vasculitis leucitoclástica
DESCRIPTION
una vision clinica e histopatológica de la vasculitis.TRANSCRIPT

Dr. Winston Maldonado GómezDermatólogo Dermatopatólogo
Docente Universidad Nacional Pedro Ruiz Gallo

S
SINDROME PURPÚRICO
VASCULITIS
VASCULITIS DE
PEQUEÑOS VASOS
V ASCULITIS
DE HIPERSENS
IBILIDAD

SINDROME PURPÚRICO
El sindrome purpúrico se debe a la extravasación de glóbulos rojos.
Esto podria deberse a: Daño inflamatorio de los vasos :
Vasculitis. Al teración del sistema de
coagulación o de las plaquetas. Procesos vasooclusivos.

TIPOS DE PURPURA
PETEQUIAS EQUIMOSIS PÚRPURA PALPABLE PÚRPURA RETIFORME
INFLAMATORIA PÚRPURA RETIFORME NO
INFLAMATORIA.

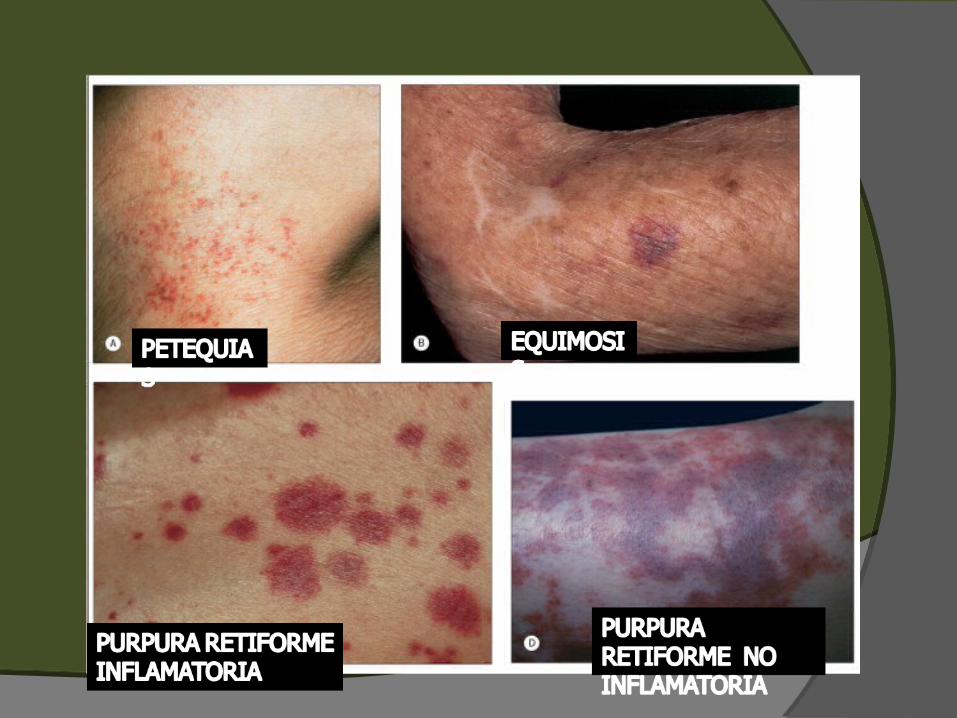

PETEQUIASPETEQUIAS
TROMBOCITOPENIA TROMBOCITOPENIA (<50 000/mm3)[:
1. Purpura Trombocitopénica Idiopática 2. Purpura Trombocitopénica Trombótica. 3. TrombocItopenias, inducidas por medicamentos..
FUNCIÓN PLAQUETARIA ANORMALFUNCIÓN PLAQUETARIA ANORMAL 1. Defectos de función plaquetarios congénitos.
2. Defectos de función plaquetarios adquiridos: AINES, Insuficiencia renal.
ETIOLOGÍA NO PLAQUETARIA:ETIOLOGÍA NO PLAQUETARIA: 1. Elevación brusca de la presión venosa central: maniobra
de Valsalva( vómitos, parto, tos paroxística) 2. Trauma. 3. Perifollicular (deficiencia de vitamina C ).

EQUIMOSIS DEFECTOS DE LA COAGULACIÓN: 1. Uso de anticoagulantes.
2. Insuficiencia hepática. 3. Deficiencia de vitamina K. 4. Coagulación intravascular diseminada.
POBRE SOPORTE DE LOS VASOS SANGUINEOS: 1. Púrpura senil. Actinic (solar, senile) purpura
2. Terapia corticoidea tópica y sistémica. 3. Deficiencia de vitamina C (escorbuto)

PÚRPURA PALPABLE I. Vasculitis leucocitoclastica debido a depósito de complejos inmunes
A. Pequeños vasos:
1. Idiopatica, asociada a infecciones o drogas por complejos IgG , IgM o Ig A(HSP). 2. Púrpura hipergammaglobulinémica de Waldenström 3. Urticaria vasculitis.
B. Vasos medianos o pequeños. 1. Crioglobulinemia mixta
2. Vasculitis Reumática(LE, AR)
II. Vasculitis leucocitoclástica pauci-immune(no es la forma más
frecuente) A. ANCA-associated
1. Wegener's 2. Microscopic polyangiitis 3. Churg–Strauss syndrome
Bolognia, J. Jorizzo, J. Rapini, R. Dermatology. 2º Ed. Ed Elsevier. 2008.España

PURPURA INFLAMATORIA RETIFORMEVasculitis de vasos medianos y pequeños
1. Vasculitis por enfermedades del tejido conectivo (LE, RA) 2. Poliarteritis nodosa
3. Vasculitis pauciinflamatorias (ANCA) Poliangiitis microscópica Granulomatosis de Wegener Sindrome de Churg–Strauss

PÚRPURA RETIFORME NO INFLAMATORIA
Oclusión por tapones plaquetarios(Necrosis por Heparina, Trombocitosis . Aglutinación relacionada al frío (Crioglobulinemia, Criofibrinogenemia) .
Oclusión debido a organismos :Hongos (mucormycosis, Aspergillus), ectima gangrenoso .
Alteración sistémica en el control de la coagulación. a)Relacionado a proteina C- y S (necrosis por Coumarina , Purpura fulminans) b)Sindrome Antifosfolipídico.
Coagulopatía vascular: Vasculopatía Livedoide. Embolization: Embolo de colesterol, Depósito de Oxalate.
Hemoglobinopatías: anemia de células falciformes, anemia hemolítica.
Neoplasias: Linfoma angiocéntrico T/NK

DEFINICION: VASCULITIS
Inflamación dirigida hacia los vasos sanguíneos, identificada mediante examen histológico

Sistemas de clasificación Patogénesis
Mediada por ANCAMediada por complejos inmunes
Compromiso anatómicoTamaño de vasos afectados y órganos
afectados Patrón histopatológico
Tipo de inflamación y distribución vascular Manifestaciones clínicas
Sindromes clínicos agrupados por evolución o respuesta a la terapia

Clasificación de vasculitis cutáneas basada en el tamaño de los vasos afectados
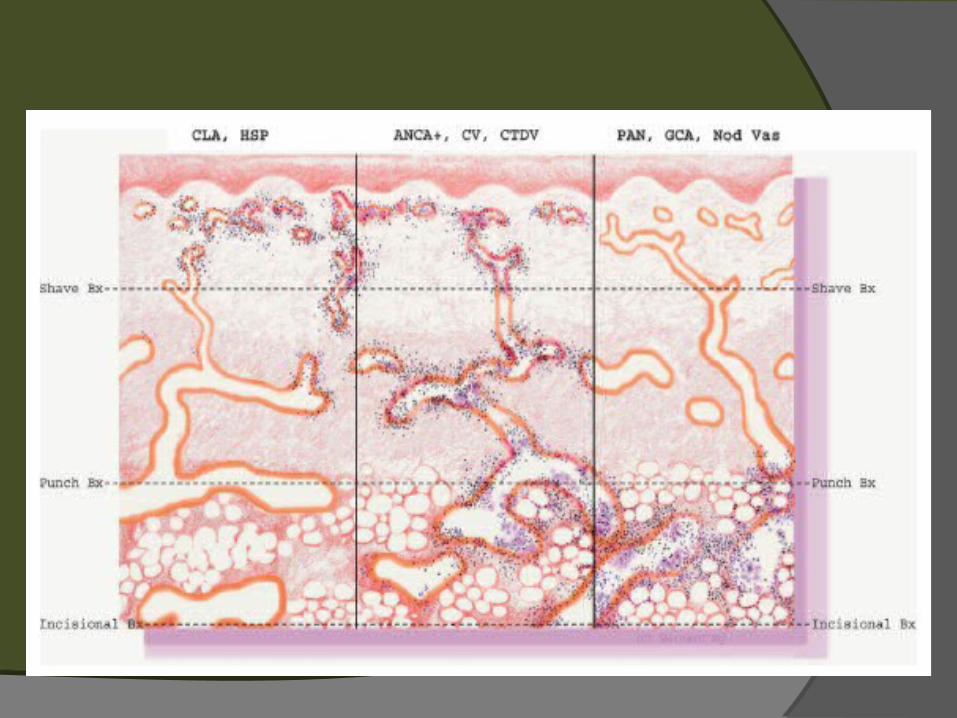

Patogénesis
Vasculitis alérgica: Sindrome de Churg-Strauss. Vasculitis urticariana.
Vasculitis mediada por anticuerpos (ANCA): Granulomatosis de Wegener. Poliangeitis microscópica.
Vasculitis mediada por complejos inmunes Vasculitis leucocitoclástica o de hipersensibilidad. Púrpura de Henoch Schonlein. Panarteritis nodosa.
Vasculitis mediada por hipersensibilidad de células T Arteritis de células gigantes.


Sin embargo…
No existe un sistema ideal para la clasificación de vasculitis
Los más relevante para el enfoque diagnóstico es:Identificación del tipo de vaso sanguíneo comprometidoDistinción entre vasculitis primaria y secundariaUso de IFD y ANCA
Clasificaciones basadas sólo en el tamaño del vaso comprometido son inexactas, sobretodo en las vasculitis ANCA+ con leucocitoclasia

VASCULITIS DE HIPERSENSIBILIDAD (Chapel Hill) Edad mayor de 16 años al comienzo de la
enfermedad. Antecedente de ingesta de fármaco al
inicio de la enfermedad. Púrpura palpable. Rash máculo papular. Biopsia que incluye anormalidad en
arteriola y vénula.
Número de criterios requeridos: 3.

Signos Físicos
VASCULITIS DE PEQUEÑOS VASOS Urticaria Púrpura Pápulas purpúricas Eritema infiltrado
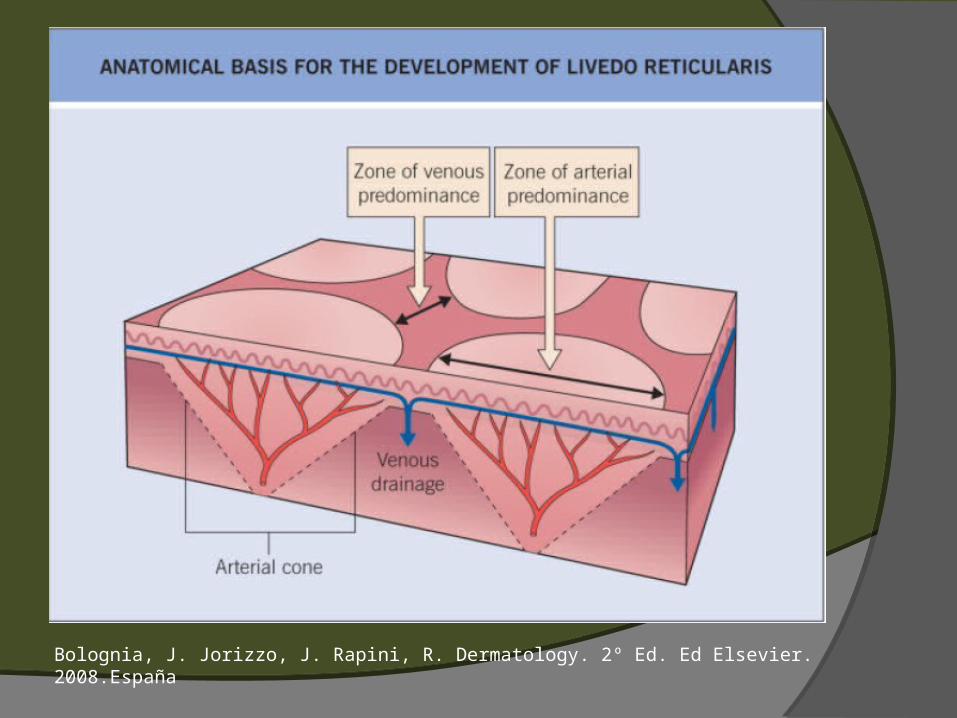
VASCULITIS DE VASOS MEDIANOS Úlceras Infartos Livedo reticularis Nódulos (eritema indurado, púrpura de HS,
Wegener)

MANIFESTACIONES CLINICAS En ocasiones se acompaña de fiebre,
artralgias, eritrosedimentasión elevada y compromiso sistémico.
El compromiso sistémico variable: articular, renal, hepático, pulmonar, cardiaco, y del SNC.
En la relacionada a fármacos curso variable con episodios recurrentes separados por periodos de meses a años.

VASCULITIS DE HIPERSENSIBILIDAD
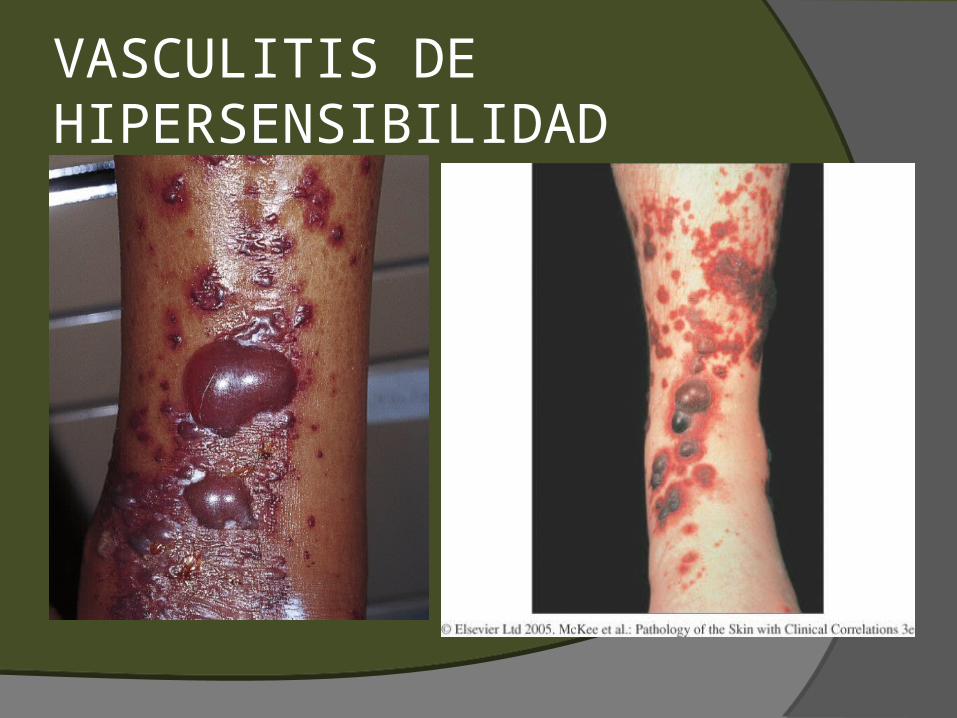
VASCULITIS DE HIPERSENSIBILIDAD

Tipos
Primaria. Sin causa o asociación conocida
SecundariaDrogasInfeccionesEnfermedad sistémicaTrauma

FACTORES ASOCIADOS
Medicamentos: betalactámicos, sulfas, AINEs.
Infecciones: Hepatitis B, HIV, Estreptococos, Estafilococos, Clamidia, Micobacterias. Candida.
Neoplásicos: Hematológicos: linfomas, mieloma múltiple,
macroglobulinemia de Waldestrom. Sólidos: Pulmón, colón, mama, renal, próstata.
Inflamatorio: Enfermedades del tej conectivo: LES, AR, SS. Enfermedad inflamatoria intestinal. Criglobulinemia.

Biopsia Un diagnóstico definitivo de vasculitis
requiere confirmación histológica.
Un diagnóstico definitivo de vasculitis no puede ser hecho sólo por biopsia sin un adecuado correlato de historia clínica, hallazgos físicos, análisis de laboratorio o angiografía

Interpretación de la biopsia Tipo de biopsia. Longitud y profundidad
Antiguedad de la lesión (entre 18-48 hrs)
Efectos de tratamientos previos
Experiencia del patólogo

SIGNOS HISTOLÓGICOS DE VASCULITIS
Signos histológicos de vasculitis aguda Cambios secundarios de vasculitis
activa Secuelas histológicas de vasculitis Cambios adyacentes indicadores de
subtipo o etiología Criterios diagnósticos de vasculitis

Signos histológicos de vasculitis aguda
Vasos de la dermis (vénulas o arteriolas)Infiltrados inflamatorios angiocéntricos y/o
angioinvasivosDisrupción y/o destrucción de la pared vascular por
el infiltrado inflamatorioDepósitos intraluminales y/o intramurales de fibrina
Vasos musculares dérmicos/subcutáneosInfiltración de la pared vascular muscular por
células inflamatoriasDepósitos intramurales y/o intraluminales de fibrina

NECROSIS FIBRINOIDE
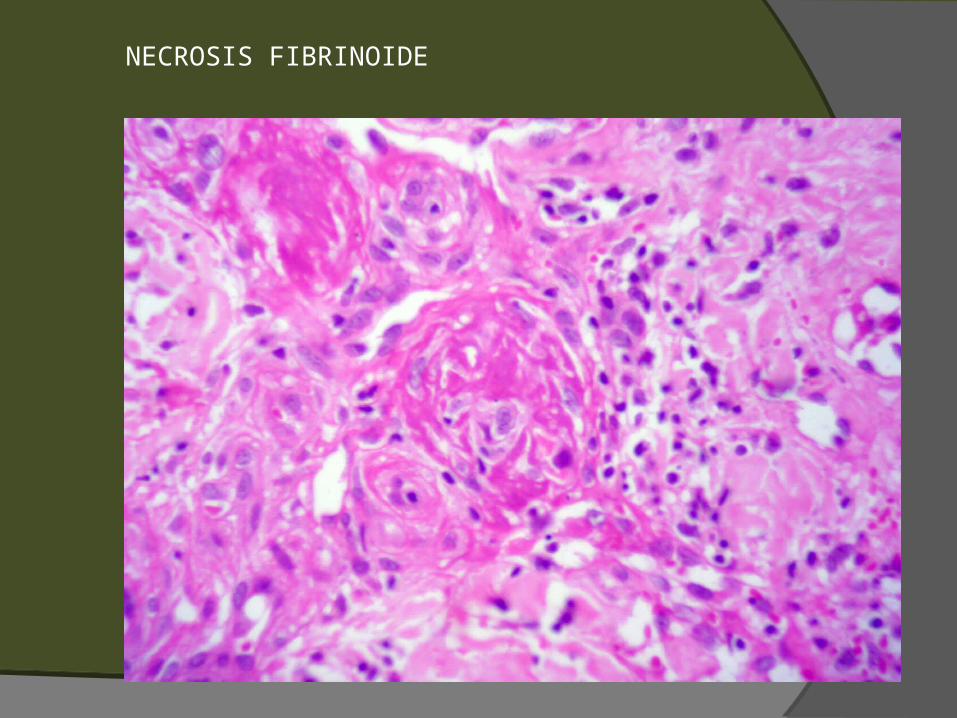
NECROSIS FIBRINOIDE
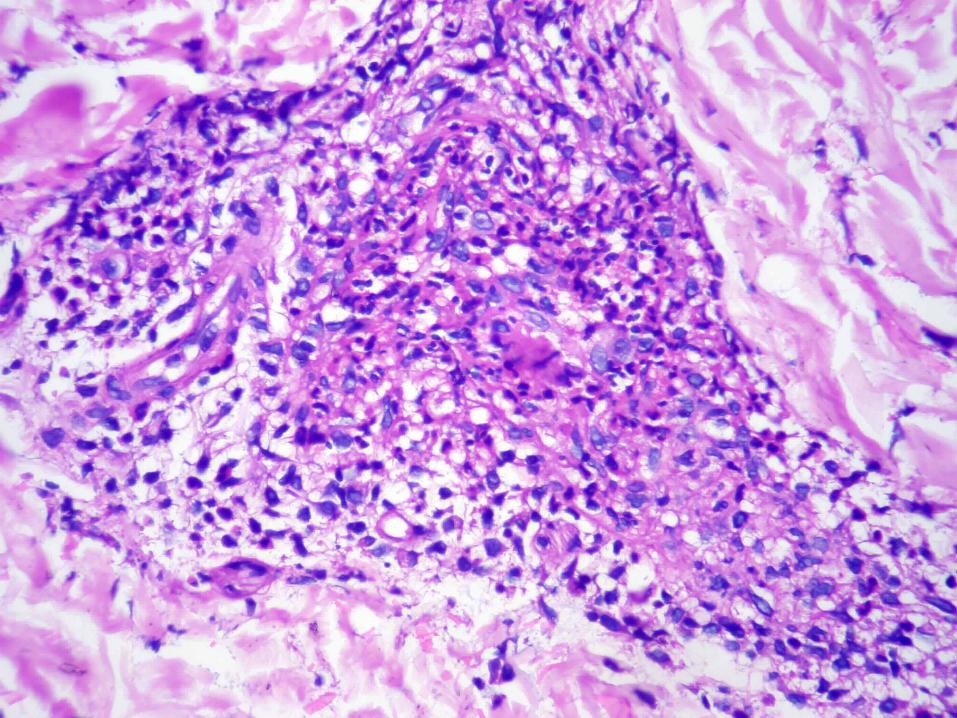

VASCULITIS DE ARTERIA MUSCULAR

Cambios secundarios de vasculitis activa
Sugestivos, pero no diagnósticos Extravasación de GR (petequias, púrpura,
hematoma) Polvo nuclear perivascular (leucocitoclasia) Edema endotelial, balonamiento o necrosis Necrosis de glándulas ecrinas (o regeneración con
hiperplasia de células basales) Ulceración Necrosis/infarto
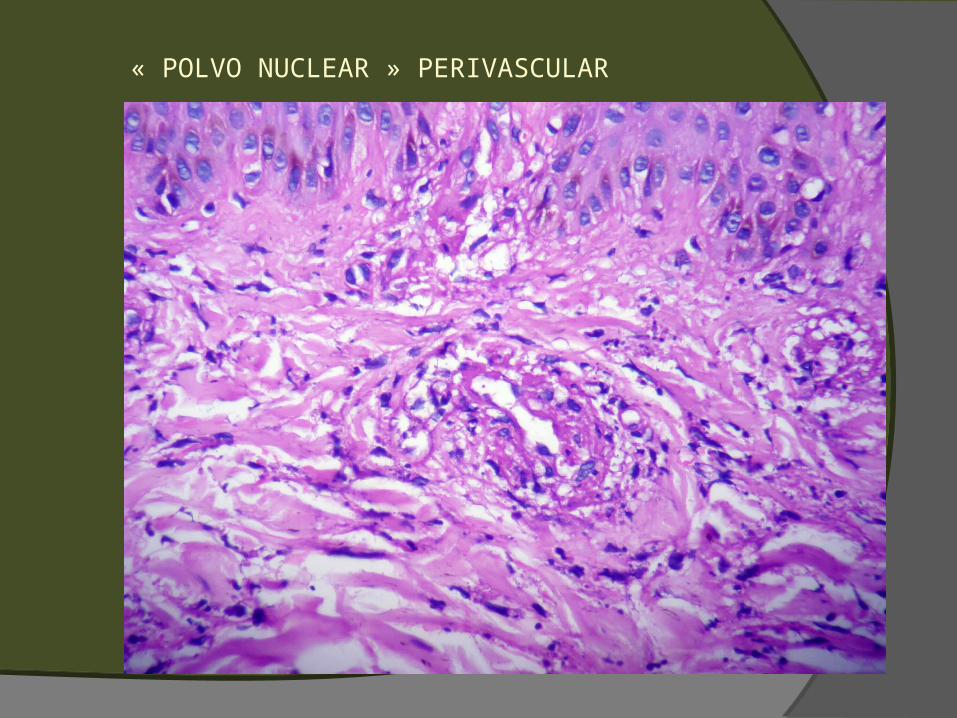
« POLVO NUCLEAR » PERIVASCULAR
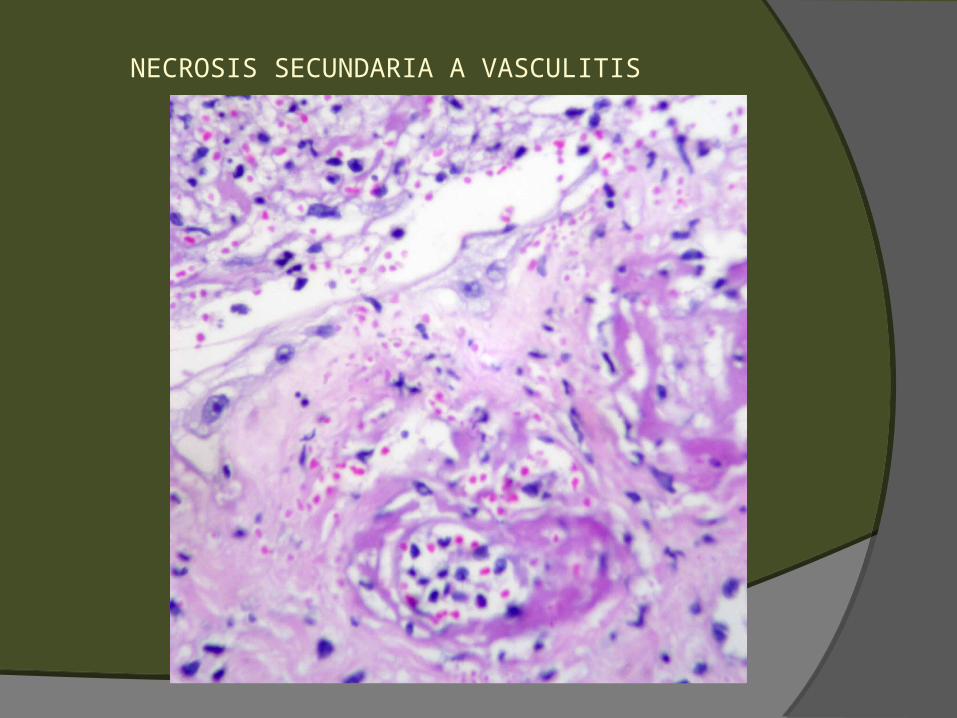
NECROSIS SECUNDARIA A VASCULITIS
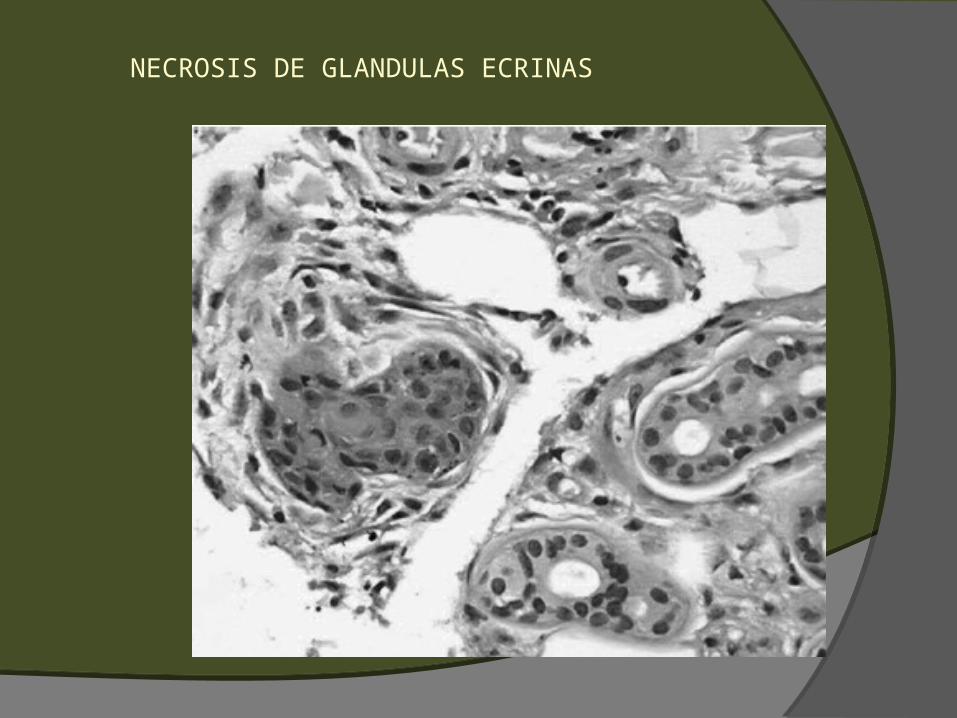
NECROSIS DE GLANDULAS ECRINAS

Secuelas histológicas de vasculitis
Laminación (en «hojas de cebolla») de los constituyentes de la pared del vaso (proliferación de pericitos y fibras musculares lisas)
Obliteración luminal (endarteritis obliterans). Proliferación de elementos celulares de la íntima o media asociados a oclusión luminal con preservación de la lámina elástica interna
Pérdida segmentaria o completa de la lámina elástica en vasos medianos y grandes, asociada con cicatriz acelular
Angioendoteliomatosis reactiva Neovascularización de la adventicia

LAMINACIÓN EN « HOJAS DE CEBOLLA »
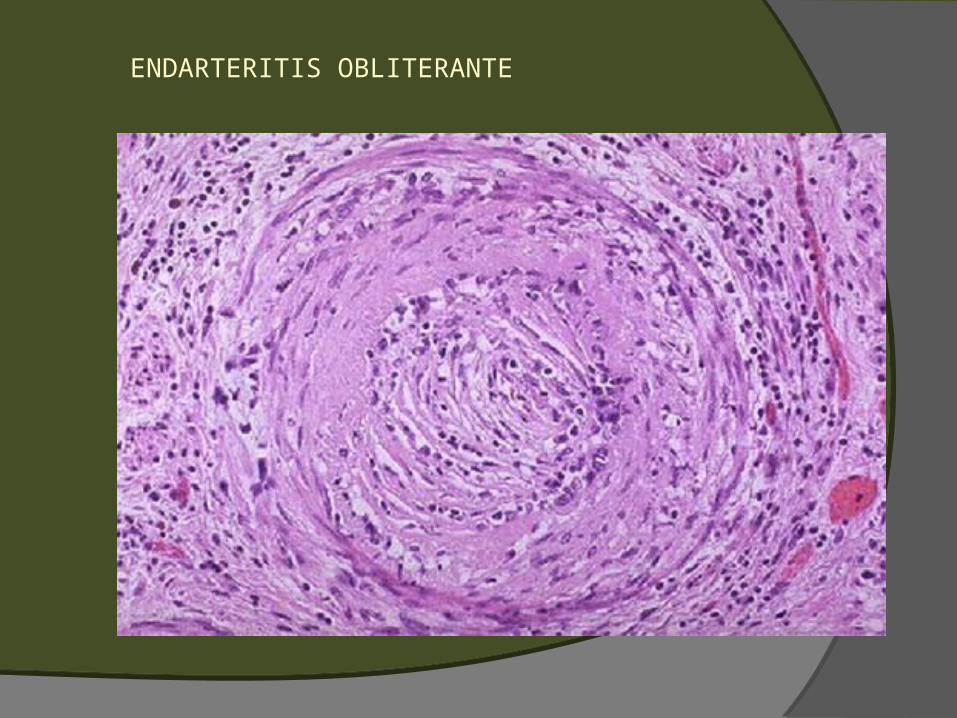
ENDARTERITIS OBLITERANTE

CICATRIZ DE ARTERITIS
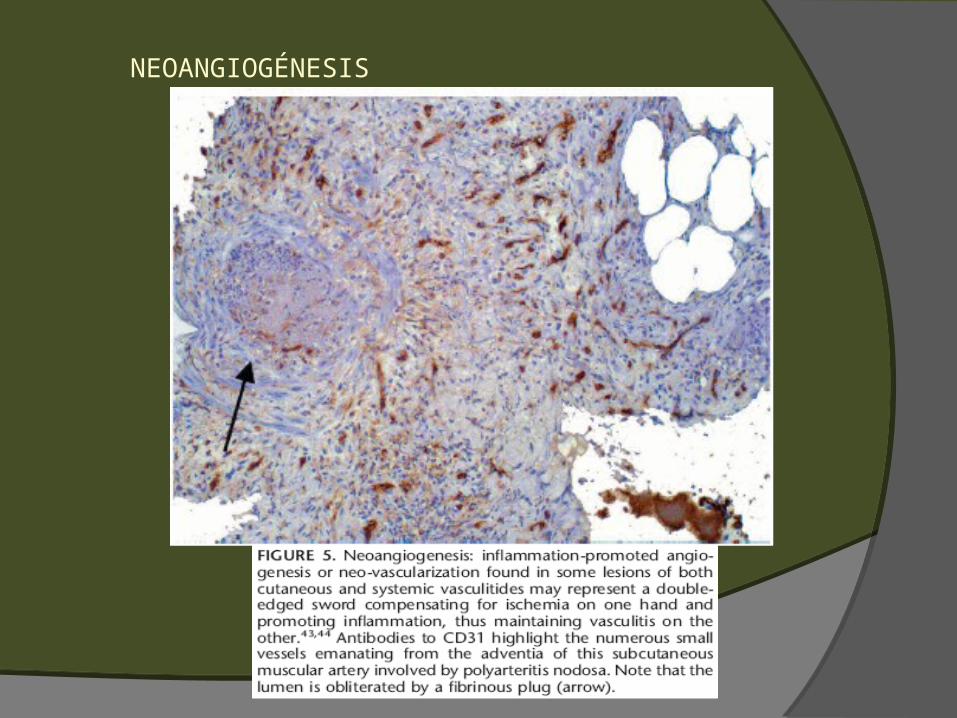
NEOANGIOGÉNESIS

ANGIOENDOTELIOMATOSIS REACTIVA

Cambios adyacentes, indicadores de subtipo o etiología
Fibrosis lamelar o estoriforme. Eritema elevatum diutinum, granuloma facial, pseudotumor inflamatorio
Dermatitis granulomatosa en empalizada (granuloma de Winkelmann) Granuloma extravascular «rojo» (eosinófilos, figuras en
flama). Sindrome de Churg-Strauss Granuloma extravascular «azul» (neutrófilos, polvo nuclear).
Granulomatosis de Wegener, vasculitis reumatoide, Churg-Strauss (raro)
Dermatitis vacuolar de interfase (a veces depósitos de mucina en la dermis). Enf. de tejido conectivo (LES, dermatomiositis)
Dermatosis pustular con abcesos neutrofílicos intraepidérmicos o subepidérmicos (infección)

GRANULOMAS COLAGENOLÍTICOS EXTRAVASCULARES

FIBROSIS


Vasculitis leucocitoclástica: Dx Diferencial
Enfermedad del suero Granuloma facial Eritema elevatum diutinum Vasculitis asociadas a IgA: PHS Vasculitis de hipersensibilidad Crioglobulinemia y macroglobulinemia Déficit hereditario de complemento Déficit de IgA Inducida por alimentos, drogas o químicos Enfermedades sistémicas: AR, EMTC, AR, LES, SS,
policondritis recidivante, Wegener, Churg-Strauss, EII
Infecciones

Púrpura de Henoch-Schonlein
Exantema purpúrico en extremidades inferiores
Asociada a artritis, dolor abdominal, hematuria, otras
Precipitación de inmunocomplejos de IgA
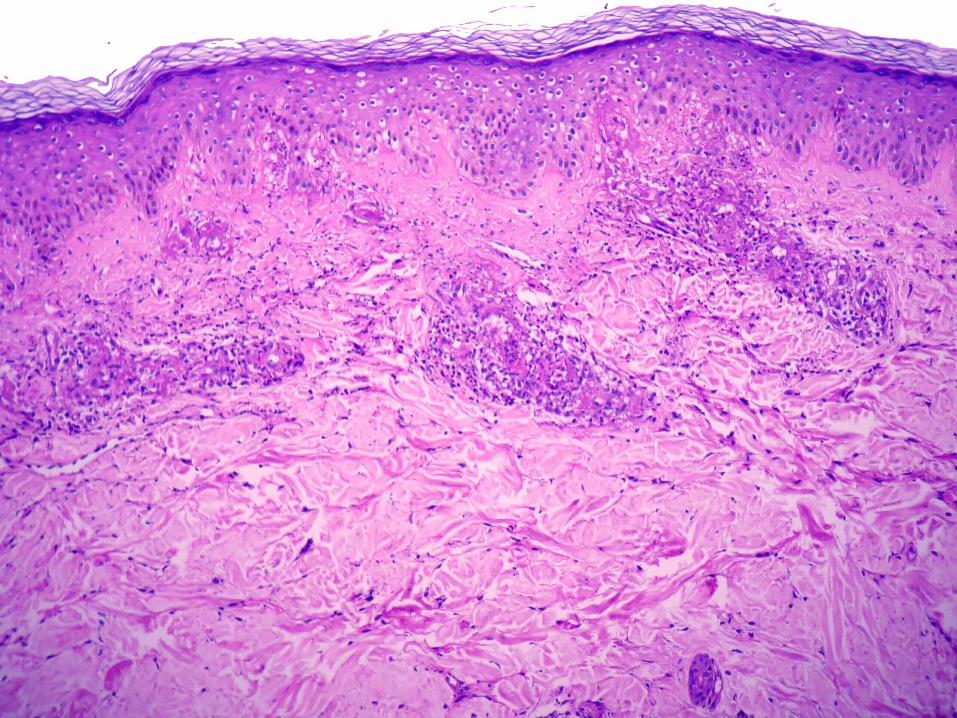
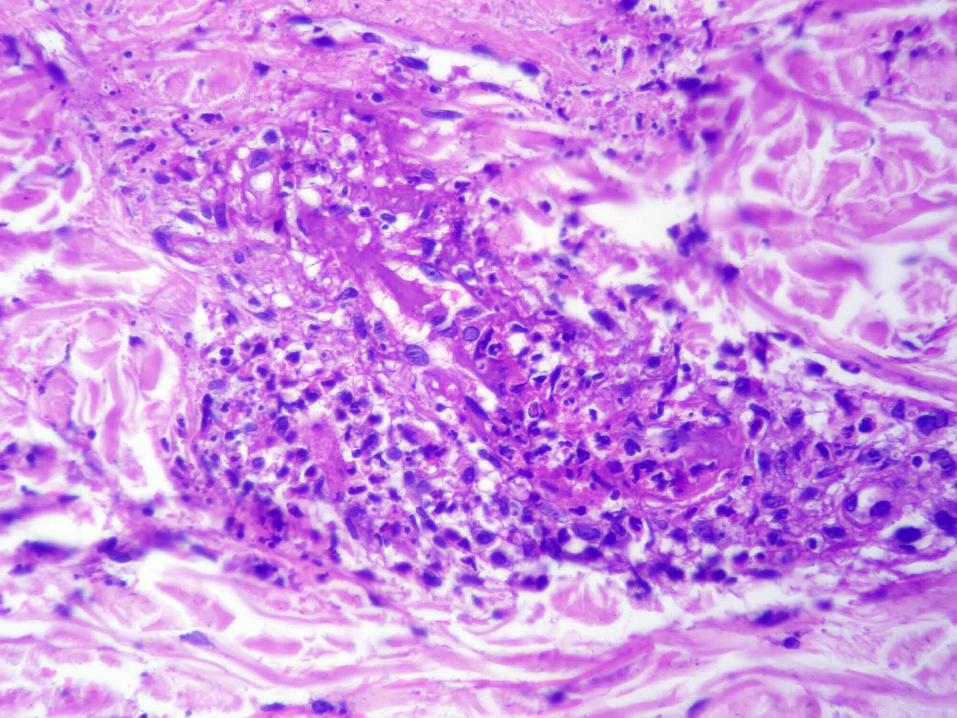


Vasculitis urticariana
Episodios recurrentes de pápulas edematosas con eritema “blanqueable”
Típicamente dura 24 a 72 horas Máculas pigmentadas residuales y
artritis episódica Asociada a: ETC (LES y SS), déficit
de inhibidor de esterasa, cáncer, complejos inmunes por drogas
Infiltrado neutrofílico perivascular e intersticial con ligera leucocitoclasia y leves indicios de lesión vascular

Urticaria crónica (Weedon)
Tipos: física, colinérgica, angioedema, agentes liberadores de histamina, mediadas por IgE, mediadas por IC
Signos histológicos:EdemaDilatación vascularInfiltrado perivascular de linfocitos y algunos
eosinófilos. Neutrófilos en fases precoces



Poliarteritis nodosa
Formas macroscópica y microscópica (poliangiitis)
Formas cutánea y sistémica Nódulos eritematosos, pulsátiles Ulceración y livedo reticularis Arterias medianas y pequeñas Infiltrado de polimorfonucleares,
necrosis fibrinoide, engrosamiento de la intima
Eosinófilos son raros. No granulomas Paniculitis focal asociada Dilataciones aneurismáticas
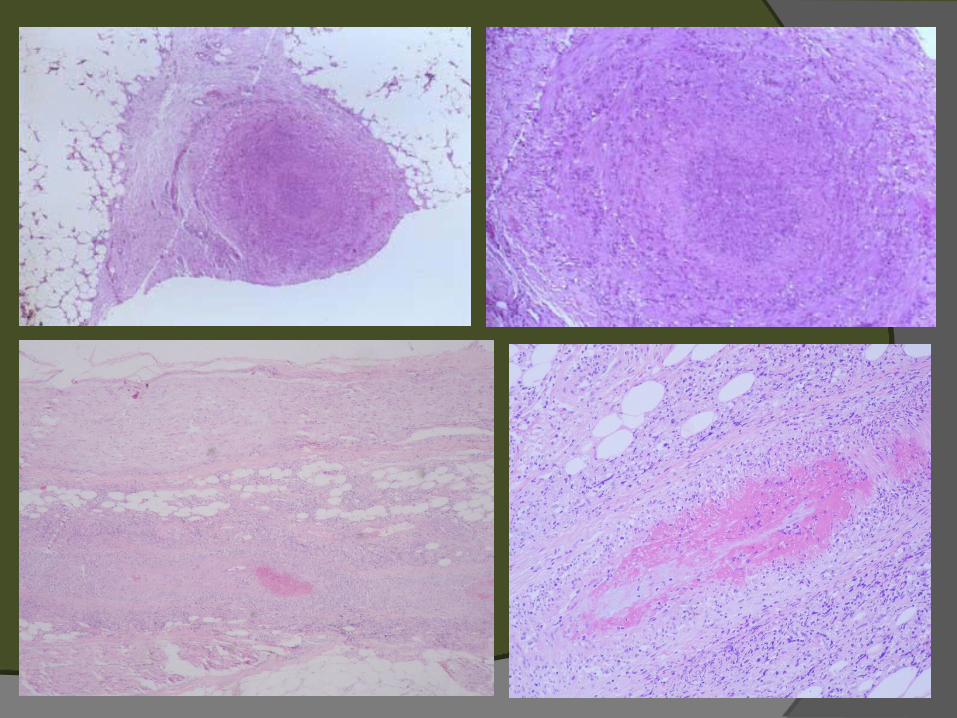

Sindrome de Churg-Strauss Vasculitis pulmonar y sistémica de vasos
pequeños asociada a granulomas extravasculares e hipereosinofilia
Lesiones cutáneas en 2/3 de pacientes. Púrpura palpable, placas eritematosas urticariformes, nódulos subcutáneos, placas equimóticas, livedo reticularis, lesiones parecidas a eritema multiforme
Histología: infiltrado polimorfonuclear y polvo nuclear (inicio). Infiltrado linfohistiocítico con células gigantes y eosinófilos. Granulomas alérgicos en empalizada
Diferencial: Wegener, PAN, granuloma necrobiótico


VASCULITIS «INCIDENTAL»
Hallazgos histológicos de vasculitis fuera de contexto clínico
Asociada a trauma o úlceras La vasculitis se considera un epifenómeno en el
contexto de las dermatosis neutrofílicas (Sweet 29%)
Vasculitis focal de vasos pequeños en una dermis fibrótica puede estar relacionada con granuloma facial, eritema elevatum diutinium o pseudotumor inflamatorio
Dermatitis de interfase con vasculitis focal: perniosis o enfermedad de tejido conectivo

VASCULITIS INCIDENTAL

DERMATITIS DE INTERFASE Y VASCULITIS

Enfermedades relacionadas inciertamente con vasculitis

En relación al correlato clinico patológico, las características clínicas depende del tamaño del vaso comprometido:Vasos pequeños: púrpura palpable,
ampollas, pústulas.Vasos medianos: nódulos, úlceras, livedo
reticularis.

ESTUDIOS DE LABORATORIO A. Necesarios: 1. Biopsia cutánea2. Hemograma.3. Velocidad de sedimentación globular4. Examen de orina5. Crioglobulinas.6. Anticuerpos contra hepatitis C, antigeno de superficie Hepatitis
B , anticuerpos VIH 7. ANCA 8. Función hepática y renal. B. Opcional
1. C3,C4,complemento total.2. ANA, anti-Ro (SS-A) 3. Biopsia cutánea para inmunofluorescencia.

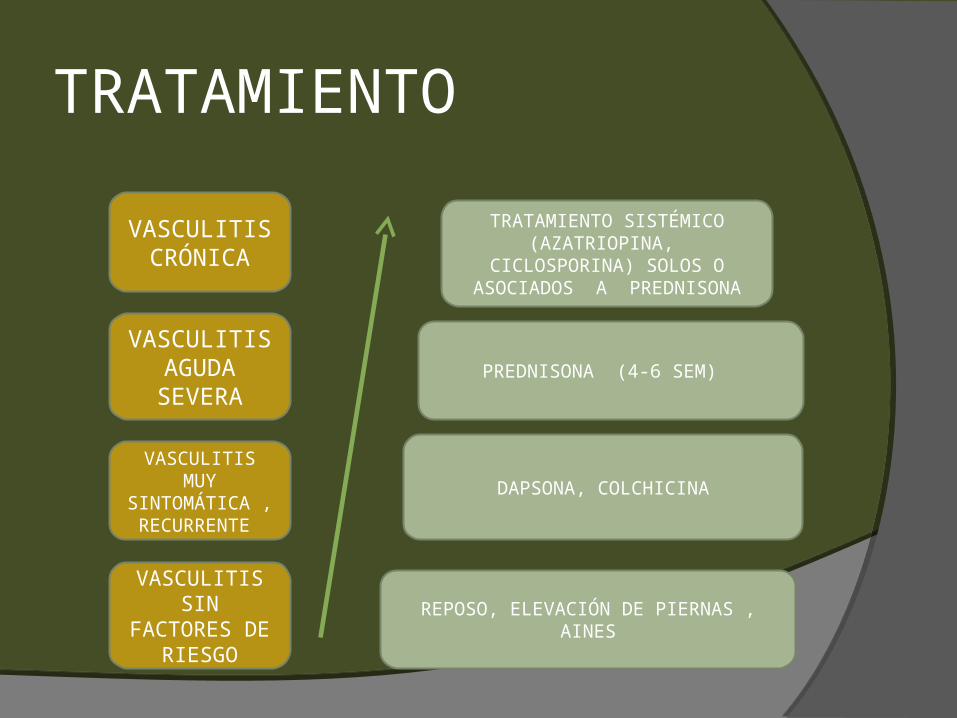
TRATAMIENTO Es basado en severidad de síntomas y extensión de
compromiso cutáneo.
El enfoque inicial debe de ser conservador: Reposo en cama. Elevación de extremidades inferiores. AINEs, analgésicos y antihistamínicos.
Si la erupción en significativamente sintomática, recurrente o progresa a forma nodular ulcerativa entonces terapia sistémica.
En caso de terapia sistémica reevaluación cada 6-12 meses.

TRATAMIENTO Colchicina (0.6-1.8 mg/d) :
resolución en 1-2 sem.Molestias GI.
Dapsona (100-200mg/d):Efecto antineutrofílico.Necesita evaluación de G6PDH.
Coadministración de Colchicina o pentoxifilina con Dapsona.

TRATAMIENTO Prednisona (0.5-1 mg/d):
Con retiro prolongado (4-6 semanas).En episodios agudos o únicos.
En casos crónicos, recurrentes o asintomáticos se recomienda el uso de fármacos ahorradores de esteroides, solos o acompañados de prednisona a altas dosis.
Azatriopina (100 mg/d): dosar TPMT. Evaluar infecciones.Riesgo de mielosupresión.Riesgo de tumores (linfomas).
Ciclosporina (2.5-5 mg/kg/d): preferible en tratamiento de infección aguda. Riesgo de nefrotoxicidad e hipertensión.
TRATAMIENTO
VASCULITIS CRÓNICA
VASCULITIS AGUDA SEVERA
VASCULITIS MUY
SINTOMÁTICA , RECURRENTE
VASCULITIS SIN
FACTORES DE RIESGO
TRATAMIENTO SISTÉMICO (AZATRIOPINA, CICLOSPORINA)
SOLOS O ASOCIADOS A PREDNISONA
PREDNISONA (4-6 SEM)
DAPSONA, COLCHICINA
REPOSO, ELEVACIÓN DE PIERNAS , AINES

GRACIAS