ł ść ł ść ń ż ś ł ż ą ł ż č -...
TRANSCRIPT

1. Podstawy i podział spektroskopii..................................................................................... 5
1.1 Podział spektroskopii według zakresu promieniowania........................................... 5 1.2 Podział spektroskopii według rodzajów układów materialnych................................... 9 1.3 Podział spektroskopii według metod otrzymywania widma. ..................................... 12
2. Spektroskopia a spektrofotometria................................................................................. 14 3. Prawa absorpcji .............................................................................................................. 15
1.1 Współczynnik absorpcji.............................................................................................. 17 1.1 Addytywność absorbancji........................................................................................... 20 3.1 Odstępstwa od praw absorpcji. ................................................................................... 22 3.2 Molowy współczynnik absorpcji i przekrój czynny na absorpcję. Intensywność pasm elektronowych....................................................................................................................... 25 3.3 Dokładność oznaczeń fotometrycznych ..................................................................... 30
3.3.1 Zakresy absorbancji użyteczne w analizie ilościowej.......................................... 33 4. Analiza ilościowa ........................................................................................................... 35
4.1 Krzywa wzorcowa (kalibracyjna) ............................................................................... 35 4.2 Analiza układów wieloskładnikowych ....................................................................... 37 4.3 Badanie równowag chemicznych (punkt izozbestyczny) ........................................... 37
5. Parametry elektronowych pasm absorpcyjnych............................................................. 38 6. Analiza złożonych widm metodą rozkładu na pasma składowe [] ................................ 42
6.1 Matematyczny rozkład złożonych widm jako problem poszukiwania minimum funkcji celu............................................................................................................................ 42 6.2 Uogólniona metoda najmniejszych kwadratów.......................................................... 43 6.3 Algorytm Slaviča ........................................................................................................ 46
7. Spektrofotometria z wykorzystaniem pochodnych widma. ........................................... 48 7.1 Otrzymywanie widm pochodnych .............................................................................. 52
8. Literatura ........................................................................................................................ 55

2

3
Spektroskopowe metody analityczne, wykład, IV Chemia
Uniwersytet Opolski, dr hab. Krzysztof Kurzak, Prof. UO
Literatura podstawowa: 1. A.Cygański, Metody spektroskopowe w chemii analitycznej., wydanie drugie,
WN-T, W-wa 1997. 2. T.Nowicka-Jankowska, E.Wieteska, K.Gorczyńska, A.Michalik, Spektrofotome-
tria UV/VIS w analizie chemicznej., PWN, W-wa 1988. 3. Z.Kęcki, Podstawy spektroskopii molekularnej., wydanie trzecie zmienione i roz-
szerzone, PWN, W-wa 1992; wydanie czwarte, PWN, W-wa 1998 (1972, 1975, 1992).
4. A.Bartecki, J.Myrczek, Z.Staszak, K.Waśko, M.Sowińska, K.Kurzak, Widma
elektronowe związków kompleksowych. Metody analizy, przetwarzania i groma-dzenia przy użyciu mikrokomputera., WN-T, W-wa 1987.
5. A.Bartecki, Barwa związków metali., Wydawnictwo Politechniki Wrocławskiej,
Wrocław 1993. 6. G.W.Ewing, Metody instrumentalne w analizie chemicznej., PWN, W-wa 1980. 7. K.Danzer, E.Than, D.Match, Analityka. Ustalanie składu substancji., WN-T, W-
wa 1980.

4
Wybrane definicje
Absorpcja Przekształcenie energii promieniowania w inne formy energii, wskutek oddziaływania z materią (pochłanianie); zjawisko fi-zyczne.
Absorbancja Wielkość absorpcji, wartość absorpcji; A [bezwymiarowa]
Transmisja Przejście promieniowania przez dane środowisko, bez zmiany promieniowania (przepuszczalność); zjawisko fizyczne.
Transmitancja Wielkość transmisji, wartość transmisji; T [%].
Widmo Uporządkowana zależność pomiędzy energią promieniowania przepuszczanego, absorbowanego lub emitowanego, a liczbą falową, długością fali elektromagnetycznej lub częstością; wy-stępują odpowiednio: w. transmisji, w. absorpcji, w. emisji.
Promieniowanie monochromatycz-ne
Wiązka promieniowania o jednakowych kwantach energii, tj. ściśle o jednej długości falowej, czy też częstości; w praktyce monochromatyczność ograniczona jest zdolnością rozdzielczą spektrofotometru.
Gęstość promieniowania
Energia lub proporcjonalna do niej liczba fotonów monochro-matycznych zawarta w 1 m3 układu napromieniowanego, czyli jest to stężenie objętościowe fotonów; ρ = I/c (I = ρc, w próżni).
Natężenie promieniowania
Energia przechodząca w ciągu 1 s przez 1 m2 powierzchni ⊥ do kierunku biegu promieniowania; w przypadku p. monochroma-tycznego może być rozumiane jako liczba fotonów przechodzą-cych w przeciągu 1 s przez 1 m2 powierzchni; I.
Spektroskopia Zajmuje się oddziaływaniem, między promieniowaniem elek-tromagnetycznym a materią, które polega na pochłanianiu części energii (absorpcja) przez materię lub oddawaniu przez materię części energii w postaci promieniowania (emisja); istotne jest określenia rodzaju przejść i właściwe ich przyporządkowanie odpowiednim pasmom czy liniom.
Spektrofotometria Zajmuje się określaniem stężenia lub zawartości atomów lub cząsteczek w danym układzie absorbującym czy emitującym, tj. stanowi podstawę ilościowej analizy chemicznej. W tym przy-padku nie jest istotny rodzaj oddziaływania z materią, lecz poda-nie dokładnej funkcji określającej natężenie widma od stężenia.

5
1. Podstawy i podział spektroskopii Szybki i wszechstronny rozwój spektroskopii jako nauki teoretyczno-doświadczalnej spowodował, że przy omawianiu zarówno jej podstaw jak i specyficznych działów, konieczne jest stosowanie różnych kryteriów podziału. Najczęściej za podstawę po-działu spektroskopii przyjmuje się następujące kryteria:
1. zakres promieniowania elektromagnetycznego, 2. właściwości układów materialnych, 3. metoda otrzymywania widma, związana z formą wymiany energii między pro-
mieniowaniem i materią. 1.1 Podział spektroskopii według zakresu promieniowania − spektroskopia kosmiczna 10-5 - 10-3 Å − spektroskopia gamna 10-3 - 1 Å − spektroskopia rentgenowska 1 - 102 Å
− spektroskopia optyczna a) w bliskim i próżniowym nadfiolecie 100 - 300 nm b) w zakresie widzialnym 360 - 830 nm c) w bliskiej (2.5-15 µm), średniej, dalekiej podczerwie-
ni
12000-50 cm-1
− radiospektroskopia a) w zakresie mikrofalowym 0.03 - 100 cm b) w zakresie krótkofalowym 10 - 100 m c) w zakresie długofalowym 100 - 4000 m
Parametrem decydującym o takim podziale jest zakres spektralny. Promieniowanie możne określić podając jego: − długość fali λ, − liczbę falową = 1/λ, − częstość ν. Częstotliwość podaje się w Hz [s-1] lub jednostkach stanowiących wielokrotność her-ców. Długość fali w obszarze nadfioletu i światła widzialnego wyraża się w:
a) mikrometrach: µm = 10-4 cm = 10-6 m, (dawniej mikron: µ); b) nanometrach: nm = 10-7 cm = 10-9 m ( = 1 mµ); c) milimikrometrach: mµ = 10-7 cm = 10-9 m ( = 1 nm); d) angstremach: Å = 10-8 cm = 10-10 m = 0.1 nm;
liczby falowe wyrażają się zwykle w cm-1 lub w µm-1.

6
λν = c; zależność obowiązująca przy rozchodzeniu się promieniowania w próżni. Wyraża ona ścisły związek pomiędzy długością fali a częstością drgania i pozwala zamienia jedną wielkość na drugą. Zależności pomiędzy częstością a długością fali są następujące.
c = λν [m] [s-1] [m s-1] = [m s-1] ν = c/λ [m s-1] [m] [m s-1 m-1] = [s-1] / [Hz]
νcν = [m s-1] [m-1] [m s-1 m-1] = [s-1] / [Hz] ν/cν = [s-1] [m s-1] [s-1 m-1 s] = [m-1] 1/λν = [m]
[cm] [nm]
100 [cm] =
10-7 [cm] =
10-2/λ [cm-1] 1/λ [cm-1] 107/λ [cm-1]
Podstawowymi jednostkami energii są:
1eV (elektronowolt): jednostka energii używana w fizyce (głównie atomowej i jądrowej) równa energii uzyskanej przez elektron w polu elektrycznym o różnicy potencjałów 1V.
1J (dżul): jednostka pracy i energii, określana jako praca, którą wykonuje na dro-
dze 1 m siła 1 N (niutona), inaczej określany jako 1 niutonometr; lub (np. dla energii elektrycznej) jako energia zużywana przy mocy 1 W (wata) w ciągu 1 sekundy, inaczej określany jako 1 watosekunda (1 Wsek); 1 J=107 ergów.
Zależność pomiędzy energią pochłoniętą a częstotliwością ν lub długością fali wywo-łującej przejście przedstawia się następująco:
λυ∆
hc=h=E , [m
smsJ -1⋅⋅⋅ ] = J
gdzie h - stała Plancka, c - prędkość światła, h = 6.62618⋅10-34 [J⋅s] (SI) oraz 6.62618⋅10-27 [erg⋅s] (zwyczajowo); c = 2.9979×108 [m s-1] ∆E jest energią pochłoniętą przy przejściu w cząsteczce ze stanu o niższej energii (stan podstawowy) do stanu o energii wyższej (stan wzbudzony). Energia pochłaniania za-leży od różnicy energii w stanie podstawowym i wzbudzonym; im mniejsza różnica energii tym większa długość fali. Zauważmy, że wprost proporcjonalna do ∆E jest częstość ν, a tym samym, liczby falowe, a nie długość fali. Dlatego w spektroskopii często wyraża się energię w cm-1, mimo że nie są to jednostki energii, lecz częstości, a energia jest do niej tylko proporcjonalna.

7
Tabela 1.3. Współczynniki konwersji jednostek energii.
Jednostki pod-
stawowe J⋅mol-1 J⋅cząsteczkę-1 cal⋅mol-1 cm-1 eV
J⋅mol-1 1 1.66×10-24 0.23901 0.083594 1.0364×10-5 J⋅cząsteczkę-1 6.022×102
3 1
cal⋅mol-1 4.184 1 0.34974 4.3363×10-5 cm-1 11.963 2.8591 1 1.2398×10-4 eV 96487.0 23.061 8065.7 1 Aby wielkość wyrażoną w jednostkach pierwszej kolumny zamienić na jednostki w górnym rzędzie kolumny należy ją pomnożyć przez mnożnik znajdujący się na prze-cięciu tych rubryk. Przykłady: 1 eV jest równoważny 8065.7 cm-1 . Widmo promieniowania elektromagnetycznego można przedstawić na schemacie. Po-dział spektroskopii według zakresu promieniowania, związany jest w istocie z różnymi metodami eksperymentalnymi. W zależności, bowiem od badanego zakresu stosuje się różne źródła promieniowania, detektory oraz urządzenia dyspersyjne. W spektroskopii optycznej najczęściej stosuje się metody optyczne, jako urządzenia dyspersyjne-pryzmaty i siatki dyfrakcyjne.

8
λ, cm 104 102 1 10-2 10-4 10-6 10-8 10-10 10-12
telewizja
fale radiowe
Radar
mikrofale
Podczer-wień ↓ ↓ da- blis-leka ka ↓ średnia
wi d z i a l n e
nadfiolet(UV)
↓ ↓ blis- da-ki leki
promie-niowanierentge-nowskie
pro-mie-nio-wanie
γ
promieniowanie kosmiczne
(vis)
ν, Hz 106 108 1010 1012 1014 1016 1018 1020 1022 Rys. 1.1. Zakresy promieniowania elektromagnetycznego.
← 200 - 800 nm → ← obszar widzialny →
nadfiolet próżniowy
nadfiolet bliski
(UV)
fi ol et
n i e b i e s k i
z i e l o n y
żółty
p o m a r a ń c z.
c z e r w o n y
bliska podczerwień
(NIR)
(vis)
λ, nm 100 200 300 400 500 600 700 800 900 Rys. 1.2. Nadfioletowy i widzialny zakres promieniowania elektromagnetycznego.
Rys. 1.3. Poszczególne barwy w widzialnym (vis) zakresie promieniowania elektro-magnetycznego.

9
1.2 Podział spektroskopii według rodzajów układów materialnych Podział spektroskopii według rodzajów układów, których widma badamy, jest znacz-nie ważniejszy, ponieważ dotyczy istoty, badanych w spektroskopii procesów. Z tego punktu widzenia, można rozróżnić następujące działy:
1) Spektroskopia jądrowa 2) Spektroskopia atomowa 3) Spektroskopia molekularna, ze szczególnym uwzględnieniem spektroskopii ukła-
dów skondensowanych obejmujących złożone związki organiczne, związki kom-pleksowe itp.
Podział ten jest związany z określonymi poziomami energetycznymi biorącymi udział w przejściach charakterystycznych dla poszczególnych układów materialnych. Scha-rakteryzujmy bliżej te poziomy energetyczne. Będą się one różniły między sobą za-równo rodzajem ruchu w tych układach jak i wielkością różnicy energii między dwo-ma najbliższymi kolejnymi poziomami. W układach atomowych i molekularnych można rozróżnić następujące rodzaje po-ziomów.
1. Poziomy elektronowe - wynikające z ruchu elektronów względem jąder. Energie przejść między poziomami wewnętrznych powłok elektronowych są rzędu dzie-siątków i tysięcy eV, o powstające widmo jest widmem rentgenowskim (elektro-ny powłok K, L). Natomiast rząd wielkości energii przejść między poziomami powłok zewnętrznych jest równy tylko kilku eV, a powstające widmo optyczne przypada na zakresy: widzialny i nadfioletu (częściowo także bliskiej podczer-wieni).
2. Poziomy oscylacyjne cząsteczek - związane z oscylacyjnym ruchem jąder w czą-steczkach wokół położeń równowagowych (ruch ten jest w przybliżeniu ruchem harmonicznym). Energie tych drgań wynoszą 0.02 - 0.05 eV, a więc odpowied-nie przejścia zachodzące pomiędzy poziomami oscylacyjnymi występują w za-kresie podczerwieni. Przejścia te bada się metodami spektroskopii w podczer-wieni ramanowskiej. Poziomy te można również określić pośrednio, badając przejścia elektronowo oscylacyjne (tzw. widma wibronowe) w zakresach wi-dzialnych i nadfioletu. Struktura elektronowo - oscylacyjna tych widm jest zwią-zana z faktem, że przejściom elektronowym w cząsteczce towarzyszy zmiana energii oscylacyjnej (a także, choć w znacznie mniejszym stopniu, zmiana ener-gii rotacyjnej).
3. Poziomy rotacyjne cząsteczek - związane z obrotowym ruchem cząstek. W przy-bliżeniu ruch ten można (dla cząsteczki nieliniowej) rozważać jako ruch swo-bodny ciała dookoła trzech wzajemnie prostopadłych osi, charakteryzujących się trzema momentami bezwładności. W zależności od wielkości momentów bez-władności wyróżnia się trzy rodzaje układów (układ sferyczny, układ symetrycz-ny i układ asymetryczny). Zmiany energii w tych przypadkach są rzędu ułamków eV, czyli mieszczą się w granicach około 100 - 0.1 cm-1. Badania widm rotacyj-nych prowadzi się więc w zakresie dalekiej podczerwieni, a nawet w zakresie mikrofalowym. Poziomy rotacyjne można też określić w widmach oscylacyjno-

10
rotacyjnych w zakresie bliskiej podczerwieni (tzw. obszar nadtonów) oraz w szczególnych przypadkach, w widmach elektronowo- rotacyjnych.
4. Poziomy struktury subtelnej - związane z obecnością spinu elektronowego. Róż-nice energii mogą się wahać od dziesiętnych części do tysięcy cm-1. Przejścia te badane są w związku z tym metodami radiospektroskopii (zwłaszcza dla lekkich atomów) lub w zakresie widzialnym a nawet nadfioletowym (w przypadkach, gdy powstaje w tych zakresach tzw. struktura multipletowa).
5. Poziomy struktury nadsubtelnej - związane z obecnością spinu jądrowego. Po-wstają w wyniku sprzężenia elektrycznych i magnetycznych momentów jąder. Występujące w tych przypadkach różnice energii są bardzo małe i wynoszą od tysięcznych do dziesięciotysięcznych części cm-1. Odpowiednie przejścia bada się metodami radiospektroskopowymi, a mianowicie metodami rezonansu ma-gnetycznego i rezonansu kwadrupolowego. W obszarze mikrofalowym rozszcze-pienia te bada się metodą elektronowego rezonansu paramagnetycznego.
6. Poziomy energetyczne - powstające w wyniku rozszczepienia poziomów swo-bodnych atomów i cząstek w zewnętrznym polu magnetycznym. Rozszczepieniu ulegają poziomy elektronowe i rotacyjne, a także poziomy struktury subtelnej. W pierwszym przypadku rozszczepienia wynoszą kilka cm-1. Bezpośrednie obser-wacje prowadzi się metodami rezonansu magnetycznego. Zjawisko dla zakresu UV-VIS nosi nazwę zjawiska Zeemana.
7. Poziomy energetyczne - powstające przy rozszczepienia poziomów swobodnych atomów i cząstek w zewnętrznym polu elektrycznym; tzw. efekt Starka. Rozsz-czepieniu ulegają poziomy elektronowe i rotacyjne wykazujące moment dipolo-wy. Wielkość rozszczepienia zależy od natężenia pola. Rozszczepienia te bada się metodami radiospektroskopowymi (rezonans elektronowy).
W obrębie spektroskopii cząsteczkowej możemy dokonać dalszego podziału ze wzglę-du na formę energii cząsteczek: − spektroskopia elektronowa , − spektroskopia oscylacyjna, − spektroskopia rotacyjna, − spektroskopia rezonansu paramagnetycznego (EPR - Electron Proton Resonance,
ESR – Electron Spin Resonance), − spektroskopia jądrowego rezonansu magnetycznego (NMR – Nuclear Magnetic
Resonance).

11
Podsumowanie: Nr Rodzaj poziomów Orientacyjny zakres 1. Poziomy elektronowe - dziesiątki i tysiące eV
- kilka eV 2. Poziomy oscylacyjne cząsteczek 0.02 - 0.05 eV 3. Poziomy rotacyjne cząsteczek ułamki eV (100 - 0.1 cm-1) 4. Poziomy struktury subtelnej dziesiętne części do tysięcy cm-1 5. Poziomy struktury nadsubtelnej tysięczne do dziesięciotysięcznych
części cm-1 6. Poziomy energetyczne w zewnętrz-
nych polach magnetycznych kilka cm-1
7. Poziomy energetyczne w zewnętrz-nych polach elektrycznych
zależy od siły zewnętrznego pola elektrycznego

12
1.3 Podział spektroskopii według metod otrzymywania widma. W zależności od metody otrzymywania rozróżniamy trzy rodzaje widm: absorpcyjne, emisyjne i Ramanowskie. W związku z tym rozróżniane są działy spektroskopii:
1) Spektroskopia absorpcyjna, 2) Spektroskopia emisyjna, 3) Spektroskopia Ramanowska (rozpraszania).
Widma absorpcyjne - można określić jako zbiór wszystkich przejść z niższych pozio-mów na wyższe. Odpowiadają, więc one zwiększeniu energii układu (pochłonięcie fotonu). Najprostszy typ widma absorpcyjnego powstaje, gdy obsadzony jest najniższy poziom energetyczny, tj. podstawowy. Obsadzenie poziomów energetycznych zwią-zane jest z równowagą termodynamiczną, która określa temperatura układu. Przyjmuje się, że w temperaturze pokojowej obsadzany jest tylko poziom podstawowy. Widma emisyjne - można określić jako zbiór wszystkich przejść z poziomów wyż-szych na niższe. Przejścia w widmach emisyjnych odpowiadają zmniejszeniu energii, czyli wypromieniowaniu fotonu. W bardziej ogólnym przypadku (np. w wyższych temperaturach) należy przyjąć, że także wyższe poziomy energetyczne są obsadzone przynajmniej częściowo, co ozna-cza, że struktura widma staje się bardziej złożona. Widma Ramanowskie - cechą charakterystyczną tego rodzaju widm jest zmiana czę-stości promieniowania rozproszonego (νr) w stosunku do częstości promieniowania padającego (νp) .
νr = νp ± ν gdzie ν jest częstością przejść dla układu rozpraszającego.
a) b)
ν1 ν2 ν3
ν1 ν2 ν3 ν4 ν5 ν6
Rys. 1.4. Przejścia w widmach: absorpcyjnych w temperaturze pokojowej (a), emisyj-nych (b).

13
λ, 1 m 1 cm 1 µm 1 nm 1A
ν, cm-1 10-4 10-2 1 102 104 106 108 1010 1012
a)
| rotacyjne | | powłoki walenc. | | przejścia jądrowe orientacje spinu przejścia cząsteczk. przejścia elektrono. |oscylacyjne| | powłoki | | przemiany | wewnętrzne | | jądrowe
b) magnetyczny rezo-nans jądrowy
elektronowy rezo-nans paramagne-tyczny
emisyjna spek-trometria atomowa
spektro-metria rentgen.
absorpcyjna spektrometria atomowa
spektro-skopia fotoelekt-ronowa
Spektros-kopia Mössbauera
spektroskopia mi-krofalowa
Spek-tro-skopia w pod-czer-wieni
spektrofoto-metria UV/VIS
spektro-skopia elektro-nowa Augera 1
analiza aktywa-cyjna
Spektro-metria mas
E, kJ mol
10-6 10-4 10-2 1 102 104 106 108
Rys. 1.5. a) Efekt wzajemnego oddziaływania promieniowania elektromagne-tycznego z atomami i cząsteczkami; b) spektroskopowe metody instrumentalne.
1 Spektroskopia elektronów Augera (AES) – Auger Electron Spectroscopy. Pomiar energii kinetycznej elektro-nów po uprzedniej fotojonizacji (tzw. elektronu wtórne), na skutek bombardowania wiązką elektronów pierwot-nych o energii 70 – 5000 eV).

14
2. Spektroskopia a spektrofotometria. Różnice pomiędzy obu dyscyplinami mogą być łatwo określone poprzez sprecyzowa-nie dla nich zadań. I tak, spektroskopia zajmuje się badaniami podstawowymi doty-czącymi cząsteczek. Obejmują one:
1. Eksperymentalne otrzymywanie różnych typów widm; Ai=f(νi), Ai=f(λi). 2. Przeprowadzanie ich analizy. 3. Zaproponowanie schematu poziomów energetycznych charakteryzujących ba-
dany układ. 4. Obliczanie (w tych przypadkach, gdy jest to możliwe) teoretycznych energii
przejść i porównanie z danymi doświadczalnymi. 5. Otrzymywanie danych dotyczących rozkładu natężeń (oraz takich wartości jak
np. moc oscylatora), zarówno teoretycznych jak i eksperymentalnych. 6. Określanie (wyznaczanie) parametrów spektrochemicznych w oparciu o zwe-
ryfikowane przez obliczenia teoretyczne dane eksperymentalne (punkt 4), np. parametry: PK, OM, AOM., i inne.
7. (Analiza) stereochemia badanego układu chemicznego w oparciu o użyteczne chemicznie parametry spektralne (punkt 6).
Spektrofotometria zajmuje się natomiast określaniem stężenia lub zawartości ato-mów lub cząsteczek w danym układzie absorbującym czy emitującym, tj. stanowi podstawę ilościowej analizy chemicznej. W tym przypadku nie jest szczególnie interesujące określenie rodzaju przejścia lub właściwe przyporządkowanie im danych linii czy pasm, natomiast istotne jest podanie dokładnej funkcji określającej zależności natężenia widma od stężenia. Na podkreślenie zasługuje również znaczenie metod spektrofotometrycznych w badaniu różnego typu równowag.

15
3. Prawa absorpcji Jednym z wielu możliwych oddziaływań promieniowania z materią jest absorpcja. Stanowi ona podstawę spektrofotometrii absorpcyjnej w nadfiolecie i zakresie wi-dzialnym. Pomiary w obszarze UV\VIS przeprowadza się najczęściej dla substancji ciekłych (rozpuszczalniki) lub rozpuszczonych (roztwory próbek stałych w rozpusz-czalnikach), rzadziej w fazie gazowej lub stałej (widma refleksyjne). Wielkością mie-rzoną jest zwykle transmitancja lub absorbancja. Są one zdefiniowane następująco:
II=To
(3.1)
T1log
IIlogA o == (3.2)
gdzie: Ιo - natężenie promieniowania padającego Ι - natężenie promieniowania przechodzącego przesz ośrodek absorbujący. Rozważmy pojemnik szklany (kuweta) o płaskich równoległych powierzchniach zewnętrznych, przez które przechodzi promieniowanie monochromatyczne. Przyjmij-my, że kuweta jest napełniona substancją absorbującą rozpuszczoną w nie absorbują-cym rozpuszczalniku. Natężenie promieniowania padającego Ιo, ulega osłabieniu przy przejściu przez ośrodek absorbujący do wartości Ι. Osłabienie padającej wiązki światła może być powodowane:
1) odbiciami na powierzchniach kuwety na granicy z powietrzem i roztworem; 2) rozpraszaniem przez rysy na kuwecie lub pojedyncze cząstki czy ich skupiska w
próbce (takie efekty mogą być powodowane, np. zmętnieniem roztworu niewi-docznym gołym okiem);
3) absorpcją promieniowania przez próbkę. Część wpływów zakłócających eliminuje się prowadząc odpowiednie pomiary po-równawcze względem odnośnika (kuweta zawierająca rozpuszczalnik lub ślepą pró-bę). W ten sposób staramy się stworzyć warunki, w których absorpcja światła jest główną przyczyną osłabienia padającego promieniowania. Ilościowy opis absorpcji energii promieniowania przez materię opiera się na ogól-nym prawie, zwanym prawem Beera∗. Zaobserwowano, że natężenie promieniowania zmniejsza się w miarę przenikania w głąb homogenicznego (jednorodnego) ośrodka oraz w miarę zwiększania stężenia rozpuszczonej substancji absorbującej. Nie zależy natomiast od natężenia wiązki padającej Ιo. Bardziej ogólnie można stwierdzić, że zmniejszenie natężenia promieniowania jest proporcjonalne do liczby absorbujących cząstek, znajdujących się na drodze równoległej i monochromatycznej wiązki promie-niowania.
∗ Zależność ta czasem nazywana jest prawem Beera-Lamberta lub Bouguera-Beera, ponieważ Bouguer i Lambert (jak i inni badacze) przyczynili się do jego ustalenia.

16
Ilościowym wyrazem tej zależności jest prawo Beera:
Kolejne przyrosty liczby, identycznych absorbujących cząstek na drodze wiązki promieniowania monochromatycznego, absorbują takie same części energii promieniowania przez nie przechodzącego.
W formie różniczkowej można to przedstawić w następujący sposób:
IkdndI
−= (3.3) gdzie: dΙ jest częścią całkowitego natężenia padającego promieniowania Ι, która zo-staje zaobserwowana wskutek przyrostu dn liczby cząstek absorbujących. Przekształcenie i scałkowanie w podanych granicach
∫ ∫−=I
I
N
0o
dnkI
dI (3.4) prowadzi do wyrażenia
NkIIlno
−= (3.5) N jest liczbą absorbujących cząstek, znajdujących się na takiej drodze wiązki promie-niowania o jednostkowym przekroju poprzecznym, na której natężenie promieniowa-nia zmniejsza się do poziomu Ι. W przypadku wiązki, której powierzchnia przekroju wynosi s [cm2], prawy człon równania musi być pomnożony przez s:
sNkIIln '
o
−= (3.6) Wielkość Ns jest miarą liczby cząstek, które biorą czynny udział w absorpcji promie-niowania. Dogodniejszą miarą tej liczby jest iloczyn stężenia c i długości drogi b. Mo-żemy, więc zapisać
cbkIIln ''
o
−= (3.7)Stała k’’ ma wymiar jednostki masy na jednostkę powierzchni przekroju poprzeczne-go. Innymi słowy, dla danego przekroju poprzecznego, absorpcja jest proporcjonalna do masy absorbującej substancji znajdującej się na drodze promieniowania. Dla wygody zastępuje się stałą k’’ inną stałą tj. a, która zawiera współczynnik wy-nikający z zamiany logarytmu naturalnego na dziesiętny, i zmienia się stosunek Ι/Ιo na Ιo/Ι (w celu usunięcia ujemnego znaku). W ten sposób otrzymujemy najprostsze sformułowanie prawa Beera:

17
cbaAIIlog o == ; gdzie
T1log=A (3.8)
Treść tego prawa możemy wyrazić następująco:
dla równoległej, ściśle monochromatycznej wiązki promieniowania elek-tromagnetycznego, absorbancja A jest proporcjonalna do stężenia roztworu c i grubości warstwy absorbującej b.
Ponieważ natężenie promieniowania przepuszczanego Ι może zmieniać się w grani-cach od zera do Io, więc absorbancja (log Ιo/Ι) może zmieniać się teoretycznie od zera do nieskończoności. W praktyce jednak, wartości absorbancji większe niż 2 nie są sto-sowane, a zakres, który umożliwia otrzymanie odpowiednio precyzyjnych wyników analitycznych, jest jeszcze bardziej ograniczony. Dokładne granice dopuszczalnych wartości absorbancji określane są częściowo przez typ przyrządu pomiarowego. 1.1 Współczynnik absorpcji
Współczynnik absorpcji jest wielkością charakterystyczną dla danej substancji, w danym rozpuszczalniku i przy danej długości fali (liczbie falowej). Jednostka współ-czynnika absorpcji zależy od jednostek, w jakich wyrażamy towarzyszące wielkości: stężenie c i grubość warstwy b. W związku z tym, występują również różne określenia współczynnika absorpcji. Wielkość b zwykle podaje się w cm, więc a uzależnione jest od c.
Stężenie sym-bol
określenie określenia dawne
c [g dm-3] a współczynnik absorp-cji
współczynnik ekstynkcji lub ab-sorpcja właściwa
cM [mol dm-3] ε molowy współczynnik absorpcji
molowy współczynnik ekstynkcji lub absorpcja molarna
Współczynnik absorpcji a stosowany jest, gdy rodzaj substancji absorbujące, a za-
tem i jego masa cząsteczkowa, jest nieznana. Molowy współczynnik absorpcji ε jest bardziej wskazany w przypadkach, gdy po-
równywana jest ilościowo absorbancja różnych substancji o znanych masach molo-wych.
Wielkości a i ε związane są z właściwościami substancji absorbującej, tj. są nieza-leżne od warunków pomiaru: stężenia i grubości warstwy, podczas gdy absorbancja A odzwierciedla właściwości określonej próbki.

18
0.0 0.2 0.4 0.6 0.8 1.0
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
12000 16000 20000 24000 28000
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
A
c, [mol dm-3 ]
b)
A
ν, cm-1
1
2
3
4
5
6
a)
Rys. 3.1. Widma Ai = f(νi) danego związku (chromoforu) w danym rozpuszczalniku o różnych stężeniach (a): 1-0.1, 2-0.09, 3-0.08, 4-0.07, 5-0.06, 7-0.05 M; zależność ab-sorbancji od stężenia (b).
19
12000 16000 20000 24000 28000
1
2
3
4
5
6
7
8
9
10
11ε,
M-1cm
-1
ν, cm-1
1,2,3,4,5,6
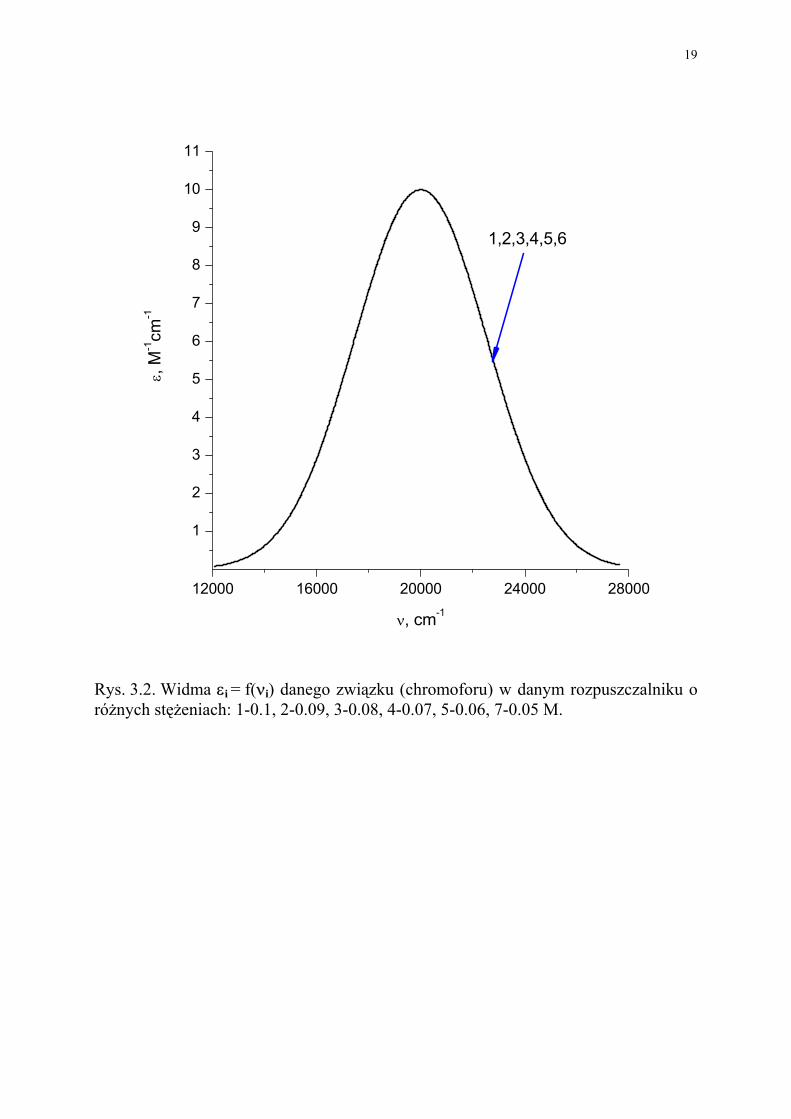
Rys. 3.2. Widma εi = f(νi) danego związku (chromoforu) w danym rozpuszczalniku o różnych stężeniach: 1-0.1, 2-0.09, 3-0.08, 4-0.07, 5-0.06, 7-0.05 M.

20
Widmo można, więc określić jako funkcję: εi = f(νi) (lub εi = f(λi)), która jest charakte-rystyczna dla substancji absorbującej (chromofora) w danym rozpuszczalniku. W przypadku krzywych Ai = f(νi) (Rys. 3.1) mamy do czynienia z 5 widmami różnymi o wzrastającej intensywności w maksimum. W przypadku krzywych εi = f(νi) (Rys. 3.2), wszystkie krzywe powinny się nałożyć, pod warunkiem, że układ spełnia prawo Lamberta-Beera. W przypadku otrzymania różnych krzywych, oznacza to, że zachodzą procesy chemiczne np. równowagowe z upływem czasu (zwróćmy uwagę, że krzywe 1-5 są przesunięte w czasie, chociażby o czas trwania pomiaru każdej z nich). Wielkość T% = 100 (I / Io) jest przydatna, w tych przypadkach, gdy zainteresowani jesteśmy promieniowaniem przepuszczonym, a nie chemicznymi właściwościami sub-stancji absorbującej, np. w badaniu rozpuszczalników. Absorbancja (lub molowy współczynnik absorpcji) jest użyteczne, gdy jest miarą stop-nia absorpcji promieniowania przez próbkę. Interesuje nas, ’co’ i ’ile’ absorbuje. 1.1 Addytywność absorbancji Prawo to dotyczy roztworów i mieszanin wieloskładnikowych. Wyraża ono absorban-cję poszczególnych składników (A1, A2, ..., An), co można przedstawić w następujący sposób:
A = A1 + A2 + .... + AM = ∑M
1=iiA
(3.9)czyli
A = a1 bc1 + a2 bc2 + ... + an bcn (3.10)lub
A = ε1 bcM1 + ε2bcM
2 + ... + εnbcMn
(3.11)
Oczywiście addytywność absorbancji jest spełniona, jeśli pomiędzy składnikami śro-dowiska absorbującego nie ma żadnych oddziaływań chemicznych. Oznacza ona, że każde indywiduum absorbuje tak, jakby inne były nieobecne. Addytywność absorbancji może być wykorzystana w różny sposób. 1) Pozwala na odejmowanie od mierzonej absorbancji jej części pochodzącej od roz-
puszczalnika (odnośnik) lub odczynnika (ślepa próba) Przykł. techniki pomiaru: Podczas wykonywania pomiarów absorpcji substancji w roztworze, wykonujemy
zwykle następujące czynności: − pomiar układu, względem którego będziemy wykonywać pomiar zasadniczy
tzw. autokorekta; w zależności od potrzeb mogą to być odpowiednio: (a) po-wietrze, (b) kuweta (spektrofotometr jednowiązkowy) lub para kuwet (sp. dwuwiązkowy), (c) kuweta (lub para) z rozpuszczalnikiem;

21
− pomiar zasadniczy, układ: kuweta z rozpuszczalnikiem (odnośnik) i kuweta z roztworem próbki absorbującej;
− odjęcie wartości absorbancji autokorekty od wartości absorbancji pomiaru zasadniczego (współczesne spektrofotometry mają taką opcję automatyczną).
2) Umożliwia także odejmowanie od widma nieznanej substancji (mieszaniny), ab-
sorbancji pochodzącej od chromoforu, o którym wiemy, że jest obecny w układzie i znamy jego widmo, w celu zidentyfikowania tego drugiego chromoforu.
3) Odgrywa ważną rolę w analizie wieloskładnikowej, tzn. jednoczesnym oznaczeniu
w tym samym roztworze dwu lub więcej substancji absorbujących.
12000 16000 20000 24000 28000
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
1.2
1.3
1.4
A
ν, cm-1
a
b
c
Rys. 3.3. Widmo mieszaniny 2-składnikowej (kontur sumaryczny, c), widmo substan-cji składowej (znane, b) i widmo substancji składowej (nieznane, a) jako wynik odej-mowania.

22
3.1 Odstępstwa od praw absorpcji. Z prawa Beera wynika, że współczynnik absorpcji jest wielkością stałą, niezależną od stężenia, grubości warstwy absorbującej i natężenia promieniowania padającego. Prawo to nie dostarcza żadnych informacji o wpływie temperatury, rodzaju rozpusz-czalnika lub długości fali. 1) W praktyce stwierdzono, że temperatura ma jedynie drugorzędny wpływ, o ile nie
zmienia się w niezwykle szerokim zakresie; − ze zmianą temperatury zmienia się natomiast stężenie, ze względu na zmianę
objętości, − można oczekiwać mniejszych lub większych zmian absorbancji ze zmianą tem-
peratury, jeżeli substancja absorbująca znajduje się w roztworze w stanie rów-nowagi z inną substancją, lub z nie rozpuszczoną substancją stałą (np. roztwór nasycony).
2) Natomiast nie można przewidzieć, w jakiś ogólny sposób, wpływu zmiany roz-puszczalnika na absorpcję danej substancji rozpuszczonej. W UV powstają ograni-czenia ze względu na zakres przepuszczalności danego rozpuszczalnika.
Jak wynika z omówionych równań, zależność absorpcji od stężenia powinna mieć cha-rakter liniowy. Niekiedy, nawet przy zachowaniu stałej temperatury i określonego roz-puszczalnika, stwierdza się, że współczynnik absorpcji nie ma wartości stałej, ale od-chyla się w kierunku mniejszych lub większych wartości; typowe odchylenia przed-stawione są na Rys. 3.4.
A
c
I
II
a)
A
c
I II
c)
A
c
I
II
b)
Rys. 3.4. Typowe odstępstwa od prawa Beera: I - prosta dla układu spełniającego pra-wa absorpcji, II - krzywe dla układów nie spełniających praw absorpcji. Główne przy-padki odstępstw charakteryzują się zmianą współczynnika absorpcji (ε): (a) zmniej-szanie się ze wzrostem stężenia; (b) wartości różne od rzeczywistej niezależnie od stę-żenia; (c) wzrost ze wzrostem stężenia.

23
Można wyróżnić dwie grupy czynników zakłócających absorpcję promieniowania przez układ: 1) związane z próbką, 2) instrumentalne.
Głównym warunkiem spełnienia praw absorpcji jest monochromatyczność działają-cego promieniowania (i niezbyt wysokie natężenie), stąd równanie (3.8) powinno być zapisane raczej w postaci następującej:
Aλ = ελ b cM (3.12) Odstępstwa związane z próbką zależą od charakteru środowiska. Ważny jest właściwy dobór stężenia badanego roztworu z trzech zasadniczych powodów:
a) współczynnik absorpcji zależy od współczynnika załamania promieniowania w danym środowisku; dla stosunkowo niewielkich stężeń roztworów współczynnik załamania jest stały i identyczny ze współczynnikiem załamania czystego roz-puszczalnika; w roztworach o większych stężeniach zmiany wartości współczyn-nika załamania mogą być przyczyną odstępstw od praw absorpcji;
b) zależność podstawowa (3.8) jest słuszna tylko dla przypadku istnienia w roztwo-
rze jednej formy cząstek; jeżeli w trakcie wzrostu stężenia, kiedy rośnie siła wza-jemnego oddziaływania składników, część z nich ulega zmianom czy deforma-cjom (w wyniku np. solwatacji, hydrolizy, asocjacji, polimeryzacji itp.), zmienia się również wartość molowych współczynników absorpcji odpowiednich cząstek i stąd w tym obszarze stężeń wystąpi zaburzenie prostoliniowego charakteru za-leżności A=ƒ(c);
c) każdemu rodzajowi cząstek przypisuje się funkcję zależną od długości fali, tzw.
przekrój czynny na zderzenia z fotonami; podczas zwiększania stężenia, po prze-kroczeniu pewnej jego wartości może następować wzajemne nakładanie się prze-krojów czynnych cząstek; w tych warunkach wartość współczynnika absorpcji maleje ze wzrostem stężenia.
Z zakłócających przyczyn instrumentalnych do pierwszoplanowych należy: − brak ścisłej monochromatyzacji wiązki promieniowania, − jeżeli w przyrządzie zachodzi rozpraszanie promieniowania; wywiera ono tym
większy wpływ, im większe są wartości mierzonych absorpcji, − zbyt duża szerokość spektralna wiązki promieniowania przechodzącego przez
próbkę. Istotną rolę odgrywa czułość detektora oraz charakter jego reakcji; korzystny jest brak odchyleń od liniowej zależności między wielkością sygnału detektora i natęże-niem, padającego nań promieniowania. Ogólnie biorąc ważna jest klasa przyrządu. Im

24
niższej klasy jest aparat, tym większe bywają odstępstwa od praw absorpcji; przy czym nie zawsze wiążą się one z zaburzeniami prostoliniowego przebiegu zależności A=ƒ(c), może wystąpić jedynie zmniejszenie kąta nachylenia prostej II (rys.3.4b) w porównaniu z prostą I, dla wiązki ściśle monochromatycznej. Ponadto w czasie pomia-rów absorbancji powinna być zachowana stałość temperatury, przynajmniej w grani-cach kilku stopni. Podobne przyczyny mogą powodować odchylenia od prawa addy-tywności absorpcji. Jeżeli jeden lub kilka składników układu nie spełnia prawa Beera - Waltera, prowa-dzi to do odstępstw od prawa addytywności. Natomiast stosowanie się poszczególnych składników od prawa Beera - Waltera nie gwarantuje spełnienia przez cały układ pra-wa addytywności absorpcji. Zmieszanie, bowiem poszczególnych składników absor-bujących w jednym układzie może prowadzić do wzajemnych oddziaływań, powodu-jących powstawanie nowych związków o odmiennej zdolności pochłaniania promie-niowania elektromagnetycznego. Odstępstwa od praw absorpcji są niepożądane w pracach analitycznych ograniczają, bowiem zakres stężeń roboczych, zmuszają do stosowania poprawek, a niekiedy w istotny sposób utrudniają badania lub nawet uniemożliwiają je. Przeciwnie, w pracach poznawczych mogą nawet stanowić dogodną podstawę do badania zjawisk i procesów powodujących te odstępstwa.

25
3.2 Molowy współczynnik absorpcji i przekrój czynny na absorpcję. Intensywność pasm elektronowych.
Współczynnik absorpcji cząsteczki zależy od jej rozmiarów. Aby wyprowadzić tę zależność rozważymy warstwę roztworu o powierzchni S i grubości dx (Rys. 3.5). Jeśli stężenie molowe cząsteczek w roztworze wynosi c, to liczba tych cząsteczek w cm3 roztworu będzie równa:
][mol1002205.6Ν;1000Νc
Ν 1-230
0 ⋅== (3.13) (gdzie N0 jest liczbą Avogadra). Stąd liczba cząsteczek w rozważanej warstwie wynie-sie:
dxS1000Νc
Ν 0W = (3.14)
Jeśli promień rozważanych cząsteczek jest r, to część przekroju warstwy, zajmowana przez te cząsteczki wyrazi się następująco:
dx1000
Ncrπ=dxS
1000Nc
Srπ
f 0202
max = (3.15)
świat ło
próbka detektor
dx
Rys. 3.5. Interpretacja przekroju czynnego cząsteczek w roztworze.

26
Gdy każda z cząsteczek absorbuje każdy kwant światła, który pojawi się na jej dro-dze, wówczas proces absorpcji będzie maksymalnie intensywny i będziemy mieli sy-tuację przedstawioną na Rys. 3.5 - stosuje się tu wyrażenie (3.15). Na ogół jednak czą-steczki nie są doskonałymi absorbantami światła. Oznaczmy przez P prawdopodo-bieństwo, że kwant, który pojawi się na drodze danej cząsteczki, zostanie zaabsorbo-wany. Wówczas zaabsorbowana część wiązki padającej wyniesie: fmax P. Udział w tym procesie danej cząsteczki, można wyrazić poprzez charakterystyczny dla niej tzw. przekrój czynny na absorpcję, σ:
σ = P π r2 (3.16) Wykorzystując σ, względne osłabienie wiązki padającej, w wyniku procesu absorpcji można zapisać w postaci:
dx1000
NcσI
dI 0=− (3.17) Uwzględniając, że osłabienie wiązki promieniowania padającego na warstwę dx jest proporcjonalne do grubości tej warstwy (dx) i stężenia roztworu (c) oraz biorąc pod uwagę, że k = 2.303 εM:
dxckI
dI=− (3.18)
Nσε2.303dx;c1000
Nσ=dxcε2.303=dxck
IdI
00 ==−
otrzymamy ostatecznie:
ε1082.3εN
2303σ 21
0
−⋅== (3.19)
Typowy pierścień aromatyczny ma promień nieco większy od 0.1 nm. Stąd maksy-malny możliwy (P = 1 we wzorze 3.16) molowy współczynnik absorpcji, obliczony z wyrażenia (3.19), wynosić będzie około: 105 [dm3 ⋅ mol-1 ⋅ cm-1]. Zwykle bardziej ty-pową wartością dla tego typu związków jest wielkość 104 [dm3⋅mol-1⋅cm-1], co ozna-cza, że jedynie 10% światła przechodzącego przez daną cząsteczkę ulega absorpcji. Rozważamy teraz oddziaływanie strumienia fotonów o energii odpowiadającej róż-nicy poziomów podstawowego 0 i wzbudzonego i, z cząsteczkami w roztworze (Rys. 3.6). Gdy gęstość padającego strumienia fotonów wynosi ρ (ρ = I/c, gdzie I jest natężeniem, a c prędkością światła), wówczas liczba wzbudzeń w jednostce czasu (= liczbie kwantów pochłanianych w jednostce czasu z padającego strumienia) wyno-si:

27
hν; ρ(ν)
i
0
Rys. 3.6. Oddziaływanie strumienia fotonów hν o gęstości ρ(ν) z cząsteczką o dwóch poziomach energetycznych: podstawowym - 0 oraz wzbudzonym - i
( ) [ ] ( )νρBNBNνhdtνdI
i0i0i0 −=− (3.20) gdzie B0i i Bi0 są odpowiednio współczynnikami Einsteina, określającymi prawdopo-dobieństwo stymulowanego przejścia z poziomu podstawowego (0) do wzbudzonego (i) i odwrotnie, natomiast N0 i Ni są obsadzeniami rozważanych poziomów. W warun-kach normalnych (temperaturach zbliżonych do temperatury otoczenia) niemal wszystkie cząsteczki znajdują się w stanie podstawowym, stąd możemy przyjąć, że Ni = 0 i podstawiając ρ(ν) = I(ν)/c oraz dt = dx /c, a wyrażenie (3.20) można zapisać:
)ν(IcBN
νhdx
c)νdI( 0i0=−
( )( )
)A(dxcBN
hννIνdI
eksp20i0 ≡=− (3.21)
Wzór (3.21) wyraża, tzw. intensywność integralną przejścia (Adośw, Kęcki: Ateor). Uwzględniając zależność: cνν = , alternatywny wzór na intensywność integralną przejścia ekspA ma postać:
( )( )
)A(dxcBN
νhνIνdI
eksp0i0 ≡=− (3.21a)
Porównanie stronami zależności (3.21) z wyrażeniem (3.18) pozwala obliczyć, z po-miarów eksperymentalnych, wartość współczynnika Einsteina dla przejścia ze stanu podstawowego do wzbudzonego badanych cząsteczek.

28
( )( ) dxcε2.303dxckdx
cBNh
IdI
MM20i0 ===− ν
νν
Odpowiednie wyrażenie ma postać:
cνhN
cε2.303Blubc
hνNcε2.303
B0
MM0i
2
0
MM0i == (3.22)
Ze względu na to, że obserwowane pasma mają skończoną szerokość, do poprawnego oszacowania wartości B0i niezbędne jest całkowanie równania (3.22) po całym pa-śmie, stąd ostateczne wyrażenie na B0i przybierze postać (c jest prędkością światła):
( )∫= νd
ννε
Nhc2.303B M
00i (3.23)
Jednocześnie z teorii Einsteina, określającej prawdopodobieństwo przejść pomiędzy poziomami energetycznymi, wynika zależność współczynnika B0i z momentem przej-ścia 01µ : (B0i jest skorelowane z momentem przejścia elektronowego, D0i, zależnością 1)
2
0i2
3
0i µh3π8B = (3.24)
gdzie ∫=∞
∞−dτµψψµ i
*00i
0ie2
22
0i νmπ8he3
µ = (3.25)
0ie
2
0ie2
2
2
3
0i νmheπ
νmπ8he3
h3π8B == (3.26)
Korzystając z wyrażenia (3.24), nie trudno otrzymać:
( )( ) c
NνhBAlub
cNhν
BAνIνdI 00i
0iteor200i
0iteor ===−
Po podstawieniu wzoru 3.26
[ ]1A2
e
2
2A0i
0ie
2
teor molmNcmeπ
cNhν
νmheπA −⋅== (3.27)

29
W ten sposób teoretyczna intensywność integralna może być obliczona po podstawie-niu wartości stałych fizycznych. e = 1.5189×10-14 [kg1/2⋅m3/2⋅s-1];
lub e = 1.6021773×10-19 [C], gdzie 1 C = 9.4802×104 [kg1/2⋅m3/2⋅s-1]; me = 9.10939×10-31 [kg]; N0 = NA = 6.022137×1023 [mol-1]; c = 2.998×108 [m⋅s-1]; h = 6.626×10-34 [J⋅s];
[ ]mmol1033.5101875.81036464.4
smkgmolsmkg
10988.81010939.910022137.6102.3073.14159
cmNe
A
1914
4
22
123
1631
23-28
2e
02
teor
−−
−
−
−−
−
×=××
=
×⋅×
×⋅×⋅==
π
Wyznaczona doświadczalnie integralna intensywność pasma jest zdefiniowana jako:
( ) [ ]mmolνdνε2.303A 1
pasmakonturzepo
'Meksp
−∫= (3.28)
Przy czym stężenie jest wyrażone w [mol m-3], grubość warstwy w [m], a częstotli-wość ν w [m-1]. Stosunek intensywności integralnej pasma, zmierzonej doświadczalnie, do teoretycz-nej intensywności integralnej nazywamy mocą oscylatora f: (2)
( ) ( )∫×=∫×
== −
pasmakonturzepo
'M
10
pasmakonturzepo
'M9
teor
eksp νdνε104.33νdνε105.33
2.303AA
f (3.29)
Wielkość f jest bezwymiarowa, jest specjalnym sposobem wyrażania intensywności pasma elektronowego. Jeżeli moc oscylatora jest bliska jedności oznacza to, że przej-ście elektronowe jest dozwolone. Jeśli natomiast jest bardzo mała, to przejście jest za-sadniczo wzbronione i tylko naruszenie reguł wyboru powoduje pojawienie się jego pasma w widmie.

30
3.3 Dokładność oznaczeń fotometrycznych Nawet dla układów, które nie wykazują odchyleń od prawa Beera, zakres stężeń stosowanych w analizie spektrofotometrycznej jest ograniczony zarówno od strony dużych jak i małych wartości. Przy dużych stężeniach substancji absorbującej zostanie przepuszczona taka ilość energii promienistej, że czułość spektrofotometru staje się nie wystarczająca. Natomiast przy małych stężeniach błąd wskazań rejestratora staje się zbyt duży w porównaniu z wielkością mierzoną. W wielu przyrządach fotoelek-trycznych wskazanie potencjometru kompensacyjnego jest wprost proporcjonalne do natężenia promieniowania padającego na fotokomórkę. Oznacza to, że najmniejsza wykrywalna zmiana natężenia ∆I będzie stała, niezależnie od bezwzględnej wartości samego natężenia. W celu uzyskania największej dokładności pomiaru absorbancji A, przyrost ∆A odpowiadający, zmianie natężenia ∆I, musi być możliwie najmniejszą częścią mierzoną absorbancji A; inaczej mówiąc wielkość ∆A/A musi być zredukowa-na do minimum. W celu wyznaczenia transmitancji, przy której ∆A/A osiąga mini-mum, należy dwukrotnie zróżniczkować równanie opisujące prawo Beera i przyrów-nać drugą pochodną do zera. Wygodne jest przedstawienie prawa Beera w następują-cej formie:
Ilog -I log=A 0 (3.30)2
następnie
( ) dII1elog-0 =dA (3.31)
skąd
dI10I1
A0.4343-=dI
IA0.4343-=dA
A1
A0
−⋅ (3.32)
Zamieniając różniczkę na przyrost skończony otrzymujemy:
A-0 10A
1I
I∆4343.0AA∆
⋅−= (3.33)
lub w postaci:
−=
logTT1
II∆4343.0
A∆A
0 (3.33a)3
2 A = log(I0/I); 10A = I0/I ⇒ I = I0 10-A ; 10-A = T. 3 Ta postać równania (3.33) wykorzystana jest do wykresu przedstawionego na rysunku 3.7a

31
Różniczkując ponownie (pamiętając, że ∆I jest stałe) otrzymujemy:
( )
−−=
20 A
10Aln1010
II∆4343.0
dAA/A∆d AA
(3.34)
Warunek uzyskania minimalnej wartości ∆A/A jest spełniony wówczas, gdy prawa strona równania (3.7) jest równa zeru. Stąd czynnik w nawiasie musi być równy zeru, a więc:
4343.0A;3026.210lnA1;
A10
Aln1010
2
A
====A
(3.35) Oznacza to, że optymalna wartość absorbancji wynosi 0.4343, co odpowiada prze-puszczalności 36.8%.
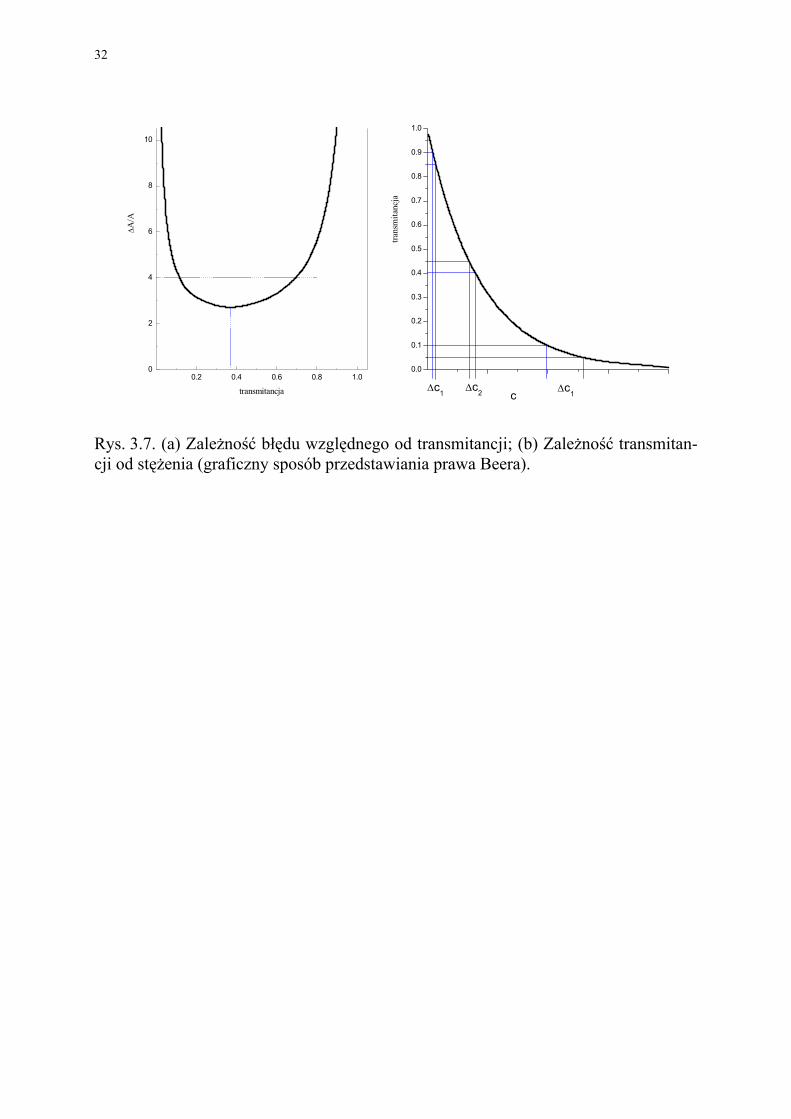
Na Rys. 3.7a przedstawiono graficznie błąd względny analizy, wynikający z 1%-owego błędu w pomiarze spektrofotometrycznym w zależności od zmieniającej się transmitancji. Z rysunku wynika jasno, że choć najmniejszy błąd uzyskuje się przy T=37%, to błąd ten nie będzie dużo większy dla zakresu transmitancji od około 15% do 65% (tj. dla absorbancji odpowiednio od 0.8 do 0.2).
Sytuacje tą można prześledzić na Rys. 3.7b, który przedstawia wykres prawa Beera wyrażony w formie I/I0=10-abc. Dowolnie przyjęta wartość ∆T=1% wykreślona została w trzech pozycjach przy transmitancji wynoszącej: 15%, 37% i 90%. Odpowiadająca tym położeniom niepewność określenia stężenia jest największa przy T=10%, z czego wynika mała precyzja oznaczeń. Przy drugiej krańcowej wartości T=90% niepewność oznaczenia stężenia jest o wiele mniejsza, ale jednocześnie odpowiada ono znacznej części całkowitego stężenia, co daje ponownie małą precyzję oznaczeń. Oczywiście, gdzieś pomiędzy tymi wartościami transmitancji, musi znajdować się taki punkt, gdzie obie tendencje równoważą się i uzyskuje się błąd minimalny. Punkt ten odpowiada przepuszczalności wynoszącej 37%.
32
0.2 0.4 0.6 0.8 1.00
2
4
6
8
10
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
∆A
/A
transmitancja c∆c1
∆c2∆c1tra
nsm
itanc
ja
Rys. 3.7. (a) Zależność błędu względnego od transmitancji; (b) Zależność transmitan-cji od stężenia (graficzny sposób przedstawiania prawa Beera).

33
3.3.1 Zakresy absorbancji użyteczne w analizie ilościowej Każdy pomiar jest obarczony pewnym błędem, na który składa się błąd przypad-kowy i odchylenie stanowiące błąd systematyczny. Zakładamy, że wskutek starannego wzorcowania przyrządu, otrzymana wartość pomiarowa nie jest obarczona błędem systematycznym lub też, że błąd ten jest korygowany za pomocą tablicy poprawek. Natomiast nie jest możliwe całkowite wyeliminowanie błędu przypadkowego, zwane-go, także błędem precyzji, a jedynie jego zmniejszenie. Poprzednio (3.3) omówiono zakresy absorbancji, dla których błąd pomiaru najmniej wpływa na błąd wyznaczenia stężenia w zależności od stosowanego przyrządu i uży-tego w nim detektora. Na kształt krzywej błędu fotometrycznego może także wpływać liczba wykonanych pomiarów, a także właściwości stosowanego przyrządu i warunki pomiarowe. Obecnie instrukcje obsługi spektrofotometrów polecają często jako najlepszy zakres absorbancji 0,8-1,3 lub podobne. W miarę możliwości należy się dostosować do zale-conego zakresu absorbancji, zwłaszcza w przypadku wyznaczania współczynników absorpcji. Nie należy nigdy przekraczać górnej granicy zaleconego zakresu absorban-cji zawsze, bowiem można badany roztwór odpowiednio rozcieńczyć lub użyć kuwety o mniejszej grubości warstwy. Nie należy natomiast sugerować się zbytnio granicą dolną zalecanego zakresu absorbancji, ponieważ w wielu przypadkach musimy posłu-giwać się znacznie niższymi wartościami absorbancji, zwłaszcza podczas oznaczeń śladowych. Podczas oznaczania śladów często nie jest, bowiem możliwe uzyskanie wyższego zakresu stężeń oznaczanego składnika (np. ze względu na małą ilość do-stępnej próbki). Niekiedy udaje się uzyskać wyższe wartości absorbancji przy tym sa-mym stężeniu oznaczanego składnika, posługując się pośrednią metodą spektrofoto-metryczną i w ten sposób podwyższając współczynnik absorpcji, lub też zmieniając analityczną długość fali na taką (np. z zakresu widzialnego do zakresu nadfioletu), przy której współczynnik absorpcji jest wyższy.
Powiększenie współczynnika absorpcji podwyższa czułość metody, tzn. pozwala oznaczać mniejsze stężenia oznaczanego składnika. Miarą czułości metody jest stosu-nek ∆A/∆c, to znaczy stosunek przyrostu absorbancji do przyrostu stężenia wywołują-cego tę zmianę. Im wyższy jest współczynnik absorpcji, tym stężenia odpowiadające dopuszczalnej dolnej granicy absorbancji są niższe. Precyzja i dokładność a czułość metody: Czułość – najmniejsza, dająca się oznaczyć ilość (stężenie) substancji, którą można określić za pomocą danej metody. Również: najmniejsza różnica stężeń lub zawartości składnika oznaczanego, które można określić za pomocą danej metody. Wielkość ta jest mniej związana z przyrządem pomiarowym, niż precyzja i dokład-ność. Natomiast jest ściśle powiązana z molowym współczynnikiem absorpcji. Celem pomiaru jest otrzymanie widma absorpcji cząsteczki. Dlatego zwykle mie-rzymy różnicę absorpcji pomiędzy próbką zawierającą roztwór danych cząsteczek a próbką zawierającą sam rozpuszczalnik (odnośnik). Najdogodniej cel ten realizuje się

34
w spektrofotometrze dwuwiązkowym, który różnicę tę rejestruje automatycznie. Aby przekonać się o tym wystarczy zapisać wyrażenie (3.9) w postaci:
s0s
0s IlogIlog
IIlogA −== (3.36)
r0r
0r IlogIlog
IIlogA −== (3.37)
gdzie: I0 - natężenie wiązki światła padającej zarówno na próbkę z badanym roztwo-rem jak i na kuwetę z rozpuszczalnikiem, Is i Ir - natężenie światła wiązki przechodzą-cej odpowiednio przez kuwetę z badanym roztworem oraz z rozpuszczalnikiem, As i Ar - absorbancja odpowiednio kuwety z próbką i rozpuszczalnikiem. Odejmując stronami wyrażenia (3.27) i (3.28) otrzymamy:
s
rsrrs I
IlogIlogIlogAA =−=− (3.38)
skąd wynika jednoznacznie, że pomiar logarytmu stosunku natężeń Ir do Is stanowi różnicę absorbancji próbki stanowiącej cel pomiaru i próbki odniesienia. Wszystko, co uprzednio napisano, dotyczy klasycznych spektrofotometrów. Obec-nie, stosując przyrządy z mikrokomputerem, można za pomocą wielokrotnego skano-wania (przemiatania) widma i nakładania poszczególnych widm na siebie wykrywać sygnały słabsze od poziomu szumów aparatu. Wynika to z faktu, że szumy dają sygna-ły przypadkowe, natomiast sygnał absorbującego składnika występuje stale, po nało-żeniu, więc szeregu widm na siebie szumy zostają wygładzone, a sygnał analityczny wielokrotnie wzmocniony.

35
4. Analiza ilościowa 4.1 Krzywa wzorcowa (kalibracyjna)
W celu utworzenia, krzywej wzorcowej należy przygotować roztwory podstawowe substancji wzorcowej wychodząc, co najmniej z dwóch naważek, a następnie przygo-tować roztwory o różnych stężeniach. Następnie należy wykonać pomiary absorbancji (pomiar w punkcie) całej serii (10 próbek) przy analitycznej długości fali substancji wzorcowej, np. 1. oznaczanie chromu metodą chromianową, =− )2
4(CrOε 1400 dm3mol-1cm-1 przy
(26801 cm-1) tj. 373 nm); metoda mało czuła, lecz precyzyjna; 2. oznaczanie niklu metodą dimetyloglioksymową =− )2
3(Ni(Dm)ε 1.5⋅104 dm3mol-1cm-1
przy (22472 cm-1) tj. 445 nm), metoda bardziej czuła.
Warunki należy dobrać tak, aby wartości absorbancji mieściły się w całym dopusz-czalnym zakresie, np. 0.2 – 1.0. Następnie należy dokonać naniesienia wyników na wykres: 1. Ai = f (ci), analiza: odczyt stężenia molowego w zależności od wyznaczonej ab-
sorbancji; lub 2. Ai = f (mi) (gdzie m – masa składnika) w [mg], analiza: odczyt masy składnika w
próbce w zależności od wyznaczonej absorbancji. Skuteczniejszą metodą jest zastosowanie do uzyskanych danych tzw. regresji liniowej i dokonywanie obliczeń bezpośrednio ze wzoru (po przekształceniu równania liniowe-go z wyznaczonymi współczynnikami).
36
0.001 0.002 0.003 0.004 0.0050.0
0.2
0.4
0.6
0.8
1.0ab
sorb
ancj
a
stężenie jonów Ni4+, mg/cm3
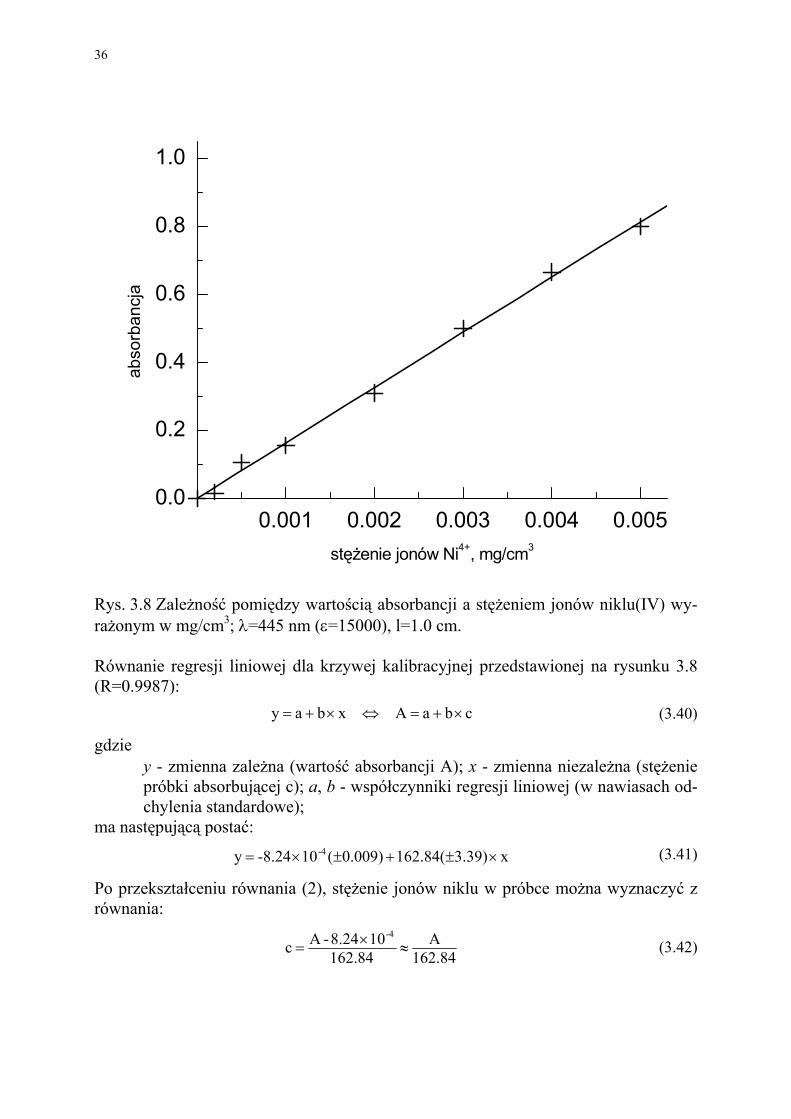
Rys. 3.8 Zależność pomiędzy wartością absorbancji a stężeniem jonów niklu(IV) wy-rażonym w mg/cm3; λ=445 nm (ε=15000), l=1.0 cm. Równanie regresji liniowej dla krzywej kalibracyjnej przedstawionej na rysunku 3.8 (R=0.9987):
cbaAxbay ×+=⇔×+= (3.40)
gdzie y - zmienna zależna (wartość absorbancji A); x - zmienna niezależna (stężenie
próbki absorbującej c); a, b - współczynniki regresji liniowej (w nawiasach od-chylenia standardowe);
ma następującą postać:
x3.39)162.84()009.0(10-8.24y -4 ×±+±×= (3.41)
Po przekształceniu równania (2), stężenie jonów niklu w próbce można wyznaczyć z równania:
84.162A
84.162108.24-Ac
-4
≈×= (3.42)

37
4.2 Analiza układów wieloskładnikowych 4.3 Badanie równowag chemicznych (punkt izozbestyczny)

38
5. Parametry elektronowych pasm absorpcyjnych Widma absorbcyjne związków kompleksowych metali składają się z szeregu pasm o różnym natężeniu. Oczywiste jest, więc traktowanie rejestrowanego widma ekspe-rymentalnego jako sumy nakładających się pasm. Jednym z najważniejszych proble-mów interpretacji elektronowych widm kompleksów metali przejściowych jest analiza liczby oraz oszacowanie położeń i intensywności pasm składowych. Wynika to z fak-tu, że eksperymentalnie otrzymany kontur w postaci NP punktów, tzn. zbioru nie cią-głych danych yi w funkcji xi, jest superpozycją nakładających się pasm, należących często do blisko siebie położonych poziomów energetycznych, których maksima są na konturze doświadczalnym nierozróżnialne. Ponieważ kształt pojedynczego pasma określa rozkład natężeń jako funkcję długości fali (liczby falowej, częstości) i zależy od pewnego wektora P - g(xi,P)
pojedyncze pasmo - g(xi,P) (5.1) a widmo doświadczalne rejestrowane jest jako zbiór nieciągłych danych (wartości dys-kretnych) yi w funkcji xi, to przyjmując kontur doświadczalnego widma jako sumę składowych pasm, otrzymujemy:
( ) ( )∑==
N
n 1nini ,xg,xy PP
(5.2)
gdzie: Pn - w ogólnym przypadku wektor parametrów Pn = [pn1, pn2, pn3,...pnLP] N - liczba pasm składowych złożonego widma. LP - liczba parametrów opisujących pojedyncze pasmo. Stopień nakładania się pasm składowych związany jest z ich liczbą w danym zakre-sie a pośrednio z symetrią otoczenia jonu centralnego w kompleksie. Silne nakładanie się pasm składowych (bliskie polożenia sąsiednich pasm) będzie powodowało, że po-szczególne pasma składowe nie będą dawały informacji o swoim istnieniu na złożo-nym konturze spektralnym, czy też nawet „przegięć”. Sytuacja taka jest charaktery-styczna dla związków kompleksowych w szczególności o niższych symetriach. Rodzaje przejść elektronowych w związkach kompleksowych:
1. Przejścia pomiędzy rozszczepionymi poziomami energetycznymi, powstającymi w wyniku obniżenia sferycznej symetrii w jonie gazowym do realnej symetrii działającego w kompleksie elektrostatycznego pola krystalicznego (przejścia d-d).
2. Przejścia wynikające z oddziaływania orbitali i jonu centralnego (przejścia z przeniesieniem elektronu, tzw. CT (charge transfer).
3. Przejścia w samym ligandzie (przejścia wewnątrz-cząsteczkowe, tzw. intramole-cular).

39
Parametry pasm odpowiadających przejściom elektronowym; 1. Położenie maksimum pasma - νo (energia przejść) 2. Natężenie pasma - εo 3. Szerokość połówkowa pasma - δ
Podstawowym problemem w elektronowej spektroskopii absorpcyjnej związków kompleksowych staje się problem wyodrębniania (znajdowania) pasm składowych ze złożonego konturu spektralnego, jakim jest widmo doświadczalne. Jest to problem matematyczny. Dla jego jednoznacznego rozwiązania niezbędne jest posiadanie in-formacji odnośnie liczby pasm składowych oraz kształtu i parametrów pojedynczego pasma. Stąd waga zagadnienia kształtu pojedynczego pasma absorpcyjnego oraz jego aproksymacji funkcjami analitycznymi. Dane literaturowe wskazują na powszechność stosowania krzywej Gaussa do opisu profilu pasma składowego elektronowych widm związków kompleksowych, która znajduje uzasadnienie w mechanizmie absorpcji światła. Jørgensen wykazał, że gaussowski rozkład natężeń w paśmie absorpcyjnym wynika z zależności kwantowo mechanicznej. Krzywa rozkładu Gaussa ma postać:
( ) ( )
−−= ln2
δνν
expενε2
2oi
oi (5.3)
gdzie: ε(νi) - molowy współczynnik absorpcji przy liczbie ftalowej νi εo - molowy współczynnik absorbcji w maksimum δ - szerokość połówkowa Widmo, będące efektem nakładania się takich pojedynczych pasm jest sumą:
( ) ( )∑
−−=
=
n
1j 2
2oji
oji ln2δνν
expενε (5.4)
gdzie: n - liczba pasm składowych j - odnosi się do j-tej składowej. Innymi funkcjami stosowanymi do aproksymacji pojedynczego pasma są: 2. funkcja Lorentza, 3. funkcja iloczynowa Gaussa i Lorentza, 4. kombinacja liniowa funkcji Gaussa i Lorentza. Wszystkie te funkcje analityczne opisane są podstawowymi parametrami: νo, εo i δ.

40
15000 20000 250000.0
0.2
0.4
0.6
0.8
1.0ν0
δ ε0/2
abso
rban
cja
liczby falowe
ε0
Rys. 3.9. Parametry pasma gaussowskiego: νo - pałożenie maksimum, εo - wysokość, δ - szerokość połówkowa. Funkcja Gaussa:
( ) ( ) ( )i2
2oi
oi g4ln2exp ν=
δν−ν−
ε=νε (5.5)
Funkcja Lorentza:
( ) ( ) ( )i
1
2
2oi
oi l4ln21 ν=
δν−ν
+ε=νε−
(5.6)
Iloczyn funkcji Gaussa i Lorentza:
( ) ( ) ( ) 1
2
2oi
2
2oi
oi 14ln2exp−
δν−ν
+
δν−ν
−ε=νε (5.7)
Kombinacja liniowa funkcji Gaussa i Lorentza:
( ) ( ) ( ) ( )ν⋅−+ν⋅=νε lR1gRi (5.8)
41
15000 20000 250000.0
0.2
0.4
0.6
0.8
1.0
abso
rban
cja
liczba falowa
f. Gaussa f. Lorentza f. iloczynowa
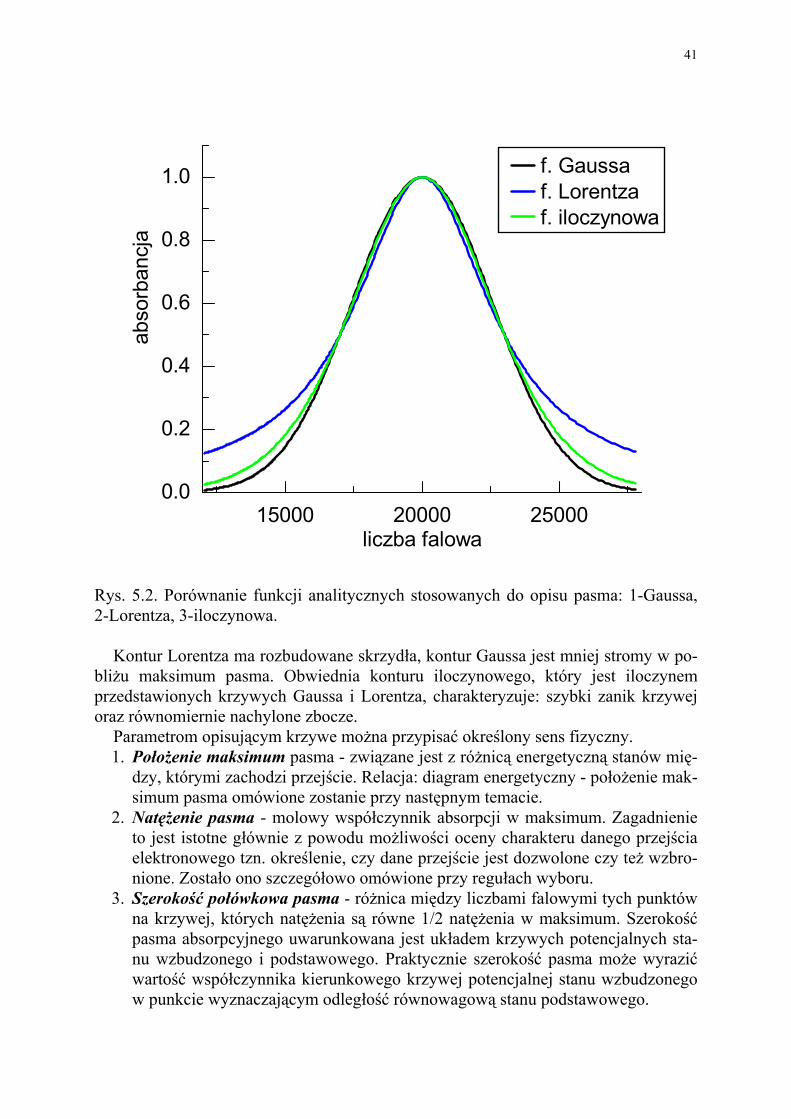
Rys. 5.2. Porównanie funkcji analitycznych stosowanych do opisu pasma: 1-Gaussa, 2-Lorentza, 3-iloczynowa. Kontur Lorentza ma rozbudowane skrzydła, kontur Gaussa jest mniej stromy w po-bliżu maksimum pasma. Obwiednia konturu iloczynowego, który jest iloczynem przedstawionych krzywych Gaussa i Lorentza, charakteryzuje: szybki zanik krzywej oraz równomiernie nachylone zbocze.
Parametrom opisującym krzywe można przypisać określony sens fizyczny. 1. Położenie maksimum pasma - związane jest z różnicą energetyczną stanów mię-
dzy, którymi zachodzi przejście. Relacja: diagram energetyczny - położenie mak-simum pasma omówione zostanie przy następnym temacie.
2. Natężenie pasma - molowy współczynnik absorpcji w maksimum. Zagadnienie to jest istotne głównie z powodu możliwości oceny charakteru danego przejścia elektronowego tzn. określenie, czy dane przejście jest dozwolone czy też wzbro-nione. Zostało ono szczegółowo omówione przy regułach wyboru.
3. Szerokość połówkowa pasma - różnica między liczbami falowymi tych punktów na krzywej, których natężenia są równe 1/2 natężenia w maksimum. Szerokość pasma absorpcyjnego uwarunkowana jest układem krzywych potencjalnych sta-nu wzbudzonego i podstawowego. Praktycznie szerokość pasma może wyrazić wartość współczynnika kierunkowego krzywej potencjalnej stanu wzbudzonego w punkcie wyznaczającym odległość równowagową stanu podstawowego.

42
Zrozumienie sensu fizycznego szerokości połówkowej pozwala na przewidywanie jego wielkości dla różnych typów przejść elektronowych i konfiguracji stanu podsta-wowego. I tak: 1. Przejścia spinowo-dozwolone: stosunkowo szerokie. 2. Przejścia spinowo-wzbronione (interkombinacyjne): pasma w niektórych przypad-kach są bardzo wąskie.
6. Analiza złożonych widm metodą rozkładu na pasma składowe [3] 6.1 Matematyczny rozkład złożonych widm jako problem poszukiwania minimum
funkcji celu Jednym z istotnych problemów dla badań w wielu dziedzinach chemii jest dostosowanie parametrów, przewidzianych przez modele teoretyczne, do danych uzyskanych z eksperymentu [1]. Zwykle kryterium w oparciu, o które dokonywana jest ocena dopasowania, wymaga obliczenia sumy kwadratów różnic pomiędzy obserwowanymi i obliczonymi danymi. Problem ogranicza się do minimalizacji tej sumy. W przypadku widm związków chemicznych, funkcja teoretyczna, którą przybliża się dane doświadczalne, ma postać wykładniczą (np. funkcja Gaussa, Lorentza). Minimalizacja sumy kwadratów odchyleń prowadzi do układu równań nieliniowych, który nie może być dokładnie rozwiązany, tzn. analityczne rozwiązanie jest niemożliwe. Wynika stąd konieczność posługiwania się iteracyjnymi metodami programowania nieliniowego. W iteracyjnych metodach poszukiwania minimum wymagane jest określenie trzech zasadniczych elementów [1]: 1. Punktu początkowego x0. 2. Kryterium zakończenia obliczeń. 3. Reguły generowania ciągu xk, takiego że d = x 0
kklim
∞→, gdzie d0 jest poszukiwanym
rozwiązaniem. Wstępne oszacowanie parametrów może pochodzić z wcześniejszego badania układu chemicznego, przez zastosowanie prostych funkcji modelowych lub można je po prostu odgadnąć kierując się intuicją. Jednym z najważniejszych problemów interpretacji widm elektronowych kompleksów metali przejściowych jest analiza liczby, położeń oraz oszacowanie intensywności pasm składowych. Wynika to z faktu, iż eksperymentalnie otrzymany kontur w postaci NP dopasowanych punktów, tzn., zbioru nieciągłych danych yi w funkcji xi, jest superpozycją nakładających się pasm. Należą one często do blisko położonych poziomów energetycznych, których maksima są na konturze doświadczalnym nierozróżnialne. W praktycznych obliczeniach z reguły ogranicza się do wyznaczenia punktu
0d ,
leżącego wystarczająco blisko punktu d0. Kryterium zakończenia obliczeń podaje

43
wtedy jak należy rozumieć określenie "wystarczająco blisko". Punkt 0
d jest przybliżonym rozwiązaniem zadania. Do rozwiązania punktu (3) stosowanych jest wiele matematycznych metod obliczeniowych. Jedną z nich jest metoda najmniejszych kwadratów. Wszystkie one jednak podlegają ograniczeniu polegającemu na tym, że są one zbieżne do lokalnego minimum, tzn. do pierwszego napotkanego minimum, nawet wówczas, gdy w przestrzeni parametrów istnieje minimum bardziej zadowalające. 6.2 Uogólniona metoda najmniejszych kwadratów Do rozkładu widm można stosować dowolną, z wielu znanych, matematyczną metodę optymalizacji (np.: m. najszybszego spadku Cauche, m. Newtona-Raphsona, m. simpleksu, m. sprzężonego kierunku Powell’a, itp.). Niemniej jednak, w przypadku widm wygodnie jest korzystać z metody, która uwzględnia specyficzną postać badanej funkcji i redukuje w ten sposób liczbę wymaganych oszacowań funkcji. Właściwości te posiada metoda najmniejszych kwadratów. W metodzie tej badana funkcja przedstawiana jest w postaci NP dopasowywanych punktów, zbioru wartości dyskretnych yi w funkcji xi, z funkcją opisującą kontur teoretyczny y(xi,Pn). Kontur absorpcyjnego pasma elektronowego może być aproksymowany za pomocą określonej funkcji analitycznej. Najczęściej aproksymuje się go krzywą rozkładu Gaussa, opisaną trzema parametrami:
2))/pp((x1i
32i2pP),g(x −−⋅= 6.1
Przyjmując kontur widma doświadczalnego jako sumę składowych gaussowskich otrzymuje się:
∑ ⋅=∑==
−−
=
NB
1n
2))/pp((xn1
NB
1nini
3n2nipP)g(x)Py(x 2,, 6.2
Algorytm najmniejszych kwadratów pozwala znaleźć dokładne wartości zbioru parametrów:
NB,1,2,j]p,,p,p[p jLPj2j1j KK ==
które są źródłem optymalnego dopasowania funkcji y(xi,pj) do zbioru nieciągłych danych yi, w sensie minimalizacji sumy kwadratów odchyleń. Można to przedstawić następująco:
minimum ]y-)P,y(x[ SNP
1i
2ini =∑=
= 6.3
gdzie: y(xi,pj) - funkcja opisująca kontur teoretyczny (obliczany);

44
yi = {yi, xi} - zbiór dyskretnych danych; kontur widma doświadczalnego; NP - liczba punktów; NB - liczba pasm; LP - liczba zmiennych (parametrów) opisujących pojedyncze pasmo (dla funkcji Gaussa LP=3). Po założeniu początkowych wartości parametrów:
NB,1,2,j]p,,p,p[p 0jLP
0j2
0j1
0j KK ==
zamiast wyszukiwania parametrów najlepszego przybliżenia, w metodzie tej oblicza się poprawki dla parametrów pj, takie aby:
NBLP,1,2,j,ppp j0jj ⋅=∆+= K
tzn. minimalizujących funkcję [1,3]:
∑ −∆+==
NP
1i
2ij
0ji ]ypp,y(x[S
6.4
Jeśli przyjęte wartości 0
jp są dobre (tzn. dostatecznie bliskie wartościom pj) to funkcja y może być aproksymowana przy pomocy skróconego szeregu Taylora (do członu liniowego) rozwiniętego względem poprawek ∆pj [1,4]:
p∆y+ ) P,x(y = ) P∆ +P,x(y jij
NBLP
1=j
0nin
0ni ×∑
× 6.5
gdzie j
i,ij p
yy
∂∂
= są pochodnymi cząstkowymi y względem parametrów pj obliczanymi
dla każdego punktu xi. W takim przybliżeniu liniowym minimalizacja w równaniu (6.4) przyjmuje postać:
] y -p∆y+ )p,x(y [= S 2ij
'ij
NBLP
1=j
0ji
NP
1=i×∑∑
×
6.6 Warunkiem koniecznym istnienia minimum jest zerowanie się pierwszych pochodnych cząstkowych funkcji S. Różniczkując S względem ∆pj otrzymujemy [4,5]:
,,ijikik
NB*LP
1=k
0ji
NP
1=ijy] y- py+ )p,(xy [2 =
pS
∆⋅∑∑⋅∆δ
δ
6.7

45
Po przyrównaniu do zera mamy:
0 =]y y-py+ )p,[y(x, NB*LP1,2,...,=j ijikikNB*LP
1=k
0ji
NP
1=i
,, ∆⋅∑∑∀ 6.8 stąd
pyy= y] )p,y(x-[yNB,*LP1,2,...,=j kijikNB*LP
1=k
NP
1=iij
0jii
NP
1=i∆⋅⋅∑∑∑∀ ,,,
6.9
Sumowanie po i odpowiada wartościom pochodnych cząstkowych i-tego punktu widma. Wskaźniki k oraz j (podobnie jak n) odpowiadają pasmom składowym. Jeśli NB jest liczbą pasm składowych w widmie, to otrzymuje się NB takich równań uzyskanych w wyniku różniczkowania funkcji S względem zmiennych pn1 (patrz równanie 6.2), podobnie otrzymuje się NB takich równań dla zmiennych pn2 oraz pn3. Ogólnie otrzymujemy układ LP*NB równań liniowych z LP*NB niewiadomymi, który można zapisać w postaci macierzowej następująco:
BA = P B= PA 1 ⋅∆∆⋅ − lub 6.10 a elementy macierzy A i wektora B wyrażają się wzorami:
,,ikij
NB*LP
1=k
NP
1=ijk yy=a ⋅∑∑ 6.11
,ij
0ji
NP
1=ij y)(pD=b ⋅∑ 6.12
gdzie:
)p,y(xy)(pD 0jii
0ji −= 6.13
jest odchyleniem konturu eksperymentalnego yi od konturu teoretycznego y(xi,P) w punkcie xi. Macierz A można, zatem przedstawić następująco:

46
∑∑∑
∑∑∑
∑∑∑
===
===
===
NP
1i
,Mi,
,Mi,
NP
1i
,i2
,Mi,
NP
1i
,i1
,Mi,
NP
1i
,Mi,
,i2
NP
1i
,i2
,i2
NP
1i
,i1
,i2
NP
1i
,Mi,
,i1
NP
1i
,i2
,i1
NP
1i
,i1
,i1
yyyyyy
yyyyyy
yyyyyy
L
OM
L
L
6.14
gdzie M=LP*NB. Z rozwiązania układu równań (6.10) uzyskuje się pierwszy zbiór poprawek, który dołączony do początkowych parametrów p0
j , daje poprawiony zbiór parametrów do obliczeń w następnej iteracji [1,4]. Procedura ta jest kontynuowana aż obliczone poprawki staną się odpowiednio małe. Opisany wyżej algorytm ma tę wadę, iż może okazać się, że wyliczona w n-tym kroku iteracyjnym wartość S(n) jest większa niż S(n-1), wtedy mamy proces rozbieżny. Aby temu zapobiec do przybliżonej wartości P(n) nie dodaje się pełnej, wyliczonej poprawki ∆P(n) lecz jedynie jej część (DM * ∆P(n)) tak, aby spełniony był warunek zbieżności, tzn. by [6]:
0 > )/SS- (S = DRMS 1)-(n(n)1)-(n Wymuszenia zbieżności iteracji dokonuje się, dobierając parametr tłumienia DM<1 z zadanego zbioru liczb. W przypadku, gdy po wyczerpaniu wszystkich możliwych wartości DM, w dalszym ciągu okaże się, że DRMS<0, proces dopasowywania parametrów zostaje przerwany. Z wzorów (6.10 - 6.13) wynika wprost jawna postać rozwiązania:
∑∑
∑ −=∆
==
=NB*LP
1kikij
NP
1i
NP
1iij
0nii
jyy
y)]P,y(xy[p
,,
,
6.15
6.3 Algorytm Slaviča Układ równań (6.8) można rozwiązać wykorzystując jedną z wielu metod rozwiązywania układów równań liniowych lub też odwracając macierz A. Algorytm najmniejszych kwadratów z użyciem metody odwracania macierzy podał Schwarz [4]. Zarówno jednak jedna jak i druga droga uzyskania poprawek w przypadku dużych macierzy jest dość czasochłonna i kłopotliwa. Dlatego też często wykorzystuje się uproszczony algorytm najmniejszych kwadratów, zaproponowany przez Slaviča [7],

47
polegający na pominięciu pozadiagonalnych elementów macierzy A oraz wprowadzeniu wag punktowych ( odwrotnie proporcjonalnych do wartości yi). Po tym uproszczeniu równanie na wyznaczanie poprawek Dpj do parametrów przyjmuje następującą postać:
d ) /y1 ( )(y
) /y1 ( )(PDy = p
i2
ijNP
1=i
i0jiij
NP
1=ij ⋅
∑
∑∆
,
,
6.16
gdzie d - współczynnik tłumienia, a (1/yi) - wagi punktowe. Ta uproszczona metoda najmniejszych kwadratów pozwala na wyeliminowanie czasochłonnej procedury odwracania macierzy A. Nie wystarcza ona jednak do analizy błędów poszukiwanych parametrów, dla której konieczna jest pełna forma tej macierzy. Pochodne wymagane dla wyznaczenia elementów macierzowych (w obu metodach) mają postać:
,i1
ln2))/pp((x
n1
i y2py 2
n3n2i == −−
δδ
6.17
,,i2
2n3n2in1i1
n2
i y)/(p)px(2lnp2ypy
=−⋅⋅=δδ
6.18
,,i3n3n2ii2
n3
i y/p)px(ypy
=−=δδ 6.19

48
7. Spektrofotometria z wykorzystaniem pochodnych widma. Spektrofotometria z wykorzystaniem pochodnych widma (spektrofotometria po-chodna) powstała w latach 50-tych, ale jej rozwój nastąpił w drugiej połowie lat 70-tych [4]. Podstawy spektrofotometrii pochodnej wywodzą się ze spektrofotometrii kla-sycznej. Zakładając stałość natężenia promieniowania padającego I0 w całym zakresie długości fal, występującym we wzorze wyrażającym prawo Lamberta-Beera:
clII
logA o ε==
można zróżniczkować to równanie względem długości fal lub liczb falowych.
D ,n
n
n
n
cldd
dAd
λλε
λ xn
== (7.1a)
D ,n
n
n
n
cldd
dAd
ννε
ν xn
== (7.1b)
gdzie: D ,νxn oznacza wartość pochodnej n (n – rząd pochodnej) widma absorpcji sub-
stancji X przy długości fali λ, ν - liczba falowa (wzór 7.1b). Wynik różniczkowania absorbancji względem długości fali lub liczby falowej jest pochodną widma absorpcji. Krzywa pochodna widma absorpcji (widmo pochodne – derivative spectrum) jest to graficzne przedstawienia wyniku różniczkowania absor-bancji względem długości fal lub liczb falowych.
Widmo absorpcji i pochodne widma różnych rzędów wyrażają zależności:
)(d
AdD ,
00
0
λλ λ fx ==
widmo rzędu zerowego, widmo absorpcji, widmo podstawowe A = f(λ),
(7.2)
)(d
AdD
1 λλ λ f==
widmo rzędu pierwszego (7.3)
)(d
AdD
22
2
λλ λ f==
widmo rzędu drugiego (7.4)
)(d
AdD ,n
n
λλ λ fx
n==
widmo rzędu n (7.5)

49
W klasycznej spektrofotometrii miarą stężenia substancji jest wartość absorbancji. W spektrofotometrii pochodnej do stężenia i grubości warstwy jest proporcjonalna war-tość pochodnej widma absorpcji. Spektrofotometria pochodna przejmuje też od spek-trofotometrii klasycznej właściwość addytywności. Pochodna widma mieszaniny składników równa się sumie wartości pochodnych widm poszczególnych składników
DDDD k21k1nnnn
+++=+ KK (7.6)
Właściwością specyficzną dla spektrofotometrii pochodnej jest zależność wartości pochodnej od kształtu pasma a ściślej od jego szerokości połówkowej δ1/2
LAPD n-maxn
n= (7.6)
gdzie Amax- maksymalna absorbancja pasma, Pn – wielomian zależny od rzędu pochodnej. Wartość pochodnej wzrasta, gdy zmniejsza się szerokość połówkowa pasma, np.:
δδ B1/2
A1/2BA gdyDD <>
nn (7.6)
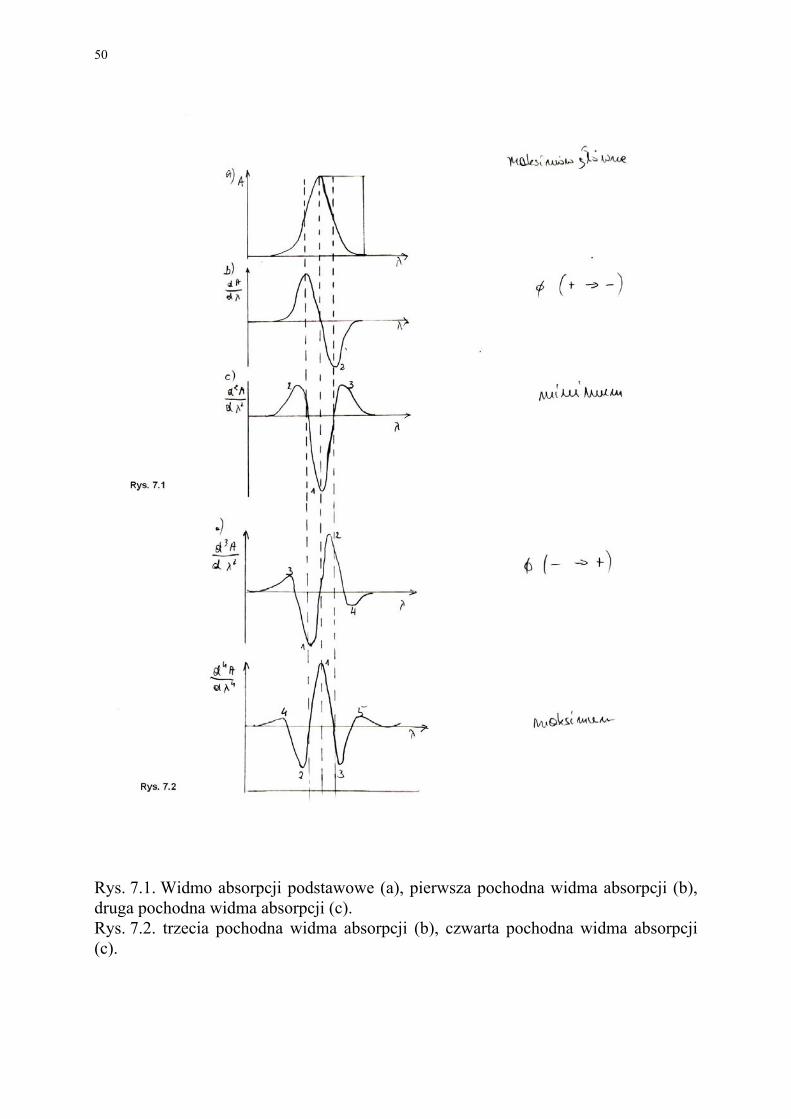
Rysunek 7.1 przedstawia pasmo absorpcji w postaci krzywej Gaussa oraz krzywe
pochodne pierwszego i drugiego rzędu. Wykres pochodnej pierwszego rzędu ma dwa piki (dodatni - odpowiadający maksymalnemu zwiększaniu się wartości absorbancji, ujemny - odpowiadający maksymalnemu zmniejszaniu się wartości absorbancji) i przecina linię zerową przy wartości odciętej odpowiadającej występowaniu maksi-mum na krzywej absorpcji. Podobny przebieg mają wszystkie pochodne wyższe rzę-dów nieparzystych. Pochodne, rzędów parzystych, mają pik główny podobny do kon-turu krzywej wyjściowej, lecz wyraźnie węższy. Po obu strona piku głównego wystę-pują piki uzupełniające zwane skrzydłami. Liczba pików w widmie n-tej pochodnej pojedynczego pasma typu krzywej Gaussa wynosi (n+1).
Rysunek 7.2 przedstawia krzywe pochodne trzeciego i czwartego rzędu. W prakty-ce skuteczna okazuje się praca z pochodnymi 3-5, a podczas silnie nakładających się pasm przydatne okazują się pochodne rzędów jeszcze wyższych.
Spektrofotometria pochodna jest bardziej selektywna i czuła w porównaniu s kla-syczną. Zwiększenie selektywności wynika z tego, że podczas różniczkowania widma podstawowego pasma ulegają zwężeniu, co powoduje, że pasma nakładające się w widmie podstawowym i nie obserwowane w postaci maksimów na wykresie pochod-nej ulegają rozdzieleniu. Im wyższy jest rząd pochodnej tym mniejsza jest szerokość głównej części pasma.
50
Rys. 7.1. Widmo absorpcji podstawowe (a), pierwsza pochodna widma absorpcji (b), druga pochodna widma absorpcji (c). Rys. 7.2. trzecia pochodna widma absorpcji (b), czwarta pochodna widma absorpcji (c).

51
Przykładowe widmo symulowane (15 pasm składowych) oraz przebieg 2-giej i 4-pochodnej złożonego konturu.

52
Zwiększanie czułości w spektrofotometrii pochodnej wynika przede wszystkim z odpowiedniej amplifikacji (elektronicznej lub numerycznej) wartości sygnału przy jed-noczesnym eliminowaniu szumów. Czułość zależy także od kształtu pasm w widmie podstawowym, a w szczególności od szerokości połówkowej pasma. W spektroskopii pochodnej zwiększa się natężenie pików ostrych, a zmniejsza łagodnych. Wysokość piku głównego na krzywej pochodnej zależy od nachylenia wstępującej i zstępującej części krzywej absorpcji i jest proporcjonalna do natężenia wyjściowego pasma. 7.1 Otrzymywanie widm pochodnych Stosowane są dwie techniki trzymywania widm pochodnych: analogowa i nume-ryczna. W technice analogowej wykorzystuje się odpowiedni układ elektroniczny umiesz-czony między detektorem a rejestratorem. Od parametrów tego układy a także parame-trów pracy, takich jak szybkość skanowania, czas odpowiedzi zależy kształt widma pochodnej. Metoda numeryczna polega na zapisaniu widma w pamięci i przetworzenie danych metodami matematycznymi. Ten sposób otrzymywania pochodnych umożliwia spek-trofotometr Specord M40 wyposażony w odpowiednie oprogramowanie. W literaturze (Savitsky i Golay [5]) zostały opracowane i przedstawione aproksy-macyjne sposoby obliczania pochodnych w postaci gotowych wzorów. Polegają one na tym, że: − Fragment krzywej (pewna liczba punktów doświadczalnych, np. 9) jest opisywany
odpowiednim wielomianem (wyznaczonym metodą najmniejszych kwadratów). Wielomian jest różniczkowany i obliczana jest wartość pochodnej dla środkowego punktu z wybranego przedziału.
− Następnie od wybranego przedziału (zbioru) punktów odrzuca się pierwszy a do-daje kolejny i całą procedurę obliczania powtarza się aż do uzyskania całego wid-ma. Przesuwamy wybrany przedział wzdłuż całego widma podstawowego.
Kształt widma pochodnej zależy od rzędu pochodnej, liczby punktów stopnia wie-
lomianu, odległości między punktowej oraz od tzw. wygładzania widma, czyli takiego przetwarzania matematycznego widma w wyniku, którego otrzymuje się krzywą o regularniejszym kształcie (pozbawionego tzw. szumów). Optymalizację parametrów różniczkowania numerycznego można przeprowadzić stosując spektrofotometry umożliwiające zapis rejestrowanych widm w pamięci komputera, a następnie stoso-wanie do obliczenia odpowiedniej pochodnej programu komputerowego.
Równania poniżej przedstawiają przykładowe wzory stosowane do obliczania od-
powiednio 1-szej, 2-giej i 4-tej pochodnej dla widma w postaci zbioru wartości funkcji rozmieszczonych w równoodległych odstępach (h).
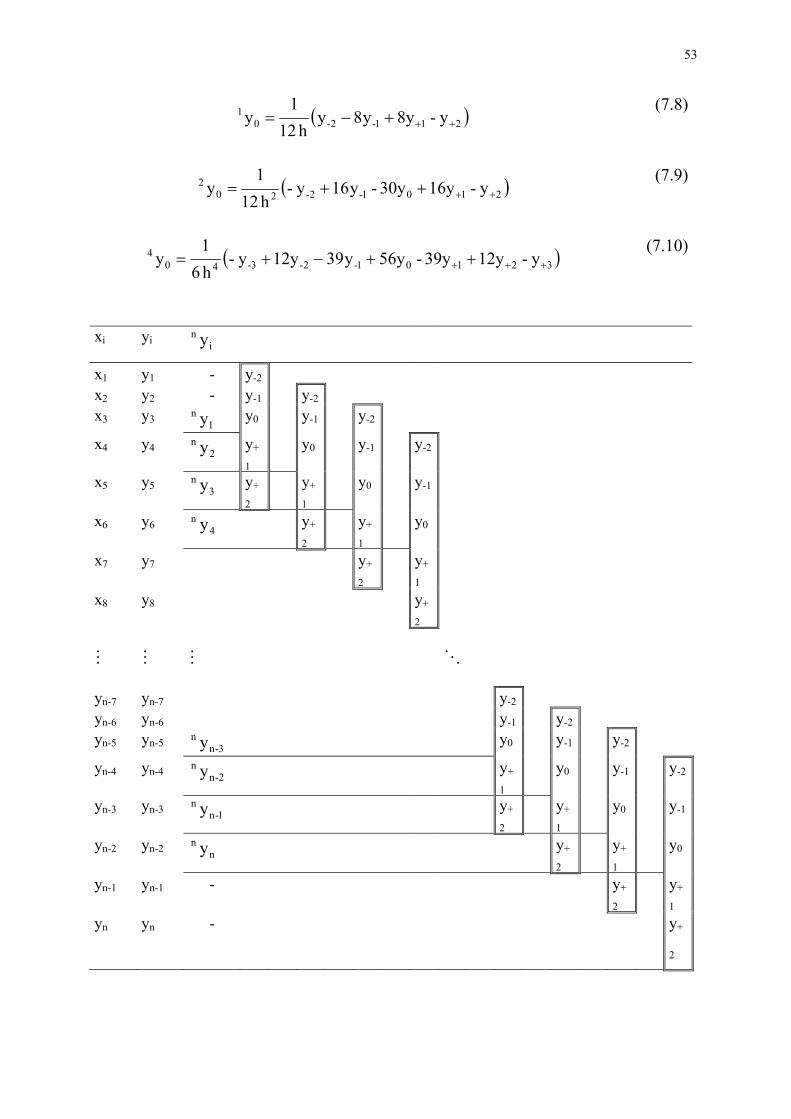
53
( )211-2-01 y-8yy8y
h121
y +++−= (7.8)
( )2101-2-202 y-16y30y-y16y-
h121
y ++++= (7.9)
( )32101-2-3-404 y-12y39y-56yy3912yy-
h61
y +++ ++−+= (7.10)
xi yi i
n y
x1 y1 - y-2 x2 y2 - y-1 y-2 x3 y3 1
n y y0 y-1 y-2
x4 y4 2n y y+
1
y0 y-1 y-2
x5 y5 3n y y+
2
y+
1
y0 y-1
x6 y6 4n y y+
2
y+
1
y0
x7 y7 y+
2
y+
1
x8 y8 y+
2
M M M O
yn-7 yn-7 y-2 yn-6 yn-6 y-1 y-2 yn-5 yn-5 3-n
n y y0 y-1 y-2
yn-4 yn-4 2-nn y y+
1
y0 y-1 y-2
yn-3 yn-3 1-nn y y+
2
y+
1
y0 y-1
yn-2 yn-2 nn y y+
2
y+
1
y0
yn-1 yn-1 - y+
2
y+
1
yn yn - y+
2

54
Stosowanie pochodnych widm umożliwia:
1. Wzrost selektywności i czułości w porównaniu spektrofotometrii klasycznej, 2. Wyznaczenie dokładnych wartości położeń pasm λmay, a także punktów przegięcia. 3. Wykorzystanie nawet bardzo słabych pasm, które na krzywej absorpcji, obserwo-
wane są w postaci przegięć lub zmian kąta nachylenia (asymetria).

55
8. Literatura
1. W.L.Masterton, L.J.Słowiński, Chemical principles with quantitative analysis,
W.B.Sander Company, Philadelphia-London-Toronto 1978.
2. C.R.Cantor, P.R.Schimmel, Biophysical Chemistry. Part II. Techniques for study of
biological structure and function., W.H.Freeman Company, San Francisco 1980.
3. J.B.Czermiński, A.Iwasiewicz, Z.Paszek, A.Sokorski, Metody statystyczne dla chemi-
ków., PWN, Warszawa 1992.; Rozdz. 5.3. Regresja nieliniowa niekinearyzowana, Me-
toda Marquardta: program B3 (Pascal).
4. A.Cygański, Metody spektroskopowe w chemii analitycznej, WN-T, Warszawa 1997.
5. A.Savitsky, M.J.E.Golay, Anal.Chem., 36 (1964) 1627.