review of disease modifying anti rheumatic · pdf filereview of disease‐modifying anti...
TRANSCRIPT

1
18th Expert Committee on the Selection and Use of Essential Medicines
(21 to 25 March 2011)
Section 2 Analgesics, antipyretics, NSAIMs, DMARDs
2.4 Disease-modifying agents used in rheumatoid disorders
Review of Disease‐Modifying Anti Rheumatic Drugs in Paediatric Rheumatic disease
September 2010 Prepared by: Peter Gowdie Rheumatology and Clinical Pharmacology Fellow 2009 Royal Children’s Hospital Melbourne, Australia

2
Contents 1. Intent of review 2. Identification of priority conditions 3. Review of priority rheumatic disease
1. Juvenile Idiopathic arthritis 1. Epidemiology 2. Disease burden and outcome 3. Clinical manifestations 4. Complications
• macrophage activation syndrome • uveitis • amyloidosis
2. Idiopathic Inflammatory Myopathies (Juvenile Dermatomyositis) 1. Epidemiology 2. Clinical Manifestations 3. Complications 4. Course and Outcome 5. Overview of management
3. Systemic Lupus Erythematosus 1. Epidemiology 2. Clinical Manifestations 3. Course and outcome 4. Overview of management
4. DMARDs
1. Methotrexate 1. Mechanism of action and Pharmacology 2. Efficacy in Juvenile Idiopathic Arthritis 3. Efficacy in Juvenile Dermatomyositis 4. Dose and administration 5. Drug interaction and folate supplementation 6. Safety 7. Monitoring and supervision 8. Formulary 9. Summary recommendations
2. Leflunomide 1. Mechanism of action and Pharmacology 2. Efficacy in Juvenile Idiopathic Arthritis 3. Dose and administration 4. Safety 5. Drug interaction 6. Monitoring and supervision 7. Formulary 8. Summary recommendations

3
3. Sulphasalazine 1. Mechanism of action and Pharmacology 2. Efficacy in Juvenile Idiopathic Arthritis 3. Dose and administration 4. Safety 5. Drug interaction 6. Monitoring and supervision 7. Formulary 8. Summary recommendations
4. Cyclosporin 1. Mechanism of action and Pharmacology 2. Efficacy in Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome
3. Efficacy in Juvenile Dermatomyositis 4. Dose and administration 5. Safety 6. Drug interaction 7. Monitoring and supervision 8. Formulary 9. Summary recommendations
5. Azathioprine 1. Mechanism of action and Pharmacology 2. Efficacy in SLE 3. Efficacy in JIA 4. Dose and administration 5. Safety 6. Drug interaction 7. Monitoring and supervision 8. Formulary 9. Summary recommendations
6. Hydroxychloroquine 1. Mechanism of action and Pharmacology 2. Efficacy in SLE 3. Dose and administration 4. Safety 5. Drug interaction 6. Monitoring and supervision 7. Formulary 8. Summary recommendations

4
1. Intent of review
• To identify priority rheumatological conditions in children • To outline the treatment options for these conditions • To outline the role of DMARDs in the treatment of priority rheumatological
conditions • To review the literature and collate the evidence for the efficacy of DMARDs in
priority conditions • To review the safety of DMARDs and outline monitoring and supervision required • To give recommendations for the inclusion of DMARDs on the WHO Essential
Medicines List 2. Identification of priority conditions Paediatric Rheumatology encompasses a broad range of inflammatory disorders involving the joints and connective tissues in children. Juvenile idiopathic arthritis is perhaps the most well recognised of the rheumatic diseases of childhood however the specialty’s scope includes conditions such as acute rheumatic fever, post‐streptococcal reactive arthritis, Kawasaki disease and Lyme disease as well as chronic systemic conditions including Systemic Lupus Erythematosus (SLE), Juvenile Dermatomyositis (JDM), and the vasculitides. The most common rheumatic disease affecting children is chronic arthritis. The functional impact of this disease can be significant and the timely administration of appropriate therapy, including DMARDs, can be effective in improving outcome. While less common, SLE and JDM are also are potentially devastating conditions and DMARD therapy plays an equally important role in their management. 3. Review of priority conditions 3.1 Juvenile Idiopathic Arthritis Chronic arthritis is a complex group of disorders comprising a number of clinical entities with the common feature of arthritis. Each type is characterised by a different mode of presentation and different disease course and outcome. The three main groups of chronic arthritis are: those affecting few joints (oligoarticular); those affecting many joints (polyarticular); and those systemic in onset. The classification of chronic arthritis has been problematic over the past few decades especially in terms of universally agreed upon definitions. This, in part, largely reflects the complex and heterogeneous nature of this group of conditions and the as yet not clearly defined immunogenetic factors contributing to their onset. It is also important to remember the population (mostly Caucasian) in which each of the major classification criteria have been described. For the purposes of this paper, the International League of Association for Rheumatology (ILAR) criteria for classification of juvenile idiopathic arthritis (JIA) has been used.[1]

5
3.1.1 Epidemiology JIA remains an uncommon but by no means rare condition affecting children worldwide. [2] Estimates of incidence and prevalence however have been difficult to ascertain for many reasons including: variation in diagnostic criteria; differences in ascertainment (community vs clinical based studies); differences in study design; low frequency of disease and small study numbers. [3] In 2002 Manners and Bower summarised the most recent epidemiological studies to that point and found a reported prevalence of 0.07 to 4.01 per 1000 children and an annual incidence of 0.008 to 0.226 per 1000 children. [3] The large difference in reported prevalence is considered to be largely due to varying study characteristics. The highest prevalence was reported in community based studies where children were examined in classrooms or homes. In 1993, Meilants performed an epidemiological study using a questionnaire followed by clinical examination and found a prevalence of definite JIA of 1.67 per 1000.[4] In 1996 Manners et al found a significantly higher prevalence (4.01 per 1000) in an urban Australian community.[5] Similarly, Tayel et al found a prevalence of JIA amongst 10‐15 year old school children in Alexandria, Egypt, of 3.3 per 1000.[6] Clinic based studies on the other hand appear to report lower prevalence rates ‐ perhaps reflecting that many clinicians fail to recognise JIA and therefore these children do not make their way to medical care in large study centres, therefore underestimating the true prevalence.[7] There is very little comparable data outlining the prevalence of JIA in populations other than those of European descent. In fact, in the most heavily populated areas of the world epidemiological data is very scarce. Outside of the US, UK and Canada some studies reveal very different data. Lower frequencies of JIA have been reported in children in Japan (annual incidence 0.0083 per 1000) and Costa Rica (annual incidence 0.068 per 10000 Arguedes 1998 and 0.054 per 1000 Arguedes 1995). In 1983 Hochberg reported an annual incidence of 0.066 per 1000 children and a prevalence of 0.26 per 1000 in urban black children in the USA however other studies suggest that this is an underestimate. [8] Many of these studies are ultimately limited by the small sample size and selection bias. One retrospective study undertaken by Kurahara et al in 2002 demonstrated lower frequency of JIA in Hawaiians of Filipino, Japanese and Samoan descent compared with Caucasians.[9] In a similar population Kurahara also found increased prevalence of JIA in rural areas compared with urban areas.[10] The extent of juvenile arthritis in the developing world and the epidemiological impact of ethnicity and geography needs further consideration especially in considering the impact and burden of disease of this group of conditions. 3.1.2 Burden of Disease and outcome The disease outcome and prognosis in JIA is variable and to some degree predictable based on the different disease subtypes. For example, sero‐positive polyarticular JIA has a relatively poor outcome compared with oligoarticular JIA whose outcome is generally very good. [11, 12] However, the latter group are also the most at risk of debilitating uveitis as a complication of their disease. Many children with JIA have an excellent prognosis and for the

6
most part remain free from significantly debilitating disease. However, 50‐70% of patients with systemic and polyarticular disease and 40‐50% of oligoarticular arthritis continue into adulthood with active disease [Laxer/Hashkes]. It has been estimated that up to 20% of children transition to adulthood with moderate to severe functional disabilities [13] and an even higher percentage (30‐40%) have significant long‐term disabilities including unemployment.[14] Major surgery, including joint replacement is required in 25‐50%. [14] Delay in referral and initiation of acceptable therapy is associated with poorer outcome. [2] The prognosis in JIA is frequently assessed with three main outcome measures: frequency of remission, functional impairment and structural damage. Similar to the outlining of epidemiology, outcome studies are also difficult to draw conclusions especially given the prevailing use of three different classification systems and the variable outcome measures without agreed upon definitions. For example, there remains no international agreement on validated remission criteria for JIA Remission rates vary between subgroups however, despite differing approaches and definitions across studies, there is consistent reporting of higher remission rates in oligoarticular disease. In the oligoarticular disease this ranged from 36% to 84%. [15] Remission rates for polyarticular disease ranged from 12.5% to 65% whilst systemic disease was 0 to 76%. [15] The large range can be explained by the relative infrequency of these subtypes compared with oligoarticular disease and therefore the small sample sizes potentially more influenced by chance. Similarly, functional outcome was better in the oligoarticular subtype and the frequency of severe disability was low. By contrast, systemic and polyarticular disease both have been documented to have significantly worse functional outcome. Systemic JIA in particular has a large proportion of patients with severe disability. [16] Whilst estimates of the burden of disease at an individual level has been documented to some extent, there remains considerable difficulty in acquiring data relating to estimates of the global burden of this group of diseases. The global impact of juvenile arthritis on disability and handicap, the educational and vocational disadvantages, life expectancy and quality of life as well as the cost of medical care remains to be defined. 3.1.3 Clinical Manifestations of JIA The common feature of all the subtypes of JIA is arthritis. Systemic symptoms typically occur in systemic and polyarticular subtypes and include: fatigue, loss of weight, anaemia, anorexia and fever. Joint inflammation results in pain and discomfort and at times considerable morning stiffness. Large joints are the most frequently affected, however any joint can be involved including cervical spine, thoraco‐lumbar spine and temporo‐mandibular joint. Growth abnormalities are not uncommon and can result in short stature or localised growth disturbance such as bony overgrowth, prematurely fused epiphyses and limb length discrepancies. Other extra‐articular manifestations include: osteopenia, rheumatoid nodules and muscle atrophy. Cardiopulmonary disease is also not uncommon particularly in systemic onset disease. Pericarditis occurs in up to 9% [17] however tamponade is rare. Myocarditis and endocarditis have also been documented to have occurred.

7
3.1.4 Complications of JIA Macrophage Activation Syndrome Macrophage Activation Syndrome (MAS) is potentially the most devastating sequelae of JIA. MAS is similar in many ways to reactive haemophagocytic lymphohistiocytosis (HLH) and the term MAS has come to refer to HLH secondary to rheumatic disease. MAS is most often associated with systemic onset JIA, however, has been recognised with other rheumatic diseases such as: polyarticular JIA, SLE, JDM and chronic infantile neurologic cutaneous and articular syndrome (CINCA). MAS contributes to a significant amount of the morbidity and mortality associated with SoJIA. Estimates of the incidence of MAS in SoJIA vary. Sawhney et al report an incidence of 6.7%.[18] However, it is believed by many that MAS is integral to the pathogenesis of SoJIA and that in fact occult MAS is common in patients with SoJIA.[19] MAS is often difficult to distinguish clinically from SoJIA. Its features include: fever (often more sustained that SoJIA), hepatosplenomegaly, anaemia, LFT abnormalities, rash, coagulopathy and central nervous system dysfunction. Laboratory markers suggestive of diagnosis include: decreasing white cell count and platelets, elevated ferritin, hypertriglyceridemia, hypofibrinogenemia and evidence of haemophaocytosis on bone marrow aspirate. Preliminary guidelines for the diagnosis of MAS in association with SoJIA have been suggested. [20] MAS is potentially fatal especially in those patients with multisystem involvement or when diagnosis is delayed. Early diagnosis and vigorous treatment is necessary. Corticosteroids and supportive care are the first line therapies, however, agents such as cyclosporin, etoposide and IVIG . The use of cyclosporin in MAS is discussed in section 4.4.2. Uveitis One of the most important complications of JIA is uveitis. This is a chronic non‐granulomatous inflammation affecting the iris and ciliary body the end result of which can be devastating. In particular, band keratopathy and cataracts occur in 42‐58% whilst glaucoma occurs in 19‐22%.[21] Its activity does not correlate with the course of the arthritis and its own course is usually insidious and often asymptomatic mandating frequent ophthalmological surveillance. Rates of uveitis vary between subtypes of arthritis with the most frequent at‐risk group being oligoarticular JIA. Uveitis has been described in most racial groups. JIA is the most frequent non‐infectious systemic disease associated with uveitis in children. Other less common non‐infectious causes include: ulcerative colitis, tubulo‐interstitial nephritis syndrome, Behcet’s disease and chronic infantile neurologic cutaneous and articular syndrome (CINCA). A high proportion of paediatric patients with uveitis do not have an underlying cause found. Kump et al found no associated systemic disease in 58% of patients with uveitis.[22]

8
The management of uveitis should be supervised by an ophthalmologist in conjucttion with a paediatrician or pediatric rheumatologist. Uveitis is usually managed with topical corticosteroids with a mydriatic agent. Systemic prednisolone administered orally or intravenously may be required in an attempt to achieve short term relief of inflammation. In children in whom uveitis is difficult to control by these measures, additional immunosuppressive agents have been used including increasing use of biological agents. The efficacy of methotrexate is discussed in section 4.1.4. Amyloidosis Amyloidosis is the tissue deposition of the protein amyloid and can occur as a complication of JIA. It is a rare complication in North America but occurs more frequently in parts of Europe. It manifests as proteinuria, nephritic syndrome, hepatosplenomegaly or anaemia and can eventually lead to renal failure. Amyloidosis and resultant renal failure as a complication of JIA was, in the past, the major cause of death. The other main cause is infection. Death rates have improved over recent decades and now the disease associated death rate in Europe is <1% and less than 0.3% in North America. [2] 3.2 Idiopathic inflammatory myopathies (Juvenile Dermatomyositis and Juvenile Polymyositis) Idiopathic inflammatory myopathies (IIM) of childhood are rare conditions often associated with significant disability. Of the inflammatory myopathies, Juvenile dermatomyositis is by far the most common in children making up 85% of cases of IIM in childhood. [23, 24] Juvenile polymyositis accounts for approximately 2‐8% and overlap syndromes with features of other autoimmune diseases accounts for approximately 3‐10%. [25] The other types of IIMs are rare in children and mostly documented in case reports only. Juvenile dermatomyositis is a systemic vasculopathy characterised histologically by perivascular B‐cell predominant inflammation and capillary loss. The pathogenesis is not completely understood but it is thought to be an autoimmune process occurring in a genetically susceptible child in response to an environmental stimuli. 3.2.1 Epidemiology Juvenile dermatomyositis is a rare autoimmune myositis of childhood. It has an estimated incidence in the United States of 3.2 per million children. [26] Mendez et al captured data over a 4 year period between 1995 and 1998 and found the annual incidence ranged between 2.5 and 4.1 per million children. Incidence rates varied slightly between racial groups ‐ 3.4 for white Caucasian, 3.3 for African‐American and 2.7 for Hispanics. [26] Symmons et al estimated an incidence in the UK and Ireland of 1.9 per million children (95% confidence intervals 1.4‐2.6). [27] The ratio of girls to boys was reported in these two studies to be 2.3:1 by Mendez et al and 5:1 by Symmons et al. [26, 27] The age of peak incidence in boys and girls is similar. Pachman et al demonstrated a bimodal peak incidence in both boys (6 and 11 years) and girls (6 and 12 years). [28]

9
3.2.2 Clinical Manifestations Juvenile dermatomyositis (JDM) is a multisystem inflammatory condition ‐ it can affect the muscle, skin, gastrointestinal tract, heart, lungs, kidneys and eyes. JDM can manifest in a heterogeneous fashion, however, the main characteristics of the disease include: symmetrical proximal muscle weakness, pathognomic skin involvement (eg Gottron’s papules, heliotropic rash), and raised muscle enzymes. The diagnosis of JDM is made through the application of diagnostic criteria established by Bohan and Peter in 1975: 1) presence of characteristic rash ‐ heliotropic rash with periorbital oedema and Gottron’s papules; 2) symmetrical proximal muscle weakness; 3) raised serum muscle enzymes (at least one of CK, LDH, AST); 4) myopathic EMG; 5) muscle biopsy demonstrating necrosis and inflammation.[29] Patients with a rash and two other of the criteria are considered to have possible JDM, while rash with three additional criteria are considered to have definite JDM. In practice, muscle biopsy and EMG are used less frequently today than they once were. They are both invasive investigations and MRI is now considered a sensitive test for myositis. The clinical course and progression of the disease is also varied. Most commonly JDM presents with an insidious evolution over a period of 3 to 6 months, however it can present acutely. Ramanan et al reviewed the clinical features and outcomes of a large case series of patients treated at the Hospital for Sick Children in Canada. [23] Of 120 patients with IIM, 105 had JDM. The most common clinical features at presentation were: Gottronʹs rash (91%), heliotrope rash (83%), malar/facial rash (42%), nailfold capillary change (80%), myalgia/arthralgia (25%), dysphonia or dysphagia (24%), anorexia (18%), fever (16%).[23] Muscle weakness is progressive and frequently results in the inability to ambulate, sit upright or attend to activities of daily living. Weakness may be accompanied by significant pain. The pharyngeal and palatal muscles may also be involved resulting in difficulty swallowing and dysphonia. This occurred in up to 24% of patients in Ramanan’s series [23] and puts the patient at risk of aspiration 3.2.3 Complications Calcinosis occurs in up to 40% of patients[30] and can result in significant disability. Deposits in the subcutaneous tissues can be painful and lead to ulceration. Early and aggressive control of inflammation may minimise the degree of calcinosis.[31] Cutaneous ulcerative disease is another skin complication which is not infrequently seen. It is thought to be associated with more severe and prolonged JDM. [32] Lipodystrophy has also been described with JDM. Cardiopulmonary abnormalities are also described however clinically significant involvement in children with dermatomyositis is unusual. The most frequent cardiac problems include cardiomegaly and nonspecific murmurs. More serious cardiac involvement is unusual but potentially life threatening. Restrictive pulmonary disease due to poor chest wall compliance and respiratory muscle weakness is common and has been reported in up to 78% patients. [33] Gastrointestinal vasculitis is rare but is a severe complication which can lead to death. It manifests as abdominal pain, haematemesis and melaena.

10
3.2.4 Course and Outcome of JDM JDM typically follows a uniphasic course although the duration is quite variable. Stringer et al followed the course of 84 patients with JDM. They found that the majority (60%) of patients had a chronic disease course defined as no remission within 3 years of diagnosis. [34] They also found that a small percentage of patients had a relapse following remission. The outlook for patients with JDM has significantly improved over the past decades with the use of more aggressive immunosuppressant therapies. Before the use of corticosteroids, this illness was devastating. Many patients died or suffered long‐term progressive and disabling disease. Functional outcome today is usually excellent with 65‐80% of patients achieving a good outcome [2] Optimal outcome seems to be achieved if diagnosis is made shortly after onset and treatment is vigorous. [35] Huber et al found that of 65 children with JDM, a favourable outcome was predominant. Eight percent were left with moderate to severe disability and there was one death. [36] Contractures and mild atrophy occur in approximately 25% of patients[2] however in one study cohort none of the patients were thought to be educationally or vocationally impaired due to their illness. [23] Although deaths are now less frequent, chronically active disease occurs in substantial number of patients and, in addition, side effects such as growth failure from prolonged steroid use are not infrequent. 3.2.5 Overview of Management Corticosteroids and general supportive care in a multidisciplinary team setting are the mainstays of treatment of JDM. The use of corticosteroids has dramatically improved to outlook for patients with JDM. High suppressive doses are used early and then tapered gradually over one to two years. The degree of supportive care is dependent upon the degree of disability. In patients with bulbar dysfunction or significant respiratory muscle weakness, care needs to be directed to prevention of aspiration and ventilatory support. Physiotherapy and occupational therapy is advised to avoid loss of motion and contractures in the first instance and then later to strengthen in order to regain normal function. Other immunosuppressive agents have not been subjected to randomised controlled trials however they are now accepted as an important part of the management of JDM. Four agents have been described in the treatment of JDM and the evidence for their use will be reviewed within this document. They are: methotrexate, cyclosporine, azathioprine and cyclophosphamide. 3.3 Systemic Lupus Erythematosus Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterised by inflammation of the blood vessels and connective tissues resulting in damage to numerous organ systems and associated with antinuclear antibodies. The diagnosis of SLE is based on well described clinical and laboratory features and is supported by the use of the American College of Rheumatology (ACR) adult SLE classification criteria first developed in 1982 and then revised in 1997. However, there are well described differences in the clinical features, serology and outcome of paediatric patients with lupus compared with adult patients [37]

11
and therefore the applicability of these diagnostic criteria have been questioned. There have been few large scale validation studies of these criteria in the paediatric population. Ferraz et al applied the ACR criteria to 103 paediatric patients with lupus and demonstrated a sensitivity and specificity of 96% and 100% respectively.[38] The aetiology of SLE is unknown. Susceptibility to the development of SLE is considered multifactorial perhaps contributed by the interaction of genetic, acquired and perhaps environmental factors. The pathogenesis involves disordered immunity with autoreactive T and B cells and antibody and immune complex deposition.[2] The clinical manifestations and course of SLE are extremely varied but can be associated with significant morbidity and mortality. 3.3.1 Epidemiology Paediatric SLE is a rare disease and is reported to have an annual incidence estimated at between 0.36 – 0.9 per 100,000 per year depending on the population in which it is studied.[2] There is no accurate prevalence data. Data from Rheumatology clinic surveys report that SLE accounts for between 1% and 4.5% of clinic population.[2] Approximately 15% of patients with SLE have there disease onset in childhood. Onset is rare before the age of 5 and it is more common in girls than boys with a ratio of approximately 5:1.[39, 40] There are limited population based studies reporting data on the distribution of SLE across racial groups. SLE has been observed disproportionately in people of African‐American, Hispanic and Asian background. [2] 3.3.2 Clinical and Serological Manifestations SLE is a multisystem inflammatory condition and can present with insidious onset or as an acute life‐threatening illness. The disease can manifest with renal, CNS, cardiac, pulmonary, musculoskeletal, cutaneous, gastrointestinal, hepatic and haematological features. One large cohort of 256 patients with paediatric SLE reported that the most common clinical manifestations were: arthritis (67%), malar rash (66%), nephritis (55%) and CNS disease (27%).[40] Constitutional symptoms such as fever, fatigue and weight loss are common in paediatric onset SLE. [41] Clinical features such as ulcers, alopecia, Raynaud’s phenomenon and photosensitivity less common in paediatric SLE.[41] One study reported more severe disease onset in paediatric patients compared to adult patients and that renal and haematological features were more common. [37] Silverman et al supported this finding in their study comparing paediatric with adult SLE. They found that children had more active disease at onset with higher frequency of renal disease and lower frequency of cardiopulmonary disease. [42] Typical serological findings include: positive antinuclear antigen (ANA), positive double stranded DNA (dsDNA), antibodies positive to extranuclear antigens (ENA), low complement levels and positive lupus anticoagulant and antiphospholipid antibodies.

12
3.3.3 Course and Outcome SLE is characterised by a chronic relapsing and remitting course. Flares can occur at any time in the course of the illness and are frequently precipitated by infection. Although overall outcome for morbidity and mortality has improved in recent times with current therapeutic options, SLE remains a serious and life‐threatening illness with an unpredictable prognosis. In 1968, Meislin and Rothfield reported a 5 year survival in patients with renal involvement and without renal involvement of 42% and 72% respectively.[43] In contrast, in 1998, Yang et al reported a 5 year survival of 91% in patients with nephritis. [44] 3.3.4 Overview of Management SLE is a complex, multisystem, chronic disease with potential for significant morbidity and mortality and is therefore best managed by a multidisciplinary team experienced in the management paediatric SLE. General aspects to management include: avoidance of excessive exposure to sunlight and protection against UVB with sunscreen and appropriate skin cover with clothing and headwear; prevention of infection with immunisation. Corticosteroids remain the first line therapy for the management of paediatric SLE. NSAIDs are frequently used to manage musculoskeletal complaints and hydroxychloroquine is used as an adjunct to corticosteroids in the treatment of fatigue, mucucutaneous features and arthritis. Immunosuppressive therapy including cyclophosphamide, azathioprine and mycophenolate are generally considered early in the course of the disease if there is evidence of diffuse proliferative glomerulonephritis, CNS involvement or pulmonary haemorrhage.[45] Other immunosuppressants considered include methotrexate and cyclosporin. The choice of drug remains controversial. In other active forms of the disease there is no agreement on the timing of initiating immunosuppressant therapy. Many of these patients are at risk of developing irreversible organ damage and, in addition, potentially face many years of high dose corticosteroid therapy with its associated toxic effects and may therefore warrant the addition of immunosuppressant therapy. Mycophenolate initially showed promising results in maintaining disease control in adult patients with resistant or relapsing lupus nephritis. It has since then been subject to meta‐analysis and found to have no difference over azathioprine in terms of response rates or development of end stage renal disease.[46] It appears to have a safer profile than azathioprine especially regards to haematological complications. Whilst current therapies have dramatically improved survival, there is hope that newer agents including cytotoxic and biologic therapies may provide superior management and ultimately lead to curative treatment. [47] 4. Disease‐Modifying Anti‐Rheumatic Drugs (DMARDs) DMARDs are not used for immediate analgesic or anti‐inflammatory effect but rather for their long term beneficial effects in controlling disease activity. These medications also play an important role in reducing the long term exposure to medications such as prednisolone

13
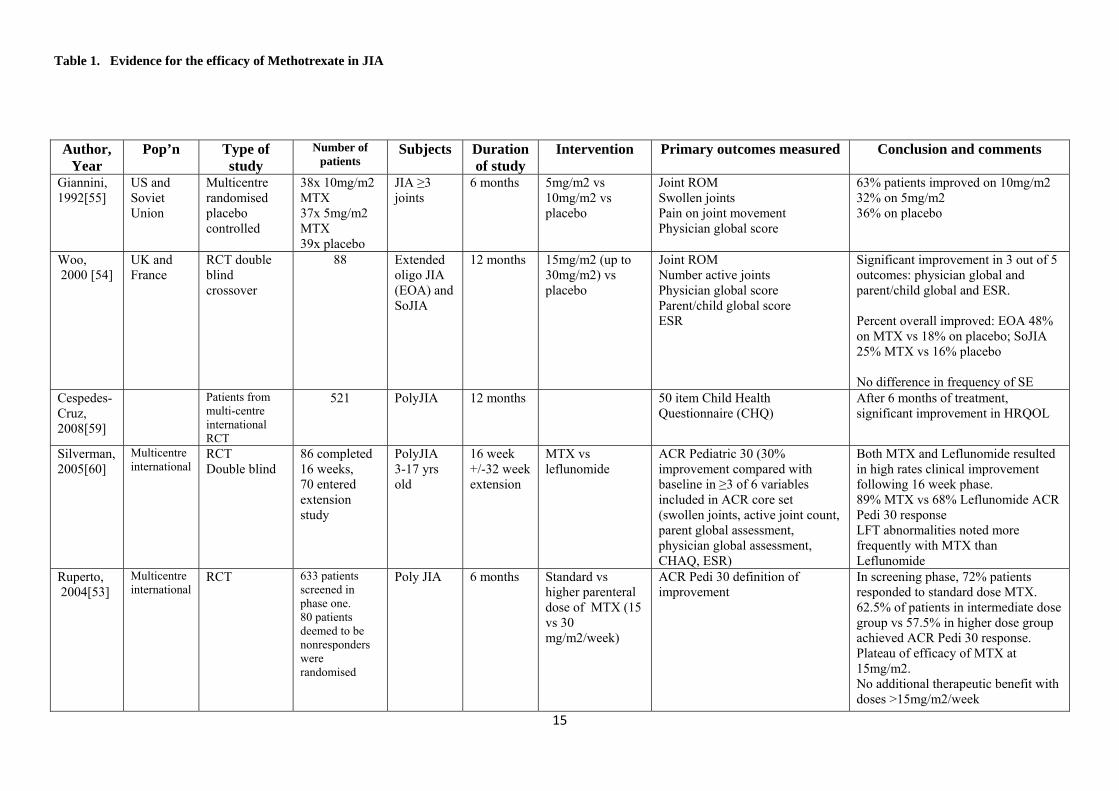
and Non‐Steroidal Anti‐Inflammatory Drugs (NSAIDs). Historically, DMARDs were used late in the course of disease progression as there were initial questions regarding toxicity and safety. Moreover, the illnesses treated were not often considered life threatening and therefore DMARD therapy was considered not warranted. It is now recognised, however, that not only are many of these medications safe and effective for use in children but also that their use early in the course of the disease may prevent irreversible damage and decrease the burden of disease. There are a number of DMARDs used in the treatment of rheumatic illnesses in children. These include: methotrexate, sulfasalazine, azathioprine, leflunomide, hydroxychloroquine and cyclosporine. Methotrexate is often considered as the first choice and it is used as treatment in many diseases such as: JIA, JDM, SLE, vasculitis, uveitis and localised scleroderma. 4.1 Methotrexate 4.1.1 Mechanism of action and Pharmacology Methotrexate is a folic acid analogue and an inhibitor of dihydrofolate reductase and thereby interferes with DNA synthesis by reducing the purine and pyrimidine supply in rapidly dividing cells. This anti‐proliferative effect is achieved with high‐dose regimens and is used for the treatment of tumours. In low‐dose DMARD therapy, methotrexate also has an immunomodulatory and anti‐inflammatory effect. Although the exact mechanism of this action remains unknown, Methotrexate is thought to effect the cellular production of a variety of cytokines and thereby acts to inhibit cell mediated immunity. There is significant variability in the pharmacokinetics of methotrexate between individuals and also specific pharmacokinetic qualities that impact on the dosing and route of administration. Methotrexate is absorbed by the gastrointestinal tract by a saturable process. The average oral bioavailability is 0.7 but this can range from 0.25 to 1.49.[48] A number of factors are thought to impact on this including: age, sex, dose, creatinine clearance, and fed vs. fasting state. [49] There is debate in the literature as to whether meals have an impact on the absorption of oral methotrexate. Absorption of orally administered methotrexate has been shown in adult studies to be reduced at doses beyond 15mg [50, 51] and some groups advocate for the use of parenteral methotrexate for doses higher than this. Once absorbed, peak serum levels are reached in approximately 1.5 hours with the elimination half life being 7 hours. The majority of elimination is through renal excretion and circulating levels fall rapidly as the drug is distributed and eliminated. [2] 4.1.2 Efficacy of Methotrexate in Juvenile Idiopathic Arthritis Methotrexate is the most frequently used second line agent in Juvenile Idiopathic Arthritis and there is good evidence of its efficacy. The evidence for its use has been demonstrated in randomised placebo controlled trials as well as from a large number of retrospective and

14
uncontrolled studies. Furthermore, these studies have shown that Methotrexate is safe. Table 1 summarises studies addressing the efficacy of methotrexate in JIA. Giannini et al published a meta‐analysis in 1993 comparing methotrexate with hydroxychloroquine, penicillamine and auranofin in JIA. A total of 520 patients were enrolled in all the three studies included in the analysis and outcome measures included physician’s global scale and ESR. Only methotrexate at 10mg/m2 showed improvement in these domains. [52] In 2004, Paediatric Rheumatology International Trials Organisation (PRINTO) published the results of a trial which ultimately compared intermediate with higher dose methotrexate in patients who had failed to respond initially to standard doses. The initial screening phase describes a total of 633 patients with JIA of which 72% responded to a standard dose of methotrexate (8‐12.5mg/m2/week) after 6 months of therapy.[53] The efficacy of methotrexate in the treatment of JIA has been demonstrated in prospective controlled trials. A systematic Cochrane review in 2001 set out to evaluate the effects of methotrexate on a number of functional domains, including functional ability, range of motion, quality of life, overall well being and pain. Only two studies [54, 55], with a total of 165 patients with JIA, fulfilled the inclusion criteria for the Cochrane review of placebo‐controlled randomised clinical trials. Based on these studies Takken et al concluded that methotrexate has a clinical effect on patient centred disability.[56] Since then there has not been any other published RCTs addressing the efficacy of methotrexate vs. placebo. Despite the evidence for the efficacy of methotrexate there are many questions relating to its use that remain unanswered or without sufficient evidence to enable concrete recommendations. For example, the response to methotrexate between the various subtypes of JIA has not been compared in a rigorous fashion. Woo et al found no difference in response to methotrexate between patients with extended oligoarticular and systemic JIA although overall there was a significant improvement of both groups compared with placebo.[54] In contrast, Ravelli et al found that when compared to systemic JIA and polyarticular JIA, extended oligoarticular JIA was the most likely to respond to methotrexate. [57] The optimal timing of discontinuation of methotrexate therapy following remission is also unclear. As documented in table 1, Gottleib et al found that after a mean of 8 months in complete remission (SD= 4 months) before discontinuation of methotrexate, relapse occurred in 52% at a mean of 11 months.[58] Most patients respond when methotrexate is restarted.[58] In summary, methotrexate is efficacious in JIA however further studies are required to further outline the duration of therapy before discontinuation.
15
Author,
Year Pop’n Type of
study Number of
patients Subjects Duration
of study Intervention Primary outcomes measured Conclusion and comments
Giannini, 1992[55]
US and Soviet Union
Multicentre randomised placebo controlled
38x 10mg/m2 MTX 37x 5mg/m2 MTX 39x placebo
JIA ≥3 joints
6 months 5mg/m2 vs 10mg/m2 vs placebo
Joint ROM Swollen joints Pain on joint movement Physician global score
63% patients improved on 10mg/m2 32% on 5mg/m2 36% on placebo
Woo, 2000 [54]
UK and France
RCT double blind crossover
88 Extended oligo JIA (EOA) and SoJIA
12 months 15mg/m2 (up to 30mg/m2) vs placebo
Joint ROM Number active joints Physician global score Parent/child global score ESR
Significant improvement in 3 out of 5 outcomes: physician global and parent/child global and ESR. Percent overall improved: EOA 48% on MTX vs 18% on placebo; SoJIA 25% MTX vs 16% placebo No difference in frequency of SE
Cespedes-Cruz, 2008[59]
Patients from multi-centre international RCT
521 PolyJIA 12 months 50 item Child Health Questionnaire (CHQ)
After 6 months of treatment, significant improvement in HRQOL
Silverman, 2005[60]
Multicentre international
RCT Double blind
86 completed 16 weeks, 70 entered extension study
PolyJIA 3-17 yrs old
16 week +/-32 week extension
MTX vs leflunomide
ACR Pediatric 30 (30% improvement compared with baseline in ≥3 of 6 variables included in ACR core set (swollen joints, active joint count, parent global assessment, physician global assessment, CHAQ, ESR)
Both MTX and Leflunomide resulted in high rates clinical improvement following 16 week phase. 89% MTX vs 68% Leflunomide ACR Pedi 30 response LFT abnormalities noted more frequently with MTX than Leflunomide
Ruperto, 2004[53]
Multicentre international
RCT 633 patients screened in phase one. 80 patients deemed to be nonresponders were randomised
Poly JIA 6 months Standard vs higher parenteral dose of MTX (15 vs 30 mg/m2/week)
ACR Pedi 30 definition of improvement
In screening phase, 72% patients responded to standard dose MTX. 62.5% of patients in intermediate dose group vs 57.5% in higher dose group achieved ACR Pedi 30 response. Plateau of efficacy of MTX at 15mg/m2. No additional therapeutic benefit with doses >15mg/m2/week
Table 1. Evidence for the efficacy of Methotrexate in JIA

16
Ravelli, 1999[57]
Italy Retrospective analysis
80
SoJIA 37 polyJIA 20 oligo JIA 23
6 months 6 month clinical response, complete disease control, disease relapse, hepatotoxicity, GI toxicity
oligo JIA sybtype was best predictor for short term clinical response and more likely to have relapse after discontinuation
Al-Sewairy, 1998 [61]
Saudi Arabia
Retrospective analysis
18 SoJIA Mean 18 months
MTX Range 2.5-15 mg/week
Joint evaluation Fever, rash LAD and splenomegaly Serositis ESR, Hb, WCC and plts
89% improved active joint count and functional class 61% significant improvement in ESR, Hb and plts No withdrawals due to toxicity
Gottleib, 1997[58]
USA Retrospective analysis 2 centres
25 patients in remission for a mean duration of 8 months
Poly, pauci and SoJIA
Mean follow up of patients post discontinu-ation was 15 months
MTX range 5.2 – 15.9 mg/m2/dose
Length of time to relapse or continued remission following discontinuation of MTX
Relapse in 52% after a mean of 11 months. Of those relapsed patients most (10/13) achieved remission again within 7 months. Optimal time for discontinuing MTX unknown
Huang, 1996[62]
Taiwan Retrospective chart review
26 JIA Mean follow up 3 years
Mean weekly dose MTX 10-15 mg/m2
73% responded 39% achieved remission Prednisolone weaned in 93% 6 patients experienced SE, 1 patient ceased MTX due to LFT abn
Reiff, 1995[63]
USA Retrospective chart review
21 (13/21 SoJIA)
Refractory JIA
Mean duration 15.2 months
Mean weekly dose MTX 27 mg
Disease activity score based on changes in concomitant therapy, labs, physician global, radiological
33% improved 5% complete remission MTX well tolerated but limited role in refractory JIA
Ravelli, 1994[64]
Italy Retrospective chart review
19 SoJIA 6 months therapy
MTX dose range 7.5-11 mg/m2/week
Response defined as ≥50% reduction in number of joints with active arthritis
63% responded 37% no response to therapy
Harel, 1993[65]
USA 23 (5 SoJIA)
17/23 responders 6/23 non responders
Median 2.5 yrs
MTX 7.5-10mg/m2
Assessed radiological progression on serial wrist XR
11/17 responders had improved carpal length All 6 nonresponders had progressive loss of carpal length Conc: radiological improvement in those responded to MTX
Wallace, 1993[66]
USA 49
PolyJRA (16/49 SoJIA)
At least 1 year
MTX 15mg/m2 45% remission on MTX
Table 1. Evidence for the efficacy of Methotrexate in JIA

17
4.1.3 Efficacy of methotrexate in Juvenile Dermatomyositis Methotrexate is the most frequently used of the immunosuppressive agents described in the treatment of JDM. A recent survey of Paediatric Rheumatologists demonstrated considerable variation in the management of JDM. [34] This variance reflects the lack of data available on which to base treatment. More recently three consensus protocols were developed at a consensus meeting in Toronto involving 12 Paediatric Rheumatologists. Each of these protocols involved the use of corticosteroids and methotrexate. [67] Methotrexate has not been subjected to a prospective randomised controlled trial and therefore evidence for its use is derived from observational studies only. In 2005 Ramanan et al demonstrated in an inception cohort study the effectiveness of methotrexate. Thirty one patients with JDM were commenced methotrexate as first line therapy at a mean dose of 15mg/m2 and their steroids aggressively tapered. These patients were compared to 22 historical controls treated with prednisolone alone. Patients in the study group were found to have a shorter time to discontinuation of corticosteroids and also reduced cumulative dose. [68] However, this study failed to demonstrate whether aggressive taper of steroids alone without the addition of methotrexate had a similar outcome. Improved outcome with the early initiation of prednisolone and methotrexate was also demonstrated by Al‐Mayoub et al in a small series of patients with JDM and associated cutaneous ulceration and/or dysphagia. [69] In a retrospective chart review, initiation of methotrexate in patients who had failed to respond to steroid therapy within 6 weeks was thought to be beneficial in reducing the overall risk of long term complications including calcinosis. [31] There is currently an international multi‐centre prospectively randomised trial co‐ordinated by PRINTO (Paediatric Rheumatology International Trials Organisation) which is comparing prednisolone alone with prednisolone/methotrexate combination and prednisolone/ cyclosporine combination in patients newly diagnosed with JDM. The results of this trial are eagerly awaited in order to guide the management of JDM.

18
Author/ Year
Pop’n Type of study
Number of
patients
Subjects Duration of study
Groups/Intervention Outcomes measured Conclusion and comments
Ramanan, 2005[68]
Single centre, Canada
Prospective cohort study with historical controls
53 31 patients prospectively studied with 22 controls
Control patients followed for 4 years; study patients followed for average 34.6 months
2 groups. Study group: MTX 10‐20mg/m2/week (max 25mg/week) and prednisolone 2mg/kg/d (max 75mg) with pred aggressively tapered every 2 weeks
Time to discontinuation of steroids, cumulative steroid dose and steroid SE. Disease activity: CHAQ, muscle strength, rash, calcinosis, time to flare.
Use of MTX and aggressively tapered steroids may be as effective traditional long‐term steroid and decreases the cumulative steroid dose
Al‐Mayouf, 2000[69]
Saudi Arabia
Prospective cohort study
12 newly diagnosed
JDM.
Patients with dysphagia, dysphonia, GI bleeding, cutaneous ulceration or resp muscle involvement were started MTX early
Mean duration of therapy was 23.5 months
2 groups: early (<6 wks) and late (5‐72 months post diagnosis) MTX therapy. 6 patients per group
Resolution of clinical indications for MTX, decreased disease activity (graded muscle strength and enzyme levels) and discontinuation of steroids
Improved muscle weakness and lack of development of calcinosis in group treated early. One patient ceased MTX due to abdominal pain
Fischer, 1979[31]
US Case series 4 3 patients wih JDM, 1 patient with JPM
9‐31 months Pred plus MTX 1mg/kg/wk intravenously
Steroid dose reduction, muscle strength, enzyme levels, EMG changes
Improvement in all parameters in 3 patients.
4th patient initially improved then relapsed
and died of bowel perforation and sepsis
Miller, 1992[70]
US Retrospective chart review
16 16 patients with recalcitrant JDM
6 year follow up MTX plus prednisolone Muscle strength, muscle enzymes, prednisolone dosage, medication toxicity
All patients treated for >8 months had clinical remission and therefore concluded that MTX is useful adjunct to prednisolone. 31% patients discontinued MTX due to SE.
Table 2. Evidence for the efficacy of Methotrexate in JDM

19
4.1.4 Efficacy of methotrexate in Uveitis Methotrexate has been used in children with uveitis for many years. Its use is supported by evidence largely from retrospective cohort studies. A retrospective chart review found methotrexate to be an effective agent in the treatment of uveitis. [71] Twenty five patients with uveitis received methotrexate (mean dose of 15.6mg/m2) and remission was achieved after a mean of 4.25 months. Six patients had methotrexate ceased after remission for 12 months and of these 4 patients were still in remission after 7.5 months. Other case series have demonstrated the effectiveness of methotrexate in uveitis resistant to topical corticosteroids.[72, 73] 4.1.4 Dose and administration in JIA and JDM Methotrexate is administered weekly. Although, as previously mentioned, the serum elimination half‐life of methotrexate is 6‐7 hours its metabolites can be measured intracellularly over 1 week. Methotrexate can be administered orally or parenterally. The standard guideline for methotrexate use is an initial dose of 10 mg/m2/week grading up to 15mg/m2/week.[2] Oral methotrexate should be given on an empty stomach as food decreases its bioavailability. Ruperto et al demonstrated that there was no additional benefit of parenteral doses above 15mg/m2. [53] Parenteral administration is recommended for doses >15mg/m2/week because of better bioavailability and gastrointestinal tolerability. Parenteral dosing should also be considered in those children who have a poor response to oral methotrexate or where poor compliance impacts on disease control.[2] There is no difference in the bioavailability of subcutaneous and intramuscular therapy. [74] 4.1.5 Drug interaction and folate supplementation A number of medications increase the bioavailability of methotrexate. These include: phenytoin, oral contraceptive pill, tetracycline, barbiturates and tranquilizers. Co‐trimoxazole should be avoided with methotrexate as it can cause severe bone marrow suppression and skin ulcerations. In addition a number of medications, including penicillins and cyclosporine, can lower the elimination of methotrexate by decreasing renal clearance. Methotrexate is contraindicated in renal failure. There is good evidence to support the use of folic acid supplementation to alleviate the common gastrointestinal and oral mucosal side effects associated with methotrexate without impacting on the beneficial anti‐inflammatory effects. [75] The use of methotrexate is safe in combination with NSAIDS and corticosteroids. There is some experience of combined DMARDs however controlled systematic studies are lacking to support this strategy in the treatment of JIA.

20
Author, Year
Type of study
Duration of study
Subjects and
numbers
Drug and monitoring
Hepatotoxicity Haematological abnormality
GI toxicity other Conclusion
Ortiz‐Alvarez, 2004 [76]
Pro‐spective study
89 JIA MTX Monthly blood tests
14% patient had significantly elevated LFTs (>2x ULN) 52% had mild elevated liver func tests
26% patients had abnormal FBE (low granulocyte or lymphocyte count or Hb) 95% of patients with abn had viral infection at the time of blood test.
No diff noted in terms of duration and cummulative dose MTX and concurrent meds
Few significantly abn blood tests beyond 2 months. Blood tests every 4‐8 weeks unnecessarily frequent
Lahdenne, 2002 [77]
Retro‐spective review. Liver bx correlated with lab findings
Patients on MTX at least 2.4 yrs
34 JIA MTX (20‐30mg/m2) for duration >2.4 yrs
All 24 pts treated with low dose MTX had grade I Roenigk changes. Of 10 patients on >20mg/m2, 4 pts grade II histology and 5 grade I. No fibrosis or cirrhosis
Aggressive MTX therapy with concomitant meds may contribute to minor reversible liver abn
Graham, 1992 [78]
Retro‐spective review
84 – 296 weeks
62 poly JIA
MTX (5‐10mg/m2), 3 monthly blood tests, PFTs on 46 pts
Transient liver function abn occurred in 9/62 (14%) 12 patients had liver biopsy – no fibrosis or cirrhosis
Macrocytic anaemia in one patient on co‐trimoxazole No cases on TP or neutropenia
Nausea occurred in 14/62 patients. Peptic ulcer in 4/62 patients also on concomitant NSAID
No PFT abn No stomatitis or rash
Suggest that well tolerated
Giannini, 1992 [55]
US/Soviet Multi‐centre RCT
6 months 89 JIA ≥3 joints
MTX 5 or 10 mg/m2
One patient had LFT abn 8 patients had GI upset
Woo, 2000 [54]
DBRCT cross over
12 months
88 EOA JIA and SoJIA
15mg/m2 (up to 30) vs placebo
Total 20 patients on MTX had AST abn vs 16 placebo patients
Bone marrow failure did not occur
Nausea and GI upset occurred in similar numbers of patients
No difference in SE between MTX and placebo
Table 3. Adverse Effects of Methotrexate

21
Author, Year
Type of study
Duration of study
Subjects and
numbers
Drug and monitoring
Hepatotoxicity Haematological abnormality
GI toxicity other Conclusion
Hashkes, 1999 [79]
Liver biopsies compared with RF for hepatotox
25 patients, 33 biopsies
2 biopsies showed grade IIIa Roenigk changes 4 showed grade II changes and 27 grade I. No significant fibrosis
No significant fibrosis. Biochem abn correlated with changes
Hashkes, 1997 [80]
Liver biopsies compared with RF for hepatotox
14 JIA 13/14 grade I Roenigk changes 1/14 grade II changes No bx with significant fibrosis. 13/14 LFT abn but only 5 had >3x ULN
No significant clinical consequences despite minor histo changes
Kugathasan, 1996 [81]
Liver biopsies
>3 years of MTX
9 JIA 10mg/m2/wk No clinical or biochemical evidence of liver injury All biopsies were normal
Table 3. Adverse Effects of Methotrexate

22
4.1.6 Safety Methotrexate is generally well tolerated in children, however, it has a number of potentially serious adverse effects. The most common side effect is nausea and vomiting for which folic acid has been reported to be beneficial. [75] Most of the other side effects are mild and reversible. Oral ulceration, alopecia, mood changes, rash, peptic ulcer and headache have been reported to occur. Table 3 summarises reported adverse effects of methotrexate. Hepatic toxicity is one of the main potentially serious adverse reactions with methotrexate use. The exact mechanism is not clearly understood. [82] Children are thought to have a reduced risk of methotrexate associated hepatotoxicity compared with adults as they generally have fewer co‐existing diseases and fewer environmental exposures such as alcohol. Acute mild liver function abnormalities occur in approximately 9% of children on low dose therapy. [83] These changes are usually transient and improve with a period of cessation. It is difficult to assess the true effect of methotrexate as it is often used in combination with NSAIDs which may contribute to a transaminase rise. The potential for long term liver damage with fibrosis and cirrhosis has raised concerns in the past however there are only a few reports of fibrosis in children and no reports of cirrhosis secondary to methotrexate. Refer to table 2 for summary of series addressing liver fibrosis and cirrhosis. Haematological abnormalities are rare with the use of methotrexate. The main toxic effects described include: macrocytic anaemia, leukopenia, thrombocytopenia and pancytopenia. [2] Only case reports of children with haematological side effects of MTX exist, however, the adult literature reports a frequency of 1‐3%. [84] In patients with mild bone marrow suppression, spontaneous recovery is usual within 2 weeks.[2] Malignancy due to methotrexate remains an area of controversy. There have been a few cases described in children with JIA on methotrexate of Hodgkins and Non‐Hodgkins lymphoma. [85, 86] Methotrexate has been reported to have caused foetal death and congenital abnormalities and therefore its use is not recommended for use in pregnancy. It is suggested that pregnancy is avoided for a minimum of 3 months after completion of therapy in male patients and for at least one ovulatory cycle in female patients. 4.1.7 Monitoring and supervision Children with JIA on long term methotrexate require regular clinical and laboratory monitoring both for the response to the medication and for its potential toxicities. Hashkes et al examined liver biopsies in children with JIA on methotrexate and found a relationship between biochemical liver function changes and histopathological changes consistent with mild fibrosis.[79] Whilst the degree of fibrosis in this series was not considered significant, the potential hepatotoxic effects of methotrexate have prompted the introduction of guidelines for the monitoring of liver toxicity in patients taking methotrexate. These guidelines were developed in 1994 and represent an expert consensus on the

23
monitoring of liver toxicity with methotrexate use in adult Rheumatoid Arthritis. Although developed based on adult data, these guidelines form the basis for monitoring of children with JIA on methotrexate.[87] It is generally recommended that liver function, renal function and full blood examination be performed at baseline for all patients. Monitoring of LFTs 4‐8 weekly was recommended by Kremer et al in the initial guidelines published in 1994.[87] Since then, Ortiz‐Alvarez et al monitored 89 patients with JIA prospectively and found that LFT abnormalities were usually mild and resolved spontaneously and that more severe abnormalities settled rapidly with discontinuation of methotrexate. [76] When there are no other co‐existing risk factors, 3 monthly monitoring following initial bimonthly for a few months seems to be appropriate. 4.1.8 Formulary Australian brand name: Methoblastin: Tablets: yellow and uncoated, 2.5mg (round), 10mg (capsule shaped); Injection: 25mg/ml, 1000mg/10ml UK brand name: Maxtrex: 2.5mg, 10mg tablets; Metoject: prefilled syringe 10mg/ml (7.5mg, 10mg, 15mg, 20mg, 25mg) US brand names: Rheumatrex: 2.5mg; Trexall: tablets 2.5mg, 5mg, 7.5mg, 10mg, 15mg Canadian brand names: Apo‐Methotrexate; Ratio‐Methotrexate 4.1.9 Summary recommendations Methotrexate has been shown to be an effective and safe therapy in the treatment of JIA. It is associated with a number of potentially serious adverse effects and therefore its use requires monitoring with blood tests and with close clinical supervision from a medical specialist familiar with the potential risks. It is not recommended for use if access to this supervision is unavailable. It is recommended for inclusion on the WHO complementary list of essential medicines. 4.2 Leflunomide 4.2.1 Mechanism of action and Pharmacology Leflunomide is an immunomodulatory medication that inhibits pyrimidine synthesis through its inhibition of the enzyme dihydro‐orotate dehydrogensase. Lymphocyte proliferation depends on pyrimidine synthesis for proliferation and leflunomide therefore has both anti‐proliferative and anti‐inflammatory effects. The active metabolite is A77‐1726 to which leflunomide is actively converted via hepatic metabolism. A77‐1726 is highly protein bound and has a half‐life of up to 18 days reaching a steady state after approximately 20 weeks. [88] The pharmacokinetics of leflunomide and its active metabolites are not affected by food intake or by gender. It is excreted almost equally in urine and faeces. The pharmacokinetics

24
of oral leflunomide was studied in 73 paediatric patients aged 3‐17 years with polyarticular JIA. [89] This analysis demonstrated that paediatric patients with bodyweights <40kg have a reduced clearance compared to adults and therefore require dose adjustment (see “Dose and administration” below). 4.2.2 Efficacy of Leflunomide in JIA Leflunomide has been shown to be effective in a number of studies in adults with Rheumatoid Arthritis. (See table 4) There are fewer studies in children with JIA, however, Sliverman et al demonstrated in an open label study in children with refractory polyarticular disease a good response to leflunomide. [90] In a randomised controlled trial of 94 patients, Silverman compared leflunomide with methotrexate in polyarticular disease. This trial concluded that treatment with leflunomide does result in clinical improvement but that the rate of improvement was not as high as that seen with methotrexate (89% Leflunomide vs 68% methotrexate met ACR Pedi 30 response).[60] Gao et al [91] addressed the potential for combining leflunomide with methotrexate by comparing combination therapy with methotrexate alone. They found a significantly higher response rate in those patients on combined therapy. Whilst there does appear to be some evidence for the use of leflunomide in the paediatric population, the exact role is yet to be defined. There appears to be a role in patients who are intolerant to methotrexate or have failed to respond to methotrexate. 4.2.3 Dose and administration Adult studies suggest the benefit of loading doses of 100mg/day for 3 days to rapidly achieve steady state. This is not required and may result in significant side effects[2]. In their study, Silverman et al used 100mg loading dose for one day in children <20kg, for two days in children between 20 and 40kg, and 100mg for three days in children >40kg. This was followed by maintenance dose of 10mg/day if <40kg and 20mg/d if >40kg. A recent population pharmacokinetic study recommended that leflunomide doses be adjusted for paediatric patients as follows: 10 mg/d for 10‐20 kg, 15 mg/d for 20‐40 kg, and 20 mg/d for > 40 kg.[89] The use of a loading dose may increase toxicity in children. (MIMS 2009) 4.2.4 Safety The most commonly reported adverse effect is gastrointestinal upset which is reported to occur in 17% of patients. This includes: abdominal pain, dyspepsia, diarrhoea and gastritis. Other common side effects include: headache, rash and alopecia. Liver function abnormalities are less frequently reported than with the use of methotrexate. [60] There appears to be a particular risk when used in combination with methotrexate. [92] In studies of leflunomide in adults with Rheumatoid arthritis, liver function abnormalities are reported in approximately 5% of patients. These changes are mild and reversible. More

25
serious liver dysfunction is uncommon and is associated with concomitant alcohol consumption, viral hepatitis or other pre‐existing liver disease. Leflunomide is teratogenic and therefore it is recommended that patients have a negative pregnancy test before starting treatment and use reliable contraception during therapy. 4.2.5 Drug interactions The enzymes involved in the metabolism of leflunomide are not entirely known. In vitro studies indicate that leflunomide inhibits cytochrome p450 and therefore caution should be taken when co‐administered with other drugs involved in cytochrome p450 metabolic pathways. 4.2.6 Monitoring and supervision Monitoring is recommended with baseline and monthly full blood count and liver function for 6 months, which can be reduced to 6‐8 weekly if stable. 4.2.7 Formulary Because safety and efficacy in children has not been clearly established with the existing evidence, most manufacturers do not list leflunomide with paediatric advice. Australian brand names: Arava® (tablets 10mg, 20mg, 100mg); Arabloc® (tablets: 10mg, 20mg) US brand names: Canadian brand names: 4.2.8 Summary recommendations In summary, despite limited studies in the paediatric population, leflunomide has been shown to be effective for the treatment of JIA both as a monotherapy[60] and in combination with methotrexate [91]. Its use in the treatment of rheumatic disease requires monitoring with blood tests and close clinical supervision to ensure that the potential side effects are minimised. It is not recommended for use if access to this supervision is unavailable. It is recommended for inclusion on the WHO complementary list of essential medicines.

26
Author, Year
Pop’n Type of study
Duration of study
Number of patients
Subjects Study groups Primary outcomes measured
Conclusion and comments
Silverman, 2005[60]
Multicentre international
DBRCT
16 week +/‐32 week extension
86 completed 16 weeks 70 entered extension study
Polyarticular JIA 3‐17 yrs old
MTX vs leflunomide Percentage of patients with ‘ACR Pedi 30’ (30% improvement compared with baseline in ≥3 of 6 variables included in ACR core set (swollen joints, active joint count, parent global assessment, physician global assessment, CHAQ, ESR)
Both MTX and Leflunomide resulted in high rates clinical improvement following 16 week phase. 89% MTX vs 68% Leflunomide ACR Pedi 30 response LFT abnormalities noted more frequently with MTX than Leflunomide
Silverman, 2005[90]
Open‐label study
26 weeks 27 patients Only 63% patients completed study
Refractory polyarticular JIA (failed response or intolerant of Methotrexate)
Leflunomide loading dose ~100mg followed by 10mg/day (+/‐ increase to 20mg/d at 8 wks)
ACR Pedi 30 52% met ACR Pedi 30 response 44% met ACR Pedi 50 and 19% met ACR pedi 70. 44% patients responded as early as 4 weeks. AE reported in 30% of patients , ceased in 2 patients
Gao, 2003[91]
China 26 weeks 40 patients Polyarticular JRA
Leflunomide (loading dose of 1mg/kg/day) D1‐3 then 0.2‐0.4 mg/d) plus MTX (0.3mg/kg iv every 2 wks until remission then 0.2mg/kg weekly orally) vs MTX alone
Tender and swollen joints, parent and physician evaluation score, general function score, ESR, CRP SE documented
Significant difference in improvement on combination compared with MTX alone. No significant difference in rate of SE. Combination therapy safe and well tolerated
Table 4. Evidence for the efficacy of leflunomide in JIA

27
Author
, Year
Pop’n Type of
study
Duration of study
Number of
patients
Subjects Study groups Outcomes Conclusion and comments
Van Rossum, 1998[93]
multicentre
DBRCT 24 weeks 69 Oligo and Polyarthritis
SSZ 50mg/kg/d (max 2g/d) Placebo
ACR Pediatric 30 (without functional measure) AEs
44% improved in SSZ group compared with 21% placebo group. More AEs with SSZ resulting in withdrawal of 10 patients in total from study (all from SSZ group). All AEs were transient and reversible. LFT abn n=2, Leukopenia n=2, hypoimmunoglobinaemia n=3, anorexia/diarrhoea/ haematoma n=1 each.
Burgos‐Vargas, 2002[94]
DBRCT 26 weeks 33 ERA SSZ 30‐60mg/kg/d (max 2g) Placebo
Reduction in active joints
46% in SSZ group compared with 42% in placebo group. Significant differences only seen in physician and patient assessments
Ansell, 1991[95]
multicentre
Pilot study, Un‐controlled
52 weeks 51 JIA (oligo, poly and SoJIA)
SSZ 40mg/kg/d increased incrementally over 6 weeks.
Active joint count, patient assessment, ESR AEs
24% good response, 16% some response, 45% no response. 8 patients withdrew due to AEs
Huang, 1998[96]
Single clinic
Retrospective chart review
Mean duration therapy 2.5 yrs
36 ERA and JIA
SSZ 50mg/kg/d (max 2g)
Active joint count, ESR Remission or improvement (defined as 25% reduction in joint number for two consecutive visits)
39% patients in remission further 25% improved 11% patients some AE. One patient ceased SSZ. No difference between ERA and JIA
Van Rossum, 2007[97]
multicentre
Pro‐spective cohort
Median 9 years (range 7‐10)
61 Patients initially managed with SSZ (32) vs those managed with placebo (29)
ACR Pediatric 30 Effective suppression of disease activity early with SSZ has beneficial effects that persist for many years
Hoza, 1991[98]
DBRCT 26 weeks 39 Oligo and polyarthriti
SSZ 20‐30mg/kg/d
Any improvement in 4 criteria:
48% SSZ group improved compared with 28% Chloroquine group
Table 5. Evidence for the efficacy of Sulfasalazine in JIA

28
Author,
Year
Pop’n Type of
study
Duration of study
Number of
patients
Subjects Study groups Outcomes Conclusion and comments
s Chloroquine 3‐4mg/kg/d
Active joints, pain, morning stiffness, ESR, functional capacity
More AEs with SSZ
Ozdogan, 1985[99]
4‐14 months
18 All types JIA
ESR Joint tenderness Active joint count
Significant improvement in ESR, joint tenderness and count
Table 5. Evidence for the efficacy of Sulfasalazine in JIA

29
4.3 Sulfasalazine 4.3.1 Mechanism of action and Pharmacology The exact mechanism of action of sulfasalazine is not clearly understood. Its role in the treatment of JIA is possibly due to its immunoregulatory and anti‐inflammatory effect including, amongst other things: the inhibition of prostaglandin synthesis; inhibition of bacterial growth; inhibition of leukotriene formation; modulation of leucocyte function; inhibition of DNA synthesis; impairment of folate metabolism and effects of lymphocyte number and function. Sulfasalazine is poorly absorbed in the small intestine. In the colon, sulfasalazine is reduced to two major active metabolites (5‐aminosalicylic acid and sulfapyridine) by colonic flora. Sulfapyridine is rapidly absorbed and metabolised by acetylation in the liver. The rate of this process varies between individuals and is genetically determined. Both 5‐aminosalicylic acid and sulfapyridine have an affinity for collagen rich tissue and concentrate in the synovial fluid as well as pleural spaces, intestinal wall and peritoneum. 4.3.2 Efficacy of Sulfasalazine in JIA The use of sulfasalazine in the treatment of JIA was first reported in 1986 [99], although it had been used for many years prior to this for the treatment of adult Rheumatoid arthritis. Table 4 summarises the studies addressing efficacy of sulfasalazine in JIA and as is illustrated in the table, whilst there are few controlled studies, there is some evidence to support its use in JIA. In a randomised double blinded placebo controlled trial of 69 patients, Van Rossum et al found that sulfasalazine reduced the overall articular severity score, global assessments and laboratory parameters. [ref] Adverse events were more frequent however these were found to be transient and reversible. Ozdogans study in 1986 [99]was a small uncontrolled trial (n=18) which demonstrated benefits of SSZ. Eleven patients had an excellent response with only 3 patients showing no response. Similarly in another open label study by Joos et al [100], 21 of 41 patients achieved remission and significant improvement was seen in 12. In this study, no change was seen in one patient and the condition worsened in three. Imundo and Jacobs [100, 101] observed that the best response rate was ANA positive young girls with oligoarticular JIA, and that the worse response (indicated by a high failure rate) was in systemic JIA. Only 28% had complete remission. Other open label studies have shown that sulfasalazine is most effective in boys older than 9 and adolescents with oligoarticular JIA, raising the question of its use in enthesitis related arthritis (ERA). The other controlled studies include a placebo‐controlled randomised blinded study of patients (n=33) with ERA. [94] Burgos demonstrated no significant difference in response rate however some improvement in physician and patient global assessments. Hoza et al also

30
[98] demonstrated no significant difference when sulfasalazine was compared to chloroquine in oligoarticular and polyarticular JIA. Sulfasalazine does not have consistent efficacy across subtypes of JIA. There is a broad experience in open, uncontrolled trials which suggest the benefit of sulfasalazine in JIA, and this is supported by the aforementioned blinded controlled Van Rossum trial. Some open label trials found an increase response in older HLAB27 positive children with oligoarticular disease, however this finding was not convincingly supported in a small blinded control study. [94] Based on best available evidence, sulfasalazine is most effective in poly and oligo articular disease and there does not appear to be a difference in response rates between these groups. On the current evidence, sulfasalazine is not effective in the management of SoJIA and, in fact, its use is potentially complicated by an increased risk of toxicity. 4.3.3 Dose and administration Sulfasalazine is administered orally in children with JIA. Initiation of therapy should begin with a small dose followed by a grading up at weekly intervals. The recommended starting dose is 5mg/kg twice daily for one week, then 10mg/kg twice daily for one week, then 20mg/kg twice daily for one week, then a maintenance dose of 20‐25mg/kg twice daily. The maximum recommended dose in children is 2g/day. Sulfasalazine should be administered after meals or with food and should not be concurrently taken with antacids. 4.3.4 Safety Intolerance and adverse events are frequently associated with sulfasalazine administration. Adverse reactions frequently reported include: rash, gastrointestinal symptoms and haematological abnormalities. One study [93] reports a discontinuation rate of approximately 30%. They found that 86% of patients on sulfasalazine reported at least one adverse event and this resulted in 10 patients (28.5%) requiring withdrawal from the study. One patient was considered as having a serious adverse event but the authors noted that all of the adverse events were reversible with discontinuation of therapy. [93] Gastrointestinal intolerance is the most frequently reported side effect. Symptoms include: anorexia, nausea, abdominal discomfort and diarrhoea. Liver function abnormalities also occur commonly estimated to occur in approx 4% patients. More severe (and potentially fatal) hepatotoxicity has been reported in association with DRESS syndrome (Drug Reaction with Eosinophilia and Systemic Symptoms) – a hypersensitivity reaction thought due to the sulfapyridine metabolite. Features include: fever, rash, raised liver function tests, eosinophilia and lymphadenopathy. Corticosteroid therapy is frequently required to treat DRESS syndrome. Haematological side effects such as leucopenia have an incidence across the literature of 3% [102]. Whilst leucopenia is reversible with cessation of the drug, other more rare but severe haematological side effects such as agranulocytosis have been reported. Agranulocytosis is

31
said to occur in 1% of patients and usually begins within 5‐12 weeks of onset of therapy [103] and is potentially fatal. Skin rash is also a common side effect and occurred in half of the patients in the Van Rossum trial [93]. Sulfasalazine has also been associated with other rarer complications such as drug induced lupus erythematosus. This may occur after several or even years of therapy and resolves following withdrawal. 4.3.5 Drug interaction Sulfasalazine is generally well tolerated with other medications. Sulfasalazine may decrease the bioavailability of cyclosporin. There are also reports of possible additive hepatotoxicity when used in combination with methotrexate. Care should be taken when prescribing sulfasalazine as numerous other drug interactions have been reported. 4.3.6 Monitoring and supervision Full blood count and Liver function tests should be performed frequently following initiation of sulfasalazine. Product information suggests baseline tests then 2 weekly for 3 months, monthly for 3 months and then monthly thereafter. It is recommended that patients be followed up clinically at least 3 monthly to ensure ongoing tolerance of sulfasalazine. 4.3.7 Formulary Sulfasalazine is FDA approved for use in children for JIA polyarticular course. It is not licensed for use in the UK. Australian brand names: Pyralin, enteric coated tablets: 500mg; Salazopyrin and Salazopyrin EN‐tabs tablets: 500mg UK brand name: Salazopyrin and Salazopyrin EN‐tabs 500mg; Salazopyrin suspension 250mg/5ml and suppositories 500mg. US brand names: Azulfidine, Azulfidine EN‐tabs, Sulfazin and Sulfazin EC, 500mg Canadian brand names: Alti‐Sulfasalazine and Salazopyrin 4.3.8 Summary recommendations The current literature suggests that sulfasalazine has a role as disease modifying therapy in the treatment of JIA and particularly oligoarticular and polyarticular disease. It is not the first DMARD of choice especially given the evidence supporting methotrexate. The consideration of sulfasalazine as an alternative DMARD in JIA should be under specialist supervision. Sulfasalazine therapy also requires specialist supervision particularly with respect to the frequent risk of adverse effects. In addition, regular monitoring with blood tests is recommended. Sulfasalazine is recommended for inclusion on the complimentary WHO essential medicine list provided these resources are available.

32
4.4 Cyclosporin A 4.4.1 Mechanism of action and Pharmacology Cyclosporin A (CyA) is a potent immunomodulatory agent used to inhibit the adaptive immune response and therefore effective in diseases known to be of autoimmune origin. It is used to prevent rejection in solid organ transplant. The effects on the immune system are likely multifactoral. Calcineurin, an important protein in the process of T and B cell activation, is bound and inhibited by CyA. This results in impaired production of a number of cytokines important in the proliferation of T cells. The effects of CyA on the T cell appear to be specific and reversible. Cyclosporin may have other effects including effects on antigen presentation. CyA is incompletely absorbed form the gastrointestinal tract. Microemulsion preparations are thought to provide improved absorption and bioavailability. It has multiple hepatic metabolic pathways and is metabolised to many metabolites before excreted in the bile. Only a small percentage is excreted in the urine. The half‐life is approximately 18 hours. 4.4.2 Efficacy of Cyclosporin in JIA and MAS The role of cyclosporin in the treatment of JIA has not been clearly defined. It may be particularly important in the treatment of SoJIA especially in association with MAS. As illustrated in Table 6, there is little strong evidence supporting its efficacy and there are no controlled trials. Evidence includes observational studies and case series. Gerloni et al described a group of 34 patients with SoJIA in a prospective open trial [104] in whom there was benefit in control of fever and in reducing steroid exposure. Twenty six percent of patients withdrew from CyA treatment though due to adverse events. In addition, 50% withdrew due to lack of efficacy or flare of disease. Stephan et al reported the effectiveness of CyA in 12 patients with reactive haemophagocytic syndrome. In five patients CyA was used as first line therapy and in seven patients it was used when steroids had failed. [105] Reiff et al described 22 patients (17 with JIA of which 14/17 had SoJIA) and concluded that CyA resulted in improvement in the majority of patients. [106] Joint count improved in 70% patients and in patients with SoJIA, fever resolved in 91%, anaemia improved in 33% and concomitant prednisolone therapy was reduced in 77% of patients, [106] supporting the hypothesis that CyA is more beneficial in the treatment of systemic symptoms (such as fever and anaemia) than the control of arthritis. Mouy et al looked at a small group of patients with MAS treated with CyA and demonstrated rapid improvement in all patients. [107] 4.4.3 Efficacy of Cyclosporin in JDM There are a number of small retrospective studies and case series reporting the benefits of CyA in patients with JDM. These studies are summarised in Table 7. Similar to JIA, there are currently no controlled trials however there is currently an international multi‐centre

33
prospectively randomised trial co‐ordinated by PRINTO (Pediatric Rheumatology International Trials Organisation) which is comparing prednisolone alone with prednisolone/methotrexate combination and prednisolone/cyclosporine combination in patients newly diagnosed with JDM. The results of this trial are eagerly awaited in order to guide the management of JDM. Heckmatt et al studied 14 patients with JDM who had not fully responded to corticosteroids or other immunosuppressants (methotrexate, azathioprine or cyclophosphamide) and had a chronically active course averaging three years. [108] Many of these patients had serious complications of the disease or of their previous treatment. Most patients were treated with 2.5‐7.5mg/kg/d (divided twice daily) and all patients were found to have improved muscle strength and steroid dose reductions. 4.4.4 Dose and administration The generally accepted dose for the management of paediatric rheumatic diseases is 3‐5mg/kg/day [2] orally and is usually divided twice daily. This is similar to the recommended dosing in adult patients with rheumatoid arthritis. In one study of children with JDM, a dose ranging between 2.5 to 7.5mg/kg/day was sufficient. [108] 4.4.5 Safety Cyclosporin is associated with potentially considerable toxicity and requires careful monitoring and supervision. Most side effects are dose dependent and responsive to dose adjustment. Cyclosporin doses are significantly higher in the treatment of solid organ transplantation compared to rheumatological indications and therefore the rates of side effects is comparatively higher. Renal function abnormalities and hypertension are the most concerning side effects and responsible for the majority of withdrawals of therapy. Rises in creatinine and urea due to a reduced glomerular filtration rate have been demonstrated. The exact mechanism for this is unclear but these changes are usually reversible. In one series of 22 patients with JIA and JDM on CyA, [106] serum creatinine doubled in 6 patients and GFR was reduced in one of 14 patients. Ostensen studied 14 patients on CyA and found marked rise in serum creatinine in 5 patients necessitating withdrawal. [109] These findings necessitate close monitoring of renal function. Interstitial fibrosis and tubular damage may also occur[110] and may be irreversible. Cyclosporin induced hypertension is also frequently seen in children and in adults has been estimated to cause hypertension in approximately 10% of adults treated. A recent Cochrane review demonstrated statistically significant increases in blood pressure with CyA in a dose‐related fashion. They concluded that prescribers should find the lowest effective dose in those patients requiring long term use. [111]
Other encountered side effects include: hypertrichosis, gingival hyperplasia, gastrointestinal disturbance, tremor and paresthesias, hepatic dysfunction and bone marrow suppression. These adverse effects are mostly reversible with reduction or cessation of therapy. In one series of 22 patients, doses less then 3.5mg/kg/d were unlikely to cause hepatotoxicity and

34
bone marrow effects and that hypertension, gum hyperplasia and hypertrichosis were not observed, suggesting that in children these side effects may be less common. [106] Since CyA is an immunosuppressive agent, children administered CyA are at increased risk of infection. 4.4.6 Drug interaction Cyclosporin should be used with caution when administered in combination with other potentially nephrotoxic medications. In particular, NSAIDs are frequently used in paediatric rheumatic disease and may exacerbate the toxic effects of CyA. In this situation close monitoring of renal function is recommended. The bioavailability of CyA increases when the drug is taken with grapefruit juice. 4.4.7 Monitoring and supervision It is recommended that blood pressure be monitored at least weekly for the first month of therapy and then monthly thereafter. [2] In addition, laboratory monitoring is required monthly particularly screening for renal, bone marrow or hepatic effects. Dose adjustment is recommended if serum creatinine increases by >30%. The need for monitoring of CyA levels in non‐transplant patients is unclear. It is generally recommended that whole blood trough levels be maintained between 125 and 175μg/ml. [2] Monitoring may be particularly important in patients with unexpected toxicity. 4.4.8 Formulary Australian Brand names: Neoral® (caps 10, 25, 50 and 100mg, solution 100mg/ml), Sandimmun®; Cicloral®(caps 25, 50 and 100mg) U.S. Brand Names: Gengraf® (caps 25 and 100mg, solution 100mg/ml); Neoral® caps 25 and 100mg, solution 100mg/ml); Sandimmune® caps 25 and 100mg, solution 100mg/ml) Canadian Brand Names: Apo‐Cyclosporine; Neoral®; Rhoxal‐cyclosporine; Sandimmune® I.V.; Sandoz‐Cyclosporine 4.4.9 Summary recommendations Cyclosporin has a role in the management of paediatric rheumatic disease. Several observational studies provide evidence for its efficacy in treating JIA and in particular SoJIA and associated MAS and also in the treatment of JDM. Cyclosporin is a potent immunosuppressive agent and is associated with a number of potentially serious adverse effects. The administration of cyclosporin should be under specialist supervision with the facility to frequently monitor with blood tests. It is recommended for the WHO essential medicines complimentary list.
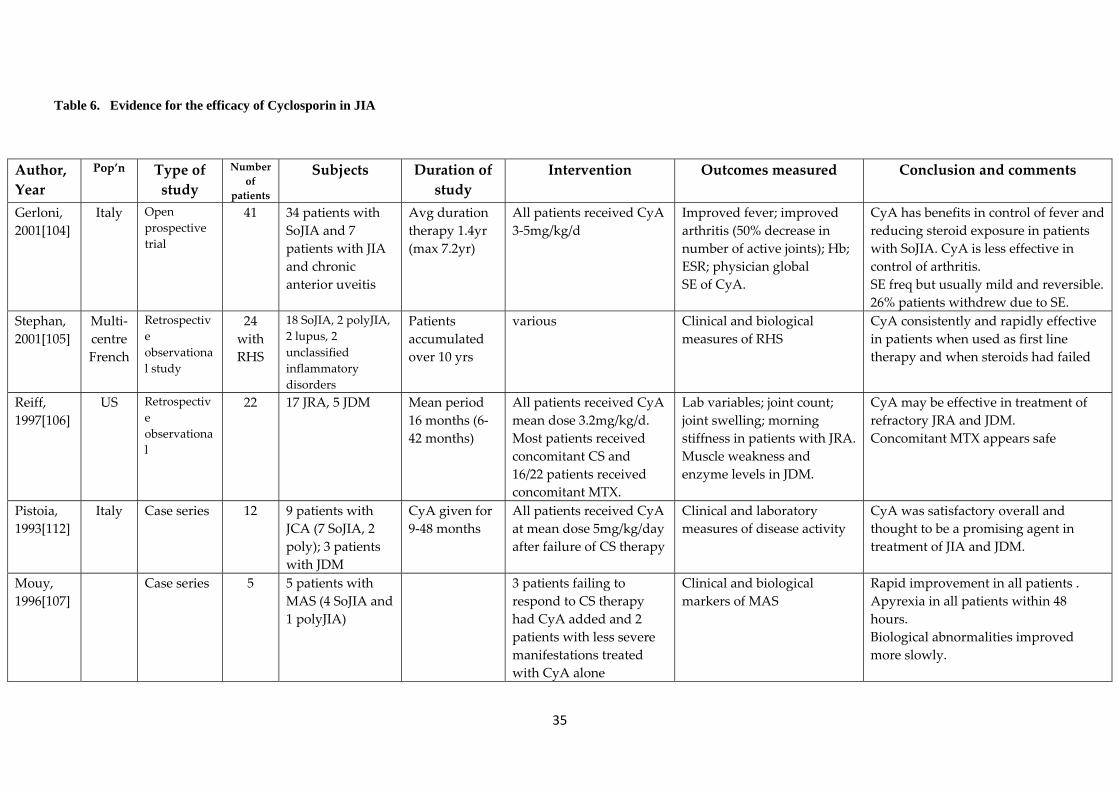
35
Author, Year
Pop’n Type of study
Number of
patients
Subjects Duration of study
Intervention Outcomes measured Conclusion and comments
Gerloni, 2001[104]
Italy Open prospective trial
41 34 patients with SoJIA and 7 patients with JIA and chronic anterior uveitis
Avg duration therapy 1.4yr (max 7.2yr)
All patients received CyA 3‐5mg/kg/d
Improved fever; improved arthritis (50% decrease in number of active joints); Hb; ESR; physician global SE of CyA.
CyA has benefits in control of fever and reducing steroid exposure in patients with SoJIA. CyA is less effective in control of arthritis. SE freq but usually mild and reversible. 26% patients withdrew due to SE.
Stephan, 2001[105]
Multi‐centre French
Retrospective observational study
24 with RHS
18 SoJIA, 2 polyJIA, 2 lupus, 2 unclassified inflammatory disorders
Patients accumulated over 10 yrs
various Clinical and biological measures of RHS
CyA consistently and rapidly effective in patients when used as first line therapy and when steroids had failed
Reiff, 1997[106]
US Retrospective observational
22 17 JRA, 5 JDM Mean period 16 months (6‐42 months)
All patients received CyA mean dose 3.2mg/kg/d. Most patients received concomitant CS and 16/22 patients received concomitant MTX.
Lab variables; joint count; joint swelling; morning stiffness in patients with JRA.Muscle weakness and enzyme levels in JDM.
CyA may be effective in treatment of refractory JRA and JDM. Concomitant MTX appears safe
Pistoia, 1993[112]
Italy Case series 12 9 patients with JCA (7 SoJIA, 2 poly); 3 patients with JDM
CyA given for 9‐48 months
All patients received CyA at mean dose 5mg/kg/day after failure of CS therapy
Clinical and laboratory measures of disease activity
CyA was satisfactory overall and thought to be a promising agent in treatment of JIA and JDM.
Mouy, 1996[107]
Case series 5 5 patients with MAS (4 SoJIA and 1 polyJIA)
3 patients failing to respond to CS therapy had CyA added and 2 patients with less severe manifestations treated with CyA alone
Clinical and biological markers of MAS
Rapid improvement in all patients . Apyrexia in all patients within 48 hours. Biological abnormalities improved more slowly.
Table 6. Evidence for the efficacy of Cyclosporin in JIA

36
Author/ Year
Pop’n Type of study
Number of
patients
Subjects Duration of study
Intervention Outcomes measured Conclusion and comments
Reiff, 1997[106]
US Retrospective
observational
22 17 JRA, 5 JDM Mean period 16 months (6‐42 months)
All patients received CyA mean dose 3.2mg/kg/d. Most patients received concomitant CS and 16/22 patients received concomitant MTX.
Lab variables; joint count; joint swelling; morning stiffness in patients with JRA.Muscle weakness and enzyme levels in JDM.
CyA may be effective in treatment of refractory JRA and JDM. Concomitant MTX appears safe
Pistoia, 1993[112]
Italy Case series 12 9 patients with JCA (7 SoJIA, 2 poly); 3 patients with JDM
CyA given for 9‐48 months
All patients received CyA at mean dose 5mg/kg/day after failure of CS therapy
Clinical and laboratory measures of disease activity
CyA was satisfactory overall and thought to be a promising agent in treatment of JIA and JDM.
Heckmatt, 1989[108]
UK Case series 14 14 patients with JDM not responded fully to CS and other immuno‐suppressants with chronically active disease avg 3 yrs
Average 12 months therapy to time of publication (5‐28 months)
CyA starting at 2.5 mg/kg/d divided bd and increased according to clinical response. Max 12mg/kg/d
Muscle weakness, extent of rash, serum CK, Monitoring of SE icluding BP, serum creatinine, CyA levels.
All patients improved on CyA. Recovery of muscle strength, resolution of complications and reduction or cessation of steroids. No serious SE observed. HT easily controlled with dose reduction.
Zeller, 1996[113]
Retrospective case review
6 Patients with refractory JDM
Mean follow up 51.5 months
CyA Clinical markers of disease. Medication SE, Steroid doses
CyA seems effective and should be considered in refractory cases. SE rare/minor
Table 7. Evidence for the efficacy of Cyclosporin in JDM

37
4.5 Azathioprine 4.5.1 Mechanism of action and Pharmacology Azathioprine is a purine analogue metabolised in the liver to its active metabolite 6‐mercaptopurine (6‐MP). 6‐MP is then further metabolised via three major pathways, the metabolites of which inhibit DNA synthesis through the suppression of guanine and adenine synthesis. Azathioprine inhibits cell mediated immunity through inhibition of T cell growth and results in reduced antibody production. The bioavailability of azathioprine is approximately 50%. [114] It is rapidly metabolised in the liver and its metabolite, 6‐MP, has a half life of 0.7 – 3 hours. Small amounts are eliminated as unchanged drug and metabolites are predominantly renally excreted. 4.5.2 Efficacy of Azathioprine in SLE Azathioprine has been used for the management of SLE in children for decades. Despite this long experience, there are no controlled trials addressing its efficacy in paediatric lupus and therefore its use remains controversial. Azathioprine is used in a variety of clinical circumstances in SLE but most frequently in combination with corticosteroids. The addition of azathioprine as second line therapy may be indicated to permit steroid dose reduction in patients requiring unacceptably high doses of corticosteroids. [2] In adult patients this was found to be associated with fewer hospitalisations.[115] The role of azathioprine in the management of paediatric lupus complicated by nephritis is even less well defined. There is conflicting evidence in the adult literature about its use in this context. Some studies are in favour of its use. A retrospective cohort study of 26 adult patients with SLE and associated proliferative lupus nephritis were managed long‐term with prednisolone and azathioprine. [116] This study concluded that azathioprine was well tolerated and that 5‐10 year survival rates were similar to those of cyclophosphamide‐based therapies. [116] One outcome study of children with lupus nephritis concluded that long‐term outcome was excellent with 94% survival at mean followup of 11 years. In this group of patients, azathioprine was the most frequently prescribed agent. [117] Although there is limited evidence for the efficacy of azathioprine in SLE and in particular paediatric SLE, many rheumatologists acknowledge that it does have a role in therapy. In particular, it is used to control serologic disease flares, allow reduction of steroid doses and maintain disease course after parenteral cyclophosphamide. [45]

38
4.5.3 Efficacy of Azathioprine in JIA The evidence for the efficacy of azathioprine is based on case series, case reports and uncontrolled trials. To date there is only one randomised double blind placebo controlled trial addressing the efficacy of azathioprine in the treatment of 32 JIA [118] Statistically significant improvement was only seen in two disease activity measurements and it is therefore not considered to have greater efficacy than placebo. It is not used routinely in the treatment of JIA. See table 8 for details of studies. An uncontrolled prospective trial involving 129 patients with refractory JCA demonstrated improvement in both clinical and laboratory outcomes with acceptable side effect profile. [119] A further retrospective study of 24 patients demonstrated clinical improvement in 62.5% of patients and remission in 37.5%. [120] Whilst the strength of this evidence is not as compelling as that of other DMARDs used in the treatment of JIA, these studies suggest that there may be a role of azathioprine in JIA. 4.5.4 Dose and administration Azathioprine should be started at 1‐1.5 mg/kg/day increasing to a maximum dose of 2‐2.5mg/kg/day. It is generally considered that a minimum of 12 weeks is required to achieve adequate therapeutic response. Azathioprine should be administered with food to reduce gastrointestinal disturbance. 4.5.5 Safety Gastrointestinal side effects manifesting as nausea, vomiting, diarrhoea and epigastric pain are common with azathioprine. These side effects can be diminished if the dose divided or if administered with meals. Up to 12 % of patients experience these symptoms and in one study 10% of adult patients with rheumatoid arthritis withdrew from therapy due to gastrointestinal intolerance. [121] Other less frequently observed side effects include: pancreatitis, liver toxicity (abnormal liver function tests), interstitial pneumonitis and rash. It is difficult to estimate the incidence of these side effects in the paediatric rheumatic disease population as studies of the use and safety of azathioprine are frequently in the adult population and in conditions other than rheumatic diseases. Dose dependent effects on the bone marrow have also been well described in association with azathioprine causing granulocyte arrest and leukopenia and, less frequently, anaemia and thrombocytopenia. This is thought to be due to reduced activity of thiopurine methyltransferase (TPMT). TMPT activity is low or absent in 0.3% of the population and causes these haematological effects shortly after initiation of therapy. Lower TMPT activity has been observed in African‐American individuals compared to Caucasian‐Americans. [122] Distinct TMPT mutations have been identified and are detectable on PCR‐based technique.

39
4.5.6 Drug interaction A number of medications, used concomitantly, have been reported to enhance the myelosuppressive effect of azathioprine including trimethoprim. 5‐ASA derivatives may decrease the metabolism of thiopurine analogs. The toxic effects of leflunomide may also be enhanced when used with azathioprine and require more stringent monitoring of bone marrow toxicity. Azathioprine may also decrease plasma cyclosporin levels. 4.5.7 Monitoring and supervision Toxicity monitoring should occur throughout treatment. It is generally recommended that clinical evaluation occur at 1‐2 months then 3 monthly thereafter or earlier if disease severity warrants. Laboratory monitoring should include: weekly FBE with differential until stable then monthly thereafter and monthly LFTs and UEC. [2] Analysis of TMPT genotype should be considered for patients who develop severe leukopenia on azathioprine. Azathioprine should be ceased if there is significant leukopenia, thrombocytopenia or elevated liver enzymes. 4.5.8 Formulary Australian brand name: Imuran® (tablets 25mg, 50mg, powder for reconstitution 50mg) UK brand names: Imuran® (tablets 25mg, 50mg, powder for reconstitution 50mg) US brand names: Imuran® (tablets 25mg, 50mg, powder for reconstitution 50mg); Azasan® Canadian brand names: Alti‐Azathioprine; Apo‐Azathioprine®; Gen‐Azathioprine; Imuran®; Mylan‐Azathioprine; Novo‐Azathioprine 4.5.9 Summary recommendations Azathioprine appears to have a role in the management of both paediatric SLE and JIA although conclusive strong evidence with RCTs is lacking. Azathioprine is associated with potentially serious side effects in particular relating to bone marrow suppression. Administration of azathioprine should be under the supervision of specialist care with access to appropriate clinical and laboratory monitoring. Azathioprine is recommended for inclusion on the complementary WHO essential medicines list.
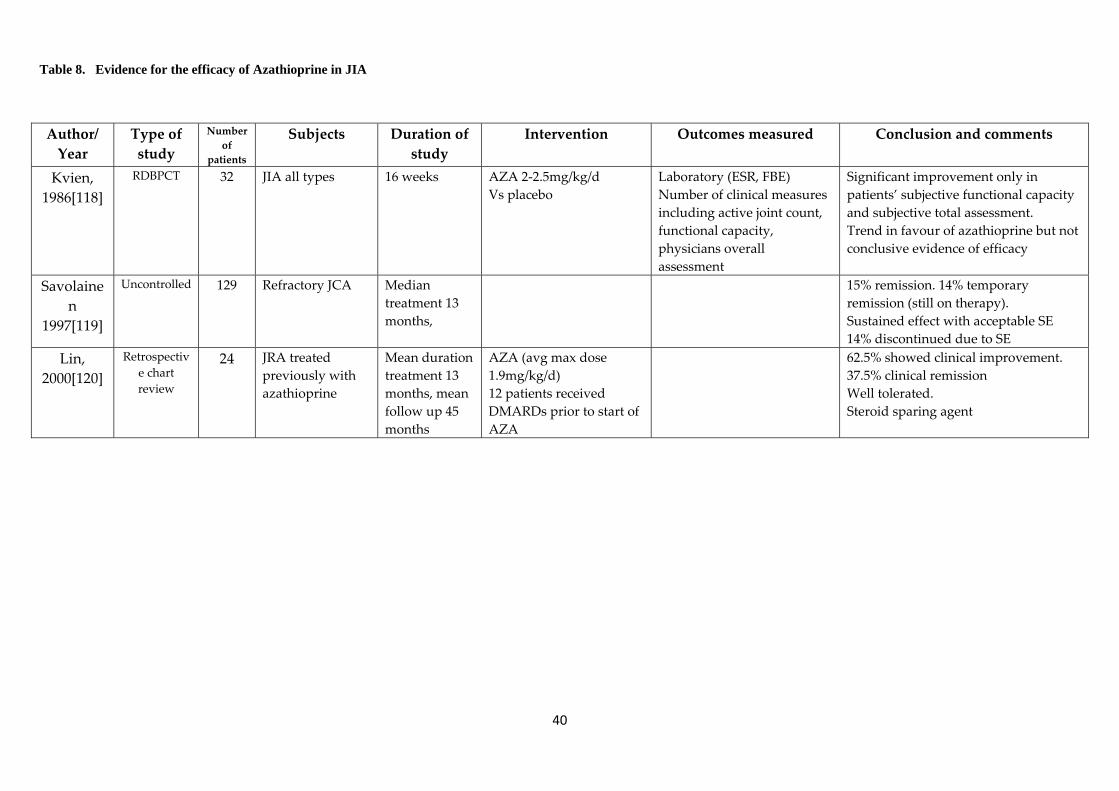
40
Author/ Year
Type of study
Number of
patients
Subjects Duration of study
Intervention Outcomes measured Conclusion and comments
Kvien, 1986[118]
RDBPCT 32 JIA all types 16 weeks AZA 2‐2.5mg/kg/d Vs placebo
Laboratory (ESR, FBE) Number of clinical measures including active joint count, functional capacity, physicians overall assessment
Significant improvement only in patients’ subjective functional capacity and subjective total assessment. Trend in favour of azathioprine but not conclusive evidence of efficacy
Savolainen
1997[119]
Uncontrolled 129 Refractory JCA Median treatment 13 months,
15% remission. 14% temporary remission (still on therapy). Sustained effect with acceptable SE 14% discontinued due to SE
Lin, 2000[120]
Retrospective chart review
24 JRA treated previously with azathioprine
Mean duration treatment 13 months, mean follow up 45 months
AZA (avg max dose 1.9mg/kg/d) 12 patients received DMARDs prior to start of AZA
62.5% showed clinical improvement. 37.5% clinical remission Well tolerated. Steroid sparing agent
Table 8. Evidence for the efficacy of Azathioprine in JIA

41
4.6 Hydroxychloroquine 4.6.1 Mechanism of action and Pharmacology Hydroxychloroquine is an antimalarial agent used in the treatment of rheumatic disease. Hydroxychloroquine inhibits the synthesis of DNA, RNA and protein by interacting with nucleic acid. Its immunosuppressive effect is via a number of mechanisms including: alteration in lysosomal pH and interference in antigen processing; stabilization of lysosomal membranes; inhibition of antigen‐antibody reactions; suppression of lymphocyte response and inhibition of neutrophil chemotaxis. [2] Hydroxychloroquine is rapidly absorbed from the intestine with an oral bioavailability of 74%. [123] The half‐life is 40 days, steady state is reached in 2 to 6 months and it is primarily renally excreted. [123] 4.6.2 Efficacy of Hydroxychloroquine in JIA Hydroxychloroquine has not been convincingly shown to be effective in the treatment of JIA. There is one placebo controlled trial addressing the efficacy of hydroxychloroquine in the treatment of JIA. [124] This randomised double blinded trial of 162 patients with polyarticular JIA compared hydroxychloroquine with penicillamine or placebo. There was no significant difference between groups. Kvien et al demonstrated in an open randomised trial (not placebo controlled) of 72 patients with oligo and polyarticular JIA, that hydroxychloroquine was not more efficacious than gold or penicillamine.[125] Interestingly, a meta‐analysis in 2000 of all RCTs and CCTs comparing hydroxychloroquine to placebo in adult patients with rheumatoid arthritis revealed an overall moderate effect and with a low toxicity profile. [126] 4.6.3 Efficacy of Hydroxychloroquine in paediatric SLE Hydroxychloroquine is frequently used as an adjunct to steroid therapy at the onset of treatment of juvenile SLE. However, there are no trials addressing the effectiveness of hydroxychloroquine in the paediatric population. Evidence for its efficacy was demonstrated in a randomised, double blinded, placebo controlled withdrawal trial by the Canadian Hydroxychloroquine Study Group. [127] Patients assigned to the placebo group had a significantly higher relative risk of flare and that the time to flare was shorter compared to those patients who continued HCQ. There have since been many other studies supporting its use in SLE. Recent systematic review demonstrated high levels of evidence that hydroxychloroquine prevents SLE flares an increases long term survival.[128] There was moderate evidence of protection against irreversible organ damage, thrombosis and bone mass loss. [128] Recommendations are that hydroxychloroquine should be commenced in most patients and continued throughout the course of the disease.

42
4.6.4 Dose and administration The usual dose of hydroxychloroquine in children is 3‐5 mg/kg kg/day divided 1‐2 times per day with a maximum of 400mg per day. It should be administered with food to reduce the occurrence of gastrointestinal intolerance. 4.6.5 Safety Hydroxychloroquine is generally considered to be a very safe medication. The frequency and severity of adverse effects is low even after prolonged use. There is a large amount of supporting evidence for this in the adult literature.[128] Gastrointestinal disturbance occurs in approximately 10% of adult patients. It is considered less common in children. Central nervous system side effects are common and include: headache, lightheadedness, insomnia, nervousness, nightmares and confusion. The most concerning side effect is ocular toxicity. This occurs rarely but can cause blindness. Retinal toxicity is thought not to occur in adults if the dose remains less than 6.5mg/kg/day.[129] Protocols and practices vary world wide, however it is generally recommended that ophthalmological examinations are performed yearly. Local protocols should be established between the prescribing physician and the treating ophthalmologist. Retinal abnormalities should prompt immediate cessation of the medication. Hydroxychloroquine is considered safe in pregnancy and breast feeding. 4.6.6 Drug interaction Hydroxychloroquine levels may be decreased by Echinacea. In addition there are a number of medications that should be avoided or monitored carefully or increased or decreased effect when used concomitantly with hydroxychloroquine. These include: beta blockers, anti‐psychotics and others. 4.6.7 Monitoring and supervision Whilst hydroxychloroquine is a safe medication, it does need regular monitoring. Full blood examination is suggested on a regular basis and ophthalmology examination should occur at least yearly. 4.6.8 Formulary Hydroxychloroquine is not FDA approved for the use in paediatric SLE. In the UK hydroxychloroquine is licensed for use in JIA, SLE and discoid LE and other dermatological conditions caused by or aggravated by the sun. Australian brand names: Plaquenil® (200mg tablets) UK brand names: Plaquenil® (200mg tablets) US brand names: Plaquenil®

43
Canadian brand names: Apo‐Hydroxyquine®; Gen‐Hydroxychloroquine; Plaquenil®; Pro‐Hydroxyquine 4.6.9 Summary recommendations Hydroxychloroquine appears to be a safe and effective treatment in the management of juvenile SLE. It has no proven role in the management of JIA. Hydroxychloroquine is recommended for the use in juvenile SLE provided patients have access to specialist care including ophthalmological and laboratory assessments. It is recommended for inclusion on the WHO essential medicines complementary list.

44
BIBLIOGRAPHY 1. Petty, R.E., et al., International League of Associations for Rheumatology classification of juvenile idiopathic
arthritis: second revision, Edmonton, 2001. J Rheumatol, 2004. 31(2): p. 390‐2.
2. Cassidy J.T., P., R. E., Laxer R.M., Lindsley C.B, Textbook of Pediatric Rheumatology. 5th ed. 2005: Elsevier Saunders.
3. Manners, P.J. and C. Bower, Worldwide prevalence of juvenile arthritis why does it vary so much? J Rheumatol, 2002. 29(7): p. 1520‐30.
4. Mielants, H., et al., Prevalence of inflammatory rheumatic diseases in an adolescent urban student population, age 12 to 18, in Belgium. Clin Exp Rheumatol, 1993. 11(5): p. 563‐7.
5. Manners, P.J. and D.A. Diepeveen, Prevalence of juvenile chronic arthritis in a population of 12‐year‐old children in urban Australia. Pediatrics, 1996. 98(1): p. 84‐90.
6. Tayel, M.Y. and K.Y. Tayel, Prevalence of juvenile chronic arthritis in school children aged 10 to 15 years in Alexandria. J Egypt Public Health Assoc, 1999. 74(5‐6): p. 529‐46.
7. Petty, R.E., Frequency of uncommon diseases: is juvenile idiopathic arthritis underrecognized? J Rheumatol, 2002. 29(7): p. 1356‐7.
8. Hochberg, M.C., M.S. Linet, and E.M. Sills, The prevalence and incidence of juvenile rheumatoid arthritis in an urban Black population. Am J Public Health, 1983. 73(10): p. 1202‐3.
9. Kurahara, D., et al., Ethnic differences in risk for pediatric rheumatic illness in a culturally diverse population. J Rheumatol, 2002. 29(2): p. 379‐83.
10. Kurahara, D.K., et al., Visiting consultant clinics to study prevalence rates of juvenile rheumatoid arthritis and childhood systemic lupus erythematosus across dispersed geographic areas. J Rheumatol, 2007. 34(2): p. 425‐9.
11. Cassidy, J.T., et al., A study of classification criteria for a diagnosis of juvenile rheumatoid arthritis. Arthritis Rheum, 1986. 29(2): p. 274‐81.
12. Oen, K., et al., Early predictors of longterm outcome in patients with juvenile rheumatoid arthritis: subset‐specific correlations. J Rheumatol, 2003. 30(3): p. 585‐93.
13. Zak, M. and F.K. Pedersen, Juvenile chronic arthritis into adulthood: a long‐term follow‐up study. Rheumatology (Oxford), 2000. 39(2): p. 198‐204.
14. Hashkes, P.J. and R.M. Laxer, Medical treatment of juvenile idiopathic arthritis. JAMA, 2005. 294(13): p. 1671‐84.
15. Adib, N., A. Silman, and W. Thomson, Outcome following onset of juvenile idiopathic inflammatory arthritis: I. frequency of different outcomes. Rheumatology (Oxford), 2005. 44(8): p. 995‐1001.
16. Packham, J.C. and M.A. Hall, Long‐term follow‐up of 246 adults with juvenile idiopathic arthritis: functional outcome. Rheumatology (Oxford), 2002. 41(12): p. 1428‐35.
17. Goldenberg, J., et al., Symptomatic cardiac involvement in juvenile rheumatoid arthritis. Int J Cardiol, 1992. 34(1): p. 57‐62.
18. Sawhney, S., P. Woo, and K.J. Murray, Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child, 2001. 85(5): p. 421‐6.
19. Behrens, E.M., et al., Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol, 2007. 34(5): p. 1133‐8.

45
20. Ravelli, A., et al., Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr, 2005. 146(5): p. 598‐604.
21. Kanski, J.J. and G.A. Shun‐Shin, Systemic uveitis syndromes in childhood: an analysis of 340 cases. Ophthalmology, 1984. 91(10): p. 1247‐52.
22. Kump, L.I., et al., Analysis of pediatric uveitis cases at a tertiary referral center. Ophthalmology, 2005. 112(7): p. 1287‐92.
23. Ramanan, A.V. and B.M. Feldman, Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin North Am, 2002. 28(4): p. 833‐57.
24. McCann, L.J., et al., The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)‐‐clinical characteristics of children recruited within the first 5 yr. Rheumatology (Oxford), 2006. 45(10): p. 1255‐60.
25. Feldman, B.M., et al., Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet, 2008. 371(9631): p. 2201‐12.
26. Mendez, E.P., et al., US incidence of juvenile dermatomyositis, 1995‐1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis Rheum, 2003. 49(3): p. 300‐5.
27. Symmons, D.P., J.A. Sills, and S.M. Davis, The incidence of juvenile dermatomyositis: results from a nation‐wide study. Br J Rheumatol, 1995. 34(8): p. 732‐6.
28. Pachman, L.M., et al., Juvenile dermatomyositis at diagnosis: clinical characteristics of 79 children. J Rheumatol, 1998. 25(6): p. 1198‐204.
29. Bohan, A. and J.B. Peter, Polymyositis and dermatomyositis (first of two parts). N Engl J Med, 1975. 292(7): p. 344‐7.
30. Blane, C.E., et al., Patterns of calcification in childhood dermatomyositis. AJR Am J Roentgenol, 1984. 142(2): p. 397‐400.
31. Fisler, R.E., et al., Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol, 2002. 47(4): p. 505‐11.
32. Crowe, W.E., et al., Clinical and pathogenetic implications of histopathology in childhood polydermatomyositis. Arthritis Rheum, 1982. 25(2): p. 126‐39.
33. Pachman, L.M. and N. Cooke, Juvenile dermatomyositis: a clinical and immunologic study. J Pediatr, 1980. 96(2): p. 226‐34.
34. Stringer, E., D. Singh‐Grewal, and B.M. Feldman, Predicting the course of juvenile dermatomyositis: significance of early clinical and laboratory features. Arthritis Rheum, 2008. 58(11): p. 3585‐92.
35. Laxer, R.M., L.D. Stein, and R.E. Petty, Intravenous pulse methylprednisolone treatment of juvenile dermatomyositis. Arthritis Rheum, 1987. 30(3): p. 328‐34.
36. Huber, A.M., et al., Medium‐ and long‐term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthritis Rheum, 2000. 43(3): p. 541‐9.
37. Tucker, L.B., et al., Adult‐ and childhood‐onset systemic lupus erythematosus: a comparison of onset, clinical features, serology, and outcome. Br J Rheumatol, 1995. 34(9): p. 866‐72.
38. Ferraz, M.B., et al., Evaluation of the 1982 ARA lupus criteria data set in pediatric patients. Committees of Pediatric Rheumatology of the Brazilian Society of Pediatrics and the Brazilian Society of Rheumatology. Clin Exp Rheumatol, 1994. 12(1): p. 83‐7.

46
39. Norris, D.G., A.R. Colon, and G.B. Stickler, Systemic lupus erythematosus in children: the complex problems of diagnosis and treatment encountered in 101 such patients at the Mayo Clinic. Clin Pediatr (Phila), 1977. 16(9): p. 774‐8.
40. Hiraki, L.T., et al., Clinical and laboratory characteristics and long‐term outcome of pediatric systemic lupus erythematosus: a longitudinal study. J Pediatr, 2008. 152(4): p. 550‐6.
41. Tucker, L.B., Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus, 2007. 16(8): p. 546‐9.
42. Brunner, H.I., et al., Difference in disease features between childhood‐onset and adult‐onset systemic lupus erythematosus. Arthritis Rheum, 2008. 58(2): p. 556‐62.
43. Meislin, A.G. and N. Rothfield, Systemic lupus erythematosus in childhood. Analysis of 42 cases, with comparative data on 200 adult cases followed concurrently. Pediatrics, 1968. 42(1): p. 37‐49.
44. Yang, L.Y., W.P. Chen, and C.Y. Lin, Lupus nephritis in children‐‐a review of 167 patients. Pediatrics, 1994. 94(3): p. 335‐40.
45. Tucker, L.B., Controversies and advances in the management of systemic lupus erythematosus in children and adolescents. Best Pract Res Clin Rheumatol, 2002. 16(3): p. 471‐80.
46. Lee, Y., et al., Induction and maintenance therapy for lupus nephritis: a systematic review and meta‐analysis. Lupus.
47. Macdermott, E.J., A. Adams, and T.J. Lehman, Systemic lupus erythematosus in children: current and emerging therapies. Lupus, 2007. 16(8): p. 677‐83.
48. Ravelli, A., et al., Plasma levels after oral methotrexate in children with juvenile rheumatoid arthritis. J Rheumatol, 1993. 20(9): p. 1573‐7.
49. Godfrey, C., et al., The population pharmacokinetics of long‐term methotrexate in rheumatoid arthritis. Br J Clin Pharmacol, 1998. 46(4): p. 369‐76.
50. Hoekstra, M., et al., Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol, 2004. 31(4): p. 645‐8.
51. Hamilton, R.A. and J.M. Kremer, Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. Br J Rheumatol, 1997. 36(1): p. 86‐90.
52. Giannini, E.H., et al., Comparative efficacy and safety of advanced drug therapy in children with juvenile rheumatoid arthritis. Semin Arthritis Rheum, 1993. 23(1): p. 34‐46.
53. Ruperto, N., et al., A randomized trial of parenteral methotrexate comparing an intermediate dose with a higher dose in children with juvenile idiopathic arthritis who failed to respond to standard doses of methotrexate. Arthritis Rheum, 2004. 50(7): p. 2191‐201.
54. Woo, P., et al., Randomized, placebo‐controlled, crossover trial of low‐dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum, 2000. 43(8): p. 1849‐57.
55. Giannini, E.H., et al., Methotrexate in resistant juvenile rheumatoid arthritis. Results of the U.S.A.‐U.S.S.R. double‐blind, placebo‐controlled trial. The Pediatric Rheumatology Collaborative Study Group and The Cooperative Childrenʹs Study Group. N Engl J Med, 1992. 326(16): p. 1043‐9.
56. Takken, T., J. Van Der Net, and P.J. Helders, Methotrexate for treating juvenile idiopathic arthritis. Cochrane Database Syst Rev, 2001(4): p. CD003129.
57. Ravelli, A., et al., The extended oligoarticular subtype is the best predictor of methotrexate efficacy in juvenile idiopathic arthritis. J Pediatr, 1999. 135(3): p. 316‐20.

47
58. Gottlieb, B.S., et al., Discontinuation of methotrexate treatment in juvenile rheumatoid arthritis. Pediatrics, 1997. 100(6): p. 994‐7.
59. Cespedes‐Cruz, A., et al., Methotrexate improves the health‐related quality of life of children with juvenile idiopathic arthritis. Ann Rheum Dis, 2008. 67(3): p. 309‐14.
60. Silverman, E., et al., Leflunomide or methotrexate for juvenile rheumatoid arthritis. N Engl J Med, 2005. 352(16): p. 1655‐66.
61. al‐Sewairy, W., et al., Methotrexate therapy in systemic‐onset juvenile rheumatoid arthritis in Saudi Arabia: a retrospective analysis. Clin Rheumatol, 1998. 17(1): p. 52‐7.
62. Huang, J.L., Methotrexate in the treatment of children with chronic arthritis‐‐long‐term observations of efficacy and safety. Br J Clin Pract, 1996. 50(6): p. 311‐4.
63. Reiff, A., et al., High dose methotrexate in the treatment of refractory juvenile rheumatoid arthritis. Clin Exp Rheumatol, 1995. 13(1): p. 113‐8.
64. Ravelli, A., et al., Factors associated with response to methotrexate in systemic‐onset juvenile chronic arthritis. Acta Paediatr, 1994. 83(4): p. 428‐32.
65. Harel, L., et al., Effects of methotrexate on radiologic progression in juvenile rheumatoid arthritis. Arthritis Rheum, 1993. 36(10): p. 1370‐4.
66. Wallace, C.A., et al., Predicting remission in juvenile rheumatoid arthritis with methotrexate treatment. J Rheumatol, 1993. 20(1): p. 118‐22.
67. Huber, A.M., et al., Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Childrenʹs Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res (Hoboken). 62(2): p. 219‐25.
68. Ramanan, A.V., et al., The effectiveness of treating juvenile dermatomyositis with methotrexate and aggressively tapered corticosteroids. Arthritis Rheum, 2005. 52(11): p. 3570‐8.
69. Al‐Mayouf, S., A. Al‐Mazyed, and S. Bahabri, Efficacy of early treatment of severe juvenile dermatomyositis with intravenous methylprednisolone and methotrexate. Clin Rheumatol, 2000. 19(2): p. 138‐41.
70. Miller, L.C., et al., Methotrexate treatment of recalcitrant childhood dermatomyositis. Arthritis Rheum, 1992. 35(10): p. 1143‐9.
71. Foeldvari, I. and A. Wierk, Methotrexate is an effective treatment for chronic uveitis associated with juvenile idiopathic arthritis. J Rheumatol, 2005. 32(2): p. 362‐5.
72. Weiss, A.H., C.A. Wallace, and D.D. Sherry, Methotrexate for resistant chronic uveitis in children with juvenile rheumatoid arthritis. J Pediatr, 1998. 133(2): p. 266‐8.
73. Malik, A.R. and C. Pavesio, The use of low dose methotrexate in children with chronic anterior and intermediate uveitis. Br J Ophthalmol, 2005. 89(7): p. 806‐8.
74. Jundt, J.W., et al., A comparison of low dose methotrexate bioavailability: oral solution, oral tablet, subcutaneous and intramuscular dosing. J Rheumatol, 1993. 20(11): p. 1845‐9.
75. Ortiz, Z., et al., The efficacy of folic acid and folinic acid in reducing methotrexate gastrointestinal toxicity in rheumatoid arthritis. A metaanalysis of randomized controlled trials. J Rheumatol, 1998. 25(1): p. 36‐43.
76. Ortiz‐Alvarez, O., et al., Guidelines for blood test monitoring of methotrexate toxicity in juvenile idiopathic arthritis. J Rheumatol, 2004. 31(12): p. 2501‐6.
77. Lahdenne, P., et al., Hepatotoxicity in patients with juvenile idiopathic arthritis receiving longterm methotrexate therapy. J Rheumatol, 2002. 29(11): p. 2442‐5.

48
78. Graham, L.D., et al., Morbidity associated with long‐term methotrexate therapy in juvenile rheumatoid arthritis. J Pediatr, 1992. 120(3): p. 468‐73.
79. Hashkes, P.J., et al., The relationship of hepatotoxic risk factors and liver histology in methotrexate therapy for juvenile rheumatoid arthritis. J Pediatr, 1999. 134(1): p. 47‐52.
80. Hashkes, P.J., et al., The long‐term effect of methotrexate therapy on the liver in patients with juvenile rheumatoid arthritis. Arthritis Rheum, 1997. 40(12): p. 2226‐34.
81. Kugathasan, S., et al., Liver biopsy findings in patients with juvenile rheumatoid arthritis receiving long‐term, weekly methotrexate therapy. J Pediatr, 1996. 128(1): p. 149‐51.
82. West, S.G., Methotrexate hepatotoxicity. Rheum Dis Clin North Am, 1997. 23(4): p. 883‐915.
83. Singsen, B.H. and R. Goldbach‐Mansky, Methotrexate in the treatment of juvenile rheumatoid arthritis and other pediatric rheumatoid and nonrheumatic disorders. Rheum Dis Clin North Am, 1997. 23(4): p. 811‐40.
84. Niehues, T. and P. Lankisch, Recommendations for the use of methotrexate in juvenile idiopathic arthritis. Paediatr Drugs, 2006. 8(6): p. 347‐56.
85. Padeh, S., et al., Hodgkinʹs lymphoma in systemic onset juvenile rheumatoid arthritis after treatment with low dose methotrexate. J Rheumatol, 1997. 24(10): p. 2035‐7.
86. Londino, A.V., Jr., J. Blatt, and A.S. Knisely, Hodgkinʹs disease in a patient with juvenile rheumatoid arthritis taking weekly low dose methotrexate. J Rheumatol, 1998. 25(6): p. 1245‐6.
87. Kremer, J.M., et al., Methotrexate for rheumatoid arthritis. Suggested guidelines for monitoring liver toxicity. American College of Rheumatology. Arthritis Rheum, 1994. 37(3): p. 316‐28.
88. Rozman, B., Clinical pharmacokinetics of leflunomide. Clin Pharmacokinet, 2002. 41(6): p. 421‐30.
89. Shi, J., et al., Population pharmacokinetics of the active metabolite of leflunomide in pediatric subjects with polyarticular course juvenile rheumatoid arthritis. J Pharmacokinet Pharmacodyn, 2005. 32(3‐4): p. 419‐39.
90. Silverman, E., et al., Long‐term open‐label preliminary study of the safety and efficacy of leflunomide in patients with polyarticular‐course juvenile rheumatoid arthritis. Arthritis Rheum, 2005. 52(2): p. 554‐62.
91. Gao, J.S., H. Wu, and J. Tian, [Treatment of patients with juvenile rheumatoid arthritis with combination of leflunomide and methotrexate]. Zhonghua Er Ke Za Zhi, 2003. 41(6): p. 435‐8.
92. Cannon, G.W. and J.M. Kremer, Leflunomide. Rheum Dis Clin North Am, 2004. 30(2): p. 295‐309, vi.
93. van Rossum, M.A., et al., Sulfasalazine in the treatment of juvenile chronic arthritis: a randomized, double‐blind, placebo‐controlled, multicenter study. Dutch Juvenile Chronic Arthritis Study Group. Arthritis Rheum, 1998. 41(5): p. 808‐16.
94. Burgos‐Vargas, R., et al., A 26 week randomised, double blind, placebo controlled exploratory study of sulfasalazine in juvenile onset spondyloarthropathies. Ann Rheum Dis, 2002. 61(10): p. 941‐2.
95. Ansell, B.M., et al., A multicentre pilot study of sulphasalazine in juvenile chronic arthritis. Clin Exp Rheumatol, 1991. 9(2): p. 201‐3.
96. Huang, J.L. and L.C. Chen, Sulphasalazine in the treatment of children with chronic arthritis. Clin Rheumatol, 1998. 17(5): p. 359‐63.
97. van Rossum, M.A., et al., Long‐term outcome of juvenile idiopathic arthritis following a placebo‐controlled trial: sustained benefits of early sulfasalazine treatment. Ann Rheum Dis, 2007. 66(11): p. 1518‐24.
98. Hoza, J., et al., Sulphasalazine and Delagil‐‐a comparative study in patients with juvenile chronic arthritis. Acta Univ Carol Med (Praha), 1991. 37(1‐2): p. 80‐3.

49
99. Ozdogan, H., et al., Sulphasalazine in the treatment of juvenile rheumatoid arthritis: a preliminary open trial. J Rheumatol, 1986. 13(1): p. 124‐5.
100. Joos, R., et al., Sulfasalazine treatment in juvenile chronic arthritis: an open study. J Rheumatol, 1991. 18(6): p. 880‐4.
101. Imundo, L.F. and J.C. Jacobs, Sulfasalazine therapy for juvenile rheumatoid arthritis. J Rheumatol, 1996. 23(2): p. 360‐6.
102. Brooks, C.D., Sulfasalazine for the management of juvenile rheumatoid arthritis. J Rheumatol, 2001. 28(4): p. 845‐53.
103. Marouf, E.S. and I.M. Morris, Neutropenia in patients with rheumatoid arthritis, treated with sulphasalazine. Br J Rheumatol, 1990. 29(5): p. 407‐9.
104. Gerloni, V., et al., Efficacy and safety profile of cyclosporin A in the treatment of juvenile chronic (idiopathic) arthritis. Results of a 10‐year prospective study. Rheumatology (Oxford), 2001. 40(8): p. 907‐13.
105. Stephan, J.L., et al., Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology (Oxford), 2001. 40(11): p. 1285‐92.
106. Reiff, A., et al., Preliminary evidence for cyclosporin A as an alternative in the treatment of recalcitrant juvenile rheumatoid arthritis and juvenile dermatomyositis. J Rheumatol, 1997. 24(12): p. 2436‐43.
107. Mouy, R., et al., Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr, 1996. 129(5): p. 750‐4.
108. Heckmatt, J., et al., Cyclosporin in juvenile dermatomyositis. Lancet, 1989. 1(8646): p. 1063‐6.
109. Ostensen, M., H.M. Hoyeraal, and E. Kass, Tolerance of cyclosporine A in children with refractory juvenile rheumatoid arthritis. J Rheumatol, 1988. 15(10): p. 1536‐8.
110. Palestine, A.G., et al., Renal histopathologic alterations in patients treated with cyclosporine for uveitis. N Engl J Med, 1986. 314(20): p. 1293‐8.
111. Robert, N., G.W. Wong, and J.M. Wright, Effect of cyclosporine on blood pressure. Cochrane Database Syst Rev, (1): p. CD007893.
112. Pistoia, V., et al., Cyclosporin A in the treatment of juvenile chronic arthritis and childhood polymyositis‐dermatomyositis. Results of a preliminary study. Clin Exp Rheumatol, 1993. 11(2): p. 203‐8.
113. Zeller, V., et al., Cyclosporin a therapy in refractory juvenile dermatomyositis. Experience and longterm followup of 6 cases. J Rheumatol, 1996. 23(8): p. 1424‐7.
114. Sandborn, W.J., Azathioprine: state of the art in inflammatory bowel disease. Scand J Gastroenterol Suppl, 1998. 225: p. 92‐9.
115. Ginzler, E., et al., Long‐term maintenance therapy with azathioprine in systemic lupus erythematosus. Arthritis Rheum, 1975. 18(1): p. 27‐34.
116. Nossent, H.C. and W. Koldingsnes, Long‐term efficacy of azathioprine treatment for proliferative lupus nephritis. Rheumatology (Oxford), 2000. 39(9): p. 969‐74.
117. Hagelberg, S., et al., Longterm followup of childhood lupus nephritis. J Rheumatol, 2002. 29(12): p. 2635‐42.
118. Kvien, T.K., H.M. Hoyeraal, and B. Sandstad, Azathioprine versus placebo in patients with juvenile rheumatoid arthritis: a single center double blind comparative study. J Rheumatol, 1986. 13(1): p. 118‐23.
119. Savolainen, H.A., et al., Azathioprine in patients with juvenile chronic arthritis: a longterm followup study. J Rheumatol, 1997. 24(12): p. 2444‐50.

50
120. Lin, Y.T., et al., Long‐term effects of azathioprine therapy for juvenile rheumatoid arthritis. J Formos Med Assoc, 2000. 99(4): p. 330‐5.
121. Singh, G., et al., Toxicity profiles of disease modifying antirheumatic drugs in rheumatoid arthritis. J Rheumatol, 1991. 18(2): p. 188‐94.
122. McLeod, H.L. and C. Siva, The thiopurine S‐methyltransferase gene locus ‐‐ implications for clinical pharmacogenomics. Pharmacogenomics, 2002. 3(1): p. 89‐98.
123. Tett, S.E., et al., Bioavailability of hydroxychloroquine tablets in healthy volunteers. Br J Clin Pharmacol, 1989. 27(6): p. 771‐9.
124. Brewer, E.J., et al., Penicillamine and hydroxychloroquine in the treatment of severe juvenile rheumatoid arthritis. Results of the U.S.A.‐U.S.S.R. double‐blind placebo‐controlled trial. N Engl J Med, 1986. 314(20): p. 1269‐76.
125. Kvien, T.K., H.M. Hoyeraal, and B. Sandstad, Slow acting antirheumatic drugs in patients with juvenile rheumatoid arthritis‐‐evaluated in a randomized, parallel 50‐week clinical trial. J Rheumatol, 1985. 12(3): p. 533‐9.
126. Suarez‐Almazor, M.E., et al., Antimalarials for treating rheumatoid arthritis. Cochrane Database Syst Rev, 2000(4): p. CD000959.
127. A randomized study of the effect of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. The Canadian Hydroxychloroquine Study Group. N Engl J Med, 1991. 324(3): p. 150‐4.
128. Ruiz‐Irastorza, G., et al., Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis. 69(1): p. 20‐8.
129. Easterbrook, M., Current concepts in monitoring patients on antimalarials. Aust N Z J Ophthalmol, 1998. 26(2): p. 101‐3.