hpcs2015 大規模量子化学計算プログラムsmashの開発と公開(石村)
TRANSCRIPT

大規模並列量子化学計算プログラムSMASHの開発と公開
石村 和也分子科学研究所
計算分子科学研究拠点(TCCI)
HPCS20152015年5月19-20日

SMASHプログラム
• 大規模並列量子化学計算プログラムSMASH (Scalable Molecular Analysis Solver for High performance computing systems)
• http://smash-qc.sourceforge.net/• Apache 2.0 オープンソースライセンス
• 2014年9月1日公開
• 対象マシン:スカラ型 (PCクラスタから京コンピュータまで)
• 言語はFortran90/95
• 超並列化(ハイブリッド並列)向け、モジュール化されたスッキリとした実装でポスト京での開発の基盤として利用可能
• 化学の実用に特化
– エネルギー微分、構造最適化計算を重点的に整備
– 現時点で、Hartree-Fock, DFT(B3LYP), MP2計算が可能

元素と分子
• 原子の種類(元素)は110程度• 現在9000万以上の分子が確認されている (例:H2O, CH4)
• 原子間の結合は電子が担っている1 2
H He3 4 5 6 7 8 9 10
Li Be B C N O F Ne11 12 13 14 15 16 17 18
Na Mg Al Si P S Cl Ar19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54
Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe55 56 *1 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86
Cs Ba Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn87 88 *2 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118
Fr Ra Rf Db Sg Bh Hs Mt Ds Rg Cn Uut Uuq Uup Uuh Uus Uuo
*1 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71
Lanthanoid La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu*2 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103
Actinoid Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr
周期表

• 電子構造計算:分子の電子分布とエネルギーを計算し、分子の反応性、物性などを求める
• 構造最適化計算:エネルギー微分値から分子の構造を計算する
量子化学計算
原子軌道 (C,H)分子構造
Hartree-Fock計算
H
H
H
H H
H
分子軌道 (原子軌道の線形結合, ベンゼン(C6H6))
エネ
ルギ
ー
A+B
C+D
化学反応 A+B → C+D
反応エネルギー
活性化エネルギー
Cl
Cl
ClCl
室温で白色の固体衣類用防虫剤
室温で無色の液体殺虫剤、農薬の原料
座標
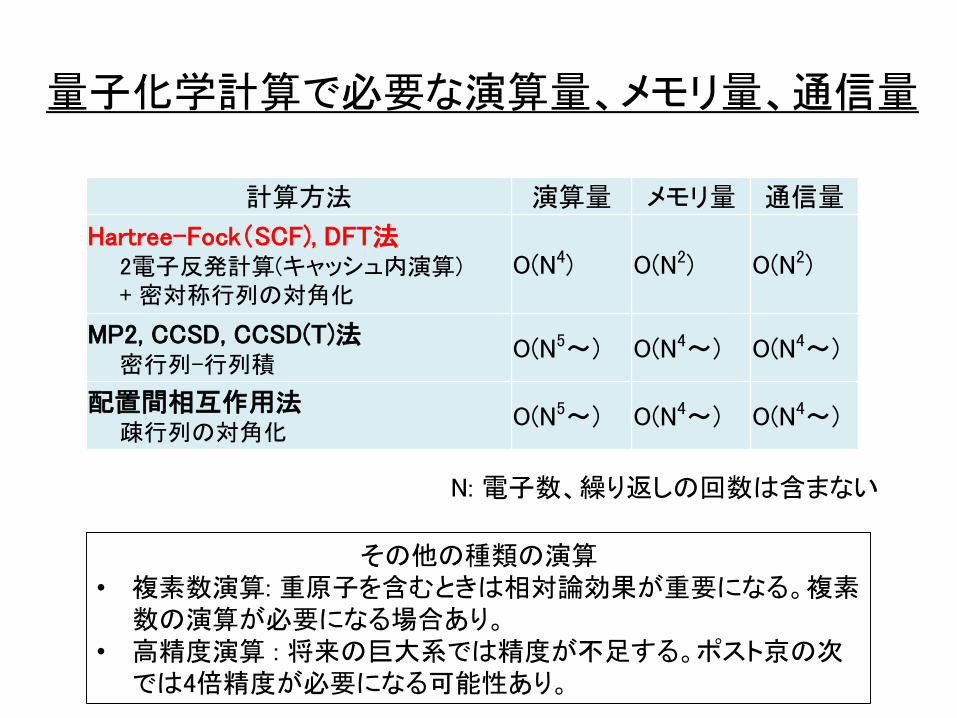
量子化学計算で必要な演算量、メモリ量、通信量
計算方法 演算量 メモリ量 通信量
Hartree-Fock(SCF), DFT法2電子反発計算(キャッシュ内演算)+ 密対称行列の対角化
O(N4) O(N2) O(N2)
MP2, CCSD, CCSD(T)法密行列-行列積
O(N5~) O(N4~) O(N4~)
配置間相互作用法疎行列の対角化
O(N5~) O(N4~) O(N4~)
N: 電子数、繰り返しの回数は含まない
その他の種類の演算• 複素数演算: 重原子を含むときは相対論効果が重要になる。複素
数の演算が必要になる場合あり。• 高精度演算 : 将来の巨大系では精度が不足する。ポスト京の次
では4倍精度が必要になる可能性あり。

,,
||22i
ii
XC CCdVHF
DFTエネルギー計算
2電子反発積分
εSCFC
DFT計算
Hamiltonian行列
F: Hamiltonian行列, C: 分子軌道係数S: 基底重なり行列, e: 分子軌道エネルギー
𝜇𝜈|𝜆𝜎 = 𝑑𝒓1 𝑑𝒓2𝜙𝜇 𝒓1 𝜙𝜈 𝒓11
𝑟12𝜙𝜆 𝒓2 𝜙𝜎 𝒓2
𝜙𝜇 𝒓1 : 原子軌道Gauss関数
• 対角化したい行列Fに固有ベクトルCが含まれているため、繰り返し法で解く
• 計算内でSを消去し、実際はF’C’=eC’の固有値問題を解く• 行列の次元は電子数に比例
• 系の情報がすべて入っている(原子の電子分布、原子核座標など)• 電子分布をGauss関数で扱うため、2電子反発積分項の計算が複雑
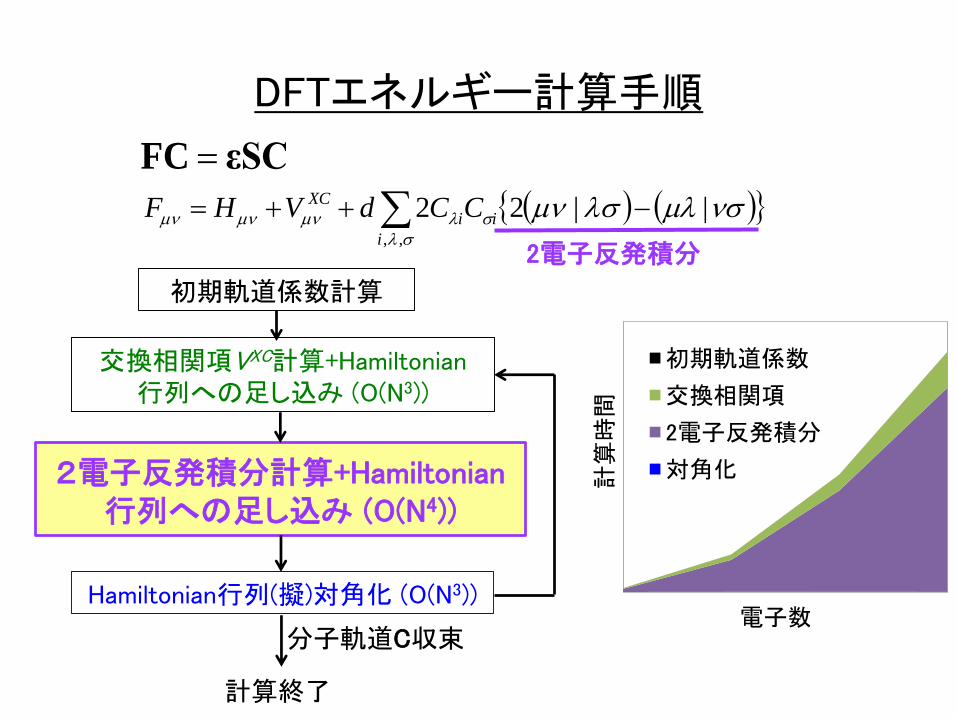
,,
||22i
ii
XC CCdVHF
DFTエネルギー計算手順
2電子反発積分
初期軌道係数計算
2電子反発積分計算+Hamiltonian行列への足し込み (O(N4))
Hamiltonian行列(擬)対角化 (O(N3))
計算終了
分子軌道C収束
交換相関項VXC計算+Hamiltonian行列への足し込み (O(N3))
εSCFC
初期軌道係数
交換相関項
2電子反発積分
対角化
電子数計
算時
間

,,,,,,
|222
aDDDDd
a
SW
a
V
a
hD
a
E
i
XC
elec
DFT構造最適化計算手順
エネルギー計算
2電子反発積分微分計算(O(N4))
計算終了
エネルギー1次微分値が閾値以下
交換相関項VXC微分計算(O(N3))
i
iiCCD C: 分子軌道係数 (固有ベクトル)e: 分子軌道エネルギー(固有値)
新規構造計算(O(N2))
i
iii CCW e
1電子積分項微分計算(O(N2))エネルギー計算
1電子積分項
交換相関項
2電子反発積分項
電子数計
算時
間

2電子反発積分計算分散アルゴリズム
!$OMP parallel do schedule(dynamic,1) reduction(+:Fock)do n, 1, -1 <----- OpenMPによる振り分け
do =1, =*(+1)/2+start=mod(+mpi_rank,nproc)+1do =start, ,nproc <----- MPIランクによる振り分け
do =1, 2電子反発積分|計算+Hamiltonian行列に足し込み
enddoenddo
enddoenddocall mpi_allreduce(Fock)
• OpenMPの分散を最外ループで行うことにより、スレッド生成などのオーバーヘッドを削減
• OpenMPの分散を動的に行い、コア間の均等な負荷分散を実現
• IF文を使わない振り分けでMPI分散コスト削減
K. Ishimura, K. Kuramoto, Y. Ikuta, S. Hyodo, J. Chem. Theory Comp. 2010, 6, 1075.初期軌道係数計算
2電子反発積分計算+Hamiltonian行列への足し込み (O(N4))
Hamiltonian行列(擬)対角化 (O(N3))
計算終了
分子軌道C収束
交換相関項VXC計算+Hamiltonian行列への足し込み (O(N3))

2電子反発積分計算ルーチンのインターフェイス
• 2電子反発積分ルーチン
– ほぼすべての量子化学計算で必要
– データはすべて引数で受け渡し
– 容易にこのルーチンのみ抜き出し可能
• call int2elec(twoeri, exijkl, coijkl, xyzijkl, nprimijkl, nangijkl, nbfijkl, maxdim, mxprsh, threshex)
twoeri 2電子反発積分(|)値 (Output)
exijkl primitive基底関数の指数 (Input)
coijkl primitive基底関数の係数
xyzijkl xyz座標
nprimijkl primitive基底関数の数
nangijkl 軌道角運動量(s=0, p=1, d=2,...)
nbfijkl 基底関数の数(s=1, p=3, d=5or6,...)
maxdim 最大twoeriの次元数
mxprsh 最大primitive基底関数の数
threshex exp(-x2)計算のxの閾値

プログラム開発の効率化
• 2電子反発積分ルーチンはHartree-Fock,DFT計算以外の高精度電子相関計算でも利用
• 2電子反発積分の微分は角運動量の異なる2電子反発積分の線形結合になるため、2電子積分ルーチンを再利用可能
• 適切な係数と角運動量を引数で渡し、2電子反発積分ルーチンをcallした後適切な要素へ結果を足しこむだけで実装完了
call int2elec(twoeri, exijkl, coijkl, xyzijkl, nprimijkl, nangijkl, nbfijkl, maxdim,
mxprsh, threshex)
例:(𝒑𝒙𝑠|𝑠𝑠)の微分計算の場合
𝜕 𝑝𝑥𝑠 𝑠𝑠 /𝜕𝑋𝑎 = 𝟐𝜶𝒂 𝒅𝒙𝒙𝑠 𝑠𝑠 − (𝒔𝑠|𝑠𝑠)
𝜕 𝑝𝑥𝑠 𝑠𝑠 /𝜕𝑌𝑎 = 𝟐𝜶𝒂 𝒅𝒙𝒚𝑠 𝑠𝑠
𝜕(𝑝𝑥𝑠|𝑠𝑠)/𝜕𝑍𝑎 = 𝟐𝜶𝒂(𝒅𝒙𝒛𝑠|𝑠𝑠)

0%
20%
40%
60%
80%
100%
6188 12288 24576 49152 98304
その他
初期軌道
(擬)対角化
交換相関項
2電子反発積分
CPUコア数
LAPACK
DFTエネルギー並列計算性能• 10万コアで154秒、5万倍のスピードアップ、実行性能13%• 行列対角化3回分(LAPACK,分割統治法)の時間は35.3秒
計算機 : 京コンピュータ分子 : (C150H30)2 (360原子)基底関数 : cc-pVDZ (4500基底)計算方法 : B3LYPSCFサイクル数 : 16
0
24576
49152
73728
98304
0 24576 49152 73728 98304
2電子反発積分
相関交換項
Total
CPUコア数
並列加速率 各ステップの割合

DFTエネルギー微分計算性能
計算機 : 京コンピュータ分子 : C150H30 (180原子)基底関数 : cc-pVDZ (2250基底)計算方法 : B3LYP
• エネルギー計算と異なり対角化計算が無いため、並列化効率はほぼ100%
0.0
4096.0
8192.0
12288.0
16384.0
0 4096 8192 12288 16384
Total
CPUコア数
並列加速率 各ステップの割合
0%
20%
40%
60%
80%
100%
1024 4096 8192 16384
1電子積分
交換相関項
2電子反発積分
CPUコア数
エネルギー微分計算

まとめ
• MPI/OpenMPハイブリッド並列量子化学計算プログラムSMASHをApache 2.0ライセンスで公開した
• 360原子系のDFTエネルギー計算は10万CPUコアで2分半、並列加速率は5万倍であった
• エネルギー計算でO(N4)の2電子反発積分の並列加速率は10万コアで9万倍以上であるが、 O(N3)の行列対角化は現時点ではノード内並列のみのため、ノード数が増加すると計算全体の並列化効率は低下している
• 180原子系のDFTエネルギー微分計算は16,300CPUコアで25秒、並列加速率は16,100倍であった
• モジュール化されたスッキリとした実装であり、今後の効率的なプログラム開発の基盤を作成した
• エネルギー計算のさらなる並列化効率向上のためには、高性能固有値ソルバの導入が重要である