rnaseqによる変動遺伝子抽出の統計: a review
TRANSCRIPT

2
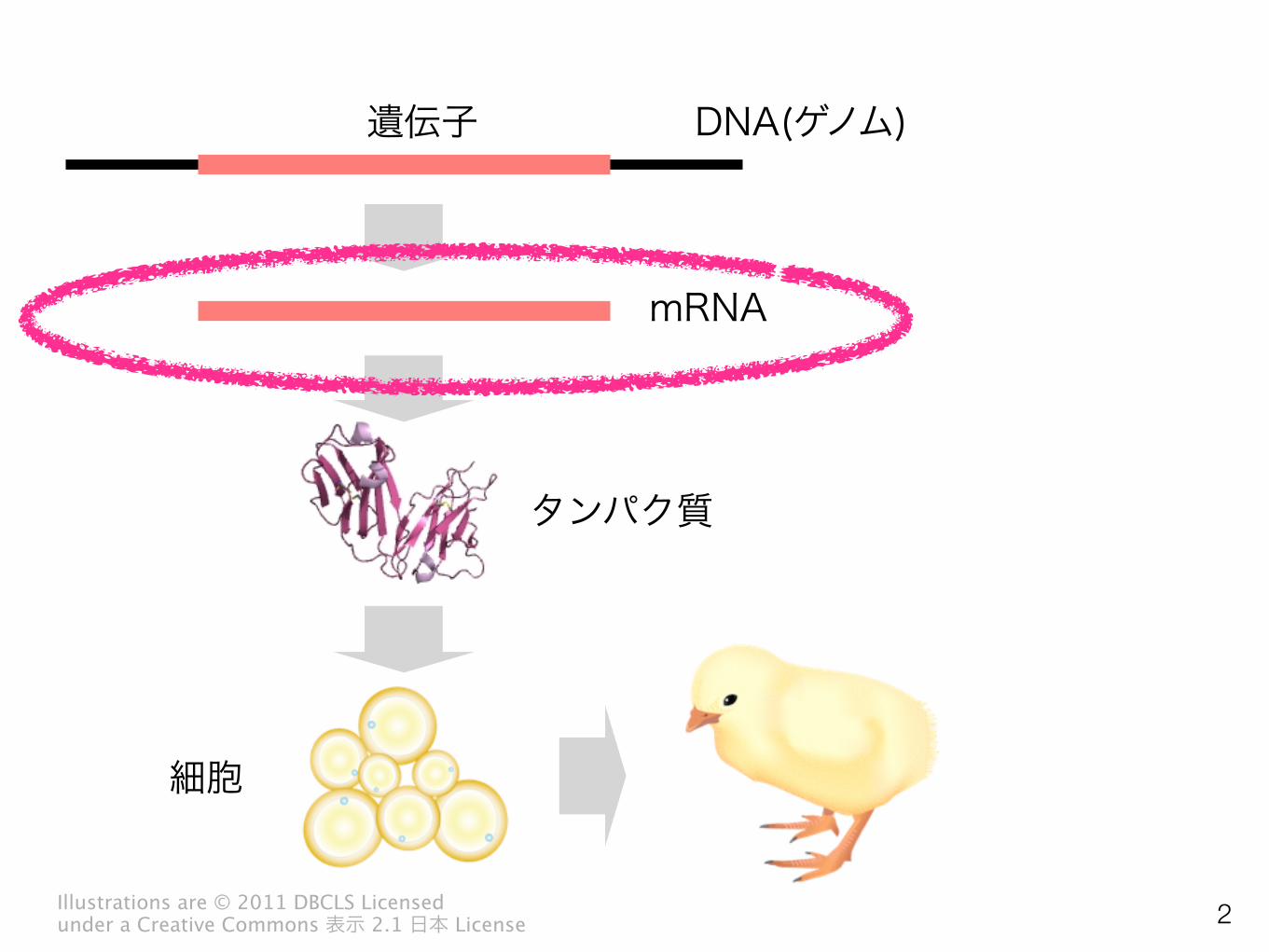
DNA(ゲノム)
mRNA
遺伝子
タンパク質
細胞
Illustrations are © 2011 DBCLS Licensed under a Creative Commons 表示 2.1 日本 License

大規模発現量観測の小史✓各遺伝子由来のタグの計数 ✓ランダムに抽出したmRNA中の,特定の部位の配列を特定し,遺伝子発現量の推定を行う方法
✓ SAGE [Velculescu et al. 1995], BodyMap [Kawamoto et al. 2000] ✓ 2003年頃の段階で,定量性を得るために,一つの組織から100万タグを超えるデータを取ることを目標にしていた.
✓ CAGE [Shiraki et al. 2003], 5’-SAGE [Hashimoto et al. 2004] ✓ RNA-seq [Ryan et al., 2008, Maher et al. 2009]
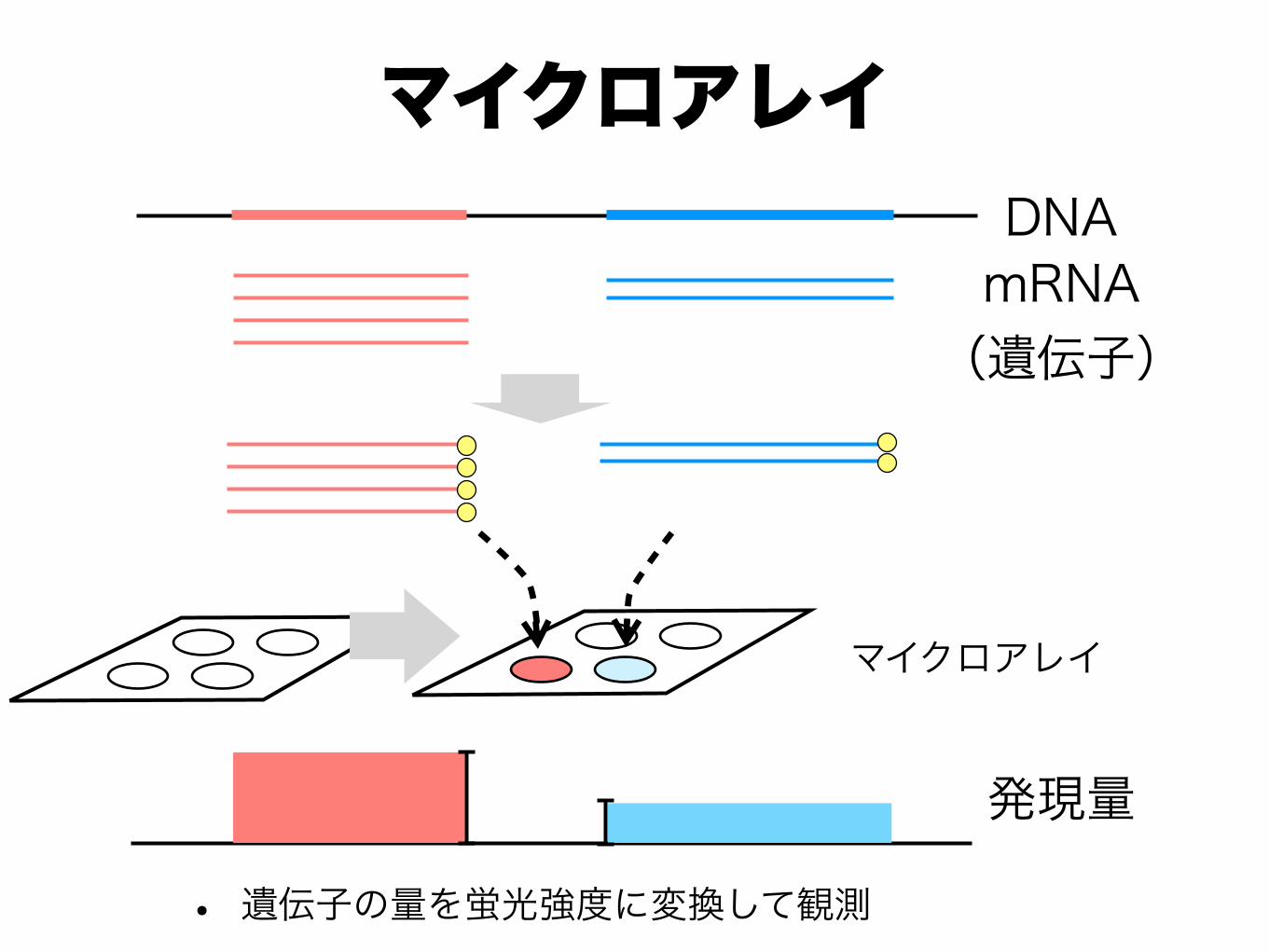
✓ハイブリベースの方法 ✓予め,各遺伝子に対応したプローブを用意しておき,そこにハイブリしたmRNAタグの量を,蛍光量などに変換して,観測する
✓マイクロアレイ [Tse-Wen, 1983, Schena et al. 1995]
マイクロアレイ
• 遺伝子の量を蛍光強度に変換して観測
マイクロアレイ
DNAmRNA (遺伝子)
発現量
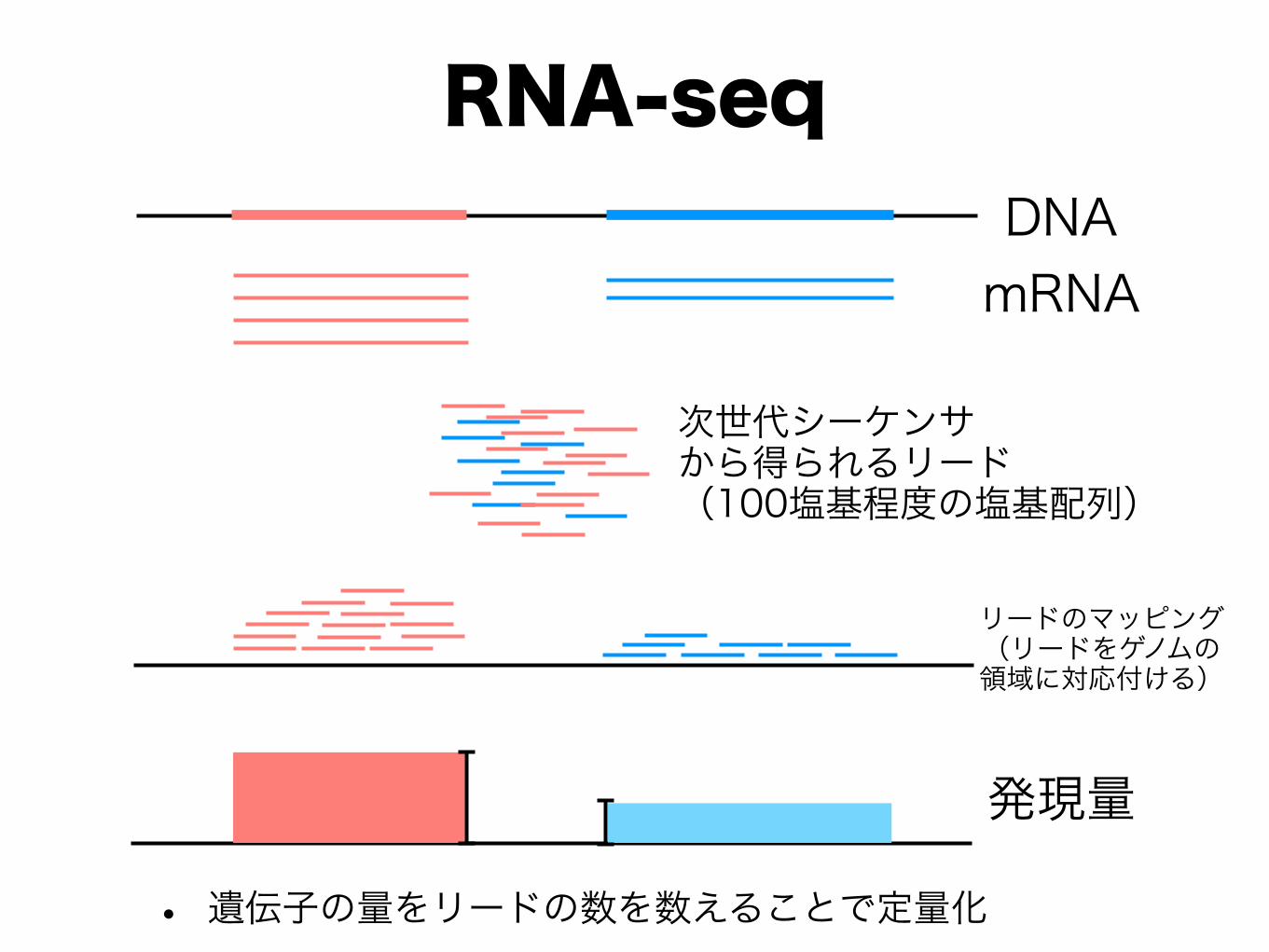
RNA-seq
• 遺伝子の量をリードの数を数えることで定量化
DNAmRNA
次世代シーケンサ から得られるリード (100塩基程度の塩基配列)
リードのマッピング (リードをゲノムの 領域に対応付ける)
発現量
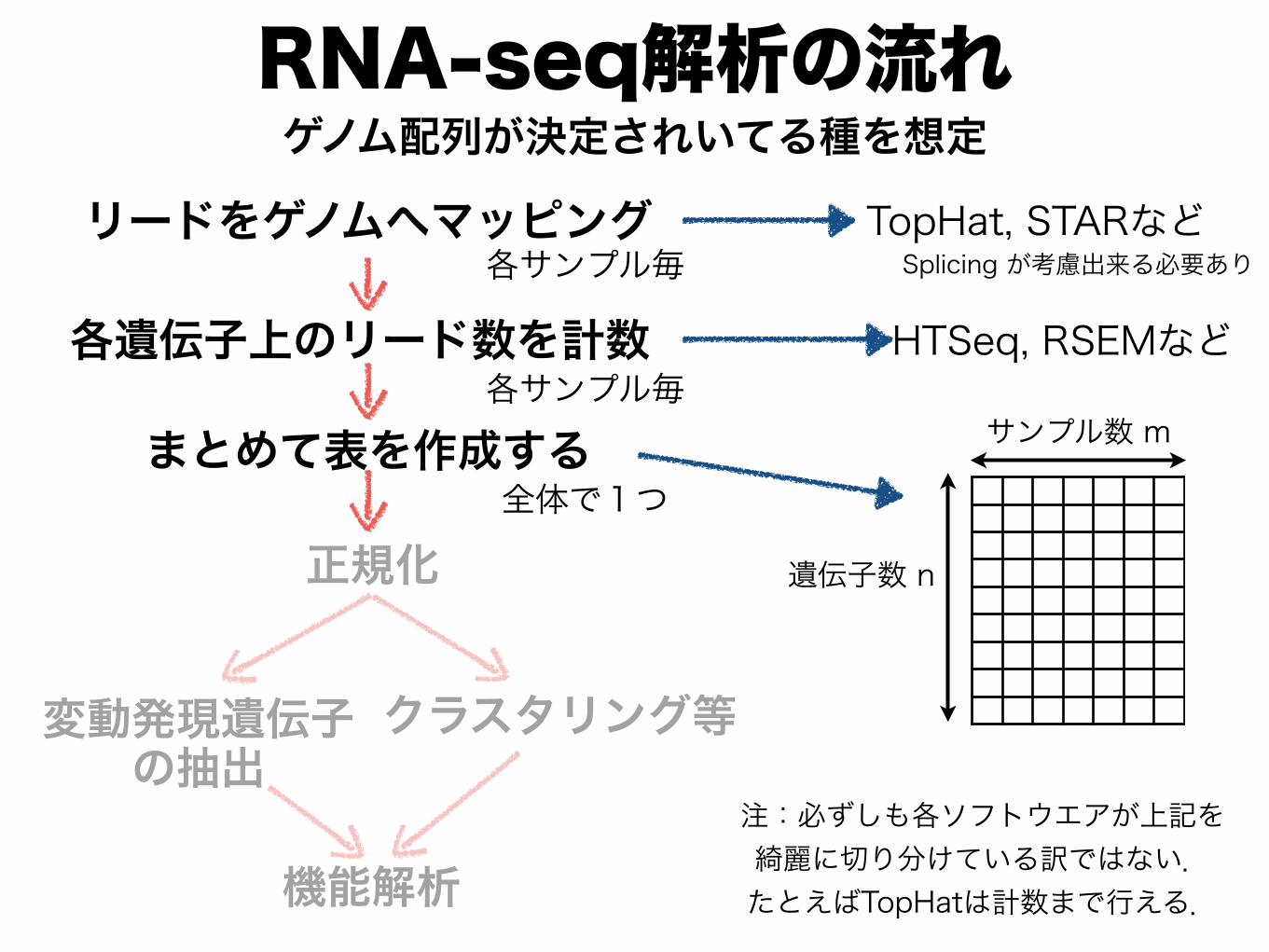
RNA-seq解析の流れ
リードをゲノムへマッピング TopHat, STARなど
各遺伝子上のリード数を計数 HTSeq, RSEMなど
遺伝子数 n
サンプル数 m
各サンプル毎
各サンプル毎
まとめて表を作成する全体で1つ
ゲノム配列が決定されいてる種を想定
変動発現遺伝子 の抽出
クラスタリング等
正規化
機能解析
Splicing が考慮出来る必要あり
注:必ずしも各ソフトウエアが上記を 綺麗に切り分けている訳ではない. たとえばTopHatは計数まで行える.
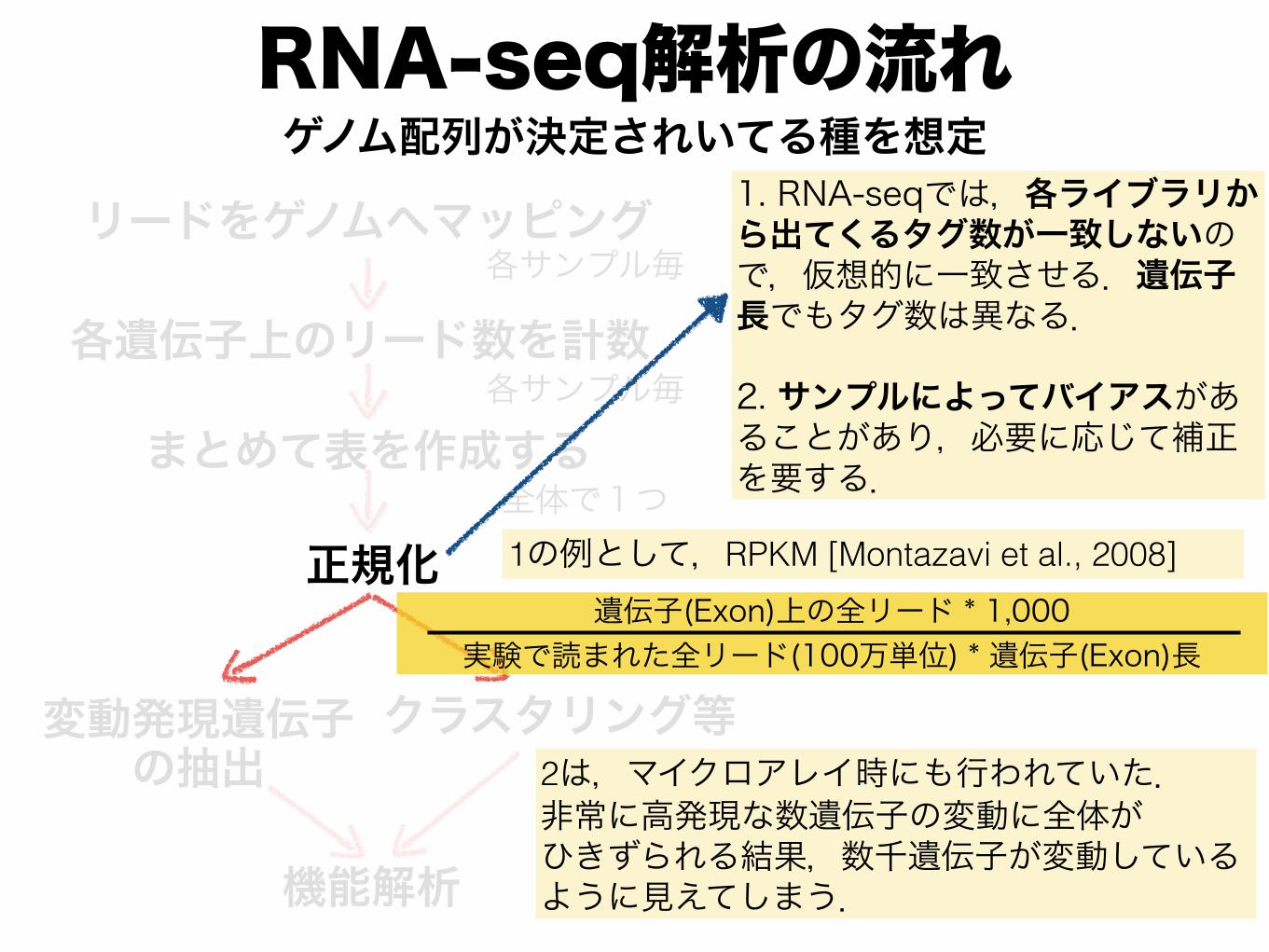
RNA-seq解析の流れ
リードをゲノムへマッピング
各遺伝子上のリード数を計数各サンプル毎
各サンプル毎
まとめて表を作成する全体で1つ
ゲノム配列が決定されいてる種を想定
変動発現遺伝子の抽出
クラスタリング等
正規化
機能解析
1. RNA-seqでは,各ライブラリから出てくるタグ数が一致しないので,仮想的に一致させる.遺伝子長でもタグ数は異なる.
2. サンプルによってバイアスがあることがあり,必要に応じて補正を要する.
1の例として,RPKM [Montazavi et al., 2008]遺伝子(Exon)上の全リード * 1,000
実験で読まれた全リード(100万単位) * 遺伝子(Exon)長
2は,マイクロアレイ時にも行われていた. 非常に高発現な数遺伝子の変動に全体が ひきずられる結果,数千遺伝子が変動している ように見えてしまう.
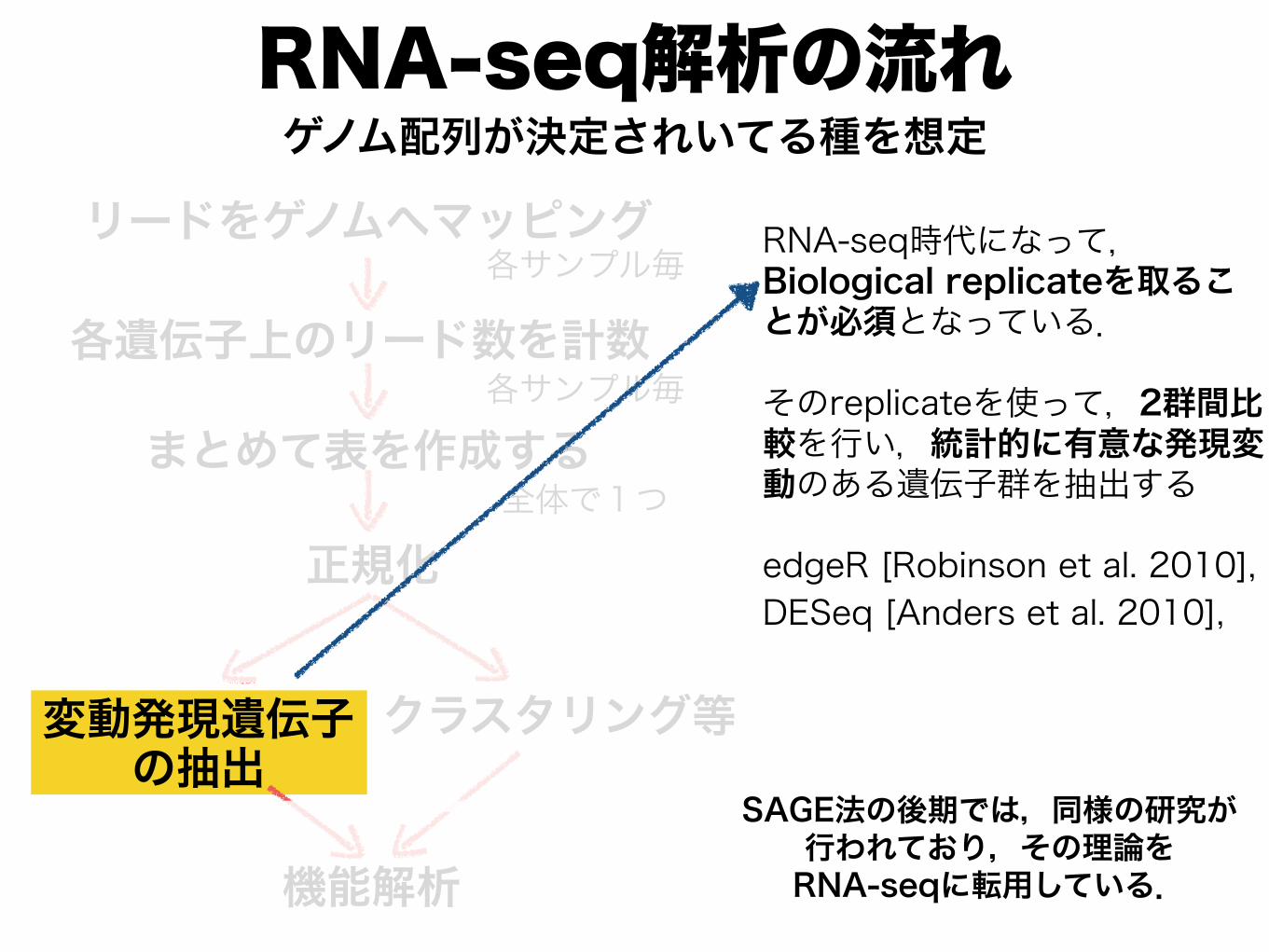
RNA-seq解析の流れ
リードをゲノムへマッピング
各遺伝子上のリード数を計数各サンプル毎
各サンプル毎
まとめて表を作成する全体で1つ
ゲノム配列が決定されいてる種を想定
変動発現遺伝子の抽出
クラスタリング等
正規化
機能解析
RNA-seq時代になって,Biological replicateを取ることが必須となっている.
そのreplicateを使って,2群間比較を行い,統計的に有意な発現変動のある遺伝子群を抽出する
edgeR [Robinson et al. 2010], DESeq [Anders et al. 2010],
SAGE法の後期では,同様の研究が 行われており,その理論を RNA-seqに転用している.

発現比率と統計的有意差• MA plot
Robinson M D et al. Bioinformatics 2010;26:139-140
© The Author(s) 2009. Published by Oxford University Press.
(平均)発現量
発現差
Fig

分割表による検定• 一般に,Fisherの正確確率検定,カイ二乗検定など. • タグ発現解析では,ポアソン分布を用いた検定が使われる
• 二項分布を考えた場合でもpが小さい場合に相当し,ポアソン分布で良く近似できる
150 100
1750 1900
1900 2000
Case Ctrl
Total
Gene1由来
Gene1以外由来
2000回のサンプルで,100回起きる事象があるとき,1900回のサンプルで,150回事象が起こる
p=100/2000=0.05の確率で表が出るコインを1900回投げ150回表が出る
単位時間tあたり,0.05t 回事象が起こるとき,(150/1900)t回事象が起こる確率
二項分布
ポアソン分布

Biological replicate はどうするか• CaseもControlも3回ずつ取られている状況を考える
• ポアソン分布の枠組みでは,Biological replicateを直接は扱えない.
• 例えば,20回ずつ取られていれば,各遺伝子毎にt検定も有効かもしれないが,3回では検定の検出力が足りない
• ましてやt検定の前提条件が満たされているか,確かめられる回数でもない.
• 実際には,統計検定が最終的な目標ではないので,「費用の問題」「それだけ実験をするなら,他の条件を観測したい」などで,大量のreplicate が取られることは無い.
• とはいえ,ある程度の有意差検定を行いたい
遺伝子数
n
Control Case

本当に二項分布/ポアソン分布なのだろうか?• 二項分布の分散は np(1-p).ポアソン分布は λ(~np) • 黒線が理論線.青点が実際の分散 • 理論値よりも分散が遥かに大きい.特に発現量が大きい時に顕著 • ポアソン分布で検定すると,発現量が大きい時,殆ど発現量に変化がないのに,有意差が生まれてしまう←モデルが誤っている
Anders, S., et al. (2013). Nature Protocols
Fig

負の二項分布を用いたモデル化• ポアソン分布に分散を表す変数を加えたい
• 負の二項分布を用いて表すモデル化が採用されている (edgeR, DESeq, cuffdiff2など) • 負の二項分布は,ポアソン分布に変数を1つ加えたもの,あるいは,複数のポアソン分布の混合分布として計算することが可能.
• 計測点が3点のみでは計算した分散の値が信頼出来ない問題は解消されていないことに注意.
P (Y = y) =
✓n
y
◆py(1� p)n�y二項分布:
負の二項分布:
匠IPSJ SIG Technical Report
ここで,E(i, j) = (n(A, j)+n(B, j)) ·n(i)/(n(A)+n(B))
である.カイ二乗検定では,この値が近似的に自由度 1のカイ二乗値に従う事を利用する.数表より,この値を読み取り,有意水準以下であれば,サンプルが独立である仮説が棄却され,発現が異なると考えられる.ポアソン分布,カイ二乗検定共に非常に頻繁に用いられる分布や検定方法であるが,生物サンプルを扱うために必要な複製実験が直接的には扱えないことが問題となる可能性がある.特にカイ二乗検定では,サンプル数増大が検出力の増加につながり,発現が多い遺伝子は多少の発現変動でも軒並み有意になる可能性があり注意を要する.
3. 2群間の発現変動遺伝子前節で刺激の前後の様な 2サンプル間の比較を扱ったが,遺伝子発現量は,常に一定の値をとっているわけではなく,細胞周期,実験時の環境など,様々な状態を反映しているため,実験を行う毎に完全に一致した値になることは無いと考えられる.そこで,RNA-seqを利用した多くの 2群間比較解析においては,各群から複数回のサンプル(生物学的複製サンプル)を取り,2群間の比較が行われる.各遺伝子に着目した場合,対応の無い 2群間比較の問題と考えることができるが,各群で行われる実験の回数は,実験費用の問題,サンプルの用意の難しさから,各群の実験が 3
回から 5回程度と非常に少ないことも多い.この少ない実験回数が,検定の際に問題となる.
3.1 t検定の利用2 群間比較で頻繁に利用される検定として t 検定(スチューデントの t検定)が挙げられる.t検定では,2群間の平均が同一の母集団由来かを検定する.問題 2 群 Aから a回の RNA-seqを行ったとする.それぞれの実験を A1, A2, ..., Aa とする.同様に群 B から b
回の RNA-seqを行い,それぞれ B1, B2, ..., Bb とする.これら 2群の間の平均に差がないことを,t検定を用いて検定する.n(i, g)を実験 iにおける遺伝子 g 由来の断片の数とする.統計量 T を以下の式で計算する
T =A− B!"1a − 1
b
#UAB
.
ここで,
A =1
a
a$
i=1
n(Ai, g),
B =1
b
b$
i=1
n(Bi, g),
UAB =
%ai=1(n(Ai, g)− A)2 +
%bi=1(n(Bi, g)− B)2
m+ n− 2
である.ただし,スチューデントの t検定は,各群から得られた分布が正規分布に従う.また,各群の分散が等分散であることが仮定されており,本来はこれらを判断した上で,検定する必要がある.しかし,先に述べた通りサンプルの数は 3つあるいは 5つなどと,分布や分散を知るために十分な数は無く,現実的には困難である.
3.2 負の二項分布を用いたモデル化現在,RNA-seq解析で多く用いられている手法が負の二項分布を用いた検定である.ポアソン分布を基本とし,パラメータを一つ増やすことで,多サンプルを得た場合に明らかになる過分散を考慮にいれることが可能である.RNA-seqによるランダムサンプリングの分布が定式化 2
の通りポアソン分布に従うと仮定すると,分散は λとなることが知られている.一方,実際にデータを調べると,λが大きい所では,分散が λより大きな値を取っている事が知られている ([4]の Figure 1,あるいは [2]の Supplemnetal
Text Figure 2.).このため,ポアソン分布を用いて検定を行うと,特に発現量が大きい遺伝子に対して,本来の値以上に低い P 値を算出する可能性がある.過分散が起きた場合に,適用されるモデルが負の二項分布である.負の二項分布を用いた検定は,以下のように定式化される定式化 4 確率変数を Y として,パラメータ pと rを用いると,負の二項分布は
P (Y = y) =
&y + r − 1
r
'py(1− p)r
と表せる.また,ガンマ関数 Γ(x) =(∞0 e−ttx−1dtが,x
が自然数の時,Γ(x) = (x− 1)!である事を用いると,
P (Y = y) =Γ(y + r)
Γ(r)Γ(y + 1)py(1− p)r
となる.期待値は pr/(1− p),分散は pr/(1− p)2 である.
系 1 変数 rを無限に飛ばすと,負の二項分布はポアソン分布に近似できる.証明 1 期待値を表す新たな変数として λ = pr
1−p を導入すると,p = λ
r+λ である.これを,負の二項分布の式に代入して,変形する.
f(y; k, r) = P (Y = y)
=Γ(y + r)
Γ(r)Γ(y + 1)py(1− p)r
=λy
y!· Γ(y + r)
Γ(r)(r + λ)r· 1"1 + λ
r
#r
ここで rを無限に飛ばすと,第 1項は rに依存せず,第 2
項は 1,第 3項は指数関数に収束するので,
limr→∞
f(y; k, r) =λy
y!
1
eλ
c⃝ 2014 Information Processing Society of Japan 2
P (Y = y) =
✓y + r � 1
r
◆py(1� p)r
=�(y + r)
�(r)�(y + 1)py(1� p)r正規化の時点で離散値では
なくなっているので, 連続値が扱えて嬉しい.

変数 rを無限に飛ばすと,負の二項分布はポアソン分布に近似できる.
期待値を表す新たな変数として λ = pr1−p を導入すると,p = λ
r+λ である.これを,負の二項分布の式に代入して,変形する.
f(y; k, r) = P (Y = y)
=Γ(y + r)
Γ(r)Γ(y + 1)py(1− p)r
=λy
y!· Γ(y + r)
Γ(r)(r + λ)r· 1!1 + λ
r
"r
ここで rを無限に飛ばすと,第 1項は rに依存せず,第 2項は 1,第 3項は指数関数に収束するので,
limr→∞
f(y; k, r) =λy
y!
1
eλ
これは,期待値 λのポアソン分布である.
1
証明
■

各遺伝子の発現量の分散を推定する• 経験的に,分散は発現量に依存する • 発現量が近い場合,分散も類似すると考えて回帰問題を考えることで,分散の推定を行っている.
• DESeqの例:サンプルi, 遺伝子gに対し,分散を次の式で推定する.
Anders, S., et al. (2013). Nature Protocols
µ(i, g) + t(i)2⌫(g)
正規化後の 推定発現量
サンプルの 総タグ数 パラメータ
遺伝子毎の値. この値を回帰で 求める
過分散を表す項
Fig

分布は推定できた.検定はどうする?• 分布が複雑で,解析的にはp値が求まらない.
• 求めた負の二項分布に従った乱数を発生させ,シミュレーションでp値を求める (DESeq)
• あるいは,フィッシャーの正確確率検定の様に,観測された値以上に極端な場合を数え上げる (edgeR)
• 例えばDESeqの戦略では • 遺伝子g由来のタグがControl から NA回,CaseからNB回が観測されたとすると
• Control と Caseは独立だと仮定するしPr(Y=NA)Pr(Y=NB)を計算 • 負の二項分布から乱数を2個(N1, N2)発生させ Pr(Y=N1)Pr(Y=N2)を計算 • 元の値より,p値が小さくなるような乱数の割合がp値
• 最後は,False Discovery Rate (FDR)によって,多重検定補正を行う

Cuffdiffについて• Cuffdiff(2)は,edgeRやDESeqと違い,RNA-seq,特にSplicing
variant を定量化する話が根本にある. • 1つの遺伝子が複数のスプライシングバリアントを保つ場合,各リードがどのスプライシングバリアントに属するかを,最尤推定を用いて定式化
• 発現量の分散モデルに関しては,DESeqのモデルを踏襲 • 但し,各exonを負の二項分布で表して,その混合分布(ベータ負の二項分布)を発現のモデルにしている
• 最近は,edgeRやDESeqも,スプライスバリアントの定量に力を入れているようである.

まとめと今後の課題• RNA-seqの導入によって,マイクロアレイに比べて定量性が高まっただけでなく,タグをランダムサンプリングするモデルが導入でき,統計的なモデル化が進んだ
• 現在まで,(SAGE法の延長による)過分散を考慮した発現差の統計解析(DESeq, edgeR)と,RNA-seqから生まれた選択的スプライシング解析(cuffdiff)の2つの道で研究が進んでおり,これらの融合が進んでいる.
• これ以上モデルを複雑にすることは,オーバーフィットとの戦いになるのではないかと思われる.
• 今後の方向性として • アリル特異性の解析 [Akama et al. NAR 2014] • 多サンプルに対する解析 • クラスタ分析との融合など,が考えられるだろう. • RNA-seqが,PacBioなどを利用した全長観測可能なものになると,スプライシングのモデル化が容易になる可能性がある.