfurt, Telefon: 0361 / 7 81-43 01
Prof. Dr. med. Hartwig Kosmehl
Chefarzt, Klinik für Pathologie, HELIOS Klinikum Erfurt,
Telefon: 0361 / 7 81-27 51
Prof. Dr. med. Jens-Gerd Scharf
Chefarzt, 2. Medizinische Klinik, HELIOS Klinikum Erfurt,
Telefon: 0361 / 7 81-24 71
Prof. Dr. med. Thomas Steiner
Chefarzt, Klinik für Urologie, HELIOS Klinikum Erfurt,
Telefon: 0361 / 7 81-22 01
Dr. med. Jörg Weniger
Hämatologe und internistischer Onkologe,
Geschwister-Scholl-Straße 6, 99085 Erfurt,
Telefon: 0361 / 5 66 78 19

* Indikation: Prophylaxe invasiver Pilzinfektionen bei Hochrisikopatienten mit allogener hämatopoetischer Stammzelltransplantation (HSZT)1 Herbrecht R et al. New Engl J Med 2002; 347:408-152 Herbrecht R., Patterson TF, Slavin M et al. Clin Infect Dis. 2014 Nov 19. pii: ciu911. [Epub ahead of print]3 Vfend® Fachinformation (Aktueller Stand unter: www.pfizermed.de/medikamente.htm)4 DRG – Entgeltkatalog 2015, G-DRG Version 2015
VFEND® 50 mg Filmtabletten; VFEND® 200 mg Filmtabletten; VFEND® 40 mg / ml Pulver zur Herstellung einer Suspension zum Einnehmen; VFEND® 200 mg Pulver zur Herstellung einer Infusionslösung; VFEND® 200 mg Pulver und Lösungsmittel zur Herstellung einer Infusionslösung; Wirkstoff: Voriconazol. Zusammensetzung: Wirkstoff: Filmtbl.: 1 Filmtbl. enth. 50 mg / 200 mg Voriconazol. Pulver (Suspension): Nach Rekonstitution m. Wasser enth. 1 ml Suspension z. Einnehmen 40 mg Voriconazol. Jede Fl. enth. 3 g Voriconazol. Pulver (Inf.-lösung): 1 ml enth. nach Rekonstitution 10 mg Voriconazol. Nach Rekonstitution ist e. weitere Verdünnung nötig, bevor appliziert werden kann. 1 Durchstechfl. enth. 200 mg Voriconazol. Sonst. Bestandteile: Filmtbl.: Lactose-Monohydrat, vorverkleisterte Stärke aus Mais, Croscarmellose-Natrium, Povidon, Magnesiumstearat, Hypromellose, Titandioxid (E 171), Triacetin. Pulver (Suspension): Sucrose, hochdisperses Siliciumdioxid, Titandioxid (E 171), Xanthangummi, Natriumcitrat, wasserfreie Citronensäure, Natriumbenzoat (E 211), natürlicher Orangengeschmack. Pulver (Inf.-lösung): Natrium-beta-cyclodex - trin-sulfobutylether (SBECD). Lösungsmittel z. Herstell. e. Inf.-lösung: 0,9%ige Natriumchloridlösung i. Wasser f. Inj.-zwecke. Anwendungsgebiete: invasive Aspergillose, Candidämie b. nicht neutropenischen Pat., Fluconazol-resistente, schwere invasive Candida-Infekt. (einschl. C. krusei), schwere Pilzinfekt. durch Scedosporium spp. u. Fusarium spp. b. Erw. u. Kdrn. ab 2 J.. I. erster Linie b. Pat. m. progressiven, mögl.-weise lebensbedrohl. Infekt.. Prophylaxe invasiver Pilzinfekt. b. Hochrisikopat. m. allogener hämatopoetischer Stammzelltransplantation (HSZT). Gegenanzeigen: Überempfindlichk. gg. d. Wirkstoff od. e.d. sonst. Bestandteile; Komedikation m. Terfenadin, Astemizol, Cisaprid, Pimozid, Chinidin, Rifampicin, Carbamazepin, Phenobarbital, Mephobarbital, hochdos. Efavirenz od. Ritonavir (ab 400 mg tägl.), Ergotalkaloiden (Ergotamin, Dihydroergotamin), Sirolimus, Johanniskraut. Nebenwirkungen: Sehr häufig: periph. Ödeme; Kopfschm.; Sehverschlechterung (einschl. verschwommenen Sehens, Chromatopsie u. Photophobie); Atemnot; Bauchschm., Übelk., Erbrechen, Durchfall; abnormale Leberfunktionstests (einschl. AST, ALT, alkalischer Phosphata- s e, Gamma-Glutamyltranspeptidase [GGT], Lactatdehydrogenase [LDH], Bilirubin); Hautausschlag; Fieber. Häufig: Gastroenteritis, Sinusitis, Gingivitis; Agranulozytose, Panzytopenie, Thrombozytopenie, Anämie; Überempfindlichk.-reakt.; Hypoglykämie, Hypokaliämie, Hyponatriämie; Depressionen, Halluzinationen, Ängstlichk., Schlaflosigk., Unruhe, Verwirrth.; Krampfanfall, Tremor, Parästhesie, Hypertonus, Schläfrigk., Synkope, Benommenh.; Netzhautblutungen; supraventrikuläre Arrhythmie, Tachykardie, Bradykardie; Hypotonie, Phlebitis; akutes Atemnotsyndr., Lungenödem; Dyspepsie, Verstopfung, Cheilitis; Gelbsucht, cholestat. Gelbsucht, Hepatitis; exfoliat. Dermatitis, makulopapulöser Hautausschlag, Pruritus, Alopezie, Hautrötung; Rückenschm.; akute Niereninsuff., Hämaturie; Brustschm., Gesichtsödem, Asthenie, Grippesympt., Schüttelfrost; Erhöh. d. Kreatininspiegels. Gelegentlich: pseudomembranöse Kolitis, Lymphangitis, Peritonitis; Verbrauchskoagulopathie, Knochenmarkversagen, Leukopenie, Lymphadenopathie, Eosinophilie; anaphylaktoide Reakt.; Nebennierenrindeninsuff., Hypothyreose; Hirnödem, Enzephalopathie, extrapyra- midale Stör., periph. Neuropathie, Ataxie, Hypästhesie, Geschmacksstör., Nystagmus; okulogyre Krisen, Stör. d. Sehnervs (einschl. optischer Neuritis), Papillenödem, Skleritis, Blepharitis, Doppeltsehen; Hypakusis, Schwindel, Tinnitus; Kammer flimmern, ventrikuläre Extrasystolen, supraventrikuläre Tachykardie, ventrikuläre Tachykardie; Thrombophlebitis; Pankreatitis, Duodenitis, Glossitis, Zungenödem; Leberversagen, Lebervergrößerung, Cholezystitis, Gallensteine; tox. epidermale Nekrolyse, Stevens-Johnson-Syndrom, Erythema multiforme, Angioödem, Psoriasis, Urtikaria, allerg. Dermatitis, Phototoxizität, makulöser Ausschlag, papulöser Ausschlag, Purpura, Ekzem; Arthritis; Nierentubulusnekro- se, Proteinurie, Nephritis; Reakt. an d. Inj.-stelle; QTc-Verlängerung i. EKG, Erhöh. d. Harnstoffwerts i. Blut, Hypercholesterinämie. Selten: Hyperthyreose; hepatische Enzephalopathie, Guillain-Barré-Syndrom; N.-opticus-Atrophie, Horn-hauttrübungen; Torsade de pointes, kompl. AV-Block, Schenkelblock, AV-Rhythmus; Pseudoporphyrie, fixes Arzneiexanthem. Häufigkeit nicht bekannt: Plattenepithelkarzinom; kutaner Lupus erythematodes; Periostitis. D. Erfahr. nach d. Markteinführ. lassen vermuten, dass Hautreakt. (v. a. Erytheme) b. Kdrn. häufiger auftr. können als b. Erw.. Warnhinweise und Vorsichtsmaßnahmen: m. VFEND beh. Pat. müssen sorgf. auf Lebertoxizität überwacht werden u. VFEND muss ggf. abgesetzt werden. Alle Pat. einschl. Kdr. sollten Sonnenlichtexposition vermeiden u. Schutzmaßnahmen wie entspr. Bekleidung u. Sonnenschutzm. m. hohem LSF anw.. Plattenepithelkarzi-nome auf d. Haut wurden b. Pat. beobachtet, v. denen einige üb. frühere phototox. Reakt. berichtet haben. Daher ist d. Notwendigk. e. Verringerung der VFEND-Exposition zu erwägen. Filmtbl.: enthält Lactose. Pulver (Suspension): enthält Sucrose (Zucker). Pulver (Inf.-lösung) u. Lösungsmittel z. Herstell. e. Inf.-lösung: enthält Natrium. Weitere Informationen s. Fach- u. Gebrauchsinformation. Abgabestatus: Verschreibungspflichtig. Pharmazeutischer Unternehmer: Pfizer Limited, Ramsgate Road, Sandwich, Kent CT13 9NJ, Vereinigtes Königreich. Repräsentant in Deutschland: PFIZER PHARMA GmbH, Linkstr. 10, 10785 Berlin. Stand: Juni 2014.
Ihr starker Partner in der Therapie von invasiven Pilzinfektionen und in der Prophylaxe. 3*
signifikanter Überlebensvorteil in der Therapie 1,2
breites Wirkspektrum 3
einfache Anwendung in drei Darreichungsformen (i. v., Tabletten, Suspension)
ZE-refinanziert 4
NEU – Auswertung der Studie Voriconazol vs. Amphotericin B zur Therapie der Invasiven Aspergillose (Herbrecht 2002 1) mit den 2008 EORTC/MSG-Kriterien bestätigt. 2
© p
rudk
ov –
Fot
olia
.com
Voriconazol i.v. /oral
www.pfizermed.de
b-4v
23vf
e-0-
0
Überlebensvorteil bestätigt! 1