prediction of toxicity and metabolism of chemicals networks gmbh henkestraße 91 91052 erlangen,...
TRANSCRIPT

Molecular Networks GmbHHenkestraße 9191052 Erlangen, Germanywww.molecular-networks.com
Prediction of Toxicity and Metabolism of Chemicals
Johann Gasteiger

2
Outline
REACH
Representation of chemical structures
Modeling of toxicity
Prediction of metabolism
Risk assessment workflow
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

3
Risk Assessment of Chemicals
REACH – Registration, Evaluation, Authorization and restriction of CHemicals
Only those chemicals used with more than 1 ton/year are allowed to be manufactured or imported into the European Union that are registeredRegistration has to provide a dossier with many data and might need a safety reportLaw since June 1, 2007Chemicals have to be accepted until Dec 1, 2013Applies to about 35,000 chemicals
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

REACH Dossier
For compounds used in more than 10 t/a a Chemical Safety Report is needed
Harmful effects on human healthHarmful effects on the environmentDetermination of Persistence, Bioacumulation and Toxicity (PBT)Evaluation of exposition
Testing is time-consuming, expensive and might need many animals
4
Use chemoinformatics methods for ranking of chemicals
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

5
Japanese Translation2005
byK. Funatsu, H. Satoh, H. Masui
J. Gasteiger, T. Engel(Editors)
Chemoinformatics - A Textbook -
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

Handbook of Chemoinformatics
J. Gasteiger (Editor)
65 authors73 contributions
4 volumes1900 pages
Wiley-VCH, Weinheim(August 2003)
From Data to Knowledge
6Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
7
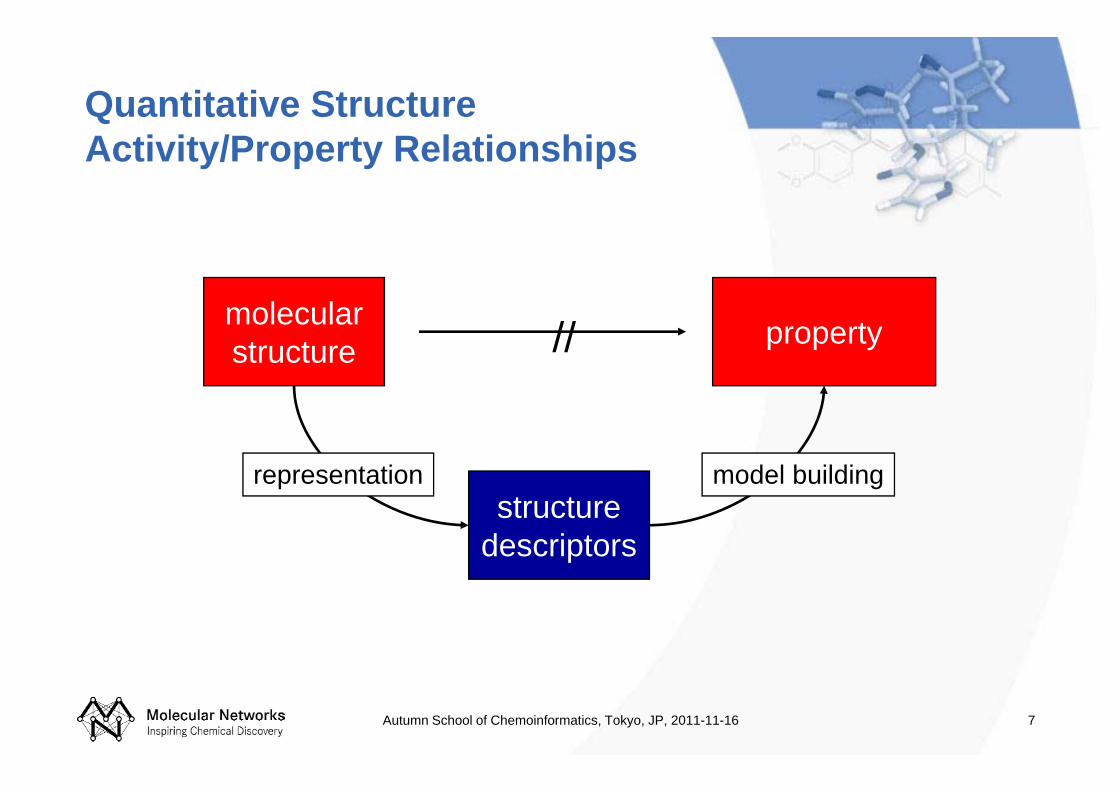
Quantitative Structure Activity/Property Relationships
molecularstructure property
structuredescriptors
//
representation model building
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
8
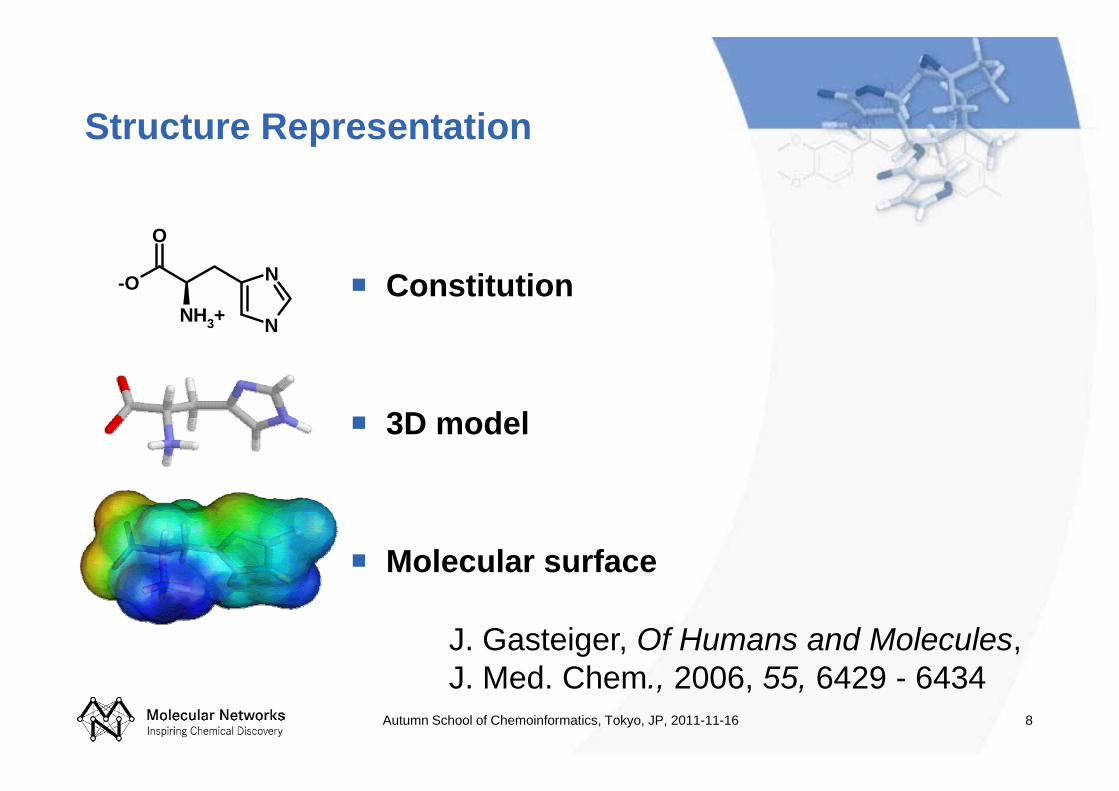
Structure Representation
Constitution
3D model
Molecular surface
N
N
O
-ONH3+
J. Gasteiger, Of Humans and Molecules, J. Med. Chem., 2006, 55, 6429 - 6434
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
9
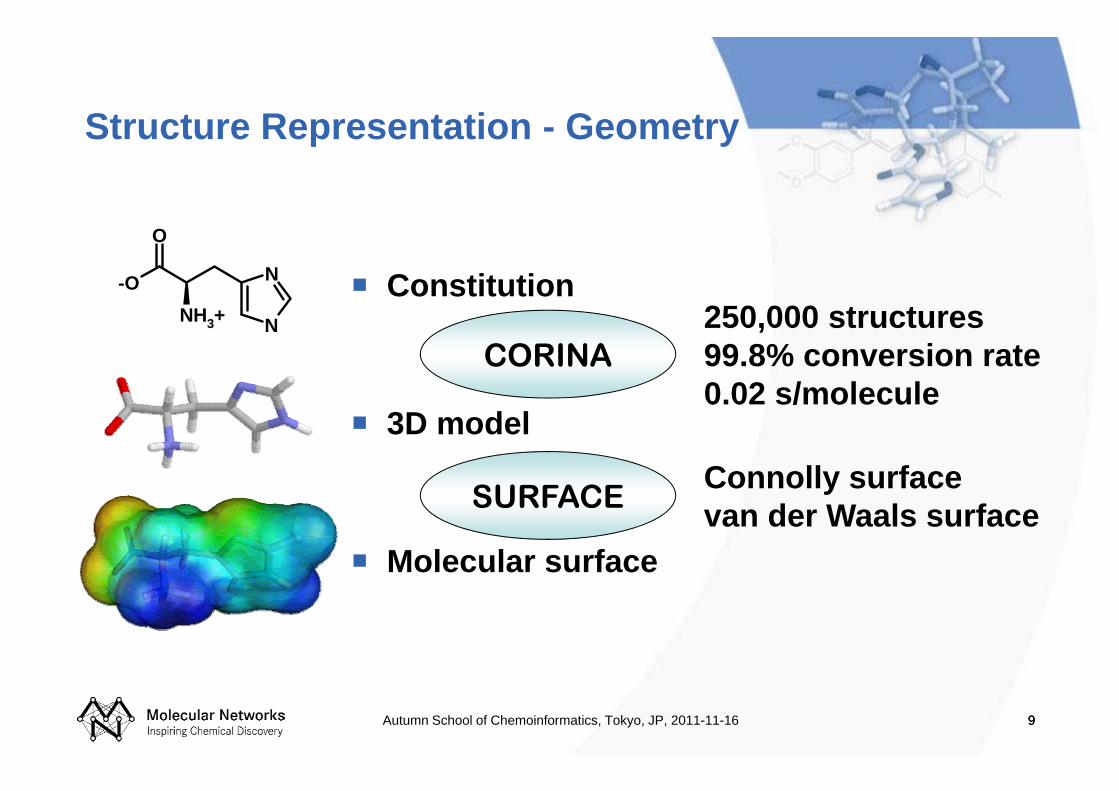
Structure Representation - Geometry
Constitution
3D model
Molecular surface
9
N
N
O
-ONH3+
CORINA
SURFACE
250,000 structures99.8% conversion rate0.02 s/molecule
Connolly surfacevan der Waals surface
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

Structure Representation -Physicochemistry
Charge distributionJ. Gasteiger, M. Marsili, Tetrahedron 36, 3219 (1980)
Inductive effectJ. Gasteiger, M. G. Hutchings, Tetrah. Lett. 24, 2541 (1983)
Resonance effectJ. Gasteiger, H. Saller, Angew. Chem. Int. Ed. Engl. 24, 687 (1985)
Polarizability effectJ. Gasteiger, M. G. Hutchings, J. Chem. Soc. Perkin 2, 559 (1984)
10Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
11
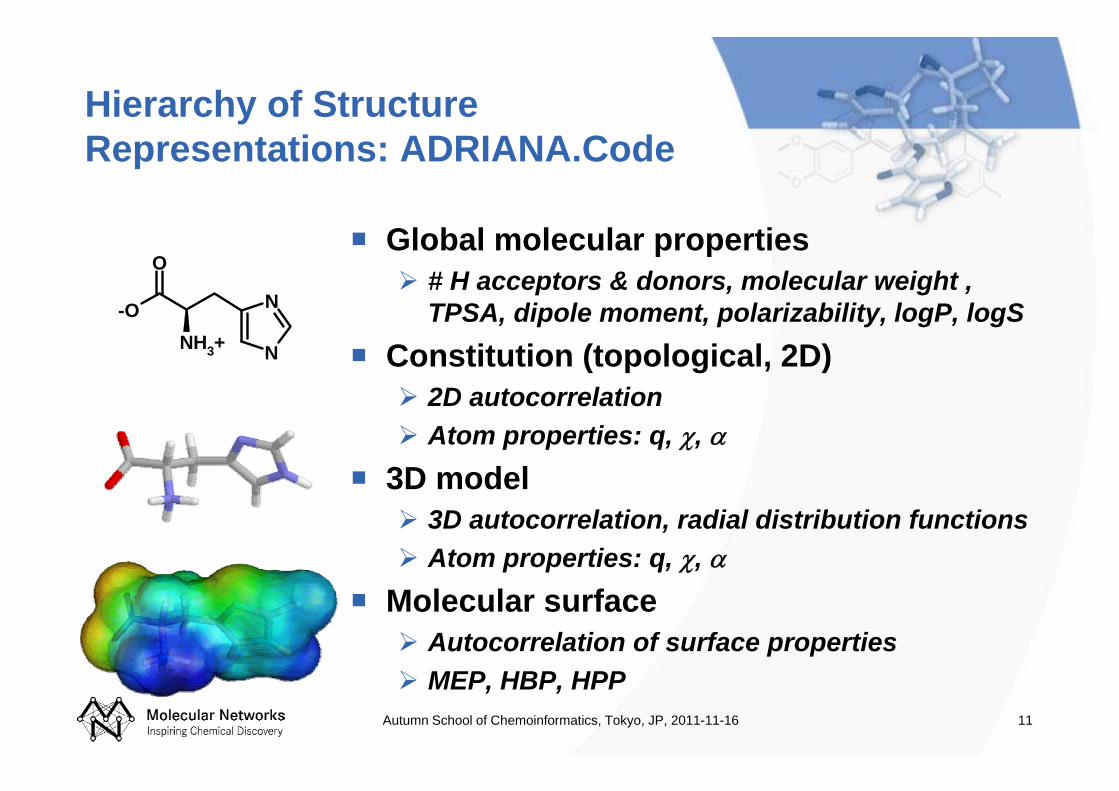
Hierarchy of Structure Representations: ADRIANA.Code
Global molecular properties# H acceptors & donors, molecular weight , TPSA, dipole moment, polarizability, logP, logS
Constitution (topological, 2D)2D autocorrelationAtom properties: q, χ, α
3D model3D autocorrelation, radial distribution functionsAtom properties: q, χ, α
Molecular surfaceAutocorrelation of surface propertiesMEP, HBP, HPP
N
N
O
-ONH3+
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

MOSES.Descriptors -Community Edition
Most structure descriptors of ADRIANA.Code are now freely available as
MOSES.Descriptors - Community Edition
http://www.molecular-networks.com/ services/mosesdescriptors
12Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

Methods for Data Analysis
Inductive learning methodsMachine learningData miningStatisticsPattern recognitionChemometricsNeural networksSupport vector machine
13Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

14
J. Zupan J. Gasteiger
Neural Networksfor Chemists
Japanese EditionMaruzen, 1996
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

15
MOSES.Descriptors – Areas of Application
Drug designClustering of compounds according to their biological activityLocating biologically active compounds in sets of diverse chemical compoundsQuantitative prediction of biological activitiesAnalysis of results of high-throughput screening...
Prediction of ADME/Tox propertiesAqueous solubility of organic compoundspKa valuesPrediction of major metabolizing CYP450 isoformClassification of toxic mode of action…
Prediction of infrared and 1H NMR spectraDye design...
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

16
MOlecular Structure Encoding System
C++ based Chemoinformatics toolkithigh performance available for many platforms (Windows, Linux, Unix)
Python interface provides easy access to the full functionality of MOSESideally suited for the development of client / server solutions
under active development since 2001Computer-Chemie-Centrum, Universität Erlangen-NürnbergMolecular Networks GmbH
300,000 lines of codewell documented and tested
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

17
Modeling toxicity of chemicals
Classification of toxic Mode of Action
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
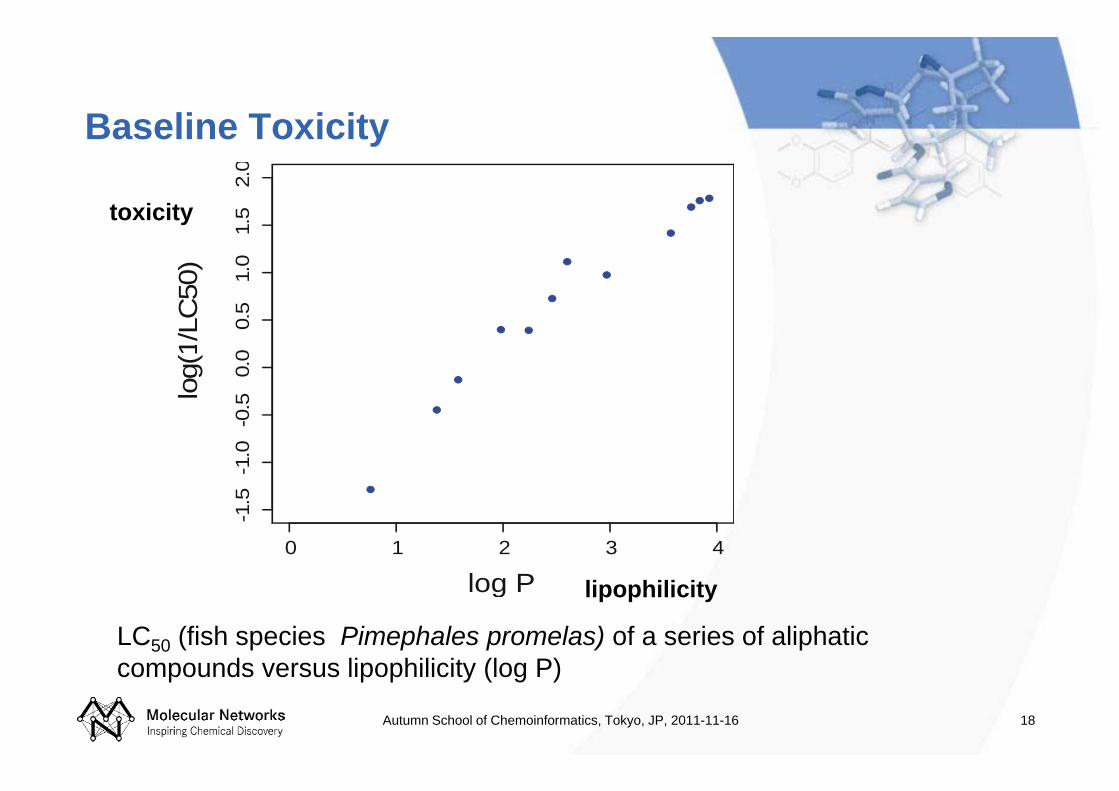
Baseline Toxicity
18
0 1 2 3 4
-1.5
-1.0
-0.5
0.0
0.5
1.0
1.5
2.0
log P
log(
1/LC
50)
LC50 (fish species Pimephales promelas) of a series of aliphatic compounds versus lipophilicity (log P)
lipophilicity
toxicity
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
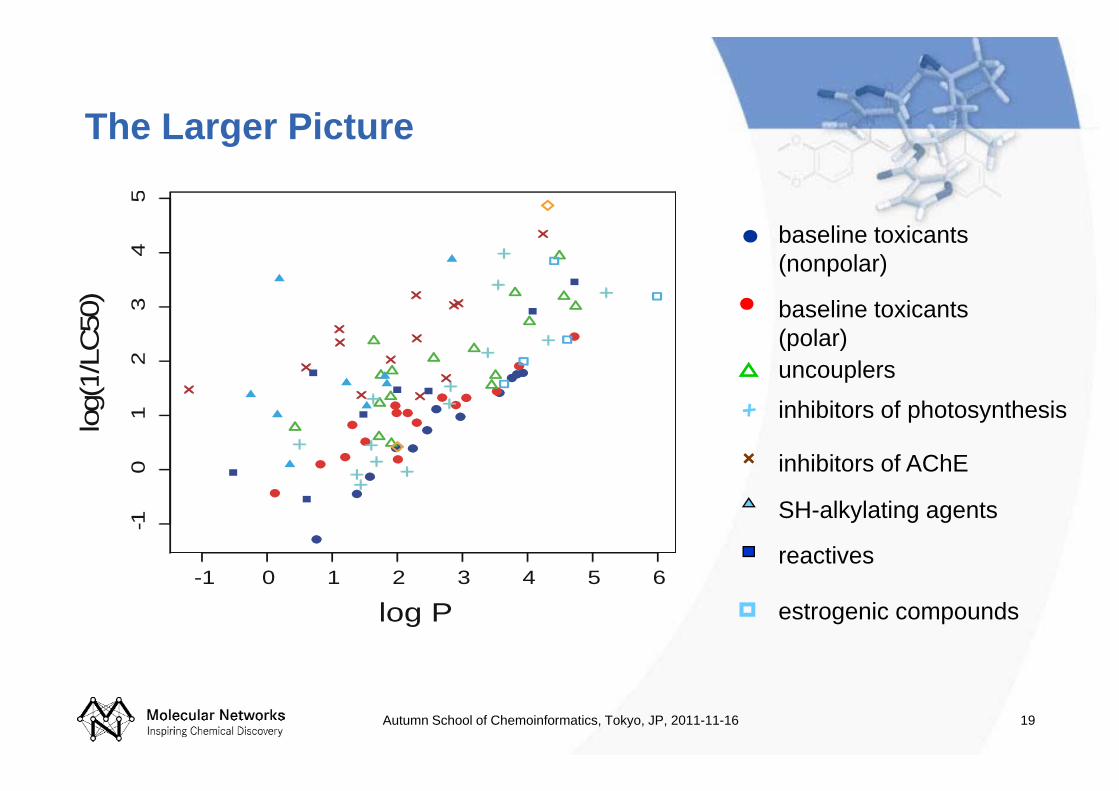
The Larger Picture
19
-1 0 1 2 3 4 5 6
-10
12
34
5
log P
log(
1/LC
50)
baseline toxicants (nonpolar)
inhibitors of AChE
SH-alkylating agents
baseline toxicants (polar)uncouplers
inhibitors of photosynthesis
reactives
estrogenic compounds
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

20
Prediction of Toxicity
Global QSAR models are of limited predictive power because of different toxic modes of action (MOA)
First classify compounds according to toxic MOA
Then develop a local QSAR model for this MOA
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

21
Why Prediction of Toxic Mode of Action (MOA)?
however: and
require different QSAR-equations.
most QSARs in toxicology focus ona certain class of compounds
OH
ClCl
OHCl
ClCl
Cl
polar narcotic uncoupler of oxidative phosphorylation
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

22
Dataset: MOA of Phenols
1. polar narcotics (156 cpds)2. uncouplers of oxidative phosphorylation (19 cpds)3. precursors to soft electrophiles (24 cpds)4. soft electrophiles (22 cpds)
221 cpds
S.Spycher, E.Pellegrini, J.Gasteiger, J. Chem. Inf. Model.,2005, 45, 200-208
A.O.Aytula, T.I.Netzeva, I.V.Valkova, M.T.D.Cronin, T.W.D.Schultz, R.Kühne, G.Schüürmann, Quant. Struct.-Act. Relat. 2002, 21, 12-22.
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

23
Counterpropagation Network Modelsfor Classification of MOA
Estimate of predictive power with 5-fold cross-validation:
RDF(α, q) 2x32 77.4%RDF(χσ) 32 85.5%
RDF(χLP, χσ) 2x32 85.1%
NHdonor, RDF(χLP, χσ) 1 + 2x32 88.7%
RDF(χ LP, χ σ ), HBP surface AC 2x8 + 12 95.9%
S.Spycher, E.Pellegrini, J.Gasteiger, J. Chem. Inf. Model., 2005, 45, 200-208
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
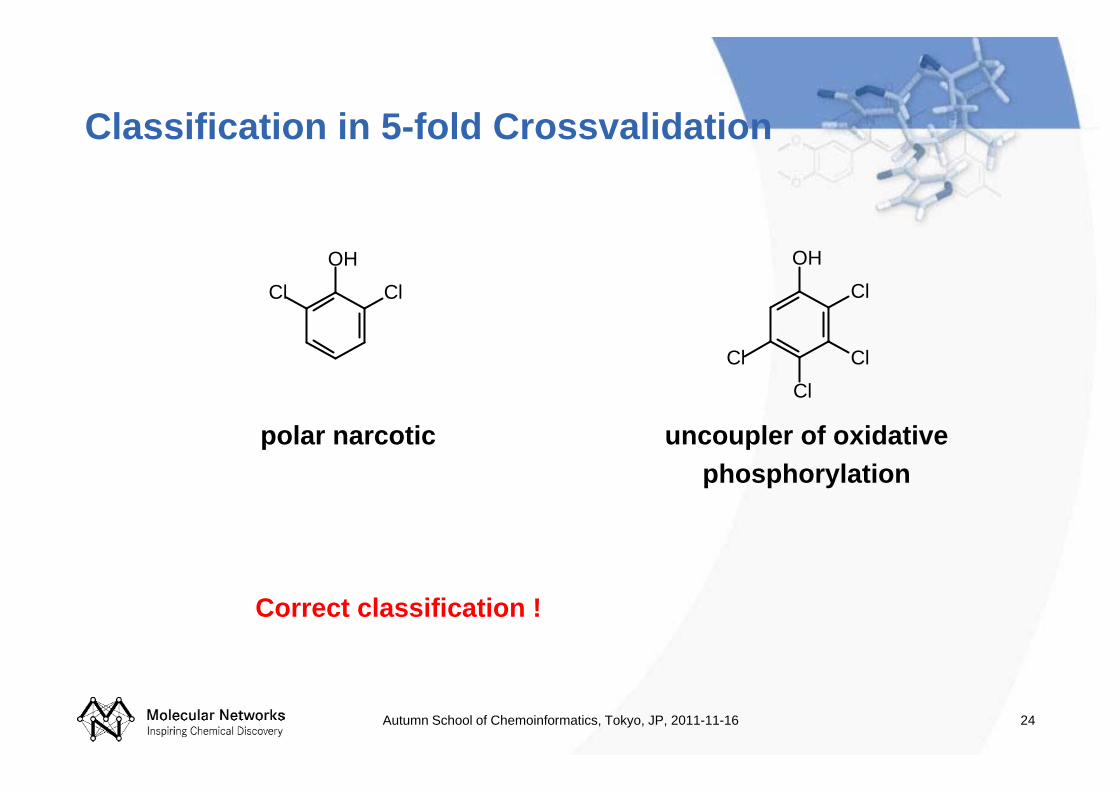
24
Classification in 5-fold Crossvalidation
polar narcotic
OHClCl
OHCl
ClCl
Cl
uncoupler of oxidative phosphorylation
Correct classification !
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

25
Metabolism of Xenobiotics
Drugs, agrochemicals, food additives
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
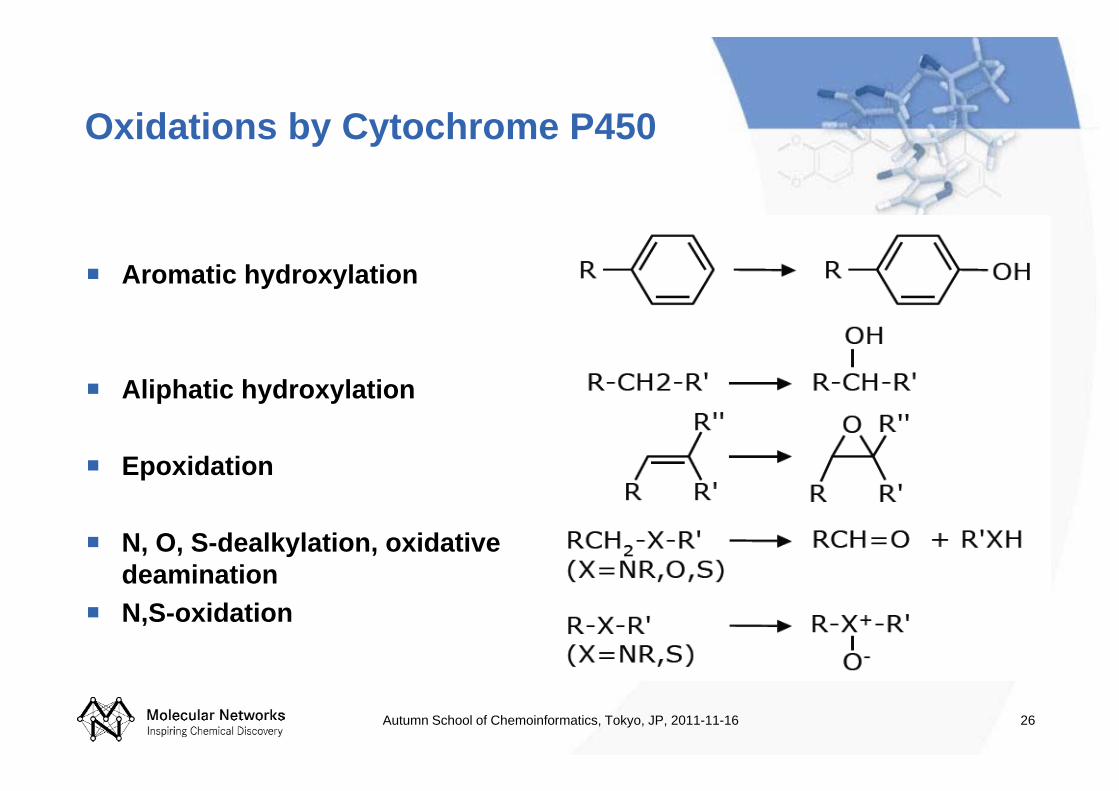
26
Oxidations by Cytochrome P450
Aromatic hydroxylation
Aliphatic hydroxylation
Epoxidation
N, O, S-dealkylation, oxidative deaminationN,S-oxidation
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

27
Development of MOSES.Metabolism
Selectivity between different cytochrome P450 isozymesin particular 3A4, 2C9, 2C19, 2D6, 1A2
Selectivity between different reaction typeschemoselectivity
Selectivity between different reaction sitesregioselectivity
Modeling different Selectivities
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

28
Development of MOSES.Metabolism
Selectivity between different cytochrome P450 isozymesin particular 3A4, 2C9, 2C19, 2D6, 1A2
Selectivity between different reaction typeschemoselectivity
Selectivity between different reaction sitesregioselectivity
Modeling different Selectivities
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

29
Data Set of 3A4, 2D6, and 2C9 Substrates
Training set: 146 drugs, substrate for 3A4, 2D6 or 2C9*
major isoform specified
*Manga, N. et al. SAR and QSAR in Env. Res. 2005, 16, 43-61.
Bufuralol Tramadol Felodipine
O OH
N
OOHN
NH
O
O
O
O
Cl
Cl
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

30
Support Vector Machine (SVM) Model
Training set: 146 drugsDescriptors (242 descriptors by ADRIANA.Code)Automatic variable selection: 12 components
2D-ACidentity(5), 2D-ACqπ(3), 2D-ACqπ(6), 2D-ACχπ(5), 2D-ACqσ(1), 2D-ACqσ(2), 2D-ACχσ(6), 3D-ACidentity([5.8-5.9[Å), nacid_groups, naliphatic_amino ,nbasic_n , r3
PredictabilityTraining: 90.4%5-fold CV: 87.8%
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

31
Validation of the Support Vector Machine Model
External validation set: 233 substrates from the Metabolite database
Predictability: 82.8%
remember: some drugs are metabolized by several isoforms
L. Terfloth, B. Bienfait, J. Gasteiger, J. Chem. Inf. Model. 2007, 47, 1688-1710
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

32
isoCYP Webservice
http://www.molecular-networks.com/online_servicesL. Terfloth, B. Bienfait, J. Gasteiger, J. Chem. Inf. Model. 2007, 47, 1688-1710
Prediction of major metabolizing CYP450 isoform(2D6, 3A4, 2C9)
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

33
Development of MOSES.Metabolism
Selectivity between different cytochrome P450 isozymesin particular 3A4, 2C9, 2C19, 2D6, 1A2
Selectivity between different reaction typeschemoselectivity
Selectivity between different reaction sitesregioselectivity
Modeling different Selectivities
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

A Data-Driven Approach to Metabolism Prediction
Extract reaction types from a metabolic reaction database (Metabolite by MDL/Symyx/Accelrys)
For each reaction type develop a statistical evaluation based on the number of observed reactions /
the number of conceivable reactíons
Use this ratio for assigning a likelihood to a reaction type
34
L.Ridder, M.Wagener, ChemMedChem, 2008, 3, 821-832
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

35
MOSES.Metabolism Reaction Rules
117 reaction rulesReaction types covered:
Aromatic hydroxylationAliphatic hydroxylationN- and O-dealkylationHydrolysis (ester, amides)Conjugation reactions (glucuronidation, sulphation, glycination, acetylation)Oxidation reactions (alcohols, aldehydes, etc.)
Empirical score for likeliness of a reaction based on literature data
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

36
Derivation of a Rule Base for Metabolite Prediction
Define reaction rules, e.g. for an acetylation
Calculate reaction probabilities based on a reaction database (Metabolite, MDL-Symyx)
Conceivable metabolites 1223Observed metabolites 122Probability 122/1223 = 0.10
RNH2
RNH
O
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

3737
Rules Relevant for Atorvastatin Metabolism
Rules were derived forAromatic hydroxylationHydroxylation of aromatic aminesAromatic hydroxylation of 1,4-substituted phenyl ringsN-dealkylation of substituted pyrroleHydrolysis of amides
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
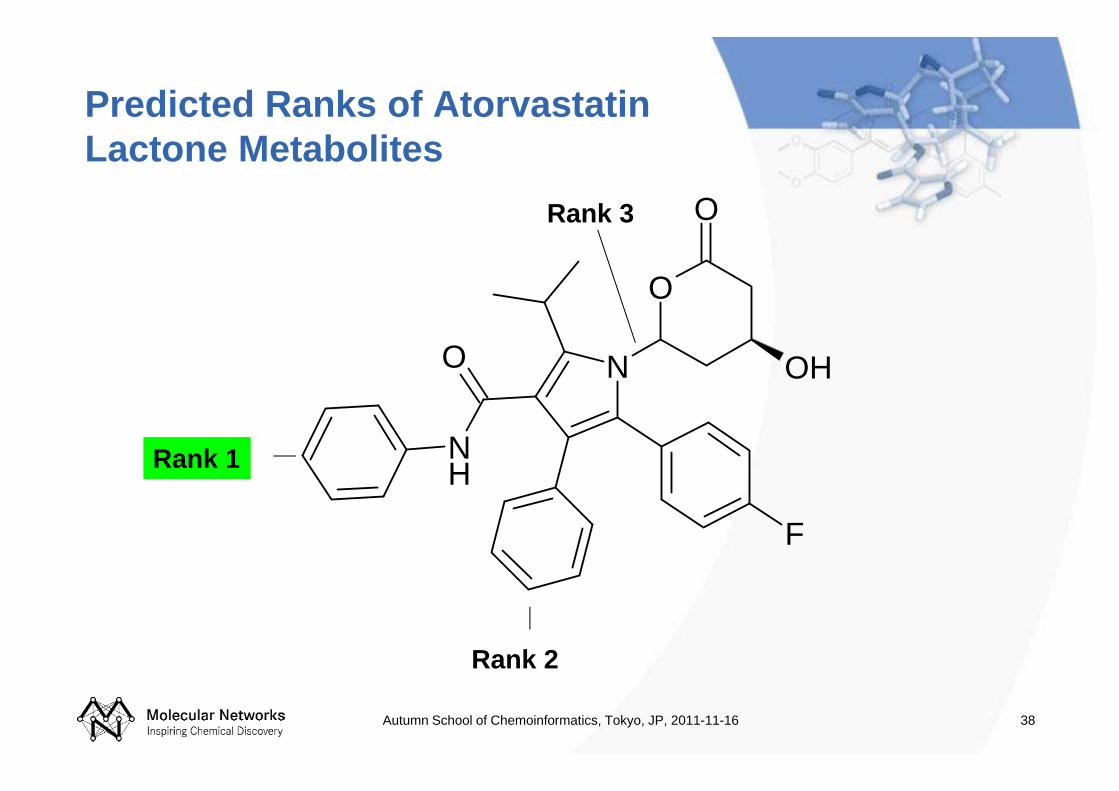
38
Predicted Ranks of Atorvastatin Lactone Metabolites
N
F
O
NH
O
O
OH
Rank 3
Rank 1
Rank 2
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

Experimentally observed Metabolite of Atorvastatin Lactone
39
Metabolite predicted for atorvastatin with highest rank corresponds to the experimental observations
N
F
O
NH
O
O
OH N
F
O
NH
O
O
OH
OH
CYP450 3A4
Rank 1
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
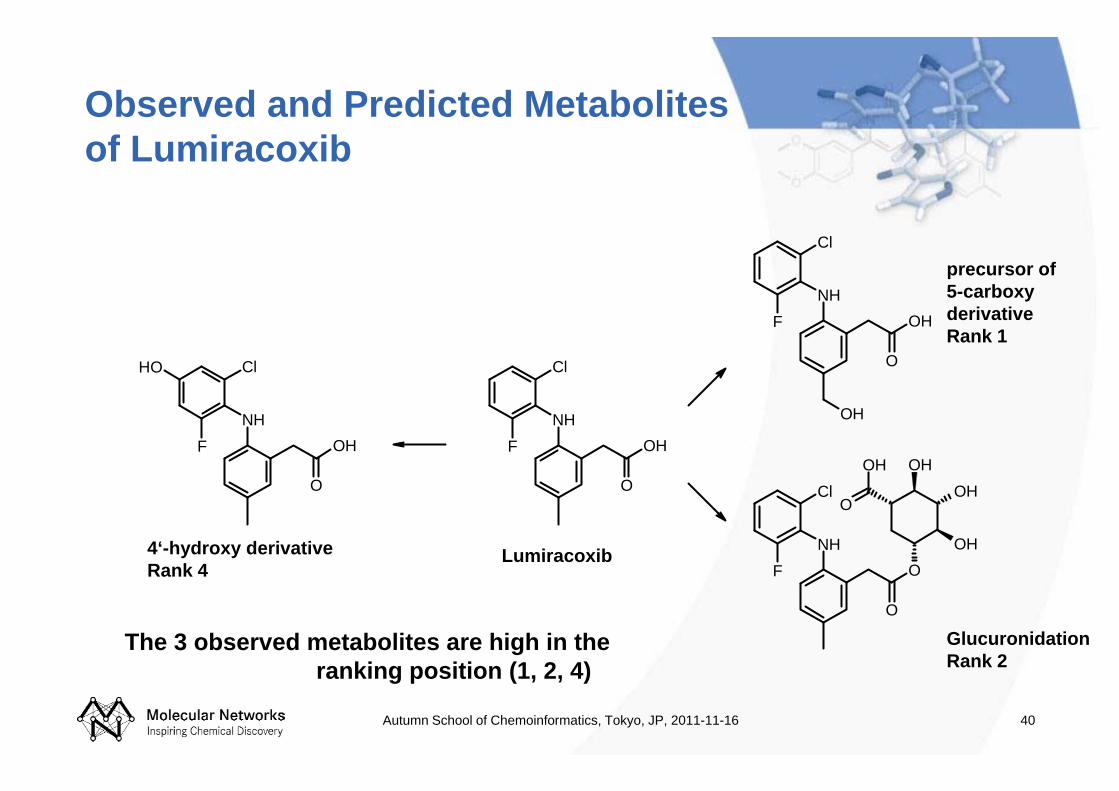
Observed and Predicted Metabolites of Lumiracoxib
40
NHOH
O
F
ClOH
NHOH
O
F
Cl
NHOH
O
F
Cl
OH
NH
O
F
Cl
OOH
OHOH
OH
O
Lumiracoxib4‘-hydroxy derivativeRank 4
precursor of 5-carboxy derivativeRank 1
GlucuronidationRank 2
The 3 observed metabolites are high in the ranking position (1, 2, 4)
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

41
In silico Toxicity and Metabolism Predictionin the Risk Assessment Workflow Using the
Chemoinformatics Platform
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

42
Areas of Applications
Hazard and risk assessment of chemicalsProduct safety of pharmaceuticals, cosmetics, food ingredients and other chemicalsComputational toxicologyRegistration of chemical substances, e.g., REACH initiativeCompound profiling
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
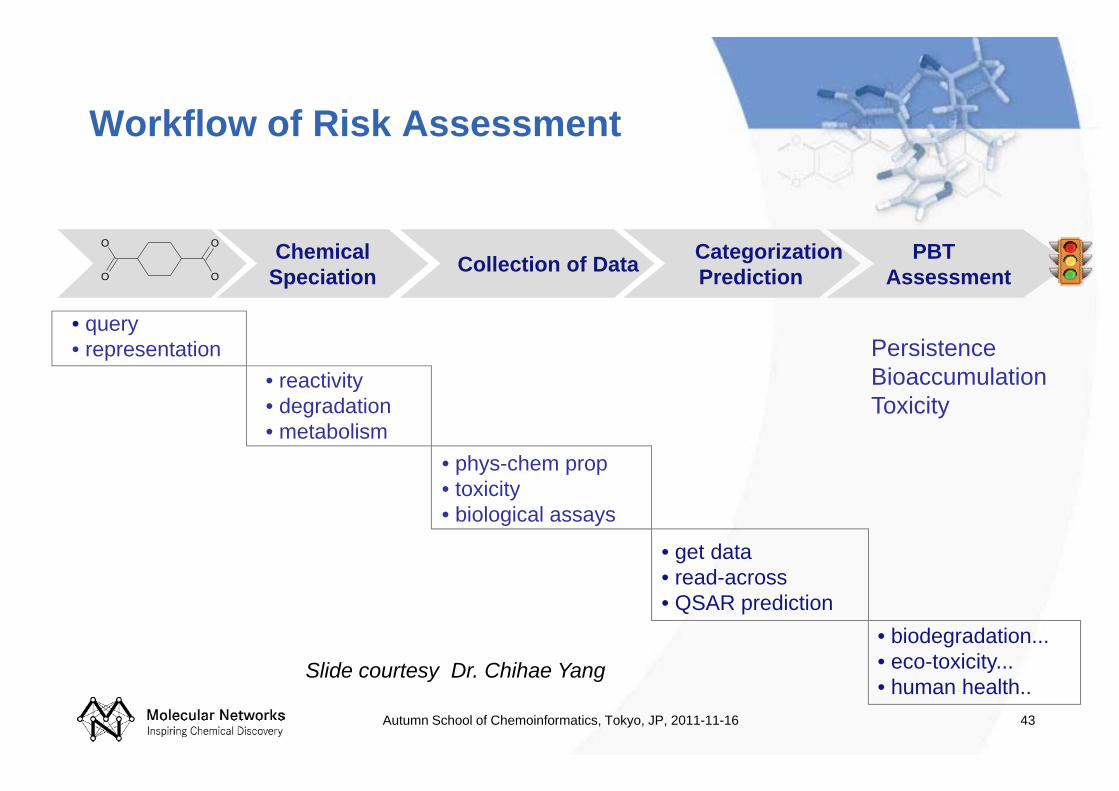
43
Workflow of Risk Assessment
Collection of Data CategorizationPrediction
PBT Assessment
ChemicalSpeciation
O
O O
O
• get data• read-across• QSAR prediction
• phys-chem prop• toxicity• biological assays
• reactivity• degradation• metabolism
• biodegradation...• eco-toxicity... • human health..
• query• representation
Slide courtesy Dr. Chihae Yang
PersistenceBioaccumulationToxicity
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16
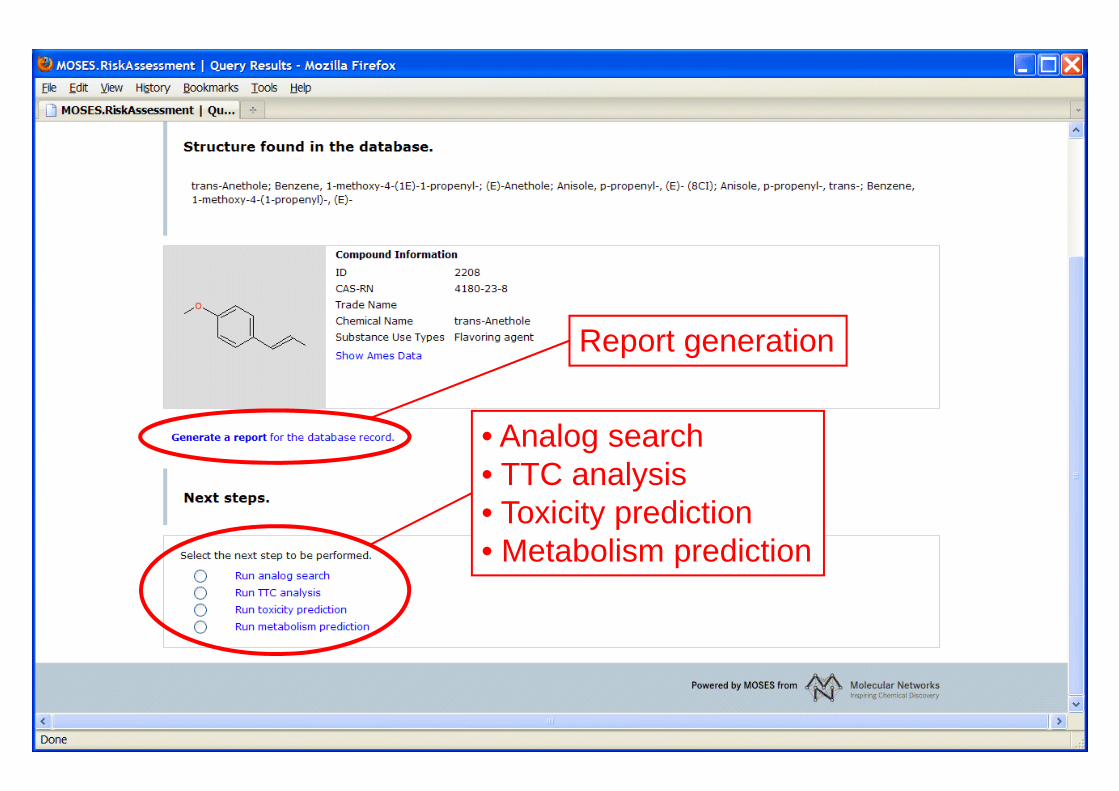
44
• Analog search• TTC analysis• Toxicity prediction• Metabolism prediction
Report generation

45
Analog searching
Specify cut off
Similarity criteria – MDL fingerprints
Run search

46
Analogs with data
List with analogs
47
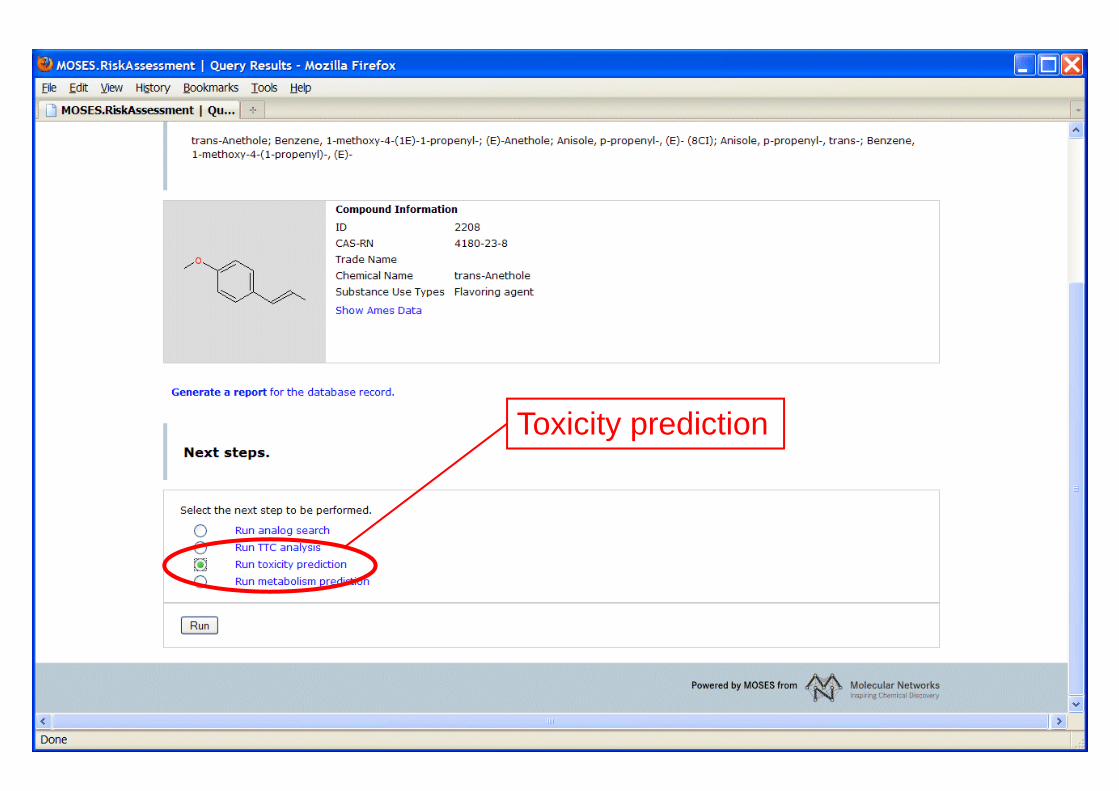
Toxicity prediction
48
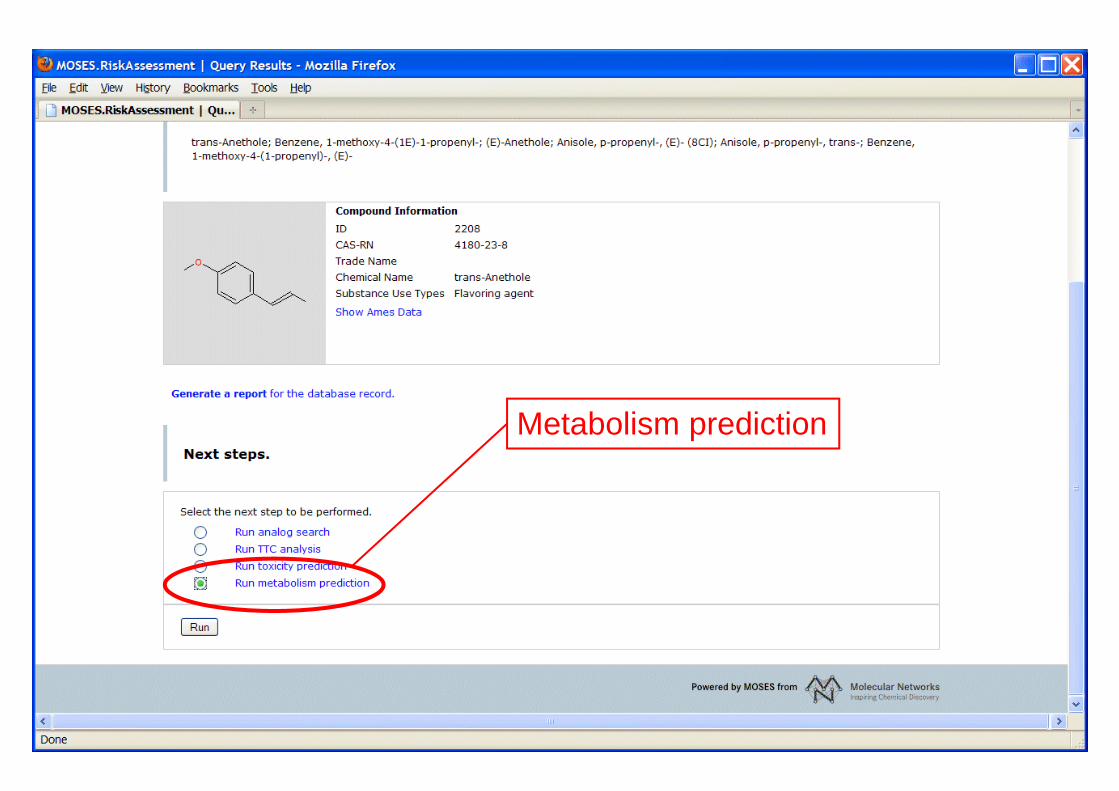
Metabolism prediction
49
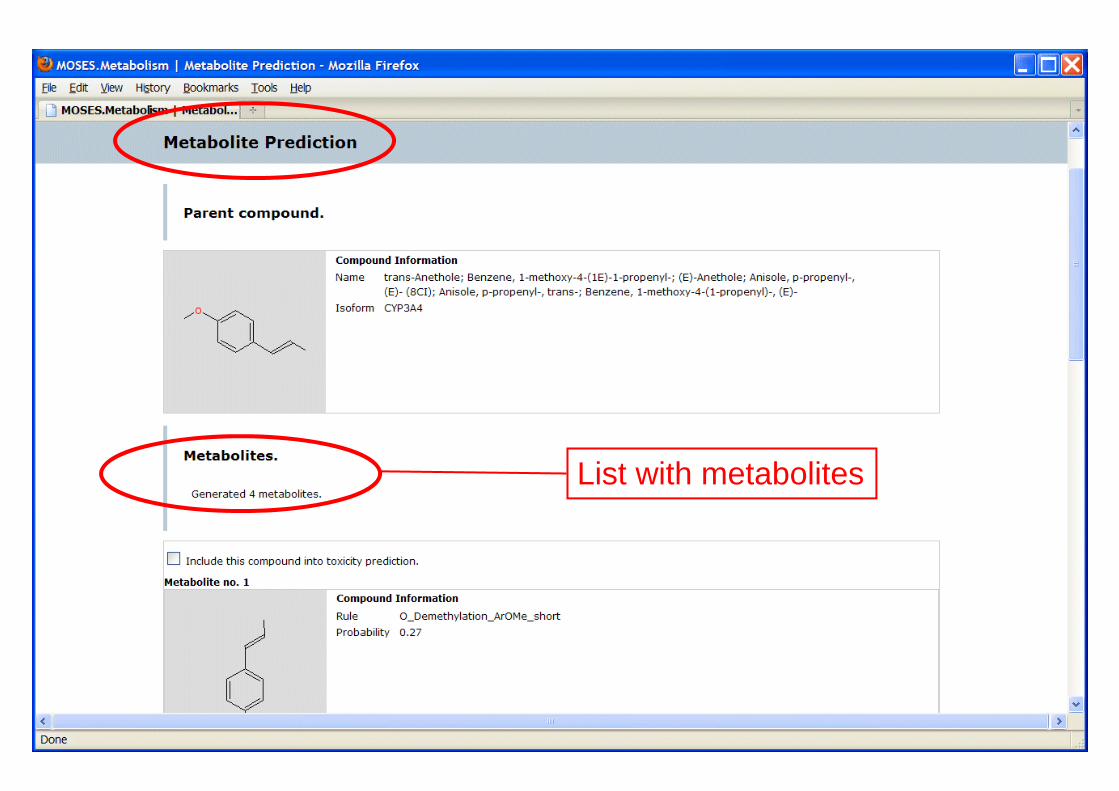
List with metabolites
50
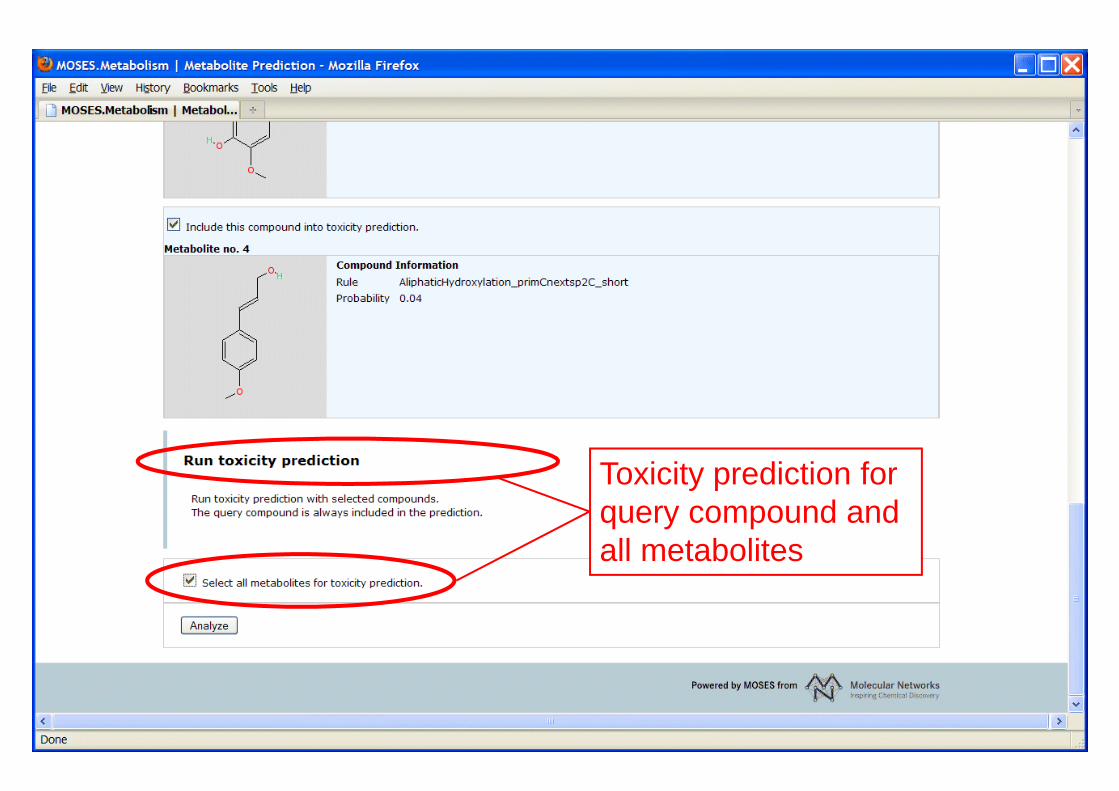
Toxicity prediction for query compound andall metabolites

51
Features & Functionality
Knowledge base for hazard and risk assessment of chemicals Database lookup by text-based, analog and similarity searches Retrieval of available study information for query compound and analogs Generation and evaluation of metabolites of query and analogs (including CYP isoform specificity) Analysis tools for query, analogs and their metabolites QSAR predictions of toxicity endpoints (e.g., Ames mutagenicity) Report generation Fully web-based, easy-to-use user interface
Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

Summary
Chemoinformatics can help us better understand chemistry
We can learn from data about the relationships between chemical structure and toxicityInformation in reaction databases can help us model metabolismRisk assessment of chemicals can profit from chemoinformatics methods
52Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16

Acknowledgements
eTOX project funded by EU-IMI (Innovative Medicine Initiative)7 academic groups5 SMEs11 pharmaceutical companies
FDA – CFSAN (Center for Food Safety and Nutrition)Development of the CERES systemCOSMOS project funded by EU and COLIPA (The European Cosmetics Association)CollaborationDr. Chihae Yang, Altamira LLC, USA
53Autumn School of Chemoinformatics, Tokyo, JP, 2011-11-16