solid-state physics crystallization andvitrification · university, sendai 980-8577, japan....
TRANSCRIPT

SOLID-STATE PHYSICS
Crystallization and vitrificationof electrons in a glass-formingcharge liquidS. Sasaki,1* K. Hashimoto,1*† R. Kobayashi,1 K. Itoh,1 S. Iguchi,1 Y. Nishio,2
Y. Ikemoto,3 T. Moriwaki,3 N. Yoneyama,4 M. Watanabe,5 A. Ueda,6 H. Mori,6
K. Kobayashi,7 R. Kumai,7 Y. Murakami,7 J. Müller,8 T. Sasaki1
Charge ordering (CO) is a phenomenon in which electrons in solids crystallize into aperiodic pattern of charge-rich and charge-poor sites owing to strong electroncorrelations. This usually results in long-range order. In geometrically frustrated systems,however, a glassy electronic state without long-range CO has been observed. We foundthat a charge-ordered organic material with an isosceles triangular lattice shows chargedynamics associated with crystallization and vitrification of electrons, which can beunderstood in the context of an energy landscape arising from the degeneracy of variousCO patterns. The dynamics suggest that the same nucleation and growth processesthat characterize conventional glass-forming liquids guide the crystallization ofelectrons. These similarities may provide insight into our understanding of theliquid-glass transition.
The physics of glassy materials represents afascinating problem in solid-state theory (1).Although progress has been made over thepast several decades toward clarifying the dy-namical aspects of the glass transition, the
processes by which liquids acquire the glassy stateupon cooling are not fully understood (2). Themostfundamental nonequilibriumdynamic phenomenaassociated with the liquid-glass transition processare crystallization and vitrification. These phenomenaarecompetingandmutuallyexclusive but are closelyrelated to each other (3, 4). In glass-forming liquids,crystallization below the melting point Tm can beavoidedwhen the system is cooled quickly enough,leading to a supercooled liquid state accompaniedby an increase in viscosity (Fig. 1A) (2, 5). Uponfurther cooling, owing to the viscous retarda-tion of crystallization, the supercooled liquidstate freezes into a glassy state; that is, vitrificationoccurs at the glass transition temperature Tg.Thus, the relationship between crystallizationand vitrification is key to the understanding ofthe liquid-glass transition. Here, we demonstratethat this general picture can be extended to crystal-
lization and vitrification of strongly correlated elec-trons realized in a geometrically frustrated charge-ordered organic system, qm-(BEDT-TTF)2TlZn(SCN)4(6), where BEDT-TTF denotes bis(ethylenedithio)tetrathiafulvalene. Surprising similarities betweenour system and conventional glass formers areobserved in the crystallization and vitrificationprocesses, which highlight the universal nature ofthe liquid-glass transition.The quasi–two-dimensional (quasi-2D) organic
materials q-(BEDT-TTF)2X consist of alternatingstacks of conducting BEDT-TTF and insulatinganion X layers; the BEDT-TTF molecules form atriangular lattice (6–8). The charge transfer be-tween these two layers leads to a 2D quarter-filledhole band system (that is, one hole per two BEDT-TTF molecules), in which the intersite Coulombrepulsions give rise to an instability toward chargeordering (CO) (9). Indeed, q-(BEDT-TTF)2RbZn(SCN)4 (henceforth q-RbZn), for example, under-goes a CO transition at 190 K (6, 7, 10, 11), wherethe charge carriers are localized periodically witha horizontal stripe pattern (see the phase diagramin fig. S1C). Such aperiodicCOstate canbe regardedas a “charge-crystal” state (11). In contrast, above theCO transition temperature, the charge of +0.5 peroneBEDT-TTFmolecule is distributed uniformlyin space; therefore, such a delocalized state canbe referred to as a “charge-liquid” state.Inq-RbZn,when the sample is cooled faster than
a critical cooling rate (~5 K/min), charge crystal-lization is kinetically avoided, leading to a “charge-glass” statewhere the charge is randomlyquenched.For comparison, in q-(BEDT-TTF)2CsZn(SCN)4(q-CsZn), which has a more isotropic triangularlattice than q-RbZn, the critical cooling rate be-comesmuch slower (fig. S1, C andD). As a result, thecharge-liquid state inevitably results in a charge-glass state evenuponvery slowcooling (<0.1K/min)(12). However, themechanism of formation of the
glassy electronic state—which has been discussedexperimentally (11–14) and theoretically (15) interms of the geometrically frustrated triangularlattice—still remains rather elusive.The system q-(BEDT-TTF)2TlZn(SCN)4, which
exists in two different crystal forms with ortho-rhombic and monoclinic symmetries (fig. S1, Aand B) (6, 16), may play a key role in the under-standing of the charge-glass state. Orthorhombicqo-(BEDT-TTF)2TlZn(SCN)4 (qo-TlZn), which hasthe same structural symmetry as q-RbZn andq-CsZn, exhibits a CO transition with a horizon-tal chargemodulation at 240 K (16). Because theanisotropy of the triangular lattice is large, thecritical cooling rate for glass formation remainsquite high (13, 14). In contrast, the monoclinicqm-(BEDT-TTF)2TlZn(SCN)4 (qm-TlZn) shows aCO transitionwith a diagonal chargemodulationat Tm = 170 K (Fig. 1, B to D) (16). The differentCO patterns of the two systems can be relatedto a difference in the strength of electron-latticecoupling, as pointed out by theoretical studies(17). Because the lattice distortion of qm-TlZnat the CO transition is much smaller than thatof qo-TlZn, and because most of the entropychange of qm-TlZn is of electronic origin (fig.S2), qm-TlZn more likely approximates a sys-tem where the observed effects are purely elec-tronic in nature. This is consistent with theextendedHubbardmodel (EHM) in the absenceof electron-phonon coupling, in which the di-agonal CO pattern rather than the horizontal oneis expected (17).The temperature-dependent resistivity r(T) of
qm-TlZn shows a strong dependence on sweep-ing rate below Tm (Fig. 1B), although the triangu-lar lattice for qm-TlZn ismore anisotropic than thatfor qo-TlZn. In qm-TlZn, the long-range CO transi-tion can be avoided by rapid cooling (≥50K/min),and charge vitrification occurs through a super-cooled charge-liquid state (Fig. 1, B and E). In thecooling/heating cycle of the r-T profile, a clearhysteresis loop associated with the glass tran-sition is observed at 145 to 165 K (Fig. 1B, inset),quite similar to what is observed in q-CsZn (12).We tentatively define the glass transition tem-perature Tg as the temperature above which theresistivity starts to branch off.To clarify the origin of charge-glass formation
in qm-TlZn, we performed resistance noise mea-surements, which are a powerful probe to detectthe slow dynamics associated with electronic glas-siness (11, 12, 18). Figure 2A shows a typical normal-ized noise power spectral density of the resistancefluctuations SR/R
2. The baseline of SR/R2 fits
well to generic 1/f with a slightly deviating fre-quency exponent, yielding 1/f a with a ~ 0.8 to0.9. For clarity, f a × SR/R
2 is plotted in Fig. 2B,which clearly shows that the resistance fluctu-ations exhibit a broad peak structure. The dataarewell fitted to the distributed Lorentzianmodel(11, 12) with a characteristic center frequency fc(=
ffiffiffiffiffiffiffiffiffiffiffiffiffif c1 f c2
p, where fc1 and fc2 are the high- and
low-cutoff frequencies, respectively), from whichwe derived the temperature evolution of therelaxation time tc = 1/(2pfc). The peak struc-ture in f a × SR/R
2 becomes broader and more
RESEARCH
Sasaki et al., Science 357, 1381–1385 (2017) 29 September 2017 1 of 5
1Institute for Materials Research, Tohoku University, Aoba-ku,Sendai 980-8577, Japan. 2Department of Physics, Facultyof Science, Toho University, Funabashi, Chiba 274-8510,Japan. 3Japan Synchrotron Radiation Research Institute,SPring-8, Sayo, Hyogo 679-5198, Japan. 4Graduate Facultyof Interdisciplinary Research, University of Yamanashi,Kohu, Yamanashi 400-8511, Japan. 5Institute ofMultidisciplinary Research for Advanced Materials, TohokuUniversity, Sendai 980-8577, Japan. 6Institute for SolidState Physics, University of Tokyo, Kashiwa, Chiba 277-8581, Japan. 7CMRC and Photon Factory, Institute ofMaterials Structure Science, High Energy AcceleratorResearch Organization (KEK), Tsukuba, Ibaraki 305-0801,Japan. 8Institute of Physics, Goethe-University Frankfurt,60438 Frankfurt, Germany.*These authors contributed equally to this work. †Correspondingauthor. Email: [email protected]
on May 16, 2020
http://science.sciencem
ag.org/D
ownloaded from
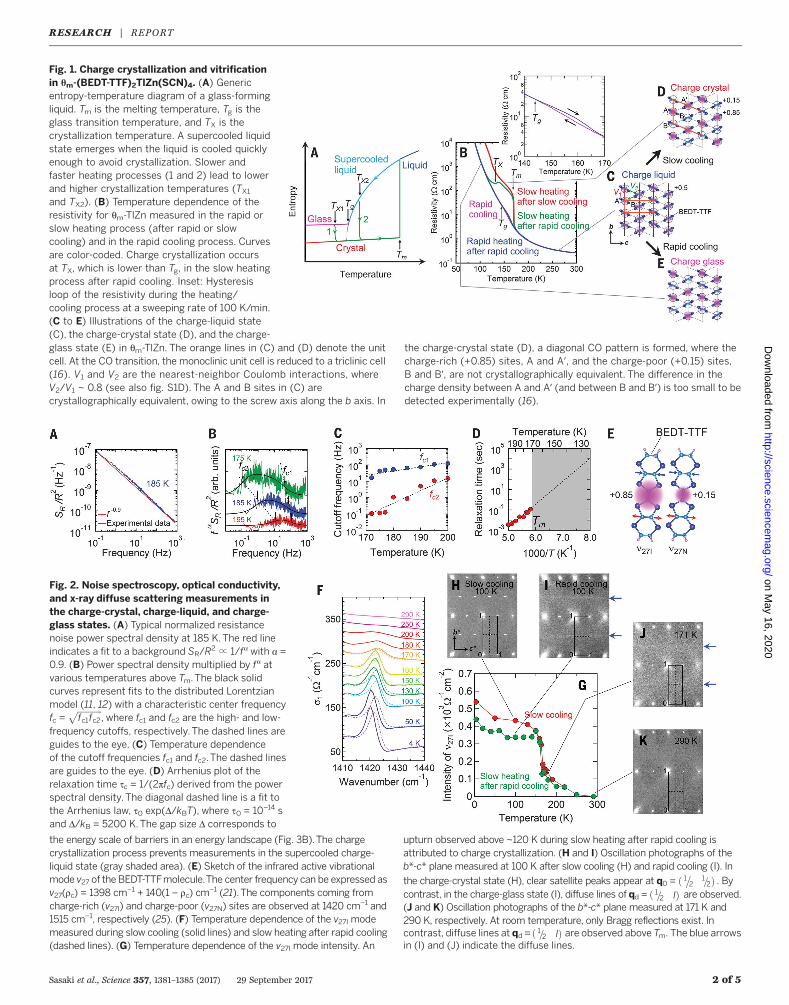
Sasaki et al., Science 357, 1381–1385 (2017) 29 September 2017 2 of 5
Fig. 2. Noise spectroscopy, optical conductivity,and x-ray diffuse scattering measurements inthe charge-crystal, charge-liquid, and charge-glass states. (A) Typical normalized resistancenoise power spectral density at 185 K.The red lineindicates a fit to a background SR/R
2 º 1/fa with a =0.9. (B) Power spectral density multiplied by fa atvarious temperatures above Tm.The black solidcurves represent fits to the distributed Lorentzianmodel (11, 12) with a characteristic center frequencyfc =
ffiffiffiffiffiffiffiffiffiffiffiffifc1fc2
p, where fc1 and fc2 are the high- and low-
frequency cutoffs, respectively. The dashed lines areguides to the eye. (C) Temperature dependenceof the cutoff frequencies fc1 and fc2.The dashed linesare guides to the eye. (D) Arrhenius plot of therelaxation time tc = 1/(2pfc) derived from the powerspectral density. The diagonal dashed line is a fit tothe Arrhenius law, t0 exp(D/kBT), where t0 = 10–14 sand D/kB = 5200 K.The gap size D corresponds to
the energy scale of barriers in an energy landscape (Fig. 3B).The chargecrystallization process prevents measurements in the supercooled charge-liquid state (gray shaded area). (E) Sketch of the infrared active vibrationalmode v27 of the BEDT-TTFmolecule.The center frequency can be expressed asv27(rc) = 1398 cm−1 + 140(1 – rc) cm
−1 (21).The components coming fromcharge-rich (v27I) and charge-poor (v27N) sites are observed at 1420 cm−1 and1515 cm−1, respectively (25). (F) Temperature dependence of the v27I modemeasured during slow cooling (solid lines) and slow heating after rapid cooling(dashed lines). (G) Temperature dependence of the v27I mode intensity. An
upturn observed above ~120 K during slow heating after rapid cooling isattributed to charge crystallization. (H and I) Oscillation photographs of theb*-c* plane measured at 100 K after slow cooling (H) and rapid cooling (I). In
the charge-crystal state (H), clear satellite peaks appear at q0 = ð 1 212= Þ�
. Bycontrast, in the charge-glass state (I), diffuse lines of qd = ð 1 2 l= Þ are observed.(J and K) Oscillation photographs of the b*-c* plane measured at 171 K and290 K, respectively. At room temperature, only Bragg reflections exist. Incontrast, diffuse lines at qd = ð 1 2 l= Þ are observed above Tm. The blue arrowsin (I) and (J) indicate the diffuse lines.
Fig. 1. Charge crystallization and vitrificationin qm-(BEDT-TTF)2TlZn(SCN)4. (A) Genericentropy-temperature diagram of a glass-formingliquid. Tm is the melting temperature, Tg is theglass transition temperature, and TX is thecrystallization temperature. A supercooled liquidstate emerges when the liquid is cooled quicklyenough to avoid crystallization. Slower andfaster heating processes (1 and 2) lead to lowerand higher crystallization temperatures (TX1
and TX2). (B) Temperature dependence of theresistivity for qm-TlZn measured in the rapid orslow heating process (after rapid or slowcooling) and in the rapid cooling process. Curvesare color-coded. Charge crystallization occursat TX, which is lower than Tg, in the slow heatingprocess after rapid cooling. Inset: Hysteresisloop of the resistivity during the heating/cooling process at a sweeping rate of 100 K/min.(C to E) Illustrations of the charge-liquid state(C), the charge-crystal state (D), and the charge-glass state (E) in qm-TlZn. The orange lines in (C) and (D) denote the unitcell. At the CO transition, the monoclinic unit cell is reduced to a triclinic cell(16). V1 and V2 are the nearest-neighbor Coulomb interactions, whereV2/V1 ~ 0.8 (see also fig. S1D). The A and B sites in (C) arecrystallographically equivalent, owing to the screw axis along the b axis. In
the charge-crystal state (D), a diagonal CO pattern is formed, where thecharge-rich (+0.85) sites, A and A′, and the charge-poor (+0.15) sites,B and B′, are not crystallographically equivalent. The difference in thecharge density between A and A′ (and between B and B′) is too small to bedetected experimentally (16).
RESEARCH | REPORTon M
ay 16, 2020
http://science.sciencemag.org/
Dow
nloaded from

asymmetric with decreasing temperature(Fig. 2, B and C), showing that the dynamicsbecome more heterogeneous. In addition,tc slows drastically over several orders ofmagnitude (Fig. 2D); extrapolating ourdata under the assumption that tc obeysan Arrhenius law [as observed for q-CsZn(12) and as expected from recent MonteCarlo simulations (15)], we find that tc maybe as high as 102 s around Tg. These resultsindicate the emergence of slow dynamicsaccompanied by increasing dynamic het-erogeneity upon approaching the charge-glass transition. We note that the chargevitrification in the present case is distinct-ly different from the drastic slowing downof charge carrier dynamics and onset ofnon-Gaussian fluctuations observed innoise measurements as a precursor ofmetal-insulator transitions (MITs) (18).Electronic glassiness inMIT systems seem-ingly only becomes stabilized by disorderin the presence of strong electronic cor-relations (18, 19) and is not observed forclean samples.Clarifying the relationship between the
heterogeneous slow dynamics and localcharge configurations may be key to theunderstanding of charge-glass formation(3, 20). To this end, we investigated theimbalance of charge distribution—thatis, charge disproportionation—at a micro-scopic level bymeans of a charge-sensitive vibra-tional mode v27 of the BEDT-TTF molecule. Thev27 mode is known as a local probe of the mo-lecular charge rc (21) and splits into two modes,v27I and v27N, in the presence of charge dispro-portionation between the A and B sites in theunit cell (Figs. 1C and 2E), where the subscriptsI and N denote the hole-rich and hole-poor sites,respectively. Figure 2F shows the temperaturedependence of the polarized optical conductivityspectra. A clear peak around 1420 cm−1 is assignedto the v27I mode (rc ~ 0.85) (16). There is a slightdifference in the peak frequency of 1.5 cm−1 be-tween the slow and rapid cooling processes, whichcorresponds to 0.01 in charge distribution on theBEDT-TTF molecule.A sizable intensity of v27I was observed above
Tm (Fig. 2G), indicating the presence of chargedisproportionation above Tm. Because the A andB sites are crystallographically equivalent aboveTm owing to the screw axis along the b axis (16),the time-averaged charge distribution above Tmis +0.5 per one BEDT-TTF molecule (Fig. 1C).Therefore, the splitting of v27 above Tm impliesthat charge disproportionation above Tm is notstatic but dynamically fluctuates on a time scaleslower than that of the molecular vibrational v27motion. Because the intensity of v27I is consideredto reflect the volume of dynamically fluctuatingcharge clusters switching between the locally or-dered and charge-liquid states, its increase withdecreasing temperature suggests that the heter-ogeneous slow dynamics observed in the noisemeasurements are caused by the dynamicallyfluctuating charge clusters.
To obtain insights into the structural origin ofthe heterogeneous slow dynamics, we performedx-ray diffuse scatteringmeasurements. Oscillationphotographs measured at various temperaturesare shown in Fig. 2, H to K. At room tempera-ture, only Bragg reflections are observed (Fig. 2K),whereas in the charge-crystal state, clear satellitepeaks appear at q0 =
�12
12=��, compatible with
the diagonal CO pattern (Fig. 2H), which is at-tributed to the periodicity of charge-rich andcharge-poor sites accompanied by a periodicalchange of the C=C double bond length of theBEDT-TTF molecules. Theoretical calculationsfor the q-type materials based on the EHM (22)have suggested that the diagonal CO pattern ismost stable when V1 > V2 (where V1 and V2 arethenearest-neighborCoulomb interactions),whichis consistent with the observations in the charge-crystal state of qm-TlZn. In contrast, in the charge-glass state, diffuse lines atqd=
�12 l=
�areobserved
(Fig. 2I). The diffuse lines can be ascribed to ge-ometric frustration.The classical ground states of the t-V model,
which is the spinless version of the EHM, on anisosceles triangular lattice are known to be dis-ordered owing to geometric frustration when V1
≥ V2 (23–25). For V1 = V2, the ground state is“macroscopically” disordered with a degeneracyof ~2N–1 (where N is the number of lattice sites).On the other hand, for V1 > V2, V1 preferentiallydetermines the two-fold periodic striped CO pat-tern along the b direction, but the geometric frus-tration along the diagonal directions arising fromthe isosceles triangular lattice gives rise to a“semimacroscopic” degeneracy of 2Lc , where Lc
is the system length in the c direction(Fig. 3A). However, introducing a smallquantum hopping term or a long-rangeCoulomb potential lifts the degeneracy,which drives the system to the diagonalCO pattern (15, 22), although the degen-eracy is preserved in a wide temperaturerange above the ordering temperature(15). This situation may induce an energylandscape with multiple local minima, asillustrated in Fig. 3B—that is, ametastablestate with an amorphous stripe-glass struc-ture as proposed in (15)—which in turncauses the heterogeneous slow dynamics inqm-TlZn. Indeed, frustration is a key con-cept for understanding glass transitions ina variety of systems (3). For example, cry-stallization inmetallic glasses is preventedif locally favored structures such as icosa-hedral order do not match the symmetryof the system (3, 26). Likewise, in qm-TlZn,locally favored short-range electronic order-ing with qd =
�12 l=
�induced by geometric
frustrationmay hinder long-range COwithq0 =
�12
12=��, thereby causing the slow
dynamics. Our results provide an experi-mental demonstration of recent theoret-ical considerations that frustration, incombination with strong quantum effects,plays an important role in the realizationof quantum charge-glass states in cleansystems, essentially free from impurities
and defects (27–30).We next examine the charge crystallization
process in detail to clarify the relationship be-tween crystallization and vitrification of elec-trons in qm-TlZn. Figure 4A displays the timeevolution of the resistivity during the chargecrystallization process from the supercooledcharge-liquid or charge-glass state (25). Themagnitude of the resistivity, which is a measureof the crystallization progress, increases withtime and then saturates. The relaxation timebecomes faster with decreasing temperature,and then slower below 157 K, which is referredto as the “nose temperature”; this characteristictemperature dependence of the relaxation timecan be explained by the theory of nucleation andgrowth at a first-order liquid-crystal phase tran-sition (25).To quantitatively evaluate the CO volume frac-
tion from the resistivity,we used the effectivemedi-um percolation theory (31). This theory describesa percolating current passing through an inhomo-geneous mixture of conducting and insulatingmedia (25). Through a generalized effectivemedi-um equation (32), we derived the time evolutionof the CO volume fraction, f(t), from the time-dependent resistivity data at various tempera-tures (Fig. 4, B and C). In the high-temperatureregion, f(t) can be fitted over the whole time rangeby the Johnson-Mehl-Avrami-Kolmogorov (JMAK)formula describing a conventional nucleation andgrowth process, where f(t) = 1 – exp(–ktn) (here,k and n are the JMAK parameters) (25, 33) (Fig.4E). By contrast, near the nose temperature, f(t)can be fitted to the JMAK formula only in the
Sasaki et al., Science 357, 1381–1385 (2017) 29 September 2017 3 of 5
Fig. 3. Semimacroscopic degeneracy of striped CO pat-terns and energy landscape. (A) Schematics of variousstriped CO patterns. The magenta circles represent the charge-rich sites. V1 and V2 (V1 > V2) are the nearest-neighborCoulomb interactions. Because all these states are degeneratein the classical limit of the t-V model, the classical groundstate can be described by the superposition of these states,which has a degeneracy of 2Lc, where Lc is the system length inthe c direction. (B) Illustration of an energy landscape withmultiple local minima separated by barriers having an energyscale of the hopping integral thopping and/or the long-rangeCoulomb interaction V, which are on the order of ~1 eV.
RESEARCH | REPORTon M
ay 16, 2020
http://science.sciencemag.org/
Dow
nloaded from
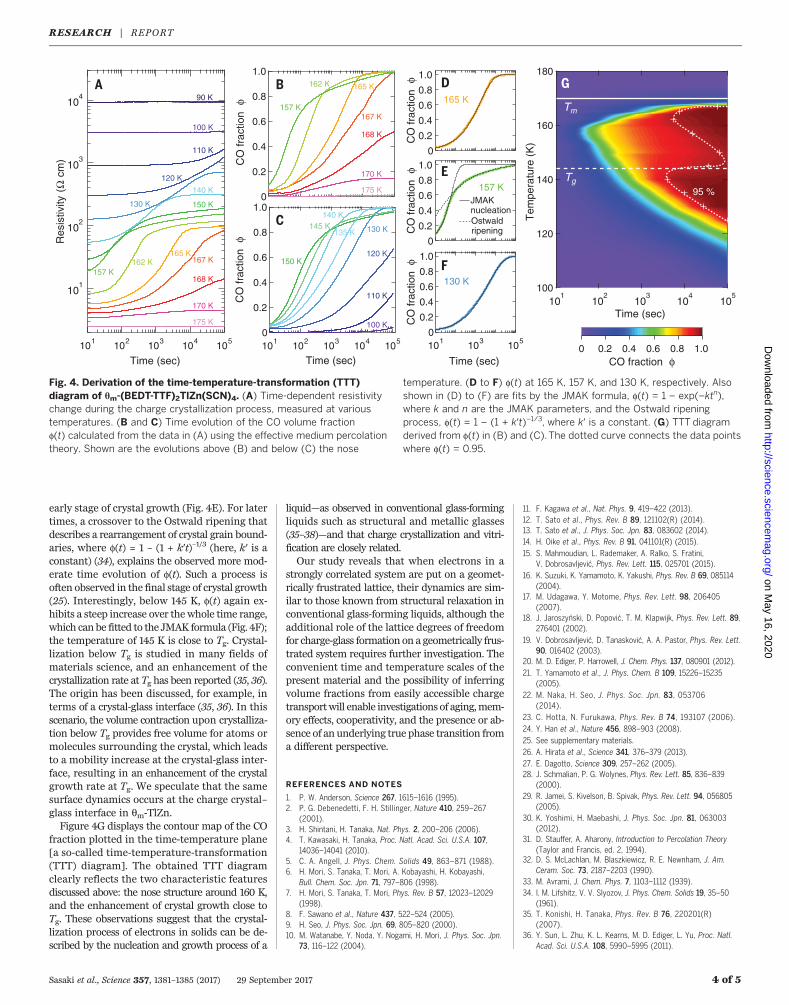
early stage of crystal growth (Fig. 4E). For latertimes, a crossover to the Ostwald ripening thatdescribes a rearrangement of crystal grain bound-aries, where f(t) = 1 – (1 + k′t)–1/3 (here, k′ is aconstant) (34), explains the observed more mod-erate time evolution of f(t). Such a process isoften observed in the final stage of crystal growth(25). Interestingly, below 145 K, f(t) again ex-hibits a steep increase over thewhole time range,which can be fitted to the JMAK formula (Fig. 4F);the temperature of 145 K is close to Tg. Crystal-lization below Tg is studied in many fields ofmaterials science, and an enhancement of thecrystallization rate at Tg has been reported (35, 36).The origin has been discussed, for example, interms of a crystal-glass interface (35, 36). In thisscenario, the volume contraction upon crystalliza-tion below Tg provides free volume for atoms ormolecules surrounding the crystal, which leadsto a mobility increase at the crystal-glass inter-face, resulting in an enhancement of the crystalgrowth rate at Tg. We speculate that the samesurface dynamics occurs at the charge crystal–glass interface in qm-TlZn.Figure 4G displays the contour map of the CO
fraction plotted in the time-temperature plane[a so-called time-temperature-transformation(TTT) diagram]. The obtained TTT diagramclearly reflects the two characteristic featuresdiscussed above: the nose structure around 160 K,and the enhancement of crystal growth close toTg. These observations suggest that the crystal-lization process of electrons in solids can be de-scribed by the nucleation and growth process of a
liquid—as observed in conventional glass-formingliquids such as structural and metallic glasses(35–38)—and that charge crystallization and vitri-fication are closely related.Our study reveals that when electrons in a
strongly correlated system are put on a geomet-rically frustrated lattice, their dynamics are sim-ilar to those known from structural relaxation inconventional glass-forming liquids, although theadditional role of the lattice degrees of freedomfor charge-glass formation on a geometrically frus-trated system requires further investigation. Theconvenient time and temperature scales of thepresent material and the possibility of inferringvolume fractions from easily accessible chargetransportwill enable investigations of aging,mem-ory effects, cooperativity, and the presence or ab-sence of an underlying true phase transition froma different perspective.
REFERENCES AND NOTES
1. P. W. Anderson, Science 267, 1615–1616 (1995).2. P. G. Debenedetti, F. H. Stillinger, Nature 410, 259–267
(2001).3. H. Shintani, H. Tanaka, Nat. Phys. 2, 200–206 (2006).4. T. Kawasaki, H. Tanaka, Proc. Natl. Acad. Sci. U.S.A. 107,
14036–14041 (2010).5. C. A. Angell, J. Phys. Chem. Solids 49, 863–871 (1988).6. H. Mori, S. Tanaka, T. Mori, A. Kobayashi, H. Kobayashi,
Bull. Chem. Soc. Jpn. 71, 797–806 (1998).7. H. Mori, S. Tanaka, T. Mori, Phys. Rev. B 57, 12023–12029
(1998).8. F. Sawano et al., Nature 437, 522–524 (2005).9. H. Seo, J. Phys. Soc. Jpn. 69, 805–820 (2000).10. M. Watanabe, Y. Noda, Y. Nogami, H. Mori, J. Phys. Soc. Jpn.
73, 116–122 (2004).
11. F. Kagawa et al., Nat. Phys. 9, 419–422 (2013).12. T. Sato et al., Phys. Rev. B 89, 121102(R) (2014).13. T. Sato et al., J. Phys. Soc. Jpn. 83, 083602 (2014).14. H. Oike et al., Phys. Rev. B 91, 041101(R) (2015).15. S. Mahmoudian, L. Rademaker, A. Ralko, S. Fratini,
V. Dobrosavljević, Phys. Rev. Lett. 115, 025701 (2015).16. K. Suzuki, K. Yamamoto, K. Yakushi, Phys. Rev. B 69, 085114
(2004).17. M. Udagawa, Y. Motome, Phys. Rev. Lett. 98, 206405
(2007).18. J. Jaroszyński, D. Popović, T. M. Klapwijk, Phys. Rev. Lett. 89,
276401 (2002).19. V. Dobrosavljević, D. Tanasković, A. A. Pastor, Phys. Rev. Lett.
90, 016402 (2003).20. M. D. Ediger, P. Harrowell, J. Chem. Phys. 137, 080901 (2012).21. T. Yamamoto et al., J. Phys. Chem. B 109, 15226–15235
(2005).22. M. Naka, H. Seo, J. Phys. Soc. Jpn. 83, 053706
(2014).23. C. Hotta, N. Furukawa, Phys. Rev. B 74, 193107 (2006).24. Y. Han et al., Nature 456, 898–903 (2008).25. See supplementary materials.26. A. Hirata et al., Science 341, 376–379 (2013).27. E. Dagotto, Science 309, 257–262 (2005).28. J. Schmalian, P. G. Wolynes, Phys. Rev. Lett. 85, 836–839
(2000).29. R. Jamei, S. Kivelson, B. Spivak, Phys. Rev. Lett. 94, 056805
(2005).30. K. Yoshimi, H. Maebashi, J. Phys. Soc. Jpn. 81, 063003
(2012).31. D. Stauffer, A. Aharony, Introduction to Percolation Theory
(Taylor and Francis, ed. 2, 1994).32. D. S. McLachlan, M. Blaszkiewicz, R. E. Newnham, J. Am.
Ceram. Soc. 73, 2187–2203 (1990).33. M. Avrami, J. Chem. Phys. 7, 1103–1112 (1939).34. I. M. Lifshitz, V. V. Slyozov, J. Phys. Chem. Solids 19, 35–50
(1961).35. T. Konishi, H. Tanaka, Phys. Rev. B 76, 220201(R)
(2007).36. Y. Sun, L. Zhu, K. L. Kearns, M. D. Ediger, L. Yu, Proc. Natl.
Acad. Sci. U.S.A. 108, 5990–5995 (2011).
Sasaki et al., Science 357, 1381–1385 (2017) 29 September 2017 4 of 5
180
160
140
120
100
Tem
pera
ture
(K
)
Time (sec)10
110
210
310
410
5
Tm
1.00.80.60.40.20CO fraction φ
Tg95 %
101
102
103
104
Res
istiv
ity (
Ω c
m)
101 102 103 104 105
Time (sec)
175 K
170 K
168 K
167 K165 K
162 K157 K
150 K
140 K
130 K
120 K
110 K
100 K
90 K
1.0
0.8
0.6
0.4
0.2
0
CO
frac
tion
φ
175 K
170 K
168 K
167 K
165 K162 K
157 K
1.0
0.8
0.6
0.4
0.2
0
CO
frac
tion
φ
101 102 103 104 105
Time (sec)
150 K
140 K
130 K
120 K
110 K
100 K
145 K135 K
1.0
0.8
0.6
0.4
0.2
0
CO
frac
tion
φ
165 K
1.0
0.8
0.6
0.4
0.2
0
CO
frac
tion
φ
157 K
Ostwald ripening
JMAK nucleation
1.0
0.8
0.6
0.4
0.2
0
CO
frac
tion
φ
101 103 105
Time (sec)
130 K
Fig. 4. Derivation of the time-temperature-transformation (TTT)diagram of qm-(BEDT-TTF)2TlZn(SCN)4. (A) Time-dependent resistivitychange during the charge crystallization process, measured at varioustemperatures. (B and C) Time evolution of the CO volume fractionf(t) calculated from the data in (A) using the effective medium percolationtheory. Shown are the evolutions above (B) and below (C) the nose
temperature. (D to F) f(t) at 165 K, 157 K, and 130 K, respectively. Alsoshown in (D) to (F) are fits by the JMAK formula, f(t) = 1 – exp(–ktn),where k and n are the JMAK parameters, and the Ostwald ripeningprocess, f(t) = 1 – (1 + k′t)–1/3, where k′ is a constant. (G) TTT diagramderived from f(t) in (B) and (C). The dotted curve connects the data pointswhere f(t) = 0.95.
RESEARCH | REPORTon M
ay 16, 2020
http://science.sciencemag.org/
Dow
nloaded from

37. Y. J. Kim, R. Busch, W. L. Johnson, A. J. Rulison, W. K. Rhim,Appl. Phys. Lett. 68, 1057–1059 (1996).
38. E. B. Moore, V. Molinero, Nature 479, 506–508 (2011).
ACKNOWLEDGMENTS
We thank K. Yoshimi, T. Kato, M. Naka, H. Shiba, C. Hotta, andK. Yamamoto for fruitful discussions, and J. Kudo and M. Kurosu fortechnical assistance. Synchrotron radiation measurements wereperformed at SPring-8 with the approvals of the Japan SynchrotronRadiation Research Institute (2014B1340, 2014B1752, 2015A1777,
2015B1752, 2015B1756, 2016A0073, and 2016B0073). X-raydiffraction study was performed under the approval of the PhotonFactory Program Advisory Committee (proposal 2014S2-001).Supported by the Deutsche Forschungsgemeinschaft within theTransregional Collaborative Research Center SFB/TR49 (J.M.);Grants-in-Aid for Scientific Research (grants 24340074, 25287080,26610096, 26102014, 15H00984, 15H00988, 15K13511, 15K17688,16H04010, 16K05430, 16K05744, 17H05138, and 17H05143) fromMEXT and JSPS; a Grant-in-Aid for Scientific Research on InnovativeAreas “p-Figuration” (grant 26102001); and the Canon Foundation.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/357/6358/1381/suppl/DC1Materials and MethodsSupplementary TextFigs. S1 to S7References (39–54)
31 October 2016; accepted 23 August 201710.1126/science.aal3120
Sasaki et al., Science 357, 1381–1385 (2017) 29 September 2017 5 of 5
RESEARCH | REPORTon M
ay 16, 2020
http://science.sciencemag.org/
Dow
nloaded from

Crystallization and vitrification of electrons in a glass-forming charge liquid
Ueda, H. Mori, K. Kobayashi, R. Kumai, Y. Murakami, J. Müller and T. SasakiS. Sasaki, K. Hashimoto, R. Kobayashi, K. Itoh, S. Iguchi, Y. Nishio, Y. Ikemoto, T. Moriwaki, N. Yoneyama, M. Watanabe, A.
DOI: 10.1126/science.aal3120 (6358), 1381-1385.357Science
, this issue p. 1378, p. 1381Scienceglasses.allowed to crystallize. The dynamics of crystallization showed similarities to the analogous processes in conventional an electronic or charge crystal. When the materials were cooled rapidly, a charge glass was formed instead and then
−−a triangular in-plane lattice. These materials can assume a state in which their charge distribution has a regular pattern studied layered organic materials with et al. and Sasaki et al.detail into analogous dynamics in electronic systems. Sato
When a liquid is cooled rapidly, it can form a glass, a state stuck between liquid and solid. Two groups looked inOrdering and disordering electrons
ARTICLE TOOLS http://science.sciencemag.org/content/357/6358/1381
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2017/09/28/357.6358.1381.DC1
CONTENTRELATED http://science.sciencemag.org/content/sci/357/6358/1378.full
REFERENCES
http://science.sciencemag.org/content/357/6358/1381#BIBLThis article cites 52 articles, 5 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.ScienceScience, 1200 New York Avenue NW, Washington, DC 20005. The title (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
Science. No claim to original U.S. Government WorksCopyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of
on May 16, 2020
http://science.sciencem
ag.org/D
ownloaded from