気管支喘息発症と疾患感受性の解析 気道リモデリ...
TRANSCRIPT

165
●ファイザーフェローシップ報告
はじめに
気管支喘息の基本病態は気道の慢性炎症であるというパラダイムはほぼ定着しつつあり,長期管理治療のための抗炎症療法がガイドラインの普及とともに広く用いられるようになった.90 年代には好酸球性気道炎症であることが解明され,その後のTh1�2 セオリーを経て,重症・難治化の病態と気道リモデリングの関与が重要視されるようになった.小児喘息において早期ステロイド吸入療法の導入が推奨されるようになったことも,気道リモデリング制御の重要性を反映していると考えられる.気道リモデリングの病理学的所見は気道局所への炎症
細胞浸潤の他,気道上皮の剝離と線維芽細胞・平滑筋の増生・肥厚や基底膜下へのコラーゲンの沈着である.喘息の病態において基底膜下組織の肥厚は治療難治性を誘導するため,その機序の解明・制御は喘息治療の最も重要なストラテジーの一つになりうると考えられる.Dr. D. Davies & Pro. ST. Holgate のグループは,喘息
の 包 括 的 病 態 理 解 の た めEMTU(Epithelial-Mesenchymal-Trophic-Unit)を提唱している1).傷害の主たる場である上皮系のみならず,線維芽細胞・平滑筋細胞を含めた気道構成組織による炎症の協調作用に注目し精力的に研究を推進している.このEMTU(Epithelial-Mesenchymal-Trophic-Unit)の概念は喘息研究者の間で広く受け入れられている.喘息発症において,環境因子のみならず遺伝的な背景の関与は疑いようもなく,発症のみならず,治療面における新たなブレイクスルーのため新規喘息関連遺伝子の発見・同定が切望されている.2002 年 Pro ST. Holgate のグループとシェーリングプラウ社は,新規喘息関連遺伝子としてADAM33(ADisintegrin And Metalloprotease 33)を Nature 誌に発
表し2),現在その機能解析を進めている.その中で特に線維芽細胞との関連性が明らかになり,喘息の病態における(myo)fibroblast:(筋)線維芽細胞の増生・分化の解析の重要性が明らかとなった.それらのバックグラウンドより,私の研究テーマとして,喘息・非喘息患者から採取した気道・気管支線維芽細胞をモチーフとし,増殖とそれに関わる細胞内シグナルを解析し,喘息・非喘息間での差違を検討することから始めた.
サンプルと方法
ヒト気管支線維芽細胞:症状・気道過敏性により気管支喘息と診断,重症度分類はGINA(Global Initiative forAsthma)ガイドライン(1995 年)を使用していた.サザンプトン大学及び病院の倫理委員会の承認後,サンプル提供者に実験の方法・目的を説明し承諾書を得てからこのスタディーを行った.全例非喫煙者で,重症患者のみ高用量の吸入ステロイドが使用されていた.経気管支鏡下にて気管分岐部・第一分岐部の上皮を擦過後,鉗子にて組織採取.顕微鏡下にて平滑筋を除いた後,線維芽細胞と思われる組織を細断し,細胞培養液下のディッシュに移し,CO2インキュベーターで数日培養する.その後,平滑筋細胞のコンタミの有無をチェックするため,細胞の一部を免疫染色する.こうして得られた線維芽細胞を 8継代まで使用した.線維芽細胞は I型コラーゲンでコートしたフラスコ上にまき 10%FBS DMEM(50IU�l penicillin, 50 µg�ml streptomycin, 2mM L-glutamine,1×Non-Essential aminoacids,1mM So-dium Pyruvate)で培養した.無血清下での実験にはUl-traculture(Cambrex 社)を使用した.試 薬,抗 体:EGF,TGFβ2(PeproTech EC社),
AG1478,U0126,LY294002,Rapamycin(LC Laborato-ries 社),Phosphotyrosine Agarose(BD Bioscieces 社),Mouse biotinylated anti-phosphotyrosine(PY20)Anti-body(Transduction Laboratories 社),Phospho-EGFR(Tyr1173)Antibody(Santa Cruz Biotechnology 社),Anti-PI3-Kinase p85,N-SH2 domain Antibody(upstatebiotechnology 社),Phospho-HER2�ErbB2(Tyr1248)
気管支喘息発症と疾患感受性の解析
気道リモデリングの制御をめざして
松本 健
日本大学呼吸器内科・サザンプトン大学病院アレルギー炎症研究部門(2002 年 8 月~2004 年 7 月)〒173―8610 東京都板橋区大谷口上町 30―1日本大学医学部附属板橋病院第一内科
日呼吸会誌 46(2),2008.

日呼吸会誌 46(2),2008.166
グラフ1 Fibroblast proliferation in serum free medium.
Antibody,Phospho-p44�42 MAP kinase(Thr202�Tyr204)(E10)Monoclonal antibody,p44�42 MAPkinase antibody,Phospho-AKT(Ser473)(587F11)Mon-oclonal Antibody,AKT Antibody,Phospho-p70S6Kinase(Thr389)(1A5)Monoclonal Antibody(Cell Sig-naling 社),Monoclonal anti-αSmooth Muscle Actin anti-body(Sigma 社),Anti-M3 receptor Antibody(Re-search&Diagnostic 社),Anti-HC Myosin Antibody(Sigma 社).細胞増殖(メチレンブルーアッセイ):I型コラーゲ
ンでコートした 24 穴ディッシュに 5×104cells�ml,0.5ml�well 細胞をまき,6時間 10%FBS DMEMにて培養し細胞をディッシュに接着させる.その後,EGFや in-hibitor を添加したUltraculture に置換し,1,2,4,6日間培養する.目的の時間培養後,培養液を 500µl�wellformal saline(4%formaldehyde,0.9%NaCl),に置換し,細胞を固定する.1時間以上かけて固定後,formalsaline を破棄しディッシュを乾燥させ,メチレンブルー液を加え細胞を染色する.30 分以上かけて染色後,メチレンブルー液を取り除き,ディッシュを乾燥させる.1:1 0.1%HCL-ethanol でメチレンブルーを細胞内から溶解させ吸光度 630nmを測定する7).1 つの条件につき3well 用意し,その平均値を使用した.免疫沈降法:刺激後の細胞をプロテアーゼ阻害剤添加
PBSで 2 回洗い,その後RIPA buffer を加えてサンプルを回収した.サンプルをソニケートした後,4℃下16,000xg 遠心,上清のみ回収した.使用する抗体と同種の IgG,その後 Protein A�G agarose を加え 1回プレクリアする.BCA法でタンパク定量し,1サンプル当たり 500µg になるよう調整し,5µg 目的の抗体を添加し,4℃ overnight でローテーションさせる.翌日,Pro-tein A�G agarose を 25~50µl 加え 4℃ 1~2 時間ローテーションさせる.遠心後,ペレットをRIPA buffer で3 回洗い,2×SDS sample buffer を加えて,95℃ 5 分間サンプルをボイルする.その後下記のウエスタンブロット法を行った.ウエスタンブロット法:刺激後の細胞をプロテアーゼ
阻害剤添加 PBSで 2 回洗い,その後RIPA buffer を加えてサンプルを回収した.BCA法にてタンパク定量を行い,SDS-Sample buffer を加えた後,1レーン当たり10µg のタンパクを SDS-PAGEした.PVDFメンブレンにタンパクを転写後,スキムミルクでブロッキングし,上記抗体を用いて immunoblot し,HRPで発色させた.フィルム上のバンド強度はNIHイメージで定量し,リン酸化�非リン酸化タンパク量で標準化した.
結果と考察
1)ヒト気管支線維芽細胞は無血清下においても増殖する.喘息の病理学的所見の特徴は,気道上皮細胞の剝離,
粘液栓,好酸球を中心とした炎症細胞浸潤,粘膜・粘膜下の浮腫といった可逆性の病変と,線維芽細胞・平滑筋の増生・肥厚や基底膜下へのコラーゲンの沈着,気道上皮での杯細胞の過形成,血管新生などの不可逆性の器質的病変からなる3).気道リモデリングが形成されると,気道壁の肥厚による不可逆性の気流制限が誘導され,喘息を難治化させ治療薬への反応も低下する.病理学的に観察される基底膜下組織肥厚のメカニズムを in vitro で解析するため,まず得られた喘息・非喘息線維芽細胞を無血清培地(Ultraculture)にて数日培養し,増殖曲線を作成した(グラフ 1).健常・喘息軽症例・喘息重症例を無血清下 6日間まで
培養した.その結果,いずれの細胞も無血清下にて増殖することが可能であったが,健常・喘息重症例では培養開始時の 2倍に増殖した後,増殖カーブはプラトーに達した.しかし,喘息軽症例では培養 2日後までは他のサンプルと同様の増殖カーブであったが,培養 4日以降,急峻な増殖を続け,6日では培養開始時の 3.5 倍以上に増殖した.喘息軽症例線維芽細胞において,何らかの増殖因子の新たな合成,そして分泌・消費がこのHyper
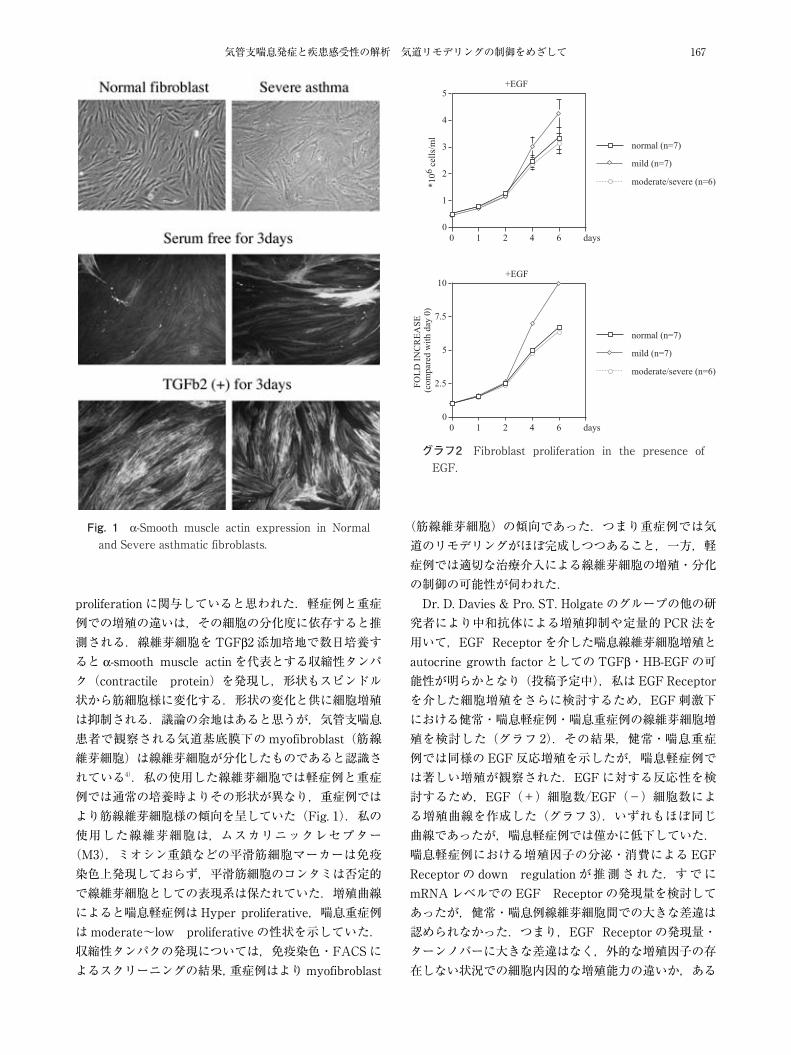
気管支喘息発症と疾患感受性の解析 気道リモデリングの制御をめざして 167
Fig. 1 α-Smooth muscle actin expression in Normal and Severe asthmatic fibroblasts.
グラフ2 Fibroblast proliferation in the presence of EGF.
proliferation に関与していると思われた.軽症例と重症例での増殖の違いは,その細胞の分化度に依存すると推測される.線維芽細胞をTGFβ2 添加培地で数日培養すると α-smooth muscle actin を代表とする収縮性タンパク(contractile protein)を発現し,形状もスピンドル状から筋細胞様に変化する.形状の変化と供に細胞増殖は抑制される.議論の余地はあると思うが,気管支喘息患者で観察される気道基底膜下のmyofibroblast(筋線維芽細胞)は線維芽細胞が分化したものであると認識されている4).私の使用した線維芽細胞では軽症例と重症例では通常の培養時よりその形状が異なり,重症例ではより筋線維芽細胞様の傾向を呈していた(Fig. 1).私の使用した線維芽細胞は,ムスカリニックレセプター(M3),ミオシン重鎖などの平滑筋細胞マーカーは免疫染色上発現しておらず,平滑筋細胞のコンタミは否定的で線維芽細胞としての表現系は保たれていた.増殖曲線によると喘息軽症例はHyper proliferative,喘息重症例はmoderate~low proliferative の性状を示していた.収縮性タンパクの発現については,免疫染色・FACSによるスクリーニングの結果,重症例はよりmyofibroblast
(筋線維芽細胞)の傾向であった.つまり重症例では気道のリモデリングがほぼ完成しつつあること,一方,軽症例では適切な治療介入による線維芽細胞の増殖・分化の制御の可能性が伺われた.Dr. D. Davies & Pro. ST. Holgate のグループの他の研
究者により中和抗体による増殖抑制や定量的 PCR法を用いて,EGF Receptor を介した喘息線維芽細胞増殖とautocrine growth factor としてのTGFβ・HB-EGFの可能性が明らかとなり(投稿予定中),私はEGF Receptorを介した細胞増殖をさらに検討するため,EGF刺激下における健常・喘息軽症例・喘息重症例の線維芽細胞増殖を検討した(グラフ 2).その結果,健常・喘息重症例では同様のEGF反応増殖を示したが,喘息軽症例では著しい増殖が観察された.EGFに対する反応性を検討するため,EGF(+)細胞数�EGF(-)細胞数による増殖曲線を作成した(グラフ 3).いずれもほぼ同じ曲線であったが,喘息軽症例では僅かに低下していた.喘息軽症例における増殖因子の分泌・消費によるEGFReceptor の down regulation が推測された.すでにmRNAレベルでのEGF Receptor の発現量を検討してあったが,健常・喘息例線維芽細胞間での大きな差違は認められなかった.つまり,EGF Receptor の発現量・ターンノバーに大きな差違はなく,外的な増殖因子の存在しない状況での細胞内因的な増殖能力の違いか,ある

日呼吸会誌 46(2),2008.168
Fig. 2 Erk1/2 phosphorylation of human bronchial primary fibroblasts in serum free condition for several days.normal: normal subject's fibroblastmild: mild asthmatic fibroblastsevere: severe asthmatic fibroblast
グラフ3 The response to EGF in human primary bron-chial fibroblasts proliferation.*EGF-stimulated cells number/Serum Free cultured-cells number
いは細胞自身が分泌・消費する何らかの autocrinegrowth factor の 存 在 が 考 え ら れ た(TGFβ・HB-EGF?).ELISA法にてTGFβ・HB-EGFの産生についてすでに検討してあり,TGFβについては健常>喘息,HB-EGFについては健常・喘息例ともに測定感度以下であった. 喘息例でのTGFβの消費亢進が疑われていた.HB-EGFは細胞外に分泌された後,細胞外基質や細胞膜表面にアンカーされるため,検出困難な状況であった.上記については他研究者により検討中.2)無血清下におけるヒト気管支線維芽細胞のHyper-
proliferation にMAPKが関与する.すでに我々(日本大学,橋本 修ら)は,気道炎症・
リモデリングにおけるMAPK family の重要性についてin vitro での疾患モデルにて報告してきた5)6).今回,喘
息・非喘息検体を使用し,細胞増殖に関連するセリン・スレオニンキナーゼであるMAPK family 及び PI3Kfamily の関わりについて検討した.MAPK family 分子は 2つのMAPK(Erk1,Erk2),3つの JNK,4つの p38及び Erk5 からなる.増殖刺激により活性化されるErk1�2,炎症性サイトカイン・細胞外ストレスにより活性化される JNK,p38MAPKに分類される.PI3K familyは,細胞膜に近接してレセプターのシグナルを細胞質内の標的分子へと伝える PI3K,その下流に存在するAKT,p70S6K,mTORなどが存在する.PI3Kはインスリン応答シグナルとしての作用のみならず,その後の研究により増殖・分化・アポトーシスなど多岐にわたる細胞現象に関与することが明らかになっている.軽症例線維芽細胞のHyper proliferation に上記の増
殖関連シグナルが関わっているか否か,western blot 法により,Erk1�2,AKTのリン酸化を検討した.無血清下で数日培養し,タンパクを回収・定量したのち,SDS-PAGE・Immunoblot を行った.リン酸化AKTは無血清条件下では認められなかったが,リン酸化Erk1�2 は僅かながら認められた(Fig. 2).健常 4例,軽症 5例,重症例 4例の線維芽細胞について検討した.健常例では無血清 6日培養にてリン酸化Erk1�2 はほとんど認められなくなっていたが,喘息例では低血清 6日培養にてもリン酸化Erk1�2 が認められ,持続的なErk1�2 の活性が推測された(グラフ 4).この持続的リン酸化Erk1�2と喘息線維芽細胞のHyper proliferation との関わりを検討するため,MEK1�2(Erk1�2 を活性化させる直接の分子)阻害剤であるU0126 を使用し,細胞増殖に対する効果を検討した(グラフ 5).その結果,U0126 濃
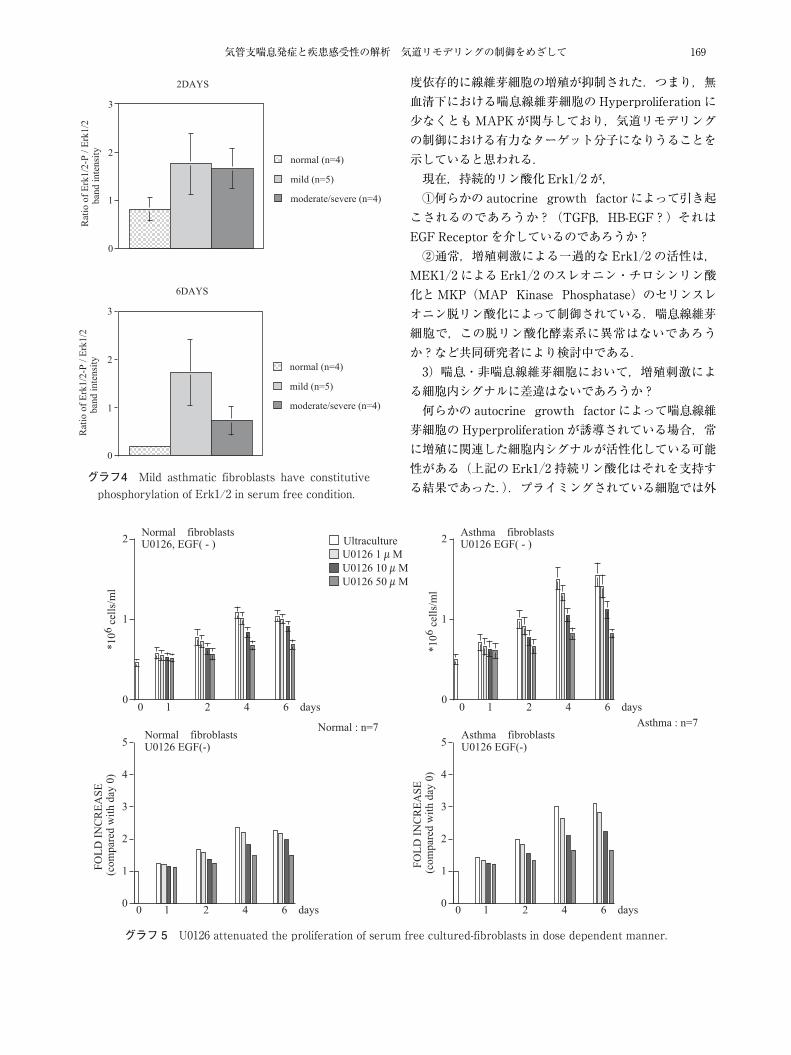
気管支喘息発症と疾患感受性の解析 気道リモデリングの制御をめざして 169
グラフ4 Mild asthmatic fibroblasts have constitutive phosphorylation of Erk1/2 in serum free condition.
グラフ5 U0126 attenuated the proliferation of serum free cultured-fibroblasts in dose dependent manner.
度依存的に線維芽細胞の増殖が抑制された.つまり,無血清下における喘息線維芽細胞のHyperproliferation に少なくともMAPKが関与しており,気道リモデリングの制御における有力なターゲット分子になりうることを示していると思われる.現在,持続的リン酸化Erk1�2 が,①何らかの autocrine growth factor によって引き起
こされるのであろうか?(TGFβ,HB-EGF?)それはEGF Receptor を介しているのであろうか?②通常,増殖刺激による一過的なErk1�2 の活性は,
MEK1�2 による Erk1�2 のスレオニン・チロシンリン酸化とMKP(MAP Kinase Phosphatase)のセリンスレオニン脱リン酸化によって制御されている.喘息線維芽細胞で,この脱リン酸化酵素系に異常はないであろうか?など共同研究者により検討中である.3)喘息・非喘息線維芽細胞において,増殖刺激によ
る細胞内シグナルに差違はないであろうか?何らかの autocrine growth factor によって喘息線維
芽細胞のHyperproliferation が誘導されている場合,常に増殖に関連した細胞内シグナルが活性化している可能性がある(上記のErk1�2 持続リン酸化はそれを支持する結果であった.).プライミングされている細胞では外

日呼吸会誌 46(2),2008.170
Fig. A EGF stimulated tyrosine phosphorylation of EGFR family in human primary bronchial fibroblasts.
Fig. B EGF induced tyrosine phosphorylation of EGFR in human primary bronchial fibroblasts.
Fig. C EGF induced tyrosine phosphorylation of erbB2 (neu) in human primary bronchial fibroblasts.
Fig. D EGF induced tyrosine phosphorylation of EGFR and erbB2 in human primary bronchial fibroblasts. AG1478 blocked tyrosine phosphorylation of EGFR and erbB2 completely.
来の刺激に対して過剰に反応を示す場合と,逆に反応が鈍化している場合が考えられる.喘息線維芽細胞ではEGFレセプターを介したHyperproliferation の可能性が明らかになってきているため8),私は EGF ligand(EGF)による細胞内シグナルの反応を正常例と軽症喘息例で検討した.EGFレセプターファミリーはEGFR,HER2�neu
(ErbB2),HER3(ErbB3),HER4(ErbB4)から構成されており,非刺激下では細胞膜表面上にそれぞれモノマーで存在している.EGF ligand(EGF,TGFα,am-phireguline etc)の結合によりレセプターはヘテロダイマーを形成し,細胞内ドメインに存在するチロシンリン酸化を経てその下流に存在する分子を活性化させる.PI3K-AKT pathway,Ras-Raf-MAPK(Erk1�2)は細胞増殖・細胞生存に関連する代表的な細胞内シグナルである.よって,喘息・非喘息線維芽細胞でのEGFによるEGFRフ ァ ミ リ ー・PI3K-AKT pathway,Ras-Raf-MAPK(Erk1�2)の活性化を検討した.I型コラーゲンでコートしたフラスコに 1.0×104cells�
cm2になるように細胞を蒔き,90%~confluent に生育した後,24 時間無血清下で培養する.その後,EGF(10µg�ml)で刺激し,目的の時間培養後,サンプルを回収
した.PY20(チロシンリン酸化抗体)で免疫沈降・ウエスタンブロットを行った.その結果,分子量 180 Kda周囲に EGFRファミリーと思われるチロシンリン酸化を認め,正常例・喘息例ともEGF(10µg�ml)刺激後 5~10 分にmaximal band が見られた(Fig. A).正常例・喘息例でBand の多少の強弱はあるものの,kinetics は同様の傾向であった.次にEGFにより,どのEGFRファミリーの活性化が誘導されるか検討するため,チロシンリン酸化EGFR抗体,チロシンリン酸化ErbB2 抗体を用いて検討した.また,EGFRファミリー特異的チロシンリン酸化阻害剤(AG1478 : Tyrphostin)の作用を併せて検討した.その結果,EGFR,ErbB2 チロシンリン酸化ともEGF(10µg�ml)刺激後 5~10 分に最大に達し,AG1478 前処理によってEGFR,ErbB2 チロシンリン酸化は完全に抑制されていた(Fig. B~D).EGF刺激により,ヒト気管支線維芽細胞においてEGFR-ErbB2 のヘテロダイマーが形成され,細胞内ドメインに存在

気管支喘息発症と疾患感受性の解析 気道リモデリングの制御をめざして 171
Fig. E PI3K, through SH2 domains of its adaptor p85 subunit, associates with proteins phosphorylated on tyrosine residues in human primary bronchial fibroblast.P: Positive control (EGF treated A431)
Fig. F Time course of Erk1/2 phosphorylation in EGF-stimulated human primary bronchial fibroblast.
Fig. G EGF induced phosphorylation of AKT and p70S6K in human primary bronchial fibroblasts.
するチロシンリン酸化が誘導された.PI3Kは細胞質にて catalytic subunit である p110 と
regulatory subunit である p85 のヘテロダイマーを形成しており,増殖刺激に反応して p85 の SHドメインを介してレセプターチロシンキナーゼと結合し,活性化する.ヒト気管支線維芽細胞におけるこの結合を確認するためEGF刺激後,サンプルを PY20 にて免疫沈降,SDS-PAGE後 p85 PI3K 抗体にて Immunoblot を行った.Positive control として EGF刺激したA431(human epi-dermoid carcinoma cell)を使用した.その結果,EGF刺激 1分後に p85 の SHドメインとEGFレセプターチロシンキナーゼとの結合が認められた(Fig. E).筋線維芽細胞と同様に気道リモデリングの責任病巣の一つである気管支平滑筋においては,EGFR-EGF Ligand による細胞増殖・細胞内シグナルについてすでにいくつか報告されている.気管支平滑筋ではmRNAレベルにおいてすべてのEGFR family(ERFR,ErbB2~4)を発現しているが,ErbB3,ErbB4 は EGFR-EGF Ligand Bind-ing に呼応する活性化は認められない9).mRNA発現は確認しなかったが,EGF刺激による気管支線維芽細胞でのErbB3 リン酸化を検討したものの,上記の報告と同様に機能的に不活性化している傾向であった(datanot shown).A431 細胞では EGF Ligand による PI3Kの活性化に
ErbB3 が重要とされている一方,PC12,A549 では認められない.EGF Ligand による PI3Kの活性化の過程で,どのEGFR family が関与しているかは細胞特異的な現象のようである.
次に PI3Kの下流に存在するAKT(PKB),p70S6K,及び EGFRの下流に存在するErk1�2(classical MAPK)の EGF刺激下におけるセリン�スレオニン・チロシンリン酸化を検討した.EGF刺激にて上記のいずれの分子もリン酸化が認められ,喘息・非喘息線維芽細胞でのTime course に差違は認められなかった(Fig. F,G).しかし,喘息・非喘息線維芽細胞両者ともErk1�2 のスレオニン・チロシンリン酸化の遷延傾向が見られた.EGF刺激によって線維芽細胞より何らかの増殖因子が分泌され,それが autocrine 的に Erk1�2 を活性化している可能性も考えられた.PI3K pathway(AKT,p70S6K)及び Erk1�2 ともそ
れぞれ選択的・特異的な阻害剤が存在する(LY294002-PI3K,Rapamycin-p70S6K,U0126-Erk1�2).増殖亢進が認められる細胞では,増殖シグナル阻害剤に耐性を示す可能性が考えられる.よってEGF(-)�(+)下における喘息・非喘息線維芽細胞の増殖・細胞内シグナル

日呼吸会誌 46(2),2008.172
Fig. H AG1478 suppressed AKT phosphorylation completely, ERK1/2 partially in EGF-stimulated fibroblast.
Fig. I LY294002 suppressed AKT, p70S6K phosphorylation. Rapamycin suppressed p70S6K phosphorylation.を上記阻害剤の共存・非共存下で検討した.また,
AG1478(EGFR Tyr inhibitor)の作用も併せて検討した(Fig. H,I).Erk1�2 スレオニン・チロシンリン酸化:喘息・非喘
息線維芽細胞において,無血清条件でも僅かにリン酸化を認める.EGF刺激により big band が誘導され,AG1478 前処理により部分的にその減弱が認められる.U0126 前処理では,完全にEGF刺激によるErk1�2 リン酸化が抑制されるが,LY294002,Rapamycin 前処理では全く抑制効果は認められない.LY294002-PI3K,Rapamycin-p70S6K,U0126-Erk1�2 pathway の特異的抑制効果を示している.AG1478 前処理によりEGF刺激によるErk1�2 リン酸化が部分的にしか抑制されないのは,EGFRの過剰活性化か,もしくはEGFR以外の経路を介したErk1�2 リン酸化の存在を伺わせる.AKTセリンリン酸化:EGFにより喘息・非喘息線維
芽細胞においてリン酸化が認められる.AG1478,LY294002 前処理によりEGFによるAKTリン酸化は完全に抑制されるが,U0126 前処理では全く影響を受けない.AKTの下流に存在する p70S6Kの阻害剤であるRa-pamycin 前処理にても全く影響を受けない.ヒト線維芽細胞でのEGF刺激による細胞内シグナルの階層性を証明している.上述したAG1478 前処理によりEGF刺激によるErk1�2 リン酸化が部分的にしか抑制されない現象に比べ,AKTリン酸化は完全に抑制される.つまりEGFR-EGF Ligand による細胞内へのシグナル伝達
は,Erk1�2 よりむしろ PI3K pathway(AKT,p70S6K)の方が特異性が高いのかもしれない.p70S6Kスレオニンリン酸化:使用した抗体は
p70S6K(Thr389)のみならず p85S6K(Thr412)リン酸化も認識するため,2つバンドが認められる.p85S6Kは p70S6Kと同一の遺伝子由来であり,N末端に核局在シグナルである 23 残基をもっている.p85S6K,p70S6Kとも PI3Kの下流に存在しており,各種増殖刺激により活性化される10).p70S6Kには主に 3つのリン酸化部位(Thr229,Thr389,Thr421&Thr424)が存在する.p70S6K(Thr229,Thr389)の部位は活性化 PI3K path-way によりリン酸化を受け,特にThr389 リン酸化はp70S6Kの酵素活性を反映している11).よって,下段のバンドが p70S6K活性化を反映している.LY294002,Rapamycin 前処理にてEGF刺激による p70S6Kスレオニンリン酸化はほぼ完全に抑制される.一方U0126 前処理では全く影響を受けない.AG1478 前処理によりEGF刺激による p70S6Kスレオニンリン酸化は部分的に減弱される.Erk1�2 の場合と同様に alternative path-way が存在するのかもしれない.EGF,inhibitors 存在・非存在下における PI3K path-
way(AKT,p70S6K)及び Erk1�2 のセリン�スレオニン・チロシンリン酸化について,喘息・非喘息線維芽細
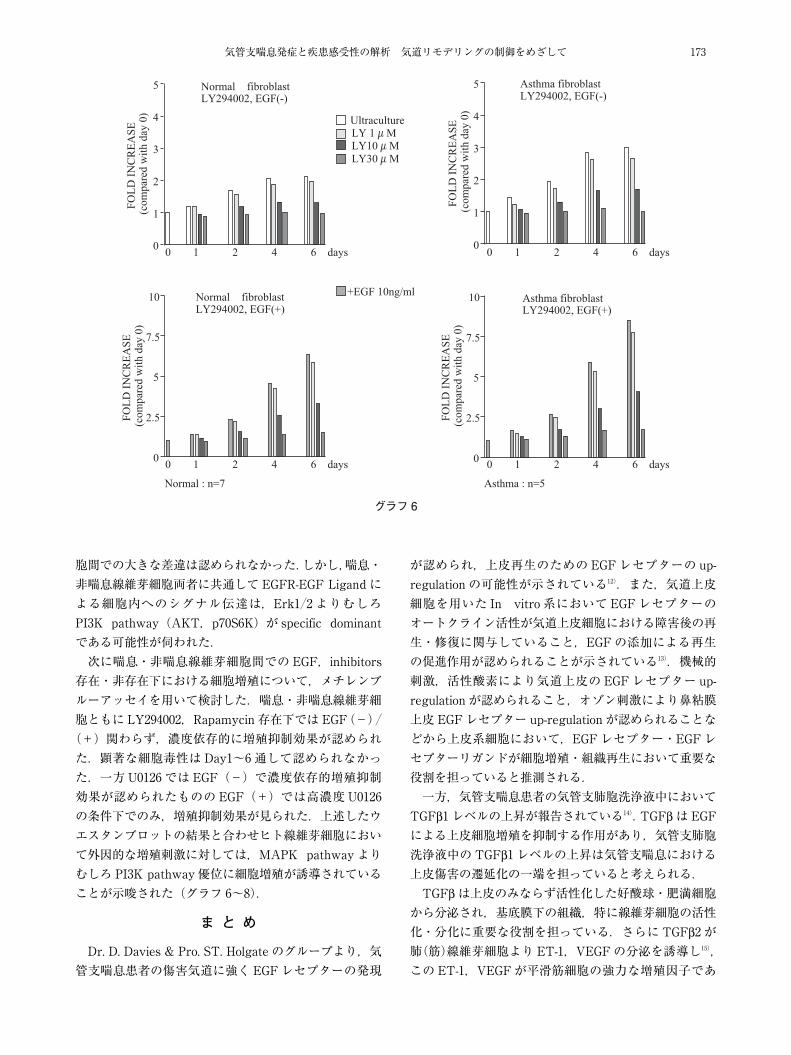
気管支喘息発症と疾患感受性の解析 気道リモデリングの制御をめざして 173
グラフ6
胞間での大きな差違は認められなかった.しかし,喘息・非喘息線維芽細胞両者に共通してEGFR-EGF Ligand による細胞内へのシグナル伝達は,Erk1�2 よりむしろPI3K pathway(AKT,p70S6K)が specific dominantである可能性が伺われた.次に喘息・非喘息線維芽細胞間でのEGF,inhibitors
存在・非存在下における細胞増殖について,メチレンブルーアッセイを用いて検討した.喘息・非喘息線維芽細胞ともに LY294002,Rapamycin 存在下ではEGF(-)�(+)関わらず,濃度依存的に増殖抑制効果が認められた.顕著な細胞毒性はDay1~6 通して認められなかった.一方U0126 では EGF(-)で濃度依存的増殖抑制効果が認められたもののEGF(+)では高濃度U0126の条件下でのみ,増殖抑制効果が見られた.上述したウエスタンブロットの結果と合わせヒト線維芽細胞において外因的な増殖刺激に対しては,MAPK pathway よりむしろ PI3K pathway 優位に細胞増殖が誘導されていることが示唆された(グラフ 6~8).
ま と め
Dr. D. Davies & Pro. ST. Holgate のグループより,気管支喘息患者の傷害気道に強くEGFレセプターの発現
が認められ,上皮再生のためのEGFレセプターの up-regulation の可能性が示されている12).また,気道上皮細胞を用いた In vitro 系においてEGFレセプターのオートクライン活性が気道上皮細胞における障害後の再生・修復に関与していること,EGFの添加による再生の促進作用が認められることが示されている13).機械的刺激,活性酸素により気道上皮のEGFレセプター up-regulation が認められること,オゾン刺激により鼻粘膜上皮EGFレセプター up-regulation が認められることなどから上皮系細胞において,EGFレセプター・EGFレセプターリガンドが細胞増殖・組織再生において重要な役割を担っていると推測される.一方,気管支喘息患者の気管支肺胞洗浄液中において
TGFβ1 レベルの上昇が報告されている14).TGFβはEGFによる上皮細胞増殖を抑制する作用があり,気管支肺胞洗浄液中のTGFβ1 レベルの上昇は気管支喘息における上皮傷害の遷延化の一端を担っていると考えられる.TGFβは上皮のみならず活性化した好酸球・肥満細胞
から分泌され,基底膜下の組織,特に線維芽細胞の活性化・分化に重要な役割を担っている.さらにTGFβ2 が肺(筋)線維芽細胞よりET-1,VEGFの分泌を誘導し15),この ET-1,VEGFが平滑筋細胞の強力な増殖因子であ
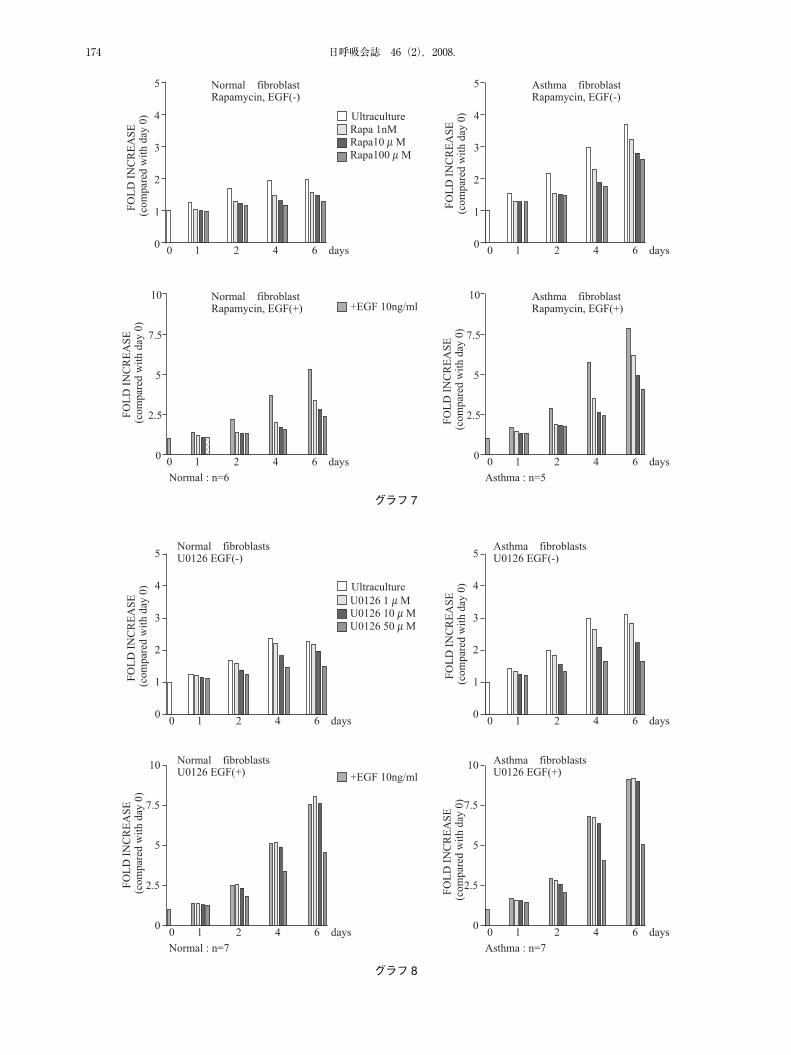
日呼吸会誌 46(2),2008.174
グラフ7
グラフ8

気管支喘息発症と疾患感受性の解析 気道リモデリングの制御をめざして 175
ることから,気管支喘息病態における上皮―線維芽細胞―平 滑 筋 細 胞 の 協 調 作 用:EMTU(Epithelial-Mesenchymal-Trophic-Unit)の概念を強く支持するサイトカインネットワークを形成していると思われる.EGF�TGFβは個々の細胞に対しては相反する作用を持つものの,気管支喘息の様々な病期において協調するように作用した結果(生体側としては不適切),上皮傷害や上皮下組織の組織改変(リモデリング)が進行し喘息の難治化を誘導する.EMTUを構成している個々の組織には外的・内的刺
激に対するシグナル伝達系の多様性が存在し,病態に応じて変化していることが推測される.今回我々が着目したMAPK pathway,PI3K pathway は,細胞増殖に関与する細胞内シグナルであると供に,抗アポトーシス・細胞死において重要な役割を演じていることが報告されている16)~18).私が行ったヒト気管支線維芽細胞の増殖及び細胞内シグナルの検討においては,無血清下では持続的MAPKリン酸化が喘息線維芽細胞の hyperprolifera-tion に関与している可能性が明らかになり,外因的な増殖刺激下では喘息・非喘息線維芽細胞において PI3Kpathway の重要性が明らかになった.多彩な細胞外ストレス下では,単一のシグナル path-
way のみに細胞現象が寄与しているとは考えがたく,細胞増殖を例にとってもMAPK,PI3K pathway の協調作用によってホメオスターシスが保たれていると考える方が妥当である.無血清下での喘息線維芽細胞 hyper-proliferation という結果は,細胞増殖及び抗細胞死によってもたらされていると思われ,cell cycle,cell sur-vival の解析が今後必要となる.また,今回の結果は細胞内シグナル特異的阻害剤の喘息気道リモデリング制御薬への可能性を示唆するものであり,患者サンプルを使用した実験結果は,より疾患特異的細胞現象を解析するという点では有益であったと思われる.謝辞:これらの実験・ディスカッションを指導していただいたUniversity of Southampton. S.T. Holgate 教授,D.E.Davies 教授に深謝します.また留学の機会を与えて頂いた日本大学医学部呼吸器内科の諸先生方および,日本呼吸器疾患研究基金ファイザーフェローシップに心から感謝致します.
参考文献
1)Holgate ST, Davies DE, Lackie PM, et al. J AllergyClin Immunol 2000 ; 105 : 193―204.
2)Van Eerdewegh P, Little RD, Dupuis J, et al. Nature2002 Jul 25 ; 418 : 426―430. Epub 2002 Jul 10.
3)Benayoun L, Druilhe A, Dombret MC, et al. Am JRespir Crit Care Med 2003 May 15 ; 167 : 1360―1368. Epub 2003 Jan 16.
4)Jeffery PK. Am J Respir Crit Care Med 2001 Nov15 ; 164 : S28―38.
5)Hashimoto S, Matsumoto K, Gon Y, et al. Am JRespir Crit Care Med 1999. Feb ; 159 : 634―640.
6)Hashimoto S, Gon Y, Takeshita I, et al. Am J RespirCrit Care Med 2001. Jan ; 163 : 152―157.
7)Oliver MH, Harrison NK, Bishop JE, et al. J Cell Sci-ence 1989 ; 92 : 513―518.
8)Chaudhary NI, Richter A, Young A, et al. Am JRespir Crit Care Med 2003 ; 167 : A958 (abstract).
9)Krymskaya VP, Hoffman R, Eszterhas A, et al. Am JPhysiol 1999 ; 276 (Lung. Cell. Mol. Physiol. 20) :L246―L255.
10)Pullen N, Thomas G. FEBS Lett 1997 ; 410 : 78―82.11)Weng QP, et al. J Biol Chem 1998 ; 273 : 16621―
16629.12)Polosa R, Puddicombe SM, Krishna MT, et al. J Al-
lergy Clin Immunol 2002 Jan ; 109 : 75―81.13)Puddicombe SM, Polosa R, Richter A, et al. FASEB J
2000 Jul ; 14 : 1362―1374.14)Redington AE, Madden J, Frew AJ, et al. Am J
Respir Crit Care Med 1997 Aug ; 156 (2 Pt 1) : 642―647.
15)Richter A, Puddicombe SM, Lordan JL, et al. Am JRespir Cell Mol Biol 2001 Sep ; 25 : 385―391.
16)Force T, Bonventre JV. Hypertension 1998 ; 31 :152―161.
17)Datta SR, Brunet A, Greenberg ME. Gene Dev1999 ; 13 : 2905―2927.
18)Vanhaesebroeck B, Alessi DR. Biochem J 2000 ; 346Pt 3 : 561―576.