monog. vi/it (page 90)
TRANSCRIPT

COAGULAZIONE
Tiziano BarbuiMonica Galli
VI
Sindrome da anticorpi antifosfolipidi


Tiziano Barbui, Monica Galli
Sindrome da anticorpi antifosfolipidi


3Indice
Sindrome da anti-fosfolipidi primaria e secondaria: due facce della stessa medaglia? Pag. 7
Fisiopatologia e possibili meccanismi di trombogenesi degli anticorpi antifosfolipidi Pag. 15
Aspetti clinici e terapeutici delle trombosi arteriose e venose Pag. 23
Aspetti clinico-terapeutici delle complicanze ostetriche Pag. 33
Diagnosi di laboratorio degli anticorpi antifosfolipidi.
1. Anticoagulante tipo lupus: diagnosi di laboratorio Pag. 46
2. Anticorpi anticardiolipina: diagnosi di laboratorio Pag. 53
3. Anticorpi anti-ß2glicoproteina 1: diagnosi di laboratorio Pag. 63
4. Anticorpi antiprotrombina: diagnosi di laboratorio Pag. 81


5Prefazione
L’esistenza di anticorpi antifosfolipidi fu per la prima volta provata nel 1941cimentando il siero di pazienti con sifilide ed estratti di cuore bovino. Il sierointeragiva con la cardiolipina ed il test fu ritenuto specifico per la diagnosi di sifilide e chiamato VDRL (Veneral Disease Research Laboratory).La specificità diagnostica fu peraltro resa insicura con le osservazioni di una positività dalla VDRL nei pazienti con malattie autoimmmuni sistemiche(Lupus Eritematoso Sistemico,LES), in assenza di malattia venerea.Nel 1983 Harris et al. (Lancet 2: 1211, 1983) pubblicarono un immunoassayper la misura quantitativa degli anticorpi anticardiolipina e rapidamente si confermò che la loro presenza si associava a manifestazioni cliniche di trombosi venosa e arteriosa e ad aborti ripetuti (sindrome da anticorpi antifosfolipidi; APA).Una tappa fondamentale per il meccanismo d’azione degli anticorpi fu il 1990. Due gruppi indipendenti dimostrarono che il target antigenico non era, nei casi associati a malattie autoimmuni e non a sifilide, il fosfolipide ma la beta2 glicoproteina I che si trovava complessata ai fosfolipidi anionici(Galli et al., Lancet 335: 1544, 1990; McNeil et al. PNAS 87: 4120, 1990). Fu poi confermato che in alcuni casi gli anticorpi interagiscono con la beta2glicoproteina I in assenza di fosfolipidi (Arvieux et al., J. Immunol. Methods 143: 223, 1991) e queste osservazioni spinsero i ricercatori a sviluppare tests in cui l’antigene non era il fosfolipide ma le proteine legate ai fosfolipi-di alimentando nuove ipotesi patogenetiche della sindrome. Le trombosi sarebbero la conseguenza delle azioni degli anticorpi sul pathway della proteina C, dell’antitrombina III, sulla protrombina, sugli endoteli, sulle pia-strine, sulle cellule apoptotiche, sulle LDL ossidate. Così ciascun ricercato-re, di volta in volta, proponeva (e propone) questi tests e li correlava con glieventi clinici nell’ambito peraltro di studi il cui disegno non consentiva di valutarne il significato e quindi la loro utilizzazione nel processo diagnosti-co rivolto al singolo paziente.Fortunatamente gli esperti del settore hanno riconosciuto la necessità di fare ordine nella diagnosi e nella definizione di sindrome da APA che rischiava di comprendere quadri clinici disparati non facilmente ascrivibili agli anticorpi in questione. Il consenso internazionale per la diagnosi di sin-drome da anticorpi antifosfolipidi comprende criteri clinici e criteri di labora-torio. I primi riguardano le trombosi vascolari (uno o più episodi nelle arte-rie, vene o piccoli vasi di qualsiasi tessuto od organo) e le complicanze della gravidanza (una o più morti di feto normale che avvengono dopo la 10° settimana di gestazione; una o più nascite premature di neonati normaliprima della 34° settimana di gestazione; tre o più aborti consecutivi sponta-nei prima della 10° settimana di gestazione). Per i criteri di laboratorio gli anticorpi anticardiolipina IgG o IgM devono essere a titolo elevato o intermedio in due o più occasioni a distanza di almeno 6 settimane. La definizione di livelli moderati di ACA è insicura (piùdi 20-30 unità internazionali?).Per il lupus anticoagulante si devono seguire le linee guida della Società Internazionale dell’Emostasi e Trombosi (Thromb. Haemost. 74: 1185, 1995). La diagnosi viene formulata quando vi è presenza di almeno un criterio clinico e di un criterio di laboratorio.Pertanto gli ACA IgA, gli anti beta2-glicoproteina1, gli antoprotrombina, gli antiproteina C o S non fanno parte dei criteri di laboratorio.La continua produzione di informazioni sui meccanismi d’azione nella clini-ca, nella profilassi e nella terapia rende necessario aggiornare le cono-scenze. In questo volume si è inteso appunto offrire e discutere le risposte per alcune tra le principali domande che la pratica clinica pone in questa condizione clinica la cui frequenza sta diventando sempre più rilevante nell’ambito degli stati trombofilici.


7
PL Meroni, *A Tincani, *G Balestrieri
Unità di Allergologia e Immunologia Clinica, Dipartimento di MedicinaInterna, Università degli Studi di Milano, IRCCS Istituto Auxologico Italiano;
* Servizio di Reumatologia, Allergologia e Immunologia Clinica, Spedali Civili, Brescia
Sindrome da anti-fosfolipidi primaria e secondaria: due facce della stessa medaglia?

8
L identificazione di anticorpi anti-fosfolipidi (aPL) risale a più di 40 annifa come condizione responsabile delle false positività croniche per itest sierologici della sifilide (FBP-STS) e genericamente correlata ad
autoimmunità (1). Negli anni ’60 l’identificazione del fenomeno del Lupus Anticoagulant (LA), la sua dipendenza dalla presenza di aPL, la sua asso-ciazione con le FBS-STS e soprattutto con manifestazioni trombotiche, trombocitopenia ed abortività configurarono per la prima volta l’esistenza di un subset di pazienti caratterizzati da un quadro clinico peculiare (2). A dispetto del nome, fondamentalmente dovuto al tipo di patologia in cui il LA venne individuato inizialmente, fu chiaro sin dall’inizio che non tutti i pazienti con LA presentavano un lupus eritematoso sistemico (LES). In altreparole, la dizione Lupus Anticoagulant risultò fuorviante non solo perché in realtà rappresentava una condizione di rischio trombofilico ma anche per-ché non necessariamente risultava essere strettamente legata alla malattia lupica. L’utilizzo di un test in fase solida per la determinazione degli aPL (test dell’anti-cardiolipina [aCL]) ha permesso di estendere gli studi epidemiolo-gici e di confermare l’esistenza della sindrome da anticorpi anti-fosfolipidi (APS) non solo in pazienti con LES (Secondary APS) ma anche in pazienti in cui non era diagnosticabile alcuna chiara malattia autoimmuni sistemica (Primary APS) (3,4,5). Le forme secondarie, seppur prevalentemente identificabili in pazienti con LES, sono state descritte in quasi tutte le altre malattie autoimmune sistemi-che. Addirittura, se inizialmente la APS fu più frequentemente diagnosticatain pazienti con LES, risultò successivamente che le forme primitive risulta-vano essere altrettanto se non più frequenti. Lo studio multicentrico più recente coordinato da R. Cervera per l’EuropeanaPL Forum su una casistica di 1000 pazienti ha chiaramente confermato questa tendenza (Tabella 1) (6).
Introduzione
Tabella 1: Classificazione dei pazienti con APS
Malattia sottostante No. %PAPS 531 53LES 370 37Lupus-like 47 4Sindrome di Sjogren 1a 23 2Artrite Reumatoide 20 2Sclerodermia 8 1Vasculiti sistemiche 7 1Dermatomiosite 2 0.2
’

9
La APS è formalmente caratterizzata dalla presenza persistente di aPL e di trombosi arteriose e/o venose e/o abortività ricorrente (7). La letteratura ha tuttavia indicato negli ultimi anni la possibilità di uno spettro di presentazionidella APS (Tabella 2)(8). L’insieme di queste segnalazioni suggerisce che i pazienti con APS rappresentino un gruppo eterogeneo e che essi costitui-scano modalità diverse di presentazione di una stessa malattia piuttosto che entità cliniche distinte. In quest’ottica vanno considerati i report di casi in cui un Lupus Eritematoso Sistemico (LES) conclamato si è sviluppato neltempo in pazienti diagnosticati inizialmente come forme primitive (9).
Forme primitive e secondarie
APS: un’unica malattia con diverso spettro clinico o entità cliniche diverse?
Tabella 2: Spettro delle presentazioni cliniche della APS
1. APS associata ad una malattia autoimmune sistemica, prevalentemente LES (Secondary APS);
2. pazienti con APS ma senza una malattia autoimmune sistemica diagnosticabile (Primary APS);
3. pazienti con APS e con “lupus-like disease”, che in altre parole manifestano segni di interessamento sistemico ma per i quali non è formalmente possibile soddisfare i criteri di classificazione per il LES;
4. presenza di aPL legati ad altre cause, quali farmaci, neoplasie, processi infettivi. La maggior parte di questi pazienti non presentano le manifestazioni tipiche della sindrome ma solo un titolo elevato di aPL. In taluni report è stata anche descritta la comparsa di manifestazioni cliniche (solitamente trombosi), ma questi casi sembrano rappresentare più l’eccezione che non la regola.
Quadro clinicoDall’analisi della letteratura emerge che essenzialmente il quadro clinico di presentazione della APS sia in corso di LES (o di altre malattie autoimmuni sistemiche) sia nelle forme primitive è sostanzialmente sovrapponibile. La tabella 3 riporta la prevalenza delle diverse manifestazioni cliniche nellePAPS e nelle APS associate a LES riscontrate nella casistica di 1000 pazienti dell’European aPL Forum (6).Va sottolineato che quanto descritto più recentemente coincide con i risul-tati riportati in varie casistiche numericamente inferiori e pubblicate negli anni antecedenti (8).Sono tuttavia riscontrabili alcune differenze che appaiono per la maggior parte imputabili all’esistenza della malattia di fondo delle forme secondarie.Questo vale soprattutto per la maggiore prevalenza di artriti franche, di epi-lessia, di osteonecrosi, di interessamento renale, di valvulopatie cardiache,di anemia emolitica e leucopenia. Tutte le sopracitate manifestazioni non solo fanno parte del contesto clinico del LES - patologia prevalentemente associata – ma costituiscono anche criteri classificativi noti per la malattia lupica stessa (10). La stessa associazione con il sesso femminile (rapporto maschi/femmine di 7:1 nella APS associata LES vs 3.5:1 in corso di PAPS) o con l’età di insorgenza (più giovane nella APS associata al LES rispetto alla PAPS) sono chiaramente influenzate dalla malattia lupica.Sovrapponibili appaiono le caratteristiche delle principali manifestazioni cliniche (abortività e trombosi) nelle due forme.
10
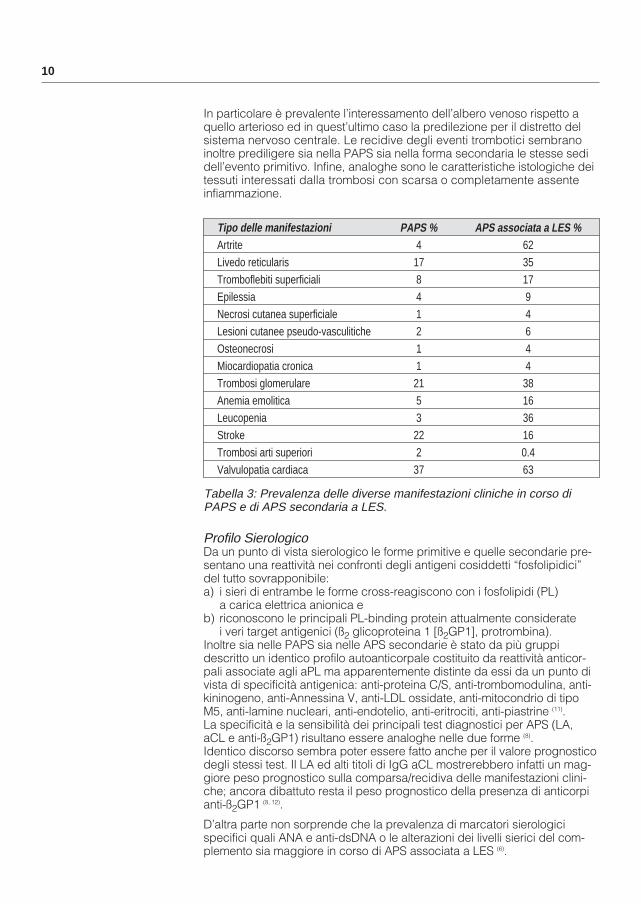
In particolare è prevalente l’interessamento dell’albero venoso rispetto a quello arterioso ed in quest’ultimo caso la predilezione per il distretto del sistema nervoso centrale. Le recidive degli eventi trombotici sembrano inoltre prediligere sia nella PAPS sia nella forma secondaria le stesse sedi dell’evento primitivo. Infine, analoghe sono le caratteristiche istologiche dei tessuti interessati dalla trombosi con scarsa o completamente assente infiammazione.
Tabella 3: Prevalenza delle diverse manifestazioni cliniche in corso diPAPS e di APS secondaria a LES.
Tipo delle manifestazioni PAPS % APS associata a LES %Artrite 4 62Livedo reticularis 17 35Tromboflebiti superficiali 8 17Epilessia 4 9Necrosi cutanea superficiale 1 4Lesioni cutanee pseudo-vasculitiche 2 6Osteonecrosi 1 4Miocardiopatia cronica 1 4Trombosi glomerulare 21 38Anemia emolitica 5 16Leucopenia 3 36Stroke 22 16Trombosi arti superiori 2 0.4Valvulopatia cardiaca 37 63
Profilo SierologicoDa un punto di vista sierologico le forme primitive e quelle secondarie pre-sentano una reattività nei confronti degli antigeni cosiddetti “fosfolipidici” del tutto sovrapponibile: a) i sieri di entrambe le forme cross-reagiscono con i fosfolipidi (PL)
a carica elettrica anionica e b) riconoscono le principali PL-binding protein attualmente considerate
i veri target antigenici (ß2 glicoproteina 1 [ß2GP1], protrombina). Inoltre sia nelle PAPS sia nelle APS secondarie è stato da più gruppidescritto un identico profilo autoanticorpale costituito da reattività anticor-pali associate agli aPL ma apparentemente distinte da essi da un punto di vista di specificità antigenica: anti-proteina C/S, anti-trombomodulina, anti-kininogeno, anti-Annessina V, anti-LDL ossidate, anti-mitocondrio di tipo M5, anti-lamine nucleari, anti-endotelio, anti-eritrociti, anti-piastrine (11).La specificità e la sensibilità dei principali test diagnostici per APS (LA, aCL e anti-ß2GP1) risultano essere analoghe nelle due forme (8). Identico discorso sembra poter essere fatto anche per il valore prognosticodegli stessi test. Il LA ed alti titoli di IgG aCL mostrerebbero infatti un mag-giore peso prognostico sulla comparsa/recidiva delle manifestazioni clini-che; ancora dibattuto resta il peso prognostico della presenza di anticorpi anti-ß2GP1 (8, 12).
D’altra parte non sorprende che la prevalenza di marcatori sierologici specifici quali ANA e anti-dsDNA o le alterazioni dei livelli sierici del com-plemento sia maggiore in corso di APS associata a LES (6).

11
Una diagnosi di PAPS può essere formulata se il paziente soddisfa i criteri classificativi della APS (7) e se può essere esclusa la contemporanea pre-senza di una malattia autoimmune sistemica. La stragrande maggioranza delle forme secondarie sono state riportate in corso di LES conclamato. Quando la patologia sistemica associata non può essere formalmente clas-sificabile come tale a causa della mancanza di un numero sufficiente di cri-teri classificativi, la maggior parte degli autori parla di sindromi lupus-like. Appare quindi essenziale per una diagnosi di PAPS poter escludere un tipodi patologia simile. In pratica è esperienza comune come ciò sia difficile inconsiderazione del fatto che molte manifestazioni cliniche della APS costi-tuiscono esse stesse criteri classificativi (o manifestazioni) del LES. Queste considerazioni hanno spinto alcuni autori a suggerire specifici crite-ri di esclusione, in presenza dei quali la diagnosi di PAPS non sarebbe pos-sibile (Tabella 4) (13). Anche seguendo questi suggerimenti rimarrebbe tutta-via aperta la possibilità che il paziente possa avere una forma di passaggioquale una lupus-like APS e pertanto solo un follow-up sufficientemente lungo potrà dirimere la questione in ultima istanza.
Quando e come formulare la diagnosi di PAPS
Esiste veramente una forma primitiva?
Un follow-up maggiore di 5 anni dopo la comparsa delle prime manifestazioni è necessario per escludere un’eventuale comparsa di LES.
Tabella 4: Criteri di esclusione per la diagnosi di PAPS (13)
• Rash malare • Rash discoide• Fotosensibilità• Ulcere orali o naso-faringee (con l’esclusione di un’ulcerazione del setto nasale)• Artrite• Pleurite (in assenza di un’embolia polmonare od uno scompenso sinistro)• Pericardite (in assenza di un infarto miocardio o di un’uremia)• Proteinuria persistente (> 0.5 gr/die) dovuta a glomerulonefrite da immunocomplessi• Anticorpi anti-dsDNA (individuati con tecnica di Farr o CLIFTI• Anticorpi anti-ENA• ANA a titolo>1:320• Terapia con farmaci noti per indurre aPL• Linfopenia < 1.0/10 g/l
Sebbene vi siano solide evidenze che più della metà dei pazienti possa all’esordio manifestare solo sintomi associati agli aPL (6), è altrettanto noto in letteratura che nel lungo decorso un numero consistente sviluppi pro-gressivamente manifestazioni che consentano di formulare una diagnosi di LES o di lupus-like disease (8, 9, 13).
Vi è accordo che ciò avvenga in un lasso di tempo lungo, giustificando la necessità di un follow-up di almeno 5 anni (8, 9, 13). Mancano tuttavia studi multicentrici e sufficientemente ampi per poter documentare l’entità numeri-ca di questo fenomeno.

12
In linea con l’ipotesi che la presenza degli aPL sia espressione di una forma autoimmune potenzialmente evolventesi nel tempo, vi è la recente osservazione della comparsa di manifestazioni cliniche della sindrome in quasi la metà di pazienti con aPL e trombocitopenia idiopatica nell’arco di 38 mesi (14).
Il trattamento delle forme primitive ricalca in linea di massima quello attuatonelle forme associate ad altre malattie autoimmune sistemiche sia per quanto riguarda la terapia delle manifestazioni acute sia per quanto con-cerne la profilassi. Ciò è vero tanto per le manifestazioni trombotiche quan-to per l’abortività e le complicanze gravidiche. La differenza fondamentale risiede nell’uso di steroidi e/o di farmaci immunosoppressori necessario per il controllo della malattia di fondo nelle forme secondarie.
La descrizione relativamente recente delle forme primitive non consente al momento attuale una valutazione oggettiva della loro prognosi. Tuttavia questo sembra essere possibile nelle forme secondarie a LES. In effetti, alcuni anni fa era stato suggerito da più gruppi che la mortalità tra i pazientilupici con aPL fosse maggiore rispetto a quella dei pazienti senza aPL. Eventi tromboembolici (arteriosi e/o venosi), trombocitopenia ed anemia emolitica furono riportati quali fattori responsabili della maggiore mortalità (15, 16, 17, 18). Pazienti con LES e positività per LA avrebbero una probabilità del 50% di manifestare un evento trombotico arterioso e/o venoso in un follow-up di 20 anni (19). La terapia profilattica con aspirina sarebbe d’altra parte in grado di ridurre significativamente questo rischio (20, 21). Che l’approccio tera-peutico sia capace di migliorare la prognosi è suggerito anche da una recente analisi di Alarcon Segovia et al. in un’ampia casistica seguita per un lungo periodo di tempo (22). Questi autori hanno infatti riportato una dimi-nuzione dell’incidenza delle manifestazioni legate alla APS nel tempo ed hanno messo in relazione questo dato alla terapia profilattica con antiag-greganti e/o anticoagulanti orali. L’attuale approccio terapeutico sarebbe inoltre responsabile di una sopravvivenza a 15 anni maggiore rispetto a quella riportata per il LES in generale (22-25). Alternativamente non si può tut-tavia escludere che la prognosi migliore possa essere in relazione ad un’associazione tra APS e forme meno aggressive di LES (22).Più recentemente, la presenza di aPL è stata anche associata ad un interessamento renale su base vasculopatica e caratterizzato da ipertensione arteriosa e fibrosi interstiziale; l’interessamento renale condizionerebbe una maggiore morbidità dei pazienti con LES e aPL (23).
Una terapia diversa per la forma primitiva?
La presenza di aPL influenza il decorso delle forme secondarie?

13Bibliografia
1. Moore JE, Mohr CF. Biological false positive serologic test for syphilis,type incidence and cause. JAMA 150: 467-73; 1952.
2. Bowie WEJ, Thompson JH, Pascuzzi CA, Owen CA. Thrombosis in SLEdespite circulating anticoagulant. J Clin Invest 62: 413-30; 1963.
3. Alarcon Segovia D, Sanchez-Guerriero J. Primary antiphospholipid syndrome. J Rheumatol 16: 482-88;1989
4. Asherson RA, Khamashta MA, Ordi-Ros J, Derksen RHWM, Machin SJ, Barquinero J, Out HH, Harris EN, Villaredell-Torres M, Hughes GRV. The “primary” antiphospholipid syndrome: major clinical and serologicalfeatures. Medicine 68:366-74;1989.
5. Mackworth-Young CG, Loizou S, Walport MJ. Primary antiphospholipid syndrome: features of patients with raised anticardiolipin antibodies and no other diseases. Ann Rheum Dis 48: 362-67;1989.
6. Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, Jacobsen S, Lakos G, Tincani A, Kontopoulou-Griva I, Galeazzi M, Meroni PL, Derksen RHWM, de Groot F, Gromnica-Ihle E, Baleva M, Bombardieri S, Houssiau F, Quéré I, Gris JC, Hachulla E, Vasconcelos C,Roch C, Fernández-Nebro A, Boffa MC, Hughes GRV, and Ingelmo M on behalf of the “Euro-Phospholipid Project Group”. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthtritis Rheum in press 2002.
7. Wilson WA, Gharavi AE, Koike T, Lockshin MD, Branch DW, Piette JC, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome. Report of an international workshop. Arthritis Rheum 42: 1309-11;1999.
8. Vincent T, Mackworth-Young C. The primary antiphospholipid syndromein Hughes syndrome, antiphospholipid syndrome. Ed Khamashta MA; Springer, London, 2001; pp.111.
9. Carbone J. Orera M. Rodriguez-Mahou M. Rodriguez-Perez C. Sanchez-Ramon S. Seoane E. Rodriguez JJ. Zabay JM. Fernandez-Cruz E. Immunological abnormalities in primary APS evolving into SLE: 6 years follow-up in women with repeated pregnancy loss. Lupus. 8:274-78, 1999.
10. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725-31; 1997.
11. Tincani A, Franceschini F, Spunghi M, Panzeri P, Balestrieri G, Meroni PL.Immunological abnormalities in the antiphospholipid syndrome in the antiphospholipid syndrome; 2002 in press.
12. Merril JT. Which antiphospholipid antibody test should be used? Rheum Dis Clin North Am 27: 525-50; 2001.

14
13. Piette JC, Wechsler B, Frances C, Godeau P. Systemic lupus erythematosus and the antiphospholipid syndrome: reflection about the relevance of ARA criteria. J Rheumatol 19: 1835-37; 1993.
14. Diz-Kucukkaya R. Hacihanefioglu A. Yenerel M. Turgut M. Keskin H. Nalcaci M. Inanc M. Antiphospholipid antibodies and antiphospholipid syndrome in patients presenting with immune thrombocytopenic purpura: a prospective cohort study. Blood 98:1760-64, 2001.
15. Glueck HI, Kant KS, Weiss MA, Pollak VE, Miller MA, Coots M. Thrombosis in systemic lupus erythematosus. Relation to the presence of circulating anticoagulants. Arch Intern Med 145:1389-95, 1985.
16. Jouhikainen T, Stephansson E, Leirisalo-Repo M. Lupus anticoagulant as a prognostic marker in systemic lupus erythematosus. Br J Rheumatol 32:568-73, 1993.
17. Gulko PS. Reveille JD. Koopman WJ. Burgard SL. Bartolucci AA. Alarcon GS. Anticardiolipin antibodies in systemic lupus erythematosus: clinical correlates, HLA associations, and impact on survival. J Rheumatol 20:1684-93, 1993.
18. Drenkard C, Villa AR, Alarcon-Segovia D, Perez-Vazquez ME. Influence of the antiphospholipid syndrome in the survival of patients with systemic lupus erythematosus. J Rheumatol 21: 1067-72; 1994.
19. Petri M. Epidemiology of the antiphospholipid syndrome. J Autoimmun 15: 145-51; 2000.
20. Erkan D, Merrill JT, Yazici Y, Sammaritano L, Buyon JP, Lockshin MD. High thrombosis rate after fetal loss in antiphospholipid syndrome: effective prophylaxis with aspirin. Arthritis Rheum 44:1466-67; 2001.
21. Wahl DG, Bounameaux H, de Moerloose P, Sarasin FP. Prophylactic antithrombotic therapy for patients with systemic lupus erythematosus with or without antiphospholipid antibodies: do the benefits outweigh the risks? A decision analysis. Arch Intern Med 160:2042-48; 2000.
22. Alarcon Segovia D, Perez-Ruiz A, Villa AR. Long-term prognosis of antiphospholipid syndrome in patients with systemic lupus erythematosus.J Autoimmun 15: 157-61; 2000.
23. Nochy D, Daugas E, Du Le Thi Huong, Piette JC, Hill G. Kidney involvement in the antiphospholipid syndrome. J Autoimmun 15: 127-32; 2000.
24. Urowitz MB, Gladman DD. How to improve morbidity and mortality in systemic lupus erythematosus. Rheumatology 39: 238-44; 2000.
25. Drenkard C, Alarcon Segovia D. The new prognosis of systemic lupus erythematosus and decreased mortality and morbidity. Israel Med Assoc 2: 232-34; 2000.
Corrispondenza: PL Meroni Unità di Allergologia e Immunologia Clinica, IRCCS Istituto Auxologico ItalianoVia L. Ariosto, 13 - 20145 Milano - Fax 02-58211-559 e-mail:[email protected]

15
Monica Galli
U.S. Emostasi e Trombosi, U.O. Ematologia, Ospedali Riuniti, Bergamo
Fisiopatologia e possibili meccanismi di trombogenesi degli anticorpi antifosfolipidi

16
La Sindrome da anticorpi antifosfolipidi (APS) è un disordine acquisitodi origine ignota, caratterizzato da trombosi arteriose e/o venose ecomplicanze della gravidanza che si associano alla presenza nel
sangue degli anticorpi antifosfolipidi (aPL) (1). Gli aPL allungano i tempi di coagulazione dei tests fosfolipide-dipendenti della coagulazione (si parla in questo caso di anticoagulante tipo lupus, LAC) (2), oppure sono evidenziati mediante metodiche ELISA che utilizzano la cardiolipina o altri fosfolipidi a carica netta negativa come antigeni in fase solida (anticorpi anticardiolipina, aCL) (3). In realtà, gli aPL non reagiscono direttamente con i fosfolipidi, bensì sono diretti contro proteine plasmatiche che hanno affinità per le superfici a cari-ca netta negativa. Tra queste proteine, le più importanti sono la ß2-glicopro-teina 1 (ß2GP1) (4) e la protrombina (PT) (5), che sono il bersaglio antigenicodella maggior parte degli aPL. Queste proteine sono trattate estensivamen-te in altre parti di questo libro. In questo capitolo ci occuperemo degli altri bersagli antigenici degli aPL, dei meccanismi di trombogenesi e dei modelli sperimentali di APS.
Introduzione

17
Antigeni degli aPL
Proposti meccanismi di trombogenesi
Gli antigeni degli aPL sono indicati nella Tabella 1 (6-12). Poichè la maggior parte di queste proteine sono coinvolte nella regolazionedella coagulazione del sangue, è verosimile che anticorpi capaci di ridurre la loro concentrazione plasmatica e/o di interferire con le loro funzioni possano produrre uno squilibrio dei sistemi pro- ed anti-coagulanti.Questo rappresenta il razionale dell’aumentato rischio trombotico dei pazienti con aPL. I dati disponibili riguardo alla prevalenza e al significato clinico di anticorpi diversi dal LAC, aCL, anti-ß2GP1 ed aPT sono piuttosto limitati e derivano, in genere, da studi retrospettivi su piccoli gruppi di pazienti. Uno degli studi più ampi è stato recentemente pubblicato da Nojima e col-laboratori (13) su 168 pazienti affetti da lupus eritematoso sistemico: la preva-lenza degli anticorpi diretti contro la proteina C, la proteina S e l’annessina V era compresa da 21 e 56% quando i tests ELISA erano eseguiti con pia-stre gamma-irradiate. La rilevanza clinica di questi dati era, peraltro, mode-sta, poiché solo gli anticorpi anti-proteina S risultavano associati alle trom-bosi venose.
Le ipotesi via via suggerite per spiegare la trombogenesi nell’APS sono indicate nella Tabella 2.
Interferenza con il sistema anticoagulante della proteina CIl sistema della proteina C è uno dei principali sistemi di controllo della coagulazione del sangue. Difetti qualitativi e/o quantitativi della proteina C e del suo cofattore, la proteina S, sono associati ad aumentato rischio di trombosi venose ed embolie polmonari (14). Gli aPL sono in grado di inibire l’inattivazione del fattore V attivato da parte della proteina C attivata (aPC) su una superficie fosfolipidica (15). Il termine “resistenza acquisita” all’aPC identifica questa condizione, che potrebbe spiegare, almeno in parte, l’aumentato rischio di trombosi venose dei pazienti con aPL.Il nostro gruppo (15) ha studiato l’inattivazione del fattore Va nel plasma di 42pazienti con aPL, dimostrando che in 26 di loro (62%) rimanevano livelli piùalti di fattore Va rispetto ai controlli. In un sistema plasmatico abbiamo, inol-tre, dimostrato la capacità degli anti-ß2GP1 di inibire l’inattivazione del fatto-re Va da parte del sistema della proteina C endogena.
Tabella 1. Antigeni degli aPL
• ß2 glicoproteina 1• Protrombina• Proteina C (attivata)• Proteina S• Trombomodulina• Annessina V• Attivatore tissutale del plasminogeno• Chininogeni a basso ed alto peso molecolare• Fattore XII della coagulazione• Lipoproteine a bassa densita’ ossidate (ox-LDL)

18
Interazione con l’annessina VL’annessina V è un potente inibitore fisiologico della coagulazione che, in presenza in presenza di ioni calcio, forma una struttura cristallina bidimensio-nale sulla superficie fosfolipidica. In tal modo riesce a dislocare i fattori della coagulazione ed esercita anche un ruolo di protezione dei meccanismi di apoptosi. Gli aPL in presenza di ß2GP1 sono risultati in grado di dislocarel’annessina V dalla superficie fosfolipidica, rendendola, pertanto, nuovamentedisponibile per i fattori della coagulazione (16). Inoltre, alcuni aPL che reagisco-no con l’annessina V inducono apoptosi delle cellule endoteliali (17).
Effetti sulle cellule endotelialiGli aPL sono in grado di riconoscere, danneggiare e/o attivare le cellule endoteliali (18). Cellule endoteliali incubate con aPL e ß2GP1 esprimono livelli aumentati di molecole di adesione (19), possono aumentare l’adesione leuco-citaria e stimolare i processi di flogosi e trombosi. Questi effetti sono verosi-milmente mediati da molecole quali ICAM-1, VICAM-1, P-selettina, come sug-gerito da un modello di topo carente di ICAM-1 e P-selettina. Pazienti aPL-positivi con trombosi arteriosa esprimono livelli aumentati di endotelina-1 (20). Si tratta di una molecola il cui ruolo fisiologico non è ancora ben definito, mache potrebbe essere implicata nei processi di regolazione del tono arteriosoe del vasospasmo. Anticorpi LAC sono risultati capaci di stimolare il “release”di miscrovescicole da parte delle cellule endoteliali (21).
Induzione del fattore tissutaleGli aPL sono in grado di stimolare la sintesi leucocitaria di fattore tissutale (22).Dopo appropriata stimolazione, i monociti isolati da pazienti con APS, ma nonquelli di pazienti aPL-positivi senza complicanze trombotiche, producevano livelli aumentati di fattore tissutale (22). Ciò richiedeva la presenza di linfociti CD 4+ e molecole del sistema maggiore di istocompatibilita’ di classe II. In uno studio, la capacità degli aPL di stimolare l’espressione di fattore tissu-tale era associata a ridotti livelli di proteina S libera ed aumento di alcuni mar-catori di stato protrombotico (23). Gli aPL possono aumentare il fattore tissutaleanche inibendo l’attività del TFPI (tissue factor pathway inhibitor) (24).
Tabella 2. Proposti meccanismi di trombogenesi nell’APS
• Interferenza con meccanismi antitrombotici dipendenti dai fosfolipidi
Interferenza con il sistema anticoagulante della proteina C;
Inibizione del TFPI (“tissue factor pathway inhibitor”);
Esposizione di fosfolipidi anionici a seguito della dislocazione dell’annessina V;
Ridotta fibrinolisi a seguito della riduzione dell’autoattivazione del fattore XII della coagulazione fosfolipide-dipendente;
Inibizione dei complessi eparina-antitrombina.
• Stimolazione della sintesi/esposizione del fattore tissutale su monociti e cellule endoteliali
• Danno vascolare/induzione di apoptosi
• Promozione dell’adesione cellulare a superfici vascolari
• Attivazione piastrinica/Rilascio di miscrovescicole
• Cross-reattivita’ con ox-LDL
• Aumento dell’endotelina 1
• Alterazione della sintesi degli eicosanoidi
• Aumento del PAI-I (“plasminogen activator inhibitor-1”)

19
Effetto sulle piastrine e sul metabolismo degli eicosanoidiAlcuni ricercatori hanno dimostrato la presenza di piastrine attivate nel san-gue dei pazienti con APS (25), e che gli aPL possono stimolare l’aggregazio-ne piastrinica (26) e favorirne l’agglutinazione (27). Gli aPL possono alterare l’equilibrio della sintesi degli eicosanoidi in senso protrombotico, come sug-gerito dall’aumentata escrezione urinaria dei metaboliti del trombossano (28).Peraltro, altri ricercatori non hanno confermato questi dati (29).
Inibizione dell’antitrombinaL’antitrombina e’ un inibitore fisiologico della coagulazione, la cui azione è accelerata dall’eparina. I pazienti congenitamente carenti di antitrombina sono ad elevato rischio di complicanze trombotiche venose. E’ stato dimostrato che in alcuni casi gli aPL cross-reagiscono con l’eparinae con sostanze eparino-simili, in tal modo inibendone il loro effetto di acce-lerazione dell’azione dell’antitrombina (30).
Altri effettiGli aPL possono cross-reagire con le ox-LDL (7), stimolando, in tal modo, il processo di aterogenesi. Inoltre, è stato suggerito anche che gli aPL posso-no interferire con la fibrinolisi. Infatti, livelli aumentati di PAI-I (“plasminogenactivator inhibitor-I”) sono stati trovati in donne aPL-positive (31). Inoltre, gli anti-ß2GP1 possono inibire l’autoattivazione del fattore XII (32), per-ciò riducendo la callicreina e l’urochinasi. Infine, mutazioni genetiche - qualila mutazione G1691A del gene del fattore V e la mutazione G20210A del gene della protrombina, la cui presenza aumenta il rischio di trombosi venose – possono contribuire a definire il rischio trombotico nell’APS (33).
Diversi modelli murini di APS suggeriscono che gli aPL possano svolgere un ruolo causativo nello sviluppo delle trombosi e delle complicanze ostetri-che. Infatti, l’immunizzazione con ß2GP1 (34) o con aPL (35) comporta un aumentato riassorbimento fetale (l’equivalente murino della poliabortività), mentre la somministrazione di un anticorpo monoclonale umano derivato da un paziente con APS provoca trombosi nei topi (36). Topi sottoposti ad infusione endovenosa di aPL e successivamente a danno della vena femo-rale sviluppano trombi nella sede di ingiuria vascolare, che sono di dimen-sioni maggiori rispetto agli animali di controllo (37). In un modello di atero-sclerosi murina (topi “knock out” per il gene del recettore delle LDL) l’immu-nizzazione con aCL umani accelera i processi di aterosclerosi (38), fornendoun’ulteriore prova della patogenicità degli aPL.Una relazione causa-effetto diretta tra gli aPL e le complicanze trombotichee ostetriche non è ancora stata data negli esseri umani. Tuttavia, la recentecaratterizzazione della ß2GP1 nello scimpanzé, insieme al riscontro di un’elevata prevalenza di anticorpi anti-ß2GP1 in questi animali (39) offre la possibilità di studiare l’APS in un modello animale più simile all’uomo.
In condizioni di laboratorio particolari, gli aPL esercitano numerosi effetti a causa dei molteplici processi biologici che coinvolgono i fosfolipidi e le mem-brane fosfolipidiche. E’, peraltro, difficile stabilire quali di questi effetti siano biologicamente rilevanti, basti pensare, ad esempio, all’effetto paradosso delLAC sui tests fosfolipide-dipendenti della coagulazione. Perciò, la rilevanza clinica di ogni proposto meccanismo d’azione basato su studi “in vitro” deve essere validato mediante modelli animali e studi clinici ben disegnati.
Modelli animali di APS
Conclusioni

20
1. Wilson WA, Gharavi AE, Koike T et al. International consensus on preliminary classification criteria for definite antiphospholipid syndrome. Arthritis Rheum 1999;42:1309-1311
2. Mackie IJ, Donohoe S, Machin SJ. Lupus anticoagulant measurement. In: Khamashta MA ed. Hughes’ syndrome. London: Springer, 2000, 214-222
3. Loizou S, McCrea JD, Rudge AC et al. Measurement of anticardiolipin antibodies by an enzyme-linked immunosorbent assay: standardization and quantitation of results. Clin Exp Immunol 1985;62:739-744
4. Galli M, Comfurius P, Maassen C et al. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990;335:1544-1547
5. Bevers EM, Galli M, Barbui T, Comfurius P, Zwaal RFA. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost 1991;66:629-632
6. Oosting JD, Derksen RHWM, Bobbink IWG et al. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C or protein S: an explanation for their pathogenic mechanism. Blood 1993;81:2618-2625
7. Vaarala O, Alfthan G, Jauhianen M et al. Cross-reaction between antibodies to oxidized lipoprotein and to cardiolipin in systemic lupus erythematosus. Lancet 1993;341:923-925
8. Matsuda J, Saitoh N, Goihci K et al. Anti-annexin V antibody in systemic lupus erythematosus patients with lupus anticoagulant and/or anticardiolipin antibodies. Am J Hematol 1994; 47:56-58
9. Carson CW, Comp PC, Esmon NL et al. Thrombomodulin antibodies inhibit protein C activation and are found in patients with lupus anticoagulant and unexplained thrombosis [Abstract]. Arthritis Rheum 1994;37:S296
10. Sugi T, McIntyre JA. Autontibodies to phosphatidylethanolamine (PE) recognize a kininogen-PE complex. Blood 1995;86:3083-3089
11. Jones DW, Gallimore MJ, Harris SL, Winter M. Antibodies to factor XII associated with lupus anticoagulant. Thromb Haemost 1999;81:387-390
12. Cugno M, Dominguez M et al. Antibodies to tissue-type plasminogen activator in plasma from patients with primary antiphospholipid syndrome.Br J Haematol 2000;108:871-875
13. Nojima J, Kuratsune H, Suehisa E et al. Association between the prevalence of antibodies to ß2-glycoprotein I, prothrombin, protein C, protein S, and annexin V in patients with systemic lupus erythematosus and thrombotic and thrombocytopenic complications. Clin Chem 2001;47:1008-1015
Bibliografia

21
14. De Stefano V, Finazzi G, Mannucci PM. Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood 1996;87:3531-3544
15. Galli M, Ruggeri L, Barbui T. Differential effects of anti-ß2-glycoprotein 1 and antiprothrombin antibodies on the anticoagulant activity of activatedprotein C. Blood 1998;91:1999-2004
16. Rand JH, Wu XX, Giesen P. A possible solution to the paradox of the “lupus anticoagulant”: antiphospholipid antibodies accelerate thrombin generation by inhibiting annexin-V. Thromb Haemost 1999; 82:1376-1377
17. Rand JH. Molecular pathogenesis of the antiphospholipid syndrome. Circ Res 2002;90:29-37
18. Dueymes M, Levy Y, Ziporen L et al. Do some antiphospholipid antibodies target endothelial cells? Ann Med Interne (Paris) 1996;147 (suppl1):22-23
19. Meroni PL, Raschi E, Camera M et al. Endothelial activation by aPL: a potential pathogenetic mechanism for the clinical manifestations of the syndrome. J Autoimmun 2000;15:237-240
20. Atsumi T, Khamashta MA, Haworth RS et al. Arterial disease and thrombosis in the antiphospholipid syndrome: a pathogenic role for endothelin 1. Arthritis Rheum 1998;41:800-807
21. Combes V, Simon AC, Grau GE et al. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest 1999;104:93-102
22. Martini F, Farsi A, Gori Am et al. Antiphospholipid antibodies (aPL) increase the potential monocyte procoagulant activity in patients with systemic lupus erythematosus. Lupus 1996;5:206-211
23. Visvanathan S, Gecy CL, Harmer JA, McNeil HP. Monocyte tissue factorinduction by activation of ß2-glycoprotein I-specific T lymphocytes is associated with thrombosis and fetal loss in patients with antiphospholipidantibodies. J Immunol 2000;165:2258-2262
24. Salemink I, Willems GM, Galli M et al. Antibodies to ß2-glycoprotein 1 from patients with antiphospholipid syndrome suppress the inhibitory activity of tissue factor pathway inhibitor. Thromb Haemost 2000; 84:653-656
25. Emmi L, Bergamini C, Spinelli A et al. Possible pathogenetic role of activated platelets in the primary antiphospholipid syndrome involving the central nervous system. Ann N Y Acad Sci 1997;823:188-200
26. Campbell AL, Pierangeli SS, Welhausen S, Harris EN. Comparison of the effects of anticardiolipin antibodies from patients with the antiphospholipid syndrome and with syphilis on platelet activation and aggregation. Thromb Haemost 1995;73:529-534

22
27. Wiener MH, Burke M, Fried M, Yust I. Thromboagglutination by anticardiolipin antibody complex in the antiphospholipid syndrome. A possible explanation of immune-mediated thrombosis. Thromb Res 2001;103:193-199
28. Lellouche F, Martinuzzo M, Said P et al. Imbalance of thromboxane/prostacyclin byosinthesis in patients with lupus anticoagulant. Blood 1991; 78:2894-2899
29. Hasselaar P, Derksen RHWM, Blokzijl L, de Groot PG. Thrombosis associated with antiphospholipid antibodies cannot be explained by effects on endothelial and platelet prostanoid synthesis. Thromb Haemost 1988;59:80-85
30. Shibata S, Harpel PC, Gharavi A et al. Autoantibodies to heparin from patients with antiphospholipid antibody syndrome inhibit formation of antithrombin III-thrombin complexes. Blood 1994;83:2532-2540
31. Ames PR, Tommasino C, Iannaccone L et al. Coagulation activation and fibrinolytic imbalance in subjects with idiopathic antiphospholipid antibodies – a crucial role for acquired free protein S deficiency. Thromb Haemost 1996; 76:190-194
32. Schousboe I, Rasmussen MS. Synchronized inhibition of the phospholipidmediated autoactivation of factor XII in plasma by ß2-glycoprotein 1 and anti-ß2 glycoprotein 1. Thromb Haenost 1995; 73: 798-804
33. Galli M, Finazzi G, Duca F et al. The G1691A mutation of factor V gene, but not the G20210A mutation of factor II gene and the C677T mutation of the methylenetetrahydrofolate reductase gene, is associated with venous thrombosis in patients with lupus anticoagulants. Br J Haematol 2001;108:865-870
34. Garcia CO, Kanbour-Shakir A, Tang H et al. Induction of experimental antiphospholipid antibody syndrome in PL/J mice following immunizationwith ß2GP1. Am J Reprod Immunol 1997;37:118-124
35. Blank M, Faden D, Tincani A et al. Immunization with anticardiolipin cofactor (ß2-glycoprotein I) induces experimental antiphospholipid syndrome in naïve mice. J Autoimmun 1994;7:441-455
36. Olee T, Pierangeli SS, Handley HH et al. A monoclonal IgG anticardiolipinantibody from a patient with the antiphospholipid syndrome is thrombo-genic in mice. Proc Natl Acad Sci USA 1996;93:8606-8611
37. Pierangeli SS, Harris EN. In vivo models of thrombosis for the antiphospholipid syndrome. Lupus 1996;5:451-455
38. George J, Afek A, Gilbrud B et al. Atherosclerosis in LDL-receptor knockout mice is accelerated by immunization with anticardiolipin antibodies. Lupus 1997;6:723-729
39. Sanghera DK, Nestelrode CS, Ferrell RE, Kamboh MI. Chimpanzee apolipoprotein H (ß2-glycoprotein 1). Report of the gene structure, a common polymorphism, and a high prevalence of antiphospholipid antibodies. Hum Genet 2001;109:63-72

23
Guido Finazzi
U.S. Emostasi e Trombosi, U.O. Ematologia, Ospedali Riuniti, Bergamo
Aspetti clinici e terapeutici delle trombosi arteriose e venose

24
Le trombosi arteriose e venose rappresentano l’evento clinico più fre-quente e clinicamente rilevante della sindrome da anticorpi antifosfoli-pidi (aPL) (1). Dati epidemiologici indicano che circa il 30-40% dei
pazienti con gli anticorpi ha una storia di trombosi, venosa nel 70% e arte-riosa in circa il 30% dei casi (2). Le trombosi profonde degli arti inferiori, con o senza embolia polmonare, sono gli eventi venosi più frequenti, mentre il circolo cerebrale è la sede piùcomune delle occlusioni arteriose. Le trombosi tendono a recidivare, tipicamente nello stesso distretto del primo evento, e pertanto richiedono una attenta valutazione prognostica e terapeutica. In questo capitolo, prenderemo in esame i fattori di rischio cli-nici e di laboratorio per lo sviluppo di eventi vascolari e discuteremo le opzioni terapeutiche per il trattamento di questi pazienti.
Introduzione
25
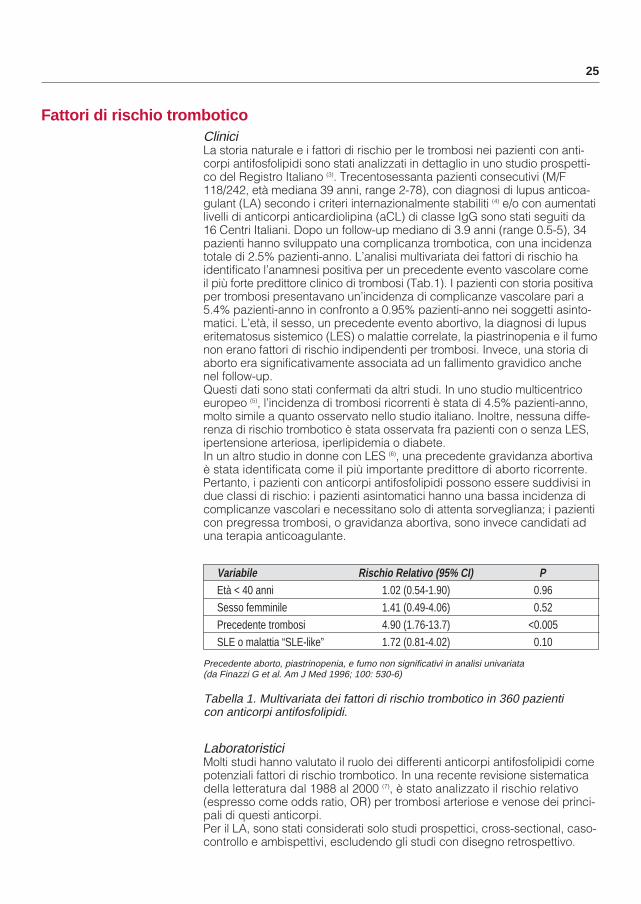
Clinici La storia naturale e i fattori di rischio per le trombosi nei pazienti con anti-corpi antifosfolipidi sono stati analizzati in dettaglio in uno studio prospetti-co del Registro Italiano (3). Trecentosessanta pazienti consecutivi (M/F 118/242, età mediana 39 anni, range 2-78), con diagnosi di lupus anticoa-gulant (LA) secondo i criteri internazionalmente stabiliti (4) e/o con aumentatilivelli di anticorpi anticardiolipina (aCL) di classe IgG sono stati seguiti da 16 Centri Italiani. Dopo un follow-up mediano di 3.9 anni (range 0.5-5), 34 pazienti hanno sviluppato una complicanza trombotica, con una incidenza totale di 2.5% pazienti-anno. L’analisi multivariata dei fattori di rischio ha identificato l’anamnesi positiva per un precedente evento vascolare come il più forte predittore clinico di trombosi (Tab.1). I pazienti con storia positivaper trombosi presentavano un’incidenza di complicanze vascolare pari a 5.4% pazienti-anno in confronto a 0.95% pazienti-anno nei soggetti asinto-matici. L’età, il sesso, un precedente evento abortivo, la diagnosi di lupus eritematosus sistemico (LES) o malattie correlate, la piastrinopenia e il fumonon erano fattori di rischio indipendenti per trombosi. Invece, una storia di aborto era significativamente associata ad un fallimento gravidico anche nel follow-up. Questi dati sono stati confermati da altri studi. In uno studio multicentrico europeo (5), l’incidenza di trombosi ricorrenti è stata di 4.5% pazienti-anno, molto simile a quanto osservato nello studio italiano. Inoltre, nessuna diffe-renza di rischio trombotico è stata osservata fra pazienti con o senza LES, ipertensione arteriosa, iperlipidemia o diabete.In un altro studio in donne con LES (6), una precedente gravidanza abortivaè stata identificata come il più importante predittore di aborto ricorrente. Pertanto, i pazienti con anticorpi antifosfolipidi possono essere suddivisi in due classi di rischio: i pazienti asintomatici hanno una bassa incidenza di complicanze vascolari e necessitano solo di attenta sorveglianza; i pazienticon pregressa trombosi, o gravidanza abortiva, sono invece candidati ad una terapia anticoagulante.
Fattori di rischio trombotico
Tabella 1. Multivariata dei fattori di rischio trombotico in 360 pazienti con anticorpi antifosfolipidi.
Variabile Rischio Relativo (95% CI) PEtà < 40 anni 1.02 (0.54-1.90) 0.96Sesso femminile 1.41 (0.49-4.06) 0.52Precedente trombosi 4.90 (1.76-13.7) <0.005SLE o malattia “SLE-like” 1.72 (0.81-4.02) 0.10
Precedente aborto, piastrinopenia, e fumo non significativi in analisi univariata(da Finazzi G et al. Am J Med 1996; 100: 530-6)
Laboratoristici Molti studi hanno valutato il ruolo dei differenti anticorpi antifosfolipidi comepotenziali fattori di rischio trombotico. In una recente revisione sistematica della letteratura dal 1988 al 2000 (7), è stato analizzato il rischio relativo (espresso come odds ratio, OR) per trombosi arteriose e venose dei princi-pali di questi anticorpi. Per il LA, sono stati considerati solo studi prospettici, cross-sectional, caso-controllo e ambispettivi, escludendo gli studi con disegno retrospettivo.

26
L’analisi di 12 studi in 1608 pazienti ha dimostrato che l’associazione del test con la trombosi era sempre statisticamente significativa con un odds ratio compreso fra 7.3 e 10.7. Peraltro, il LA è un fenomeno di laboratorio eterogeneo in termini di bersagli antigenici, di meccanismi di interferenza con i fosfolipidi della cascata coagulatoria e quindi anche di test coagulativi necessari per la diagnosi. Ne consegue che nessun singolo test coagulati-vo è al 100% sensibile e specifico per la diagnosi di LA. Ci si può pertantochiedere quale sia il singolo test coagulativo che, nell’ambito della diagnosidi LA, è il miglior predittore del rischio trombotico.Il Russell’s Viper Venom Time diluito (dRVVT) e il Kaolin Clotting Time (KCT)sono due test di coagulazione comunemente usati per la diagnosi di LA. In uno studio recente, questi due test si sono dimostrati differentemente associati al rischio di sviluppare eventi vascolari (8). Infatti, nessuna correla-zione è stata osservata tra la storia trombotica dei pazienti e il grado di positività del KCT, indipendentemente dal laboratorio dove è stato eseguitoil test e dal tipo di strumentazione usata. La stessa mancanza di associa-zione è stata osservata analizzando separatamente le trombosi arteriose e venose. Invece, la maggior parte dei tests basati sul dRVVT erano associaticon una storia di eventi vascolari quando si considerava un dRVVT ratio >1.5. Questi risultati sono in accordo con un altro studio di 100 pazienti LA-positivi seguiti per una mediana di circa 3 anni: più alto era il valore di dRVVT ratio, più alta la frequenza di trombosi (9). Peraltro, bisogna conside-rare un punto importante circa l’associazione fra grado di positività al dRVVT e rischio trombotico. I vari tipi di dRVVT commercialmente disponi-bili o preparati “in-house” dai singoli ricercatori sono diversi fra loro per composizione e concentrazione dei reagenti fosfolipidici (10). Questo può modificare la loro specificità e sensibilità diagnostica e può spiegare i risultati di alcuni studi che hanno dimostrato l’associazione fra dRVVT e trombosi con alcuni tipi di reagenti ma non con altri (11). E’ pertanto necessario un ulteriore lavoro di standardizzazione del test prima di raggiungere conclusioni definitive. Certamente, comunque, il dRVVT appare oggi come il più promettente candidato per identificare i pazienti con LA ad alto rischio trombotico.Per quanto riguarda gli anticorpi aCL, l’analisi sopra citata (7) ha valutato 14 studi (esclusi quelli retrospettivi) che hanno riportato l’OR del test nei con-fronti del rischio trombotico. L’associazione risultava statisticamente signifi-cativa solo nel 50% degli studi (OR compreso fra non significativo e 4.9). L’associazione degli aCL con la trombosi era comunque strettamente dipendente dal titolo anticorpale. Infatti, negli studi nei quali erano inclusi solo pazienti con aCL >40 U GPL l’associazione con gli eventi vascolari raggiungeva la significatività statistica con un OR compreso fra 1.5 e 10.Il significato clinico degli anticorpi anti-ß2-glicoproteina 1 (anti-ß2GP1) e anti-protrombina (aPT) è stato analizzato principalmente in studi retrospetti-vi (7). La maggioranza degli studi relativi agli anti-ß2GP1 ha dimostrato una significativa associazione con le trombosi: l’analisi cumulativa di 1506 pazienti ha dato un OR di 5.3 (4.06-6.98), indipendentemente dall’isotipo dell’anticorpo e dalla sede di trombosi. Quando le trombosi arteriose e venose venivano analizzate separatamente, solo uno studio riportava una significativa associazione tra anti-ß2GP1 di tipo IgM e trombosi arteriose (peraltro non confermata in analisi multivariata), mentre cinque studi mostravano che anticorpi anti-ß2GP1 di classe IgG conferivano un OR statisticamente significativo per trombosi venose. Dieci studi retrospettivi o caso-controllo hanno valutato l’associazione tra aPT e trombosi. Sette studi hanno trovato un OR statisticamente significativo in analisi uni-variata, ma solo in due l’associazione si è confermata in analisi multivariata.In conclusione, l’insieme di questi studi indica che il LA è il più potente pre-
27
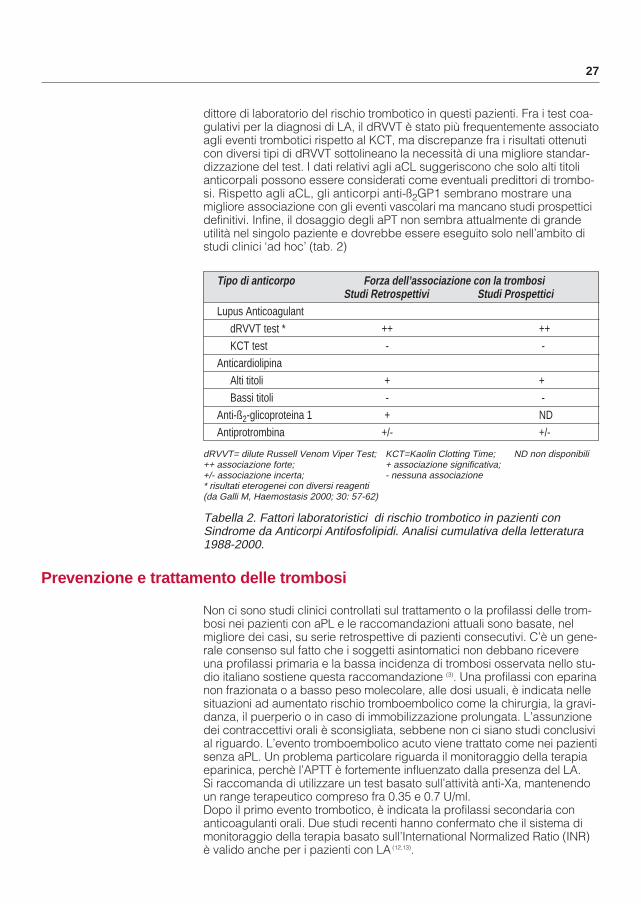
dittore di laboratorio del rischio trombotico in questi pazienti. Fra i test coa-gulativi per la diagnosi di LA, il dRVVT è stato più frequentemente associatoagli eventi trombotici rispetto al KCT, ma discrepanze fra i risultati ottenuti con diversi tipi di dRVVT sottolineano la necessità di una migliore standar-dizzazione del test. I dati relativi agli aCL suggeriscono che solo alti titoli anticorpali possono essere considerati come eventuali predittori di trombo-si. Rispetto agli aCL, gli anticorpi anti-ß2GP1 sembrano mostrare una migliore associazione con gli eventi vascolari ma mancano studi prospetticidefinitivi. Infine, il dosaggio degli aPT non sembra attualmente di grande utilità nel singolo paziente e dovrebbe essere eseguito solo nell’ambito di studi clinici ‘ad hoc’ (tab. 2)
Non ci sono studi clinici controllati sul trattamento o la profilassi delle trom-bosi nei pazienti con aPL e le raccomandazioni attuali sono basate, nel migliore dei casi, su serie retrospettive di pazienti consecutivi. C’è un gene-rale consenso sul fatto che i soggetti asintomatici non debbano ricevere una profilassi primaria e la bassa incidenza di trombosi osservata nello stu-dio italiano sostiene questa raccomandazione (3). Una profilassi con eparinanon frazionata o a basso peso molecolare, alle dosi usuali, è indicata nelle situazioni ad aumentato rischio tromboembolico come la chirurgia, la gravi-danza, il puerperio o in caso di immobilizzazione prolungata. L’assunzionedei contraccettivi orali è sconsigliata, sebbene non ci siano studi conclusivial riguardo. L’evento tromboembolico acuto viene trattato come nei pazientisenza aPL. Un problema particolare riguarda il monitoraggio della terapia eparinica, perchè l’APTT è fortemente influenzato dalla presenza del LA. Si raccomanda di utilizzare un test basato sull’attività anti-Xa, mantenendo un range terapeutico compreso fra 0.35 e 0.7 U/ml. Dopo il primo evento trombotico, è indicata la profilassi secondaria con anticoagulanti orali. Due studi recenti hanno confermato che il sistema di monitoraggio della terapia basato sull’International Normalized Ratio (INR) è valido anche per i pazienti con LA (12,13).
Tabella 2. Fattori laboratoristici di rischio trombotico in pazienti conSindrome da Anticorpi Antifosfolipidi. Analisi cumulativa della letteratura1988-2000.
Tipo di anticorpo Forza dell’associazione con la trombosi Studi Retrospettivi Studi Prospettici
Lupus AnticoagulantdRVVT test * ++ ++KCT test - -
Anticardiolipina Alti titoli + +Bassi titoli - -
Anti-ß2-glicoproteina 1 + NDAntiprotrombina +/- +/-
dRVVT= dilute Russell Venom Viper Test; KCT=Kaolin Clotting Time; ND non disponibili++ associazione forte; + associazione significativa;+/- associazione incerta; - nessuna associazione* risultati eterogenei con diversi reagenti (da Galli M, Haemostasis 2000; 30: 57-62)
Prevenzione e trattamento delle trombosi

28
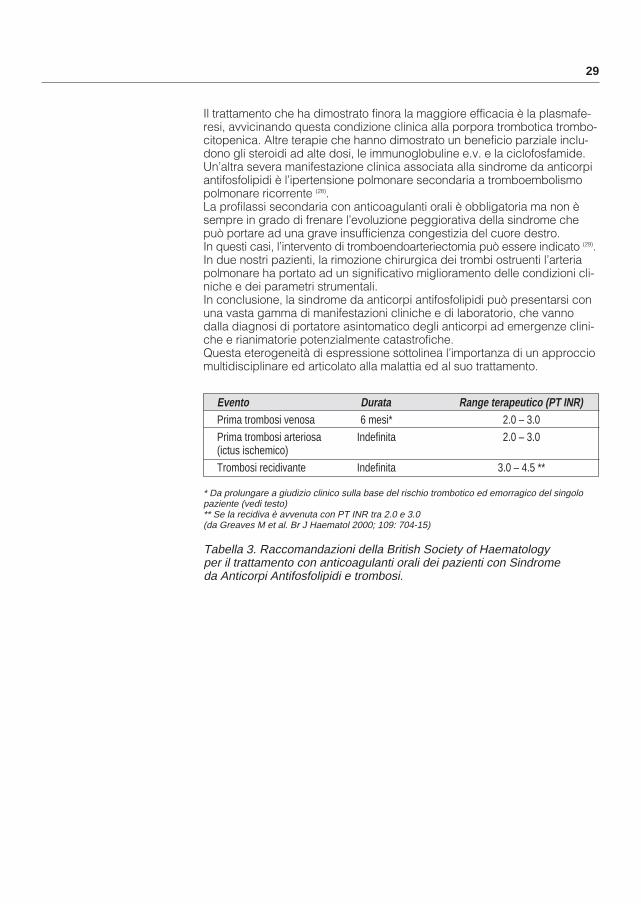
Si raccomanda, però, attenzione nell’uso di alcune tromboplastine ricombi-nanti (Innovin e Thromborel R) che hanno prodotto significative sovrastime dell’INR. La durata e l’intensità del trattamento anticoagulante orale in que-sti pazienti non sono chiaramente stabiliti. Derksen et al. (14) hanno riportato che la probabilità di non avere ricorrenze di trombosi in un periodo di otto anni era 100% nei pazienti trattati con anticoagulanti orali con INR tra 2.5 e4.0 contro 22% tra i pazienti che avevano sospeso la warfarina. Rosove et al. (15) hanno osservato in 70 pazienti che la terapia anticoagulante orale ad alta intensità (PT INR >3) conferiva una migliore protezione antitrombotica (0 eventi per anno) rispetto a quella ad intensità intermedia (INR 2-2.9; 7% eventi per anno), bassa (INR<1.9; 57% eventi per anno), all’aspirina (32% eventi per anno) o a nessun trattamento (19% eventi per anno), ma cinque pazienti hanno presentato eventi emorragici rilevanti con la warfarina ad alte dosi (3.1% eventi per anno). Questi risultati sono stati confermati in uno studio di 147 pazienti (16). Il trattamento con warfarina ad alta intensità(INR>3), con o senza aspirina, è stato significativamente più efficace che il trattamento con warfarina a bassa intensità (INR<3) o con aspirina da sola nella prevenzione di ulteriori eventi tromboembolici (incidenza di recidive 1.3%, 23% e 18% per anno, rispettivamente). Emorragie gravi sono state osservate in 29 pazienti durante la terapia anticoagulante orale (7.1% per anno). Questi studi suggerirebbero che i pazienti con aPL che abbiano presentatoun evento trombotico maggiore dovrebbero ricevere una terapia anticoagu-lante orale a lungo termine e ad alte dosi (INR >3.0). Peraltro, diversi Autorihanno sollevato serie preoccupazioni circa il rischio di raccomandare tale terapia sulla base solo di studi retrospettivi e non randomizzati (17-19).Emorragie cerebrali fatali sono possibili in corso di terapia anticoagulante orale e la probabilità cumulativa di sanguinamenti gravi aumenta sia con la durata che con l’intensità del trattamento. Inoltre, in studi più recenti, anchese su casistiche più limitate, il trattamento ad intensità convenzionale (PT INR 2.0–3.0) è risultato efficace, soprattutto nella prevenzione del trom-boembolismo venoso (20-24).Pertanto, il rischio-beneficio della terapia anticoagulante orale ad alte dosi e a lungo termine in questi pazienti è ancora incerto e richiede studi pro-spettici e controllati prima che venga raccomandata. Uno studio clinico controllato multicentrico in grado di dare una risposta a questo problema si è recentemente concluso e i dati dovrebbero essere disponibili entro l’anno 2002 (studio WAPS, Warfarin in AntiPhospholipid Syndrome) (25). Nel frattempo, la decisione sulla durata e l’intensità della profilassi con anticoagulanti orali deve essere presa su base individuale tenendo in considerazione i fattori di rischio trombotico e quelli di compli-canze emorragiche del singolo paziente. Le raccomandazioni suggerite dalla British Society of Haematology a questo proposito sono riassunte nella Tabella 3 (26).
Due complicanze particolarmente gravi della sindrome da aPL meritano di essere segnalate a parte. La prima è la rara “catastrophic antiphospholipid syndrome” (27), caratterizzata da una microangiopatia trombotica multidi-strettuale, che si associa ad insufficienza cardiaca e respiratoria ed una mortalità di circa il 50%. Circa il 30% dei pazienti presenta anche evidenza di coagulazione intravascolare disseminata. La patogenesi di questa complicanza è incerta, mentre sono noti alcuni fat-tori scatenanti fra i quali, infezioni, traumi chirurgici, farmaci o la sospensio-ne del trattamento anticoagulante.
Aspetti clinici particolari
29
Il trattamento che ha dimostrato finora la maggiore efficacia è la plasmafe-resi, avvicinando questa condizione clinica alla porpora trombotica trombo-citopenica. Altre terapie che hanno dimostrato un beneficio parziale inclu-dono gli steroidi ad alte dosi, le immunoglobuline e.v. e la ciclofosfamide.Un’altra severa manifestazione clinica associata alla sindrome da anticorpi antifosfolipidi è l’ipertensione polmonare secondaria a tromboembolismo polmonare ricorrente (28). La profilassi secondaria con anticoagulanti orali è obbligatoria ma non è sempre in grado di frenare l’evoluzione peggiorativa della sindrome che può portare ad una grave insufficienza congestizia del cuore destro. In questi casi, l’intervento di tromboendoarteriectomia può essere indicato (29).In due nostri pazienti, la rimozione chirurgica dei trombi ostruenti l’arteria polmonare ha portato ad un significativo miglioramento delle condizioni cli-niche e dei parametri strumentali.In conclusione, la sindrome da anticorpi antifosfolipidi può presentarsi con una vasta gamma di manifestazioni cliniche e di laboratorio, che vanno dalla diagnosi di portatore asintomatico degli anticorpi ad emergenze clini-che e rianimatorie potenzialmente catastrofiche. Questa eterogeneità di espressione sottolinea l’importanza di un approcciomultidisciplinare ed articolato alla malattia ed al suo trattamento.
Tabella 3. Raccomandazioni della British Society of Haematology per il trattamento con anticoagulanti orali dei pazienti con Sindrome da Anticorpi Antifosfolipidi e trombosi.
Evento Durata Range terapeutico (PT INR)Prima trombosi venosa 6 mesi* 2.0 – 3.0Prima trombosi arteriosa Indefinita 2.0 – 3.0(ictus ischemico)Trombosi recidivante Indefinita 3.0 – 4.5 **
* Da prolungare a giudizio clinico sulla base del rischio trombotico ed emorragico del singolo paziente (vedi testo)** Se la recidiva è avvenuta con PT INR tra 2.0 e 3.0(da Greaves M et al. Br J Haematol 2000; 109: 704-15)

30
1. Greaves M. Antiphospholipid antibodies and thrombosis. Lancet 1999; 353: 1348-53.
2. Finazzi G. The epidemiology of the antiphospholipid syndrome: who is at risk ? Curr Rheumat Rep 2001; 3: 271-6
3. Finazzi G, Brancaccio V, Moia M et al. Natural history and risk factors for thrombosis in 360 patients with antiphospholipid antibodies. A four-year prospective study from the Italian Registry. Am J Med 1996; 100: 530-6.
4. Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants:an update. Thromb Haemost 1995; 74:1185-90
5. Vianna JL, Khamashta MA, Ordi-Ros J et al. Comparison of the primary and secondary antiphospholipid syndrome: a European multicenter study of 114 patients. Am J Med 1994; 96: 3-9.
6. Ramsey-Goldman, R, Kulzer JE, Kuller LH, Guzick D, Carpenter AB, Medsger TA jr. Pregnancy outcome and anti-cardiolipin antibody in women with systemic lupus erythematosus. Am J Epidemiol 1993; 138: 1057-69.
7. Galli M. Which antiphospholipid antibodies should be measured in the antiphospholipid syndrome ? Haemostasis 2000; 30 (suppl.2): 57-62.
8. Galli M, Dlott J, Norbis F, Ruggeri L, Cler L, Triplett DA, Barbui T. Lupus anticoagulant and thrombosis: clinical association of different coagulation and immunologic tests. Thromb Haemostas 2000; 84: 1012-6.
9. Galli M, Finazzi G, Norbis F, Marziali S, Marchioli R, Barbui T. The risk of thrombosis in patients with lupus anticoagulants is predicted by their specific coagulation profile. Thromb Haemost 1999; 81: 695-700
10. Foster WM, Triplett DA, Barna LK. Evaluation of 7 dilute Russell’s viper venom time (dRVVT) kits. Lupus 1994; 3:351
11. Norbis F, Barbui T, Galli M. Comparison of two Russell’s viper venom time (dRVVT) for distinguishing the phospholipid-dependent inhibitors of coagulation. Thromb Haemostas 1997 (suppl.) 331
12. Robert A, Le Querrec A, Delahousse B et al. Control of oral anticoa-gulation in patients with the antiphospholipid syndrome. Influence of the lupus anticoagulant on international normalized ratio. Thromb Haemost 1998; 80: 99-103.
13. Tripodi A, Chantarangkul V, Clerici MG et al. Laboratory control of oral anticoagulant treatment by the INR system in patients with the antiphospholipid syndrome and lupus anticoagulant. Results of a collaborative study involving nine commercial thromboplastins. Br J Haematol 2001; 115: 672-8.
Bibliografia

31
14. Derksen RHWM, de Groot PG, Kater L et al. Patients with antiphospholipidantibodies and venous thrombosis should receive long term anticoagu-lant treatment. Ann Rheum Dis 1993; 52: 689-92.
15. Rosove MH, Brewer PMC. Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Int Med 1992; 117: 303-8.
16. Khamashta MA, Cuadrado MJ, Mujic F et al. The management of thrombosis in the antiphopsholipid syndrome. N Engl J Med 1995; 332: 993-7.
17. Nasr SZ, Parke AL. Thrombosis in the antiphospholipid syndrome. N Engl J Med 1995; 333: 666 (letter).
18. Slivka A, Walz E. Thrombosis in the antiphospholipid syndrome. N Engl J Med 1995; 333: 665-6 (letter).
19. Al-Sayegh FA, Ensworth S, Huang S, Stein HB, Klinkhoff AV. Hemorrhagic complications of longterm anticoagulant therapy in 7 patients with systemic lupus erythematosus and antiphospholipid syndrome. J Rheumatol 1997; 24: 1716-8.
20. Ginsberg JS, Wells PS, Brill-Edwards P, Donovan D, Moffatt K, Johnston M, Stevens P, Hirsh J. Antiphospholipid antibodies and venous thromboembolism. Blood 1995; 86: 3685-91.
21. Prandoni P, Simioni P, Girolami A. Antiphospholipid antibodies, recurrent thromboembolism and intensity of warfarin anticoagulation. Thromb Haemostas 1996; 75: 859.
22. Rance A, Emmerich J, Fiessinger JN. Antiphospholipid antibodies and recurrent thromboembolism. Thromb Haemostas 1997; 77: 221-2.
23. Schulman S, Svenungsson E, Granqvists et al. Anticardiolipin antibodiespredict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Am J Med 1998; 104: 332-338.
24. Gonzales-Trujillo JL, Villegas-Jimenez A, Rios-Luna N, Cabral AR, Cesarman G, Alarcon-Segovia D. Conventional intensity may be as effective as high intensity oral anticoagulation in the antiphospholipid syndrome. J Autoimmun 2000; 15: A22
25. Finazzi G The Italian Registry of antiphospholipid antibodies. Haematologica 1997; 82: 101-105.
26. Greaves M, Cohen H, Machin SJ, Mackie I. Guidelines on the investigationand management of the antiphospholipid syndrome. Br J Haematol 2000; 109: 704-15
27. Asherson RA. The catastrophic antiphospholipid syndrome, 1998. A review of the clinical features, possible pathogenesis and treatment. Lupus 1998; 7 (Suppl.2): S55-S62.

32
28. Martinuzzo ME, Pombo G, Forastiero RR, Cerrato GS, Colorio CC, Carrerars LO. Lupus anticoagulant, high levels of anticardiolipin, and anti-ß2 glycoprotein 1 antibodies are associated with chronic thromboembolic pulmonary hypertension. J Rheumatol 1998; 25: 1313-9.
29. Sandoval J et al. Primary antiphospholipid syndrome presenting as chronic thromboembolic pulmonary hypertension. Treatment with thromboendoarteriectomy. J Rheumatol 1996; 2: 772-5.
Corrispondenza: Dr. Guido FinazziUnità Semplice Emostasi e Trombosi, Unità Operativa Ematologia, Ospedali Riuniti, Largo Barozzi 1, 24128 Bergamo, Italy. Tel 035 269492. Fax: 035 266667. E-mail: [email protected]

33
Tincani A., Taglietti M., Biasini Rebaioli C., Frassi M., Gorla R., Balestrieri G.,Meroni P.L.*, Motta M.#, Zatti S, Lojacono A.°, Faden D.°
Servizio di Reumatologia, Allergologia e Immunologia Clinica; # Divisione di Neonatologia e Terapia Intensiva Neonatale; ° Divisione di Ostetricia e Ginecologia, Spedali Civili di Brescia; * Unità di Allergologia e Immunologia Clinica,
Dipartimento di Medicina Interna, Università degli Studi di Milano, IRCCS Istituto Auxologico Italiano, Milano.
Aspetti clinico-terapeutici delle complicanze ostetriche

34
Storicamente la prima descrizione di Sindrome da Antifosfolipidi (APS)risale a più di 20 anni fa, quando fu segnalata la positività del LupusAnticoagulant (LA) in pazienti con episodi trombotici e ripetute perdite
fetali nel periodo centrale della gravidanza(1) .Dagli anni ’80 a oggi sono state approntate varie metodiche per lo studio dei così detti anticorpi antifosfolipidi (aPL) basate su metodi ELISA e non funzionali come il LA: il test per anticardiolipina (aCL)(2), per anti-ß2 glicopro-teina 1 (anti-ß2GP1)(3), per anti-protrombina (aPT)(4) ed altri ancora. Il test per gli anticorpi anticardiolipina con le sue caratteristiche di metodo a largo impiego ha svolto una funzione “trainante” rendendo possibile studiepidemiologici numericamente significativi. Attraverso questi studi è statopossibile identificare la sindrome, che comprendeva nei criteri clinici la patologia ostetrica fino dalla sua prima descrizione(5).In questi 20 anni, le numerose osservazioni raccolte, gli studi clinici, i proto-colli applicati hanno portato al concetto moderno che gli anticorpi antifosfo-lipidi rappresentino una causa curabile di aborto o di perdita fetale(6).
Introduzione

35
I dati della letteratura sono oggi concordi nel ritenere che la presenza di aPL, comunque determinati, sia associata a patologia ostetrica. In effetti, sin dalla sua prima identificazione il LA è stato descritto come un prolungamento dei test di coagulazione fosfolipido-dipendenti in pazienti con storia di ripetuti aborti del 2° trimestre di gravidanza, associato o menoa fatti trombotici, spesso nell’ambito di malattie sistemiche come il lupus eri-tematoso sistemico (da cui il nome “lupus anticoagulant”, LA) (7-8).D’altra parte, negli anni ’80, l’introduzione del test aCL (2) ha confermato come altamente significativa l’associazione con la patologia abortiva, sia nell’ambito della malattia lupica che in donne altrimenti sane (aborto idiopa-tico) (9-10). Per questo motivo la presenza di aPL, indagati con il LA e/o con gli aCL, in donne con taluni problemi ostetrici (più di 3 aborti, o morte endouterina del feto, o parti pretermine, accompagnati da preeclampsia o severa insufficienza placentare) è andata configurando una popolazione di pazienti originariamente definite come “Lupus Ostetrico” (11). Questa defini-zione è stata successivamente abbandonata, dal momento che la patologiaostetrica è considerata uno dei due criteri clinici per la diagnosi e la classifi-cazione della Sindrome da Antifosfolipidi (12).
E’ di interesse che la presenza di aCL in donne affette da Lupus eritemato-so sistemico si sia dimostrata un fattore di rischio significativo per incidenti ostetrici (9) e che d’altra parte sia risultata molto compromessa la prognosi riproduttiva (50% di insuccessi) di donne con riscontro di aCL e/o LAC al 2° trimestre di gravidanza, senza che le pazienti avessero precedentianamnestici significativi (10).Non è ancora oggi chiaro se i titoli, l’isotipo dell’anticorpo o la sua specifi-cità siano influenti nel determinare il livello del rischio. Per quanto riguarda il titolo dell’anticorpo è interessante notare come taluni trials controllati, tra quelli che hanno codificato il trattamento, includano pazienti con bassi titoli di aCL (13); al contrario altri autori associano il rischio di insuccessi gravidici con l’isotipo IgG a titolo >20 unità GPL (o con LA positivo) (14).
Anche sul ruolo degli anticorpi anti-ß2GP1 nell’inquadrare le pazienti con problemi ostetrici i pareri non sono concordi. In effetti secondo taluni autoriquesto test non aggiungerebbe informazioni rispetto a quelle fornite dai testclassici (aCL e LA) (15) mentre per altri il test avrebbe una specificità mag-giore per taluni subset di patologia ostetrica (16). Probabilmente queste discordanze sono da imputarsi alla mancata standardizzazione delle meto-dologie applicate per la determinazione degli anticorpi anti-ß2GP1.
Nonostante le numerose segnalazioni di associazione tra anticorpi antifo-sfolipidi e perdite fetali, la dimostrazione che gli anticorpi causino le perditefetali si è avuta soltanto attraverso la osservazione dell’effetto che la pre-senza di aPL è in grado di esercitare sull’andamento della gravidanza in animali da esperimento.
In effetti è stato rilevato che ceppi di animali lupus prone, caratterizzati dalla presenza di aCL (MRL/lpr), hanno un minor numero di nati vivi rispettoad altri ceppi, pure lupus prone, ma senza aCL (NZB x W F1). D’altra parte, l’infusione di anticorpi in topi naive (BALB/c, ICR, CD1) durante la gravidanza risulta in un’alta percentuale di riassorbimento fetale,equivalente a quello che nell’uomo si definisce aborto/morte endouterina del feto (17).
Antifosfolipidi e Patologia Ostetrica
Modelli sperimentali: patogenesi della sindrome.

36
Anche se genericamente il meccanismo patogenetico degli antifosfolipidi viene ricondotto alla trombofilia, è risultato chiaro nel corso degli anni che i fenomeni trombotici non sono sufficienti a giustificare la particolare influen-za di questi anticorpi sullo sviluppo della gravidanza.
E’ infatti ipotizzabile che gli anticorpi si leghino direttamente al trofoblasto, dal momento che: 1) è stato dimostrato che alla sua superficie viene esposta, durante la sua
differenziazione in sinciziotrofoblasto, la fosfatidilserina (18) e inoltre 2) nelle placente di pazienti poliabortive con sindrome da antifosfolipidi
è stata rilevata da metodiche di immunoistochimica la presenza di ß2GP1(19).
Pertanto il trofoblasto verrebbe ad assemblare i possibili bersagli degli anti-corpi circolanti. A conferma di questa ipotesi sta la dimostrazione che, in vitro, gli aPL sono in grado di legare cellule trofoblastiche e di modularne l’attività interferendo con la formazione del sinciziotrofoblasto, con la sintesidi gonadotropina corionica e con la capacità di invasione. In sintesi è stato dimostrato un possibile effetto degli anticorpi sullo svilup-po e l’impianto della placenta, giustificando da un lato un danno non necessariamente legato a fenomeni trombotici e dall’altro l’associazione tra aPL ed aborti precoci (20).
D’altra parte, recentemente è stato focalizzato anche il ruolo della annessi-na V o placental anti-coagulant protein I (PAP-I) nel mantenimento della integrità della placenta. La annessina V, fortemente espressa sulla superfi-cie apicale dei microvilli del sinciziotrofoblasto (21, 22), è dotata di una potenteattività anticoagulante in quanto ha la capacità di legare le superfici di fosfolipidi a carica elettrica negativa, formando uno strato protettivo che impedisce l’avvio di reazioni coagulative. Nelle placente di pazienti con sindrome da antifosfolipidi è stata riscontratauna diminuita quantità di annessina V a livello placentare. Inoltre in vitro è stato dimostrato che gli aPL, probabilmente complessati con ß2GP1, ridu-cono il livello di annessina V di cellule trofoblastiche in coltura (23). Questa potrebbe essere una seconda via patogenetica di danno aPL-mediato della gravidanza, basata su fenomeni trombotici a livello delle strutture (trofoblasto e/o endotelio) che esprimono annessina V.
Molto è stato scritto riguardo al tipo di perdita gravidica associata agli aPL.In questo senso i criteri internazionali della conferenza di consenso tenutasia Sapporo (Giappone) nel 1998(12), hanno permesso un accordo sulle mani-festazioni da prendere in considerazione:
1) una o più morti dopo la 10 settimana di gestazione di un feto altrimenti sano, e con normale morfologia;
2) uno o più parti pretermine ( = 34 settimane) conseguenti a preeclampsia severa o insufficienza placentare;
3) tre o più aborti spontanei prima della 10 settimana di gestazione, con escluse altre cause di aborto (genetiche, anatomiche, infettive, endocrine, anomalie placentari, etc.).
I criteri di consenso hanno fatto chiarezza anche in termini di nomenclatura.In effetti i dati talora discordanti dei vari reports potrebbero essere dipesi anche dalla diversa nomenclatura applicata. Ad esempio alcuni gruppi fis-savano la differenza tra aborti e morti del feto a 20 settimane mentre altri a 16 settimane di gestazione.
La Clinica della Gravidanza nelle Pazienti con la Sindrome.

37
Tuttavia non è ancora chiaro se siano più tipiche della sindrome le perdite fetali (dopo la 10 settimana di gestazione), come sostenuto da un certo numero di autori tra quelli che ne hanno inizialmente disegnato il profilo (24-
26), o le perdite preembrioniche/embrioniche (prima della 10a settimana di gestazione), come risulterebbe da altre più recenti segnalazioni (13). Oltre agli aborti spontanei e alle perdite fetali del 2° e 3° trimestre, numero-se complicanze ostetriche materno-fetali sono associate alla sindrome: la preeclampsia-HELLP, la insufficienza uteroplacentare, lo IUGR (intrauterinegrowth retardation) ed il parto pretermine (27).
In questo ambito, il rapporto tra aPL e preeclampsia è uno degli argomenti più dibattuti. Infatti numerosi studi hanno sottolineato come l’incidenza di preeclampsia sia particolarmente elevata in pazienti con classica Sindromeda Antifosfolipidi, primaria o secondaria (25, 28). D’altra parte, anche nella popolazione ostetrica con preeclampsia è stata osservata una elevata frequenza di pazienti con anticorpi antifosfolipidi (29,
30), anche se non in modo univoco (31). Peraltro, uno studio recente (32), che esamina 317 pazienti gravide con un episodio di preeclampsia in una precedente gravidanza e quindi ritenute arischio di recidive, concludeva che anticorpi di classe IgG antifosfolipidi non classici (anti-fosfatidilserina) erano associati con la preeclampsia seve-ra, gli aCL di classe IgG erano associati con lo IUGR, ma entrambi i test avevano uno scarso valore predittivo per le citate complicazioni. In apparente contrasto con questo dato, abbiamo recentemente dimostratouna significativa associazione tra LA e preeclampsia in una casistica di 132gravidanze seguite prospetticamente in 92 pazienti con lupus eritematososistemico, con un valore predittivo estremamente significativo (RR 9.2) (33).
D’altra parte, anche per quanto riguarda la frequenza dello IUGR o la pre-dittività degli aPL verso questa complicazione si ricavano dalla analisi dellaletteratura voci contrastanti. In effetti lo IUGR è riportato con frequenze chevariano dal 30 al 12% in una serie di studi che comunque concordano almeno su un significativo aumento di prevalenza (27, 34, 35), contrariamente adaltri che non la confermano affatto (29, 31). I diversi risultati sulla associazione dei problemi ostetrici agli aPL possonotrovare varie spiegazioni.Come sopra ricordato, una certa responsabilità nel creare risultati discor-danti è probabilmente attribuibile alla non sempre rigorosa classificazione degli esiti sfavorevoli della gravidanza. Inoltre in questa ottica è necessarionon dimenticare un esame attento delle metodologie applicate dai singoli studi alla determinazione degli aPL. In effetti con questo nome si intende una famiglia di anticorpi eterogenei, identificabili con test diversi e con diversa specificità diagnostica per la Sindrome; pertanto i risultati ottenuti sulla stessa casistica con test diversi non sono sempre paragonabili.Dall’altro è utile ricordare che i 50 anni trascorsi dalla introduzione del LA non ne hanno appianato completamente i problemi metodologici (36), così come i 19 anni trascorsi dalla introduzione degli aCL non sono bastati a completarne il lavoro di standardizzazione (37). Pertanto risultati ottenuti in Centri diversi non sempre sono completamente sovrapponibili.
Se comunque, come è generalmente accettato, nella APS si registra un aumento di complicanze ostetriche, la conseguenza logica dovrebbe esse-re un aumento dei parti pretermine. In effetti nelle casistiche di gravidanze in pazienti con APS la frequenza del parto pretermine è stata stimata attor-no al 30% (28).

38
Fortunatamente però, la prematurità grave (prima della trentesima settima-na) è oggi infrequente e comunque i mezzi e la capacità raggiunte nei reparti di terapia intensiva neonatale trasformano queste nascite preterminein esiti globalmente favorevoli nella larga maggioranza dei casi.
Infine non si può trascurare, tra i problemi clinici delle gravidanze in pazien-ti con APS, la possibilità di un evento tromboembolico. Va al riguardo sottolineata la potenziale sinergia tra il rischio trombofilico proprio della gravidanza e/o del puerperio e quello rappresentato dagli aPL.
I primi tentativi terapeutici degli anni ’80 utilizzarono un trattamento steroi-deo a dosaggio immunosoppressivo associando aspirina a basse dosi come antiaggregante (38) . Benché questo trattamento si sia mostrato allora e anche in studi successivi(39) certamente efficace nel migliorare la prognosi fetale, uno studio control-lato del 1992 (40) ha mostrato come altrettanto efficace fosse l’utilizzo di epa-rina a dosaggio profilattico (20.000 U), anche in questo caso in associazio-ne a basse dosi di aspirina (81 mg), con il vantaggio non indifferente che il trattamento con eparina sembra indurre un minor numero di complicanze ostetriche (parto pretermine, ipertensione, etc.) (40-42) rispetto ai corticosteroidi.
A partire da queste osservazioni il trattamento di pazienti gravide con APS è stato codificato come associazione di basse dosi di aspirina ed eparina dapprima non frazionata e poi a basso peso molecolare. In questo atteg-giamento, comunque, ci sono ancora dei punti non uniformemente interpre-tati. Per esempio non è chiaro il timing dell’inserimento dell’eparina soprat-tutto per le pazienti in cui la sindrome è stata diagnosticata sulla base di trombosi anamnestiche e che sono quindi in trattamento con anticoagulantiorali. In effetti il problema si pone in quanto gli anticoagulanti orali sono teratogeni in un periodo tra la settima e la dodicesima settimana di gesta-zione e pertanto una conversione ad eparina in periodo preconcezionale viene considerata da taluni autori più sicura. D’altra parte, il periodo intercorrente prima del verificarsi del concepimentonon è facilmente valutabile e può prolungarsi nel tempo, pertanto, se è già stata instaurata terapia eparinica, questo periodo può essere gravato da effetti collaterali non trascurabili.
Il ruolo preciso della aspirina a basso dosaggio è anche fino ad oggi discusso. In questo ambito è interessante considerare i risultati di 2 studi randomizzati relativamente recenti, che includevano solo pazienti poliabor-tive, escludendo quelle con malattia autoimmune e con precedenti trombo-si. I due studi differivano tuttavia dal momento che in uno le pazienti mostravano la sola positività per aCL (essendo state escluse le donne gra-vide LA positive)(43) e nell’altro erano in larga maggioranza LA positive (13). Entrambi questi trials attribuiscono maggiore efficacia al trattamento epari-nico associato all’antiaggregante (acido acetilsalicilico a basso dosaggio: 75-100 mg/die), rispetto a quello basato sul solo antiaggregante anche se in realtà Rai et al. (13) puntualizzano che il vantaggio della associazione con eparina si esaurisce alla tredicesima settimana di gestazione.
Comunque, nonostante le differenze citate, entrambi questi 2 gruppi di lavoro dimostrano una percentuale di nati vivi nel braccio della sola aspiri-na intorno al 40%.
La terapia

39
Questo dato è in stridente contrasto con il valore di sopravvivenza emerso da altri studi che riportano in pazienti con APS trattate in gravidanza con lasola aspirina una percentuale di successi attorno all’80%. Infine un recentestudio controllato dimostrerebbe un effetto della aspirina paragonabile al placebo (44) in una casistica di poliabortive genericamente positive ai test per aCL (senza cioè necessariamente includere solo le positività medio-alte,come suggerito dai criteri classificativi) in cui erano prevalenti gli aborti embrionici o pre-embrionici. In effetti, in questa casistica, anche le pazienti in placebo avevano un out-come ostetrico assolutamente favorevole (intorno all’80% di esiti favorevoli).È utile in questo contesto ricordare che l’uso della aspirina ha un suo razio-nale riconducibile allo stimolo sulla produzione di IL-3 (45). Infatti quest’ultimacostituisce un fattore di crescita per il trofoblasto e, come tale, contribuisce al mantenimento di una gravidanza normale (46). I livelli sierici di IL-3 sono ridotti in donne gravide con APS (47) e, nel modello sperimentale, l’aggiunta di IL-3 esogena è stata in grado di abrogare completamente le complican-ze ostetriche correlate agli aPL (48). In contrasto con questi riscontri in vivo, stanno alcuni esperimenti in vitro, ove l’IL-3 non sembra in grado di impedi-re l’effetto degli aPL sul trofoblasto (49).
Un’altra fonte di dibattito è stata a lungo considerata l’impiego delle immu-noglobuline umane endovena come trattamento profilattico della perdita della gravidanza in pazienti con APS. Recentemente, tuttavia, uno studio controllato condotto su un numero significativo di pazienti ha dimostrato che il trattamento con immunoglobuline in vena aggiunto al trattamento classico con eparina aspirina non migliora la prognosi riproduttiva delle pazienti con APS (50). Questo trattamento quindi sarebbe da riservare a pazienti in cui la terapia convenzionale sia fallita o a gravidanze che neces-sitino di un trattamento complementare estemporaneo per superare proble-mi intercorrenti. In questi casi le immunoglobuline in vena potrebbero esse-re ragionevolmente impiegate anche senza che ci sia l’evidenza della loro reale efficacia.A queste indicazioni di “supporto” si aggiunge anche, ovviamente, quella della piastrinopenia, complicanza non rara nelle pazienti gravide con APS, e che trova un trattamento generalmente pronto ed efficace nell’uso delle immunoglobuline in vena.
Infine, se l’atteggiamento condiviso per la profilassi della gravidanza nelle pazienti con APS è basato sull’impiego di aspirina a basse dosi associata ad eparina, conviene non dimenticarne i possibili effetti collaterali. La concomitanza del trattamento eparinico con l’effetto della gravidanza stessa può diventare un fattore di rischio non trascurabile per osteoporosi,soprattutto quando, per una concomitante patologia autoimmune, la paziente abbia assunto o assuma corticosteroidi. L’incidenza di osteoporosi associata all’uso di eparina in gravidanza può assestarsi tra lo 0.2 (eparina a basso peso molecolare) e il 2% (eparina nonfrazionata) e per questo motivo è generalmente consigliata un’adeguata integrazione calcica (51).
La conoscenza della sindrome e l’applicazione di un trattamento ad perso-nam (evidentemente evolutosi nel corso degli anni in seguito alle informazioniprogressivamente acquisite), ha radicalmente mutato la prognosi ostetrica delle donne con APS.E’ comunque opinione condivisa che i risultati in questo ambito non siano esclusivamente legati ai trattamenti farmacologici applicati.
Conclusioni
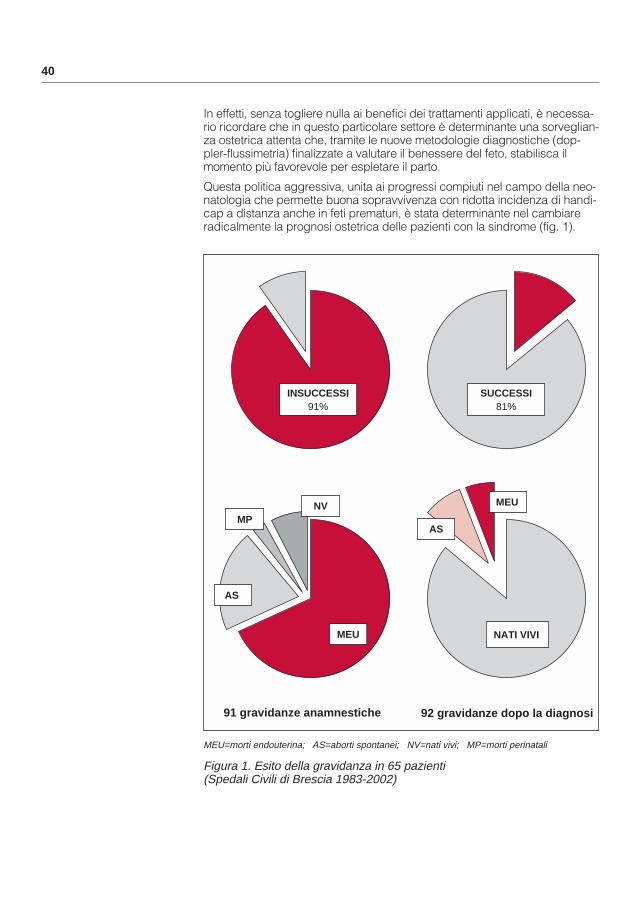
40
In effetti, senza togliere nulla ai benefici dei trattamenti applicati, è necessa-rio ricordare che in questo particolare settore è determinante una sorveglian-za ostetrica attenta che, tramite le nuove metodologie diagnostiche (dop-pler-flussimetria) finalizzate a valutare il benessere del feto, stabilisca il momento più favorevole per espletare il parto.
Questa politica aggressiva, unita ai progressi compiuti nel campo della neo-natologia che permette buona sopravvivenza con ridotta incidenza di handi-cap a distanza anche in feti prematuri, è stata determinante nel cambiare radicalmente la prognosi ostetrica delle pazienti con la sindrome (fig. 1).
INSUCCESSI91%
91 gravidanze anamnestiche 92 gravidanze dopo la diagnosi
MEU NATI VIVI
MEU
AS
ASMP
NV
SUCCESSI81%
Figura 1. Esito della gravidanza in 65 pazienti (Spedali Civili di Brescia 1983-2002)
MEU=morti endouterina; AS=aborti spontanei; NV=nati vivi; MP=morti perinatali

41Bibliografia
1) Soulier JP, Boffa MC: Avortments à repetition, thromboses et anticoagu-lant circulant antithromboplastin. Nouv. Presse Med. 1980; 9: 859-864
2) Harris EN, Gharavi AE, Boey ML, et al.: Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 1983; 2: 1211-1214.
3) Balestrieri G, Tincani A, Spatola L.: Anti-ß2-glycoprotein 1: a marker of antiphospholipid syndrome? Lupus 1995; 4:122.
4) Arvieux J, Darnige L, Caron C.: Development of an ELISA for autoanti-bodies to prothrombin showing their prevalence in patients with lupus anticoagulants. Thromb Haemost 74: 1120-1125, 1995.
5) Hughes GRV: Thrombosis, abortion, cerebral disease and the lupus anticoagulant. Br. Med. J. 1983; 287: 1088-89.
6) Geis W, Branch W: Obstetric implications of antiphospholipid antibodies: pregnancy loss and other complications. Clin Obstet Gynecol 2001 March; 44(1): 2-10.
7) Laurell AB, Nilsson IM. Hypergamma-globulinemia, circulating anticoagulant and biologic false positive Wassermann reaction: a study of 2 cases. J Lab Clin Med 1957;49:694-707.
8) Lee SL, Sanders M: A disorder of blood coagulation in systemic lupus erythematosus. J Clin Invest 1955, 34: 1814-1822.
9) Lockshin MD, Druzin ML, Goei S., et al.: Antibody to cardiolipin as a predictor of fetal di stress or death in pregnant patients with systemic lupus erythematosus. N Engl J Med 1985; 313: 152-6 1985.
10) Lockwood CJ, Romero R, Feinberg RF., et al.: The prevalence and biologic significance of lupus anticoagulant and anticardiolipin antibodiesin a general obstetric population. Am J Obstet Gynecol. 1989 Aug;161(2):369-73.
11) Lubbe WF, Palmer SJ, Butler WS, et al.: Fetal survival after prednisone suppression of maternal lupus anticoagulant. Lancet 1983 Jun 18;1(8338):1361-3.
12) Wilson W, Garavi A, Koike T., et al.: International consensus statement on preliminary classification criteria for definite Antiphospholipid yndrome: report of an international workshop. Arthritis Rheum 1999; 42: 1309-1311.
13) Rai R. Cohen H, Dave M, et al.: Randomized controlled trial of aspirin and aspirin plus heparin in pregnant women with recurrent miscarriagesassociated with phospholipid antibodies. BMJ. 1997; 314: 253-257.
14) Silver RM, Porter TF, Van Leeuween I, et al: Anticardiolipin antibodies: clinical consequences of “low titres”. Obstet Gynecol. 1996; 87: 494-500.

42
15) Lee RM, Emlen W, Scott JR, et al.: Anti-ß2 glycoprotein 1 antibodies in women with recurrent spontaneous abortion, unexplained fetal deathand antiphospholipid syndrome. Am J Obstet Gynecol. 1999; 181: 642-648.
16) Faden D, Tincani A, Tanzi P, et al.: Anti-ß2 glycoprotein 1 antibodies in a general obstetric population: preliminary results on the prevalence and correlation with pregnancy outcome. Anti-ß2 glycoprotein 1 antibodies are associated with some obstetrical complications, mainly preeclampsia-eclampsia. European Journal of Obstetrics & Gynecology and reproductive biology 73 (1997), 37-42.
17) Tincani A, Spatola L, Cinquini M et al.: Animal models of antiphospholipidsindrome. Revue de Rheumatisme, 1998; 11: 614-618.
18) Rote NS, Vogt E, DeVere G, Obringer AR: The role of placental trophoblast in the pathophysiology of the Antiphospholipid antibody syndrome. Am J. Reproduct Immunol 1998; 38: 125-36.
19) La Rosa L, Meroni PL, Tincani A, et al.: ß2 glycoprotein 1 and placental anticoagulant protein I in placentae from patients with antiphospholipid sindrome. J Rheumatol 1994 Sep; 21(9): 1684-93.
20) Di Simone N, Meroni PL, Del Papa N et al.: Antiphospholipid antibodies affect trophoblast gonadotropin secretion and invasiveness by binding directly and through adhered ß2 Glicoprotein 1. Arthr Rheum 2000; 43: 140-150.
21) Krikun G, Lockwood Cj, Wu XX et al.: The expression of the placental anticoagulant protein, annexin V, by villous trophoblasts: immunolocali-zation and in vitro regulation. Placenta 1994;15:601-612.
22) Andree HAM: Displacement of factor Va by annexin V in Phospholipid binding and anticoagulant action of annexin V. 1992 Ed. Universitaire Pers Maastricht, The Netherlands, pp. 73-85.
23) Rand JH: The pathogenic role of annexin-V in the antiphospholipid syndrome. Curr Rheumatol Rep 2000 Jun; 2(3): 246-51. ????
24) Branch DW.: Immunologic disease and fetal death. Clin Obstet Gynecol. 1987;30:295-311.
25) Branch DW., Dudley DJ Scott JR, et al.: Antiphospholipid antibodies and fetal loss. N Engl J Med. 1992; 326:952.
26) Oshiro BT, Silver RM, Scott JR, et al.: Antiphospholipid antibodies and fetal death. Obstet. Gynecol. 1996;87: 489-493.
27) Yasuda M, Takakuwa K, Tokunaga A, Tanaka K. Prospective studies of the association between anticardiolipin antibody and outcome of pregnancy. Obstet Gynecol 1995; 86:555-559.
28) Lima F, Khamashta MA, Buchanan NM, et al.: A study of sixty pregnancies in patients with the antiphospholipid sindrome. Clin Exp Rheumatol. 1996; 14:131-136.

43
29) Pattison NS, Chamley LW, McKay EJ, et al.: Antiphospholipid antibodies in pregnancy : prevalence and clinical association. Br J Obstet Gynaecol. 1993; 100: 909-913.
30) Moodley J, Bhoola V, Duursma J, et al.: The association of antipho-spholipid antibodies with severe early-onset preeclampsia. S Afr Med J. 1995; 85: 105-107.
31) Lynch A, Marlar R, Murphy J, et al.: Antiphospholipid antibodies in predicting adverse pregnancy outcome. A prospective study. Ann Intern Med. 1994; 120: 470-75.
32) Bch DW, Porter TF, Rittenhouse L, et al.: Antiphospholipid antibodies in women at risk for preeclampsia. Am J Obstet Gynecol 2001 Apr; 184(5): 825-32.
33) Tincani A, Doria A.: Clinical predictor of obstetrical outcome in SLE pregnancy. submitted
34) Branch DW, Silver RM, Blackwell JL, et al.: Outcome of treated pregnancies in women with antiphospholipid syndrome: an update of Utah experience. Obstet Gynecol. 1992; 80: 614-620.
35) Kutteh WH , Ermel LD: A clinical trial for the treatment of antiphospholipidantibody-associated recurrent pregnancy loss with lower dose heparin and aspirin. Am J Reprod Immunol. 1996; 35: 402-407.
36) Triplett DA: Many faces of Lupus. Lupus 1998;7 Suppl 2:S18-22.
37) Tincani A, Allegri F, Sanmarco M, et al.: Anticardiolipin antibody assay: a methodological analysis for a better consensus in routine determina-tions: a cooperative project of the European Antiphospholipid Forum. Thromb Haemost 2001 Aug; 86(2): 575-83.
38) Lubbe WF, Butler WS, Palmer SJ, Liggins GC: Fetal survival after prednisone, suppression of maternal lupus anticoagulant. Lancet 1983, 18; 1(8338): 1361-63.
39) Hasegawa I, Takakuwa K, Goto S, et al.: Effectiveness of prednisolone/ aspirin therapy for recurrent aborters with antiphospholipid antibody. Hum Reprod 1992; 7: 203-207.
40) Cowchock FS, Reece EA, Baldan D, et al.: Repeated fetal losses associated with antiphospholipid antibodies: a collaborative randomisedtrial comparing prednisone with low dose heparin treatement. Am J Obstet Gynecol 1992; 166: 1318-27.
41) Petri M., Howard D, Repke J: Risk factors for preterm birth in lupus pregnancy: a prospective study. Proceedings of the third International Conference on SLE. Lupus, 1992: 1(suppl.): 159.
42) Backos M, Rai R, Baxter N, et al. : Pregnant complications in women with recurrent miscarriage associated with antiphospholipid antibodies treated with low dose aspirin and heparin. Br J Obstet Gynaecol 1999; 106:102-107.

44
43) Kutteh WH. Antiphospholipid antibody associated recurrent pregnancy loss: treatment with heparin and low-dose aspirin is superior to low-dose aspirin alone. Am J Obstet Gynecol 1996;174:1584-1589.
44) Pattison NS et al.: Does aspirin have a role in improving pregnancy outcome for women with antiphospholipid syndrome? Am. J. Obst. Gynecol, 2000; 183(4): 1008-12
45) Fishman P. Aspirin modulates IL-3 production: additional explanation for the preventive effects of aspirin in antiphospholipid syndrome. J. Rheumatol. 1995 (22), 1086.
46) Wegman TG: The cytokine basis for cross-talk between the maternal immune and reproductive systems. Curr. Opin. Immunol. 1990 (2), 765.
47) Fishman P: Aspirin-IL-3 interrelationships in patients with antiphospholipidsyndrome. Am J Reprod. Immunol. 1996 (35), 80.
48) Fishman P: Prevention of fetal loss in experimental antiphospholipid syndrome by in vivo administration of recombinant interleukin-3. J. Clin. Invest. 1993 (91), 1834.
49) Chamley LW, Konarkowska B, Duncalf AM, et al.: Is Interleukin-3 important in antiphospholipid antibody-mediated pregnancy failure? Fertility and Sterility 2001 Oct; 76(4), 700-06.
50) Branch DW, Peaceman AM, Druzin M, et al.: A multicenter, placebo-controlled pilot study of intravenous immune globulin treatment of antiphospholipid syndrome during pregnancy. The Pregnancy Loss Study Group. Am J Obst Gynecol. 2000 Jan; 182 (1 Pt 1): 122-27.
51) Ruiz-Irastorza G, Khamashta MA, Nelson-Piercy C., et al.: Lupus pregnancy: is heparin a risk factor for osteoporosis? Lupus 2001; 10(9): 597-600.
Corrispondenza: Dr.ssa Angela TincaniServizio di Reumatologia, Allergologia, Immunologia Clinica, Spedali Civili di BresciaP.le Spedali Civili, 1 25123 BRESCIATel: 030-3996449; fax: 030-3995085; e-mail: [email protected]

45
Diagnosi di laboratorio degli anticorpi antifosfolipidi.

46
Considerata l’impossibilità di identificare con un singolo test le diverse classi di anticoagulante tipo lupus (LA), il Comitato Scientifico e di Standardizzazione (SSC) della Società Internazionale per l’Emostasi e Trombosi (ISTH), ha disegnato una strategia diagnostica basata su tre cri-teri (1). Il primo prevede che uno (o più) dei test dipendenti dai fosfolipidi sia prolungato oltre i limiti della norma (test di screening). Bisogna poi dimostrare che il prolungamento sia effettivamente dovuto alla presenza di un anticoagulante circolante e non ad una carenza di uno (o più) dei fattori della coagulazione (test della miscela per il secondo criterio).Per soddisfare il terzo criterio bisogna provare che l’anticoagulante siadiretto contro i fosfolipidi, o complessi proteine-fosfolipidi e non contro i sin-goli fattori della coagulazione (test di conferma).
Introduzione
1. Anticoagulante tipo lupus:diagnosi di laboratorio
Armando Tripodi
Centro Emofilia e Trombosi Angelo Bianchi Bonomi, Dipartimento di Medicina Interna, Università e IRCCS Ospedale Maggiore, Milano.

47
In teoria, qualsiasi test dipendente dai fosfolipidi, che esplori globalmente, o in parte, la cascata coagulatoria potrebbe essere idoneo a svelare la pre-senza del LA. In pratica, il test più usato per ragioni storiche e di praticità è il tempo di tromboplastina parziale attivato (APTT). Tuttavia, nonostante lasua popolarità, l’APTT non è in generale idoneo allo screening dei pazienticon sospetto di LA, a causa della scarsa sensibilità, che dipende essen-zialmente dal tipo e concentrazione dei fosfolipidi (2). Test più sensibili sonoil tempo di coagulazione al caolino (KCT) (3) e il test al veleno di Vipera Russell diluito (dRVVT) (4). Il primo è un test globale della coagulazione, che ha dimostrato una elevata sensibilità, dovuta probabilmente al fatto che nella sua formulazione i fosfolipidi sono (quasi) assenti, rendendo la presenza del LA molto più evidente anche quando esso è presente a basso titolo. Gli svantaggi del KCT sono la necessità di eseguirlo con tecnica manuale, i tempi di coagulazione piuttosto lunghi e la variabilità dei risultati, che impongono una rigida standardizzazione ed una accura-ta definizione del range di normalità.
Il dRVVT è anch’esso un test globale dipendente dai fosfolipidi, che esplorala porzione di cascata coagulatoria a valle del fattore X attivato. La sua semplicità di esecuzione,anche con strumenti automatici e la sua buona sensibilità, ne hanno favorito la rapida diffusione in tutti i laboratori. Anche iltempo di protrombina (PT), se eseguito con tromboplastina opportunamen-te diluita, si può considerare per la diagnostica del LA (5). Come per l’APTT anche per il PT i risultati dipendono in larga misura dalla tromboplastina adoperata. Risultati soddisfacenti sono stati segnalati con l’uso di trombo-plastine ricombinanti (6).
Si esegue con uno qualunque dei test di screening precedentemente esaminati e consiste nella ripetizione del test su una miscela plasma paziente/plasma normale. La persistenza del prolungamento del tempo di coagulazione eseguito sulla miscela (mancata correzione), suggerisce la presenza di un anticoagulante circolante.
Il test della miscela deve essere molto ben standardizzato ed i risultati devovo essere interpretati secondo criteri ben precisi. Di solito il test sulla miscela, in parti uguali (plasma paziente/plasma normale), è eseguito senzaincubazione. Titoli di anticorpo molto bassi potrebbero richiedere miscele più ricche in plasma paziente ed alcuni anticorpi tempo-dipendenti potreb-bero richiedere una incubazione a 37°C. Non esistono criteri ben definiti per giudicare in maniera univoca se la miscela “corregge”. Un criterio semplice potrebbe essere quello di giudicare “corrette” quelle miscele i cui tempi di coagulazione rientrino nei limiti della norma stabiliti per quel test, in quel laboratorio.
Sono basati sull’incremento, o la diminuzione della concentrazione dei fosfolipidi, o sull’uso di fosfolipidi a conformazione particolare. Il tempo di coagulazione di un test dipendente dai fosfolipidi, prolungato per la pre-senza del LA, si accorcia sensibilmente fino a correggere quasi completa-mente il difetto, se ripetuto aumentando la concentrazione dei fosfolipidi. Alternativamante, il test si prolungherà se viene diminuita la concentrazionedei fosfolipidi. Esistono numerosi test di conferma, almeno tanti quanti sonoi test di screening, ed esistono diverse fonti possibili di fosfolipidi.
Test di screening
Test della miscela
Test di conferma

48
Fra i test di conferma piu’ usati ricordiamo l’APTT con aggiunta di lisato pia-strinico quale fonte di fosfolipidi (5). Esso è di semplice esecuzione, più complessa è però la preparazione delle piastrine lisate, ottenute per lavaggio e ripetuti congelamenti/scongelamenti di un plasma ricco in pia-strine. I risultati dipendono dalla preparazione della piastrine e vi possono essere notevoli differenze fra una preparazione e la successiva. Il test di conferma può anche essere eseguito con il dRVVT con aggiunta di fosfoli-pidi concentrati.
Esistono dei kit commerciali che consentono di eseguire in maniera integra-ta l’iter diagnostico di screening e conferma, avendo tutto il materiale necessario già pronto. L’uso di silice micronizzata in combinazione con fosfolipidi a bassa concentrazione può essere un buon sostituto del KCT nella procedura di screening (7). La ripetizione del test con fosfolipidi a più alta concentrazione consente di disporre di un test di conferma facilmente automatizzabile e di semplice esecuzione (8). La Textarina, veleno di rettile capace di attivare la protrombina in presenza di fattore V e fosfolipidi può essere usata per disegnare un test di screening per il LA. La ripetizione della procedura con Ecarina, altro veleno di rettile, che attiva la protrombi-na senza fattore V e fosfolipidi, consente di disporre di un test di conferma basato sul rapporto Textarina/Ecarina (9). Infine, fra le procedure di confermabisogna ricordare l’APTT eseguito mediante fosfolipidi a conformazione esagonale (10) per il quale esiste anche un kit commerciale. La conformazio-ne esagonale renderebbe i fosfolipidi più disponibili a legare il LA che ver-rebbe, pertanto, riconosciuto con una maggiore specificità. Tutte queste procedure, non hanno tuttavia risolto definitivamente il problema della spe-cificità. False positività in plasmi con inibitori diretti contro il fattore V o VIII,o in presenza di eparina, si possono occasionalmente riscontrare anche con l’uso di questi test. La storia clinica del paziente, che dovrà sempre di necessità accompagnare la provetta in laboratorio, aiuterà a risolvereeventuali dubbi.
Se è vero che nessuno dei test di screening e di conferma può in assoluto garantire il successo nella diagnosi di laboratorio per il LA, è però altrettanto vero che qualunque dei test sopra menzionati ha buone probabilità di succes-so se eseguito su un plasma raccolto tenendo presente alcune precauzioni.
Contaminazione da eparina. Un test prolungato, il cui plasma è stato raccolto al di fuori della responsabilità diretta del Centro che esegue le indagini di laboratorio, dovrebbe essere considerato con sospetto e prima di intraprende-re lunghe e costose procedure analitiche (test di miscela, test di conferma), èopportuno eseguire almeno un tempo di trombina per escludere la presenza di eparina.
Residuo piastrinico. Poiché il LA è diretto contro i fosfolipidi (o complessi pro-teine-fosfolipidi), la presenza nel plasma di piastrine residue può, mascheran-do quegli anticorpi a basso titolo, ridurre la capacità diagnostica dei test di screening (11) e di conferma (12). L’effetto può essere marcato soprattutto nei campioni di plasma che saranno conservati congelati prima della esecuzione dei test. Il congelamento e lo scongelamento facilitano la frammentazione pia-strinica, con esposizione dei fosfolipidi di membrana. La doppia centrifugazio-ne è in genere sufficiente a rimuovere la maggior parte delle piastrine. La filtra-zione del plasma attraverso filtri di adatta porosità (0.22 µm), assicura l’elimi-nazione di tutte le piastrine. Quando possibile, è consigliabile di eseguire i testsu plasma fresco.
L’influenza della fase preanalitica e la sua standardizzazione

49
Plasma normale. Particolare cura deve essere posta nella scelta del plasma normale da usare per il test della miscela. Esso deve avere un contenuto nor-male per tutti i fattori della coagulazione e non deve contenere piastrine resi-due, o loro frammenti . Non tutti i plasmi liofilizzati commerciali soddisfano questi requisiti. Un pool di plasmi normali preparato in casa, filtrato, congela-to rapidamente e conservato a -70° C può essere una alternativa efficace e a basso costo.
Un problema di rilevanza pratica è costituito dai pazienti il cui plasma giungeall’osservazione del laboratorio quando essi sono già stati trattati con eparina(trattamento dell’evento acuto), o con anticoagulanti orali (prevenzione secon-daria del tromboembolismo). In tali condizioni il tempo di coagulazione di tuttii test dipendenti dai fosfolipidi è più o meno prolungato, rendendo di fatto problematica l’interpretazione dei risultati dei test di miscela e di conferma.Sebbene la diagnosi di laboratorio possa essere più comodamente effettuataalla fine delle terapia, vi possono essere delle ragioni per richiedere una inda-gine di laboratorio anche in corso di terapia. Gli approcci che si possono seguire saranno diversi a seconda che la terapia sia eparinica, o anticoagu-lante orale.
Terapia eparinica. Quello che si può fare è tentare di neutralizzare l’eparina mediante aggiunta di sostanze anti-epariniche, quali il polibrene o l’eparinasi.Scegliendo con cura le proporzioni plasma/inibitore, mediante esperimenti eseguiti con i propri metodi e reagenti, è possibile identificare le condizioni ottimali per neutralizzare l’eparina senza alterare i risultati del test. E’ però richiesta una notevole perizia nel maneggiare questi reagenti, soprat-tutto il polibrene. Una alternativa apparentemente più praticabile è la neutra-lizzazione dell’eparina mediante una resina (Ecteola), che mescolata al pla-sma, dopo incubazione è capace di adsorbire quantitativamente l’eparina, lasciando il plasma supernatante apparentemente inalterato. Per informazionipiù dettagliate sull’uso dell’Ecteola si può consultare la letteratura (13).
Anticoagulanti orali. Nel caso che il paziente sia anticoagulato con dicumaroli-ci, il problema può essere affrontato in due modi. Il primo è l’esecuzione dei test previa miscela del plasma paziente con una aliquota equivalente di pla-sma normale. Il plasma normale dovrebbe correggere interamente il difetto indotto dai dicumarolici e, pertanto, sulla miscela si può successivamente eseguire la comune diagnostica per il LA. Un’alternativa valida è quella di eseguire direttamente un test di conferma a due diverse concentrazioni di fosfolipidi, senza correggere preventivamente il difetto coagulatorio indotto dai dicumarolici. Anche se i tempi di coagulazionedi base sono prolungati, l’entità della correzione dopo aggiunta di fosfolipidi concentrati consente di effettuare la diagnosi con modeste interferenze. Recentemente, abbiamo avuto modo di validare questa strategia. Plasmi da pazienti in terapia anticoagulante e positivi per LA, raccolti in diversi centri ita-liani, sono stati sottoposti alla diagnostica centralizzata presso il nostro labo-ratorio mediante due procedure di conferma basati sull’SCT (7) ed il dRVVT adue diverse concentrazioni di fosfolipidi, senza aggiunta di plasma normale. I risultati sono stati paragonati con quelli ottenuti con l’APTT con fosfolipi-di esagonali e aggiunta di plasma normale (Staclot LA, Stago, Asnieres, Francia), da noi considerato come standard d’oro, proprio per la presen-za del plasma normale. La sensibilità e specificità dell’SCT e dRVVT, da noi disegnati, rispetto allo Staclot LA è risultata molto vicina, o superiore al 90% (14). L’SCT e il dRVVT hanno rispetto allo Staclot LA il vantaggio di un costo nettamente inferiore e la possibilità di essere eseguibili con qualsiasi tipo di coagulometro.
Diagnosi di laboratorio nei pazienti anticoagulati

50
Una alternativa ulteriore nella diagnostica del LA nei pazienti anticoagulati potrebbe essere costituita dal test ecarina/textarina (9), ma la sua validità resta ancora da dimostrare.
Nessuna strategia diagnostica per quanto elaborata assicura il successo nella totalità dei casi. Un solo test potrebbe non essere adeguato in tutti i casi. D’altro canto, molti test renderebbero difficile l’interpretazione e non giustificherebbero i costi. La responsabilità della scelta della strategia dia-gnostica più adeguata spetta al laboratorio, noi possiamo suggerire le seguenti regole.
1. Selezione accurata dei pazienti, riservando l’indagine di laboratorio ai casi con pregressa storia. Questo si realizza curando i rapporti fra laboratorio e medici prescrittori dei test, specialmente gli specialisti d’organo (ematologi, angiologi, ginecologi, immunologi clinici, internisti, ecc.), che sono fra i maggiori fruitori della diagnostica per il LA.
2. Esecuzione del prelievo sotto la diretta responsabilità del laboratorio, o concordandone le modalità.
3. Preparazione del plasma secondo le indicazioni di cui sopra, special-mente se esso sarà congelato per indagini future.
4. Esecuzione di almeno due test di screening scelti fra KCT (o test equiva-lente) e dRVVT. L’APTT, a meno di certezze sulla sua sensibilità, dovrebbe essere eseguito insieme, non in alternativa ai primi due.
5. Esclusione che il test di screening sia prolungato per la presenza di eparina.
6. Esecuzione del test della miscela, ponendo cura nella scelta del plasmanormale e nei criteri di interpretazione.
7. Esecuzione del test di conferma. E’ buona norma che questo sia scelto sulla base del test di screening.
8. Per una corretta interpretazione dei risultati dei test della miscela e di conferma, è opportuno acquisire esperienza mediante prove simulate con plasmi test sicuramente negativi e positivi per LA, o altri inibitori confondenti (es. inibitori contro il fattore VIII).
9. Misura del titolo degli anticorpi in fase solida (anticardiolipina). La misuradegli anticorpi in fase solida non è da considerarsi alternativa, ma com-plementare alla ricerca del LA. La positività per LA e anticardiolipina non è necessariamente coesistente in tutti i pazienti.
10.In caso di positività, è necessario riconfermare la diagnosi a distanza di 3-4 mesi, soprattutto là dove essa è stata riscontrata occasionalmente(screening pre-chirurgico). Uno dei criteri diagnostici per la sindrome da anticorpi antifosfolipidi è la persistenza degli anticorpi. Talvolta gli anti-corpi antifosfolipidi insorgono in seguito a infezioni virali o batteriche, o inseguito all’assunzione di farmaci e non hanno di solito rilevanza clinica. Un caso esemplare di anticorpi transitori senza storia clinica di trombosi è costituito dai pazienti in età pediatrica, che vengono trovati positivi al LA in occasione dello screening pre-chirurgico che precede la tonsil-lectomia. Gli anticorpi spariscono invariabilmente dopo la rimozione della causa.
Considerazioni conclusive

51
1. Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria for the diagnosis of lupus anticoagulants: an update. Thromb Haemostas 1995; 74: 1185-90.
2. Kelsey IR, Stevenson KJ and Poller L. The diagnosis of lupus anticoagulants by the activated partial thromboplastin time. The central role of phosphatidyl serine. Thromb Haemostas 1984; 52:172-75.
3. Exner T, Rickard KA, Kronemberg KA. A sensitive test demonstrating lupus anticoagulants and its behavioural patterns. Brt J Haematol 1978; 40: 143-51.
4. Exner T, Papadopulos G, Koutts J. Use of a simplified dilute Russell’s viper venom time (DRVVT) confirms heterogeneity among lupus anticoagulants. Blood Coag Fibrinol 1990; 1: 259-66.
5. Triplett DA, Brandt JT, Kaczor D, Schaeffer J. Laboratory diagnosis of lupus inhibitors: a comparison of the tissue thromboplastin inhibition procedure with a new platelet neutralization procedure. Am J Clin Path 1983; 79: 678-82.
6. Arnout J, Vanrusselt M, Huybrechts E, Vermylen J. Optimization of the dilute prothrombin time for the detection of the lupus anticoagulant by use of a recombinant tissue factor. Br J Haematol 1994; 87: 94-9.
7. Chantarangkul V, Crippa L, Tripodi A, Mannucci PM. Colloidal silica clotting time (CSCT) in the detection of lupus anticoagulant. Thromb Res 1991; Suppl XIII: 79.
8. Chantarangkul V, Tripodi A, Arbini A, Mannucci PM. Silica clotting time (SCT) as a screening and confirmatory test for detection of lupus anticoagulants. Thromb Res 1992; 67: 355-65.
9. Triplett DA, Stocker KF, Unger GA, Barna LK. The textarin/ecarin ratio: a confirmatory test for lupus anticoagulants. Thromb Haemostas 1993; 70: 925-32.
10. Triplett DA, Barna LK, Unger GA. A hexagonal (II) phase phospholipid neutralization assay for lupus anticoagulant identification. Thromb Haemostas 1993; 70: 787-93.
11. Sletness KE, Gravem K, Wisloff F. Preparation of plasma for the detection of lupus anticoagulants and phospholipid antibodies. Thromb Res 1992; 66: 43-53.
12. Chantarangkul V, Tripodi A, Clerici M, Bressi C, Mannucci PM. Laboratory diagnosis of lupus anticoagulants. Effect of residual platelets in plasma, assessed by Staclot-LA® and silica clotting time. Thromb Haemost 2002. In press
13. Van den Besselaar AMHP, Meeuwisse-Braun J. Enzymatic elimination of heparin from plasma for activated partial thromboplastin time and prothrombin time testing. Blood Coag Fibrinol 1993; 4: 635-8.
Bibliografia

52
14. Tripodi A, Chantarangkul V, Clerici M, Mannucci PM. Laboratory diagnosis of lupus anticoagulants for patients on oral anticoagulant treatment. Performance of dilute Russell viper venom test and silica clottingtime in comparison to Staclot-LA®. Submitted
Corrispondenza: A. TripodiVia Pace, 9 - 20122 MilanoTel.: 02 55035437 - FAX: 02 5516093e-mail: [email protected]

53
2. Anticorpi anticardiolipina:diagnosi di laboratorio
Angela Tincani*, Massimo Cinquini*, Michela Spunghi*, Flavio Allegri*,Genesio Balestrieri*, Pierluigi Meroni°
* Reumatologia, Allergologia e Immunologia Clinica, Spedali Civili di Brescia° Allergologia e Immunologia Clinica, Istituto Auxologico Milano
Gli anticorpi anticardiolipina (aCL) costituiscono uno dei due criteri labora-toristici per la diagnosi di Sindrome da Antifosfolipidi (APS) (1), che è carat-terizzata, come è noto, dall’associazione di trombosi artero-venose, patolo-gia gravidica e anticorpi anti fosfolipidi (aPL) (2,3). La frequenza della APS, sia primaria che secondaria ad altre malattie autoimmuni sistemiche, giustifica ampiamente la diffusione dei test per la ricerca di aPL (4).
Introduzione
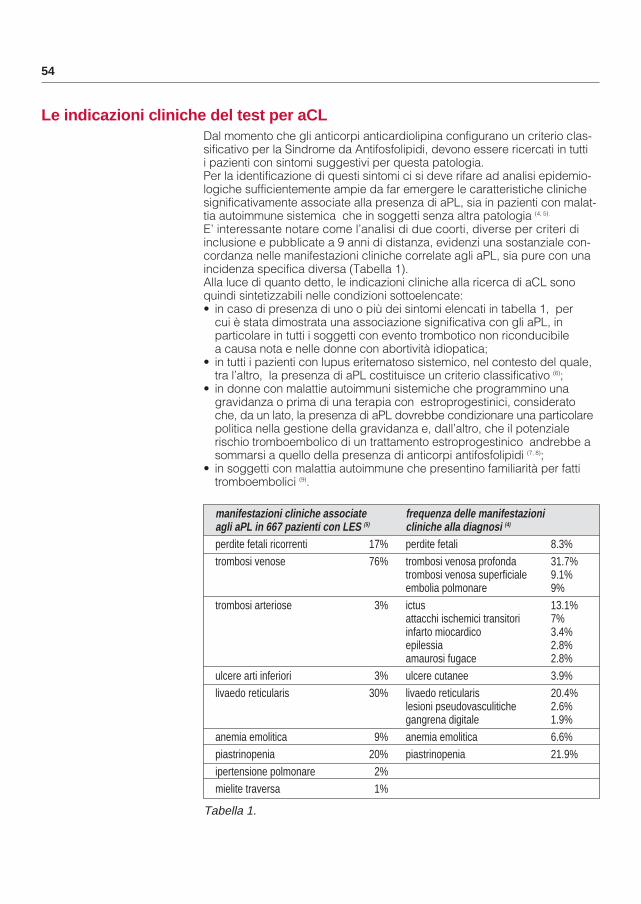
54
Dal momento che gli anticorpi anticardiolipina configurano un criterio clas-sificativo per la Sindrome da Antifosfolipidi, devono essere ricercati in tutti i pazienti con sintomi suggestivi per questa patologia.Per la identificazione di questi sintomi ci si deve rifare ad analisi epidemio-logiche sufficientemente ampie da far emergere le caratteristiche cliniche significativamente associate alla presenza di aPL, sia in pazienti con malat-tia autoimmune sistemica che in soggetti senza altra patologia (4, 5).
E’ interessante notare come l’analisi di due coorti, diverse per criteri di inclusione e pubblicate a 9 anni di distanza, evidenzi una sostanziale con-cordanza nelle manifestazioni cliniche correlate agli aPL, sia pure con una incidenza specifica diversa (Tabella 1). Alla luce di quanto detto, le indicazioni cliniche alla ricerca di aCL sono quindi sintetizzabili nelle condizioni sottoelencate: • in caso di presenza di uno o più dei sintomi elencati in tabella 1, per
cui è stata dimostrata una associazione significativa con gli aPL, in particolare in tutti i soggetti con evento trombotico non riconducibile a causa nota e nelle donne con abortività idiopatica;
• in tutti i pazienti con lupus eritematoso sistemico, nel contesto del quale, tra l’altro, la presenza di aPL costituisce un criterio classificativo (6);
• in donne con malattie autoimmuni sistemiche che programmino una gravidanza o prima di una terapia con estroprogestinici, considerato che, da un lato, la presenza di aPL dovrebbe condizionare una particolarepolitica nella gestione della gravidanza e, dall’altro, che il potenziale rischio tromboembolico di un trattamento estroprogestinico andrebbe a sommarsi a quello della presenza di anticorpi antifosfolipidi (7, 8);
• in soggetti con malattia autoimmune che presentino familiarità per fatti tromboembolici (9).
Le indicazioni cliniche del test per aCL
Tabella 1.
manifestazioni cliniche associate frequenza delle manifestazioni agli aPL in 667 pazienti con LES (5) cliniche alla diagnosi (4)
perdite fetali ricorrenti 17% perdite fetali 8.3%trombosi venose 76% trombosi venosa profonda 31.7%
trombosi venosa superficiale 9.1%embolia polmonare 9%
trombosi arteriose 3% ictus 13.1%attacchi ischemici transitori 7%infarto miocardico 3.4%epilessia 2.8%amaurosi fugace 2.8%
ulcere arti inferiori 3% ulcere cutanee 3.9%livaedo reticularis 30% livaedo reticularis 20.4%
lesioni pseudovasculitiche 2.6%gangrena digitale 1.9%
anemia emolitica 9% anemia emolitica 6.6%piastrinopenia 20% piastrinopenia 21.9%ipertensione polmonare 2%mielite traversa 1%

55
Il test degli anticorpi anti cardiolipina è a tutt’oggi probabilmente il mezzo più usato per la diagnosi di Sindrome da Antifosfolipidi. Le ragioni della suafortuna risiedono in motivazioni sia storiche che concettuali. In effetti, il ruolo della falsa positività della reazione di Wassermann (che uti-lizza la cardiolipina come substrato) in pazienti con malattie autoimmuni sistemiche in stadio clinico o preclinico (10) talvolta associata al LAC (11) è noto dagli anni ’50. E, d’altra parte, il fenomeno del LAC è stato definito come un allungamento dei tempi di coagulazione fosfolipide dipendente, dovuto ad anticorpi diretti contro fosfolipidi a carica negativa, quale appun-to la cardiolipina (così chiamata perchè originariamente estratta dal musco-lo cardiaco bovino) (12, 13). Su queste basi nel 1983 fu concepito un nuovo test in fase solida, dapprima radioimmunologico (14), poi immunoenzimatico (15) per la rilevazione degli aCL. La applicazione dell’ELISA aCL su vaste casistiche, consentì di definire la APS così come la conosciamo oggi (2), dimostrando, nei fatti, la validità di questo test. Nel 1990 venne fornita la dimostrazione (16-18), che anticorpi così detti “anticar-diolipina”, erano in realtà in larga maggioranza diretti contro un cofattore pla-smatico che lega la CL e i fosfolipidi a carica elettrica negativa: la ß2-glico-proteina 1 (ß2GP1). Si può pertanto paradossalmente affermare che il test degli anticorpi anticardiolipina risulta essere basato su un equivoco. Questa scoperta consentì di interpretare quello che era già risultato chiaro dal primoworkshop collaborativo per la standardizzazione della metodica e cioè che iltest risultava più stabile ed efficiente se veniva aggiunto ai tamponi di reazio-ne siero bovino, che in effetti, a posteriori, risultò essere fonte di ß2GP1 (19). La qualità più interessante del test aCL ELISA classico è quella di essere ingrado di rilevare, almeno in via teorica, diverse popolazioni anticorpali che potrebbero tutte essere comprese nella così detta famiglia degli “anti fosfo-lipidi” (figura 1).
Anti cardiolipina: perché nel 2002?
Figura 1. Il test aCL ELISA classico può rilevare anticorpi diretti verso la cardiolipina, verso la ß2GP1 e verso il complesso CL-ß2GP1. Il test ELISAanti-ß2GP1 umana rileva esclusivamente anticorpi anti-ß2GP1; gli anticor-pi aCL evidenziati con questa metodica sono definiti aCL-ß2GP1 dipen-denti, principalmente presenti nelle malattie autoimmuni sistemiche. Gli aCL rilevabili con un test ELISA in assenza di ß2GP1 rilevano anticorpi principalmente presenti nelle malattie infettive.
aCL ELISA CLASSICO
Cardiolipina (CL)
ß2 Glicoproteina 1 bovina (ß2GP1)
ß2GP1 Umana
principalmentein malattie autoimmuni
anti ß2GP1
principalmentein malattie infettive
veri aCL che reagisconocon il fosfolipide

56
1. Innanzitutto, data l’alta affinità della ß2GP1 per i fosfolipidi a carica negativa, quali appunto la CL, la ß2GP1 si legherà al fosfolipide copulato alla micropiastra. In questo modo il sistema è in grado di concentrare la ß2GP1 sulla superficie delle micropiastre, ottenendo un effetto simile a quello della plastica attivata o “high antigen binding”. Un sistema di questo genere diventa adatto alla rilevazione di anticorpi anti ß2GP1, che oggi sembrano rappresentare la maggior parte degli anticorpi antifosfolipidi dei pazienti con la sindrome (20).
2. Anche nell’ipotesi, sostenuta da taluni autori (21), che il target dell’anti-corpo sia non tanto la ß2GP1 di per sé, ma piuttosto il complesso ß2GP1-CL, è facilmente comprensibile che il test aCL ELISA classico che include sulla micropiastra sia la CL che la ß2GP1 viene ad essere il mezzo più adeguato di osservazione.
3. Infine, almeno teoricamente, il test aCL ELISA è in grado di legare gli anti CL “veri”, cioè realmente diretti verso la molecola fosfolipidica. Il significato di questi anticorpi è ancora discusso. In effetti è comune-mente accettato che questi anticorpi siano caratteristici nel corso di malattie infettive, anche se recentemente è stato ipotizzato un loro possi-bile ruolo quali fattori indipendenti di rischio per fatti tromboembolici (21, 22).
Una parte rilevante delle interpretazioni sopra riportate, si basa sul fatto chela ß2GP1 ha una struttura altamente conservata nelle diverse specie, cosic-ché fra molecola umana e bovina esiste una omologia superiore al 90% (23). In questo modo gli anticorpi rilevabili in un sistema in cui è presente ß2GP1 bovina sono in gran parte sovrapponibili agli anticorpi rilevabili utilizzando ß2GP1 umana. Esistono comunque talune eccezioni. In effetti, la scoperta del cofattore ha condotto alla creazione di un test ELISA per anti ß2GP1 umana (24), ed alla conseguente dimostrazione che la popolazione anticorpale così rilevabile mostrava una associazione più significativa con le manifestazioni cliniche della APS (25). Inoltre è stata dimo-strata l’esistenza di anti ß2GP1 in assenza di aCL-ß2GP1 dipendenti e\o di LAC (26). I motivi per cui i due tipi di test ELISA (aCL classico e anti-ß2GP1) rilevano popolazioni anticorpali solo parzialmente sovrapponibili sono da imputarsi al fatto che taluni anticorpi presenti in pazienti con sindrome sem-brano riconoscere solo la ß2GP1 umana e pertanto reagiscono solo con il test degli anti ß2GP1, mentre altri sembrano diretti verso la CL di per sè o verso un complesso CL-ß2GP1 e pertanto vengono rilevati solo dal test classico della cardiolipina. E’ comunque utile sottolineare che entrambi questi casi rappresentano eccezioni.
Certamente, l’analisi della letteratura indica nel LAC il maggiore fattore di rischio conosciuto per la patologia trombotica in corso di patologia autoim-mune sistemica e il maggior fattore di rischio per recidiva trombotica in casodi sindrome primaria (9, 27). Tuttavia i vantaggi offerti dai test ELISA, quali la valutazione semi-quantitativa e la identificazione dell’isotipo dell’anticorpo, non sono trascurabili. Un innegabile vantaggio è inoltre quello di poter effet-tuare la ricerca di anticorpi anti fosfolipidi in pazienti già in trattamento anti-coagulante. L’impiego contemporaneo del LAC e del test ELISA aCL con-sente d’altra parte di accrescere la sensibilità diagnostica per la APS. Esiste infatti una quota non trascurabile, sebbene variabile nelle diverse casi-stiche (28), di pazienti con APS rilevabili solo grazie alla positività per aCL. La motivazione è da ricercare nel fatto che i test ELISA consentono di rileva-re anche concentrazioni anticorpali basse, non sufficienti a determinare una modifica dei processi coagulativi in un test funzionale come il LAC, ma in grado di identificare pazienti con un rischio trombotico, che possono speri-mentare eventi clinicamente significativi, soprattutto in caso di variazione deltitolo e/o di sovrapposizione di altri fattori di rischio trombotico.
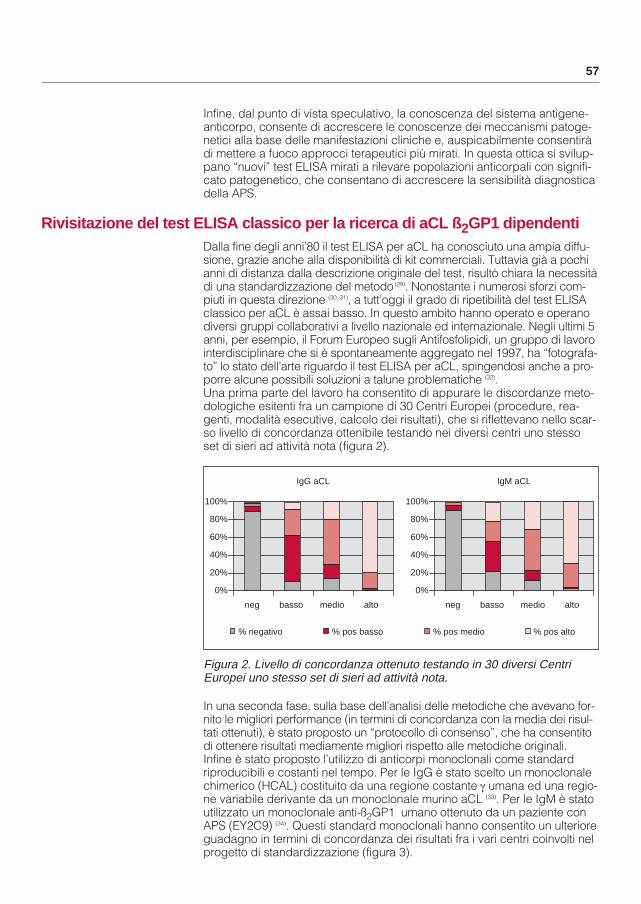
57
Infine, dal punto di vista speculativo, la conoscenza del sistema antigene-anticorpo, consente di accrescere le conoscenze dei meccanismi patoge-netici alla base delle manifestazioni cliniche e, auspicabilmente consentirà di mettere a fuoco approcci terapeutici più mirati. In questa ottica si svilup-pano “nuovi” test ELISA mirati a rilevare popolazioni anticorpali con signifi-cato patogenetico, che consentano di accrescere la sensibilità diagnosticadella APS.
Dalla fine degli anni’80 il test ELISA per aCL ha conosciuto una ampia diffu-sione, grazie anche alla disponibilità di kit commerciali. Tuttavia già a pochi anni di distanza dalla descrizione originale del test, risultò chiara la necessitàdi una standardizzazione del metodo (29). Nonostante i numerosi sforzi com-piuti in questa direzione (30, 31), a tutt’oggi il grado di ripetibilità del test ELISA classico per aCL è assai basso. In questo ambito hanno operato e operano diversi gruppi collaborativi a livello nazionale ed internazionale. Negli ultimi 5anni, per esempio, il Forum Europeo sugli Antifosfolipidi, un gruppo di lavoro interdisciplinare che si è spontaneamente aggregato nel 1997, ha “fotografa-to” lo stato dell’arte riguardo il test ELISA per aCL, spingendosi anche a pro-porre alcune possibili soluzioni a talune problematiche (32).Una prima parte del lavoro ha consentito di appurare le discordanze meto-dologiche esitenti fra un campione di 30 Centri Europei (procedure, rea-genti, modalità esecutive, calcolo dei risultati), che si riflettevano nello scar-so livello di concordanza ottenibile testando nei diversi centri uno stesso set di sieri ad attività nota (figura 2).
In una seconda fase, sulla base dell’analisi delle metodiche che avevano for-nito le migliori performance (in termini di concordanza con la media dei risul-tati ottenuti), è stato proposto un “protocollo di consenso”, che ha consentito di ottenere risultati mediamente migliori rispetto alle metodiche originali.Infine è stato proposto l’utilizzo di anticorpi monoclonali come standardriproducibili e costanti nel tempo. Per le IgG è stato scelto un monoclonale chimerico (HCAL) costituito da una regione costante γ umana ed una regio-ne variabile derivante da un monoclonale murino aCL (33). Per le IgM è stato utilizzato un monoclonale anti-ß2GP1 umano ottenuto da un paziente con APS (EY2C9) (34). Questi standard monoclonali hanno consentito un ulterioreguadagno in termini di concordanza dei risultati fra i vari centri coinvolti nel progetto di standardizzazione (figura 3).
Rivisitazione del test ELISA classico per la ricerca di aCL ß2GP1 dipendenti
Figura 2. Livello di concordanza ottenuto testando in 30 diversi Centri Europei uno stesso set di sieri ad attività nota.
% negativo
neg basso
IgG aCL
medio alto
0%
20%
40%
60%
80%
100%
neg basso
IgM aCL
medio alto
0%
20%
40%
60%
80%
100%
% pos basso % pos medio % pos alto
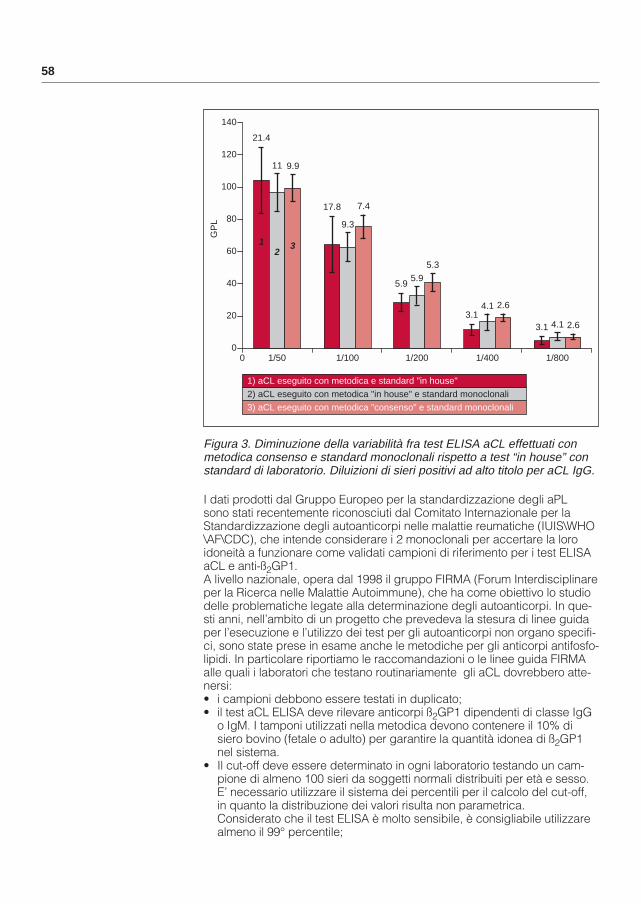
58
Figura 3. Diminuzione della variabilità fra test ELISA aCL effettuati con metodica consenso e standard monoclonali rispetto a test “in house” con standard di laboratorio. Diluizioni di sieri positivi ad alto titolo per aCL IgG.
0 1/50
21.4
11 9.9
1/100 1/200 1/400 1/800
GP
L
0
20
40
60
80
100
120
140
17.8
9.3
7.4
5.95.9
5.3
3.14.1 2.6
3.1 4.1 2.6
12
3
3) aCL eseguito con metodica "consenso" e standard monoclonali
2) aCL eseguito con metodica "in house" e standard monoclonali
1) aCL eseguito con metodica e standard "in house"
I dati prodotti dal Gruppo Europeo per la standardizzazione degli aPL sono stati recentemente riconosciuti dal Comitato Internazionale per la Standardizzazione degli autoanticorpi nelle malattie reumatiche (IUIS\WHO\AF\CDC), che intende considerare i 2 monoclonali per accertare la loro idoneità a funzionare come validati campioni di riferimento per i test ELISA aCL e anti-ß2GP1. A livello nazionale, opera dal 1998 il gruppo FIRMA (Forum Interdisciplinareper la Ricerca nelle Malattie Autoimmune), che ha come obiettivo lo studio delle problematiche legate alla determinazione degli autoanticorpi. In que-sti anni, nell’ambito di un progetto che prevedeva la stesura di linee guida per l’esecuzione e l’utilizzo dei test per gli autoanticorpi non organo specifi-ci, sono state prese in esame anche le metodiche per gli anticorpi antifosfo-lipidi. In particolare riportiamo le raccomandazioni o le linee guida FIRMA alle quali i laboratori che testano routinariamente gli aCL dovrebbero atte-nersi: • i campioni debbono essere testati in duplicato;• il test aCL ELISA deve rilevare anticorpi ß2GP1 dipendenti di classe IgG
o IgM. I tamponi utilizzati nella metodica devono contenere il 10% di siero bovino (fetale o adulto) per garantire la quantità idonea di ß2GP1 nel sistema.
• Il cut-off deve essere determinato in ogni laboratorio testando un cam-pione di almeno 100 sieri da soggetti normali distribuiti per età e sesso. E’ necessario utilizzare il sistema dei percentili per il calcolo del cut-off, in quanto la distribuzione dei valori risulta non parametrica. Considerato che il test ELISA è molto sensibile, è consigliabile utilizzare almeno il 99° percentile;

59
• I risultati devono essere espressi in unità GPL o MPL (mg/ml di anticorpo),introducendo in ogni test una curva di calibrazione di almeno 5 punti, creata con gli standard di Harris o con standard secondari calibrati su questi. Sono attualmente in corso di valutazione anticorpi monoclonali umani o umanizzati.
• E’ consigliabile una valutazione semiquantitativa dei risultati: negativo, se sotto il cut-off; positivo basso, se >del cut-off e < 30 GPL/MPL; positivo medio, se >30 e <80 GPL/MPL; positivo alto, se > 80GPL/MPL;
• Utile uno scambio periodico interlaboratori di sieri, per valutare la riproducibilità della metodica.

60Bibliografia
1. Wilson W, Gharavi A, Lockshin MD, Branch DW, Piette JC, Brey R, Derksen R, Harris EN, Hughes GR, Triplett DA, Khamashta MA. International consensus statement on preliminary classification criteria for definite Antiphospholipid Syndrome: report of an international workshop. Arthritis Rheum 1999 42; 1309-1311
2. Hughes GRV. Thrombosis, abortion, cerebral disease and lupus anticoagulant. Br Med J 1987; 287: 1088-1089
3. Roubey RAS. Antiphospholipid antibody sindrome. In Koopman WJ, ed, Arthritis and allied conditions: a textbook of rheumatology, 13th edition 1997; 1393-1406.
4. Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, Jacobsen S, Lakos G, Kontopoulou-Griva I, Galeazzi M, Meroni PL, Derksen RHWM, de Groot PG, Gromnica-Ihle E, Baleva M, Mosca M, Bombardieri S, Houssiau F, Gris JC, Quere I, Hachulla E, Vasconcelos C,Roch B, Fernandez-Nebro A, Boffa MC, Hughes GRV, Ingelmo M. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; in press
5. Alarcon-Segovia d, Perez-Vazquez ME, Villa AR, Drenkard C, Cabiedes J. Preliminary classification criteria for the antiphospholipid syndrome within systemic lupus erythematosus. Seminars Arthritis and Rheum 1992; 21: 275-285
6. Piette JC. Updating the American College of Rheumatology criteria for systemic lupus erythematosus: comment on the letter by Hochberg. Arthritis Rheum 1998; 41(4):751
7. Lockshin MD. Pregnancy loss and antiphospholipid antibodies. Lupus 1998; 7 (Suppl 2):S86-S89
8. Lahita RG. Hormonal contraception and replacement and the use of androgens in the antiphosphoòipid syndrome. Journal of autoimmunity 2000; 15:213-216
9. Ginsberg JS, Wells PS, Brill-Edwards P, Donovan D, Moffatt K, Johnston M, Stevens P, Hirsh J. Antiphospholipid antibodies and venous thromboembolism. Blood 1995; 86: 3685-91
10. Moore JE, Lutz WB. The natural history of systemic lupus erythematosus: an approach to its study trough chronic biologic false positive reactors. J Chronic Dis 1955; 1: 297-316
11. Laurell AB, Nilsson IM. Hypergammaglobulinemia, circulating anticoa-gulant, and biologic false positive Wasserman reaction. J Lab Clin Med 1957; 49: 694-707
12. Feinstein DI, Rapaport SI. Acquired inhibitors of blood coagulation. Prog Haemostas Thromb 1972; 1: 75-95
13. Pangborg MC. Isolation and purification of a serologically active phospholipid from beef heart. J Biol Chem 1942; 143: 247-256

61
14. Harris EN, Gharavi AE, Boey ML, Patel BM, Mackworth-Young CG, Loizou S, Hughes GRV. Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 1983; 2: 1211-1214
15. Loizou S, Mc Crea JD, Rudge AC, Reynolds R, Boyle CC, Harris EN. Measurement of anticardiolipin by an enzyme linked immunosorbent assay (ELISA): standardization and quantitation of results. Clin Exp Immunol 1985: 62:738
16. Galli M, Comfurius P, Maassen, Hemker HC, De Baets MH, Van Breda-Vrieman PJC, Barbui T, Zwaal RFA, Bevers EM. Anticardiolipin antibodiesdirected not to cardiolipin but to a plasma protein cofactor. Lancet 1990; 335: 1544-1547
17. Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K, Koike T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet 1990 ; 336 :177-178
18. McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Antiphospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: b2glycoprotein I (apolipoprotein H). Proc Natl Acad Sci USA 1990; 87:4120-4124
19. Harris EN, Gharavi AE, Patel SP, Hughes GRV.Evaluation of the anti-cardiolipin antibody test: report of an international workshop held 4 april 1986. Clin Exp Immunol 1987; 68: 215-22
20. Roubey RA. Immunology of the antiphospholipid antibody syndrome. Arthritis Rheum 1996; 39: 1444-54
21. Tincani A, Balestrieri G, Spatola L, Cinquini M, Meroni PL, Roubey RA.Anticardiolipin and anti-ß2 glycoprotein 1 immunoassays in the diagnosis of antiphospholipid syndrome. Clin Exp Rheumatol. 1998; 16: 396-402
22. Woehrle R, Oppermann M, Matthias T, Helmke K. Different antiphospholipid-antibodies are indipendent risk factors for thrombotic events and cerebrovascular insult in SLE-patients.Autoimmunity Reviews 2002; 37
23. Tincani A, Spatola L, Prati E, Allegri F, Ferremi P, Cattaneo R, Meroni PL,Balestrieri G. The anti-ß2 glycoprotein 1 activity in human anti-phospho-lipid syndrome sera is due to monoreactive low-affinity autoantibodies directed to epitopes located on native b2-glycoprotein I and preserved during species’ evolution. J 1996; 157: 5732-38
24. Arvieux J, Roussel B, Jacob MC, Colomb MG. Measurement of antiphospholipid antibodies by ELISA using ß2 glycoprotein 1 as antigen. J Immunol Methods 1991; 143:223-229
25. Balestrieri G, Tincani A, Spatola L, Allegri F, Prati E, Cattaneo R, Valesini G, Del Papa N, Meroni PL. Anti-ß2 glycoprotein 1: a marker of antiphospholipid sindrome? Lupus 1995; 4:122-130

62
26. Alarcon Segovia D, Mestanza M, Cabiedes, Cabral A. The antiphospholipid/cofactor syndromes. II. A variant in patients with systemic lupus erythematosus with antibodies to ß2 glycoprotein 1 but not antibodies detectable in standard antiphospholipid assays. J Rheumatol 1997; 24: 1545-51
27. Horbach DA, Voort E, Donders RCJM, Derksen RHWM, De Groot P. Lupus anticoagulant is the strongest risk factor for both venous and arterial thrombosis in patients with systemic lupus erythematosus. Thromb Haemost 1996; 76: 916-924
28. Petri M. Epidemiology of the antiphospholipid syndrome. In The antiphospholipid syndrome 1996; 2:13-28
29. Harris EN. Special report. The Second International Anti-cardiolipin Standardization Workshop/the Kingston Anti-Phospholipid Antibody Study (KAPS) group. Am J Clin Pathol 1990; 94: 476-484
30. Tincani A, Spatola L, Cattaneo R, Balestrieri G. Antiphospholipid ELISA: the state of the art. Lupus 1996; 5: 477-479
31. Tincani A, Balestrieri G, Allegri F, Cinquini M, Vianelli M, Taglietti M, Sanmarco M, Ichikawa K, Koike T, Meroni PL, Boffa MC. Overwiev on anticardiolipin ELISA standardization. J of Autoimmunity 2000; 15:195-197
32. Tincani A, Allegri F, Sanmarco M, Cinquini M, Taglietti M Balestrieri G, Koike T, Meroni PL, Boffa MC. Anticardiolipin antibody assay: a Methodological analysis for a better consensus in routine determinations.Thromb Haemost 2001; 86: 576-583
33. Ichikawa K, Tsutsumi A, Atsumi T, Matsuura E, Kobayashi S, Hughes GRV, Khamashta M, Koike T. A chimeric antibody with the human g1 constant region as a putative standard of assays to detect IgG ß2 glycoprotein 1 antibodies. Arthritis Rheum 1999; 42:2461-2470
34. Ichikawa K, Khamashta M, Koike T, Matsuura E, Hughes GRV. ß2 glycoprotein 1 reactivity of monoclonal anticardiolipin antibodies from patients with the antiphospholipid syndrome. Arthritis Rheum 1994; 37:1453-1461
Indirizzo per la corrispondenza: Dr.ssa Angela TincaniReumatologia Allergologia e Immunologia ClinicaP.le Spedali Civili 1, 25123 BresciaTel: 030 3996449 - 030 3995448Fax: 030 382796 - 030 3995085e-mail: [email protected]

63
3. Anticorpi anti-ß2glicoproteina 1: diagnosi di laboratorio
A. Biasiolo, V. Pengo
Servizio Prevenzione e Terapia TrombosiDipartimento medicina Clinica e SperimentaleOspedale “ex Busonera” Padova
Gli anticorpi antifosfolipidi (aPL) associati a manifestazioni cliniche partico-lari come trombosi venose ed arteriose o aborti ricorrenti, caratterizzano la cosiddetta Sindrome da Anticorpi antifosfolipidi (APS). Il ruolo fisiopatologi-co di questi anticorpi nel provocare le trombosi non è attualmente noto nonostante le numerose ipotesi proposte.Nell’ultimo decennio il significato degli aPL è stato completamente rivoluzio-nato dall’osservazione che tali anticorpi non sono diretti contro i fosfolipidi anionici, bensì riconoscono particolari proteine plasmatiche legate ad altre superfici anioniche (es. fosfolipidi).E’ stato abbondantemente dimostrato inoltre, che la maggior parte degli anticorpi presenti nel siero di pazienti affetti da APS sono diretti principal-mente contro due proteine leganti i fosfolipidi: la ß2 glicoproteina 1 (ß2GP1)e la protrombina.Gli anticorpi anti-ß2 glicoproteina 1 (anti-ß2GP1) sono responsabili della reattività anticardiolipina presente nel siero dei pazienti con APS, mentre l’attività Lupus Anticoagulant (LA) spesso co-presente è attribuibile sia ad anti-ß2GP1 che ad anticorpi anti-protrombina.La diagnosi di laboratorio degli anti-ß2GP1 si basa su un test ELISA che uti-lizza come antigene la ß2GP1 umana purificata.Tuttavia la mancanza di standardizzazione di questo metodo e quindi i dif-ferenti risultati ottenuti su pazienti aventi uguali patologie, rende per alcuni dubbia l’utilità clinica della determinazione degli anti-ß2GP1.
Introduzione

64
La ß2GP1 è una glicoproteina conosciuta da molto tempo: è stata isolata da Schultze e collaboratori più di 40 anni fa (1). Ha un peso molecolare di 50 KDa ed è presente nel plasma alla concentrazione di 0,2 mg/ml. E’ una singola catena polipeptidica costituita da 326 amminoacidi (in prevalenza prolina, cisteina e triptofano) e da 5 oligosaccaridi contenenti glicosammina(2). Mediante tecniche di clonaggio e sequenziamento del c-DNA (3,4) è stata stabilita la completa e corretta sequenza amminoacidica della proteina la quale risulta altamente conservata per più dell’ 80% fra le diverse specie animali (umana, bovina, murina). La molecola della ß2GPI è organizzata in 5 unità omologhe ripetute di circa 60 residui amminoacidici dette”sushi domains”(5) ciascuna con due ponti disolfuro, a parte il V° dominio che ne possiede tre. Numerosi studi, identificando il V° dominio della ß2GP1 comeimportante nel legame della proteina ai fosfolipidi (6-9) suggerivano che il sito di legame fosse localizzato fra gli aminoacidi Cys 281-288 (10).
Recentemente, interessanti studi cristallografici, facendo luce sulla strutturatridimensionale della proteina, hanno fornito utili informazioni circa il funzio-namento di questa molecola. La ß2GP1 appare come una struttura lunga 130 e larga 80 Åmstrong (Fig.1) simile ad una J allungata, organizzata in 5 domini (11,12). Il ripiegamento spaziale del V° dominio devia fortemente da quello standard osservato negli altri 4 domini.
Anticorpi anti-ß2glicoproteina 1: antigene
Figura 1. Struttura tridimensionale della ß2GP1 umana (Bouma B. et al., EMBO J 1999; 18:5166-5177).

65
Il gene che codifica per la ß2GP1 umana è localizzato sul cromosoma 17 (q23-qter) (14) e a tutt’oggi sono stati identificati ben 4 polimorfismi, dovutia mutazioni puntiformi, responsabili di precise sostituzioni amminoacidiche:Ser/Asn 88, Leu/Val 247, Cys/Gly 306, Trp/Ser 316. La differenza anche di un solo amminoacido, può portare ad anomale modificazioni conformazio-nali della proteina, in seguito ad alterate interazioni con i fosfolipidi di mem-brana. Polimorfismi sul sito di legame per i fosfolipidi, oppure sul sito anti-genico della ß2GP1 potrebbero influenzare la produzione di anticorpi anti-ß2GP1 e lo sviluppo della APS. Atsumi e collaboratori hanno osservato che la mutazione in posizione 247, cioè quella localizzata fra i siti di legame ai fosfolipidi nel dominio V° e il potenziale epitopo per gli anticorpi anti-ß2GP1,è più frequente in soggetti bianchi affetti da APS con gli anti-ß2GP1, rispet-to ai pazienti con APS, ma senza tali anticorpi (15). Hirose e colleghi, consi-derando la stessa mutazione fra diversi gruppi razziali con APS, ha osser-vato l’elevata frequenza dell’allele valina in tutti i gruppi considerati (asiatici,bianchi, neri) e una netta predominanza nei soggetti sani bianchi (16). Questi dati potrebbero spiegare l’alta prevalenza della APS nella popolazio-ne dei bianchi. La ß2GP1 mutata in posizione 316, è invece incapace di riconoscere i fosfolipidi anionici. Ciò ha fatto pensare che la presenza di
La sequenza carica positivamente CKNKEKKC presente nel V° dominio e il vicino uncino idrofobico (Ser 311-Lys 317) sembrano essere coinvolti rispettivamente nel legame della proteina ai fosfolipidi anionici e al suo ancoraggio sulle membrane cellulari (Fig.2). Per quanto riguarda gli altri domini sembra che il III° ed il IV°, fortemente glicosilati, siano protetti dalle interazioni proteina–proteina, mentre i domini I° e II° rappresentino i siti di legame riconoscuti dagli anti-ß2GP1 (13).
Figura 2. Schema rappresentativodell’interazione fra ß2GP1 umana e mem-brana fosfolipidica (Bouma B. et al., EMBO J 1999; 18:5166-5177).

66
questa mutazione potesse proteggere in qualche modo i soggetti dalla pro-duzione di anticorpi anti-ß2GP1. Horbach e collaboratori hanno dimostrato che il difetto in forma eterozigote non è protettivo nei pazienti con LES e APS considerati e che forse potrebbe esserlo in forma omozigote (17).L’osservazione della frequenza della mutazione Trp/Ser 316 nella popola-zione ha portato a risultati discordanti, Gushinken ha osservato che la seri-na in questa posizione è associata a trombosi in un piccolo gruppo di pazienti con LES (18), mentre Kamboh, nello stesso anno, ha dimostrato chequesta mutazione è meno comune nei pazienti con aPL rispetto ai controlli (19).Con i dati attualmente in nostro possesso è difficile stabilire i fattori di rischio genetico correlati alla comparsa di anticorpi antifosfolipidi e alle manifestazioni cliniche della APS. Possiamo dedurre che la maggior parte dei pazienti con APS o con solo gli aPL positivi, hanno la forma più comunedi ß2GP1.La ß2GP1 è sintetizzata principalmente dagli epatociti (3), ma anche da altritipi cellulari come cellule endoteliali, neuroni e linfociti (20). Tra le principali proprietà della ß2GP1 ricordiamo la sua capacità di legarsi ai fosfolipidi anionici (21), alle piastrine (22), all’eparina (23), al DNA (24) ed ai mito-condri (25). “In vitro” possiede attività anticoagulante infatti è capace di inibi-re la fase di contatto della coagulazione (26), l’attività protrombinasica delle piastrine normali ed attivate (27) e l’aggregazione indotta da ADP (28). La ß2GP1 ha anche attività procoagulante, infatti inibisce l’attività anticoa-gulante del più importante inibitore della coagulazione: la proteina C attiva-ta (29,30). Tuttavia l’importanza fisiologica della ß2GP1 nella coagulazione rimane in discussione, infatti persone carenti di questa proteina non hanno né segni di sanguinamento né di trombosi (31,32).In passato è stato proposto il suo coinvolgimento nel metabolismo lipidico, come cofattore della lipoproteina lipasi (33). E’ stata definita apolipoproteina H perché presente nei chilomicroni, VLDL, HDL, e soprattutto nella frazionelipoproteica pesante (d>1.23 g/ml) (34), oggi sembra che solo piccole quan-tità di proteina siano legate alle lipoproteine (35). La ß2GP1 sembra anche coinvolta nella rimozione di particelle “non-self” e nel processo dell’apopto-si (36,37). Recentemente è stata dimostrata la suscettibilità della ß2GP1 al trat-tamento con plasmina ed altre proteasi. Nel 1999 Horbach ha dimostrato “in vivo” il clivaggio proteolitico della proteina durante la fibrinolisi e l’aumento della concentrazione plasmatica di una forma di ß2GP1 clivata che possiede minore affinità per i fosfolipidi anionici (38). L’anno successivo Matsuura ha dimostrato che il trattamento con plasmina induce una modifi-cazione conformazionale del V° dominio, che sembra riflettersi in un’ altera-ta esposizione degli epitopi criptici presenti sul IV° dominio responsabili dellegame fra ß2GP1 ed anticorpi specifici. Tutto ciò si traduce in una minore antigenicità della proteina clivata (39). Poiché nel plasma di pazienti con CID (38) e APS (40) si sono dimostrate ele-vate concentrazioni di ß2GP1 clivata si ritiene questo processo, importante nella down-regulation dei fenomeni atero-trombotici autoimmuni nella APS.
La ß2GP1 è il principale “cofattore” degli anticorpi antifosfolipidi di tipo autoimmune. Per molti anni questi anticorpi ed in particolare gli anticorpi aCL sono stati ritenuti diretti contro i fosfolipidi anionici. E’ ormai ampiamen-te dimostrato che gli aCL, nel test ELISA utilizzato per individuarli, ricono-scono la ß2GP1 umana presente nel campione in esame o la ß2GP1 bovina presente nei tamponi di lavaggio e bloccaggio. E’ l’interazione fra la superfi-cie fosfolipidica con le sue cariche negative (nel caso specifico rappresen-tata dalla cardiolipina) e la ß2GP1 a favorire il legame con gli anticorpi (41-43).
Interazioni antigene-anticorpo

67
Esistono comunque anticorpi aCL di tipo infettivo che riconoscono diretta-mente la cardiolipina.Gli anticorpi anti-ß2GP1 invece riconoscono la ß2GP1 in assenza di fosfoli-pidi anionici. Una superficie di plastica particolarmente idrofilica come il polivinilcloruro o il polistirene irradiato con raggi γ è sufficiente per rendere antigenica la molecola.La necessità di una superficie idrofilica adatta, per indurre l’antigenicità della proteina è stata spiegata in due diversi modi. Secondo alcuni, l’intera-zione tra la ß2GP1 e la superficie carica negativamente indurrebbe una modificazione conformazionale nella proteina tale da esporre alcuni epitopicriptici sulla molecola, accessibili agli anticorpi anti-ß2GP1 (44,45).Altri invece ritengono che solo un’elevata densità di superficie della ß2GP1 (come si verifica su una piastra ELISA oppure su una superficie fosfolipidi-ca) possa stabilizzare il legame con l’anticorpo, che ha una bassa affinità (46).Sembra che gli anticorpi anti-ß2GP1 monoclonali riconoscano bene epito-pi localizzati sul IV° dominio (47), mentre pare che gli anticorpi policlonali pre-feriscano epitopi localizzati sul I° dominio (13).
Il coinvolgimento degli anticorpi anti-ß2GP1 nella patogenesi delle manife-stazioni cliniche che caratterizzano la APS è più recente e pertanto meno documentato se paragonato con i dati a disposizione in letteratura circa l’associazione fra aPL in generale e APS (48,49).I meccanismi fisiopatologici attraverso i quali gli anticorpi anti-ß2GP1 indur-rebbero le trombosi (venose e/o arteriose) e gli aborti ripetuti non sono ancora conosciuti. La purificazione per affinità di questi anticorpi, cioè mediante una matrice solida inerte a cui viene legata stabilmente la ß2GP1 umana, è sicuramentel’approccio migliore per studiarne il comportamento “in vitro”(50). Tuttavia in letteratura non sono molti i lavori basati su questa logica, spesso gli studi vengono condotti su plasma di pazienti positivi per gli anticorpi anti-ß2GP1,oppure su IgG totali o IgG isolate mediante liposomi di cardiolipina, quindi con sistemi non completamente puri.E’ ormai accettato che gli anticorpi anti-ß2GP1 possiedono un’attività anti-coagulante nei comuni test fosfolipide-dipendenti (es. dRVVT).Nel 1999 il nostro gruppo ha dimostrato che sei preparazioni di anticorpi anti-ß2GP1, isolate per affinità mostravano un’attività procoagulante “in vitro” tale da accorciare consistentemente il Tempo di Protrombina (PT). Tale accorciamento dipendeva dalla concentrazione di anticorpo nel siste-ma e dalla presenza o meno di ß2GP1, mentre non era influenzato dal tipo di tromboplastina utilizzata. L’accorciamento del PT si potrebbe spiegare con la maggior o più rapida produzione di trombina in relazione ad una maggiore produzione di fattore X attivato (Xa). Salemink e collaboratori infatti hanno dimostrato che gli anticorpi anti-ß2GP1 presenti nel plasma di pazienti con APS, inducono un aumento di fattore Xa, dipendente dalla presenza di ß2GP1 e TFPI. Secondo gli autori i complessi anticorpo anti-ß2GP1 - ß2GP1 formati in presenza di PL, potreb-bero inibire il TFPI impedendo la formazione del complesso quaternario TFPI/FXa/FVIIa/TF con conseguente maggior produzione di fattore Xa (51). Nel tentativo di spiegare il ruolo degli anti-ß2GP1 nella APS, altri autori hanno dimostrato che il plasma di pazienti con aPL, così come le IgG totali e le IgG purificate con liposomi di CL impediscono l’inattivazione del fattoreVa, suggerendo come spiegazione alla patogenesi degli eventi tromboem-bolici l’ipotesi che gli anti-ß2GP1 possano indurre una acquisita resistenza alla proteina C attivata (52).
Anticorpi anti-ß2GP1: patogenesi

68
L’ipotesi invece proposta da Rand e Wu (53) circa lo spiazzamento dell’an-nexina V (proteina con attività anticoagulante) dalle cellule endoteliali e dai trofoblasti placentari indotta da anticorpi è stata recentemente smentita da Willems e Bevers. Gli esperimenti di Willems e colleghi, su modelli di mem-brana, hanno dimostrato che gli anticorpi aCL/ anti-ß2GP1 in presenza di ß2GP1, non sono in grado di alterare lo scudo formato dall’annexina V sullemembrane di queste cellule (54).Bevers e collaboratori, peraltro, non riscontrano alcun ostacolo da parte degli anticorpi anti-ß2GP1 sull’inibizione della produzione di trombina indot-ta dall’annexina V (55). Fino ad oggi l’attenzione dei ricercatori è stata rivolta principalmente alla capacità della ß2GP1 di stimolare i linfociti B nella produzione di autoanti-corpi, tralasciando o considerando marginale l’importanza che questo anti-gene potrebbe avere nell’immunità di tipo cellulare. Già nel 1994 Kornberg aveva riportato che gli aPL erano capaci di stimolare i monociti a produrre un’attività procoagulante (PCA) simile al fattore tissutale (TF) (56). Più recen-temente Visvanathan e McNeil riportano che nel 44% dei pazienti con APS esistono dei linfociti T CD4+ ß2GP1 specifici che proliferano e secernono interferone se stimolati con ß2GP1(57). Gli stessi ricercatori dimostrano con un successivo lavoro che se si stimolano i monociti di pazienti affetti da APS con ß2GP1 in presenza di linfociti T CD4+ e molecole di classe II (MHC) si assiste ad un esposizione di TF sulla superficie cellulare (58). Poiché questo comportamento è esclusivo dei monociti di pazienti con APSe non riguarda i monociti di pazienti con aPL, ma senza la sindrome, gli autori suggeriscono che la diatesi procoagulante nella APS potrebbe esse-re dovuta ad una up-regulation dell’espressione del TF sui monociti indottadalla continua stimolazione dei linfociti T-ß2GP1 specifici ad opera della ß2GP1. Anche altri ricercatori (Hattori e coll.) hanno identificato cellule T-ß2GP1 specifiche. Al contrario del precedente gruppo, Hattori dimostra tuttavia la presenza di queste cellule sia nelle APS che nel gruppo di con-trollo e osserva la capacità di proliferare in risposta alla stimolazione con ß2GP1 ridotta e non nella forma nativa (59).Attualmente le ipotesi proposte sono parimenti accettabili pur non essendoprive di elementi opinabili. Riteniamo molto probabile che gli anticorpi a-ß2GP1 possano essere coinvolti in più di un meccanismo fisiologico nel provocare lo stato di ipercoagulabilità tipico della APS.
Gli anticorpi anti-ß2GP1 sono identificabili mediante test ELISA nei quali la ß2GP1 umana viene adsorbita sulla plastica di piastre da microtitolazionein assenza di fosfolipidi. Il test, proposto per la prima volta nel 1991 dalla Dott.ssa Arvieux, è un test ELISA di tipo indiretto (60). Gli anticorpi anti-ß2GP1presenti nel campione da analizzare reagiscono con lo specifico antigene (ß2GP1) fissato alla plastica. Per impedire i legami aspecifici al sistema, solitamente si impiega una soluzione al 10% di siero fetale bovino in tampo-ne fosfato. Dopo ripetuti lavaggi per allontanare i materiali non legati speci-ficamente all’antigene, l’isotipo (IgG, IgM o IgA) del complesso antigene-anticorpo viene rivelato, come nell’ELISA aCL, mediante un antisiero anti-immunoglobuline umane marcato con un’enzima (fosfatasi alcalina o peros-sidasi). L’aggiunta del substrato specifico (p-nitrofelilfosfato o perossido di idrogeno) innesca una reazione cromogenica, misura dell’attività enzimati-ca e proporzionale alla quantità di anticorpo specifico presente nel campio-ne originale.La determinazione degli anticorpi anti-ß2GP1 in questo test è fortemente correlata al tipo di piastra utilizzata. Superfici molto idrofiliche, come le pia-
Diagnosi di laboratorio

69
stre di polivinilcloruro (45) o di polistirene ossidate dal trattamento con raggi γ (44)
rendono antigenica la ß2GP1 favorendo il legame dell’anticorpo specifico. Il plasma dei pazienti affetti da APS, così come gli anti-ß2GP1 purificati per affinità da questi soggetti, non sono in grado di riconoscere la ß2GP1 né immobilizzata su piastre in polistirene normale, né in fase fluida. Il comportamento eterogeneo degli anti-ß2GP1 nel riconoscere l’antigene impiegando supporti solidi di diversa composizione è stato dimostrato recentemente da Tsutsumi e colleghi. Il siero di 10 pazienti con APS e 3 anticorpi monoclonali anti-ß2GP1 diretti contro epitopi localizzati su domini diversi, sono stati testati utilizzando ben 20 differenti tipi di piastre ELISA. Nonostante la maggior parte delle piastre fosse in grado di misurare gli anti-ß2GP1, significative differenze si verificavano utilizzando gli anticorpi monoclonali (61).E’ auspicabile che i risultati di questo lavoro, unitamente all’esito del proget-to europeo di standardizzazione condotto da Arvieux e Reber, portino all’identificazione di un gold-standard per l’ELISA anti-ß2GP1.Infatti nella determinazione degli anti-ß2GP1 la maggior parte dei laboratori utilizza kit del commercio rapidi e di facile esecuzione, pur essendo altresì numerosi i gruppi che impiegano invece metodi “fatti in casa”. L’enorme variabilità interlaboratorio dei risultati ha spinto i ricercatori alla realizzazione del progetto multicentrico di standardizzazione i cui risultati preliminari sono stati presentati all’ 8° Simposio Internazionale degli aPL (Sapporo 1998). Dallo studio è emerso che le differenze nei risultati sono dovute principal-mente ai diversi valori di cut-off calcolati nei laboratori partecipanti e che l’uso di uno standard di riferimento unico permetterebbe di migliorare la standardizzazione del test (62). Il processo di standardizzazione non è sicu-ramente un compito facile, infatti il test ELISA è caratterizzato da tanti passaggi ed è particolarmente ricco di variabili. Nel tentativo di una sua
Figura 3. SDS-PAGE di due pre-parazioni di ß2-GP1 umana. In entrambe le preparazioni sono evidenti solo le bande a 50kDa relative alla proteina in esame.
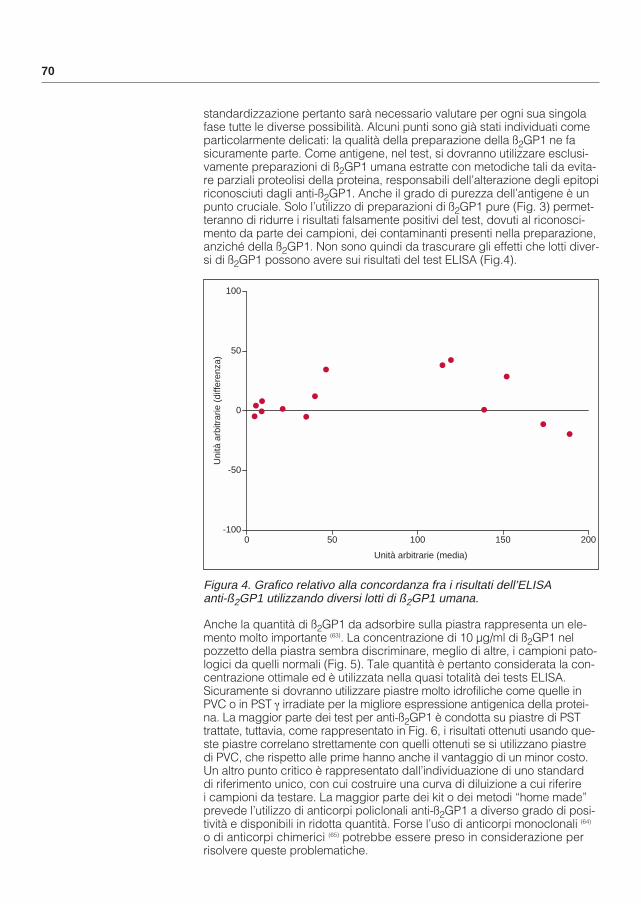
70
standardizzazione pertanto sarà necessario valutare per ogni sua singola fase tutte le diverse possibilità. Alcuni punti sono già stati individuati come particolarmente delicati: la qualità della preparazione della ß2GP1 ne fa sicuramente parte. Come antigene, nel test, si dovranno utilizzare esclusi-vamente preparazioni di ß2GP1 umana estratte con metodiche tali da evita-re parziali proteolisi della proteina, responsabili dell’alterazione degli epitopiriconosciuti dagli anti-ß2GP1. Anche il grado di purezza dell’antigene è un punto cruciale. Solo l’utilizzo di preparazioni di ß2GP1 pure (Fig. 3) permet-teranno di ridurre i risultati falsamente positivi del test, dovuti al riconosci-mento da parte dei campioni, dei contaminanti presenti nella preparazione,anziché della ß2GP1. Non sono quindi da trascurare gli effetti che lotti diver-si di ß2GP1 possono avere sui risultati del test ELISA (Fig.4).
Figura 4. Grafico relativo alla concordanza fra i risultati dell’ELISA anti-ß2GP1 utilizzando diversi lotti di ß2GP1 umana.
0
Unità arbitrarie (media)
Uni
tà a
rbitr
arie
(di
ffere
nza)
50 100 150 200-100
-50
0
50
100
Anche la quantità di ß2GP1 da adsorbire sulla piastra rappresenta un ele-mento molto importante (63). La concentrazione di 10 µg/ml di ß2GP1 nel pozzetto della piastra sembra discriminare, meglio di altre, i campioni pato-logici da quelli normali (Fig. 5). Tale quantità è pertanto considerata la con-centrazione ottimale ed è utilizzata nella quasi totalità dei tests ELISA.Sicuramente si dovranno utilizzare piastre molto idrofiliche come quelle in PVC o in PST γ irradiate per la migliore espressione antigenica della protei-na. La maggior parte dei test per anti-ß2GP1 è condotta su piastre di PST trattate, tuttavia, come rappresentato in Fig. 6, i risultati ottenuti usando que-ste piastre correlano strettamente con quelli ottenuti se si utilizzano piastre di PVC, che rispetto alle prime hanno anche il vantaggio di un minor costo.Un altro punto critico è rappresentato dall’individuazione di uno standard di riferimento unico, con cui costruire una curva di diluizione a cui riferire i campioni da testare. La maggior parte dei kit o dei metodi “home made” prevede l’utilizzo di anticorpi policlonali anti-ß2GP1 a diverso grado di posi-tività e disponibili in ridotta quantità. Forse l’uso di anticorpi monoclonali (64)
o di anticorpi chimerici (65) potrebbe essere preso in considerazione per risolvere queste problematiche.
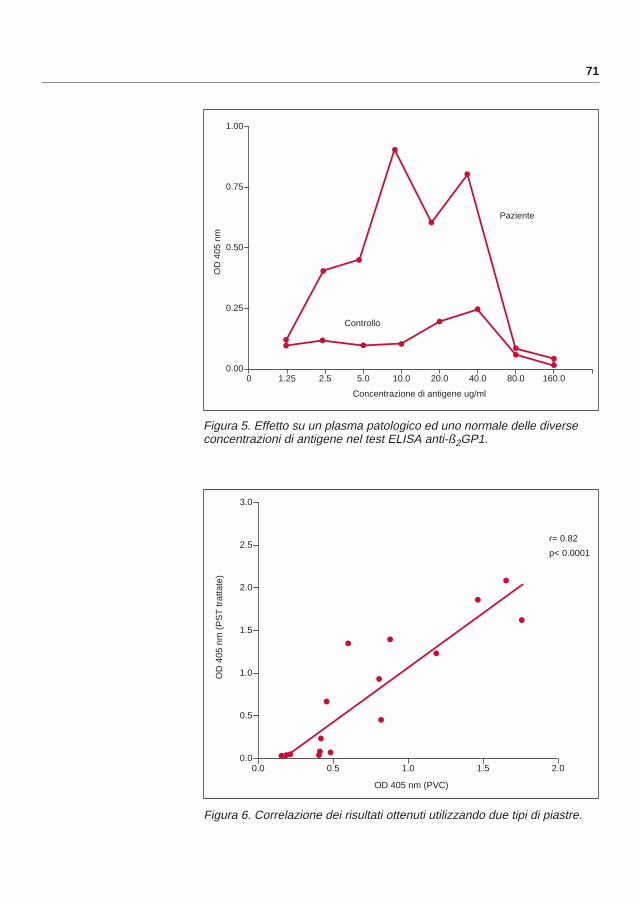
71
0
Concentrazione di antigene ug/ml
Controllo
PazienteO
D 4
05 n
m
1.25 2.5 5.0 10.0 20.0 40.0 80.0 160.00.00
0.25
0.50
0.75
1.00
Figura 5. Effetto su un plasma patologico ed uno normale delle diverse concentrazioni di antigene nel test ELISA anti-ß2GP1.
0.0
r= 0.82
p< 0.0001
OD 405 nm (PVC)
OD
405
nm
(P
ST
trat
tate
)
0.5 1.0 1.5 2.00.0
0.5
1.0
1.5
2.0
2.5
3.0
Figura 6. Correlazione dei risultati ottenuti utilizzando due tipi di piastre.

72
E’ accettato che il Lupus Anticoagulant (LA) rappresenti, tra i test che misu-rano gli aPL, il più importante fattore di rischio tromboembolico. Un sottogruppo di anti-ß2GP1 si comporta “in vitro” come un classico LA: questi anticorpi sono capaci infatti di interferire con i più comuni test coa-gulativi fosfolipide-dipendenti. E’ probabile che essi, formando dei com-plessi bivalenti stabili con la ß2GP1 sulla superficie fosfolipidica (66) o poten-ziando la concentrazione di ß2GP1 sulla superficie stessa (67), impediscano illegame dei fattori della coagulazione (68). Nel 1997 abbiamo dimostrato che la concentrazione dei fosfolipidi è cruciale per l’espressione dell’attività LA di anti-ß2GP1 purificati per affinità nel test dRVVT (69). Tale attività tende a scomparire in presenza di un eccesso o in caso di completa assenza di fosfolipidi nel sistema. Pertanto l’esecuzione del dRVVT in queste due con-dizioni potrebbe a nostro avviso rappresentare un buon test di screening per individuare gli anti-ß2GP1 con attività LA. Test privi di PL esogeni comeil Tempo di coagulazione al caolino (KCT) o che impiegano reagenti con bassissime concentrazioni di fosfolipidi come il Tempo di inibizione della tromboplastina (TTI) sono da considerare invece inadeguati (70).Galli e colleghi hanno proposto due profili coagulativi diversi per distingue-re anticorpi differenti aventi attività LA. I pazienti positivi per LA, che pro-lungano più il dRVVT rispetto al KCT (profilo dRVVT) hanno un maggior rischio per le trombosi rispetto a quelli che appartengono all’altro profilo. Sembra che il profilo dRVVT, associato agli anticorpi ß2GP1 dipendenti sia più strettamente correlato agli eventi tromboembolici mentre il profilo KCT sarebbe più indicativo della presenza di anticorpi anti-protrombina (71). Poter individuare la prevalenza di un tipo di anticorpo rispetto all’altro mediante un profilo coagulativo sarebbe di enorme importanza dal punto di vista clinico.Per misurare l’effetto degli anti-ß2GP1 sull’inibizione della produzione di trombina Sheng e collaboratori hanno recentemente proposto un metodo cromogenico più sensibile rispetto ai convenzionali test coagulativi normal-mente impiegati nella diagnosi del LA e capace di distinguere gli anti-ß2GP1 caratteristici delle APS da quelli prodotti da cause diverse (72).
Gli anti-ß2GP1 sono più fortemente associati alle manifestazioni cliniche della APS rispetto agli aCL (73). Numerosi sono ormai gli studi che stabiliscono una stretta correlazione fra anti-ß2GP1 e trombosi arteriose e venose (45,74-76).Balestrieri e colleghi hanno trovato anche un’associazione fra questi anti-corpi e perdite fetali ricorrenti (63).La domanda che ci si pone da qualche anno è la seguente: gli anti-ß2GP1 si possono considerare un marker per fare la diagnosi di APS? Allo stato attuale, disponendo solo di dati relativi a studi retrospettivi, condotti su un numero esiguo di pazienti e ottenuti impiegando metodi non ben standar-dizzati, rispondere è difficile.Tuttavia alcuni ricercatori hanno dimostrato che gli anti-ß2GP1 hanno un valore predittivo per le trombosi più alto rispetto agli aCL (77,78).L’importanza di questi anticorpi è stata solo parzialmente riconosciuta a livello internazionale, tanto che i criteri per fare diagnosi certa di APS sono stati leggermente rivisti e modificati. Fra i criteri di laboratorio compare la definizione di positività per IgG e/o IgM a medio-alto titolo persistente, di anticorpi anticardiolipina-ß2dipendenti. Tale definizione ci sembra tuttavia confondente, perché non stabilisce con precisione il test da eseguire.
Proprietà anticoagulante degli anticorpi anti-ß2GP1
Significato clinico degli anticorpi anti-ß2GP1

73
Infatti nel test ELISA aCL classico, in cui l’antigene è rappresentato dalla CL, la ß2GP1 è presente nel siero fetale bovino del tampone bloccante e di diluizione. Pertanto, con questa tecnica si possono evidenziare tantissimianticorpi: quelli che riconoscono la ß2GP1(bovina o umana), quelli diretti con-tro la CL (da noi definiti aCL autentici) presenti nelle patologie infettive e quellidiretti contro altre proteine leganti la CL (protrombina, fattore C4, fattore H) (79).Nel fare la diagnosi di APS l’utilizzo del test ELISA per anti-ß2GP1 (descrittoprecedentemente), offre a nostro avviso alcuni vantaggi:1. permette di quantificare gli anti-ß2GP1 specie-specifici, cioè capaci
di riconoscere la ß2GP1 umana e non quella bovina2. non rileva gli aCL autentici che non sono associati alle APS ma a
patologie di tipo infettivo.Nel nostro laboratorio solitamente il plasma positivo in ELISA aCL standardviene testato anche in ELISA per anti-ß2GP1 umana soprattutto se il cam-pione è risultato LA negativo secondo lo schema riportato nella Fig.7. Se il dosaggio risulta positivo a medio-alto titolo (>40 unità) anche a distanza di 6 settimane, facciamo diagnosi di APS.Nonostante la buona correlazione esistente fra ELISA anti-ß2GP1 ed aCL spesso troviamo campioni che mostrano dei risultati discordanti.
aCL + (LA -)
ELISA anti-ß2GP1umana
ELISA anti-ß2GP1bovina
ELISA a CLmodificato
ELISA proteine leganti la CL(protrombina, Fattore H,
Fattore C4, etc.)
negativo positivo
Figura 7. Schema per lo studio degli anticorpi antifosfolipidi in presenza di aCL positivi e LA negativo.

74
Figura 8. A) SDS-PAGE relativo alle proteine leganti la CL, oltre all’albu-mina (66kDa), alla ß2-GP1 (50kDa) ed alle IgG (150kDa), sono evidenti 3 bande ad elevato peso molecolare; B) SDS-PAGE delle tre proteine isola-te; C) identificazione mediante Western blot del componente C4 del com-plemento (1) e del Fattore H (2).
Se il test ELISA anti-ß2GP1 risulta negativo, procediamo dapprima nell’escludere la presenza di anticorpi aCL di tipo infettivo testando il cam-pione con un’ ELISA aCL modificato (cioè utilizzando tamponi privi di ß2GP1). Ripetiamo poi l’ELISA a-ß2GPI utilizzando come antigene la ß2GP1 bovina, poiché sono state dimostrate in letteratura anche positività specie-specifiche (80). Infine procediamo nella ricerca di anticorpi diretti con-tro altre proteine leganti i fosfolipidi (Fig. 8). Per fare la diagnosi di APS, la nostra esperienza ci suggerisce di affiancare il test ELISA anti-ß2GP1 ai testconvenzionali (ELISA aCL, LA) e di testare anche i soggetti che possiedonole caratteristiche cliniche tipiche della sindrome, ma che risultano negativi ai test classici. La diagnosi basata solamente sulla positività in ELISA aCL,in mancanza di anti-ß2GP1 ed LA, potrebbe infatti indurre ad una sovrasti-ma dei pazienti con APS (81).Sicuramente la realizzazione della standardizzazione del test e di studi pro-spettici longitudinali aiuterà a chiarire il ruolo di questi anticorpi nella praticaclinica.

75
1) Schultze HE, Heide K, Haupt H. Über ein bisher unbekanntes niedermolekulares ß2-Globulin des humanserum. Naturwissenschaften 1961;48:719.
2) Lozier J, Takahashi N, Putnam FW. Complete amino acid sequence of human plasma ß2-glycoprotein 1. Proc Natl Acad Sci 1984;81:3640-4.
3) Steinkasserer A, Estaller C, Weiss EH, Sim RB, Day AJ. Complete nucleotide and deduced amino acid sequence of human ß2-glycoprotein 1.Biochem J 1991;277:387-91.
4) Kristensen T, Schousboe I, Boel E et al FEBS Letter 1992;289:183-6.
5) Kato H, Enjyoji K. Amino acid sequence and location of the disulfide bonds in bovine ß2-glycoprotein 1 : the presence of five sushi domains. Biochemistry 1991:30:11687-94.
6) Hunt JE, Simpson RJ, Krilis SA. Identification of a region of ß2-glycopro-tein 1 critical for lipid binding and anti-cardiolipin antibody cofactor activity. Proc Natl Acad Sci 1993;90:2141-5.
7) Kertesz Z, Yu B, Steinkasserer A, Haupt H, Benham A, Sim RB, Characterization of binding of human ß2-glycoprotein 1 to cardiolipin. Biochem J 1995;310:315-21.
8) Igarashi M, Matsuura E, Igarashi Y, Nagae H, Ichikawa K, Triplett DA, Koike T. Human ß2-glycoprotein 1 as an anticardiolipin cofactor determined using deleted mutants expressed by a baculovirus system. Blood 1996;87:3262-70.
9) Hagihara Y, Goto Y, Kato H, Yoshimura T. Structure and function of ß2-glycoprotein 1: special reference to the interaction with phospholipid. Lupus 1995;4 suppl 1:S3-5.
10) Hunt J, Krilis S. The fifth domain of ß2-glycoprotein 1 contains a phospholipid binding site (Cys 281-Cys 288) and a region recognized by anticardiolipin antibodies. J Immunol 1994;152:653-9.
11) Bouma B, de Groot PG, van den Elsen JM, Ravelli RBG, Schouten A, Simmelink M, Derksen RHWM, Kroon J, Gros P. Adhesion mechanism of human ß2-glycoprotein 1 to phospholipid based on its crystal structure.EMBO J 1999;18:5166-74.
12) Schwarzenbacher R, Zeth K, Diederichs K, Gries A, Kostner GM, Laggner P, Prassi R. Crystal structure of human ß2-glycoprotein 1: implication for phospholipid binding and the antiphospholipid syndrome. EMBO J 1999;18:6228-39.
13) McNeely PA, Victoria EJ, Marquis D, Crisologo JF, Tuyay DC, Linnik MD. APS patient sera preferentially recognize the first domain of ß2-glycoprotein 1. Lupus 1998;7:S176 (abstract).
14) Steinkasserer A, Cockburn DJ, Black DM, Boyd Y, Solomon E, Sim RB. Assignment of apolipoprotein H (APOH: ß2-glycoprotein 1) to human chromosome 17q23qter; determination of the major expression site. Cytogenet Cell Genet 1992;60:31-3.
Bibliografia

76
15) Atsumi T, Tsutsumi A, Amengual O, Khamashta MA, Hughes GRV, Miyoshi Y, Ichikawa K, Koike T. Correlation between ß2-glycoprotein 1 valine/leucine 247 polymorphism and anti-ß2-glycoprotein 1 antibodies in patients with primary antiphospholipid syndrome. Rheumatology 1999;38:721-3.
16) Hirose N, Williams R, Alberts AR, Furie RA, Chartash EK, Jain RA, Sison C,Lahita RG, Merrill JT, Cucurull E, Gharavi AE, Sammaritano LR, Salmon JE,Hashimoto S, Sawada T, Chu CC, Gregersen PK, Chiorazzi N. A role for the polymorphism at position 247 of the ß2-glycoprotein 1 gene in the generation of anti-ß2-glycoprotein 1 antibodies in the antiphospholipid syndrome. Arthritis Rheum 1999;42:1655-61.
17) Horbach DA, van Oort E, Tempelman MJ, Derksen RHWM, de Groot PG.The prevalence of a non-phospholipid-binding form of ß2-glycoprotein 1in human plasma. Thromb Haemost 1998;80:791-7.
18 Gushiken FC, Arnett FC, Ahn C, Thiagarajan P. Polymorphism of ß2-glycoprotein 1 at codons 306 and 316 in patients with systemic lupus erythematosus and antiphospholipid syndrome. Arthritis Rheum 1999;42:1189-93.
19) Kamboh MI, Manzi S, Medhi H, Fitzgerald S, Sanghera DK, Kuller LH, Atson CE. Genetic variation in apolipoprotein H (ß2-glycoprotein 1) affects the occurrence of antiphospholipid antibodies and apolipoproteinH concentrations in systemic lupus erythematosus. Lupus 1999;8:742-50.
20) Caronti B, Calderaro C, Alessandri C, Conti F, Tinghino R, Paladini G, Valesini G. ß2-glycoprotein 1 (ß2-GP1) mRNA is expressed by several cell types involved in anti-phospholipid syndrome-related tissue damage. Clin Exper Immunol 1999;115:214-9.
21) Wurm H. ß2-Glycoprotein 1 (Apolipoprotein H) interactions with phospholipid vesicles. Int J Biochem 1984;16:511-5.
22) Schousboe I. Binding of ß2-Glycoprotein 1 to platelets: effect of adenilate cyclase activity. Thromb Res 1980;19:225-37.
23) Polz E, Wurm H, Kostner GM. Investigation on ß2-glycoprotein 1 in the rat: Isolation from serum and demonstration in lipoprotein density fractions. Int J Biochem 1979;11:265-70.
24) Kroll J, Larsen JK, Loft H, Ezban M, Wallevik K, Faber M. DNA-binding proteins in Yoshida ascites tumor fluid. Biochim biophys Acta 1976; 434:490-501.
25) Schousboe I. Purification, characterization and identification of an agglutinin in human serum. Biochim biophys Acta 1979;579:393-408.
26) Schousboe I. ß2-glycoprotein 1: a plasma inhibitor of the contact activa-tion of the intrinsic blood coagulation pathway. Blood 1985;66: 1086-91.
27) Nimpf J, Bevers EM, Bomans PHH, Till U, Wurm H, Kostner GM, Zwaal RF.Prothrombinase activity of human platelets is inhibited by ß2-glycopro-tein 1. Biochim biophis Acta 1986;884:142-9.
28) Nimpf J, Wurm H, Kostner GM. Interaction of ß2-glycoprotein 1 with human blood platelets: influence upon the ADP-induced aggregation. Thromb Haemost 1985;54:397-401.

77
29) Mori T, Takeya H, Nishioka J, Gabazza EC, Suzuki K. ß2-Glycoprotein 1 modulates the anticoagulant activity of activated protein C on the phospholipid surface. Thromb Haemost 1996;75:49-55.
30) Matsuda J, Gohchi K, Kawasugi K, Gotoh M, Saitoh N, Tsukamoto M. inhibitory activity of anti-ß2-glycoprotein 1 antibody on factor Va degradation by activated-protein C and its cofactor protein S. Am J Hematol 1995;49:89-91.
31) Bancsi LFJMM, van der Linden IK, Bertina RM. ß2-glycoprotein 1 deficiency and the risk of thrombosis. Thromb Haemost 1992;67:649-53.
32) Lane DA, Mannucci PM, Bauer KA, Bertina RM, Bochkov NP, Boulyjienkov V, Chandy M, Dahlbäck B, Ginger EK, Miletich JP, et al. Inherited thrombophilia: part 1. Thromb Haemost 1996;76:651-62.
33) Nakaya Y, Schaefer EJ, Brewer BH Jr. Activation of human post heparin lipoprotein lipase by apolipoprotein H (ß2-glycoprotein 1). Biochem Biophys Res Commun 1980;95:1168-1172.
34) Polz E, Kostner GM. The binding of ß2-glycoprotein 1 to human serum lipoproteins: distribution among density fractions. FEBS Letter 1979; 102:183-6.
35) Gambino R, Ruiu G, Pagano G, Cassader M. The binding of apolipo-protein H (ß2-glycoprotein 1) to lipoproteins. Prostaglandins Other Lipid Mediat 1999;57:351-359.
36) Chonn A, Semple SC, Cullis PR. ß2-glycoprotein 1 is a major protein associated with very rapidly cleared liposomes in vivo, suggesting a significant role in the immune clearance of “non-self” particles. J Biol Chem 1995;270:25845-9.
37) Manfredi AA, Rovere P, Heltai S, Galati G, Nebbia G, Tincani A, BalestrieriG, Sabbadini MG. Apoptotic cell clearance in systemic lupus erythema-tosus -II. Role of ß2-glycoprotein 1. Arthritis Rheum 1998; 41:215-23.
38) Horbach DA, van Oort E, Lisman T, Meijers JCM, Derksen RHWM, de Groot PG. ß2-Glycoprotein 1 is proteolytically cleaved in vivo upon activation of fibrinolysis. Thromb Haemost 1999;81:87-95.
39) Matsuura E, Inagaki J, Kasahara H, Yamamoto D, Astumi T, Kobayashi K,Zhao D, Ichikawa K, Tsutsumi A, Yasuda T, Triplett DA, Koike T. Proteolytic cleavage of ß2-glycoprotein 1: reduction of antigenicity and the structural relationship. Int Immunol 2000;12:1183-92.
40) Kato H, Ohkura N, Hagihara Y, Yoshimura T, Goto Y. Generation of nicked form of domain V in ß2-glycoprotein 1 by plasmin: its in vivov significance. Lupus 1998;7 (suppl. 2):S174 (Abstr.).
41) Mc Neil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid binding inhibitor of coagulation: ß2-Glycoprotein 1 (Apolipoprotein H). Proc Natl Acad Sci 1990;87:4120-4.
42) Galli M, Comfurius P, Maassen C, Hemker HC, De Baets MH, Van Brieda-Vriesman PJC, Barbui T, Zwaal RFA, Bevers EM. Anticardiolipin antibodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990;335:1544-7.

78
43) Matsuura E, Igarashi Y, Fujimoto M, Ichikawa K, Koike T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet 1990;336:177-8.
44) Matsuura E, Igarashi Y, Yasuda T, Triplett DA, Koike T. Anticardiolipin antibodies recognize ß2-glycoprotein 1 structure altered by interacting with an oxygen modified solid phase surface. J Exp Med 1994;179:457-62.
45) Pengo V, Biasiolo A, Fior MG. Autoimmune antiphospholipid antibodies are directed against a cryptic epitope expressed when ß2-glycoprotein 1is bound to a suitable surface. Thromb Haemost 1995:73:29-34.
46) Roubey RAS, Eisemberg RA, Harper MF, Winfield JB. “Anticardiolipin” autoantibodies recognized ß2-glycoprotein 1 in the absence of phospholipids. J Immunol 1995;154:954-60.
47) Ichikawa K, Khamashta MA, Koike T, Matsuura E, Hughes GRV. ß2-glycoprotein 1 reactivity of monoclonal anticardiolipin antibodies from patients with the antiphospholipid syndrome. Arhritis Rheum 1994;37:1453-61.
48) Pengo V, Biasiolo A, Brocco T, Tonetto S, Ruffatti A. Autoantibodies to phospholipid-binding plasma proteins in patients with thrombosis and phospholipid-reactive antibodies. Thromb Haemost 1996;75:721-4.
49) Viard JP, Amoura Z, Bach JF. Association of anti-ß2-glycoprotein 1 antibodies with lupus-type circulating anticoagulant and thrombosis in systemic lupus erythematosus. Am J Med 1992;93:181-6.
50) Pengo V, Brocco T, Biasiolo A, Rampazzo P, Carraro P, Zamarchi R. Procoagulant effect of anti-ß2-glycoprotein 1 antibodies with Lupus Anticoagulant activity. Blood 1999; 94:3814-9.
51) Salemink I, Blezer R, Willems GM, Galli M, Bevers EM, Lindhout T. Antibodies to ß2-glycoprotein 1 associated with antiphospholipid syndrome suppress the inhibitory activity of tissue factor pathway inhibitor. Thromb Haemost 2000;84:653-6.
52) Galli M, Ruggeri L, Barbui T. Differential effects of anti-ß2-glycoprotein 1and antiprothrombin antibodies on the anticoagulant activity of activatedprotein C. Blood 19998;91:1999-2004.
53) Rand JH, Wu X-X. Antibody-mediated disruption of the annexin-V antithrombotic shield: a new mechanism for thrombosis in the antiphospholipid syndrome. Thromb Haemost 1999;82:649-55.
54) Willems GM, Janssen MP, Comfurius P, Galli M, Zwaal RF, Bevers EM. Competition of annexin V and anticardiolipin antibodies for binding to phosphatidylserine containing membranes. Biochemistry 2000; 39:1982-9.
55) Bevers EM, Janssen MP, Willems GM, Zwaal FA. No evidence for enhanced thrombin formation through displacement of annexin V by antiphospholipid antibodies [letter]. Thromb Haemost 2000;83:792-4.
56) Kornberg A, Blank M, Kaufman S, Shoenfeld Y. Induction of tissue-factor like activity in monocytes by anticardiolpin antibodies. J Immunol 1994:153:1328-32.

79
57) Visvanathan S, McNeil HP. Cellular Immunity to ß2-glycoprotein 1 in patients with the antiphospholipid syndrome. J Immunol 1999; 162:6919-25.
58) Visvanathan S, Geczy CL, Harmer JA, McNeil HP. Monocyte tissue factor induction by activation of ß2-glycoprotein 1 -specific T lymphocytesis associated with thrombosis and fetal loss in patients with antiphospholipis antibodies. J Immunol 2000;165:2258-62.
59) Hattori N, Masataka K, Junichi K, Tsuneyo M, Yasuo I, Yutaka K. T cell that are autoreactive to ß2-glycoprotein 1 in patients with antiphospholipidsyndrome and healthy individuals. Arhtritis Rheum 2000;43:65-75.
60) Arvieux J, Roussel B, Jacob MC, Colomb MG. Measurement of antiphospholipid antibodies by ELISA using ß2-glycoprotein 1 as an antigen. J Immunol Methods 1991;143:223-9.
61) Tsutsumi A, Ichikawa K, Matsuura E, Sawada KI, Koike T. Heterogeneous behavior of anti-ß2-glycoprotein 1 antibodies on various commercially available enzyme immunoassay plates coated with ß2-glycoprotein 1. J Rheumatol 2000;27:391-6.
62) Arvieux J, Schousboe I, Boffa MC. On behalf of the participants to the European Forum on the antiphospholipid antibodies (aPL): multicenter evaluation of the Elisa for anti-ß2-glycoprotein 1 antibodies. Lupus 1998;7:S183(abstract).
63) Balestrieri G, Tincani A, Spatola L. Anti-ß2-glycoprotein 1 antibodies: a marker of antiphospholipid sindrome? Lupus 1195;4:122-30.
64) Arnout J, Meijer P, Vermylen J. Lupus anticoagulant testing in Europe: an analysis of results from the first European Concerted Action on Thrombophilia (ECAT) survey using plasmas spiked with monoclonal antibodies against human ß2-glycoprotein 1. Thromb Haemost 1999; 81:929-34.
65) Ichikawa K, Tsutsumi A, Atsumi T, Matsuura E, Kobayashi S, Hughes GR, Kamashta MA, Koike T. A chimeric antibody with the human 1 constant region as a putative standard for assay to detect IgG ß2-glycoprotein 1 -dependent anticardiolipin and anti-ß2-glycoprotein 1 antibodies. Arthritis Rheum 1999;42:2461-70.
66) Arnout J, Wittevrongel C, Vanrusselt M, Hoylaerts M, Vermylen J. ß2-glycoprotein I dependent lupus anticoagulants form stable bivalent antibody-ß2-glycoprotein 1 complexes on phospholipid surfaces. Thromb Haemost 1998;79:79-86.
67) Takeya H, Mori T, Gabazza EC, Kuroda K, Deguchi H, Matsuura E, Ichikawa K, Koike T, Suzuki K. Anti-ß2-Glycoprotein 1 monoclonal antibodies with lupus anticoagulant activity enhance the ß2-Glycoprotein 1 binding to phospholipid. J Clin Invest 1997;99:2260-8.
68) Willems GM, Janssen MP, Pelsers MMAL, Comfurius P, Galli M, Zwaal RFA, Bevers EM. Role of divalency in the high affinity binding of anti-cardiolipin antibody-ß2-Glycoprotein 1 complexes to lipid membranes. Biochemistry 1996;35:13833-42.

80
69) Pengo V, Balestrieri G, Tincani A, Spatola L, Biasiolo A, Brocco T. Utilization of dilute Russell’s viper venom time to detect autoantibodies against ß2-Glycoprotein 1 which express anticoagulant activity in the presence but not in the absence of exogenous phospholipids. Thromb Haemost 1997;77:123-6.
70) Pengo V, Biasiolo A, Ramazzo P, Brocco T. dRVVT is more sensitive than KCT or TTI for detecting lupus anticoagulant activity of anti-ß2-glycoprotein 1 antibodies. Thromb Haemost 1999;81:256-8.
71) Galli M, Finazzi G, Bevers EM, Barbui T. Kaolin clotting time and dilute Russell’s viper venom time distinguish between prothrombin-dependent and ß2-Glycoprotein 1 -dependent antiphospholipid antibodies. Blood 1995;86:617-23.
72) Sheng Y, Hanly GJ, Reddel SW, Kouts S, Guerin J, Koike T, Ichikawa K, Sturgess A, Krilis SA. Detection of “antiphospholipid” antibodies: a single chromogenic assay of thrombin generation sensitively detects lupus anticoagulants, anticardiolipin antibodies, plus antibodies bindingß2-glycoprotein 1 and prothrombin. Clin Exp Immunol 2001;124:502-8.
73) McNally T, Mackie IJ, Machin SJ, Isenberg DA. Icreased levels of ß2-glycoprotein 1 antigen and ß2-glycoprotein 1 binding antibodies are associated with a history of thromboembolic complications in patients with SLE and primary antiphospholipid syndrome. Br J Rheumatol 1995;34:1031-6.
74) Cabral AR, Cabiedes J, Alarcon-Segovia D. Antibodies to phospholipid-free ß2-glycoprotein 1 in patients with primary antiphospholipid syndrome. J Rheumatol 1995;22:1894-8.
75) Amengual O, Atsumi T, Khamashta MA, Koike T, Hughes GRV. Specificity of ELISA for antibody to ß2-glycoprotein 1 in patients with antiphospholipid syndrome. Br J Rheumatol 1996;35:1239-43.
76) Galli M. Which antiphospholipid antibodies should be measured in the Antiphospholipid Syndrome? Haemost 2000;30:57-62.
77) Roubey RAS, Maldonado MA, Byrd S. Comparison of an enzyme-linked immunosorbent assay for antibodies to ß2-glycoprotein 1 and a conven-tional anticardiolipin immunoassay. Arthritis Rheum 1996;39:1606-7.
78) Tsutsumi A, Matsuura E, Ichikawa K, Fujisaku A, Mukai M, Kobayashi S, Koike T. Antibodies to ß2-glycoprotein 1 and clinical manifestation in patients with systemic lupus erythemotosus. Arthritis Rheum 1996; 39:1466-74.
79) Rampazzo P, Biasiolo A, Garin J, Rosato A, Betterle C, Ruffatti A, Pengo V. Some patients with antiphospholipid sindrome express hitherto undescribed antibodies to cardiolipin-binding proteins. Thromb Haemost 2001;85:57-62.
80) Arvieux J, Regnault V, Hachulla E, Darnigel L, Roussel B, Bensa JC. Heterogeneity and immunochemichal properties of anti-ß2-glycoprotein 1antibodies. Thromb Haemost 1998;80:393-8.
81) Pengo V, Biasiolo A. The risk of overdiagnosis of antiphospholipid antibody syndrome. Thromb Haemost 2001;86:933 (letter).

81
La prima descrizione degli anticorpi antiprotrombina (aPT) risale al 1959, quando Loeliger riportò il caso di un paziente il cui anticoagulante tipo lupus (LAC) era più evidente nella miscela con plasma normale di controlloche nel plasma del paziente stesso (1). Il livello plasmatico della protrombinaera ridotto. Loeliger dimostrò che la protrombina era necessaria per l’espressione dell’attività LAC del paziente. Successivamente, fu descritto un altro caso di LAC associato a severa ipoprotrombinemia e a manifesta-zioni emorragiche importanti (2). Negli anni successivi furono descritti nume-rosi casi di LES con manifestazioni emorragiche, associate alla presenza di LAC e di ipoprotrombinemia.Tipicamente, si osservava la riduzione sia dell’antigene, sia dell’attività dellaprotrombina.Negli anni ‘80 fu dimostrato che l’ipoprotrombinemia dei pazienti con LAC era causata dalla presenza di anticorpi non neutralizzanti, che legavano la protrombina senza inibirne la conversione in trombina (3). Fu ipotizzato che l’ipoprotrombinemia fosse conseguente alla rapida eliminazione dal circolo dei complessi protrombina-antiprotrombina. Nel 1984 Edson et al. (4) dimo-strarono la presenza di anticorpi antiprotrombina (aPT) nel plasma di pazienti LAC-positivi senza ipoprotrombinemia severa. Fleck et al. (5) studia-rono 42 pazienti LAC-positivi, riscontrando la presenza di aPT in 31 di essi (74%), 15 dei quali presentavano l’allungamento del tempo di protrombina.Questi ricercatori conclusero, sulla base di esperimenti di assorbimento delplasma con protrombina insolubile, che questi anticorpi LAC erano polispe-cifici, poichè reagivano sia con la protrombina, sia con i fosfolipidi a carica netta negativa.
4. Anticorpi antiprotrombina:diagnosi di laboratorio
Monica Galli
U.S. Emostasi e Trombosi, U.O. Ematologia, Ospedali Riuniti, Bergamo
Introduzione

82
Gli aPT si legano alla protrombina immobilizzata su piastre di polistirene gammairradiate (6), oppure di PVC altamente attivate (7, 8), ma non su piastre di polistirene normale. In queste condizioni, la loro prevalenza è di circa il 50% dei pazienti con anticorpi antifosfolipidi (aPL). Sia la protrombina umana, sia quella bovina sono abitualmente riconosciute, anche se quella umana è un antigene migliore (6, 9). La protrombina è riconosciuta in modo più efficiente quando è legata, mediante ioni calcio, alla fosfatidilserina immobilizzata sulle piastre ELISA: la prevalenza degli aPT sale a circa il 90% (7).Ciò può avere diverse spiegazioni. In primo luogo, il legame alla fosfatidil-serina può permettere un orientamento ed una concentrazione migliori, favorendo, perciò, il legame anticorporale. Inoltre, la fosfatidilserina in fase solida può legare, mediante gli ioni calcio, i complessi protrombina-aPT eventualmente circolanti. Infine, non si può escludere che gli aPT interagi-scono con neoepitopi che la protrombina espone solo a seguito del legamecon la fosfatidilserina.Attualmente, non è del tutto chiaro se gli aPT siamo anticorpi a bassa affi-nità oppure siano diretti contro neo-epitopi della protrombina. A favore della prima possibilità sta l’osservazione che la protrombina modi-fica la sua conformazione tridimensionale a seguito del legame calcio-mediato con superfici contenenti fosfatidilserina. A favore della seconda vi sono i dati di Field et al. (10), che hanno dimostratoche il legame delle IgG aPT alla protrombina è divalente, e richiede la pre-senza di entrambe le porzioni Fab. Esperimenti di cinetica di legame con-dotti da Willems et al. (11) confermano che IgG aPT (ma non il loro frammentoFab1) formano complessi divalenti (o multivalenti) con la protrombina su una membrana contenente fosfatidilserina, aumentando, in tal modo, gran-demente l’affinità della protrombina per la superficie fosfolipidica.Rauch et al. (12) hanno dimostrato che gli aPT riconoscono anche la protrom-bina legata alla fosfatidiletanolamina in fase esagonale, e che questo com-plesso neutralizza l’attività LAC. Nel nostro laboratorio abbiamo dimostrato che l’interazione della protrombina con la prevalenza di anticorpi IgG e IgMdiretti contro il complesso calcio-mediato protrombina-fosfatidiletanolaminain fase esagonale è di circa il 70% (13). Il legame era strettamente dipenden-te dalla presenza del calcio.L’epitopo riconosciuto dagli aPT non è ancora stato chiaramente definito. In due casi LAC-positivi con severa ipoprotrombinemia, Bajaj et al. (14) hannodimostrato che il plasma reagiva non solo con la protrombina, ma anche con la pretrombina I (ovvero, il segmento carbossi-terminale della protrom-bina) e con la DIP-alfa-trombina (ovvero, il segmento carbossi-terminale della pretrombina 1). Al contrario, non venne rilevata reattività con il seg-mento amino-terminale della protrombina, nè della pretrombina 1, nè con la trombina. Malia et al. (15) hanno confermato che gli aPT reagiscono con la protrombina ed il suo frammento 1-2, ma non con la trombina.È probabile che la maggioranza degli aPT sia di natura poli-od oligo-clonale.Puurunen et al. (16) hanno dimostrato che gli aPT cross-reagiscono con ilplasminogeno in un gruppo di pazienti con infarto miocardico. Studi di inibi-zione hanno dimostrato che il legame anticorporale alla protrombina potevaessere impedito dalla protrombina, dal plasminogeno e da peptidi sintetici di entrambe le proteine in fase fluida. Questi ricercatori hanno ipotizzato che gli aPT, cross-reagendo con il pla-sminogeno, possono interferire con la via fibrinolitica.
Proprietà immunologiche e siti di riconoscimento antigenico

83
Inizialmente, vennero usate metodiche di doppia immunodiffusione o di cross-immunoelettroforesi, che hanno il vantaggio di identificare i comples-si protrombina/aPT. Questi tests, tuttavia, non danno nessuna stima quanti-tativa degli aPT. Inoltre, in alcuni casi il titolo o l’affinità anticorpale sono troppo bassi per dare linee di precipitazione chiare.I test ELISA sono attualmente i più usati per l’identificazione degli aPT (6-8). Di seguito viene indicata la metodica di impiego comune nel nostro labora-torio per il dosaggio degli aPT di isotipo G e M. Si sottolinea che si tratta diuna metodica “in-house”.
Materiali:• piastre ELISA Titertek in PVC altamente attivato (ICN-Flow);• protrombina umana (Stago);• anticorpi di coniglio coniugati con perossidasi anti-IgG ed anti-IgM
umane (DAKO);• tampone carbonato, pH 9.6;• tampone di lavaggio (Tris 50 mM, NaCl 120 mM, pH 7.4,
contenente 0.05% Tween 20);• tampone di diluizione dei campioni e degli anticorpi coniugati
(Tris 50 mM, NaCl 120 mM, pH 7.4, contenente 0.05% Tween 20 e BSA 1%);
• soluzione cromogenica: preparare una soluzione di substrato cromogenicoTMB (Sigma) 6 mg/ml in DMSO, diluirne 418 µl in 25 ml di tampone acetato,a cui si aggiungono 10 µl di acqua ossigenata 10 vol;
• acido solforico 4N;• lettore di piastre ELISA.
Metodo:• seminare in ogni pozzetto di una piastra ELISA 50 µl di soluzione di
protrombina 10 µl/ml in tampone carbonato ed incubare per tutta la notte a + 4 C°. I successivi passaggi sono eseguiti tutti a temperatura ambiente;
• lavare la piastra 1 volta con tampone di lavaggio (100 µl/pozzetto);• incubare la piastra per 1 ora con tampone di diluizione (150 µl/pozzetto)
per il blocco del legame aspecifico;• lavare la piastra 2 volte con tampone di lavaggio (100 µl/pozzetto);• incubare per 1 ora i campioni (50 µl/pozzetto, in doppio) diluiti 1:100
in tampone di diluizione;• lavare la piastra 4 volte con tampone di lavaggio (100 µl/pozzetto);• incubare per 1 ora gli anticorpi coniugati diluiti in tampone di diluizione
(50 µl/pozzetto) (per la diluizione degli anticorpi coniugati debbono essereseguite le indicazioni della ditta produttrice);
• lavare la piastra 4 volte con tampone di lavaggio (100 µl/pozzetto);• seminare il substrato cromogenico (100 µl/pozzetto) ed incubare finchè
un controllo positivo abbia raggiunto una OD di circa 1;• bloccare la conversione del substrato cromogenico con acido solforico
(50 µl/pozzetto);• leggere a 450 nm di lunghezza d’onda.
Poichè non esistono standard di riferimento, si consiglia di seminare in ognipiastra almeno 8 controlli normali ed almeno 3 controlli patologici. I campioni vengono considerati positivi quando la loro OD supera di alme-no 2 deviazioni standard la media degli 8 controlli negativi inseriti in ogni piastra.Attualmente, non sono ancora a disposizione kits commerciali per l’identifi-cazione di questi anticorpi.
Dosaggio aPT

84
L’attività LAC nel plasma è, in genere, causata dall’effetto combinato degli aPT e di un altro tipo di anticorpo antifosfolipide (aPL), gli anticorpi anti-ß2glicoproteina 1 (17). Tuttavia, in una minoranza di pazienti, essa è prodotta esclusivamente dalla presenza degli aPT. L’attività LAC degli aPT si eserci-ta nel plasma umano, ma non in quello animale (18). L’origine di questa spe-cie-specificità non è chiara.Studi condotti in sistemi purificati della coagulazione hanno dimostrato che gli aPT interferiscono con l’attivazione della protrombina da parte del com-plesso del fattore Xa e Va attivato su una superficie fosfolipidica anionica artificiale (18) o piastrinica (19) in presenza di ioni calcio. L’attività anticoagu-lante si esercitava sulla protrombina di origine umana ma non di quella bovina, ed era indipendente dalla fonte umana o bovina dei fattori Xa e Va. In presenza di fosfolipidi, calcio e protrombina gli aPT interferiscono anche con l’attivazione del fattore X da parte del complesso dei fattori IX e VIIIa (20).Alcuni aPT non hanno attività anticoagulante e, quindi, possono essere evi-denziati solo mediante tests immunoenzimatici. Fino ad ora, non è ancora stata data spiegazione all’esistenza di aPT che differiscono per le loro pro-prietà anticoagulanti.
Per rispondere alla domanda se la misurazione degli aPT debba essere inserita nello screening dei pazienti con sindrome da anticorpi antifosfolipi-di, abbiamo eseguito una ricerca della letteratura pubblicata in inglese dal 1992 al 2001 (21). Abbiamo trovato 17 studi che fornivano o permettevano di calcolare l’Odds Ratio con il 95% intervallo di confidenza degli aPT per le trombosi venose ed arteriose in 2352 casi e 611 controlli.Nonostante il grande numero di pazienti e di associazioni con le trombosi, il disegno retrospettivo della maggior parte degli studi disponibili non ci ha permesso di giungere ad una chiara conclusione rispetto alla rilevanza cli-nica degli aPT. La mancanza di una documentazione oggettiva della trom-bosi, della sequenza temporale tra la misurazione degli anticorpi e il verifi-carsi dell’evento, e di un gruppo di controllo riducono grandemente il livellodi evidenza degli studi retrospettivi. Inoltre, gli studi erano troppo eteroge-nei per permettere una meta-analisi.Con queste limitazioni, la nostra rivisione sistematica ha dimostrato che 18 associazioni con le trombosi su 43 (42%) erano significative. In particolare, lo erano 3 di 12 associazioni con le trombosi arteriose, 7 di 16 con le trom-bosi venose ed 8 di 15 con qualunque tipo di trombosi. L’insieme di questi dati non sembra sostenere l’utilità di misurare gli aPT nei pazienti aPL-posi-tivi a particolare rischio trombotico.
Gli aPT aumentano la generazione di trombina sulle cellule endoteliali (22)
così come in un sistema dinamico (23). Ciò è probabilmente causatodall’effetto stabilizzante il legame della protrombina alle superfici procoagu-lanti e può suggerire la possibilità che gli aPT con attività anticoagulante abbiano un significato protrombotico. Tuttavia, in un altro sistema purificatogli aPT non si sono dimostrati capaci di aumentare la generazione di trom-bina in presenza di monociti e piastrine(24).Apparentemente, gli aPT non interferiscono con due reazioni coagulative fosfolipide-dipendenti che regolano la cascata coagulativa: l’inibizione da parte del TFPI (tissue factor pathway inhibitor) della generazione del fattore
Attività anticoagulante
Rilevanza clinica
Fisiopatologia della trombosi

85
Xa indotta dal complesso fattore VIIa/fattore tissutale (25); 2. l’inattivazione del fattore Va da parte del sistema della proteina C/proteina S (26). Tuttavia, questi esperimenti sono stati condotti in sistemi plasmatici ed i loro risultati non sono stati confermati in sistemi purificati della coagulazione.In conclusione, non è ancora chiaro se gli aPT siano rilevanti rispetto all’eziopatogenesi delle trombosi.
In generale, gli aPT non richiedono trattamento, tranne che nei casi di mani-festazioni emorragiche severe. Le condizioni che possono richiedere tera-pia sono gli interventi chirurgici, ed il sanguinamento muco-cutaneo o geni-to-urinario. La terapia di scelta sono gli steroidi (27, 28). Sono stati usati con successo il metilprednisolone, 30 mg/kg/die per 3 giorni di seguito da pred-nisone, 2 mg/kg/die per 14 giorni (28); la ciclofosfamide, 1 gr al giorno uno in combinazione con il prednisone, 1 mg/kg/die per un mese (27). In caso di insuccesso, il danazolo (29), le gammaglobuline ad alte dosi (30), e la ciclofo-sfamide (29) sono state riportate avere un variabile grado di efficacia.Il trattamento delle trombosi solleva due tipi di problemi nei pazienti con aPT. L’eparina, gli anticoagulanti orali, gli antiaggreganti piastrinici possonoaumentare il rischio emorragico causato dall’ipoprotrombinemia. Perciò, è necessario essere cauti nel somministrare questi trattamenti. Sono in via di completameno diversi studi clinici multicentrici randomizzati, che stabiliranno la durata e l’intensità ottimali della terapia antitrombotica nei pazienti con PL (esempio, studio WAPS, ref. 31).L’altro aspetto è legato al controllo della terapia anticoagulante orale. La presenza degli aPT, associata ad un grado variabile di ipoprotrombine-mia, potrebbe influenzare il risultato del tempo di protrombina calcolato mediante l’INR (International Normalized Ratio). In effetti, è stata riportata una variabile risposta delle tromboplastine del commercio alla presenza degli anticorpi antifosfolipidi, ed alcune di esse, in particolare quelle ricom-binanti, sono risultate molto sensibili al LAC (32-34). Uno studio multicentrico ha, però, dimostrato che la presenza di questi anticorpi non interferisce in modo significativo con il tempo di protrombina misurato con le trombopla-stine comunemente usate in Italia nei pazienti LAC-positivi in terapia anti-coagulante orale (35).
Trattamento

86
1. Loeliger A. Prothrombin as co-factor of the circulating anticoagulant in systemic lupus erythematosus? Thromb Diath Haemorrh 1959; 3:237-256
2. Rapaport SI, Ames SB, Duval BJ. A plasma coagulation defect in systemic lupus erythematosus arising from hypoprothrombinemia combined with antiprothrombinase activity. Blood 1960; 15:212-227
3. Lechner K. Acquired inhibitors in nonhemophilic patients. Hemostasis 1974; 3:65-93
4. Edson JR, Vogt JM, Hasegawa DK. Abnormal prothrombin crossed immunoelecrophoresis in patients with lupus inhibitors. Blood 1984; 64:807-812
5. Fleck RA, Rapaport SI, Rao LVM. Anti-prothrombin antibodies and the lupus anticoagulant. Blood 1988; 72:512-519
6. Arvieux J, Darnige L, Reber G, Bensa JC, Colomb MG. Development of an ELISA for autoantibodies to prothrombin showing their prevalence in patients with lupus anticoagulants. Thromb Haemost 1995; 74:1120-1125
7. Galli M, Beretta G, Daldossi M, Bevers EM, Barbui T. Different anticoagulant and immunological properties of anti-prothrombinantibodies in patients with antiphospholipid antibodies. Thromb Haemost 1997; 77:487-491
8. Pengo V, Biasiolo A, Brocco T, Tonello S, Ruffatti A. Autoantibodies to phospholipid-binding plasma proteins in patients with thrombosis and phospholipid-reactive antibodies. Thromb Haemost 1996; 75:721-724
9. Rao LVM, Hoang AD, Rapaport SI. Differences in the interactions of lupus anticoagulant IgG with human prothrombin and bovine prothrombin. Thromb Haemost 1995; 73:668-674
10. Field SL, Chesterman CN, Day Y-P, Hogg PJ. Lupus antibody bivalency is required to enhance prothrombin binding to phospholipid. J Immunol 2001; 166:6118-6125
11. Willems GM, Janssen MP, Comfurius P, Galli M, Zwaal RFA, Bevers EM. Kinetics of prothrombin-mediated binding of lupus anticoagulant antibodies to phosphatidylserine containing phospholipid membranes: an ellipsometric study. (submitted)
12. Rauch J. Lupus anticoaulant antibodies: Recognition of phospholipid-biding protein complexes. Lupus 7:S29, 1998 (suppl 2)
13. Galli M, Barbui T. Antiprothrombin antibodies: detection and clinical significance. Blood 1999; 93:2149-2157
14. Bajaj SP, Rapaport SI, Barclav S, Herbst KD. Acquired hypoprothrombinemia due to nonneutralizing antibodies to prothrombin: mechanism and management. Blood 1985; 65:15381544
Bibliografia

87
15. Malia RG, Brooksfield C, Bulman I, Greaves M. Prothrombin fragment F 1-2: the epitope for antiphospholipid antibody expression. XVIth Congress of the International Society of Thrombosis and Haemostasis, Florence, Italy, june 6-12, (abstract 689), 1997
16. Puurunen M, Manttari M, Manninen V, Palosuo T, Vaarala O, Antibodies to prothrombin crossreact with plasminogen in patients developing myocardial infarction. Br J. Haematol 1998; 100:374-380
17. Horbach DA, van Oort E, Derksen RHWM, de Groot PG. The contribution of antiprothrombin antibodies to lupus anticoagulant activity. Thromb Haemost 1998; 9:790-796
18. Bevers EM, Galli M, Barbui T, Comfurius P, Zwaal RFA. Lupus anticoagulant IgG’s (LA) are not directed to phospholipids only, but to a complex of lipid-bound human prothrombin. Thromb Haemost 1991; 66:629-632
19. Galli M, Bevers EM, Comfurius P, Barbui T, Zwaal RFA. Effect of antiphospholipid antibodies on procoagulant activity of activated platelets and platelet-derived microvesicles. Br J Haematol 1993; 83:466-472
20. Permpikul P, Rao LVM, Rapaport SI. Functional and binding studies of the roles of prothrombin and ß2-glycoprotein 1 in the expression of lupus anticoagulant activity. Blood 1994; 83:2878-92
21. Galli M.Should we include antiprothrombin antibodies in the sceenig for the antiphospholipid syndrome? J Autoimm 2000;15 (suppl 2): 101-106
22. Rao VM, Hoang AD, Rappaport SI. Mechanism and effects of the binding of lupus anticoagulant IgG and prothrombin to surface phospholipid. Blood 1996; 88:4173-4182
23. Field SL, Hogg P, Daly EB, Daly Y-P, Murray B, Owens D, Chesterman CN.Lupus anticoagulants form immune complexes with prothrombin and phospholipid that can augment thrombin production in flow. Blood 1999; 94:3421-3428
24. Hoffmann M, Monroe DM, Roubey RSA. IgG from two patients with the antiphospholipid syndrome increase thrombin generation in an “in vitro” cell-based model of coagulation. XVIth Congress of the International Society of Thrombosis and Haemostasis, Florence, Italy, june 6-12, (abstract 2), 1997
25. Salemink I, Willems GM, Galli M, Bevers EM, Lindhout T. Antibodies to ß2-glycoprotein 1 from patients with antiphospholipid syndrome suppress the inhibitory activity of tissue factor pathway inhibitorThromb Haemost 2000; 84:653-656
26. Galli M, Ruggeri L, Barbui T. Differential effects of anti-ß2glycoprotein 1 and antiprothrombin antibodies on the anticoagulant activity of activated protein C. Blood 1998; 91:1999-2004

88
27. Simel DL, St Clair EW, Adams J, Greenberg CS. Correction of hypoprothrombinemia by immunosuppressive treatment of the lupus anticoagulant-hypoprothrombinemia syndrome. Am J Med 1987; 83:563-566
28. Bernini JC, Buchanan GR, Ashcraft J. Hypoprothrombinemia and severe hemorrhage associated with a lupus anticoagulant. J Pediatr 1993; 123:937-939
29. Williams S, Linardic C, Wilson O, Comp P, Grainick HR. Acquired hypoprothrombinemia: effects of Danazol treatment. Am J Hematol 1996; 53: 272-276
30. Pernod G, Arvieux J, Carpentier PH, Mossuz P, Bosson JL, Polack B. Successful treatment of lupus anticoagulant-hypoprothrombinemia syndrome using intravenous immunoglobulins. Thromb Haemost 1997; 78:969 (letter)
31. Finazzi G, Barbui T for the Provisional Steering Committee of the WAPS Study. Feasibility of a randomized clinical trial for the prevention of recurrent thrombosis in the antiphospholipid syndrome: the WAPS project. Annales de Medicine Interne 1996; 147 (suppl 1): 38-41
32. Della Valle P, Crippa L, Safa O, Tomassini L, Pattarini E, Viganò-D’Angelo S,Sabbadini MG, D’Angelo A. Potential failure of the International Normalized Ratio (INR) system in the monitoring of oral anticoagulant in patient with lupus anticoagulants. Annales de Medicine Interne 1996; 147 (suppl 1):10-14
33. Moll S, Ortel TL. Monitoring warfarin therapy in patients with lupus anticoagulants. Ann Int Med 1997; 127:177-182
34. Laurie AS, Purdy G, Mackie IJ, Machin SJ. Monitoring of oral anticoagulanttherapy in lupus anticoagulant positive patients with the anti-phospholipidsyndrome. Br J Haematol 1997; 98:887-92
35. Tripodi A, Chantarangkul V, Clerici M, Negri B, Galli M, Mannucci PM. Laboratory control of oral anticoagulation treatment in patients with antiphospholipid syndrome and lupus anticoagulant. Results of a collaborative study involving nine commercial thromboplastins. Br J Haematol 2001; 115:672-678
Tutti i diritti riservati - Stampato in Italia - Speed 2000 - 07/02


Par
t. N
o. 9
8090
-46