multiples sequenz alignment
DESCRIPTION
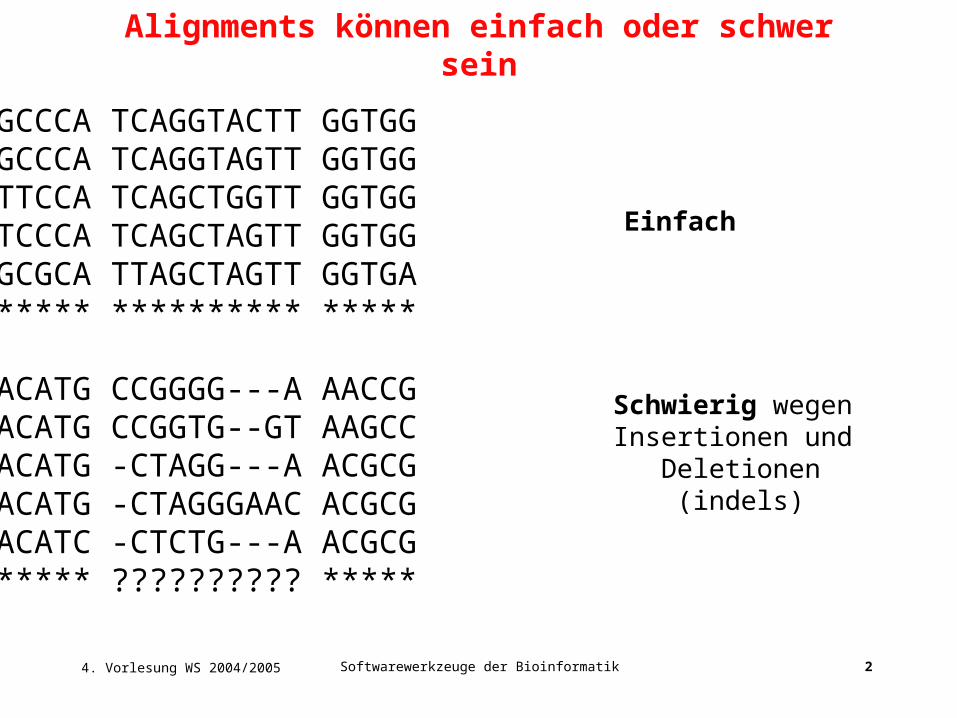
Literatur: Kapitel 4 in Buch von David Mount Thioredoxin-Beispiel heute aus Buch von Arthur Lesk. Multiples Sequenz Alignment. Alignments können einfach oder schwer sein. GCGGCCCA TCAGGTACTT GGTGG GCGGCCCA TCAGGTAGTT GGTGG GCGTTCCA TCAGCTGGTT GGTGG GCGTCCCA TCAGCTAGTT GGTGG - PowerPoint PPT PresentationTRANSCRIPT

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 1
Multiples Sequenz Alignment
Literatur: Kapitel 4 in Buch von David Mount
Thioredoxin-Beispiel heute aus Buch von Arthur Lesk
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 2
Einfach
Schwierig wegen Insertionen und
Deletionen(indels)
Alignments können einfach oder schwer sein
GCGGCCCA TCAGGTACTT GGTGGGCGGCCCA TCAGGTAGTT GGTGGGCGTTCCA TCAGCTGGTT GGTGGGCGTCCCA TCAGCTAGTT GGTGGGCGGCGCA TTAGCTAGTT GGTGA******** ********** *****
TTGACATG CCGGGG---A AACCGTTGACATG CCGGTG--GT AAGCCTTGACATG -CTAGG---A ACGCGTTGACATG -CTAGGGAAC ACGCGTTGACATC -CTCTG---A ACGCG******** ?????????? *****
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 3
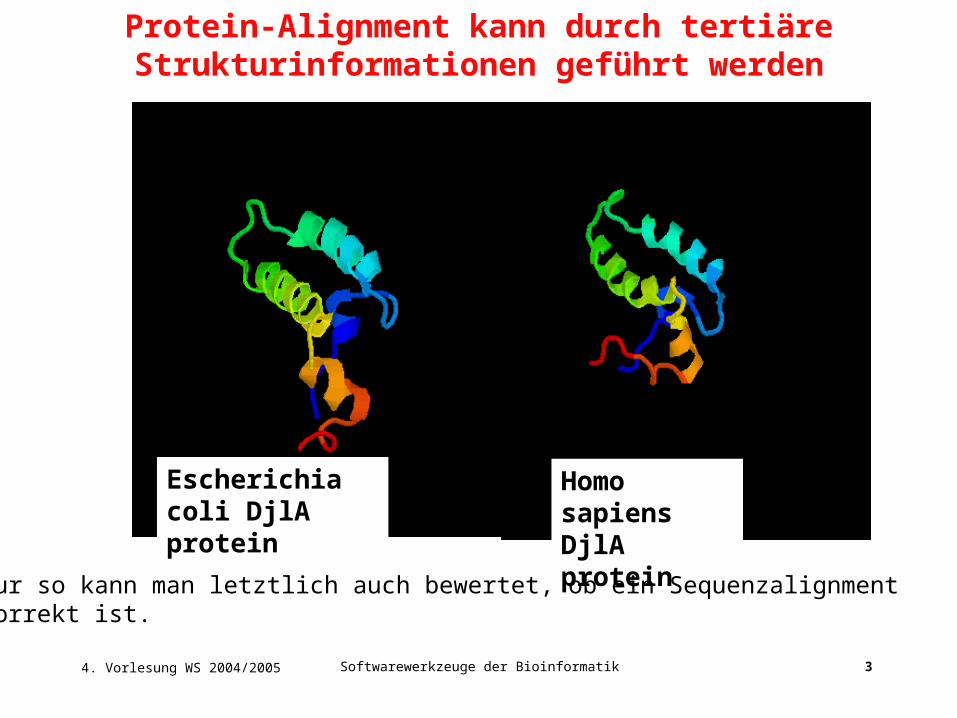
Homo sapiens DjlA protein
Escherichia coli DjlA protein
Protein-Alignment kann durch tertiäre Strukturinformationen geführt werden
nur so kann man letztlich auch bewertet, ob ein Sequenzalignmentkorrekt ist.

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 4
• Homologie: Ähnlichkeit, die durch
Abstammung von einem gemeinsamen
Ursprungsgen herrührt –
die Identifizierung und Analyse von
Homologien ist eine zentrale Aufgabe
der Phylogenie.
• Ein Alignment ist eine Hypothese
für die positionelle Homologie
zwischen Basenpaaren bzw.
Aminosäuren.
Definition von “Homologie”
http://www.cellsignal.com

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 5
MSA für Thioredoxin-FamilieFarbe Aminosäuretyp Aminosäurengelb klein, unpolar Gly, Ala, Ser, Thrgrün hydrophob Cys, Val, Ile, Leu
Pro, Phe, Tyr, Met, Trpviolett polar Asn, Gln, Hisrot negativ geladen Asp, Glublau positiv geladen Lys, Arg

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 6
Infos aus MSA von Thioredoxin-Familie
Thioredoxin: aus 5 beta-Strängen bestehendes beta-Faltblatt, das auf beiden Seiten von alpha-Helices flankiert ist.
gemeinsamer Mechanismus: Reduktion von Disulfidbrücken in Proteinen

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 7
Infos aus MSA von Thioredoxin-Familie
1) am stärksten konservierte Abschnitte entsprechen wahrscheinlich dem
aktiven Zentrum.
Disulfidbrücke zwischen Cys32 und Cys35 gehört zu konserviertem WCGPC[K
oder R] Motiv
Andere konservierte Sequenzabschnitte, z.B. Pro76Thr77 und Gly92Gly93 sind
an der Substratbindung beteiligt.

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 8
Infos aus MSA von Thioredoxin-Familie
2) Abschnitte mit vielen Insertionen und Deletionen entsprechen vermutlich
Schleifen an der Oberfläche. Eine Position mit einem konservierten Gly oder
Pro läßt auf eine Wendung der Kette (‚turn‘) schließen.
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 9
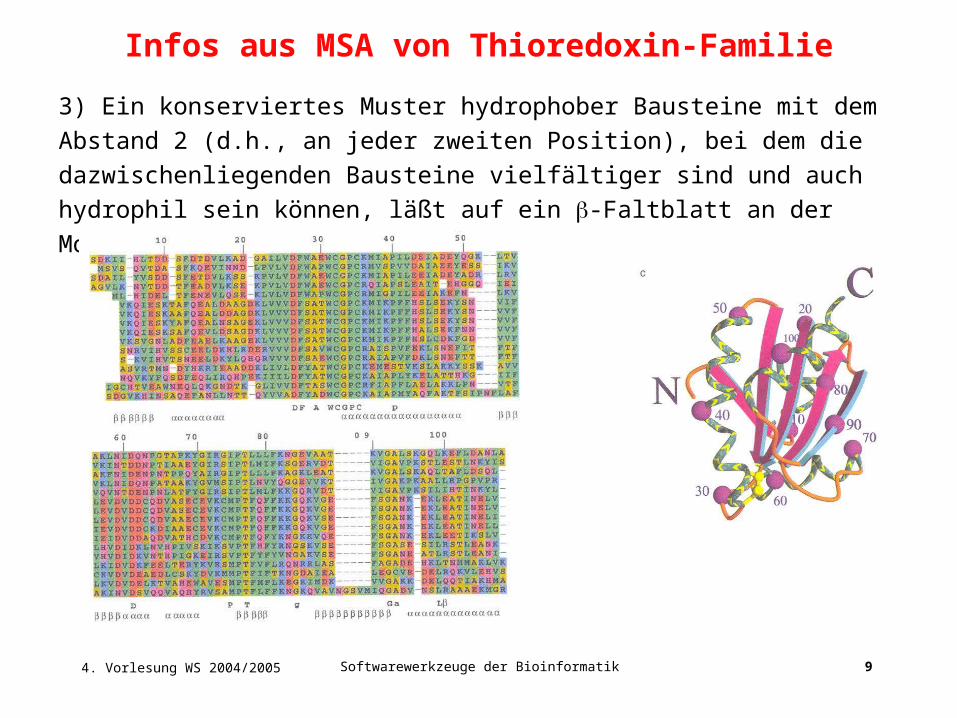
Infos aus MSA von Thioredoxin-Familie
3) Ein konserviertes Muster hydrophober Bausteine mit dem Abstand 2 (d.h.,
an jeder zweiten Position), bei dem die dazwischenliegenden Bausteine
vielfältiger sind und auch hydrophil sein können, läßt auf ein -Faltblatt an der
Moleküloberfläche schließen.
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 10
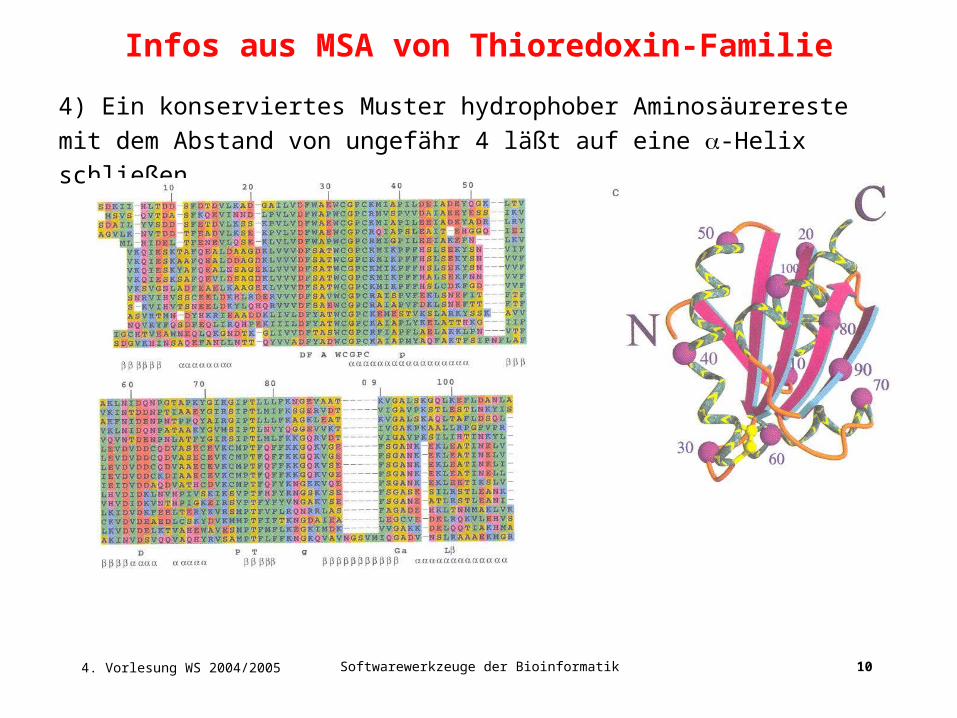
Infos aus MSA von Thioredoxin-Familie
4) Ein konserviertes Muster hydrophober Aminosäurereste mit dem Abstand
von ungefähr 4 läßt auf eine -Helix schließen.

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 11
Infos aus MSA von Thioredoxin-Familie
Die Thioredoxine sind Teil einer Superfamilie, zu der auch viele weiter entfernte
homologe Protein gehören,
z.B. Glutaredoxin (Wasserstoffdonor für die Reduktion von Ribonukleotiden bei
der DNA-Synthese)
Protein-Disulfidisomerase (katalysiert bei der Proteinfaltung den Austausch
falsch gefalteter Disulfidbrücken)
Phosducin (Regulator in G-Protein-abhängigen Signalübertragungswegen)
Glutathion-S-Transferasen (Proteine der chemischen Abwehr).
Die Tabelle des MSAs für Thioredoxinsequenzen enthält implizit auch Muster,
die man zur Identifizierung dieser entfernteren Verwandten nutzen kann.

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 12
Es gibt im wesentlichen 3 unterschiedliche Vorgehensweisen:
(1) Manuell
(2) Automatisch
(3) Kombiniert
Multiples Sequenz-Alignment - Methoden

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 13
manuelles Alignment bietet sich an falls
– Alignment einfach ist.
– es zusätzliche (strukturelle) Information gibt.
– automatische Alignment –Methoden in lokalen Minima feststecken.
– ein automatisch erzeugtes Alignment manuell “verbessert” werden kann.
Gründe für manuelles Alignment

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 14
2 Methoden:
• Dynamische Programmierung
– betrache 2 Proteinsequenzen von 100 Aminosäuren Länge.
- wenn es 1002 Sekunden dauert, diese beiden Sequenzen erschöpfend
zu alignieren, dann wird es
1003 Sekunden dauern um 3 Sequenzen zu alignieren,
1004 Sekunden für 4 sequences und
1.90258x1034 Jahre für 20 Sequenzen.
• Progressives Alignment
multiples Sequenzalignment

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 15
berechne zunächst paarweise Alignments
für 3 Sequenzen wird Würfel aufgespannt:
D.h. dynamische Programmierung hat nun Komplexität n1 * n2 * n3mit den Sequenzlängen n1, n2, n3.
Sehr aufwändig! Versuche, Suchraum einzuschränken.
dynamische Programmierung mit MSA Programm

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 16
dynamische Programmierung mit MSA Programm
Links: Baum für 5 Sequenzen ohne Paarung von Sequenzen.
Neighbour-joining Methode: berechne Summe aller Kantenlängen
S = a + b + c + d + e (Kantenlängen sind bekannt)
In diesem Fall seien sich A und B am nächsten. Konstruiere daher den Baum rechts.
Generell: Verbinde die Sequenzpaare mit den kürzesten Abständen …
Man erhält den Baum mit der kleinsten Summe der Kantenlängen.
Konstruiere anhand phylogenetischem Baum ein versuchsweises Multiples Sequenz Alignment.

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 17
Dieses Alignment dient dazu, den möglichen Raum inmitten des Würfels
einzugrenzen, in dem das beste MSA zu finden sein sollte.
Grosse Rechenersparnis!
dynamische Programmierung mit MSA Programm

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 18
• wurde von Feng & Doolittle 1987 vorgestellt
• ist eigentlich eine heuristische Methode. Daher ist nicht garantiert, das
“optimale” Alignment zu finden.
• benötigt (n-1) + (n-2) + (n-3) ... (n-n+1) paarweise Sequenzalignments
als Ausgangspunkt.
• weitverbreitete Implementation in Clustal
(Des Higgins)
ClustalW ist eine neuere Version, in der den Parameter für Sequenzen
und Programm Gewichte (weights) zugeteilt werden.
Progressives Alignment
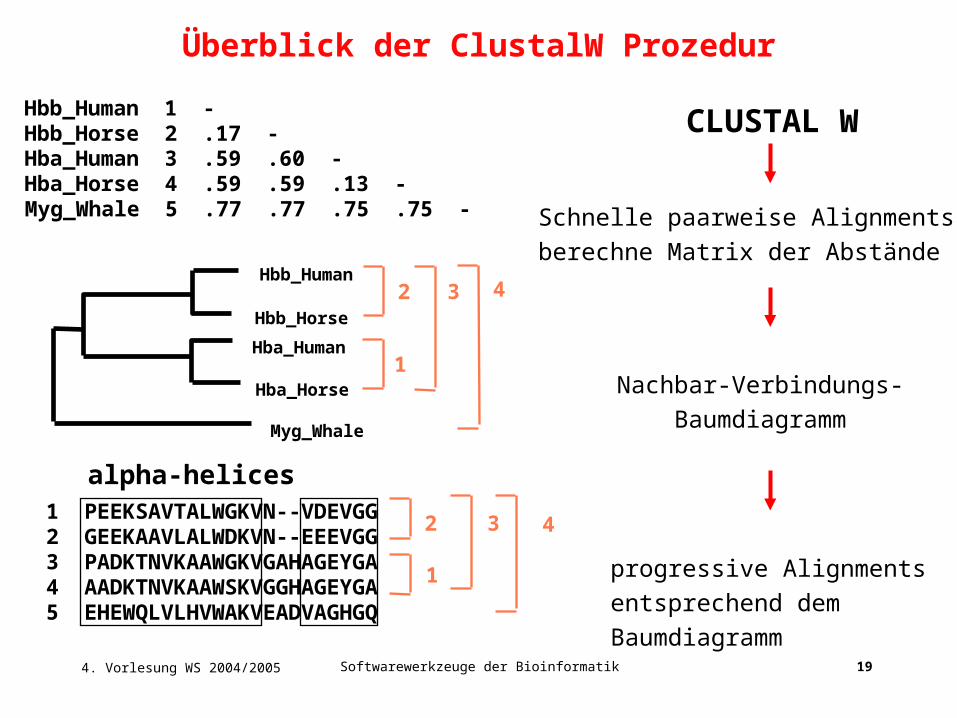
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 19
Schnelle paarweise Alignments:
berechne Matrix der Abstände
1 PEEKSAVTALWGKVN--VDEVGG2 GEEKAAVLALWDKVN--EEEVGG3 PADKTNVKAAWGKVGAHAGEYGA4 AADKTNVKAAWSKVGGHAGEYGA5 EHEWQLVLHVWAKVEADVAGHGQ
Hbb_Human 1 -Hbb_Horse 2 .17 -Hba_Human 3 .59 .60 -Hba_Horse 4 .59 .59 .13 -Myg_Whale 5 .77 .77 .75 .75 -
Hbb_Human
Hbb_Horse
Hba_Horse
Hba_Human
Myg_Whale
2
1
3 4
2
1
3 4
alpha-helices
Nachbar-Verbindungs-
Baumdiagramm
progressive Alignments
entsprechend dem
Baumdiagramm
CLUSTAL W
Überblick der ClustalW Prozedur

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 20
• Berechne alle möglichen paarweisen Alignments von Sequenzpaaren.
Es gibt (n-1)+(n-2)...(n-n+1) Möglichkeiten.
• Berechne aus diesen isolierten paarweisen Alignments den “Abstand”
zwischen jedem Sequenzpaar.
• Erstelle eine Abstandsmatrix.
ClustalW- Paarweise Alignments

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 21
Clustal W - Nachbarschaftsbaum
• aus den paarweisen Distanzen wird ein Nachbarschafts-Baum erstellt
• Dieser Baum gibt die Reihenfolge an, in der das progressive Alignment
ausgeführt werden wird.

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 22
• aligniere die beiden ähnlichsten Sequenzen zuerst.
• dieses Alignment ist dann “fest” und wird nicht mehr angetastet. Falls
später ein GAP eingeführt werden muss, wird er in beiden Sequenzen
an der gleichen Stelle eingeführt.
• Deren relatives Alignment bleibt unverändert.
Multiples Alignment - Erstes Paar
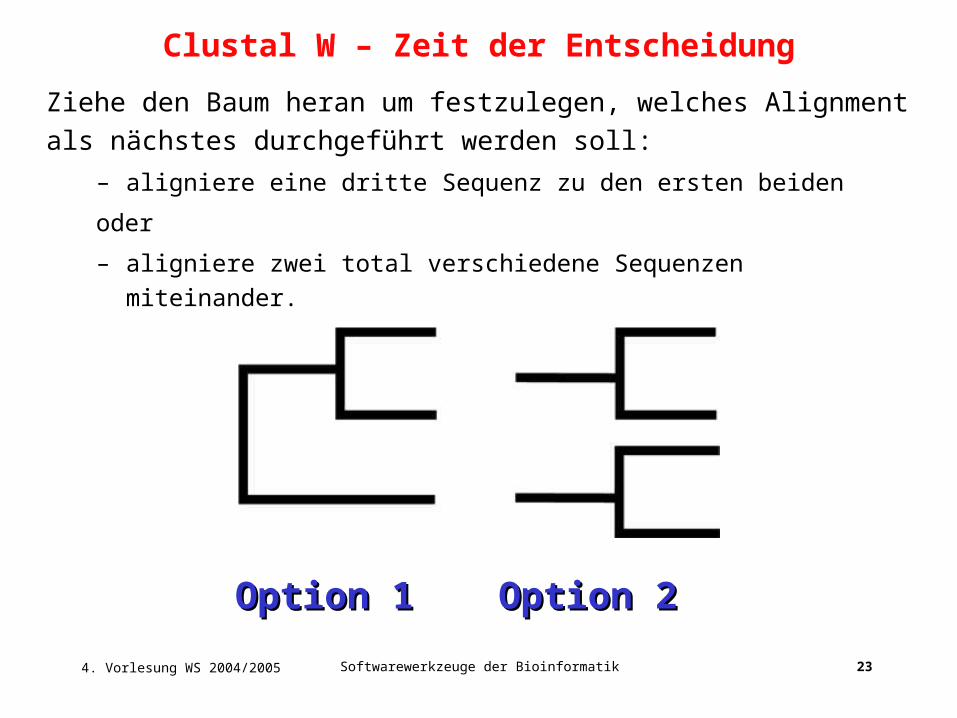
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 23
Ziehe den Baum heran um festzulegen, welches Alignment als nächstes
durchgeführt werden soll:
– aligniere eine dritte Sequenz zu den ersten beiden
oder
– aligniere zwei total verschiedene Sequenzen miteinander.
Option 1Option 1 Option 2Option 2
Clustal W – Zeit der Entscheidung

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 24
Wenn beim Alignment einer dritten Sequenz mit
den ersten beiden eine Lücke eingefügt werden
muss um das Alignment zu verbessern, werden
beide als Einzelsequenzen betrachtet.
+
ClustalW- Alternative 1

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 25
Falls, andererseits, zwei getrennte Sequenzen aligniert
werden müssen, werden diese zunächst miteinander
aligniert.
+
ClustalW- Alternative 2
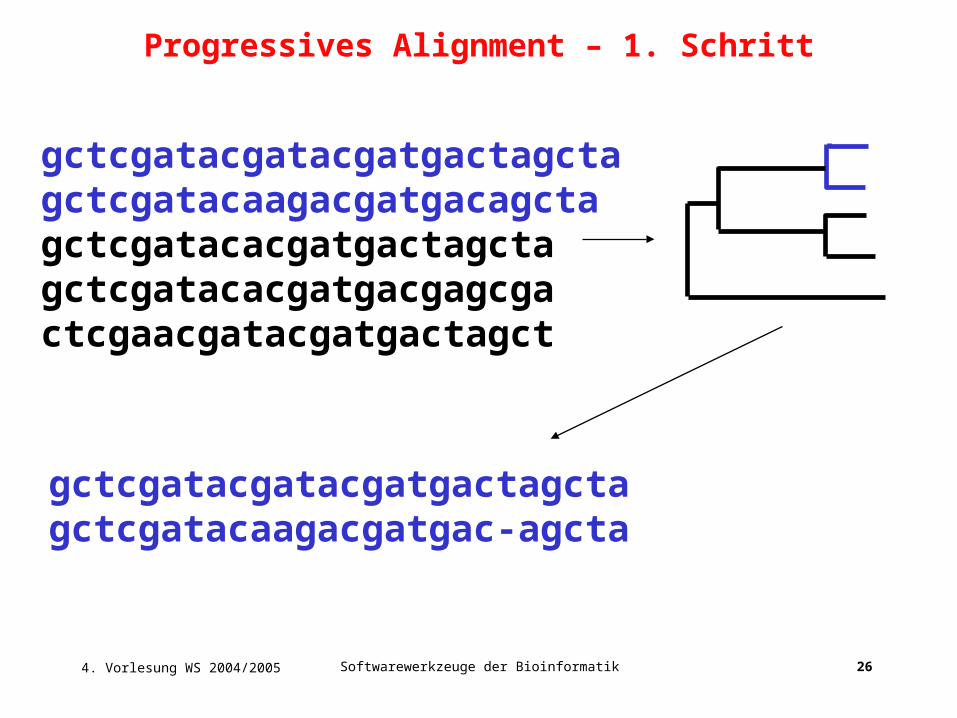
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 26
gctcgatacgatacgatgactagctagctcgatacaagacgatgacagctagctcgatacacgatgactagctagctcgatacacgatgacgagcgactcgaacgatacgatgactagct
gctcgatacgatacgatgactagctagctcgatacaagacgatgac-agcta
Progressives Alignment – 1. Schritt
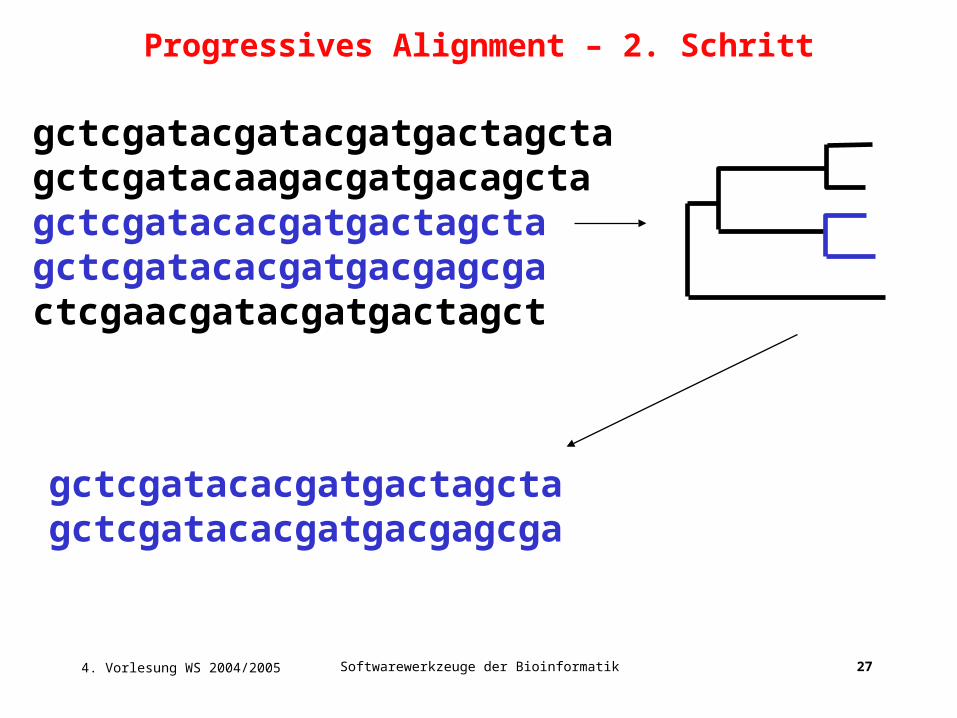
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 27
gctcgatacgatacgatgactagctagctcgatacaagacgatgacagctagctcgatacacgatgactagctagctcgatacacgatgacgagcgactcgaacgatacgatgactagct
gctcgatacacgatgactagctagctcgatacacgatgacgagcga
Progressives Alignment – 2. Schritt
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 28
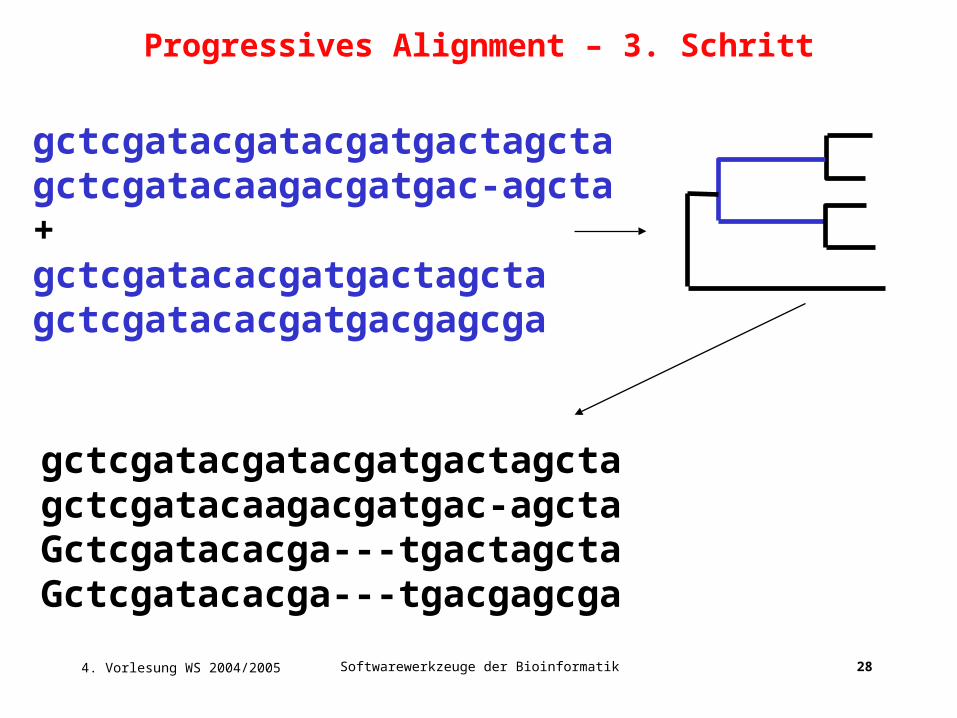
gctcgatacgatacgatgactagctagctcgatacaagacgatgac-agcta+gctcgatacacgatgactagctagctcgatacacgatgacgagcga
gctcgatacgatacgatgactagctagctcgatacaagacgatgac-agctaGctcgatacacga---tgactagctaGctcgatacacga---tgacgagcga
Progressives Alignment – 3. Schritt
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 29
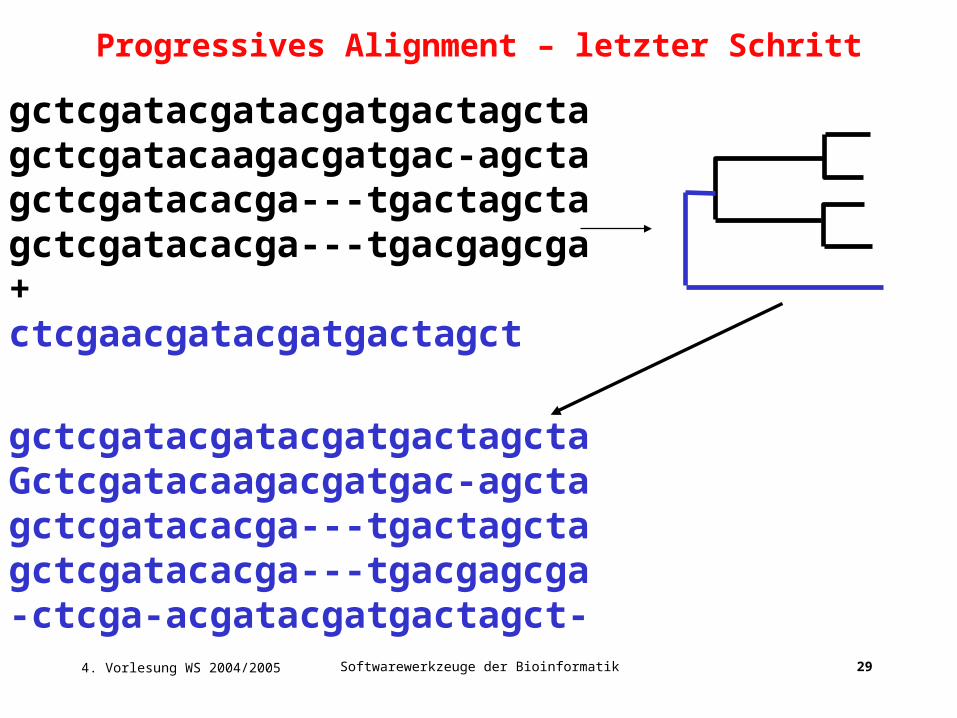
gctcgatacgatacgatgactagctagctcgatacaagacgatgac-agctagctcgatacacga---tgactagctagctcgatacacga---tgacgagcga+ctcgaacgatacgatgactagct
gctcgatacgatacgatgactagctaGctcgatacaagacgatgac-agctagctcgatacacga---tgactagctagctcgatacacga---tgacgagcga-ctcga-acgatacgatgactagct-
Progressives Alignment – letzter Schritt

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 30
Vorteil:
– Geschwindigkeit.
Nachteile:
– keine objektive Funktion.
– Keine Möglichkeit zu quantifizieren ob Alignment gut oder schlecht ist.
– Keine Möglichkeit festzustellen, ob das Alignment “korrekt” ist.
ClustalW- Vor- und Nachteile

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 31
Mögliche Probleme:
– in ein lokales Minimum zu geraten.
Falls zu einem frühen Zeitpunkt ein Fehler im Alignment eingebaut
wird, kann dieser später nicht mehr korrigiert werden.
– Zufälliges Alignment.
ClustalW - Lokales Minimum

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 32
• Sollen all Sequenzen gleich behandelt werden?
Obwohl manche Sequenzen eng verwandt und andere entfernt
verwandt sind?
• Sollen alle Positionen der Sequenzen gleich behandelt werden?
Obwohl sie unterschiedliche Funktionen und Positionen in der
dreidimensionalen Strukturen haben können?
Genauigkeit des Alignments verbessern

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 33
• Sequenzgewichtung
• Variable Substitutionsmatrizen
• Residuen-spezifische Gap-Penalties und verringerte Penalties in hydrophilen
Regionen (externe Regionen von Proteinsequenzen), bevorzugt Gaps in
Loops anstatt im Proteinkern.
• Positionen in frühen Alignments, an denen Gaps geöffnet wurden, erhalten
lokal reduzierte Gap Penalties um in späteren Alignments Gaps an den
gleichen Stellen zu bevorzugen
ClustalW- Besonderheiten

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 34
• Zwei Parameter sind festzulegen (es gibt Default-Werte, aber man sollte
sich bewusst sein, dass diese abgeändert werden können):
• Die GOP- Gap Opening Penalty ist aufzubringen um eine Lücke in
einem Alignment zu erzeugen
• Die GEP- Gap Extension Penalty ist aufzubringen um diese Lücke um
eine Position zu verlängern.
ClustalW- vom Benutzer festzulegende Parameter

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 35
• Bevor irgendein Sequenzpaar aligniert wird, wird eine Tabelle von GOPs
erstellt für jede Position der beiden Sequenzen.
• Die GOP werden positions-spezifisch behandelt und können über die
Sequenzlänge variieren.
• Falls ein GAP an einer Position existiert, werden die GOP und GEP
penalties herabgesetzt – und alle anderen Regeln treffen nicht zu.
• Daher wird die Bildung von Gaps an Positionen wahrscheinlicher, an
denen bereits Gaps existieren.
Positions-spezifische Gap penalties

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 36
• Solange kein GAP offen ist, wird GOP hochgesetzt falls die Position innerhalb von 8
Residuen von einem bestehenden Gap liegt.
• Dadurch werden Gaps vermieden, die zu eng beieinander liegen.
• An jeder Position innerhalb einer Reihe von hydrophilen Residuen wird GOP
herabgesetzt, da diese gewöhnlich in Loop-Regionen von Proteinstrukturen liegen.
• Eine Reihe von 5 hydrophilen Residuen gilt als
hydrophiler stretch.
• Die üblichen hydrophilen Residuen sind:
D Asp K Lys P Pro
E Glu N Asn R Arg
G Gly Q Gln S Ser
Dies kann durch den Benutzer geändert werden.
Vermeide zu viele Gaps
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 37
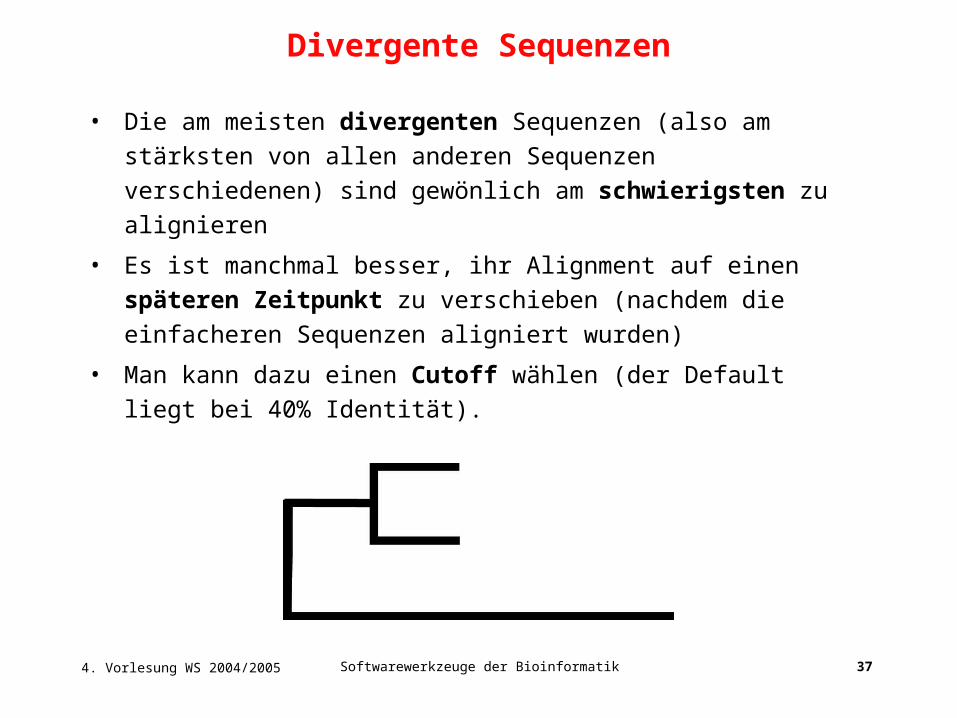
• Die am meisten divergenten Sequenzen (also am stärksten von allen
anderen Sequenzen verschiedenen) sind gewönlich am
schwierigsten zu alignieren
• Es ist manchmal besser, ihr Alignment auf einen späteren Zeitpunkt
zu verschieben (nachdem die einfacheren Sequenzen aligniert
wurden)
• Man kann dazu einen Cutoff wählen (der Default liegt bei 40%
Identität).
Divergente Sequenzen

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 38
• Progressives Alignment ist ein mathematischer Vorgang, der völlig unabhängig von
der biologischen Realität abläuft.
• Es kann eine sehr gute Abschätzung sein.
• Es kann eine unglaublich schlechte Abschätzung sein.
• Erfordert Input und Erfahrung des Benutzers.
• Sollte mit Vorsicht verwendet werden.
• Kann (gewöhnlich) manuell verbessert werden.
• Es hilft oft, farbliche Darstellungen zu wählen.
• Je nach Einsatzgebiet sollte der Benutzer in der Lage sein, die zuverlässigen
Regionen des Alignments zu beurteilen.
• Für phylogenetische Rekonstruktionen sollte man nur die Positionen verwenden, für
die eine zweifelsfreie Hypothese über positionelle Homologie vorliegt.
Tips für progressives Alignment

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 39
• Es macht wenig Sinn, proteinkodierende DNS-Abschnitte
zu alignieren!
ATGCTGTTAGGGATGCTCGTAGGG
ATGCT-GTTAGGGATGCTCGT-AGGG
Das Ergebnis kann sehr unplausibel sein und entspricht eventuell nicht dem
biologischen Prozess.
Es ist viel sinnvoller, die Sequenzen in die entsprechenden Proteinsequenzen
zu übersetzen, diese zu alignieren und dann in den DNS-Sequenzen an den
Stellen Gaps einzufügen, an denen sie im Aminosäure-Alignment zu finden
sind.
Alignment von Protein-kodierenden DNS-Sequenzen

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 40
GDE- The Genetic Data Environment (UNIX)
CINEMA- Java applet available from:
– http://www.biochem.ucl.ac.uk
Seqapp/Seqpup- Mac/PC/UNIX available from:
– http://iubio.bio.indiana.edu
SeAl for Macintosh, available from:
– http://evolve.zoo.ox.ac.uk/Se-Al/Se-Al.html
BioEdit for PC, available from:
– http://www.mbio.ncsu.edu/RNaseP/info/programs/BIOEDIT/bioedit.html
Software für manuelle Alignments
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 41
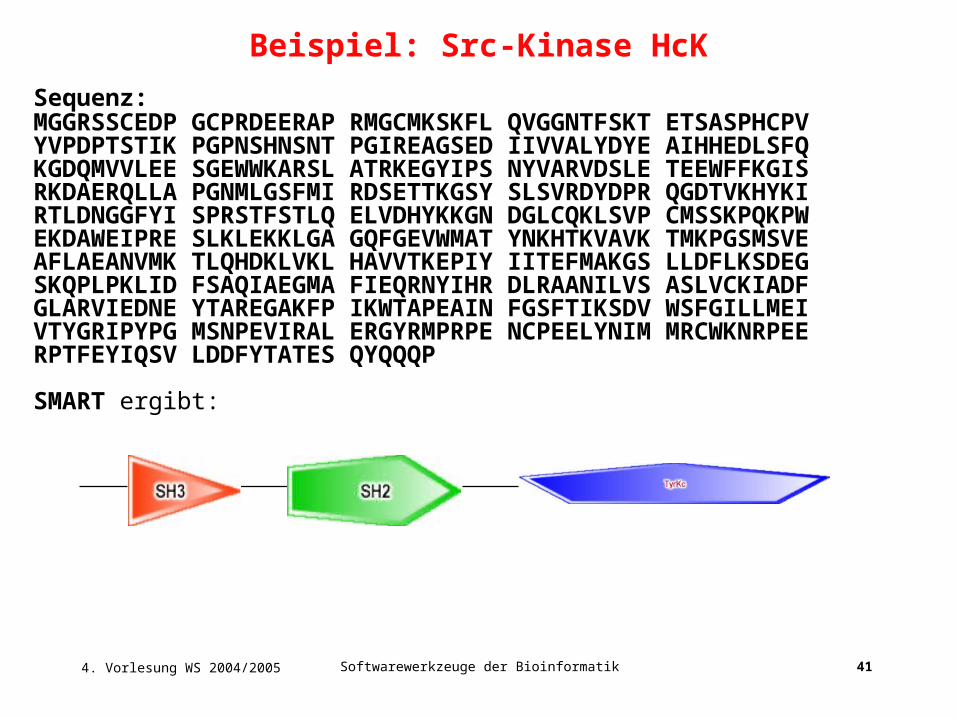
Sequenz: MGGRSSCEDP GCPRDEERAP RMGCMKSKFL QVGGNTFSKT ETSASPHCPVYVPDPTSTIK PGPNSHNSNT PGIREAGSED IIVVALYDYE AIHHEDLSFQKGDQMVVLEE SGEWWKARSL ATRKEGYIPS NYVARVDSLE TEEWFFKGISRKDAERQLLA PGNMLGSFMI RDSETTKGSY SLSVRDYDPR QGDTVKHYKIRTLDNGGFYI SPRSTFSTLQ ELVDHYKKGN DGLCQKLSVP CMSSKPQKPWEKDAWEIPRE SLKLEKKLGA GQFGEVWMAT YNKHTKVAVK TMKPGSMSVEAFLAEANVMK TLQHDKLVKL HAVVTKEPIY IITEFMAKGS LLDFLKSDEGSKQPLPKLID FSAQIAEGMA FIEQRNYIHR DLRAANILVS ASLVCKIADFGLARVIEDNE YTAREGAKFP IKWTAPEAIN FGSFTIKSDV WSFGILLMEIVTYGRIPYPG MSNPEVIRAL ERGYRMPRPE NCPEELYNIM MRCWKNRPEERPTFEYIQSV LDDFYTATES QYQQQP
SMART ergibt:
Beispiel: Src-Kinase HcK
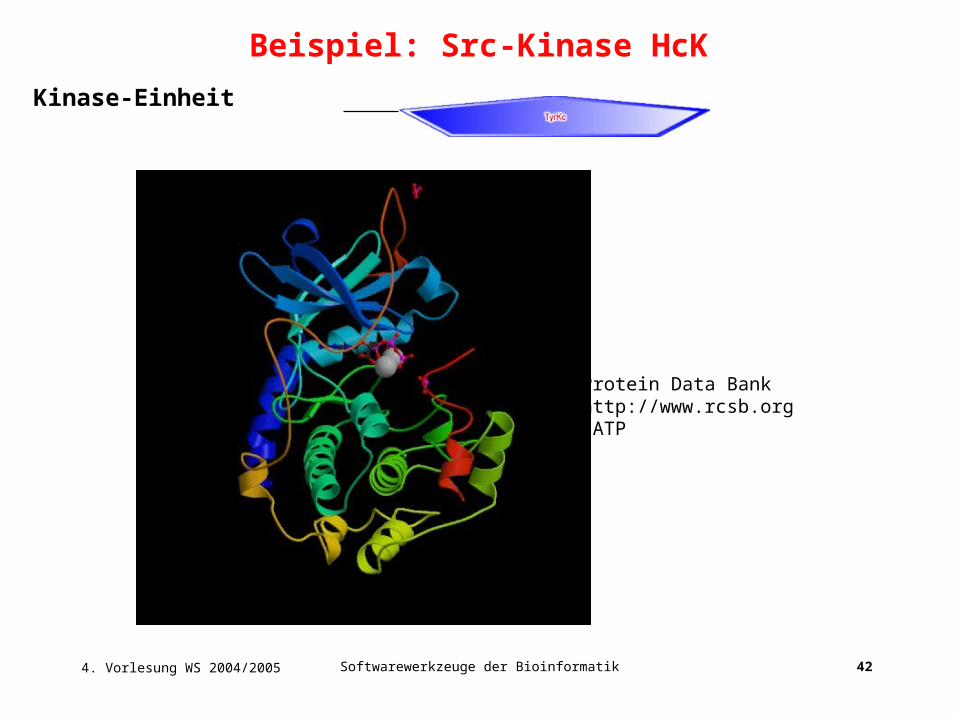
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 42
Kinase-Einheit
Beispiel: Src-Kinase HcK
Protein Data Bankhttp://www.rcsb.org1ATP

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 43
SH3 Domäne
Src homology 3 (SH3) Domänen binden an Zielproteine mit Sequenzen, die Proline
und hydrophobe Aminosäuren enthalten. Pro-enthaltende Polypeptide können an
SH3 in zwei verschiedenen Orientierungen binden. SH3 Domänen sind kleine
Proteinmodule von ungefähr 50 Residuen Länge. Man findet sie in vielen
intrazellulären oder Membran-assoziierten Proteinen …
Beispiel: Src-Kinase HcK
CATH: 1abo

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 44
SH2 Domäne
Die Src homology 2 (SH2) Domäne ist eine Proteindomäne mit etwa 100
Aminosäuren. SH2 Domänen funktionieren als Regelmodule von intrazellulären
Signalkaskaden indem sie mit grosser Affinität an Phospho-Tyrosin enthaltende
Peptide binden. SH2 Domänen findet man oft zusammen mit SH3 Domänen …
Ihre Struktur ist alpha+beta …
Beispiel: Src-Kinase HcK
CATH: 1g83 1fbz 1aot
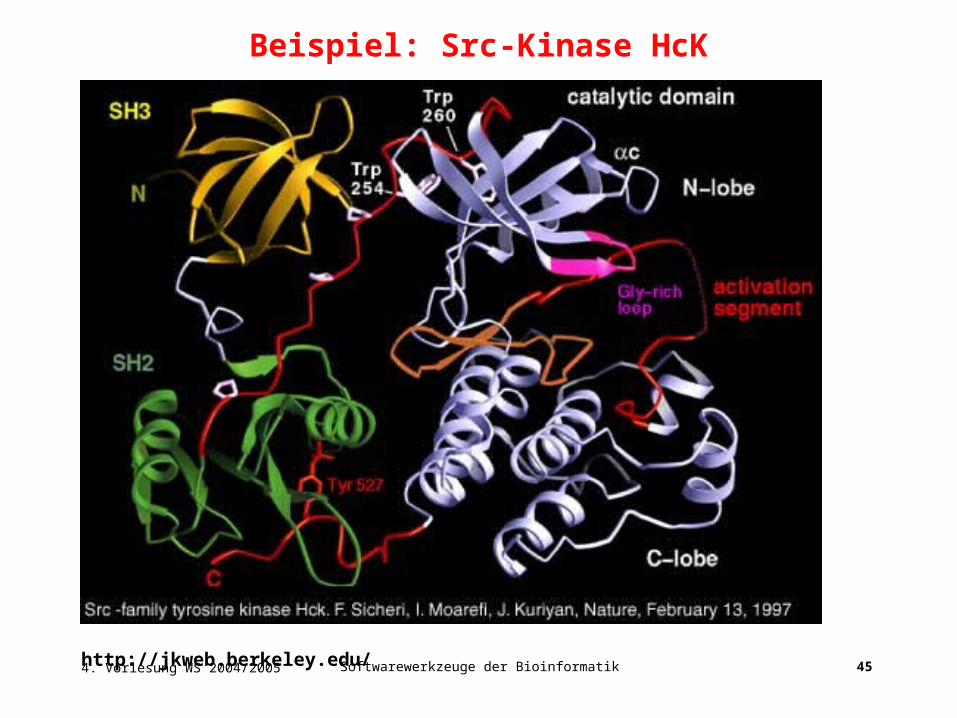
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 45http://jkweb.berkeley.edu/
Beispiel: Src-Kinase HcK
4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 46
http://www.cellsignal.com
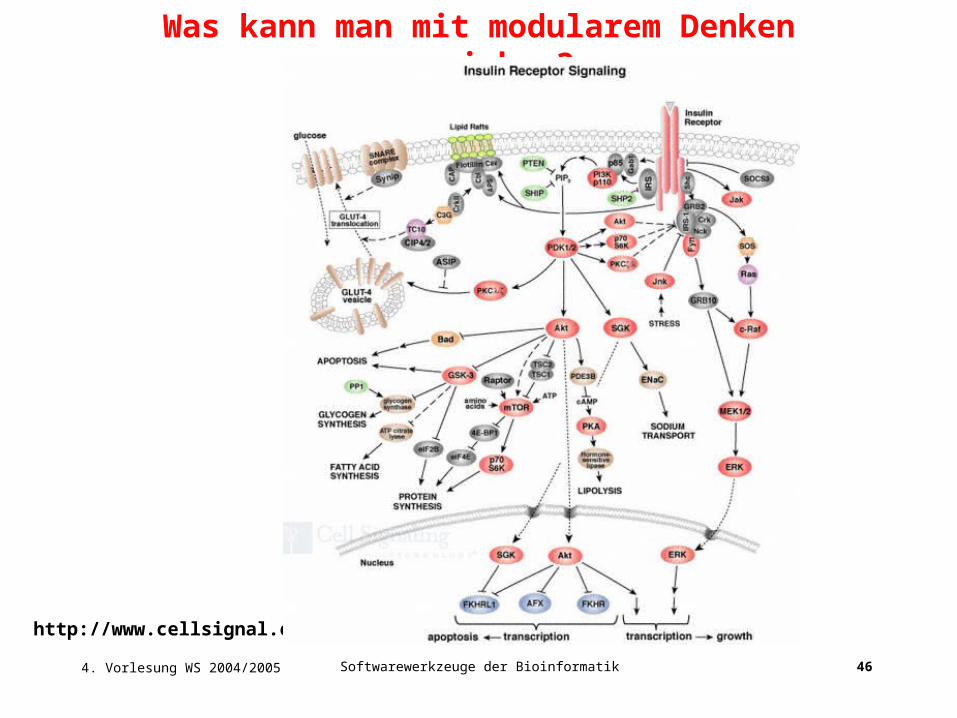
Was kann man mit modularem Denken erreichen?

4. Vorlesung WS 2004/2005 Softwarewerkzeuge der Bioinformatik 47
Progressive Alignments sind die am weitesten verbreitete Methode für
multiple Sequenzalignments.
Sehr sensitive Methode ebenfalls: Hidden Markov Modelle (HMMer)
Multiples Sequenzalignment ist nicht trivial. Manuelle Nacharbeit kann in
Einzelfällen das Alignment verbessern.
Multiples Sequenzalignment erlaubt Denken in Proteinfamilien und –
funktionen.
Zusammenfasusng