herencia mitocondrial
DESCRIPTION
Generalidaes, conceptos basicos y Neuropatia optica de LeberTRANSCRIPT

Contenido
1. INTRODUCCIÓN.....................................................................................3
2. OBJETIVOS.............................................................................................4
2.1 Objetivo General...................................................................................4
2.2 Objetivos Específicos............................................................................4
3. ¿Qué es mitocondria?..............................................................................4
3.1 Anatomía de la mitocondria..................................................................4
3.2 División de las mitocondrias y segregación mitótica.............................5
3.3 Función de las mitocondrias.................................................................5
4. ¿Qué es Genética Humana?...................................................................5
5. ¿Qué es Herencia?..................................................................................5
6. ¿Qué es un gen?.....................................................................................5
7. ADN mitocondrial.....................................................................................6
7.1 Generalidades......................................................................................6
7.2 Herencia del ADN mitocondrial humano...............................................6
7.3 Tasa de mutación del ADN mitocondrial...............................................6
7.4 Heteroplasmia en el ADN mitocondrial.................................................6
8. Herencia Mitocondrial:.............................................................................8
9. Las enfermedades mitocondriales...........................................................9
9.1 ¿Qué son las enfermedades mitocondriales?......................................9
9.2 Características del genoma mitocondrial:...........................................10
10. PRESENTACION DE LA ENFERMEDAD POR HERENCIA
MITOCONDRIAL.......................................................................................................11
10.1 Neuropatía óptica hereditaria de Leber (NOHL)..............................11
10.1.1 Antecedentes históricos..............................................................11
10.1.2 Genética.....................................................................................11
10.1.3 Manifestaciones clínicas.............................................................12

10.1.4 Diagnostico.................................................................................13
10.1.5 Conclusiones del caso................................................................14
11. ANEXOS................................................................................................15
11.1 Formas de segregación de la herencia mitocondrial.......................15
12. BIBLIOGRAFÍA:.....................................................................................17
Página | 2

HERENCIA MITOCONDRIAL
1. INTRODUCCIÓN
Todos los tipos de herencia corresponden a segmentos de ADN nuclear y se
guían por las reglas de Mendel o por las de la herencia multifactorial. Sin embargo,
las células poseen numerosas mitocondrias, cada una de las cuales posee un
diminuto genoma de ADN.
Los gametos no aportan en igual cantidad los genes mitocondriales al cigoto
porque la porción intermedia del espermatozoide, que lleva las mitocondrias
degenera sin aportarlas al óvulo, mientras que el ovocito presenta una gran cantidad
de mitocondrias que son las únicas que tendrá el cigoto.
Por lo tanto, la herencia mitocondrial se hereda únicamente por la línea
materna. Además no se aporta un solo genoma mitocondrial sino varios millares,
aunque no todos iguales, dada la gran frecuencia de mutación. Esta particularidad
se denomina heteroplasmia, que es la heterogeneidad de los genomas
mitocondriales contenidos en las células de un mismo organismo. A medida que se
desarrolla el organismo y aumenta su número de células el genoma de las
mitocondrias puede ir cambiando en diferentes tejidos y a distintas edades, por esta
razón la población de mitocondrias en un momento dado puede ser diferente en
diversos tejidos y la identificación de mutaciones en el genoma mitocondrial debe
referirse a una edad y un sitio determinado. La tasa de mutación del ADN
mitocondrial es 10 veces más elevada que la del ADN nuclear, debido a que los
sistemas de reparación de errores en la replicación de ADN son mucho menos
sofisticados en las mitocondrias.
En general las mutaciones que afectan el genoma mitocondrial y se expresan
en el fenotipo, lo hacen en tejidos con alto consumo de energía (ATP) como el
sistema nervioso y el tejido muscular estriado.
Página | 3

2. OBJETIVOS2.1 Objetivo General
Obtener los conocimientos concretos sobre la herencia mitocondrial con el fin
de enriquecer nuestro aprendizaje para mantener un régimen correcto de
estudio.
2.2 Objetivos Específicos
Reforzar los conocimientos adquiridos en clase para mejorar nuestro
aprendizaje.
Estudio de la variabilidad mitocondrial en sus diversos ámbitos de aplicación
en genética humana
MARCO TEÓRICO
3. ¿Qué es mitocondria?
Las mitocondrias son organelos subcelulares que se encuentran en el
citoplasma de las células eucariotas, cuya función principal es la producción de la
energía celular en forma de trifosfato de adenosina (ATP, por sus siglas en inglés).
3.1 Anatomía de la mitocondria
El tamaño y la forma de las mitocondrias varían en grado considerable, en
función de su origen y su estado metabólico. La mitocondria está rodeada por una
fina membrana externa y contiene una membrana interna muy invaginada. El
número de las invaginaciones, llamadas crestas, varia con la actividad respiratoria
de un tipo particular de célula, esto se debe a que las proteínas que median la
cadena de transporte de electrones y la fosforilación oxidativa están unidas a la
membrana mitocondrial interna, por lo tanto, la velocidad de respiración varía con el
área de la superficie de la membrana.
El compartimento interno mitocondrial presenta una sustancia tipo gel de
agua, llamada matriz que tiene altas concentraciones de enzimas solubles del
metabolismo oxidativo.
La matriz también contiene la maquinaria genética mitocondrial, que expresan
solo unas pocas proteínas de la membrana mitocondrial interna.
Página | 4

3.2 División de las mitocondrias y segregación mitótica
Las mitocondria no se sintetizan nunca de novo, sino que siempre surgen por
crecimiento y división de otras ya existentes.
Durante la división celular, las mitocondrias se distribuyen de forma aleatoria
entre las células hijas. Si en una célula que se va a dividir existen moléculas de
ADNmt diferentes, la proporción de moléculas mutantes que pasara a las células
hijas pueden variar mucho, y en consecuencia también el fenotipo. Este fenómeno
se conoce como segregación mitótica y puede tener muchas consecuencias,
especialmente desde un punto de vista genético.
3.3 Función de las mitocondrias
Las mitocondrias cumplen la función adicional de almacenar calcio intracelular
junto con el retículo endoplasmatico y también desempeñan un papel importante en
la muerte celular programada o apoptosis.
4. ¿Qué es Genética Humana?
La Genética humana es la ciencia que estudia los fenómenos de la herencia y
la variación.
5. ¿Qué es Herencia?
Todos los seres vivos, animales y vegetales, tienen la propiedad de transmitir
a sus descendientes una serie de caracteres biológicos que les hacen semejantes a
ellos.
A este conjunto de caracteres transmisibles a los descendientes es lo que se llama
herencia biológica.
Las unidades fundamentales de la herencia biológica reciben el nombre de
genes.
6. ¿Qué es un gen?
El gen es un segmento corto de ADN que se encarga de codificar una
proteína específica.
Página | 5

7. ADN mitocondrial7.1 Generalidades
Una de las principales características de las mitocondrias es la de poseer un
sistema genético propio y la maquinaria necesaria para sintetizar el ADN, ARN y las
proteínas que codifica.
Cada mitocondria contiene entre 2 y 10 copias de la molécula de ADN y a su
vez cada célula contiene cientos de mitocondrias, con lo que el número de copias de
este genoma en una célula oscilará entre 1000 y 10000 dependiendo de cada
órgano y tejido.
El ADN mitocondrial presenta toda una serie de características propias que lo
diferencian del ADN nuclear.
7.2 Herencia del ADN mitocondrial humano
Este se hereda como un patrón no mendeliano, durante muchos años, el
hecho de que el ADN mitocondrial presentaron una herencia estrictamente materna
y ausente de recombinación se ha considerado un dogma, a pesar de que en otros
organismos como plantas, hongos, mejillones o peces sí que se produce
recombinación del ADN mitocondrial.
7.3 Tasa de mutación del ADN mitocondrial
La tasa de mutación depende de la velocidad a la que surgen y se fijan las
mutaciones en los linajes, permite introducir una escala temporal en la evolución
molecular y así hacer estimas temporales sobre el ancestro común más reciente
entre dos linajes, es decir, estimar el tiempo de divergencia entre ellos.
7.4 Heteroplasmia en el ADN mitocondrial
La heteroplasmia es la heterogeneidad de los genomas mitocondriales
contenidos en células de un mismo organismo, también se define como el estado en
el que un individuo, célula o mitocondria presenta más de un genotipo de ADN
mitocondrial. En muchos individuos no hay evidencia de heteroplasmia, pero hay
evidencias que indican que el ADNmt está mutando constantemente.
El ADN humano actual es una molécula de 16.569 pares de bases formada
por dos cadenas complementarias. Utiliza un código genético ligeramente distinto al
del genoma nuclear o universal.
Página | 6

Como estas mutaciones ocurren al azar, todas las mutaciones adquiridas
estarán presentes en muy bajo nivel y puede que no se detecten en una muestra de
tejido o de sangre.
Utilizamos el término homoplasmia para describir el estado en el que no
podemos detectar esas mutaciones adquiridas.
La heteroplasmia se puede observar en:
Un individuo puede presentar más de un tipo de ADNmt en un solo tejido
Un individuo puede tener un tipo de ADNmt en un tejido y otro en tipo en otro
tejido.
Un individuo puede ser heteroplasmico en un tejido y homoplasmico en otro
tejido.
Cuando una célula heteroplasmica se divide, la herencia mitocondrial de las
células hijas es una cuestión de azar.
Durante la transmisión se produce un cuello de botella, y como resultado de
ello, en la ovogénesis un número reducido de moléculas de ADNmt determinará el
genotipo citoplasmático de la siguiente generación. Después de varios ciclos de
división la proporción de ADNmt mutante y normal en una célula puede derivar hacia
el mutante puro o hacia el normal puro (homoplasmia) es un proceso conocido como
segregación replicativa. Este proceso puede suceder durante la replicación de la
célula somática o durante la proliferación de las células germinales femeninas.
Existen dos tipos de heteroplasmias: puntuales y de longitud
Las heteroplasmias puntuales se deben a un único cambio de base y en los
primeros años fueron escasamente reportadas debido fundamentalmente a la
tecnología de secuenciación utilizada, mucho menos sensible y limpia que la
usada actualmente.
Las heteroplasmias de longitud son mucho más frecuentes que las puntuales
y se deben a la variación en el número de bases en un tracto homopolimerico,
cuando se presenta esta circunstancia es muy difícil poder caracterizar la
población de variantes de longitud por secuencia directa y en estos casos es
necesaria la secuencia de las dos cadenas de ADNmt y el uso de primers
alternativos para poder determinar el tracto homopolimerico.
Página | 7

8. Herencia Mitocondrial:
No hay que confundir herencia mitocondrial con enfermedad mitocondrial,
muchas de las alteraciones mitocondriales tienen su origen en proteínas codificadas
por genes del genoma nuclear.
Cualquier expresión de los genes mitocondriales se rige por pautas
claramente diferentes de las pautas que rigen la expresión de los genes nucleares.
En primer lugar los gametos no aportan igualitariamente genes mitocondriales al
cigoto porque la porción intermedia del espermatozoide, que lleva las mitocondrias,
degenera sin aportarlas al huevo, mientras que el ovocito presenta gran cantidad de
mitocondrias, que son las únicas que tendrá el cigoto. No se aporta un solo genoma
mitocondrial sino varios millares, aunque no todos iguales, dada la gran frecuencia
de mutación.
No todos los genes son heredados por igual de ambos padres. El genoma
extracelular es heredado solamente a través de la madre.
Las mitocondrias masculinas no contribuyen a la formación de los nuevos
cigotos. El patrón da lugar a pedigrees en los cuales todos los niños de una madre
afectada pueden estar afectados y ninguno de los niños de un padre afectado pueda
estar afectados.
La expresión de las enfermedades heredadas a través de las mitocondrias es
a menudo variable y puede ser de penetrancia incompleta. Esta observación es
posible, debido en parte al hecho que una población de mitocondrias, que puede en
sí misma ser genéticamente heterogénea, es en el hecho transmitida por la madre.
La expresividad dependerá del número de mitocondrias que contengan el gen
anómalo.
Si todas las mitocondrias transmitidas por la madre son del mismo genotipo,
esto se llama homoplasmia. Si existen diferencias genéticas entre ellas, se llama
heteroplasmia.
Muchas de las alteraciones mitocondriales tienen su origen en proteínas
codificadas por genes del genoma nuclear.
Página | 8

Se refiere a enfermedades causadas por alteraciones del ADN o genoma
mitocondrial. Las mitocondrias son organelas probablemente descendientes de
células procariotas que se incorporaron en estado simbiótico a las células
eucariotas primitivas.
La mayor parte del genoma de estas células procariotas precursoras de las
mitocondrias se fue trasladando, a través de fenómenos de transferencia génica, al
núcleo a lo largo de la evolución. Dentro de las mitocondrias queda tan solo una
pequeña porción que sería el actual genoma mitocondrial, también llamado genoma
citoplasmático, por el compartimento celular en el que se encuentra.
El genoma mitocondrial codifica para 2 ARN ribosómicos, 22 ARN de
transferencia y 13 ARN mensajeros. Estos últimos se traducirán a proteínas en el
interior de la propia mitocondria para a su vez unidas a otras proteínas importadas
del citoplasma, formar parte de los complejos multienzimáticos de la cadena
respiratoria mitocondrial.
El ADNmt humano actual es una molécula de 16.569 pares de bases formada
por dos cadenas complementarias. Utiliza un código genético ligeramente distinto al
del genoma nuclear o universal. Se encuentra en un medio muy rico en radicales
libres, productos secundarios de la cadena respiratoria mitocondrial, y carece de
histonas protectoras.
Existe evidencia de transmisión ocasional y parcial del genoma mitocondrial
paterno.
9. Las enfermedades mitocondriales 9.1 ¿Qué son las enfermedades mitocondriales?
Las enfermedades mitocondriales empezaron a describirse en la década de
los 60 en pacientes con intolerancia al ejercicio, estudios histoquímicos en tejido
muscular de estos individuos revelaron la presencia de acúmulos de mitocondrias,
tanto de aspecto normal como anormal.
Las mutaciones o delecciones en el ADNmt del ser humano causan un grupo
numeroso complejo y heterogéneo de enfermedades. El espectro clínico y la edad
de comienzo de las enfermedades mitocondriales varían ampliamente. Los órganos
Página | 9

con requerimientos energéticos altos son en especial vulnerables a las
enfermedades mitocondriales: el cerebro, el corazón, el músculo esquelético, el ojo,
el oído, el hígado, el páncreas y el riñón.
Las áreas con acumulaciones de mitocondrias aparecían de color púrpura y
estas fibras de aspecto anormal se denominaron ragged-red-fibers y se convirtieron
en un marcador patológico de las miopatías mitocondriales. Sin embargo, pronto fue
evidente que en muchos pacientes con RRF la miopía estaba asociada con signos y
síntomas que implican al sistema nervioso central y se denominaron
encefalomiopatías mitocondriales, y también pronto quedó claro que la ausencia de
RRF no excluye una etiología mitocondrial.
El término enfermedad mitocondrial se refiere solo a los desórdenes de la
cadena respiratoria mitocondrial o sistema de fosforilación oxidativa, que engloba la
cadena de transporte electrónico y la fosforilación oxidativa que representa el
objetivo final del metabolismo oxidativo, que es ser la principal fuente celular de
ATP.
Las enfermedades mitocondriales se presentan en ambos sexos pero se
transmiten exclusivamente por vía materna.
9.2 Características del genoma mitocondrial:
Es una pequeña molécula de ADN circular, cuya secuencia de nucleótidos se
conoce en su totalidad.
Codifica 13 polipéptidos, dos ARN ribosómicos y 22 ARN de transferencia.
El genoma mitocondrial codifica solamente una pequeña parte de los
componentes necesarios para la función de la mitocondria.
El genoma nuclear codifica los componentes restantes, que a su vez,
sintetizados son transportados a la mitocondria.
Puesto que el ADN mitocondrial se encuentra confinado en la mitocondria, su
replicación, transcripción y traducción tienen lugar en el interior de esta.
La traducción mitocondrial utiliza un código genético propio, es decir, el
significado de algunos codones cambia con respecto al código genético
universal y el número de ARNt utilizado es menor (solamente los 22
codificados por el ADNmt).
Página | 10

10. PRESENTACION DE LA ENFERMEDAD POR HERENCIA
MITOCONDRIAL
10.1 Neuropatía óptica hereditaria de Leber (NOHL)
La neuropatía óptica hereditaria de Leber (NOHL) es una enfermedad de
transmisión materna, que fue descrita por primera vez en 1871 por Theodor Leber.
Se demostró un siglo después que se debía a mutaciones puntuales del ADN
mitocondrial (ADNmt), aunque se sospecha que otros factores ambientales y
genéticos están relacionados. El hallazgo de mutaciones específicas en el ADNmt
en pacientes con similar fenotipo ha permitido garantizar el diagnóstico que hasta
hace algunos años se basaba en la herencia de tipo mitocondrial, las características
clínicas y el aspecto del fondo de ojo que en ocasiones puede resultar similar a otras
neuropatías ópticas. Este tipo de neuropatía por lo regular se sospecha tardíamente,
sobre todo si no se han precisado antecedentes de herencia mitocondrial.
10.1.1 Antecedentes históricos
En 1988, Wallace y otros identificaron por primera vez, el primer punto de
mutación en el ADNmt ligado a una enfermedad hereditaria. Describieron la
sustitución de un simple nucleótido, adenina por guanina, en la posición 11.778 del
ADNmt, en ND4 del complejo I de la cadena respiratoria.
En 1991, Huoponen y otros encontraron un nuevo tipo de mutación en la
posición 3 460, que codifica para la subunidad I ND1, del complejo I, que ha sido
confirmada por otros autores. Fue propuesta otra mutación en la posición 14.484 por
Howell y otros. A estas tres mutaciones se les ha designado como marcadores de la
enfermedad o primarias, porque con solo una presente los pacientes con NOHL
tienen un alto riesgo de padecer la enfermedad.
10.1.2 Genética
Dado que las mitocondrias de una persona derivan del citoplasma del óvulo,
la herencia de esta enfermedad es materna. Cada mitocondria contiene de dos a
diez copias de ADN circular donde se localizan los genes, que codifican para 13 de
las 67 proteínas que componen la cadena respiratoria.
Las mutaciones primarias de NOHL 11.778, 14.484 y 3.460 afectan el
complejo I del ADN mitocondrial o los genes de la ubiquinona oxidoreductasa. La
neuropatía óptica hereditaria de Leber es el resultado de un defecto en la cadena
Página | 11

respiratoria. Las mutaciones mitocondriales afectan los tejidos altamente
dependientes del metabolismo oxidativo, tales como la retina, el sistema nervioso
central, el corazón y el riñón. Se han hecho estudios para comprender la bioquímica
de las células afectadas por las mutaciones que causan NOHL, usando híbridos
citoplasmáticos sin mitocondrias a las cuales se les introducen mitocondrias de
individuos afectados. Estas investigaciones han permitido determinar que las
mutaciones de la posición 14.484 producen una reducción en la transferencia de
electrones y en la síntesis de ATP del complejo I, por su parte la mutación 11.778
induce un desdoblamiento de la respiración en el híbrido, mientras que la mutación
3.460 afecta la velocidad del consumo del oxígeno. El estrés oxidativo induce la
apoptosis en las células portadoras de las mutaciones. Además se ha visto una
disminución en las defensas antioxidantes (superóxido dismutasa, glutatión
peroxidasa y glutatión reductasa) en las células portadoras de mutaciones de esta
enfermedad. La apoptosis celular que se produce en esta neuropatía óptica, es la
responsable de las manifestaciones clínicas de la enfermedad y el deterioro visual.
El nervio óptico es dependiente de ATP, para garantizar el constante flujo
axoplásmico, necesario para el transporte de la información a lo largo de la vía
visual.
El 85 % de los pacientes con NOHL presentan mutaciones ADNmt
homoplásmicas (un solo tipo de ADNmt), el restante 15 % son heteroplásmicas
(presencia de ADNmt mutante y no mutante en diferentes proporciones). Las
mutaciones mitocondriales son generalmente heteroplásmicas. El grado de
heteroplasmia de un paciente determina si va a tener o no la enfermedad. La
relación entre los niveles de heteroplasmia y la condición clínica fue estudiada por
Man y otros, quienes mostraron que pacientes heteroplásmicos con alto grado de
mutación en ADNmt tenían mayor probabilidad de desarrollar pérdida visual.18
Diferentes estudios han demostrado que con cerca de un 70 % del ADNmt mutado
se expresa la enfermedad. 8,24 Por otra parte la presencia de una de las
mutaciones primarias de la enfermedad, como la 11.778, 3.460 y 14.484, no significa
que se vaya a padecer la misma, porque se ha observado que sujetos con la
mutación de forma homoplásmica no siempre llegan a padecer los síntomas.
Página | 12

10.1.3 Manifestaciones clínicas
Clínicamente, hay un comienzo agudo de pérdida visual, bilateral y simétrica.
Esto finalmente evoluciona a una atrofia óptica muy severa y una disminución
permanente de agudeza visual. En la fase aguda, que dura algunas semanas, el ojo
afectado muestra una apariencia edematosa de la capa de fibras nerviosas y vasos
peripapilares inflamados (microangiopatía). Estas características principales se ven
en un examen del fondo ocular justo antes o después de comenzar la pérdida de
visión. Un examen revela agudeza visual reducida, pérdida de visión del color y un
escotoma cecocentral en una prueba de campo visual.
La neuropatía óptica hereditaria de Leber se puede manifestar en otras tres
diferentes formas aunque atípicas, además de la clásica forma subaguda de días o
semanas de evolución.
Subclínica
De desarrollo lento
De estadio agudo clásico pero con recuperación espontánea.
En la neuropatía óptica hereditaria de Leber subclínica, la agudeza visual
disminuye poco. El inicio de la enfermedad de desarrollo lento es por lo regular en la
niñez y el resultado visual es favorable.
10.1.4 Diagnostico
El diagnóstico se basa en un examen oftalmoscópico. También son signos de
la LHON: inflamación de la papila óptica, tortuosidad vascular, telangiectasias
peripapilares, microangiopatía y escotomas centrales en estudio del campo visual.
Página | 13

Una tomografía de coherencia óptica (TCO) confirma la inflamación de la capa de
fibras nerviosas de la retina. También se observa discromatopsia rojo-verde en la
prueba de visión de color y pseudopapiledema durante la angiografía con
fluoresceína. En el test de Snellen, es normal una puntuación de 20/200 o inferior.
Puesto que la LHON se hereda por vía materna, las madres portadoras
pasarán la mutación a todos sus hijos, mientras que los padres portadores no lo
harán. La mutación puede detectarse mediante un análisis genético, pero esto no
implica que la enfermedad vaya a manifestarse.
10.1.5 Conclusiones del caso
La neuropatía óptica hereditaria de Leber es una de las neuropatías ópticas
hereditarias más frecuentes, que afecta predominantemente a adultos jóvenes. En
Cuba hasta hace alrededor de dos décadas, se comportaba como una enfermedad
extremadamente rara según la notificación de casos. El número de pacientes y de
familias confirmadas con estudios de ADNmt, actualmente sobrepasa el de 1/40 000
referido en los países de mayor índice. Esto sucedió en un período determinado
coincidente con la gran epidemia de neuropatía sufrida por los habitantes de Cuba
en la que se demostró que la población estaba sometida a grave estrés oxidativo.
Cada día se producen más avances en el campo de la genética, que permite
identificar un número mayor de mutaciones asociadas a la NOHL, lo que unido al
conocimiento de las características clínicas de la enfermedad ha permitido identificar
las familias afectadas y actuar sobre los factores de riesgo.
Página | 14

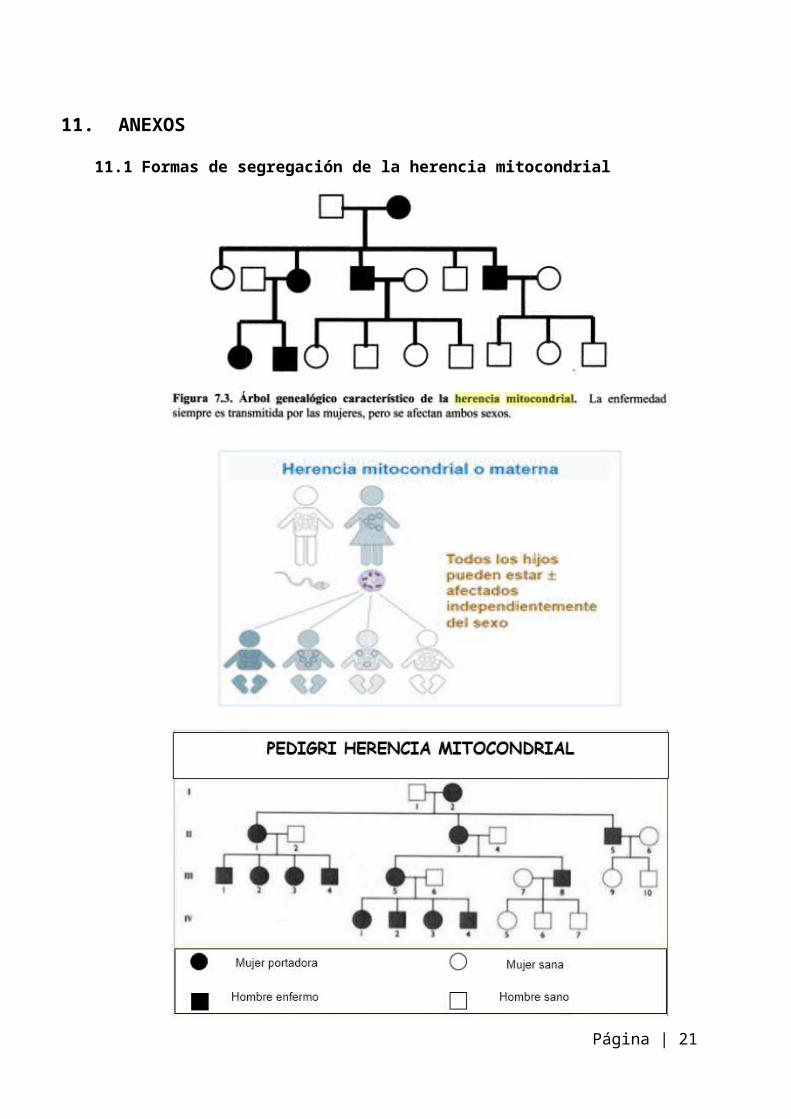
11. ANEXOS11.1 Formas de segregación de la herencia mitocondrial
Página | 15

Página | 16

12. BIBLIOGRAFÍA:
1. Solari Alberto Juan. Genética humana: fundamentos y aplicaciones en medicina
[en línea]. Buenos Aires. Medica Panamericana. [Fecha de acceso 20 julio de
2015]. URL disponible en: https://books.google.com.ec/books?id=e-
slX7S1KdsC&printsec=frontcover&hl=es&source=gbs_atb#v=onepage&q&f=false
2. Oliva Rafael. Genetica medica [en línea]. Barcelona. Ediciones Universitarias
Barcelona; 2004. [Fecha de acceso 20 julio de 2015]. URL disponible en:
https://books.google.com.ec/books?
id=9sCJ80bEsRsC&printsec=frontcover&hl=es&source=gbs_atb#v=onepage&q&f
=false
3. Gafo Javier, Abrisqueta Jose Antonio. Deficiencia mental y comienzo de la vida
humana [en línea]. Universidad Pontifica Comillas; 1999. [Fecha de acceso 20
julio de 2015]. URL disponible en: https://books.google.com.ec/books?
id=4ZLTqMhk27cC&printsec=frontcover&dq=jose+gafo&hl=es&sa=X&ved=0CCY
Q6AEwAmoVChMIpeaw37zwxgIVhKkeCh2bFgfL#v=onepage&q=jose
%20gafo&f=false
4. Perez Arellano Jose Luis. Manual de patología genera [en línea]. España.
Elsevier España; 2010. [Fecha de acceso 20 julio de 2015]. URL disponible en:
https://books.google.com.ec/books?id=HdOrVw-
0h0UC&printsec=frontcover&dq=manual+de+patologia+general&hl=es&sa=X&ve
d=0CBsQ6AEwAGoVChMIq5ni-L3wxgIVS6keCh08YQv5#v=onepage&q=manual
%20de%20patologia%20general&f=false
5. Álvarez Iglesias Vanessa. Estudio multidisciplinar de la variabilidad del ADN
mitocondrial en poblaciones humanas [en línea]. Universidad Santiago de
Compostela. [Fecha de acceso 20 julio de 2015]. URL disponible en:
https://books.google.es/books?
id=wdqlIslzHKMC&printsec=frontcover&dq=estudio+multidisciplinar&hl=es&sa=X
&ved=0CB4Q6AEwAGoVChMIh4jYt6jxxgIVC1oeCh2TUALA#v=onepage&q=estu
dio%20multidisciplinar&f=false
Página | 17

6. Passarge Eberhard. Genética texto y atlas [en línea]. Editorial Médica
Panamericana; 2009. [Fecha de acceso 20 julio de 2015]. URL disponible en:
https://books.google.es/books?
id=bgQ_xyJYkigC&printsec=frontcover&dq=passarge&hl=es&sa=X&ved=0CCEQ
6AEwAGoVChMI7ozBuanxxgIVSHYeCh1FBwo1#v=onepage&q=passarge&f=fals
e
7. Jimenez Adriano. Manual de Neurogenetica [en línea]. Ediciones Diaz de Santos;
2003. [Fecha de acceso 17 julio de 2015]. URL disponible en:
https://books.google.es/books?
id=E6XFouOOv38C&dq=manual+de+neurogenetica&hl=es&source=gbs_navlinks
_s
8. Michelli Federico. Tratado de neurología clínica [en línea]. Buenos Aires: editorial
Medica Panamericana, 2002. [Fecha de acceso 17 julio de 2015]. URL disponible
en: https://books.google.es/books?
id=E6XFouOOv38C&printsec=frontcover&dq=manual+de+neurogenetica&hl=es&
sa=X&ved=0CB4Q6AEwAGoVChMInba-
pqrxxgIVBiweCh19Tw6p#v=onepage&q=manual%20de
%20neurogenetica&f=false
9. Lisker Ruben. Introducción a la genética humana [en línea]. Mexico. Editorial el
manual moderno; 2013. [Fecha de acceso 17 julio de 2015]. URL disponible en:
https://books.google.es/books?
id=sfEWCQAAQBAJ&pg=PT25&dq=introduccion+a+la+genetica+humana&hl=es
&sa=X&ved=0CB4Q6AEwAGoVChMI5Yn7uq3xxgIVRiweCh17HwLB#v=onepage
&q=introduccion%20a%20la%20genetica%20humana&f=false
10.Romeo Casabona Carlos María. Genoma humano; fundamentos para el estudio
de los efectos sociales de las investigaciones sobre el genoma humano. 1ra
edición. Universidad de Deusto. [Fecha de acceso 17 julio de 2015].
11.Bioquímica. Voet Donald, Voet Judith G. Editorial Ed. Médica Panamericana,
2006. [Fecha de acceso 17 julio de 2015].
12.Chinnery PF, Howell N, R Andrews, Turnbull DM: la genética mitocondrial
clínicos. J Med Genet 36: 425-436, 2005. [Fecha de acceso 17 julio de 2015].
Página | 18

13.Gilbert-Barness E, Barness L: Enfermedades metabólicas. Fundamentos de la
Gestión clínica, Genética y Patología. Eaton Publishing. Natrick, Massachusetts,
2006. [Fecha de acceso 17 julio de 2015].
14.Harper PS: Consejo Genético práctica. 6ta edicion Edward Arnold, London, 2007.
[Fecha de acceso 17 julio de 2015].
15.Wallace DC: Las enfermedades mitocondriales en el hombre y los ratones.
Ciencia 283: 1482-1488, 2007.
16.Wallace DC et al: mitocondrial y enfermedades neuro-oftalmológica. En El
metabólica y bases moleculares de la enfermedad hereditaria. 8va edición.
McGraw-Hill, Nueva York, 2011. [Fecha de acceso 17 julio de 2015].
17.Wallace DC, Lott MT: genes mitocondriales en las enfermedades degenerativas,
el cáncer y el envejecimiento. En: Emery y Principios de Rimoin y Práctica de
Genética Médica. Edición 4ta. DL Rimoin et al. Ediciones Churchill-Livingstone,
Edimburgo, 2012. [Fecha de acceso 17 julio de 2015].
18.Lewin B. Genes VIII, Nueva Jersey. Prentice Hall, 2010. [Fecha de acceso 17
julio de 2015].
19.Brown TA. Genomas. Oxford: Wiley. 2012. [Fecha de acceso 17 julio de 2015].
20.Griffiths AF. Wessler SR. Lewontin RC, Gelbart WM, introducción al análisis
genético. Edicion 8va. Nueva York, 2005. [Fecha de acceso 17 julio de 2015].
21.Strachan T, Lee AP. genética molecular humana. 3ra edicion Oxford, 2012.
[Fecha de acceso 17 julio de 2015].
22.Anderson. secuencia y organización del genoma mitocondrial humano.
Naturaleza, 2010. [Fecha de acceso 17 julio de 2015].
23.Andrews RM, Hewell N, reanálisis y revisión de la secuencia de referencia de
Cambridge para mitocondrial humano ADN, 2009. [Fecha de acceso 17 julio de
2015].
24.Chinnery, DM Turnbull. genética mitocondrial clínicos, 2008. [Fecha de acceso 17
julio de 2015].
Página | 19

25.DiMauro, Davidzon, mitocondrial ADN, Genet, 2008. [Fecha de acceso 17 julio de
2015].
26.MITOMAP. Mitocondrial accedido base de datos del genoma humano, 2009.
[Fecha de acceso 17 julio de 2015].
27.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of
optic neuropathies. Prog Retin Eye Res. 2004 [citado 15 jul 2015];23(1).
Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/14766317
28.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, et al.
Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy.
Science. 1988 [citado 15 jul 2015];242(4884). Disponible
en:http://www.sciencemag.org/cgi/content/abstract/sci;242/4884/1427
29.Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML. A new mtDNA
mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum
Genet. 1991 Jun [citado 15 jul 2015];48(6). Disponible
en:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1683111/
30.Johns DR, Neufeld MJ, Park RD. An ND-6 mitochondrial DNA mutation
associated with Leber hereditary optic neuropathy. Biochem Biophys Res
Commun. 1992 [citado 15 jul 2015];187(3). Disponible
en:http://linkinghub.elsevier.com/retrieve/pii/0006291X92904795
31.Howell N, McCullough D, Bodis-Wollner I. Molecular genetic analysis of a
sporadic case of Leber hereditary optic neuropathy. Am J Hum Genet. 1992
[citado 15 jul 2015];50(2). Disponible
en:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1682448/
32.Sadun AA, Carelli V, Salomao SR, Berezovsky A, Quiros PA, Sadun F, et al.
Extensive investigation of a large Brazilian pedigree of 11778/haplogroup Leber
hereditary optic neuropathy. Am J Ophthalmol. 2003 [citado 15 jul 2015];136(2).
Disponible en: http://linkinghub.elsevier.com/retrieve/pii/S0002939403000990
33.Santiesteban-Freixas R, Rodriguez-Hernandez M, Mendoza-Santiesteban CE,
Carrero-Salgado M, Francisco-Plasencia M, Mendez-Larramendi I, et al.
Manifestaciones clínicas e identificación molecular de pacientes con Neuropatía
Página | 20
Óptica Hereditaria de Leber en centro de referencia nacional para la
neuroftalmología en Cuba. Rev Neurol. 1999;29(5):408-15.
34.Hudson G, Carelli V, Horvath R, Zeviani M, Smeets HJ, Chinnery PF. X-
inactivation patterns in females harbouring mtDNA mutations that cause Leber
hereditary optic neuropathy. Molecular Vision. 2007 [citado 15 jul 2015];13.
Disponible en: http://www.molvis.org/molvis/v13/a265/
35.Yen MY, Wang AG, Wei YH. Leber's Hereditary Optic Neuropathy: a Multifactorial
Disease. Prog Retin Eye Res. 2006 [citado 15 jul 2015];25(4):381-96. Disponible
en:http://linkinghub.elsevier.com/retrieve/pii/S1350946206000152
36.Carelli V, Ross-Cisneros FN, Sadun AA. Optic Nerve Degeneration and
Mitochondrial Dysfunction: Genetic and Acquired Optic Neuropathies. Neurochem
Int. 2002 [citado 15 jul 2015];40(6). Disponible
en:http://www.ncbi.nlm.nih.gov/pubmed/11850115
37.Carvajal Cuenca A, Fernández Morales H, Heyden Cordero M. Reporte de la
primera familia costarricense con neuropatía Optica Hereditaria de Leber y una
revisión del tema. Neuroeje. 2006 [citado 15 jul 2015];20(1). Disponible
en: http://www.binasss.sa.cr/revistas/neuroeje/20n1/art4.pdf
38.Man PYW, Turnbull DM, Chinnery PF. Leber Hereditary Optic Neuropathy. J Med
Genet. 2002 [citado 15 jul 2015];39. Disponible
en: http://www.ncbi.nlm.nih.gov/pmc/articles /PMC1735056/pdf/v039p00162.pdf
39.Ghelli A, Porcelli AM, Zanna C, Martinuzzi A, Carelli V, Rugolo M. Protection
against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary
optic neuropathy cybrids. Invest Ophthalmol Vis Sci. 2008 [citado 15 jul
2015];49(2):6716. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/18235013
40.Floreari M. Antioxidant defenses in cybrids harbouring mtDNA mutations
associated with Leber´s hereditary optic neuropathy. FEBS J. 2005 [citado 15 jul
2015];3(272):1124-35. Disponible
en:http://www.bentham.org/open/tobioj/articles/V003/53TOBIOJ.pdf
41.Genasetti A, Valentino ML, Carelli V, Vigetti D, Viola M, Karousou EG, et al.
Assessing heteroplasmic load in Leber´s hereditary optic neuropathy mutation
Página | 21

3460G A/MT-ND1 with A real-time PCR quantitative approach. J Mol Diagn. 2007
[citado 15 jul 2015]. Disponible
en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1975109/
Página | 22