importance of syndecan-4 and syndecan -2 in osteoblast cell adhesion and survival mediated by a...
TRANSCRIPT

E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /yexc r
Research Article
Importance of syndecan-4 and syndecan -2 in osteoblastcell adhesion and survival mediated by a tissuetransglutaminase−fibronectin complex
Z. Wang1, D. Telci1,2, M. Griffin⁎
School of Life and Health Sciences, Aston University, Aston Triangle, Birmingham B47ET, UK
A R T I C L E I N F O R M A T I O N
⁎ Corresponding author. Fax: +44 121 204 514E-mail address: [email protected] (M. G
1 These authors contributed equally to this w2 Present address: Department of Genetics an
0014-4827/$ – see front matter © 2010 Elseviedoi:10.1016/j.yexcr.2010.10.015
A B S T R A C T
Article Chronology:
Received 10 August 2010Revised version received
23 September 2010Accepted 16 October 2010Available online 28 October 2010
Tissue transglutaminase (TG2) has been identified as an important extracellular crosslinkingenzyme involved in matrix turnover and in bone differentiation. Here we report a novel celladhesion/survival mechanism in human osteoblasts (HOB) which requires association of FN
bound TG2 with the cell surface heparan sulphates in a transamidase independent manner. Thisnovel pathway not only enhances cell adhesion on FN but also mediates cell adhesion and survivalin the presence of integrin competing RGD peptides.We investigate the involvement of cell surfacereceptors and their intracellular signalling molecules to further explore the pathway mediated bythis novel TG-FN heterocomplex. We demonstrate by siRNA silencing the crucial importance of thecell surface heparan sulphate proteoglycans syndecan-2 and syndecan-4 in regulating thecompensatory effect of TG-FN on osteoblast cell adhesion and actin cytoskeletal formation in thepresence of RGD peptides. By use of immunoprecipitation and inhibitory peptides we show thatsyndecan-4 interacts with TG2 and demonstrate that syndecan-2 and the α5β1 integrins, but notα4β1 function as downstreammodulators in this pathway. Using function blocking antibodies, weshow activation of α5β1 occurs by an inside out signalling mechanism involving activation and
binding of protein kinase PKCα and phosphorylation of focal adhesion kinase (FAK) at Tyr861 andactivation of ERK1/2.
© 2010 Elsevier Inc. All rights reserved.
Keywords:
Tissue transglutaminaseOsteoblastsIntegrinSyndecanCell adhesion
Introduction
The importance of cell and extracellular matrix (ECM) interactionmediated by the cell surface receptors and ECM proteins, has beenwell demonstrated. The amino acid sequence Arg–Gly–Asp (RGD)commonly found within the ECM proteins of bone, includingfibronectin (FN), type I collagen, and vitronectin [1,2], is the mostwidely occurring cell adhesive motif recognized by about half of allknown integrins, such asα2β1,α3β1,α5β1,αVβ1,αVβ3, andαVβ5
2.riffin).ork.d Bioengineering, Yeditepe
r Inc. All rights reserved.
[3,4]. Matrix turnover occurs regularly in bone, during both boneremodelling and following injury [5]. During this process cell surfaceMMPs are secreted, which digest the matrix proteins during theremodelling process. During matrix degradation, RGD containingpeptides are released, which can result in loss of cell adhesion viablocking the binding between integrins and ECM and ultimatelyleading to cell death (anoikis) [6]. It has been reported that thesynthetic RGD peptides can block the integrin-regulated celladhesion on RGD containing ECM proteins and result in anoikis.
University, 34755 Istanbul, Turkey.

368 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
Although collagen I is the most abundant ECM protein in bone, thecell adhesion protein fibronectin (FN), which is found in tightassociation with collagen, is also essential for cell adhesion duringbone formation. Within FN, in addition to RGD, at least two otherregions have been reported to be involved inmediating cell adhesionwhich include the synergy site PHSRN (Pro–His–Ser–Arg–Asn) andthe heparin binding sites Hep 1 and Hep 2 [7]. Of the FN-relatedintegrins, α5β1 is probably the major cell surface integrin thatinteracts with the RGD cell binding sites on FN, initiating the celladhesion process by associationwith the synergy binding site. Otherkey cell surface receptors found on osteoblasts are the heparansulphate proteoglycans referred to as syndecans, ofwhich syndecan-4 is probably the most predominant. Syndecan-4 acts as a co-receptor in β1 integrin-mediated cell adhesion and belongs to afamily composed of 4 members (syndecans1–4). Syndecan-4 isinvolved in actin cytoskeleton organization via its interaction withtheheparin binding sites of FN [8]. Another syndecan familymemberwhose role is probably lesswell understood is syndecan-2 [9], whichis also involved in actin stress fibre formation.
A further ECMassociated protein also thought to be key tomatrixformation during bone mineralization is the protein crosslinkingenzyme tissue transglutaminase (TG2) [10]. TG2 when activated byCa2+ stabilizes proteins via intermolecular crosslinking through ε(γ-glutamyl) lysine bridges. Although mainly localized in the intracel-lular environment where its Ca2+-mediated activity is tightlyregulated by the binding of GTP/GDP, the enzyme is also secretedonto the cell surface and ultimately into the extracellular matrix(ECM) by an unknown mechanism not involving the classical ER/Golgi route [11]. Once deposited into ECM, TG2 forms a hetero-complexwith its high affinity binding partner FN (TG-FN), onwhichit binds to the N-terminal 42 kDa fragment [12]. Once bound to FN,the enzyme becomes more resistant to proteolytic degradation andits transamidating activity is reduced [6]. The importance of the TG-FN heterocomplex in enhancing human osteoblasts (HOB) celladhesion and spreading was first reported in 2001 by Griffin andcolleagues [13], which triggered subsequent investigations of thenatural occurrence of this TG-FNmatrix complex in the ECM of HOBcells [14]. It was later shown that this heterocomplex of TG-FN aswell as enhancing cell adhesion could also compensate for the RGDpeptide-induced loss of cell adhesion, which could be inhibited bythe digestion of cell surface heparan sulfates by heparanasetreatment, suggesting the involvement of cell surface heparansulfates [6]. Importantly the compensatory effect of this matrixcomplex is not influenced by the transglutaminase activity inhibi-tors, indicating the function of TG2 as amatrix scaffold protein ratherthan as crosslinking enzyme. Given the importance of osteoblasts inbone healing and matrix turnover [15], this report further exploresthe cellular mechanisms, including the importance of different cellsurface receptors and intracellular signaling pathways, involved inthis RGD-independent cell adhesion/survival pathway in relation toHOB cell function.
Materials and methods
Reagents and antibodies
Human plasma fibronectin was purchased from Sigma-Aldrich(UK). The FN synthetic peptides GRGDTP, GRADSP, and Rho kinase(ROCK) inhibitor Y27632 were obtained from Calbiochem. The
synergy peptide (H-PHSRN-OH) and CS-1 (EILDVPST) peptidewere from Bachem. The GK21 peptide (GENPIYKSAVTTVVN-PIYEGK) and the scrambled control peptide (GTAKINEPYSVTVPY-GEKNKV) in tandem with the antennapedia third helix sequence(PQIKIWFQNRRMKWKK) and the A5-1 peptide (VILVLF) [16] werechemically synthesized by Peptide Protein Research, UK. TG2inhibitor 1,3-dimethyl-2 [(oxopropyl)thio] imidazolium deriva-tive R283 was synthesized at Aston University. Purified guinea pigliver TG2 (gplTG) was purified according to Leblanc et al. [17]. TheFITC-labelled phalloidin was purchased from Sigma-Aldrich, UK.The mouse anti-phosphorlated-ERK1/2, mouse anti-ERK1/2 anti-body, rabbit anti-β1 integrin, mouse anti-human FAK, and mouseanti-PKCα antibodies were from Santa Cruz; the mouse anti-α-tubulin antibody was from Sigma-Aldrich; and anti-human Tyr (p)397 and Tyr (p)861 were from Upstate Cell Signalling Solutionsand Biosource UK, respectively. The rabbit polyclonal anti-syndecan-4 and syndecan-2 antibodies, which recognize theintracellular domains in the core proteins of these receptors,were from Zymed Invitrogen Immunodetection. The FITC-conju-gated anti-mouse IgG and the swine anti-rabbit secondaryantibody were from Dako, UK.
Cell lines
Human osteoblasts were a kind gift by Prof. S. Downes (Universityof Nottingham, Nottingham, UK) and cultured in Dulbecco'smodified Eagle's medium (DMEM) as previously described [6].TG2-transfected HOB cells were established and cultured aspreviously [18].
Cell adhesion assay
Cell adhesion assays were performed as described before [6].Following serum starving for 16 h, the cells were pre-treated with100 μg/ml of the GRGDTP peptides or its control peptides GRADSP,according to previous toxicity findings, unless specified in certainexperiments [6]. Cells were then seeded onto 96-well plates(2.5×104/well) coated with human plasma FN (5 μg/ml, FNmatrix)with or without immobilized guinea pig liver TG (20 μg/ml, gplTG)(TG-FNmatrix) as previously described [6]. Briefly, following certaintreatments, the cells were allowed to adhere on FN or TG-FN matrixfor around 20–40min (tominimize the secretion of any endogenousproteins) in the presence of RGD or RAD peptides. After the fixationand permeabilization, the cells were stained via May-Grunwald andGiemsa co-staining. Digital images of three non-overlapping fieldscovering the centre of the well were acquired using a video digitalcamera (Olympus DP10) at 40× magnification (total cells analysednormally between 400 and 500 cells). The cell attachment andspreading were quantified using the Scion image analysisprogramme (http://www.scioncorp.com), which is developed atthe National Institute of Health (Washington DC, USA). The numberof cells per image was assessed through threshold and particleanalysis setting with a minimum particle size of 50 pixels. Spreadcells were discriminated from non-spread cells by their two-colourappearance (dark purple for nucleus and pink for cytoplasm). Cellswere fixed, permeabilized and stained and digital images capturedper each sample. Attachment and spreadingwere quantified and thenumber of cells per image was assessed as stated previously [6]. Insome experiments, cell adhesion was performed on a heparin-blockedmatrix. 300 μg/ml of heparin in 50 mMTris–HCl, pH7.4, was

369E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
used to block the heparan binding sites on FN or on TG2 before orafter the immobilization of TG2 on the FN matrix, respectively aspreviously described [19]. The co-treatment of cells with the RGDand the synergy (H-PHSRN-OH) synthetic peptides or the CS 1peptides was performed at the concentration of 75 μg/ml of eachpeptide, to minimize the cell toxicity, while 150 μg/ml of RADpeptides was used as the control treatment.
siRNA transfection
The HP GenomeWide siRNA sequences targeting human synde-can-4 (SI00046816) and syndecan-2 (SI00046795) and the non-silencing (NS) control siRNA were obtained from Qiagen (UK). Thetarget sequences are non-homologues for any other syndecantypes or cell surface receptors. The transfection was performedusing HiPerfect transfection reagent (Qiagen) according to themanufacturer's protocol.
Cytosol and membrane fractionation
HOB cells grown to 70% confluency and serum starved for 16 hwere pretreated with PKCα inhibitor Go6976 or DMSO (thevehicle control treatment) and trypsinized and then neutralized byaddition of soybean trypsin inhibitor, while phorbol 12-myristate13-acetate (PMA), a well-known activator of PKCα, was used as apositive control treatment for PKCα activation. Following theirtreatment with RAD or RGD containing peptides, cells were platedonto 6-well plates coatedwith FN or TG-FNmatrices at a density of6×105 cells per well and allowed to attach for 30 min. PKCαtranslocation to the membrane was analysed as previouslydocumented [19]. Briefly, following incubating the cells on FN orTG-FN matrix, cells were washed once with ice-cold PBS, pH 7.4and collected into pre-chilled homogenization buffer containing20 mM Tris–HCl, pH 7.4, 10 mM EGTA, 2 mM EDTA, 1 Mm NaF,1 mM Na3VO4, 50nM okadaic acid, 0.1 mM PMSF and 1% (v/v)protein inhibitor cocktail (Sigma-Aldrich, UK). After homogeniza-tion via 4 cycles of freezing and thawing (one cycle includesincubating in liquid nitrogen for 2 min and incubating in waterbath at 37 °C for 7 min, the cell lysates were pre-cleared at 1300gfor 10 min to pellet the nuclei and unbroken cells. The super-natants were collected and centrifuged at 100,000g for 60 min toseparate cytosolic (supernatant) and membrane fractions. Themembrane fractions were further washed once with ice-cold PBS,pH7.4. The cytosol and membrane fractions of samples wereWestern Blotted and probed with PKCα antibody (80 kDa).Measurement of the presence lactate dehydrogenase (LDH)activity in the membrane fraction was used to assess the presenceof any cytosol proteins in this fraction.
Fluorescence staining
Sub-confluent HOB cells were serum-starved for 16 h, harvestedand pre-treated with 100 μg/ml of GRADSP or GRGDTP peptides asdescribed above. Cell adhesion was performed as introducedearlier. For the actin stress fibre staining, the cells were incubatedwith FITC-labelled phalloidin (20 μg/ml) in blocking buffer (3%heat-inactivated BSA in PBS, pH7.4). For phosphotyrosine proteinsafter blocking the cells they were incubated with rabbit polyclonalanti-phosphotyrosine antibody (1:100 in blocking buffer) fol-lowed by anti-mouse IgG-FITC diluted 1:200 in the blocking buffer.
Slides were mounted with Vectashield mountant (Vector Labora-tories) and examined as mentioned above.
Biotinylation of cell surface proteins
Biotinylation of cell surface protein with EZ-link Sulfo-NHS-Biotin(Thermo, UK) was performed according to manufacturer's proto-col. Briefly the cells were incubated with 0.2 mg/ml of Sulfo-NHS-biotin in PBS, pH 8.0 for 15 min at 4 °C. After washing once with50 mM Tris–HCl, pH8.0 and twice with PBS, pH8.0, the cells werelysed with 1% SDS in PBS, pH 8.0 and cell lysates containing 600 μgof total protein (denatured by boiling for 5 min) was incubatedwith 50 μl of resin beads slurries (Thermo, UK) at 4 °C over night.After washing once with PBS, pH 8.0, the beads were boiled with30 μl of Laemmli buffer for 5 min and stored at−80 °C forWesternblotting to detect the cell surface proteins by using specificantibodies.
Western blotting and co-immunoprecipitation
As introduced above, after the desired treatment cells werecollected and lysed by addition of 70 μl of cell lysis buffer (SantaCruz). After centrifugation at 300×g for 10 min, the supernatantswere stored at −70 °C until use. SDS gel electrophoresis andWestern blotting were performed as described previously todetect the presence of the target antigens by using specificantibodies [6]. For the co-immunoprecipitation, cells seeded ontoheparin treated TG-FN or non-treated FN matrices, and werecollected in lysis buffer as previously described [19]. The targetedproteins in the immuno-complex were detected via Westernblotting by using their specific antibodies.
Statistics
Data were expressed as mean±S.D. The data shown are derivedfrom a representative experiment (n>3) undertaken in triplicate.The comparisons between the data sets were performed usingStudent's t test (two-tailed distribution with equal variance).Statistical significant difference between data sets was defined inthe text by p<0.05 (two-sided).
Results
Matrix bound TG2 and not cell surface associated TG2 isrequired for compensating the RGD-induced loss of celladhesion mediated by TG-FN heterocomplex
In our previous work we demonstrated that the TG-FN hetero-complex could both enhance cell adhesion and compensate for theloss of cell adhesion induced by the blockage of integrins by RGDpeptides. This mechanism was shown to be mediated by thebinding of the TG-FN complex to cell surface heparan sulphateproteoglycans [6]. Previous work has also proposed that cellsurface associated TG2 can interact with cell surface β integrinsand is able to support cell adhesion on FN by acting as a FN co-receptor [20]. To investigate whether the cell surface TG2 asopposed to FN bound TG2 once deposited into the matrix isinvolved in the RGD-independent cell adhesion process, HOB cellswhich contain only trace levels of endogenous TG2 were stably

370 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
transfected with TG2 to establish the tg2-HOB cell line [18]. Over-expression of TG2 in HOB cells (Fig. 1A and B), which leads toincreased cell surface enzyme [18], did not compensate for RGD-induced loss of cell adhesion on a FN matrix, when compared towild type HOB cells. This is in contrast the compensation given
Fig. 1 – Effects of cell surface TG2 and PHSRN cell binding domain oattached cells or the percentage of spread cells±S.D. shown are fromin triplicate The ordinate in the top graphs represents the mean wtattachment to FN (which represents 100%). The mean percentage omean percentage of wt attachment to FN±S.D in the presence of coand TG2 transfected HOB cells (tg2-HOB) to FN and TG-FN matricesimportance of the PHSRN cell binding domain within FN molecule75 μg/ml PHSRNpeptide for 30 min and then seededon FNor TG-FNmRAD or 75 μg/ml RGD peptide performed as described in Materials
when the TG2 is prebound to FN which occurs when it is depositedinto the matrix (Fig. 1A and B). Hence only the pre-deposited TG-FN complex can rescue the loss of cell adhesion induced by theRGD peptide, with no significant differences (p>0.05) observedbetween wild type and tg2-HOB cells.
f FN on the RGD independent cell adhesion. The percentage ofa representative of four separate experiments, each performedcell attachment expressed as the mean percentage of controlf cell attachment for tg2-transfected cells was expressed as thentrol RAD peptide. A and B, cell adhesion of wild type (wt HOB)in the presence of 100ug/ml of RAD or RGD peptide. C, thein RGD-independent cell adhesion. HOB cells were treated withatrices and the cell adhesion assay in the presence of 150 μg/ml
and methods.

371E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
The PHSRN cell binding domain of FN is not required for thecell adhesion mechanism mediated by the TG-FNheterocomplex in the presence of RGD peptides
HOB cells which express α5β1 integrins [6] anchor on FN not onlyby recognizing the RGD sequence but also with the association oftheir binding to the synergy sequence (PHSRN). Wild type HOBcells were treated with RGD and/or synergy peptide PHSRN(Fig. 1C). Compared to RGD peptide treatment (40% loss of celladhesion), the PHSRN peptide only caused around 30% loss of celladhesion at 75 μg/ml. Only when seeded on a TG-FN matrix, cancells accomplish cell attachment and spreading comparable tocontrol levels in the presence of either peptides.
Compensation of the RGD-induced loss of HOB cell adhesionon TG-FN matrix is dependent on the interaction of TG2 withthe heparan sulphate proteoglycan syndecan-4 via itsheparan sulfate chains
Our earlier data showed that the compensation of the RGD-induced loss of cell attachment and spreading on TG-FN could beinhibited by heparinase digestion of the sulphate heparans on theHOB cell surface [6]. To extend this observation, we firstinvestigated which component of the TG-FN complex compen-sates for the RGD peptide induced loss of cell adhesion. Heparin,which has a high affinity for TG2 and which also binds to theheparin binding site on FN [21], was used to coat the TG-FNmatrixpreparation. The heparin coatingwas applied both before and afterimmobilizing TG2 onto the FN.When heparinwas used to block FNprior to application of TG2, it did not have any effect on the bindingof TG2 [19]. Similarly the function of the TG-FN complex was notsignificantly (p>0.05) affected compared to the non-treatedmatrix (Fig. 2A). However, when heparin treatment was appliedafter formation of the TG-FN complex the heparin treated complexcould not compensate for the loss of cell adhesionmediated by theRGD peptides (Fig. 2B).
To confirm syndecan-4 as amajor heparan sulphate proteoglycaninvolved in the TG-FNmediated cell adhesion process its expressionwas reduced to around 50% (Fig. 2C) using siRNA. Syndecan-4 siRNAtreatment led to a significant loss (p<0.05) of cell attachment andspreading on the FN matrix compared to the non-treated HOB cellsor cells treated with the non-silencing (NS) control siRNA.Importantly the TG-FN complex lost its compensatory cell adhesioneffect in syndecan-4 siRNA-treated HOB cells in the presence of theRGD peptide. Compared to cell attachment, cell spreading wasaffected more by the syndecan-4 siRNA treatment, further confirm-ing the importance of syndecan-4 in actin stress fibre formation(Fig. 2D and E). Immunoprecipitation of TG2 using an anti-syndecan4 antibody (Fig. 2F), which detects the intracellular domain of thesyndecan-4 core protein, indicated a direct interaction between thetwo proteins, further supporting the hypothesis that cell surfacesyndecan-4 in HOB cells is the cell surface receptor that directlyinteracts with TG2 in the TG-FN complex.
PKCα is the intracellular link between the syndecan-4 andβ1 integrin signalling pathway in the RGD-independent celladhesion by TG-FN
In fibroblasts, we previously demonstrated that PKCα is the cruciallink between the syndecan-4 and β1 integrin co-signalling, when
these cells are seeded on TG-FN in the presence of RGD peptide.Use of the GK21 peptide, which mimics the intracellular PKCαbinding site within β1 integrin cytoplasmic domain and in turninhibits the interaction between β1 integrins and PKCα, was usedto block the interaction between PKCα and the β1 integrinintracellular domain as reported previously [22]. This peptidereduced cell attachment and spreading of HOB cells on FNcompared to control cells. More importantly, GK21 inhibited thecompensatory function of the TG-FN complex in RGD-treated cells,while in the control scrambled peptide group the TG-FN complexrestored the loss of cell adhesion and spreading caused by the RGDpeptide (Fig. 3A).
The role of PKCα was further investigated by detecting itstranslocation from the cytosol to membrane. HOB cells were pre-treated with Go6976, which has been demonstrated to be aspecific inhibitor for PKCα, in the presence of RAD or RGDpeptides. Cells pre-treated with 50nM PMA and RAD peptideseeded on FN were used as the positive control, since it has beenshown that PMA is a general PKCα activator. The presence of LDHin the membrane fraction was used to rule out any contaminationof the membrane fraction with the cytosol (Fig. 3B). Westernblotting showed that PMA promoted the translocation of PKCαfrom cytosol to membrane in the presence of RAD peptide. In thecells treated with DMSO vehicle alone, translocation of PKCα wassignificantly inhibited by the treatment of HOB cells with RGDpeptides but this inhibition could be compensated by the TG-FNcomplex (Fig. 3B). This compensatory effect of the TG-FN complexon the RGD-dependent PKCα activation was blocked by the PKCαinhibitor Go6976, further confirming the importance of PKCα inthe RGD-independent cell adhesion mediated by TG-FN.
Compensation of RGD induced loss of cell adhesion in HOBcells requires the involvement of the cell surface receptorsyndecan-2 and its downstream signalling molecule ROCK
Since syndecan-2 like syndecan-4 is highly expressed in osteo-blasts [23], we hypothesized that syndecan-2 can also participatein the RGD-independent cell adhesion in response to the TG-FNheterocomplex.
siRNA silencing of syndecan-2 reduced its expression to around45%−50% compared to its expression in non-treated and NS siRNAcontrol groups (Fig. 4A), while the expression of both syndecan-4and β1 integrins were not significantly affected. The syndecan-2siRNA treatment of HOB cells led to 25% loss in both cellattachment and cell spreading on FN alone, and in the presenceof RGD peptide, around a 50% reduction of cell attachment andspreading was achieved compared to controls. Importantly TG-FNfailed to compensate the RGD peptide induced loss of celladhesion, strongly suggesting the requirement of syndecan-2 inthis novel adhesion mechanism (Fig. 4B and C).
Fluorescent visualization of the actin stress fibres by confocalmicroscopy indicated that syndecan-2 siRNA-treated HOB cellscould not form well-organized actin cytoskeleton structures com-pared to the cells treated with the NS siRNA (Fig. 4D). Since it hasbeen reported that Rho and ROCK are the downstream signallingmolecules in the syndecan-2 signalling pathway [24], cell adhesionassays were undertaken in the presence o the ROCK inhibitorY27632. Treatmentwith Y27632 inhibited more than 30% (p<0.05)cell attachment and spreading on FN (Fig. 4E), compared to thecontrol cells treated with DMSO. On the contrary to the observed

372 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
compensatory effect for the RGD peptide-induced loss of celladhesion in the control cells, in the presence of the RGD peptides,the Y27632-treated HOB cells completely lost their spreading abilityon the FN matrix, which could not be compensated by the TG-FNcomplex (Fig. 4E), thus confirming the importance of ROCK. To studywhether there is a direct interaction between syndecan-2 and TG2,anti-syndecna-2 antibody recognizing the cytoplasmatic domain ofsyndecan-2 core protein was used to pull down the syndecan-2immunocomplex. Interestingly unlike the discovery of the directinteraction between TG2 and syndecan-4, Western blotting for TG2in the immunocomplex revealed the absence of the TG2 antigen inthis complex (Fig. 4F), suggesting the interactionbetween syndecan-2 and TG2 is likely to be indirect.
The α5β1 integrin inside-out signalling pathway is crucialfor TG-FN to compensate the loss of cell adhesion mediatedby the RGD peptides
We next undertook experiments to investigate the role of β1integrin and its major FN binding companion α5 in the RGDindependent cell adhesion on TG-FN. Treatment of HOB cells withthe β1 integrin functional blocking antibody—HMβ1-1 [25] led to a46% decrease in cell attachment and blocked 56% of cell spreadingon FN compared to non-treated cells seeded on FN, while itsisotype control antibody showed no effect on both cell attachmentand spreading. In contrast cells seeded on the TG-FN hetero-complex restored the loss of cell attachment and spreading back to~90% (Fig. 5A). HMβ1-1 is thought to exert its blocking effect byinhibiting the signalling transduction of the β1 integrin outside-inpathway [25], which was confirmed via detecting the phosphor-ylation of FAK, a downstream signallingmolecule of β1 integrin. Asshown in Fig. 5B, in HMβ1-1 treated HOB cells, the antibodyinhibited the phosphorylation of FAK at Tyr397 compared toisotype antibody treated cells on a FN matrix, while thephosphorylation of Tyr861 was not significantly affected. Giventhat FAK is phosphorylated at Tyr861 residue in response tointegrin clustering without an integrin−ligand engagementthrough inside-out signalling [26], this result indicates that theinside-out signal transduction was not influenced by the HMβ1-1function blocking antibody. Co-immunoprecipitation assays indi-cated that there was no direct interaction between syndecan-4 and
Fig. 2 – The importance of heparan sulfate chains of syndecan-4 inA, Heparin(300 μg/ml) was incubated with the FN matrix for 30 miimmobilization of TG2 onto this matrix as described in Materials anmatrices in the presence of the RAD or RGD peptides (100ug/ml) forTG-FN matrices are expressed as the mean percentages of attachmtreatment±S.D. B, Heparin (300 μg/ml) was applied after the formnon-treated and heparin treated-TG-FN matrices. Percentages of cepresence of RAD peptide were set as 100%. C, HOB cell monolayers w1:10 siRNA to transfection reagent ratio and non silencing control (transfection, syndecan-4, syndecan-2 and β1 integrin protein expreassess equal loadings. D and E, HOB cell monolayers were treatedwias described inMaterials andmethods. Transfected andnon-transfecand the percentage of cells adhering to FN and TG-FNmatrices was dand methods. The percentage of attached cells or the percentage ofseparate experiments, each performed in triplicate. F, co-immunopwere immunoprecipitated with anti-syndecan-4 andWestern blottisolated from guinea pig liver (gplTG) was used as the internal stan
β1 integrin (Fig. 5C), supporting our observations above that inHOB cells the β1 integrin is activated by syndecan-4 in anintracellular manner via PKCα [19].
To investigate the involvement α5β1 integrin, an inhibitingpeptide of this integrin, A5-1was utilized in HOB cell adhesionexperiments [16]. It has been reported that the A5-1 peptideblocks the α5β1 integrin-related cell adhesion, proliferation,migration and angiogenesis [16]. The peptide A5-1 inhibited cellattachment and spreading in a dose-dependent manner, and at theconcentration of 10 μM it blocked cell attachment and spreadingby 50%. Importantly use of the peptide A5-1 led to the failure of theTG-FN complex to restore RGD peptide-induced loss of celladhesion, indicating the importance of the α5β1 integrin in thisprocess (Fig. 5D).
α4 integrin is not involved in the ability of TG-FN tocompensate the loss of cell adhesionmediated by RGD peptides
In addition to its cell binding domain, fibronectin also containsactive binding sites CS-1 and CS-5 for α4β1 integrin within thealternatively spliced V region [27]. This integrin is expressed inHOB cells [28] (Supplemental Fig. 2A). Because TG2 is also knownto be a ligand for α4β1 integrin [29,30], we explored whether thecompensatory effect of TG-FN in RGD peptide-induced loss of celladhesion requires the involvement of α4β1 integrins. The celladhesion of HOB cells on FN and/or TG-FN was not significantly(p>0.05) affected with either control mouse IgG treatment orfunction blocking anti-α4 integrin antibody (P4C2) treatments at30 μg/ml (Fig. 6A and Supplementary Fig. 2B) in the presence ofRAD peptide. P4C2 antibody did not induce further loss of cellattachment and spreading caused by the RGD peptide. In bothmouse IgG and P4C2 antibody treated cells, TG-FN rescued the lossof cell adhesion induced by RGD peptides, suggesting thecompensatory effect of this heterocomplex is independent of thepresence of cell surface α4β1 integrin. CS-1 peptide which blocksthe interaction between α4β1 integrin and FN did not affect HOBcell adhesion in the presence of RAD or RGD peptide on FN whencompared to the control cells, respectively. No significantdifference in the compensation effect of TG-FN on RGD-inducedloss of cell adhesion was observed in the control and CS-1-treatedcells (Fig. 6B).
RGD-independent cell adhesion on TG-FN mediated by PKCα.n to block the heparin binding sites on FN before thedmethods. HOB cells (2.5×104/well) were then seeded onto thethe cell adhesion assay. Cell attachment and spreading on FN orent of RAD-treated cell on the FN matrix without heparination of the TG-FN complex and cell adhesion observed onll adhesion and spreading on non-treated FN matrix in theere treated with 5 μg syndecan-4 siRNA (A) at themost effectiveNS) siRNA was applied as the negative control. Followingssions were analysed by Western Blotting. Tubulin was used toth 5 μg syndecan-4 specific siRNA or non silencing control siRNAtedHOB cellswere pre-incubatedwith RGDpeptide (100 μg/ml)etermined in a 40 min adhesion assay as described in Materialsspread cells±S.D. shown are from a representative of fourrecipitation of syndecan-4 and TG2 in HOB. Cell lysate samplesed for TG2 detection as described in Materials and methods. TG2dard.


Fig. 3 – The role of PKCα in RGD-independent cell adhesion on the TG-FN matrix. A, the interaction of PKCα with β1 integrin isrequired by TG-FN to exert its function. GK21 peptide was utilized in RGD-independent cell adhesion process on FN or TG-FNmatrix,while DMSO (0.1%) and scrambled peptide were used as control. The percentage of attached cells or the percentage of spread cells±S.D. shown are from a representative of four separate experiments, performed in triplicate. B, HOB cells were treated with 50 nMPMA (an activator of PKCα) in the presence of the RAD peptide, as the positive control treatment for PKCα activation, or the PKC-αinhibitor Go6976 (dissolved in DMSO). Cells were allowed to attach to FN and TG-FN matrices in the presence of RAD (a) or RGD (g)peptides as described in the in Materials andmethods. DMSOwas used as the vehicle control treatment. 5 independent experimentswere undertaken, Western blotted and immunoprobed with mouse monoclonal PKC-α antibody. The upper blot represents thelevels of PKC-α in the cytosolic fraction, while the lower blot indicates the levels of PKC-α translocated to membrane. LDH was alsodetected in both fractions by using anti-LDH antibody in Western blotting to rule out the cross-contamination of the membranefractions with the cytosol. Percentage of PKCα in the membrane fractions was calculated after densitometric measurement of theWestern blotting signals, in which (a) represents the RAD peptides and (g) the RGD peptides.
374 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
Intracellular signalling in HOB cells after plating on theTG-FN heterocomplex
Using immunofluorescence, phosphotyrosine containing proteinscan easily be detected in the focal contacts of the HOB cells seeded
Fig. 4 – The role of syndecan-2 and its downstream signalingmolecuA, HOB cell monolayers were treated with 5 μg syndecan-2 targettinratio and non-silencing (NS) control siRNA was applied as negativeintegrinproteinexpressionswereanalysedbyWesternblotting. Tubuwere treated with syndecan-2 siRNA and non-silencing (NS) controleither seeded on FN or TG-FN matrices in the presence of 100 μg/mlnon-treated attachedand spread cells onFN in thepresenceofRAD±detected in syndecan-2 siRNA (SDC-2 siRNA) or control siRNA-treatepeptide. As described in Materials and methods, after siRNA treatmeusing confocalmicroscopy. Bar, 20 μm. E, afterROCK inhibitor Y27632orTG-FNmatrices in thepresenceofRADorRGDpeptide. Thepercentaas 100%. F, co-immunoprecipitationof syndecan-2 andTG2assayswerfor TG2. gplTG was loaded as the internal standard.
on FN without RGD pre-treatment (Fig. 7A). In HOB cells whichwere attached and spread on FN, tyrosine phosphorylated proteinsshowed a clear distribution from cytosol to membrane, regardlessof the presence of RAD peptide. These cells exhibited a flatmorphology and initial formation of focal adhesion contacts. Cells
le ROCK in regulating RGD-independent cell adhesion on TG-FN.g siRNA at the most effective 1:10 siRNA to transfection reagentcontrol. Following transfection, syndecan-4, syndecan-2 and β1linwasused toassess equal loadings.B andC,HOBcellmonolayerssiRNA as described in Fig. 2. Following transfection the cells wereRGD or RAD synthetic peptides. Mean percentage value ofS.D. (control)wasusedas 100%.D, actin stress fiber formationwasd (NS siRNA) HOB cells in the presence of 100ug/ml RAD or RGDnt, the fluorescence-stained actin stress fiber was visualized by(10 mM)orDMSO treatment (0.1%),HOB cellswere seededonFNgesvalueofnon-treated attached and spreadcells onFNwasusedeperformedwithanti-syndecan-2antibody andWesternblotting


Fig. 5 – The importance of α5β1 integrin in RGD-independent cell adhesionmediated by TG-FN. A, pre-treated HOB cell suspensions(2.5×105/ml) with 25 μg/ml HMβ1-1 β1 integrin functional blocking antibody or its isotype control antibody were seeded on FN orTG-FNmatrix as described inMaterials andmethods. B, HOBcells treatedwithHMβ1-1β1 integrin functional blocking antibodyor itsisotype control antibody were seeded on FN and TG-FN matrices. The samples were collected and used in the detection of FAK-Y397
and FAK-Y861 via Western blotting. α-Tubulin was used to ensure equal loading of each samples. C, detection of the interaction ofsyndecan-4 and β1 integrins. HOB cells sampleswere prepared and used in immunoprecipitation as described in Fig. 2 legend. Rabbitpolyclonal anti-β1 integrin antibody was used to detect β1 integrin bands by Western blotting. The first lane shows the β1 integrinstandard (β1 INT standard). D, HOB cells treated with an α5β1 integrin targeting peptide A5-1 at the concentrations of 2.5, 5, 10, 25and 50 μM were used in the cell adhesion assay as introduced in Materials and methods, while RAD and RGD peptides at theconcentration of 100 μg/ml were used as the negative and positive control treatments, respectively.
376 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
seeded on TG-FN matrix without RGD were spread with fullyformed focal adhesion points, which were positively stained forphosphotyrosine proteins. This phosphotyrosine staining patternwas lost from focal contacts in cells plated on FN in the presence ofRGD peptide which showed a very weak and condensedphosphotyrosine staining pattern in the cytoplasm of these non-spread cells. In contrast, on TG-FN, HOB cells incubated with RGDpeptide appeared to maintain control levels of phosphorylatedtyrosine proteins, although cells appeared slightly less spread and
more rounded than control cells seeded on FN without RGD. Thismaintenance of tyrosine phosphorylation although slightly re-duced is not unexpected since our previous work indicated thatwhen seeded on TG-FN, the phosphorylation of FAK at Tyr397 tookplace in HOB cells even in the presence of RGD peptides [6]. Theautophosphorylation of Tyr397 can afterwards promote thephosphorylation at the other sites within the FAK molecule. Asshown in Fig. 7B, the RGD peptides blocked the phosphorylation ofTyr861 in cells plated on FN alone but not when plated on TG-FN.

Fig. 6 – The role of α4β1 integrin in the cell adhesion process on TG-FNmatrix. A, HOB cells in suspension (2.5×105) were pre-treatedwith 30 μg/ml of anti-α4 integrin antibody (P4C2) or control mouse IgGs (Isotype Ab) in the presence of RGD and RAD (100 μg/ml)peptides before seeding on either FN or TG-FN. B, HOB cells were treated with 75 μg/ml α4β1 integrin blocking peptide CS-1 with orwithout addition of 75 μg/ml RGD peptide before seeding on the FN or TG-FNmatrix. Data, expressed as percentage of control valueson FN in the presence of RAD peptide (150 μg/ml), are from one of four separate experiments each undertaken in triplicate.
377E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
This data confirms the result shown in Fig. 5B in that the TG-FNcomplex rescues the loss of cell adhesion via phosphorylation ofFAK at Tyr861, an indicator of the ligand- independent activation ofβ1 integrin signalling. Next, we tested the involvement of thedownstream MEK-ERK survival pathway in TG-FN compensatedRGD-induced loss of cell adhesion. In HOB cells seeded on BSAmatrix, tyrosine phosphorylation levels of ERK1 and ERK2 (p-ERK1/2) were reduced to around 40% and 60% respectively of thatof the FN control (Fig. 6C). In comparison to control levels, thecontent of p-ERK1/2 antigen was not significantly (p>0.05)different in the cells seeded on FN and TG-FN with or withoutRAD peptide. The effect of RGD peptide on the amount of p-ERK1/2antigen was apparent, considering the 50% reduction in p-ERK1and 30% reduction in p-ERK2 antigen levels observed in cellsseeded on FN with RGD peptides. In response to TG-FN, the cellcontent of ERK1 and ERK2 phosphorylation was found to beapproximately 85% and 95% of that of control cells seeded on FNwithout RGD peptide. Immunoblots of the same cell extractsprobed for whole ERK1/2 proteins showed no statisticallysignificant (p>0.05) difference in the levels of total ERK1/2.
Taken together, these data strongly suggests that FAK Tyr861
phosphorylation and activation of ERK1/2 is part of themechanism
by which adhesion signals are transduced by TG-FN in a RGD-independent manner.
Discussion
The importance of osteoblasts in the formation of a mineralizedextracellular matrix during bone growth and bone healing is welldocumented [31]. During this process the cell surface receptorsα5β1 integrin and syndecan-4 co-signalling pathways play animportant role in regulating cell adhesion and actin cytoskeletonformation [22]. In our previous work we demonstrated that celladhesion and spreading of HOB cells was markedly enhanced bybinding to a TG2–FN heterocomplex [13], which was capable ofmaintaining cell adhesion even in the presence of integrincompeting RGD peptides [6], which are frequently released duringmatrix turnover [31]. As both a cell surface andmatrix protein, TG2may also be involved in the cell adhesion on FN by acting as anintegrin coreceptor [20]. Hence the first task of this study was todistinguish between the different roles of cell surface and matrixTG2 in this RGD-independent cell adhesion process by using aTG2-overexpressing HOB cell line which has abundant cell surface
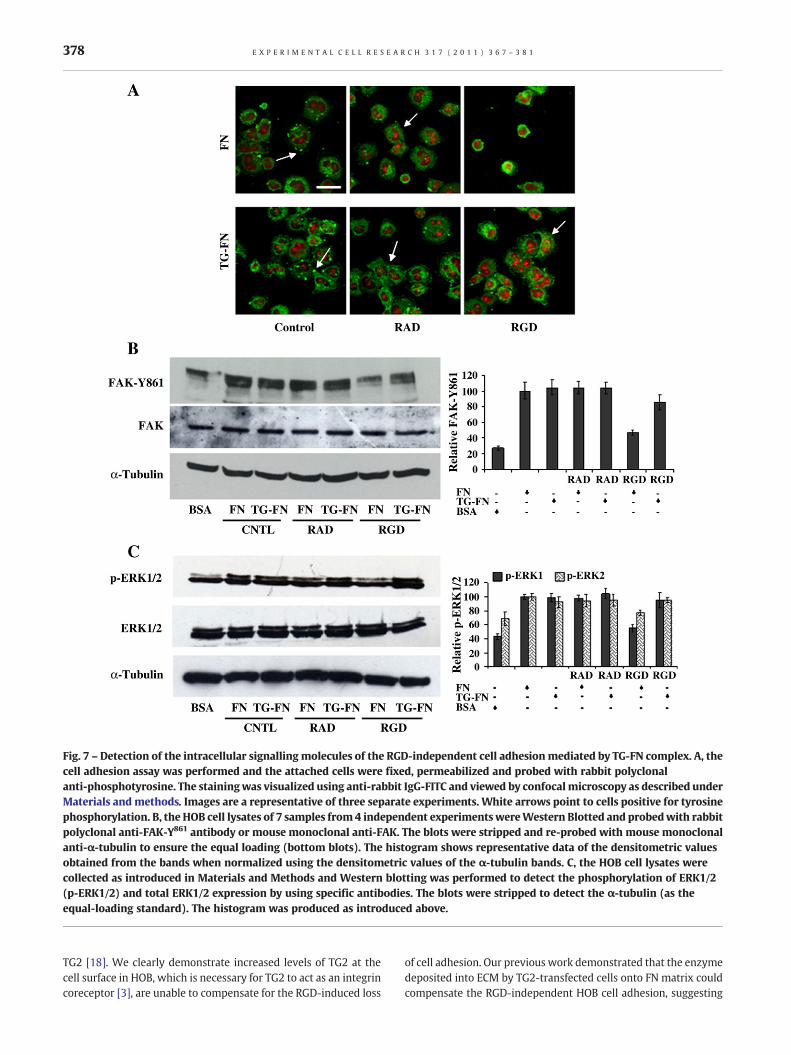
Fig. 7 – Detection of the intracellular signalling molecules of the RGD-independent cell adhesionmediated by TG-FN complex. A, thecell adhesion assay was performed and the attached cells were fixed, permeabilized and probed with rabbit polyclonalanti-phosphotyrosine. The stainingwas visualized using anti-rabbit IgG-FITC and viewed by confocalmicroscopy as described underMaterials and methods. Images are a representative of three separate experiments. White arrows point to cells positive for tyrosinephosphorylation. B, the HOB cell lysates of 7 samples from 4 independent experiments wereWestern Blotted and probedwith rabbitpolyclonal anti-FAK-Y861 antibody or mouse monoclonal anti-FAK. The blots were stripped and re-probed with mouse monoclonalanti-α-tubulin to ensure the equal loading (bottom blots). The histogram shows representative data of the densitometric valuesobtained from the bands when normalized using the densitometric values of the α-tubulin bands. C, the HOB cell lysates werecollected as introduced in Materials and Methods and Western blotting was performed to detect the phosphorylation of ERK1/2(p-ERK1/2) and total ERK1/2 expression by using specific antibodies. The blots were stripped to detect the α-tubulin (as theequal-loading standard). The histogram was produced as introduced above.
378 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
TG2 [18]. We clearly demonstrate increased levels of TG2 at thecell surface in HOB, which is necessary for TG2 to act as an integrincoreceptor [3], are unable to compensate for the RGD-induced loss
of cell adhesion. Our previous work demonstrated that the enzymedeposited into ECM by TG2-transfected cells onto FN matrix couldcompensate the RGD-independent HOB cell adhesion, suggesting

379E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
the function of this enzyme as a matrix protein in regulating cellbehavior(s) [6]. As introduced earlier, the role of PHSRN motif hasalso been reported to be involved in cell attachment [32]. Hencesynthesized PHSRN peptides were used in the cell adhesion assayin the presence or absence of the RGD peptides, which showed noeffect on the cell attachment and spreading on either FN or TG-FNduring around 30 min incubation time, ruling out the involvementof this cell binding site of FN on the RGD-independent celladhesion on TG-FN. The heparin binding sites within FN can bindto both cell surface syndecans andα4β1 integrin. However neitherthe anti-α4 integrin function blocking antibody nor the CS-1peptide have any effect on HOB cell adhesion on the TG-FN matrixunder the conditions of our assay, since cells plated on TG-FN inthe presence of either of these agents not only attached but alsospread in the presence of the RGD peptide, which ruled out theinvolvement of α4β1 integrin. We also demonstrate that theblocking of the heparin binding sites on FN by heparin [33], did notaffect the compensation of TG-FN in RGD-induced loss of celladhesion, suggesting that the heparin binding sites of FN are notrequired by TG-FN to function. In contrast, the heparin binding site(s) on TG2 [21], which when blocked with treatment by heparin ofthe FN immobilized TG2was found to be crucial for this complex toexert its compensatory effect, indicating that TG2 is the functionalcomponent in the heterocomplex mediating the cell adhesionprocess in the presence of the RGD peptides.
Our previous study demonstrated that in HOB cells thedegradation of cell surface heparan sulfates by heparinasetreatment led to the failure of TG-FN in rescuing the RGD-
Fig. 8 – Schematic showing role for the TG-FN heterocomplex in bonor growthosteoblast TG2 is upregulatedwhich leads to the secretionTG-FN complex. Another influence of osteoblasts inmatrix remodellmetalloproteinases (MMPs) leading to the release of the RGD contacompeting for cell surface integrin binding sites ultimately leadingsurface heparan sulphate proteoglycan syndecan-4, TG2 within thevia PKCαwhich in turn triggers the inside-out signal transduction ofthus maintaining cell adhesion and homeostasis.
independent cell adhesion [6]. Among the cell surface heparansulphate proteoglycans, syndecan-4 is the most widely expressedmember and is abundant in osteoblasts [34]. Reducing theexpression of syndecan-4 in HOB by siRNA silencing led to theloss of the compensatory effect of TG-FN in RGD impaired celladhesion. Moreover the co-immunoprecipitation assay using antisyndecan-4 antibody to pull down TG2 [19], strongly suggests adirect interaction betweenHOB cell surface syndecan-4 andmatrixTG2, given the high affinity of interaction between these twoproteins (Kd of 15.92+/−0.96) [35]. Hence our data indicates thatin HOB cells syndecan-4 is one of the major heparan sulphateproteoglycans that participate in the signal transduction processwhen cells are bound to the FN-TG complex in the presence of RGDcontaining peptides.
We next turned our attention to elucidating the downstreamsignalling molecules that mediate this novel cell adhesion process.The role of PKCα, as an important signallingmolecule of syndecan-4 and a crucial link between syndecan-4 and β1 integrin co-signaltransduction via its direct interaction with β1 integrin, was firstsuggested by using its inhibitor Go6976 in HOB cells [6]. Toconfirm and expand on our previous discovery, the GK21 peptide,which blocks the interaction between PKCα and β1 integrin [36],was used in the cell adhesion assay. The loss of the compensatoryeffect of TG-FN in the GK21-treated HOB cells in the presence ofthe RGD peptides, suggested not only the requirement of theintracellular activation of β1 integrins by PKCα for TG-FN to exertits function but also the potential involvement of the intracellularactivation of β1 integrins (also known as the β1 integrin inside-out
e remodelling. Duringmatrix deposition following bone healingand depositionof the enzyme into ECMand the formation of theing is the degradation of thematrix proteins by release ofmatrixining peptides, which can induce the loss of cell adhesion viato cell death (anoikis). Via its direct interaction with the cellTG-FN heterocomplex activates syndecan-4 mediated signallingα5β1 integrins and actin cytoskeleton formation by syndecan-2,

380 E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
signalling). This hypothesis was tested by using a β1 integrinfunction blocking antibody HMβ1-1 in the cell adhesion assay. Wenext demonstrated that the effect of HMβ1-1 on cell adhesion isdependent on its inhibition of the outside-in signalling withoutinfluencing the inside-out one. This was undertaken by detectingthe phosphorylation of FAK at Tyr861 and Tyr397 and by showingthat HMβ1-1 only reduced the p-FAK397 but not the p-FAK861. Asone of the major intracellular signaling molecules in the β1integrin and FAK-related cell survival pathway, the role of ERK1/2in the RGD-independent cell adhesion on TG-FN matrix wasinvestigated by studying the phosphorylation of ERK1/2. The effectof TG-FN complex on restoring the loss of the phosphorylation ofERK1/2 caused by the RGD peptides indicated that ERK1/2 isrequired by TG-FN to compensate the RGD-independent celladhesion, which could also be important in the rescuing the cellsfrom RGD-induced anoikis. The close association of α5 integrinwith β1 integrin in cell adhesion on FN is well demonstrated[37,38]. It has also been reported that this integrin complex is oneof the most important integrins in osteoblasts [39]. By using aspecific α5β1 integrin-targeting peptide A5-1, which blocks thefunction ofα5β1, we successfully blocked the compensatory effectof TG-FN on the peptide-treated HOB cells and showed a dose-dependent reduction on both the FN and TG-FN groups withoutany significant difference between the two groups. Anothermember of the syndecan family reported to be involved inregulating actin cytoskeleton formation is the syndecan-2 whichcan act as an associate receptor for both syndecan-4 and β1integrin [9,24].
Considering the abundant presence of syndecan-2 in osteo-blasts [23], we tested the potential involvement of syndecan-2 inthe RGD-independent osteoblast cell adhesion on the TG-FNmatrix. Inhibition of syndecan-2 expression by specific siRNAsilencing led to the loss of compensation of cell adhesion in thepresence of RGD peptides as well as the loss of actin skeletonformation. Unlike syndecan-4, no direct interaction betweensyndecan-2 and FN bound TG2 could be detected via co-immunoprecipitation. Hence we demonstrate that syndecan-2does not interact with the TG-FN matrix directly but acts as adownstream effector in the RGD-independent cell adhesionmediated by TG-FN. The failure of TG-FN in restoring the loss ofcell adhesion mediated by RGD in the presence of the ROCKinhibitor Y27632 in HOB cells, suggests that ROCK, whose activityis associatedwith syndecan-2 driven cell adhesion [24], is requiredby TG-FN in its signalling transduction mechanism by most likelyacting as a downstream signalling molecule of syndecan-2.
The role of osteoblasts inmatrix turnover in bone has been verywell reported [40]. During this process, TG2 is secreted into theECM, where it has been implicated in matrix deposition and bonemineralization [10]. An important finding in this report is that TG2regulates RGD-independent cell adhesion via cell surface heparansulphate proteoglycans. However these receptors also have awider role in regulating osteoblast-related matrix remodelling[41] and in a rat limb model of bone healing syndecan-2, -4 and -3have a been shown to be upregulated [34]. It has been previouslydemonstrated [31,41] that during the wound healing process,matrix remodelling leads to the release of matrix RGD peptideswhich can induce the loss of cell adhesion and trigger cell death viaanoikis. The reported [10,42,43] increased expression and depo-sition of TG2 into the ECM during bone remodelling would lead tothe formation of the TG-FN heterocomplex, which could both
enhance cell adhesion and maintain osteoblast survival duringmatrix turnover, thus facilitating tissue homeostasis during thisperiod (see Fig. 8). Our work therefore demonstrates anothernovel important function of the cell surface heparan sulfateproteoglycans, syndecan-4 and syndecan-2 in bone growth andhealing via their interaction with TG2 (Fig. 8).
Conclusion
The above work demonstrates a molecular mechanism for theRGD-independent osteoblast adhesion process mediated by theTG-FN heterocomplex, within which TG2 is the functionalcomponent. Through its direct interaction with cell surfacesyndecan-4, TG2 activates the co-receptors of the syndecan-4pathway—α5β1 integrin and syndecan-2. Via activation of PKCα, adownstream signaling molecule of syndecan-4, TG-FN triggers theinside-out signaling of α5β1 integrin leading to the phosphory-lation of FAK and ERK1/2, while activation of syndecan-2 and ROCKsignaling leads to formation of the actin cytoskeleton. Theinvolvement of α4β1 integrin and the role of TG2 as a cell surfaceintegrin coreceptor within this process was ruled out.
Acknowledgments
This work was in part supported by Marie Curie TRACKS RTN withgrant number MRTN-CT-2006-036032.
Appendix A. Supplementary data
Supplementary data to this article can be found online atdoi:10.1016/j.yexcr.2010.10.015.
R E F E R E N C E S
[1] J. Takagi, Structural basis for ligand recognition by RGD(Arg-Gly-Asp)-dependent integrins, Biochem. Soc. Trans. 32(2004) 403–406.
[2] N.C. Avery, A.J. Bailey, The effects of the Maillard reaction on thephysical properties and cell interactions of collagen, Pathol. Biol.54 (2006) 387–395.
[3] U. Hersel, C. Dahmen, H. Kessler, RGD modified polymers:biomaterials for stimulated cell adhesion and beyond, Biomaterials24 (2003) 4385–4415.
[4] J.M. Lemieux, M.C. Horowitz, M.A. Kacena, Involvement ofintegrins alpha(3)beta(1) and alpha(5)beta(1) and glycoproteinIIb in megakaryocyte-induced osteoblast proliferation, J. Cell.Biochem. 109 (2010) 927–932.
[5] S.S. Smyth, E.D. Reis, H. Vaananen, W. Zhang, B.S. Coller, Variableprotection of beta 3-integrin-deficient mice from thrombosisinitiated by different mechanisms, Blood 98 (2001) 1055–1062.
[6] E.A. Verderio, D. Telci, A. Okoye, G. Melino, M. Griffin, A novelRGD-independent cel adhesion pathway mediated byfibronectin-bound tissue transglutaminase rescues cells fromanoikis, J. Biol. Chem. 278 (2003) 42604–42614.
[7] H. Tran, R. Pankov, S.D. Tran, B. Hampton, W.H. Burgess, K.M.Yamada, Integrin clustering induces kinectin accumulation, J. CellSci. 115 (2002) 2031–2040.
[8] A. Woods, J.R. Couchman, Signaling from the matrix to the

381E X P E R I M E N T A L C E L L R E S E A R C H 3 1 7 ( 2 0 1 1 ) 3 6 7 – 3 8 1
cytoskeleton: role of cell surface proteoglycans in matrixassembly, Kidney Int. Suppl. 54 (1996) S64–S67.
[9] Y. Kusano, K. Oguri, Y. Nagayasu, S. Munesue, M. Ishihara, I. Saiki, H.Yonekura, H. Yamamoto, M. Okayama, Participation of syndecan 2in the induction of stress fiber formation in cooperation withintegrin alpha5beta1: structural characteristics of heparan sulfatechains with avidity to COOH-terminal heparin-binding domain offibronectin, Exp. Cell Res. 256 (2000) 434–444.
[10] M.T. Kaartinen, S. El-Maadawy, N.H. Rasanen, M.D. McKee, Tissuetransglutaminase and its substrates in bone, J. BoneMiner. Res. 17(2002) 2161–2173.
[11] Z. Balklava, E. Verderio, R. Collighan, S. Gross, J. Adams, M. Griffin,Analysis of tissue transglutaminase function in the migration ofSwiss 3T3 fibroblasts: the active-state conformation of theenzyme does not affect cell motility but is important for itssecretion, J. Biol. Chem. 277 (2002) 16567–16575.
[12] S.S. Akimov, A.M. Belkin, Cell-surface transglutaminase promotesfibronectin assembly via interaction with the gelatin-bindingdomain of fibronectin: a role in TGF beta-dependent matrixdeposition, J. Cell Sci. 114 (2001) 2989–3000.
[13] D.J. Heath, P. Christian, M. Griffin, Involvement of tissue transglutaminase in the stabilisation of biomaterial/tissue interfacesimportant in medical devices, Biomaterials 2 (23) (2002)1519–1526.
[14] D.J. Heath, S. Downes, E. Verderio, M. Griffin, Characterization oftissue transglutaminase in human osteoblast-like cells, J. BoneMiner. Res. 16 (2001) 1477–1485.
[15] C.H. Damsky, Extracellular matrix−integrin interactions inosteoblast function and tissue remodelinG, Bone 25 (1999)95–96.
[16] E.Y. Kim, J.Y. Bang, S.I. Chang, I.C. Kang, A novel integrinalpha5beta1 antagonistic peptide, A5-1, screened by Protein Chipsystem as a potent angiogenesis inhibitor, Biochem. Biophys. Res.Commun. 377 (2008) 1288–1293.
[17] A. Leblanc, N. Day, A. Menard, J.W. Keillor, Guinea pig livertransglutaminase: a modified purification procedure affordingenzyme with superior activity in greater yield, Protein Expr. Purif.17 (1999) 89–95.
[18] E. Verderio, A. Coombes, R.A. Jones, X. Li, D. Heath, S. Downes, M.Griffin, Role of the cross-linking enzyme tissue transglutaminasein the biological recognition of synthetic biodegradable polymers,J. Biomed. Mater. Res. 54 (2001) 294–304.
[19] D. Telci, Z. Wang, X. Li, E.A. Verderio, M.J. Humphries, M. Baccarini,H. Basaga, M. Griffin, Fibronectin-tissue transglutaminase matrixrescues RGD-impaired cell adhesion through syndecan-4 andbeta1 integrin co-signaling, J. Biol. Chem. 283 (2008)20937–20947.
[20] S.S. Akimov, D. Krylov, L.F. Fleischman, A.M. Belkin, Tissuetransglutaminase is an integrin-binding adhesion coreceptor forfibronectin, J. Cell Biol. 148 (2000) 825–838.
[21] S. Gambetti, A. Dondi, C. Cervellati, M. Squerzanti, F.S. Pansini, C.M.Bergamini, Interaction with heparin protects tissuetransglutaminase against inactivation by heating and byproteolysis, Biochimie 87 (2005) 551–555.
[22] M. Parsons, M.D. Keppler, A. Kline, A. Messent, M.J. Humphries, R.Gilchrist, I.R. Hart, C. Quittau-Prevostel, W.E. Hughes, P.J. Parker,T. Ng, Site-directed perturbation of protein kinase C−integrininteraction blocks carcinoma cell chemotaxis, Mol. Cell. Biol. 22(2002) 5897–5911.
[23] D.Modrowski, M. Basle, A. Lomri, P.J. Marie, Syndecan-2 is involvedin the mitogenic activity and signaling of granulocyte-macrophagecolony-stimulating factor in osteoblasts, J. Biol. Chem. 275 (2000)9178–9185.
[24] J.R. Whiteford, V. Behrends, H. Kirby, M. Kusche-Gullberg, T.Muramatsu, J.R. Couchman, Syndecans promote integrin-mediatedadhesion of mesenchymal cells in two distinct pathways, Exp. CellRes. 313 (2007) 3902–3913.
[25] K. Noto, K. Kato, K. Okumura, H. Yagita, Identification and
functional characterization of mouse CD29 with a mAb, Int.Immunol. 7 (1995) 835–842.
[26] Q. Shi, D. Boettiger, A novelmode for integrin-mediated signaling:tethering is required for phosphorylation of FAK Y397, Mol. Biol.Cell 14 (2003) 4306–4315.
[27] J.L. Sechler, A.M. Cumiskey, D.M. Gazzola, J.E. Schwarzbauer, Anovel RGD-independent fibronectin assembly pathway initiatedby alpha4beta1 integrin binding to the alternatively spliced Vregion, J. Cell Sci. 113 (2000) 1491–1498.
[28] S. Nakayamada, Y. Okada, K. Saito, M. Tamura, Y. Tanaka, Beta1integrin/focal adhesion kinase-mediated signaling inducesintercellular adhesion molecule 1 and receptor activator ofnuclear factor kappaB ligand on osteoblasts and osteoclastmaturation, J. Biol. Chem. 278 (2003) 45368–45374.
[29] T. Isobe, H. Takahashi, S. Ueki, J. Takagi, Y. Saito,Activity-independent cell adhesion to tissue-typetransglutaminase is mediated by alpha 4 beta 1 integrin, Eur.J. Cell Biol. 78 (1999) 876–883.
[30] H. Takahashi, T. Isobe, S. Horibe, J. Takagi, Y. Yokosaki, D.Sheppard, Y. Saito, Tissue transglutaminase, coagulation factorXIII, and the pro- polypeptide of von Willebrand factor are allligands for the integrins alpha(9)beta(1) and alpha(4)beta(1),J. Biol. Chem. 275 (2000) 23589–23595.
[31] L. Liaw, H.C. Crawford, Functions of the extracellular matrix andmatrix degrading proteases during tumor progression, Braz. J.Med. Biol. Res. 32 (1999) 805–812.
[32] S.E. Ochsenhirt, E. Kokkoli, J.B. McCarthy, M. Tirrell, Effect of RGDsecondary structure and the synergy site PHSRN on cell adhesion,spreading and specific integrin engagement, Biomaterials 27(2006) 3863–3874.
[33] Z. Mostafavi-Pour, J.A. Askari, J.D. Whittard, M.J. Humphries,Identification of a novel heparin-binding site in the alternativelyspliced IIICS region of fibronectin: roles of integrins andproteoglycans in cell adhesion to fibronectin splice variants,Matrix Biol. 20 (2001) 63–73.
[34] S.J. Song, S.M. Cool, V. Nurcombe, Regulated expression ofsyndecan-4 in rat calvaria osteoblasts induced by fibroblastgrowth factor-2, J. Cell. Biochem. 100 (2007) 402–411.
[35] A. Scarpellini, R. Germack, H. Lortat-Jacob, T. Muramtsu, T.S.Johnson, E. Billett, E.A. Verderio, Heparan sulphate proteoglycansare receptors for the cell-surface trafficking and biological activityof transglutaminase-2, J. Biol. Chem. 284 (2009) 18411–18423.
[36] A.C. Parsons, G. Yosipovitch, D.J. Sheehan, O.P. Sangueza, C.S.Greenberg, D.C. Sane, Transglutaminases: the missing link innephrogenic systemic fibrosis, Am. J. Dermatopath. 29 (2007)433–436.
[37] S. Johansson, G. Svineng, K. Wennerberg, A. Armulik, L. Lohikangas,Fibronectin−integrin interactions, Front. Biosci. 2 (1997)d126–d146.
[38] E. Cukierman, R. Pankov, D.R. Stevens, K.M. Yamada, Takingcell−matrix adhesions to the third dimension, Science 294(2001) 1708–1712.
[39] G.B. Schneider, R. Zaharias, C. Stanford, Osteoblast integrinadhesion and signaling regulate mineralization, J. Dent. Res. 80(2001) 1540–1544.
[40] E.S. Roberts, M.A. Zandonatti, D.D. Watry, L.J. Madden, S.J.Henriksen, M.A. Taffe, H.S. Fox, Induction of pathogenic sets ofgenes in macrophages and neurons in neuro AIDS, Am. J. Pathol.162 (2003) 2041–2057.
[41] J. Vanwauwe, G. Vannyen, M.C. Coene, P. Stoppie, W. Cools, J.Goossens, P. Borghgraef, P.A.J. Janssen, Liarozole, an inhibitor ofretinoic acid metabolism, exerts retinoid-mimetic effects invivo,J. Pharmacol. Exp. Ther. 261 (1992) 773–779.
[42] E.A. Verderio, T. Johnson, M. Griffin, Tissue transglutaminase innormal and abnormal wound healing: review article, AminoAcids 26 (2004) 387–404.
[43] D. Telci, M. Griffin, Tissue transglutaminase (TG2)—a woundresponse enzyme, Front. Biosci. 11 (2006) 867–882.