the role of nibrin in doxorubicin-induced apoptosis and ...the role of nibrin in doxorubicin-induced...
TRANSCRIPT

The Role of Nibrin in Doxorubicin-Induced Apoptosis andCell Senescence in Nijmegen Breakage SyndromePatients LymphocytesOlga Alster1, Anna Bielak-Zmijewska1, Grazyna Mosieniak1, Maria Moreno-Villanueva2, Wioleta Dudka-
Ruszkowska3, Aleksandra Wojtala1, Monika Kusio-Kobiałka3, Zbigniew Korwek1, Alexander Burkle2,
Katarzyna Piwocka3, Jan K. Siwicki4, Ewa Sikora1*
1 Laboratory of the Molecular Bases of Aging, Nencki Institute of Experimental Biology, Polish Academy of Sciences, Warsaw, Poland, 2Molecular Toxicology Group,
Department of Biology, University of Konstanz, Konstanz, Germany, 3 Laboratory of Cytometry, Nencki Institute of Experimental Biology, Polish Academy of Sciences,
Warsaw, Poland, 4Department of Immunology, Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw, Poland
Abstract
Nibrin plays an important role in the DNA damage response (DDR) and DNA repair. DDR is a crucial signaling pathway inapoptosis and senescence. To verify whether truncated nibrin (p70), causing Nijmegen Breakage Syndrome (NBS), isinvolved in DDR and cell fate upon DNA damage, we used two (S4 and S3R) spontaneously immortalized T cell lines fromNBS patients, with the founding mutation and a control cell line (L5). S4 and S3R cells have the same level of p70 nibrin,however p70 from S4 cells was able to form more complexes with ATM and BRCA1. Doxorubicin-induced DDR followed bycell senescence could only be observed in L5 and S4 cells, but not in the S3R ones. Furthermore the S3R cells onlyunderwent cell death, but not senescence after doxorubicin treatment. In contrary to doxorubicin treatment, cells from allthree cell lines were able to activate the DDR pathway after being exposed to c-radiation. Downregulation of nibrin innormal human vascular smooth muscle cells (VSMCs) did not prevent the activation of DDR and induction of senescence.Our results indicate that a substantially reduced level of nibrin or its truncated p70 form is sufficient to induce DNA-damagedependent senescence in VSMCs and S4 cells, respectively. In doxorubicin-treated S3R cells DDR activation was severelyimpaired, thus preventing the induction of senescence.
Citation: Alster O, Bielak-Zmijewska A, Mosieniak G, Moreno-Villanueva M, Dudka-Ruszkowska W, et al. (2014) The Role of Nibrin in Doxorubicin-InducedApoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients Lymphocytes. PLoS ONE 9(8): e104964. doi:10.1371/journal.pone.0104964
Editor: Arianna L. Kim, Columbia University Medical Center, United States of America
Received March 26, 2014; Accepted July 16, 2014; Published August 13, 2014
Copyright: � 2014 Alster et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability: The authors confirm that all data underlying the findings are fully available without restriction. Relevant data are included within the paper.
Funding: This work was supported by National Center of Science in Poland (grants: 2011/01/M/NZ1/01597, UMO-2011/01/B/NZ3/02137 and 0728/B/P01/2011/40). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: The authors have declared that no competing interests exist.
* Email: [email protected]
Introduction
Nijmegen Breakage Syndrome (NBS) is a rare autosomal
recessive disorder characterized by genomic instability and
increased risk of haematopoietic malignancies observed in more
than 40% of the patients by the time they are 20 years old [1].
NBS is caused by mutations in the NBN gene (originally
designated as NBS1) encoding nibrin. More than 90% of the
patients are homozygous for the same mutation (c.657-661del5)
what results in the formation of two truncated fragments of the
95 kDa nibrin: 26 kDa N-terminal fragment (p26-nibrin) and
70 kDa C-terminal fragment (p70-nibrin), which are produced by
alternative initiation of translation at a cryptic upstream start
codon. This mutation is actually hypomorphic as the truncated
p70-nibrin is able to retain some of the vital cellular functions of
the full-length protein. The truncated p70-nibrin can form the
MRN (Mre11-Rad50-Nbs1) complex with two other proteins,
Mre11 and Rad50 [2,3]. However, null mutation of the Nbn gene
is lethal in mice [4].
Stress-induced premature senescence (SIPS) is a relatively fast,
telomere erosion independent, process. Among its characteristic
features we can distinguish irreversible growth arrest, altered cell
morphology, DNA foci formation, activation of senescence-
associated b-galactosidase (SA-b-Gal) and senescence associated
secretory phenotype-SASP (reviewed in [5]). Recently, it was
shown that double-strand DNA breaks (DSBs), after induction of
the DNA damage response (DDR), are crucial for cellular
senescence [6]. Briefly, upon DSB induction ataxia telangiectasia
mutated (ATM) kinase is activated. The activated kinase
phosphorylates nibrin at its Ser 343 residue and H2AX histone,
at its Ser 139 residue (cH2AX). Phosphorylated nibrin forms a
trimeric complex (MRN) along with Mre11 and Rad50, which is
recruited to the vicinity of DSBs where nibrin interacts with
cH2AX [7]. Ultimately, Chk1, Chk2 (checkpoint kinase 1 and 2,
respectively) and p53 are activated. p53 promotes senescence
(when DNA damage is irreparable) via transactivation of
CDKN1A, which encodes the cyclin dependent kinase inhibitor
p21 [5].
DDR activation, not only can lead to senescence but also to
transient cell cycle arrest and DNA repair or apoptosis.
Improperly functioning DDR often results in increased radiosen-
PLOS ONE | www.plosone.org 1 August 2014 | Volume 9 | Issue 8 | e104964

sitivity, genomic instability and cancer development. Since NBS1
deficient cells are characterized by genomic instability and NBS
patients suffer from haematopoietic malignancies, we hypothesized
that the molecular pathways leading to DNA damage-induced
senescence might be impaired in patients affected with this disease.
Most cell lines derived from NBS patients were established
following transformation with viral oncogenes, which inhibit key
regulatory genes such as the tumor suppressor gene proteins p53
and pRb, thus allowing the cell to bypass the senescence program
and become immortal [8]. Accordingly, spontaneously immortal-
ized T cell lines, S3R and S4, carrying the same mutation within
the NBN gene, but with a seemingly functional p53/p21 response
after gamma irradiation [9], are a very useful cellular model in
studying the mechanisms of DNA damage-induced senescence.
Therefore we used two cell lines derived from NBS patients (S3R
and S4) and the control, L5 cell line (spontaneously immortalized
spleenocytes obtained from a healthy donor) to examine if they are
prone to DNA damage-induced senescence. To induce DNA
damage and DDR activation we used doxorubicin, which is a
DNA damaging agent acting through different mechanisms. It can
lead to the formation of direct and indirect DNA damage through:
intercalation into DNA, DNA binding and alkylation, DNA cross-
linking, interference with DNA unwinding or DNA strand
separation, helicase activity as well as inhibition of topoisomerase
II and generation of free radicals [10].
Materials and Methods
1. Cell linesThe spontaneously immortalized T cell lines: S3R and S4 were
established from peripheral blood mononuclear cells (PBMC)
derived from NBS patients homozygous for the 657del5 mutation
of the NBN gene [9] and the L5 cell line was established from the
spleen of a healthy donor as described previously [9,11]. All of the
cell lines were cultured in the RPMI 1640 medium (Gibco, Life
Technologies, Warsaw, Poland) supplemented with 10% FCS
(Biochrom, Biomibo, Warsaw, Poland), 50 mg/ml gentamycin
(Sigma, Poznan, Poland), 2 mM glutamine (Sigma, Poznan,
Poland) and 20 U/ml of IL-2 (R&D, Biokom, Warsaw, Poland).
Human vascular smooth muscle cells (VSMCs) were obtained
from Lonza (Basel, Switzerland). hVSMC were grown in SmBM
medium (Lonza, Basel, Switzerland). S3R, S4 and L5 cells were
seeded at a density of 0,26106/ml 24 h before doxorubicin
(Sigma, Warsaw, Poland) treatment. VSMCs were seeded at a
density of 26103/cm2 24 h before transfection.
2. DNA content and cell cycle analysisFor DNA analysis the cells were fixed in 70% ethanol and
stained with PI solution (3,8 mM sodium citrate, 50 mg/ml RNAse
A, 500 mg/ml PI in PBS). All of the used agents were purchased at
Sigma Aldrich (Poznan, Poland). DNA content was assessed using
flow cytometry and analyzed with the CellQuest Software. 10000
events were collected per sample (FACSCalibur, Becton Dick-
inson, Warsaw, Poland).
3. ImmunoprecipitationS3R and S4 cells were lysed with modified RIPA buffer [12].
Equal amounts of protein (750 mg) were taken for immunopre-
cipitation. The supernatants were precleared by adding Protein A/
G PLUS-Agarose Immunoprecipitation Reagent (Santa Cruz
Biotechnology, Inc., Dallas, Texas, USA) and incubated with IP
antibody-IP matrix complexes overnight using NBS1 rabbit
polyclonal antibody (Sigma Aldrich, Poznan, Poland), Mre11
rabbit monoclonal antibody (Cell Signaling, Lab-JOT Ltd.,
Warsaw, Poland), ATM rabbit monoclonal antibody (Epitomics,
Burlingame, California, USA) according to the manufacturer’
protocol (Santa Cruz Biotechnology, Inc., Dallas, Texas, USA).
Beads were washed with PBS and immune complexes were eluted
with sodium dodecyl sulphate (SDS)-containing buffer and boiled.
Mre11, BRCA1, ATM and NBS1 were detected using the
Western blotting technique with the following antibodies: BRCA1
mouse monoclonal (R&D, Biokom, Warsaw, Poland), ATM rabbit
monoclonal antibody (Epitomics, Burlingame, California, USA),
Mre11 rabbit monoclonal antibody (Cell Signaling, Lab-JOT Ltd.,
Warsaw, Poland), NBS1 rabbit polyclonal antibody (Sigma
Aldrich, Poznan, Poland) and secondary rabbit polyclonal
antibody conjugated with horseradish peroxidase (Dako, Poland).
4. Western blotting analysisWhole cell protein extracts were prepared according to the
Laemmli method [13]. Equal amounts of protein were separated
electrophorectically in 8, 12 or 15% SDS-polyacrylamide gels and
afterwards transferred to nitrocellulose membranes. Membranes
were blocked in 5% non-fat milk dissolved in TBS containing
0,1% Tween-20 (Sigma Aldrich, Poznan, Poland) for 1 h at RT
and incubated with one of the primary monoclonal or polyclonal
antibodies: anti-ATM (1:500) (Millipore, Merck, Warsaw, Poland),
anti-p-ATM Ser 1981 (1:1000), H2AX (1:500) and anti-cH2AX
(1:1000) (Abcam, Cambridge, UK), anti-p16 (1:500), anti-p53
(DO-1) (1:500), anti-p21 (C-19) (1:500) (Santa Cruz Biotechnology
Inc., Dallas, Texas, USA), anti-p-p53 Ser 15, anti-Chk1, anti-p-
Chk1 Ser 317, anti-Chk2, anti-p-Chk2 Thr 68, anti-NBS1 Ser 343
(Cell Signaling, Lab-JOT Ltd., Warsaw, Poland), anti-PARP1
(1:1000) (Becton Dickinson, Diag-med, Warsaw, Poland) anti-
NBS1 (1:500), anti-b-actin (1:50000) (Sigma Aldrich, Poznan,
Poland) and anti-GAPDH (1:50000) (Millipore, Merck, Warsaw,
Poland). The proteins were detected with appropriate secondary
antibodies conjugated with horseradish peroxidase and ECL
reagents (GE Healthcare, Buckinghamshire, UK), according to the
manufacturer’s protocol.
5. Gamma irradiation procedureAsynchronously growing cells were treated with 4 Gy of c-
irradiation. Immediately after irradiation the cells were diluted to a
concentration 0,256106 cells/ml and cultured for 3 h. Cells were
collected after 3 h (untreated and treated with 4 Gy of irradiation).
Whole cell extracts were prepared for Western blotting analysis.
6. Silencing of the NBN geneTo downregulate NBN expression the cells were seeded in 6 or
12-well plates (26104 or 86103 cells per well, respectively) and
transfected with 60 nM siRNA (NBN or negative) (Life Technol-
ogies, Warsaw, Poland) using Lipofectamine 2000 (Life Technol-
ogies, Warsaw, Poland). Transfection was performed according to
the manufacturer’s protocol. About 20 h after transfection
medium was replaced with fresh one and cells were cultured for
three days in the presence of doxorubicin (100 nM) (Sigma
Aldrich, Poznan, Poland).
7. Detection of SA-b-GalDetection of SA-b-Gal was performed according to Dimri et al.
(1995) [14]. Briefly, cells were fixed with 2% formaldehyde, 0,2%
glutaraldehyde in PBS, washed and exposed overnight at 37uC to
a solution containing: 1 mg/ml 5-bromo-4-chloro-3-indolyl-b-D-
galactopyranoside, 5 mM potassium ferrocyanide, 5 mM potassi-
um ferrycyanide, 150 mM NaCl, 2 mM MgCl2 and 0,1 M
phosphate buffer, pH 6,0. All of the used agents were purchased at
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 2 August 2014 | Volume 9 | Issue 8 | e104964

Sigma Aldrich (Poznan, Poland). Photos were taken using the
Evolutions VF digital CCD camera (Media Cybernetics, Rock-
ville, Maryland, USA).
8. Apoptosis detectionThe level of apoptosis was measured by flow cytometry
(FACSCalibur) using the annexin V/7-AAD assay (Becton
Dickinson, Diag-med, Warsaw, Poland). Externalization of phos-
phatidylserine (PS) to the outer layer of the cell membrane was
examined by binding of annexin V in the presence of 7-AAD, a
dye which stains dead cells. Briefly, cells were washed with PBS,
suspended in the annexin V binding buffer, stained for 15 min
with annexin V conjugated with PE and 7-AAD. Analysis was
performed with FACSCalibur using the CellQuest Software (BD
Biosciences, Warsaw, Poland). 10000 events were collected per
sample.
9. Bromodeoxyuridine labeling assayTo evaluate DNA synthesis BrdU (Sigma Aldrich, Poznan,
Poland) was added to the medium (10 mM) and cells were cultured
for 24 h. Afterwards the cells were fixed in ethanol. BrdU was
detected using a primary antibody against BrdU (Becton
Dickinson, Warsaw, Poland) and a secondary Alexa 488 antibody
(Life Technology, Warsaw, Poland). The cells were observed
under a fluorescence microscope (Nikon, Tokyo, Japan) with the
use of 450–490 nm -excitation wavelength. Photos were taken
using the Evolutions VF digital CCD camera (Media Cybernetics,
Rockville, Maryland, USA).
10. ImmunocytochemistryFor immunofluorescence the cells were fixed with 2% parafor-
maldehyde (Sigma Aldrich, Poznan, Poland) at RT for 20 minutes
and afterwards were incubated on slides with the anti-53BP1
monoclonal antibody (Novus, Cambridge, USA). Secondary anti-
rabbit Alexa 488-conjugated IgG antibody was used (Life
Technology, Warsaw, Poland). Cells were observed under a
fluorescence microscope (Nikon, Tokyo, Japan) and photos were
taken using the Evolutions VF digital CCD camera (Media
Cybernetics, Rockville, Maryland, USA).
11. Fluorimetric Detection of DNA unwinding (FADU)methodA modified and automated version of the FADU (Fluorimetric
Detection of alkaline DNA Unwinding) method was used to
measure the percentage of double-stranded DNA after treatment
with doxorubicin (1 and 10 mM). The percentage of DNA damage
was analyzed 30, 60 and 90 min after treatment with a DNA
damaging agent as described previously by Moreno-Villanueva et
al. [15,16]. The method is based on partial denaturation
‘‘unwinding’’ of double-stranded DNA under controlled alkaline
and temperature conditions. DNA strand breaks are sites where
DNA unwinding can start. Briefly, after infliction of DNA damage
cell lysis was performed. DNA unwinding was terminated by
adding a neutralization solution. SybrGreen, a commercially
available dye, which only binds to double stranded DNA, was used
to determine the amount of double-stranded DNA. The lower the
fluorescence the less double-stranded DNA in the sample.
12. Statistical analysisStudent’s T test was used to calculate statistical significance: *,
p,0,05; **, p,0,01; ***, p,0,001.
Results
1. FADU analysis of L5, S3R and S4 cells treated withdoxorubicinDoxorubicin is a DNA-damaging agent which is widely used in
chemotheraphy. It has been shown that cytostatic doses of
doxorubicin can lead to the induction of cellular senescence. Cells
sensitivity to this agent can vary between different types of cells.
Therefore the first step was to analyze the cells sensitivity to
treatment with this agent. To do this we used the FADU method.
FADU enables to measure, in an automatic way, the percentage of
double-stranded DNA [15,16], which accounts for 100% in
control cells (Fig. 1). SybrGreen, a fluorescent dye used in this
method, binds only to double-stranded DNA. Therefore, the less
intensive fluorescence the less double-stranded DNA can be
observed. To this end we treated all of the cell lines: with the
mutated form of nibrin (S3R and S4) and spontaneously
immortalized cells from a healthy donor (L5) with two concen-
trations of doxorubicin (1 and 10 mM) and analyzed the
percentage of double-stranded DNA, after short periods of time
(30, 60 and 90 min). The most sensitive, to treatment with
doxorubicin, were the S3R cells. In the case of this cell line, a
significantly lower amount of double-stranded DNA could be
found at all of the analyzed time points after treatment with both
concentrations of doxorubicin, in comparison with the untreated
cells. In the case of S4 cell line a statistically significant decrease of
the percentage of double-stranded DNA, in comparison with
control cells, could be observed 90 min after treatment with the
lower (1 mM) concentration of doxorubicin and in all of the time
points after treatment with the higher (10 mM) concentration of
this agent. This shows that even though S3R and S4 cell lines
possess the same NBN mutation their sensitivity to treatment with
doxorubicin is different. Furthermore, it turned out that, the
control (L5) cells are more sensitive to doxorubicin treatment than
the S4 cells, however less sensitive than the S3R cells. The
obtained results allowed us to speculate that different concentra-
tions of doxorubicin could be cytostatic for particular cell lines and
different doses could be needed for the induction of doxorubicin-
induced senescence.
2. Cell cycle arrest and apoptosis in doxorubicin-treatedL5, S3R and S4 cellsOne of the hallmarks of senescence is cell cycle arrest. Cells
undergoing senescence can be arrested in the G1/S or G2/M
phases of the cell cycle, however stress-induced premature
senescence (SIPS) is predominantly associated with cell cycle
arrest in the G2/M phase of the cell cycle. We treated L5, S3R
and S4 cells with various concentrations of doxorubicin, ranging
from 10 to 250 nM and analyzed DNA content using flow
cytometry. As it is shown in Figure 2A and in Table 1 treatment
with doxorubicin arrested cells from all of the cell lines in the G2/
M phase of the cell cycle. In case of the control (L5) cell line the
majority of cells were arrested after treatment with 50 nM
doxorubicin (approximately 30%). The largest fraction of S4 cells
(almost 50%) arrested in the G2/M, was observed after treatment
with 100 nM doxorubicin. In the S3R cell population the majority
of cells were found in the G2/M phase of the cell cycle after
treatment with 10 and 50 nM doxorubicin (about 35%). The
subG1 fraction which represents apoptotic cells did not exceed
11% in the case of the L5 cell line and 12% in the case of S4 cells.
S3R cells were much more prone to spontaneous apoptosis and
about 30% of the cells were found in the subG1 fraction. A
concentration dependent increase in the level of apoptosis could be
observed after treatment with doxorubicin in all of the cell lines.
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 3 August 2014 | Volume 9 | Issue 8 | e104964

The DNA content analysis can underestimate the level of
apoptosis due to the fact that cells with 4C DNA undergoing
apoptosis, may have $2C DNA and cannot be distinguished from
the cells found in the S and G1 phases of the cell cycle. Therefore,
to estimate more accurately the percentage of cells undergoing
apoptosis, we performed the Annexin V/7-AAD cytometric
analysis. As expected this method revealed more apoptotic cells
in all of the analyzed cell lines in comparison with the DNA
content analysis. However, in the S4 cells, concentration
dependence after treatment with doxorubicin still could not be
observed. In the case of S3R cells about half of the cell population
underwent cell death after treatment with 50 and 100 nM
doxorubicin, i.e. significantly more than control cells (Fig. 2B).
These results show that S3R cells are very prone to both
spontaneous and doxorubicin induced apoptosis and generally
more sensitive to the treatment than the S4 cells. Nonetheless, a
substantial fraction of cells from all of the cell lines can be arrested
in the G2/M phase of the cell cycle upon treatment with different
concentrations of doxorubicin.
Since DNA-damage induced senescence is associated with
persistant activation of the DNA-damage response (DDR)
pathway, which can be observed 24–48 h after treating the cells
with a DNA damage inducing agent, we decided to analyze the
activation of this pathway after treating the cells with the selected
concentrations of doxorubicin. Such concentrations of doxorubi-
cin were selected which led to the accumulation of the most cells in
the G2/M phase of the cell cycle and relatively low level of cell
death. S3R cells were treated with 10 nM and S4 cells with
100 nM doxorubicin. For comparison we used spontaneously
immortalized spleenocytes obtained from a healthy donor (L5),
treated with 50 nM doxorubicin (Fig. 2C). In the case of the L5
cell line, we observed the presence of p-ATM (Ser 1981), p-p53
(Ser 15) and p-Chk1 (Ser 317), even in untreated cells. After
treatment with doxorubicn (24 h) increased levels of these proteins
and the presence of Chk2 and p-Chk2 (Thr 68) was observed
proving the presence of an active DDR. In untreated S4 cells the
phosphorylated form of Chk1 (Chk1 Ser 317) was detected.
Interestingly, after treatment with doxorubicin we noticed a
significant increase in the level of p-ATM (Ser 1981), p-Chk1 (Ser
317), p-Chk2 (Thr 68), p-p53 (Ser 15) and cH2AX. Our results
show that upon treatment with doxorubicin the DDR pathway is
only activated in the L5 and S4 cell lines, however this process
can’t be observed in the S3R cell line.
3. Doxorubicin-induced senescence of L5 and S4, but notS3R cellsThere is data showing that immortalized and cancer cells retain
the ability to undergo senescence including that induced by DNA
damage [17,18]. We have previously shown [19] that treatment of
human colon cancer HCT116 cells with a low dose of doxorubicin
for one day, followed by culture in a drug-free medium, led to the
induction of senescence. Therefore, we decided to use the same
experimental approach and treated the L5, S3R and S4 cells with
the chosen concentrations of doxorubicin (50 nM for L5, 10 nM
for S3R and 100 nM for S4) for 24 hours and afterwards cultured
the cells for four days in a drug free medium (1+4). We observed a
time-dependent increase in the number of SA-b-Gal positive cells
in L5 and S4, but not in S3R cell line (Fig. 3A, B). In case of the
L5 cell line the majority of SA-b-Gal-positive cells (approximately
95%) were observed on day 1+4. In case of the S4 cell line the
most SA-b-Gal positive cells were observed on day 1+3(approximately 50%). The presence of SA-b-Gal positive cells
was accompanied by an increase in the level of p53 (Ser 15) in
both cell lines, however a time dependent increase in the level of
p21 was only observed in the S4 cell line. Surprisingly, in the L5
cell line, a time dependent decrease in the level of this protein was
found (Fig. 3C). Two crucial pathways play an important role in
senescence: p53-p21 and p16-pRb. Sometimes these pathways
overlap therefore we also decided to check the level of p16, which
is a key protein in the p16-pRb pathway.
Figure 1. FADU analysis in L5, S3R and S4 cells treated withdoxorubicin. The percentage of double-stranded DNA, was measuredin all of the cell lines using the FADU method. All of the cell lines weretreated with 1 and 10 mM doxorubicin and the measurements wereperformed 30, 60 and 90 min after treatment with this agent. Data iscalculated as the percentage of control. The values are means 6 SDobtained from three independent experiments. Statistical significancewas estimated using the Student’s T test.doi:10.1371/journal.pone.0104964.g001
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 4 August 2014 | Volume 9 | Issue 8 | e104964

Figure 2. Dose-dependent influence of doxorubicin on cell cycle arrest, apoptosis and activation of the DNA damage response(DDR). A. DNA content, analyzed by flow cytometry, 24 h after treatment with different concentrations of doxorubicin (0–250 nM). Representative
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 5 August 2014 | Volume 9 | Issue 8 | e104964

The p16-mediated senescence acts mainly through the retino-
blastoma (pRb) pathway by inhibiting the action of the cyclin
dependent kinases and leads to G1 cell cycle arrest [20]. We did
not observe any changes in the level of this protein in the S3R and
S4 cell lines, however, a time dependent decrease in the level of
p16 was observed in the L5 cell line (Fig. 3C). The observation
made in the L5 cell line requires further elucidation. It seems that
untreated S3R cells might have the p53/p21 pathway already
active, which is further slightly activated after treatment with
doxorubicin, but this is not accompanied by an increase in SA-b-Gal activity. In the L5 and S4 cells stronger activation of the p53/
p21 pathway correlated with an increase in SA-b-Gal activity.
This encouraged us to investigate whether the lack of induction of
senescence in the S3R cell line was due to the fact that the cells
underwent cell death. Using the annexin V/7-AAD assay, we
measured the level of apoptosis a day after treatment with
doxorubicin and on subsequent days after transferring the cells to
fresh medium (Fig. 3D). In all of the cell lines we observed a time-
dependent increase in the level of apoptosis. Three days after
culturing the cells in drug free medium (1+3) approximately 55%
of cells underwent apoptosis in all of the cell lines, however it
should be underlined that the cells were treated with different
concentrations of doxorubicin. The S4 cells were treated with a
ten times higher concentration of doxorubicin (100 nM) than the
S3R cells (10 nM). Moreover, in the case of the L5 and S3R cell
lines a high basal level of apoptosis could be observed. Despite the
high level of cell death, a fraction of S4 (more than 40% of SA-b-Gal positive cells on day 1+4) and L5 (about 95% of SA-b-Gal
positive cells on day 1+4) of cells, that survived, were able to
undergo senescence.
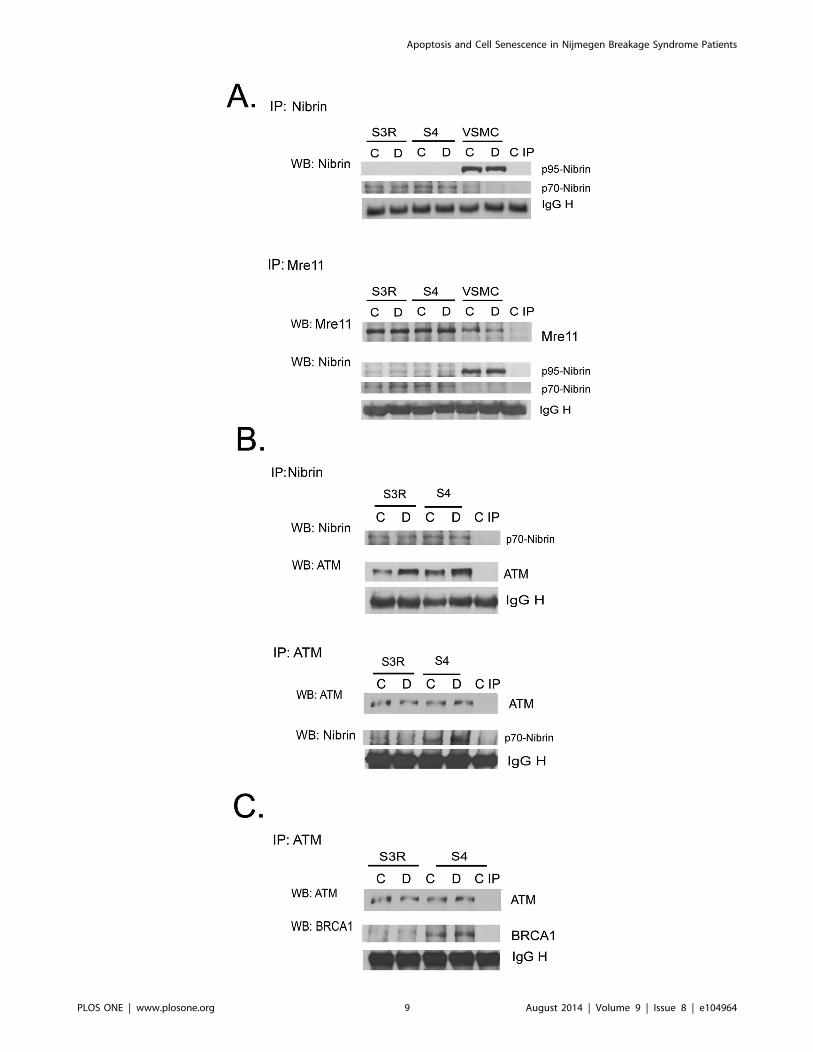
4. The level of p70-nibrin in S3R and S4 cellsWe were interested whether the differences in the cells
susceptibility to doxorubicin treatment and cell fate were due to
a different level of the truncated form of nibrin (p70), which is
present in the S3R and S4 cells. To elucidate this we performed an
immunoprecipitation assay which showed the same level of p70-
nibrin in both untreated and doxorubicin treated S3R and S4 cells
(Fig. 4A). The p95 form of nibrin was not detected neither in S3R
nor in S4 cells, however it was observed in VSMC cells, which
were used as a positive control. To confirm the above observation
and to exclude the possibility that the amount of immunoprecip-
itated protein was not equal, we verified the p70-nibrin level also
after IP, using the anti-Mre11 antibody. In this case we checked
the level of Mre11 by WB, as a loading control, and followed with
analysis of nibrin. Also this time we did not detect any differences
histograms from one of three independent experiments. B. Concentration-dependent apoptosis measured 24 h after treatment with doxorubicin (0–250 nM). The percentage of apoptotic cells was estimated by the Annexin V/7-AAD flow cytometry assay in three independent experiments. The barsshow means 6 SD values. Data was analyzed using the CellQuest software. Statistical significance was estimated using the Student’s T test. C.Expression of the DDR proteins analyzed by Western blotting in control (C) and treated with doxorubicin (D) S3R, S4 and L5 cells. Whole cell extractswere prepared 24 h after cell treatment with the following cytostatic concentrations of dox: 50 nM (L5), 10 nM (S3R), 100 nM (S4) and b-actin wasused as a loading control.doi:10.1371/journal.pone.0104964.g002
Table 1. DNA content (%) in L5, S3R and S4 cells treated with different concentration of doxorubicin.
L5
% untreated 10 nM 50 nM 100 nM 250 nM
SubG1 11,4765,32 7,1865,85 13,8365,45 20,4865,42 24,6469,37
G1 59,5765,61 65,1963,24 49,6764,6 57,49610,15 59,2865,42
S 7,4964,86 6,2763,92 3,9260,21 2,9260,92 3,3360,24
G2/M 19,5263,46 21,7365,98 30,1611,14 17,9566,36 11,7362,99
S3R
% untreated 10 nM 50 nM 100 nM 250 nM
SubG1 27,9364,14 29,2365,9 31,6465,49 33,6365,23 32,6461,28
G1 25,8364,1 19,0463,9 17,1864,01 19,7764,56 25,8762,45
S 15,5763,72 12,5361,12 10,761,22 15,2861,12 16,0160,59
G2/M 25,0862,09 34,8464,49 35,2464,4 26,0763,57 1965,57
S4
untreated 10 nM 50 nM 100 nM 250 nM 500 nM
SubG1 6,4364,87 7,5964,07 6,5662,74 6,7261,57 11,3763,39 10,6760,73
G1 42,01615,9 40,37612,17 31,6369,31 20,0768,12 18,1562,18 35,5361,49
S 17,1365,69 16,6765,85 14,1265,28 1164,73 7,6863,56 20,7864,08
G2/M 20,866,52 24,1166,18 35,5465,18 48,4865,64 39,7969,42 31,0265,35
The cells were treated with doxorubicin for 24 h and DNA content was measured by flow cytometry. The percentage of DNA in all phases (including subG1) of the cellcycle is shown. Data obtained after treatment with the selected concentrations of doxorubicin, chosen for the induction of senescence, is shown in bold.doi:10.1371/journal.pone.0104964.t001
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 6 August 2014 | Volume 9 | Issue 8 | e104964

Figure 3. The induction of cellular senescence and apoptosis upon doxorubicin treatment. A. SA-b-Gal activity. Cells were treated for 1day with doxorubicin (L5 - 50 nM, S3R - 10 nM and S4 - 100 nM) and then cultured in doxorubicin-free medium (1+n; n- are days of culture withoutdoxorubicin). The bars show means 6 SD. The percentage of SA-b-Gal positive cells from at least three independent experiments and representativeimages from one of three independent experiments. B. Magnification 200x. C. Expression of protein markers of senescence (p-p53, p53, p21 and p16)analyzed by Western blotting in untreated (0) cells and on the following days after culturing in doxorubicin-free medium (1+n), b-actin was used as aloading control. D. The percentage of apoptotic cells after treatment with doxorubicin estimated by the AnnexinV/7-AAD flow cytometry assay. Thebars show means 6 SD values. Data were obtained from three independent experiments. Statistical significance was estimated using the Student’s Ttest.doi:10.1371/journal.pone.0104964.g003
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 7 August 2014 | Volume 9 | Issue 8 | e104964

in the level of p70-nibrin. To verify the functionality of the
truncated nibrin we analyzed its binding to ATM. After
immunoprecipitating either ATM or nibrin it was observed that
p70-nibrin was able to form a complex with ATM in both NBS1
deficient cell lines (Fig. 4B). However, the IP revealed that more
ATM immunoprecipitated with p70-nibrin in S4 than S3R cells.
This may suggest that formation of DNA damage-induced ATM-
nibrin complex is more efficient in S4 cells. This difference was
already found in untreated cells and correlated with the observed
higher phosphorylation of ATM in response to doxorubicin
treatment of S4 cells (Fig. 2C). The possible better function of the
DNA damage/repair response in S4 cells was confirmed by a
further IP experiment showing that in these cells more BRCA1
was immunoprecipitated with ATM (Fig. 4C) suggesting that S4
are more efficient in DNA repair than the S3R cells.
5. Radiation induced activation of the DDR pathway inL5, S3R and S4 cellsDespite the fact that Nijmegen Breakage Syndrome was caused
by the same mutation in the S3R and S4 cell lines their
susceptibility to doxorubicin treatment differed. To verify whether
this was a characteristic feature of only doxorubicin, we used a
different DNA-damaging agent. Therefore the cells ability to
activate the DDR pathway after being exposed to c-radiation (4Gy
and cultured for 3 h) was analyzed (Fig. 5). Interestingly, exposure
to c-radiation of both S4 and S3R as well as control (L5) cells led
to an efficient induction of DDR. An increase in the level of the
following proteins was observed in all of the analyzed cell lines: p-
ATM (Ser 1981), p-Chk1 (Ser 317), p-p53 (Ser 15) and cH2AX.
The phosphorylated form of Chk2 (Thr 68) was only noticed upon
exposure to c-radiation of the S4 cells. This indicated that these
cells retain the capacity to upregulate the components of the DDR
pathway, at least for a short period of time.
6. The role of nibrin in DNA damage induced senescenceof human Vascular Smooth Muscle CellsSince a relatively low level of the truncated nibrin (p70-nibrin)
in S4 cells was sufficient for activation of the DDR signaling
pathway followed by senescence, we asked whether downregula-
tion of the NBS1 protein in normal cells would influence DDR
activation and senescence upon treatment with doxorubicin.
Transfection of L5 cells, using the nucleofection method, turned
out to be unsuccessful. Only 25% of the transfected cells were
viable 24 h after transfection. Therefore to analyze the effect of
downregulation of nibrin on the induction of senescence, we used
vascular smooth muscle cells (VSMCs) which were shown by us to
undergo senescence after treatment with doxorubicin [21]. Before
treatment with doxorubicin, the cells were transfected with
negative siRNA or NBN siRNA with 85% transfection efficiency
measured a day after transfection (not shown). As shown in
Figure 6A the level of NBS1 in cells transfected with NBN siRNA
and cultured in the presence of doxorubicin for three days was
reduced from two to four times. Moreover the levels of p-NBS1
(Ser 343) and p-ATM (Ser 1981) were substantially reduced in
these cells. However, there were no differences in the level of p53
and p21 proteins between cells transfected with negative siRNA
and NBN siRNA (Fig. 6A). Next we decided to verify whether the
downregulation of nibrin would affect the formation of 53BP1 foci,
after treatment with doxorubicin. Recently 53BP1 has been
recognized as a convenient marker of DSBs [22]. We observed
that the formation of 53BP1 foci was not affected when the level of
NBS1 was reduced (Fig. 6B, 6C). This could suggest that
senescence was also not affected in cells with reduced level of
NBS1. Indeed, the percentage of SA-b-Gal positive cells was
substantially increased already two days after treatment with
doxorubicin in both types of cells and accounted for 100% on day
3 of treatment with doxorubicin (Fig. 6D, 6E). These results were
confirmed using the BrdU incorporation assay, which showed
complete inhibition of proliferation in cells which were transfected
with negative siRNA and NBN siRNA and subsequently treated
with doxorubicin (Fig. 6F).
Taken together, the obtained results performed on human
VSMCs indicate that a substantially reduced level of NBS1 did not
influence doxorubicin-induced DDR and senescence in these cells.
Discussion
The aim of our study was to investigate the role of nibrin in
doxorubicininduced senescence.
Cellular senescence is associated withpermanent growth arrest.
We can distinguish two types of cellular senescence: replicative
which is telomere shortening dependent and stress-induced
premature senescence, which is telomere shortening independent.
Replicatively senescing cells are believed to activate the G1
restriction point. However, it was recently documented that
replicative senescence can stop the cells in both the G1/S and G2/
M phases of the cell cycle [23] while SIPS is mainly associated with
cell cycle arrest cells in the G2/M phase of the cell cycle.
NBS1 deficient cells have improperly functioning cell cycle
checkpoints [24], including a defect of the DNA damage induced
intra-S-phase checkpoint which is responsible for the radio-
resistant DNA synthesis (RDS)- a continuation of DNA synthesis
despite the presence of radiation-induced DNA damage [25].
However, the reports concerning the status of cell cycle
checkpoints in NBS deficient cells are discrepant, since both
impaired and normal G1/S or G2/M arrest after cell irradiation
have been reported (reviewed by [26]). Previously it was
documented that S3R cells had a reduced capacity to undergo
G1 arrest and showed a marked accumulation of cells in the G2/
M phase of the cell cycle 24 h after 4Gy c-irradiation, though to a
lesser extent than the S4 cells [27]. We have shown that treatment
with doxorubicin of L5, S3R and S4 cells led to an arrest in the
G2/M phase of the cell cycle. However, we proved that S3R cells
had a less efficient G2 checkpoint than S4 cells. Treatment of S3R
cells with 100 nM doxorubicin, the concentration which halted
most of the S4 cells in the G2/M phase of the cell cycle, led to
massive cell death of S3R cells. Nevertheless the percentage of
S3R cells which were arrested in the G2/M phase of the cell cycle,
after treatment with the selected, cytostatic concentrations of
doxorubicin, was comparable to the one observed in control L5
cells.
The higher propensity of S3R than S4 cells to undergo
apoptosis was connected with a decrease in the level of double-
stranded DNA as revealed using the FADU method. One should
keep in mind that doxorubicin is a DNA-damaging agent which
acts through different mechanisms. Amongst all, it induces the
formation of cross-links which prevent DNA from unwinding. The
FADU method enables to measure DNA susceptibility to unwind,
which is a function of the number of chromatin modifications.
Therefore the FADU method, in the context of this particular
agent, can only be used as a screening method which enables to
verify the cells susceptibility to treatment with different concen-
trations of doxorubicin. Nevertheless, the decrease in the amount
of double-stranded DNA was observed with the increasing
concentrations of doxorubicin and time of treatment in all of the
three examined cell lines, proving that at least a portion of DNA
acquires double strand breaks upon treatment with doxorubicin.
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 8 August 2014 | Volume 9 | Issue 8 | e104964
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 9 August 2014 | Volume 9 | Issue 8 | e104964

Generally, NBS1 deficient cells have impaired DNA repair. This
process seems to be more severe in the S3R than in the S4 cells
due to the lower level of the BRCA1 protein, which doesn’t
interact with ATM in the S3R cells. It was reported that
downregulation of the NBS1 protein level by siRNA led to an
increase in irradiation-induced mutation frequency in human
lymphoblastoid cells [26]. Moreover, it is worth to note that null
mutation of Nbs is lethal in mice [4].
Interestingly, the presence of less double-stranded DNA, after
treatment with doxorubicin, in the S3R cells, than in the S4 and
L5 cells, was not linked to ATM activation. However, we observed
increased levels of p-ATM and its downstream targets such as p-
Chk1, p-p53 and cH2AX 24 h after treatment with doxorubicin
in control (L5) and S4 cells. Moreover in the S4 cells upon
doxorubicin treatment, a substantial increase in the level of p-
Chk2 (Thr 68) could be seen. Surprisingly all of the cell lines
retained the ability to activate DDR upon exposure to c-radiation.Several studies showed severe impairment of the DDR activation
in NBS1 deficient cells. Namely, cells from NBS patients have
been reported to be deficient in ATM phosphorylation of p53,
Chk2 and other substrates following DNA damage. Other studies
showed that the C-terminal fragment of nibrin was sufficient to
stimulate ATM activation at early times after irradiation. In
contrast, nuclear expression of a nibrin transgene lacking the C-
terminal 100 amino acids was unable to stimulate ATM activation
under the same conditions ([28] and literature there). This was
most likely due to the lack of the ATM binding domain. We have
shown that despite the presence of the same NBN gene mutation,
DDR is only activated in the S4 cells. Furthermore, this pathway
was also activated in the L5 cells. In S3R cells some elements of
the DDR (p-p53, p-Chk1 and p-Chk2) were already present in
untreated cells and 24 h treatment with doxorubicin did not lead
to an increase in the level of these proteins. It is tempting to
speculate that the different response of the two NBS1 deficient cell
lines to treatment with doxorubicin is caused by the presence of a
lower level and/or nonfunctional truncated form of nibrin (p70-
nibrin) in S3R cells. Indeed, it has been shown that the level of
p70-nibrin can vary in cells obtained from NBS patients [2].
However, our results showed the same amount of p70-nibrin in S4
and S3R cells. Moreover, in both cell lines p70-nibrin co-
immunoprecipitated with ATM. Nevertheless we observed that a
higher level of p70-nibrin precipitated with ATM in S4 cells than
in the S3R cells. In contrast to the results obtained using the S4
cells and the L5 cells with wild-type NBN gene, we did not observe
ATM phosphorylation after treatment with doxorubicin in S3R
cells. On the other hand, a low level of the phosphorylated form of
p53 (p-p53 Ser 15) was detected in untreated, S3R cells and its
level increased after treatment with doxorubicin. Others [29]
showed impaired, but still detectable, ATM and p53 phosphor-
ylation in doxorubicin-treated NBS fibroblasts. Interestingly, in
NBS fibroblasts the p26 instead of the p70 fragment of nibrin
could be found, which doesn’t possess the ATM binding domain.
This discrepancy could be explained by the fact that p53 can be
phosphorylated on Ser 15 not only by ATM, but also by DNA-
PK, which plays a vital role in DSB repair as well as in driving cells
to apoptosis [30]. Nonetheless, the results obtained by Hou et al.
[29] allowed to conclude that NBS1 is acting upstream of ATM.
On the other hand, ATM phosphorylates nibrin at its Ser 343
residue [7]. We showed that nibrin can act both downstream and
upstream of ATM, as downregulation of nibrin affected
phosphorylation of both nibrin and ATM. These results suggested
that DDR could be compromised in cells with a diminished level
of nibrin. However, in VSMCs, in which the level of nibrin was
substantially reduced, the p53/p21 pathway was practically not
affected which suggests that in normal cells there must be a
redundancy of this protein. Surprisingly, despite the presence of
the same amount of p70-nibrin in both cell lines, the p53/p21
pathway was only activated in the S4 cells. This could imply a
failure in DDR activation downstream of ATM in the S3R cells.
However, these cells had much less ATM bound to nibrin in the
IP assay.
Moreover, we detected a higher basal level of apoptosis in
control S3R cells, but a substantially lower level of the BRCA1
protein in comparison with S4 cells in the IP assay. This indicates
that S3R cells could have a limited capacity for DNA repair what
could be reflected by a very high rate of spontaneous apoptosis in
these cells. Indeed, also the basal level of p-p53 was higher in S3R
than in S4 cells indicating p53-dependent apoptosis.
It seems that DDR can be a culprit of cell senescence, therefore
we wondered whether S3R cells would be able to senesce after
treatment with doxorubicin. Indeed, in both L5 and S4 cells we
Figure 4. Levels of nibrin, p70-nibrin, MRE11, ATM and BRCA1 in the DDR complex estimated by immunoprecipitation assay. A.Level of nibrin: wild-type (p95) and the truncated form (p70) in control (C) and doxorubicin treated (D, 1 mM/1 h) S3R, S4 and VSMCs. Expression ofnibrin was analyzed by immunoprecipitation using an anti-NBS1 antibody followed by Western blotting with anti-NBS1 (upper panel). Alternatively,IP using anti-MRE11 antibody was performed followed by WB with anti-NBS1 (lower panel). MRE11 was used as a loading control. The last lane (C IP)shows the negative IP control. Note that p95 is only present in VSMCs, in which there is no p70-nibrin. B. ATM binding to nibrin in control anddoxorubicin-treated S3R and S4 cells analyzed by immunoprecipitation using anti-NBS1 antibody (upper panel) or anti-ATM antibody (lower panel).Levels of ATM and p70 were detected by WB. Loading controls were performed in both variants of IP. C. Expression of BRCA1 in control and dox-treated S3R and S4 cells was analyzed by immunoprecipitation using anti-ATM antibody followed by WB using an anti-BRCA1 antibody. Loading andnegative IP controls were performed as above.doi:10.1371/journal.pone.0104964.g004
Figure 5. Activation of the DNA damage response pathwayupon c-irradiation. Expression of the DDR proteins analyzed byWestern blotting in control (C) and exposed to radiation (IR) S3R, S4 andL5 cells. Whole cell extracts were prepared 3 h after exposure to 4 Gy ofc-radiation, b-actin was used as a loading control.doi:10.1371/journal.pone.0104964.g005
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 10 August 2014 | Volume 9 | Issue 8 | e104964

Figure 6. The role of nibrin in doxorubicin-induced senescence of human Vascular Smooth Muscle Cells (VSMCs). Cells weretransfected with negative siRNA (2) or NBN siRNA (+) and afterwards cultured for three days in the presence of doxorubicin (100 nM). A.Downregulation of the NBS1 protein level in VSMCs using specific siRNA (60 nM). Whole cell extracts were prepared at indicated time points aftertreatment with doxorubicin. Expression of the indicated proteins was estimated by Western blotting, b-actin was used as a loading control. Theamount of the protein in cells transfected with NBN siRNA was calculated by densitometry as a fraction of that present in cells transfected with
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 11 August 2014 | Volume 9 | Issue 8 | e104964

observed the appearance of the common and widely used marker
of senescence, which is increased SA-b-Gal activity. The presence
of this marker of senescence is common in adherent cells [5]
however data concerning senescence of lymphoid cells and the
presence of this hallmark is scarce [31]. Additionally increased
activity of SA-b-Gal in the S4 cells was accompanied by a time-
dependent increase in the level of p21, which is a cdk inhibitor.
Thus, we can conclude that L5 and S4 cells, contrary to S3R cells,
are able to activate the DDR and undergo senescence. Moreover
VSMCs with a highly reduced level of nibrin were also able to
undergo senescence just like cells with the proper level of this
protein. We can speculate that there is a minimal amount of nibrin
or its truncated p70 form which is indispensable for the activation
of DDR and the subsequent induction of senescence. Interestingly,
it has been shown very recently that doxorubicin treated ATM-
deficient human fibroblasts underwent Akt-dependent SIPS
without DDR activation [32]. It seems that S3R cells are unable
to activate such a program and, most likely any senescence
pathway.
We showed that S3R cells are generally more sensitive to
doxorubicin treatment than the S4 and L5 cell lines. Also others
showed extreme variations in the propensity to undergo DNA
damage-induced apoptosis (40-fold) amongst lymphoid cells
derived from the NBS patients [33]. The authors did not find a
correlation between the propensity to undergo apoptosis and the
level of the truncated form of nibrin-p70. The mechanisms of cell
death in these cells is still awaiting elucidation.
It seems that, despite the presence of a similar level of p70-
nibrin, in the S3R and S4 cell lines, the differences in ATM
phosphorylation and its ability to bind nibrin were crucial for the
efficient activation of DDR and the induction of senescence. We
observed that some proteins which are involved in the DNA
damage/repair pathway (ATM, BRCA1) were more efficiently
recruited to the DNA damage-induced complex in S4 than in S3R
cells what might explain the differences in the cell fate after
treatment with doxorubicin.
Moreover it cannot be excluded that the described in this paper
differences in the S3R and S4 cell phenotype, may result from
genomic instability of patients with Nijmegen Breakage Syndrome
or the immortalization process. It has also been previously shown
that NBS patients with the same genotype may vary in the
phenotypic expression [34]. It is worth to note that unsupervised
clustering of whole genome gene expression arrays of S3R and S4
cells indicated that common gene expression changes, between the
two lines, also exist [35].
Acknowledgments
Experiments using flow cytometry were performed at the Laboratory of
Cytometry at the Nencki Institute of Experimental Biology. We would like
to thank Mrs. Anna Leonowicz for her skillful technical assistance.
Author Contributions
Conceived and designed the experiments: OA ABZ GM ES. Performed
the experiments: OA ABZ GM WDR AW MKK. Analyzed the data: OA
ABZ GM MMV ZK KP ES. Contributed reagents/materials/analysis
tools: MMV KP AB JKS. Contributed to the writing of the manuscript:
OA ABZ GM ES.
References
1. Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA, Digweed
M (2012) Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis 7: 1–19.
2. Kruger L, Demuth I, Neitzel H, Varon R, Sperling K, et al. (2007) Cancer
incidence in Nijmegen breakage syndrome is modulated by the amount of a
variant NBS protein. Carcinogenesis 28: 107–111.
3. Maser RS, Wong KK, Sahin E, Xia H, Naylor M, et al. (2007) DNA-dependent
protein kinase catalytic subunit is not required for dysfunctional telomere fusion
and checkpoint response in the telomerase-deficient mouse. Mol Cell Biol 27:
2253–2265.
4. Zhu J, Petersen S, Tessarollo L, Nussenzweig A (2001) Targeted disruption of
the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality
in mice. Curr Biol 11: 105–109.
5. Sikora E, Arendt T, Bennett M, Narita M (2011) Impact of cellular senescence
signature on ageing research. Ageing Res Rev 10: 146–152.
6. d’Adda di Fagagna F (2008) Living on a break: cellular senescence as a DNA-
damage response. Nat Rev Cancer 8: 512–522.
7. Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, et al. (2002)
NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT
domain. Curr Biol 12: 1846–1851.
8. Freedman DA (2005) Senescence and its bypass in the vascular endothelium.
Front Biosci 10: 940–950.
9. Siwicki JK, Degerman S, Chrzanowska KH, Roos G (2003) Telomere
maintenance and cell cycle regulation in spontaneously immortalized T-cell
lines from Nijmegen breakage syndrome patients. Exp Cell Res 287: 178–189.
10. Gewirtz DA (1999) A critical evaluation of the mechanisms of action proposed
for the antitumor effects of the anthracycline antibiotics adriamycin and
daunorubicin. Biochem Pharmacol 57: 727–741.
11. Siwicki JK, Hedberg Y, Nowak R, Loden M, Zhao J, et al. (2000) Long-term
cultured IL-2-dependent T cell lines demonstrate p16(INK4a) overexpression,
normal pRb/p53, and upregulation of cyclins E or D2. Exp Gerontol 35: 375–
388.
12. Keeshan K, Mills K, Cotter TG, McKenna SL (2001) Elevated Bcr-Abl
expression levels are sufficient for a haematopoietic cell line to acquire a drug-
resistant phenotype. Leukemia 15: 1823–1833.13. Laemmli UK (1970). Cleavage of structural proteins during the assembly of the
head of bacteriophage T4. Nature. 227: 680–685.14. Dimri GP, Lee X, Basile G, Acosta M, Scott G, et al. (1995). A biomarker that
identifies senescence human cells in culture and in aging skin in vivo. Proc NatlAcad Sci USA 92: 9363–9367.
15. Moreno-Villanueva M, Eltze T, Dressler D, Bernhardt J, Hirsch C (2011) The
automated FADU-assay, a potential high-throughput in vitro method for earlyscreening of DNA breakage. Altex 28: 295–303.
16. Moreno-Villanueva M, Pfeiffer R, Sindlinger T, Leake A, Muller M (2009) Amodified and automated version of the ‘Fluorimetric Detection of Alkaline DNA
Unwinding’ method to quantify formation and repair of DNA strand breaks.
BMC Biotechnol 9: 39.17. Sherman MY, Meng L, Stampfer M, Gabai VL, Yaglom JA (2011) Oncogenes
induce senescence with incomplete growth arrest and supress the DNA damageresponse in immortalized cells. Aging Cell 10(6): 949–961.
18. Shay JW, Roninson IB (2004) Hallmarks of senescence in carcinogenesis and
cancer therapy. Oncogene 23: 2919–2933.19. Sliwinska MA, Mosieniak G, Wolanin K, Babik A, Piwocka K, et al. (2009)
Induction of senescence with doxorubicin leads to increased genomic instabilityof HCT116 cells. Mech Ageing Dev 130: 24–32.
20. Rayess H, Wang MB, Srivatsan ES (2012) Cellular senescence and tumorsuppressor gene p16. Int J Cancer 130: 1715–1725.
21. Bielak-Zmijewska A, Wnuk M, Przybylska D, Grabowska W, Lewinska A, et al.
(2014) A comparison of replicative senescence and doxorubicin-inducedpremature senescence of vascular smooth muscle cells isolated from human
aorta. Biogerontology 15: 47–64.22. Schultz LB, Chehab NH, Malikzay A, Halazonetis TD (2000) p53 binding
protein 1 (53BP1) is an early participant in the cellular response to DNA double-
strand breaks. J Cell Biol 151: 1381–1390.
negative siRNA (1). B. 53BP1 staining in doxorubicin-treated control cells and cells with silenced nibrin. Representative images from one of threeindependent experiments. Magnification 100x. C. 53BP1 staining in doxorubicin treated control cells and cells with silenced nibrin. Cells with DNAdamage were divided into four groups based on the number of 53BP1 foci: cells without 53BP1 foci, with one focus, with 2–5 foci, with more than 5foci. Means from three independent experiments. D. SA-b-Gal activity in doxorubicin treated VSMC cells. Representative images from one of threeindependent experiments, magnification 100x. E. The percentage of SA-b-Gal positive cells (a mean 6 SD) from three independent experiments. F.BrdU incorporation assay. Control cells and cells transfected with negative siRNA or NBN si RNA were cultured with BrdU for 24 h. Data presented asmeans 6 SD from three independent experiments.doi:10.1371/journal.pone.0104964.g006
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 12 August 2014 | Volume 9 | Issue 8 | e104964

23. Mao Z, Ke Z, Gorbunova V, Seluanov A (2012) Replicatively senescent cells are
arrested in G1 and G2 phases. Aging 4: 431–435.24. Shiloh Y (1997) Ataxia-telangiectasia and the Nijmegen breakage syndrome:
related disorders but genes apart. Annu Rev Genet 31: 635–662.
25. Falck J, Petrini JH, Williams BR, Lukas J, Bartek J (2002) The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet
30: 290–294.26. Zhang Y, Zhou J, Lim CU (2006) The role of NBS1 in DNA double strand
break repair, telomere stability, and cell cycle checkpoint control. Cell Res 16:
45–54.27. Siwicki JK, Berglund M, Rygier J, Pienkowska-Grela B, Grygalewicz B, et al.
(2004) Spontaneously immortalized human T lymphocytes develop gain ofchromosomal region 2p13–24 as an early and common genetic event. Genes
Chromosomes Cancer 41: 133–144.28. Cerosaletti K, Wright J, Concannon P (2006) Active role for nibrin in the
kinetics of atm activation. Mol Cell Biol 26: 1691–1699.
29. Hou YY, Toh MT, Wang X (2012) NBS1 deficiency promotes genomeinstability by affecting DNA damage signaling pathway and impairing telomere
integrity. Cell Biochem Funct 30: 233–242.
30. Hill R, Lee PW (2010) The DNA-dependent protein kinase (DNA-PK): More
than just a case of making ends meet? Cell Cycle 9: 3460–3469.
31. Chebel A, Bauwens S, Gerland LM, Belleville A, Urbanowicz I, et al. (2009)
Telomere uncapping during in vitro T-lymphocyte senescence. Aging Cell 8:
52–64.
32. Park J, Jo YH, Cho CH, Choe W, Kang I, et al. (2013) ATM-deficient human
fibroblast cells are resistant to low levels of DNA double-strand break induced
apoptosis and subsequently undergo drug-induced premature senescence.
Biochem Biophys Res Commun 430: 429–435.
33. Thierfelder N, Demuth I, Burghardt N, Schmelz K, Sperling K, et al. (2008)
Extreme variation in apoptosis capacity amongst lymphoid cells of Nijmegen
breakage syndrome patients. Eur J Cell Biol 87: 111–121.
34. Nijmegen breakage syndrome (2000) The International Nijmegen Breakage
Syndrome Study Group. Arch Dis Child. 82, 400–406.
35. Degerman S, Siwicki JK, Osterman P, Lafferty-Whyte K, Keith WN, et al.
(2010) Telomerase upregulation is a postcrisis event during senescence bypass
and immortalization of two Nijmegen breakage syndrome T cell cultures. Aging
Cell 9: 220–235.
Apoptosis and Cell Senescence in Nijmegen Breakage Syndrome Patients
PLOS ONE | www.plosone.org 13 August 2014 | Volume 9 | Issue 8 | e104964