s contribution a l'étude de la complexation du cérium et
TRANSCRIPT

Hirtn
Su
3.1
CEA-R-3521PREMIER MINISTRE
COMMISSARIAT A L'ENERGIE ATOMIQUE
CONTRIBUTION A L'ÉTUDEDE LA COMPLEXATION DU CÉRIUM
ET DES URANIDES PAR L'ACIDEDIÉTHYLÈNETÉTRAMINEPENTAACÉTIQUE
(D.T.P.A.)
par
HAFEZ Badreldin Mohamed
g DIRECTION DE LA PROTECTION ET DE LA SURETE RADIOLOGIQUES
Centre d'Etudes Nucléaires de Fontenay-aux-Roses
Rapport CEA - R - 3521
1968 S E R V I C E C E N T R A L DE D O C U M E N T A T I O N DU C.E.AKa!
C.E.N-SACLAY B.P. n'2, 91-GIF-sur-YVETTE-France

(TH.)
CEA-R-3521 - HAFEZ Mohamed Badreldin
CONTRIBUTION A L'ETUDE DE LA COMPLEXATION DUCERIUM ET DES URANIDES PAR L'ACIDE DIETHYLENE-TETRAMINEPENTAACETIQUE (DTPA)
Sommaire. - Etude spectrophotométrique de la complexationdu cérium et des uranides (uranium, neptunium, plutonium etaméricium) dans leurs différents états d'oxydation par l'aci-de diéthylènetriaminepentaacétique (DTPA). Deux états phy-sicochimiques ont retenu notre attention, l'état ionique etl'état précipité d'hydrolyse. Dans le premier cas , nous avonsétudié l'influence du pH sur la formation du complexe, etdans tous les cas où le complexe est stable, nous avons dé-terminé le rapport moléculaire (5) [élément]/[chélatant] ducomplexe formé et nous avons calculé leurs constantes destabilité. Dans le deuxième cas, l'état de précipité
CEA-R-3521 - HAFEZ Mohamed Badreldin
A SPECTROPHOTOMETRIC STUDY OF THE COMPLEXESOF CERIUM AND URANIDES WITH DIETHYLENETETRA-MINEPENTAACETIC ACID (DTPA)
Summary. - A spectrophotometric determination was madeof the complexes of cerium and uranides in their differentdegrees of oxidation with diethylenetriaminepentaacetic acid(DTPA). Two physico-chemical states, the ionic and hydro-lysis ones, were considered. In the former case, we stu-died the influence of the pH on the formation of the complex,and whenever the complexes were stable, we determined themolecular ratio and calculated their stability constants. Inthe latter case, we studied the conditions of solubilization

(suite CEA-R-3521)
d'hydrolyse, nous avons étudié les conditions de solubilisa-tion du précipité , par formation du complexe, en fonction dupH, de l'âge du précipité et du temps de contact précipité -chélatant.
1968 140 pages
Commissariat à l'Energie Atomique - France
of the precipitate by formation of complexes soluble inwater as a function of pH, age of the precipitate and timeof precipitate-chelate contact.
1968 140 pages
Commissariat à l'Energie Atomique -' France

A partir de 1968, les rapports CEA sont classés selon les catégories qui figurent dans le plan de classi-fication ci-dessous et peuvent être obtenus soit en collections complètes, soit en collections partiellesd'après ces catégories.
.Ceux de nos correspondants qui reçoivent systématiquement nos rapports à titre d'échange, et quisont intéressés par cette diffusion sélective, sont priés de se reporter à la lettre circulaire CENS/DOC/67/4690du 20 décembre 1967 que nous leur avons adressée, et qui précise les conditions de diffusion.
A cette occasion nous rappelons que les rapports CEA sont également vendus au numéro par la Directionde la Documentation Française, 31, quai Voltaire, Paris 7e.
PLAN DE CLASSIFICATION
1. APPLICATIONS INDUSTRIELLES DESISOTOPES ET DES RAYONNEMENTS
2. BIOLOGIE ET MEDECINE
2. 1 Biologie générale2. 2 Indicateurs nucléaires en biologie2. 3 Médecine du travail2. 4 Radiobiologie et Radioagronomie2. 5 Utilisation des techniques nucléaires en
médecine
3. CHIMIE
3. 1 Chimie générale3. 2 Chimie analytique3. 3 Procédés de séparation3. 4 Radiochimie
4. ETUDES DU DOMAINE DE L'ESPACE
5. GEOPHYSIQUE, GEOLOGIE,MINERALOGIE ET METEOROLOGIE
6. METAUX, CERAMIQUESET AUTRES MATERIAUX
6. 1 Fabrication, propriétés et structure desmatériaux
6. 2 Effets des rayonnements sur les matériaux6. 3 Corrosion
7. NEUTRONIQUE, PHYSIQUE ETTECHNOLOGIE DES REACTEURS
7. 1 Neutronique et physique des réacteurs7. 2 Refroidissement, protection, contrôle et
sécurité7. 3 Matériaux de structure et éléments
classiques des réacteurs
8. PHYSIQUE
8. 1 Accélérateurs8. 2 Electricité, électronique, détection des
rayonnements8. 3 Physique des plasmas8. 4 Physique des états condensés de la matière8. 5 Physique corpusculaire à haute énergie8. 6 Physique nucléaire8. 7 Electronique quantique, lasers
9. PHYSIQUE THEORIQUEET MATHEMATIQUES
10. PROTECTION ET CONTROLE DESRAYONNEMENTS. TRAITEMENT DESEFFLUENTS
10. 1 Protection sanitaire10. 2 Contrôle des rayonnements10. 3 Traitement des effluents
11. SEPARATION DES ISOTOPES
12. TECHNIQUES
12. 1 Mécanique des fluides - Techniques duvide
12. 2 Techniques des températures extrêmes12. 3 Mécanique et outillage
13. UTILISATION ET DEVELOPPEMENTDE L'ENERGIE ATOMIQUE
13. 1 Centres d'études nucléaires, laboratoireset usines
13. 2 Etudes économiques, programmes
13. 3 Divers (documentation, administration,législation, etc...)
Les rapports du COMMISSARIAT A L'ENERGIE ATOMIQUE sont, à partir du n° 2200, en vente a laDocumentation Française, Secrétariat Général du Gouvernement, Direction de la Documentation, 31, quaiVoltaire, PARIS VIIe.
The C.E.A. reports starting with n° 2200 are available at the Documentation Française, SecrétariatGénéral du Gouvernement, Direction de la Documentation, 31, quai Voltaire, PARIS VIIe.
ORSAYSÉRIE A,
N- D'ORDREL_ _
~~ ^r
322
THÈSESPRÉSENTÉES
A LA FACULTÉ DES SCIENCESDE L'UNIVERSITÉ DE PARIS
CENTRE D'ORSAY
POUR OBTENIR
LE GRADE DE DOCTEUR ÈS-SCIENCES
PAR
HAFEZ Mohamed
PREMIÈRE THÈSE
Contribution à l'étude de la complexation du cérium et des uranides
par l'acide diéthylèiietétraminepentaacétique (DTPA)
DEUXIÈME THÈSE
Propositions données par la Faculté
Soutenues le 5 Mars 1968, devant la Commission d'Examen
MM. LEFORT
BOUISSIERESGUILLAUMONTLAFUMA
Président
Examinateurs

- Rapport CEA-R-3521 -
Centre d'Etudes Nucléaires de Fontenay-aux-RosesDépartement de la Protection Sanitaire
CONTRIBUTIONA L'ÉTUDE DE LA COMPLEXATION
DU CÉRIUM ET DES URANIDESPAR L'ACIDE
DIÉTHYLÈNETÉTRAMINEPENTAACÉTIQUE(DTPA)
par
Mohamed Badreldin HAFEZ
A mes parents,
A ma fiancée,
A ma famille.
Juin 1968

Ce travail a été effectué sous la direction de Monsieurle Professeur BOUISSIERBS, à qui je tiens à exprimer ma sincèrereconnaissance pour l'intérêt bienveillant et les éminents con-seils qu'il m1a prodigués.
Monsieur GUILLAUMONT, Assistant à la Faculté des Sciences,a dirige' ces recherches avec une attention constante et m'a fourniune aide précieuse à la fois scientifique et morale„ Qu'il veuillebien trouver ici le témoignage de ma profonde gratitude.
L'ensemble de ces travaux a été réalisé au Centre d'EtudesNucléaires de Fontenay-aux-Roses, du Commissariat à l'Energie Ato-mique (C.:']oA.). Je remercie bien vivement Monsieur le DocteurJAMMET, Chef du Département de la Protection Sanitaire (D.P.S0),qui m'a accueilli dans son Service.
J'adresse aussi mes remerciements à Monsieur le DocteurLAFUMA, Chef du Groupe de Radio-Pathologie interne qui in'a proposéle sujet de ces recherches et a mis à ma disposition son laboratoire.
Monsieur PATTI, Ingénieur au C.E.A., m'a aidé de ses con-seils et m'a grandement facilité la tâche, je l'en remercie sincè-rement .
Que Monsieur CHESNE, Chef de la Section d'Etudes ChimiquesRadioactives du Département de Chimie du C.E.Ao qui m'a fourni lapossibilité d'effectuer certaines expériences dans son laboratoiresoit également remercié.
Que Monsieur le Professeur LEFORT qui me témoigne l'hon-neur de présider ce jury, veuille bien accepter mes remerciaientsdéférents et respectueux.

Je ne saurais oublier d'exprimer mon amicale reconnais-
sance à Messieurs KOEHLY, JEANMAIRE, R3MY et particulièrement àMadame BEAU, pour les services qu'ils ne m'ont jamais refucés.
Enfin je tiens à remercier le Bureau. dfEducation de laRépublique Arabe Unie, le Département des Relations Extérieuresdu C.EoA., Monsieur le Docteur F6 SOUROUR et Monsieur COURTEIX
qui se sont largement employés à faciliter mon séjour en France0
INTRODUCTION
Le rapide développement de l'énergie nucléaire a con-
duit à la production d'importantes quantités de radionuclides„Corrélativement les risques de contamination interne se sont
accrus et les problèmes de décontaminâtion sont devenus de pre-
mière importance.
Pour les résoudre, il convient d'administrer à un or-
ganisme vivant un agent susceptible d'accélérer l'éliminationnaturelle de l'élément contaminant. Le choix d'un tel agent chi-
mique est délicat. Il dépend de nombreux facteurs, notamment deson aptitude à donner des complexes stables et solubles avecl'élément qu'il est vital d'éliminer (1) (2)0 Or ce dernier se
trouve dans l'organisme à l'état ionique ou dans un état colloï-
dal, états oui dépendent essentiellement des propriétés chimiquesde l'élément et du pH de l'organe dans lequel il se concentre
(4).
Ce sont les recherches in vivo qui permettent de testerl'efficacité dfun agent décontaminant (5), mais ce sont les re-cherches in vitro qui guident le choix de celui-ci pour une
application biologique particulière. En effet à partir de ces

dernières, il est souvent possible d'établir quel agent donneradans des conditions proches de celles de la matière vivante, lerésultat escompté.
Parmi les complexants utilisés pour la decontaminationinterne, il convient de donner, à côté de divers chélatant orga-niques comme la parathonaone (6), le citrate de zirconium (7)et le 2-3-dimercaptopropanol (B.A.L.) (8), une place particulièreaux acides aminopolycarboxyliquespeu toxiques (9). Ainsi l'éthy-lènediaminetétraacétique (ïï.D.T.A.) est couramment utilisé pourremédier aux diverses intoxications industrielles (10) (11).Cependant l'acide diéthylènetriaminepentaacétique (T3eT.P.A.)
découvert en 1957, semble offrir d'excellentes conditions de dé-cont amination (12).
Ce chélatant a déjà fait l'objet de nombreuses appli-cations biologiques (13) (14) mais n'a donné lieu par contrequ'à peu d'études "in vitro" (16) (17) (18) (19).
Dans le cadre des recherches "in vitro", il nous a
paru intéressant d'étudier la complexation par le D.T.P.Ao, d1élé-ment s de grande toxicité tels que le cerium, l'uranium, le neptu*-
niura, le plutonium et I'américium0 Nous avons choisi d'étudierces éléments, car, parmi les produits de fission, Ce, émetteurP~, a une longue période effective et les éléments transuraniens
considérés, émetteur ot , ne sont pas éliminés naturellement par
l'organisme (20). Par ailleurs, il était également souhaitablede comparer pour ces mômes éléments, le pouvoir complexant dece chélatant avec celui d'autres acides aminopolycarboxyliques,
comme l'E.D.T.A., l'acide triéthylènetétraaminohexaacétique
(Ï.T.H.A.), l'acide 2-2 (bis) di-carboxyméthylaminodiéthyléther-tétraacétique (BoA.E.T.A.), et le méthanesuifonate de la desfer-rioxamine B (desféral).
La chélation des éléments envisagés a été étudiée pourdifférents états d'oxydation, à l'état ionicue et à l'état d'by-droxyde ou de colloïde. Dans le premier cas nous avons recherchél'influence du pH et de la concentration du chélatant sur laformation des complexes. Nous en avons déterminé le rapport mo-léculaire et la constante de stabilité. Les conditions de solubi-lisation des hydroxydes ou des colloïdes ont été étudiées dansle deuxième cas. Nous nous sommes attaché , en particulier ànoter l'influence du pH et de divers paramètres : âge du préci-pité, temps du contact précipité-chélatant.
Pour réaliser ce travail nous avons utilisé la spectro-photométrie d'absorption dans l'ultra-violet et dans le visible.Cette teohnioue permet, en effet, par des méthodes d'analyse
relativement simples de déterminer la composition des complexesdfun élément dans un état de valence donné et de calculer les
constantes de stabilité de ceux-ci.
Nous exposerons dans le premier chapitre les méthodes
expérimentales mises à profit. Le deuxième chapitre sera consa-cré à l'étude des complexes formés entre le D.T.P.A. et le cérium
tri- et tétra-valent à l'état ionique et colloïdal. Dans le troi-
sième chapitre, nous présenterons les résultats concernant lap^c
complexation par ce chélatant de l'uranium ( U), du neptunium
(?57Np), du plutonium (259Pu) et de l'américium (241Am) dansleurs divers états d'oxydation. Après l'étude de chaque élément,
nous donnerons sous une forme succincte les résultats obtenus avecles autres chélatants mentionnés précédemment. Enfin, nous discu-terons l'ensemble des résultats dans un quatrième chapitre.

PREMIER CHAPITRE
MÉTHODES EXPÉRIMENTALES
I - GENERALITES
1 - Réactifs
Les différents acides, bases, sels ou réactifs n'ontsubi aucun traitement préalable avant leur enroloi. Ils étaientde qualité Merck, sauf indication spéciale.
Le chlorure et le sulfate de cérium, ainsi que le chlo-rure d'uranyle étaient de pureté "Analar". Les oxydes tétrava-
lents de neptunium, de plutonium et d'américium fournis par le
Département de Métallurgie du G'0E.A, renfermaient inoins clé 0,2 C3de thorium et d'uranium. L'acide orthochloroplatinique était
rigoureusement pur et les différents chélatants, provenant des
laboratoires "K and K", avaient la pureté "Analar". Snfin,l'amalgame de zinc servant aux réductions a été obtenu en mettant
en contact pendant 20 minutes, des pastilles de zinc de "500 mgpréalablement attaquées par une solution chlorhydrique, avec une
solution de chlorure mercurique à 5 %0
11

2 - Formules et constantes de dissociation des chélatants
L'abréviation usuelle des acides aminopolycarboxyliques
que nous avons utilisés est la suivante :
- E.D.T.Ao pour l'acide éthylènediaminetétraacétique :
HOOC -
HOOC -
IT - CH2 - CH2 - ïïf
CH2 - COOH
- COOH
B.A.3.T.A, pour l'acide 2~2(bis)-di-carboxyméthyleaminodiéthyléthertétraacétiquei
HOOC - \' N - 0 - CH2 -
HOOC - H2 C
CH2 - COOH
- COOH
- D.T.P.A. pour l'acide diéthylènetriaminepentaacétique:
HOOC - H2C
HOOC -
CH2 COOHCH2 - COOH
- N - - CH2 -
C!I2 - COOH
- T.T.II.A. pour l'acide triéthylènetétraminehexaacétique :
HOOC - H2C.
HOOC -
CH2COOH CH2COOHCH2COOH
CH2 COOH
12
Le DESFERAL, métanesulfonate de la desferrjocamine B,peut être considéré comme un acide tri-hydroxamique.
CH,
-N - C-(CH0)0-C-NH-(CH0)(--N - C-(CH9)O-C-M-(CH0)I--N - CI II 2 2 || 2 5 II 2 II 5 l IIOH 0 0 OH 0 0 OH 0
Les pK de dissociation que nous avons rassemblés dansle tableau 1 ont été déterminés par différents auteurs, à 20°C.
Tableau 1
PKp= - log Kp
PK,
PK2
pK3
PK4
pK5
PK6
EDTA (21)
1,99
2,67
6,16
10,26
BAETA (22)
1,75
2,76
8,84
9,47
DTPA (23)
1,80
2,55
4,33
8,60
10,58
TTHA (24)
2,46
2,52
4,00
5,98
9,35
10,33
3 - Techniques spectrophotométriques
Nous avons opéré avec un spectrophotomètre Jobin etYvon, type I-IAROC, dans le cas du cérium et de l'uranium et avecun spectrophotomètre automatique JOUAÏÏ pour les solutions deneptunium, de plutonium et d!américium.
13

Les solutions étaient contenues dans des cuves de quartz,parallélépipédiques, de section carrée de 1 cm de côté, de 5 cnrde volume et bouchées éinerio Le relevé des spectres a été réali-sé lorsque la solution le permettait, entre 210 mu. , limite de
détection des deux appareils, et 1.100 my- , tous les 2 m^L au-tour des maxima et tous les 4 mu. entre ceux-ci. Toutes les me-sures d'absorption ont été effectuées à environ 25°C, par rapportà un témoin de même composition que l'échantillon à mesurer, mais
exempt de l'élément étudié.
Les solutions ont été conservées dans des tubes de
plexiglass, hermétiquement fermés, à l'abri de la lumière et àla température ambiante. Les pH des solutions ont été mesurés avecun pHnètre "BIOLYON" type 71 avec des électrodes BN et BNA.
Mentionnons pour terminer que toutes les manipulationsconcernant la préparation des solutions de neptunium, de pluto-nium, d'américium et les mesures spectrophotométriques ont étéréalisées en boîte à gants étanche ; celle-ci était blindée dans.
241le cas de Am.
II - PREPARATION DES SOLUTIONS ET DOSAGE DES ELEMENTS DANS LEURS
DIFFERENTS ETATS D'OXYDATION
1 - Préparation des solutions des éléments étudiés
a) Préparâtion_des solutions_de_CeCe x_U_et^
Les solutions de cérium trivalent et tétravalent, et
d'uranium hexavalent, ont été préparées par dissolution dans de
l'eau distillée d'une quantité exactement pesée de Cl^Ce, de
14
(S0,)p Ce ou de UOp Cl , de manière à obtenir des concentrationsrespectivement égales à : 1 ,4 0 10~
4 M ; 0,8 „ 10~4 M et—21,6 o 10" Mo La dissolution complète de (S0,)p Ce a nécessité
l'addition de quelques gouttes d'acide sulfurique.
Les solutions d'uranium tétravalent ont été obtenues— o
en réduisant une solution de chlorure d'uranyle 1,6 0 10" M ,HC1 1 M, par l'hydrogène naissant en présence d'acide orthochlo-
roplatinique (25), ce dernier étant utilisé dans la proportionde 5 % en poids du chlorure d'uranyle traité. La solution ini-tialement jaune prend au bout de 3 heures la coloration verte
IVcaractéristique de U tandis que le catalyseur donne lieu à laformation d'un précipité noir. L'efficacité de la méthode deréduction a été vérifiée par spectrophotométrie à 645 ma. et à
420 m LC , longueurs d'ondes caractéristiques d'absorption deet de U02
+20 Le rendement est de 100 #.
b) Préparation des solutions de neptunium, de plutonium et•m »yii— "r B^ •• ••• «^ ••• »•• » »j« » » » ^ m »r» •• » ij» •• » •^^_^*»i^»p«M__— H—B. A.-»*JJ- — — »^ — m—— •—^»^— •ihu^^ •• — — — »•— •— -^— — — —
Les solutions mères de neptunium, de plutonium etPd'américium de concentration de l'ordre de 2 0 10 M ont étépréparées par attaque perchlorique (3 N) à chaud d'une quantité
pesée 'd'oxyde HOg. La dissolution de l'oxyde de neptunium anécessité l'addition d'une goutte d'acide fluorhydrique. Nousavons fait bouillir à reflux les solutions de neptunium et de
plutonium, pendant 12 à 15 heures afin d'éliminer éventuellementles polymères de la valence IV (26).
Nous avons ensuite utilisé la méthode coulométrique
(27) pour préparer successivement, en milieu perchlorique 2N leséléments dans leurs différents états de valence : III - IV" - Vet VI o
15

2 - Dosages des elements en solution
Etant donné le mode de préparation des solutions decérium et d'uranium les concentrations en Cesont connues.
a) Dosages des solutionsd'américium
III CeIV, , ulv
KOEHLY (28) a mis au point une méthode electro chimiquepermettant de doser l'américium par coulométrie ; nous l'avonsappliquée au dosage du neptunium et du plutonium.
La durée de l'opération est de 90 minutes. On calculela concentration en élément de la solution d'après la relationsuivante :
C = Q / U
où : C est la molarité de la solution à doser; Q est la quantitéd'électricité , exprimée en coiilombs, mise en jeu pour passer del'état de valence N à l'état N + 1 ; U est le volume de
•7la prise d1 essai en cm . La précision de cette méthode de dosage
est de 0,3 %.
b)
Ce dosage a été utilisé pour confirmer les résultatsobtenus avec la méthode précédente. Il consiste à dissoudrel'oxyde dans un mélange d'acide nitrique 1 1 N et f luorhydri que0,1 N.
16
Le neptunium, le plutonium ou l'américium sont oxydésau moyen d'oxyde argentique, jusqu'à la valence VI, puis réduitsjusqu'à la valence IV en présence d'acide sulfaminue par unequantité connue de Pe en excès, qui est titrée en retour par lesulfate cérique. Le point équivalent est determine par potentio-métrie à intensité constante0 Cette méthode donne une précision
de 0,5 #.
3 - Préparation des mélanges élément-chélatant.
Nous avons préparé les solutions complexantes des élé-ments étudiés de deux façons différentes :
- Pour étudier la complexation des éléments à l'état ionique,les solutions mères acides, perchloriques ou chlorhydriques1 à 3 N» de l'élément et C-:-.\ chélatant sont mélangées volume à
•zvolume (2 cm ) et agitées vigoureusement » Puis, par additionde quelques gouttes de NaOH ou Î\ŒLOH 1 ou 0,1 N, le pH dumélange est ajusté à une valeur déterminée comprise entre 1et 11 . On amène finalement le volume à 5 cm avec de l'eaubidistillée et on vérifie le pH«
- Pour étudier la chélation des éléments à l'état de précipitéd'hydrolyse ou à l'état de colloïde on ajuste indépendammentle pH de la solution de l'élément et celui du chélatant avantde réaliser le mélange à volume égalo
Dans les deux cas, les mesures sont effectuées aprèsavoir éliminé, s'il y a lieu, le précipité par une centrifuga-tion de 30 minutes à 5«000 tours par minute.
17

Enfin pour suivre l'influence du temps de contact surla complexation, le surnageant est rerais aussitôt après la mesureen contact avec le précipité ; on l'agite vigoureusement et lasuspension ainsi obtenue est conservée à la température ambiante0
III - METHODES ANALYTIQUES
Nous avons utilisé différentes méthodes pour déterminerla stoechiométrie de l'équilibre schématique :
raM + nB \Bn (1)
traduisant la formation d'un complexe M Bn à partir de l'élémentà l'état de cation MN+ et du complexant B = H Y"~p.x-p
1 - Méthode de variation moléculaire
Le principe de cette méthode consiste à mélanger uncertain volume constant d'une solution de concentration donnéeen M ou B avec un même volume d'une gamme de solutions de con-centration variable en B ou M. Dans un premier temps, la concen-tration totale de l'élément ["M], est constante et celle duchélatant [B] varie ; dans un deuxième temps on maintient [B] .constante tandis que M varie.
Cette méthode permet de déterminer le rapport molécu-laire pour lequel la réaction a lieu. Pour cela on construit legraphique représentant la densité optique à la longueur d'onded'un maximum caractéristique du complexe, en fonction du rapportdes concentrations [w]t / [B]t . Si la réaction est quantita-
18
tive, celui-ci présente deux portions linéaires dont l'intersec-tion est le point équivalent. Dans le cas contraire, les deuxportions linéaires se raccordent par une courbe, c'est pourquoiil est souvent nécessaire de confirmer le rapport moléculaire ducomplexe par une autre méthode.
2 - Méthode basée sur le rapport des pentes
Cette méthode introduite par HARVEY et MOOTING (30)permet de connaître la composition du complexe à partir des pen-tes des courbes obtenues en portant en ordonnée la densité opti-que au maximum d'absorption du complexe et en abscisse la concen-tration de l'un des composants tandis que l'autre est en excès etvice-versa.
Le rapport des pentes de ces deux droites donne lerapport moléculaire du complexe formé. Cette méthode s1 appliquedans le cas où le système étudié ne donne pas lieu à la forma-tion de plus d'un ou de deux complexes.
3 - Méthode de variations continues
Cette méthode a été développée initialement par J03(31). Elle permet à la fois de déterminer le rapport r = -Tr-et la constante K de l'équilibre (3)t généralement dans le casoù un seul complexe prédomine. Pour cela on fait varier à pHconstant le rapport des concentrations totale de M et B de tellemanière que leur somme [M]. + , = G reste constante.
Lorsque [B], = [B] + n [ M B ] = x G augmente vis-à-vis de [M] = (1 - x) G - n [M B ] , la concentration du cora
19

plexe, et par la même la densité optique à une longueur d'ondecaractéristique de celui-ci, varie linéairement en fonction de
[B],x = —-z , avec une pente positive avant le point équivalent
°oet avec une pente négative après ce point. La concentration ducomplexe est maximale pour :
x = nm + n 1 + r
réactionnulle
On obtient ainsi la valeur
de r «
Sur la figure 1 nous avons re-présenté une courbe réelle(32) des variations de [ Bavec x . L'axe des abscissescorrespond à une réaction ri-goureusement nulle et les
0%B 100 %B
droites D. etquantitative.
à une réaction
Figure 1 - voir textePour x = ——— au point A surm + nla courbe réelle, correspondentdeux écarts, l'un £Crt par rap-
port à la réaction quantitative, l'autreréaction nulle. La valeur de Kl'une des deux relations :
•CL par rapport à lase déduit de £ ou £ ' d'après
™ v,m n1
m + n
m+n mm nn ( 1m + n
m + n - 1
-_£•)t'
m + n(2)
20
La constante de formation K que l'on détermine expéri-
mentalement correspond à l'équilibre
n Hx-p n (3)
qui est valable au pH donné où prédomine le coordinat HX Y~p
libéré par le chélatant.
Généralement on introduit la constante de stabilité, K',qui correspond à la formation du complexe écrit sous la forme
Mm Yn (Nm-nx) à .partir des constituants MN+ et Y-x
m MN+ + n M (Nm-nx)m n (4)
II existe une relation entre K et K 1 . Elle s'établit
en tenant compte des équilibres (3), (4) et des constantes Kde dissociation du chélatant :
= K / (Kp+, .
Signalons pour terminer que la méthode des variationscontinues peut être généralisée au cas où plusieurs complexes destabilité suffisamment différente se forment (33) (34) (35).
Dans toute notre étude nous avons exprimé les constan-tes de stabilité en fonction des concentrations et des coeffi-cients d'extinction molaire £ en mole" .cm" .1.
21

DEUXIEME CHAPITRE
ÉTUDE DES COMPLEXES CÉRIUM-DTPA
Nous exposerons en premier dans ce chapitre les condi-'tions de formation de certains complexes de cerium tri- et têtra-valent avec le DTPA, l'élément se trouvant soit à l'état ioniquesoit sous forme d'un précipité d'hydrolyse.
On sait que le cérium trivalent est en milieu non com-plexant à l'état de cation incolore, Ce , jusqu'à pH 5 > 2 ('56).La formation de l'hydroxyde de cérium à pH 7,5 pour fCeJ ~10~ M,est précédée de celle de cations hydrolyses ("57) tels que CegCOH)^Cet hydroxyde donne en présence cl1 air un mélange de composition :2 Ce ( O H ) , , Ce (OH)., qui s'oxyde en Ce (OH) , avec le temps etcela d'autant plus rapidement que le pH est élevé.
Le cérium tétravalent est beaucoup plus acide que Ce .Il existe en solution aqueuse acide sous forme cationique
*2T j *"> i
Ce (OH) jaune et Ce (OH)? orange (35). L'hydroxyde, jaune
clair précipite vers pl-î 1 même en milieu suifurique.
23

I - ETUDE DE LA COMPLEXATION DU CERIUM TRI VALENT A L'ETAT IONIQUE
Nous avons initialement relevé les spectres d'absorptionde deux solutions chlorhydriques à pH 3,5:l'une 2,8.10" M en DTPA,
—4 +3l'autre 7.10 M en Ce . Nous avons également mesuré l'absorptiond'un mélange Ce5"1" = 7.10" M et DTPA = 2,8.10"5 M au même pH0
La figure 2 représente les trois spectres obtenus. Celuide Ce (spectre 1) présente quatre maxima à 212 (£= 100),222 (6 = 170), 240 (£ = 450) et 252 (£= 00) mu, ce qui est enaccord avec les données de la littérature (38). Le DTPA n'absorbepas au delà de 270 mu (spectre 2) et par conséquent n'interférerapas avec les maxima d'absorption du ou des chélates qui dans lesconditions définies ci-dessus se situent à 270 mu. et 298 mu. 0
1 - Variation du spectre d'absorption en fonction du pE
Nous avons étudié l'évolution du spectre d'absorptiondu mélange ci-dessus en fonction du pH (figure 3)o La figure 4représente les variations de la densité optique à 270 et 298 mu.nvec le pH0 Elle indique que pour un tel mélange, où[Ce111] /[DTPA] = 1/4, la totalité de Ce111 est complexée à par-tir de pli 3,5 ; par ailleurs elle montre que la densité optique
TTT
à 298 mu. x>ermet de suivre l'apparition des complexes Ce -DTPA.
2 - Détermination du rapport moléculaire des complexes
a) Méthode_devariationmoléculaire à
Si la concentration du chélatant est fixée à 7.10"4 M—4. —4-et que la concentration du cérium varie de 10 à 20010 M, on
24
observe à 298 mu. une augmentation rapide de l'absorption avecl'augmentation de la concentration du cérium jusqu'à un rapport[Ce111]/[DTPA] égal à 1/1 (figure 5). Au delà de ce rapport,la densité optique augmente plus lentement jusqu'à un rapportrCeIIIl/[DTPA] égal à 2/1 ; ensuite elle ne varie plus.
Inversement, les variations de la densité optique à298 myu. en fonction de [DTPA] , lorsque [ceI3:I]= 7.10"4 M sontreprésentées sur la figure 6. On observe des discontinuités del'absorption pour [Ce111]/[DTPA] = 2/1 et 1/1. Les spectresd'absorption correspondant à ces différents rapports de concen-tration sont réunis sur les figures 7 et 8.
On a préparé à pH 3,5, d'une part des solutions decérium à différentes concentrations (10*"4 à 10""-5 M) auxquelles<2on a ajouté un excès de DTPA (4.10 M) et d'autre part dessolutions de DTPA (10 à 10" M) contenant chacune un excès de
—pcérium (4.10 M). La figure 9 représente les variations de ladensité optique à 298 min. , des solutions Ce - DTPA, en fonc-tion de [ce111] et de [DTPA].
Pour ICe I variable et F DTPA] fixé en excès, lar ni Tvariation est linéaire en fonction de [Ce J (courbe 1). Dans
le cas où [DTPA] varie et où |_Ce J est constante, elle estreprésentée par une courbe comprenant deux parties linéaires(courbe 2). Les rapports entre la pente de la première droiteet celles des parties linéaires de la seconde courbe indiquentla formation de deux complexes, caractérisés par des rapportsr = Ce111 / DTPA égaux respectivement à 2/1 et 1/1.
25

c) Méthode de variâtions_continues
Nous avons confirmé les résultats obtenus, par la mé-thode de variations continues. La concentration totale du cériumet du DTPA était égale à 1,1.10 M pour toutes les solutions ;nous avons travaillé à pH 3,5 et effectué les mesures à 298 myuu ,La courbe de la figure 10 met en évidence deux maxima pour desrapports [cei:CI]/[DTPA] égaux à 1/1 et 2/1.
d) Calcul des constantes de stabilité des complexes
Les valeurs des constantes de formation des complexesr = 2/1 et 1/1 ont été déterminées à partir de la relation (2)et des valeurs des densités optiques lues sur la courbe de lafigure 10. On donne dans le tableau 2, log K, log K1 et lesvaleurs des coefficients d' extinction molaire de ces complexes»
Tableau 2
r
2/1
1/1
X mu,
298
298
L
150
195
log K
9,5
4,5
log K'
15,7
20,3
Pour calculer log K et log K1 nous avons admis qu'à_•*
pH 3,5 l'ion Hp Y J est prépondérant et que le cérium trivalentest complexé selon les équilibres î
2 Ce5+ + H2 Y""5
Ce5+ + H2 Y"5 Ce
2 H
2 H
+
26
rt1L'existence du complexe Ge^ Y n'avait pas été prou-vée jusqu'à ce jour, par contre MOELLER (23) avait déjà calculé
-2la constante de stabilité du complexe r = 1/1, Ce Y , et trouvélog K1 = 20,5 j nous obtenons Log K1 =20,3.
En résumé pour les pH supérieurs à 3,5, en présence deDTPA, Ce existe à l'état de deux complexes dont les rapportsmoléculaires sont successivement 2/1 et 1/1, du moins pour10~4 M < [cei:EI]<2.10~5 M . Ces complexes absorbent à lamême longueur d'onde, mais leurs coefficients d'extinction molai-res sont différentSo
Des mesures complémentaires ont montré qu'en présenced!un excès de DTPA et pour les pH supérieurs à 8 , le complexede rapport 2/1 n'est pas stable, il se forme uniquement le com-plexe de rapport r = 1/1» A ces pH le coordinat prédominantlibéré par le DTPA dans les solutions est
II - ETUDE DE LA COMPLEXATION DU CURIUM TETRAVALENT A L'ETAT IONIQUE
1 - Variation des spectres d'absorption en fonction du pH
Le spectre d'absorption d'une solution suifurique 0,2 N,2.10""4 M en cérium tétravalent présente un maximum à 320 mu.( <1 = 5 770),(figure 11, spectre 1 )0 La position de celui-ci etle coefficient dfextinction molaire que nous avons calculé sont enaccord avec les valeurs publiées (39).
L'absorption d'une solution [ceIV] = 2.10""4 M , [DTPA] =1.10~5 M, caractérisée par un rapport [ceIV]/[DTPAJ =1/5 , aété mesurée en fonction du pH, 24 heures après sa préparation(figure 11). La solution de CeIV était initialement à pH 0,8»
27

Aux pli inférieurs à 2, le spectre présente des maxima
vers 240 et 252 m/*. c'est-à-dire à des longueurs d'onde où Ce^+
absorbe; à pH 2,5 un nouveau maximum apparaît à 270 H A. ; à pH 3on l'observe toujours mais on note aussi l'apparition d'un épau-
lement vers 290 nu*.,,
Au delà de pH 4,5 le spectre présente deux maxima à270 et 298 m x. et se confond avec celui d'une solution Ce -DTPA
de même concentration en élément et en chélatant.
Après les mesures, les solutions sont stockées et leur
absorption est mesurée à intervalles de temps réguliers „ Lesspectres observés après trois semaines, révèlent au delà de pH 3,deux maxima à 270 et 298 myut ; aux pH inférieurs, ils présentent
deux bandes d'absorption à 254 et 294 m u. 0
II paraît logique de déduire de ces résultats que le
pH le plus favorable pour mettre en évidence la présence de com-TTT #
plexe Ce -DTPA est pH 2,5. En effet, lorsque le pH atteint 3,l'apparition d'un maximum à 270 mu, et d!un épaulement à 290 mu.
IV IIIindique un effet de réduction de Ce en Ce , que confirme
le spectre obtenu aux pH supérieurs à 4,5» qui est identique à
or7A270= 200. Le vieil-celui du complexe CeIi:E-DTPA 1/1 £
lissement des solutions favorise cet effet de réduction, jusqu'à
pH 2,5 , puisqu'un mélange où |CeIVl/PDTPAl = 1/1 , âgé de 4TTT
mois, absorbe la lumière comme le complexe Ce -DTPA-1/1.
2 - Détermination du rapport moléculaire du complexe à pH 2.5
a) Méthode de variation moléculaire
JVNous avons préparé des mélanges à pH 2,5 Ce"4 < > 1 0 M, [DTPA! variable de façon à obtenir des rapports
* dans les solutions d'âge inférieur à 24 heures,,
28
de concentration [ceIV]/[DTPAJ allant de 4/1 à 1/4, En admettantqu'à ce pH les mesures de densité optique à 270 mu- , effectuées
le plus tôt possible après la préparation des solutions sont
caractéristiques de la complexation de Ce , on peut déterminer
un rapport moléculaire égal à 1/1 comme le montre la figure 12.
b ) Méthode_des_variations__continues
A ce pH cette méthode conduit au même résultat (figure
13) et permet de calculer la constante de formation du complexe
1/1 : log K = 4, et sa constante de stabilité : log K = 22,06 ;
pour ce calcul on a supposé que le coordinat KL Y~ participe
seul à la formation du complexe.
Soulignons que le complexe Ce -DTPA 1/1 absorbe à la
même longueur d'onde que celui de Ce , 270 mu. , mais son coef-
ficient d'extinction molaire est bien plus élevé (£=2 774)0Par ailleurs la bande d'absorption est environ quatre fois plus
large à mi-hauteur que celle attribuée au complexe Ce -DTPA 1/1,
3 - Influence de la concentration du DTPA sur 1'effet réducteur
II était intéressant de préciser l'effet réducteur du
chélatant. Pour cela nous avons opéré à pH 3,5 et 6, pH les plus
favorables pour mettre en évidence et étudier l'influence de laconcentration du DTPA sur la réduction de Ce
A pH 6 (figure 14), pour une faible proportion deDTPA, l'hydroxyde de Ce précipite ; il se dissout ensuite pro-
gressivement lorsqu'on augmente la concentration en chélatant
et disparaît complètement pour un rarmort [Ce J/[DTPA] = 2/1.
Le spectre présente alors des épaulements vers 250 et 300 mu.
et un maximum à 270 mu.. Lorsque [ce J/[DTPA] =1/1 le spectre
29

obtenu trois heures après la préparation de la solution est celui
du complexe de CeIi:E-DTPA-2/1. ( t = 150 à 270 et 298
A pH 3,5 , on constate les mêmes phénomènes mais leur
apparition est plus lente. Le spectre d'absorption présente24 heures après la préparation du mélange un maximum à 270 mju.et un épaulèrent à 294 m u. , la solution est jaune pâle ; toutle cérium est complexé pour un rapport [Ce J /[DTPA] =1/1. Lors-
que le rapport de concentration devient inférieur à 1/1, l'ab-sorption diminue sur tout le spectre et le maximum à 294 ma,apparaît nettement. Le spectre tend vers celui du complexe CeDTPA-2/1 et devient identique à celui-ci après une semaine.
Ces observations montrent que le chélatant introduiten faible quantité dans les solutions de Ce agit comme un ré-
ducteur pour pH>3. Il est employé en totalité pour la réductionde Ce , car on ne décèle dans ces conditions que la présence
de Ge')+0 Nous avons effectivement vérifié que de l'anhydridecarbonique se dégageait, ce qui traduit une dégradation du DTPA0Si on augmente le rapport des concentrations chélatant-élément,le cérium est alors engagé dans un mélange de complexes Ce -DTPA et Ce -DTPA-1/1, mais en fonction du temps, le complexede Ce disparaît et seul subsiste celui de Ce de rapport
r = 2/1 ( £ = 155 à 270 et 298 m^c). Par ailleurs l'effet de
réduction est favorisé par l'augmentation du pli.
III - ETUDE DE LA COMPLEXATION DU CERIUM TRIVALENT A L'ETAT D'HYDROXYDE
L'absorption des solutions obtenues comme il a été ditdans le premier chapitre, est mesurée en fonction de l'âge du
précipité d'hydrolyse et en fonction du pH. Les concentrations dessolutions sont respectivement 1,4.10 M et 5,2.10 M pour lecérium et le DTPA.
30
1 - Influence de l'âge du -précipité et du pH sur la formationdu complexe
Les solutions de cérium à différents pH sont conservéespendant des temps allant de 5 minutes à 24 heures, avant l'addi-tion du chélatanto On procède aux mesures d'absorption 24 heuresaprès avoir ajouté du DTPA, dans les conditions déjà décrites.Dans tous les cas le spectre relevé présentait des maxima à 270et 298 m u. 7 aussi avons-nous reporté sur la figure 15 les varia-tions de la densité optique à cette dernière longueur d'onze, en
fonction du pH, pour des âges différents de l'hydroxyde de Ce .
Si le DTPA est introduit dans les solutions de cériumtrivalent dès que leur pH est ajusté à la valeur appropriée, lacourbe obtenue est rigoureusement identique à celle observée n-mcle cas de Ce- pour 3,5 < pH < 11, (figure 15 courbe 1)0
Ce est totalement complexé par le DTPA lorsquecelui-ci est ajouté 5 minutes après la préparatior de l'hydroxyde
pour pH < 6. Entre pH 6 et pH 7, on constate une légère augmen-tation de l'absorption. On n'observe pas de variation entre
pH 7 et pH 9, mais une augmentation progressive de la densitéoptique s'amorce au delà de pH 9 (courbe 2).
Dans le cas d'un hydroxyde colloïdal âgé de 10 minuteson remarque entre pH 6 et pH 7 que 1' absorption est légèrementplus importante que pour un temps de vieillissement de 5 minutes,,Cette différence s'accentue fortement entre pH 7 et pH 12 (cour-
be 3)o Au delà de pH 12, une partie de l'hydroxyde n'est pas com-plexé e0
Lorsque le DTPA est ajouté après 30 minutes, l'évolu-tion se fait toujours dans le même sens, avec cette fois une
31

augmentation très nette de l'absorption entre pH 7 et pH 10,5 ;mais au delà de pH 11, la formation du complexe n'est pratique-
ment plus possible (courbe 4)o
Si le DTPA a été ajouté en vue de complexer un hydro-xyde de Ce âgé de 3 heures, l'absorption augmente toujoursentre pH 6 et pH 7, devient importante entre pH 7 et pH 8 etdiminue entre pH 8 et pH 9 (courbe 5)<>
Pour un hydroxyde âgé de 5 heures, on observe une absorp-tion très importante entre pH 6 et pH 7 ; pour des pH supérieurs,la chélation est très faible (courbe 6). Enfin, lorsque le DTPA
n'a été ajouté qu'après 24 heures, la quantité de cérium complexéest minime et même nulle au delà de pH 7 (courbe 7)»
2 - Discussion et interprétation des résultats
Les différences d1absorption observées en fonction dupH et du temps de vieillissement de 1'hydroxyde de Ce peuvent
s'expliquer ainsi :
- Jusqu'à pH 5,5 quel que soit l'âge de la solution deCe111, l'addition de DTPA en excès conduit au complexe Ce -DTPA,r = 1/1 que nous avons mis en évidence précédemment.
- Entre pH 7,2 et pH 12, l'augmentation rapide de ladensité optique observée en.fonction de l'âge de l'hydroxyde deCe résulte de l'oxydation de celui-ci en hydroxyde de Ce etde l'apparition dans la solution de complexe Ce - Ce -
DTPA-1/1» Effectivement, nous avons mis en évidence la présencede Ce par un dosage de 1' orthophénantroline. Le complexe Ce -
DTPA-1/1 absorbant comme nous l'avons vu entre 220 et 360 m Lu ,IIImais beaucoup plus que celui de Ce contribue donc à l'augmen-
tation rapide de la densité optique vers 298 m »*- „ On observe
32
également dans cette zone de pH que la complexation de l'hydroxydecolloïdal s!effectue d'autant plus difficilement que celui-ciest plus âgé. Cela indique une transformation de celui-ci versune forme non complexable,
La figure 15 montre en outre que pour des pH comprisentre 5,5 et 7,2 le vieillissement d'une solution de Ce entraî-ne aussi une augmentation de la densité optique mais proportion-
nellement moins importante que pour lespH plus élevés auxquelsnous venons de nous intéresser.
Dans ce dernier domaine de pH, selon MIRONOV (40) et .
TANANAEV (41) l'hydroxyde de cérium III aurait pour formuleC0p (OH)j- Clo Au contact de l'air et en fonction du temps, ilse transformerait en Ce (OH), qui à son tour s'oxyderait à
Ce (OH),. Ainsi s'expliquerait le retard constaté dans la forma-IV
tion du complexe de Ce -DTPA-1/1» Après 15 heures de vieillis-sement du colloïde de Ce , ce serait donc principalement cecomplexe que 1' on rencontrerait dans la solution ; mais après24 heures, la chute brutale de l'absorption serait due à l'appa-
rition de la forme non complexable de l'hydroxyde de CeIV
IV - ETUDE DE LA COMPLEXATION DE CERIUM TETRAVALENT A L'ETAT D»HYDROXYDE
Pour effectuer cette étude, on ajuste le pH d'une solu-tion de cérium tétravalent 4.10" M, de façon à obtenir des va-
leurs comprises entre 1,5 et 10, toutes les 0,5 unités. Aprèsdes temps de vieillissement du colloïde de 30 minutes à 27 jours,
on ajoute un égal volume d'une solution de DTPA, 2,8.10"" M, au
même pïï. Les mesures d1absorption sont effectuées 24 heures après,le mélange étant centrifugé s'il y a lieu»
33

1 - Influence de l'âge du précipité et du pH sur la formationdes complexes
Pour un temps de vieillissement de l'hydroxyde de 30-mi-nutes, à pH 1,5, le spectre d'absorption présente un maximumvers 255 n u,. La solution est jaune pâle0 A pH 2, l'absorptionaugmente et le maximum se trouve déplacé vers 264 muo La solu-tion est jaune. A pH 2,5 l'absorption continue à croître et lemaximum se situe à 270 mu.; la solution est alors jaune trèsintense» A pH 3, le spectre présente, outre ce dernier maximum,un épaulement vers 298 mutinais l'absorption diminue à toutes leslongueurs d'onde. La solution est moins colorée que précédemment.A pH 3,6, l'absorption est plus faible qu'à pH 3, la solution estincolore0 A pH 4, l'hydroxyde n'est pas totalement complexé ; lespectre d'absorption relevé sur le surnageant ressemble à celuidu complexe Ce -DTPA-1/1. Aux pH supérieurs l'hydroxyde n'estplus du tout complexée L'ensemble de ces observations est traduitpar les spectres de la figure 160
Nous avons répété cette série de mesures pour des préci-pités âgés de 1 à 30 jours» Les courbes présentées sur la figure17 montrent le comportement du chélatant en présence de l'hydro-xyde pour différents pH, en fonction de l'âge de celui-ci. Leevaleurs notées correspondent à l'absorption à 270 m/4, longueurd'onde d'absorption du complexe Ce -DTPA-1/10 (£ = 2770)
Ainsi quel que soit l'âge de l'hydroxyde et pour un temps
de contact de 24 heures, la complexation est totale jusqu'à pH3,6 , mais on obtient selon le pH un complexe de degré d'oxyda-tion différent.
34
Pour un hydroxyde précipité depuis 30 minutes, lecérium est complexé à l'état tétravalent jusqu'à pH 2,5 , commele confirme l'absorption importante à 270 m M. . La solution estjaune.
De pH 2,5 à pH 3,6 tout le cérium est engagé dans Lesohélates de Ce 1/1 et de Ce 1/1 . En effet un maximum appa-raît à 298 m.t* et l'absorption diminue à 270 m^» A pH 2,5 lasolution est jaune clair et à pH 3,6 elle est incolore.
Entre pH 3,6 et pH 4,6 , le colloïde n'est que partiel-lement complexable et au delà de pH 4,6 on ne remarque plus aucuneaction du chélatant.
Ce pH de complexation limite est inférieur pour deshydroxydes plus âgés.
2 - Influence du temps de contact du précipité d'hydroxyde avecle chélatant
II nous a paru intéressant d'étudier l'influence dutemps de contact entre le chélatant et le précipité d'hydroxyde,dans le but d'une part de mettre en évidence un éventuel effetde dépolymérisation et d'autre part de vérifier l'effet réducteur
IVdu DTPA, déjà observé pour Ce ionique»
Comme nous l'avons fait après 24 heures de contact,nous avons relevé au cours du temps, le spectre d'absorption d'unmélange effectué après 30 minutes de vieillissement de l'hydroxyde,Pour cela, après chaque mesure de l'absorption du surnageant,nous avons remis celui-ci en contact avec le précipité d'hydro-xyde et conservé la suspension ainsi obtenue à la températureambiante.

- Mesures après 7 jours de contact
Le spectre relevé accuse une diminution générale del'intensité pour pH < 3,6 et une augmentation pour pH > 3,6 ;corrélativement la quantité de cérium non complexé diminue.
JVAun'estdelà de pH 7, l'absorption est nulle ; 1'hydroxyde de Ce"
pas complexé.
- Mesures après 12 jours de contact
On observe par rapport au cas précédent une diminutiongénérale de l'absorption, plus ou moins importante selon le pH.Les densités optiques obtenues entre pH 5,5 et pH 6,5 sont plusélevées qu'entre pH 5 et pH 5,5. Au delà de pH 7 la chélationest toujours nulle.
- Mesures après 21 jours de contact
D'une façon générale, l'absorption diminue : tout l'hy-droxyde est bien solubilisé jusqu'à pH 4, mais au-delà, une partieseulement est en solution et pour pH } 1 il n'y a toujours pasde chélation.
- Mesures après 2 mois de contact
La diminution de l'absorption par rapport au cas pré-cédent est importante aux pH faibles. De pH 4 à pH 7 au contraire,l'absorption est notablement augmentée et la fraction de Ce àl'état de précipité a beaucoup diminué. Au delà de pH 7, l'ab-sorption est maintenant sensible mais la solubilisation n'estque partielle et elle devient nulle aux alentours de pH 9.
- Mesures après 8 mois de contact
Jusqu'à pH 8,8 tout le cérium est complexé et les spec-tres d'absorption obtenus sont identiques qualitativement etquantativement à ceux de Ce111 - DTPA-1/1 . Au delà de pH 8,8une partie du précipité d'hydroxyde n'est pas solubilisée.
Nous avons également étudié l'influence du temps decontact du DTPA sur des hydroxydes d'âge allant jusqu'à 27 jours»Ils sont d'autant plus difficiles à solubiliser qu'ils sont âgés.Par exemple dans le cas d'un, hydroxyde âgé de 27 jours, un tempsde contact de sept mois avec le DTPA, n'est pas suffisant poursolubiliser 1'hydroxyde au delà de pH 7.
La figure 18 représente la variation de la densitéoptique à 270 mu. en fonction du pH, pour des solutions obtenues
IVpar action du DTPA sur des hydroxydes de Ce âgés de 30 minutesà 27 jours et pour différents temps de contact précipité-chélatant.
Ces derniers résultats conduisent aux conclusions sui-vantes
Lorsque l'on augmente le temps de contact hydroxyde-chélatant, la diminution continue observée sur tous les spectresentre pH 1 et 3,6 peut s'expliquer par la réduction de Ce encomplexe de Ce d'où la diminution de l'absorption à 270 m
Pour les pH compris entre 4 et 8,8, l'augmentation dutemps de contact entraîne une disparition du précipité d'hydro-xyde de cérium tétravalent ; elle s'accompagne d'une augmentationde la densité optique qui est due à la formation du complexe de
37
Ce -DTPA-1/1. Celui-ci est par la suite réduit, en fonctiondu temps, en complexe de Ce -DTPA-1/1.
V - CONCLUSION
Les résultats que nous avons exr>osés dans ce chapitremontrent que le cerium trivalent forme deux complexes solubleset stables avec le DTPA entre pH 3,5 et pH 9 dans les rapportsmoléculaires 2/1 et 1/1. Les logarithmes de leurs constantes destabilité K1 sont respectivement 15,7 et 20,3.
A l'état têtravalent, la formation d'un complexe 1/1 aprincipalement lieu à pH 2,5 (log K' = 22,06). Ce complexe estréduit par le DTPA en complexe Ce -DTPA-1/1. Deux facteursfavorisent cet effet : l'augmentation du pH, l'augmentation dela concentration en chélatant.
L'hydroxyde de Ce frais qui précipite à partirIIIde pH 5,5 est totalement solubilisé sous forme de complexe Ce
DTPA-1/1 en présence d'une quantité suffisante de DTPA. L'aug-mentation du pH et de l'âge de l'hydroxyde rend la complexationde plus en plus difficile par suite de l'oxydation de Ce enCe au sein du précipité et de l'évolution de l'hydroxyde deCeIV.
Ces derniers résultats ont été confirmés par l'étude dela complexation de l'hydroxyde de Ce en fonction du pli, de sone et du temps de contact chélatant-hydroxyde. Celui-ci est
totalement complexé par DTPA jusqu'à pH 3,6 ; au delà de ce pHla fraction complexée diminue. Aucun effet n'est observé pour24 heures de contact, mais avec le temps, le DTPA conduit aucomplexe Ce -DTPA-1/1 qui est par la suite réduit en complexeCem-DTPA-1/1.
38
VI - COMPLEXATIOI DU CERIUM PAR DIFFERENTS CHELATANTS
III IVNous avons étudié la chélation de Ce et Ce par
d'autres acides polyamines carboxyliques. Les résultats que nousavons obtenus pour ce qui concerne l*état ionique sont résumés
ci-dessous, dans le tableau 3<>
Tableau 3
Chélatant
EDTA
BAETA
2THA
Ce111
GeIV
Ce111
Ce1'"
Oe111
Ce1'"'
iDSSPERAL! ce111
iOe1»
pH de for-mation ducomplexe
2,8
3,0
3, S
6
Rapportmolé-
culaire
1/1
1/1
2/1
1/1
2/3
Maximumd'absorp;ion m tu
270
290
280280
•443
£
200
i
250
180
260
20004
i
log K
6,20
4,15
10,20
6,50
1~ ,25
log K'
14,80
16,90
15,4519,20
1F,25
Remarques c oncernant le complexe
de CeIV
Complexe instableréduit en complexe
III Vde Ce à tousles pE
Complexe instableau coi-irs du tempsréduit en complexe
HIde Ce à tousles pli
jiffet de réc'uctioren complexe de
ITTCe a tous lespH
Effet de réductioren cor pi exe deCe , reductionimmédiate pours^^mçp.s
39

Des études quantitatives antérieures de la complexation
du cerium trivalent par l'EDTA (42) (43) (44) (45) ont déjà misen évidence l'existence du complexe de composition 1/1. Elles ont
permis de calculer log K1 = 15,9& ï nous avons obtenu la valeurlog K1 = 14,80 pour ce complexe. A notre connaissance aucun autretravail ne se rapporte à la chélation de Ce et Ce par BAETA,
TTHA, et Desferal.
40
TROISIÈME CHAPITRE
Nous présentons dans ce chapitre les conditions de for-mation de certains complexes d'uranium, de neptunium, de plutoniumet d'américium avec le D T F A.
PREMIERE PARTIE
ETUDE DEÏ3 COMPLEXES URAJTIUM-D T P A
L'uranium tdtravalent est;en absence de complexant, à
l'état de cations U , dans les milieux aqueux suffisamment acides.Le premier stade de l'hydrolyse de cet ion conduit à U (OH) (46),
la constante d'acidité étant égale à ô.10~2 K ( i-.= 0,5) (47), Parailleurs les ions monomères hydrolyses, U (Oil)n oeuventconduire à la formation de polymères. L'hydroxycie, U (OH). , vert,
peu soluble en milieu acide précipite vers prl 1 si la concentra-tion en élément est 10~2 M (48).
Dans l'acide chlorhydrique, l'uranium hexavalent se2+trouve principalement sous forme d'ion uranyle U0? vers pH 2 - 3 .

La précipitation de l'hydroxyde a lieu autour de pH 4,2 ; ellepeut être suivie de celle d'uranates et de polyuranates alcalinsjaunes, peu solubles (49) (50).
Nous avons relové les spectres d'absorption de solutionschlorhydriques 2 N d'uranium tétra— et hexavalent, 8.10 M enélément. Les positions des principaux maxima sont mentionnées dansle tableau 4, ci-après.
Tableau 4
V
U.VI
Couleur dela solution
verte
jaune
Longueur d'onde desprincipaux maxima
d'absorption
468 , 492 , 548 , 645
420 , 428
10 - 1 2 - 8 - 2 0
11 - 13
Ces valeurs sont en accord avec celles déjà publiées
(46) (51).
I - ETUDE DE LA COMPLEXATION DE L'URANIUM TETRAVALENT A L'ETAT IONIQUE
1 - Variation du spectre d'absorption en fonction du pli et du
temps de réaction
L'absorption d'une solution chlorhydrique [U J = 8.10""^ M
-2et [DTPA] = 6,4.10 M, pour laquelle = 1/8, a été mesuréeCDTPAJen fonction du pH0 Pour la gamme de pH choisie, les mesures ont étéréalisées immédiatement et jusqu'à 4 mois après la préparation du
42
mélange, effectué dans les conditions précisées au chapitre I,à partir de solutionr chlorhydriques normales.
Le spectre des solutions fraîchement préparées estcelui de U jusqu'à pH 1,5. Au-delà de ce pH des modificationsont lieu. Ainsi de pH 2 à pH 9, le spectre présente des maximavers 474, 538, 599 m y- et deux autres vers 623 et 650 mix .
Lorsqu'on effectue les mesures 1 heure après la prépa-ration du mélange, le spectre relevé entre pH 2 et 4,8 est iden-tique à celui obtenu ci-dessus dans les mômes conditions. Pourles pH supérieurs à 4,8 et dans la zone de 600 à 700 mu. , l'ab-sorption de la solution considérée diminue, d'autant plus quele pH est plus élevé.
Après 3 heures le spectre observé (figure 19) de pH 2 àpH 4,8 ne présente pas de modifications par rapport aux précédentsmais, pour les pH alcalins, l'absorption continue de décroîtreentre 600 et 700 ny*. . Ce phénomène s'accentue avec l'augmentationdu pH ; à pH 9 on remarque la disparition du maximum d'absorptionà 623 miL .
Sept heures après, on ne constate pas jusqu'à pH 4,8de différences par rapport aux spectres relevés après 3 heures.Au-delà de ce pïï, la diminution de l'absorption dans la zone de600 à 700 my. est d'autant plus marquée que le milieu est plusalcalin. Dans la zone de 400 à 450 nyx , l'absorption augmente enfonction du pH, niais le maximum d'absorption à 538 mix. diminueet se décale vers les longueurs d'onde plus courtes.
Après 4 mois, on constate une similitude des spectresd'absorption entre pH 2 et 3 ; aux pH plus élevés le spectre
43

présente les mêmes changements que précédemment mais à des pHplus bas.
L'examen de ces premiers résultats montre que l'absorp-tion à 623 et 650 m^ est caractéristique de la complexation deIF par le DTPA. Ainsi complexé U est stable même en présenced'air durant au moins 4 mois, entre pH 1,8 et 3. Aux pH supérieursà 4,8 l'instabilité du complexe croît avec le pE. Cesobservations nous ont permis de poursuivre nos recherches et nousavons tout d'abord choisi de travailler à pH 2,2»
2 - Détermination du rapport moléculaire des complexes
a) Méthode de variation moléculaire
On a fait varier le rapport des concentrations de U—•*
et de DTPA, en fixant à 8.10 J M la concentration de l'uraniumtétravalent. Les premières mesures d'absorption ont été effectuéesà 650 imj 3 heures et 7 jours après la réalisation du mélange. Lafigure 20 présente dans ces conditions les variations de la den-sité optique à cette longueur d'onde en fonction du rapport [U v] / [DTPA].On constate que tout le U lv est complexé pour [U ]/[DTPA] = 1/1.
•Z
Aux concentrations en DTPA inférieures à 8.10~"- M, l'uranium tétra-valent non complexé est à l'état de précipité. En effet à ce pHIF ne reste pas en solution en milieu non complexant.
Si on répète les mesures sur les solutions âgées de3 semaines, on n'observe pas de changement de l'absorption . Parcontre au-delà de ce temps, la densité optique diminue très len-tement à toutes les longueurs d'onde, bien que le spectre restequalitativement inchangé. Elle diminue plus rapidement pour unrapport [ U1 ] /[DTPA] = 1/1 que pour un rapport [UIV]/[DTPA] = 1/2.
44
L'étude de l'absorption de solutions de U - DTPA àpH 3 t 4 et 5,5 toute chose restant égale par ailleurs indiquequ'un complexe U - DTPA se forme comme précédemment pour unrapport 1/1, En fonction du temps le complexe 1/1 est moins sta-ble que pour un rapport de concentration lu J/[DTPAJ = 1/20
b) Méthode de variations continues
La concentration totale des solutions d'uranium et deP
chélatant a été maintenue à 1,6.10" M. Les mesures ont été effec-tuées à 650 nyu f 3 heures après la préparation du mélange. Ellessont présentées figure 21 et indiquent bien la formation d'uncomplexe U - DTPA-1 /1 « Cette méthode appliquée à pH 3 et 4 con-duit à des résultats identiques.
La méthode de variations continues n' est applicableau calcul des constantes de stabilité que lorsque la concentrationtotale des réactifs est constante. Or nous avons observé qu'audelà d'un rapport de concentration [u j/[DTPA] = 1/1 l'uraniumprécipite ; la partie pointillée de la courbe de la figure 21n'est donc pas valable,, Pour calculer la constante de formationdu complexe 1/1 nous avons opéré sur la partie descendante ettrouvé log K = 4,2. On en déduit log K» = 21,60 puisqu'à pH 2,2ce sont les ions H,Y qui prédominent. Les mesures à pH 3 et 4,conduisent à la môme valeur.
L'ensemble de ces résultats montrent, que le complexede rapport moléculaire 1/1 ( £ ,cn „ = 20) se forme à partir deopu mi».pH 1,5 ; il est stable entre pH 1 ,8 et 3 pendant au moins quatremois, néanmoins sa stabilité diminue avec l'augmentation du pHet du temps o L'excès de DTPA dans la solution assure sa stabili-sation ; en effet pour un rapport [ïï J/(DTPAj = 1/2, on remarque
45

que la diminution de l'absorption en fonction du temps, sur l'en-
semble du spectre est moins importante que pour un rapport 1/10 Enabsence d'un excès de DTPA, l'uranium têt ravalent s'oxyde, ce quidéplace l'équilibre de complexation vers la gauche» Cela explique
l'instabilité du complexe UIV - DTPA-1 /1 dans de telles conditions.
II - ETUDE DE LA COMPLEXATION DE L'URANIUM HEXAVALENT A L'ETAT IONIQUE
1 - Variation du spectre d'absorption en fonction du
Nous avons relevé les spectres d1 absorption d'une solu-
tion de I)VI, M et de DTPA , 6,4. 10"2K à diffé"rents pH. Les mesures ont été effectuées 24 heures après avoir
réalisé le mélange.
De pH 1,5 à 3, l'ion uranyle donne avec le DTPA un pré-
cipité blanc granuleux. Le surnageant n'absorbe pas. A pH 3 le
précipité commence à se solubiliser, il est totalement dissousà pH 3,5 et la solution est jaune, son spectre d'absorption pré-
sente un maximum à 422 mu. entouré de deux épaulements à 410 m -
et 430 ma. . Un minimum d'absorption se situe à 388 myx6
Aux pH supérieurs à 3,5, le spectre d'absorption subit
des modifications successives comme le montre l'examen de la fi-
gure 22.
Ainsi à pH 4,5 l'épaulement de 410 myu. a presque dis-
paru, le minimum est passé de 388 à 398 mi*, et la densité optique
a augmenté sensiblement à toutes les longueurs d'onde. Vers pH 5,6
le spectre présente deux maxima à 422 et 432 mu. et un minimum à
403 mu., La densité optique continue à croître.
46
A pH 6 l'intensité du spectre augmente toujours et
le maximum de 432 myu. s'est déplacé à 430 mi*.. Il s'atténue àpH 6,5 et le minimum se situe à 392 mu.0
Le spectre d'absorption reste inchangé jusqu'à pH 8 ;au-delà on observe la précipitation d'hydroxyde d'uranium hexa-valent. Enfin, à pH 12 tout l'uranium précipite.
En résumé le DTPA forme avec l'uranium hexavalent un
composé solide entre pH 1,5 et 3, qui est soluble entre pH 3,5et 8,5o Aux pH supérieurs lr ne donne pas lieu à la formationde complexe et précipite.
Les variations du spectre d'absorption des mélangesétudiés sont dues soit à l'existence de différents complexes,
soit à l'instabilité d'un ou plusieurs d'entre eux en fonction
du pH. Les méthodes de variation moléculaire et de variation con-tinue ont permis de préciser ce point.
2 - Détermination du rapport moléculaire des complexes
a) Méthode__de__variation^moléculaire
On a choisi une concentration en uranium hexavalentégale à 8.10~"- M et [DTPA] a varié de manière à obtenir un rap-
[ T7"T*U J/[DTPA] . Les mesures ont été effectuées 24 heu-
res après la préparation du mélange,,
A pH 3,5 (figure 23 A) pour un rapport [uVIJ/[DTPA]= 8/1
le spectre est celui de l'ion uranyle. Lorsqu'on augmente la
concentration de DTPA un précipité apparaît, tout l'uranium est
à l'état de précipité pour [UVI]/[DTPA] = 2/1. Il passe en solu-
47

tion dès que ce rapport devient 1/1 et sa solubilité augmente avecDTPA ; pour un rapport 1/4, U est entièrement soluble. Les
maxima du spectre d'absorption de la solution se situent alorsà 412, 422 et 456 m/x. Au delà de [uVI] / [DTPA] = 1/4 les den-sités optiques augmentent bien que l'allure du spectre change peu0On note cependant la .disparition du maximum à 456 mu. .
La figure (25 B) représente à pH 4,5 les différentsspectres d'absorption obtenus en fonction du pH. Pour un rapport[il ] /[DTPA] = 2/1, U est partiellement engagé dans un inso-luble qui disparaît progressivement avec l'augmentation de laconcentration en DTPA. Sa solubilité est totale lorsque U ] /[DÏPA]= 1/1. La solution présente alors un spectre dont les maximasont situés à 422 et 452 my^ ; ils sont précédés d'un épauleraentà 410 nyx. Pour des rapports [ïïVI] / [DTPA] inférieurs à 1/1 ,1/2 et 1/3, l'absorption des solutions augmente légèrement, etlorsqu'il devient inférieur à 1/4 le spectre d'absorption subitde nouvelles modifications qui conduisent à la disparition dumaximum à 432 mit quand [uVI] /[DTPA] <1/6.
On a reporté sur la figure 23 0 les spectres correspon-dant à différents rapports LU ]/[DTPA] pour pH 5,5» Ils ressem-blent à ceux obtenus à pH 4,5, mais sont plus intenses. U donneavec le DTPA un précipité légèrement soluble tant que [ïï ] /[DTPA] > 2/1. Quand [UVI]/[DTPA] =1/1, UVI est en solution et lespectre de celle-ci présente les deux maxima de 422 et 432 m l*.et l'épaulement de 410 mu. . Pour un rapport inférieur, l'absorp-tion augmente de façon inégale, le maximum à 432 my^ croît plusrapidement que celui à 422 mu., puis l'absorption diminue lors-qu'il dépasse 1/6.
A pH 7, les divers spectres d'absorption obtenus sontreprésentés figure 23 D. Ils montrent bien que le complexe entiè-
48
rement soluble ne commence à se former que pour un rapport[ïï ]/[DTPA] =1/1 ; le spectre qui lui correspond présente alorsle maximum à 422 mw. , et l'épaulement à 410 ny*. . Pour un rapportinférieur, l'absorption augmente, un maximum apparaît à 430 mu.,puis l'allure du spectre reste sensiblement la même jusqu'à[u ]/[DTPA] = 1/4 . Pour des concentrations en DTPA supérieures,la densité optique augmente, le maximum de 432 m . est alors net.
La méthode de variation moléculaire, permet donc de met-tre en évidence doux complexes U -DTPA de composition différente,l'un U /DTPA = 2/1, est insoluble pour les pH compris entre 1et 3, le second soluble de pH 4 à pH 7,5 est caractérisé par unrapport moléculaire U /DTPA égal à 1/1. La longueur d'ondede 422 m . est la mieux adaptée pour déceler la présence de cescomplexes en solution. La suite de modifications spectrales ob-servées avec l'augmentation de la concentration en DTPA est pro-
VT ""bablement due à une instabilité du complexe U -DTPA-1/1. Uneinstabilité de même ordre a été mise en évidence sur le complexeUVI-EDTA-1/1 (52) et UVI-citrate-1/1 (53).
k) Méthode de^variations^continues
Nous avons opéré à pH 5,2 en maintenant \JJ ] + [DÏPA] =1,6010" M et effectué les mesures à 422 myu., 24 heures après laconfection du mélange. La courbe de la figure 24 indique l'exis-tence de deux complexes correspondant aux rapports moléculairesr = 2/1 et 1/1. Les logarithmes de leurs constantes de forma-tion sont respectivement de l'ordre de 3,7 et 7,2. Il s'ensuitque log K» S 13 pour UVI / DTPA = 2/1 et log K' =15,5 pourU / DTPA = 1/1. Les deux complexes absorbent à 422 nnw. avecles coefficients d'extinction molaire £p/i
= 51 et = 36.
49

Ill - ETUDE DE LA COMPLEXATION DE L'HYDROXYDE D ' URANIUM TETRA VALENT
Pour cette étude nous avons ajusté isolément le pH desolutions rjf^\ = 1,6010"
2 M et le pH de solutions DTPA ,1,28JO~1
avant d'effectuer leur mélange volume à volume„ On constate dansces conditions que le début de précipitation de l'hydroxyde d'ura-nium tétravalent se situe vers pH 1,2 ; la précipitation est to-tale à pH 2,3' » le précipité est blanc jusqu'à pH 3, il prend uneteinte grise puis noire au-delà de pH 5.
Pour les mélanges U - DTPA, effectués entre pH 2,3 et4, U n'est pas complexé même après 6 mois de contact hydroxyde-chélatant.
Entre pH 4 et 8,5 la solubilisation de l'hydroxyde estprogressive en fonction du pH ; nous avons tracé le spectred'absorption des surnageants de 350 à 1 000 m c, 24 heures aprèsla préparation du mélange, tous ces spectres présentent des ban-des d'absorption aux longueurs d'onde caractéristiques du com-
JVplexe U - DTPA - 1/1, déjà mises en évidence = 468 , 492 ,548 , 645 mu.» Toutefois, un epaulement supplémentaire apparaîtà 420 mic-dès pH 4<> L'absorption diminue d'une façon importanteavec l'augmentation du pH, dans les domaines de longueur d'ondeassociés à U - DTPA- 1/1 ; corrélativement la densité optiqueà 420 mu. augmente.
48 heures après la confection du mélange tout l'hydro-xyde est solubilisé entre pH 7,5 et 9, la solution passe progres-sivement du noir au brun puis au jaune, son spectre présente uneabsorption très importante vers 420 mu- .
Pour des pH compris entre 5,5 et 7,5 le même résultatest obtenu pour un temps de contact hydroxyde-chélatant pluslong : 21 jours.
M
50
Nous avons poursuivi ces mesures pendant 4 mois ; après30 jours on ne constate pas de modifications du spectre des solu-tions de pH supérieur à 4.
Finalement on obtient une solution jaune dont le spec-tre d'absorption présente une intense bande d'absorption à 420Il est difficile dans l'état actuel de nos mesures d'attribuerce spectre à un complexe U - DTPA bien que nous ayons remarquéque le spectre d'une solution absorbant à 420 m p. (£ = 200)acidifiée jusqu'à 1 N puis réajustée au pH initial 5,5 est iden-
VItique à celui d'un milieu renfermant le complexe U -DTPA-1/1 :
IV - ETUDE DE LA COMPLEXATION DES FORMES HYDROLYSEES D'URANIUM HEXAVALENT
Nous avons amené indépendamment au pH désiré, des solu-VI —2tions fraîchement préparées de U 1,6.10 M et des solutions
de DTPA 1,28.10" M puis procédé au mélange. Les lectures auspectrophotomètre ont été faites 24 heures après, la figure 25représente les différents spectres d'absorption en fonction du pH.
Aux pH inférieurs à 4,5 la totalité de l'uranium forneun précipité granuleux blanc, comme nous l'avons vu. De pH 5 à7,5 , U est complètement soluble ; la couleur des solutionsest jaune et leurs spectres sont identiques à ceux observés lorsde l'étude de la complexation de U à l'état ionique. Cependanton retrouve à pH 5 le spectre relevé précédemment à pH 3,5 et àpH 7, celui que l'on avait obtenu à pH 5,5. Au delà de pH 7,5l'hydroxyde d'uranium hexavalent n'est pas complexé dans lesconditions étudiées.
Afin de déterminer l'effet de la concentration duchélatant sur la complexation des formes hydrolysées de l'ion
51

uranyle, nous avons appliqué à différents pH et sur un hydroxydeâgé de 30 minutes, la méthode de variation moléculaire en fixant[uVI] à 1,6.10~2 M et en faisant varier [DTPA]. Jusqu'à pH 4 lecomplexe qui se forme, avec un rapport moléculaire 2/1, est inso-luble. Aux pH supérieurs U se dissout en donnant lieu à la for-mation de complexes pour des rapports [u ]/[DTPA~| égaux respec-tivement à 1/1 , 1/2 et 1/4. Les spectres d'absorption de cessolutions sont identiques à celui du complexe U - DTPA 1/1précédemment observé. Cela implique à pH > 5,5 la consommationde plus d*un molécule de DTPA par atome d'uranium pour complexer1'hydroxyde d'uranium hexavalent.
1 - Influence de l'âge de l1hydroxyde sur la formation de complexe
Nous avons étudié la complexation de l1hydroxyde d'ura-nium hexavalent par le DTPA, en fonction de l'âge du précipité quia varié entre 10 minutes et 5 jours» Pour cela, à des solutions_pd'uranyle 1,6.10 M, ajustées à un pH variant entre 5,2 et 10,on ajoute après des temps différents une solution de DTPA2,8010 M au même pHo
Lorsqu'on effectue la mesure après 24 heures de contact,la solubilité des hydroxydes d'âge compris entre 10 minutes et3 heures est plus faible entre pH 5,2 et 6 qu'entre pH 6 et 80Pour des hydroxydes plus âgés la quantité d'uranium complexédiminue avec l'augmentation du pH et devient nulle à pH 100 Cesobservations sont traduites sur la figure 26.
Si on répète ces mesures pour des temps de contact com-pris entre 1 et 97 jours, on constate que dans le domaine depH 5 - 6 la chélation de 1'hydroxyde est plus lente qu'aux pHprécédents. La complexation de la totalité du précipité nécessitetrois semaines. Au delà de pH 8 après 3 mois de contact avec leDTPA, une partie de l'hydroxyde n'est encore pas complexée.
52
V - CONCLUSION
L'uranium tétravalent forme avec le DTPA le complexeU -DTPA-1/1 stable entre pH 1,8 et 4 au moins pendant 4 mois.Le logarithme de sa constante de stabilité, log K1, est 21,60.
Il absorbe principalement à 650 nyt avec £ = 200 Sastabilité diminue avec l'augmentation du pH au delà de pH 4.
A l'état hexavalent, l'uranium conduit avec le DTPAaux complexes U -DTPA-2/1 et 1/1 dont les constantes de forma-tion K sont à pH 5,2 respectivement 103'7 (log K« - 13) et 107'2
(log K' - 15',5). A 422 ny-t le coefficient d'extinction molaire deces deux complexes est du môme ordre de grandeur £ = 300 Enprésence d'un excès de DTPA le complexe 1/1 est d'autant plusinstable que le pH est élevé.
L'hydroxyde d'uranium IV n'est solubilisé par le DTPAqu'aux pH supérieurs à 4* Cette mise en solution a lieu en pré-sence d'un excès de chélatant, par l'intermédiaire du complexeinstable U -DTPA-1/1. Quant à l'hydroxyde d'uranium hexavalent,il n'est complexé qu'entre pH 5 et pH 7,5. Les solutions obtenuesà pH 5,5 renferment alors le complexe U -DTPA-1/1 si la quantitéde chélatant est suffisante,.
VI - COMPLEXATION DE L»URANIUM PAR DIFFERENTS CHSLATANTS
Nous avons étudié la chélation du U et U par d'au-tres acides polyamines carboxyliques. Les résultats que nous avonsobtenus sont groupés au tableau 5.
53
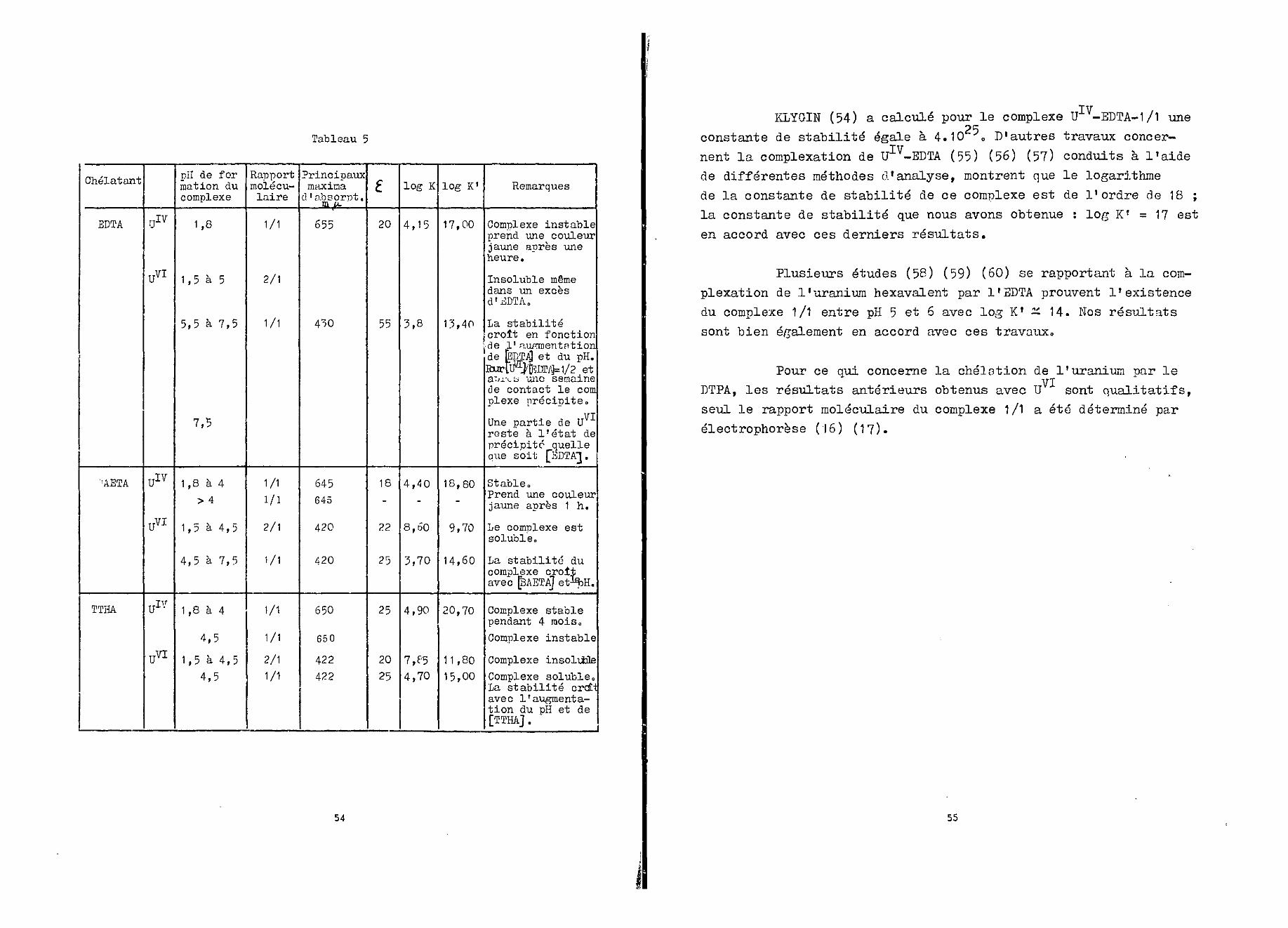
Tableau 5
Chélatant
EDTA
";AETA
TTHA
rjiv
UVI
UIV
UVI
UIV
UVI
pH de format ion ducomplexe
1,8
1,5 à 5
5,5 à 7,5
7,5
1,8 à 4> 4
1,5 à 4,5
4,5 à 7,5
1,8 à 4
4,5
1,5 à 4,54,5
Rapportmolécu-laire
1/1
2/1
1/1
1/1
1/1
2/1
1/1
1/1
1/1
2/1
1/1
Principauxmaxima
cl 'absorpt.m u.
655
430
645645
420
420
650
650
422422
t
20
55
18
22
25
25
20
25
log K
4 ,15
3,8
4 ,40
8,60
3,70
4,90
7,f;54,70
log K 1
17,00
13,40
18,80
9,70
14,60
20,70
11,8015,00
Remarques
Complexe instableprend une couleurjaune après uneheure .
Insoluble mêmedans un excèsd'SDTA.
La stabilitécroît en fonctionde 1' augmentationde fepTAj et du pH.leur Dj Ô3HP/i}= 1/2 eta^rLb une semainede contact le cornplexe précipite»
Une partie de Ureste à l'état deprécipite quelleque sbit [ïïDTA],
Stable»Prend une couleurjaune après 1 h.
Le complexe estsoluble»
La stabilité ducomplexe croîtavec [BAETAj et-^H.
Complexe stablependant 4 mois»Complexe instable
Complexe insolttQeComplexe soluble 0La stabilité crcllavec l'augmenta-tion du pH et de[TTHA] .
54
KLYGIN (54) a calculé pour le complexe UIV-EDTA-1/1 uneOR
constante de stabilité égale à 4.10 o D'autres travaux concer-
nent la complexation de UIV-EDTA (55) (56) (57) conduits à l'aidede différentes méthodes d'analyse, montrent que le logarithmede la constante de stabilité de ce complexe est de l'ordre de 18 ;la constante de stabilité que nous avons obtenue : log K' = 1 7 esten accord avec ces derniers résultats.
Plusieurs études (58) (59) (60) se rapportant à la com-plexation de l'uranium hexavalent par l'SDTA prouvent l'existencedu complexe 1/1 entre pH 5 et 6 avec log K1 — 14. Nos résultats
sont bien également en accord avec ces travaux0
Pour ce qui concerne la chélation de l'uranium par leVIDTPA, les résultats antérieurs obtenus avec U sont qualitatifs,
seul le rapport moléculaire du complexe 1/1 a été déterminé parélectrophorèse (16) (17).
55

DEUXIEME PARTIE
ETUDE DES COMPLEXES NEPTUNIUM-DTPA
L'ion Np- crue l'on trouve en solution perchlorique 1
à 10 M s'oxyde à l'air en neptunium têt ravalent ; celui-ci existe
à l'état d'ion Np en ïailiexi perchlorique 0,1 - 10 M, pour les
pH supérieurs à 1 il s'hyc^rolyse (26) .
Au degré d'oxydation 5 correspond l'ion NpO- stable
pour 10"'< fPïClO,] < 1 M. Enfin on rencontre Np à l'état de2+ ~ —2NpOp dans les solutions perchloriquesd'acidité supérieure à 10 M.
La position des maxima des spectres d'absorption de—2solution perchlorique 2 N, 1,07.10 M en neptunium, que nous
avons relevés est donnée dans le tableau 6 pour les différents
états de valence de neptunium,,
Tableau 6
AT HINp
TT IVNp
NpV
NPVI
Couleur dela solution
violet
jaune vert
vert
rosé
Longueur d'onde desprincipaux maxima
ri 'absorption en m
552 , 602 , 661 , 785
504 , 724 , 825 , 956
476 , 957 , 983
467 , 557
£
38 , 25 , 28 , 42
19 , 57 , 17 , 61
17 , 19 , 94
19 , 17
Ces valeurs sont en accord avec celles déjà publiées (61).
56
1 - Etude de la complexation du neptunium t rivaient à l'étationique
A pH 0,5 le spectre d'absorption relevé sur un mélangeà volume égal, d'une solution de Np 2,14.10"" M fraîchementpréparée et d'une solution de DTPA 8,56.10~1 M, est celui deNp (figure27A) . En fonction du temps on note une dim .finitionde la densité optique des maxima à 552, 605, 661 et 785 ny- 0
II en apparaît un nouveau à 750 m te • Nous verrons ultérieurementIVqu'il est caractéristique d'un complexe Np - DTPA.
Si on augmente le pH ce phénomène a lieu plus rapide-
ment qu'à pH 0,5.
Nous avons renouvelé ces expériences en présence d'amal-game de zinc, le spectre (1) obtenu (figure 27B)à pH 0,5 présenteles maxima caractéristiques de Np^+ et est stable au cours dutemps. Par contre lorsque le pH croît, il se forme un précipitéblanc; corrélativement la densité optique diminue sur l'ensembledu spectreo
L'existence d'un complexe Np - DTPA, semble peu pro-bable puisque nous n'avons observé aucun changement immédiat dansle spectre des solutions de neptunium en présence de ce chélatant,
III en complexeLe DTPA, par contre, accélère l'oxydation de NpNp - DTPA. Nous avons observé que celle-ci est complète en15 minutes alors que ïïp seul est oxydé par l'air en Npbeaucoup plus lentement en milieu non complexanto
G-EL'MAN et ses collaborateurs (62) ont obtenu des résul-tats analogues en étudiant la chélation de Np par l'EDTA.
57

II'- ETUDE DE LA COMPLEXATION DU NEPTUNIUM TETRAVALENT A L'ETAT
IONIQUE
1 - Variation du spectre d'absorption en fonction du pH
IVL'absorption d'une solution perchlorique 2 N de Np"1,07.10~2 M etd%TPA 4,28.10""2 M a été mesurée en fonction du pH.
Le spectre d'absorption d'un mélange, étudié à pH 0,5,présente de nettes modifications par rapport à celui de Np(figure 28). En particulier le maximum de 724 au*, se déplace à750 nyu et celui de 956 mu. disparaît* Les nouveaux maxima obser-vés se situent à : 438 , 51 5 , 563 , 750 , 790 , 800 , 852 ,936 et 985 mu.. Ce spectre reste inchangé jusqu'à pH 5,80
Au-delà de ce pH, il subit de nouvelles modifications :l'intensité de la bande d'absorption à 750 mu- diminue, les maximaà 790 et 800 nyt- disparaissent, tandis qu'apparais sent de nou-velles bandes à 795 et 930 m *. 0 Le maximum à 995 nyw. n'existeplus» L'absorption de la solution envisagée conserve ces carac-téristiques jusqu'à pH 8,5»
Pour des pH compris entre 8,5 et 11 l'intensité duspectre d'absorption diminue, notamment à 750 m/*., et de nou-veaux pics apparaissent à 715 et 970
Ainsi la complexation de Np par le DTPA commence dèspH 0,5. Les modifications spectrales observées avec l'augmenta-tion du pH peuvent être dues à l'existence de différents complexesou à l'instabilité d'un complexe. Nous avons choisi pour en déci-der de poursuivre notre étude aux pH qui correspondent à desspectres d'absorption différents : 0,5 - 2,2 - 7,5 et 9*
58
2 - Détermination du rapport moléculaire des complexes à diffé-rents
On a réalisé à l'aide d!une solution perchlorique 2 N—_ p
de neptunium tétravalent 1,07.10 M et d'une solution de DTPAde concentration variable, des mélanges de pH approprié, où lerapport [ Np1 V] / [DTPA] a varié de 4/1 à 1/4. Les mesures ont étéeffectuées 24 heures après.
A pH 0,5 (figure 29) pour [NpIV] /[DTPA] = 4/1 le maximumà 724 mw. caractéristique de Np diminue tandis que se dessine
~ ivcelui de 750 nyt- caractéristique du mélange ÏÏp -DTPA» LorsqueL^P ]/ [DTPA] diminue, ces modifications s'accentuent, ceci jus-qu'à la valeur 1/1, pour laquelle le spectre est déjà celui observéentre pH 1,5 et 5,8. Enfin pour des valeurs defaibles le spectre ne subit plus de modification.
1 V] / [DTPA] plus
A pH 2,2, Np est entièrement complexé pour un rapport[NpIV]/[DTPA] = 1/1 mais à pH 7,5 et 9 un rapport 2/3 est néces-saire pour obtenir le même résultat (figure 30). Pour les concen-trations en DTPA supérieures à celles correspondant à ces rapports,le spectre du mélange ne change pas, et pour des concentrationsen DTPA inférieures Np qui n'est pas complexé précipite.
b) Méthode de variations continues
IVLa concentration totale du mélange Np -DTPA a été fixéeà 2,14.10"* M. Nous avons travaillé à pH 2,2 , 7,5 et 9. La densitéoptique optique aux longueurs d'onde des maxima caractéristiques du
complexe aug-
59
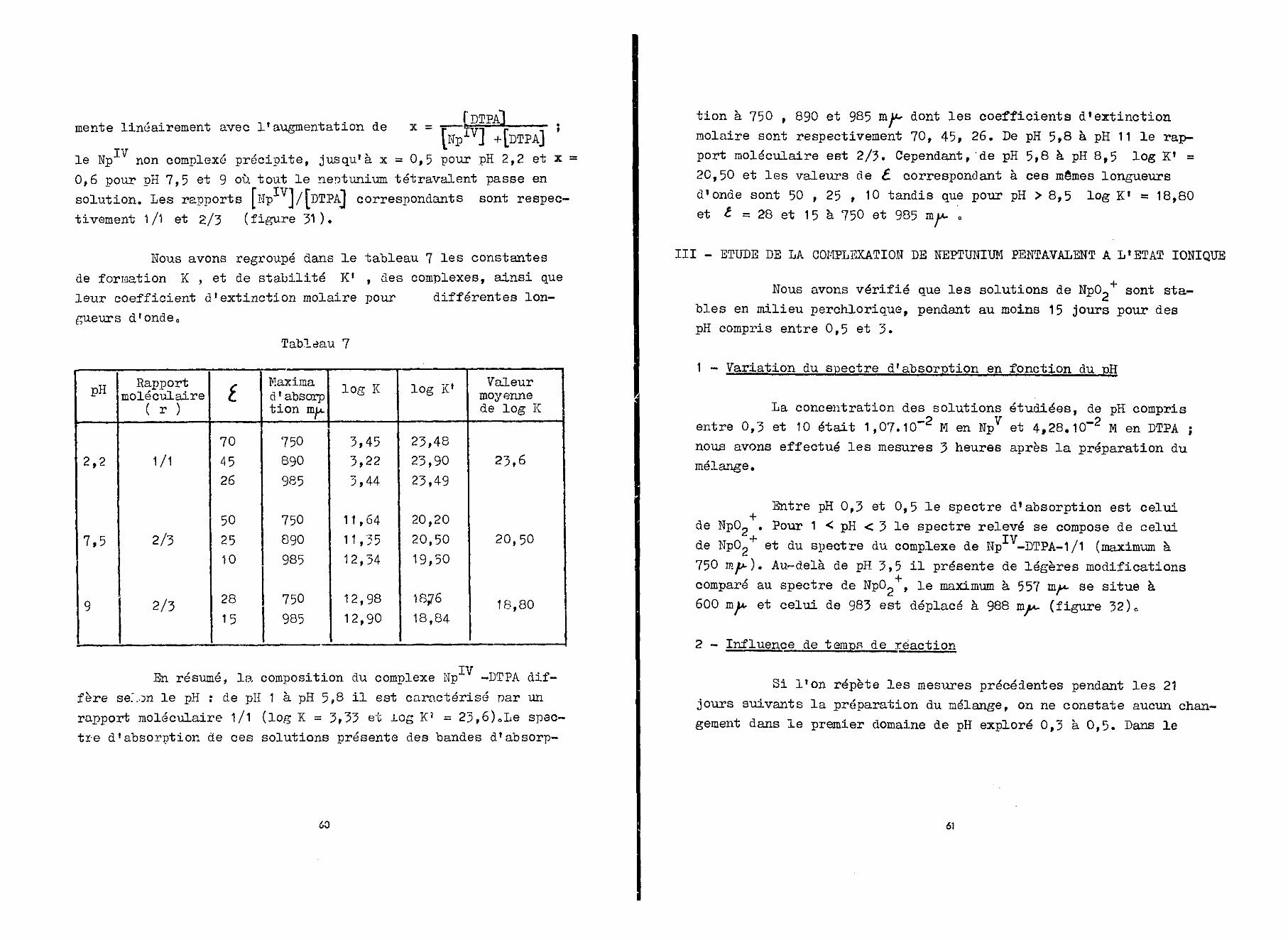
mente linéairement avec l'augmentation de
IV
x = [DTPA]^T7\[NÏ» ] +[DTPA]
le Npav non complexé précipite, jusqu'à x = 0,5 pour pH 2,2 et x0,6 pour pH 7,5 et 9 où tout le neptunium tétravalent passe ensolution. Les rapports [Hp ]/[DTPA] correspondants sont respec-
tivement 1/1 et 2/3 (figure 31).
Nous avons regroupé dans le tableau 7 les constantesde formation K , et de stabilité K1 , des complexes, ainsi queleur coefficient d'extinction molaire pour
gueurs d'onde.»
Tableau 7
différentes Ion-
pH
2,2
7,5
9
Rapportmoléculaire
( r )
1/1
2/3
2/3
£
704526
502510
2815
Maximad ' absorption mu
750890985
750890985
750985
log K
3,453,223,44
11,6411,3512,34
12,9812,90
log K'
23,4823,9023,49
20,2020,5019,50
18?618,84
Valeurmoyennede log K
23,6
20,50
18,80
En résumé, la composition du complexe Np -DTPA dif-fère ser.on le pH : de pH 1 à pH 5,8 il est caractérisé par unrapport moléculaire 1/1 (log K = 3,33 et log K' = 23,6)0Le spec-tre d'absorption de ces solutions présente des bandes d'absorp-
60
tion à 750 , 890 et 985 m to dont les coefficients d'extinctionmolaire sont respectivement 70, 45, 26. De pH 5,8 à pH 11 le rap-port moléculaire est 2/3. Cependant, de pH 5,8 à pH 8,5 log K1 =20,50 et les valeurs de £ correspondant à ces mêmes longueursd'onde sont 50 , 25 , 10 tandis que pour pH > 8,5 log K1 = 18,80et t = 28 et 15 à 750 et 965 mix- 0
III - ETUDE DE LA COMPLICATION DE NEPTUNIUM PENTAVALENT A L'ETAT IONIQUE
Nous avons vérifié que les solutions de NpO-* sont sta-bles en milieu perchlorique, pendant au moins 15 jours pour despH compris entre 0,5 et 3.
1 - Variation du spectre d'absorption en fonction du
La concentration des solutions étudiées, de pH comprisentre 0,3 et 10 était 1,07.10""2 M en NpV et 4,28. 10~2 M en DTPA ;nous avons effectué les mesures 3 heures après la préparation dumélange.
Entre pH 0,3 et 0,5 le spectre d'absorption est celuide NpOp . Pour 1 < pH < 3 le spectre relevé se compose de celui
+ TVde NpOg et du spectre du complexe de Np -DTPA- 1/1 (maximum à750 m^O. Au-delà de pH 3,5 il présente de légères modificationscomparé au spectre de Np02 , le maximum à 557 m^ se situe à600 mA, et celui de 983 est déplacé à 988 ny*. (figure 32 K
2 - Influence de temps de réaction
Si l'on répète les mesures précédentes pendant les 21jours suivants la préparation du mélange, on ne constate aucun changement dans le premier domaine de pH exploré 0,3 à 0,5. Dans le
61

second (pH 1 à 3) par contre, le facteur temps joue un rôle impor-tant. Le spectre du mélange aussitôt après sa préparation est celuide Np00 , mais trois heures après on observe, comme nous venons de
IVle voir le maximum à 750 mit, caractéristique du complexe Np -DTPA-1/1 » L'absorption à cette longueur d'onde est maximum à pH 2,2,elle disparaît 'à pH 3, tandis que l'intensité du maximum d'absorp-
tion de ÎTpOp à 983 mu- diminue.
Après 24 heures les phénomènes observés précédemments'accentuent, bien qu'à pH 2,2 le maximum d'intensité d'absorption
se situe encore à 983
Après 4 jours et plus, le spectre de la solution estqualitativement et quantitativement celui du complexe Np -DTPA-1/1, pour des pH compris entre 1,8 et 2,4. De part et d'autre decette zone de pH, le spectre se compose de ceux de Np -DTPA-1/1
et de NpOp+ (figure 33), en effet au delà de pH 2,4, on observe
l'importante absorption à pH 3»5 à 10, le spectre des solutionsn'est pas modifié en fonction du temps, ce qui est une indicationde l'existence d'un, ou de, complexes Np -DTPA.
La détermination de la composition moléculaire de celui-ciou de ceux-ci et le calcul de leur constante de stabilité est dif-ficile car l'absorption à 988 ny- ne suit pas la loi de Beer-Lambert (61). Par ailleurs l'absorption à 600 mu. est trop faible.
Ces résultats montrent néanmoins l'existence d'un effetde réduction de Np en complexe de Np ' par le DTPA, qu'il nous a
paru intéressant de confirmer.
62
3 - Influence de la concentration en DTPA sur 1' effet de réduction
Nous avons opéré pour cette étude à pH 2,2, valeur dupour laquelle l'effet de réduction est maximum, et ajouté
V -?à un volume connu d'une solution de Np de concentration 2,14o10 Mun volume identique de solutions de DTPA, de différentes concen-
trations,de façon à faire varier le rapport [Np ] / [DTPA] de 4/1 à 1/4.
Les lectures ont été effectuées 48 heures après la con-fection du mélange. On remarque que la bande d'absorption carac-téristique de Np00
+ diminue lorsque la concentration du DTPATVaugmente tandis que se détache celle du complexe Np - DTPA-1/1
à 750 my . Pour un rapport [NpV]/[DTPA] égal ou inférieur à 1/2
le spectre d'absorption est celui de Np - DTPA-1/1 , tandis quedans le cas d'un rapport de concentration supérieur, les spectresindiquent la présence, dans le mélange, de complexe Np - Np -DTPAo Ces mômes mesures répétées au cours du temps n'apportentpas de modifications aux observations que l'on vient de rappor-ter.
En résumé de pH 1 à 3, DTPA réduit NpV à la valencequatre, puis donne lieu à la formation du complexe Np - DTPA -1/1. Le pH optimum pour cette réduction est 2,2 , la totalité duneptunium est réduite après 96 heures pour [ ïïp ] /[DTPA] - </.?,
Ainsi une molécule de DTPA est dégradée en libérant de l'anhy-dride carbonique pour la réduction d'un ion NpOp+.
IV - ETUDE DE LA COMPLEXATION DU NEPTUNIUM HEXAVALENT A L'ETAT IONIQUE
En l'absence de DTPA, une solution perchlcrique 2 N de
ajustée entre pH 0,5 et 3, reste stable pendant au moins15 jours.
63

L'addition d'un excès de DTPA sur de telles solutions"-2fraîchement préparées 2,14.10 M en élément, fait immédiatement
virer la solution du rosé au vert, couleur caractéristique de Np02 .Le spectre d'absorption confirme cette observation qualitative(figure 34). Le DTPA agit donc comme un réducteur puissant» L'étudede l'influence du pH sur cette réduction conduit aux mêmes résul-tats que ceux obtenus avec NpOp : entre pH 1 et 3 il y a forma-tion du complexe NpIV - DTPA 1/1.
V - ETUDE DE LA COMPL}!XATION DE NEPTUNIUM A L'ETAT D'HïDROXïDE
Nous avons procédé ici, suivant la technique habituelleen ajustant indépendamment le pH du DTPA et le pH des solutionsde neptunium, dont les concentrations respectives étaient
? 12,12.10 M et 1,06.10 M. Nous avons ensuite effectué le mélangepour différents âges du précipité et les mesures spectrophotomé-triques ont été poursuivies dans le temps, pendant 4 mois.
- NpIII
Lfhydroxyde commence à précipiter à partir d'e pH 1, laprécipitation est totale à pH 2. Jusqu'à pH 2 le spectre obtenusur le surnageant correspond à celui du complexe Np - DTPA 1/1Pour les pH supérieurs à 2, le DTPA est sans action sur le préci-pité pendant toute la durée des observations0
IV
A pH 1,5 tout le Np reste à l'état de précipité quel-que soit le temps de contact avec le DTPA. Durant 4 mois nousn'avons pas observé la formation de complexe»
64
- Np1
Le neptunium pentavalent reste ionique à pH 8 - 9 etest complexé par le DTPA. Cependant au sein de solutions de nep-tunium-DTPA plus âgées que 5 jours, il apparaît un précipitéinsoluble»
VI- Np
Lfhydroxyde de neptunium hexavalent précipite aux pHsupérieurs à 3,7. Le précipité frais, en contact avec le DTPAdonne une solution verte présentant le spectre d'absorption duou des complexes Np -DTPA. La complexation d'un hydroxyde plus âgéest plus difficile. Elle devient irréalisable après une semaine.
VI - CONCLUSION
IIIL'existence d'un complexe de Np -DTPA est peu probable,Par contre nous avons mis en évidence plusieurs complexes Np -DTPA de rapport moléculaire différent selon le pH : de pH 0,5 àpH 5,8, r = 1/1 (log K' = 23,6) ; de pH 5,8 à pH 8,9, r = 2/3(log K' = 20P50) ; enfin pour pH > 8,5 , r est encore égal à 2/3mais log K' = 18,80. Le neptunium pentavalent est réduit par leDTPA, entre pH 1 et 3 ï c'est le complexe NpIV-DTPA-1/1 qui seforme alors. Cependant pour pH>3,5 Np est complexé. Le DTPAréduit immédiatement le neptunium hexavalent à tous les pH.
Nous avons constaté qu'il n'était pas possible de solu-TV VTbiliser avec le DTPA les hydroxydes de Np et ceux de Np
âgés de plus d'une semaine, même après quatre mois de contact.
65

VII - COMPLEXATION DU NEPTUNIUM PAR DIFFERENTS CHELATANTS
Nous présentons dans le tableau 8 les principaux résul-
tats que nous avons obtenus sur la chélation du neptunium parle BAETA et le TTHA.
t- '
Les seules études concernant la chélation du neptuniumpar les acides polyaminecarboxyliques ont trait à l'EDIA. Nousrappelons les principaux résultats obtenus.
GEL1MAN et ses collaborateurs (62) qui ont étudié, parspectrophotometrie la complexation de Np par l'EDTA n'ont pasmis en évidence la formation de complexe. Par contre JENKINS (63)a montré, en utilisant une méthode polarographique, la présenced'un complexe Npi:CI-EDTA-1/1 (log K» = 23) dans HCI 1 M. Dans lesmômes conditions cet auteur a calculé log K1 =22,8 pour le com-plexe Np -EDTA-1/1 . On remarquera combien les valeurs des cons-tantes de stabilité des complexes 1/1 de Np et Np avecd'EDTA sont proches.
La complexation du Np par l'EDTA a été prouvée par
plusieurs auteurs (64) (65) (66). Ils ont montré la présence ducomplexe 1/1, NpCLY"5 pour pH 3,2 (log K1 = 10,2). Pour ce qui
VIconcerne la complexation du Np , aucun complexe n'a pu être misen évidence (67).
66
Tableau fc
Chéla-tant
BAETA ITp111
1TPIV
NPV
IIpVI
ÏTHA Hp111
Np17
ÎJlp
ÎTp
pH de for-nation ducomplexe
0,5 à 5,6
pH>5 ,6
pH >2,5
pH<2,5
'
0,5 à 10
pH < 3,5
PH > 3,5
Kapraortmolé-
culaire
1/1
2/3
1/1
Principauxmaxima d'absorption
mu.
750, 890
750, 890
£
70, 45
7?, 43
log K
4,50
1 ,'^0
5,00
los K»
13,30
16,50
??,77
Remarnues
Pas mis en évidence.
Complexe stable au cours dutemps
Difficile à étudier quanti-tativementAucun changement du spectre
du IÏB , at>rès addition deBAETÀ
Réduction immédiate
Pas mis en évidence
Complexe stable
;-]ff et de réduction à la va-lence IV ar>rè£ 4 jours decontact
Complexe difficile 'à éturiiertsar snectro'ohotO'Tu'trie
;';ffot de réduction immédiat
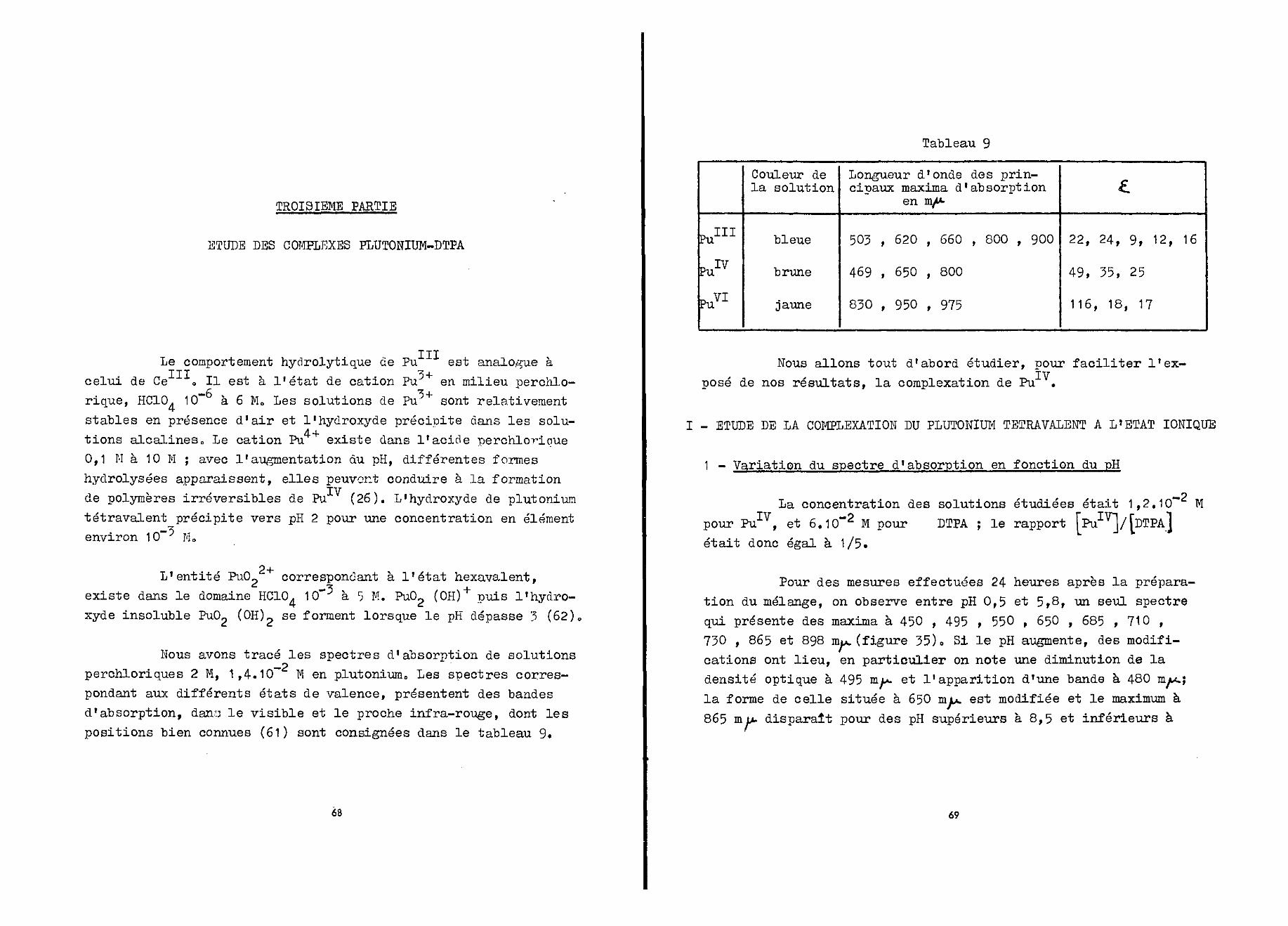
TROISIEME PARTIE
ETUDE DES COMPLEXES FLUTONIUM-DTPA
Le comportement hydrolytique de Pu est analogue à
celui de Ce o II est à l'état de cation Pu en milieu perchlo-
rique, HC10, 10" à 6 M0 Les solutions de Pu + sont relativement
stables en présence d'air et l'hydroxyde précipite dans les solu-
tions alcalineSo Le cation Pu existe dans l'acide perchlorioue
0,1 M à 10 M ; avec l'augmentation du pH, différentes formes
hydrolysées apparaissent, elles peuvent conduire à la formation
de polymères irréversibles de Pu (26). L'hydroxyde de plutonium
tétravalent précipite vers pH 2 pour une concentration en élément
environ 10" M0
L'entité PuO. 2-fcorrespondant à l'état hexavalent,
.0. 10~5 à r; M. Pu02 (OH) + puis l'h;
xyde insoluble PuOp (OH)p se forment lorsque le pH dépasse 3 (62)
existe dans le domaine HC10. WfJ à c} M. Pu09 (OH) puis 1»hydro-
Nous avons tracé les spectres d'absorption de solutions—2perchloriques 2 M, 1,4.10 M en plutonium,. Les spectres corres-
pondant aux différents états de valence, présentent des bandesd'absorption, dano le visible et le proche infra-rouge, dont lespositions bien connues (61) sont consignées dans le tableau 9.
68
Tableau 9
Pu111
PuIV
PuVI
Couleur dela solution
bleue
brune
jaune
Longueur d'onde des prin-cipaux maxima d'absorption
en m/*-
503 , 620 , 660 , 800 , 900
469 , 650 , 800
830 , 950 , 975
£
22, 24, 9, 12, 16
49, 35, 25
116, 18, 17
Nous allons tout d'abord étudier, pour faciliter l'ex-posé de nos résultats, la complexation de Pu .
I - ETUDE DE LA COMPLEXATION DU PLUTONIUM TETRAVALENT A L'ETAT IONIQUE
1 - Variation du spectre d'absorption en fonction du pH
La concentration des solutions étudiées était 1,2.10" Mpour PuIV, et 6.10"2 M pour DTPA ; le rapport [PUIV]/[DTPA]était donc égal à 1/5»
Pour des mesures effectuées 24 heures après la prépara-tion du mélange, on observe entre pH 0,5 et 5,8, un seul spectrequi présente des maxima à 450 , 495 , 550 , 650 , 685 , 710 ,730 , 865 et 898 mix (figure 35)„ Si le pH augmente, des modifi-cations ont lieu, en particulier on note une diminution de ladensité optique à 495 my . et l'apparition d'une bande à 480 my^c;la forme de celle située à 650 mi*, est modifiée et le maximum à865 mu. disparaît pour des pH supérieurs à 8,5 et inférieurs à
69
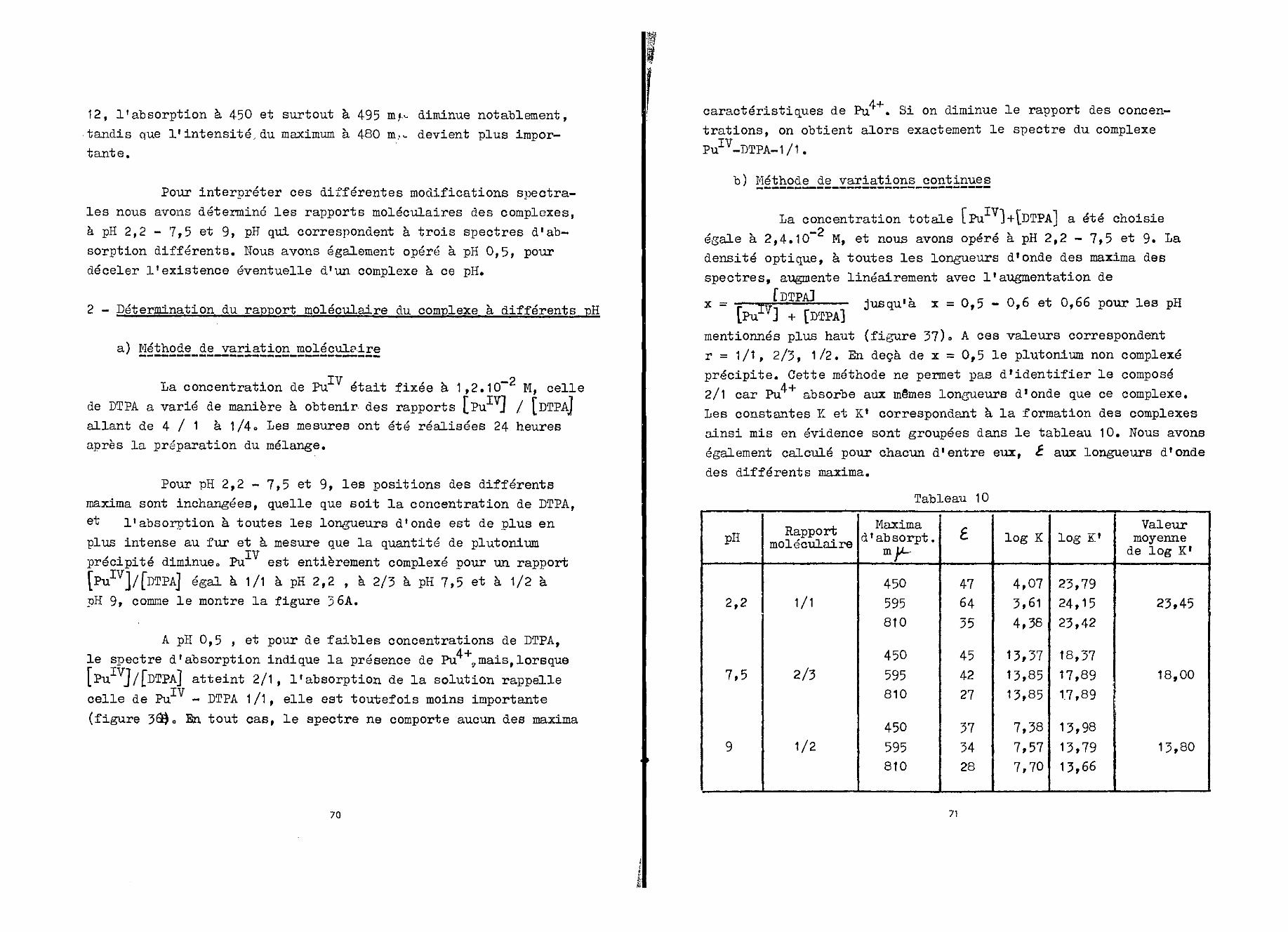
12, l'absorption à 450 et surtout à 495 m^ diminue notablement,• tandis que l'intensité, du maximum à 480 m..», devient plus impor-tante.
Pour interpréter ces différentes modifications spectra-les nous avons déterminé les rapports moléculaires des complexes,à pH 2,2 - 7,5 et 9, pH qui correspondent à trois spectres d'ab-sorption différents. Nous avons également opéré à pH 0,5, pourdéceler l'existence éventuelle d'un complexe à ce pH.
2 - Détermination du rapport moléculaire du complexe à différents pH
a) Méthode_de__variation moléculaire
iv ?La concentration de Pu était fixée à 1,2.10""* M, cellede DTPA a varié de manière à obtenir des rapports [PU J / [DTPAjallant de 4 / 1 à 1/4» Les mesures ont été réalisées 24 heuresaprès la préparation du mélange.
Pour pH 2,2 - 7,5 et 9, les positions des différentsmaxima sont inchangées, quelle que soit la concentration de DTPA,et l'absorption à toutes les longueurs d'onde est de plus enplus intense au fur et à mesure que la quantité de plutoniumprécipité diminuée Pu est entièrement complexé pour un rapport[puIV]/fDTPA] égal à 1/1 à pH 2,2 , à 2/3 à pH 7,5 et à 1/2 àpH 9, comme le montre la figure 36A.
A pH 0,5 , et pour de faibles concentrations de DTPA,le spectre d'absorption indique la présence de Pu „ mais, lors que[PUIV]/[DTPA] atteint 2/1, l'absorption de la solution rappellecelle de Pu(figure
DTPA 1/1, elle est toutefois moins importanteEn tout cas, le spectre ne comporte aucun des maxima
70
caractéristiques de Pu . Si on diminue le rapport des concen-trations, on obtient alors exactement le spectre du complexePuIV-DTPA-1/1.
b) Méthode^de variations_gontinues
La concentration totale [PU V]+\DTPA] a été choisieégale à 2,4.10~2 M, et nous avons opéré à pH 2,2 - 7,5 et 9. Ladensité optique, à toutes les longueurs d'onde des maxima desspectres, augmente linéairement avec l'augmentation de
x = Y ^DTPAj jusqu'à x = 0,5 - 0,6 et 0,66 pour les pH[PU VJ 4- [DTPA]
mentionnés plus haut (figure 37)» A ces valeurs correspondentr = 1/1, 2/3, 1/2. En deçà de x = 0,5 le plutonium non complexéprécipite. Cette méthode ne permet pas d'identifier le composé2/1 car Pu absorbe aux mômes longueurs d'onde que ce complexe.Les constantes E et K1 correspondant à la formation des complexesainsi mis en évidence sont groupées dans le tableau 10. Nous avonségalement calculé pour chacun d'entre eux, £ aux longueurs d'ondedes différents maxima.
Tableau 10
pH
2,2
7,5
9
Rapportmoléculaire
1/1
2/3
1/2
Maximad'absorpt.
m IU-
450
595810
450
595810
450
595810
£
476435
4542
27
373428
log K
4,073,614,38
13,3713,8513,85
7,38
7,577,70
log £'
23,7924,1523,42
18,3717,891.7,89
13,98
13,7913,66
Valeurmoyennede log K1
23,45
18,00
13,80
71

En résumé, selon le pH, la composition du complexe varie.C'est ainsi que de pH 1 à 5,8 le rapport moléculaire est égal à1/1 (log K1 = 23,45). Le spectre d'absorption de ce complexe pré-sente des bandes caractéristiques à 450, 495 et 810 nu«- avec£= 47, 64 et 35o De pH 5,8 à pH 8,5 r = 2/3 (log K' = 18,00) etles valeurs de £ correspondant aux mêmes longueurs d'onde sont45, 42 et 27. Enfin, pour pH 8,5 la composition du complexe est1/2 (log K» = 13,81) et £ à 450, 459 et 810 myw. est égal à 37, 34et 28.
II - ETUDE DE LA COMFLEXATION DU PLUTONIUM TRIVALENT A L'ETAT IONIQUE
1 - Variation du spectre d'absorption en fonction du pH
III —2On a mélangé un volume d'une solution de Pu 2,4*10 Mavec un égal volume d'une solution de DTPA 1,2.10~1 M, [Pu111]/[DTPA] = 1/5o Les mesures d'absorption ont été effectuées immédia-tement, 2 heures, 24 heures et une semaine après, à des pH comprisentre 0,2 et 100
Dans le cas de mesures effectuées immédiatement aprèsla préparation de la solution (figure 38), le spectre d'absorption
3+est celui de Pu , à pH 0,20 II présente à pH 0,5 , comme celuid'une solution de Pu - DTPA de même concentration en Pu etDTPA, deux maxima d'absorption à 450 et 500 ny- . On note égale-ment, mais légèrement modifiés, les deux maxima caractéristiquesde Pu5* à 650 et 800 m u- 0 De pH 0,5 à pH 6,2 , on note sur lespectre d'absorption les maxima à 450 , 500 , 550 , 650 , 685 ,710 , 730 , 865 et 898 mu. que nous avons déjà trouvés lors de
IVl'étude de la complexation de Pu .Au delà de pH 6,2 la solutionprend une couleur verte, son spectre rappelle alors celui duPu , mais présente toutefois, par rapport à celui-ci certaines
72
modifications importantes : le pic à 560 ny*. est plus aigu et lemaximum à 620 m>n* est déplacé à 595 mi*.. Ce dernier spectre tra-
III _duit la complexation de Pu " par uTPA0
Gi l'on effectue après deux heures, de nouvelles lec-tures, on constate que jusqu'à pH 6,2 , le spectre de la solutionest celui du complexe PuIV - DTPA-1/1. Entre pli 6,2 et pH 8,3 onnote à la fois une diminution de 1' absorption à 560 mu. et 620 m>u-
IVet une augmentation de la concentration du complexe Pu - DTPA1/10 Par contre de pH 8,3 à 12 le spectre correspond à celuiobtenu précédemment aux mêmes pH.
Des mesures reprises 24 heures et une semaine plus tardmontrent l'existence du complexe Pu - DTPA-1/1, jusqu'à pH 8,3.Au delà de ce pH le spectre est celui du. ou des complexes Pu -DTPA. Nous avons donc poursuivi notre étude en milieu basique0
2 - Détermination du rapport moléculaire des complexes
a) Methode^de^variation_moléculaire
On a mélangé à volume égal une solution de Pu1,2o10"" H et des solutions de DTPA de concentrations variables.Le mélange a été ajusté à pH 9 et les mesures ont été effectuéesaux longueurs d'onde des maxima caractéristiques du complexe,immédiatement après avoir effectué le mélange.
Pour de faibles concentrations en DTPA, Pu non com-plexe précipite. Avec l'augmentation de la concentration du ché-latant la quantité de plutonium trivalent précipité diminue, lespectre d'absorption devient plus intense, et atteint un maximum
73

pour [PU ]/[DTPA] = 2/3 0 Aux concentrations de chélatant plusélevées tout le plutonium est complexé (figure 39).
b ) Méthode_de_^variations__c ontinue s
Elle a été appliquée pour [Pu111] + DTPA]= 2,4»10"2 Mà pH 9. Les mesures ont été effectuées comme précédemment. Lesrésultats obtenus (figure 40) montrent bien que le complexe seforme pour [PU ]/[DTPA] = 2/3 (x = 0,6). On a calculé log Ket log K' dont les valeurs moyennes déduites des mesures à 560,595 et 870 mu. sont égales respectivement à 13,70 et 18,00. Cecomplexe absorbe à 560, 595 et 870 avec des coefficients d'extinc-tion molaire égaux à 36, 30 et 26.
III - ETUDE DE LA COMPLEXATION DU PLUTONIUM HEXAVALENT A L'ETAT IONIQUE
Aucun complexe Pu -DTPA suffisamment stable, n'a étédécelé par spectrophotométrie. Par contre nos mesures montrentque Pu est réduit par le DTPA0 Nous avons donc seulement étudiél'effet de réduction en fonction du pH, de l'âge des solutionset de la concentration en chélatant.
1 - Variation du spectre flabsorption en fonction du
VI — ?A pH acide on a mélangé les solutions de Pu 2,4.10 Met de DTPA 1 ,2»10"1 M [puVIJ /[DTPA] = 1/5, puis le pH a été ajusté.La figure 41 représente les variations du spectre d'absorption dumélange obtenu en fonction du pH, 24 heures après sa conf ectiona
A pH 0,5 on observe des maxima à 450, 495, 550, 580,650, 685, 710, 730, 898 mju. caractéristiques de PuIV - DTPA-1/1
O TJ I
et les maxima à 830 et 950 mu. caractéristiques de Pu 0 .
74
A pPI 1 la concentration du complexe Pu - DTPA-1/1augmente tandis que diminue celle de Pu
Avec l'augmentation du pH ces modifications s'accentuentet aux pH supérieurs à 3,3 le spectre obtenu correspond uniquementà celui du complexe Pu - DTPA-1/1. Deux jours après la prépara-tion du mélange toutes les solutions renferment le complexePuIV - DTPA-1/1 quel que soit leur pH.
2 - Influence de la concentration en DTPA sur la réduction
Nous avons travaillé à pH 3 et 7 en fixant la concentra-tion de Pu 1 à 1,2o10~2 M et en faisant varier [DTPA] de façonà obtenir des rapports de concentration compris entre 4/1 et 1/40Les mesures ont été effectuées 48 heures après avoir réalisé lemélange.
La figure 42 représente les spectres d'absorption obte-nus à pH 3 pour différentes concentrations en DTPA. Le spectred'absorption de Pu est,à ce pH, identique à celui relevé enHC10. 1 N, il présente bien les maxima à 850 et 950 mu. 0 Pourun rapport de concentration [pu j/[DTPAl = 4/1 on observe à la
IVfois les bandes d'absorption caractéristiques du complexe Pu -DTPA-1/1 à 450 , 495 , 550 , 650 mu. et les maxima d'absorption
Oj /
de PuO. 2+ bi l'on augmente la concentration de DTPA, on noteIV
une augmentation de la concentration du complexe Pu - DTPA-1/1
et une diminution de celle de Pu 0 2+ . Pour [puVI] / [DTPA 1 = 1/2IVle spectre correspond à celui de Pu - DTPA-1/1.
C'est donc le complexe Pu - DTPA-1/1 qui se formejusqu'à pH 5,8 quand on mélange Pu et DTPA dans un rapport 1/2.
75

Des mesures effectuées 3 jours après la préparationdu mélange conduisent à des résultats identiques,
A pli 7, des essais analogues ont montré l'existencedu complexe Pu - OTPA-2/3o II se forme à partir d'un rapport[puVIJ/[DTPA] = 2/5 o
Ces résultats montrent qu'une molécule de DTPA est uti-2-flisée à la réduction d'un groupement Pu 09 quel que soit le
\T"T c-pHo Par ailleurs la réduction de Pu" croît avec l'augmentationdu pH et avec le temps.
IV - ETUDE .DE LA COÏ-ÎPLEXA.TION DES HÏDROXÏD33 DE PLUTONIUM
-1Nous avons mis en contact une solution de DTPA 1,2.10 Mde pH donné, avec une solution ou une suspension fraîche ou âgée_ pde plutonium 2,4.10 M, ajustée au même pH. Les mesures d'absorp-tion ont été répétées pendant plusieurs mois.
III- Pu'
L'hydroxyde de plutonium III qui précipite à pH 1,5n'est pas complexable par le DTPA quel que soit le temps de con-
tact avec le chélatanto
-Pu17
L'hydroxyde précipite dès pH 1, le DTPA est sans action sur
celui-ci, même après plusieurs mois0
76
- PuVI
Pour un pH inférieur à 5,5, un hydroxyde âgé de 10 mi-nutes donne pour un temps de contact de 24 heures, le complexePu - DTPA-1/1» Par contre aux pH supérieurs à 5,5 il n'est quepartiellement complexé sous cette forme, et la fraction qui resteà l'état de précipité croît avec l'âge de 1'hydroxyde.
V - CONCLUSIONS
Ces résultats permettent de conclure à l'existence d'uncomplexe Pu -DTPA-2/3 instable dans le temps pour les pH infé-rieurs à 8,2. Il est par contre stable aux pH supérieurs (log K! =18,00).
Le DTPA forme quatre complexes avec le plutonium tétra-valent qui existent chacun dans une zone de pH bien déterminée.Ils sont caractérisés par les rapports moléculaires suivants :r = 1/2 pour pH 0,5 ; r = 1/1 entre pH 1 et 5,3, (log K» = 23,45)r = 2/3 entre pH 5,8 et 8,5 (log K« = 18,00) et r = 1/2 pour pHsupérieur à 8,5 (log K' = 13,80).
Pu est réduit par le DTPA, suivant le pH on obtient.IVles complexes Pu -DTPA 1/2 (pH < 5,8) ou 2/3 (pH ? 7). Seul
1'hydroxyde de Pu est solubilisé à l'état de complexe Pu -DTPA-1/1, à condition toutefois que le pH reste inférieur à 5,5.
VI - COMPLICATION DU PLUTONIUM PAR DIFFERENTS CHELATANTS
Parallèlement à cette étude sur la chélation du pluto-nium par le DTPA, nous avons étudié certains des complexes quecet élément forme avec d'autres acides polyaminecarboxyliquea
77

Le tableau 10 résume les principaux résultats obtenuspour ce qui concerne la complexation à l'état ionique.
Les seules études concernant la chélation du plutoniumpar les acides polyaminecarboxyliques se rapportent à l'EDTA (68)(69). Nous en rappelons les principales conclusions.
FOREMAN et SMITH (70) ont utilisé la méthode d'échanged'ions pour étudier la nature des espèces ioniques formées entrele plutonium et l'EDTA. Dans le domaine de pli 1 à 4 ces auteursont montré l'existence du complexe Pu -EBTA-1/1 : Pu Y"~
(K* = 1 ,3.1018). A pH "3,3 PuIV donne lieu à la formation de Pu Y°et Pu0 Y
4+ respectivement avec K» = 4,5.1017 et K' = 1,6.1024.VI 2Au même pH, Pu serait probablement à l'état Pu Op Y (K1 =
D'autres travaux (71) (72) (73) plus récents, n'ont paspporté de résultats quantitatif
78
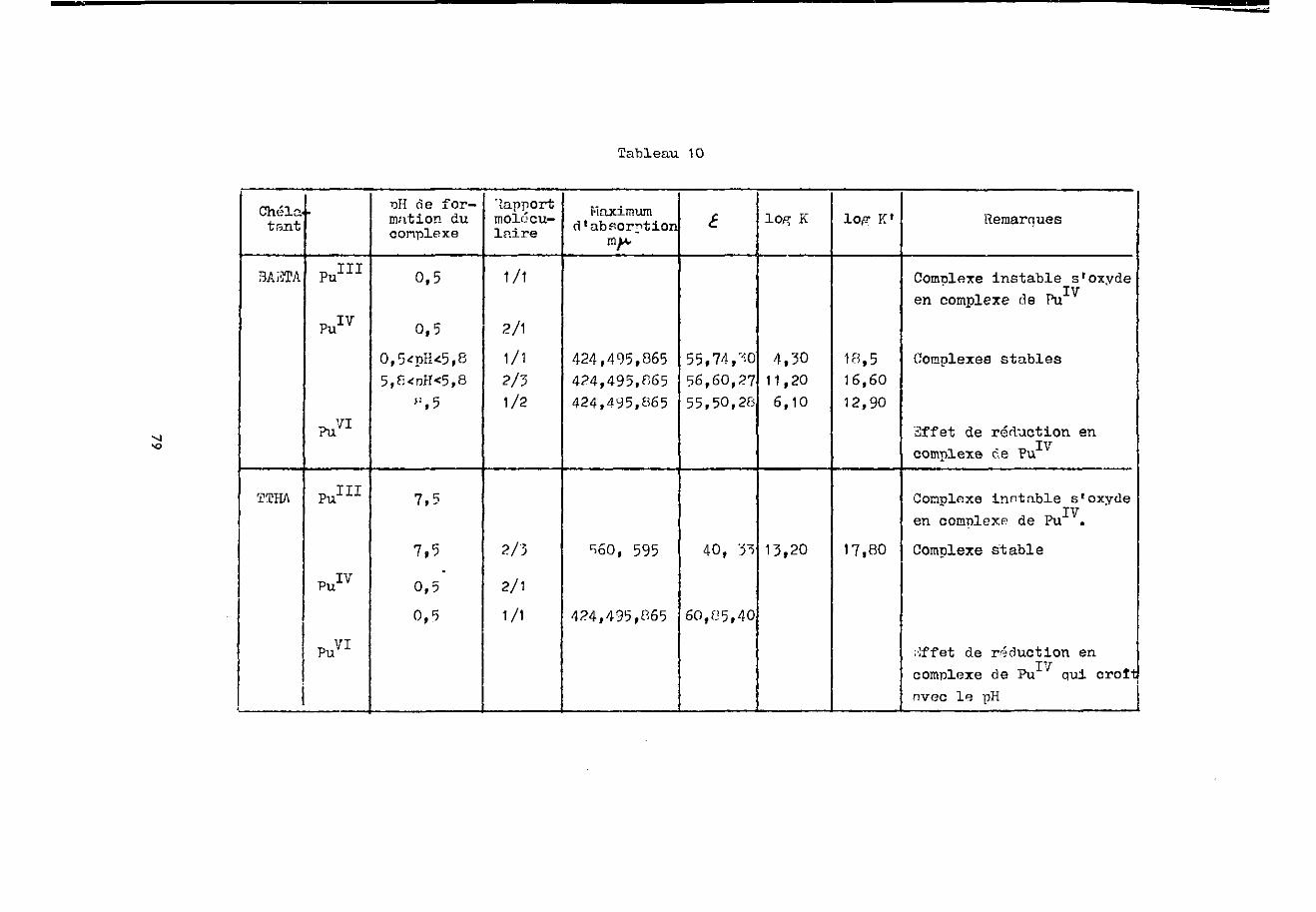
Tableau 10
Chélatant
3ASTA
TTHA
•
Pu111
PuIV
PuVI
Pu111
PuIV
PuVI
r>H de for-mation ducomplexe
0,5
0,5
0,5<pH<5,85,£<r>H<5,8
>J,5
7,5
7,5
0,5
0,5
Rapportmolécu-laire
1/1
2/1
1/12/3
1/2
2/3
2/1
1/1
Maximumd' absorption
424,495,8654?4,495,B65
424,495,865
560, 595
424,495,865
<
55,74,'<0
56,60,??55,50,26
40, 33
60,85,40
lo K
4,3011,20
6,10
13,20
iOfr K1
18,5
16,60
12,90
17,80
Remarques
Complexe instable s'oxydeen complexe de Pu
Complexes stables
Effet de réduction enIVcomplexe c.e Pu
Complexe instable s'oxyde
en complexe de Pu .
Complexe stable
;-;ffet de réduction en
complexe de Pu qui croît
nvec le pH
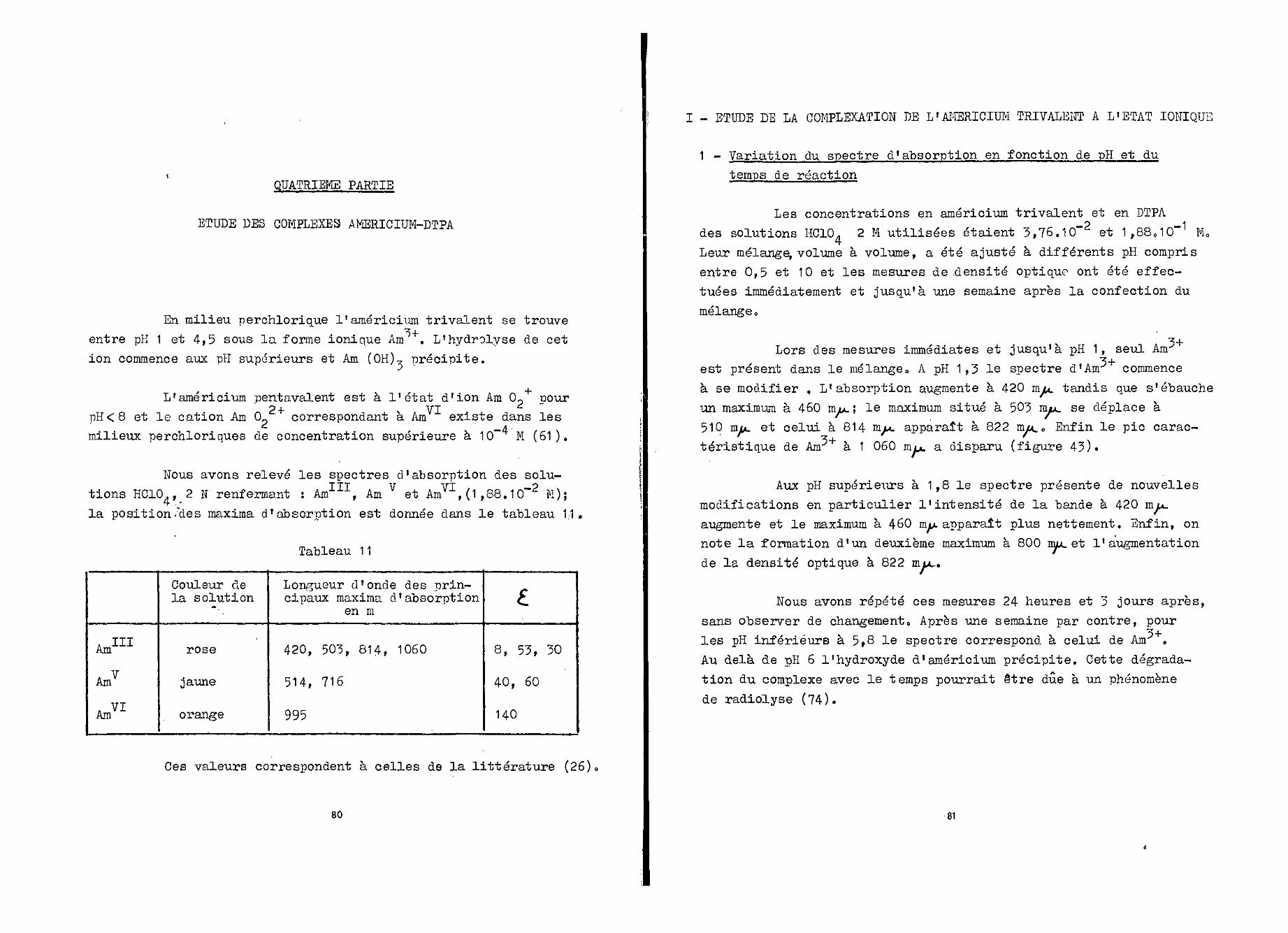
QUATRIEME PARTIE
ETUDE DES COMPLEXES AMERICIUM-DTPA
En milieu perchlorique l'américium trivalent se trouve
entre pH 1 et 4,5 sous la forme ionique Am . L'hydrolyse de cetion commence aux pïï supérieurs et Am (OH), précipite.
L'américium pentavalent est à l'état d'ion Am 00 nour24- VIpH<8 et le cation Am 00 correspondant à Am existe dans les
—4.milieux perchloriques de concentration supérieure à 10 M (61).
Nous avons relevé les spectres d'absorption des solu-tions HC104, 2 N renfermant : Am
111, Am V et AmVI,(1,88.10~2 M);
la posit ion/de s maxima d'absorption est donnée dans le tableau 1.1.
Tableau 11
Am111
AmV
A VIAm
Couleur dela solution
"°* .
rosé
jaune
orange
Longueur d'onde des prin-cipaux maxima d'absorption
en m
420, 503, 814, 1060
514, 716
995
£
8, 53, 30
40, 60
140
Ces valeurs correspondent à celles de la littérature (26)
80
I - ETUDE DE LA COMPLEXATION DE L'AMERICIUM TRIVALENT A L'ETAT IONIQUE
1 - Variation du spectre d'absorption en fonction de pH et du
temps de réaction
Les concentrations en américium trivalent et en DTPA
des solutions HC10, 2 M utilisées étaient 3,76.10~2 et 1,88010"1 M0
Leur mélange, volume à volume, a été ajusté à différents pH comprisentre 0,5 et 10 et les mesures de .densité optique ont été effec-tuées immédiatement et jusqu'à une semaine après la confection du
mélangée
3+Lors des mesures immédiates et jusqu'à pH 1, seul Anr3+est présent dans le mélange» A pH 1,3 le spectre d'Am commence
à se modifier , L'absorption augmente à 420 nyc tandis que s'ébaucheun maximum à 460 rai*. ; le maximum situé à 503 m/u. se déplace à
.510 m u. et celui à 814 niyuc apparaît à 822 m .» Enfin le pic carac-téristique de Anr+ à 1 060 mu. a disparu (figure 43)•
Aux pH supérieiirs à 1,8 le spectre présente de nouvellesmodifications en particulier l'intensité de la bande à 420 m *.augmente et le maximum à 460 mym. apparaît plus nettement. Enfin, on
note la formation d'un deuxième maximum à 800 nu*, et l'augmentation
de la densité optique à 822 mix..
Nous avons répété ces mesures 24 heures et 3 jours après,sans observer de changement„ Après une semaine par contre, pourles pH inférieurs à 5,8 le spectre correspond à celui de Am'5 .Au delà de pH 6 l'hydroxyde d1américium précipite. Cette dégrada-
tion du complexe avec le temps pourrait être due à un phénomène
de radiolyse (74).
81

Nous avons nar la suite effectué les mesures de la densité optique à 822 m^u car les maxima de 460 à 1060 mv~ ne sontpas assez intenses et la loi de Béer n'est pas vérifiée à 510
2 - Détermination du rapport moléculaire des complexes
a ) Wét ho de^d e__variat i on^molé culaire
•7 , n
On a mélangé à volume égal une solution d'Ain , 3,76,10 Met des solutions de DTPA de concentration variable, ajusté le plià 3,5 et effectué les mesures aussitôt après.
La figure 44 représente la variation de la densité opti-que à 822 my- en fonction de [AmII][J/[DTPA3. 311e traduit l'exis-tence de deux complexes caractérisés par r = 2/1 et 1/1.
Nous avons effectué de nouvelles mesures à pH 8, 5, pourun rapport moléculaire supérieur à 1/1, c'est-à-dire pour les fai-bles concentrations de DTPA. Dans ces conditions "Am non complexéprécipite. Par contre pour un rapport de concentration égal ouinférieur à .1/1 tout l'américium est complexé. Le spectre corres-pond alors à celui du complexe Am -DTPA-1/U
Met hod edsvari at ions continues
Pour une concentration (.Am ] + [DTPA] = 3,76.10~2 M,l'absorption augmente, à pH 7, avec l'augmentation de
x: = -—Ytf-i— f T jusqu'à 0,33 (figure 45 A)»Am non complexé[Am J + [DTPA]
précipite. Au delà de cette valeur l'absorption décroît. Par con-tre à pH 9 le maximum est atteint pour x = 0,5 (figure 45 A). Lafigure 45 B correspond aux mesures effectuées à pH 3,5. Elle montrebien l'existence de deux complexes Am -DTPA-2/1 et -1/1.
82
IIIEn résumé deux complexes Am - DTPA se forment entrepH 3,5 et 9 avec les rapports moléculaires 2/1, £ = 25 et 1/1,t = 30. Toutefois le complexe 2/1 n'est pas stable aux pH supé-rieurs à 7. Les constantes de formation et de stabilité sont con-signées dans le tableau 120
Tableau 12
pH
7
9
Rapportmoléculaire
(r)
2/1
1/1
1/1
Maximad'absorp-tion m/ju
822
822
822
t
25
30
30
log K
5,89
3,02
3,00
log K »
20,00
23,20
23,20
II - ETUDE DE LA COMPLICATION DE L'AMERICIUM PENTA et HEXAVALENT A
L'ETAT IONIQUE
Lorsqu'on mélange des solutions d'américium penta ouhexavalent 3,76.10~2 M et des solutions de DTPA 1,88.10""1 M, onobserve immédiatement un changement de couleur de la solution,du jaune pâle au rosé, couleur caractéristique de l'américiumtrivalento Le spectre d'absorption de ces solutions confirme bienen effet que le mélange renferme le complexe Am - DTPA 1/1. LeDTPA agit comme un réducteur.
III - ETUDE DE LA COMPLEXATION' D'AI-IERICIUM TRI PENTA-ET HEXAVALENT
A L'ETAT D'HYDROXYDE
- AmIII
Nous avons mis en contact pendant 24 heures, une solu-tion de DTPA,1,88.10"" M avec des hydroxydes d'américium trivalent
83

pen suspension, préparés à partir de solutions 3,76.10 î-î en élé-ment» L'âge de ces hydroxydes a varié entre 10 minutes et 7 jours.
On constate dans ces conditions que DTPA peut formerimmédiatement le complexe Am - DTPA-1/1 avec des précipitésâgés de moins de 5 jours. Pour des hydroxydes plus âgés, la com-plexation devient de plus en plus difficile et n'est plus possibleaprès 7 jours0
Si 1 !on poursuit ces mesures en fonction du temps, onconstate que l'intensité du spectre du complexe Am - DTPA-1/1,formé à partir d'un hydroxyde de 5 jours, reste constante pendant5 jours, puis diminue. Après une semaine la solution n'absorbeplus0
- AmV et AmVI
Avec des hydroxydes â#és de 5 jours au moins, DTPA con-duit au complexe Am -DTPA-1/1« La formation de celui-ci devientde plus en plus difficile et même impossible avec des hydroxydesplus âgés. Les résultats obtenus en fonction du temps de contact
IIIsont identiques à ceux exposés dans le cas de Am .
IV - CONCLUSION
Nous avons mis en évidence entre pH 1 et pH 9 deux cora-IIIplexes Am -DTPA solubles et stables entre pH 1,8 et pH 6, de
rapport moléculaire 2/1 (£ = 23 à 822 mfO et 1/1 ( £ = "50 àon
822 mn>) leurs constantes de stabilité sont respectivement 10e)'z <^r\
et 10 •*' .Le complexe 2/1 n'est pas stable pour les pH supé-rieurs à 5,8 ou dans un excès de chélatant.
84
Pour ce qui concerne l'américium penta- et hexavalent,nous avons constaté en présence de DTPA un effet de réductioninstantané à l'état d'oxydation trois.
ô
V - COMPLICATION DE L'ATOICIUM PAR DIFFERENTS CHELATANTS
Nous n'avons étudié que la complexation de l'américiumpar le TTHA. Le spectre d'absorption obtenu à pH 1,8 sur un mélangeAm - TTHA, ne présente par rapport au spectre d'Am seulque de très légères modifications ": apparition d'une bande d'absorp-tion à 814 m/c et d'un épaulement à 805 m/c . Le complexe de rap-port 2/1 est stable entre pH 1,8 et 6 (log K = 7,78, log K1 = 18,20et celui correspondant à r = 1/1 est stable jusqu'à pH 10 (log K =
4,9, log K» = 21,10).
Nous avons observé un effet de réduction immédiat avecl'américium penta- et hexavalent.
D'après les données de HJG-3R (76) (77), le pK de disso-ciation du complexe Am -EDTA-1/1 mesuré par échange ionique surDowex 50 entre pH 2 et 3,3 est 18,16. Les expériences du mêmetype effectuées par MOSKVIN et coll. (75) confirment ce résultat ;K1 = 9,7.101 , pour pH compris entre 1 et 2,20 De plus, ces auteurs
~ Q
ont mis en évidence le complexe neutre HAmY K = 5.10^ aux mêmes pH.
85

QUATRIEME CHAPITRE
CONCLUSION ET DISCUSSION GÉNÉRALE
Les recherches exposées dans ce mémoire se rapportentà l'étude de la complexation du cérium et des uranides, dans leursdifférents degrés d'oxydation, par l'acide diéthylènetriaminepenta-acétique (DTPA), Deux états physico-chimiques ont retenu notre
attention pour ces éléments : l'état ionique et l'état de précipitéd'hydrolyse0 Dans le premier cas, nous avons déterminé : l'influencedu pH sur la complexation, les rapports moléculaires, r = élément /
chélatant des complexes formés et lorsque ces derniers étaientstables, nous avons calculé leurs constantes de stabilité. Dans ledeuxième cas, nous avons étudié les conditions de solubilisation
des précipités d'hydrolyse par formation des complexes, en fonction
du pH, de l'âge des hyciroxydes et du temps du contact de ceux-ciavec le chélatant:.
L'action du DTPA sur le cérium trivalent conduit à laformation à partir de pH 3,5 de deux complexes caractérisés par
r = 1/1 et 2/1 dont les logarithmes des constantes de stabilité, K1,sont respectivement égaux à 15»70 et 20g3. Le complexe 2/1 n'estpas stable dans un excès de chélatant, ni pour des valeurs de pH
supérieures à 6,5.
87

Aux pH inférieurs à 2,3 , Ce * ne donne pas lieu à laformation de complexe avec DTPA, il est réduit au degré d'oxyda-tion 3, en fonction du temps. Par contre pour 2,3 pH«~2,7 lecérium tétravalent forme un complexe stable 1/1 : log Kf = 22,06 ;ce complexe est cependant réduit progressivement avec le temps etl'augmentation du pH en complexe Ce - DTPA - 2/1. L'ensemble deces observations est schématisé figure 46 A.
L'étude de la complexation de l'uranium tétravalent parle DTPA révèle l'existence entre pH 1,8 et 4, d'un complexe sta-ble : \fV - DTPA - 1/1 log K1 = 21,75. Aux pH supérieurs à 4celui-ci n'est pas stable. La couleur de la solution vire du vertau jaune, mais son spectre ne peut cependant être attribué à laprésence d'un complexe U - DTPA, même après 4 mois. Par contre,si on acidifie les solutions de pH 4 et si on les réajuste au pHinitial, elles renferment le complexe U - DTPA 1/1.
L'uranium hexavalent forme deux complexes, l'un carac-térisé par r = 2/1, insoluble entre pH 2 et 4, l'autre solublepour pH> 4 : r = 1/1 log K' = 15,5. Ce dernier présente une ins-tabilité, d'une part pour des rapports de concentration [il ]/[DTPA] inférieurs à 1/1 et d'autre part avec l'augmentation du pH0
Pour ce qui concerne le neptunium triyalent nous n'avonsIIIpu mettre en évidence l'existence de complexe Np - DTPA car
Np est oxydé en Np . Selon le pH, Np forme deux complexesavec le DTPA. Entre pH 0,5 et 5,8 le complexe 1/1 se forme,(log K» = 23,6); entre pH 5,8 et 8,5 , r = 2/3 (log K1 = 20,49)et pour pH> 8,5 le logarithme de la constante de stabilité de cecomplexe est 18,8 . Vis-à-vis de Np , DTPA a selon le pH, troiscomportements différents. Jusqu'à pH 0,5 il est sans action ; pour1,8<pH<3 il réduit Np : par exemple, après 48 heures dans unesolution telle que [NpVJ/[DTPA] soit égal à 1/2, le complexe
88
NpIV-DTPA-1/1 est présent. Enfin au delà de pH 3,5 le DTPA formeavec NpV un complexe Np - DTPA. Np est immédiatement réduit enNpV par DTPA à tous les pH.
Le plutonium trivalent est transitoirement complexé parle DTPA jusqu'à pH 6,8. Après un certain temps la solution ren-ferme le complexe Pu -DTPA-1/1. Par contre pour les pH ) 6,8l'espèce PuIi:[-.DTPA-2/3 (log K' = 18,00) est stable vis-à-visde l'oxydation. Le plutonium tétravalent et le DTPA forment quatrecomplexes différents en fonction du pH. A pH 0,5, r = 2/1 ; pourles pH compris entre 0,5 et 5,8, r = 1/1 (log K» = 23,90) puisr = 2/3 (log K» = 18,06) entre pH 5,8 et 8,5, enfin aux pH supé-rieurs à 8,5 r égale 1/2 (log K' = 13,8). Quant au plutoniumhexavalent, il est réduit à l'état tétravalent par le DTPA et con-duit, ainsi, au complexe PuIV-DTPA-1/1. L'effet réducteur croîtavec l'augmentation du pH0
Enfin, l'américiuin trivalent forme deux complexes pourpH > 1,8 avec le DTPA : Amm-DTPA-2/1 (log K' = 20,1) etAmIi:i:-DTPA-1/1 (log K' = 23). Le complexe de rapport 2/1 n'estpas stable dans un excès de DTPA, pour les pH supérieurs à 6,8.Le DTPA a un effet réducteur immédiat sur Am et Ara , il conduitdirectement à la formation du complexe Am -DTPA-1/1.
L'ensemble des observations concernant les élémentstransuraniens est schématisé dans la figure 46 B.
L'hydroxyde de Ce fraîchement préparé est solubilisépar le DTPA à l'état de complexe Ce111 - DTPA-1/1 tandis quel'action de ce chélatant sur un hydroxyde âgé aboutit à la forma-tion d'un mélange de complexes Ce111 - CeIV - DTPA. La totalité
89
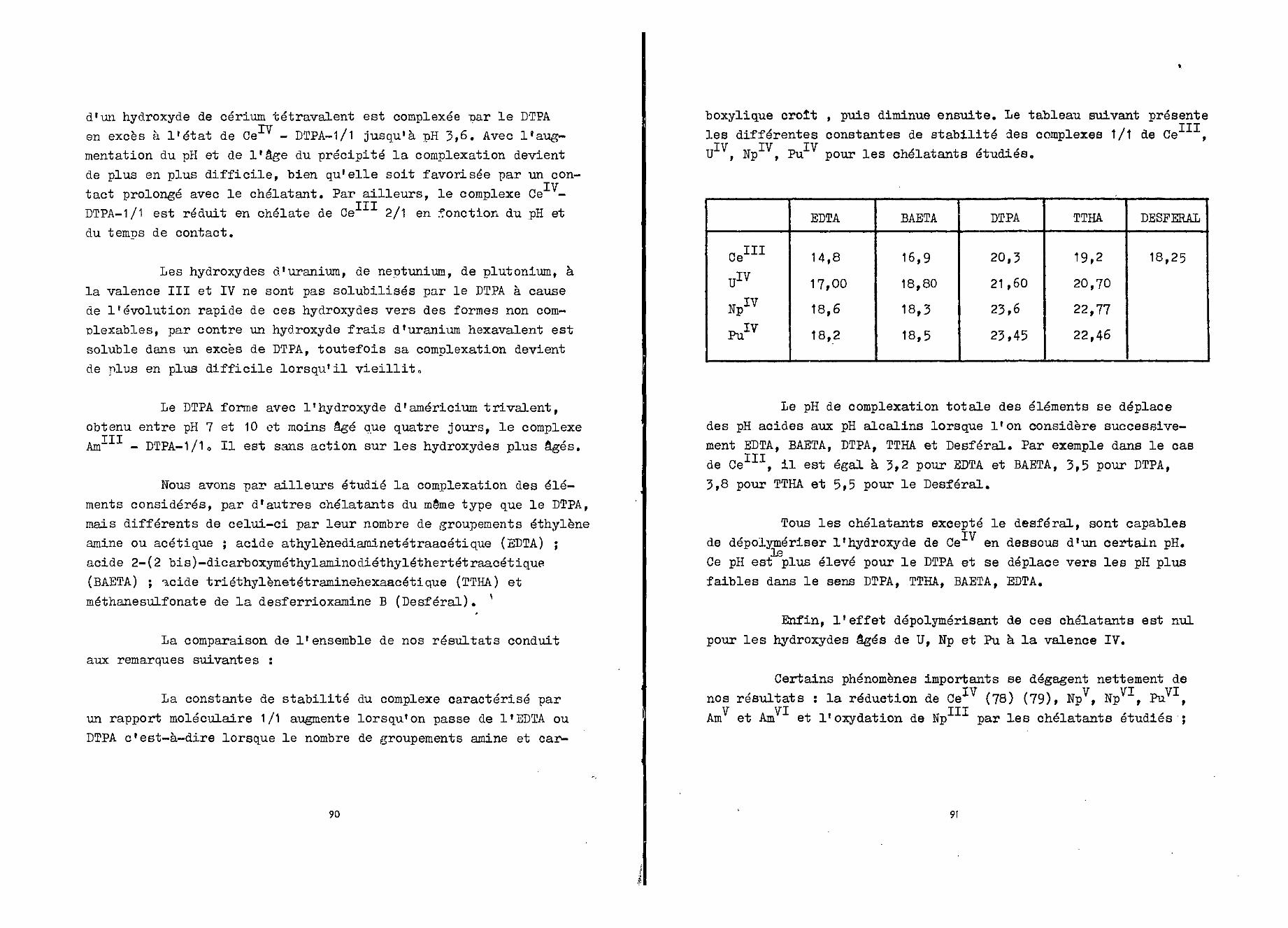
d'un hydroxyde de cérium tétravalent est complexée par le DTPAen excès à l'état de CeIV - DTPA-1/1 jusqu'à pH 3,6. Avec l'aug-mentation du pH et de l'âge du précipité la complexation devientde plus en plus difficile, bien qu'elle soit favorisée par un con-tact prolongé avec le chélatant. Par ailleurs, le complexe Ce -DTPA-1/1 est réduit en chélate de Ce 2/1 en fonction du pH etdu temps de contact.
Les hydroxydes d'uranium, de neptunium, de plutonium, àla valence III et IV ne sont pas solubilisés par le DTPA à causede l'évolution rapide de ces hydroxydes vers des formes non com-plexables, par contre un hydroxyde frais d'uranium hexavalent estsoluble dans un excès de DTPA, toutefois sa complexation devientde plus en plus difficile lorsqu'il vieillit,,
Le DTPA forme avec 1'hydroxyde d'américium trivalent,obtenu entre pH 7 et 10 et moins âgé que quatre jours, le complexeAm - DTPA-1/1o II est sans action sur les hydroxydes plus âgés.
Nous avons par ailleurs étudié la complexation des élé-ments considérés, par d'autres chélatants du même type que le DTPA,mais différents de celui-ci par leur nombre de groupements éthylèneaminé ou acétique j acide athylènediaminetétraacétique (EDTA) ;acide 2-(2 bis)-dicarboxyméthylaminodiéthyléthertétraacétique(BAETA) ; icide triéthylènetetraminehexaacétique (TTHA) etméthanesuifonate de la desferrioxamine B (Desféral). ^
f
La comparaison de 1'ensemble de nos résultats conduitaux remarques suivantes :
La constante de stabilité du complexe caractérisé parun rapport moléculaire 1/1 augmente lorsqu'on passe de l'EDTA ouDTPA c'est-à-dire lorsque le nombre de groupements aminé et car-
90
boxylique croît , puis diminue ensuite. Le tableau suivant présenteITTles différentes constantes de stabilité des complexes 1/1 de Ce ,
U , Np , Pu pour les chélatants étudiés.
Ce111
0IV
HpIV
PUIV
EDTA
14,8
17,00
18,6
18,2
BAETA
16,9
18,80
18,3
18,5
DTPA
20,3
21,60
23,6
23,45
TTHA
19,2
20,70
22,77
22,46
DESPERAL
18,25
Le pH de complexation totale des éléments se déplacedes pH acides aux pH alcalins lorsque l'on considère successive-ment EDTA, BAETA, DTPA, TTHA et Desféral. Par exemple dans le casde Ce111, il est égal à 3,2 pour EDTA et BAETA, 3,5 pour DTPA,3,8 pour TTHA et 5,5 pour le Desféral.
Tous les chélatants excepté le desféral, sont capablesde dépolymériser l'hydroxyde de Ce en dessous d'un certain pH.
leCe pH est plus élevé pour le DTPA et se déplace vers les pH plusfaibles dans le sens DTPA, TTHA, BAETA, EDTA.
Enfin, l'effet dépolymérisant de ces chélatants est nulpour les hydroxydes âgés de U, Np et Pu à la valence IV.
Certains phénomènes importants se dégagent nettement derésultats : la réduction de CeIV (78) (79), NpV, NpVIf Pu
VI,Am et Am et l'oxydation de Np par les chélatantB étudiés ;nos
91

la stabilisation de la valence IV de U, Np et Pu, vis-à-vis del'oxydation (80) (81) (82) par la formation des complexes ; etenfin l'instabilité en fonction du pH et de la concentration enchélatant des complexes formés avec l'uranium hexavalent.
Dans tous les cas où il y a diminution du degré d'oxyda-tion des éléments étudiés, nous avons mis en évidence la dégradationdu DTPA, l'effet de réduction étant accéléré lorsque le pH croît(83) (84) (85). On peut admettre qu'il est dû aux électrons duchélatant libérés par la dégradation de ce dernier. Par ailleurs,comme on ne constate pas de réduction dans le cas de NID lorsqu'ilest complexé (pour pH > 3,5), il est probable que la réduction alieu sur l'élément non encore engagé dans le complexe. Ceci estconfirmé par le fait que Ce peut être réduit en Ce-5 aux pH<1,5»pH pour lesquels il n'est pas complexé. Ainsi le chélatant réduitCeIV, NpV (pH<3,5), NpVI, PuVI, AmV et AmVI respectivement enCe111, NpIV, Pu et Am qui sont à leur tour complexés parles acides polyaminecarboxyliques non dégradés.
Nous avons vu au cours de ce travail que les spectresd'absorption des solutions U - DTPA présentent une suite de modi-fications en fonction du pH et de la concentration en chélatant,que nous avons attribuées à une instabilité du complexe UDTPA-1/1.
Récemment MIKHAILOV et coll (86) ont formulé l'hypothèsesuivante» L'uranium hexavalent présente, après la formation deliaisons covalentes ordinaires avec l'oxygène et môme après'laformation de liaisons secondaires avec le chélatant, deux couchesnon saturées (5 f et 6 d) (87) (88) dans le groupement U0?. Lesliaisons secondaires sont perpendiculaires à l'axe 0 = U = 0 etprovoquent un déplacement des charges négatives du chélatant versl'uraniumo D'autre part les atomes d'oxygène dans l'ion uranyle,ne seraient pas seulement divalents, mais pourraient former desétats de valence supérieure par déplacement de leurs électrons non
92
•cartages vers les couches non saturées de l'uranium. Il y auraitdonc compétition entre les liaisons additionnelles datives et lesliaisons secondaires, ce nui pourrait être un facteur d'empêchementà la formation de complexes stables de l'ion uranyle avec des grou-pements possédant deps caractères donneurs d'électrons comme l'amo-niac (89), les aminés (90) et les acides polyaminecarboxyliquescomme l'EDTA (52) (91) (92). Nous pensons qu'on peut admettrecette hypothèse, pour expliquer l'instabilité des autres complexespolyaminecarboxylioues de l'ion uranyle.
L'ensemble de notre étude montre que le DTPA est parmiles divers acides aminopolycarboxylioues étudiés, celui qui offrele plus d'intérêt pour le décontamination des éléments considérés,lorsqu'ils ne sont pas à l'état d'hydroxydes. En effet, la cons-tante de stabilité des complexes ou'il forme avec ceux-ci esttoujours la plus élevée.
93

BIBLIOGRAPHIE
1 - KETY S.S.
J. Biol. Chem. 142, 181, 1942
2 - "Radioactive Metal Mobilization in Medicine"
par CATSCH A., SPRINGFIELD, Ch. C. THOMAS, p. 10, 1964
3 - AEBERHARDT A.
Thèse Paris. Rapport C.E.A. n° 1856, 1961
4 - CHIADOT P. et LAFUMA J.
Rev. d'Hygiène et de Médecine Sociale, 5, 391, 1962
5 - HELLER H.J. et CATSCH A.
Strahlenther. 109, 464, 1959
6 - AUB J.C., EVANS R.D., GALLAGHER D.M. et TIBBETTS D.M.
Ann. Int. Med. 11, 1443, 1938
7 - SCHUBERT J.
Science, 105, 389, 1947
8 - PETERS R.A., STOCKEN L.A. et THOMSON R.H.S.
Nature 156, 601,1945

9 - SCHUBERT J.
Ann. Rev. Nucl. Soi. 5, 369, 1955
10 - BELKNAP E.L.Industr. Med. Surg. 21, 305, 1952
11 - CATSCH A., PANY H.E. et TREGUBENKO I.P.Pond. Ural. Filial Akad. Nauk USSR, Sverdlovsk, 1951
12 - KROLL H., KORMAN S., SPIEGEL E., HART H.E., ROSOFF B.,
SPENCER H. et LASZLO D.Nature, 180, 919, 1957
13 - RAZUMOVSKY N.O., TORCHINSKAYA O.L. et BALABUKHA V.S.
Biofizika, 6, 610, 1961
14 - TAYLOR D.M. et SOWBY P.D.Phys. Med, Biol. 7,83, 1962
15 - SMITH V.H.Nature, 181 , 1792, 1958
16 - RAZBITNAYA L.M.
Radiokhimlya, 6, 2, 193, 1964
17 - RAZBITNAYA L.M. et KORONINA I.A.
Radiokhimiya 6, 5, 593, 1961
18 - MOELLER T. et THOMPSON L.C.
J. Inorg. Nucl. Chem. 24, 499, 1962
96
19 - BAYBARZ R.D.
J. Inorg. Nucl. Chem. 27, 1831, 1965
20 - "Report of Commitee II on permissible dose for internationalradiation.Recommendation of the International Commission on
tiRadiological Protection. Pergamon Press, 1959.
21 - HOLLECK L. et ECKARDT D.
Naturwiss 8A, 660, 1953
22 - SCHWARZENBACH G. et WILLI A.
Stability Constant part 1. Organic Ligands par J. BJERRUMLondon Pergamon Press 1960
23 - MOELLER T. et FERRUS
Inorg. Chem. 1, 49, 1962
24 - GRIMES J.H., HUGGARD M.J. et WILFORD S,P.
J. Inorg. Nucl. Chem. 25, 1225, 1963
25 - GOGLIATI G., DE LEONE R. et LANZ R.
RT/CHI (64) 14 Roma Giugno, 1964
26 - KATZ J.J. et SEABORG G.T.
"The chemistry of the actinide elements"London,Nethuen Co Ltd, p. 300, 1957
27 - CHARLOT.G. et BEZIER D.
"L'analyse qualitative et les réactions en solution",Troisième Edition»Paris, Masson Editeur, p. 323, 1955
28 - KOEiïLY G,
Anal. Chinu Acta, 33, 418-425, 1965
97

29 - CORPEL J. et REGNAUD P.
Anal. Chim. Acta, 35, 508,1966
30 - HARVEY A.E. et MANNING D.L.J. Amer. Chem. Soc. 72, 4488, 1950
31 - JOB P.Ann. Chim. 9, 113, 1928 \ 16, 97, 1936
32 - CHARLOT G. et GAUGUIN R.Les méthodes d'analyse des réactions en solutionParis, Masson Editeur, p. 74, 1951
33 - KLAUSBNK.S. et LANGMYHR F.J.Anal. Chim. Acta 28, 335, 1963
34 - V/ATKINS K.O. et JONES M.M.J. Inorg. Nucl. Chem. 24, 809, 1962
35 - WATKINS K.O. et JONES M.M.
J. Inorg. Nucl. Chem. 24, 1235, 19b2
36 - Consulter : "Nouveau Traité de Chimie Minéraie"-PASCAL P.Tome VII, Premier-Fascicule, Masson et C , 1962
37 - "The rare earth elements" par TRIFONOV D.N.London, Pergamon Press, p. 57-72, 1963
38 - JOLANDA M. HOJMAN
Bull. Inst. Nucl. Soi. 5, 89, 1955
98
39 - TROMBE F."Nouveau traité de Chimie Minérale"-PASCAL P.Tome VII, Deuxième Fascicule, Paris, Masson et C1962
, p. 1201,
40 - MIRONOV N.N. et CHERNYAEV N.P.Russ. J. Inorg. Chem. 6, 9, 1104, 1961
41 - TANANAEV I.V. et BOKMEL'DER M. Ya
Russ. J. Inorg. Chem. 5, 3, 337, 1960
42 - IRVING H.M.N.H. et CONESA J.P.
J. Inorg. Nucl. Chem. 26, 1945, 1964
43 - KOROBOVA V.A. et PRIK G.A.Russ. J. Inorg. Nucl. Chem. 10, 4, 456, 1965
44 - KUMOK V.N. et SEREBRENNIKOV V.V.
Russ. J. Inorg. Nucl. Chem. 10, 9, 1095, 1965
45 - THOMPSON L.C. et LORAAS J.A.Inorg. Chem. 2, 1, 89, 1963
46 - Consulter : "Nouveau Traité de Chimie Minéraie"-PASCAL p.Tome XV, Deuxième Fascicule, Paris, Masson Editeur, 1961
47 - CHARLOT G."Analyse Quantitative Minérale"Paris, Masson Editeur, p. 200, 1957
48 - KRAUS K.A. et NELSON F0J. Amer. Chem. Soc. 72, 3901, 1950
99

49 - SUTTON JoJ. Inorg. Nucl. Chem. 1-2, 68, 1955
50 - CHERNYAEV I.J., GOLOVNYA V.A. et ELLERT G.V.Russ. J. Inorg. Chem. 5, 7, 719, 1960
51 - KOMAR N.P. et TRET'YAK Z.A.J. Anal. Chem. 10, 236, 1955
52 - RYABCHBCOV D.I., PALEI P.N. et MIKHAILOVA Z.K.Zhur. Anal. Khim. 14, 581, 1959
53 - NEUMAN W.F. et TISHKOFF G.H."Pharmacology and toxicology of uranium»New York,Me Graw Hill, p. 1087, 1951
54 - KLYGIN I.D., SMIRNOV et NIKOL'SKAYA N.A.
Russ. J« Inorg. Chem. 4, 1279, 1959
55 - KROT N.N., ERMOLAEV N.P. etGEL'MANA.D.
Russ. J. Inorg. Chem. 7, 9, 1062, 1962
56 - SMITH T.D.
J. Inorg. Nucl. Chem. 11, 314, 1959
57 - PALEI P.N, et HSÎJ LI YUANRuss. J. Inorg. Chem. 6, 12, 1337, 1961
58 - KOZLOV A.G. et KROT N.N.
Russ. J. Inorg. Chem. 5, 9, 954, 1960
59 - BHAT T.R. et KRISHNAMURTHY M.
J. Inorg . Nucl. Chem. 26, 587, 1964
100
60 - JAMES D.B., POWELL J.E. et SPEDDING F.H.J. Inorg. Nucl. Chem. 19, 133, 1961
61 - Consulter : "Nouveau Traité de Chimie Minérale"-PASCAL P.Tome XV, Troisième Fascicule, Paris, Masson et Cie, 1962
62 - Consulter : "Complex Compounds of Transuranium Elements"par GEL1MAN A.D., Traduit du russe, Consultants Bureau,New-York, 1962
63 - JENKINSRapport anglais AERE G/A 2721, 1958
64 - ZOLOTOV YUoA. et NOVIKOV YTJ.P.Zhur. Neorg. Khim, 4, 7, 1693, 1959
65 - GEL'MAN A.D., MOSKVIN A.I. et ARTYUKHIN P.I.
Atomnaya Energiya 7, 2, 162, 1959
66 - GEL'MAN A.D. et MEFOD'EVA M. P.
Dok. Akad. Nauk 124, 815, 1959
67 - HINDMAN J.C., MAGNUSSON L.B. et LACHAPELLE T.J.
"The Transuranium Elements" Me Graw-Hill,New York,
Paper 15, 2, p. 1039, 1949
68 - KREVINSKAYA M.E.
Radiokhimiya 1, 5, 548, 1959
69 - NEWTON T.W; and BAKER F.B.
J. Phys. Chem. 61, 934, 1957
101

70 - FOREMAN J.K. et SMITH T.D.
J. Chem. Soc. 1752, 1957
71 - GEL «MAN A.D. et MOSKVIN A.I.
Atomnaya Energiya 3, 10, 314, 1957
72 - FUGER J. et CUNNINGHAM B.B.
J. Inorg. Nucl. Chem. 27, 1079, 1965
73 - GEL'MAN A.D., ARTYUKHIN P.I. et MOSKVIN A.I.
Russ. J. Inorgo Chem. 4, 6, 599, 1959
74 - ASPREY L.B., STEPHANOU S.E. et PENNEMAN R.A.
J, Am. Chem. Soc. 72, 1425, 1950
75 - MOSKVIN A.I.
Radiokhimiya 1, 4, 430, 1959
76 - FUGER J.
J. Inorg. Nucl. Chem. 5, 4, 332, 1958
77 - FUGER J.
J. Inorg. Nucl. Chem. 18, 263, 1961
78 - PALEI P.N. and UDAL'TSOVA N.I.
Zhurnal Anal. Khim. 15, 6, 668, 1960
79 - BHAT T.R., RADHAMMA D.
Indian J. Chem. 3, 151, 1965
SO - Consulter s "An Introduction to the Chemistry of Complexcompound" par GRINBERG A.A.London,Pergamon Press, 1962
102
81 - Consulter : "Chemistry of metal Chelate Compounds"par MA.RTELL A.ïï. et CALVIN M.New-York, Prentice Hall, 1962
82 - CUNNINGHAM B0B.
"Rare Earth Research" Macmillan Company, New- York,
Po 127, 1961
83 - Consulter : "The Sequestration of metals" par SMITH R0London, Chapman & Hall Ltd, 1959
84 - PELLERIN F.
Ann. Biol. Clin. 20, 7/9, 707, 1962
85 - Consulter : "Complexometric titrations" par SCHWARZENBACH G.London, Methuen & Co Ltd, 1960
86 - DYATKiNA'MoB., MARKOV v.p.f TSAPKINA i.v. et MIKHAILOV YU<NRusse J. Inorgo Chem. 6, 5, 293, 1961
87 - MOELLER T.
Handbook of chemistry and physics45 th edition, 1965
88 - TRZEBIATOWSKA B0J0 et BUKIETYNSKA K0
Jo Inorgo Nucl. Chem. 19, 38, 1961
103

89 - ZOLOTAVIN V.L., BEZRUKOV I. YA et SANNIKOV YU.I.
Russ. J. Inorg. Chem. 6, 3, 295, 1961
90 - SACCONI L.
Atti. Accad. Nazi. Lincie Classe Soi. Fis0 Mat. Nat.8, 6, 639, 1949
91 - CABELL M.J.
Analyst 77, 859, 1952
92 - MACEK K. et PRIBIL P.
Collection, Czech, chem. Communications 20, 715, 1955
104
PLAN
- INTRODUCTION
- PREMIER CHAPITRE - TECHNIQUES ET METHODES EXPERIMENTALES
Pages
7
11
I - Techniques expérimentales ' 11A
1 - Réactifs 11
2 - Formules et constante de dissociation des 12chélatants
3 - Techniques speotrophotometriqu.es }3
II - Préparation des solutions et dosages des élémentstf 14dans leurs différents états dfoxydation
1 — Préparation des solutions des éléments étudiés<e 14
a) Préparation de solutions de Ce , Ce , 14UTI et UW
b) Préparation des solutions de neptunium, deplutonium et d'amériçium dans leurs différentsétats de valence 15
105

2 - Dosages des éléments en solution
a) Dosage des solutions de neptunium, de pluto-nium et d'américium par coulométrie
b) Dosage des solutions de neptunium et de plu-tonium par titrage avec Ce4+
3 - Préparation des mélanges élément-chélatant ....
III - Méthodes analytiques
1 - Méthode de variation moléculaire
2 - Méthode basée sur le rapport des pentes
3 - Méthode de variation continue
16
16
16
17
18
18
19
19
- DEUXIEME CHAPITRE - ETUDE DES COMPLEXES CERIUM, D.T.P.A. 23
I - Etude de la complexation du cérium trivalent à l!étationique 24
1 - Variation du spectre d'absorption en fonction du pH 24
2 - Détermination du rapport moléculaire des complexes 24
a) Méthode de variation moléculaire à pH 3,5 24b) Méthode basée sur le rapport des pentes.. 25c) Méthode de variations continues 26d) Calcul des constantes de stabilité des complexes 26
II - Etude de la complexation du cérium tétravalent à l'étationique 27
1 - Variation du spectre d'absorption en fonction du pH 27
2 - Détermination du rapport moléculaire du complexe àPH 2,5 28
106
3 -
a) Méthode de variation moléculaireb) Méthode de variations continues
Influence de la concentration du DTPA sur l'effetréducteur
2829
29
III - Etude de la complexation du cérium trivalent à l'étatd'hydroxyde
1 - Influence de l'âge du précipité et du pH sur laformation du complexe .
2 - Discussion et interprétation des résultats
30
31
32
IV - Etude de la complexation du cérium tétravalent à l'étatd'hydroxyde 33
1 - Influence de l'âge du pr^oipité colloïdal et du pHsur la formation des complexes
2 -.Influence du temps du contact du précipité d'hy-droxyde. avec le chélatant
V - Conclusion
VI - Complexation du cérium par différents chélatants
34
35
38
39
- TROISIEME CHAPITRE - 41
Première Partie : Etude des complexes uranium-DTPA .. .„. 41
I - Etude de la complexation de l'uranium tétravalent àl'état ionique 42
1 - Variation du spectre d'absorption en fonction dupH et du temps de réaction 42
107

2 - Détermination du rapport moléculaire des complexes 44
a) Méthode de variation moléculaire 44b) Méthode de variations continues 45
II - Etude de la complexation de l'uranium hexavalent à1'état ionique
1 - Variation du spectre d'absorption en fonction du pH
2 - Détermination du rapport moléculaire des complexes
a) Méthode de variation moléculaireb) Méthode de variations continues
III - Etude de la complexation dé l'hydroxyde d'uraniumtêtravalent
IV - Etude de la complexation dc-s formes hydrolyséesd'uranium hexavalent
- Influence de l'âge de l'hydroxyde sur la formationde complexes "
V - Conclusion
VI - Complexation de l'uranium par différents chélatants
Deuxième Partie : Etudes des complexes neptunium D.T.P.A.
I - Etude de la complexation du neptunium trivalent àl'état ionique
46
46
47
47
49
50
51
52
53
53
56
57
II - Etude de la complexation du neptunium têt ravalent àl'état ionique 58
108
1 - Variation du spectre d'absorption en fonction du pH 58
2 - Détermination du rapport moléculaire des complexesà différents pH 59
«
a) Méthode de variation moléculaire 59b) Méthode de variations continues 59
III - Etude de la complexation de neptunium pentavalent àl'état ionique 61
1 - Variation du spectre d'absorption en fonction du pH gi
2 - Influence de temps de réaction 61
3 - Influence de la concentration en D.T.P.A. sur l'effetde réduction 63
IV - Etude de la complexation de neptunium hexavalent à l'état
ionique 63
V - Etude de la complexation de neptunium à l'état d'hydroxyde 64
VI - Conclusion
VII - Complexation du neptunium par différents chélatants
Troisième Partie : Etude des complexes plutonium-LTPA
I - Etude de la complexation du plutonium tétravalent à1'état ionique
1 - Variation du spectre d'absorption en fonction du pH
2 - Détermination du rapport moléculaire du complexeà différents pH
a) Méthode de variation moléculaireb) Méthode de variations continues
65
66
68
69
69
70
70
71
109

II - Etude de la complexation du plutonium tri valent, à
1 ' état ionique 72
III -
1 - Variation du spectre d'absorption en fonction du pH 72
2 - Détermination du rapport moléculaire des complexes 73
a) Méthode de variation moléculaire 73"b) Méthode de variations continues 74
Etude de la complexation du plutonium hexavalent à
l1 état ionique 74
1 - Variation du spectre d'absorption en fonction du pH 74
2 - Influence de la concentration en D.T.P.A, sur laréduction 75
IV - Etude de la complexation des hydroxydes de plutonium 76
V - Conclusions
VI - Complexation du plutonium par différents chélatants
Quatrième Partie : Etude des complexes américium-DTP£ . . e
I - Etude de la complexation de l'américium trivalent àl'état ionique
1 - Variation du spectre d'absorption en fonction dupH et du temps de réaction
2 - Détermination du rapport moléculaire des complexes
a) Méthode de variation moléculaireb) Méthode de variations continues
77
77
80
81
81
82
82
82
110
II - Etude de la complexation de l1américium penta ethexavalent à l1état ionique
III - Etude de la complexation de l'américium tri, penta ethexavalent à l1état d'hydroxyde
83
83
IV - Conclusion 84
V - Complexation de l'américium par différents chélatants 85
- QUATRIEME CHAPITRE - CONCLUSION ET DISCUSSION GENERALE 87
- BIBLIOGRAPHIE 95
Manuscrit reçu le 18 mars 1968
111
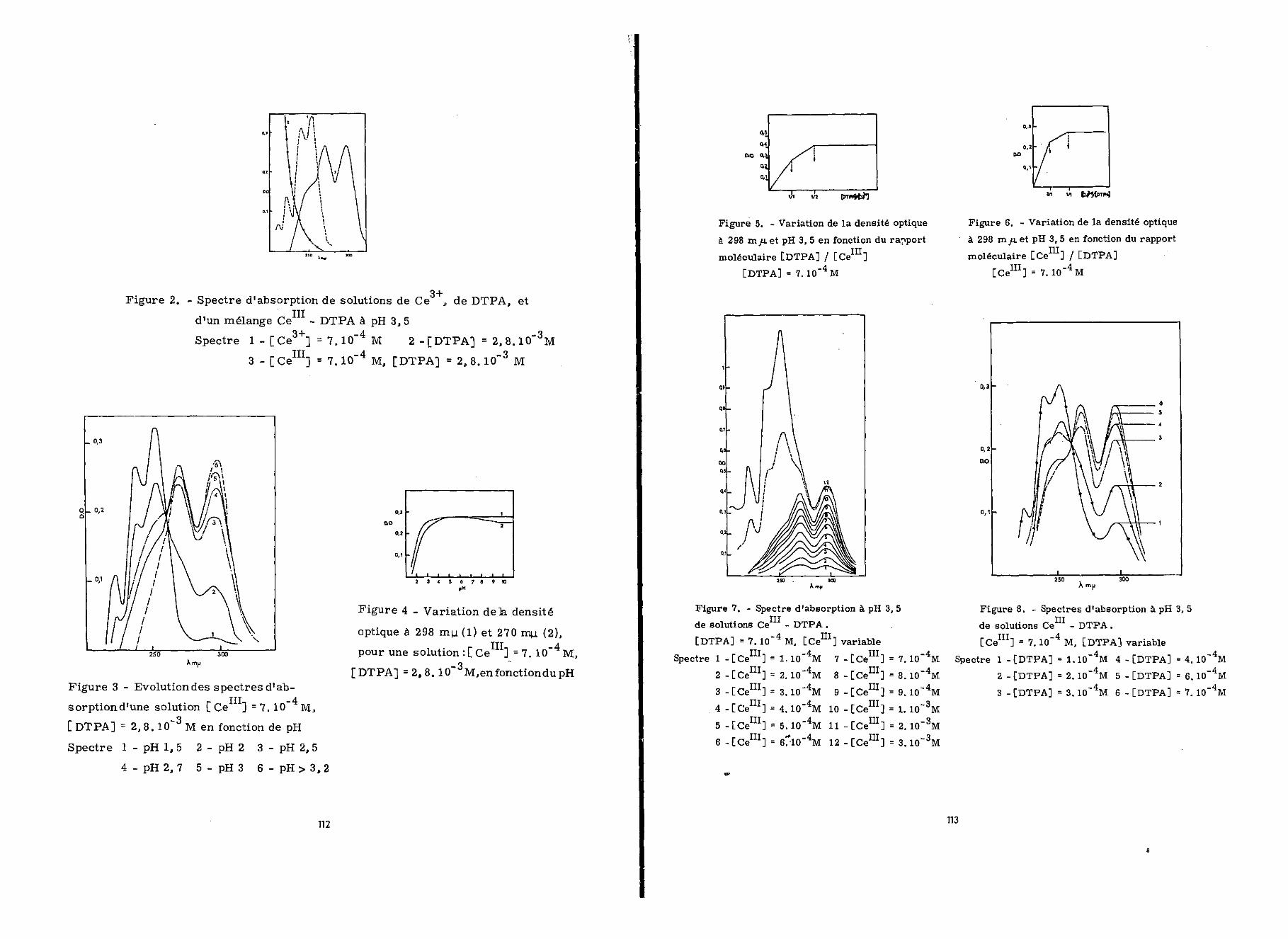
- 0,3
- 0,2
-0,1
3+Figure 2. - Spectre d'absorption de solutions de Ce , de DTPA, et
d'un mélange Ce111 - DTPA à pH 3, 5
Spectre 1 - [Ce3+] = 7.10"4 M 2 -[DTPA] = 2,8.10TTT -4 q
3 - [Ce 3 = 7.10 * M, [DTPA] =2 ,8 .10 J M
-3,
250 300
0,3
0.0
0,2
0,1
2 3 < 5 6 7 8 » 1 0pH
Figure 3 - Evolution des spectres d'ab-
sorptiond'une solution [ Ce ] =7.10" M,
[DTPA] = 2,8.10"3 M en fonction de pH
Spectre 1 - pH 1, 5 2 - pH 2 3 - pH 2, 5
4 - pH 2, 7 5 - pH 3 6 - p H > 3 , 2
112
Figure 4 - Variation dek densité
optique à 298 my (1) et 270 n^ (2),
pour une solution : [ Ce ] = 7. 10 M,
[DTPA3 = 2, 8.10~3 M,en fonction du pH
txo o,\o-l
Figure 5. - Variation de la densité optique
à 298 m/i et pH 3, 5 en fonction du rapport
moléculaire [DTPA] / [CeIE][DTPA] = 7. 10"4M
Figure 7. - Spectre d'absorption à pH 3, 5de solutions Ce - DTPA .
[DTPA] = 7. 10"4 M, [Ce111] variable
Spectre 1 - [Ce111] = 1. 10"4M 7 - [Ce111] = 7. 10~4M
2 - [Ce111] = 2. 10"4M 8 - [Ce™] = 8. 10"4M3 - [Ce111] = 3. 10'4M 9 - [Ce™] = 9. 10"4M
4 -[Ce111] = 4. 10~4M 10 -[Ce111] = 1. 10"3M
5 -[Ce111] = 5. 10"4M 11 -[Ce111] = 2. 10"3M
6 -[Ce111] = 6r-10- 12 -[Ce111] = 3. 10~3M
0,300
0,1
i/l
Figure 6. - Variation de la densité optiqueà 298 m /i et pH 3, 5 en fonction du rapport
111moléculaire [ C e ] / [DTPA]
[Ce111] = 7. 10 ~4 M
0,3
0,2
00
0,1
250 300
Figure 8. - Spectres d'absorption à pH 3, 5de solutions Ce - DTPA.
[Ce111] = 7.10"4 M, [DTPA] variable
Spectre 1 -[DTPA] = 1.10"4M 4 -[DTPA] = 4. 10~4M2 - [DTPA] = 2. 10"4M 5 - [DTPA] = 6. 10~4M
3 -[DTPA] = 3. 10"4M 6 - [DTPA] = 7. 10"4M
113
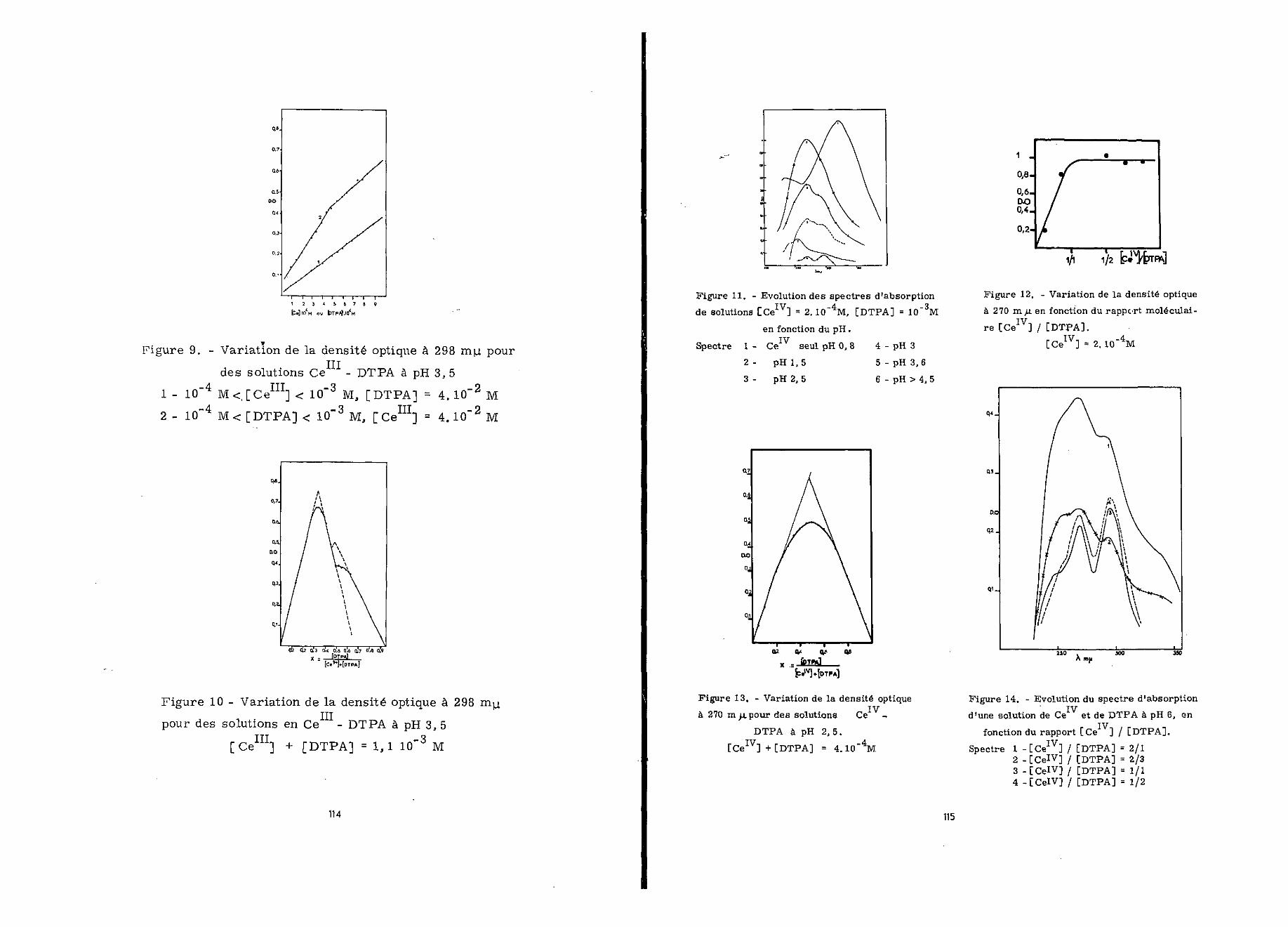
o,».
0,7.
0,6
0.5
OO
Q<
2 3 4 5 6 7 8 9
Figure 9. - Variation de la densité optique à 298 mu pour
des solutions Ce - DTPA à pH 3, 5-4 TTT -^ 9
1 - 10 * M<[Ce ] < 10 ° M, [DTPA] = 4.10"^ M-4 -^ TTT 9
2 - 10 * M< [DTPA] < 10 0 M, [ Ce ] = 4.10"^ M
0,4.
0,7.
an
cxo
0,1
0,1
0.1
eu eu a'j o/< ois o!a a? o> 0)9x . [PTPA!
" [Ct*J.[DTPA]
Figure 10 - Variation de la densité optique à 298
pour des solutions en Ce - DTPA à pH 3, 5TTT -S
[Ce ] + [DTPA] = 1,1 10 ° M
114
Figure 11. - Evolution des spectres d'absorption
de solutions [CeIV] = 2. 10~4M, [DTPA] = 10"3M
Spectre 1 -
2 -
3 -
en fonction du pH.
CeIV seul pH 0, 8
p H l . 5
pH 2,5
4 - pH 3
5 - pH 3, 6
6 - pH > 4, 5
Q7
a»
cxo
o,
°A
°i
02 CV
X .= •
46
Figure 13. - Variation de la densité optique
à 270 m )*. pour des solutions
DTPA à pH 2,5.IV
CeIV-
[Ce ]+[DTPA] = 4. 10 M.
1 .
0,8-
0,6.DO0,4.
0,2-
1/2
Figure 12. - Variation de la densité optique
à 270 m;u. en fonction du rapport moléculai-
re [CeIV] / [DTPA].TV 4
[Ce ] = 2. 10 M
Q3-
D.O
ÎSO 100 ISO
Figure 14. - Evolution du spectre d'absorptionIVd'une solution de Ce et de DTPA à pH 6, en
fonction du rapport [CeIV] / [DTPA].
Spectre 1 -[CeIV] / [DTPA] = 2/12 -[CelV] / [DTPA] = 2/33 -[CeIV] / [DTPA] = 1/14 -[CeIV] / [DTPA] = 1/2
115
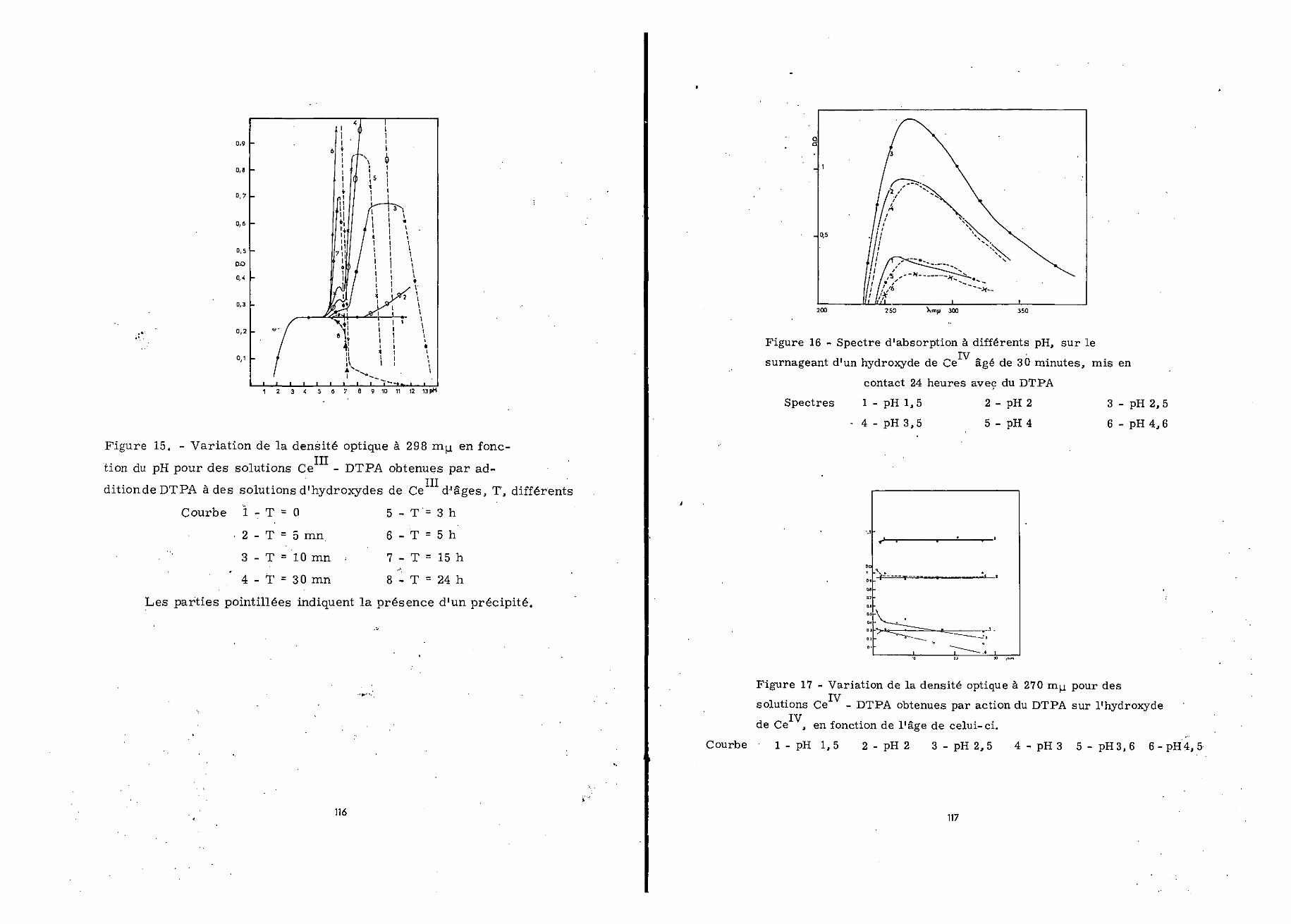
0,9
0,8
0,7
0,6
0,5
DO
0,<
0,3
0,2
0,1
3 ^ 5 û 7 8 9 10 11 12 13 pH
Figure 15. - Variation de la densité optique à 298 m.|j en fonc-
tion du pH pour des solutions Ce - DTPA obtenues par ad-
ditiondeDTPA à des solutions d'hydroxydes de Ce d'âges, T, différents
Courbe 1 - T = 0 5 - T = 3 h
• 2 - T = 5 mn 6 - T = 5 h
3 - T = iO mn ; 7 - T = 15 h
4 - T = 30 mn 8 - T = 24 h
Les parties pointillées indiquent la présence d'un précipité.
116
0,5
200 250 Xmji 300 350
Figure 16 - Spectre d'absorption à différents pH, sur le
surnageant d'un hydroxyde de Ce âgé de 30 minutes, mis en
contact 24 heures avec du DTPA
Spectres 1 - pH 1,5 2 - pH 2 3 - pH 2,5
- 4 - p H 3 , 5 5 - p H 4 6 - p H 4 , 6
r—- T-—
Figure 17 - Variation de la densité optique à 270 m|j pour des
solutions Ce - DTPA obtenues par action du DTPA sur l'hydroxydeIV
de Ce , en fonction de l'âge de celui-ci.
Courbe 1 - pH 1, 5 2 - pH 2 3 - pH 2, 5 4 - pH 3 5 - p H 3 , 6 6-pH4, 5
117
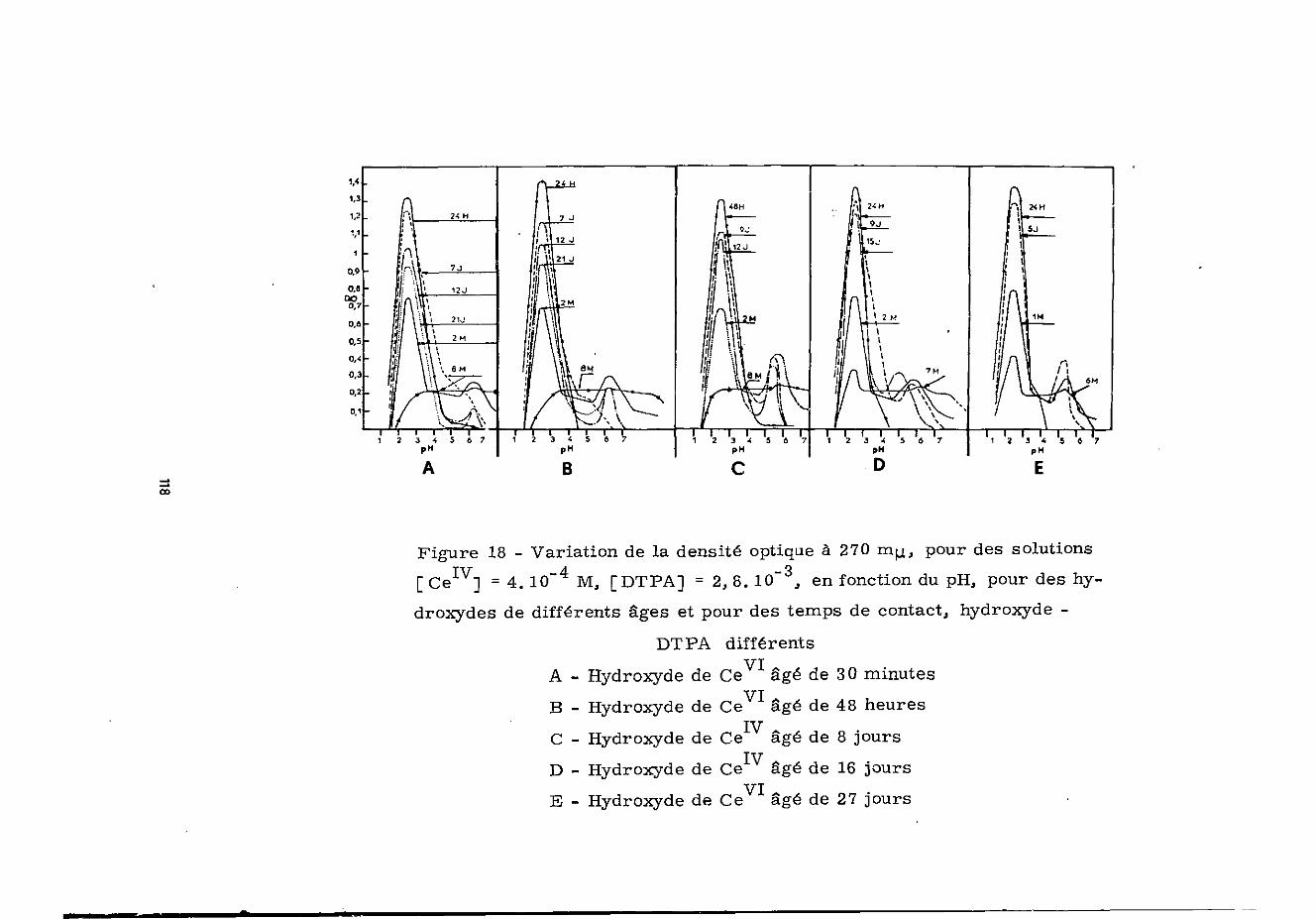
1,3
1,2
V
1
0.9
0.9000,7
0.6
0,5
0,<
0,3
0,2
0.1
I I1 2 3 4 5 6 7
pH
c3 <pH
D
Figure 18 - Variation de la densité optique à 270 rajj, pour des solutions
[CeIV] = 4. 10~4 M, [DTPA] = 2, 8. 10~3, en fonction du pH, pour des hy-
droxydes de différents âges et pour des temps de contact, hydroxyde -
DTPA différentsVIA - Hydroxyde de Ce âgé de 30 minutesVIB - Hydroxyde de Ce âgé de 48 heures
C - Hydroxyde de Ce âgé de 8 jours
D - Hydroxyde de Ce âgé de 16 jours
E - Hydroxyde de CeV âgé de 27 jours
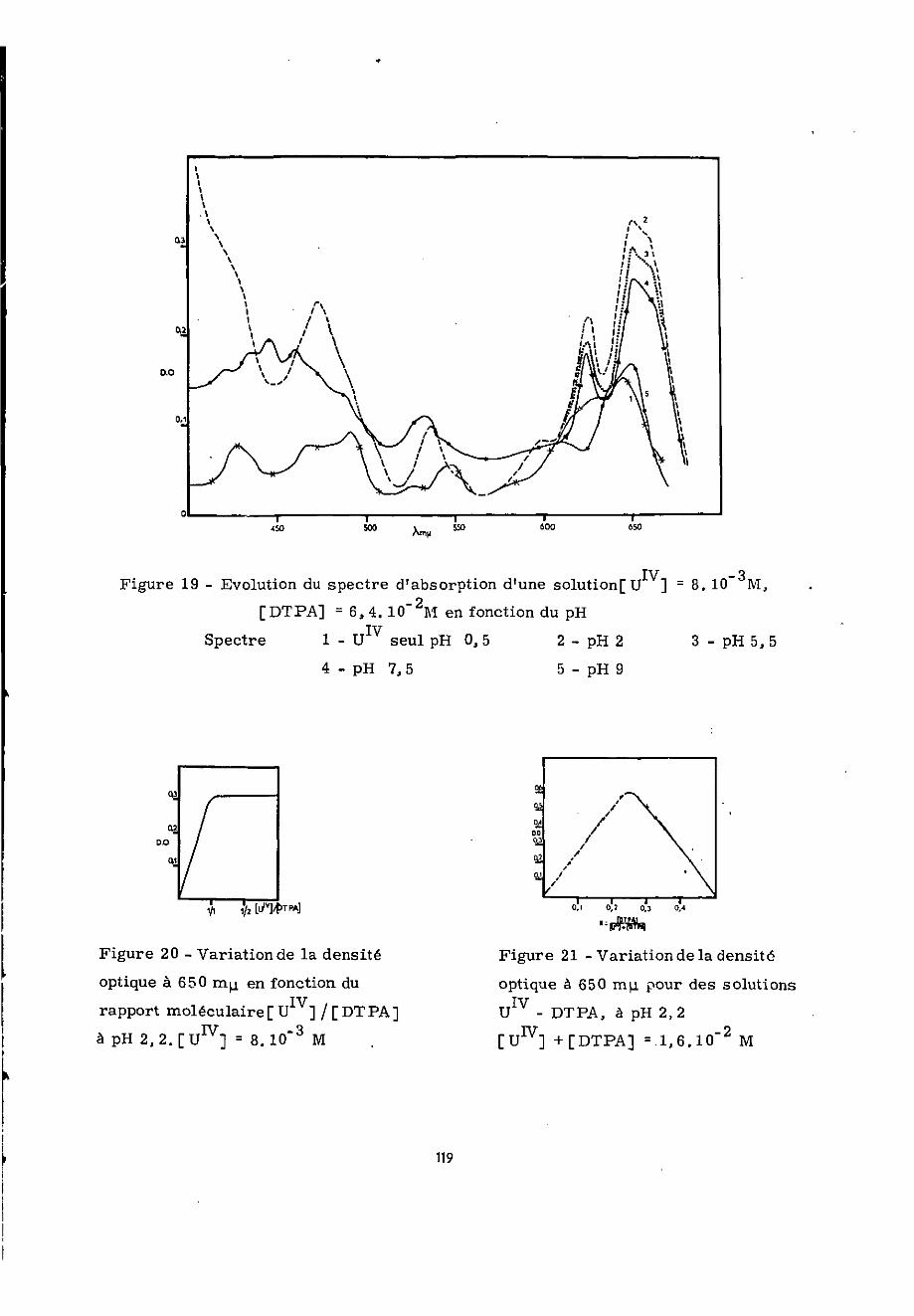
Q3
0,2
0.0
0,1
450 500 550 600 650
Figure 19 - Evolution du spectre d'absorption d'une solution[U ] =
[DTPA] = 6,4. 10"2M en fonction du pH
Spectre 1 - UIV seul pH 0, 5 2 - pH 2
8.10~3M,
1 - U seulpH 0,5
4 - pH 7, 5 5 - pH 9
3 - pH 5, 5
03
Q2o.o
Figure 20 - Variation de la densité
optique à 650 m^ en fonction du
rapport moléculaire[ U ] / [DTPA]TV -9
à pH 2 , 2 . [ U V] = 8. 10 d M
SE
0,1 0,7 0.3 0,4
Figure 21 -Variation de la densité
optique à 650 mu pour des solutions
UIV - DTPA, à pH 2,2TV -9
[U ] +[DTPA] = 1,6.10 * M
119
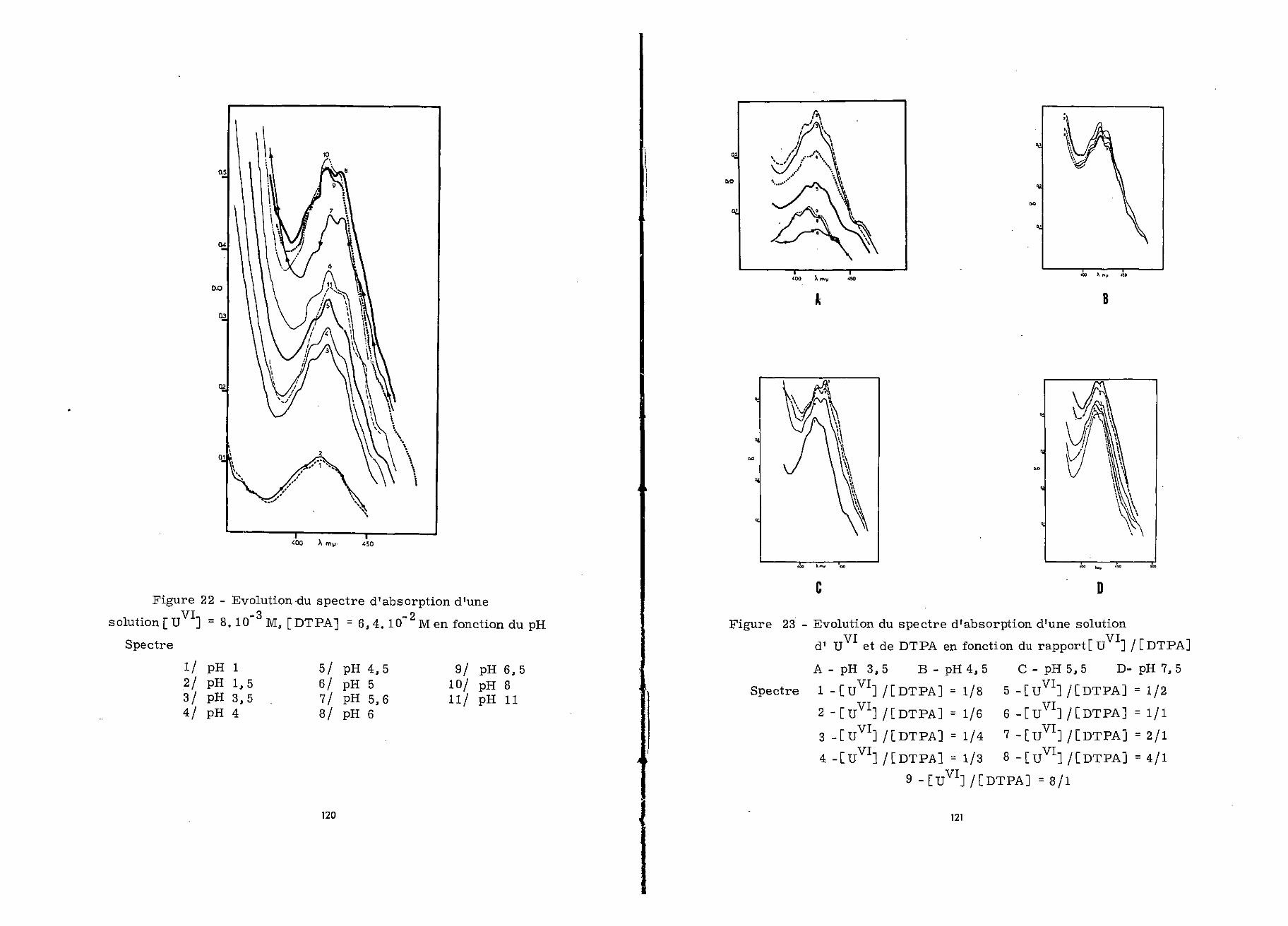
as
03
10
400 m\i
Figure 22 - Evolution-du spectre d'absorption d'une
>n[ l
Spectre
solution [UVI] = 8. 10"3 M, [DTPA] = 6,4. 10"2Men fonction du pH
I/ pH 12/ pH 1,53/ pH 3,54/ pH 4
5/ pH 4,56/ pH 57/ pH 5,68/ pH 6
9/ pH 6,510/ pH 8ll/ pH 11
120
<u
<00 X mu 4SO<00 X mu <W
400
c D
Figure 23 - Evolution du spectre d'absorption d'une solution
d UVI et de DTPA en fonction du rapport[UVI] /[DTPA]
C - pH 5,5 D- pH 7,5
5 -[UVI] /[DTPA] = 1/2
6 -[UVI] /[DTPA] = 1/1
7 -[UVI]/[DTPA] = 2/1
4 -[Uv i] /[DTPA] = 1/3 8 -[UVI] /[DTPA] = 4/1
9 -[UV I]/[DTPA] = 8 / 1
A - pH 3, 5 B - pH 4, 5
Spectre 1 -[UVI] /[DTPA] = 1/8
2 -[UVI] /[DTPA] = 1/6
3 -[UVI] /[DTPA] = 1/4
121
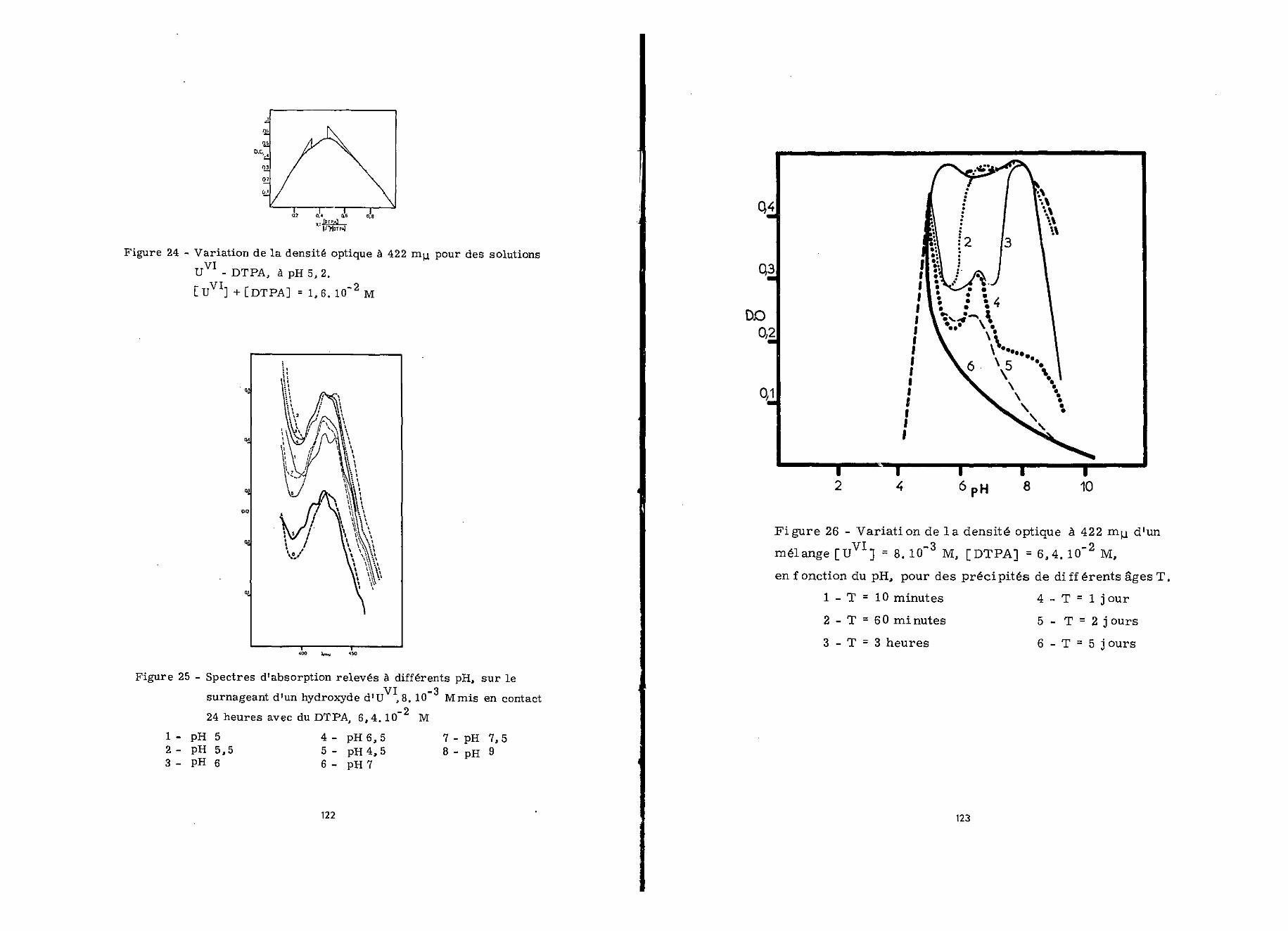
asO.C.
03
Figure 24 - Variation de la densité optique à 422 m^ pour des solutionsVI
U - DTPA, à pH 5, 2.VT 9iu v i] +CDTPA] = 1,6.10"^ M
Figure 25 - Spectres d'absorption relevés à différents pH, sur leVI -3
surnageant d'un hydroxyde d'U ,8. 10 Mmis en contact-2
24 heures avec du DTPA, 6,4.10 M
1 - pH 52 - pH 5,53 - PH 6
4 - pH6,55 - pH 4, 56 - pH 7
122
7 - pH 7, 58 - pH 9
Q3
DO0,2
0,1
I2
I4
I6 PH 8
I10
Figure 26 - Variation de la densité optique à 422 m^ d'unVT -3 9
mélange [Uv ] = 8. 10 ° M, [DTPA] = 6,4.10 * M,
en fonction du pH, pour des précipités de différents âges T.
1 - T = 10 minutes 4 - T = 1 j our
2 - T = 6 0 mi nutes 5 - T = 2 j ours
3 - T = 3 heures 6 - T = 5 3 ours
123
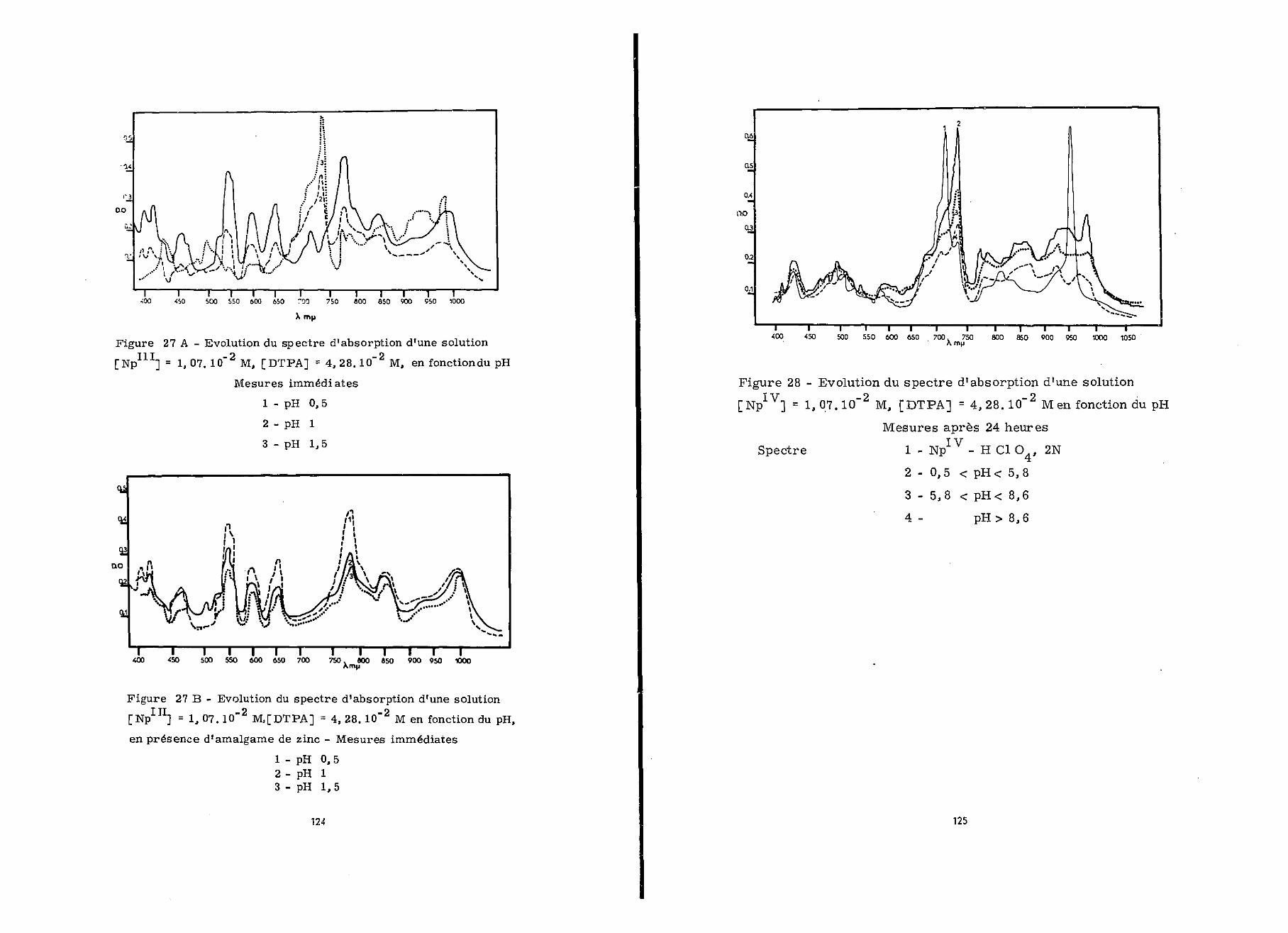
D.O
\j
l*. x-~vx' 1v/|v\y~\! ^
•JOOi
<50i i r i500 S50 600 650 "00 750
i i r i i800 850 900 ÇSO 1000
Figure 27 A - Evolution du spectre d'absorption d'une solutionTT T *) -*)
[Np ] = 1, 07. 10 M, [DTPA] = 4, 28. 10 ' M, en fonction du pH
Mesures immédiates
1 - pH 0, 5
2 - pH 1
3 - pH 1,5
cxo
400 450I I I I I
500 550 600 650 700 750. 800 850 900 950 1000Amp
Figure 27 B - Evolution du spectre d'absorption d'une solution
[Np111] = 1, 07.10"2 M,[DTPA] = 4, 28.10"2 M en fonction du pH,
en présence d'amalgame de zinc - Mesures immédiates
1 - pH 0, 52 - pH 13 - pH 1, 5
124
0,6
05
OX
O.O
0,3
0,1
450 500 550 600 650 700. 750 800 850 900 950 1000 1050A mp
Figure 28 - Evolution du spectre d'absorption d'une solutionTV -2 - 9
[Np ] = 1, 07. 10 M, [DTPA] = 4, 28. 10 * M en fonction du pH
Spectre
Mesures après 24 heures
1 . NPIV - H CIO , 2N
2 - 0 , 5 < pH< 5,8
3 - 5 , 8 < pH< 8,6
4 - pH > 8, 6
125
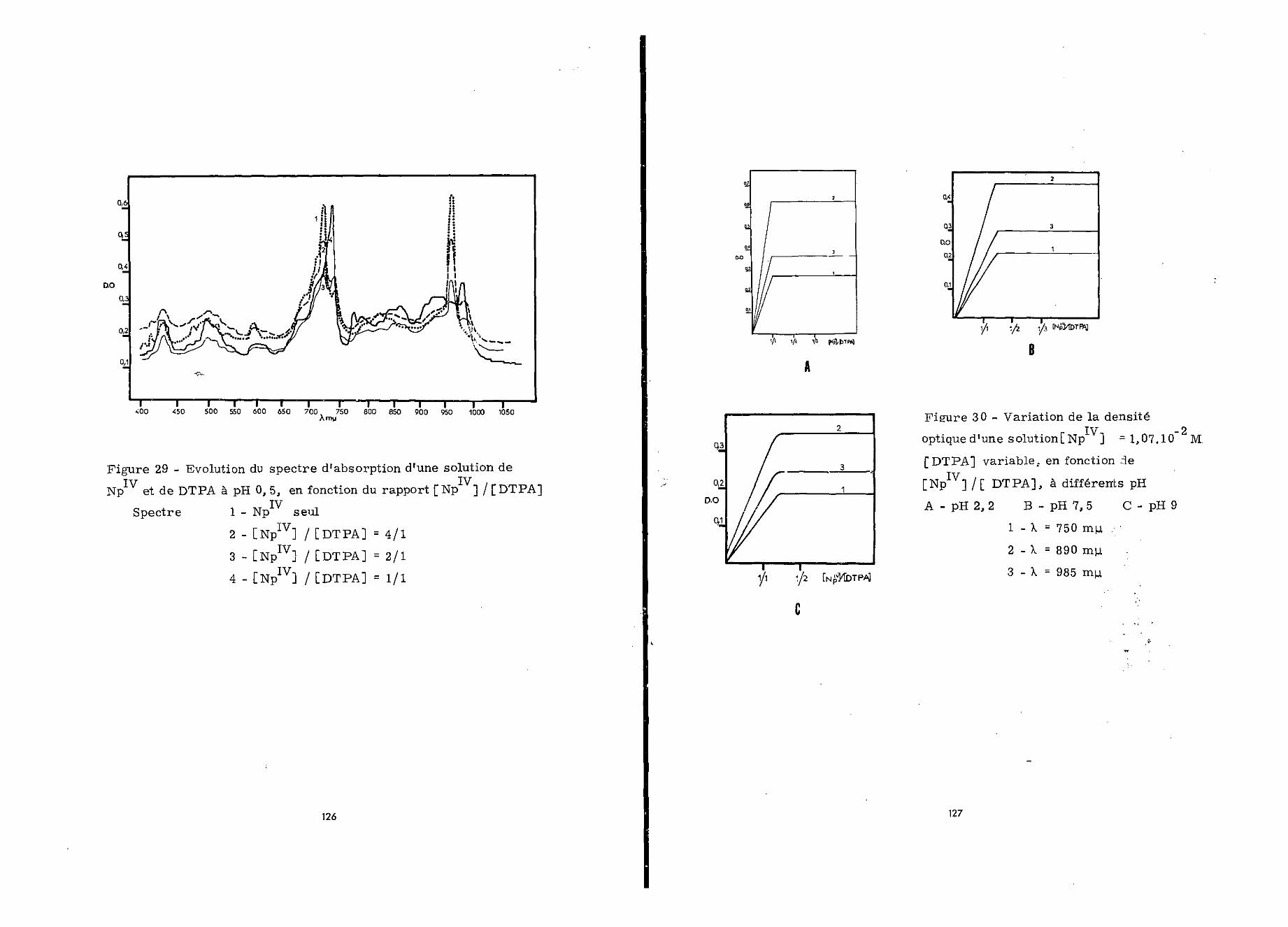
0,6
0,5
D.O
0,3
o,:
^00I
450I I I I
500 550 600 650 700 750 aoo asoi i
900 950 1000 1050
Figure 29 - Evolution du spectre d'absorption d'une solution de
NpIV et de DTPA à pH 0, 5, en fonction du rapport [Np ] / [DTPA]IVSpectre 1 - Np seul
2 - [NpÏV] / [DTPA] = 4 / 1IV3 - [Np ] / [DTPA] = 2/1
4 - [Np 1 V ] /[DTPA] = 1/1
V1 'A M>l<l>Tfi»i
126
Q3
°
D.O
Oi
1/1 ',/2
C
03
no0,2]
0,1
:/, \,B
Figure 30 - Variation de la densitéIV -2
optique d'une solution[Np ] =1,07.10 M.
[DTPA] variable, en fonction .de
[NpIV] /[ DTPA], à différents pH
A - pH 2, 2 B - pH 7, 5 C - pH 9
1 - X = 750 mn .
2 - X = 890 m|a
3 - \ = 985
127
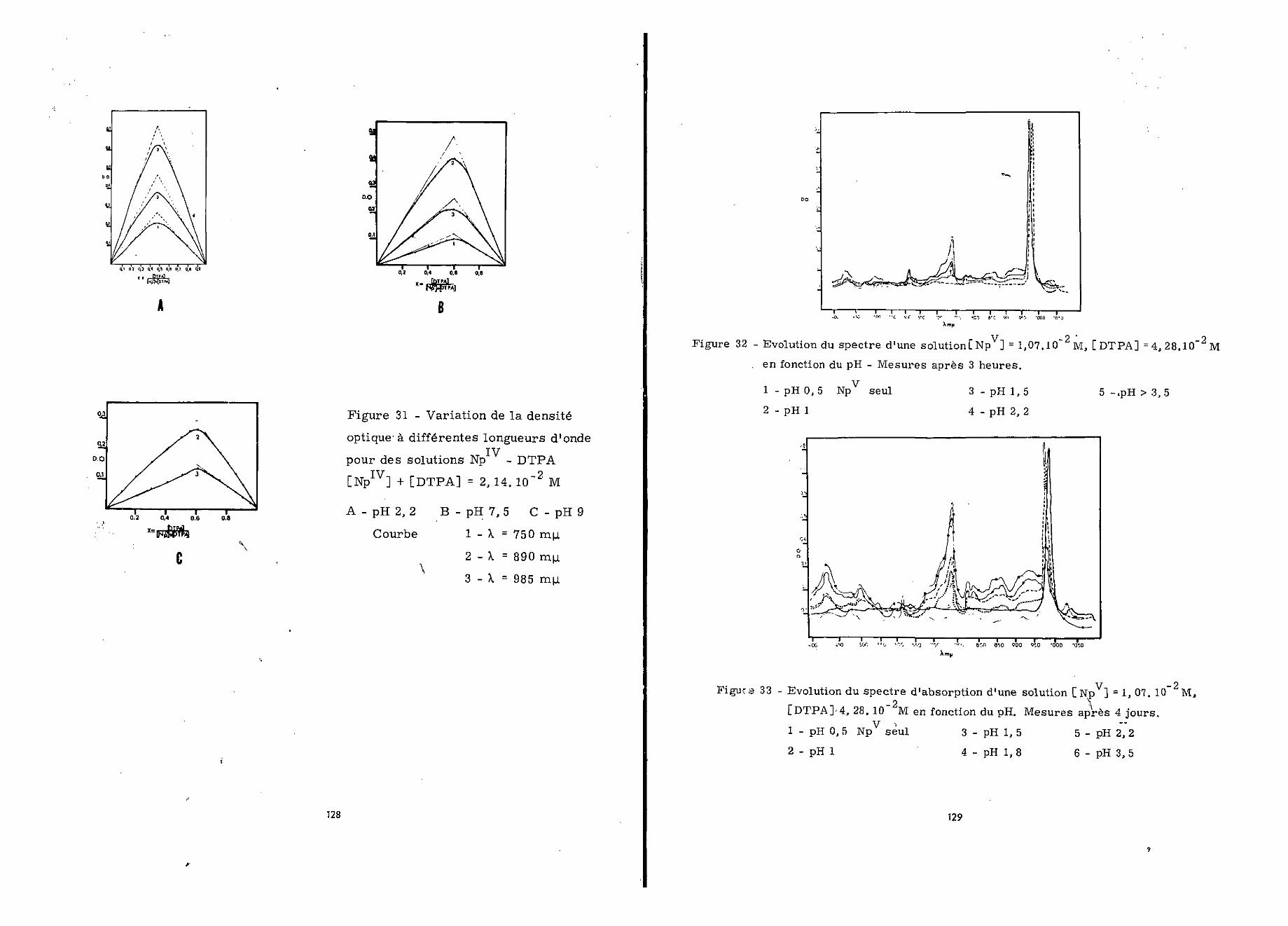
uDO
0,1 o j 0,3 a* 0,4 0,6 0,7 0,6 0,9
D.O
0.1
0,2 0,6
X=
C
0.8
\
03
D.O
02
0.1
0,4 0,8
*Â]
0,8
B
Figure 31 - Variation de la densité
optique1 à différentes longueurs d'ondeIV
pour des solutions Np - DTPATV 9
[Np ] + [DTPA] = 2, 14. 10 M
A - pH 2,2
Courbe
B -
X
pH 7, 5 C - pH 9
1 - X = 750 m|a
2 - X = 890 m|j
3 - X = 985 m|a
128
V 2 ' 9Figure 32 - EvoUrtion du spectre d'une solution[Np ] = 1,07.10" M, [DTPA] =4, 28.10 M
. en fonction du pH - Mesures après 3 heures.
1 - pH 0, 5 NpV seul
2 - pH 1
3 - pH 1,5
4 - pH 2, 2
5 -,pH > 3, 5
Xmp800 850 900 9ÎO '000 -QÎO
V 2Figura 33 - Evolution du spectre d'absorption d'une solution [Np ] = 1, 07. 10" M,
2 \[DTPA]'4, 28. 10~ M en fonction du pH. Mesures après 4 jours.
1 - pH 0, 5 NpV seul
2 - pH 1
3 - pH 1, 5
4 - pH 1, 8
5 - pH 2, 2
6 - pH 3, 5
129
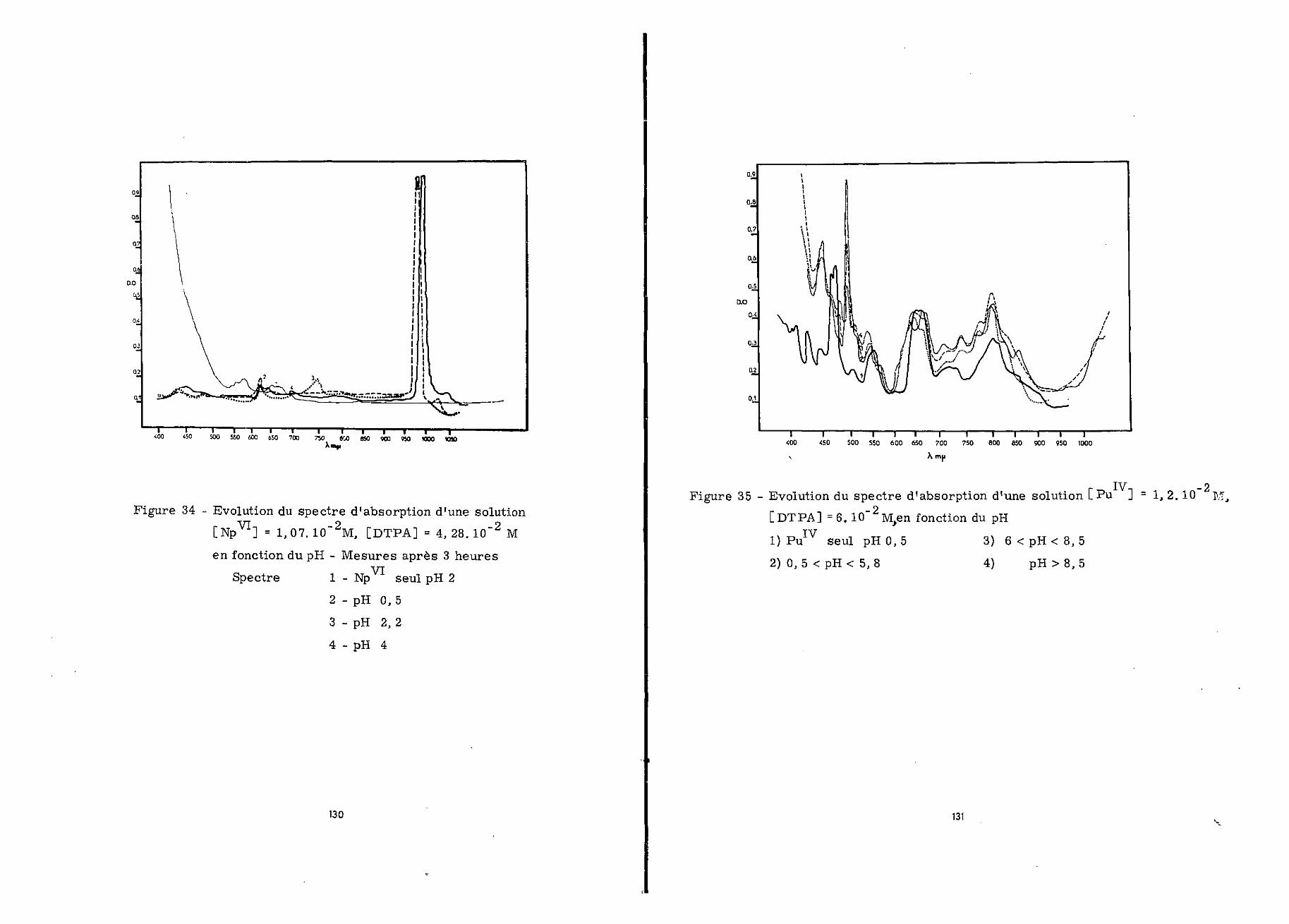
07
06
0.0
0,1
1 r-400 «0
"T 1 I I I500 550 600 650 700
"1750
1 I I I I I800 a» «JO 950 XX» XHO
Figure 34 - Evolution du spectre d'absorption d'une solutionTTT p n
[Npv ] = 1,07. 10 M, [DTPA] = 4,28.10 M
en fonction du pH - Mesures après 3 heures
Spectre 1 - Np^1 seul pH 2
2 - pH 0, 5
3 - pH 2,2
4 - pH 4
130
0,9
0,8
0,7
0,6
0,5
D.O
0,3
0,2
I400
I I I I I I I I I I I I450 500 550 600 650 700 750 600 650 900 950 1000
Figure 35 - Evolution du spectre d'absorption d'une solution [Pu ]
[DTPA] = 6. 10"2M>en fonction du pHIV " - - ~ - 3) 6 < pH < 8, 5
1,2.10~2M,
1) Pu seul p H O , 5
2) 0, 5 < pH < 5, 8 4) pH > 8, 5
131
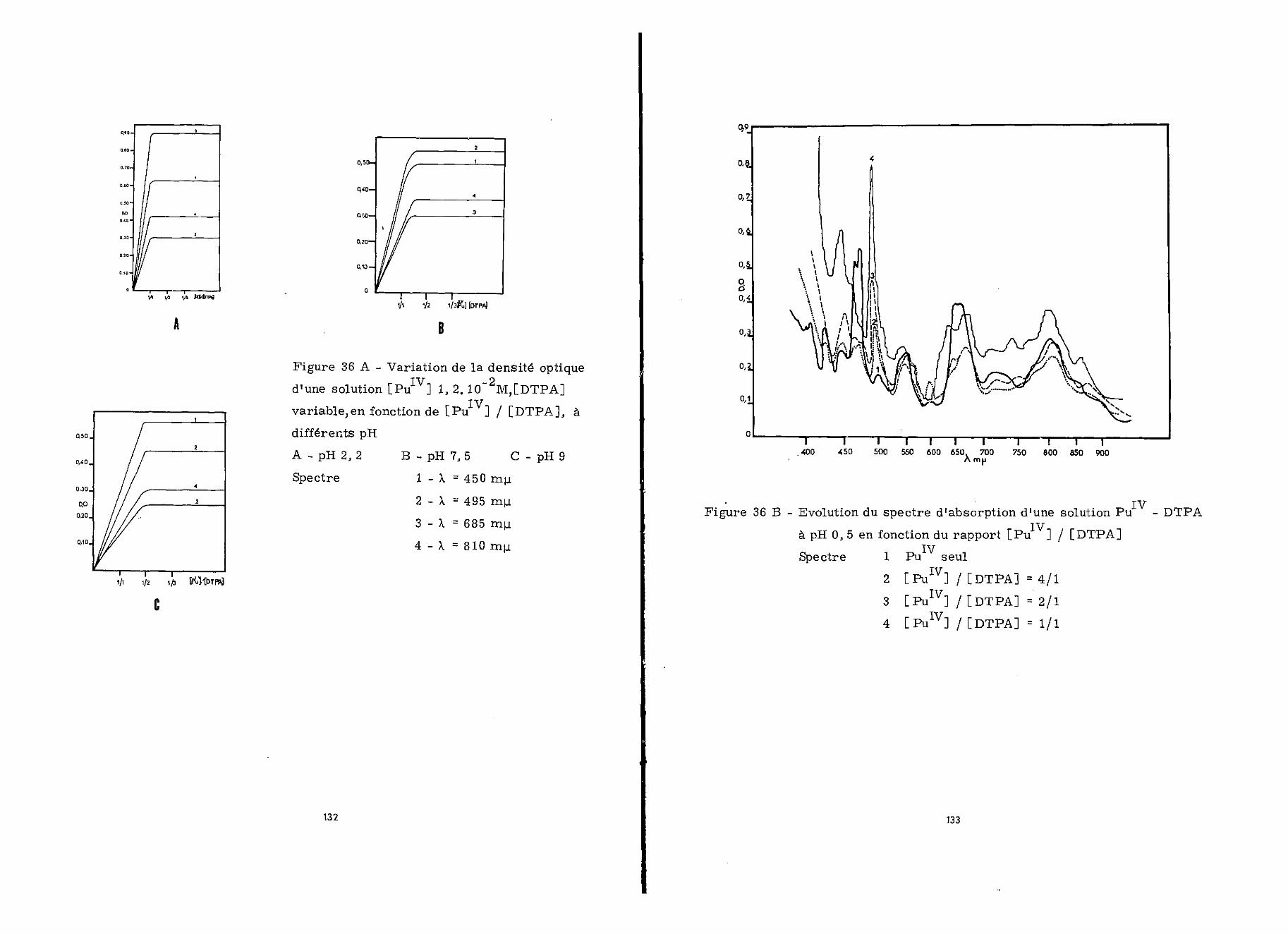
0,90*
0,50—
Q<0—
0,20-
0,10-
•fi 1/aftl (DTPA)
A
0,50.
0/0.
0,30.
DP
0,20.
0,10.
Figure 36 A - Variation de la densité optiqueTV 2
d'une solution [Pu ] 1, 2. 10 M,[DTPA]
variable,en fonction de [PuIV] / [DTPA], à
différents pH
A - pH 2, 2 B - pH 7, 5 C - pH 9
Spectre 1 - X = 450 m(j
2 - X = 495 m|j
3 - X = 685
4 - X = 810
1/1 1/2 1/3
C
132
0,7
QO
I I I I I I i I.400 450 500 550 600 650. 700 750
! I I800 850 900
Figure 36 B - Evolution du spectre d'absorption d'une solution Pu
à pH 0, 5 en fonction du rapport [Pu1 ] / [DTPA]
Spectre 1
IV - DTPA
Pu seulIV
2 [Pu ] / [DTPA] = 4/1IV
3 [Pu V] /[DTPA] = 2/1IV4 [Pu V] /[DTPA] = 1/1
133
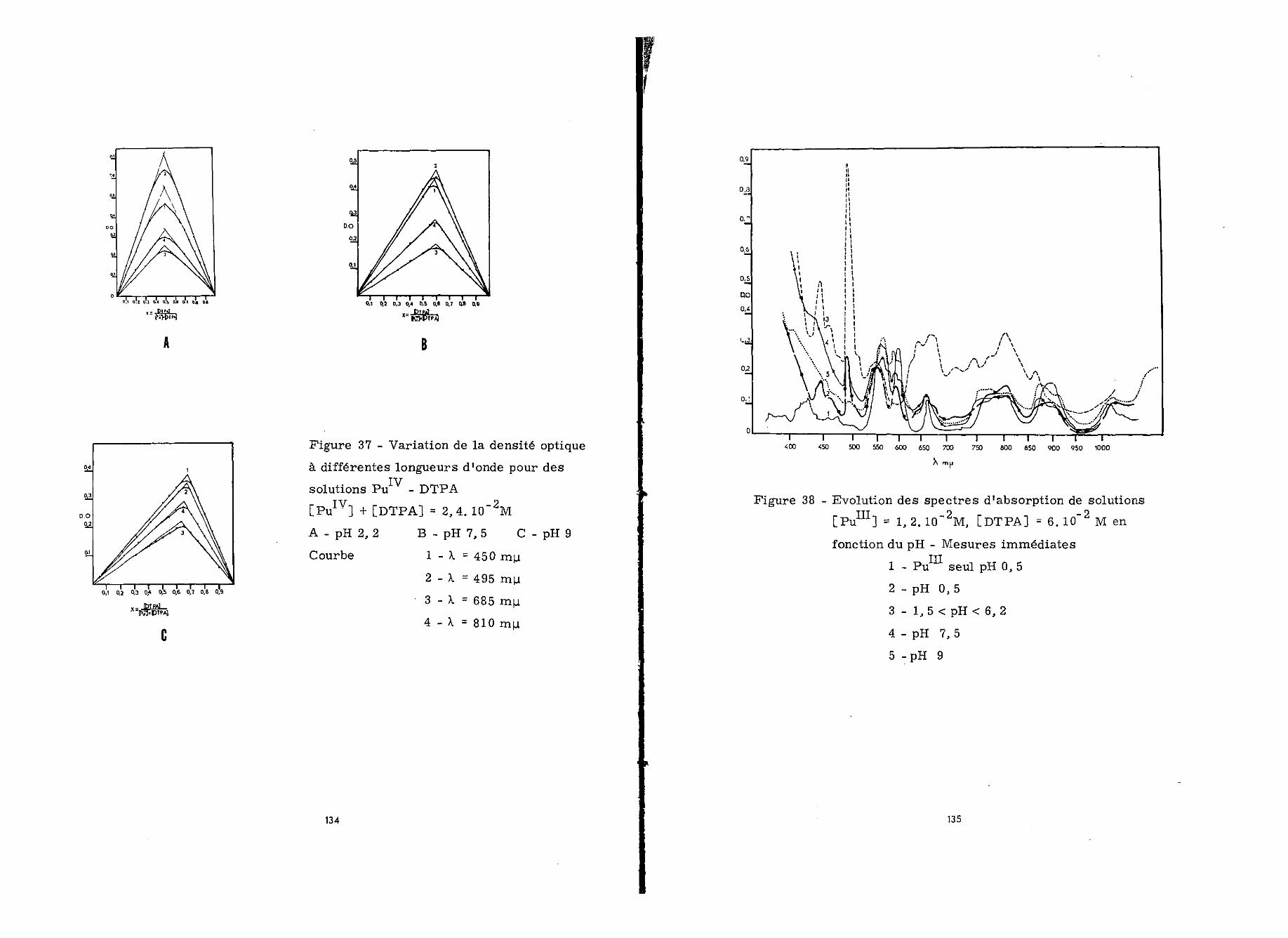
0!l O.'j KJ <X4 Kt a< 0-7 04 0*a_btr
A
0|i
92
D.O
0,1 0,2 Q3 0,4 0,5 0,6 0,7 0,8 0,9
C
D.o
wj
0.1
0,1 0,2 0,3 0,4 0,5 0,6 0.7 <U> 0.9
X=
0,9
O.iî
0,5_
DO
Figure 37 - Variation de la densité optique
à différentes longueurs d'onde pour des
solutions PuIV - DTPATV 9
[Pu ] + [DTPA] = 2,4. 10 M
0,2
o.:
i<50
I I ! I500 550 600 650
I700 750
I I I I 1800 850 900 950 1000
'A - pH 2, 2
Courbe
B - pH 7, 5 C - pH 9
1 - X = 450 mu
2 - X = 495 m|i
3 - X = 685 m|j
4 - X = 810 m|a
Figure 38 - Evolution des spectres d'absorption de solutions
[Pu111] = l, 2. 10"2M, [DTPA] = 6. 10"2 M en
fonction du pH - Mesures immédiates
1 - Pu111 seul pH 0, 5
2 - pH 0,5
3 - 1,5 < p H < 6, 2
4 - pH 7,5
5 -pH 9
134 135
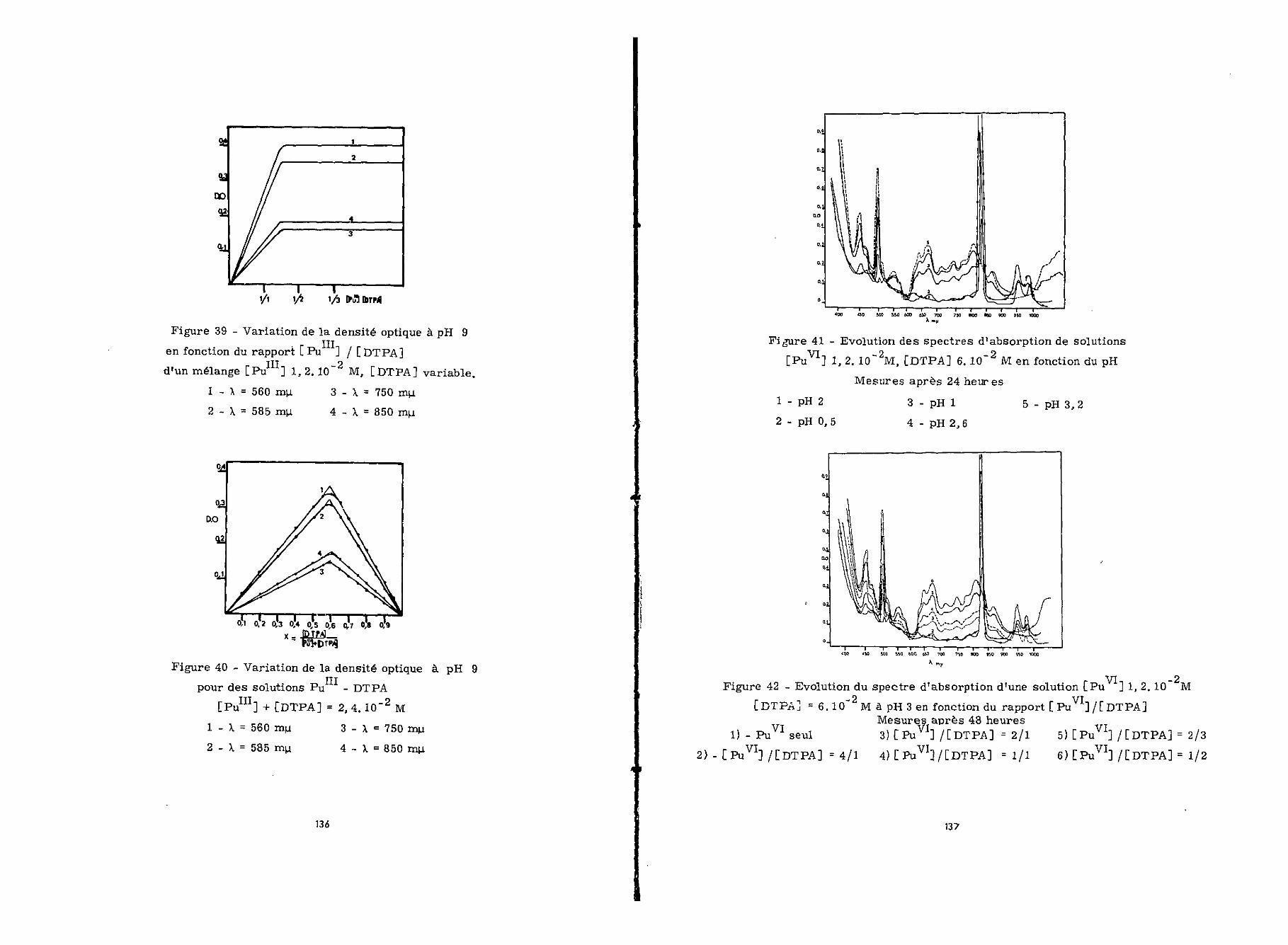
Ii/t
I0
Figure 39 - Variation de la densité optique à pH 9
en fonction du rapport [Pu ] / [DTPA]III 2
d'un mélange [Pu ] 1, 2. 10~ M, [DTPA] variable.
1 - X = 560 mia 3 - X = 750 mu
2 - X = 585 mu 4 - X = 850 nip.
D.o
qj
ai 0.2 0*3 0)4 0,5 0,6 0,7 0>0?9X -"
Figure 40 - Variation de la densité optique à pH 9
pour des solutions Pu - DTPATTT 9
[Pu ] + [DTPA] = 2,4. 10~* M
1 - X = 560
2 - X = 585
3 - X
4 - X
750
850
136
0,7
o,î
0.2
0.2
0,1
400 «0 500 SSO «00 «SO TOO 750 «00 «J «00 MO 1000
Figure 41 - Evolution des spectres d'absorption de solutions
[Pu^1] 1, 2. 10~2M, [DTPA] 6. 10~2 M en fonction du pH
Mesures après 24 heures
1 - PH 2 3 - pH 1 5 - pH 3, 2
2 - pH 0, 5 4 - pH 2, 6
<00 <SO MO UO &00 «SO 700 7*0 «00 KO «0
VI -2Figure 42 - Evolution du spectre d'absorption d'une solution [Pu ] 1, 2. 10 M
p "XT"![DTPA] = 6. lO" M à pH 3 en fonction du rapport [Pu ]/[DTPA]
1) - PuVI seulMesures après 48 heures3) [PuVI] /[DTPA] = 2 / 1 5) [PuVI] /[DTPA] = 2/3
2) - [PuVI] /[DTPA] = 4 / 1 4)[PuVI]/[DTPA] =1/1 6) [PuVI]/[DTPA] = 1/2
137
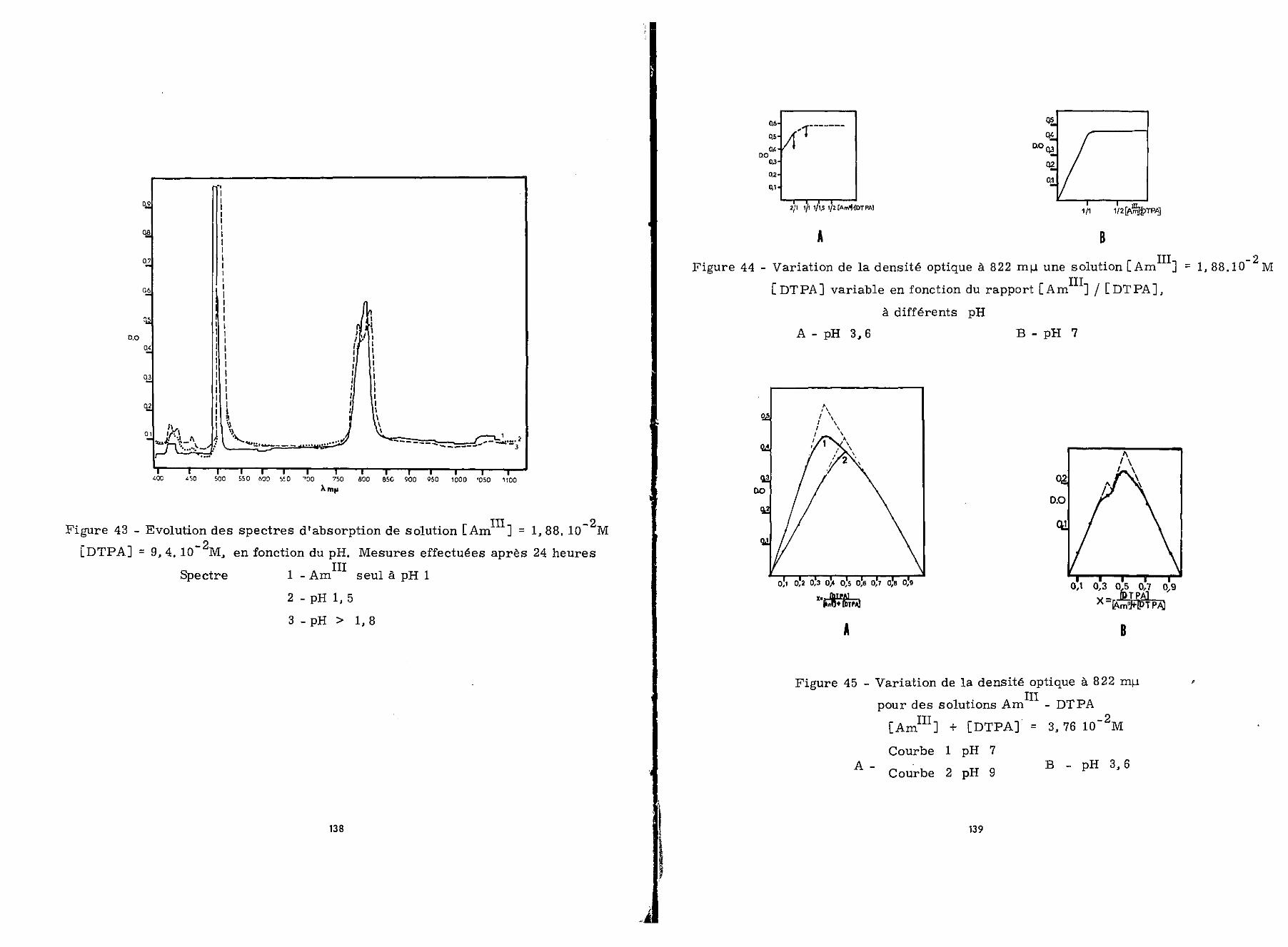
12
Q8,
0.7
D.O
a<
Q3
01
<OC ^50 500 550 ftOO iSO ''OO 750 800 850 900 950 1000 '050 1100
III 2Figure 43 - Evolution des spectres d'absorption de solution [Am ] = 1,88. 10~ M
o[DTPA] = 9,4. 10~ M, en fonction du pH. Mesures effectuées après 24 heures
Spectre 1 - Am seul à pH 1
2 - pH 1, 5
3 - pH > 1, 8
138
D.O
ao
2/1 1/1 1/1,5 1/2 [ArrdlDTPAl
A
1/1 1/2[AmHbTPÂ|
B
Figure 44 - Variation de la densité optique à 822 mu une solution [Am ] =
[DTPA] variable en fonction du rapport [Am ] / [DTPA],
à différents pH
A - pH 3, 6 B - pH 7
, 88.10"2M
42cxo
42
02]
D.O
oîi 0,2 0,3 0*4 o's o;e 0*7 o;e o> 0,1 0,3 0,5 0,7 0.9
A
Figure 45 - Variation de la densité optique à 822 m|j
pour des solutions Am - DTPA
[Am111] + [DTPA] = 3,76 10~2M
A -Courbe 1 pH 7
Courbe 2 pH 9B - pH 3,6
139
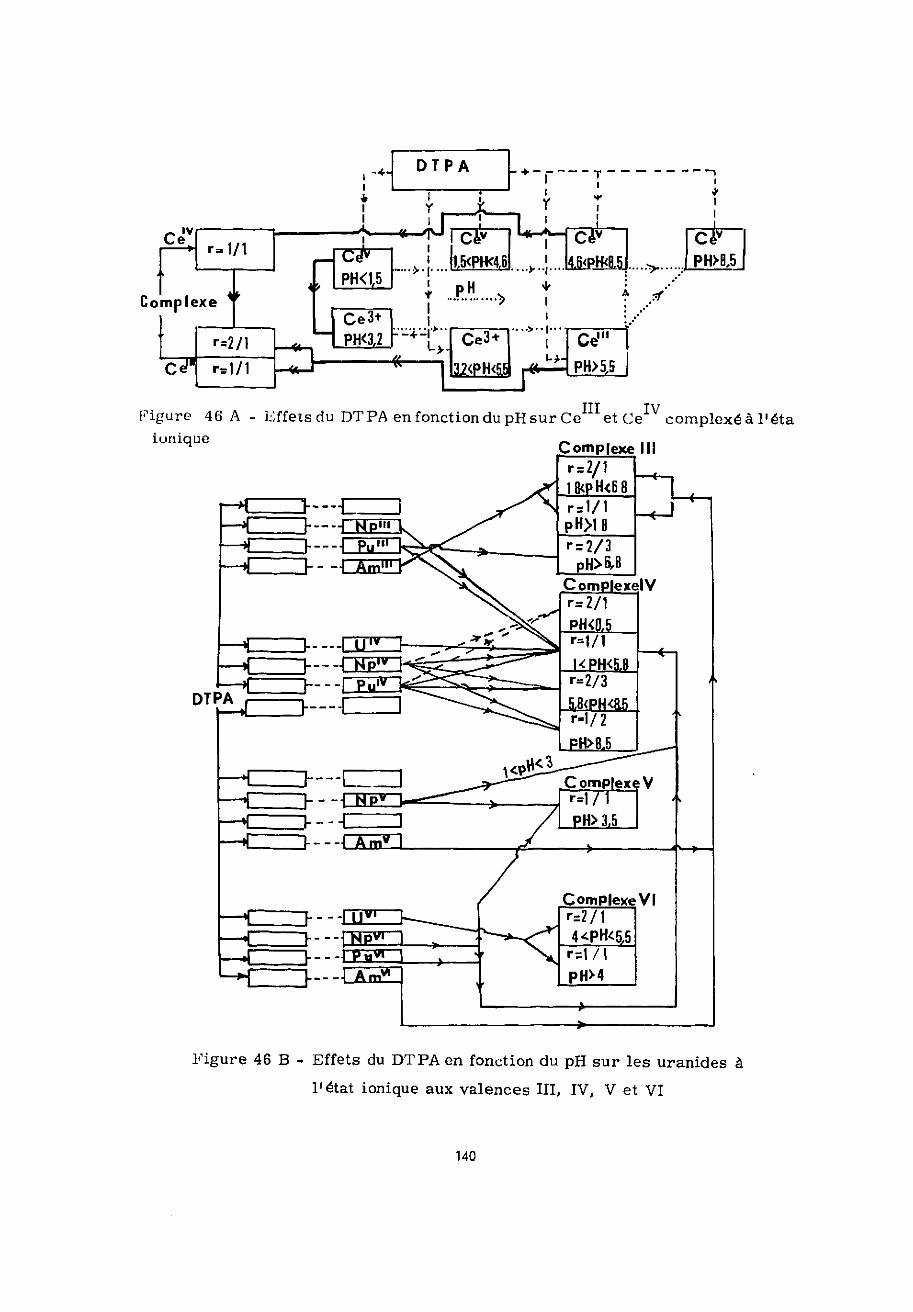
à
Co
'
•up le
'
C^"
r ir= i
4
r=?
rs|
i\f I
k
ni\
t
i-tu i
• •M
V
• ^
1 ~*~I
«11j^
1
C^PH<1,5
Ce3tPH<3,2
DT
iY1
i1_....>. |. .ifIi
I U.\_*u>
«
P A
ii
f*ip
15<PH<4,6nllP." N/
Ce3*
32<PH<&S
* I
r~i i
iiiiii(_*-
r*-1r
~ T
TiiCev
46<PH<8.5 .
A.
PH>5,5
11
jciv
,_ PH>8,5_••"
^f
Figure 46 A - Effets du DTPA en fonction du pH sur Ce111 et CeIV complexée l'étaionique
Complexe III
DTPA
Figure 46 B - Effets du DTPA en fonction du pH sur les uranides à
l'état ionique aux valences III, IV, V et VI
140
