6446€¦ · 依三期臨床統計分析結果,進 行藥證申請...
TRANSCRIPT

1
藥華醫藥股份有限公司
簡式公開說明書
簡式公開說明書為公開說明書之重點摘錄,投資人投資前應至金融監督管理
委員會指定之資訊申報網站詳閱公開說明書之內容。
公開說明書網址:公開資訊觀測站(http://mops.twse.com.tw)→基本資料→
電子書
本公司網址:http://www.pharmaessentia.com
查詢電話:886-2-2655-7688
依「公司募集發行有價證券公開說明書應行記載事項準則」第三條第三項各
款之記載:
1.本次現金增資所發行之股票,為因應證券市場價格之變動,證券承銷商必
要時得依規定進行安定操作。
2.本公司普通股股票面額為每股新台幣壹拾元。
一、本次募集與發行有價證券之計畫及其預計可能產生效益概述:
(一)本次發行之有價證券:
本次現金增資發行新股 20,000 仟股,每股面額新台幣 10 元,競
價拍賣最低承銷價格係以中華民國證券商業同業公會申報競價拍賣
約定書前興櫃有成交之 30 個營業日其成交均價扣除無償配股除權(或
減資除權)及除息後簡單算術平均數之七成為為上限,定為每股新台幣
132.5 元,依投標價格高者優先得標,每一得標人應依其得標價格認
購;公開申購承銷價格則以各得標單之價格及其數量加權平均所得之
價格新台幣 173.82 元為之,惟均價高於最低承銷價格之 1.2 倍,故公
開申購承銷價格每股以新台幣 159 元溢價發行,募集總金額為新台幣
3,393,387 仟元。
(二)發行計畫及預計可能產生效益概述:
單位:新臺幣仟元
計畫項
目
預定
完成
日期
所需資
金總額
預定資金運用進度
105 年度 106 年度
第三季 第四季 第一季 第二季 第三季 第四季
充實營
運資金
暨改善
財務結
106
年底 3,393,387 397,428 152,572 548,391 717,436 625,287 952,273
股票代號:6446
2
構
預計可
能產生
效益
本次現金增資預計共募集新台幣 3,393,387 仟元用以充實營運資金,透過長期穩定資金的挹注,順利支應新藥 P1101 及各新藥開發專案之研發、實驗室耗材及臨床試驗等資金需求;另亦可進一步提升營運規模及公司價值,進而強化財務結構,降低營運風險,提升公司整體之競爭力。
二、最近三年度及最近期會計師查核簽證或核閱財務報告之查核或核閱意見:
簽證會計師事務所名稱 簽證會計師 簽證或核閱
財務報告年季 查核或核閱意見
安永聯合會計師事務所 林素雯、黃建澤 102年 無保留意見
安永聯合會計師事務所 林素雯、黃建澤 103年 無保留意見
安永聯合會計師事務所 林素雯、黃建澤 104年 無保留意見
安永聯合會計師事務所 林素雯、林麗凰 105年第一季 保留意見(註)
註:105 年第一季有關子公司相關資訊未經會計師核閱。
三、最近三年度及最近期簡明資產負債表及綜合損益表:
(一) 簡明資產負債表
1. 國際財務報導準則(合併) 單位:新台幣仟元
年度 項目
最 近 三年 度 財 務 資 料(註) 105 年截至 3月 31日 102 年 103 年 104 年
流 動 資 產 1,854,598 1,190,786 600,635 1,153,171
不動產、廠房及設備 258,336 373,554 343,266 337,809
無 形 資 產 42,413 34,313 20,378 19,908
其 他 資 產 52,266 60,160 76,320 71,982
資 產 總 額 2,207,613 1,658,813 1,040,599 1,582,870
流 動 負 債 76,527 247,305 196,917 191,701
非 流 動 負 債 1,475 109,464 103,733 102,235
負 債 總 額 78,002 351,038 299,152 296,704 歸屬於母公司業主之 權 益 2,129,611 1,468,565 689,561 1,283,718
股 本 1,877,310 1,891,822 1,902,832 1,954,583
資 本 公 積 2,437,489 429,849 201,709 911,061
保 留 盈 餘 (2,185,188) (853,947) (1,415,909) (1,582,894)
其 他 權 益 - 841 929 968
庫 藏 股 票 - - - -
非 控 制 權 益 - - - -
權 益 總 額 2,129,611 1,468,565 689,561 1,283,718
註:各年度財務報表均經會計師查核簽證,105年第一季合併財務資料業經會計
3
師核閱完成
2. 國際財務報導準則(個體) 單位:新台幣仟元
年 度
項 目
最 近 三 年 度 財 務 資 (註)
102 年 103 年 104 年
流 動 資 產 1,854,598 1,172,741 582,164
採 權 益 法 之 投 資 - 18,995 16,776
不動產、廠房及設備 258,336 373,554 343,266
無 形 資 產 42,413 34,313 20,378
其 他 資 產 52,266 59,210 76,320
資 產 總 額 2,207,613 1,658,813 1,038,904
流 動 負 債 76,527 80,784 245,610
非 流 動 負 債 1,475 109,464 103,733
負 債 總 額 78,002 190,248 349,343
股 本 1,877,310 1,891,822 1,902,832
資 本 公 積 2,437,489 429,849 201,709
保 留 盈 餘 (2,185,188) (853,947) (1,415,909)
其 他 權 益 - 841 929
庫 藏 股 票 - - -
權 益 總 額 2,129,611 1,468,565 689,561
註:各年度財務報表均經會計師查核簽證
(二) 簡明綜合損益表 1. 國際財務報導準則(合併)
單位:新台幣仟元
年 度 項目
最 近 三 年 度 財 務 資 料(註) 105 年截至
3 月 31 日 102 年 103 年 104 年
營業收入 72,327 13,356 11,589 512
營業毛利 67,241 7,368 (30,466) (900)
營業損益 (601,071) (863,472) (861,853) (388)
營業外收入及支出 7,921 10,366 8,652 99
稅前淨利 (593,150) (853,106) (853,201) (166,985)
繼續營業單位本期淨利 (593,150) (853,106) (853,201) (166,985)
停業單位損失 - - - -
本期淨利(損) (593,150) (853,106) (853,201) (166,985)
本期其他綜合損益(稅後淨額)
68 - (130) 39
4
年 度 項目
最 近 三 年 度 財 務 資 料(註) 105 年截至
3 月 31 日 102 年 103 年 104 年
本期綜合損益總額 (593,082) (853,106) (853,331) (166,946)
淨利歸屬於母公司業主 (593,150) (853,106) (853,201) (166,985)
淨利歸屬於非控制權益 - - - -
綜合損益總額歸屬於母公司業主
(593,082) (853,106) (853,331) (166,946)
綜合損益總額歸屬於非控制權益
- - - -
每股盈餘 (3.4) (4.54) (4.5) (0.88)
註:各年度財務報表均經會計師查核簽證,105年第一季合併財務資料業經會計師核
閱完成
2. 國際財務報導準則(個體) 單位:新台幣仟元
年 度
項 目
最 近 三 年 度 財 務 資 料(註)
102 年 103 年 104 年
營業收入 72,327 13,356 11,589
營業毛利 67,241 7,368 (30,466)
營業損益 (601,071) (863,472) (858,935)
營業外收入及支出 7,921 10,366 5,734
稅前淨利 (593,150) (853,106) (853,201)
繼續營業單位本期淨利
(593,150) (853,106) (853,201)
停業單位損失 - - -
本期淨利(損) (593,150) (853,106) (853,201)
本期其他綜合損益(稅後淨額)
68 - (130)
本期綜合損益總額 (593,082) (853,106) (853,331)
每股盈餘 (3.40) (4.54) (4.50)
註:各年度財務報表均經會計師查核簽證
5
四、特別記載事項:
項目 名稱 摘要意見 簽名或蓋章
(一)證券承銷
商評估總
結意見
凱基證券股
份有限公司
代表人:許道
義
承銷部門主
管:林能顯
依本承銷商之意
見,藥華醫藥股份
有限公司本次募集
與發行有價證券符
合「發行人募集與
發行有價證券處理
準則」及相關法令
之規定,暨其計畫
具可行性及必要
性,其資金用途、
進度及預計可能產
生效益亦具合理
性。
(二)律師法律
意見書
六合法律事
務所
律師:陳宏杰
本律師意見,該公
司本次向財團法人
中華民國證券櫃檯
買賣中心申請股票
上櫃,並未發現有
違反法令致影響股
票上櫃之情事。
(三)由發行人
填寫並經
會計師複
核之案件
檢查表彙
總意見
不適用 不適用 不適用
(四)發行人委
請專家,
就其目前
營運狀況
及本次發
行有價證
券後之未
來發展,
不適用 不適用 不適用

6
進行比較
分析並出
具之意見
(五)合併、分
割、收購
或股份受
讓發行新
股案,獨
立專家就
換股比例
合理性之
意見書
不適用 不適用 不適用
7
五、其他必要記載事項:
依財團法人中華民國櫃檯買賣中心 105 年 3 月 21 日證櫃審字
第 1050100384 號函除依「初次申請有價證劵上櫃用之公開說明書
應記載事項準則」之揭露事項外,尚應於公開說明書特別記載乙
節中揭露下列事項:
(一) 風險事項乙節
1. 本公司目前尚無新藥上市銷售,然為執行臨床試驗將持續發生
龐大研發支出,且研發專案可能因臨床試驗失 敗而無法上市,
有關本公司對未來可能面臨營運資金短絀及新藥無法上市之
風險所採具體因應措施之說明,暨推薦證券商之評估意見。
公司說明: 新藥研發公司具有高資金需求、產品或技術開發時程長、風
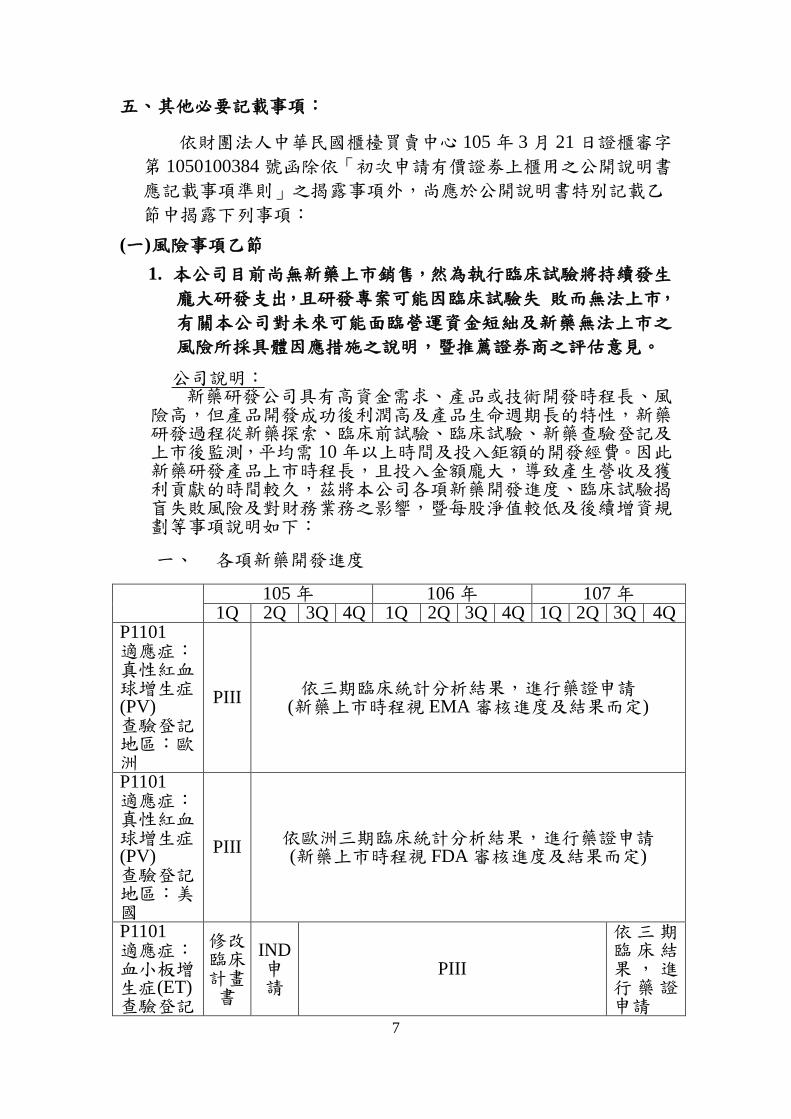
險高,但產品開發成功後利潤高及產品生命週期長的特性,新藥研發過程從新藥探索、臨床前試驗、臨床試驗、新藥查驗登記及上市後監測,平均需 10 年以上時間及投入鉅額的開發經費。因此新藥研發產品上市時程長,且投入金額龐大,導致產生營收及獲利貢獻的時間較久,茲將本公司各項新藥開發進度、臨床試驗揭盲失敗風險及對財務業務之影響,暨每股淨值較低及後續增資規劃等事項說明如下:
一、 各項新藥開發進度
105 年 106 年 107 年 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q
P1101 適應症:真性紅血球增生症(PV) 查驗登記地區:歐洲
PIII 依三期臨床統計分析結果,進行藥證申請
(新藥上市時程視 EMA 審核進度及結果而定)
P1101 適應症:真性紅血球增生症(PV) 查驗登記地區:美國
PIII 依歐洲三期臨床統計分析結果,進行藥證申請 (新藥上市時程視 FDA 審核進度及結果而定)
P1101 適應症:血小板增生症(ET) 查驗登記
修改 臨床 計畫書
IND 申請
PIII
依三期臨床結果,進行藥證申請
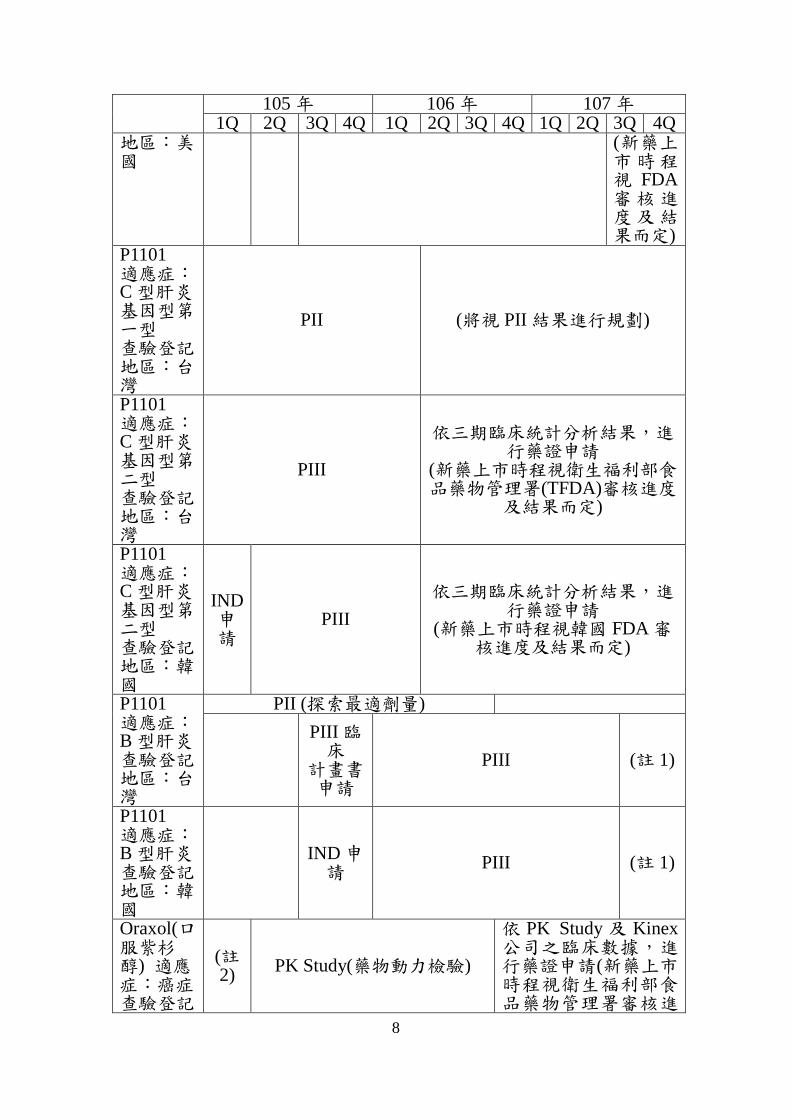
8
105 年 106 年 107 年 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q
地區:美國
(新藥上市時程視 FDA審核進度及結果而定)
P1101 適應症:C 型肝炎基因型第一型 查驗登記地區:台灣
PII (將視 PII 結果進行規劃)
P1101 適應症:C 型肝炎基因型第二型 查驗登記地區:台灣
PIII
依三期臨床統計分析結果,進行藥證申請
(新藥上市時程視衛生福利部食品藥物管理署(TFDA)審核進度
及結果而定)
P1101 適應症:C 型肝炎基因型第二型 查驗登記地區:韓國
IND 申請
PIII
依三期臨床統計分析結果,進行藥證申請
(新藥上市時程視韓國 FDA 審核進度及結果而定)
P1101 適應症:B 型肝炎 查驗登記地區:台灣
PII (探索最適劑量)
PIII 臨床
計畫書申請
PIII (註 1)
P1101 適應症:B 型肝炎 查驗登記地區:韓國
IND 申請
PIII (註 1)
Oraxol(口服紫杉醇) 適應症:癌症 查驗登記
(註2)
PK Study(藥物動力檢驗)
依 PK Study 及 Kinex公司之臨床數據,進行藥證申請(新藥上市時程視衛生福利部食品藥物管理署審核進

9
105 年 106 年 107 年 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q
地區:台灣
度及結果而定)
KX01 適應症:牛皮癬 查驗登記地區:台灣
PI (將視 PI 結果進行規劃)
P1101 + PD1 適應症:B 型肝炎
臨床前試驗 IND 申請
PI PII
註 1:依三期臨床統計結果進行藥證申請,新藥上市時程視 TFDA 及韓國 FDA審核進度及結果而定。
註 2:以授權(Licensing-in)對象 Kinex 公司於韓國及南美等區域執行的臨床數據(共 200 位病人),加上本公司於台灣執行之 PK study 數據,向 TFDA 申請新劑型新藥(505b(2)新藥)的開發計劃。
(一) P1101 治療 PV 本公司於 98 年在加拿大進行第一期人體臨床試驗,98年獨家授權奧地利專業孤兒藥開發公司 AOP Orphan Pharmaceuticals AG(下稱 AOP 公司)於歐洲地區、中東及前蘇聯獨立國(CIS)進行以 P1101 用於治療血液疾病之臨床試驗及市場開發權利。AOP 公司於 99 年啟動歐洲 P1101 for PV 第一/二期人體臨床試驗,並於 101 年完成;102 年啟動 P1101 for PV 第三期人體試驗,並在 104 年 2 月完成 260 餘名受試者之收案,臨床設計治療期間為 12 個月,於 105 年第一季完成第三期人體臨床試驗,105 年第三季整理臨床數據後即可向EMA 申請藥證;美國部分係依據歐洲臨床試驗數據(再加部分數據報告)向美國 FDA 提出藥證申請。 本公司目前於台中廠自行生產製造 P1101 原料藥,然原料藥仍需經由稀釋、充填、確效、品質分析、安定性測試、組裝及最後包裝等成為商品,為提高病患使用方便性,本公司與 AOP 公司共同決定將 P1101 最終產品設計成筆型(PEN)注射器,將 P1101 充填於專用卡式管(cartridge),並組裝於筆型注射器中,爰於 103 年 4 月與 AOP 公司簽定筆型劑型研究開發合約,由 AOP 公司協助筆型注射器之開發及尋找相關委託開發廠商,故歐洲地區於 105 及 106 年度尚須投入相關費用 213,014 千元。另本公司係以歐洲第三期臨床數據向 FDA申請藥證,並委由AOP公司擔任美國藥證申請之CRO公司,預估 105 至 107 年度 CRO 相關顧問費用、申請藥證及上市後監測(PMS)等支出共計 377,527 千元。
(二) P1101 治療 ET 本公司於 104 年向 FDA 溝通 ET 臨床試驗相關事宜,預計於 105 年第二季~第三季提出 IND 申請,第三季獲准開始美國第三期臨床試驗,經參酌 PV 於歐洲之收案速度,並考量 ET 長時間未有新藥出現,本公司預期 6~12 個月可以完成收案,加以臨床設計治療期間為 12 個月,故預計於 107 年第

10
二季可完成第三期人體試驗。 本公司擬自行主導 ET 產品在美國的第三期臨床試驗,參酌 AOP 公司於歐洲進行 PV 第三期臨床試驗之經驗,預估105 至 107 年度自行投入 ET 臨床之試驗用藥品、CRO 相關研發支出、申請藥證及上市後監測(PMS)等支出共計 826,932千元。
(三) P1101 治療 C 型肝炎 1.基因型第一型(HCV GT1) 本公司於100年取得TFDA台灣第二期臨床試驗許可,
因 HCV GT1 治療期間(48 週)較 HCV GT2(24 週)為長,考量台灣 C 型肝炎病患以 GT2 型為多數,且研發預算及時間成本等因素,故臨床實驗進度較 HCV GT2 為慢,惟本公司已與多家醫院簽訂委託醫院臨床試驗,預計 106 年第一季將可完成二期臨床試驗,將視第二期臨床試驗結果進行後續規劃。另預估 105 至 106 年度尚須投入臨床試驗用藥品及CRO/CRC 等委託研究相關服務費用共計 22,195 千元。
2.基因型第二型(HCV GT2) 本公司於101年取得TFDA台灣第二期臨床試驗許可,
103 年完成台灣第二期臨床試驗收案,於 104 年 5 月取得TFDA 第三期人體試驗同意函,已於 105 年 1 月正式啟動三期收案,該臨床設計治療期間為 24 週,台灣及韓國第三期臨床試驗病人數預計共收足 240 人,故預計於 106 年第一季可完成第三期人體試驗;韓國部分已於 105 年 3 月取得韓國食品藥物管理署(Ministry of Food and Drug Safety 簡稱MFDS)核准同意進行該人體臨床試驗。另預估 105 至 107 年度尚須投入臨床試驗用藥品、CRO/CRC 等委託研究相關服務費用、藥證申請及上市後監測等費用共計 174,445 千元。
(四) P1101 治療 B 型肝炎 本公司於 101 年取得 TFDA 台灣第二期臨床試驗許可,以 P1101 單獨使用進行治療 B 型肝炎的臨床試驗,目的係探索 P1101 之最適劑量,惟因預算考量,故臨床試驗進度較慢,預計於 106 年第三季完成試驗。預估 105 至 106 年度尚須投入臨床試驗用藥品及 CRO/顧問等相關費用共計 61,951 千元。 另本公司已規劃預計於 105 年第三季同時向 TFDA 及韓國 FDA 申請以複方治療方式(以小分子抗病毒藥物與 P1101搭配的療法)進行治療 B 型肝炎之第三期臨床試驗,以加快產品上市時程,預計於 106 年第一季進入第三期臨床試驗,試驗治療期間為 48 週/24 針,故預計於 107 年第三季完成第三期試驗報告。預估105至107年度尚須投入臨床試驗用藥品、CRO/顧問費用、藥證申請及上市後監測等費用共計 318,685千元。
(五) 口服紫杉醇(Oraxol) 依 104 年 10 月 30 日財團法人醫藥品查驗中心(CDE)業務諮詢會議記錄,CDE 原則接受本公司以授權方 Kinex 公司於韓國及南美等區域執行之臨床數據(含第一、第二、第三期臨床試驗共計 200 位病人),加上在本公司在台灣執行的人體

11
藥物動力研究及反應(Pharmacokinetics Study)數據(24位/期間一年),向 TFDA 提出新劑型新藥(505b(2)新藥)的開發計劃。本公司預計於 105 年第二季向 TFDA 提出申請新劑型新藥的開發計劃,核准後將開始人體藥物動力研究及反應試驗。該臨床設計治療期間為 12 個月,故預計於 106 年第四季完成PK 數據報告並取得 Kinex 提供之臨床試驗報告後,併同向TFDA 提出藥證申請。預估 105 至 107 年度尚須投入 PK 臨床支出共計 16,878 千元。
(六) 牛皮癬(Psoriasis)用藥 由於美國紐約州生技公司 Kinex 公司所開發的嶄新化合物分子 KX01 已證明對於癌症細胞的增生有顯著的抑制效果,並已進入美國人體臨床試驗第二期。本公司認為 KX01 化合物作用的抑制增生之生物機轉,應可適用於非惡性細胞增生之頑固性牛皮癬疾病,因此向 Kinex 公司取得 KX01 化合物在台灣、中國大陸、港澳、星馬等地區的授權,以針對牛皮癬適應症開發皮膚外用劑型,屬於新成份新藥。本公司 KX01產品已於 104 年 10 月開始進行第一期臨床試驗,預計於 105年 10 月完成第一期人體臨床試驗,由於臨床試驗仍在早期階段(第一期),且為新適應症之研發,考量第二期及第三期臨床試驗成本昂貴,故本公司將視第一期試驗結果再評估後續規劃。預估 105 年度尚須投入臨床試驗醫院及管理師費用、CRO 費用共計 5,682 千元。
(七) P1101+PD1 治療 B 型肝炎 本公司 Anti-PD1 抗體的新藥開發案,係利用人體慢性 B型肝炎小鼠模型來研究以下幾個治療方式(1)PD-1 抗體單獨使用、(2)PD-1 抗體與 P1101 合併使用之各種方式對治療 B型肝炎的效果。目前抗體藥物分子已完成設計並準備進行臨床前動物毒理試驗,預計於106年申請第一期人體臨床試驗。預估 105 至 107 年度尚須投入顧問諮詢費、試驗用動物及試驗分析報告費用、臨床試驗等費用共計 419,800 千元。 綜上,本公司 105 至 107 年度各新藥開發之費用合計約為
2,437,109 千元。另本公司目前開發進度最快之 P1101 應用於適應症 PV,已於 105 年第一季完成第三期人體臨床試驗,將於 105年下半年度分別向 EMA 及 FDA 申請藥證。未來 P1101 如能獲准上市銷售,並不必然表示本公司能立即轉虧為盈,本公司未來損益情形除視歐美 P1101 於 PV 適應症之銷售情形外,亦因尚有多項臨床計畫進行,相關研發費用持續支出,如來自 P1101之營收無法支應其他臨床計畫之研發支出,短期將有可能持續發生虧損。
二、 臨床試驗揭盲(unblinded)失敗風險及對財務業務之影響
本公司開發中新藥 P1101 係新一代長效型干擾素,醫學界已確認干擾素之療效(市面上已經有 Pegasys 及 Peg- intron 等干擾素),但副作用一直困擾著病人及醫生,也可能也是基於這些因素,目前應用於治療 PV 疾病的干擾素藥物(Pegasys)僅能以仿單

12
外治療(off-label use)的方式進行,但有些州或國家病人就必須自行負擔費用,另外市面上不久前才核准的小分子藥物 Jakafi 則僅能用於第二線用藥;亦即目前並無主管機關正式核准用於治療PV 疾病的第一線藥物。本公司 P1101 是第一個以符合法規規範並將進行註冊的創新長效型干擾素新藥,係目前市面上長效型PEG 干擾素中,純度最高,病人耐受度最好且副作用更低的最長效型干擾素(臨床已證明可以 2-4 週施打一次,其他二個都是一週施打一次),依已在世界知名醫藥刊物 Blood 所發表的第二期臨床試驗獲得的結果,本公司認為第三期試驗應可達到預期之結果。
第三期臨床試驗目標係比較 P1101 與 Hydroxyurea(HU/係目前優先使用的化療方式)藥物治療 PV 之療效與安全性,篩選參與試驗者為尚未接受任何治療或使用 HU 治療不到三年之 PV 患者,臨床試驗係以開始接受治療 12 個月後的疾病反應率是否優於(superiority) HU 為主要試驗指標(primary endpoint),本疾病反應率(disease response rates)係指受試者中對於以下療效指標皆有反應的病患(successfully treated patients)人數比例,療效指標包括血球容積比小於 45%且無需放血(自最近一次放血至少三個月無需再放血)、血小板每公升小於 400 克、白血球每公升小於 10 克及脾臟大小正常等,本公司預期 106 年第三季應可得知臨床試驗之結果,茲說明若統計分析結果不能證明 P1101 之疾病反應率明顯優於 HU,對本公司財務業務之影響如下:
情況 因應措施 對財務業務之影響
EMA 有條件
的同意
(conditional
approval)
P1101 上市
若統計分析結果不能
證明 P1101 明顯優於
HU,為使產品盡快上
市,則 AOP 公司及本
公司將於申請藥證前
之 會 議 (Pre-BLA
meeting)與 EMA 討論
以縮減仿單適應症病
患族群(例如建議不適
用於 PV 初期患者、年
輕患者或孕婦)的方式
申請藥證的可能性,並
將持續進行臨床試驗
(例如治療期間更長),
以更多的臨床數據證
明 P1101 之療效並申
請增加仿單適應症病
若 EMA 同意於仿單
(package insert)中註明
適 應 症 的 病 患 族 群
(subgroup),則短期內可
能影響潛在病患總人
數,亦可能影響產品上
市時程。
另由於前述情況只是於
仿單中縮減適應症的病
患族群,P1101 仍為符合
法規審查並進行註冊治
療 PV 的長效型干擾素
(第一線用藥),故有關產
品行銷規劃的影響應該
不大。

13
情況 因應措施 對財務業務之影響
患族群或療效。
EMA 不同意
P1101 上市
臨床數據顯示干擾素
的長期治療效果優於
短期,本臨床試驗係以
12 個月的治療期間作
為有效性的觀察,若
EMA 不同意 P1101 上
市,本公司將持續進行
臨床試驗,以更多的臨
床數據證明 P1101 之
療效。
若 P1101 不能上市,將
對本公司業務發展造成
嚴重影響,本公司將須
再投入更多資金於產品
開發。
本公司原臨床試驗的設計(名稱為 PROUD-PV)是以將來市埸最大化來設計,以儘快取得藥證為優先考量,所以假設數據不如原指標,本公司會先接受於仿單中縮減適應症病患族群之方式,爭取 P1101 及早上市,同時因目前的”Conti-PV”仍屬為繼續再進行的臨床試驗,往後會再用更多的臨床數據爭取更廣更優的療效。
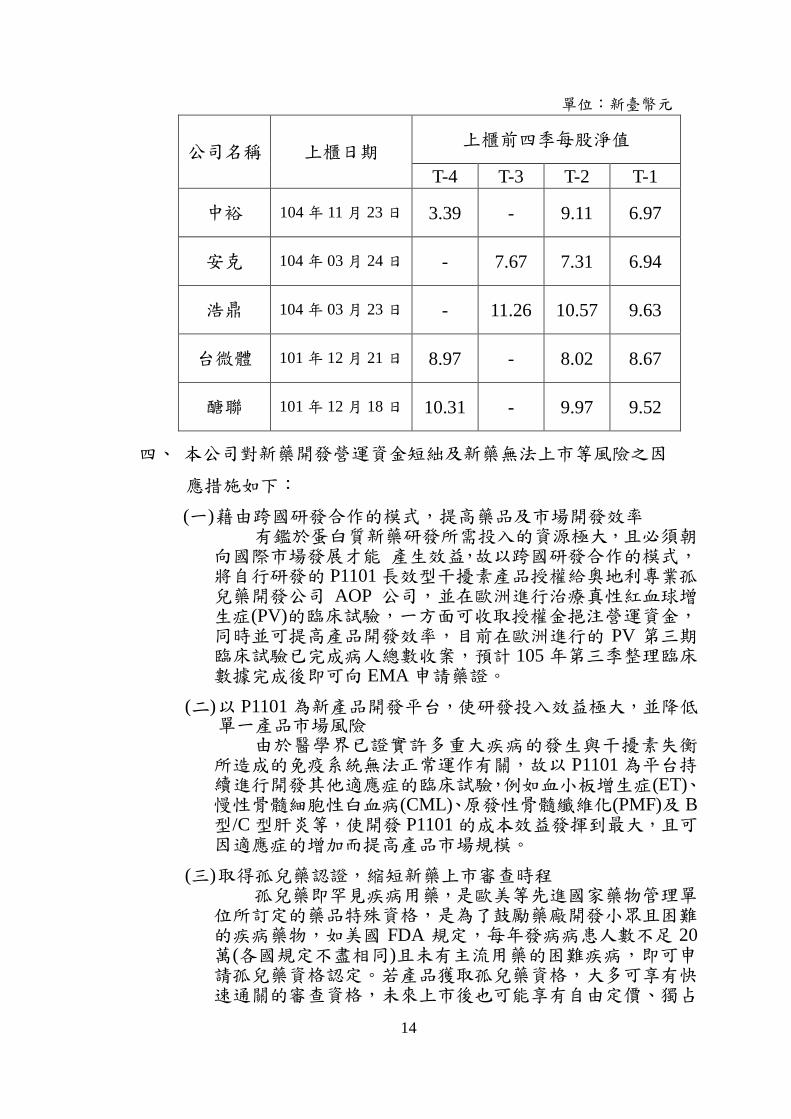
三、 每股淨值較低及後續增資規畫
本公司 104 年 9 月底、12 月底及 105 年 3 月底每股淨值分別為 5.06 元及 3.62 元及 6.57 元,由下表,同業公司於上櫃掛牌前之每股淨值亦有逐期遞減情況,此乃行業特性,通常新藥產業在歷經增資後,如尚無新藥上市銷售或再次增資,在研發費用持續支出下,每股淨值多呈現逐期下降之情形。
本公司開發中新藥 P1101 尚未上市銷售,且相對於同業本公司有較多項新產品研發計畫持續進行,致持續產生虧損使淨值降低。本公司於申請上櫃前向主管機關申報之現金增資發行新股5,000 千股業於 105 年 1 月 5 日申報生效,並於 105 年 3 月募集資金 750,000 千元,致 105 年 3 月底之每股淨值略為上升至 6.57元,預期上櫃掛牌前最近一季財務報告之每股淨值仍能維持於 5元以上;另將於上櫃前辦理現金增資(預計發行 20,000 千股,假設每股 150 元,預計可募集 3,000,000 千元),充實營運資金並維持資本結構之健全。
14
單位:新臺幣元
公司名稱 上櫃日期 上櫃前四季每股淨值
T-4 T-3 T-2 T-1
中裕 104 年 11 月 23 日 3.39 - 9.11 6.97
安克 104 年 03 月 24 日 - 7.67 7.31 6.94
浩鼎 104 年 03 月 23 日 - 11.26 10.57 9.63
台微體 101 年 12 月 21 日 8.97 - 8.02 8.67
醣聯 101 年 12 月 18 日 10.31 - 9.97 9.52
四、 本公司對新藥開發營運資金短絀及新藥無法上市等風險之因
應措施如下:
(一) 藉由跨國研發合作的模式,提高藥品及市場開發效率 有鑑於蛋白質新藥研發所需投入的資源極大,且必須朝
向國際市場發展才能 產生效益,故以跨國研發合作的模式,將自行研發的 P1101 長效型干擾素產品授權給奧地利專業孤兒藥開發公司 AOP 公司,並在歐洲進行治療真性紅血球增生症(PV)的臨床試驗,一方面可收取授權金挹注營運資金,同時並可提高產品開發效率,目前在歐洲進行的 PV 第三期臨床試驗已完成病人總數收案,預計 105 年第三季整理臨床數據完成後即可向 EMA 申請藥證。
(二) 以 P1101 為新產品開發平台,使研發投入效益極大,並降低單一產品市場風險
由於醫學界已證實許多重大疾病的發生與干擾素失衡所造成的免疫系統無法正常運作有關,故以 P1101 為平台持續進行開發其他適應症的臨床試驗,例如血小板增生症(ET)、慢性骨髓細胞性白血病(CML)、原發性骨髓纖維化(PMF)及 B型/C 型肝炎等,使開發 P1101 的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模。
(三) 取得孤兒藥認證,縮短新藥上市審查時程 孤兒藥即罕見疾病用藥,是歐美等先進國家藥物管理單
位所訂定的藥品特殊資格,是為了鼓勵藥廠開發小眾且困難的疾病藥物,如美國 FDA 規定,每年發病病患人數不足 20萬(各國規定不盡相同)且未有主流用藥的困難疾病,即可申請孤兒藥資格認定。若產品獲取孤兒藥資格,大多可享有快速通關的審查資格,未來上市後也可能享有自由定價、獨占

15
市場等優惠條件。P1101 用於治療 PV 已分別於 100 年及 101年取得歐洲 EMA 及美國 FDA 的孤兒藥認證,且用於治療ET及 PMF等疾病亦於 103年獲得美國 FDA的孤兒藥認證,因此有助於縮短新藥上市審查時程。
(四) 已辦理現金增資充實營運資金,加計股票初次上櫃現金增資及 P1101 上市銷售產生之現金流入,資金狀況應無虞
本公司依目前帳上現金及已辦理現金增資發行新股5,000 千股,已於 105 年 3 月底募集資金 750,000 千元,可支應 105 年度之營運資金需求;而本公司預計於股票初次上櫃前辦理現金增資募集資金 3,000,000 千元,加上新藥 P1101倘陸續於歐美地區上市銷售,其上市後之營業活動現金流入亦可支應以後各年度營運資金及各項研發計畫,故本公司對於未來營運資金需求已有具體因應措施。
有關公司對未來可能面臨營運資金短絀及新藥無法上市之風險所採具體因應措施之說明,推薦證券商之評估意見:
1. 推薦證券商有關該公司新藥無法上市之風險所採具體因應措施之評估意見:
長效型干擾素係已核准用於治療感染性疾病的藥物(例如默克藥廠的 PEG-Intron 治療 C 型肝炎,羅氏藥廠的 Pegasys 治療 C型肝炎及 B 型肝炎),但受限於病人最大耐受劑量及副作用等因素,目前尚無經核准用於治療血液疾病的干擾素藥物,而目前 Incyte公司的 Jakafi小分子藥物係用於對HU(愛治膠囊)有抗藥性或不耐受之 PV 病人為主,故目前並無正式核准用於治療 PV 的第一線用藥。該公司 P1101 在歐洲進行治療 PV 的臨床試驗,其第二期臨床試驗數據(於 2013 及 2014 美國血液疾病協會(ASH/AJH)發表,41位病患PV病患的資料顯示,整體疾病反應率超過90%,45~50%的病患在血液學參數正常化下顯示完全反應,且脾臟大小亦有正常化趨勢,一年之後所有病患完全無須放血),其試驗結果顯示P1101 相較於目前市面上長效型干擾素(PEG-Intron 及 Pegasys)擁有較高的純度、病人最高耐受劑量、更長效及更低副作用的優勢,因此該公司開發 P1101 長效型干擾素的風險較低(非首次用於人體之藥物);另因目前尚無被正式核准用於治療 PV 的長效干擾素藥物,若 P1101 第三期臨床試驗數據結果無法證明 P1101 明顯優於 HU,該公司與 AOP 公司擬以縮減仿單適應症病患族群的方式申請藥證,由於 P1101 為 EMA 及 FDA 認證的孤兒藥,且目前尚無正式核准的 PV 第一線用藥,故 EMA 有可能同意以 conditional approval 方式核准 P1101 上市,其因應措施應屬合理。 對財務業務影響方面,假使該公司以仿單縮減適應症病患族群之方式申請P1101上市,短期內雖可能影響其潛在病患總人數,惟該公司將持續進行臨床試驗(例如治療期間更長),以更多的臨床數據證明 P1101 之療效並申請增加仿單適應症病患族群或療效,故長期而言,對其業務發展之影響應屬有限;而藥品售價方面,由於目前尚無核准用於治療 PV 的第一線用藥,該公司預估 P1101售價係以第一線用藥之定位,綜合考量病人接受度及已上市類似

16
藥物 Jakafi 之售價後保守訂定(實際售價須視當時市場狀況而定),故對銷售價格之影響應屬有限。另假使 EMA 不同意 P1101 上市,則對該公司業務財務將造成嚴重影響,將使其營運獲利轉正時間延後,營運資金來源亦可能受到影響,惟該公司 P1101 除授權給AOP 公司於歐洲進行治療 PV 的臨床試驗外,尚自行於台灣及韓國進行治療 C 型肝炎及 B 型肝炎的臨床試驗,該產品開發計畫應不受 PV 臨床試驗結果之影響。整體而言,該公司對於 PV 臨床試驗揭盲失敗的風險所擬定之因應措施尚屬合理。
2. 推薦證券商有關該公司營運資金短絀所採具體因應措施之評估說明:
該公司係以整合研發、生產及銷售一條龍為經營模式之蛋白質新藥公司,為降低新藥開發風險,該公司新藥選題以干擾素為主要產品,可提高產品開發成功機會,且因適應症多,使產品開發平台得以延伸,目前進行中之適應症包含 PV 及肝炎,並已開始進行腫瘤疾病的研發計畫,故可降低單一產品市場風險。另為縮短新藥上市審查時程分別於 100 年度及 101 年度取得 P1101 用於治療 PV 之歐洲 EMA 及美國 FDA 的孤兒藥認證,103 年度亦取得P1101 用於治療 ET 及 PMF 等疾病之美國 FDA 的孤兒藥認證,且以授權開發方式將P1101於歐洲市場的開發權授權給AOP公司,該公司則保留生產製造權及未來歐洲地區銷售權利金,除可收取授權金挹注營運資金,並可藉由 AOP 公司之市場開發經驗提高產品開發效率,累積行銷經驗有助於美國市場之開發。整體而言,該公司於產品研發及市場開發之規劃及採取之措施,尚屬合理。 資金方面,該公司截至 104 年 12 月可動用資金為 563,909 千元,並已辦理現金增資發行新股 5,000 千股,於 105 年 3 月底募集資金 750,000 千元,該公司為籌措足夠之營運資金以支應新藥研發之資金需求,積極推動公司申請上櫃,預計股票初次上櫃前辦理現金增資可籌措 3,000,000 千元,且該公司新藥 P1101 陸續於歐美地區上市銷售,其上市後之營業活動現金流入亦可支應以後各年度營運資金及各項研發計畫,整體而言,該公司對未來可能面臨營運資金短絀及新藥無法上市之風險所採之具體因應措施尚屬合理。
2. 本公司自行設廠之必要性、決議程序、投資計畫內容及自行設廠
可能產生之風險與相關因應措施之說明,暨推薦證券商之評估意
見。
公司說明:
一、 自行設立蛋白質藥廠之原因及必要性
(一) 蛋白質原料藥 (Active Pharmaceutical Ingredients, API) 藥廠
因本公司授權予奧地利孤兒藥公司 AOP Orphan Pharmaceuticals(以下簡稱 AOP 公司)在歐洲進行 P1101 應用於真性紅血球增生症之人體臨床試驗,該第三期人體臨

17
床試驗原預計於 101 年開始展開,且考量下列因素,本公司於 100 年 3 月經董事會決議後,決定自行設置蛋白質藥廠,進行 P1101 原料藥(API)的生產。
A. 因本公司原本委製 P1101 試驗用藥品之生物技術開發中心(DCB),其生產設施規劃屬多產品 型的 研發型先導工廠(pilot plant),產能小且不易擴充,因此僅能生產供臨床第一期及第二期使用之藥品。第三期試驗用藥品及商業量產需求的產品數量較多,品質要求也較嚴謹,另第三期的生產還需考量日後商業化量產的銜接問題,故試驗用藥品已無法繼續委託生物技術開發中心進行生產。 B. 因本公司授權予 AOP 公司在歐洲進行 P1101 第三期人體臨床試驗,依據歐洲藥物管理局(以下簡稱歐盟 EMA)規範,用於第三期人體臨床試驗的藥品與將來上市銷售的藥品,在產製過程各項規格皆須一致,經評估與其他合格之第三期臨床試驗用藥品委製廠、爾後再轉換製造廠之可行性後,決議自行設立廠房進行製造生產 P1101 原料藥,其主要原因為製造廠之轉換所費不貲,必須另外付生產技術轉移費、分析方法轉移費及試作費等,相關經費動輒近百萬美金,且轉換過程所費時間長,較之自行設立製造場所,唯一差別只是發生時間的推遲而已,屆時恐受制於上市期之逼近,與委製廠間談判籌碼更形見絀,另需投入更多人力以克服時間不足之壓力,所需費用支出恐將更鉅。此外,台灣尚未有拿到量產許可的 GMP 蛋白質藥廠可進行委託生產製造工作。 C. PEGylation 為本公司重要核心技術,且已有專利保護,為避免核心技術外流及支應未來其他適應症用藥需求,故自行設立蛋白質藥廠的建置亦為重要考量因素之一。 D. 本公司致力於新藥開發,從事新藥創新發明、試驗發展、生產製造,到取得藥證後行銷國際市場,希望透過完整的垂直整合能打造真正研發、製造均在臺灣,臨床試驗與銷售又能與歐美及國際接軌的新藥產品。
綜上所述,故本公司建置一座能符合 EMA 與美國 FDA規範之蛋白質新藥廠實屬必要。
(二) 針劑充填廠
P1101 規劃將於 106 年申請 C 型肝炎藥證,因此需確定未來充填量產基地,以進行技術移轉、試量產、製程確效、取得安定性試驗資料等作業,於藥證申請時提供量產廠相關資訊。目前臨床用藥先前是委託永昕生物醫藥股份有限公司進行充填,但永昕的產能僅每日 4000 劑,並無供應藥證取得後市場需求的能力。量產基地之選擇,主要評估項目如下:
A. 生產設備可相容於產品包裝 C 型肝炎產品是採針劑包裝(syringe),且膠塞

18
具特殊包覆(Teflon),生產廠必須能充填針劑包裝(syringe)容器,並採用真空充填方式,以避免對膠塞的包覆造成損傷。但綜觀國內針劑商業化量產的 GMP 廠,均無針劑包裝(syringe)真空充填之商業化生產能量,僅有能力供應小批量臨床試驗。
B. 生產排程彈性 委託生產均需遷就受託廠產能與訂單,因此生
產排程較無彈性,無法因應訂單的變化調整生產,可能造成將來產品缺貨或效期不足醫院拒收等情形。
C. 委託生產與運輸費用 若委託國外公司生產,則費用高昂,每劑委託
費用約為自行充填的三倍以上(自行充填每劑成本約 55元);且國際間冷藏運輸費用估計每劑約需 10 元,運輸時發生溫度偏差的風險也高。
D. 其他效益 自建充填廠後,將來新產品如 B 型肝炎,亦可
自行生產,避免技術移轉可能發生的問題。另外,充填廠所規劃的設備,具備可充填成注射針劑(syringe)或注射筆的 cartridge 形式,目前肝病市場主要是以亞洲為主,主要的劑型是注射針劑,同時為保有未來充填劑型的彈性,在選購充填設備時,主要考慮具有可充填針劑(syringe)和cartridge 的設備為主,除未來可與歐、美市場接軌外,更可以擴大生產的彈性。
綜合考量以上事項,本公司將自行建造充填廠,預定於106 上半年完工,以承接 C 型肝炎商業化量產生產責任。
二、 決議程序及投資計畫內容
本公司相關建廠計畫均通過董事會決議後執行,說明如下:
(一) 蛋白質原料藥藥廠
A. 決議程序
係依據本公司「核決權限表」及「取得或處分資產處理程序」之規定,超過新臺幣三千五百萬元者,應經董事會通過後辦理。本公司於 100 年 3 月 18 日將設立干擾素第三期人體試驗製造場所之議案提請董事會決議,並說明另覓國外之合格製造場所或由本公司自行設立之效益評估,依該次董事會決議「將會議中報告資料彙整後補送各位董監事,並另召開臨時董事會以續行討論該案」;後經本公司 100 年 3 月 31 日臨時董事會決議通過自行設廠生產干擾素。後因配合蛋白質先導廠資金到位及廠址定案等因素,延至 101 年 5 月正式動工至 10 月始完工。
B. 蛋白質新藥建廠處所選擇考量
本公司基於竹科的積極招商及配合國家政策並經 100

19
年 3 月 31 日董事會決議申請進駐竹北生醫園區,因與行政院國家發展基金及耀華玻璃管理委員會就股權爭議尚未達成具體協議,故未能取得進駐許可。100 年 9 月 28 日董事會決議進行替代案準備,並依據建廠顧問的建議,尋得深坑科學園區,經審慎評估後於 100 年 12 月 8 日董事會決議以租賃深坑廠房。
本公司依據 100 年 12 月 8 日董事會通過於深坑設廠,並按計畫執行中,惟當時本工程承攬廠商亞翔工程股份有限公司(上市公司,下稱亞翔公司)之配合建築師表示,因深坑場址係普通工業區,內裝許可證申請將需經過「都市設計審議」,需費時二至三個月,恐無法按原訂時程完工。後經中科管理局之協助,中科均豪精密工業股份有限公司(上櫃公司,下稱均豪公司)所自建之廠房可出租 1300 坪(位於第三樓層),本公司請亞翔公司工程設計人員實地履勘後表示,完全符合本廠之技術規格要求(廠房面積大、樓高8.2M、符合室內裝修及消防許可要求、台電電力供應穩定、良好的自來水水質及廢水排放容易管理),更可大幅縮短施工時程。本公司因此提案經 101 年 2 月 21 日董事會決議將干擾素第三期人體臨床試驗製造場所改設置於中科園區均豪公司之廠房內。
C. 蛋白質新藥廠建廠主要支出如下:
類別 金額 備註 廠房建置費 109,725 千元 係無塵室建置及廠房
各式系統工程 儀器設備 150,275 千元 係生產設備及分析檢
測用儀器等 裝潢工程 3,984 千元 係廠區辦公室裝潢 辦公設備 2,918 千元 係廠區辦公室設備 顧問費用 16,155 千元 係建廠顧問及確效顧
問 合計 283,057 千元
註:100 年 12 月 8 日董事會通過「干擾素第三期人體臨床試驗製造廠所案」預算(含設備)為 700 萬美元。101 年 5 月 28 日董事會討論 101 年預算案時,將預估設備總支出增加約 115 萬美元。故預算合計約美金 815 萬元(裝潢、辦公設備及顧問費係依核決權限申請核准後支付),與上表廠房建置及儀器設備費用新臺幣 260,000千元相當。
(二) 針劑充填廠
本公司針劑充填廠係屬規劃階段,相關預算業於 104 年12 月 21 日董事會通過,預計於台中廠區進行建置,預計投入金額為約新臺幣 90,000 千元,規劃完工時程為 15 個月(包含設計規劃、設備交期、確效作業等,設備交期一般為下訂後 12 至 14 個月交機),預計於 106 年第二季完工後進行製程確效,並於 106 年第四季配合臨床試驗結果遞件申請藥品上市許可。相關預計支出概算如下:

20
類別 金額 備註
租賃改良 27,500 千元 係無塵室建置及廠房各
式系統工程
儀器設備 62,500 千元 係生產設備及分析檢測
用儀器等
合計 90,000 千元
三、 資金來源
(一) 蛋白質原料藥藥廠
本公司於 100 年 3 月 18 日經董事會通過辦理現金增資29,101 千股,預計募集 349,218 千元(以每股 12 元溢價發行),用以支應蛋白質製造場所建置所需經費,惟因股東繳款不如預期而經 100年 4月 20日董事會修正發行新股總數為 25,201千股,合計募集資金 302,418 千元。建廠過程中,本公司亦積極推展研發中產品的開發進度,故除支付建廠相關費用外,尚需支應各項臨床試驗費,故為維持公司營運不出現資金缺口,於 101 年 9 月 7 日董事會決議,「於借款總額不逾新臺幣 75,000 千元額度內、貸款年利率百分之十二(或月利率百分之一)以下之條件辦理借貸,以填補九、十月營運資金缺口」。本公司即依董事會授權之年利率 12%向關係人借款75,000 千元,用以週轉支付建廠廠商款項。對此,本公司規劃辦理現金增資,經 101 年 11 月 13 日董事會以現金增資案每股 21 元溢價發行新股 22,000 千股,合計募集 462,000 千元,增資額於 101 年 12 月收足股款資金到位後,立即償還向關係人借款並陸續支付廠商款項。
(二) 針劑充填廠
本公司預計以自有資金進行針劑充填廠房擴建。
四、 預計效益
(一) 蛋白質原料藥藥廠
A. 產能規劃
依本公司台中廠之設備產能預估,每批次約可生產11g 之 P1101 原料藥。本廠最大的產能約可生產 400g (40批次生產/三班)。依目前人員配置狀況,每批次的生產時程為 2~4 周;若以每年生產 8~13 批次估算,則可提供予每年約 25~40 萬針之產量,相當於每年可醫治 15,000 名以上之真性紅血球增生症病人(一位真性紅血球增生症病人每年約需 6~7 mg 的 P1101 進行治療((一針約 360 ug P1101;第一年治療大約每兩週打一針,第二年起的治療大約每個月打一針)。歐洲及美國的真性紅血球增生症病人分別大約是 15 萬人左右,按照 AOP 公司的銷售預測,直到 2020 年分別約有 1 萬名病人的需求,以本廠目前最大規劃產能來看,可以供應 6 萬名病人的需求,足以提

21
供初期的產能需求。為因應 P1101 取得藥證上市後之量產需求,本公司將依上市後之市場需求狀況,逐步擴充現有生產設備及產能。
B. 方便管理,掌握進度
因為一般蛋白質藥製造有不可預期的變異,若委外製造必須派專員到廠協助控管。例如:委託生物技術發展中心(DCB)製造時,其中間體必須送回本公司分析,且決定收取的樣本液範圍,做下一段的純化;若國外則無法送回分析必須全權委託,時程不僅無法控管掌握,恐影響藥品製造(例如中間體安定性問題),因此本公司在國內設立自製生產的場所是勢在必行計畫。
C. 快速經驗累積,降低營運成本
本公司透過設立製造廠可快速累積相關經驗,了解製程的關鍵因素並加以控制,降低量產失敗的風險與生產成本。此外,藉由製造廠的設立,了解生產製程與機器運轉的相關性,並建立關鍵設備的預防性保養措施,降低不可預期的風險。
綜上所述,因蛋白質藥廠生產製造模式不同於國內其他藥廠,故相關人力資源較少,製造廠的設立,有助於人才養成與經驗累積,本公司引進歐、美的藥廠顧問,藉以提升人員的視野並與國際接軌,降低國際 GMP 法規的更動所帶來的衝擊,當藉由製造廠的設立,本公司就能掌握關鍵人力、技術、法規資訊與設備運轉,對於日後若要新設另一製造廠,將可降低建製與運轉成本。
(二) 針劑充填廠
A. 產製時間排程彈性
委外加工須配合充填廠商排程時間,若本公司自行充填,生產排程握有主控權,亦能配合本公司原料藥生產及人員調度,例如原料藥廠與充填廠人力相互支援,使人力獲得充分利用。
B. 品質控管較佳
因充填廠商也有充填其他藥品,且充填廠設備在現有設備規格也不能完全符合本公司產品特殊要求,若本自公司自行充填,產品品質較易控管。
C. 充填成本較低
本公司評估自行充填成本遠低於委外加工,係因除既有委託加工費用外,免除運輸、貼標、包裝及儲存等其他加工費用。
五、 自行設廠之風險說明及相關因應措施
(一) 藥證無法取得之風險
藥證之發給取決於二項因素,新藥臨床試驗結果及生產品質(GMP)。

22
臨床方面,本公司 P1101 目前正在歐洲進行治療真性紅血球增生症的第三期臨床試驗,其第二期臨床試驗已獲得高療效低副作用的成果,且臨床數據顯示,P1101 可將導致真性紅血球增生症疾病的 JAK2 突變基因進行修複,因此德國官方主管醫藥機構特別提及 P1101 的效果能達到 Functional cure的效果(真性紅血球增生症的發生主要是因為 JAK2基因發生突變,病人接受 P1101 的治療後,所突變的基因會被修復,故稱之為 Functional cure)。此一結果乃是真性紅血球增生症疾病臨床研究上的一大突破,且因已被認定為孤兒藥,產品有很高的機率可獲得核准。
生產方面,從台中蛋白質藥廠開始規劃時,即以通過歐洲 EMA 以及美國 FDA 標準為目標。整廠設計邀請國外專業藥廠規劃公司協助,設備均採用世界知名品牌,廠房也以國際製藥標準建置。在軟體方面,持續增加人力,延攬曾經在國內外通過 EMA、FDA 查廠的藥廠工作之人才加入,聘請高階顧問輔導,所有努力都是以通過查廠為目標。台中廠亦已經通過二次 TFDA 稽核(第一次 TFDA 稽核後於 102 年 4月取得 GMP 廠證,第二次 TFDA 稽核為 104 年 11 月,並於105 年 2 月 16 日函知本公司,廠證有效期間延長至 108 年 1月 23 日),留下相當不錯的評價。對於藥華醫藥台中廠通過EMA 或 FDA 查核,本公司深具信心。若 GMP 查廠發現缺失,本公司也會積極採取因應將缺失改善,一般而言於改善後補件即可取得核准。
因應對策:
A. 本公司於 101 年 10 月完成蛋白質藥廠設廠,11 月開始試產,並於 102 年 4 月拿到 TFDA GMP 廠證,有助於未來因應申請歐洲 EMA 查廠的經驗。本公司蛋白質藥廠擁有微生物製程區,能由細菌產製蛋白質藥物。廠房內所有的製程儀器,包括發酵槽、均質機、濃縮透析儀、管柱層析儀、高壓蒸汽滅菌機及蛋白質藥廠最重要的注射用水(WFI,water for injection)生成機等,都是選用世界知名及符合EMA 及 FDA 規格的廠牌,此外,本公司亦積極加強人員標準作業流程的訓練,以符合查廠要求。本公司也聘請了歐、美的藥廠顧問,藉以培養人才並與國際接軌,品質管理顧問是前英國 MHRA 的官員,也是英國第一位 QP (Qualified Person),並擔任許多官方稽核員的訓練師,該顧問每個月至少到廠 1~2 週進行駐廠輔導;另聘 QC 顧問,其服務於美國藥廠超過 20 年以上,並曾擔任品質管控(QC)的 Director,她的豐富經驗有助於提升 QC 部門的運轉效能並符合歐美對品管的要求;亦聘一位全世界最好的充填廠Vetter 所推薦的顧問,為公司長期的駐廠顧問並協助順利通過查廠。
B. 本公司 P1101 可用於多項適應症,如血小板增生症(ET)、慢性骨髓細胞性白血病(CML)、原發性骨髓纖維化(PMF)

23
及 B 型/C 型肝炎等,使開發 P1101 的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模,可避免無法取得單一藥證而造成風險。
(二) 廠房承租之風險
本公司目前台中廠區承租情形如下:
承租
面積
租賃
期間
年租金
(千元) 出租人
租金之計算
及
支付方式
租約所定之
限制
1,493.05
坪
101.5~
106.4 13,437
均豪
公司
每月約 1,120
千元;每月電
匯
不得轉租
A. 廠房租賃期間自 101 年 5 月 1 日至 106 年 4 月 30 日,共計 5 年,依承租合約"「我 方 若擬續租,擁有優先續租權」,並於租約期滿前三個月以書面通知甲方(均豪公司)",且甲方(均豪精密)並無提早回收之權利。本公司已經提早向均豪公司提出續租之需求,積極保有承租自身權利,均豪公司也表達續租之意願(105 年 2 月 17 日已電子郵件通知本公司預計於合約到期前一年將發文詢問是否續約),雙方已經持續在討論提早簽約事宜,保障公司權益。
B. 本公司承租廠區土地為中科管理局所有,本公司亦是中科管理局合格進駐廠商,受「中部科學工業園區自建廠房轉租審核原則」,「乙方如擬將其於本約土地上所興建之建築物轉租、轉借或轉讓予他人,應事先報經甲方(中科管理局)同意。惟轉租、轉借或轉讓之對象應以經核准設立之園區事業為限」,故本公司承租權益係受中科管理局保障。
C. 土地為中科管理局所有,地上物為均豪公司所建之廠房,於 97 年落成後,3 樓廠區處於無使用情形,才經中科管理局引薦本公司向均豪公司承租。本公司探詢目前並無其它廠商提出承租需求,若均豪公司不再續租,又將呈現無使用狀況,實非均豪公司所樂見。
推薦證券商說明:
1. 該公司自行設立蛋白質藥廠之必要性
該公司係蛋白質新藥公司,其經營模式包含了新藥創新發明、試驗發展、生產製造及行銷國際市場,即所謂一條龍營運模式的新藥公司,透過完整的垂直整合打造研發、製造均在臺灣,臨床試驗及銷售與歐美及國際接軌的新藥產品,其經營模式亦常見於國際大藥廠,而國內生物製劑藥廠如永昕生物醫藥(股)公司(以生物相似藥(Bio-similar)為主)及聯亞生技開發(股)公司(以合成肽為主的人用與動物用疫苗及蛋白質/單株抗體藥品)亦皆以自有藥廠之經營模式。茲將其自行設立蛋白質原料藥藥廠並規劃建造針劑充填產線的必要性評估說明如下:
(1) 蛋白質原料藥藥廠
該公司 P1101 第一期及第二期臨床用藥係委託生物技術發展中心(DCB)製造,並由美國 Pyramid Laboratories, Inc 及聯亞生技開發(股)公司完成充填後用於臨床試驗,其第一期及第二期臨床試驗已於 99 年完成,經查閱該公司 100 年 3月 18 日董事會議事錄,主要係考量藥品審查單位(EMA)對於

24
第三期人體臨床試驗的藥品與將來上市銷售的藥品,在產製過程中各項規格需一致性的要求,及若不自行建廠其未來需面臨的製造廠所轉換的風險及成本等因素後,經董事會決議自行建置蛋白質原料藥藥廠以因應第三期臨床用藥及未來商業化產品製造的需求,故應有其必要性。另依該公司新藥研發、製造及銷售一條龍的營運模式,採自行設立蛋白質藥廠更能有助於其人員的訓練及經驗累積,故該公司自行建置蛋白質藥廠,短期而言可符合申請藥證及未來商業化量產需求,長期而言更可達到在地深耕及人才培訓目的之長遠目標,故該公司 101 年自行設立蛋白質原料藥藥廠有其必要性。
(2) 針劑充填廠
C 型肝炎產品是採針劑包裝(syringe),且膠塞具特殊包覆(Teflon)。生產廠必須能充填針劑包裝容器,並採用真空充填方式,以避免對膠塞的包覆造成損傷。綜觀國內針劑商業化量產的 GMP 廠,較缺乏針劑包裝真空充填之商業化生產能量,僅有能力供應小批量臨床試驗。故該公司規劃自行建造針劑充填產線主要係為 P1101治療C型肝炎第三期臨床用藥需求及因應 106 年申請 C 型肝炎藥證的時程進度,並可提供P1101 治療 B 型肝炎的第三期臨床試驗用藥需求,避免技術移轉可能發生的問題,經評估有其必要性。
2. 決議程序
(1) 蛋白質原料藥廠
經查閱該公司董事會議記錄(100 年 3 月 18 日、3 月 31日、9 月 28 日、12 月 8 日及 101 年 2 月 21 日、5 月 28 日),該公司對於自行建廠或委託製造之評估、建廠處所之選擇、及建廠經費預算等議案皆經董事會討論決議,因金額未達取得處分資產應公告標準,故無相關公告,尚無重大異常。
(2) 針劑充填廠
該公司針劑充填廠目前係屬規劃階段,其相關預算已於104 年 12 月 21 日董事會通過,預計於台中廠區進行建置,預計投入金額為約新臺幣 90,000 千元,其決議程序尚無重大異常。
3. 廠房合規情形
該公司於 101 年 10 月完成蛋白質藥廠設廠,11 月開始試產完成,並於 102 年 4 月拿到 TFDA 核發的 GMP 廠證(該廠證有效日期為 105 年 1 月 23 日,每三年複查)。該公司於 104 年 7 月20 日提出 GMP 複查申請,TFDA 於 104 年 11 月 9-10 日進行稽查,12 月 7 日收到 TFDA 稽核報告,該公司並於 105 年 1 月 8日函覆 TFDA 缺失改善情形,TFDA 已於 105 年 2 月 16 日函知該公司,廠證有效期間延長至 108 年 1 月 23 日。
另該公司在勞工安全及環境安全業已取得合規證明如下:
(1) 勞工安全

25
公司名稱 文件名稱 說明 藥華醫藥台中分公司 - 生產廠房
建物使用執照
行政院國家學委員會中部科學工業園區管理局核發
室內裝修許可證
行政院國家學委員會中部科學工業園區管理局核發
消防安全檢查報告書
該公司委由聯亞建築物公共安全檢查有限公司並向科技部中部科學工業園區管理局申報取得核可
勞工安全衛生工作守則及人員名單
該公司分別指派取得相關核可證照員工專任職業安全衛生業務主管及職業安全衛生管理員,並向科技部中部科學工業園區管理局申報取得核可
中科管理局安全衛生檢查函文
科技部中部科學工業園區管理局每年度例行勞工安全衛生檢查,該公司並無重大缺失
危險性機器設備清單、核可文件及檢修記錄
該公司相關危險性機器設備業已取得勞動部職業安全衛生署檢查合格證,並已定期檢修
(2) 環境安全
公司名稱 文件名稱 說明 藥華醫藥台中分公司 -生產廠房
毒物化學物質許可清單
該公司業取得台中市政府環保局核發各類毒物化學物質許可清單
固定汙染源操作許可證
該公司業已取得中部科學工業園區管理局核發固定汙染源設置及操作許可證
水汙染防治許可證
該公司業已取得中部科學工業園區管理局核發水汙染防治許可證
廢棄物清理計畫書
該公司廢棄物清理計畫書業已申報中部科學工業園區管理局,並委由取得台中市政府環保局廢棄物清理許可證之廠商進行廢棄物處置
綜上評估,該公司台中分公司廠房合乎相關法令規定,無重大異常之情事。
4. 對於該公司自行設廠風險因素及因應措施合理性
(1) 無法取得藥證之風險
該公司係以自建的蛋白質藥廠生產的 P1101 進行治療真性紅血球增生症的第三期臨床試驗,已於 104 年 2 月完成總病人數收案,預計 105 年第三季將提出藥證申請,同期間亦將申請藥品上市前的查廠,若第三期臨床數據顯示主要療效

26
指標為有效且生產藥廠也通過歐盟 EMA 查廠,則可如期取得藥證,因此能否如期取得該藥證,取決於第三期臨床數據的結果及該藥廠是否能通過 EMA 查廠要求,茲將該公司因應此風險因素之措施評估說明如下:
(a) P1101 治療真性紅血球增生症第三期臨床試驗結果不如預期的風險
i. 干擾素已被醫界用於治療真性紅血球增生症,故 P1101 第三期臨床數據不如預期可能性較低:干擾素藥物一直以來都被認為能夠治癒血液疾病,但是缺點為副作用過大難以被病人接受。該公司 P1101 改良了目前干擾素藥物副作用大而導致劑量難以調升的缺點,達到低副作用、安全且劑量調整幅度大的功效,使治療效果更加提高,故該公司 P1101 在開發風險上較全新藥物(NCE/New chemical entity )低,且 P1101用於治療真性紅血球增生症已於100年及101年分別通過歐盟 EMA 及美國 FDA 的孤兒藥認證,其藥證審查更為迅速,使產品上市核准的可能性提高。
ii. 以 P1101 為新產品開發平台,使研發投入效益極大,降低單一產品市場風險:
由於醫學界已證實許多重大疾病的發生與干擾素失衡所造成的免疫系統無法正常運作有關,故該公司以 P1101 為平台持續進行開發其他適應症的臨床試驗,例如血小板增生症(ET)、慢性骨髓細胞性白血病(CML)、原發性骨髓纖維化(PMF)及 B 型/C 型肝炎等,使開發 P1101 的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模。
(b) 蛋白質藥廠未能通過歐盟 EMA 審查(查廠)的風險
該公司蛋白質藥廠為 TFDA 認證合格的 GMP 藥廠(TFDA 於 102 年 1 月查廠,4 月核發 GMP 廠證),代表該藥廠在廠房設計、製造流程管理、品質文件系統及人員訓練皆已具備法規要求水準,另經查閱該公司回覆TFDA 於 104 年 11 月進行的週期性稽查缺失事項及改善情形,亦無大或嚴重缺失,且皆有適當之改善措施。此外,該公司為因應查廠作業,已於 103 年 9 月至 12 月進行 4 批次連續成功的確效批次生產,而為確保順利通過歐盟 EMA 的審查,該公司亦聘請相關領域之專業顧問團隊,進行駐廠輔導,並已擬定具體準備工作計劃(第一階段在 105 年 4 月前完成系統改善,第二階段在 7 月前完成狀態確認,第三階段在 8 月前進行演練完成查廠準備)。綜上評估,該公司蛋白質藥廠本已具備 TFDA GMP藥廠資格,且持續提升營運管理,而對於歐盟查廠亦已擬妥具體工作計畫,故通過歐盟 EMA 查廠應屬可期。
綜上評估,該公司對於無法取得藥證之風險所採取之因應措施應尚屬合理。

27
(2) 承租廠房未能續租之因應措施
該公司向均豪公司(其主要業務為液晶面板相關設備商)承租廠房,經檢視廠房租賃契約書,於廠房租賃契約條款第二條第二項內容提及「該公司若擬續租,該公司擁有優先續租權」,依此條款內容,故該公司於租約到期時擁有優先續租權利;又以目前面板產業供過於需的情形於短期內尚無改變的環境下,均豪公司收回該出租廠房自用的可能性不高,且該公司承租廠區土地為中科管理局所有,均豪公司僅為廠房所有權者,故依據「中部科學工業園區自建廠房轉租審核原則」第一條說明,如均豪公司擬將其於本約土地上所興建之建築物轉租、轉借或轉讓予他人,應事先報經中科管理局同意。惟轉租、轉借或轉讓之對象應以經核准設立之園區事業為限,而該公司亦是中科管理局合格進駐廠商,故承租權益係受中科管理局保障。
另該公司考量台中廠房租約係於 106 年 4 月 30 日到期,屆時若租期無法續約致廠房被收回,勢必影響該公司營運發展計畫,故該公司採取以下因應方式:
A. 現有租約到期與出租人商談續約條件,爭取長期承租或承購可能性。
B. 藉由進入 IPO 資本市場增加籌資管道,強化財務實力,提高購買或自建廠房的實力。
C. 該公司以現有租約到期日前一年與出租人商談續約條件,無論現有廠房是否能完成續約或承購,基於長期發展性因素考量,該公司都將積極尋找合適廠址,以購地或租地(科學園區)自建廠房或購買既有廠房後改建之方式,設立自有廠房。經評估由於該公司已有自建蛋白質廠經驗且亦已取得 GMP 廠證,目前正積極規劃準備歐盟EMA 產品上市前查廠作業,已累積豐富的蛋白質藥廠營運經驗,故其未來增建或搬遷新廠計劃對於目前營運尚不致造成重大影響。
綜上評估,該公司為落實新藥研究發展及生產製造及行銷一條龍的營運模式而自行設立蛋白質藥廠,並配合肝炎藥物生產需求而規劃增建充填產線有其必要性,對於廠房若不能續約而被收回的風險,亦已採取可行的因應措施,尚屬合理。
3. C 型肝炎市場規模之成長主要來自不含干擾素(IFN-Free)之小分子口服用藥,面對干擾素在 C 型肝炎用藥市占率之下降,本公司所採因應措施之說明,暨推薦證券商之評估意見。
公司說明:
依據專業健康醫療資訊技術服務公司 Decision Resources 之預測,隨著創新的高價藥物推出,C 肝藥物在主要醫藥市場可能從 99 年的17 億美元,在 104 年急遽成長到 160 億美元,顯見其龐大之市場需

28
求潛力,然小分子口服新藥每顆用藥高達千元美金,一個療程下來藥費高達 8 至 9 萬美元,高額的醫藥費將造成病人或健保無法負擔,此外,目前這些口服用藥除了對 HCV G1a 及 HCV G1b 有效,並非對所有其他的 HCV 基因型都有效。其中美國 HCV 病人大都是屬於原先長效型干擾素最沒有效的HCV G1a(根據奇美醫學中心病理臨床病理科資料顯示只有 40%左右的病人會被治癒),另外歐洲 HCV 病人則大都是屬於療效稍為好些的 HCV G1b,但也只能達到 50%左右。反觀亞洲的 HCV 病人,雖然也都是 HCV G1b 病人,但在臨床實驗結果發現,使用傳統長效型干擾素其治癒率竟然可以達到 80%以上,剛開始歐美醫界對這報告不以為然,後經研究發現,原因是和歐美人比起來,亞洲的 HCV 病人幸運的少了困擾歐美醫界的一個突變的基因,即後來才發現的 IL28b。結論是傳統長效型干擾素在亞洲 HCV基因第一型的病人還是相當有效的 (80%以上的病人會被治癒,Clinical Liver Disease, Vol 5, No1, Jan 2015),只是仍存有副作用及療程偏長等情形。此外,將傳統長效型干擾素使用在另一個基因體型的HCV G2 (Genotype 2,其中台灣及韓國約佔一半 HCV 病人數,據估算兩國需要治療病人共約近百萬人),因為療程只要 6 個月,再加上由台灣醫生作出領先全世界的發現,傳統長效型干擾素治療率達 80%,故此對本公司新藥 P1101 而言,絕對是最好切入的一塊市場,因為P1101 已改良到每兩個禮拜施打一次,而且在臨床二期已證實 P1101的副作用較傳統長效型干擾素一個禮拜施打一次要減少很多,病人接受度也高,將來售價將和羅氏的傳統長效型干擾素來作性價比,P1101有機會成為在台灣及韓國兩國肝病醫生在治療HCV G2病人的最優先選擇。
簡言之,P1101 在 HCV G2 應用上有其特殊的地域性,因 Gilead的小分子用藥 Solvadi 治療 C 肝的效果可達 90%,但是藥價昂貴只能賣到歐、美、日等高所得國家,對多數低所得國家較負擔不起,如亞洲國家,但因使用長效型干擾素在 HCV G2 也有將近 80%的治療,其療程又短(半年)、治癒率也高,價格比 Solvadi 低很多,再加上它的副作用又少,將來取得藥證應該難度相對較少;因此本公司 P1101將先以亞洲市場訂為行銷目標的主打市場。
推薦證券商說明:
依據 Decision Resources 之預測,隨著創新的高價小分子口服藥物推出,C 肝藥物在主要醫藥市場可能從 2010 年的 17 億美元,在2015 年急遽成長到 160 億美元,然考量其療程藥費高達 8 至 9 萬美元,故該公司將市場鎖定於亞洲地區,根據統計,台灣 C 型肝炎的盛行率為 4.4%,其中第二型基因型的患者佔 39.5% ,總病患數(目標市場)約為 40 萬人。韓國方面,C 型肝炎的盛行率為 1.3% (約等同於62 萬 5 千名病患),基因型 2 型患者佔 45%,總病患數(目標市場)為28 萬人,故其目標市場總病患人數約 68 萬人,整體而言,該公司將P1101 適用於 C 型肝炎基因體第二型之市場目標設定在盛行率高之亞洲市場,縱其新藥價格不及小分子口服新藥,然亦有其市場性,該公司所採之因應措施應屬合理。
(二) 特別記載事項乙節

29
1. 推薦證券商對該公司業績變化合理性及未來發展性之評估
藥華醫藥股份有限公司(以下簡稱藥華醫藥或該公司)主要專注於新藥研發、生產及銷售,其最近二年度及 104 年前三季之業績變化情形如下表,有關該公司業績變化之合理性及未來發展性為何?經洽推薦證券商評估如后:
單位:新臺幣千元
年度
項目
102 年度 103 年度 104 年前三季
金額 % 金額 % 金額 %
營業收入(淨額) 72,327 100.00 13,356 100.00 8,165 100.00
營業成本 5,086 7.03 5,988 44.83 18,400 225.35
營業毛利(損) 67,241 92.97 7,368 55.17 (10,235) (125.35)
營業費用 668,312 924.01 870,840 6,520.22 562,013 6,883.20
營業淨損 (601,071) (831.04) (863,472) (6,465.05) (572,248) (7,008.55)
營業外收入及支出 7,921 10.95 10,366 77.61 3,908 47.86
稅前淨損
歸屬於母
公司權益 (593,150) (820.10) (853,106) (6,387.44) (568,340) (6,960.69)
歸屬於非
控制權益 - - - - - -
所得稅費用 - - - - - -
本期淨損 (593,150) (820.10) (853,106) (6,387.44) (568,340) (6,960.69)
期末資本額 1,877,310 1,891,822 1,899,542
每股稅後淨損(元)
追溯前(註 1) (3.40) (4.54) (3.00)
追溯後(註 2) (3.12) (4.49) (3.00) 資料來源:該公司各期間經會計師查核簽證或核閱之合併財務報告。 註 1:係以當年度加權平均流通在外股數計算之基本每股稅後淨損(潛在普通股具反稀釋
作用)。 註 2:係以 104 年 9 月底股數 189,954 千股為基準往前追溯調整之基本每股稅後淨損。
(一) 該公司所屬行業之產業概況及所營業務之主要內容
1. 該公司所屬行業之產業概況
藥華醫藥係以自行研發的 PEG(聚乙二醇)技術平台及小分子合成藥物技術為基礎,進行研究及開發蛋白質新藥的公司,屬生技製藥產業(產品屬生物製劑 Biologics)。依其經營策略,其短期產品發展計畫主要聚焦於目前尚無合適治療方式的罕見疾病(孤兒藥)及慢性肝炎市場,以縮短新藥審查時程。該公司自行研發的新一代(Biobetter)長效型干擾素(Pegylated interferon α-2b,產品代號為P1101)已分別於100年及101年通過歐洲藥物管理局(下稱EMA)及美國食品藥物管理局(下稱 FDA)於治療真性紅血球增生症(Polycythemia Vera,下稱 PV)的孤兒藥認證,並藉由跨國研發合作的模式,授權奧地利專業孤兒藥開發公司 AOP Orphan Pharmaceuticals AG(下稱 AOP 公司)在歐洲進行治療 PV 的臨床試驗,其第二期臨床試驗結果已證實 P1101 較目前市面上產品(干擾素)具有更高的純度及較低的副作用,且第三期人體臨床試驗已於104 年 2 月完成病人總數收案,預計 105 年第三季整理臨床數據完
30
成後即可向 EMA 申請藥證。
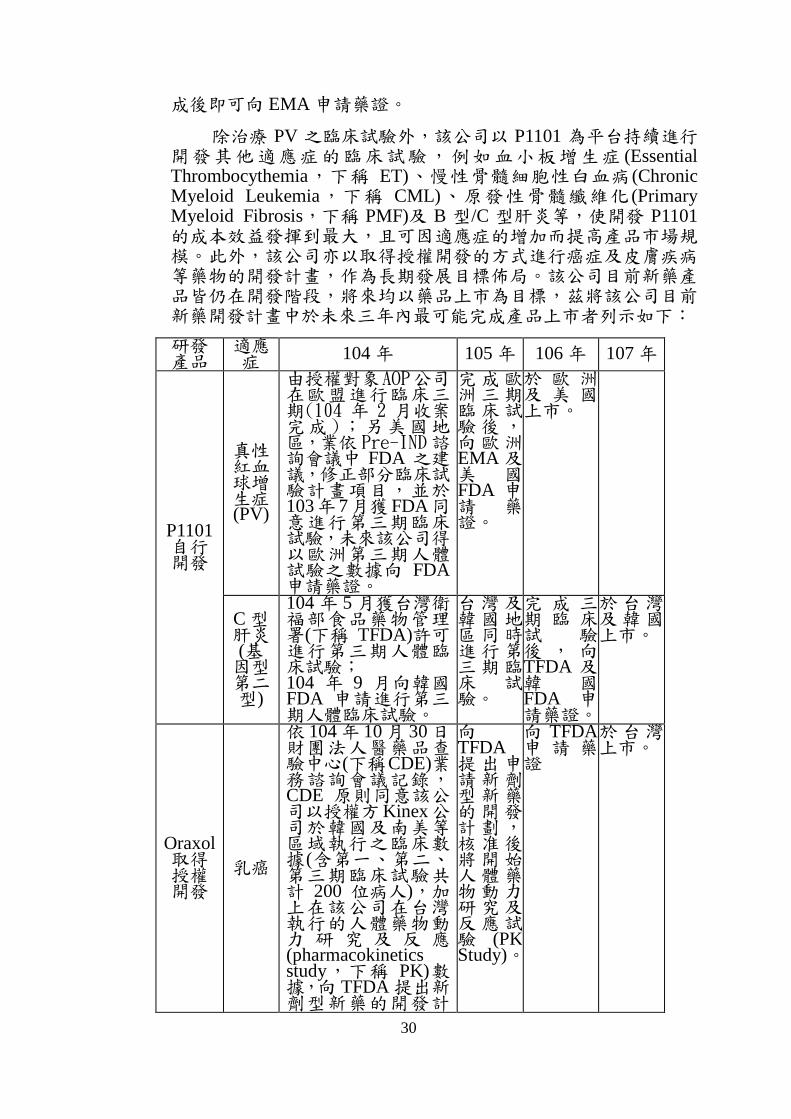
除治療 PV 之臨床試驗外,該公司以 P1101 為平台持續進行開發其他適應症的臨床試驗,例如血小板增生症 (Essential Thrombocythemia,下稱 ET)、慢性骨髓細胞性白血病(Chronic Myeloid Leukemia,下稱 CML)、原發性骨髓纖維化 (Primary Myeloid Fibrosis,下稱 PMF)及 B 型/C 型肝炎等,使開發 P1101的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模。此外,該公司亦以取得授權開發的方式進行癌症及皮膚疾病等藥物的開發計畫,作為長期發展目標佈局。該公司目前新藥產品皆仍在開發階段,將來均以藥品上市為目標,茲將該公司目前新藥開發計畫中於未來三年內最可能完成產品上市者列示如下:
研發 產品
適應症 104 年 105 年 106 年 107 年
P1101 自行 開發
真性 紅血球增生症(PV)
由授權對象AOP公司在歐盟進行臨床三期(104 年 2 月收案完成 );另美國地區,業依 Pre-IND諮詢會議中 FDA 之建議,修正部分臨床試驗計畫項目,並於103年 7月獲FDA同意進行第三期臨床試驗,未來該公司得以歐洲第三期人體試驗之數據向 FDA申請藥證。
完成歐洲三期臨床試驗後,向歐洲EMA 及美 國FDA 申請 藥證。
於 歐 洲及 美 國上市。
C 型肝炎(基因型第二型)
104 年 5 月獲台灣衛福部食品藥物管理署(下稱 TFDA)許可進行第三期人體臨床試驗; 104 年 9 月向韓國FDA 申請進行第三期人體臨床試驗。
台灣及韓國地區同時進行第三期臨床 試驗。
完 成 三期 臨 床試 驗後 , 向TFDA 及韓 國FDA 申請藥證。
於台灣及韓國上市。
Oraxol 取得 授權 開發
乳癌
依 104 年 10 月 30 日財團法人醫藥品查驗中心(下稱CDE)業務諮詢會議記錄,CDE 原則同意該公司以授權方Kinex公司於韓國及南美等區域執行之臨床數據(含第一、第二、第三期臨床試驗共計 200 位病人),加上在該公司在台灣執行的人體藥物動力 研 究 及 反 應(pharmacokinetics study,下稱 PK)數據,向 TFDA 提出新劑型新藥的開發計
向TFDA提出申請新劑型新藥的開發計劃,核准後將開始人體藥物動力研究及反應試驗 (PK Study)。
向 TFDA申 請 藥證
於台灣上市。

31
研發 產品
適應症 104 年 105 年 106 年 107 年
劃。目前正由 Kinex公司在南美洲進行第三期臨床試驗。
生物製劑(Biologics)係以生物為來源(由人體、植物、動物或微生物之來源所衍生),或利用新生物技術(例如基因工程、融合瘤技術等)所開發的產品,包括經由基因工程或細胞培養技術製造而得的蛋白質、胜肽及其衍生物,用於治療或預防人類疾病,目前多應用於治療貧血、癌症、自體免疫性疾病等。生物製劑因具高水溶性,因此較易被製成各種液態藥劑,如:口服液及針劑等。
生物製劑由於具有專一辨識性,相較於小分子化學合成藥物,生物製劑在治療上較不會有系統性毒性問題,而因生產技術困難,大分子藥物的價格約是傳統化學治療藥物價格的數倍至數十倍,因此在各大藥廠積極開發下,使全球生物藥品發展非常快速,依美國專業醫藥資訊廠商 IMS Health 統計,93 年生物藥品占全球整體藥品市場約 10.7%,至 103 年已占 17.1%,市場規模約達 1,808億美元,較 102 年成長 11.6%,且全球研發中的生物新藥將持續取得藥證上市,預估 103 至 109 年複合年成長率約可達 11.9%,市場規模約達 3,560 億美元。
由於生物藥品本質上係以生物為來源所開發的藥品,故在原適應症產品研發成功取得上市核准後,仍可以該原研發產品尋找致病機轉相似的其他適應症疾病,經各期臨床試驗後申請藥品查驗登記,取得另一適應症的藥證許可,使藥品銷售額因銷售區域及擴充的適應症增加而提高。 藥品療效類別之銷售額排名方面,依據 IMS Health 統計資料,102年全球生物藥品中以免疫疾病、糖尿病及癌症為主要療效類別,合計約占市場 50%比重,且糖尿病及免疫疾病至 107 年成長率仍可達 10%以上,而預估至 107 年銷售額成長率最高的療效類別藥物為發炎疾病,年複合成長率約可達 45%,其次為眼科產品。 在核准上市新藥方面,生技藥品於 82~102 年平均每年約有 4 件取得上市核准,而 103 年取得美國藥品評估暨研究中心(CDER)批准上市許可的生技藥品數量共有 11 件,其中有 7 件(63.6%)為以罕見疾病條件通過核准之生物製劑申請(Biological License Application,下稱 BLA)藥品,而以罕見疾病藥物資格申請上市許可之藥物中有4 件為治療癌症的抗體藥物,且多以加速審查方式通過;此外,美國生物製劑評估暨研究中心(CBER)核准上市之治療用生物藥品亦有 10 件,顯示生技藥品在 103 年呈現大幅增加的榮景。而由前述CDER 核准的罕見疾病用藥數量觀之,可預期未來開發癌症的抗體藥物以罕見疾病用藥資格上市將是未來藥廠採取的常用策略。依美國 FDA 統計資料,103 年 FDA 所核准上市的新藥有 41 件,其中 11 件為生技藥品,相較於 89 年只核准 2 件生技藥品大幅增加,且比重也由 89 年 6.9%提高至 103 年 26.83%,由於生物技術研發持續突破,生物藥品預防及治療疾病的應用也越來越廣泛,預期生物藥品占整體藥品市場比重將越來越高。

32
以下茲就該公司目前新藥開發計畫中於未來三年內最可能完成產品上市者,說明其市場概況:
(1) P1101 治療 PV PV 為一種骨髓細胞增生疾病,使患者紅血球、白血球及血
小板異常增加,為罕見血液疾病,其疾病症狀發展緩慢,有時好幾年都沒有明顯症狀,發病病人以老年人居多。因為紅血球過多,血液黏稠,血液在某些組織流動緩慢而無法供應足夠的氧氣,會造成病人頭痛、暈眩、感覺虛弱、呼吸困難等症狀。嚴重的會造成脾臟腫大、血栓,增加中風危險。目前尚未發現有效治療方法,僅能靠定期放血來維持正常血液濃度,不僅不便,還可能因為重複放血引發其他併發症,如血小板過多症等,嚴重影響病人生活品質。
罕見疾病指的就是盛行率低、少見、患病人數稀少疾病的疾病;各國對罕見疾病的定義均不相同。台灣罕病法對於罕見疾病的定義為:盛行率在萬分之一以下、遺傳性及診治的困難性,以上述三項指標為參考原則;歐洲定義一萬人中少於五個人患病,則為罕見疾病;在美國只要有少於 20 萬人患病,則歸類此疾病為罕見疾病。依世界衛生組織(WTO)罕見疾病定義為病患占總人口的 0.65-1‰之間的疾病或病變。目前全球已確認的罕見病有 7000~8000 種,約占人類疾病之 10%,而目前僅有約 1%的罕見病能夠有效治療。罕見疾病各有不同的發病過程與疾病特徵,常常是極為嚴重或是威脅到生命的。例如骨髓系增生疾病(Myeloproliferative neoplasms, MPNs)是骨髓系幹細胞發生異常變化,使得其分化的血液細胞發生過度增生所造成的一種疾病,典型的骨髓增生性血液疾病包括 PV、ET、CML 和PMF。目前對於罕見疾病僅能以藥物控制疾病症狀而無法治癒,而隨著每年新增的病患人數使用藥需求亦逐年提高,依據生技製藥產業預測機構 EvaluatePharma 預估,全球孤兒藥市場規模 104 至 109 年將以 11.7%的年複合成長率增加,至 109 年約可達美金 1,800 億元;另依遺傳疾病家庭手冊(Genetics Home Reference)與歐洲 Orphanet 期刊統計,美國及歐盟分別約有 19.3萬及 13.3 萬 PV 病患,合計約達 32.6 萬人,若以 PV 患病人數及每位病人醫療費用計算(假設以目前 PV 口服用藥 Jakafi 藥價(一個月用藥 60 顆售價美金 10,618.44 元),一年用藥費用達美金 10 萬元)估算,每年潛在市場規模將超過美金 100 億元。
(2) P1101 治療 C 型肝炎(基因型第二型) 病毒的各種基因型就如人類有不同人種,具有不同的特
性,地理分佈也有差異。現行的治療方法中,C 肝被根治的成功率高達 6 成以上,但治療成敗的關鍵之一,就是病毒的基因型,一旦患者感染某些較難纏的病毒基因型時,治療時程必須延長,才能提升治癒率。目前已知的 C 肝病毒基因型有 6 種,台灣以第一型與第二型病毒為多。C 肝病毒基因型攸關治療成效,第一型及第四型病毒通常較難治,第二型與第三型則相對好治,然 C 型肝炎疫苗仍無有效的治療方法,因此慢性 C 型肝炎未來將成為慢性肝病之主要原因。

33
依世界衛生組織(WHO)100年統計全球每年約有300至400萬人感染 C 型肝炎病毒,約有 1.3 至 1.7 億人口患有慢性 C 型肝炎,其中中國大陸約有 4,000 萬個慢性 C 肝帶原者,台灣約30 萬個病患,加上初期 C 肝感染多數無明顯症狀,且 70~80%的感染會轉變成慢性病,故檢測率低,因此實際感染的患者人數可能比上述統計數字為高,可見 C 型肝炎感染已成為全球公共衛生上之重要議題。惟目前尚未有較佳的治療藥物,因此市場對於療效佳且副作用低的 C 肝藥物潛在需求非常高。目前治療 C 型肝炎之研發方向為直接抗病毒藥物與長效型干擾素及雷巴威林(Ribavirin)合併治療,或者不同直接抗病毒藥物的組合治療而完全避免干擾素。依據專業健康醫療資訊技術服務公司Decision Resources 之預測,隨著創新的高價藥物推出,C 肝藥物在主要醫藥市場可能從 99 年的 17 億美元,在 104 年急遽成長到 160 億美元,顯見其龐大之市場需求潛力。
(3) Oraxol 治療乳癌 Oraxol 為口服劑型的紫杉醇(Paclitaxel,太平洋紫杉醇)。
紫杉醇為細胞有絲分裂抑制劑,被廣泛應用於治療晚期卵巢癌、乳癌、非小細胞及小細胞肺癌等多種癌症,紫杉醇自 81年上市至今,累計銷售額已超過 200 億美元。目前紫杉醇主要以靜脈注射方式給藥,且需要較高劑量才能在病人體內停留足夠時間達成療效,但是高劑量紫杉醇常會造成副作用,如貧血、白血球低下引發感染、神經痛等。
該公司於 102 年 12 月取得美國 Kinex Pharmaceuticals, LLC(下稱 Kinex 公司)獨家授權在台灣和新加坡進行開發Oraxol® (口服紫杉醇)的權利,其適應症主要係用以治療乳癌、肺癌及卵巢癌。目前該公司已規劃採該藥品由授權方於韓國及南美洲等區域進行之二期/三期臨床數據,搭配該公司於台灣執行之人體藥物動力檢驗(PK)數據,向 TFDA 提出新劑型新藥的開發計劃。依據 IMS Health 統計資料,103 年全球治療乳癌暢銷藥物 Herceptin(賀癌平)的銷售金額達美金 55.6 億元,較 102年增加 5.8%,顯示乳癌藥物的市場需求仍持續穩定成長,故該公司開發 Oraxol 治療乳癌應具市場發展性。
2. 該公司所營業務之主要內容 藥華醫藥專注於新藥研發、生產及銷售,目前開發中主要產
品 P1101 由合作夥伴 AOP 公司於歐洲進行治療 PV 的第三期人體臨床試驗,由於該公司新藥產品尚未核准上市銷售,故最近二年度及 104 年前三季之營業收入來源主要為臨床試驗用藥品收入(帳列研究收入)、里程碑金收入(Milestone Payment)及商品銷售收入,列示說明如下:
產品項目
重要用途及功能 102 年度 103 年度 104 年前三季
營收 淨額
% 營收 淨額
% 營收 淨額
%
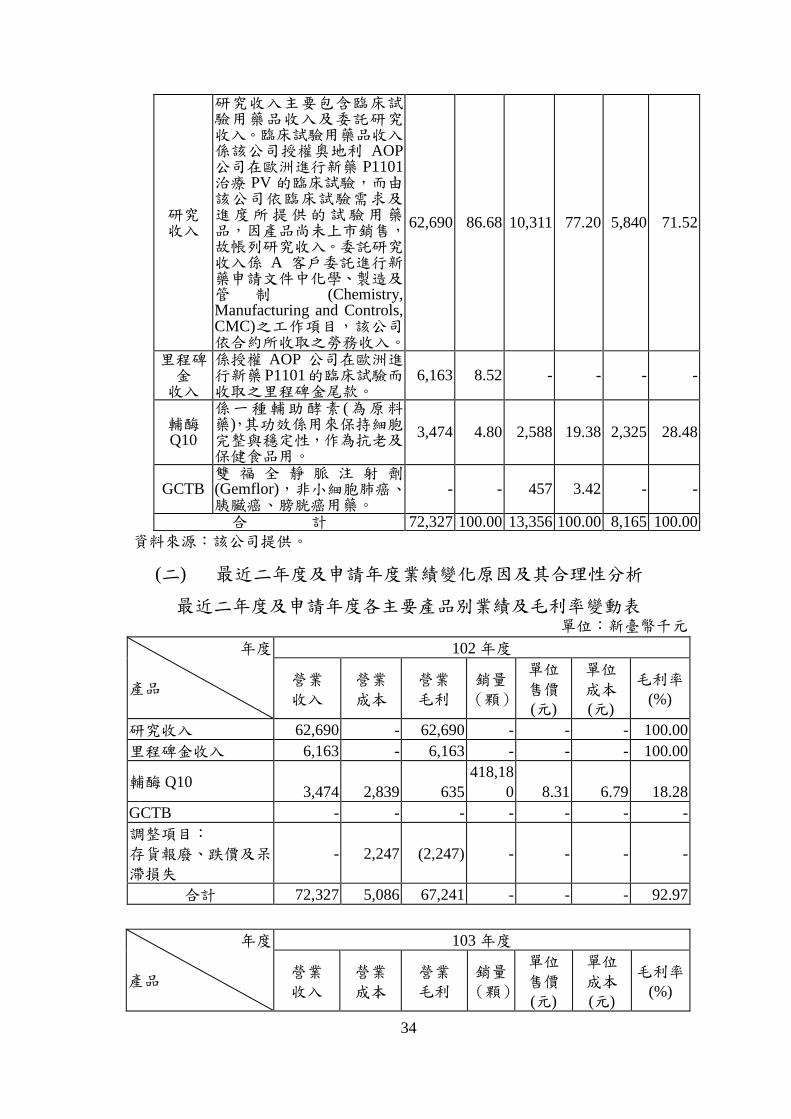
34
研究 收入
研究收入主要包含臨床試驗用藥品收入及委託研究收入。臨床試驗用藥品收入係該公司授權奧地利 AOP公司在歐洲進行新藥 P1101治療 PV 的臨床試驗,而由該公司依臨床試驗需求及進度所提供的試驗用藥品,因產品尚未上市銷售,故帳列研究收入。委託研究收入係 A 客戶委託進行新藥申請文件中化學、製造及管 制 (Chemistry, Manufacturing and Controls, CMC)之工作項目,該公司依合約所收取之勞務收入。
62,690 86.68 10,311 77.20 5,840 71.52
里程碑金 收入
係授權 AOP 公司在歐洲進行新藥P1101的臨床試驗而收取之里程碑金尾款。
6,163 8.52 - - - -
輔酶Q10
係一種輔助酵素 (為原料藥),其功效係用來保持細胞完整與穩定性,作為抗老及保健食品用。
3,474 4.80 2,588 19.38 2,325 28.48
GCTB 雙 福 全 靜 脈 注 射 劑(Gemflor),非小細胞肺癌、胰臟癌、膀胱癌用藥。
- - 457 3.42 - -
合 計 72,327 100.00 13,356 100.00 8,165 100.00
資料來源:該公司提供。
(二) 最近二年度及申請年度業績變化原因及其合理性分析
最近二年度及申請年度各主要產品別業績及毛利率變動表 單位:新臺幣千元
年度
產品
102 年度
營業
收入
營業
成本
營業
毛利
銷量
(顆)
單位
售價(元)
單位
成本(元)
毛利率(%)
研究收入 62,690 - 62,690 - - - 100.00
里程碑金收入 6,163 - 6,163 - - - 100.00
輔酶 Q10 3,474 2,839 635
418,18
0 8.31 6.79 18.28
GCTB - - - - - - -
調整項目:
存貨報廢、跌價及呆滯損失
- 2,247 (2,247) - - - -
合計 72,327 5,086 67,241 - - - 92.97
年度
產品
103 年度
營業
收入
營業
成本
營業
毛利
銷量
(顆)
單位
售價(元)
單位
成本(元)
毛利率(%)
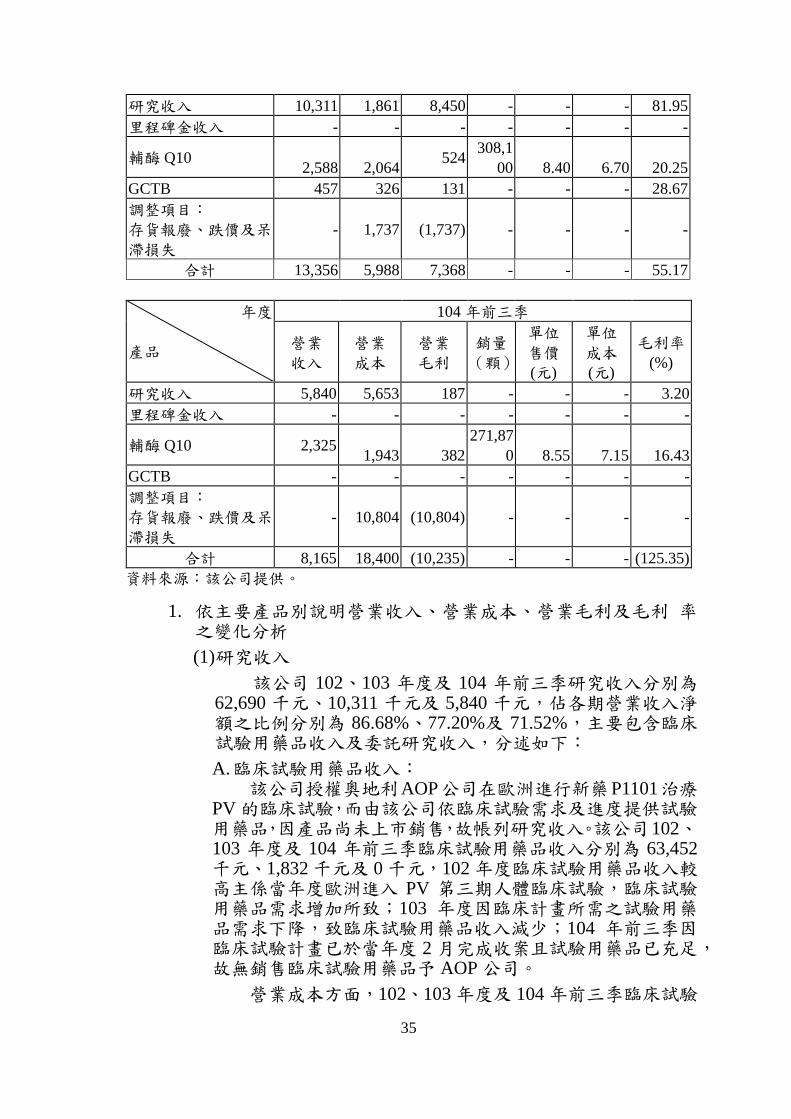
35
研究收入 10,311 1,861 8,450 - - - 81.95
里程碑金收入 - - - - - - -
輔酶 Q10 2,588 2,064
524 308,1
00 8.40 6.70 20.25
GCTB 457 326 131 - - - 28.67
調整項目:
存貨報廢、跌價及呆滯損失
- 1,737 (1,737) - - - -
合計 13,356 5,988 7,368 - - - 55.17
年度
產品
104 年前三季
營業
收入
營業
成本
營業
毛利
銷量
(顆)
單位
售價(元)
單位
成本(元)
毛利率
(%)
研究收入 5,840 5,653 187 - - - 3.20
里程碑金收入 - - - - - - -
輔酶 Q10 2,325 1,943 382
271,87
0 8.55 7.15 16.43
GCTB - - - - - - -
調整項目:
存貨報廢、跌價及呆滯損失
- 10,804 (10,804) - - - -
合計 8,165 18,400 (10,235) - - - (125.35)
資料來源:該公司提供。
1. 依主要產品別說明營業收入、營業成本、營業毛利及毛利 率之變化分析
(1) 研究收入
該公司 102、103 年度及 104 年前三季研究收入分別為62,690 千元、10,311 千元及 5,840 千元,佔各期營業收入淨額之比例分別為 86.68%、77.20%及 71.52%,主要包含臨床試驗用藥品收入及委託研究收入,分述如下:
A. 臨床試驗用藥品收入: 該公司授權奧地利AOP公司在歐洲進行新藥P1101治療
PV 的臨床試驗,而由該公司依臨床試驗需求及進度提供試驗用藥品,因產品尚未上市銷售,故帳列研究收入。該公司102、103 年度及 104 年前三季臨床試驗用藥品收入分別為 63,452千元、1,832 千元及 0 千元,102 年度臨床試驗用藥品收入較高主係當年度歐洲進入 PV 第三期人體臨床試驗,臨床試驗用藥品需求增加所致;103 年度因臨床計畫所需之試驗用藥品需求下降,致臨床試驗用藥品收入減少;104 年前三季因臨床試驗計畫已於當年度 2 月完成收案且試驗用藥品已充足,故無銷售臨床試驗用藥品予 AOP 公司。
營業成本方面,102、103 年度及 104 年前三季臨床試驗

36
用藥品收入之營業成本均為 0 千元,毛利率均為 100%,主係臨床試驗用藥品生產之成本已於早期認列費用,無相對應成本。
B. 委託研究收入:
該公司 102、103 年度及 104 年前三季委託研究收入分別為(762)千元、8,479 千元及 5,840 千元,102 年度收入呈現負值主要係認列以前年度之研究收入折讓所致;103 年度及 104年前三季委託研究收入係來自A客戶委託進行新藥申請文件中 有 關 新 成 分 新 藥 化 學 、 製 造 與 管 制 (Chemistry, Manufacturing and Controls, CMC)之工作項目,其內容包含製程放大開發、分析方法開發及確效、產品與不純物之鑑定及準備 CMC 文件等,該公司依合約收取勞務收入。
營業成本方面,102、103 年度及 104 年前三季委託研究收入之營業成本分別為 0 千元、1,861 千元及 5,653 千元;營業毛利分別為(762)千元、6,618 千元及 187 千元。102 年度受認列研究收入折讓影響呈現負毛利;103 年度及 104 年前三季該公司考量 P1101 進入第三期臨床試驗最後階段及其他產品開發計畫之人力需求,故經 A 客戶同意後,將原部分委託事項轉由 Pharmaron Inc.承接(Pharmaron Inc.為 CRO 公司,業務涵蓋新藥研發臨床前的全流程,包括化學、生物、藥物代謝及動力學、藥理、毒理等各領域),致營業成本提高,毛利率下降。
(2) 里程碑金收入
該公司 102 年度認列里程碑金收入 6,163 千元係授權AOP 公司在歐洲進行新藥 P1101 的臨床試驗而收取之里程碑金收入尾款,佔該年度營業收入淨額之比例為 8.52%,該授權合約之里程碑金總額為 200 萬歐元,截至 102 年已全數收取完畢,該公司對 AOP 公司之授權內容請詳「(二)、2.主要銷售對象變化分析」說明。
營業成本方面,因相關成本已於早期認列為費用,故無相對應成本,致營業毛利為 100%。
(3) 輔酶 Q10
輔酶(Q10)係一種輔助酵素(為原料藥,即藥品中具有醫療效用的基本成份, Active Pharmaceutical Ingredients, API),其功效係用來保持細胞完整與穩定性,作為抗老及保健食品用,係該公司創立初期開發並商品化完成之產品,非發展重心,然因市場需求穩定且產品生命週期較長,故截至目前仍有小量銷售。該公司 102、103 年度及 104 年前三季輔酶 Q10銷貨收入分別為 3,474 千元、2,588 千元及 2,325 千元,佔各年度營業收入淨額之比例分別為 4.80%、19.38%及 28.48%。102 年度銷售金額較高主係經營寵物保健食品之 B 客戶對輔酶 Q10 之採購金額上升所致;103 年度及 104 年前三季銷售金額變動不大,尚無重大異常。

37
營業成本方面,102、103 年度及 104 年前三季輔酶 Q10銷貨成本分別為 2,839 千元、2,064 千元及 1,943 千元,營業毛利分別為 635 千元、524 千元及 382 千元,營業毛利率分別為 18.28%、20.25%及 16.43%。各期毛利率變動主要受毛利較低之 B 客戶之銷貨金額增減影響,尚無重大異常。
(4) 抗癌藥 Gemcitabine (下稱 GCTB)
該公司於 95 年 7 月以雙福全靜脈注射劑(Gemflor)取得衛生署藥品許可證,並於 99 年 11 月取得製劑原料(GCTB)的藥品許可證,然因衛生署於 96 年 12 月 19 日公告「西藥製劑製造工廠實施 GMP 標準(PIC/S GMP)之時程」,規定癌症藥品之製造廠須符合 PIC/S GMP 規範,故該公司對 GCTB則需由專用凍晶注射劑生產廠進行生產,而該公司對 GCTB係採委外生產模式,原委託製造廠因無法符合 PIC/S 規範,致無法將 GCTB 原料藥製成成品,該公司考量委託國外廠商生產成本過高,目前國內符合資格且可配合生產之廠商又不易尋求,致該項產品目前尚未投入生產,103 年度小量銷售予友華生技醫藥(股)公司作為研究使用,佔該年度營業淨額3.42%。
GCTB 產品係該公司 99 年開發成功的製劑藥物原料,由於製造成本及生產廠商資格的限制,該公司並未將 GCTB列為重點發展產品,故僅 103 年度銷售予客戶作為研究使用,該年度營業成本為 326 千元,營業毛利率為 28.67%。
(5) 存貨報廢、跌價及呆滯等損失
該公司 102、103 年度及 104 年前三季提列存貨報廢、跌價及呆滯等金額分別為 2,247 千元、1,737 千元及 10,804千元。102 年度存貨報廢、跌價及呆滯等損失主要來自輔酶Q10 膠囊商品報廢損失;103 年度係依該公司存貨庫齡政策提列 GCTB 產品之備抵跌價及呆滯損失;104 年前三季金額較 102 及 103 年度為高,主係該公司於 104 年 6 月底考量GCTB 藥證即將到期,在評估過往去化速度及市場性後,已於 104 年 6 月底將 GCTB 存貨提列 100%之備抵呆滯及跌價損失。
2. 主要銷售對象變化分析
最近二年度及申請年度前十大客戶變動表
單位:新臺幣千元
102 年度 103 年度 104 年前三季
客戶名稱 銷售額 % 客戶名稱 銷售額 % 客戶名稱 銷售額 %
1 AOP 69,615 96.25 A 客戶 8,479 63.48 A 客戶 5,840 71.52
2 B 客戶 1,587 2.19 AOP 1,832 13.72 B 客戶 1,291 15.81
3 自然人 540 0.75 B 客戶 1,031 7.72 自然人 360 4.41
4 自然人 342 0.47 自然人 570 4.27 自然人 176 2.16
5 榮新診所 191 0.27 友華生技 457 3.42 榮新診所 88 1.08
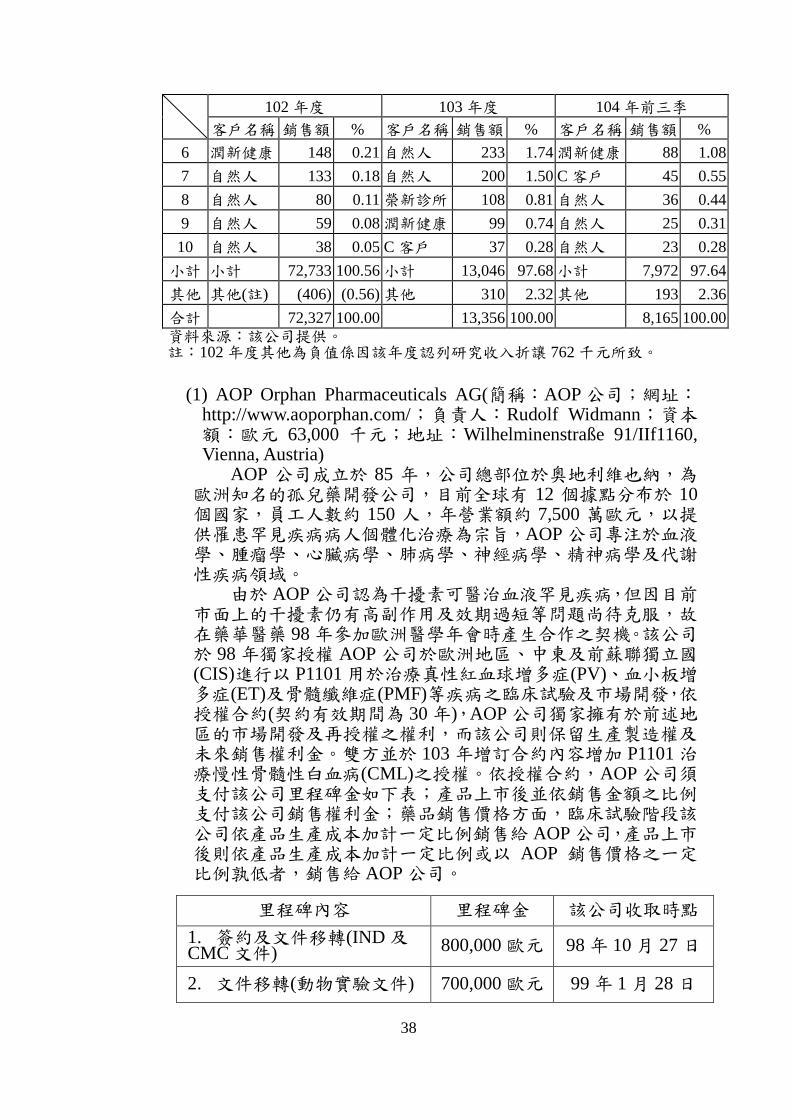
38
102 年度 103 年度 104 年前三季
客戶名稱 銷售額 % 客戶名稱 銷售額 % 客戶名稱 銷售額 %
6 潤新健康 148 0.21 自然人 233 1.74 潤新健康 88 1.08
7 自然人 133 0.18 自然人 200 1.50 C 客戶 45 0.55
8 自然人 80 0.11 榮新診所 108 0.81 自然人 36 0.44
9 自然人 59 0.08 潤新健康 99 0.74 自然人 25 0.31
10 自然人 38 0.05 C 客戶 37 0.28 自然人 23 0.28
小計 小計 72,733 100.56 小計 13,046 97.68 小計 7,972 97.64
其他 其他(註) (406) (0.56) 其他 310 2.32 其他 193 2.36
合計 72,327 100.00 13,356 100.00 8,165 100.00
資料來源:該公司提供。 註:102 年度其他為負值係因該年度認列研究收入折讓 762 千元所致。
(1) AOP Orphan Pharmaceuticals AG(簡稱:AOP 公司;網址:http://www.aoporphan.com/;負責人:Rudolf Widmann;資本額:歐元 63,000 千元;地址:Wilhelminenstraße 91/IIf1160, Vienna, Austria)
AOP 公司成立於 85 年,公司總部位於奧地利維也納,為歐洲知名的孤兒藥開發公司,目前全球有 12 個據點分布於 10個國家,員工人數約 150 人,年營業額約 7,500 萬歐元,以提供罹患罕見疾病病人個體化治療為宗旨,AOP 公司專注於血液學、腫瘤學、心臟病學、肺病學、神經病學、精神病學及代謝性疾病領域。
由於 AOP 公司認為干擾素可醫治血液罕見疾病,但因目前市面上的干擾素仍有高副作用及效期過短等問題尚待克服,故在藥華醫藥 98 年參加歐洲醫學年會時產生合作之契機。該公司於 98 年獨家授權 AOP 公司於歐洲地區、中東及前蘇聯獨立國(CIS)進行以 P1101 用於治療真性紅血球增多症(PV)、血小板增多症(ET)及骨髓纖維症(PMF)等疾病之臨床試驗及市場開發,依授權合約(契約有效期間為 30 年),AOP 公司獨家擁有於前述地區的市場開發及再授權之權利,而該公司則保留生產製造權及未來銷售權利金。雙方並於 103 年增訂合約內容增加 P1101 治療慢性骨髓性白血病(CML)之授權。依授權合約,AOP 公司須支付該公司里程碑金如下表;產品上市後並依銷售金額之比例支付該公司銷售權利金;藥品銷售價格方面,臨床試驗階段該公司依產品生產成本加計一定比例銷售給 AOP 公司,產品上市後則依產品生產成本加計一定比例或以 AOP 銷售價格之一定比例孰低者,銷售給 AOP 公司。
里程碑內容 里程碑金 該公司收取時點
1. 簽約及文件移轉(IND 及CMC 文件) 800,000 歐元 98 年 10 月 27 日
2. 文件移轉(動物實驗文件) 700,000 歐元 99 年 1 月 28 日

39
3. 第一期臨床試驗 PK 數據及研究報告,與該臨床試驗執行之相關文件交付
350,000 歐元 99 年 12 月 9 日
4. AOP公司執行第二期臨床試驗有正面結果 150,000 歐元 102 年 12 月 31 日
資料來源:該公司與 AOP 公司於 98 年 9 月 1 日簽訂之授權合約。
另依生產製造合約,P1101 產品核准上市後,AOP 公司須提供年度需求計畫(Yearly Rolling Forecast),並每三個月更新一次,供該公司做為生產排程之參考,且該需求計畫應作為未來採購單之部分文件,若實際採購數量低於該需求計畫數量,則AOP 公司應就差異數量計算差額支付該公司。
最近二年度該公司對 AOP 公司主要為銷售臨床試驗用藥品(因產品尚未上市銷售,故帳列研究收入)及收取里程碑金,其銷貨金額分別為 69,615 千元及 1,832 千元。102 年度銷貨金額較高係因 AOP 公司開始在歐洲進行 P1101 用於治療 PV 第三期人體臨床試驗,臨床試驗用藥品銷售增加,同年該公司依據合約收取里程碑金尾款(金額 6,163 千元),故營業收入大幅增加;103年度因臨床試驗用藥品需求有限,致該年度銷售金額下降;104 年前三季因無新增臨床試驗用藥需求,故無臨床試驗用藥品銷售。
(2) A 客戶 A 客戶成立於 99 年,為國內上市公司中國石油化學工業開
發(股)公司(股票代號:1314)之轉投資公司,A 客戶主要從事與癌症相關的新藥研發。 藥華醫藥於 102 年受 A 客戶委託進行 CMC 準備工作項目,工作項目依合約訂定之收款時程共分為 14 項,合約總價款為17,762 千元,分別於工作項目完成之 7~30 天內收款。該公司分別於 103 年度及 104 年前三季認列相關委託研究收入 8,479 千元及 5,840 千元。
(3) B 客戶 B 客戶自 92 年為專業寵物營養品代理商,目前代理來自
英、美、日等國與台灣本地各類型寵物營養品。藥華醫藥受 B客戶之委託研發寵物食用之 Q10 配方,B 客戶下單後,由藥華醫藥提供配方及原物料,B 客戶提供包材,藥華醫藥再委外進行客製化生產製造。
該公司最近二年度及 104 年前三季銷售寵物食用之 Q10 金額分別為 1,587 千元、1,031 千元及 1,291 千元,由於 Q10 非該公司主要產品,故未對該產品進行推廣行銷,102 年度銷貨金額增加係受到 B 客戶需求增加所致。
(4) 榮新診所(網址:http://www.reshining-clinic.com.tw/;負責人:張茂松;地址:台北巿大安區建國南路二段 276 號 9 樓)
榮新診所係提供健康照護及健康管理服務之醫療院所。 該公司最近二年度及 104 年前三季銷售輔酶 Q10 予榮新診
所,銷售金額分別為 191 千元、108 千元及 88 千元,由於 Q10

40
非該公司主要產品,故未對該產品進行推廣行銷,銷貨金額增減主要係受到榮新診所需求變化所致。
(5) 潤新健康股份有限公司 ( 簡稱:潤新健康;網址:http://www.ruenshin.com.tw/;負責人:陳明村;資本額:10,000千元;地址:台北巿大安區建國南路二段 276 號 5 樓)
潤新健康成立於 100 年,係銷售醫美保養品,提供美療療程之健康管理公司。
該公司最近二年度及 104 年前三季銷售輔酶 Q10 予潤新健康,銷售金額分別為 148 千元、99 千元及 88 千元,由於 Q10非該公司主要產品,故未對該產品進行推廣行銷,銷貨金額增減主要係受到潤新健康需求變化所致。
(6) 友華生技醫藥股份有限公司(簡稱:友華;股票代號:4120;網址:http://www.oep.com.tw/;負責人:蔡正弘;資本額:867,471千元;地址:台北市復興南路一段 368 號 7 樓)
友華成立於 71 年,擁有西藥產品、腫瘤用藥、醫學美容品、嬰幼兒營養保健及成人營養保健等事業,並成立子公司專注於新藥之研發製造,係垂直整合藥物研發、臨床試驗、製造及藥品銷售之跨國性藥廠。
103 年度該公司將 GCTB 原料藥小量銷售予友華作為研究使用,金額共計 457 千元,佔該年度營業淨額 3.42%。由於 GCTB非該公司營業重心,故 103 年度後未再進行 GCTB 銷售。
(7) C 客戶 C客戶成立於99年,專長在保健食品的研發、製造及銷售,
並成立「GCO 購物網」,網羅台灣在地本土保健食品,涵蓋生技、製藥、食品各領域的優質廠商,代理的產品以美妝保養品、健康食品、保健食品及營養補充品為主。
該公司 103 年度及 104 年前三季銷售輔酶 Q10 予 C 客戶,銷售金額分別為 37 千元及 45 千元,由於 Q10 非該公司主要產品,故未對該產品進行推廣行銷,銷貨金額減少主要係受到 C客戶終端需求變化所致。
(8) 自然人 輔酶 Q10 為健康食品,其銷售對象有自然人,主要係客戶
透過公司網站訊息及客戶介紹而直接向公司購買,包含該公司管理階層(董事、監察人及總經理)、員工、股東及一般消費者等,針對自然人客戶不另進行個別銷售對象分析。
3. 最近二年度及申請年度與二家同業財務報告損益資料分析比較 最近二年度及最近期申請公司與同業之營業收入及毛利率一覽表
單位:新臺幣千元
公司名稱
年度 項目
102 年度 103 年度 104 年前三季 金額 % 金額 % 金額 %
藥華醫藥
營業收入淨額 72,327 100.00 13,356
100.00 8,165 100.00
營業成本 5,086 7.03 5,988 44.83 18,400 225.35
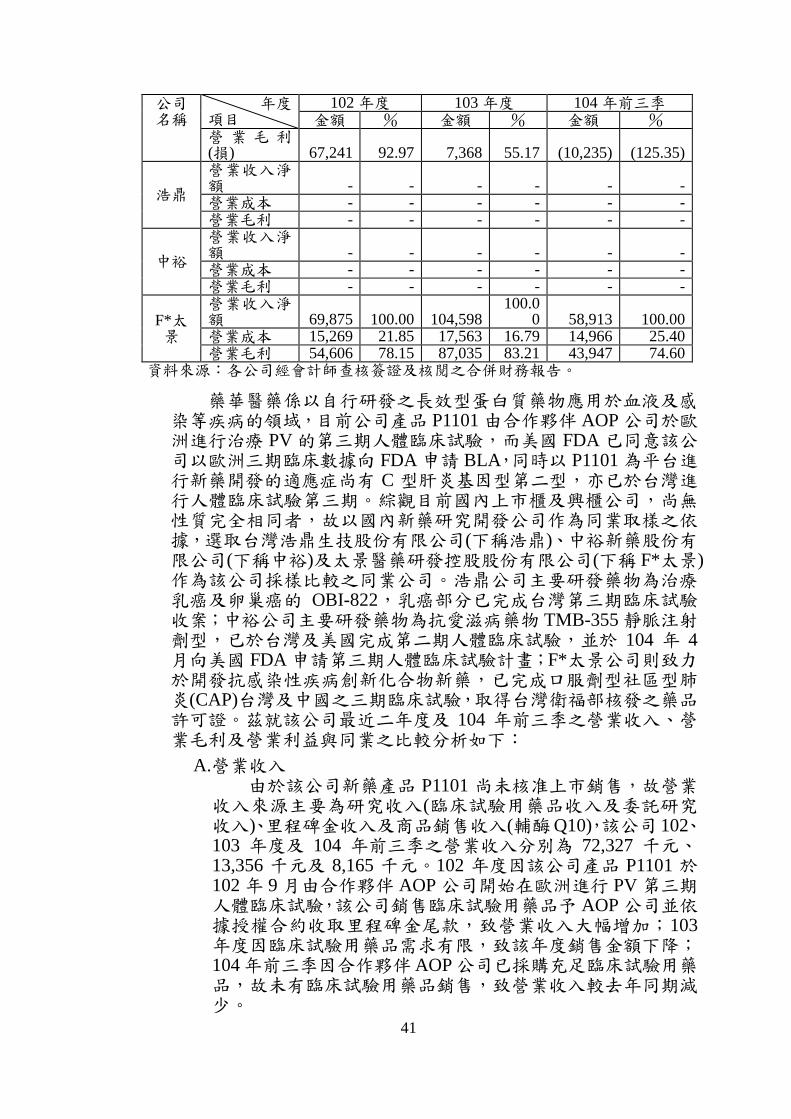
41
公司名稱
年度 項目
102 年度 103 年度 104 年前三季 金額 % 金額 % 金額 %
營 業 毛 利(損) 67,241 92.97 7,368 55.17 (10,235) (125.35)
浩鼎
營業收入淨額 - - - - - - 營業成本 - - - - - - 營業毛利 - - - - - -
中裕
營業收入淨額 - - - - - - 營業成本 - - - - - - 營業毛利 - - - - - -
F*太景
營業收入淨額 69,875 100.00 104,598
100.00 58,913 100.00
營業成本 15,269 21.85 17,563 16.79 14,966 25.40 營業毛利 54,606 78.15 87,035 83.21 43,947 74.60
資料來源:各公司經會計師查核簽證及核閱之合併財務報告。
藥華醫藥係以自行研發之長效型蛋白質藥物應用於血液及感染等疾病的領域,目前公司產品 P1101 由合作夥伴 AOP 公司於歐洲進行治療 PV 的第三期人體臨床試驗,而美國 FDA 已同意該公司以歐洲三期臨床數據向 FDA 申請 BLA,同時以 P1101 為平台進行新藥開發的適應症尚有 C 型肝炎基因型第二型,亦已於台灣進行人體臨床試驗第三期。綜觀目前國內上市櫃及興櫃公司,尚無性質完全相同者,故以國內新藥研究開發公司作為同業取樣之依據,選取台灣浩鼎生技股份有限公司(下稱浩鼎)、中裕新藥股份有限公司(下稱中裕)及太景醫藥研發控股股份有限公司(下稱 F*太景)作為該公司採樣比較之同業公司。浩鼎公司主要研發藥物為治療乳癌及卵巢癌的 OBI-822,乳癌部分已完成台灣第三期臨床試驗收案;中裕公司主要研發藥物為抗愛滋病藥物 TMB-355 靜脈注射劑型,已於台灣及美國完成第二期人體臨床試驗,並於 104 年 4月向美國 FDA 申請第三期人體臨床試驗計畫;F*太景公司則致力於開發抗感染性疾病創新化合物新藥,已完成口服劑型社區型肺炎(CAP)台灣及中國之三期臨床試驗,取得台灣衛福部核發之藥品許可證。茲就該公司最近二年度及 104 年前三季之營業收入、營業毛利及營業利益與同業之比較分析如下:
A.營業收入 由於該公司新藥產品 P1101 尚未核准上市銷售,故營業
收入來源主要為研究收入(臨床試驗用藥品收入及委託研究收入)、里程碑金收入及商品銷售收入(輔酶Q10),該公司 102、103 年度及 104 年前三季之營業收入分別為 72,327 千元、13,356 千元及 8,165 千元。102 年度因該公司產品 P1101 於102 年 9 月由合作夥伴 AOP 公司開始在歐洲進行 PV 第三期人體臨床試驗,該公司銷售臨床試驗用藥品予 AOP 公司並依據授權合約收取里程碑金尾款,致營業收入大幅增加;103年度因臨床試驗用藥品需求有限,致該年度銷售金額下降;104 年前三季因合作夥伴 AOP 公司已採購充足臨床試驗用藥品,故未有臨床試驗用藥品銷售,致營業收入較去年同期減少。

42
與採樣同業相較,同業浩鼎及中裕產品尚在研發階段,尚未產生營業收入;F*太景主要收入來源為研發中藥物授權之簽約金及諮詢服務收入,營業收入呈現逐年成長係 F*太景分別於 101 年 6 月及 103 年 1 月簽訂授權合約所致;藥華醫藥除98年將P1101用於血液相關疾病的歐洲市場獨家銷售權授權予 AOP 公司外,尚未有其他授權計畫,加以主要產品P1101 仍在進行人體臨床試驗,故 103 及 104 年前三季營收未有成長。整體而言,該公司主要產品 P1101 尚在新藥研發階段,故 102、103 年度及 104 年前三季營業收入變化主要係受臨床試驗進度影響,尚無重大異常。
B.營業成本及營業毛利 該公司 102、103 年度及 104 年前三季之營業成本分別為
5,086 千元、5,988 千元及 18,400 千元,營業毛利分別為 67,241千元、7,368 千元及(10,235)千元,營業成本主要來自輔酶 Q10之銷貨成本、存貨報廢、跌價及呆滯損失與勞務成本。102及 103 年度因臨床試驗用藥品收入及里程碑金收入等成本已於前期認列費用,故無相對應成本;104 年前三季因提列跌價及呆滯損失 10,804 千元,致當期呈現負毛利率。
與採樣同業相較,同業浩鼎及中裕產品尚在研發階段,尚未產生營業毛利;F*太景之營業毛利則因簽訂授權合約隨營業收入成長而增加。整體而言,該公司 102、103 年度及 104 年前三季營業毛利變動情形與同業相較,主要仍因各公司新藥研發進度及業務模式而有所差異,尚無重大異常。
4. 營業費用及營業利益變化原因及其合理性分析 單位:新臺幣千元
年度 項目
102 年度 103 年度 104 年前三季
金額 佔營收比率
金額 佔營收比
率 金額
佔營收比率
推銷費用 3,722 5.15 854 6.39 2,007 24.58 管理費用 134,951 186.58 104,704 783.95 82,273 1,007.63 研 究 發 展費用 529,639 732.28 765,282 5,729.88 477,733 5,850.99 營 業 費 用合計 668,312 924.01 870,840 6,520.22 562,013 6,883.20 營業淨損 (601,071) (831.05) (863,472) (6,465.05) (572,248) (7,008.55) 資料來源:該公司各期間經會計師查核簽證或核閱之合併財務報告。
(1) 推銷費用 該公司推銷費用主要包括旅費及廣告費等,102、103 年度
及 104 年前三季推銷費用分別為 3,722 千元、854 千元及 2,007千元,推銷費用佔營業收入比重分別為 5.15%、6.39%及 24.58%。102 年度金額較高主要係因 102 年度該公司派員參與罕見血液疾病研討會,旅費支出增加所致;103 年度及 104 年前三季推銷費用主要用於參加生技展所支付之相關廣告費用,尚無重大異常。 (2) 管理費用
該公司管理費用主要包括薪資費用、勞務費、租金支出、

43
旅費、保險費、折舊及各項攤提等,102、103 年度及 104 年前三季管理費用分別為 134,951千元、104,704千元及 82,273千元。102 年度管理費用較 103 年度高,係因該公司於 102 年度發行員工認股權並認列相關費用 82,825 千元所致; 104 年前三季管理費用與 103 年同期相當,尚無重大異常。
單位:新臺幣千元 年度
項目 102 年度 103 年度 104 年前三季
金額 佔管理費用比率
金額 佔管理費用比率
金額 佔管理費用比率
薪資費用 一般薪資費用 23,928 17.73 23,547 22.49 25,781 31.34 員工認股權費用 82,825 61.37 41,855 39.97 13,087 15.91 限制型員工權利新股(註)
- -
- -
4,908 5.96
小計 106,753 79.10 65,402 62.46 43,776 53.21 勞務費 11,263 8.35 11,377 10.87 9,184 11.16 其他 16,935 12.55 27,925 26.67 29,313 35.63 管理費用合計 134,951 100.00 104,704 100.00 82,273 100.00
資料來源:該公司各期間經會計師查核簽證或核閱之合併財務報告及該公司提供。 註:該公司考量限制員工權利新股於 104 年 7 月 8 日經向證期局申報生效,雖尚未
發放,然部分績效條件已達成,未來依該辦法發行限制型新股予員工之可能性極高,該公司認為對該等員工負有相當給付義務、很有可能需要流出經濟效益之資源以清償該義務,擬於取得證期局申報生效作為發行該辦法之意圖明確確立時點,以最適估計(單位數*財務報導日之公允價值)推估已達標之單位數作為應有負債數之會計處理係符合 IAS 37「負債準備、或有負債及或有資產之會計處理」公報規定,而非以「IFRS 2 股份基礎給付會計處理」認列相關酬勞成本。另依據 IAS1.69 之規定,該公司預期將於報導期間後 12 個月內到期清償該負債,故將其帳列其他流動負債-其他項下,未來此負債將於限制型新股發行時予以沖銷之。
(3) 研究發展費用 該公司研發費用主要包括薪資費用、委託研究費、耗材試
劑、臨床試驗費用、勞務費及折舊費用等,102、103 年度及 104年前三季研究費用分別為 529,639 千元、765,282 千元及 477,733千元。103 年度較 102 年度增加 235,643 千元,主要係因該公司於 103 年度配合 P1101 歐洲臨床試驗進度,於台中廠進行 P1101批次生產確效,並著手開發 P1101 未來上市後的產品設計(筆型劑型注射器 PEN/Cartridge)所致,其中 103 年度認列 AOP 公司開發筆型注射器之委託研究費用 131,104 千元(包含筆殼開發費用、內部 Cartridge 設計、充填測試、確效及產品安定性測試等費用),耗材部分則增加 34,113 千元,另臨床研究費用亦因該公司於台灣積極展開產品 P1101 用於適應症 B 型肝炎及 C 型肝炎之人體臨床試驗,致相關金額較 102 年度增加 41,111 千元,勞務費部分因該公司認列 P1101適用於 PV及ET之美國第三期人體臨床試驗 IND 及臨床試驗設計等諮詢費用,與口服新藥Oratecan 及 Oraxol 新藥 IND 及產品開發等諮詢故費用增加28,451 千元;104 年前三季因委託研究費支出減少,致當期研發費用較 103 年同期下降 84,558 千元。經比較各期研發費用變動原因尚無重大異常。
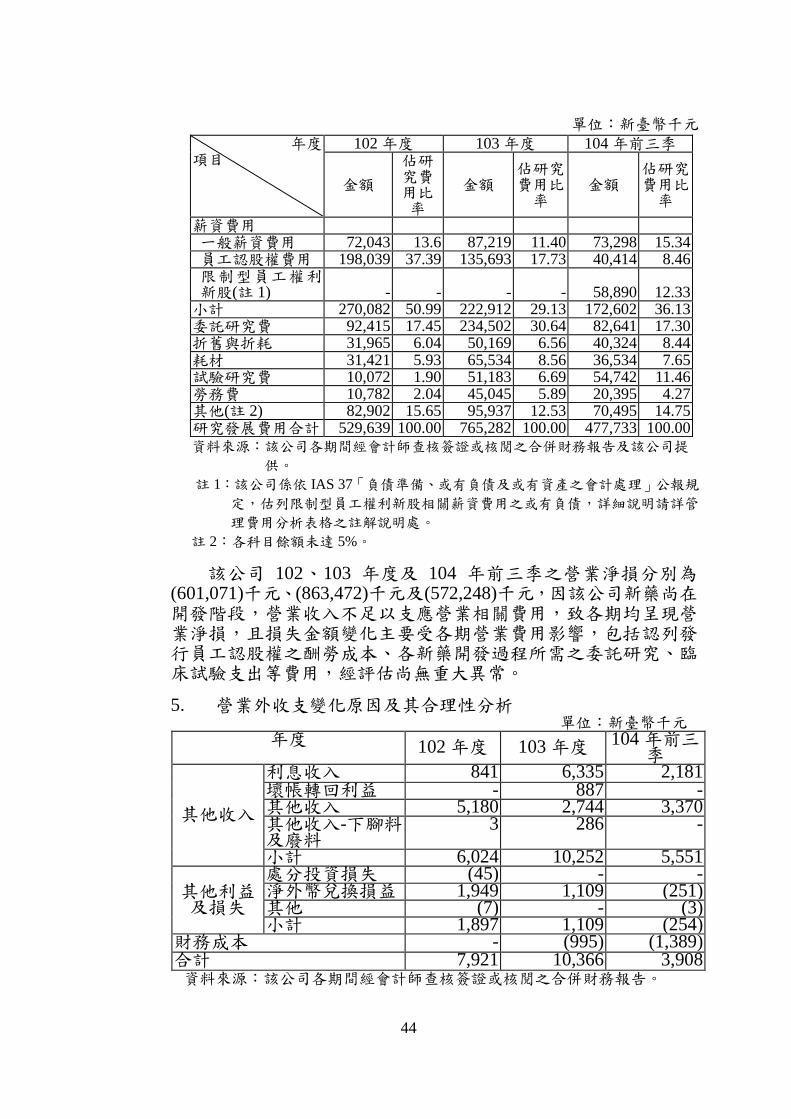
44
單位:新臺幣千元
年度 項目
102 年度 103 年度 104 年前三季
金額
佔研究費用比率
金額 佔研究費用比率
金額 佔研究費用比率
薪資費用 一般薪資費用 72,043 13.6 87,219 11.40 73,298 15.34 員工認股權費用 198,039 37.39 135,693 17.73 40,414 8.46 限制型員工權利新股(註 1) - - - - 58,890 12.33 小計 270,082 50.99 222,912 29.13 172,602 36.13 委託研究費 92,415 17.45 234,502 30.64 82,641 17.30 折舊與折耗 31,965 6.04 50,169 6.56 40,324 8.44 耗材 31,421 5.93 65,534 8.56 36,534 7.65 試驗研究費 10,072 1.90 51,183 6.69 54,742 11.46 勞務費 10,782 2.04 45,045 5.89 20,395 4.27 其他(註 2) 82,902 15.65 95,937 12.53 70,495 14.75 研究發展費用合計 529,639 100.00 765,282 100.00 477,733 100.00 資料來源:該公司各期間經會計師查核簽證或核閱之合併財務報告及該公司提
供。
註 1:該公司係依 IAS 37「負債準備、或有負債及或有資產之會計處理」公報規
定,估列限制型員工權利新股相關薪資費用之或有負債,詳細說明請詳管
理費用分析表格之註解說明處。
註 2:各科目餘額未達 5%。
該公司 102、103 年度及 104 年前三季之營業淨損分別為(601,071)千元、(863,472)千元及(572,248)千元,因該公司新藥尚在開發階段,營業收入不足以支應營業相關費用,致各期均呈現營業淨損,且損失金額變化主要受各期營業費用影響,包括認列發行員工認股權之酬勞成本、各新藥開發過程所需之委託研究、臨床試驗支出等費用,經評估尚無重大異常。
5. 營業外收支變化原因及其合理性分析 單位:新臺幣千元
年度 102 年度 103 年度 104 年前三季
其他收入
利息收入 841 6,335 2,181 壞帳轉回利益 - 887 - 其他收入 5,180 2,744 3,370 其他收入-下腳料及廢料
3 286 -
小計 6,024 10,252 5,551
其他利益 及損失
處分投資損失 (45) - - 淨外幣兌換損益 1,949 1,109 (251) 其他 (7) - (3) 小計 1,897 1,109 (254)
財務成本 - (995) (1,389) 合計 7,921 10,366 3,908 資料來源:該公司各期間經會計師查核簽證或核閱之合併財務報告。

45
(1) 其他收入
該公司 102、103 年度及 104 年前三季之其他收入分別為 6,024千元、10,252千元及 5,551千元,茲就各細項分述如下:
A. 利息收入
該公司銀行活期存款及定期存款所產生之利息收入,其中 103 年度利息收入較 102 年度及 104 年前三季為高係因該公司於 102 年下半年度現金增資共募集 1,866,000 千元致銀行存款大幅增加。
B. 壞帳轉回利益
該公司於 103 年度將溢提之備抵呆帳予以迴轉,故認列壞帳轉回利益 887 千元。
C. 其他收入
該公司其他收入主要係政府單位之補助收入,102、103年度及 104 年前三季之其他收入分別為 5,180 千元、2,744千元及 3,370 千元,其中政府單位之補助收入分別為 3,707千元、2,744 千元及 3,296 千元。102 年度金額較高係因當年度獲得台北生技獎補助收入增加所致。
D. 其他收入-下腳料及廢料
該公司其他收入-下腳料及廢料主要係出售試產時不適用之物料,102 及 103 年度之其他收入-下腳料及廢料分別為 3 千元及 286 千元。
(2) 其他利益及損失
該公司 102、103 年度及 104 年前三季之其他利益及損失分別為 1,897 千元、1,109 千元及(254)千元,茲就各細項分述如下:
A. 處分投資損失
該公司於102年度認列轉投資公司藥華生技(股)公司及立陽生技(股)公司清算後之投資損失,上開轉投資公司背景簡述如下:
(A) 藥華生技(股)公司係該公司於 93 年 1 月 9 日經董事會通過設立,主要是為申請進駐中央研究院基因體研究中心的南港育成中心。依當時育成中心規定,該公司並不符合進駐條件(須為專責研發之新公司或子公司),故另成立藥華生技(股)公司申請進駐(實收資本額為 3,000 千元,該公司投資 2,000 千元)。嗣後經該公司與中央研究院協商,自95 年 9 月起獲准由該公司直接租賃使用,故藥華生技(股)公司已無實質的運作需要,為節省行政管理費用,該公司董事會於 102 年 7 月 23 日決議解散清算藥華生技(股)公司,並於 102 年 11 月 19 日完成清算退回現金股款 654千元,總計認列投資損失 1,346 千元(含 102 年度投資損失 28 千元)。
(B) 該公司希望在不出資之原則下,尋找合作夥伴推動GCTB 及 CPTB (均為抗癌藥物之原料藥)之生產,故該公

46
司於 100 年 12 月 8 日董事會決議,以技術作價入股之方式,與上市公司立隆電子工業(股)公司之轉投資公司台灣慧聚(股)公司共同成立立陽生技(股)公司。該公司原預計授權上開二項研發技術,並以 70,000 千元分階段技術作價入股取得立陽生技(股)公司約 1/3 股權,惟因對方對產品原料來源一直無法掌握,而影響產品開發進度,故雙方決定先辦理減資後解散立陽生技(股)公司,由該公司取回原授權之技術,並於 102 年 11 月 15 日完成立陽生技(股)公司之清算退回現金股款 239 千元,總計認列投資損失61 千元(含 102 年度投資損失 17 千元)。。
B. 淨外幣兌換損益
102、103 年度及 104 年前三季兌換(損)益分別為 1,949千元、1,109 千元及(251)千元,該公司外幣兌換損益主要來自於支付設備款或委託研究費用等,淨外幣兌換利益逐年減少主要受到新臺幣自 102 年起走貶影響。
(3) 財務成本
該公司 103 年度及 104 年前三季利息支出分別為 995 千元及 1,389 千元,104 年前三季利息支出增加主要係因該公司為充實營運資金增加銀行短期借款所致。

47
(三) 該公司未來發展性之評估
1. 該公司新藥開發之營運策略
該公司係以自行研發的 PEG 技術平台及小分子合成技術為基礎,進行研發蛋白質新藥的公司,其蛋白質原料藥藥廠已於 102 年 4 月通過衛福部評鑑為 GMP 藥廠,為國內極少數自主研發及生產製造的蛋白質藥廠,其經營理念係以自有核心技術統整新藥研發、生產製造及銷售一條龍之營運模式,期望透過完整的垂直整合打造真正研發、製造在臺灣,臨床試驗及銷售與歐美及國際接軌的新藥產品。茲將其新藥開發之營運策略說明如下:
(1) 核心技術方面:自主核心技術,確保長期產品發展能力
該公司以自行研發的「聚乙二醇高分子(PEG)與蛋白質偶合技術」(改良長效型高分子(PEG)的定點連接方法),開發出高專一度連接特定胺基酸的專屬 linker,並在干擾素 α的 N 端上加入胺基酸( Proline),進行偶合反應,成功製造出高純度(95%以上為單一成分)的新一代 PEG 長效型干擾素藥物(PEG-IFN-α2b),即該公司新藥「P1101」,達到延長藥效時間(即延長注射間隔)及低副作用的目的。
聚乙二醇化(PEGylation)主要是為了使蛋白質藥物達到延長藥效的目的,該公司 PEG 定點連接技術可以改良長效型高分子(PEG)的定點連接方式,將 PEG 分子連接到蛋白質上特別設計的位置,發展出長效型蛋白質新藥品,故可持續開發其他長效型之蛋白質藥品,例如應用在血液疾病如 PEG-GCSF 長效型白血球生長激素、PEG-EPO 長效型紅血球生成素、PEG-FSH 長效型促卵泡激素等,及應用在感染疾病如 B 型肝炎及 C 型肝炎等。該公司上述核心技術已有專利保護,有效期間可至 123 年。
(2) 產品開發方面
A. 以 P1101 為新產品開發平台,使研發投入效益極大,並降低單一產品市場風險
醫學界已證實許多重大疾病的發生與干擾素失衡所造成的免疫系統無法正常運作有關,故該公司以 P1101 為平台持續進行開發除真性紅血球增生症(PV)以外之其他適應症的臨床試驗,例如血小板增生症(ET)、慢性骨髓細胞性白血病(CML)、原發性骨髓纖維化(PMF)及 B 型/C 型肝炎等,使開發 P1101 的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模。
此外在免疫療法方面,P1101 為新一代長效型干擾素,副作用相較於其他藥品較輕微,再加上劑量調整幅度高,使醫生可依照不同適應症或病情輕重有更大的用藥範圍,過去干擾素常因其副作用不能被病人忍受,所以在癌症治療上的應用非常有限。故該公司計畫研發 PD-1/PD-L-1 的單株抗體搭配 P1101 使用,因為其高療效和安全性,提升

48
病人免疫反應以對抗多種癌症,該公司將善用這一大優勢,擴展 P1101 的治療範圍,並計畫運用於惡性黑色素癌、T cell淋巴癌、毛細胞白血病等適應症。
B. 取得孤兒藥認證,縮短新藥上市審查時程
孤兒藥即罕見疾病用藥,是歐美等先進國家藥物管理單位所訂定的藥品特殊資格,是為了鼓勵藥廠開發小眾且困難的疾病藥物。若產品獲取孤兒藥資格,大多可享有快速通關的審查資格,未來上市後也可能享有自由定價、獨占市場等優惠條件。該公司以 P1101 用於治療 PV 已分別於100 年 101 年取得歐洲 EMA 及美國 FDA 的孤兒藥認證,且用於治療 ET 及 PMF等疾病亦於 103 年獲得美國 FDA 的孤兒藥認證,因此有助於縮短新藥上市審查時程。
C. 授權開發方式
該公司亦以取得授權開發的方式(Licensing-in)進行癌症藥物的開發計畫,增加研發中產品廣度,同時藉以培養研發人員累積相關經驗,例如向 Kinex 公司取得授權,在台灣及新加坡開發癌症口服劑型用藥 Oraxol 及 Oratecan 之專屬權利,及在大中華及東南亞開發 KX01 牛皮癬用藥的權利,此外,亦向中央研究院取得授權,透過其小分子化合物研究技術,開發地中海貧血及鐮刀型貧血的新藥。
(3) 市場開發方面:藉由跨國研發合作的模式,提高藥品及市場開發效率
該公司有鑑於蛋白質新藥研發所需投入的資源極大,且必須朝向國際市場發展才能產生效益,故以跨國研發合作的模式,將自行研發的 P1101 產品授權給奧地利 AOP 公司於歐洲進行治療 PV 的臨床試驗,一方面可收取里程碑金挹注該公司營運資金,同時並可提高產品開發效率。
(4) 生產製造方面:建造蛋白質廠,自主生產製造,實現一條龍之營運模式
該公司於 101 年 10 月完成蛋白質原料藥廠設廠,11月開始試產完成,並於 102 年 4 月獲得 TFDA GMP 廠證。目前在歐洲進行的 PV第三期人體臨床試驗所使用的 P1101藥品,即係由該公司自行生產製造。該公司已於 103 年 9月至 12 月進行 4 批次連續成功的確效批次生產,以因應歐洲EMA及美國FDA對未來原料藥商業化量產之查廠要求,該公司為國內極少數具備自主研發及製造能力的蛋白質藥廠。
綜上所述,該公司以自行研發的技術開發 P1101,解決目前干擾素藥品半衰期(Half-Life)太短且給藥後的副作用過大的問題,短期產品發展計畫主要聚焦於目前尚無合適治療方式(unmet medical need)的罕見疾病(孤兒藥)及慢性肝炎的市場,可縮短新藥審查時程;並藉由跨國研發合作的模式,將 P1101 授

49
權給 AOP 公司進行 PV 的人體臨床試驗,授權範圍包含產品開發權及未來在歐洲市場獨家銷售權,有助於提升產品開發效率及未來在歐洲市場行銷佈局;產品生產方面係以自有蛋白質藥廠生產,自主彈性調配產能及掌握品質控管;產品發展策略將以 P1101 為平台,延續開發其他適應症的治療試驗,縮短新產品的開發時程、降低研發風險及研究經費,拓展其未來業務發展在產品組合上之優勢。
2. 該公司各項新藥開發進度
105 年 106 年 107 年 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q
P1101 適應症:真性紅血球增生症(PV) 查驗登記地區:歐洲
PIII 依三期臨床統計分析結果,進行藥證申請
(新藥上市時程視 EMA 審核進度及結果而定)
P1101 適應症:真性紅血球增生症(PV) 查驗登記地區:美國
PIII 依歐洲三期臨床統計分析結果,進行藥證申請 (新藥上市時程視 FDA 審核進度及結果而定)
P1101 適應症:血小板增生症(ET) 查驗登記地區:美國
修改 臨床 計畫書
IND 申請
PIII
依 三 期臨 床 結果,進行藥 證 申請 ( 新藥上市 時 程視 FDA審 核 進度 及 結果而定)
P1101 適應症:C型肝炎基因型第一型 查驗登記地區:台灣
PII (將視 PII 結果進行規劃)
P1101 適應症:C型肝炎基因型第二型 查驗登記地區:台灣
PIII
依三期臨床統計分析結果,進行藥證申請
(新藥上市時程視衛生福利部食品藥物管理署(TFDA)審核進度及結果而
定)
P1101 適應症:C型肝炎基因型第二
IND 申請
PIII
依三期臨床統計分析結果,進行藥證申請
(新藥上市時程視韓國 FDA 審核進度及結果而定)
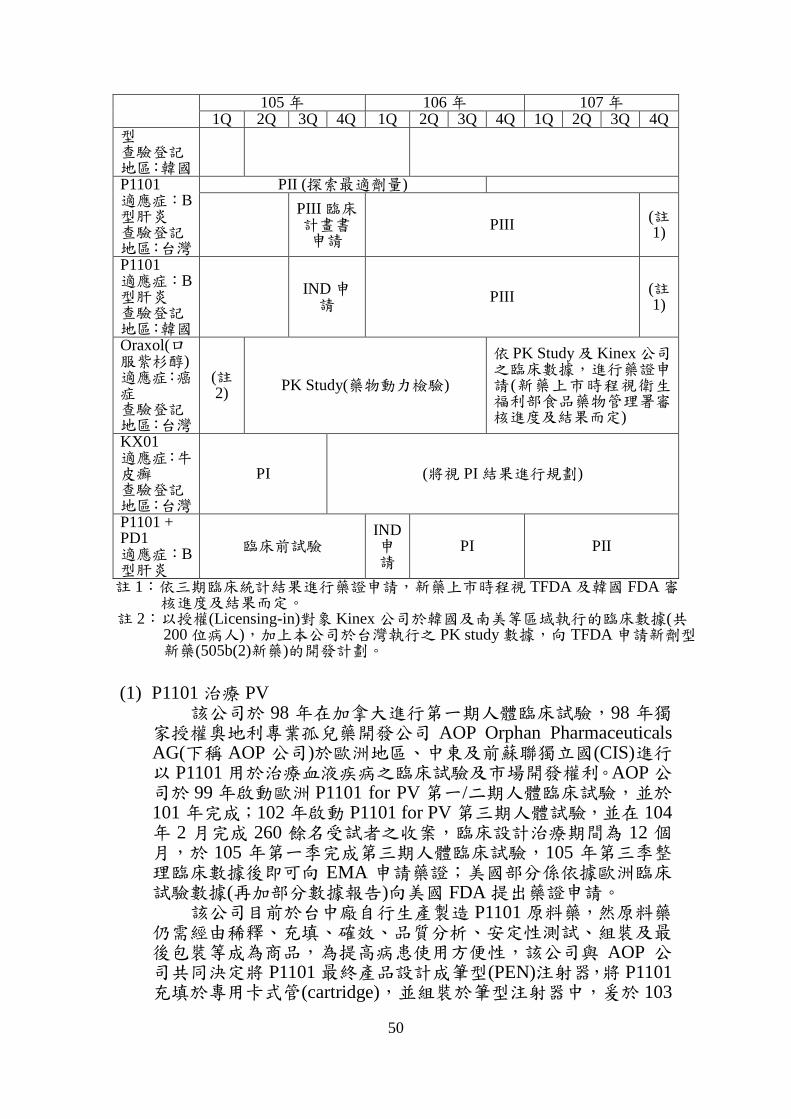
50
105 年 106 年 107 年 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q 1Q 2Q 3Q 4Q
型 查驗登記地區:韓國 P1101 適應症:B型肝炎 查驗登記地區:台灣
PII (探索最適劑量)
PIII 臨床 計畫書申請
PIII (註1)
P1101 適應症:B型肝炎 查驗登記地區:韓國
IND 申請
PIII (註1)
Oraxol(口服紫杉醇) 適應症:癌症 查驗登記地區:台灣
(註2)
PK Study(藥物動力檢驗)
依 PK Study 及 Kinex 公司之臨床數據,進行藥證申請(新藥上市時程視衛生福利部食品藥物管理署審核進度及結果而定)
KX01 適應症:牛皮癬 查驗登記地區:台灣
PI (將視 PI 結果進行規劃)
P1101 + PD1 適應症:B型肝炎
臨床前試驗 IND 申請
PI PII
註 1:依三期臨床統計結果進行藥證申請,新藥上市時程視 TFDA 及韓國 FDA 審 核進度及結果而定。
註 2:以授權(Licensing-in)對象 Kinex 公司於韓國及南美等區域執行的臨床數據(共 200 位病人),加上本公司於台灣執行之 PK study 數據,向 TFDA 申請新劑型 新藥(505b(2)新藥)的開發計劃。
(1) P1101 治療 PV 該公司於 98 年在加拿大進行第一期人體臨床試驗,98 年獨
家授權奧地利專業孤兒藥開發公司 AOP Orphan Pharmaceuticals AG(下稱 AOP 公司)於歐洲地區、中東及前蘇聯獨立國(CIS)進行以 P1101 用於治療血液疾病之臨床試驗及市場開發權利。AOP 公司於 99 年啟動歐洲 P1101 for PV 第一/二期人體臨床試驗,並於101 年完成;102 年啟動 P1101 for PV 第三期人體試驗,並在 104年 2 月完成 260 餘名受試者之收案,臨床設計治療期間為 12 個月,於 105 年第一季完成第三期人體臨床試驗,105 年第三季整理臨床數據後即可向 EMA 申請藥證;美國部分係依據歐洲臨床試驗數據(再加部分數據報告)向美國 FDA 提出藥證申請。
該公司目前於台中廠自行生產製造 P1101 原料藥,然原料藥仍需經由稀釋、充填、確效、品質分析、安定性測試、組裝及最後包裝等成為商品,為提高病患使用方便性,該公司與 AOP 公司共同決定將 P1101 最終產品設計成筆型(PEN)注射器,將 P1101充填於專用卡式管(cartridge),並組裝於筆型注射器中,爰於 103

51
年 4 月與 AOP 公司簽定筆型劑型研究開發合約,由 AOP 公司協助筆型注射器之開發及尋找相關委託開發廠商,故歐洲地區於105 及 106 年度尚須投入相關費用 213,014 千元。另該公司係以歐洲第三期臨床數據向 FDA 申請藥證,並委由 AOP 公司擔任美國藥證申請之 CRO 公司,預估 105 至 107 年度 CRO 相關顧問費用、申請藥證及上市後監測(PMS)等支出共計 377,527 千元。
(2) P1101 治療 ET 該公司於 104 年向 FDA 溝通 ET 臨床試驗相關事宜,預計於
105 年第二季~第三季提出 IND 申請,第三季獲准開始美國第三期臨床試驗,經參酌 PV 於歐洲之收案速度,並考量 ET 長時間未有新藥出現,該公司預期 6~12 個月可以完成收案,加以臨床設計治療期間為 12 個月,故預計於 107 年第二季可完成第三期人體試驗。
該公司擬自行主導 ET 產品在美國的第三期臨床試驗,參酌AOP公司於歐洲進行 PV第三期臨床試驗之經驗,預估 105至 107年度自行投入 ET 臨床之試驗用藥品、CRO 相關研發支出、申請藥證及上市後監測(PMS)等支出共計 826,932 千元。
(3) P1101 治療 C 型肝炎 i. 基因型第一型(HCV GT1)
該公司於 100 年取得 TFDA 台灣第二期臨床試驗許可,因 HCV GT1 治療期間(48 週)較 HCV GT2(24 週)為長,考量台灣 C 型肝炎病患以 GT2 型為多數,且研發預算及時間成本等因素,故臨床實驗進度較 HCV GT2 為慢,惟該公司已與多家醫院簽訂委託醫院臨床試驗,預計 106 年第一季將可完成二期臨床試驗,將視第二期臨床試驗結果進行後續規劃。另預估 105 至 106 年度尚須投入臨床試驗用藥品及 CRO/CRC等委託研究相關服務費用共計 22,195 千元。
ii. 基因型第二型(HCV GT2) 該公司於 101 年取得 TFDA 台灣第二期臨床試驗許可,
103 年完成台灣第二期臨床試驗收案,於 104 年 5 月取得TFDA 第三期人體試驗同意函,已於 105 年 1 月正式啟動三期收案,該臨床設計治療期間為 24 週,台灣及韓國第三期臨床試驗病人數預計共收足 240 人,故預計於 106 年第一季可完成第三期人體試驗;韓國部分已於 105 年 3 月取得韓國食品藥物管理署(Ministry of Food and Drug Safety 簡稱 MFDS)核准同意進行該人體臨床試驗。另預估 105 至 107 年度尚須投入臨床試驗用藥品、CRO/CRC 等委託研究相關服務費用、藥證申請及上市後監測等費用共計 174,445 千元。
(4) P1101 治療 B 型肝炎

52
該公司於 101 年取得 TFDA 台灣第二期臨床試驗許可,以P1101單獨使用進行治療B型肝炎的臨床試驗,目的係探索 P1101之最適劑量,惟因預算考量,故臨床試驗進度較慢,預計於 106年第三季完成試驗。預估 105 至 106 年度尚須投入臨床試驗用藥品及 CRO/顧問等相關費用共計 61,951 千元。
另該公司已規劃預計於 105 年第三季同時向 TFDA 及韓國FDA 申請以複方治療方式(以小分子抗病毒藥物與 P1101 搭配的療法)進行治療 B 型肝炎之第三期臨床試驗,以加快產品上市時程,預計於 106 年第一季進入第三期臨床試驗,試驗治療期間為48 週/24 針,故預計於 107 年第三季完成第三期試驗報告。預估105 至 107 年度尚須投入臨床試驗用藥品、CRO/顧問費用、藥證申請及上市後監測等費用共計 318,685 千元。
(5) 口服紫杉醇(Oraxol) 依 104 年 10 月 30 日財團法人醫藥品查驗中心(CDE)業務諮
詢會議記錄,CDE 原則接受本公司以授權方 Kinex 公司於韓國及南美等區域執行之臨床數據(含第一、第二、第三期臨床試驗共計200 位病人),加上在本公司在台灣執行的人體藥物動力研究及反應(Pharmacokinetics Study)數據(24 位/期間一年),向 TFDA 提出新劑型新藥(505b(2)新藥)的開發計劃。該公司預計於 105 年第二季向 TFDA 提出申請新劑型新藥的開發計劃,核准後將開始人體藥物動力研究及反應試驗。該臨床設計治療期間為 12 個月,故預計於 106 年第四季完成 PK 數據報告並取得 Kinex 提供之臨床試驗報告後,併同向 TFDA 提出藥證申請。預估 105 至 107 年度尚須投入 PK 臨床支出共計 16,878 千元。
(6) 牛皮癬(Psoriasis)用藥 由於美國紐約州生技公司 Kinex 公司所開發的嶄新化合物分
子 KX01 已證明對於癌症細胞的增生有顯著的抑制效果,並已進入美國人體臨床試驗第二期。本公司認為 KX01 化合物作用的抑制增生之生物機轉,應可適用於非惡性細胞增生之頑固性牛皮癬疾病,因此向 Kinex 公司取得 KX01 化合物在台灣、中國大陸、港澳、星馬等地區的授權,以針對牛皮癬適應症開發皮膚外用劑型,屬於新成份新藥。本公司 KX01 產品已於 104 年 10 月開始進行第一期臨床試驗,預計於 105 年 10 月完成第一期人體臨床試驗,由於臨床試驗仍在早期階段(第一期),且為新適應症之研發,考量第二期及第三期臨床試驗成本昂貴,故本公司將視第一期試驗結果再評估後續規劃。預估 105 年度尚須投入臨床試驗醫院及管理師費用、CRO 費用共計 5,682 千元。
(7) P1101+PD1 治療 B 型肝炎 該公司 Anti-PD1 抗體的新藥開發案,係利用人體慢性 B 型
肝炎小鼠模型來研究以下幾個治療方式(1)PD-1 抗體單獨使用、(2)PD-1 抗體與 P1101 合併使用之各種方式對治療 B 型肝炎的效果。目前抗體藥物分子已完成設計並準備進行臨床前動物毒理試驗,預計於 106 年申請第一期人體臨床試驗。預估 105 至 107 年度尚須投入顧問諮詢費、試驗用動物及試驗分析報告費用、臨床

53
試驗等費用共計 419,800 千元。
3. 該公司新藥開發之營運風險及因應措施
(1) 新藥研發屬高風險行業,投入金額高且產品研發時程不確定性高
新藥研發公司具有高資金需求、產品或技術開發時程長、風險高,但產品開發成功後利潤高及產品生命週期長的特性,新藥研發過程從新藥探索、臨床前試驗、臨床試驗、新藥查驗登記及上市後監測,平均需 10 年以上時間及投入約美金 8億元的開發經費。因此新藥研發產品上市時程長,且投入金額龐大,導致產生營收及獲利貢獻的時間較久,故可能發生營運資金不足,而無法完成新藥研發計畫之風險。
因應對策:
A. 藉由跨國研發合作的模式,提高藥品及市場開發效率
鑑於蛋白質新藥研發所需投入的資源極大,且必須朝向國際市場發展才能產生效益,故以跨國研發合作的模式,將自行研發的 P1101 長效型干擾素產品授權給奧地利專業孤兒藥開發公司 AOP 公司,並在歐洲進行治療真性紅血球增生症(PV)的臨床試驗,一方面可收取授權金挹注營運資金,同時並可提高產品開發效率,目前在歐洲進行的PV 第三期臨床試驗已完成病人總數收案,預計 105 年第三季整理臨床數據完成後即可向 EMA 申請藥證。
B. 以 P1101 為新產品開發平台,使研發投入效益極大,並降低單一產品市場風險
界已證實許多重大疾病的發生與干擾素失衡所造成的免疫系統無法正常運作有關,故該公司以 P1101 為平台持續進行開發其他適應症的臨床試驗,例如血小板增生症(ET)、慢性骨髓細胞性白血病(CML)、原發性骨髓纖維化(PMF)及 B 型/C 型肝炎等,使開發 P1101 的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模。
C. 取得孤兒藥認證,縮短新藥上市審查時程
罕見疾病用藥,是歐美等先進國家藥物管理單位所訂定的藥品特殊資格,是為了鼓勵藥廠開發小眾且困難的疾病藥物,如美國 FDA 規定,每年發病病患人數不足 20 萬(各國規定不盡相同)且未有主流用藥的困難疾病,即可申請孤兒藥資格認定。若產品獲取孤兒藥資格,大多可享有快速通關的審查資格,未來上市後也可能享有自由定價、獨占市場等優惠條件。該公司以 P1101 用於治療 PV 已分別於 100 年及 101 年取得歐洲 EMA 及美國 FDA 的孤兒藥認證,且用於治療 ET 及 PMF 等疾病亦於 103 年獲得

54
美國 FDA 的孤兒藥認證,因此有助於縮短新藥上市審查時程。
(2) 該公司蛋白質藥廠申請歐洲EMA審查(查廠)的時程及結果,亦為影響 P1101 產品上市的重要因素
該公司 P1101目前正在歐洲進行治療真性紅血球增生症(PV)的第三期臨床試驗,其第二期臨床試驗已獲得高療效低副作用的成果,使產品上市核准的可能性提高,惟該公司申請EMA審查其蛋白質藥廠商業化生產 P1101的時程及結果,亦為影響 P1101 產品上市的重要因素。
因應對策:
該公司於 101 年 10 月完成蛋白質藥廠設廠,11 月開始試產,並於 102 年 4 月拿到 TFDA GMP 廠證,有助於未來因應申請歐洲 EMA 查廠的經驗。該公司廠房內所有的製程儀器,包括發酵槽、均質機、濃縮透析儀、管柱層析儀、高壓蒸汽滅菌機及蛋白質藥廠最重要的注射用水產生機等,都是選用世界知名及符合 EMA 及 FDA 規格的廠牌,此外,該公司亦積極加強人員標準作業流程的訓練,以符合查廠要求。
(四) 綜合具體結論
係以自行研發之PEG技術平台及小分子合成藥物技術為基礎進行研發及製造長效型蛋白質新藥的公司,聚焦在血液及感染等疾病的領域,目前公司產品 P1101 由合作夥伴奧地利專業孤兒藥開發公司 AOP 公司於歐洲進行治療真性紅血球增生症的第三期人體臨床試驗,同時以 P1101 為平台進行新藥開發的適應症尚有 C 型肝炎基因型第二型及 B 型肝炎。由於該公司新藥產品 P1101 尚未核准上市銷售,故最近二年度及最近期營業收入來源主要為研究收入(臨床試驗用藥品收入及委託研究收入)、權利金收入及 Q10 商品銷售收入,該公司 102、103年度及 104 年前三季之營業收入分別為 72,327 千元、13,356千元及 8,165 千元;營業成本分別為 5,086 千元、5,988 千元及18,400千元;營業毛利分別為67,241千元、7,368千元及(10,235)千元;營業損益分別為(601,071)千元、(863,472)千元及(572,248)千元,各期均呈現營業淨損,主要係因該公司主要產品 P1101仍在歐洲進行第三期人體臨床試驗階段,產品尚未核准上市,且尚有其他開發中產品持續投入研發費用,惟此乃新藥研發及製造公司之特性。
發中產品 PV 預計於 106 年下半年度在歐洲及美國上市銷售、C 型肝炎基因型第二型產品預計於 107 年於台灣及韓國上市銷售,經評估其新藥上市進度基礎尚有依據。另營運資金方面,該公司截至 104 年 12 月底帳上現金為 548,033 千元,另該公司申請上櫃前之現金增資發行新股 5,000 千股之申報案業於 105 年 1 月 5 日經主管機關申報生效,預計於 105 年 3 月份

55
募集資金 750,000 千元,尚可支應該公司 105 年度之營運資金需求,且該公司預計於股票初次上櫃前辦理現金增資募集3,000,000 千元,倘 P1101 亦如期於 106 年下半年度上市銷售,該公司 105 至 109 年度將維持適足之營運資金。
2.本公司主要產品長效型干擾素P1101未來如獲准於歐洲及美國上市銷售,歐洲係由授權對象奧地利孤兒藥公司 AOP Orphan Pharmaceuticals AG 負責銷售,有關本公司於美國行銷布局規劃及所採具體作法之說明,暨推薦證券商之評估意見。
公司說明:
紅血球增生症(PV)為罕見血液疾病,目前並無專門治療PV之藥物,本公司 P1101 治療 PV 已被 FDA 認證為孤兒藥,新藥上市後非以藥局櫃台為銷售通路,而是藉由主治醫生之診斷後對病人之建議(目前市面上用藥如愛治膠囊(HU)及 Jakafi 之銷售方式亦是由醫生開立處方後給藥),為降低取得藥證至開始銷售的時間成本,本公司亦已規劃妥善相關行銷布局規劃,茲說明如下:
(一) 產品上市前之行銷規劃
如上所述,P1101 非以藥局櫃台為銷售通路,而是藉由主治醫生之診斷後對病人之處方,故本公司已規劃並舉辦/參與多場次的血液疾病研討會,將 P1101 治療 PV 的二期臨床試驗成果,分享給美國血液疾病權威醫生(或稱意見領袖 Key Opinion Leaders, KOLs)及各醫院血液疾病相關醫生,提高P1101 產品上市前之知名度。
具體作法:
1. 提高產品知名度方面(醫學研討會)
(1) 104 年 8 月:受邀參加並贊助美國 KOL Dr. Srdan Verstovsek在美國Virginia主辦的血液疾病醫師會議,並安排一天研討會專題討論 P1101。
(2) 104 年 9 月:受邀參加在克羅埃西亞舉辦的 Leukemia & Lymphoma (白血病及淋巴腫瘤醫學會議)。
(3) 104 年 11 月:參加血液疾病患者的討論會,並受邀參加由美國Dr. Richard Silver醫師於紐約市舉行的第八屆國際血液疾病醫師會議,會議中與競爭對手Incyte 產品再次做產品特性比較。
(4) 105 年 1 月:與中華民國血液病協會在台灣共同舉辦「MPN Asia」第一屆國際血液增生疾病年會,會中邀請了歐美亞多國的KOLs,不僅讓臨床醫師們交流,並大幅度的增加了 P1101 應用在國際上的知名度。
上述活動主要係藉由增加第一線臨床醫師對 P1101

56
的了解與使用的信心,使醫師及病人們未來更願意主動使用 P1101 來治療有需要的病患,達到相輔相成的推廣效果,本公司將持續參加或主辦相關醫學研討會議,積極將P1101 介紹給第一線治療病患的醫生,增加產品市場占有率。
2. 產品設計
本公司自行生產之 P1101 為蛋白質藥物,從蛋白質藥廠生產完成後,用於治療病人之前需經過稀釋、充填及組裝:一般注射針有分傳統針筒型(syringe)或注射筆卡式充填(cartridge)兩種形式,考量傳統針筒型注射方式,對醫生施藥有其不便性,且病人須回醫院或診所請醫生或護士施打(二週施打一針),在交通時間及投藥方式等因素下,會影響病人接受治療的意願及持續性而影響治療效果,故為使病患方便施藥,增加其持續接受治療的意願,並提高產品形象達到產品差異化之行銷目的,本公司與 AOP 公司共同商議將 P1101 最終產品設計成筆型(PEN)劑型,將 P1101 充填於專用卡式管(cartridge),並組裝於筆型注射器中,大幅提高產品利基。該筆型劑型之開發計畫已於 104 年 12 月完成 30 人(8 個國家/35 個試驗中心)之安全性及有效性臨床試驗。
3. 產品品牌設計
配合 P1101 產品上市進度,本公司歐洲合作夥伴AOP 公司已將 P1101 之商品名稱(治療血液疾病)正式命名為BESREMi○R並進行商標註冊(授權地區以外之商標權所有權歸本公司),本公司未來於美國地區銷售亦將使用相同商標。
4. 申請 EAP(Expanded/Early Access Program),使 P1101 銷售時間提前
本公司已委託知名專業顧問規劃向FDA申請EAP,預估 105 年第四季 FDA 核准後,P1101 可在美國 FDA 核准藥證之前,銷售給其他被醫生診斷可使用 P1101 治療的病人,增加產品銷售收入(售價非以產品上市價格銷售),預估美國地區因 EAP 而增加的病人約達 450~750 人,且當 P1101 取得藥證後,其 EAP 藥價將可以產品正式上市的價格銷售,提前 P1101 的銷售時間。
(二) 藉助策略合作夥伴之行銷經驗及銷售通路,縮短新藥銷售增學習曲線 雖然本公司目前尚未於美國設立銷售據點且無新藥銷售
經驗,惟本公司可藉由策略合作夥伴 AOP 公司在歐洲地區之P1101 銷售經驗,加快累積 P1101 在美國銷售的經驗,提高成功機會;另因上述研討會及行銷活動,目前已有許多國際藥廠積極與本公司洽談在美國市場的合作可能性,本公司亦將審慎評估,選擇最有利於本公司新藥長期發展的策略合作夥伴及合作方式,達成預估銷售目標。

57
(三) 設立美國子公司,建立自主行銷團隊 除藉助策略合作共同行銷模式外,本公司目前已派駐專
人(資深處長級)籌劃美國行銷推廣方案,將逐步建立產品行銷據點,預計於 106 年在美國設立子公司,為 P1101 產品之長期發展做準備。
推薦證券商說明:
該公司 P1101 治療 PV 屬罕見血液疾病用藥,已於 101 年通過美國 FDA 孤兒藥認證,其藥品上市後的銷售管道並非由藥廠直接銷售給病人,而是由主治醫生視病人情況給予藥方(故醫生對該藥品的認識程度甚為重要),本推薦證券商經評估該公司上述行銷布局規劃及採取之措施,並比較其國外相似藥物(Jakafi 治療 PV 疾病而言)廠商 Incyte 公司之銷售方式,亦是藉由參加血液疾病研討會議或舉辦醫療會議等方式,發表該藥物之特性及臨床試驗結果介紹給各權威醫生(KOL),使其應用於病人之治療,故其行銷布局及作法尚屬合理。
(三) 本次募集與發行有價證券於申報生效時經金融監督管理委員會證券期貨局通知應補充揭露之事項
本公司過去虧損之主要原因及日後改善計畫
由於本公司新藥產品 P1101 尚未核准上市銷售,故最近三年度營業收入來源主要為研究收入(臨床試驗用藥品收入及委託研究收入)、里程碑金收入及 Q10 商品銷售收入。由於新藥研發公司具有產品開發期長、投資金額龐大的特性,故在產品上市銷售前,因無穩定的收入來源,故皆呈現虧損情況,係屬行業特性,本公司同時進展多項臨床試驗並規劃自行生產 P1101,故目前營業收入尚不足以支應研發費用。惟本公司授權予 AOP 公司於歐洲地區進行之真性紅血球增生症第三期人體臨床試驗已於 104 年2 月收案完成,預計於 105 年底於歐洲及美國地區申請藥證,如新藥獲准上市,來自歐洲地區之藥品銷貨收入及權利金收入,與美國地區之藥品銷貨收入,將有助於改善虧損情形。
因應對策
A. 藉由跨國研發合作的模式,提高藥品及市場開發效率 本公司有鑑於蛋白質新藥研發所需投入的資源極大,且必須朝向國際市場發展才能產生效益,故以跨國研發合作的模式,將自行研發的 P1101 長效型干擾素產品授權給奧地利專業孤兒藥開發公司 AOP 公司,並在歐洲進行治療真性紅血球增生症(PV)的臨床試驗,一方面可收取授權金挹注營運資金,同時並可提高產品開發效率,目前在歐洲進行的 PV 第三期臨床試驗已完成病人總數收案,預計 105 年第三季整理臨床數據完成後即可向 EMA 申請藥證。
B. 以 P1101 為新產品開發平台,使研發投入效益極大,並降低單一產品 市場風險

58
由於醫學界已證實許多重大疾病的發生與干擾素失衡所造成的免疫系統無法正常運作有關,故本公司以 P1101 為平台持續進行開發其他適應症的臨床試驗,例如血小板增生症(ET)、慢性骨髓細胞性白血病(CML)、原發性骨髓纖維化(PMF)及 B 型/C 型肝炎等,使開發 P1101 的成本效益發揮到最大,且可因適應症的增加而提高產品市場規模。
C. 取得孤兒藥認證,縮短新藥上市審查時程 孤兒藥即罕見疾病用藥,是歐美等先進國家藥物管理單位所訂定的藥品特殊資格,是為了鼓勵藥廠開發小眾且困難的疾病藥物,如美國 FDA 規定,每年發病病患人數不足 20萬(各國規定不盡相同)且未有主流用藥的困難疾病,即可申請孤兒藥資格認定。若產品獲取孤兒藥資格,大多可享有快速通關的審查資格,未來上市後也可能享有自由定價、獨占市場等優惠條件。該公司以 P1101 用於治療 PV 已分別於 100年及 101 年取得歐洲 EMA 及美國 FDA 的孤兒藥認證,且用於治療 ET 及 PMF 等疾病亦於 103 年獲得美國 FDA 的孤兒藥認證,因此有助於縮短新藥上市審查時程。

59
公司印鑑:藥華醫藥股份有限公司
負責人(簽名或蓋章):詹青柳