1 - livro bioq
TRANSCRIPT

1
CAPÍTULO 1: ÁGUA
OBJETIVOS:
Conhecer as características e propriedades da água.
Conhecer a estrutura da água – pontes de hidrogênio.
Definir os pontos de dissociação da água.
Descrever a equação de Henderson – Hasselbalch.
Verificar as interações biológicas de soluções tampão.
As células vivas contêm carboidratos, lipídeos, aminoácidos, proteínas, ácidos nucleicos, nucleotídeos e compostos relacionados em quantidades variáveis. A massa de cada um desses compostos de estruturas químicas muito variadas é constituída basicamente por carbono (C), hidrogênio (H), oxigênio (O), nitrogênio (N), fósforo (P) e enxofre (S). Dois desses elementos, hidrogênio e oxigênio, combinam-se para formar o mais abundante componente celular, a água (H2O), que não se inclui em nenhuma das categorias acima mencionadas. Mais de 90% do plasma sanguíneo é H2O, o músculo contém cerca de 80% de H2O e ela constitui mais da metade da maioria dos outros tecidos animais ou vegetais.
Além de ser o mais abundante componente celular, a H2O é também indispensável à vida. Os nutrientes que a célula consome, o oxigênio usado na oxidação deles e os produtos residuais formados são todos transportados pela água. Assim é importante observar que esse composto químico tem um número excepcional de propriedades que o tornam sobremaneira peculiar e bastante apropriado para o desempenho como solvente da vida.
Propriedades da Água
Entre solventes, como: etanol, metanol, acetona, acetato de etila, clorofórmio, amônia etc., a água possui:
1. o mais alto ponto de ebulição;2. o mais alto calor específico de vaporização;3. o mais alto ponto de fusão.Isto ocorre devido às grandes forças intermoleculares que atuam entre
moléculas de água adjacentes em solução.
Uma molécula de água consiste de dois átomos de hidrogênio ligados a um átomo de oxigênio. A distância da ligação O-H é de 0,958 Ângstrons (1 Ângstrom equivale a 10-10 m), e o ângulo formado pelos três átomos é de 104,50. Os átomos de hidrogênio não estão arranjados linearmente, pois os quatro orbitais híbridos sp3 do oxigênio estendem-se aproximadamente na direção dos vértices de um tetraedro. Os átomos de hidrogênio ocupam duas

2
vértices do tetraedro, e os pares de elétrons não-ligantes do átomo de oxigênio ocupam os dois outros vértices (em uma molécula perfeitamente tetraédrica, como o metano, CH4, os ângulos de ligação são de 109,5°).
A molécula de água é altamente polarizada, pois os átomos de oxigênio eletronegativos tendem a atrair elétrons do átomo de hidrogênio, deixando uma carga positiva residual cercando o próton. Devido a essa polarização, as moléculas de água comportam-se como dipolos, uma vez que elas podem ser orientadas em ambas as direções como íons positivos e negativos. É essa propriedade que dá à água a capacidade de atuar como solvente. Os elevados pontos de ebulição e de fusão da água e seu alto calor de vaporização são resultado da interação entre moléculas de água vizinhas.
Cada molécula de água em solução tende a ficar rodeada por quatro outras moléculas com átomos de oxigênio negativamente polarizados, ficando atraídos aos prótons carregados positivamente. A atração entre o oxigênio de uma molécula e o hidrogênio de outra é representado como H.....O, e é chamada ponte de hidrogênio ou ligação de hidrogênio. Embora a energia necessária para romper esta ligação seja bem menor que a necessária para quebrar uma ligação O-H covalente, o efeito aditivo com a ponte de hidrogênio explica as propriedades incomuns da água. As pontes de hidrogênio são forças de natureza elétrica do tipo dipolo permanente, porém bem mais intensas.
INFOESCOLA.COM.BR
Dissociação da Água e seu Produto Iônico (Kw)
A água é uma molécula neutra com leve tendência a ionizar-se. Essa ionização é expressa como:
H2O H+ + OH-

3
Na verdade não existem prótons livres em solução. O próton está sim associado a uma molécula de água sob a forma de íon hidrônio, H3O+, porém continuaremos representando esses íons por H+. A ionização da água é descrita pela expressão de equilíbrio:
[H+] [OH-]
Ka =
[H2O]
K é a constante de ionização. Já que a concentração da água não dissociada é bem maior que as concentrações dos íons que a compõem, ela pode ser considerada constante e incorporada à K para produzir uma expressão para a ionização da água:
Kw = [H+] [OH-]
A água é um eletrólito fraco e a sua dissociação para formar H+ e OH- é muito reduzida.
H2O H+ + OH-
[H+] [OH-]
K =
[H2O]
A concentração da água pura é muito grande em comparação com qualquer concentração possível de solutos e pode ser considerada constante. O valor numérico é 55,5 M e pode ser obtido ao se dividir o número de gramas de água em um litro, 1000g, pelo peso molecular da água, 18 g/mol.
[H+] [OH-]
Ka = donde,
55,5
Ka x 55,5 = [H+] [OH-] = Kw
Kw é a constante do produto iônico da água, e a concentração da água está inclusa em seu valor. O valor numérico de Kw pode ser determinado de forma experimental ao se medir a concentração de íons hidrogênio em água pura. A concentração de íons hidrogênio também é igual por definição à concentração do íon hidroxila, porque a água é um ácido monoprótico (libera um único próton por molécula), a 25 °C, em água pura,

4
[H+] = 10-7 M = [OH-]
Assim, a 25 °C, o valor numérico de Kw = [H+] [OH-] = (10-7) (10-7) = 10-14.
Essa relação é válida para qualquer solução aquosa, seja ela neutra, ácida ou básica.
O valor de Kw, a constante de ionização da água é 10-14 M2 a 250 C. A água pura deve conter quantidades equimolares de H+ e OH-, de forma que:
[H+] = [OH-] = 10-7M
Soluções com [H+] = 10-7 M são ditas neutras, as com [H+] > 10-7 M são ditas ácidas, e as com [H+] < 10-7 M são ditas básicas. Os valores de [H+] para a maioria das soluções são muito pequenos e, portanto, não são práticos para fins de comparação. Uma forma mais prática é conhecida como pH. Sendo:
pH = -log10 [H+]
Quanto mais alto o pH, menor será a concentração de H+; quanto menor o pH maior será a concentração de H+. O pH da água pura é 7,0, enquanto soluções ácidas têm pH < 7,0 e soluções básicas têm pH > 7,0.
Como na água pura [H+] = 1 x 10-7 M e pH = 7,0, calcular o pH das seguintes soluções aquosas:
a) 1,1 x 10-3 M de HCl.
b) 2,1 x 10-3 M de NaOH.
Uma quantidade semelhante, o pKa, pode ser definida por analogia com a definição de pH:
pKa = -log10Ka
O valor de pKa é uma outra medida que indica a força do ácido: quanto menor seu valor, mais forte será o ácido. A situação é o inverso da observada

5
em Ka, em que maiores valores implicam ácidos mais fortes. A definição de ácido mais importante para a Bioquímica é a de Bronsted, que diz ser:
Ácido: qualquer substância que pode doar prótons.
Base: qualquer substância que pode aceitar prótons.
Ex: HCl H+ + Cl-
CH3COOH H+ + CH3COO-
NH4+ NH3 + H+
Expressão Geral: HA H+ + A-
Bases correspondentes:
Cl- + H+ HCl
CH3COO- + H+ NH4+
HA: Ácido de Brönsted (pode fornecer um próton)
A- : Base conjugada (pode aceitar o próton para formar o ácido)
Eletrólitos fortes: Em solução aquosa são quase completamente
dissociados em íons.
Ex: Na+Cl- Na+ + Cl-
HCl H+ + Cl-
HCl em H2O = ionização
HCl + H2O H3O + Cl- ou
(Ác. Conj.)1 + (Base Conj.)2 (Ác. Conj.)2 + (Base Conj.)1
Ionização de Ácidos Fracos
Um ácido fraco é apenas parcialmente ionizado em solução aquosa.
Ex: HA + H2O H3O+ + A- ou

6
(Ác. Conj.)1 + (Base Conj.)2 (Ác. Conj.)2 + (Base Conj.)1
Constante de Equilíbrio = Constante de Ionização = Kion
Keq = Kion = [H3O + ] [A - ]
[HA] [H2O]
Sendo [H2O] = Constante (55,5 moles/L), então: Ka = Kion = [H3O + ] [A - ]
[HA]
HA H+ + A- logo Keq = [H + ] [A - ]
[HA]
Ionização de Bases Fracas
BOH B+ + OH-
Keq = Kb = [B + ] [OH - ]
[BOH]
Bases Fracas: Aminas Orgânicas
RNH2 + H2O RNH3 + OH- ou
(Base Conjugada)1 + (Ácido Conjugado)2 (Ácido Conjugado)1 + (Base
Conjugada)2
A H2O serve como ácido doando prótons para RNH2. Seria então o mesmo
que:
A- + H2O HA + OH- ou
(Base. Conj.)1 + (Ác. Conj.)2 (Ác. Conj.)1 + (Base Conj.)2
Kion = [HA] [OH - ]
[A-] [H2O]
Kion e [H2O] combinados dão Kb
Kb = [HA] [OH - ]

7
[A-]
Serve para calcular a [OH-] de uma base fraca
Agora se: [OH-] = Kb = [A - ]
[HA]
E se: [H+] = Ka = [HA]
[A-]
[H+] [OH] = Kw
Ka[HA] . Kb [A - ] = Kw
[A-] [HA]
Ka . Kb = Kw = 10-14
Logo: logKa + logKb = log Kw ou
- logKa - logKb = - log Kw
Então como:
pH = log [H+]
pKa = logKa
pKb = - logKb
pKa + pKb = - log Kw = 14
Equação de Henderson – Hasselbalch
É a equação que relaciona o valor de Ka de qualquer ácido fraco ao pH da solução que contém esse ácido e sua base conjugada. Essa relação é muito usada na prática bioquímica, principalmente quando é necessário controlar o pH para condições ideais de reação. Muitas reações deixam de se processar quando o pH não está no valor ideal. Além de muitas reações não ocorrerem, algumas consequências fisiológicas drásticas podem resultar a partir de variações de pH no organismo.

8
Aplicando a lei da ação das massas à ionização dos ácidos fracos, tem-se:
HA H+ + A-
de onde tem-se: Ka = [H + ] [A - ]
[HA]
Logo: [H+] = Ka [HA]
[A -]
Aplicando-se logaritmo dos dois lados, tem-se:
log[H+] = logKa + log [HA] Multiplicando por (-1) : [A-]
- log[H+] = -logKa – log [HA] [A-]
Se: - logKa = pKa e – log [HA] = log [A - ] [A-] [HA]
Então vem que: pH = pKa + log [A - ] [HA]
pH = pKa + log [base conjugada] [ácido conjugado]
Essa é a relação conhecida como equação de Henderson-Hasselbalch e é útil para prever as propriedades de soluções tampão utilizadas para controlar o pH de misturas de reações.
Solução Tampão
Solução tampão é aquela solução que resiste a uma variação do pH quando se adiciona ácido ou base. Consiste de uma mistura de ácido fraco de Brönsted e sua base conjugada. Ex: Misturas de ácido acético e acetato de sódio ou hidróxido de amônio e cloreto de amônio. A importância da solução tampão está em sua capacidade de impedir mudanças bruscas de pH. Por exemplo, o plasma sanguíneo é a solução tampão ideal para conservar os valores do pH do sangue em 7,2 – 7,3 ± 0,2, pois fora desse intervalo não há vida.
Outro fator de importância de uma solução tampão reside no fato de que enzimas do processo metabólico apresentam máxima ação catalítica dentro de limites definidos de pH.

9
Ex: Adicionando NaOH a uma mistura de ácido acético e acetato de potássio:
OH- + CH3COOH CH3COO- + H2O
CH3COOH CH3COO- + H+ OH- H2O
Adicionando base, há dissociação adicional do CH3COOH para dar mais prótons, conservando a [H+] ou o pH sem variar. Adicionando ácido a uma solução de tampão de acetato:
H+ + CH3COO- CH3COOH
Os prótons adicionadas (HCI, por exemplo) combinam-se com o CH3COO-
presente na mistura tampão (como acetato de potássio) para formar o ácido fraco não dissociado CH3COOH. Logo o pH resultante é muito menor do que ocorreria, se a base conjugada estivesse ausente.
Eficiência ou capacidade tamponante da solução
Os fatores de eficiência de um tampão são:
Concentração molar dos camponentes do tampão. A capacidade tamponante é diretamente proporcional à concentração molar dos componentes.
Relação entre concentração da base conjugada e ácido fraco. A solução tampão mais eficiente tem: [Ácido] = [Base]
Tampões Fisiológicos
Os tampões fisiológicos dependem de vários fatores, entre os quais da concentração molar dos componentes do tampão, da relação entre [base conjugada] e [ácido fraco]. O primeiro desses fatores já exclui componentes encontrados nos metabolismos intermediários de baixa concentração, tais como ésteres fosfóricos da glicose, ácidos orgânicos do ciclo de Krebs e os aminoácidos livres. Nas plantas, alguns ácidos orgânicos, como málico, cítrico e isocítrico podem acumular-se nos vacúolos, tendo importante papel no pH da célula.
Nos animais, existe um sistema tampão complexo e vital no sangue circulante. São componentes desse sistema:
CO2 – HCO3;
NaH2PO4 – Na2HPO4;
As formas oxigenada e desoxigenada da hemoglobina; As proteínas plasmáticas.
O pKa do H2CO3 é igual a 6,1. No entanto, a razão entre base conjugada e ácido fraco é aproximadamente 20:1 no intervalo do pH do sangue: 7,35 – 7,45. Era de se esperar que esse sistema não fosse muito eficaz como tampão.

10
Porém H2CO3 – HCO3- é um tampão muito importante para o sangue, pois o
ácido fraco H2CO3 entra rapidamente em equilíbrio com o CO2 dissolvido no plasma:
H2CO3 CO2 dissolvido + H2O
O CO2 dissolvido está em equilíbrio com o CO2 da atmosfera e, dependendo da pressão parcial do CO2 da fase gasosa, escapará para o ar (como nos pulmões, onde o CO2 é expirado) ou penetrará no sangue (como nos tecidos periféricos, onde o CO2 é produzido pela respiração das células). O sistema tampão funciona não pela alteração da razão 20:1, mas mantendo essa razão e aumentando ou diminuindo a quantidade total dos componentes do tampão.
> http://qmc.ufsc.br/qmcweb/QMCWeb - Revista Eletrônica de Química
Outro tampão importante do sangue: Hemoglobina oxigenada: HhbO2 (Ác. Forte: pKa = 6,2) e Hemoglobina desoxigenada: HHb (pKa = 7,7). Nos pulmões, onde a pressão parcial de O2 é maior, HHbO2 predomina em relação à HHb, e o sangue tende a ser mais ácido.
Nos tecidos periféricos, onde a pressão parcial de O2 é mais baixa, há predominância de HHb (pKa = 7,7) e o pH tende a aumentar. Esses efeitos são compensados pela diminuição da concentração de CO2 dos pulmões em relação à dos tecidos periféricos, os dois efeitos em conjunto são responsáveis pela variação mínima do pH do sangue.
HHbO2 H+ + HbO2 - pKa = 6,2
HHb H+ + Hb- pKa = 7,7
Sistemas de Tamponamento de Importância Fisiológica
O pH da maioria dos sistemas vivos é aproximadamente 7, e o principal tampão em células é o par: H2PO4
- / HPO42-. No sangue, a concentração dos
íons fosfato é insuficiente para uma ação tamponante, por isto opera um sistema tampão diferente. No pH do sangue, a maior parte do CO2 dissolvido presente está na forma de HCO3
-. O CO2, que é transportado para os pulmões para ser expirado, encontra-se como HCO3
-. HCO3-(bicarbonato) é o composto-

11
tampão mais significativo no sangue. A capacidade tamponante do sangue depende principalmente de dois equilíbrios:
(1) CO2 + H2O H2CO3 (CO2 gasoso dissolvido no sangue e ácido carbônico)
(2) H2CO3 H+ + HCO3- (ácido carbônico e o bicarbonato formado pela
dissolução de H+)
Quando o pH do sangue diminui (devido à produção metabólica de H+), o equilíbrio entre HCO3
- e H2CO3 desloca-se na direção do ácido carbônico. Ao mesmo tempo, o H2CO3 perde H2O para se tornar CO2, que é expirado dos pulmões como CO2 gasoso. Quando o pH do sangue aumenta, relativamente mais HCO3
- é formado e a respiração é ajustada de modo que quantidades aumentadas de O2 nos pulmões possam ser reintroduzidas no sangue para conversão em H2CO3.
Distúrbios no Sistema Tamponante
Distúrbios Ácido-Base
Os distúrbios ácido-base são caracterizados como metabólicos e respiratórios. A Acidose Metabólica resulta de um aumento de produção ou acúmulo de ácidos ou por perda de excessiva de bicarbonato, através dos rins ou do trato intestinal. A Alcalose Metabólica resulta da administração ou do acúmulo de bicarbonato ou seus precursores, perda excessiva de ácido ou perda de líquido extracelular contendo mais cloreto do que bicarbonato. A Acidose Respiratória é causada pela ventilação diminuída e consequente retenção de dióxido de carbono. Isso ocorre agudamente com apnéia do sono, asma e cronicamente com a síndrome de hipoventilação da obesidade, doença pulmonar obstrutiva crônica e certas doenças neuromusculares. A Alcalose Respiratória resulta de ventilação aumentada e eliminação de dióxido de carbono. Isso pode ser mediado por ansiedade, acidente vascular cerebral, pneumonia, altas altitudes, embolia pulmonar entre outros.
HIPERTEXTO
1. Acidose: quando o pH fica tão baixo quanto 7,1.Tratamento: NAHCO3 intravenoso.Onde ocorre: Doenças pulmonares obstrutivas.Ex: Enfisema (doença que impede a expiração eficiente de CO2).
2. Alcalose: quando pH fica tão alto quanto 7,6.Tratamento: A alcalose metabólica pode ser tratada com KCL ou NACL e a alcalose respiratória melhora com respiração de uma atmosfera rica em CO2.

12
Onde ocorre: A hiperventilação acelera a perda de CO2.
Consequências Fisiológicas do Tamponamento do Sangue
O processo de respiração exerce papel importante no tamponamento do sangue. Uma elevação na [H+] pode ser corrigido com uma elevação na taxa de reação, pois:
a) H+ (aq.) + HCO3-(aq.) H2CO3(aq.)
b) uma elevação [H2CO3] aumenta os níveis de CO2 dissolvido e finalmente a quantidade de CO2(g) nos pulmões.
H2CO3 (aq) CO2(aq) + H2O (ℓ)
CO2(aq) CO2(g)
c) Uma elevação na taxa de respiração REMOVE o excesso de CO2 dos pulmões, iniciando um deslocamento no equilíbrio das reações anteriores.
d) A remoção do CO2(g) diminui a quantidade de CO2 dissolvido, fazendo o H+ reagir com HCO3: Resultado: Há diminuição na concentração de [H+] no sangue, retornando-o ao seu nível original. Assim o pH do sangue é mantido constante.
CONTEXTUALIZANDO
Hiperventilação: Ocorre quando há uma respiração muito rápida e profunda (removendo uma tal quantidade de CO2 dos pulmões que o pH do sangue sobe, levando à fraqueza e ao desmaio). Atletas usam o aumento do pH pela hiperventilação nas corridas de 100 metros rasos, pois alcalinizam o sangue antes da corrida com exercícios e, quando a produção de ácido lático começa a crescer fazendo baixar o pH do sangue, não haverá um decréscimo tão grande que venha a provocar dores musculares. Qualquer ácido que entre na corrente sanguínea, eleva a [H+], logo há abaixamento do pH, ocasionando excesso de CO2 nos pulmões. Ex: A ingestão de altas doses de aspirina pode causar “envenenamento por aspirina”. Exposição a elevadas altitudes é semelhante à hiperventilação ao nível do mar. A taxa de respiração aumenta (por causa do ar rarefeito) então mais CO2 é removido dos pulmões abaixando a concentração de H+ no sangue e, portanto, elevando o pH. Hipoventilação: Muitas vezes quando se está soluçando, as pessoas alertam para segurar a respiração. O que ocorre é uma hipoventilação que provoca aumento na [CO2] nos pulmões, resultando no abaixamento do pH.
ATIVIDADES DE AVALIAÇÃO
1) Definir ácido e base segundo Brönsted.

13
2) Caracterizar um sistema tampão e indicar os fatores que determinam sua eficência.
3) Definir pKa e descrever os procedimentos experimentais para determinar o valor do pKa do ácido acético.
4) Escrever a equação de Henderson – Hasselbalch e mostrar sua utilidade na análise de um sistema tampão.
5) Dar exemplos de tampão biológicos.6) O tampão bicarbonato (HCO3
- / H2CO3), presente no plasma em equilíbrio com CO2, apresenta pKa igual a 6,1. Descrever o funcionamento deste sistema, mostrando o efeito da adição de H+ e de CO2 sobre o pH do plasma.
7) O produto iônico da água possibilita calcular a concentração de H+ para uma dada concentração de OH- e vice-versa; portanto responda: Qual é a concentração de H+ em uma solução de NaOH 0,1M?
8) Qual é a concentração de OH- em uma solução na qual a contração de H+ é 0,00013 M?
9) Calcular o pKa do ácido láctico, sabendo-se que, quando a concentração do ácido láctico é 0,010 M e a concentração de lactato é 0,087M, o pH da solução é 4,8.
10) Calcular o pH de uma mistura que contém ácido acético 0,1M e acetato de sódio 0,2M. O pKa do ácido acético é 4,76.
11) Calcular a relação entre as concentrações de acetato e de ácido acético requerida para um sistema tampão com pH 5,3.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 1. São Paulo: Cengage Learning – 2011. Tampões e Sistemas de Tamponamento de Importância Fisiológica são comentados no livro, no Capítulo 2.
Conn, E. E.; Stumpf, P. K. Introdução à Bioquímica. 4ª Ed. São Paulo, Edgard Blucher, 1980. A Ionização de Ácidos Fracos e de Bases Fracas é comentada no Capítulo 1 desse livro.
Mahan, K.; Escott-Stump. Alimentos, nutrição & dietoterapia. 9 ed. São Paulo: Roca, 1998. Distúrbios Ácido-Base são comentados no Capítulo 8.
Voet, D.; Voet, J. G.; Pratt, C. W. Fundamentos de Bioquímica. Porto Alegre: Artmed, 2000. A Química Ácido-Base é comentada no Capítulo 2.
LEITURA, VÍDEOS E SITES RECOMENDADOS
http://qmc.ufsc.br/qmcweb/

14
FILME – UMA VERDADE INCONVENIENTE – 2006 (documentário de AL Gore)
VÍDEO – UMA VERDADE INCONVENIENTE – 2006
CAPÍTULO 2: CARBOIDRATOS
OBJETIVOS:
Observar a importância dos carboidratos para a vida humana.
Verificar a presença dos carboidratos em alimentos e vegetais.
Diferenciar os tipos de carboidratos.
Classificar os carboidratos quanto a sua estrutura.
Classificar os carboidratos quanto a sua função biológica.
Os carboidratos constituem ¾ do mundo biológico e aproximadamente 80% do aporte calórico da humanidade. A glicose é o carboidrato mais importante. É sob essa forma que a maior parte dos carboidratos da dieta é absorvida pela corrente sanguínea ou é em glicose que o fígado converte os outros açúcares. Também é a partir de glicose que todos os carboidratos do organismo são formados.
Os carboidratos classificam-se em: monossacarídeos, dissacarídeos, oligossacarídeos e polissacarídeos. Quando a palavra “carboidrato” foi inventada, referia-se originalmente aos compostos com fórmula geral Cn(H2O)n. No entanto, somente os açúcares simples, ou monossacarídeos, encaixam-se exatamente nessa fórmula. Os outros tipos de carboidratos baseiam-se em unidades de monossacarídeos e apresentam fórmulas gerais ligeiramente diferentes. Os dissacarídeos, como os polissacarídeos, não atravessam a parede intestinal. Só são aproveitados como fonte de energia se previamente hidrolisados a monossacarídeos, que passam rapidamente do trato intestinal à corrente sanguínea. Os oligossacarídeos e os polissacarídeos não hidrolisados passam ao largo do intestino delgado até o intestino grosso, onde exercem um efeito benéfico (fibra). Os oligossacarídeos podem ser atacados pela microflora intestinal gerando produtos metabólicos: ácido acético e láctico. Em grandes quantidades têm efeito laxante podendo causar diarreia.
Os carboidratos proporcionam também texturas desejáveis, palatabilidade agradável, poder edulcorante. Dos polissacarídeos do mundo biológico, o homem só digere amido, glicogênio e certas dextranas. O glicogênio é

15
semelhante à amilopectina do amido, porém mais ramificado. Constitui a reserva energética dos animais, armazenando-se principalmente no fígado e, em menor quantidade, no músculo. O amido e o glicogênio começam a ser hidrolisados na boca pela ação da α – amilase contida na saliva. Ela produz um fragmento de seis unidades de glicose que são também hidrolisados produzindo maltose, maltotreose e maltotetrose. O alimento vai para o estômago e, no duodeno, ocorre uma hidrólise. Lá, β – amilase ataca o amido e os fragmentos da ação da α – amilase, liberando unidades de maltose. A maltose é hidrolisada à glicose pela enzima maltase e é transportada à corrente sanguínea.
Os polissacarídeos diferentes do amido e glicogênio não são hidrolisados pelas enzimas gastrointestinais passando ao intestino grosso, mais ou menos intactos. São eles: celulose, hemicelulose e pectina das paredes vegetais. Eles facilitam a passagem do bolo fecal através do sistema digestivo. A eliminação rápida de produtos não absorvidos evita o aparecimento de condições propícias ao desenvolvimento de câncer, ajudam a diminuir o colesterol do sangue e retardam o aparecimento de arterioscleroses. Carboidratos digeríveis proporcionam aproximadamente 4 Kcal/g, energia equivalente à proporcionada por 1g de proteína e inferior às 9Kcal/g dos lipídeos.
Dissacarídeos
AçúcarFonte
Significado Clínico
Maltose Digerido pela amilase ou hidrólise do amido. Cereais e malte em germinação.
Lactose Leite. Pode ocorrer na urina durante a gravidez. Na deficiência de lactase. A má absorção provoca diarreia e flatulência.
Sacarose Açúcar de cana e beterraba. Sorgo. Abacaxi. Raiz de cenoura.
Na deficiência da sacarase. A má absorção provoca diarreia e flatulência.
Trealose1 Fungos e leveduras. Principal açúcar da hemolinfa de insetos.
α–D–glicopiranosil-(1→1)-α–D–glicopiranósido.
Pentoses de importância fisiológica
Açúcar Fonte Importância bioqímica Significado clínico
D-Ribose Ácidos nucleicos Elementos estruturais dos ácidos nucleicos e coenzimas, p. ex., ATP, NAD, NADP, flavoproteínas. As ribose-fosfatos são intermediárias da via das pentoses-fosfato.

16
D-Ribulose Formada nos processos metabólicos
A ribulose-fosfato é um intermediário da via das pentoses-fosfato.
D-Arabinose Goma arábica. Gomas da ameixa e da cereja.
Constituinte das glicoproteínas.
D-Xilose Gomas de madeiras, proteoglicanas, glicosaminoglicanas.
Constituinte de glicoproteínas.
D-Lixose Músculo cardíaco. Constituinte de uma lixoflavina isolada do músculo cardíaco humano.
L-Xilulose Intermediário da via do ácido urônico. Encontrada na urina
Hexoses de importância fisiológica
Açúcar Fonte Importância bioqímica Significado clínicoD-Glicose Sucos de frutas. Hidrólise do
amido, do açúcar de cana, da maltose e da lactose.
É o açúcar do organismo. O açúcar transportado pelo sangue, e o principal usado pelos tecidos.
Presente na urina (glicosúria) no diabetes mellitus devido ao aumento da glicose sanguínea (hiperglicemia).
D-Frutose Sucos de frutas. Mel. Hidrólise do açúcar de cana e da inulina (da alcachofra de Jerusalém).
Pode ser transformada em glicose no fígado e assim usada no organismo.
Intolerância hereditária a frutose conduz ao acúmulo de frutose e hipoglicemia.
D-Galactose Hidrólise da lactose. Pode ser transformada em glicose no fígado e metabolizada. Sintetizada na glândula mamária para formar a lactose do leite. Um ___________________________
A falha na metabolização conduz à galactosemia e à catarata.
Carboidratos Fontes Prod. finais da digestão
Observação
Polissacarídeos indigeríveis1. Celulose2. Hemicelulose
Talos e folhas de vegetais. Camada externa de revestimento de grãos.
-
-
3. Pectinas4. Gomas e mucilagens
FrutasSementes e secreções vegetais
-
-
5. Subst. derivadas de algas
Plantas marinhas e algas-
Polissacarídeos parcialmente digeríveis1. Inulina Alcachofra, cebola, alho,
cogumelos.Frutose
2. Galactógenos Escargot Galactose
3. Manoses Leguminosas Manose
4.Rafinose Açúcar de beterraba, feijão, feijão branco, lentilhas
Glicose, frutose e galactose
5. Estaquiose Feijão Pentoses
6.Pentosanas Frutas e gomas -Polissacarídeos digeríveis1. Amido e dextrinas
Grão; vegetais (especialmente leguminosas e tubérculos)
Glicose
2. Glicogênio Produtos de carne e frutos do Glicose

17
mar.
Carboidratos Fontes Prod. finais da digestão
Observação
Dissacarídeos e Oligossacarídeos1. Sacarose Açúcar de cana e beterraba,
melaço e xarope de bordoGlicose e Frutose
2. Lactose Leite e derivados Glicose e Galactose
3. Lactulose Produtos sintéticosNão
metabolizados
Não aparece em alimentos; é sintético, não digerível e é usado como laxativo.
4. Maltose e Maltotriose
Produtos do Malte; alguns cereais matinais
Glicose
5. Trealose Cogumelos, insetos, leveduras Glicose
Carboidratos Fontes Prod. finais da digestão
Observação
Monossacarídeos1. Glicose Sorbitol*
Frutas, mel, xarope de milhoFrutas, vegetais e produtos dietéticos
GlicoseEm frutas e vegetais, as concentrações de glicose e frutose dependem do amadurecimento da espécie e do estado de preservação.
2. Frutose Frutas e mel Frutose
3.Galactose - GalactoseEsses monossacarídeos não ocorrem na forma livre no alimento.4.Manose
Manitol*-Abacaxi, azeitona, aspargos, batata-doce, cenoura e produtos diet
Manose
Carboidratos Fontes Prod. finais da digestão
Observação
Pentoses1. Ribose - Ribose
Ribose, Xilose e Arabinose não ocorrem na forma livre nos alimentos. São derivados de pentosanas de frutas, dos ácidos nucleicos de produtos da carne e frutos do mar.
2. Xilose Xilitol* Frutas, vegetais, cereais,
cogumelos, frutos do mar, gomas de mascar dietéticas e outros produtos dietéticos
Xilose
3.Arabinose - Arabinose
Carboidratos Fontes Prod. Finais da digestão
Observação
Derivados de Carboidratos1. Álcool Etílico Licores fermentados
Absorvidos nessa forma
São produtos da quebra natural ou induzida de carboidratos.
2. Ácido Láctico Leite e produtos lácteos
3. Ácido Málico Frutas

18
Os carboidratos têm função estrutural por causa do DNA e RNA. Têm função relacionada aos receptores de membranas, que são de natureza glicoproteicas. Sua fórmula geral é Cn(H2O)n = CnH2nOn.
Fotossíntese
A fotossíntese é o processo pelo qual a energia luminosa é transformada em energia química. A equação geral da fotossíntese é:
6 CO2 + 6 H2O C6H12O6 + 6O2 ∆G0’ = + 2.870 kJ. mol-1
Como quase todos os organismos que não fazem fotossíntese dependem da energia química presente nos compostos produzidos pelos seres fotossintetizadores, pode-se dizer que toda a energia consumida pelos sistemas biológicos deriva primariamente da energia solar. A equação da fotossíntese é exatamente o inverso da equação de oxidação total da glicose que ocorre em todas as células aeróbias:
C6H12O6 + 6O2 6 CO2 + 6 H2O ∆G0’ = - 2.870 kJ. mol-1
Porém é incorreto afirmar que a fotossíntese é um processo inverso ao da respiração, definida como comumente como a oxidação da glicose a CO2 e H2O.
Monossacarídeos, Dissacarídeos, Oligossacarídeos e Polissacarídeos
Os monossacarídeos são incapazes de serem hidrolisados a uma forma mais simples. Os dissacarídeos podem ser hidrolisados, dando duas moléculas de monossacarídeos. Os carboidratos conhecidos como monossacarídeos são as Hexoses (Açúcares com 6C) e as Pentoses (açúcares com 5C). Os oligossacarídeos produzem de 3 a 10 unidades de monossacarídeos, enquanto os polissacarídeos produzem de 10 a 10.000 ou mais unidades de monossacarídeos.
Monossacarídeos
Os monossacarídeos podem ser poliidroxialdeídos (aldose) ou poliidroxicetonas (cetose). Os monossacarídeos mais simples possuem três átomos de carbono. São as trioses. O gliceraldeído é a aldose com três carbonos (uma aldotriose), e a diidroxiacetona é a cetose com três carbonos (uma cetotriose). Aldoses que possuem 4, 5, 6, e 7 átomos de carbono são chamadas aldotetroses, aldopentoses, aldoexoses e aldoeptoses, respectivamente e as cetoses correspondentes são cetotetroses, cetopentoses, cetohexoses e cetoheptoses. Os açúcares com seis carbonos são os mais abundantes na natureza, porém dois açúcares com cinco carbonos, a ribose e a desoxirribose, estão presentes nas estruturas de RNA e DNA,

19
respectivamente. Açúcares com 4 e 7 carbonos têm importantes papéis na fotossíntese e em vias metabólicas.
Algumas moléculas não podem ser superpostas em suas imagens especulares. Essas imagens são isômeros ópticos (estereoisômeros) umas das outras. Um átomo de carbono quiral (assimétrico) é o objeto da isomeria óptica. O carboidrato mais simples, que contém um carbono quiral, é o gliceraldeído, podendo existir em duas formas isoméricas que são imagens especulares uma da outra. Elas são designadas D-gliceraldeído e L-gliceraldeído. Os estereoisômeros de imagem especular são também chamados enantiômeros.
Os dois enantiômeros do gliceraldeído são os únicos estereoisômeros possíveis nos açúcares de três carbonos, mas as possibilidades aumentam conforme as estruturas possuam mais átomos de carbono. Para mostrar as estruturas das moléculas resultantes, é preciso usar-se a perspectiva bidimensional da estrutura molecular, denominada método de projeção de Fischer. Nesse método, as ligações escritas “verticalmente” no papel, que é bidimensional, representam as ligações direcionadas para trás do papel, se forem consideradas três dimensões, ao passo que as ligações escritas “horizontalmente” representam as ligações direcionadas para a frente do papel. A designação da configuração como L ou D depende do arranjo do carbono quiral com número mais alto.
Estereoisômeros de imagens não-especulares e que não podem ser sobrepostos são chamados diastereoisômeros. São chamados epímeros os diasteroisômeros que diferem uns dos outros na configuração em somente um C quiral. Ex: D – eritreose e D – treose; α – D – galactose e α – D – glicose e α – D – Manose. A maioria dos açúcares importantes encontrados na natureza possui a configuração D, baseada no D-gliceraldeído.
20
Wikipédia.org
Açúcares especialmente os que possuem cinco ou seis carbonos existem normalmente como moléculas cíclicas em vez das cadeias abertas. A ciclização é resultante da interação entre os grupos funcionais em carbonos distantes, como C-1 e C-5, para formar um hemiacetal cíclico (em aldoexoses). Outra possibilidade é a formação de um hemicetal cíclico (em cetoexoses) através da interação entre C-2 e C-5. Em ambos os casos, o carbono carbonílico torna-se um novo centro quiral chamado carbono anomérico. O açúcar cíclico pode assumir qualquer uma das duas formas diferentes α e β, e são chamados anômeros um do outro.
Além das fórmulas de projeção de Fischer, existem também as fórmulas de projeção de Haworth que representam mais precisamente o formato nas moléculas cítricas com cinco ou seis membros. O anel de cinco membros é chamado furanose, e o de seis, piranose.
H C O
H C OH
H O C H
H C OH
H C OH
CH2OH
D-Glucose (Está presente em frutas, mel, milho e raízes). É armazenada no fígado e nos músculos, como o glicogênio.
CH2OH
C O
HO C H
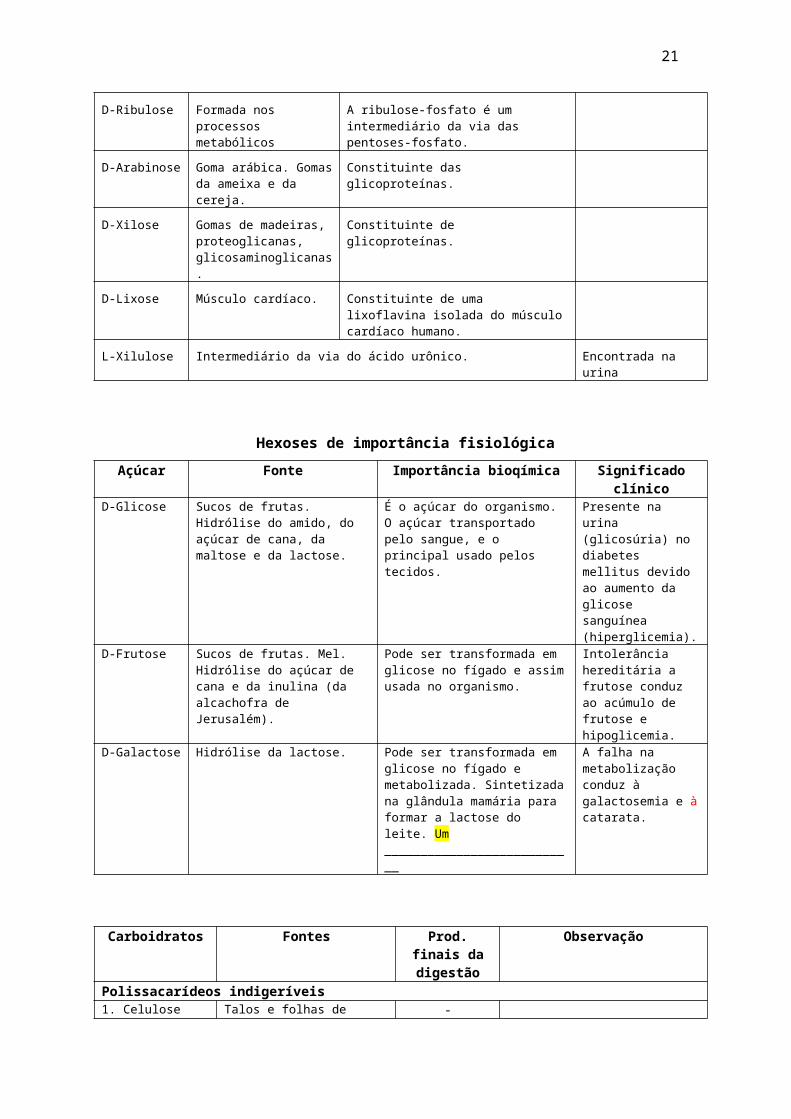
21
H C OH
H C OH
CH2OH
D- Frutose (Levulose ou açúcar da fruta. Está presente em mel e frutas).
Galactose: Não é encontrada na forma livre na natureza. É produzida a partir da lactose (açúcar do leite) por hidrólise no sistema digestório.
Reações dos Monossacarídeos
Reações de Oxidorredução
Essas reações dos açúcares são fundamentais na bioquímica. A oxidação dos açúcares é responsável pelo fornecimento de energia para que os processos vitais dos organismos sejam realizados.
Reações de Esterificação
Os grupos hidroxila dos açúcares podem reagir com ácidos e derivados para formar ésteres. Os ésteres de fosfato são muito importantes por serem intermediários na degradação de carboidratos para fornecer energia.
Formação de Glicosídeos
Ocorre a formação de glicosídeos quando um grupo hidroxila de açúcar ligado a um carbono anomérico reage com outra hidroxila formando uma ligação glicosídica. Essa ligação não é um éter, pois os glicosídeos podem ser hidrolisados aos alcoóis originais. As ligações glicosídicas entre monossacarídeos são a base para a formação de dissacarídeos, oligossacarídeos e polissacarídeos. Diferentes formas estereoquímicas são possíveis em ligações glicosídicas, com importantes consequências para a função das substâncias assim formadas.
Dissacarídeos
São formados pela união de dois monossacarídeos por ligações glicosídicas. Os mais importantes são: sacarose, lactose e maltose. A sacarose é formada por α - D- Glicose + α – D- Frutose. É o açúcar de uso comum. Está presente na cana de açúcar, açúcar de beterraba, melaço, xarope de bordo, xarope de milho, açúcar de bordo, frutas, vegetais e mel. A lactose, o açúcar presente no leite, é um dissacarídeo formado por β – D- lactose e D- glicose. A galactose é um epímero C-4 da glicose. A maltose é um dissacarídeo obtido da hidrólise do amido, consistindo em dois resíduos de D-glicose. A levedura, especialmente a da cerveja, contém enzimas que hidrolisam o amido no broto
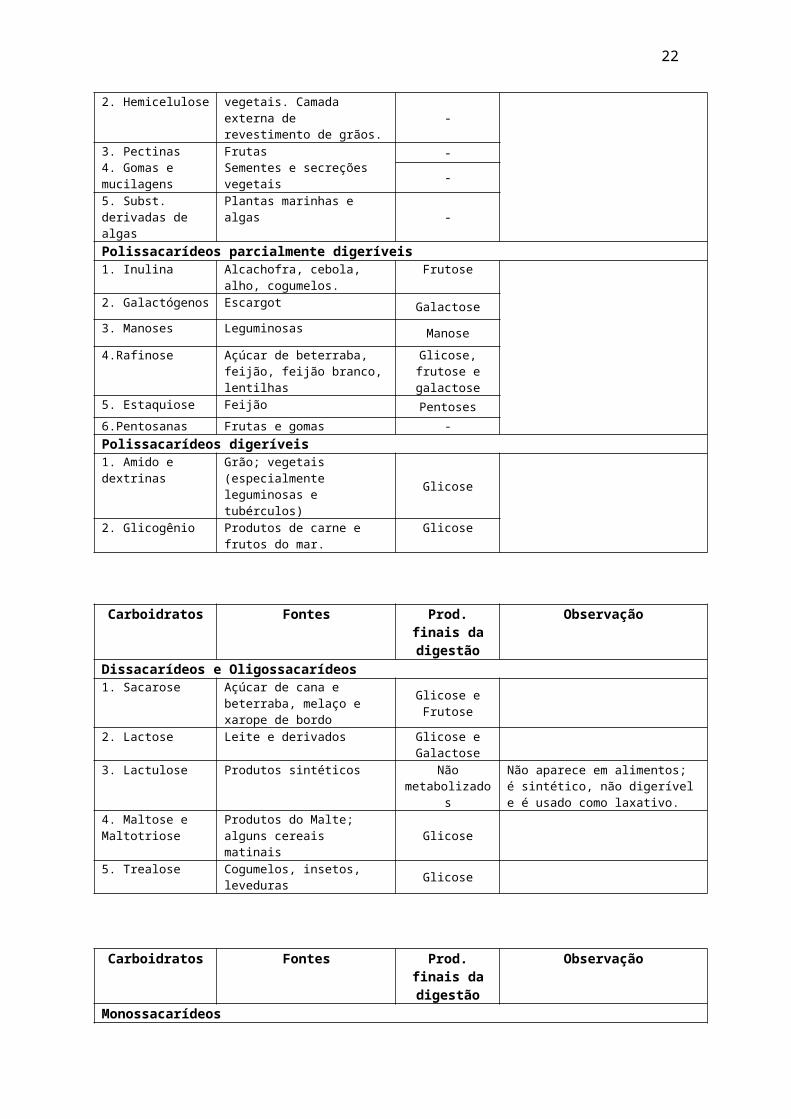
22
da cevada (malte), primeiro em maltose e depois em glicose, que é fermentada na preparação da cerveja.
: Hidrolisada por enzimas digestivas ou fervida com ácido, dando partes iguais de glicose + frutose. Esta mistura se chama açúcar invertido. (Enzima: invertase) e causa cárie dentária. Em vez de hiperatividade, o consumo de carboidrato aumenta a produção de serotonina que traz um efeito sedativo ao sistema nervoso.
: Açúcar do malte. Não é encontrado livre na natureza. É produzida durante a digestão, por enzimas que quebram grandes moléculas de amido em fragmentos de dissacarídeos, que podem ser quebrados em duas moléculas de glicose para facilitar a absorção. Isto ocorre na natureza. Quando a semente de um grão de cereal brota, suas enzimas convertem o grão em maltose. Ex.1: Malte da cevada usado como adoçante. Ex.2: Fabricação da cerveja: o amido é hidrolisado pela diástase, uma enzima obtida de grãos germinantes.
:: : Açúcar do leite. Não existe em vegetais. Está limitada quase que exclusivamente às glândulas mamárias de animais lactentes. Por hidrólise dá: galactose + glicose. Problemas com esse açúcar: 1 - Pessoas com ausência da enzima lactase, não fazem uma hidrólise eficiente. 2 – Crianças pequenas nascidas sem a enzima do fígado que converte galactose em glicose.
Polihidróxiálcoois ou Poliálcoois
Os polihidróxiálcoois ou poliálcoois inibem uma elevação rápida de açúcar no sangue.
SACAROSE forma alcoólica: SORBITOL
MANOSE forma alcoólica: MANITOL
XILOSE forma alcoólica: XILITOL
SORBITOL: É naturalmente encontrado em frutas. Tem poder adoçante igual ao da glicose. É bem absorvido e tem o mesmo valor energético da glicose.
MANITOL: Existe nas frutas, é precariamente digerido, produz metade das calorias da glicose, por grama.
SACAROSE
MALTOSE
LACTOSE
São usados em produtos para pessoas incapazes de tolerar grandes ingestões de açúcar, pois são absorvidos mais lentamente no trato digestório e, portanto, inibem uma elevação rápida do açúcar no sangue.
23
XILITOL: Absorvido apenas 1/5 tão rápido quanto a glicose. É usado em gomas de mascar sem açúcar porque as bactérias cariogênicas são incapazes de usá-lo como substrato.
sacarose
lactose
Adoçantes Alternativos
São mais doces que os açúcares naturais. São digeridos ou absorvidos. Não têm valor nutritivo.
HIPERTEXTO
Mel de Abelhas
O néctar da flor que contém sacarose é levado pela abelha para a colmeia. No favo, a abelha envolve o néctar com a enzima invertase que hidrolisa a maioria da sacarose em glicose e frutose. Após várias horas de evaporação, o mel amadurecido e concentrado é armazenado em células seladas. A doçura do mel varia com a concentração dos açúcares e grau de cristalização. Vitaminas e minerais aparecem como quantidades traços. A absorção entre açúcar e mel é quase igual. As diferenças entre mel e açúcar de mesa estão no fato de o mel conter, além de maior teor de frutose, outros componentes minoritários, como vitaminas, compostos fenólicos, flavonoides, minerais, entre outros. A frutose no sangue é principalmente convertida em glicogênio no fígado, um processo que não precisa de insulina. Porém, o alto conteúdo de glicose faz do mel um alimento que deve ser controlado para diabéticos, não insulino-dependentes.
Polissacarídeos
Os polissacarídeos de interesse são: amido, dextrina, glicogênio. São menos solúveis e mais estáveis que os açúcares mais simples. O amido e o glicogênio são geralmente completamente digeríveis.

24
: : é encontrado apenas em vegetais, em ambas as formas: a) Amilose - que possui cadeias retas e longas de unidade de glicose; b) Amilopectina - que possui cadeias ramificadas de unidades de glicose. Os grânulos de amido de vários tamanhos e formas estão encerrados dentro das células do vegetal pelas paredes de celulose. Características do amido: 1. Os grânulos são insolúveis em H2O fria; 2. O cozimento causa o intumescimento dos grânulos e a mistura se torna um gel; 3. O cozimento edemacia e rompe a célula para deixar o amido disponível para os processos digestivos enzimáticos.
Amido Alimentar Modificado (amido resistente à ação das enzimas): É agente espessante usado em alimentos preparados comercialmente, como molhos de saladas, recheios de tortas, sopas enlatadas, caldos, pudins enlatados e alimentos para bebês. A modificação permite a retenção de propriedades espessantes desejáveis, perdidas no amido comum após esfriamento e estocagem.
: Produtos intermediários que ocorrem na hidrólise do amido. São formadas durante a digestão e também como resultado de uma variedade de processos comerciais que usam ácidos, enzimas ou calor seco. Diminuindo em tamanho, as moléculas dos sacarídeos vão aumentando em solubilidade e doçura. Aplicações comerciais: em xarope de milho (rico em dextrinas). Dextrose: É a glicose produzida pela hidrólise de amido de milho.
: : Forma de armazenamento de carboidratos em humanos e animais. É a primeira e a mais prontamente disponível fonte de glicose e energia. Consiste de: cadeias ramificadas de unidades de glicose semelhantes àquelas do amido de vegetal. (~ 340g de glicogênio é armazenado no fígado e nos músculos). As pequenas quantidades de glicogênio nos alimentos animais são convertidas em ácido láctico antes de estarem disponíveis para o consumo.
: Constituem a estrutura celular dos vegetais. A C celulose lembra o amido, pois contém muitas moléculas de glicose em forma não ramificada parecida com a amilose, porém unidas de uma forma que não
AMIDO
DEXTRINAS
GLICOGÊNIO
CELULOSE E

25
são hidrolisadas pelas enzimas que hidrolisam o amido. É encontrada apenas em vegetais: polpa de frutas e vegetais, peles, talos, folhas, cobertura externa de grãos, nozes, sementes e leguminosas. As hemiceluloses são polissacarídeos não celulósicos. Diferem das celuloses na estrutura, pois têm menos unidades de glicose. Podem consistir de hexoses, pentoses e formar ácidos destes compostos. Os produtos de fibra sintéticos, como a metilcelulose e a carboximetilcelulose, são usados em laxativos, assim como na produção de alimentos de baixas calorias devido a sua propriedade de produzir volume e sociedade.
: Polissacarídeo, não celulósico, constituído de unidades de um derivado de galactose. Como absorve H2O e forma um gel, é usada para fazer geleias e gelatinas. É encontrada em maçãs, frutas cítricas, morangos e outros.
: São semelhantes à pectina, exceto pelo fato de que as unidades de galactose estão combinadas a outros açúcares (glicose) e polissacarídeos, encontrados em secreções vegetais ou sementes e são adicionadas a alimentos processados para conferir propriedades ou qualidades específicas.
: São encontrados em frutos do mar e algas. Ex.: carragenina, adicionada como agente espessante e estabilizante em muitos produtos alimentares processados.
CONTEXTUALIZANDO
Intolerância à Lactose
Mais de 75 % dos adultos do mundo inteiro sofrem com a intolerância à lactose. Isso ocorre especialmente em certas raças. Por exemplo: até 90% dos adultos com ascendência africana ou asiática são deficientes em lactase sendo, portanto, menos capazes de metabolizar lactose que os indivíduos originários do norte da Europa. O mecanismo pelo qual a enzima é perdida
PECTINA
GOMAS E MUCILAGENS
POLISSACARÍDEOS DE ALGAS

26
com a idade não está claro, mas é determinado geneticamente e representa uma redução na quantidade da proteína enzimática, e não uma enzima modificada e inativa. O tratamento para esse distúrbio é a redução do consumo de leite passando a ingerir iogurtes e queijos, além de comer vegetais verdes como brócolis, de modo a assegurar a ingestão adequada de cálcio, usar produtos com adição de lactase ou ingerir a lactase em comprimidos antes das refeições (Champe, Harvey, Ferrier, 2009, p. 88).
ATIVIDADES DE AVALIAÇÃO
1. Defina os seguintes termos:
a) polissacarídeo; b) furanose; c) piranose; d) aldose; e) cetose; f) ligação glicosídica; g) oligossacarídeo; h) glicoproteína.
2. Citar exemplos de polissacarídeos estruturais e de reserva.
3. Descrever a estrutura do glicogênio e indicar a porção da molécula que sofre alongamento ou encurtamento.
4. Quais são as principais diferenças entre as paredes celulares das plantas e das bactérias?
5. Como a quitina se difere da celulose em estrutura e função?
6. Como o glicogênio se difere do amido em estrutura e função?
7. Que são epímeros? Exemplifique.
8. O poliálcool mais frequentemente usado em chiclete e em doces dietéticos é o α-sorbitol. Boa parte desse álcool é obtido pela redução da D-glicose. Compare essas duas estruturas e explique o modo pelo qual isso pode acontecer.
9. Qual é a base metabólica para a observação de que muitos adultos não podem ingerir grandes quantidades de leite sem ter dificuldades gástricas?
10. Qual é o benefício das fibras na alimentação?
11. Pesquise e indique resumidamente o papel das glicoproteínas como determinantes antigênicos para os grupos sanguíneos.
Bibliografia Comentada

27
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol. 3. São Paulo: Thomson Learning, 2008. Monossacarídeos, Dissacarídeos, Oligossacarídeos são comentados no Capítulo 16.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. – Porto Alegre: Artmed, 2009. O Capítulo 7 da Unidade II expõe sobre Carboidratos.
Liberato, M. C. T. C. Estudo Químico e Bioprospecção de Produtos da Abelha Apis mellifera L. do Estado do Ceará. Tese. (Doutorado em Biotecnologia) – Universidade Estadual do Ceará – Renorbio. Fortaleza. 2011. O assunto Mel e os monossacarídeos nele contidos são abordados exaustivamente.
Mahan, K.; Escott-Stump. Alimentos, nutrição & dietoterapia. São Paulo: Roca, 2005. Na Parte I, que trata dos Princípios Nutricionais, no Capítulo 3, são apresentados tópicos relativos aos carboidratos.
LEITURA, VÍDEOS E SITES RECOMENDADOS
http://www.quimica2000.cjb.net/
CAPÍTULO 3: LIPÍDIOS
OBJETIVOS:
Conhecer as macromoléculas estruturais e de revestimento, que são as gorduras, chamadas quimicamente de lipídios.
Conhecer os tipos de lipídios e onde são encontrados no nosso organismo e ainda os principais alimentos que contêm grande quantidade dessas moléculas.
Classificar os vários tipos de lipídios. Aprender as funções dos lipídios, em animais e vegetais. Observar a localização celular dos diversos lipídios. Estudar a funções do colesterol e das vitaminas lipossolúveis, suas
transportadoras. Aprender sobre a deficiência das proteínas e as doenças relacionadas.
Os lipídios são compostos que ocorrem na natureza. Pode-se também defini-los como moléculas orgânicas naturais isoladas de células e tecidos por extração com solventes orgânicos não polares. As características que melhor definem os lipídeos estão relacionadas com sua solubilidade, pois são relativamente insolúveis na água e são solúveis nos solventes não polares, tais

28
como o éter, o clorofórmio e o benzeno. Gorduras e óleos são lipídeos típicos em termos de solubilidade, mas esse fato não define realmente sua natureza química. Químicamente, pode-se dizer que o lipídeo é uma mistura de compostos que compartilham algumas propriedades com base em semelhanças estruturais, principalmente uma preponderância de grupos apolares. Uma classificação que se relacione com a sua natureza química poderia ser aquela que encaixa os lipídeos em dois grupos principais. O primeiro grupo consiste em compostos de cadeia aberta com grupos de cabeça polar e longas caudas apolares, e inclui os ácidos graxos, os triacilgliceróis, os esfingolipídios, os fosfoacilgliceróis e os glicolipídios. O segundo grupo principal consiste em compostos de anéis fundidos (cadeias cíclicas), os esteroides, sendo um importante representante desse grupo o colesterol.
Também é possível classificar os lipídeos como simples e complexos. Nesse caso, são chamados Lipídeos simples os ésteres que, por hidrólise total dão origem somente a ácidos graxos e alcoóis. Podem ser: 1. Gorduras e Óleos, que são ésteres de ácidos graxos com o glicerol. São denominados triacilgliceróis (TAG). 2. Ceras que são ésteres de ácidos graxos com álcoois monohidroxílicos de pesos moleculares mais elevados e geralmente de cadeia linear. Os Lipídeos complexos (ou lipídeos compostos) são os ésteres de ácidos contendo outros grupos além de álcoois e de ácidos graxos, como 1. Fosfolipídeos (ou Fosfatídeos) que são lipídeos que contêm, além de ácidos graxos e um álcool, um resíduo de ácido fosfórico e, frequentemente, têm bases nitrogenadas e outros substituintes, como nos glicerofosfolipídeos, o álcool é o glicerol e, nos esfingolipídeos, o álcool é a esfingosina. 2. Glicolipídeos (Glicoesfingolipídeos), que são lipídeos que contêm um ácido graxo, esfingosina e carboidrato. Outros Lipídeos Complexos são lipídeos tais como Sulfolipídeos (contêm enxofre) e Aminolipídeos (contêm grupos amino). As lipoproteínas também podem ser enquadradas nesta categoria, bem como os Precursores e derivados de lipídeos que são obtidos na sua maioria por hidrólise dos lipídeos simples e composto. Incluem: ácidos graxos, glicerol, esteroides, alcoóis, além do glicerol e esteróis, aldeídos graxos e corpos cetônicos, hidrocarbonetos, vitaminas lipossolúveis, hormônios e pigmentos.
Ácidos Graxos
São ácidos carboxílicos alifáticos. São considerados compostos anfipáticos porque o grupo carboxila é hidrofílico e a cauda de hidrocarboneto é hidrofóbica. Ocorrem como ésteres nas gorduras naturais e nos óleos. Neste caso, geralmente, têm cadeia linear e número par de átomos de carbono. Podem ter cadeia saturada ou insaturada. As propriedades físicas e fisiológicas dos ácidos graxos dependem do comprimento da cadeia e do grau de insaturação. Ex.: Os pontos de fusão dos ácidos graxos, com o nº. par de átomos de carbono, aumentam com o aumento do comprimento da cadeia e

29
diminuem com a insaturação. Os ácidos graxos possuem cadeias retas de hidrocarboneto terminando em um grupo carboxila de um lado e um grupo metila no outro.
A maioria das cadeias dos ácidos graxos tem entre 4 e 22C, com aqueles de 16 e 18 carbonos, ou ácidos graxos de cadeia longa, sendo os de maior prevalência. No organismo, os ácidos graxos são uma parte importante dos Fosfolipídeos nas Membranas Celulares. Os ácidos graxos são classificados pelo nº de C, pela posição da 1ª dupla ligação e pelo nº de duplas ligações. A localização da 1ª dupla ligação, contada a partir da terminação metila do Ácido Graxo, é designada pelo nº ω (ômega).
Ex.: Ácido Linoleico: ω–6 (ômega-6) 18:2. Ou seja: possui 18C, 2 duplas, sendo a 1ª dupla no C6 contado a partir do CH3.
Ácidos Graxos Saturados
Contêm o nº máximo de H que a cadeia pode suportar. Estão concentrados em certos alimentos animais (carnes bovina, frango, porco, laticínios) e alimentos vegetais (palmeira, semente de palmeira e óleo de coco). O nível de saturação determina a consistência da gordura à temperatura ambiente. Em geral, quanto > a cadeia, mais saturada ela é e mais dura a gordura de coco será em temperatura ambiente. Exceção: óleo de coco (altamente saturado e líquido à temperatura ambiente por causa da prevalência ou predominância dos Ácidos Graxos de cadeia curta; menos que seis átomos de carbonos.
Ácidos Graxos Monoinsaturados

30
Contêm apenas uma dupla ligação. Ex.: ácido oleico (presente em azeite de oliva, óleo de canela, óleo de amendoim, amendoins, nozes pecãs, amêndoas e abacates). No organismo, o ácido oleico é formado pelo estearato através da ação da enzima dessaturase. Os ácidos graxos monoinsaturados podem desempenhar um papel no tratamento do diabetes.
Ácidos Graxos Trans
A maior parte dos ácidos graxos monoinsaturados nos alimentos ocorre na forma cis, significando que os H estão do mesmo lado da dupla ligação. No processamento dos alimentos, os ácidos graxos trans são formados quando se adiciona H2 a óleos líquidos para torná-los semi-sólidos e mais estáveis. As fontes de ácidos graxos trans na dieta são margarina dura, gordura, frituras, produtos de panificação ricos em gorduras e lanches salgados.
Acidos Graxos Saturados
Ácidos Graxos Insaturados
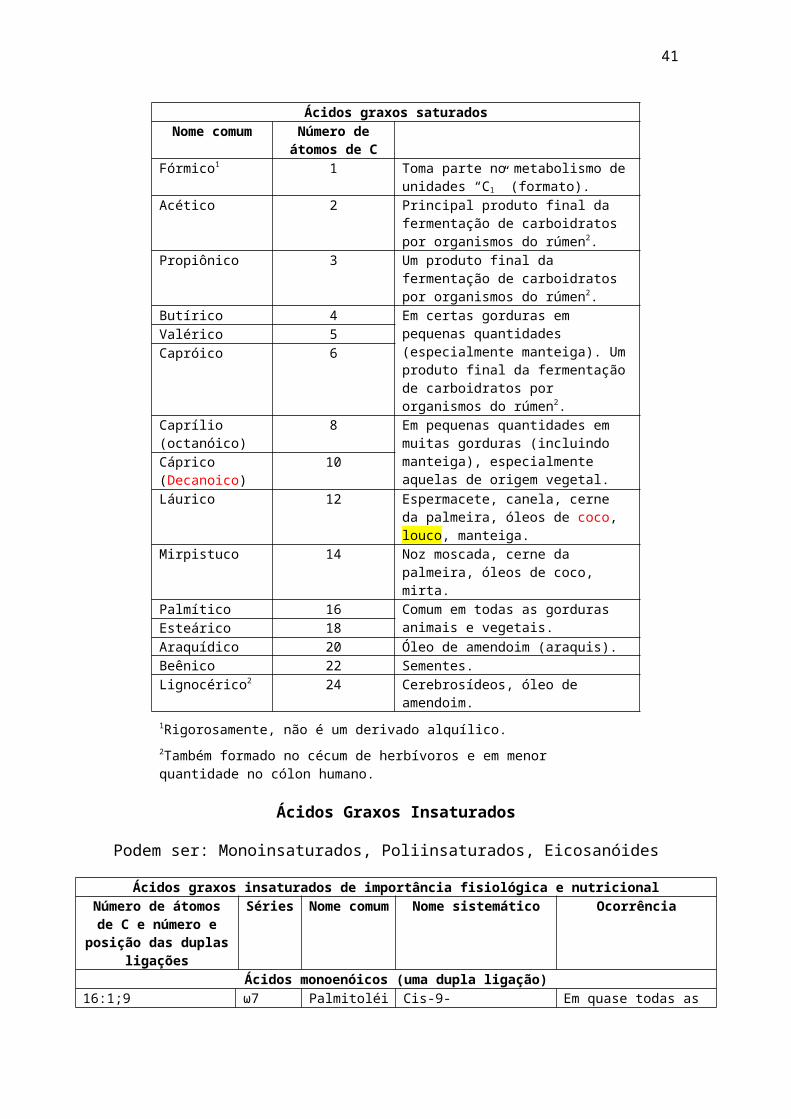
Ácidos graxos saturadosNome comum Número de
átomos de CFórmico1 1 Toma parte no metabolismo de
unidades “C1” (formato).Acético 2 Principal produto final da fermentação
de carboidratos por organismos do rúmen2.
Propiônico 3 Um produto final da fermentação de carboidratos por organismos do rúmen2.
Butírico 4 Em certas gorduras em pequenas quantidades (especialmente manteiga). Um produto final da fermentação de carboidratos por organismos do rúmen2.
Valérico 5Capróico 6
Caprílio (octanóico)
8 Em pequenas quantidades em muitas gorduras (incluindo manteiga), especialmente aquelas de origem vegetal.
Cáprico (Decanoico)
10
Láurico 12 Espermacete, canela, cerne da palmeira, óleos de coco, louco, manteiga.
Mirpistuco 14 Noz moscada, cerne da palmeira, óleos de coco, mirta.
Palmítico 16 Comum em todas as gorduras animais e vegetais.Esteárico 18
Araquídico 20 Óleo de amendoim (araquis).Beênico 22 Sementes.Lignocérico2 24 Cerebrosídeos, óleo de amendoim.
1Rigorosamente, não é um derivado alquílico.2Também formado no cécum de herbívoros e em menor quantidade no cólon humano.

31
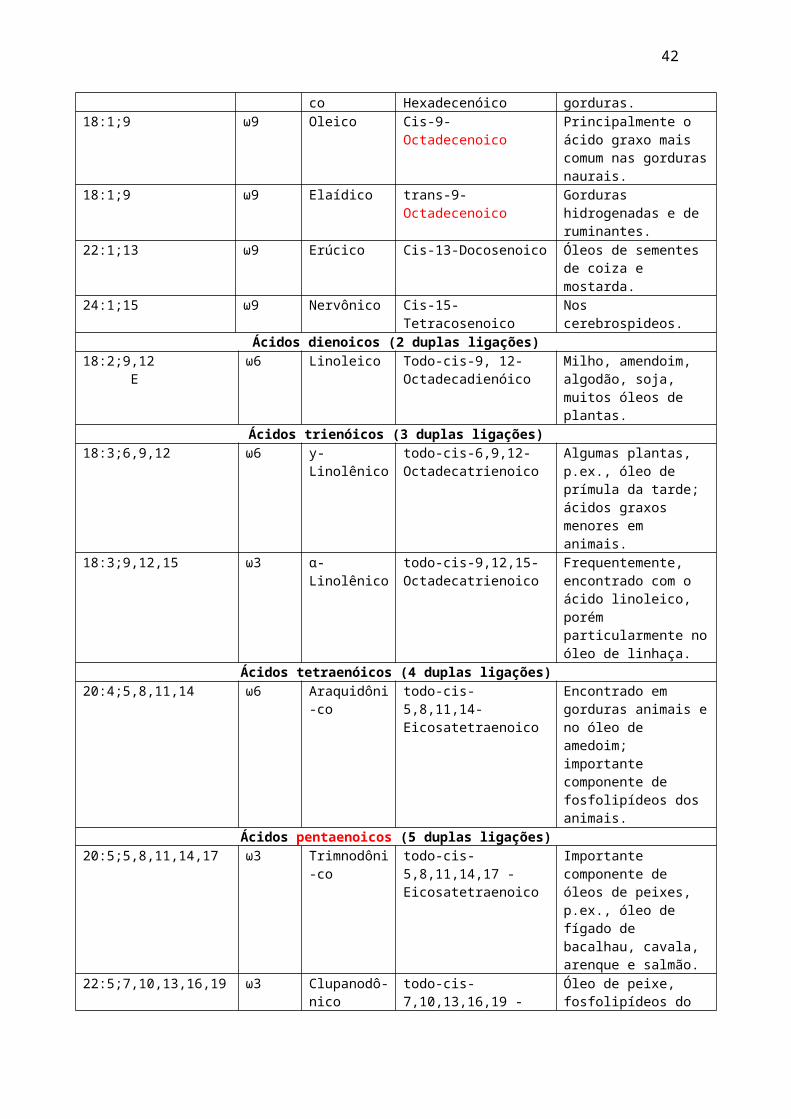
Podem ser: Monoinsaturados, Poliinsaturados, Eicosanóides
Ácidos graxos insaturados de importância fisiológica e nutricionalNúmero de átomos de C e número e posição das duplas ligações
Séries Nome comum
Nome sistemático Ocorrência
Ácidos monoenóicos (uma dupla ligação)16:1;9 ω7 Palmitoléico Cis-9-Hexadecenóico Em quase todas as
gorduras.18:1;9 ω9 Oleico Cis-9-Octadecenoico Principalmente o ácido
graxo mais comum nas gorduras naurais.
18:1;9 ω9 Elaídico trans-9-Octadecenoico Gorduras hidrogenadas e de ruminantes.
22:1;13 ω9 Erúcico Cis-13-Docosenoico Óleos de sementes de coiza e mostarda.
24:1;15 ω9 Nervônico Cis-15-Tetracosenoico Nos cerebrospideos.Ácidos dienoicos (2 duplas ligações)
18:2;9,12 E
ω6 Linoleico Todo-cis-9, 12-Octadecadienóico
Milho, amendoim, algodão, soja, muitos óleos de plantas.
Ácidos trienóicos (3 duplas ligações)18:3;6,9,12 ω6 y-Linolênico todo-cis-6,9,12-
OctadecatrienoicoAlgumas plantas, p.ex., óleo de prímula da tarde; ácidos graxos menores em animais.
18:3;9,12,15 ω3 α-Linolênico todo-cis-9,12,15-Octadecatrienoico
Frequentemente, encontrado com o ácido linoleico, porém particularmente no óleo de linhaça.
Ácidos tetraenóicos (4 duplas ligações)20:4;5,8,11,14 ω6 Araquidôni-
cotodo-cis-5,8,11,14-Eicosatetraenoico
Encontrado em gorduras animais e no óleo de amedoim; importante componente de fosfolipídeos dos animais.
Ácidos pentaenoicos (5 duplas ligações)20:5;5,8,11,14,17 ω3 Trimnodôni-
cotodo-cis-5,8,11,14,17 -Eicosatetraenoico
Importante componente de óleos de peixes, p.ex., óleo de fígado de bacalhau, cavala, arenque e salmão.
22:5;7,10,13,16,19 ω3 Clupanodô-nico
todo-cis-7,10,13,16,19 -Docosapentaenoico
Óleo de peixe, fosfolipídeos do cérebro.
Ácidos hexaenoicos (6 duplas ligações)22:6;4,7,10,13,16,19 ω3 Cervônico todo-cis-5,8,11,14-
DocosahexaenoicoÓleos de peixe, fosfolipídeos do cérebro.
Ácidos Graxos Incomuns
Os ácidos graxos incomuns contém nº. ímpar de átomos de carbono. Ex.: ácido margárico, ácido malválico.
Ácidos Graxos Essenciais

32
Os ácidos graxos essenciais produzem efeitos especiais no organismo vivo. Não podem ser produzidos pelo organismo humano. Devem ser administrados pelos alimentos. Exemplo: o ácido linoleico é transformado pelo organismo no ácido araquidônico (4 vezes mais insaturado e com 20 carbonos). O ácido araquidônico é o ácido graxo verdadeiramente “essencial” para o organismo humano. O Ácido Linoleico (ω–6) e o Ácido α – Linolênico (ω–3) são os dois ácidos graxos essenciais na dieta porque previnem sintomas de deficiência e não podem ser sintetizados pelos seres humanos. Esses dois Ácidos Graxos são os compostos de origem para os outros ácidos graxos biologicamente ativos. O Ácido Linoleico, através da ação das enzimas dessaturase, pode ser convertido em Ácido α – Linolênico e Ácido Araquidônico, e ambos podem desempenhar uma função no início do desenvolvimento cerebral. O Ácido Araquidônico pode prevenir a dermatite encontrada na deficiência de Ácido Graxo Essencial; portanto, tem atividade parcial de ácido graxo essencial. Devido ao fato do Ácido Araquidônico ser sintetizado a partir do Ácido Linoleico, ele se torna essencial, se a dieta for deficiente em Ácido Linoleico.
Na família do ω–3, o Ácido docosaexaenóico (DHA, C22: 6 ω–3), desempenha uma função principal no funcionamento da retina e desenvolvimento cerebral, e acredita-se hoje que seja essencial para bebês. Essas famílias de Ácidos Graxos são também precursoras dos eicosanoides (prostaglandinas, tromboxanos e leucotrienos), compostos como hormônios, que ajudam no controle da pressão sanguínea, frequência cardíaca, dilatação vascular, coagulação sanguínea, lipólise e resposta imunológica.
Cada família dá origem a uma série diferente de eicosanoides. Ex.: Ácido Araquidônico (família ω–6) é precursor de Prostaglandina, Tromboxano A2, que causa agregação plaquetária, formação de coágulo e vasoconstrição. Em contraste, os Ácidos Graxos ω–3 favorecem a produção de prostaciclinas (que têm efeitos opostos, isto é, previnem a formação de coágulo e causam vasodilatação). Também inibem a enzima dessaturase, que diminui a produção de ácido Araquidônico e, consequentemente, de tromboxano A2. A ingestão na dieta dos Ácidos Graxos Essenciais está positivamente relacionada com suas concentrações nos ésteres de colesterol e fosfolipídeos no sangue, por isso, essas quantidades bioquímicas são usadas para confirmar a ingestão na dieta.

33
Sintomas da deficiência de Ácido Linoleico ω–6
- Dermatite e desenvolvimento precário em bebês alimentados com uma fórmula sem gordura.
- Os animais também têm insuficiência reprodutiva e fígado gorduroso. A deficiência de Ácidos Graxos Essenciais em crianças e adultos tem sido observada durante um longo período de nutrição parenteral total sem gordura.
- Uma deficiência de Ácido Linolênico ω–3 produziu mudanças neurológicas (entorpecimento, parestesia, fraqueza, incapacidade de andar, visão embaçada) que foram revertidas quando o Ác. Linolênico foi fornecido. Outros sintomas da deficiência do ω–3 são: dificuldades de aprendizado, eletrorretinograma anormal e acuidade visual prejudicada.
Acilgliceróis
São produtos da reação de esterificação de uma molécula de glicerol com até três moléculas de ácidos graxos. Podem ser: Monoacilglicerol, Diacilglicerol e Triacilglicerol.
Triacilgliceróis ou Triglicerídeos

34
(TAG)
Os triacilgliceróis ou triglicerídeos são ésteres do álcool glicerol com ácidos graxos. Contêm uma molécula de Glicerol (um álcool triídrico) e um a três ácidos graxos na ligação do éster. São os óleos de vegetais ou as gorduras de origem animal. Suas propriedades físicas são determinadas pela proporção e estrutura química dos seus ácidos graxos constituintes. Os triacilgliceróis líquidos à temperatura ambiente são chamados em geral, óleos, e os que são sólidos são chamados gorduras. Os ácidos graxos menores e menos insaturados caracterizam as gorduras que são macias ou óleos líquidos em Tº ambiente. As gorduras sólidas (ex.: gordura da carne), contêm grandes quantidades de ácidos graxos de cadeia longa, isto é, ácidos palmítico (C16:0) esteárico (C18:0). As propriedades dos glicerídeos também são influenciadas pelo nº ω e pela posição dos ác. graxos na molécula de glicerol. Nos alimentos, os lipídeos mais abundantes são os triacilgliceróis. Os triacilgliceróis são hidrolisados dando como resultado três ácidos graxos e o glicerol.
Funções
ENERGIA: Devido a sua alta densidade energética e baixa solubilidade, os Triacilgliceróis (TAG) do tecido adiposo são a maior forma de armazenamento de energia no organismo.
Os TAG são armazenados de forma mais eficiente que o GLICOGÊNIO, pois produzem 2 ½ vezes o ATP depois da oxidação e são armazenados sem H2O. Geralmente, os seres humanos possuem algumas semanas de reservas adiposas, mas apenas o valor de cerca de um dia de glicogênio.
O tecido adiposo auxilia a manter órgãos e nervos em posição e protege-os contra choques e lesões traumáticas. (Efeito Amortecedor).
A camada subcutânea de gordura isola o organismo, preservando o calor do organismo e mantendo a temperatura do organismo (Efeito Isolamento Térmico).
As gorduras auxiliam no transporte e absorção de vitaminas lipossolúveis.

35
Deprimem as secreções gástricas e tornam mais lento o esvaziamento gástrico.
As gorduras adicionam o paladar da dieta e produzem uma sensação de saciedade após a refeição.
Hidrogenação dos Triacilgliceróis (HIPERTEXTO)
As gorduras comercializadas para alimentação são obtidas pela hidrogenação parcial de óleos vegetais. O resultado são as gorduras “parcialmente hidrogenadas” presentes em muitos produtos alimentícios. A vantagem comercial da hidrogenação parcial é a maior duração da vida útil da gordura. Há, porém, um problema com a hidrogenação parcial. O catalisador isomeriza parte das duplas ligações não hidrogenadas e substitui o arranjo natural em cis por um arranjo artificial em trans; acumulam-se, porém, provas das gorduras em trans estarem associadas a riscos crescentes de moléstias cardiovasculares.
Ácidos Graxos Poliinsaturados
Os ácidos graxos poliinsaturados contêm duas ou mais duplas ligações. O ácido graxo poliinsaturado predominante na dieta é o Ácido Linoleico. Fontes de Ác. Linoleico são as sementes de vegetais e os óleos que produzem. Óleos que não são provenientes de sementes como: óleo de coco, óleo de palmeira, manteiga de cacau são fontes pobres de ácido linoleico. Existem duas principais famílias de Ác. Graxos Poliinsaturados: ω – 3 e ω – 6. Essas famílias não são conversíveis, tendo funções bioquímicas muito diferentes.
Os ácidos graxos poliinsaturados estão relacionados com a resposta à lesão e inflamação. São classificados pelo número de suas ligações duplas: linoleico (3), araquidônico (4) e eicosapentanóico (5). Dependendo do tecido envolvido, os ácidos graxos entram ou na cascata da prostaglandina ou na do leucotrieno, levando à produção de hormônios eicosanoides. Esses hormônios podem alterar o tamanho e a permeabilidade dos vasos sanguíneos, a atividade das plaquetas e contribuir para a coagulação do sangue, modificando os processos inflamatórios. Estudos mostram a eficácia dos ácidos poliinssaturados na esclerose múltipla, nas doenças inflamatórias (artrite reumatoide e dermatite atípica) e na prevenção da aterosclerose.
PERÓXIDOS
MEMBRANAS CELULARESAGPI
PROSTAGLANDINAS

36
BAINHA DE MIELINA VÍRUS SISTEMA IMUNOLÓGICO
Ácidos Graxos Ômega – 3
São de interesse nutricional o ácido α – linolênico (C18:3) e seus derivados ácidos Eicosapentanóicos (C20:5) e o ácido Docosahexanóico (C22:6). Fontes de ácido α – linolênico são os óleos de salada, margarinas e gordura feita de óleo de canola ou de soja. Óleos de peixes e mariscos são ricos em ácidos Eicosapentanóicos e Docosahexanóico. Os ácidos graxos ω–3 afetam o metabolismo lipoproteico. Os efeitos colaterais potenciais de altas doses de ácidos graxos ω–3 incluem maior tempo de sangramento, infecções, diabetes, peroxidação lipídica.
Lipídeos Compostos
Fosfolipídeos: O 2º maior componente lipídico do organismo são os lipídeos em que um dos ácidos graxos é substituído por uma substância contendo fósforo, assim como o ácido fosfórico.
O
O CH2 – O – C – R1
R2 – C – O – CH O
CH2 – O – P – OCH2CH2N+(CH3)3
O-
Lecitina: A lecitina (fosfatidilcolina) contém ácido fosfórico e a base colina contém nitrogênio. São encontradas grandes concentrações combinadas com proteínas nas membranas celulares, onde facilitam a passagem de gorduras para dentro e para fora da célula, e no sangue, onde também agem no transporte de lipídios (como parte de lipoproteínas). Os ácidos graxos contidos na alimentação irão influenciar os ácidos graxos que aparecem nos fosfolipídios.
MICROCIRCULAÇÃO
LECITINA
SISTEMA IMUNOLÓGICO
PAREDE DOS VASOS
AGREGAÇÃO DE HEMÁCIAS
ADESIVIDADE PLAQUETÁRIA
PERMEABILIDADE DOS VASOS SANGUÍNEOS

37
1- Atua no transporte e utilização de ácidos graxos e colesterol (em lipoproteínas) através da enzima Lecitina – Colesterol Aciltransferase (LCAT).
2- A lecitina é, entre os fosfolipídeos, o mais amplamente distribuído nos alimentos. Fígado, gema de ovo, feijão de soja, amendoim, espinafre e gérmen de trigo são fontes ricas em lecitina.
3- A lecitina, porém, não é um nutriente essencial, pois o organismo produz a quantidade que é necessária. Além disso, a lecitina da alimentação é digerida antes de ser absorvida, portanto, os suplementos são de pequeno valor.
4- Devido às suas propriedades emulsificantes, a lecitina é adicionada aos produtos alimentícios. Ex: margarina, bolachas e produtos de confeitaria.
Outros Fosfolipídeos
As cefalinas são semelhantes em estrutura às lecitinas. Os lipontóis contêm inositol, um composto com atividade semelhante às vitaminas. Os esfingolipideos contêm um complexo aminoálcool no lugar do glicerol. Todos eles são encontrados em altas concentrações no tecido nervoso. A esfingosina é encontrada no cérebro e outros tecidos nervosos como um componente da bainha de mielina.
ESFINGOSINA: GLICEROL:
HO – CH – CH = CH – (CH2)12 – CH3 H2COH
CH– N – H HCOH
H H2COH
CH2 – O – H

38
Glicolipídeos
Incluem os cerebrosídeos e os gangliosídeos que contêm a esfingosina e uma cadeia muito longa de ácidos graxos (> 22C). O componente carboidrato dos cerebrosídeos é a galactose. Os gangliosídeos também contêm glicose e um composto complexo contendo um aminoaçúcar. Estruturalmente, ambos os compostos são componentes do tecido nervoso e certas membranas celulares, onde desempenham uma função no transporte de lipídeos.
Esteroides e Esteróis
Os esteroides constituem um grande grupo de compostos cíclicos que podem ser considerados derivados de um núcleo hidrocarbonético comum: o núcleo ciclopentanofenantreno. Os esteroides são, portanto, lipídeos de origem vegetal e animal, possuidores de um esqueleto de carbonos tetracíclico característico. São encontrados praticamente em todos os tecidos do organismo e causam uma grande variedade de efeitos fisiológicos. Os esteroides estão estruturalmente relacionados aos terpenos e são biossintetizados a partir do precursor lanosterol (um triterpeno). O lanosterol, por sua vez, provém da ciclização do esqualeno, um hidrocarboneto acíclico.
Os esteroides podem ser classificados como: Esteróis; Ácidos Biliares; Hormônios Sexuais Masculinos; Hormônios Sexuais Femininos; Hormônios da Gravidez; Hormônios Adrenocorticais; Vitaminas D; Saponinas; Glicosídeos Cardíacos.
Esteróis
O mais conhecido é o colesterol que está presente em todas as células animais e é particularmente abundante em tecidos nervosos. No núcleo do colesterol há oito centros de assimetria e, teoricamente, algo como 256 isômeros são possíveis. O colesterol é o precursor dos ácidos biliares, esteróis fecais e hormônios esteroides.
Colesterol

39
Os esteróis são caracterizados pela estrutura em anel, complexa com grupos laterais individuais. Além do colesterol, que é encontrado apenas em tecidos animais, os esteróis comuns incluem o ergosterol, que ocorre em leveduras, e β-sitosterol, que é encontrado em alimentos vegetais. O colesterol é um componente essencial das membranas estruturais de todas as células dos mamíferos. É o principal componente do cérebro e das células nervosas. É encontrado também em altas concentrações nas glândulas supra-renais, onde os hormônios adrenocorticais são sintetizados e, no fígado, onde é sintetizado e estocado. O colesterol é uma chave intermediária na biossíntese de uma série de esteroides importante como: ácidos biliares, hormônios adrenocorticais (aldosterona) e hormônios sexuais (estrogênios, testosterona e progesterona).
Os lipídeos sanguíneos (colesterol, triglicerídios e fosfolipídios) são transportados na corrente sanguínea ligados às proteínas. Estas partículas complexas, chamadas lipoproteínas variam em composição, tamanho e densidade. As cinco classes de lipoproteínas são: Quilomícrons; Lipoproteínas de Densidade muito Baixa (VLDL); Lipoproteínas de Densidade Intermediária (IDL), Lipoproteínas de Baixa Densidade (LDL) e Lipoproteínas de Alta Densidade (HDL). Elas possuem quantidades variáveis de triglicerídeo, colesterol, fosfolipídeo e proteína. A proporção de proteína e gordura determina a densidade. As partículas com mais proteínas são mais densas, portanto, HDL tem mais proteínas do que LDL.
Colesterol Total
É o colesterol contido em todas as funções de lipoproteína. Cerca de 60 a 70% do total é carreado com LDL, 20 a 30% do total é carreado com HDL, 10 a 15% do total é carreado com VLDL. Dentre as lipoproteínas ricas em triglicerídeos estão os Quilomícrons, VLDL e quaisquer produtos remanescentes ou intermediários formados no catabolismo.
Lipoproteínas e Metabolismo
Quílomícrons: Maiores partículas. Transportam a gordura e o colesterol da alimentação, do intestino delgado para a periferia. Na corrente sanguínea, os triglicerídeos nos quilomícrons são hidrolisados pela lipase lipoproteíca.

40
Lipoproteínas de Densidade Muito Baixa (VLDL): São sintetizadas no fígado para transportar triglicerídeos endógenos e colesterol. 60% desta partícula são triglicerídeos.
Lipoproteínas de Densidade Intermediária (IDL): São formadas com o catabolismo do VLDL e são precursoras do LDL.
Lipoproteínas de Baixa Densidade (LDL): São os transportadores primários de colesterol no sangue. Consequentemente, o Colesterol Total e a LDL – Colesterol estão altamente correlacionados.
Lipoproteínas de Alta Densidade (HDL): Contêm mais proteínas do que qualquer outra lipoproteína. A teoria mais aceita para o efeito antiaterogênico do HDL é que ele está envolvido no transporte do colesterol em excesso das membranas para as lipoproteínas ricas em triglicerídeos, que são então removidas por receptores no fígado. Este é o processo de transporte de colesterol reverso, que ajuda o organismo a livrar-se do colesterol e previne o acúmulo de lipídios na parede arterial. Nas análises clínicas laboratoriais, normalmente usa-se a fórmula: LDL – C = (Colesterol Total) – (HDL - C) – (TG/5), pois algumas vezes não é quantificado o LDL-C diretamente, principalmente quando TG < 400g/dl.
CONTEXTUALIZANDO
Vitaminas Lipossolúveis
As vitaminas lipossolúveis A, D, E, K ocorrem em alimentos de origem animal ou vegetal ricos em gorduras sendo absorvidas juntamente com os lipídios. Como os lipídios, elas são transportadas pelas proteínas plasmáticas. Estão envolvidas em diversos processos, inclusive atuando como coenzimas. A vitamina K, por exemplo, participa como cofator de reações de carboxilação de resíduos de glutamato de várias proteínas, entre as quais os fatores responsáveis pela coagulação sanguínea. A vitamina A é obtida a partir de carotenoides vegetais e está envolvida nas reações da visão e no crescimento e diferenciação de tecidos epiteliais. O colesterol e o ergosterol são precursores da Vitamina D (Calciferol). Essa vitamina é conhecida como a vitamina da luz solar porque uma modesta exposição à luz solar normalmente é suficiente para a maioria das pessoas produzir vitamina D, utilizando a luz ultravioleta e o colesterol da pele. Dois esteróis – um dos lipídeos de animais (7 – desidrocolesterol) e um dos vegetais (ergosterol) – podem servir como precursores da vitamina D. A abertura do anel de 7 – desidrocolesterol produz uma forma de provitamina de 7 – desidrocolesterol que produz Colecalciferol (Vitamina D3). A abertura do anel de ergosterol produz ergocalciferol, ou vitamina D2. As vitaminas D2 e D3 necessitam de metabolismo posterior para
41
produzir as formas metabolicamente ativas de 1,25-diidroxivitamina D2 e D3
(Calcitriol). Dessa forma, a vitamina D desempenha um papel importante, juntamente com o cálcio e o fósforo, na saúde dos ossos e dentes. A vitamina E, juntamente com as vitaminas A, C, e D, atuam como antioxidantes, bloqueando a ação lesiva dos radicais livres sobre as estruturas celulares.
ATIVIDADES DE AVALIAÇÃO
1. Caracterizar estruturalmente os ácidos graxos mais comuns na natureza.
2. Definir triacilglicerol. Descrever as vantagens para os seres vivos do armazenamento de triacilgliceróis.
3. Correlacionar a consistência das gorduras animais e óleos vegetais com a estrutura dos ácidos graxos componentes destas substâncias.
4. Definir Glicerofosfolipídio e Esfingolipídio.
5. Caraterizar as classes principais de lipoproteínas plasmáticas, indicando a sua função.
6. Definir “lipídio anfipático”.
7. Desenhe a estrutura de um fosfoacilglicerol que contenha glicerol, ácido oleico, ácido esteárico e colina.
8. Escreva a fórmula estrutural de um triacilglicerol e o nome das partes que o compõem.
9. Como as estruturas dos esteroides se diferem das de outros lipídeos?
10. Qual a semelhança estrutural entre a Vitamina D3 e o Colesterol?
11. As margarinas são feitas de óleos vegetais normalmente líquidos. Por que elas são sólidas?
12. Compare as estruturas e as propriedades físicas dos TAG, dos esfingolipídeos e dos glicerofosfolipídeos.
13. Resuma as funções dos esteroides e eicosanoides.
14. Fale sobre a doença ADENOLEUCODISTROFIA (ALD). O que a ocasiona? Como é composto o “óleo de Lorenzo”, que é usado como terapia na ALD?
15. Descreva a nomenclatura dos ácidos graxos.
16. Sobre os Triacilgliceróis, defina: a) Rancificação; b) Saponificação.
17. Esquematiza um fosfolipídeos, mostrando a ligação fosfodiéster.
42
18. Qual a importância do colesterol? De que outros compostos ele é a molécula inicial?
19. Compare as vias de degradação e biossíntese de ácidos graxos.
20. Esboce o papel da carnitina no transporte de moléculas de acil-CoA para mitocôndria.
21. O principal composto de armazenamento de energia nos animais é a gordura. Por que isso é vantajoso?
22. Por que as plantas não usam gordura/óleos como seus principais
compostos de armazenamento de energia?
23. Por que o linoleato e o linolenato são considerados ácidos graxos
essenciais?
24. Discuta o papel das unidades isoprenoides na biossíntese de colesterol.
25. Por que uma pessoa alcoólatra pode ter um fígado gorduroso?
26. Qual o papel do citrato no transporte de grupos acetilas da mitocôndria para
citossol?
27. Que característica todos os esteroides possuem? E quais implicações
biossintéticas, essa característica em comum atribui?
28. Por que o colesterol deve ser empacotado para transporte, em vez de
correr livremente no sangue?
29. Faça a diferença entre o colesterol bom e o mau colesterol.
Bibliografia Comentada
Campbell, M. K. Bioquímica. 3ª ed. Porto Alegre: Artmed Editora, 2000. Lipídeos, a natureza química dos lipídeos as vitaminas lipossolúveis e Prostaglandinas e Leucotrienos são apresentados e comentados de forma muito didática no Capítulo 6 desse livro.
Mahan, K.; Escott-Stump. Alimentos, nutrição & dietoterapia. São Paulo: Roca, 2005. Os Lipídios foram comentados no Capítulo 3, que trata dos macronutrientes carboidratos, proteínas e lipídios.

43
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª Ed. Rio de Janeiro: Ed. Guanabara Koogan S. A., 1999. Os autores apresentam lipídios na Parte 2 de seu livro no Capítulo 6, que comenta sobre estruturas de carboidratos e lipídios.
LEITURAS, FILMES E SITES RECOMENDADOS:
Filme: O Óleo de Lorenzo (1992)
Filme: O Colesterol é inimigo do Coração (Globo Vídeos)
www.arquivosdeorl.org.br
www.hepcentro.com.br/esteatose.htm
CAPÍTULO 4: AMINOÁCIDOS E PROTEÍNAS
OBJETIVOS:
Conhecer as macromoléculas estruturais do nosso organismo, as proteínas.
Conhecer os tipos de moléculas que compõe as proteínas, os aminoácidos.
Verificar os alimentos ricos em proteínas.
Estudar os tipos e funções de aminoácidos, peptídeos e proteínas.
Reconhecer as estruturas dos diferentes aminoácidos.
O nome “proteína” é de origem grega e significa “de 1ª importância”. Em sua composição estão presentes C, H e O. As proteínas também contêm aproximadamente 16% de N, juntamente com S e, algumas vezes, outros elementos, tais como fósforo (P), ferro (Fe) e cobalto (Co). Os vegetais sintetizam a proteína a partir do N, que obtêm a partir de nitratos e amônia do solo e, nas circunstâncias únicas das leguminosas, os nitratos se tornam disponíveis simbioticamente a partir do N2 atmosférico pelas bactérias nos nódulos das raízes. Já os animais obtêm o N que necessitam de alimentos proteicos seja de origem vegetal ou animal. O metabolismo animal, a excreção e morte finalmente fazem retornar o N2 ao solo numa continuação do ciclo do nitrogênio.

44
Estrutura e Classificação
A base da estrutura da proteína são os aminoácidos, dos quais 20 foram reconhecidos como constituintes da maioria das proteínas. Eles são compostos que apresentam em sua molécula um grupo amino (-NH2) e um grupo carboxila (-COOH). Apenas a Prolina, possui um grupo imino (-NH-) em vez do grupo amino. Em pH fisiológico, esses grupos estão na forma ionizada: -NH3
+, -COO-
e –NH2+ -. Os aminoácidos têm uma fórmula básica comum, na qual os grupos
amino e carboxila estão ligados ao carbono α, ao qual também se liga um átomo de hidrogênio e um grupo variável chamado cadeia lateral ou grupo R. É a estrutura da cadeia lateral R que diferencia os aminoácidos entre si.
Estrutura básica característica de um aminoácido
H R Cα COOH H3 N+

45
Os aminoácidos se combinam para formar proteínas através de uma ligação chamada peptídica que une os carbonos carboxílicos de um aminoácido ao N de outro. O composto resultante tem um grupo carboxila livre em uma das pontas e um grupo amino livre na outra, possibilitando a cadeia a continuar se ligando a outros aminoácidos em qualquer das pontas. As proteínas variam no tamanho, como, por exemplo, de polipeptídeos relativamente pequenos, como a Adrenocorticotropina (ACTH) com 23 unidades de aminoácidos até moléculas muito complexas com várias centenas de milhares de unidades de aminoácidos.
Os polipeptídeos que constituem a Estrutura Primária das proteínas podem conter de poucas até 300 unidades de aminoácidos. Muitas cadeias de polipeptídeos podem ser ligadas entre si, normalmente através de ligações S – S da cistina, em uma forma helicoidal, pregueada ou espiral randômica

46
chamada de Estrutura Secundária. As proteínas mais complexas se caracterizam por uma Estrutura Terciária, na qual a cadeia polipeptídica está enrolada sobre si mesma em uma forma globular, com toda a estrutura sendo presa rigidamente por forças interatômicas, tais como ligações de hidrogênio. As extensas possibilidades de variação oferecidas por estas estruturas resultam em milhões de proteínas diferentes com propriedades e funções biológicas específicas.
As proteínas existem nas formas fibrosa e globulares. As Proteínas Fibrosas caracterizam-se por várias cadeias peptídicas helicoidais torcidas juntas para formar uma haste rija. São caracterizadas por baixa solubilidade e alta força mecânica. Aparecem em elementos estruturais, tais como colágeno do tecido conjuntivo, queratina do cabelo e unhas e miosina do tecido muscular. As Proteínas Globulares são encontradas em líquidos teciduais. São muito solúveis e facilmente desnaturadas. As Proteínas Globulares de interesse na nutrição são: a caseína, presente no leite, a albumina presente no ovo, na carne etc. Proteínas Simples são aquelas que produzem apenas aminoácidos a partir da hidrólise. Incluem albuminas, globulinas, prolaminas entre outras. As proteínas que são solúveis em H2O, tais como as albuminas e globulinas, estão presentes nos líquidos animais, enquanto que as menos solúveis, tais como a miosina e a proteína muscular, estão presentes nos tecidos. Proteínas Conjugadas são combinações nas quais uma substância não proteica está ligada a uma molécula de proteína simples, como um grupo prostético, assim facilitando as funções que nem os próprios constituintes poderiam provavelmente realizar. As proteínas conjugadas incluem as Nucleoproteínas encontradas no Ácido Ribonucleico (RNA) e Ácido Desoxirribonucleico (DNA). As Mucoproteínas e as Glicoproteínas são aquelas que combinam proteínas com quantidades variáveis de Polissacarídeos Complexos, tais como Mucina presente nas secreções gástricas, Lipoproteínas presentes no plasma sanguíneo, Fosfoproteínas que contêm H3PO4 ligado à proteína por ligações éster, tal como na caseína do leite; Metaloproteínas, tais como ferritina e a hemossiderina nas quais metais como Fe++, Cu++ e Zn++ estão ligados às proteínas. Proteínas Derivadas: Proteoses, Peptonas e Peptídeos formam-se nos vários estágios do metabolismo proteico.

47
GLOBULAR
FIBROSA
Leguminosas como Fontes de Proteína (HIPERTEXTO)
As leguminosas são únicas no reino vegetal por causa de suas sementes, ricas em proteínas e pobres em amido, diferente das sementes de grãos de cereais, que são pobres em proteínas e ricas em amido. O seu conteúdo de Aminoácidos Essenciais é mais parecido com o das proteínas animais. Suplementadas com uma pequena quantidade de metionina dos cereais ou fontes animais, o seu padrão de aminoácido é adequado para suportar tanto a

48
vida quanto o crescimento nos humanos. Os feijões de soja são únicos pelo fato de que um produto de seu processamento, a proteína da soja isolada, é a proteína com um padrão de aminoácido capaz de suportar a vida e o crescimento. Esta característica extraordinária é talvez a consequência casual de uma habilidade rara dos vegetais leguminosos de fixar o N gasoso do ar para o seu uso. Na realidade, o vegetal por si mesmo não tem esta capacidade e, como todos os outros vegetais, deve retirar o N através das raízes ou na forma de nitrato de íons de amônio. Entretanto, localizadas nos nódulos das raízes de leguminosas estão as bactérias Rhizobium, que fixam o N da atmosfera. Numa relação simbiótica, as bactérias fornecem o N na forma de aminoácido e amidas em troca da energia fornecida pelo vegetal através da fotossíntese.
Desnaturação de Proteínas
A estrutura tridimensional de uma proteína é significativa para a sua função. As ligações que mantêm a forma tridimensional são relativamente fracas e facilmente rompidas por agitação mecânica, extremos de temperatura, acidez ou alcalinidade. Quando isso ocorre, a proteína perde sua forma, característica e não é mais capaz de desempenhar sua função neste papel particular. Este desemaranhamento irreversível da molécula de proteína, chamado desnaturação, é um evento que normalmente é prejudicial e algumas vezes desastroso para o organismo, dependendo de quantas moléculas de proteína são afetadas. Enquanto um ovo está fritando, a clara do ovo gradualmente engrossa e fica branca conforme a proteína é desnaturada pelo calor. Do mesmo modo, uma queimadura severa desnatura e, assim, destrói a proteína na pele e vasos sanguíneos. Na realidade é a suscetibilidade das proteínas ao excesso não moderado de calor, frio, ácido e álcalis que determina a limitação ambiental na qual os humanos podem sobreviver e funcionar.
Funções das Proteínas
As proteínas da dieta estão envolvidas na síntese das proteínas teciduais e outras funções metabólicas especiais; nos processos anabólicos, fornecendo os aminoácidos necessários para a construção e manutenção dos tecidos orgânicos; como uma fonte de energia, as proteínas são equivalentes aos carboidratos no fornecimento de 4 Kcal/g. Entretanto, são consideravelmente

49
mais caras, tanto em custo quanto na quantidade de energia necessária para o metabolismo. As proteínas desempenham um papel estrutural não apenas em todos os tecidos do corpo, mas também na formação de enzimas, hormônios e de vários líquidos e secreções corpóreas. Como anticorpos, as proteínas estão envolvidas na função do sistema imunológico. Na forma de lipoproteínas, as proteínas participam do transporte de triglicerídeos, colesterol, fosfolipídeos e vitaminas lipossolúveis. Muitas vitaminas e minerais são ligados a carreadores proteicos específicos para o transporte. A albumina transporta ácidos graxos livres e bilirrubina, assim como muitas drogas. As proteínas também contribuem para a homeostase através da manutenção de relações osmóticas normais entre os líquidos corpóreos, como é evidenciado pela formação de edema como uma consequência de hipoproteinemia. A albumina é particularmente importante para esta função. Devido a sua estrutura única, as proteínas são capazes de se combinar ou com substâncias ácidas ou com substâncias básicas, mantendo assim o equilíbrio ácido-base do sangue e tecidos.
Enzimas como Proteínas
O fato de todas as enzimas serem proteínas e suscetíveis à desnaturação é significante para as necessidades ambientais do organismo humano. As enzimas funcionam por se ligarem a moléculas de formas e tamanhos característicos. Sendo assim a importância da manutenção da forma original de uma enzima em particular é imprescindível. Como a desnaturação muda a forma das enzimas tornando-as não funcionais, a maioria dos organismos vivos não pode tolerar extremos de temperatura e pH. Algumas exceções são formas diminutas de vegetais que sobrevivem a temperaturas próximas da fervura de verões quentes ou no ambiente frio de bancos de neve. Devido às exigências rígidas da função enzimática de um pH ótimo, existe uma série de mecanismos para eliminar o excesso de materiais ácidos ou alcalinos do sangue. Exemplo fisiológico da desnaturação de enzima é o efeito do ácido láctico que se acumula durante exercícios vigorosos, alcançando, eventualmente, um nível suficiente para interferir com a ação enzimática normal, contribuindo para a resultante fadiga.
As enzimas em alimentos não cozidos são desnaturadas pelo ácido clorídrico (HCl) do estômago. A papaína (enzima da papaia) é rapidamente desnaturada conforme a temperatura da carne sobe durante o cozimento. Se não for destruída desta maneira, é mais tarde desnaturada e parcialmente digerida quando atinge o estômago. O mesmo fato ocorre com enzimas que são tomadas oralmente como suplemento. Neste caso, entretanto, as enzimas são empacotadas em cápsulas que não dissolvem até que alcancem o intestino delgado.

50
Aminoácidos Essenciais (AAE) e Não Essenciais
Aminoácidos Essenciais (AAE) são aqueles em que a síntese do organismo não é suficiente para suprir as necessidades metabólicas e, sendo assim, precisam ser fornecidos como uma parte da dieta. São eles: treonina, valina, triptofano, isoleucina, lisina, leucina, fenilalanina, metionina, histidina e arginina. A ausência ou a ingestão inadequada de qualquer um desses aminoácidos leva a um balanço de N negativo, perda de peso, crescimento prejudicado em bebês e crianças e sintomas clínicos. Os aminoácidos não essenciais remanescentes, como alanina, ácido aspártico, asparagina, ácidotglutâmico, glutamina, glicina, prolina e serina são igualmente importantes para a estrutura da proteína. Entretanto, se quantidades adequadas de certos aminoácidos não essenciais não estiverem presentes na hora da síntese da proteína, podem ser sintetizados a partir de AAE ou de precursores de C e N, apropriados prontamente sintetizados na célula. Os aminoácidos condicionalmente essenciais são aqueles que se tornam essenciais sob certas circunstâncias clínicas, como p. ex: taurina, cisteína e, possivelmente, tIrosina são tidos como condicionalmente essenciais nos bebês prematuros.
“Pool” Metabólico de Aminoácidos
Não há uma grande reserva de aminoácidos livres no organismo, e qualquer quantidade acima da necessária para a síntese de proteína tecidual e dos vários compostos não proteicos que contém N é metabolizada. Entretanto, nas próprias proteínas celulares um “pool” metabólico de AA existe num estado de equilíbrio dinâmico que pode ser requisitado a qualquer momento para suprir uma necessidade apropriada. A contínua dinâmica da proteína no adulto é provavelmente necessária para a manutenção de um “pool” de AA e a capacidade de suprir a necessidade por aminoácidos pelas várias células e tecidos quando são estimulados a fazer as proteínas necessárias. Os tecidos mais ativos para a dinâmica de proteína são as proteínas do plasma, mucosa intestinal, pâncreas, fígado e rins, enquanto os músculos, pele e cérebro são muito menos ativos.
Funções Especiais dos AA
Quase todos os aminoácidos têm certas funções únicas no organismo. O Triptofano, o aminoácido mais complexo, é um precursor da vitamina Niacina e do neurotransmissor Serotonina. A Metionina é o principal doador de grupos metila para a síntese de compostos tais como Colina e Carnitina. Também é um precursor da Cistina e de muitos outros compostos que contêm enxofre. A Fenilalanina é um precursor da Tirosina e juntas levam à formação de Tiroxina e Epinefrina. A Tirosina é o precursor do qual são feitos o pigmento da pele e do cabelo. A Arginina e a Citrulina estão envolvidas especificamente na síntese de Ureia no fígado. A Glicina, o mais simples e mais ubíquo dos aminoácidos,

51
se combina com muitas substâncias tóxicas, convertendo-a em formas inócuas que são então excretadas. Também é usada na síntese do núcleo Porfirina da hemoglobina e é um constituinte de um dos ácidos da bile (ácido glicocólico). A Histidina é essencial para a síntese de Histamina, que causa vasodilatação no sistema circulatório. A Creatinina, sintetizada a partir da Arginina, Glicina e Metionina, se combina com o fosfato para formar a fosfocreatinina, um importante reservatório de fosfato de alta energia na célula.
A Glutamina, formada pelo ácido Glutâmico, e a Asparagina, formada a partir do ácido aspártico, têm importantes papéis como reservatórios de grupos amino por todo o corpo. A Glutamina tem recebido atenção recentemente como uma fonte primária de combustível para o trato intestinal, especialmente no controle da síntese de glicogênio e degradação proteica, assim como para manter a integridade contra transformação bacteriana. É o aminoácido mais abundante no plasma e no músculo esquelético. O Ácido Glutâmico é um precursor do neurotransmissor Ácido Gama-Amino-Butírico (GABA).
Fontes Endógenas
Síntese de Proteínas Teciduais, Enzimas,
Proteínas, Hormônios, etc.

52
(~140g/dia)
SÍNTESE(AA não essenciais)
Fígado Amônia
Ureia
α – cetoácido Urina
Oxidação, etc.
Glicose, Corpos Cetônicos, etc.
Acetil-CoA, ciclo do Ác.Cítrico
CO2+H2O+ATP
Equação de Henderson – Hasselbalch
pH = pka + log [A] / [HA]
POOL DE AA
TRANSAMINAÇÃO DESAMINAÇÃO
Síntese e Degradação
1
Conversão
Digestão e Absorção
2Excreção Renal3
4
Fontes exógenas (~70g/dia)
Proteínas do alimento
Constituintes teciduais nitrogenados não
proteicos essenciais (Purinas, Pirimidinas,
Colina, Creatinina, Niacina, Porfirina,
Epinefrina, Tiroxina, Ác. Biliares, Melanina,
Produtos de Desintoxicação, etc.)
Excesso de AA (0,9 a 1,0g/dia)

53
Esta equação pode ser usada para calcular o pH de uma solução contendo um ácido fraco, após a adição de um ácido ou base fortes. Também pode ser usada para prever as formas iônicas dos aminoácidos. É útil para calcular como o pH de soluções fisiológicas (Ex.: sangue ou líquido intracelular) responde às alterações na concentração de ácidos fracos e/ou sua forma “salina” correspondente. Por ex.: No sistema tampão do bicarbonato, a equação de Henderson – Hasselbalch prevê como as alterações de concentração influenciam o pH. Ela poderá prever alterações no pH à medida que as concentrações de HCO-
3 ou CO2 são alteradas ou as formas iônicas das drogas. A equação também é útil para calcular a abundância de formas iônicas de drogas ácidas e básicas. Por ex.: a maioria das drogas são ácidos fracos ou bases fracas. As drogas ácidas (HA) liberam um próton (H+), levando à formação de um ânion carregado (A-).
HA H+ + A-
As bases fracas (BH+) também podem liberar um H+; entretanto, a forma protonada das drogas básicas usualmente é carregada, e a perda de um próton produz a base sem carga (B).
BH+ B + H+
Uma droga passa através das membranas mais facilmente se está sem carga. Assim, em um ácido fraco, o HA sem carga pode permear-se através das membranas, e o A- não. Em uma base fraca, a forma sem carga, B, penetra através da membrana celular, e a BH+ não. Assim, a concentração efetiva da forma permeável de cada droga em um sítio de absorção é determinada pela concentração relativa das formas carregadas e descarregadas. A proporção entre as duas formas é determinada pelo pH no sítio de absorção e pela força do ácido ou base fracos, que é representado pelo pka do grupo ionizável.
A equação de Henderson-Hasselbalch é útil para determinar qual a uantidade de droga encontrada em cada lado de uma membrana que separa dois compartimentos que diferem em pH, como, por exemplo, o estômago (pH 1,0 – 1,5) e o plasma sanguíneo (pH 7,4).
Propriedades Ácido-Básicas dos Aminoácidos e Proteínas
As proteínas adquirem propriedades iônicas em virtude das cadeias laterais dos aminoácidos que as compõem. Muitas dessas cadeias laterais podem ionizar-se e agir como ácidos fracos. Dependendo do pK do grupo funcional da cadeia lateral, essa ionização pode produzir uma carga positiva ou negativa.
Formas Ionizadas dos Aminoácidos

54
Se um grupo funcional sofre dissociação ou é protonado, depende do pH da solução. A equação de Henderson-Hasselbalch descreve a quantidade de ionização (proporção de dissociação para protonado) para cada grupo funcional individual, desde que cada um tenha seu valor de pKa e ionize-se independentemente dos outros.
A curva de titulação da alanina mostra a dissociação independente de seus dois grupos funcionais: o grupo α-amino e o grupamento α-carboxila. A curva de titulação da esquerda para a direita ilustra a mudança do estado de ionização da alanina, assim como também é descrito da direita para a esquerda na reação abaixo:
H H+ H H+ H
HOOC – C – NH3+ -OOC – C – NH3
+ -OOC – C – NH2
CH3 H+ CH3 H+ CH3
Até pH 1,0 Até pH 6,0 Até pH 11,0
Conforme os prótons vão sendo removidos da molécula, eles são primeiramente removidos do grupo carboxila, já que esse tem o menor pK (pKa = 2,3). Quando o pH aumenta em direção ao pK do grupamento amina (pKa = 9,9), esse então perde seus prótons. Cada pKa representa o ponto médio de

55
dois equilíbrios, ilustrando que os aminoácidos (e proteínas) têm capacidade de tamponamento.
No pH 7,0, as cadeias laterais ionizáveis de aminoácidos em proteínas apresentam cargas características:
Carregadas positivamente: lisina, arginina. Carregadas negativamente: aspartato, glutamato. A histidina torna-se carregada positivamente se pH for inferior a 6,0. A cisteína torna-se carregada negativamente se pH for maior que 8,0.
pH Isoelétrico
O total de cargas de um aminoácido ou de uma proteína equivale à soma de todas as cargas das cadeias laterais de cada aminoácido. O valor do pH que produz um total de carga igual a zero (neutra) na molécula é denominado pH isoelétrico, ou pI.
Se o pH > que pI, o total de cargas no aminoácido (ou proteína) será negativo.
Se o pH < que pI, o total de cargas no aminoácido (ou proteína) será positivo.
As proteínas não se deslocam em um campo elétrico quando o pH do sistema tampão é igual ao seu ponto isoelétrico, pois elas não possuem carga para serem atraídas para o catodo ou para o anodo.
CONTEXTUALIZANDO
Fenilcetonúria e Erros Inatos do Metabolismo
As mutações que levam a deficiências em enzimas são normalmente referidas como “erros inatos do metabolismo”, uma vez que envolvem defeitos no DNA dos indivíduos afetados. Os erros em enzimas que catalisam reações de aminoácidos têm frequentemente consequências desastrosas, que levam a formas graves de retardo mental. A fenilcetonúria é um exemplo bem conhecido. A fenilalanina, o fenilpiruvato, o fenilactato e o fenilacetato acumulam-se no sangue e na urina. Há evidências disponíveis que sugerem que o fenilpiruvato, que é uma fenilcetona, causa retardo mental pela sua interferência na conversão do piruvato em acetil-CoA (um intermediário importante em muitas reações bioquímicas) no cérebro. É também provável que o acúmulo desses produtos nas células cerebrais resultem em um desequilíbrio osmótico, fazendo com que a água flua para dentro das células cerebrais. Elas então aumentam de tamanho até comprimirem-se umas contra as outras no cérebro em desenvolvimento. Nos dois casos, o cérebro não está

56
apto a desenvolver-se normalmente. O teste do pezinho é como se pode fazer o diagnóstico da doença. A partir da descoberta o aminoácido fenilalanina deve ser limitado à quantidade necessária para a síntese de proteína.
(Retirado de Campbell, M. K. Bioquímica, 3ª ed. Artmed Editora LTDA, 2000.p. 113.)
ATIVIDADES DE AVALIAÇÃO
1. Escrever a fórmula básica característica de um aminoácido. Dar exemplos de:
a. aminoácido com um grupo amino e dois grupos carboxílicos.
b. aminoácido com um grupo carboxílico e dois grupos amino.
2. Definir Ponto Isoelétrico (pI) de um aminoácido.
3. Analisando o grupo R, classificar os aminoácidos em polares e apolares. Entre os polares, citar aqueles que, em pH 7, apresentam grupo com carga negativa (aminoácidos ácidos), carga positiva (aminoácidos básicos) e carga nula (polares sem carga).
4. Esquematizar a ligação peptídica.
5. Definir Proteínas Globulares e Fibrosas. Citar exemplos.
6. Definir estrutura primária.
7. Descrever as estruturas regulares – α-hélice e folha β pregueada – que compõem a estrutura secundária das proteínas globulares.
8. Definir estrutura terciária de proteínas globulares. Esquematizar os tipos de ligações que a mantêm, indicando os aminoácidos que participam dessas ligações.
9. Definir estrutura quaternária de proteínas globulares. Citar exemplos de proteínas com estrutura quaternária.
10. Verificar a posição dos radicais polares e apolares de uma proteína em solução aquosa.
11. Definir ponto isoelétrico de uma proteína e indicar como pode ser determinado.
12. Definir desnaturação de uma proteína e descrever a ação do pH extremo e temperatura sobre a estrutura das proteínas.

57
13. Citar 04 exemplos de proteínas com função não-enzimática.
14. Em que diferem as pontes de hidrogênio da estrutura secundária e terciária de proteínas globulares?
15. Discutir a seguinte afirmação: a estrutura primária de uma proteína determina a sua estrutura terciária.
16. Para o tripeptídeo: Ala – Lys – Ser:
a. Classificar os aminoácidos segundo o grupo R
b. Esquematizar as ligações que estes aminoácidos poderiam formar na estrutura terciária de uma proteína.
17. Defina proteínas ácidas e proteínas básicas. Fale sobre o pI de cada uma.
18. O que é renaturação de proteínas?
19. Fale sobre eletroforese.
20. Fale sobre o que ocasiona a doença conhecida como anemia falciforme.
21. Explique a origem do nome α-aminoácido.
22. Usando as seguintes formas para um α-aminoácido, relacione-as com os seus respectivos pHs.
NH3+ NH2 NH3
+
H – C – COO-; H – C – COO-; H – C – COOH
R R R
pH 1; pH 7 e pH 11
23. Uma cadeia peptídica possui um sentido porque seus componentes têm extremidades diferentes, isto é, os grupamentos α-carboxila. Quem, por convenção, inicia a cadeia peptídica?
24. Que são grupos prostéticos? E centros alostéricos?
25. Escreva a fórmula estrutural geral de um L-aminoácido e de um D-aminoácido.
26. Qual o primeiro e o último aminoácido encontrado?
27. Cite a classificação dos aminoácido quanto à cadeia lateral.
28. Cite as principais propriedades físicas e químicas dos aminoácidos que
concordam com sua representação zwiteriônica.

58
29. O Ácido Glutâmico possui pK1 = 2,2; pK2 = 9,7 e pKr = 4,2:
a) Represente as reações de equilíbrio entre as diversas formas do ácido glutâmico, nos pHs correspondentes aos pK1, pK2 e pKr. Determine a carga elétrica líquida para cada um desses pHs.
b) Calcule o ponto isoelétrico do ácido glutâmico.
c) Fazendo-se passar uma corrente elétrica numa solução de ácido glutâmico em pH 1,0, para que polo deverão migrar as formas iônicas presentes em solução?
30. Com relação à titulação de 50 ml de uma solução de glicina 0,2 N na forma totalmente protonada (H3NCH2COOH), pK1= 2,34 pK2 = 9,6 ) com NaOH 0,1N, responda:
a) Que volume de NaOH é necessário para titular o grupo alfa-COOC da glicina ?
b) Quanto de NaOH é necessário para titular o grupo alfa-NH3 da glicina?
c) Que volume de NaOH é necessário para que a glicina fique com carga elétrica igual a zero ? Neste ponto qual o pH da solução e qual a fórmula iônica da glicina?
31. Um peptídeo com 24 resíduos de aminoácidos está unido por quantas ligações peptídicas?
32. Descreva a estrutura do aspartame.
33. Mostre como ocorre a ligação peprtídica.
34. Faça a estrutura de um tripeptídeo constituído por uma alanina-histidina-valina.
35. Explique o processo de desnaturação das proteínas.
36. Cite três diferenças entre proteínas fibrosas e globulares.
37. Que importantes diferenças existem entre as estruturas proteicas em alfa-hélice e beta- pregueada?
38. Comenta-se que a diferença entre a seda e a lã é a diferença entre suas estruturas helicoidais e da folha pregueada. Você considera esse ponto de vista válido? Justifique.
39. Cite duas semelhanças e diferenças entre a hemoglobina e mioglobina.

59
Bibliografia Comentada
Campbell, M. K. Bioquímica. 3ª ed. Porto Alegre: Artmed Editora, 2000. Aminoácidos e Peptídeos são apresentados e comentados de forma muito didática no Capítulo 3, da Parte 2, desse livro.
Champe, P. C. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade I do livro são apresentados os tópicos: Aminoácidos (Capítulo I), Estrutura das Proteínas (Capítulo II), Proteínas Globulares (Capítulo III) e Proteínas Fibrosas (Capítulo IV). O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Mahan, K.; Escott-Stump. Alimentos, nutrição & dietoterapia. São Paulo: Roca, 2005. Aminoácidos e Proteínas foram comentados na Parte I (Princípios Nutricionais) Capítulo 3, que trata dos macronutrientes carboidratos, proteínas e lipídios.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª Ed. Rio de Janeiro: Ed. Guanabara Koogan S. A., 1999. Os autores apresentam aminoácidos e proteínas na Parte 1 de seu livro no Capítulo 2, comentando amplamente sobre o assunto.
LEITURAS, FILMES E SITE RECOMENDADOS:
o www.fotosearch.com.br/video-filme/proteínas.html
o Filme: FOME (HUNGER) – 2008
o Filme: GARAPA (JOSÉ PADILHA) - 2009
CAPÍTULO 5: ENZIMAS
OBJETIVOS:
Estudar um tipo mais específico de proteínas, as enzimas, biocatalizadores naturais, produzidos por várias glândulas do organismo animal e vegetal.
Conhecer o mecanismo de uma reação enzimática.
Caracterizar os tipos de enzimas, seus mecanismos e aplicações, de modo que o aluno saiba identificar uma enzima em um processo biológico.
Quase todas as enzimas conhecidas são PROTEÍNAS, (com exceção de moléculas de RNA que agem como enzimas, catalisando processos). A catálise

60
é a atividade mais importante das enzimas. Elas são catalisadores de sistemas biológicos, dispositivos moleculares que determinam o perfil de transformações químicas e participam na transformação de diferentes formas de energia. Na ausência de catálise, a maioria das reações nos sistemas biológicos ocorreria de forma extremamente lenta para fornecer produtos necessários ao metabolismo de um organismo.
Características mais Importantes
1. Poder catalítico
As enzimas aceleram as reações por fatores de 1.000.000 vezes, ou como catalisadores aumentam de várias ordens de grandeza a velocidade das reações que catalisam.
2. Especificidade
Por serem altamente específicas, as enzimas “selecionam”, entre todas as reações potencialmente possíveis, aquelas que efetivamente irão ocorrer. A especificidade das enzimas chega ao ponto de distinguir estereoisômeros de um determinado composto.
Atuação das Enzimas na Cinética das Reações
As enzimas atuam na cinética das reações acelerando a velocidade da reação por diminuir sua energia de ativação. A velocidade de uma reação é proporcional à concentração de uma substância.
Ex.: Conversão irreversível de A em B:
V = K [A] onde K = constante de velocidade da reação
É uma reação de 1ª ordem, já que a velocidade depende da concentração do reagente A com expoente 1. A maior parte das reações químicas nos organismos envolve no mínimo três moléculas diferentes e são geralmente reversíveis. São de 2ª ordem:
2A B + C ou A + B C + D
V = K [A]2 ou V = K [A] [B]
Teoria das Colisões
A Teoria das Colisões explica o fato acima. Para reagirem, as moléculas presentes em uma solução devem colidir com orientação apropriada e a colisão deve levá-las a adquirir certa quantidade de energia que lhes permita atingir o estado de transição. Para levar todas as moléculas de um mol de uma substância até o estado de transição, necessita-se de uma quantidade de energia chamada Energia de Ativação. Pode-se dizer que Energia de

61
Ativação é a energia que separa os reagentes dos produtos. A velocidade de uma reação será diretamente proporcional ao nº de moléculas com energia igual ou maior que a Energia do Estado de Transição.
A velocidade das reações pode ser aumentada:
1. Aumentando-se o nº de moléculas em solução, ou seja, sua concentração.
2. Aumentando-se a população de moléculas com energia necessária para reagir, o que pode ser obtido pela elevação de temperatura uma vez que um nº maior de moléculas atingirá a energia de ativação e a velocidade aumentará.
3. Diminuindo a barreira da Energia de Ativação, pois com esta diminuição, mesmo mantida a temperatura inicial, um nº maior de moléculas estará em condições de reagir. Essa redução é obtida pela presença de catalisadores. Eles são substâncias que aceleram a velocidade de uma reação, sem alterar a proporção entre reagentes e produtos encontrada no final da reação e sem serem efetivamente consumidos durante o processo.
A Eficiência da Catálise Enzimática
As enzimas, como todos os catalisadores, aceleram reações, mas não podem alterar a constante de equilíbrio ou a variação de energia livre. A velocidade de uma reação depende da energia livre de ativação, ou energia de ativação (∆G°), fornecimento da energia necessária para iniciar a reação. A energia de ativação para uma reação não catalisada é maior do que a de uma reação catalisada. Em outras palavras, pode-se dizer que uma reação não catalisada necessita de mais energia para ser iniciada e, por este motivo, sua velocidade é menor que a da reação catalisada. Dessa forma, pode-se dizer que os catalisadores criam um “novo caminho” para a reação, com um novo estado de Transição que requer uma Energia de Ativação menor. Em uma reação, no caso da catálise enzimática, os reagentes são chamados substratos. As enzimas apresentam propriedades mais interessantes para as células que os catalisadores inorgânicos.
Interação Enzima – Substrato
As enzimas são macromoléculas proteicas. Embora o total da molécula enzimática seja necessário para o papel catalítico, a ligação com o substrato dá-se apenas em uma região pequena e bem definida da enzima. Esta região chama-se Centro Ativo ou Sítio Ativo da Enzima. O Centro Ativo é formado por resíduos de aminoácidos, trazidos à proximidade uns dos outros pelos dobramentos da cadeia polipeptídica que definem a estrutura terciária da proteína. O centro ativo, assim organizado, constitui uma cavidade em forma definida que permite à enzima “reconhecer” seu substrato. De fato, uma molécula, para ser aceita como substrato, deve ter a forma espacial adequada
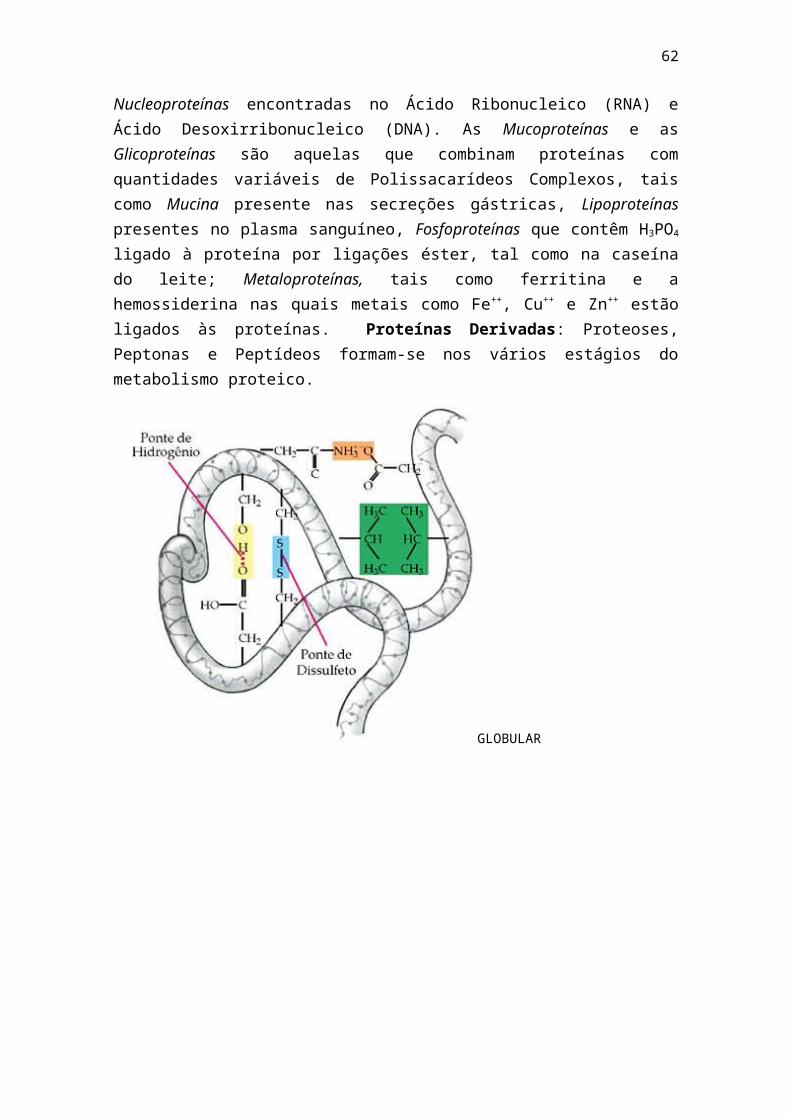
62
para alojar-se no centro ativo e grupos químicos capazes de estabelecer ligações precisas com os radicais do centro ativo.
Classificação das Enzimas e os Tipos de Reações que Catalisam
Classe Tipo de Reação
1.Óxido-Redutases Óxido-Redução
AH2 + B A + BH2
2. Transferases Transferência de Grupos
A – X + B A + B – X
3. Hidrolases Hidrólise
A – B + H2O A – H + B – OH
4. Liases Adição de grupos a duplas ou remoção de grupos deixando duplas.
X Y
A – B A = B + X – Y
5. Isomerases Rearranjos Intramoleculares
A – B A – B
Y X Y X
6. Ligases Condensação de duas moléculas, associada à hidrólise de uma ligação de alta energia (em geral, do ATP)
A + B A – B
Exemplos de Especifidade das Enzimas
Enzimas Proteolíticas são específicas para hidrólise de proteínas. A pepsina é uma enzima proteolítica que hidrolisa ligações peptídicas dos quais participam grupos carboxílicos de aminoácidos aromáticos como Triptofano, Fenilalanina e Tirosina, enquanto a tripsina é uma enzima proteolítica que reconhece apenas ligações peptídicas formadas por arginina ou lisina. Graus extremos de especificidade são encontrados entre as enzimas L-aminoxidases, que são capazes de reconhecer isômeros na configuração L, sendo inativas para os aminoácidos na forma D.
pH e Temperatura Interferem na Atividade Enzimática
A maioria das enzimas apresenta um valor de pH para o qual a sua atividade é máxima. A velocidade da reação diminui à medida que o pH se afasta desse valor ótimo, que é característico para cada enzima. O pH ótimo depende do nº e tipo de grupos ionizáveis que a enzima apresenta, e da

63
sequência em que estão organizados, ou seja, depende da sua estrutura primária. A eficiência da catálise depende de encontrarem-se, enzima e substrato com conformação e carga adequadas para permitir a interação. Com relação à Temperatura:
A 0º C: a velocidade da reação enzimática é próxima de zero. Aumentando-se a temperatura, há aumento de velocidade, porém isto só ocorre enquanto a enzima conservar sua estrutura original.
Acima de 50º - 55ºC: a maioria das proteínas globulares (enzimas também) são desnaturadas, havendo alterações nas moléculas e havendo também perda do poder de catálise.
Cinética Enzimática em Termos Matemáticos
A velocidade de uma reação química é normalmente expressa em termos de variação na concentração de um reagente ou de um produto em um determinado intervalo de tempo. Em uma reação do tipo:
A B V = K [A],
pode-se dizer que a reação catalisada ocorre em duas etapas:
1. A Enzima (E) liga-se reversivelmente ao Substrato (S), formando um complexo Enzima – Substrato (ES).
2. O Produto (P) é liberado e a enzima volta à forma livre, podendo ligar-se a outra molécula de substrato.
Outro exemplo em uma reação: A + B C + D A e B devem ligar-se simultaneamente ao centro ativo, onde ocorre a
reação, com liberação dos produtos C e D.
Estudando a Cinética Enzimática
Partindo de uma reação S P (onde há apenas um substrato e um produto:
E + S ES
ES E + P
K1 K3
E + S ES E + P K2
K1
K2
K3

64
A formação do complexo ES ocorre numa velocidade maior que a sua decomposição e as equações de velocidades correspondentes serão:
V1= K1[E] [S]
V3= K3[ES] Onde K1 > K2 > K3
Logo, a velocidade da reação global, ou seja, a velocidade da formação do produto é igual a V3, já que esta é a etapa mais lenta e limitante do processo.
K1
E + S ES K2
Quando o equilíbrio da 1ª etapa está estabelecido com 50% das enzimas sob a forma livre, haverá 50% das enzimas na forma ES. Nestas condições a V será ½ V Máxima.
Quando V=1/2 da V MÁX.
KM indica a afinidade que uma enzima apresenta pelo seu substrato.
Comparação de KM:
KM = [ S] indica que 50% dos sítios ativos estão ocupados pelo substrato.
Ex1: A enzima Lactato Desidrogenase (LDH) do músculo cardíaco tem um KM
elevado, logo tem baixa afinidade por Piruvato.
EX2: A LDH do músculo esquelético tem um KM baixo, logo tem alta afinidade por Piruvato.
Isto significa que o Piruvato será preferencialmente convertido a Lactato no músculo esquelético, mas no cardíaco será preferencialmente usado no metabolismo aeróbico, ao invés de ser convertido a Lactato.
CONSIDERAÇÕES SOBRE A EQUAÇÃO DE MICHAELIS-MENTEN
KM = [S]
Keq = __[ES]_____ = __K1____ [E] [S] K2
50% das enzimas está livre e 50% das enzimas na forma ES (o equilíbrio da 1ª etapa fica estabelecido)
Nesta situação: [S] = KM (constante de Michaelis- Menten)

65
“A constante de Michaelis-Menten é numericamente igual à [S] que determina a metade da VELOCIDADE MÁXIMA”.
O valor de KM pode identificar o grau de afinidade da ensina pelo substrato. Quando [S] é muito inferior à KM, ou seja KM + [S] é praticamente igual à KM, conclui-se que:
Com [S] pequenas, a velocidade de reação é diretamente proporcional à [S].
Quando a [S] é muito maior que KM, então:
KM + [S] = [S]
Isto indica que quando [S] é muito elevada, a velocidade da reação é constante e máxima, independendo da [S].
Enzimas Alostéricas ou Reguladoras
As propriedades cinéticas de muitas enzimas não podem ser explicadas pelo modelo de Michaelis-Menten. Um importante grupo é constituído das Enzimas Alostéricas. Nas enzimas alostéricas, a ligação do substrato a um centro ativo pode afetar as propriedades dos outros centros ativos, na mesma molécula de enzima. As enzimas alostéricas são sempre oligoméricas, com sítios reguladores e catalíticos topologicamente distintos. A característica cinética das Enzimas Alostéricas é a relação atípica entre a atividade e a concentração do substrato. Até aqui foi estudado o caso de enzima em que a ligação de uma molécula de substrato não tem efeito nas constantes de dissociação intrínsecas dos sítios vagos. Elas têm, neste caso, curvas de velocidade hiperbólicas. Porém, se a ligação de uma molécula de substrato induz modificação estrutural ou eletrônica que resulta em afinidades alteradas dos sítios vagos, a curva de velocidade não seguirá mais a cinética de Michaelis-Menten e a enzima será chamada alostérica. As enzimas alostéricas normalmente apresentam uma curva de velocidade sigmoide.
PERFEIÇÃO CATALÍTICA
Algumas enzimas estão próximas da perfeição catalítica.
Kcat = V MÁX
[ETotal]
Sendo: Kcat = A constante catalítica que mede para uma dada concentração de enzima, a eficiência máxima obtida em condições de Vmáx, quando todas as enzimas estão complexadas com o substrato.
Kcat é também conhecida como nº de renovação (turnover number) da enzima, porque equivale ao nº máximo de moléculas de substrato que um centro ativo converte em produto por segundo. Ela indica a rapidez com que

66
uma enzima opera, quando todos os centros ativos estão ocupados. Em outras palavras pode-se dizer que Kcat evidencia com que eficiência o complexo enzima-substrato origina produto.
Associando-se o valor de Kcat ao valor de KM, é possível definir-se uma nova constante Kcat/KM que pode relacionar a eficiência catalítica da enzima com a sua afinidade pelo substrato. Quando a relação Kcat/KM tiver um valor baixo significa que a enzima tem pouca afinidade pelo substrato (logo ↑ KM) ou baixa eficiência em gerar produto a partir do complexo ES, ou pelas duas razões. Quando Kcat/KM for um valor alto significa que a enzima tem alta eficiência em transformar ES em produto (logo Kcat ↑) e portanto alta afinidade pelo substrato (↓ KM)
São exemplos de enzimas com alta eficiência: Acetilcolinesterase (transmissão do impulso nervoso), Anidrose carbônica (remoção de CO2 dos tecidos) Catalase e Superóxido dismutase (remoção de radicais livres do oxigênio). Um exemplo de enzima com pouca eficiência: Pepsina.
HIPERTEXTO
Uma enzima particularmente útil para exames é a acetilcolinesterase (AChE), que é importante no controle de certos impulsos nervosos. Muitos pesticidas interferem com essa enzima, portanto, agricultores deveriam ser frequentemente testados para se ter a certeza de que eles não estariam sendo inapropriadamente expostos a essas substâncias tóxicas importantes para a agricultura. Na verdade, existem mais de 20 enzimas que são tipicamente usadas em laboratórios clínicos para o diagnóstico de doenças. Existem marcadores altamente específicos para enzimas ativas no pâncreas, nas hemácias, no fígado, no coração, no cérebro, na próstata e em muitas glândulas endócrinas. Uma vez que essas enzimas são relativamente fáceis de se medir, inclusive por meio da utilização de técnicas automatizadas, elas fazem parte dos exames de sangue “de rotina” que o médico pode requisitar (Campbell & Farrel, 2011, p. 144).
INIBIDORES ENZIMÁTICOS
São responsáveis pela diminuição da atividade enzimática. Como a ação enzimática controla o metabolismo, muitos medicamentos baseiam suas propriedades na inibição de certas enzimas, como, por exemplo, as Sulfonamidas, que combatem infecções bacterianas. O medicamento age inibindo uma enzima bacteriana que não existe no organismo do hospedeiro. As propriedades tóxicas de alguns inibidores são usadas no combate aos insetos. Os inibidores podem ser Irreversíveis e Reversíveis, segundo a estabilidade de sua ligação com a molécula de enzima.

67
Inibidores Irreversíveis
Reagem quimicamente com as enzimas, levando a uma inativação definitiva. Um bom exemplo são os compostos organofosforados. Eles formam ligações covalentes com o grupo OH de resíduos do aminoácido serina ou de iodoacetamida que reage com o grupo SH de resíduos do aminoácido cisteína.
O O
– CH2 – SH + ICH2 – C –NH2 – -CH2 – S – CH2 – C – NH2 + HI
Resíduo de cisteína Iodoacetamida
Outro exemplo de Inibidor Irreversível é a aspirina (Ácido Acetil-Salicílico). Atua como antiinflamatório, antipirético e analgésico. Transfere irreversívelmente seu grupo acetil para o grupo OH de um resíduo de serina da molécula da enzima Cicloxigenase, inativando-a. Esta enzima é a responsável pela catálise da 1ª reação da via de síntese das prostaglandinas (substâncias reguladoras de um conjunto de processos fisiológicos). Sem a ação das prostaglandinas, processos como a reação inflamatória, ficam atenuados. Outro Inibidor irreversível é a penicilina. Ela se liga a enzimas da via de síntese da parede bacteriana, inibindo-as. Desprovidas de parede, as células ficam sujeitas à lise.
Inibidores Reversíveis
Podem ser: Competitivos e Não-Competitivos. Os Inibidores competitivos competem com o substrato pelo centro ativo da enzima. Certas moléculas porque apresentam configuração espacial semelhante à do substrato, são capazes de ligarem-se ao centro ativo da enzima, produzindo um complexo enzima-inibidor semelhante ao complexo Enzima-Substrato. São os Inibidores Competitivos Ic.
E + Ic EIc
O complexo EIc jamais gera produto, logo a atividade enzimática decresce. Os inibidores competitivos são muito empregados por sua atividade terapêutica, já que inibem reações que ocorrem específicamente ou principalmente no organismo parasita, vírus ou bactéria. Um exemplo importante é o medicamento AZT, que é o inibidor da DNA polimerase (transcriptase reversa), necessária para a replicação do vírus HIV, causador da AIDS. Outros exemplos de Inibidores competitivos são também usados na quimioterapia de tumores e de leucemias. Aqui, a célula tumoral comporta-se como o agressor do organismo e tem metabolismo diferente do da célula normal sob vários aspectos, inclusive uma velocidade de multiplicação muito maior. As drogas que afetam reações enzimáticas normais e imprescindíveis às células em geral agirão preferencialmente sobre as células tumorais; porém
En
E

68
também atingirão alguns tecidos normais que se dividem rapidamente, como a medula óssea (que produz as células sanguíneas), a mucosa intestinal e os folículos capilares. O alvo de escolha para ação dessas drogas é a replicação do DNA, em suas várias etapas.
Inibidores não-Competitivos (INc)
Não têm semelhança estrutural com o substrato da reação que inibem e seu efeito deve-se à ligação a radicais que não pertencem ao centro ativo; esta ligação altera a estrutura enzimática inviabilizando a catálise. O ponto de ligação do inibidor não-competitivo (INC) é a cadeia lateral de um aminoácido, por exemplo: o grupo OH da serina ou o SH da cisteína. Têm ação inespecífica, podendo atuar sobre grande número de enzimas (diferente do que ocorre com os Ic).
E + INc E - INc
Diferença entre INC e Irreversível
Nos inibidores irreversíveis, uma molécula enzimática ligada ao inibidor está definitivamente inativada. Nos inibidores reversíveis não-competitivos, (INc) uma molécula de enzima, que, em um instante, está ligada ao inibidor (inativa), pode tornar-se ativa, ao ficar livre do inibidor, em um momento seguinte. Exemplos de INc são os metais pesados como Hg2+, Pb2+, Ag+ que reagem com os grupos SH das proteínas. São altamente tóxicos.
Antimetabólitos ou Análogos de Substratos
Têm fórmula estrutural semelhante aos substratos naturais e, ao contrário dos inibidores competitivos, ligam-se ao centro ativo e geram produtos. No entanto, os produtos gerados por eles são diferentes do produto gerado pelo substrato e não prosseguem na sequência metabólica normal, por não serem aceitos como substrato pela enzima seguinte, por serem instáveis ou por qualquer outro mecanismo. A via metabólica sobre a qual interferem fica, portanto, interrompida. Muitos antimetabólitos ocorrem naturalmente, sendo muitas vezes mecanismo de defesa de vegetais contra a ingestão de suas folhas e sementes por insetos, pássaros e mamíferos. Um grande nº de quimioterápicos é constituído por análogos de substratos, particularmente utilizados no tratamento do câncer.
Regulação da Atividade Enzimática
Ocorre basicamente por controle da disponibilidade de enzimas exercido sobre as velocidades de síntese e de degradação das enzimas, que determinam sua concentração celular e por controle da atividade da enzima, efetuado por mudanças estruturais da molécula enzimática e que redundam em alterações da velocidade de catálise. Algumas enzimas tornam-se funcionais
69
através de outros processos de ativação. Certas enzimas são sintetizadas na formas de precursores inativos, chamados zimogênios. É necessário que haja hidrólise de certas ligações peptídicas e remoção de um segmento da cadeia de aminoácidos para que um zimogênio adquira propriedades de enzima. Dessa forma a cadeia polipeptídica que resta assume uma nova estrutura espacial onde é, então, organizado um centro ativo funcional. Exemplos de enzimas sintetizadas como zimogênios são a pepsina e a quimiotripsina.
Cofatores
São denominados Cofatores moléculas ou íons que se associam a um grande número de enzimas para que elas possam exercer seu papel catalítico. Eles podem ser íons metálicos ou moléculas orgânicas, não-proteicas, de complexidade variada, que recebem o nome de coenzimas. Os íons metálicos ligam-se aos radicais de aminoácidos de cadeia proteica ou estão presentes em grupos prostéticos. As enzimas atuam como aceptores de átomos ou grupos funcionais retirados do substrato em uma dada reação e como doadores destes mesmos grupos ao participarem de uma outra reação. Por isto diz-se que são transportadoras de determinados grupos. Durante a catálise, coenzima e substrato acham-se alojados no centro ativo da enzima, consistindo a reação na remoção de determinado grupo químico do substrato e sua transferência para a enzima, ou vice-versa. Em alguns casos, a coenzima encontra-se ligada covalentemente à molécula enzimática, constituindo, portanto, um grupo prostético da proteína; em outros casos, a coenzima é uma molécula “livre”, reunindo-se à enzima apenas no momento da catálise. A estrutura química de uma coenzima é bastante variável. Algumas como a Adenosina Trifosfato (ATP) e a Guanina Trifosfato (GTP) são completamente sintetizadas pelas células. Outras apresentam um componente orgânico que não pode ser sintetizado pelos animais superiores. Deve ser obtido através da dieta, constituindo uma vitamina.
As vitaminas são necessárias na dieta em pequenas quantidades já que são precursores de coenzimas. As vitaminas são divididas em Hidrossolúveis e Lipossolúveis. As Hidrossolúveis incluem as vitaminas do complexo B: tiamina (B1), riboflavina (B2), ácido pantotênico (B3), nicotinamida (B5), piridoxina (B6); biotina (B7), ácido fólico (B9), cobalamina (B12) e o ácido ascórbico (vitamina C). As vitaminas lipossolúveis são: A, D, E, K. São as vitaminas hidrossolúveis as que têm função de coenzimas ou fazem parte de moléculas de coenzimas.
CONTEXTUALIZANDO
Algumas enzimas são encontradas somente em tecidos específicos ou em um número limitado de tecidos. A enzima lactato-desidrogenase (LDH)

70
apresenta-se de duas formas diferentes, chamadas de isoenzimas, no coração e nos músculos. As duas formas diferenciam-se discretamente na composição de aminoácidos e podem ser separadas com base nas cargas resultante dessa diferença. Uma vez que a LDH é um tetrâmero de quatro subunidades, ela também pode existir em cinco diferentes formas, dependendo da origem das subunidades. Um aumento de qualquer forma de LDH no sangue indica algum tipo de dano tecidual. Um ataque cardíaco poderá ser diagnosticado com certeza se houver um aumento da forma cardíaca do LDH. De modo similar, existem diferentes formas da creatina-quinase (CK), uma enzima que se encontra no cérebro, no coração e no músculo esquelético. O aparecimento da forma cerebral pode indicar um acidente vascular cerebral ou um tumor no cérebro, ao passo que o tipo cardíaco indica o infarto. Depois de um ataque cardíaco, a CK aparece no sangue mais rapidamente do que a LDH. A monitoração da presença de ambas as enzimas permite melhorar a possibilidade de diagnóstico, o que é muito útil, uma vez que um ataque cardíaco brando pode ser difícil de ser diagnosticado. Um nível elevado da isoenzima cardíaca no sangue é uma indicação segura de dano no tecido cardíaco (Campbell & Farrel, 2011, p. 144).
HIPERTEXTO
A rubisco ou RuBisCO (ribulose-bisfosfato carboxilase oxigenase, é a enzima mais abundante nas plantas e, por conseguinte, a proteína mais abundante no planeta. Esta enzima capta o dióxido de carbono procedente do ar e um açúcar existente na célula chamado RuDP (ribulose 1,5-difosfato ou RuBP - ribulose bis-fosfato). A reacção entre estes dois reagentes dá origem a duas moléculas do açúcar PGA (fosfoglicerato). A RuBisCO é assim responsável pelo importante primeiro passo do ciclo de Calvin e em concreto pela fixação do dióxido de carbono na sua forma orgânica. É importante dizer que a reação pode acontecer tanto com dióxido de carbono quanto com oxigênio molecular (O2). Quando o oxigênio é absorvido, o processo faz parte da respiração celular, sem absorção de carbono. Algumas plantas (por volta de 5% das plantas existentes na Terra) utilizam um processo intermédio e mais seletivo para absorção do dióxido de carbono, evitando o contato direto com o oxigênio do ar típico do "processo rubisco" (às vezes chamado C3), com o processo chamado C4 ou o CAM ("Crassulacean acid metabolism"). O C4 usa estruturas dentro da célula e um sistema bioquímico de transporte que envolve moléculas com quatro átomos de carbono (por isso C4). O CAM é usado por plantas em ambientes muito áridos, em que os estômatos só abrem à noite, a fim de economizar água, que se perderia por evaporação, se estes ficassem abertos durante o dia. Assim, à noite, absorvem o dióxido de carbono em moléculas de quatro carbonos que ficam armazenadas em vacúolos para serem utilizadas durante o dia.

71
Os processos C3, C4 e CAM têm vantagens e desvantagens. O C3 é mais direto, gastando cerca de 18 ATPs para fixar cada molécula de dióxido de carbono, mas requer concentrações mais altas de dióxido de carbono e água. O C4 requer mais estruturas e mais energia (30 ATPs), porém trabalha com concentrações de dióxido de carbono mais baixas na atmosfera e é mais apropriado para ambientes secos. O CAM é usado em ambientes predominantemente áridos, mas fixa pouco dióxido de carbono, levando a taxas de crescimento da planta baixas.
ATIVIDADES DE AVALIAÇÃO
1. Considerando a reação A → B, não catalisada:a) Definir velocidade de reação.b) Escrever a equação de velocidade em função da concentração de A.
2. Definir enzima, substrato e sítio ativo.3. Escrever as reações de formação do produto a partir de E e S. Escrever a
equação de velocidade de formação de P.

72
4. Fazer o gráfico da velocidade da reação S → P, catalizada enzimaticamente, em função da concentração de S. Descrever os procedimentos experimentais, que levariam à obtenção dos dados para a construção deste gráfico.
5. Analisando o gráfico do item 4, verificar, em cada trecho da curva, as concentrações de Enzima livre, substrato e complexo ES.
6. Definir constante de Michaelis-Menten (KM) e mostrar a relação entre seu valor e a afinidade da enzima pelo seu substrato.
7. Definir e dar exemplos de inibidor competitivo e não-competitivo.8. Definir cofator. Dar exemplos de cofatores inorgânicos (ativadores metálicos)
e orgânicos (coenzimas).9. Definir vitaminas, relacionando sua função com atividade enzimática.10. Caracterizar enzima alostérica. Definir centro alostérico. Qual a diferença
entre a curva de velocidade de uma enzima comum e a de uma enzima alostérica?
11. Definir regulação enzimática por modificação covalente.12. Representar o grupo ativo de NAD+ e de FAD nas formas reduzida e
oxidada.
13 Definir Zimogênio.
14 Descreva as enzimas LDH, CK e AChE. Após um ataque cardíaco qual enzima cardíaca aparece primeiro: a LDH ou a CK ?
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 1. São Paulo: Cengage Learning - 2011. O Comportamento das Proteínas: Enzimas é o título do Capítulo 6 do volume 1 desse livro. São apresentados os tópicos relativos ao assunto e todos comentados de forma muito didática.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade I do livro, no Capítulo 5, são apresentados os tópicos relativos às Enzimas. O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Cooper, Geoffrey M. (2000). "10.The Chloroplast Genome". The Cell: A Molecular Approach (2nd ed.). Washington, D.C: ASM Press.ISBN 0-87893-106-6.
Mahan, K.; Escott-Stump. Alimentos, nutrição & dietoterapia. São Paulo: Roca, 2005. Enzimas foram comentados no Capítulo 1 (Digestão, Absorção, Transporte e Excreção de Nutrientes).
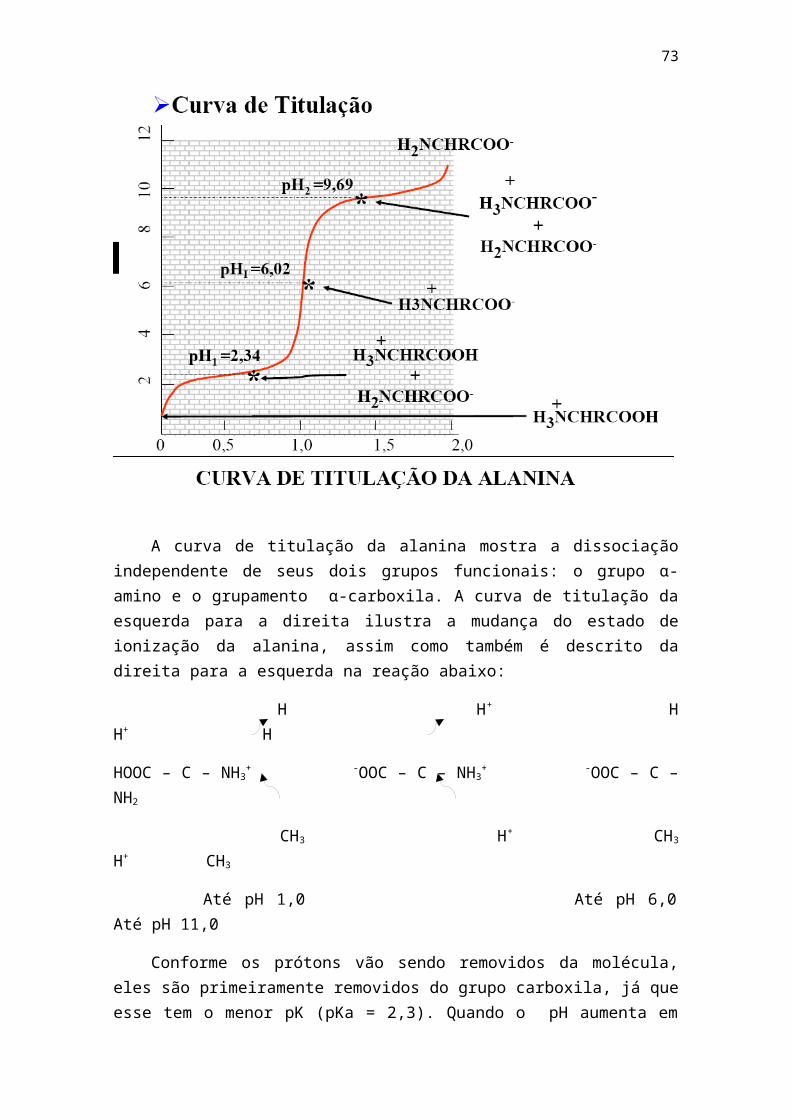
73
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª Ed. Rio de Janeiro: Ed. Guanabara Koogan S. A., 1999. Os autores apresentam Enzimas na Parte 1 de seu livro no capítulo 5, comentando amplamente sobre o assunto.
LEITURAS, VÍDEOS E SITE RECOMENDADOS
veja.abril.com.br/.../enzima-pode-ser-novo-alvo-na-luta-contra o mal de parkinsom.
www.not1.com
CAPÍTULO 6: ÁCIDOS NUCLEICOS
OBJETIVOS:
Conhecer as estruturas formadoras do DNA e RNA.
Aprender as funções de DNA e RNA.
Fazer a diferença entre DNA e RNA.
Saber o que é o genoma humano.
Conhecer a replicação do DNA.
Perceber como se dá o teste de DNA ou teste de reconhecimento de paternidade.
Os ácidos nucleicos têm sido objeto de investigação bioquímica desde que foram, pela primeira vez, isolados no núcleo da célula, há mais de 100 anos. Eles ocorrem em todas as células vivas, onde não só são responsáveis pelo armazenamento e transmissão da informação genética, mas também pela tradução dessa informação, expressa pela síntese precisa das proteínas, características de cada célula. São biopolímeros de alto peso molecular como as proteínas, só que, nos Ácidos Nucleicos, a unidade que se repete é o mononucleotídeo.
Um nucleotídeo consiste em uma base, uma ose e um ou mais grupamentos fosfato. Os nucleotídeos possuem uma variedade de papéis no metabolismo celular. São moedas energéticas nas transações metabólicas; os elos químicos essenciais na resposta das células aos hormônios e outros estímulos extracelulares e os componentes estruturais de uma coleção de cofatores enzimáticos e intermediários metabólicos, além é claro, de constituírem os Ácidos Nucleicos: Ácido Desoxirribonucleico (DNA) e Ácido

74
Ribonucleico (RNA), que são as moléculas repositórias da informação genética. A ose em um desorribonucleotídeo é a desoxirribose. O prefixo desoxi indica que nesta ose falta um átomo de oxigênio que está presente na ribose, o composto original. A base nitrogenada é um derivado de purina ou pirimidina. São as bases das moléculas de DNA que levam a informação genética enquanto seus grupamentos ose e fosfato têm um papel estrutural.
Os nucleotídeos formam uma porção de coenzimas, tais como FAD, NAD+, NADP+, Coenzima A e S–Adenosil-Metionina. As estruturas básicas do DNA e RNA consistem em cadeias nas quais Ácido Fosfórico e resíduos de açúcar alternam-se. No RNA, o açúcar é a D-ribofuranose. No DNA, o açúcar é a 2-desoxi-D-ribofuranose. Ligada a todas as unidades de açúcar, há uma base nitrogenada que pode ser tanto um derivado purínico como pirimidínico. É a sequência de bases nas longas cadeias de açúcar-fosfato que determina as propriedades biológicas da molécula.
Estrutura dos Nucleotídos
Tanto o RNA como o DNA contêm as duas purinas, Adenina e Guanina. Várias bases menos comuns foram encontradas nos RNA de transferência (RNA transportadores, tRNA). Entre elas, estão a hipoxantina, a 1-metil-hipoxantina, 1-metilguanina, e treonilcarbamoiladenina. Também a pirimidina Citosina é comum ao RNA e DNA, mas, os dois tipos de ácidos nucleicos diferem na quarta base nitrogenada, o RNA contém Uracila enquanto o DNA contém Timina. Portanto todos os ácidos nucleicos contêm adenina, guanina e citosina. O DNA (mas não os RNAs), também contém Timina, enquanto os RNAs (mas não o DNA), também contêm Uracila.

75
A estrutura das bases que contêm oxigênio foi escrita na forma cetônica (ou lactâmica). É necessário salientar que há um equilíbrio entre as formas cetônicas e enólicas (ou lactímicas) o qual depende do pH do meio. É a forma lactâmica que predomina na pH fisiológico.
Outras pirimidinas têm sido detectadas em amostras purificadas de DNA; 5-metilcitosina ocorre em DNA isolado do germe do trigo e outras fontes vegetais. Também têm aparecido traços dessa base no DNA do timo e em outras fontes, nos mamíferos. A citosina é substituída pela 5-hidrometilcitosina no DNA de certos vírus bacterianos, principalmente os bacteriófagos T que infectam E. coli. Em tRNA, foram encontrados os derivados de pirimidina - diidrouracila, pseudouridina e 4-tiouracila.
Nucleosídeos
A molécula de nucleotídeo sem o grupo fosfato é chamada de nucleosídeo. Os nucleotídeos são, portanto, ésteres de Ácido Fosfórico dos nucleosídeos.
NOMES DOS NUCLEOSÍDEOS
BASE RIBONUCLEOSÍDEO DESOXIRRIBOCUCLEOSÍDEO
ADENINA ADENOSINA 2’ – DESOXIADENOSINA
GUANINA GUANOSINA 2’ – DESOXIGUANOSINA
URACILA URIDINA 2’ – DESOXIURIDINA
CITOSINA CITIDINA 2’ – DESOXICITTIDINA
TIMINA TIMINA RIBONUCLEOSÍDEO 2’ – DESOXITIMIDINA
O resíduo de ribose de um ribonucleotídeo tem três posições (as hidroxilas dos carbonos 2’, 3’ e 5’), onde o fosfato pode ser esterificado, enquanto que o 2’-desoxirribonucleosídeo tem somente as posições 3’ e 5’ disponíveis. Todas essas possibilidades podem ser obtidas pela hidrólise parcial de Ác. Nucleicos por vários métodos. Além disso, vários componentes celulares são ésteres 5’-fosfato.
Um dos mais importantes nucleotídeos que ocorre naturalmente é a Adenosina-5’-Monofosfato (AMP). Esse composto em conjunto com dois de seus derivados, Adenosina-5’-Difosfato (ADP) e Adenosina-5’-Trifosfato (ATP) exerce um papel extremamente importante na conservação e utilização da energia liberada durante o metabolismo celular. O significado fisiológico desses componentes consiste na sua capacidade de doar e aceitar grupos fosfato em reações bioquímicas.

76
O impedimento estérico pela base heterocíclica determina que uma vez formada, não existe liberdade de rotação em torno da ligação β-N-glicosídica, que une os açúcares às purinas ou pirimidinas. Os nucleosídeos e os nucleotídeos, assim, existem como Confôrmeros estáveis, não-interconversíveis, syn e anti, que só podem ser interconvertidas pela ruptura e reformação da ligação da ligação glicosídica. Embora ambos os confôrmeros ocorram naturalmente, os confôrmeros anti predominam e é o confôrmero anti de nucleotídeos que participa no pareamento normal das bases na DNA dupla fita.
Principais Bases, Nucleosídeos e Nucleotídios
BASE
X=H
NUCLEOSÍDEO X=RIBOSE OU
DESOXIRRIBOSE
NUCLEOTÍDEO X-Ribose fosfato
ADENINA
A
ADENOSINA
A
ADENOSINA MONOFOSFATO
AMP
GUANINA
G
GUANOSINA
G
GUANOSINA MONOFOSFATO
GMP
CITOSINA
C
CITIDINA
C
CITIDINA MONOFOSFATO
CMP
URACILA
U
URIDINA
U
URIDINA MONOFOSFATO
UMP
TIMINA
T
TIMIDINA
T
TIMIDINA MONOFOSFATO
TMP
As abreviações A, G, C, T e U referem-se às bases adenina, guanina, citosina, timina e uracila, respectivamente, tanto no estado livre, como formando os nucleosídeos ou nucleotídeos. O prefixo “d” (deoxi) indica que o açúcar é 2’-dioxi-D-ribose, ex., dGTP. Os nucleosídeos fosforilados no carbono 3’- ou no 5’ da ribose são denominados nucleosídeo 3’-monofosfato e nucleopídeo 5’-monofosfato respectivamente. Ex.: adenosina 3’-monofosfato ou

77
3’-AMP. Contudo, como o 5’-hidroxil é um produto esterificado dos mais comuns, “5”- é omitido quando se refere a 5’-nucleotídeos.
As abreviações tais como “UMP” ou “AMP”, portanto, dizem respeito a nucleotídeos em que o fosfato liga-se, por um grupo éster, ao carbono 5 da pentose. Fosfatos adicionais, unidos por ligações anidridos de ácidos aos fosfatos já existentes em um mononucleotídio, formam nucleosídeos di - e trifosfatos, tais como ADP (adenosina difosfato) e ATP (adenosina trifosfato).
OBS: Um número com apóstrofo indica um átomo da ribose ou da desoxirribose, enquanto um número sem apóstrofo indica um átomo de anel de purina ou pirimidina.
Os mono, di e trifosfato de adenosina já foram descritos. Existem derivados correspondentes de guanosina, citidina e uridina, assim como de desoxiadenosina, desoxicitidina, e desoxitimidina que exercem funções importantes no metabolismo celular. Por exemplo, os nucleosídeos 5’-trifosfato servem como precursores da síntese de RNA e DNA. Derivados de certos nucleosídeos 5’-difosfato agem como coenzimas, fornecendo resíduos de açúcares em certas reações; outros derivados de adenosina – 5’-difosfato participam de reações de óxido-redução. Assim, uridina 5’-disfosfato, ligada à glucose, serve como doador de glucose. Adenosina – 5’-disfosfato, ligada à nicotinamida, forma uma coenzima de óxido-redução extremamente importante, a nicotinamida – adenina – dinucleotídeo, NAD+.
DNA e RNA
Em células procarióticas, o DNA normalmente ocorre sob forma de um anel composto de uma fita dupla extremamente torcida, parcialmente associado com a porção interna da membrana plasmática, mas livre de complexos proteicos. Ao contrário, cerca de 98% do DNA total numa célula diferenciada típica, eucariótica, é encontrado no núcleo, como um polímero composto de uma fita dupla extremamente torcida, ligado a proteínas básicas chamadas Histonas; o complexo é conhecido como Cromatina. Quantidades muito menores de DNA são sempre encontradas na matriz mitocondrial de células eucarióticas e em cloroplastos, sob a forma de pequenos anéis formados de fitas duplas, livres de complexos proteicos.
O segundo ácido nucleico componente da célula, o RNA, ocorre em múltiplas formas, todas elas servindo como elos informacionais extremamente importantes entre DNA, o veículo fundamental da informação, e as proteínas. O menor desses polímeros é chamado RNA transportador (tRNA). O tRNA corresponde a cerca de 60 diferentes espécies moleculares. Os tRNA exercem uma série de funções sendo a mais importante delas agir como transportadores

78
específicos de aminoácidos ativados para locais determinados nos moldes sintetizadores de proteínas.
Os tRNA compreendem cerca de 10-15% do RNA total da célula. Um segundo grupo de RNA inclui os RNA ribossômicos (rRNA). Esses ácidos nucleicos estão sempre associados com um grande número de proteínas num complexo altamente ordenado chamado Ribossoma. Eles perfazem cerca de 75-80% do RNA total de célula. O terceiro grupo importante de RNA é formado pelos RNA mensageiros (mRNA) que compreendem cerca de 5-10% do RNA total da célula. Em células bacterianas, os mRNA são extremamente instáveis, no sentido de serem constantemente degradados e ressintetizados. Em células eucarióticas, a velocidade de renovação é muito menor. Esses ácidos nucleicos, com uma composição básica muito semelhante àquela do DNA, estão intimamente envolvidos na transcrição e tradução da informação programada pelo DNA para a síntese das proteínas.
Determinação das Relações Molares entre as Bases nos Ácidos Nucleicos
As características fundamentais de um ácido nucleico são a composição e a sequência de bases purínicas e pirimidínicas. Dados de composição são usados para calcular a equivalência entre A e U e entre C e G no RNA; e entre A e T e entre C e G no DNA. Estudos estruturais concluíram que DNA de diferentes fontes tinham padrões de difração de RX semelhantes. Isso sugeriu um padrão molecular uniforme para todas as moléculas de DNA. Os dados também sugeriram que o DNA consistia de duas ou mais cadeias polinucleotídicas arranjadas numa estrutura helicoidal.
Com evidências baseadas no pareamento, na equivalência e em dados de titulação que sugeriam que as longas cadeias de nucleotídeos são mantidas unidas por meio de pontes de hidrogênio entre os resíduos de bases, Watson e Crick construíram seu modelo de DNA, em 1953. Nesse modelo, duas cadeias polinucleotídicas estão enroladas numa dupla hélice. As características importantes de seu modelo de DNA são:
1. Duas cadeias polinucleotídicas helicoidais estão enroladas em torno de um eixo comum. As cadeias correm em sentidos opostos.
2. As bases purínicas e pirimídicas estão do lado de dentro da hélice, enquanto as unidades fosfato e desoxirribose estão de fora. Os planos das bases são perpendiculares ao eixo da hélice. Os planos das oses são mais ou menos perpendiculares aos das bases.
3. O diâmetro da hélice é de 20 Å. Bases adjacentes são separadas por por 3,4 Å, ao longo do eixo da hélice, e com uma rotação de 36 graus. Assim a estrutura helicoidal se repete a cada 10 nucleotídeos em cada cadeia, isto é, a intervalos de 34 Å.

79
4. As duas cadeias são mantidas juntas por pontes de hidrogênio entre os pares de bases. Aadenina está sempre pareada com a timina. A guanina está sempre pareada com a citosina.
5. A sequência de bases ao longo de uma cadeia polinucleotídica não é, de nenhum modo, restrita. A sequência exata de bases leva a informação genética.
Assim, temos uma estrutura espacial formada de duas cadeias enroladas em torno de um eixo comum e mantidas unidas pelas ligações específicas de adenina com timina e citosina com guanina. As cadeias não são idênticas, mas, por causa do pareamento de bases, uma é exatamente o complemento da outra. O aspecto mais importante da dupla hélice de DNA é a especificidade do pareamento das bases. Watson e Crick deduziram que a adenina deve se parear com a timina, e a guanina com a citosina, devido a fatores estéricos e de pontes de hidrogênio. A dupla hélice de DNA pode ser reversivelmente separada. Os dois filamentos da hélice de DNA são separados quando as pontes de hidrogênio são rompidas entre suas bases pareadas.
Replicação Semiconservativa do DNA
O poder do DNA de formar uma hélice dupla é de grande importância quando se considera sua função na célula. O DNA é capaz de produzir cópias exatas de si mesmo. É a Replicação. A molécula de DNA abre-se ao meio. As duas cadeias de polinucleotídeos se separam. São formadas novas cadeias complementares, ao longo de cada polinucleotídeo matriz. O processo de duplicação do DNA é Semiconservativo, pois cada molécula recém-formada recebe uma cadeia de polinucleotídeo da original. Os fenômenos de replicação de DNA só ocorrem na presença da DNA-polimerase e a energia necessária para a replicação é obtida a partir do ATP. Embora as informações para a síntese das proteínas encontrem-se no núcleo da célula, elas são produzidas predominantemente no citoplasma O RNA é o mensageiro do DNA que comanda a síntese das proteínas. A estrutura helicoidal dupla sugere um mecanismo para a replicação perfeita da informação genética. Por causa da complementaridade da estrutura helicoidal, cada fita serve de molde para especificar a sequência de bases na síntese de uma nova fita complementar. Em consequência, na síntese de duas moléculas-filhas de DNA, cada uma será perfeitamente idêntica à do DNA-mãe. Esta distribuição de átomos parentais é chamada de semiconservativa.

80
Figura digitalizada a partir da página 39 do Livro "Color Atlas of Genetics"de Eberhard Passarge publicado pela Editora Thieme em 1995.
Representação da forquilha de replicação, onde estão mostradas as principais enzimas e cofatores que formam o complexo de replicação (figura disponível no site www.ncbi.nlm.nhi.gov livro The Cell:
A molecular Approach, de J. Cooper, Sinauer Assoc. Co.).
RNA
No RNA, a ribose substitui a Desoxirribose e a Uracila substitui a Timina (formando par com Adenina). Embora o DNA seja quase que exclusivamente nuclear (exceção: encontrado em cloroplastos e em mitocôndrias), o RNA é

81
tanto nuclear como citoplasmático. Denomina-se Transcrição ao processo mediante o qual a mensagem do DNA é transportada para o RNA.
O RNA transportador desempenha um papel-chave na síntese proteica. Cada molécula de tRNA transporta um aminoácido para os ribossomos e decifra a informação contida no RNA mensageiro em termos de aminoácidos (correspondentes ao código), dispondo-os na sequência correta. 60 – 70% da molécula dos tRNA apresenta uma estrutura helicoidal. Essa e outra evidência indicam uma estrutura em folha de trevo para todos os tRNA, com o anticódon (isto é, a trinca de nucleotídeos necessários para o ajuste do tRNA específico ao molde de mRNA, durante a síntese de proteínas) localizado na pétala central do trevo. Outros pesquisadores sugeriram uma estrutura terciária para o tRNA, o que está mais próxima à realidade.
Várias espécies de RNA ocorrem em ribossomos procarióticos e eucarióticos e é designado RNA ribossômico. O RNA ribossômico (rRNA) tem uma estrutura helicoidal resultante do dobramento de uma polímero de fita simples sobre si mesmo, em áreas onde são possíveis ligações por pontes de hidrogênio. Porém, o rRNA não ocorre como um polímero de fita dupla. Além do mais, uma vez que o rRNA não tem a estrutura helicoidal do DNA, estável e extremamente rígida, ele pode existir em várias conformações. Assim, na ausência de eletrólitos ou em altas temperaturas, pode ocorrer uma conformação de fita simples. Em forças iônicas baixas, pode aparecer em bastão compacto com regiões helicoidais regularmente arranjadas e, em forças iônicas altas, aparece uma espiral compacta.
Principalmente a concentração de Mg++ tem um papel importante nas estruturas macromoleculares do RNA, possivelmente porque o Mg++ forma ligações de coordenação com os grupos fosfato do ácido nucleico. Em baixas concentrações de Mg++, ocorre dissociação dos complexos de RNA, enquanto que em altas concentrações de Mg++ a associação dos complexos é favorecida.
Por causa da heterogeneidade e instabilidade metabólica do RNA mensageiro, só recentemente foi possível uma caracterização cuidadosa. O RNA mensageiro parece ser principalmente de fita única, e sua complementariedade em relação à sequência de bases do DNA foi demonstrada através da formação de moléculas híbridas artificiais de DNA-RNA de fita dupla.
Estruturas
A estrutura da molécula de DNA corresponde a uma escada espiralada, onde os pares de bases nitrogenadas representam os degraus e a ligação fosfato-açúcar os corrimões. Pode ser representada assim:

82
A G C T
A linha vertical refere-se à ose, enquanto A, G, C e T representam as bases. No diagrama, P dentro da linha em diagonal indica uma ligação fosfodiéster. Esta linha diagonal une o meio de uma linha vertical com a ponta da outra. Estas funções referem-se respectivamente a 3’-OH e a 5’-OH.
A C
5’ 5’
Agora se for:
A C G
3’ 3’
5’ 5’ 5’
pApCpG ou pACG
Isto mostra que uma cadeia de DNA tem polaridade, pois uma ponta de cadeia tem um grupamento 5’-OH e a outra um 3’-OH, nenhum deles ligado a outro nucleotídeo.
Estrutura do RNA
A molécula é formada por 1 única cadeia de polinucleotídeo, que se torna dupla ocasionalmente ao dobrar-se em torno de si mesma.
Diferenças entre DNA e RNA
PP
P
P P
3’ 3’
OH
OH

83
DNA RNA
Principalmente no Núcleo
Localização Principalmente no Citoplasma
Adenina
Guanina
Bases Púricas Adenina
Guanina
Timina
Citosina
Bases Pirimídicas Uracila
Citosina
Desoxirribose Pentose Ribose
Ácido Fosfórico Ligação entre Nucleotídeos
Ácido Fosfórico
Depósito de Informações Genéticas
Função Síntese de Proteínas
Desnaturação do DNA
As pontes de hidrogênio entre os pares de base são importantes para manter a estrutura da dupla-hélice. Para romper as pontes de hidrogênio e as interações do empilhamento das bases na conformação nativa do DNA é necessário que seja cedida energia a uma amostra de DNA, como por aquecimento ao DNA em solução, por exemplo. A desnaturação por calor

84
também pode ser chamada fusão. Quando o DNA sofre aquecimento, as fitas se separam. A desnaturação por aquecimento é uma maneira de obter um DNA de fita simples.
HIPERTEXTO
Retrovírus (HIV): Recebe esse nome pelo fato de sua replicação ocorrer de trás para frente, se comparado ao dogma central da Biologia Molecular (cria DNA a partir do RNA, pois a sequência de bases de RNA nesse caso é quem direciona a síntese do DNA).
Fluxo de Informações Genéticas na Célula
A sequência de bases no DNA codifica a informação genética. Uma etapa necessária sempre que uma célula se divide para produzir células filhas é a duplicação do DNA, dando origem a uma nova molécula de DNA com a mesma sequência de bases que a original. Esse processo, como já foi dito, chama-se replicação. O DNA não é o molde direto para a síntese de proteínas. Estes moldes são as moléculas de RNA. A formação de produtos dos genes requer RNA; a produção de RNA em um molde de DNA é chamada transcrição. A sequência de bases do DNA reflete-se na sequência de bases do RNA. Três tipos de RNA estão envolvidos na biossíntese de proteínas; dos três, o RNA mensageiro (mRNA) tem importância especial. Uma sequência de três bases no mRNA especifica a identidade de um aminoácido da forma direcionada pelo código genético. O processo pelo qual a sequência de bases dirige a sequência de aminoácidos é chamado tradução. Em quase todos os organismos, o fluxo

85
de informações genéticas é DNA RNA Proteínas. As exceções são os retrovírus, onde o RNA, e não o DNA, é o material genético. Os genes especificam os tipos de proteínas que são feitos pelas células. Expressam-se fenotipicamente através das proteínas que produzem. Ex.: olhos escuros: células da íris com genes para produção das proteínas responsáveis pelo pigmento escuro.
Gene → Proteína → Fenótipo
Diagnóstico pelo DNA
A genética molecular é uma ferramenta. É o estudo da expressão e herança dos genes a nível molecular. Na clínica bioquímica isso envolve o estudo das diferenças nas sequências de pares de base no DNA. A análise do DNA de um paciente e dos seus genes pode fornecer informação para o diagnóstico. Mutações em genes isolados podem causar doenças hereditárias como fibrose cística, anemia falciforme, hipercolesterolemia familiar e distrofia muscular de Duchenne, enquanto que desordens em vários genes são importantes nas doenças mais comuns tais como doença coronariana, diabetes mellitus e câncer. Duas proteínas diferentes quanto aos efeitos biológicos podem possuir os mesmos aminoácidos. Como? As características específicas de cada proteína dependem da sequência dos aminoácidos. Um bom exemplo a ser estudado é a anemia falciforme. Esse problema ocorre devido à mudança de um único aminoácido em uma molécula formada por 600 aminoácidos.
Hemoglobina normal: Val – His – Leu – Thr – Pro – Glu – Glu - etc.
Hemoglobina Anormal: Val – His –Leu – Thr – Pro – Val – Glu - etc.
Genoma Humano
A maior parte do genoma humano não tem código para proteínas funcionais, pois elas são longas regiões entre e dentro dos genes. O genoma não é idêntico entre uma pessoa e outra: uma média de 200 pares de bases serão diferentes. Quando uma mudança ocorre na região de código, poderá levar à síntese de uma proteína alterada, defeituosa em sua função. Mudanças na região de não-código são frequentemente neutras, mas podem ser úteis para o geneticista molecular. Recentemente, mudanças no não-código do DNA têm sido identificadas como causa de doenças como Síndrome X e distrofia miotônica. A Reação em Cadeia da Polimerase é um método usado para aumentar uma quantidade pequena de DNA com base na reação de enzimas isoladas.

86
Endonucleases de Restrição
O DNA pode ser fracionado em pequenos pedaços por enzimas chamadas Endonucleases de Restrição. Essas enzimas são restritas ao corte de DNA numa sequência de pares de bases específicas. A presença de sítios polimórficos, onde há diferenças na sequência dos pares de bases do DNA pode criar ou abolir um lugar de corte para uma particular enzima endonuclease de restrição. Dessa forma, os polimorfismos levam à produção de fragmentos de diferentes comprimentos depois da ação da enzima.
CONTEXTUALIZANDO
Lúpus é uma doença autoimune envolvendo o processamento de RNA. Aparece no final da adolescência ou início da fase adulta com uma erupção cutânea na fronte e nas bochechas (semelhante às faces de um lobo, daí o nome lúpus). Podem ocorrer problemas renais, artrite, acúmulo de líquido ao redor do coração e inflamação dos pulmões. 90% dos doentes são mulheres. Uma vez que o processamento do mRNA ocorre em todos os tecidos e órgãos do corpo, essa doença afeta vários sistemas e pode espalhar-se facilmente.
ATIVIDADES DE AVALIAÇÃO
1. Escreva a sequência complementar (na notação padrão 5’→3’) para:
a) GATCAA; b) ACGCGT; c) TCGAAC; d) TACCAT
2. Uma amostra de DNA contendo 30% de adenina e 20% de guanina provavelmente conterá ____% de Timina e _____% de citosina.
3. a) Compare e contraste RNA e DNA, relativamente a:
1. Composição química 2. Estrutura secundária
3. Localização nas células vivas - 4. nº dos diferentes “tipos” ou “espécies”
5. funções biológicas
b) Desenhe a fórmula estrutural e dê o nome de:
1. um ribosídeo de purina
2. um desoxirribosídeo – 5’ – difosfato de purina
3. um ribosídeo – 3’, 5’ – difosfato de pirimidina
4. um ribosídeo – 2’, 3’ – difosfato de purina

87
5. um desoxirribosídeo – 3’, 5’ – monofosfato cíclico de purina.
c) Desenhe a unidade repetitiva da molécula de DNA. Indique as posições de quebra que são: a) predominantemente nucleosídeo – 3’ – fosfato; b) predominantemente nucleosídeo – 5’ – fosfato
4. Descreva dois tipos bem diferentes de forças (ou ligações) que estão envolvidas na estabilização da estrutura de dupla hélice do DNA.
5. Descreva os tipos de RNA até agora conhecidos, explicando suas funções.
6. Defina os termos REPLICAÇÃO, TRANSCRIÇÃO E TRADUÇÃO
7. Por que se diz que a replicação do DNA é um processo semiconservativo?
8. Descreva o complexo chamado CROMATINA.
9. Definir, em termos de DNA e síntese de proteínas, o termo RETROVÍRUS.
10. Cite várias (mais de 2) doenças autoimunes relacionadas com o RNA.
11. Qual o papel das enzimas denominadas Endonucleases de Restrição?
12. Pesquise e fale sobre a “Reação em Cadeia da Polimerase” também conhecida como PCR.
13. Pesquise e fale sobre Métodos de Clonagem.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 2. São Paulo: Thomson Learning - 2007. Nos capítulos 9, 10, 11, 12, 13 e 14 do vol. 2 são apresentados tópicos relativos ao assunto e todos comentados de forma muito didática.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade VI do livro, nos Capítulos 29, 30, 31, 32 e 33 são apresentados os tópicos relativos aos Ácidos Nucleicos. O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Stryer, L. Bioquímica. 4ª Ed. Rio de Janeiro: Editora Guanabara Koogan S. A.1996. Os capítulos 4, 5 e 6 apresentam tópicos relativos aos Ácidos Nucleicos.
Murray, R. K.; Granner, D. K.; Mayes, P. A.; Rodwell, V. W. Harper: Bioquímica. 8ª ed. São Paulo: Atheneu, 1998. Os capítulos 35, 36, 37, 38, 39, 40, 41 e 42 da Seção IV, abordam tópicos detalhados sobre Ácidos Nucleicos.

88
LEITURA, VÍDEOS E SITE RECOMENDADOS:
Gattaca (Gattaca - Experiência Genética) é um filme de ficção científica produzido nos Estados Unidos em 1997).
A Experiência (Europa filmes, 2001).
www.clonesfilmes.net
O clone (Clone) - 1997
O outro eu/ eu e meu clone (the order me) - 2000
CAPÍTULO 7: BIOENERGÉTICA
OBJETIVOS:
Conhecer como os processos biológicos ocorrem e de onde vem a energia necessária para esses processos.
Relembrar o conceito de energia livre de Gibbs.
Conceituar reações de óxido-redução em um sistema biológico.
Verificar a participação de coenzimas nas reações de respiração celular.
Verificar a importância do ciclo de Krebs para a obtenção de intermediários da via glicolítica.
Moléculas consumidas como nutrientes precisam ser decompostas para a obtenção de energia a ser usada na criação de novas moléculas. Esse processo ocorre em várias etapas nas quais os doadores de elétrons transferem energia aos aceptores de elétrons. A bioenergética é, portanto o estudo da variação de energia que acompanha as reações bioquímicas, ou seja, ela descreve a transferência e utilização da energia em sistemas biológicos fazendo uso de algumas ideias básicas da termodinâmica, particularmente do conceito de energia livre. Alterações na energia livre (ΔG) fornecem uma medida da probabilidade energética da ocorrência de uma reação química, permitindo prever se uma reação irá ocorrer. O conceito de energia livre é essencial para compreender a função especial que a Adenosina Trifosfato (ATP) desempenha ao transferir energia de processos catabólicos geradores de energia para reações que requerem energia. Os sistemas biológicos são essencialmente isotérmicos e utilizam a energia química para impulsionar os processos vitais. A bioenergética interessa-se somente pelos

89
estados de energia inicial e final dos componentes iônicos, e não o mecanismo nem o tempo necessário para que ocorra a alteração química, ou seja, o que ela prevê é a possibilidade de um processo ocorrer.
O sentido e a extensão de uma reação química são determinados pelo grau em que dois fatores são alterados durante a reação. Esses fatores são a entalpia e a entropia. Entalpia (∆H) é uma medida da mudança no conteúdo de calor dos reagentes e produtos. Entropia (∆S) é uma medida da desorganização dos reagentes e produtos. Nenhuma delas é por si só suficiente para determinar se uma reação química poderá ocorrer espontaneamente no sentido em que é escrita. Porém, combinadas matematicamente a entalpia e a entropia podem ser usadas para definir a energia livre de Gibbs, que prediz o sentido em que a reação ocorrerá espontaneamente. A energia livre de Gibbs, ∆G, é talvez a forma mais adequada de medir as variações de energia nos sistemas vivos, pois mede a energia disponível para realizar um trabalho a uma pressão e temperatura constantes, o que descreve o estado vivo.
∆G = ∆H -T∆S
(onde ∆G é a variação na energia livre; ∆H corresponde à variação na entalpia e ∆S indica a variação na entropia. T é a temperatura absoluta em graus Kelvin (K): K = °C +273).
Reações Exergônicas e Endergônicas
Ao se falar de reação espontânea significa dizer que ela ocorrerá sem adição de energia e a variação da energia livre de Gibbs assume um valor negativo (- ∆G). Isto é, ela é exergônica. No entanto quando o processo é não-espontâneo, ou seja, precisa de adição de energia para que possa ocorrer, possui um valor positivo para a variação de energia livre (+∆G), isto é, ela é endergônica. Se a variação de energia livre tem apenas 1 kcal mol-1 em qualquer direção, então a reação é considerada reversível. Adicionando reagentes ou removendo produtos, a reação muda para a direita; caso os reagentes sejam removidos ou se os produtos forem adicionados, a reação muda para a esquerda.
Acoplamento dos Processos Endergônicos aos Processos Exergônicos
Um mecanismo possível de acoplamento pode ser imaginado quando um intermediário obrigatório comum (I) faz parte de ambas as reações, isto é:
A + C → I → B + D
As reações de síntese, contração muscular, condução do impulso nervoso e transporte ativo – os chamados processos vitais obtêm energia por ligação química, ou acoplamento, às reações oxidativas. A conversão do metabólito A

90
ao metabólito B ocorre com liberação de energia livre (reação exergônica), que é aplicada a outra reação, na qual a energia livre é necessária para converter o metabólito C ao metabólito D (reação endergônica). Como uma parte da energia liberada na reação de degradação é transferida para a reação de síntese em uma forma diferente de calor, os termos exotérmico e endotérmico não podem ser aplicados a estas reações. São usados os termos exergônico e endergônico para indicar que o processo é acompanhado por perda ou ganho, respectivamente, de energia livre, sem considerar a forma da energia envolvida. Na prática, um processo endergônico não existe separadamente. Ele deve ser um componente de um sistema acoplado: exergônico – endergônico, onde a variação líquida global é exergônica.
Algumas reações exergônicas e endergônicas dos sistemas biológicos são acopladas assim. Deve-se levar em conta que este tipo de sistema tem um mecanismo interno para o controle biológico da velocidade, no qual os processos oxidativos ocorrem desde que a existência de um intermediário obrigatório comum permita que a velocidade de utilização do produto da via de síntese (D) determine, pela lei da ação das massas, a velocidade na qual A é oxidado. Na verdade, estas inter-relações fornecem uma base para o conceito do Controle Respiratório, o processo que previne que um organismo perca o controle. Uma extensão deste conceito de acoplamento é proporcionado pelas reações de desidrogenação, que são acopladas às hidrogenações por um transportador intermediário.
Um método alternativo de acoplamento entre um processo exergônico e outro endergônico é a síntese de um composto de alta energia potencial na reação exergônica e incorporação deste novo composto na reação endergônica, assim efetuando uma transferência de energia livre do percurso exergônico para o endergônico. Na célula viva, o principal intermediário de alta energia ou composto transportador é Adenosina Trifosfato ou Trifosfato de Adenosina (ATP). O ATP é, portanto, formado por uma molécula de adenosina (adenina + ribose) à qual estão ligados três grupos fosfato. Com a remoção de um grupo fosfato, obtém-se ADP; se dois fosfatos forem removidos, o produto resultante é o monofosfato de adenosina (AMP). Devido a um ∆G° grande e negativo, o ATP é denominado um composto fosfatado de alta energia.
Muitas reações acopladas utilizam ATP para gerar um intermediário comum. Essas reações podem envolver a clivagem do ATP, ou seja, a transferência de um grupo fosfato do ATP para outra molécula. Outras reações levam à síntese de ATP pela transferência de fosfato de um intermediário rico em energia para o difosfato de adenosina (ADP), formando ATP. Um exemplo de reações acopladas é o caso da fosforilação da glicose ao ser acoplada para a hidrólise de um grupo fosfato do ATP. A fosforilação da glicose e a hidrólise do ATP são duas partes da mesma reação. Somando-se as duas, pode-se

91
determinar a variação de energia geral e garantir que no total ela seja exergônica.
Metabolismo
As reações exergônicas são denominadas catabolismo (geralmente a quebra ou oxidação de moléculas combustíveis), enquanto as reações de síntese, que constroem as substâncias, são denominadas anabolismo. O conjunto de processos catabólicos e anabólicos constituem o metabolismo. O catabolismo e o anabolismo são vias separadas, e não simplesmente o oposto uma da outra. No catabolismo, um processo oxidativo, há liberação de energia enquanto no anabolismo, um processo redutivo, ocorre necessidade de energia.
Reações de Óxido-Reduções Biológicas
Reações de oxirredução são aquelas em que os elétrons são transferidos de um doador a um aceptor. A oxidação é a perda de elétrons e a redução é o ganho de elétrons. A substância que perde elétrons, ou seja, a que é oxidada, é chamada de agente redutor. A substância que ganha elétrons, ou seja, a que é reduzida é chamada agente oxidante. Tanto os agentes oxidantes como os redutores são necessários para a transferência de elétrons ocorrer. Os nutrientes, ao serem oxidados, perdem prótons e elétrons (H+ + e-) e têm seus átomos de carbono convertidos a CO2. Os prótons e elétrons são recebidos por coenzimas na forma oxidada, que passam assim à forma reduzida. Várias reações de oxidação biológicas são acompanhadas da transferência de um próton (H+). A meia reação de oxidação é escrita como uma reação reversível porque a ocorrência da oxidação ou da redução depende dos outros reagentes presentes. Um exemplo de uma meia reação de oxidação é a conversão de NADH, a forma reduzida de Nicotinamida Adenina Dinucleotídeo, para a forma oxidada, NAD+. Essa coenzima é importante em várias reações.

92
Outro importante aceptor de elétrons é o FAD (Flavina Adenina Dinucleotídeo), que é a forma oxidada do FADH2.
Oxidação de Nutrientes e Produção de Energia
A oxidação dos nutrientes para fornecer energia a um organismo não acontece sem a redução de algum aceptor de elétrons. O principal aceptor de elétrons na oxidação aeróbia é o oxigênio. A redução de metabólitos desempenha um importante papel nos processos anabólicos dos organismos

93
vivos. As biomoléculas importantes são sintetizadas nos organismos por várias reações em que um metabólito é reduzido enquanto a forma reduzida de uma coenzima é oxidada.
CARBOIDRATOSLIPÍDIOS
PROTEÍNAS
CO2 (H++ e-) COENZIMAS ATP+H20 (oxidadas)
COENZIMAS (H++ e-) O2 + ADP + Pi (reduzidas)
A reoxidação das coenzimas é obtida pela transferência dos (H++ e-) para o oxigênio molecular, que é então convertido à H2O. A energia derivada desta oxidação é utilizada para sintetizar um composto rico em energia, a adenosina trifosfato (ATP) a partir da adenosina difosfato (ADP) e fosfato inorgânico (HPO4
2- a pH 7,4 – representado por Pi). É a energia química do ATP que será diretamente usada para promover os processos biológicos que consomem energia. Portanto, para que a energia derivada da oxidação dos alimentos possa ser aproveitada pelas células, ela deve estar sob a forma de ATP. O aproveitamento da energia do ATP é feito associando a retirada de seu grupo fosfato terminal aos processos que requerem energia. Desta forma, a energia química armazenada no ATP pode ser utilizada em processos químicos (biossínteses), mecânicos (contração muscular), elétricos (condução de estímulo nervoso), osmóticos (transporte ativo atraves de membranas) ou luminosos (bioluminescência).
A retirada do grupo fosfato terminal do ATP não constitui uma hidrólise simples. A reação de hidrólise tem um valor de ΔGº’ negativo, mostrando ser termodinamicamente viável.
ATP + H2O ADP + Pi + H+ ΔGº’ = -31KJ.mol-1
A velocidade desta reação é convenientemente baixa. Se fosse diferente, a ação das enzimas que catalisam a hidrólise de ATP (sob controle celular) hidrolisaria todo o ATP formado, inviabilizando a vida celular. Além disto, a energia liberada pela hidrólise seria dissipada na forma de calor, uma forma de energia que não pode ser aproveitada pelas células.
Exemplos para entender a utilização do ATP como “doador” de energia:

94
Glicose + Pi Glicose 6-Fosfato + H2O ΔGº’= + 12 KJ/mol
ΔGº’ > 0, logo a reação é inviável.
As células contornam isto, fazendo:
Glicose + ATP Glicose 6-Fosfato + ADP ΔGº’= - 17 KJ/mol
ΔGº’ < 0, logo a reação é viável.
Outro exemplo comum nas reações metabólicas são as sínteses. Vamos supor que a reação de condensação de A e B tenha um ΔGº’ desfavorável:
A + B → A – B ΔGº’ > 0
Este problema pode ser contornado pela utilização de ATP. Neste caso, um caminho possível será a transferência do grupo fosfato terminal do ATP para o composto A, e a reação será termodinamicamente viável:
ADP + Pi
OXIDAÇÃO DE NUTRIENTE PROCESSOS QUE REQUEREM ENERGIA
ATP
A + ATP A – FOSFATO + ADP ΔGº’ < 0
O composto A fosforilado pode reagir com B, em uma reação também termodinamicante viável, liberando o grupo fosfato.
A – FOSFATO + B A – B + FOSFATO ΔGº’ < 0
Da soma das duas reações vem:
A + B + ATP A – B + ADP + FOSFATO ΔGº’ < 0
Fontes Principais de Fosfato Participantes da Conservação ou Captação de Energia
1. A primeira fonte é a Fosforilação Oxidativa. É a fonte de fosfato quantitativamente mais importante, dos organismos aeróbicos. A energia livre, para manter este processo funcionando, provém da oxidação na cadeia respiratória utilizando O2 molecular dentro da mitocôndria.

95
2. Na Glicólise, a formação líquida de dois fosfatos de alta energia resulta da formação de lactato a partir de uma molécula de glicose, gerados em duas reações catalisadas pela fosfoglicerato-quinase e piruvato-quinase, respectivamente.
3. No Ciclo do Ácido Cítrico, um fosfato de alta energia é produzido, na passagem catalisada pela succinil-tioquinase.
O ATP permite que o acoplamento de reações termodinamicamente desfavoráveis tornem-se favoráveis. Um exemplo é a fosforilação da glicose a glicose – 6 – fosfato, a primeira da via glicolítica, é altamente endergônica e não poderia ocorrer isoladamente em condições fisiológicas.
(1) Glicose + Pi Glicose – 6 – fosfato + H2O (ΔGº’ = + 13,8 kj/mol)
Para que essa reação aconteça ela deve estar acoplada a uma outra que seja mais exergônica do que a fosforilação da glicose-endergônica. Tal reação é a hidrólise do fosfato terminal do ATP.
(2) ATP ADP + Pi (ΔGº’ = - 30,5 kj/mol)
Acoplando-se as reações 1 e 2, tem-se uma reação global, catalisada pela hexoquinase, altamente exergônica, e em condições fisiológicas longe do equilíbrio, assim irreversível.
Glicose + ATP HEXOQUINASE Glicose – 6 – fosfato + ADP (ΔGº’ = - 16,7 kj/mol).
Ciclo de Krebs
No metabolismo aeróbio, os nutrientes são oxidados a dióxido de carbono e água. Os organismos podem obter muito mais energia a partir dos nutrientes por meio da oxidação aeróbia do que pela oxidação anaeróbia. A glicólise produz apenas duas moléculas de ATP para cada molécula de glicose metabolizada. Na oxidação aeróbia completa de cada molécula de glicose até dióxido de carbono e água podem ser produzidas 30 a 32 moléculas de ATP. Três processos atuam no metabolismo aeróbio: o ciclo do ácido cítrico, aqui discutido, e o transporte de elétrons e a fosforilação oxidativa.
O ciclo do ácido cítrico é anfibólico, pois atua tanto no catabolismo como no anabolismo. O ciclo do ácido cítrico ocorre em local da célula diferente de onde a glicólise ocorre. Enquanto a glicólise ocorre no citosol, o ciclo do ácido cítrico ocorre na mitocôndria e a maioria das enzimas que dele participam está presente na matriz mitocondrial.

96
camilalemos.comPortal do conhecimento relacionado à saúde.
Em condições aeróbias, a oxidação do piruvato produzido pela glicólise prossegue com a formação de dióxido de carbono e água como produtos finais. O piruvato proveniente da glicose é oxidado a uma molécula de dióxido de carbono e um grupo acetila que se liga a coenzima A. A acetil-CoA entra no ciclo do ácido cítrico. Além da glicose, vários aminoácidos produzem piruvato e, portanto, acetil-CoA, ao serem degradados. Outros aminoácidos e os ácidos graxos também produzem acetil-CoA sem a formação intermediária de piruvato. A acetil-CoA constitui, portanto, o ponto de convergência do metabolismo degradativo de carboidratos, aminoácidos e ácidos graxos. Completando o catabolismo destes compostos, a acetil-CoA, qualquer que seja sua proveniência será totalmente oxidada a CO2 pelo Ciclo de Krebs, com a produção de coenzimas reduzidas. Paralelamente a esta oxidação, o ciclo de Krebs produz um grande número de compostos utilizados como precursores para biossínteses.

97
Função Anabólica do Ciclo de Krebs
A redução de coenzimas não é a única função do ciclo de Krebs. Os compostos intermediários do ciclo de Krebs podem ser utilizados como precursores em vias biossintéticas. Oxaloacetato e α-cetoglutarato formam aspartato e glutamato, respectivamente. Succinil-CoA é precursora do grupo heme. A eventual retirada desses intermediários pode ser compensada por reações que permitem o seu nível. Entre essas reações, chamadas reações anapleróticas (reações de preenchimento), a mais importante é a que leva à formação de oxaloacetato a partir do piruvato, catalisada pela piruvato carboxilase:
COO- COO-
C=O + CO2 + H2O + ATP C=O + ADP + Pi + 2H+
CH3 CH2
COO-
Piruvato Oxaloacetato
HIPERTEXTO
No Ciclo de Krebs, o oxaloacetato tem um papel até certo ponto catalítico: como não é efetivamente consumido pelas reações do ciclo, já que é reposto pela última reação, teoricamente com apenas uma molécula de oxaloacetato poder-se-ia oxidar uma quantidade qualquer de acetil-CoA. Entretanto, a velocidade com que esta oxidação ocorreria seria muito baixa, uma vez que, após a condensação de acetil-CoA com oxaloacetato, iniciando o ciclo, novas moléculas de acetil-CoA só poderiam ser oxidadas quando, ao final das reações do ciclo, o oxaloacetato fosse regenerado. O ajuste da velocidade de consumo de acetil-CoA pelo Ciclo de Krebs à sua própria concentração é feito por intervenção da reação catalisada pela piruvato carboxilase. Esta enzima é fortemente ativada pela própria acetil-CoA. Desta forma, quando, por exemplo, a glicólise é intensa e grande quantidade de piruvato é transformada em acetil-CoA, o acúmulo desta coenzima ativa a piruvato carboxilase e o piruvato passa a originar oxaloacetato. Com concentrações altas de oxaloacetato e acetil-CoA, a citrato sintase, que dá início ao ciclo, pode funcionar a velocidades altas.
CONTEXTUALIZANDO
A energia livre de Gibbs, ∆G, é provavelmente a maneira mais conveniente para se medir as variações de energia nos sistemas vivos, uma vez que mede

98
a energia disponível para realizar um trabalho à temperatura e pressão constantes, o que descreve o estado vivo. Mesmo os animais de sangue frio estão a uma temperatura e pressão constantes em um dado tempo; quaisquer mudanças da temperatura e da pressão são suficientemente lentas, de modo a não afetar as medidas de ∆G.
Espontaneidade e Reversibilidade: Espontaneidade significa que uma dada reação pode ocorrer sem adição de energia como, por exemplo, a água de uma represa no topo de uma montanha, que possui energia potencial para descer mas que não o fará a menos que se abra a barragem. Como a água sempre corre montanha abaixo, essa é a direção em que a variação de energia é negativa (-∆G); no entanto bombear a água montanha acima não é um processo espontâneo (requer energia), e tem um valor positivo para a variação de energia livre (+∆G). Se a variação de energia livre for apenas de 1kcal/mol (± 4Kj/mol) em ambas as direções, então essa reação é considerada reversível, podendo ocorrer em ambas as direções. Adicionando-se reagentes ou retirando produtos, a reação irá para a direita; se forem retirados os reagentes ou adicionados produtos a reação se deslocará para a esquerda. Esse aspecto é de muita importância para grande número de vias metabólicas; muitas reações são completamente reversíveis. Isso significa que as mesmas enzimas podem ser usadas tanto na via que leva à quebra da substância, como na via que forma a substância (Campbell & Farrel, 2008).
ATIVIDADES DE AVALIAÇÃO
1. Definir Bioenergética
2. Quem controla a velocidade de liberação de energia?
4. Como os processos vitais (reações de síntese, contração muscular, condução do impulso nervoso, transporte ativo) obtêm energia?
5. O que é um processo endergônico? E um processo exergônico?
6. Definir catabolismo e anabolismo.
7. Definir metabolismo.
8. Indique um método alternativo de acoplamento entre um processo exergônico e outro endergônico.
9. Quem é o principal intermediário de alta energia, ou melhor, o composto transportador, na célula viva?
10. Qual é a função do ATP no processo de fosforilação?
11. Como age um ciclo ATP/ADP?

99
12. Quais são as três fontes principais de fosfato de alta energia que participam da conservação ou captação de energia?
13. De onde provém a energia livre que mantém a fosforilação oxidativa funcionando?
14. Sendo a fosforilação da glicose a glicose 6-fosfato, altamente endergônica, como pode ocorrer na via glicolítica? Explique.
16. O que ocorre com os nutrientes ao serem oxidados? Explique todas as etapas até chegar ao ATP + H2O.
18. Como a energia derivada da oxidação dos alimentos pode ser aproveitada pelas células?
19. Desenhe o Ciclo de Krebs.
20. Fale sobre a função anabólica do Ciclo de Krebs.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. Nos Capítulos 15 e 16 do vol. 3 são apresentados tópicos relativos ao assunto e todos comentados de forma muito didática.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade II do livro, nos Capítulos 6 a 9 são apresentados os tópicos relativos ao assunto abordado. O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Murray, R. K.; Granner, D. K.; Mayes, P. A.; Rodwell, V. W. Harper: Bioquímica. 8ª ed. São Paulo: Atheneu, 1998. Os Capítulos 12 e 13 ,da Seção II, abordam tópicos detalhados sobreo assunto.
LEITURA, VÍDEOS E SITES RELACIONADOS:
Síndrome de leigh – anatapat-unicamp
Adams RD, Victor M, Ropper AH. Principles of Neurology, 6th Ed. McGraw-Hill, New York, 1997.
Carpenter S, Karpati G. Pathology of Skeletal Muscle. 2nd. Ed. Oxford University Press, 2001.
Johns DR, Fadic RN. Genetic Mitochondrial Disorders. in Martin JB (ed) Molecular Neurology. Scientific American, New York, 1998.

100
CAPÍTULO 8: CADEIA DE TRANSPORTE DE ELÉTRONSOxidação de Coenzimas e Síntese de ATP
OBJETIVOS:
Compreender o metabolismo aeróbico.
Observar a síntese de ATP e a geração de energia celular.
Verificar a produção de água no final da cadeia transportadora de elétrons.
Localizar os intermediários da cadeia transportadora de elétrons, na mitocôndria e suas funções particulares.
Compreender a função das coenzimas do sistema mitocondrial.
Através do metabolismo aeróbio, um organismo pode extrair energia de modo eficiente a partir dos nutrientes. Moléculas ricas em energia, como a glicose, são metabolizadas por uma série de reações de oxidação, levando por fim à produção de CO2 e água. Os processos de oxidação da glicose, de vários aminoácidos e de ácidos graxos levam à produção de Acetil-CoA, que, no ciclo de Krebs, é totalmente oxidada a CO2. Os intermediários metabólicos dessas reações cedem elétrons a coenzimas específicas NAD+ (Nicotinamida Adenina Dinucleotídeo) e FAD (Flavina Adenina Dinucleotídeo) formando as coenzimas reduzidas ricas em energia, NADH e FADH2. O ciclo de Krebs constitui o estágio final e máximo de oxidação dos átomos de carbono que compõem carboidratos, proteínas e lipídeos. A oxidação destes compostos é acompanhada da redução de grande quantidade das coenzimas NAD+ e FAD. Observe o nº de moles destas coenzimas reduzidas durante a oxidação de um mol de glicose.
Reação Moles de NADH Moles de FADH2
Glicólise
Gliceraldeído 3-fosfato→ 3-Fosfoglicerato 2
-
Piruvato → Acetil-CoA 2 -
Ciclo de Krebs
Isocitrato → α-Cetoglutarato
α-Cetoglutarato → Succinil- CoA
Malato → Oxaloacetato
Succinato → Fumarato
2
2
2
-
-
-
-
2
Total 10 2
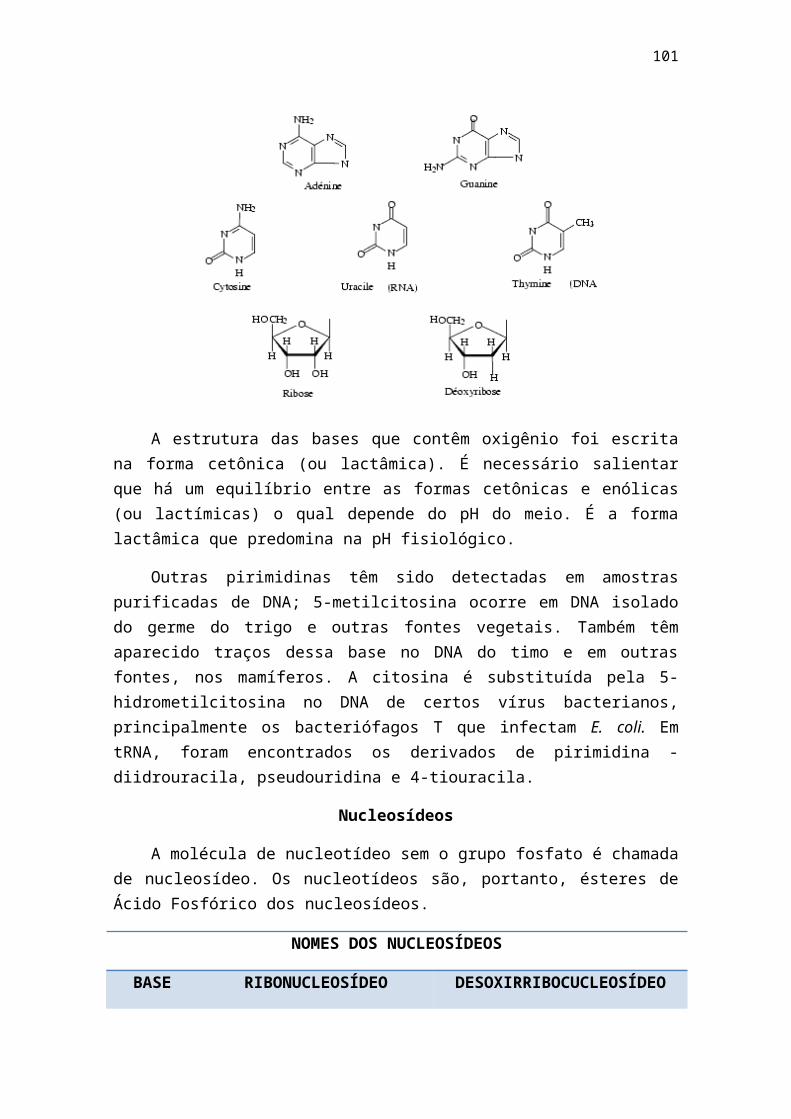
101
Do ponto de vista energético, verifica-se que da energia total disponível inicialmente na molécula de glicose, uma fração muito pequena levou à produção de ATP; a maior parte da energia inicial foi conservada nas coenzimas reduzidas. O mesmo ocorre na oxidação de aminoácidos e lipídios. As coenzimas reduzidas devem ser reoxidadas por duas razões:
1. Para que voltando à forma oxidada, possam participar outra vez das vias de degradação dos nutrientes.
2. Porque é a partir da oxidação destas coenzimas que a energia nelas é conservada e pode ser aproveitada pelas células, para sintetizar ATP.
As coenzimas reduzidas podem doar, cada uma, um par de elétrons a um conjunto especializado de transportadores de elétrons chamados “cadeia de transporte de elétrons”. A energia liberada pela oxidação de nutrientes é usada pelos organismos na forma de energia química do ATP. Enquanto os elétrons fluem através da cadeia transportadora de elétrons, eles perdem muito de sua energia livre. Parte dessa energia pode ser captada e armazenada para a produção de ATP a partir de ADP e fosfato inorgânico (Pi) e a esse processo dá-se o nome de “fosforilação oxidativa”. O restante da energia livre que não é captada para a síntese de ATP serve para impulsionar outras reações.
A produção de ATP por fosforilação oxidativa (processo endergônico) é separada do transporte de elétrons para o oxigênio (um processo exergônico), no entanto as da cadeia transportadora de elétrons estão ligadas entre si e associadas à síntese de ATP pela fosforilação de ADP. A atividade da cadeia de transporte de elétrons leva ao bombeamento de prótons (H+) pela membrana mitocondrial interna, criando um gradiente de pH, também conhecido como gradiente de prótons. A solução para as células aproveitarem energia para a síntese de ATP é exatamente transformar a energia contida nas coenzimas reduzidas em um gradiente de prótons e utilizá-lo para promover a síntese de ATP. A produção do gradiente de prótons é conseguida pela transferência dos elétrons das coenzimas para o oxigênio, através de passagens intermediárias por vários compostos que constituem uma cadeia de transporte de elétrons.
Nos organismos aeróbios, a oxidação de coenzimas é feita por transferência de seus elétrons para o oxigênio. Recebendo elétrons, o oxigênio liga-se a prótons do meio formando água. Este processo libera grande quantidade de energia, em virtude da diferença de potenciais de óxido-redução entre a coenzima e o oxigênio. A energia liberada na oxidação de um mol de NADH permite a síntese de alguns moles de ATP. Se a transferência de elétrons das coenzimas reduzidas fosse feita diretamente para o oxigênio, toda a energia do processo seria liberada como calor, portanto, inutilizável pelas células para promover os processos que requerem energia.

102
Lembrete:
A cadeia de transporte de elétrons está presente na membrana mitocondrial interna. Os compostos que compõem a cadeia de transporte de elétrons são organizados de acordo com seus potenciais de óxido-redução. Assim, os elétrons partem da coenzima reduzida, que tem potencial de óxido-redução menor que os componentes da cadeia de transporte de elétrons, e percorrem uma sequência de transportadores com potenciais de óxido-redução crescentes, até atingirem o oxigênio, que tem o maior potencial de óxido-redução. As transferências de elétrons entre estes compostos são sempre acompanhadas de queda de energia livre. O transporte de elétrons é facilitado pelo fato de tais compostos estarem organizados em membranas, com posições definidas, de modo a situar cada componente exatamente entre aquele que lhe fornecerá elétrons e aquele ao qual seus elétrons serão doados.
Enquanto se processam as passagens de elétrons, forma-se um gradiente de prótons, já que se estabelece uma concentração de prótons diferente de cada lado da membrana onde ocorre o transporte de elétrons. A síntese de ATP é possível porque aproveita a energia potencial contida no gradiente de prótons. Esta síntese consiste na fosforilação do ADP (ADP + Pi → ATP) e, como utiliza energia derivada da oxidação das coenzimas, é denominada fosforilação oxidativa. A oxidação das coenzimas reduzidas pela cadeia de transporte de elétrons processa-se na membrana interna da mitocôndria, da qual fazem parte os componentes da cadeia. Estes componentes agrupam-se em quatro complexos: I, II, III e IV.
WWW.BLOGCONSTRUINDOOCONHECIMENTO.ZIP.NET
A ÚNICA FORMA DE ENERGIA UTILIZÁVEL PELAS CÉLULAS EM TAIS PROCESSOS É A ENERGIA QUÍMICA PRESENTE NO ATP.

103
UBIQUINONA
Composição dos complexos da cadeia de transporte de elétrons
Componentes transportadores de elétrons
Número aproximado de polipeptídios
Complexo I
(NADH-CoQ redutase)
FMN
Centros Fe-S
26
Complexo II
(Succinato-CoQ redutase)
FAD
Centros Fe-S
Citocromo b
5
Complexo III
(Coenzima Q – citocromo c redutase)
Citocromos b e c1
Centros Fe-S
10
Complexo IV
(Citocromo c oxidase)
Citocromos a e a3
Íons de cobre
6-13
Embora não faça parte de Complexos dois constituintes da cadeia de transporte de elétrons têm papéis muito importantes. A coenzima Q (CoQ) conecta os Complexos I e II ao Complexo III, e o citocromo c conecta o Complexo III ao Complexo IV. Os elétrons que se encontram presentes no NADH são transferidos desta coenzima para o Complexo I, do Complexo I para, a coenzima Q, depois para o Complexo III, citocromo c, Complexo IV e finalmente para o oxigênio. Os elétrons presentes no succinato e em outros substratos têm uma entrada especial na cadeia de transporte de elétrons: são transferidos ao Complexo II e deste para a coenzima Q; então seguem a rota comum: Complexo III, citocromo c, Complexo IV e oxigênio. Estas transferências são possíveis porque todos os compostos presentes nos complexos, mais a coenzima Q e o citocromo c, podem apresentar-se nos estados reduzidos e oxidado: Ao receberem um elétron, reduzem-se; transferindo o elétron oxidam-se e podem receber elétrons novamente.

104
Excetuando-se a coenzima Q, todos os componentes da cadeia de transporte de elétrons são proteínas. A estas estão associados grupos prostéticos, como FAD, FMN e centros Ferro-Enxofre (Fe-S).
COMPLEXO I
O Complexo I (NADH-CoQ redutase) oxida o NADH, transferindo seus elétrons para a coenzima Q. É formado por 26 cadeias polipeptídicas. A estas cadeias estão associados: uma molécula de flavina mononucleotídio (FMN) e 6 ou 7 centros Fe-S. A FMN é um derivado da riboflavina com estrutura semelhante à do FAD e como este, capaz de receber 2 prótons e 2 elétrons passando à forma reduzida FMNH2. O doador de elétrons para a redução de FMN (1ª transferência de elétrons para a cadeia de transporte de elétrons) é o NADH produzido pelo metabolismo.
NADH + H+ + FMN NAD+ + FMNH2
(Complexo I) (Complexo I)
WWW.BLOGCONSTRUINDOOCONHECIMENTO.ZIP.NET
O resultado é a oxidação do NADH e a entrada dos elétrons na membrana interna da mitocôndria, de onde só saem para serem doados ao oxigênio, no final da cadeia. Os centros Fe-S não recebem prótons; são transportadores de elétrons: Fe3+ para Fe2+. Os elétrons do FMNH2 são transferidos para o primeiro centro Fe-S; depois, através de passagens por outros centros Fe-S, deixam o Complexo I, sendo entregues à coenzima Q. Na transferência de elétrons do FMNH2 para os centros Fe-S, os prótons são excluídos, sendo transferidos da membrana interna da mitocôndria para o espaço intermembranas, sendo esta a primeira etapa da formação do gradiente de prótons (da matriz mitocondrial são retirados prótons que vem de NADH + H+; no espaço intermembranas são introduzidos prótons) Ocorre então uma

105
diferença de concentração de prótons apreciável entre o interior e o exterior da mitocôndria.
HIPERTEXTO
O FMN, como o FAD, é sintetizado a partir da vitamina riboflavina. Ela contém a estrutura em anel flavina receptora de elétron, mas não a porção monofosfato de adenosina (AMP) do FAD. A deficiência grave de riboflavina diminui a habilidade da mitocôndria de produzir ATP a partir da fosforilação oxidativa devido à carência de FMN nos carreadores de elétrons.
COMPLEXO II
O Complexo II (Succinato-CoQ Redutase) oxida o succinato, transferindo seus elétrons também para a coenzima Q. Ele é a segunda porta de entrada de elétrons na cadeia de transporte de elétrons, em direção ao oxigênio. A enzima Succinato – Desidrogenase tem FAD como grupo prostético e catalisa a oxidação de succinato à fumarato: os e- e os prótons do succinato são transferidos para o FAD, que se reduz a FADH2. Os prótons presentes no FADH2 são devolvidos à matriz mitocondrial. O complexo II não contribui para a formação do gradiente de prótons. No complexo II, os elétrons provenientes do succinato são transferidos ao FAD, grupo prostético da succinato desidrogenase; a seguir são captados por centros Fe-S e pelo citocromo b560, passando, então para a coenzima Q.
A coenzima Q é o ponto de convergência de elétrons provenientes de NADH (Complexo I), succinato (Complexo II), glicerol 3-fosfato e acil-CoA. Coenzima Q ou Ubiquinona (CoQ) é uma quinona com uma longa cadeia lateral composta de unidades isoprênicas. A forma mais encontrada nos mamíferos apresenta 10 unidades. As características hidrofóbicas da CoQ permitem sua mobilidade na fase lipídica da membrana, ao contrário dos outros

106
componentes da cadeia de transporte de elétrons, que têm posições relativamente fixas na membrana mitocondrial, com exceção do citocromo c.
Citocromos de vários tipos participam da cadeia de transporte de elétrons. São proteínas transportadoras de elétrons que contêm heme como grupo prostético. O íon de ferro é o responsável pela capacidade de transferência de elétrons desta proteína: O íon pode alterar entre Fe2+ e Fe3+. Os citocromos são classificados em a, b e c, segundo o espectro de absorção que apresentam. Os três tipos estão representados na cadeia de transporte de elétrons.
Cada citocromo é constituído por uma cadeia polipeptídica com uma sequência de aminoácidos que lhe é própria. Nos citocromos dos tipos b e c, o grupo heme é idêntico ao da hemoglobina quanto aos radicais substituintes. No tipo a, aparece um grupo heme modificado, com um grupo isoprênico e um grupo formila em lugar de grupos vinila e metila, respectivamente. Os citocromos diferem também quanto à forma com que o grupo heme está ligado à cadeia proteica. Nos tipos a e b, a ligação à proteína é não-covalente e, no tipo c, é covalente (tioéter), formada com resíduos de cisteína.
WWW.BLOGCONSTRUINDOOCONHECIMENTO.ZIP.NET
COMPLEXO III
O Complexo III transfere elétrons da coenzima Q para o citocromo c. O Complexo III (Coenzima Q – citocromo c redutase) é constituído por 2 citocromos b (b562 e b566, os índices indicam o pico máximo de absorção e caracteriza cada subtipo de citocromo), por um centro Fe-S e pelo citocromo c1. As transferências de elétrons da coenzima Q para os componentes do complexo III, e deste para o citocromo c, são acompanhadas de movimento de prótons: os citocromos e o centro Fe-S recebem apenas elétrons, e os prótons da CoQ reduzida (CoQH2) são liberados no espaço intermembranas. O Complexo III promove a oxidação da coenzima Q e a redução do citocromo c, contribuindo para a criação do gradiente de prótons. O citocromo c é uma

107
proteína pequena localizada na face externa da membrana interna da mitocôndria. Seu tamanho e mobilidade fazem com que cumpra sua função na cadeia de transporte de elétrons: conectar o Complexo III, do qual recebe elétrons, ao Complexo IV ao qual doa elétrons.
WWW.BLOGCONSTRUINDOOCONHECIMENTO.ZIP.NET
COMPLEXO IV
O Complexo IV (citocromo c oxidase) transfere elétrons para o oxigênio. Ele contém 2 citocromos do tipo a (a e a3) e dois íons de cobre, cada qual associado a um dois citocromos. Os íons de cobre alternando entre Cu2+ e Cu1+
fazem parte do transporte de elétrons. O Complexo IV é responsável pela doação de 4 elétrons para a molécula de O2 que, ligando-se a prótons do meio, converte-se em 2 moléculas de H2O. A retirada de prótons da matriz mitocondrial contribui para o estabelecimento do gradiente de prótons. Além disto, o Complexo IV contribui para a produção do gradiente lançando prótons da matriz para o espaço intermembranas. A utilização de O2 pelo complexo IV é de cerca de 95% do O2 consumido pelo homem; a produção de H2O neste processo chega a cerca de 300 ml diários (água metabólica). Essa água é fundamental para a sobrevivência do indivíduo, como no caso dos animais que hibernam ou de camelos, que passam longos períodos sem ingerir água. O caminho exato percorrido pelos elétrons no Complexo IV não é ainda conhecido. Acredita-se que os elétrons do citocromo c são recebidos pelo complexo formado entre o citocromo a e um íon de cobre (CuA). Depois são transferidos para o citocromo a3 ligado ao outro íon de cobre (CuB), e, finalmente, para o O2, que se combina com prótons da matriz, reduzindo-se a água.
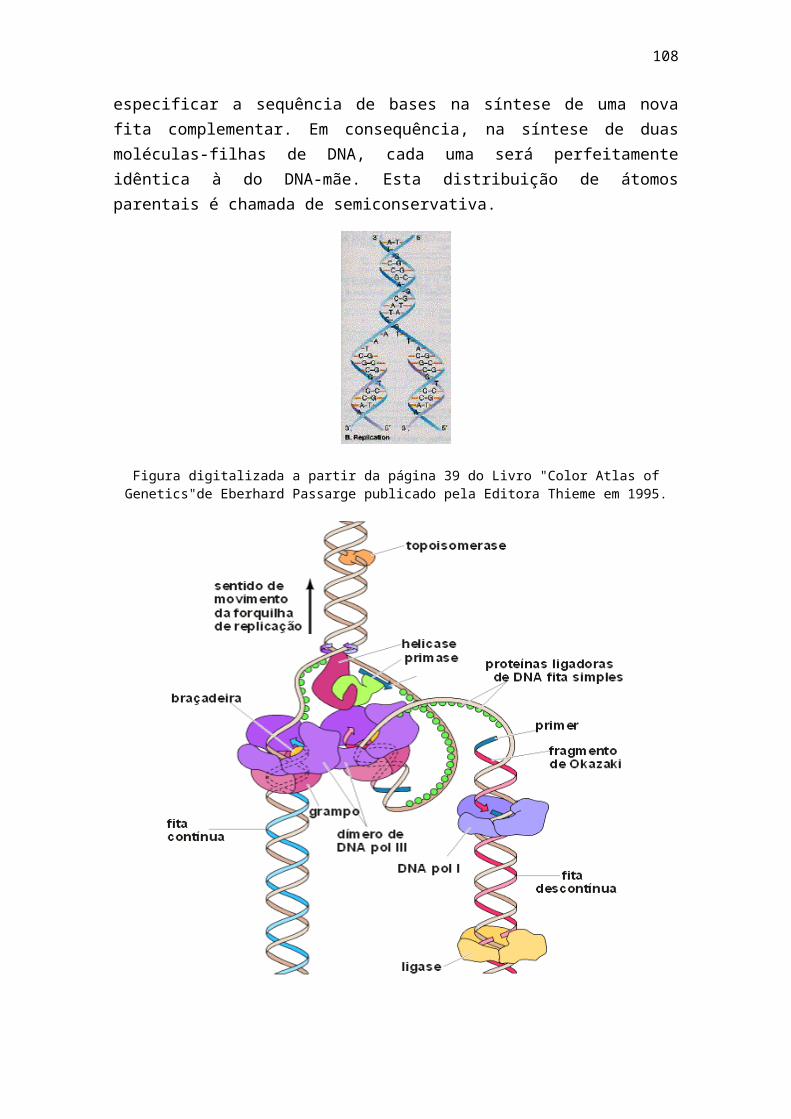
108
WWW.BLOGCONSTRUINDOOCONHECIMENTO.ZIP.NET
CONTEXTUALIZANDO
Esporte e Metabolismo
Atletas treinados, especialmente os de elite, são mais conscientes do resultado do metabolismo aeróbio e anaeróbio do que as pessoas que não são atletas. As características genéticas e o treinamento são importantes no sucesso do atleta, porém uma compreensão aguda da fisiologia e do metabolismo é igualmente importante. Para planejar a nutrição adequada para o desempenho, um atleta sério deve compreender a natureza do metabolismo e como ele se relaciona ao esporte escolhido. A musculatura, ao trabalhar, tem quatro diferentes fontes de energia disponíveis após um período de repouso:
1. A creatina fosfato, que reage diretamente com o ADP na fosforilação em nível de substrato para produzir ATP.
2. A glicose derivada dos depósitos musculares de glicogênio, consumida inicialmente pelo metabolismo anaeróbio.3. A glicose do fígado, derivada tanto de depósitos de glicogênio quanto da gliconeogênese a partir do ácido láctico produzido no músculo (ciclo de Cori), novamente consumida inicialmente pelo metabolismo anaeróbio.
4. Metabolismo aeróbio nas mitocôndrias musculares.Inicialmente as quatro fontes de energia estão disponíveis para o
músculo. Quando a creatina fosfato esgota-se, restam as outras fontes. Quando o glicogênio muscular termina, o estímulo anaeróbio fornecido por ele diminui proporcionalmente, e, quando o glicogênio hepático acaba, resta apenas o metabolismo aeróbio a dióxido de carbono e água. Atletas bem condicionados e treinados apresentam um maior número de mitocôndrias em suas células musculares. Um grande atleta americano, Greg LeMond, foi vítima

109
de um tiro de chumbo grosso nas costas durante uma caçada. Ele se recuperou, mas apesar de ganhar mais prêmios em competições, nunca se sentiu realmente bem novamente. Começou a ganhar peso e a não responder ao treinamento. Depois de alguns exames descobriu que tinha uma rara condição chamada miopatia mitocondrial. Quando ele treinava intensamente, suas mitocôndrias começavam a desaparecer. Ele era essencialmente um atleta aeróbio sem a capacidade de processar combustível aerobiamente. Daí se pode avaliar a importância da cadeia transportadora de elétrons e das mitocôndrias para o atleta (Campbell & Farrel, 2008).
ATIVIDADES DE AVALIAÇÃO1. Citar os compostos que fazem parte da cadeia de transporte de elétrons e
caracterizá-los quimicamente.2. Esquematizar a sequência dos compostos da cadeia de transporte de
elétrons, indicando os transportadores de elétrons e os transportadores de prótons e elétrons.
3. Indicar a localização celular da cadeia de transporte de elétrons.4. Indicar o número de ATP sintetizados para cada NADH e FADH2 oxidados.5. Citar exemplos de processos biológicos que utilizam ATP.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. No Capítulo 20 do vol. 3, o assunto Cadeia de Transporte de Elétrons é abordado de forma muito didática.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade II do livro, no Capítulo 6, são apresentados tópicos relativos ao assunto abordado. O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª ed. 1999. Rio de Janeiro: Editora Guanabara Koogan S. A. O Capítulo 11 da Parte 3 desse livro explana de forma bastante compreensiva o assunto Cadeia de Transporte de Elétrons.
Smith, C.; Marks, A. D.; Lieberman, M. Bioquímica Médica Básica de Marks – Uma abordagem clínica. 2ª ed. Porto Alegre: Artmed, 2007. O Capítulo 21 do livro aborda o assunto com muito detalhamento e de forma didática.
LEITURA, VÍDEOS E SITES RECOMENDADOS:
www.biotopics.co.uk/a2/electrontransportchain.html
vcell.ndsu.edu/animations/etc/movie.htm

110
CAPÍTULO 9: FOSFORILAÇÃO OXIDATIVA E LANÇADEIRAS DE ELÉTRONS
OBJETIVOS:
Conhecer as lançadeiras de elétrons e o gradiente de protóns gerados nesses processos.
Diferenciar e definir desacopladores e inibidores da fosforilação oxidativa.
Descrever a teoria de Mitchell, para o acoplamento de elétrons durante a fosforilação oxidativa.
A transferência de elétrons ao longo da cadeia de transporte de elétrons é energeticamente favorecida, já que o NADH é um forte doador de elétrons, e o oxigênio molecular é um grande aceptor de elétrons. O fluxo de elétrons do NADH para o oxigênio, porém, não resulta diretamente na síntese de ATP. Os componentes da cadeia de transporte de elétrons apresentam-se organizados em ordem crescente de potenciais de óxido-redução, desde as coenzimas reduzidas até ao Oxigênio. As transferências de elétrons se processam com liberação de energia, aproveitada para sintetizar ATP. Uma parte da energia liberada pelas reações de oxidação na cadeia transportadora de elétrons é usada para acionar a fosforilação do ADP. As reações de oxidação que liberam energia originam o bombeamento de prótons e, consequentemente, o gradiente de pH pela membrana mitocondrial interna. Além do gradiente de pH, há uma diferença de voltagem pela membrana, gerada pelas diferenças de concentração de íons nos lados interno e externo. A energia do potencial eletroquímico (queda de voltagem) pela membrana é convertida em energia química armazenada pelo ATP pelo processo de acoplamento. Como ocorre a conexão entre o Transporte de Elétrons e a Fosforilação é uma pergunta que exige resposta.
É necessário um fator de acoplamento para ligar a oxidação e a fosforilação. Esse acoplamento é explicado pela teoria quimiosmótica conhecida também como hipótese de Mitchell. O transporte de elétrons está acoplado à fosforilação do ADP pelo bombeamento de prótons através da membrana mitocondrial interna. Esses prótons são bombeados da matriz para o espaço intermembranas. Esse processo cria, através da membrana mitocondrial interna, um gradiente elétrico (com mais cargas positivas no lado externo da membrana do que no lado interno) e um gradiente de pH (o meio externo da membrana está em pH mais baixo do que o meio interno). A energia gerada por esse gradiente de prótons é suficiente para impulsionar a síntese de ATP. Assim, o gradiente de prótons funciona como o intermediário comum que acopla a oxidação à fosforilação.
Um complexo proteico oligomérico, conhecido como ATP sintase, separado dos complexos de transporte de elétrons, exerce a função de

111
sintetizar ATP. A ATP sintase compreende dois componentes, cada um, constituído por várias cadeias polipeptídicas.
ATP – Sintase
Fica nas microesferas que estão na face interna da membrana interna da mitocôndria. As microesferas são ligadas à membrana por pequenas hastes. A ATP-Sintase compreende dois componentes:
1. Porção esférica chamada: Fator de acoplamento 1 (F1) (corresponde às microesferas que contêm os sítios de síntese de ATP).
2. A porção 2 fica embebida na membrana interna constituindo um canal através do qual os prótons retornam à matriz mitocondrial. É chamada Fo
porque contém o sítio de ligação para oligomicina (um antibiótico que inibe a síntese de ATP, pois se liga ao Fo da ATP sintase, que se torna impermeável à proteína).
Em conformidade com o que propõe a teoria quimiosmótica após os prótons serem transferidos para o lado citosólico da membrana mitocondrial interna, eles retornam à matriz mitocondrial. A membrana interna da mitocôndria é impermeável a prótons em toda a sua extensão exceto na ATP sintase. É por esse canal que os prótons atravessam a membrana, de volta à matriz mitocondrial.
As velocidades do transporte de elétrons e da síntese de ATP são reguladas pela concentração de ADP. Porém as necessidades celulares de ATP variam de acordo com o estado fisiológico. Por exemplo: uma fibra muscular pode ter suas necessidades aumentadas de 100 vezes em segundos, quando passa do repouso para o exercício intenso. Para promover o ajuste da produção de ATP ao seu gasto, o transporte de elétrons e a síntese de ATP são acoplados, em outras palavras, só há oxidação de coenzimas se houver síntese de ATP, e vice-versa. Essa regulação da velocidade de oxidação das coenzimas (equivalente à velocidade de consumo de oxigênio) exercida pela concentração de ADP (tem concentração limitante) chama-se controle respiratório. O resultado do controle respiratório é um perfeito ajuste entre a velocidade de produção de coenzimas reduzidas e a velocidade de sua oxidação pela cadeia de transporte de elétrons, com produção de ATP. É esse ajuste que vai regular a produção de energia pela célula.
HIPERTEXTO
O salicilato, quel é um produto da degradação da aspirina nos humanos, é solúvel em lipídeos e tem um próton dissociável. Em altas concentrações, como no envenenamento por salicilato, ele é capaz de desacoplar parcialmente a mitocôndria. O declínio da concentração de ATP na célula e consequente aumento de AMP no citosol estimulam a glicólise. A superestimulação da rota
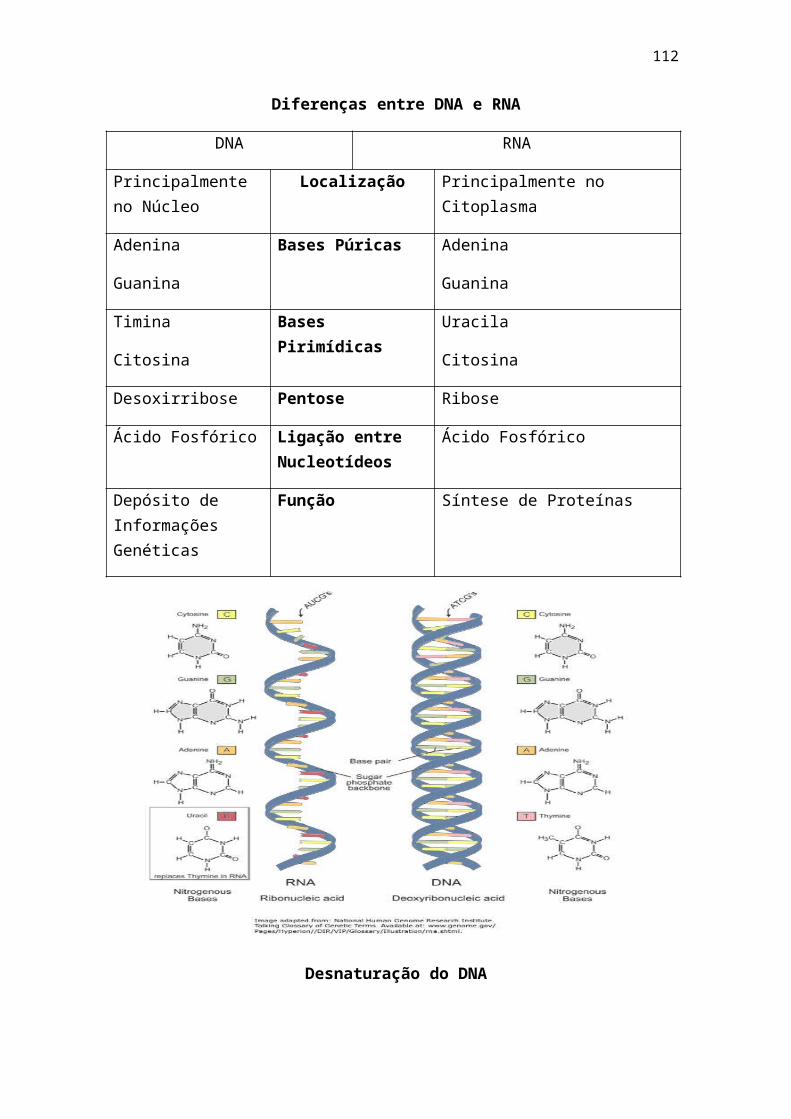
112
glicolítica resulta em níveis aumentados de ácido lático no sangue e acidose metabólica.
Desacopladores
Desacopladores são compostos que inibem a fosforilação do ADP sem afetar o transporte de elétrons. Um modo de ação razoável para os desacopladores pode ser proposto com base na existência de um gradiente de prótons. Um exemplo de desacoplador é o 2,4-dinitrofenol que é um ácido; sua base conjugada, o ânion dinitrofenolato, é o verdadeiro desacoplador, uma vez que pode reagir com prótons no espaço intermembranas, reduzindo, dessa forma, a diferença na concentração de prótons entre os dois lados da membrana mitocondrial interna. Antibióticos como valinomicina e gramicidina A são desacopladores. Eles são ionóforos, e criam um canal pelo qual íons como H+, K+ e Na+ podem atravessar a membrana. O gradiente de prótons então é anulado, levando ao desacoplamento da oxidação e da fosforilação.
Quando os processos de oxidação mitocondrial estão operando normalmente, o transporte de elétrons do NADH ou do FADH2 para o oxigênio resulta na produção de ATP. Quando um desacoplador está presente, o oxigênio ainda é reduzido a H2O, porém não se produz ATP. Se o desacoplador for removido, a síntese de ATP ligada ao transporte de elétrons é reiniciada. Portanto, desacopladores são substâncias capazes de dissociar o transporte de elétrons da fosforilação oxidativa. Quando os dois processos são desacoplados, o transporte de elétrons pode prosseguir, porém a síntese de ATP para. A energia produzida pelo transporte de elétrons é liberada em forma de calor, ao invés de sintetizar ATP. Um exemplo é a aspirina e outros salicilatos que desacoplam a fosforilação oxidativa. Superdosagem dessas substâncias dão como resultado um aumento da temperatura corporal.
Inibidores
Existem drogas capazes de agir de modo específico sobre cada um dos complexos da cadeia de transporte de elétrons, impedindo o prosseguimento da transferência de elétrons. São os inibidores. O resultado disso é a paralisação do transporte de elétrons e das vias metabólicas que dependem da cadeia para a reoxidação de coenzimas. Assim, um transportador reduzido, incapaz de passar adiante seus elétrons, é também incapaz de receber elétrons do transportador antecedente. Dessa forma os componentes da cadeia localizados antes do ponto de atuação da droga estarão reduzidos, e a cadeia torna-se inoperante. Sem o transporte de elétrons não se forma o gradiente de prótons e, consequentemente, não há síntese de ATP. Os inibidores são divididos em:
1. Inibidores característicos da cadeia respiratória.2. Inibidores da fosforilação oxidativa.
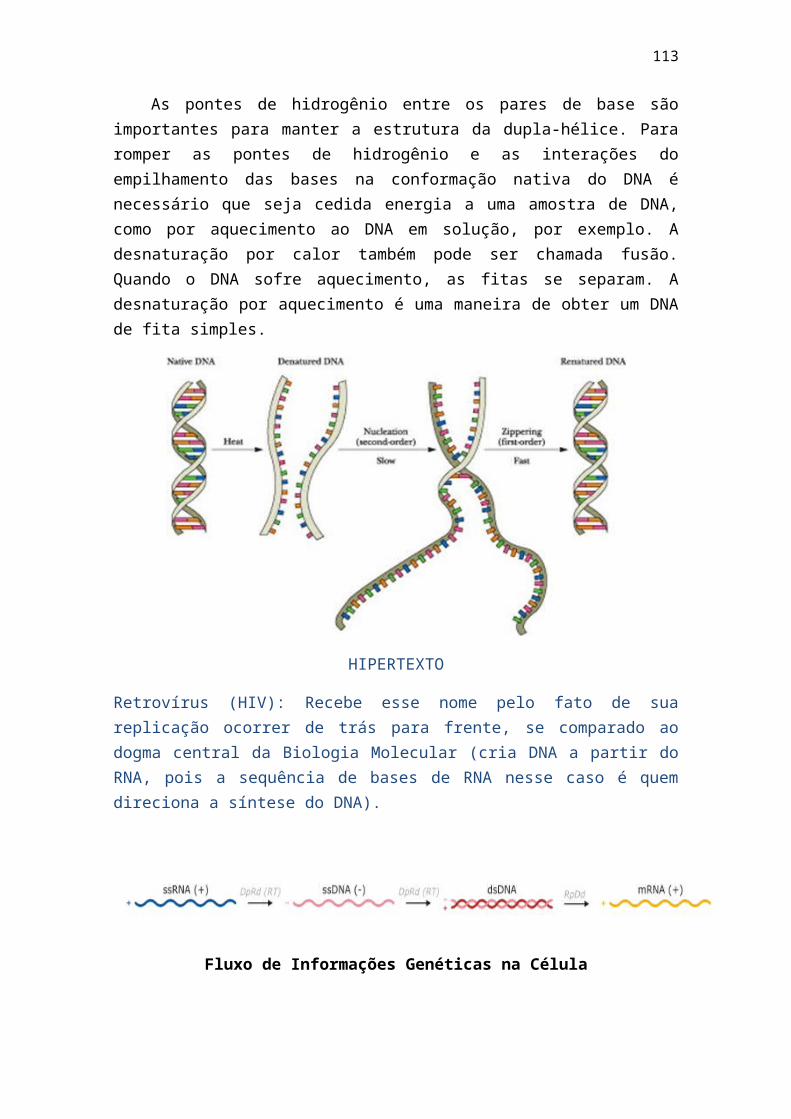
113
Inibidores que paralisam a respiração pelo bloqueio da cadeia respiratória atuam em três locais:
1. Impedindo a oxidação de substratos que se comunicam diretamente com a cadeia respiratória, via uma desidrogenase dependente de NAD, por bloquear a transferência de elétrons da Flavoproteína NADH redutase para a CoQ. Ex: Barbituratos, e Rotenona.
2. Inibindo a cadeia respiratória entre os citocromos b, CoQ e o citocromo c1. Ex.: Antimicina A (antibiótico).
3. Inibindo a transferência de elétrons do citocromo aa3 para o oxigênio (podem paralisar totalmente a respiração). São inibidores muito potentes, como, por exemplo: Azida (N3
-), Monóxido de carbono (CO) e cianeto (CN-).
Inibidores da Cadeia de Transporte de Elétrons e o complexo em que atuam
Inibidores Complexo
Barbituratos (hipnóticos) I
Rotenona (inseticida)
Malonato (análogo do succinato, inibidor competitivo da succinato desidrogenase)
II
Antimicina A III
Cianeto, monóxido de carbono, ácido sulfúrico, azida sódica
IV
Fosforilação no Nível do Substrato
É a síntese de ATP obtida diretamente em reações que fazem parte da glicólise e do ciclo de Krebs e que utilizam como substratos compostos ricos em energia: 1,3 – Bisfosfoglicerato; Fosfoenolpiruvato; Succinil – CoA. A fosforilação no nível do substrato não é afetada por desacopladores. A produção de ATP pela fosforilação no nível do substrato responde por uma pequena fração do total produzido em condições aeróbias e, por ser independente do transporte de elétrons, não é afetada por desacoplado.
Cadeias de Transporte de Elétrons Bacteriano
Nas bactérias, encontram-se cadeias de transporte de elétrons bem mais diversificadas. Nelas, além das coenzimas reduzidas, podem ser fornecedores de elétrons: NH4
+, NO2-, H2S, H2, S e Fe. O aceptor final também pode variar, e

114
além do O2, podem ter esta função: NO3-, NO2
-, SO42-, CO3
2 e substâncias orgânicas, como o fumarato. Quando o aceptor final é diferente do O2, a cadeia é dita anaeróbia e caracteriza a respiração anaeróbia.
Circuitos de Transporte ou Lançadeiras
A membrana interna da mitocôndria é impermeável a NAD+ e NADH e, portanto, a oxidação do NADH citosólico não pode ser feita diretamente pela cadeia de transporte de elétrons. Porém, as coenzimas reduzidas no citosol podem ser indiretamente oxidadas pela cadeia de transporte de elétrons; graças a sistemas designados circuitos ou lançadeiras. Nelas, os elétrons do NADH são transferidos para um composto citosólico, que, reduzido, pode atravessar a membrana interna da mitocôndria. Alternativamente, os elétrons são transferidos para um composto que, pode transferir elétrons para um componente da membrana interna. Por qualquer dos dois processos, o composto que transporta os elétrons é reoxidado; ao doá-lo, retorna ao citossol e pode participar de um novo ciclo. Há dois tipos de lançadeiras: Glicerol – Fosfato e Malato – Aspartato
Lançadeira glicerol fosfato
É um sistema transportador que foi muito estudado no músculo do vôo de insetos. Nesse mecanismo é usada uma enzima dependente de FAD presente na face externa da membrana mitocondrial interna que oxida o glicerol fosfato. O glicerol fosfato é produzido pela redução de diidroxiacetona fosfato; no curso da reação o NADH é oxidado a NAD+. Nessa reação, o agente oxidante (que também é reduzido) é o FAD, e o produto é o FADH2, que passa então os elétrons pela cadeia transportadora de elétrons, levando à produção de 1,5 mol de ATP para cada mol de NADH citosólico. Esse mecanismo também foi observado no músculo e no cérebro de mamíferos.
Lançadeira Malato-Aspartato
Nas células hepáticas, cardíacas e renais de mamíferos o NADH citossólico reduz oxaloacetato, em uma reação catalisada pela malato desidrogenase citossólica. O malato formado entra na mitocôndria, onde é oxidado pela malato desidrogenase mitocondrial, que também utiliza, NAD+
como coenzima. Este processo produz NADH mitocondrial a partir de NADH citossólico, apesar da impermeabilidade da membrana interna da mitocôndria ao NADH. O oxaloacetato formado na mitocôndria não atravesssa a membrana interna, mas pode receber um grupo amino do glutamato formando aspartato, que sai da mitocôndria e no citossol, refenera o oxaloacetato. A passagem de malato e aspartato através da membrana interna da mitocôndria é efetuada por proteínas presentes nesta membrana. Resumindo pode-se dizer que esta lançadeira é usada para transportar equivalentes redutores do NADH

115
citossólico para a matriz mitocondrial no rim, fígado e coração dos mamíferos. O malato pode transpor a membrana mitocondrial, mas o oxaloacetato não.
CONTEXTUALIZANDO
Tecido Adiposo Marrom: Um caso de ineficiência útil
Quando o transporte de elétrons gera um gradiente de prótons, parte da energia produzida assume a forma de calor. Há duas situações nas quais a dissipação da energia como calor é útil para o organismo: a termogênese sem tremores induzida pelo frio (produção de calor) e a termogênese induzida pela dieta. A termogênese sem tremores, induzida pelo frio, permite que os animais sobrevivam em baixas temperaturas após terem se adaptado a essas condições, e a termogênese induzida pela dieta evita o desenvolvimento da obesidade apesar da alimentação excessiva prolongada. (A energia é dissipada como calor na medida em que as moléculas de alimento são metabolizadas em vez de serem armazenadas como gordura.) Esses dois processos podem ser bioquimicamente iguais, ocorrendo, principalmente, se não exclusivamente, no tecido adiposo marrom (TAM), que é rico em mitocôndrias (O tecido adiposo marrom obtém sua cor por causa do grande número de mitocôndrias presentes nele, em vez das gorduras brancas usuais.) A chave para esse uso “ineficiente” de energia no tecido adiposo marrom parece ser uma proteína mitocondrial chamada termogenina, também conhecida como “proteína desacopladora”. Quando tal proteína ligada à membrana é ativada na termogênese, ela funciona como um canal de prótons pela membrana mitocondrial interna. Como todos os outros desacopladores, ela “abre um buraco” na membrana mitocondrial e diminui o efeito do gradiente de prótons. Os prótons fluem de volta para a matriz por meio da termogenina, desviando-se do complexo ATP sintase. Poucas pesquisas sobre a bioquímica ou a fisiologia do tecido adiposo marrom foram realizadas em humanos. Recentemente os pesquisadores dedicaram grandes esforços para identificar o gene que codifica a proteína desacopladora envolvida na obesidade. Alguns pesquisadores também propuseram uma ligação entre a síndrome de morte súbita do lactente e o metabolismo do tecido adiposo marrom. Acreditam que uma ausência de TAM, ou mesmo uma mudança para tecido adiposo normal muito precocemente, poderia levar ao resfriamento da temperatura corporal de um modo capaz de afetar o sistema nervoso central.
ATIVIDADES DE AVALIAÇÃO
1. Citar três inibidores da cadeia de transporte de elétrons, indicando os transportadores sobe os quais atuam.

116
2. Verificar se é possível a oxidação de malato e de succinato em presença de rotenona.
3. Descrever a hipótese do acoplamento quimiosmótico para a fosforilação oxidativa.
4. Indicar o número de ATP sintetizados para cada NADH e FADH2 oxidados.5. Definir desacoplador e citar um exemplo.6. Definir inibidor de fosforilação oxidativa e citar um exemplo.7. Definir controle respiratório.8. Definir fosforilação ao nível do substrato e citar as reações onde ocorre esta
fosforilação.9. A membrana interna da mitocôndria é impermeável a ATP e NADH. Mostrar
como:a) O NADH produzido na via glicolítica pode ser oxidado na cadeia
respiratória (lançadeiras do malato e do glicerol-fosfato).b) O ATP produzido na mitocôndria pode ser utilizado no citosol.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. No Capítulo 20, do vol. 3, o assunto Fosforilação Oxidativa e Inibidores e Desacopladores da Cadeia de Transporte de Elétrons são abordados de forma muito didática.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade II do livro, no Capítulo 6, são apresentados tópicos relativos ao assunto abordado. O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª ed. 1999. Rio de Janeiro: Editora Guanabara Koogan S. A. O Capítulo 11 da Parte 3 desse livro explana de forma bastante compreensiva o assunto Cadeia de Transporte de Elétrons e Fosforilação Oxidativa.
Smith, C.; Marks, A. D.; Lieberman, M. Bioquímica Médica Básica de Marks – Uma abordagem clínica. 2ª ed. Porto Alegre: Artmed, 2007. O Capítulo 21 do livro aborda o assunto com muito detalhamento.
CAPÍTULO 10: METABOLISMO DE CARBOIDRATOSGLICÓLISE E FORMAÇÃO DE ACETIL-CoA
OBJETIVOS: Entender o metabolismo. Da Glicose à Piruvato (Falta o verbo no objetivo) Oxidação do Piruvato: do Piruvato à Acetil-CoA (Falta o verbo no
objetivo)

117
Produção de Energia (Falta o verbo no objetivo) Glicólise Anaeróbica (Falta o verbo no objetivo)
O uso da glicose como fonte energética pode ser considerada universal, pois dos microorganismos ao homem quase todas as células são potencialmente capazes de atender suas demandas energéticas apenas a partir deste açúcar. A glicose é o principal açúcar da dieta e o carboidrato que circula no sangue para garantir que todas as células tenham um fornecimento de substrato energético contínuo. A glicose é armazenada nas células como glicogênio, o qual pode fornecer uma fonte interna de substrato energético para a glicólise em situações de emergência. Assim sendo pode-se dizer que a glicose é prontamente disponível a partir da dieta, de reservas internas de glicogênio e do sangue. Ela é imprescindível para algumas células e tecidos, como hemácias e tecido nervoso, por constituir o único substrato que estes tecidos são capazes de oxidar para obter energia. O cérebro utiliza glicose quase exclusivamente como um substrato energético.
O primeiro estágio do metabolismo da glicose é chamado glicólise. A glicólise é um processo anaeróbio que, sozinho, produz apenas duas moléculas de ATP. A oxidação aeróbia completa da glicose em dióxido de carbono e água (envolvendo a glicólise, o ciclo do ácido cítrico e a fosforilação oxidativa) produz energia equivalente a 32 moléculas de ATP. Durante atividades físicas vigorosas, o corpo metaboliza aerobiamente os carboidratos, as gorduras e as proteínas para gerar combustível; entretanto, mais carboidratos são utilizados conforme a intensidade da atividade física aumenta. Além de servir como uma fonte anaeróbica e aeróbica de ATP, a glicólise é uma rota anabólica que fornece precursores para a biossíntese. Por exemplo, no fígado e no tecido adiposo, essa rota produz piruvato como um precursor para a biossíntese de ácidos graxos. A glicólise também fornece precursores para a síntese de compostos, como aminoácidos e ribose-5-fosfato, o precursor de nucleotídeos.
Em corridas de 400 metros, em que ocorrem explosões repentinas de gasto de energia, o corpo utiliza os carboidratos mais rapidamente do que pode processá-los de modo aeróbio. A glicose será metabolizada por meio da glicólise, com o piruvato sendo o produto final. O piruvato será convertido em ácido láctico, que, consequentemente, será exportado dos músculos para o fígado. Assim, os dois ATP da glicólise anaeróbia serão uma fonte de energia adicional nessas condições. Sob condições aeróbias, o principal objetivo da glicólise é alimentar o piruvato no ciclo do ácido cítrico, em que as etapas metabólicas posteriores gerarão um aumento considerável na síntese de ATP.
Portanto, todas as células oxidam glicose a piruvato para obter ATP; o piruvato pode ser oxidado a CO2, aumentando muito a produção de ATP. Nas células anaeróbias, a degradação da glicose para no piruvato.

118
Glicose Piruvato Permite aproveitar menos de 10% da energia total da glicose (A maior parte fica conservada como piruvato).
libera ~200kj.mol-1
Glicose CO2
libera 2870kj.mol-1
Nas células aeróbias, o piruvato é subsequentemente oxidado, com grande ganho de ATP.
Esquema da oxidação completa da glicose
Na glicólise, no citosol, a glicose é convertida em frutose-1,6-bisfosfato, que gera duas moléculas de piruvato. Quando o piruvato é formado, ele pode ter vários destinos. No metabolismo aeróbio, o piruvato perde dióxido de carbono, e os dois átomos de carbono restantes ficam ligados à coenzima A como um grupo acetila para formar a acetil-CoA, que, então, entra no ciclo do ácido cítrico. No metabolismo anaeróbio existem dois destinos para o piruvato. Em organismos capazes de fermentação alcoólica, o piruvato perde dióxido de carbono, dessa vez produzindo acetaldeído, que será então reduzido a etanol. O destino mais comum para o piruvato no metabolismo anaeróbio é a sua redução a lactato, chamada glicólise anaeróbia para distinguir da conversão de glicose a piruvato que é conhecida simplesmente como glicólise. O metabolismo anaeróbio é a única fonte de energia nas hemácias dos mamíferos, assim como em diversas espécies de bactérias, como o Lactobacillus no soro do leite e o Clostridium botulinum em alimentos enlatados estragados. Em todas essas reações a conversão de glicose ao produto é uma reação de oxidação, exigindo reações de redução em que NAD+ é convertido em NADH.
Glicólise: Oxidação de Glicose a Piruvato
Na glicólise ocorre, portanto, duas fosforilações por ATP e duas fosforilações por fosfato inorgânico. Os quatro grupos fosfato são transferidos para ADP, formando quatro ATP.
A glicólise pode ser dividida em quatro etapas. Estas quatro etapas são compostas por dez reações sequenciais que compõem a glicólise.
Oxidação parcial da
glicose
Oxidação completa
da glicose

119
Etapa I: Ocorre uma dupla fosforilação da hexose, à custa de 2 ATP, originando uma hexose com 2 grupos fosfato. Na primeira reação da glicólise, os organismos efetuam a fosforilação da glicose através de uma reação que utiliza ATP como doador de grupo fosfato. A reação é essencialmente irreversível e catalisada por quinases. Quinases são enzimas que transferem um grupo fosfato de um composto de alta energia (em geral ATP) para um composto aceptor. Na maioria dos organismos e tecidos, a enzima hexoquinase é quem atua catalisando a fosforilação da glicose. No fígado, quem atua é a glicoquinase. Em seguida, ocorre a isomerização da glicose 6-fosfato a frutose 6-fosfato, por ação da isomerase, a fosfoglicoisomerase, e nova fosforilação, utilizando ATP catalisada pela fosfofrutoquinase. Forma-se então uma hexose com 2 grupos fosfato: a 1,6-bisfosfato.
Etapa II: Ocorre a clivagem da frutose 1,6 – bisfosfato, produzindo duas trioses fosforiladas: diidroxiacetona fosfato e gliceraldeído 3-fosfato. Essa reação é catalisada por aldolase. As duas trioses fosforiladas são isômeras e devem ser interconvertidas por ação de uma isomerase específica a triose fosfato isomerase. A conversão de diidroxiacetona fosfato em gliceraldeído 3-fosfato possibilita que todos os carbonos da glicose sejam oxidados a piruvato, apesar de apenas o gliceraldeído 3-fosfato ser substrato da próxima enzima e poder, portanto, prosseguir pela via glicolítica.
Etapa III: Nesta etapa ocorre oxidação e nova fosforilação, desta vez por fosfato inorgânico (Pi), das trioses fosfato, formando duas moléculas de um intermediário com 2 grupos fosfato. As duas moléculas de gliceraldeído 3-fosfato obtidas por fosforilação à custa de 2 ATP são novamente fosforiladas, agora por fosfato inorgânico, formando duas moléculas de 1,3-bisfosfoglicerato; assim, o substrato, um aldeído, é oxidado a um ácido. Esta etapa é cumprida por uma reação de óxido-redução complexa, catalisada pela gliceraldeído 3-fosfato desidrogenase.
Etapa IV: Compreende dois eventos de formação de ATP. Ocorre transferência dos grupos fosfato deste intermediário para ADP, formando 4 ATP e 2 moléculas de piruvato. A glicólise tem, portanto, um rendimento de 2 ATP: para cada molécula de glicose são produzidos 4 ATP (2 por triose), dos quais devem ser descontados os 2 ATP consumidos na Etapa I.

120
http://www.fortunecity.com
Glicólise Anaeróbia: Fermentações
O NADH produzido na glicólise pode ser oxidado anaerobiamente: o piruvato é convertido a lactado ou etanol
Equação geral da glicólise (eq. 1) é:
O
C6H12O6 + 2ADP + 2Pi + 2NAD+ 2H3C –C – COO- + 2ATP + 2H2O + 2NADH + 2H+
Glicose Piruvato
A redução de NAD+ está associada à oxidação da glicose e à produção de ATP. Como o NAD+ existe nas células em concentrações limitantes, muito inferiores às dos substratos, a manutenção do funcionamento da glicólise depende da reoxidação do NADH. Os organismos regeneram o NAD+ através de dois processos diferentes, segundo a disponibilidade de oxigênio. Quando

121
em situação de aerobiose, utilizam o oxigênio para oxidar o NADH enquanto no caso de haver anaerobiose, o próprio piruvato produzido pela glicólise serve como aceptor dos elétrons do NADH, sendo reduzido a lactato (equação 2):
O OH
2H3C – C – COO- + 2 NADH + 2 H+ 2H3C – C – COO- + 2 NAD+
H
Piruvato Lactato
HIPERTEXTO
Acidose Lática
A deficiência da enzima piruvato desidrogenase evita a oxidação do piruvato, levando a seu acúmulo no citoplasma. Isto aumenta a conversão de piruvato em lactato e produz um aumento do lactato e piruvato sanguíneos. Os prótons que acompanham esses ânions são neutralizados pelo bicarbonato sérico, criando uma acidose metabólica com uma elevada diferença de ânions. A acidose lática é uma das várias acidoses metabólicas causadas pelo acúmulo de ácidos orgânicos no sangue.
Algumas espécies de bactérias, as hemácias, fibras musculares de contração rápida (fibras brancas) e fibras musculares em geral, quando submetidas a esforço intenso, utilizam esse processo. Nestas condições, o oxigênio trazido pela circulação torna-se insuficiente para promover a oxidação da grande quantidade de NADH resultante do trabalho muscular e a fibra muscular fica submetida a uma anaerobiose relativa. A oxidação do NADH pelo piruvato gera, então, o lactato caracteristicamente produzido por músculos em anaerobiose, permitindo que, pela regeneração do NAD+, a glicólise possa prosseguir, formando ATP.
OH
C6H12O6 + 2ADP + 2Pi 2H3C – C – COO- + 2 ATP + 2H2O
H
Glicose Lactato
A equação acima é a soma da equação de conversão de glicose a piruvato (equação 1) com a de conversão de piruvato a lactato (equação 2) e chama-se equação geral da conversão de glicose a lactato.
lactato
desidrogenase

122
Em leveduras e algumas bactérias, a regeneração do NAD+ é feita por outro processo: o piruvato é descarboxilado, originando acetaldeído, que, servindo como aceptor de elétrons do NADH, reduz-se a etanol:
O O
H3C – C – COO- + H+ H3C – C – H + CO2
Piruvato Acetaldeído
O OH
H3C – C –H + NADH + H+ H3C – C – H + NAD+
H
Etanol
As fermentações diferem nas reações que regeneram o NAD+, dependendo das enzimas utilizadas no processo e no produto final da reação ou seja, o piruvato pode ser convertido a lactato, etanol, butirato, propionato, etc. Então, pode-se afirmar que existe fermentação lática, fermentação alcoólica, fermentação propiônica etc.
Conversão de Piruvato a Acetil-CoA
Em aerobiose, a conversão do piruvato em acetil-CoA é o primeiro passo para sua oxidação total. Nas células eucarióticas, o piruvato do citosol entra na mitocôndria, através de uma translocase específica, e é transformado em acetil-CoA, conectando, portanto, a glicólise e o Ciclo de Krebs. Assim sendo, o piruvato deixa de ser o aceptor dos elétrons do NADH produzido pela glicólise e esta coenzima não será regenerada no citosol; será oxidada pelo oxigênio, aceptor final de elétrons no metabolismo aeróbio. O piruvato é convertido a acetil-CoA, através de uma descarboxilação oxidativa, de acordo com a equação:
O O
H3C – C – COO- + HS – CoA + NAD+ H3C – C – SCoA + NADH + CO2
Piruvato Coenzima A Acetil-CoA
Piruvato descarboxilase
desidrogenase
álcool
DEGRADAÇÃO ANAERÓBICA DA GLICOSE =
FERMENTAÇÃO

123
CICLO DE KREBS
http://www.tudomaisumpouco.com/aula6respiração
Via das pentoses fosfato (ou rota da hexose monofosfato)
A rota da hexose monofosfato consiste em duas reações oxidativas irreversíveis, seguidas de uma série de conversões reversíveis açúcar-fosfato. Nenhum ATP é consumido ou produzido diretamente no ciclo o carbono 1 da glicose 6-fosfato é liberado como CO2, e dois NADH são produzidos para cada glicose 6-fosfato, que entra na parte oxidativa da rota. Esta via ocorre no citosol da célula, e fornece uma importante porção do NADH da célula, que funciona como um redutor bioquímico. É particularmente importante no fígado e glândulas mamárias, que são ativos na biossíntese de ácidos graxos, e no córtex adrenal, o qual é ativo na síntese de esteroides dependente de NADH. Esta via também produz ribose-fosfato, necessária para a biossíntese de nucleotídeos e dá condições para o uso de açúcares de cinco carbonos.
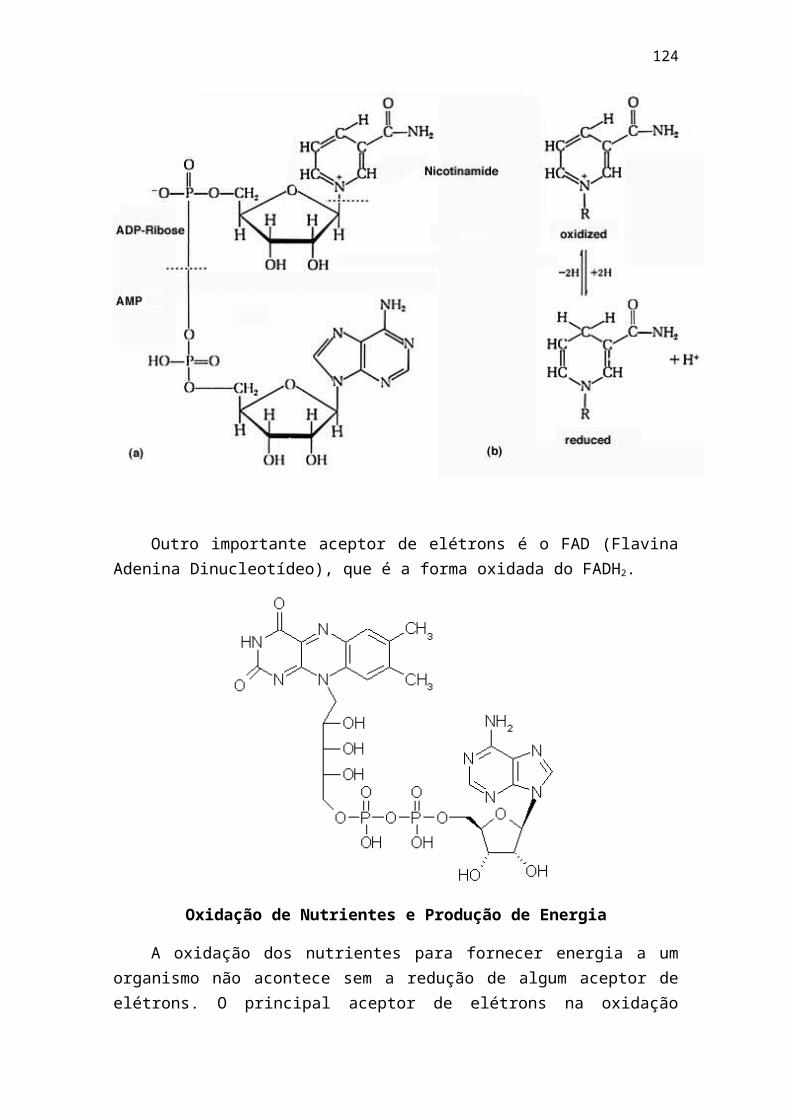
124
VIA DAS PENTOSES FOSFATO
http://www.bioq.unb.br/htm/textos_explic/via_pentoses.htm
Metabolismo de carboidratos
Monossacarídeos
Os carboidratos são entregues às células principalmente na forma de glicose, juntamente com quantidades menores de outros monossacarídeos. A frutose e a galactose são convertidas em glicose no fígado. A maior parte da glicose é oxidada pelo ciclo do ácido cítrico para atingir as necessidades de energia imediatas de todos os tecidos. Um pouco de glicose é convertida em: ribose, frutose (para espermatozoides), desoxirribose, glicosamina e galactosamina, e esqueletos de carbono para a produção de aminoácidos não essenciais. Carboidrato em excesso é convertido em glicogênio ou ácidos graxos, que são depois armazenados como triglicerídeos no tecido adiposo.
Galactose: É convertida no fígado em UDFG (uridina difosfato glicose) que pode ser incorporada no glicogênio ou convertida em glicose-1-PO4 e metabolizada pelas vias da glicose.
Frutose: Sua entrada na célula não é insulino-dependente. Pode ser convertida em glicose; aumenta a glicose ou triglicerídeos no sangue em indivíduos suscetíveis com diabetes não insulino-dependentes.

125
A frutose e a galactose não são dependentes da insulina para entrar na célula.
Glicogênio, Amido, Sacarose e Lactose
Glicogênio: É sintetizado principalmente pelo fígado e músculos quando a oferta de glicose supera as necessidades energéticas imediatas destes órgãos.
Glicogênio Hepático: Produz glicose (por degradação), que é exportada para manter a glicemia (concentração de glicose sanguínea) nos períodos entre as refeições e no jejum noturno.
Glicogênio Muscular: Provê energia apenas para a fibra muscular em contração intensa, quando a demanda energética ultrapassa o aporte de O2.
A degradação do glicogênio produz 1-fosfato. A glicose 1-fosfato é convertida pela fosfoglixomutase. A glicose 6-fosfato que pode ser degradada pela glicólise, formando lactato, no músculo. No fígado, a glicose 6-fosfato é preferencialmente hidrolisada por ação da glicose6-fosfatase, produzindo glicose, que é liberada na circulação. A síntese de glicogênio utiliza como precursor uma forma ativada de glicose e gasta 2 ATP por glicose incorporada.
GALACTOSE
GALACTOSE-1-PO4
GLICOSE-1-PO4
GLICOGÊNIO
GLICOSE
GLICOSE-6-PO4
FRUTOSE-1,6-DIPO4
GLICERALDEÍDO—3-PO4GLICOSE
FRUTOSEMÚSCULO
DIIDROXIACETONA FOSFATO
FÍGADOUTP
UPPG
ÁCIDO PIRÚVICO

126
Doenças hereditárias do metabolismo do glicogênio
Tipo Enzima deficiente Consequências
I von Gierke Glicose 6-fosfatase Acúmulo de glicogênio hepático e aumento do fígado; inabilidade de corrigir a glicemia no jejum.
II Pompe Α-1,4 Glicosidase1 Acúmulo generalizado de glicogênio, insuficiência cardiorrespiratória e morte precoce.
III Cori Enzima desramificadora Glicogênio com ramificações curtas, resultantes da ação do glicogênio fosforilase.
IV Andersen Enzima ramificadora Glicogênio com cadeias muito longas não ramificadas; morte precoce.
V McArdle Glicogênio fosforilase muscular Incapacidade de realizar exercícios intensos.
VI Hers Glicogênio fosforilase hepática Semelhantes às do tipo I, mas menos intensas.
VII - Fosfofrutoquinase muscular Semelhantes à do tipo V.
VIII Tarui Fosforilase quinase hepática Semelhantes às do tipo I, mas menos intensas.
IX - Glicogênio sintase hepática Diminuição do glicogênio hepático.
1 Enzima lisossômicas que hidrolisa os segmentos lineares do glicogênio; em indivíduos normais, constitui uma via secundária do metabolismo do glicogênio.
CONTEXTUALIZANDO
As cáries dentárias estão entre as doenças mais frequentes nos Estados Unidos e, possivelmente, no mundo, embora tratamentos modernos, como aplicações de flúor e uso do fio dental, tenham reduzido a sua incidência entre os jovens. Os fatores que contribuem para a cárie dentária são uma combinação de uma dieta com alto teor de açúcar refinado, o desenvolvimento da placa dentária e o metabolismo anaeróbico. A dieta com alto teor de açúcar permite o pronto crescimento de bactérias na boca, e a sacarose talvez seja o açúcar mais facilmente usado por elas. Por outro lado, as bactérias podem fazer a sua “cola” de polissacarídeos a partir desse açúcar não-redutor. As bactérias desenvolvem-se ampliando suas colônias pegajosas, formando placas na superfície dos dentes. As bactérias que crescem debaixo da superfície da placa fazem metabolismo anaeróbico, já que o oxigênio não se difunde pela superfície encerada da placa dental. Os dois produtos

127
predominantes, o lactato e o piruvato, são ácidos orgânicos relativamente fortes, e são esses produtos ácidos que, de fato, causam a destruição da superfície esmaltada. As bactérias, é claro, crescem melhor nos buracos das cáries, e, se o esmalte for inteiramente degradado, as bactérias crescerão ainda mais rapidamente na camada macia da dentina, embaixo do esmalte. A adição de flúor à água resulta em uma superfície de esmalte muito mais dura, e o fluoreto pode inibir o metabolismo das bactérias. O uso do fio dental diariamente rompe a placa e evita o estabelecimento de condições anaeróbicas (Campbell, 2000).
ATIVIDADES DE AVALIAÇÃO
1. Quantas moléculas de piruvato se formam a partir de uma molécula de glicose?
2. Que hexose dá origem a trioses?
3. Indicar as reações de óxido-redução.
4. Indicar os compostos ricos em energia.
5. Identificar as reações catalisadas pelos seguintes tipos de enzimas:
a) Quinase.b) Isomerase.c) Aldolase.d) Desidrogenase.
6. Considerando o nº de moléculas de ATP consumidas e formadas, estabelecer o saldo final de ATP na degradação de uma molécula de glicose pela via glicolítica.
7. Citar os compostos que devem ser fornecidos à via glicolítica para:
a) iniciá-la (haver formação de lactato).b) mantê-la em funcionamento.
8. Indicar a função da via glicolítica.
9. Esquematizar as reações de fermentação alcoólica que possibilitam a obtenção de NAD+ na forma oxidada.
10. Citar as vitaminas necessárias para as seguintes conversões:a) Glicose Lactatob) Lactato Glicose

128
11. Escrever a reação de formação de acetil-CoA a partir de piruvato e indicar: a) – As 5 coenzimas necessárias; b) – As vitaminas envolvidas; c) – A localização celular.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. No Capítulo 17, do vol. 3, o assunto Glicólise é abordado de forma muito didática.
Campbell, M. K. Bioquímica. 3ª Ed. Porto Alegre: Artmed Editora, 2000. A contextualização sobre cáries, explica de modo didático sua produção por anaerobiose.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade II do livro, no Capítulo 8, são apresentados tópicos relativos ao assunto abordado. O livro é ricamente ilustrado tornando o assunto de fácil compreensão.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª ed. 1999. Rio de Janeiro: Editora Guanabara Koogan S. A. Os Capítulos 9, 12, 13 da Parte 3 desse livro explanam de forma bastante compreensiva os tópicos explorados nesse capítulo.
Smith, C.; Marks, A. D.; Lieberman, M. Bioquímica Médica Básica de Marks – Uma abordagem clínica. 2ª ed. Porto Alegre: Artmed, 2007. O Capítulo 22 do livro aborda o assunto com muito detalhamento e de forma didática.
LEITURA, VÍDEO E SITES RECOMENDADOS
www.ebah.com.br/content/AFAAAAAkUAD/glicolise
pathmicro.med.sc.edu/portuguese/chapter_3_bp.htm
gratisvideoaulas.blogspot.com/.../biologia-glicolise-respirac
CAPÍTULO 11: GLICONEOGÊNESE
OBJETIVOS:
Verificar o catabolismo de glicose.Diferenciar a quebra e síntese de glicose no organismo.

129
Compreender o papel da glicose no fígado, como reserva de energia.Relacionar glicose com doenças como, diabetes e glaucoma.Conceituar o consumo de glicose de forma anaeróbica.
A glicose ocupa um papel central no metabolismo, tanto como combustível quanto como um precursor de carboidratos estruturais essenciais e de outras biomoléculas. O cérebro (consome 120 g de glicose por dia) e as células vermelhas do sangue (consomem 30 g de glicose, diariamente) são quase completamente dependentes da glicose como fonte de energia. É preciso que o suprimento de glicose seja ininterrupto, por isso o organismo dispõe de mecanismos destinados a manter a oferta de glicose circulante, possibilitando a captação contínua mesmo muito depois das refeições. À proporção que diminui a glicose circulante, derivada da ingestão de alimentos, a degradação do glicogênio hepático incumbe-se da manutenção da concentração adequada de glicose sanguínea. O nível de glicose no sangue é mantido nos limites normais pela liberação de glicose a partir do glicogênio do fígado (glicogenólise), por exemplo, durante o sono, ou jejum prolongado. Porém, a capacidade de armazenamento de glicogênio no fígado é suficiente para suprir o cérebro com glicose por apenas aproximadamente dez a dezoito horas na ausência de ingestão de carboidratos, após o que outra via metabólica de produção de glicose, a gliconeogênese, é acionada. Essa via requer tanto enzimas mitocondriais como citosólicas. Durante o jejum de uma noite, cerca de 90% da gliconeogênese ocorre no fígado com os rins fornecendo 10% das moléculas de glicose recém-sintetizadas. No entanto, em um jejum prolongado, os rins tornam-se importantes produtores de glicose, cerca de 40%, da produção total.
HIPERTEXTO
O principal fator envolvido na regulação dos níveis de glicose sanguínea é a própria concentração de glicose sanguínea e de hormônios, particularmente insulina e glucagon. À medida que os níveis de glicose sanguínea se elevam após uma refeição, a concentração aumentada de glicose estimula as células β do pâncreas para estimular insulina. Certos aminoácidos, particularmente arginina e leucina, também estimulam a liberação de insulina pelo pâncreas.
Os níveis sanguíneos de glucagon, que é secretado pelas células α do pâncreas, podem aumentar ou diminuir, dependendo do conteúdo da refeição. Os níveis de glucagon diminuem em resposta a uma refeição rica em carboidratos, mas podem aumentar em resposta a uma refeição rica em proteínas. Após uma refeição típica mista contendo carboidrato, proteína e gordura, os níveis de glucagon permanecem relativamente constantes enquanto os níveis de insulina aumentam (Smith, Allan & Lieberman, 2007).

130
Fonte de energia para diferentes tecidos
Composto
Tecido Glicose Ácidos graxos Corpos cetônicos
Cérebro +
Hemácias e leucócitos +
Medula renal +
Retina +
Mucosa intestinal +
Fígado + +
Adiposo + +
Músculos esqueléticos e cardíaco
+ + +
Córtex renal + + +
Fonte: Marzzoco & Torres, 1999.
A energia livre da glicose é sequestrada na forma de ATP por meio da glicólise, do ciclo do ácido cítrico e da fosforilação oxidativa. A gliconeogênese é uma rota importante do metabolismo dos carboidratos e se processa no fígado e, minoritariamente, nos rins. Ela é a rota metabólica que através de precursores que não são carboidratos, tais como lactato, piruvato, glicerol e aminoácidos, converte-os a glicose.
Glicogênio
Glicose 1-PO4
Glicose 6-PO4
Frutose 6-PO4
Frutose -1,6 – Difosfato

131
Gliceraldeído -3-Fosfato + Diidroxiacetonafosfato
2 moléculas de Piruvato
Via Aeróbia Via Anaeróbia (Ciclo de Krebs) (2 de Ácido Láctico)
Figura: Resumo simplificado das reações da glicólise e da gliconeogênese
Quando em jejum, a maior parte da glicose do organismo deve ser suprida por meio da gliconeogênese (significa síntese de glicose nova), que é a biossíntese da glicose a partir de precursores que não são carboidratos. A gliconeogênese ocorre no fígado e, em menor extensão, nos rins como já foi dito. Os precursores não-carboidratos que podem ser convertidos à glicose incluem os produtos da glicólise, lactato e piruvato, intermediários do ciclo do ácido cítrico e os esqueletos de carbono da maioria dos aminoácidos. A degradação de proteínas é um processo normal. A utilização de aminoácidos para a gliconeogênese é uma via metabólica habitual cotidiana, contribuindo para a manutenção da glicemia durante o jejum noturno.
Antes de tudo, porém, todas essas substâncias devem ser convertidas a oxalacetato, o material de partida para a gliconeogênese. O oxalacetato é um intermediário na produção de fosfoenol piruvato na gliconeogêse como será visto mais adiante. Os únicos aminoácidos que não podem ser convertidos a oxalacetato em animais são leucina e lisina, pois sua hidrólise gera apenas acetil-CoA. Da mesma forma, ácidos graxos não podem servir como precursores de glicose em animais porque a maioria dos ácidos graxos é degradada completamente a acetil-CoA. No entanto, ao contrário dos animais, as plantas possuem uma rota para a conversão de acetil-CoA a oxalacetato que é o ciclo do glioxalato, de modo que os ácidos graxos podem ser a única fonte de carbono das células vegetais. O glicerol, um produto da hidrólise de triacilgliceróis, é convertido à glicose por meio da síntese do intermediário glicolítico diidroxiacetona – fosfato.
HIPERTEXTO
Hipoglicemia Neonatal Idiopática

132
Recém-nascidos têm uma necessidade crítica da gliconeogênese. O suprimento de glicose pela placenta é interrompido e não há glicose imediatamente disponível na dieta. Uma vez que o cérebro precisa ter uma fonte prolongada de glicose proveniente do sangue, os genes para as enzimas gliconeogênicas são ativados simultaneamente ao nascimento. Ocasionalmente, esta ativação não ocorre e o recém-nascido deve ser alimentado com uma solução de glicose ou terá hipoglicemia (Fonte: Pelley, 2007).
Relação entre diferentes órgãos na gliconeogênese
A produção de glicose ocorre a partir de substratos produzidos pelo músculo como lactato no esforço intenso e alanina no jejum. O lactato origina-se dos músculos submetidos à contração intensa e de outras células que degradam glicose anaerobiamente – medula adrenal, e, principalmente, hemácias. A maior parte da glicose sintetizada destina-se ao cérebro. Os aminoácidos são provenientes de degradação de proteínas endógenas, principalmente as musculares, durante o jejum. Ainda no músculo, os aminoácidos são convertidos à alanina, a forma de transporte do aminoácido ao fígado. O glicerol é derivado da hidrólise de triacilgliceróis (TAG) do tecido adiposo, durante o jejum. O fígado é o principal órgão responsável pela gliconeogênese e, apenas em casos de jejum muito prolongado, o córtex renal chega a dar contribuição importante.
Reações da Gliconeogênese
De onze reações necessárias para converter piruvato em glicose livre, sete são reversíveis, catalisadas por enzimas glicolíticas. A gliconeogênese utiliza as reações reversíveis da glicólise e substitui por outras as reações irreversíveis. Contudo, três das enzimas glicolíticas, glicoquinase, fosfofrutoquinase (PFK) e piruvato-quinase, catalisam reações com grandes variações de energia livre negativa na direção da glicólise. Essas reações devem ser substituídas na gliconeogênese por reações que tornem a síntese de glicose termodinamicamente favorável. Assim, essas reações são contornadas pela ação das enzimas glicose-6-fosfatase, frutose-1,6-bisfosfatase e piruvato-carboxilase/PEP-carboxiquinase.
Alanina

133
Cisteína
Glicina Lactato
Serina
Treonina
Triptofano
Piruvato
Aspartato
Asparagina Oxalacetato
Malato
Citrato
Fumarato Ciclo de Krebs
Fenilalanina Isocitrato
Tirosina Succinato
α-cetoglutarato
Succinil-CoA
Arginina
Isoleucina Glutamato
Metionina Glutamina
Valina Histidina
Prolina

134
educadorfisico.esp.br
Figura: Rotas que convertem lactato, piruvato e intermediários do ciclo do ácido cítrico a oxalacetato.
Os esqueletos de carbono de todos os aminoácidos, exceto leucina e lisina, podem ser em parte convertidos a oxalacetato e, então, à glicose através das reações da figura acima.
1.
O O Piruvato-carboxilase O O O
CH3 – C – C – O- -O - C – CH2 –C – C – O-
Piruvato HCO-3 + ATP ADP +Pi Oxalacetato
2.
O O O Fosfoenolpiruvato – carboxicinase O O
-O - C – CH2 –C – C – O- H2C - C – C -O- PO32-
Oxalacetato GTP GDP + CO2 Fosfoenolpiruvato
Figura: Conversão de piruvato a oxalacetato e, então, a fosfoenolpiruvato
A transformação da alanina e do lactato, em glicose, se inicia por sua conversão ao piruvato. A alanina origina piruvato por ação da alanina aminotransferase; o lactato é convertido a piruvato por ação da lactato desidrogenase. A transformação do piruvato em glicose pela gliconeogênese processa-se no sentido oposto ao da glicólise, utilizando quase todas as suas enzimas, com exceção daquelas que catalisam reações irreversíveis. Essas

135
reações são contornadas através de outras reações, catalisadas naturalmente, por outras enzimas. As três etapas em que a gliconeogênese difere da glicólise são:
1. Conversão de piruvato a fosfoenolpiruvato.
O piruvato é convertido a oxalacetato antes da conversão a fosfoenolpiruvato. A conversão de piruvato a fosfoenolpiruvato compreende o transporte de piruvato para a mitocôndria, sua carboxilação a oxaloacetato, o transporte de oxaloacetato para o citossol e a transformação deste composto em fosfoenolpiruvato. As enzimas envolvidas são (1) a piruvato-carboxilase e (2) a fosfoenolpiruvato-carboxiquinase.
Para ser utilizado como substrato da piruvato carboxilase, uma enzima mitocondrial, o piruvato produzido no citosol entra na mitocôndria por ação da piruvato translocase. Essa enzima contém biotina (vitamina B7), como grupo prostético. Para promover a carboxilação do piruvato e produzir oxalacetato, a biotina combina-se com CO2 à custa de ATP. O oxalacetato passa para o citossol através da lançadeira do malato-aspartato e, por ação da fosfoenolpiruvato carboxiquinase, é convertido a fosfoenolpiruvato, através de descarboxilação e fosforilação à custa de GTP. O fosfoenolpiruvato então é transformado em frutose 1,6-bisfosfato pelas enzimas que também compõem a glicólise, que como catalisam reações reversíveis, podem operar a via no sentido inverso.
Piruvato + CO2 + H2O + ATP Oxalacetato + ADP + Pi + 2 H+
Oxalacetato + GTP Fosfoenolpiruvato + CO2 + GDP
2. Conversão de frutose 1,6-bisfosfato a frutose 6-fosfato.
Essa reação substitui a reação irreversível catalisada pela enzima fosfofrutoquinase. É uma reação de hidrólise do grupo fosfato do carbono 1, catalisada pela frutose 1,6-bisfosfatase.
Frutose 1,6-bisfosfato + H2O Frutose 6-fosfato + Pi
A fosfoglicoisomerase pode levar à isomerização da frutose 6-fosfato a glicose 6-fosfato.
3. Conversão de glicose 6-fosfato à glicose.
A irreversibilidade da reação catalisada pela glicoquinase, pode ser contornada substituindo-se esta reação por uma reação de hidrólise do grupo fosfato ligado ao carbono 6, catalisada pela glicose 6-fosfatase. É, portanto, uma reação semelhante à anterior.
Glicose 6-fosfato + H2O Glicose + Pi

136
O produto da reação, a glicose, ao contrário da glicose fosforilada, pode atravessar livremente a membrana plasmática. A glicose 6-fosfatase ocorre exclusivamente no fígado e rins, e é graças a ela, que estes órgãos, especialmente o fígado, podem exportar glicose para corrigir a glicemia. O glicerol para ser usado como composto gliconeogênico é fosforilado a glicerol 3-fosfato, que é oxidado a diidroxiacetona fosfato. Esse é um composto da via glicolítica e pode, portanto prosseguir em direção à glicose pelas reações da glicólise e as substitutivas (frutose 1,6-bisfosfatase e glicose 6-fosfatase).
Ao final do processo, pode-se afirmar que para cada molécula de glicose formada a partir de duas moléculas de piruvato são necessários 6 ATP, utilizados nas reações catalisadas por piruvato carboxilase, fosfenolpiruvato carboxiquinase e fosfoglicerato quinase.
Partindo do piruvato, a equação geral da gliconeogênese é:
2 Piruvato + 6ATP + 6H2O + 2NADH Glicose + 6ADP + 6Pi +2NAD+ +2H+
Caso o composto inicial seja o lactato, a equação será:
2 Lactato + 6ATP +6H2O Glicose + 6ADP +6Pi + 4H+
Partindo do glicerol, a síntese de uma molécula de glicose consome somente 2 ATP na reação catalisada pela glicerol quinase.

137
CONTEXTUALIZANDO
Alterações nos Níveis de Glicose Sanguínea após uma Refeição
Após uma refeição rica em carboidratos, a glicose sanguínea se eleva de um nível de jejum de aproximadamente 80 a 100 mg/dL para um nível de cerca de 120 a 140 mg/dL dentro de um período de 30 minutos a 1 hora. A concentração de glicose no sangue, então começa a diminuir, retornando à faixa do jejum aproximadamente duas horas após a refeição. Os níveis de glicose sanguínea aumentam quando a glicose da dieta é digerida e absorvida. Os valores não sobem mais do que aproximadamente 140 mg/dL em uma pessoa saudável normal, pois os tecidos captam a glicose do sangue, armazenando-a para uso subsequente e oxidando-a para produzir energia. Após a refeição ser digerida e absorvida, os níveis de glicose sanguínea declinam, pois as células continuam a metabolizar a glicose. Se os níveis de glicose sanguínea continuam a subir após uma refeição, a alta concentração de

138
glicose causará a liberação de água dos tecidos como resultado do efeito osmótico da glicose. Os tecidos irão se tornar desidratados, e sua função será afetada. Um coma hiperosmolar pode ocorrer por desidratação do cérebro. Inversamente, se os níveis de glicose sanguínea continuam a diminuir após uma refeição, os tecidos que dependem de glicose sofrem de uma perda de energia. Se os níveis de glicose sanguínea diminuem abruptamente, o cérebro não será capaz de produzir uma quantidade adequada de ATP. Isso resultará em tontura, seguida de sonolência e, finalmente, coma. As células sanguíneas vermelhas não serão capazes de produzir ATP o suficiente para manter a integridade de suas membranas. A hemólise dessas células diminuirá o transporte de oxigênio para os tecidos do corpo. Eventualmente, todos esses tecidos que dependem de oxigênio para a produção de energia falharão em realizar suas funções normais. Se o problema for grave o suficiente, poderá ocorrer a morte. Consequências devastadoras de excesso ou insuficiência de glicose normalmente são evitadas, pois o corpo é capaz de regular seus níveis de glicose sanguínea. À medida que a concentração de glicose sanguínea se aproxima da faixa normal de 80 a 100 mg/dL aproximadamente duas horas após uma refeição, o processo de glicogenólise é ativado no fígado. O glicogênio hepático é a fonte primária de glicose sanguínea durante as primeiras horas de jejum. Subsequentemente, a gliconeogênese começa a ter um papel como fonte adicional de glicose sanguínea. O carbono para a gliconeogênese, um processo que ocorre no fígado, é fornecido por outros tecidos. O músculo em exercício e as células sanguíneas vermelhas fornecem lactato por meio da glicólise; o músculo também fornece aminoácidos pela degradação de proteínas, e o glicerol é liberado do tecido adiposo à medida que as reservas de triacilgliceróis são mobilizadas. Mesmo durante um jejum prolongado, os níveis de glicose sanguínea não diminuem muito. Após 5 a 6 semanas de jejum, os níveis de glicose sanguínea diminuem para apenas 65 mg/dL (Smith, Marks & Lieberman, 2007).
ATIVIDADES DE AVALIAÇÃO
1. Defina Gliconeogênese, indicando quando ocorre e qual o seu balanço energético.
2. Quais são as três etapas que diferenciam a gliconeogênese da glicólise? Dê as reações que ocorrem.
3. Quais as enzimas usadas em cada etapa que diferencia a gliconeogênese da glicólise?

139
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. No Capítulo 18, do vol. 3, o assunto Gliconeogênese é amplamente discutido.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade II, do livro no Capítulo 10, são apresentados os tópicos relacionados à Gliconeogênese com muitas ilustrações, tornando o assunto de fácil compreensão.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª ed. 1999. Rio de Janeiro: Editora Guanabara Koogan S. A. O Capítulo 14 da Parte 3 desse livro explana de forma bastante compreensiva o assunto Gliconeogênese.
Pelley, J. W. Bioquímica. Rio de Janeiro: Elsevier, 2007.
Smith, C.; Marks, A. D.; Lieberman, M. Bioquímica Médica Básica de Marks – Uma abordagem clínica. 2ª ed. Porto Alegre: Artmed, 2007. O Capítulo 31 do livro aborda o assunto detalhadamente e de forma didática.
LEITURA, VÍDEOS E SITE RECOMENDADOS
http://www.ufpe.br/dbioq/portalbq04/metabolismo_de_carboidros.htm
www.ebah.com.br/content/ABAAAApsYAF/bioquimica
CAPÍTULO 12: METABOLISMO DE LIPÍDIOS
OBJETIVOS:
Compreender o metabolismo de lipídios em nosso organismo.
Verificar onde e como as gorduras ficam acumuladas em nosso corpo.
Aprender o custo energético da queima de gorduras no corpo humano.
Observar os destinos de metabólitos secundários aos lipídios principais e seu destino dentro do metabolismo.
Avaliar o quanto o acúmulo de gorduras em nosso corpo é prejudicial à saúde.
Conhecer como gorduras são fabricadas em nosso organismo e fazer a relação gasto x consumo das mesmas.

140
A maioria dos lipídeos encontrados no corpo insere-se nas categorias de ácidos graxos e triacilgliceróis; glicerofosfolipídeos e esfingolipídeos; eicosanóides; colesterol, sais biliares, e hormônios esteroides, e vitaminas lipossolúveis. Esses lipídeos têm diferentes estruturas químicas e funções, entretanto apresentam uma propriedade comum: sua relativa insolubilidade em água. Os lipídeos da dieta, absorvidos no intestino, e os que são sintetizados endogenamente são distribuídos nos tecidos pelas lipoproteínas plasmáticas, para utilização ou armazenamento.
Os triacilgliceróis (também chamados gorduras ou triglicerídeos) são os lipídios mais abundantes da dieta (cerca de 90%). Eles constituem a principal forma de armazenamento de energia metabólica nos seres humanos. Representam a maior reserva energética do organismo, em média, 20% do peso corpóreo, o que equivale a uma massa 100 vezes maior do que a do glicogênio hepático. Os triacilgliceróis são armazenados nas células adiposas, sob forma anidra. Os ácidos graxos, armazenados como triacilgliceróis servem como sua principal fonte de energia. Os glicerofosfolipídeos e os esfingolipídeos, os quais contêm ácidos graxos esterificados, são encontrados nas membranas e nas lipoproteínas plasmáticas na interface entre os componentes lipídicos dessas estruturas e a água circundante.
Os ácidos graxos poliinsaturados que contêm 20 carbonos formam os eicosanoides, que regulam muitos processos celulares. O colesterol, além de proporcionar estabilidade à bicamada de fosfolipídeos das membranas, age como precursor dos sais biliares, compostos semelhantes aos detergentes que agem no processo de digestão e absorção de lipídeos, e dos hormônios esteroides, que, entre outras ações, regulam o metabolismo, o crescimento e a reprodução. As vitaminas lipossolúveis são lipídeos que estão envolvidos em uma variedade de funções, como visão, crescimento e diferenciação (vitamina A), coagulação sanguínea (vitamina K), prevenção do dano oxidativo celular (vitamina E) e metabolismo do cálcio (vitamina D).
Degradação de Triacilgliceróis: (do depósito)
O

141
H2C – O – C – R H2C – OH O
O lipase dos H C – OH + 3R – C – O- +3H+
HC –O - C - R + 3H2O H2C - OH
O adipócitos
H2C – O - C – R
Triacilgliceróis (TAG) Glicerol Ácidos Graxos
Os TAG da dieta são transportados pelos quilomícrons (Figura 1), hidrolisados pela lipase lipoproteica e dão como produtos finais da hidrólise glicerol e ácidos graxos. O glicerol não pode ser reaproveitado pelos adipócitos, que não têm glicerol quinase e são liberados na circulação. No fígado e outros tecidos, por ação da glicerol quinase, é convertido a 1,6 Glicerol 3-Fosfato e transformado em diidroxiacetona fosfato, um intermediário da glicólise ou da gliconeogênese.
H2C – OH ATP ADP + H+ H2C – OH NAD+ NADH +H H2COH
H C – OH H C – OH C O
H2C – OH glicerol quinase H2 C – O – P Glicerol 3-fosfato H2C-O- P
Glicerol Glicerol 3-fosfato Desidrogenase Diidroxiacetona
fosfato
Figura 1 – Quilomícrons
Fonte: http://bioquimicadocolesterol.blogspot.com
Os ácidos graxos são degradados através de uma via que se processa no interior das mitocôndrias. Os ácidos graxos liberados dos adipócitos são transportados pelo sangue ligados à albumina e utilizados pelos tecidos,

142
incluindo fígado e músculos, como fonte de energia; o tecido nervoso e as hemácias são exceções, porque obtêm energia exclusivamente a partir da oxidação da glicose.
Degradação de Ácidos Graxos
Para sua oxidação, os ácidos graxos são ativados e transportados para a matriz mitocondrial. Para serem oxidados, os ácidos graxos são convertidos à uma forma ativada: Acil-CoA. Esta etapa é catalisada por acil-CoA sintetases, associadas à membrana externa da mitocôndria:
O
RCH2CH2COO- + ATP + HSCoA RCH2CH2CSCoA + AMP + PPi
Ác. Graxo Acil – CoA
A membrana interna da mitocôndria é impermeável a acetil-CoA, e
somente os radicais acila são introduzidos na mitocôndria, ligados à Carnitina. Ela é sintetizada a partir de aminoácidos e é amplamente distribuída nos tecidos animais e vegetais, sendo muito abundante nos músculos.
Sistema Utilizado para Transporte de Radicais Acila
O sistema usado para transportar os radicais acila consta das etapas 1 a 4.
1) Na face externa da membrana interna, a carnitina-acil transferase I transfere o radical acila da coenzima A para a carnitina.
2) A acil-carnitina resultante é transportada através da membrana interna por uma translocase específica.
3) Na face interna, a carnitina-acil transferase II doa o grupo acila da acil-carnitina para uma coenzima A da matriz mitocondrial, liberando a carnitina.
4) A carnitina retorna ao citossol pela mesma translocase.
É desta forma, portanto, que o radical acila dos ácidos graxos atinge o interior da mitocôndria, onde ocorre a sua oxidação.
β-Oxidação ou Ciclo de Lynen
No processo conhecido como β-oxidação ou Ciclo de Lynen, a acil-CoA presente na matriz mitocondrial é oxidada a acetil-CoA, produzindo NADH e FADH2. Esta via metabólica é composta de uma série cíclica de quatro reações, ao final das quais a acil-CoA é encurtada de dois carbonos, que são liberados sob a forma de acetil-CoA. São as seguintes as reações:

143
1. Oxidação da acil-CoA a uma enoil-CoA (acil-CoA β-insaturada) de configuração trans, à custa da conversão de FAD a FADH2 (única reação irreversível da via);
2. Hidratação da dupla ligação, formando uma 3-hidroxiacil-CoA (isômero L);
3. Oxidação do grupo hidroxila a carbonila, resultando uma β-cetoacil-CoA e NADH;
4. Cisão da β-cetoacil-CoA por reação com uma molécula de CoA, com formação de acetil-CoA e uma acil-CoA com dois carbonos a menos; esta acil-CoA refaz o ciclo várias vezes, até ser totalmente convertida a acetil-CoA.
A oxidação completa de um ácido graxo necessita não apenas do Ciclo de Lynen, mas também do Ciclo de Krebs. No Ciclo de Lynen, ocorre a conversão do ácido graxo a acetil-CoA, e, no Ciclo de Krebs, ocorre a oxidação do radical acetil a CO2. Em cada volta do Ciclo de Lynen, serão produzidos 1 FADH2, 1 NADH, 1 acetil-CoA e 1 acil-CoA com 2 átomos de carbono a menos que o ácido graxo original. Sempre que o número de átomos de carbono do ácido graxo for par, a última volta do ciclo de oxidação inicia-se com uma acil-CoA de 4 carbonos, a butiril-CoA, e, nesse caso, são produzidas 2 acetil-CoA (além de FADH2 e NADH). Para a oxidação completa de uma molécula de ácido palmítico, que tem 16 átomos de carbono, são necessárias 7 voltas no ciclo (já que na última volta são produzidas 2 moléculas de acetil-CoA), com a produção de 8 acetil-CoA.
O
β α O H3C-C-SCoA

144
R-CH2-CH2C-SCoA Acetil-CoA
Acil-CoA R-C-SCoA
(com n carbonos) Acil-CoA
(com n-2 carbonos)
HS-CoA
β
H O O α O
R-C - C- C –SCoA R-C-CH2-C-SCoA
H β-Cetoacil-CoA
Trans-∆2-enoil-CoA
H2O OH O NADH + H+
R-C-CH2-C-SCoA
H
L-Hidroxil-CoA NAD+
Figura - No Ciclo de Lynen, a acil-CoA formada no final de cada volta tem dois carbonos a menos e reinicia o ciclo.
A oxidação de ácidos graxos não é restrita à mitocôndria. A degradação de ácidos graxos ocorre em outras organelas citoplasmáticas: glioxissomos e peroxissomos. Nos vegetais, a β-oxidação é feita apenas nos glioxissomos. A acetil-CoA produzida pode ser convertida em glicose, graças à presença, nestas organelas, das enzimas do ciclo do glioxilato. Estas enzimas são ausentes de células animais, que, por isto, são incapazes de utilizar acetil-CoA como um composto gliconeogênico. Nos animais, a via de oxidação peroxissômica promove o encurtamento de ácidos graxos muito longos (com mais de 20 carbonos). Os ácidos graxos de cadeia longa penetram nos peroxissomos sem auxílio da carnitina e são convertidos nas respectivas acil-CoA. A oxidação das acil-CoA longas é catalisada por uma acil-CoA oxidase, que reduz oxigênio à água oxigenada, que é então decomposta em H2O e O2
por ação da catalase.
Oxidação de ácidos graxos com nº ímpar de carbonos
Ácidos graxos com o número ímpar de átomos de carbonos constituem uma pequena fração dos ácidos graxos da dieta e são também oxidados pela

145
β-oxidação. Neste caso, a última volta do ciclo de Lynen inicia-se com uma acil-CoA de cinco carbonos e produz uma molécula de acetil-CoA e uma propionil-CoA, em vez de duas de acetil-CoA. A propionil-CoA é convertida a succinil-CoA, um intermediário do ciclo de Krebs, para sua oxidação.
HIPERTEXTO
Deficiência de Vitamina B12
A existência da vitamina B12 foi descoberta em 1926, quando, Geoge Minot e William Murphy descobriram que a anemia perniciosa, uma doença frequentemente fatal nos idosos, caracterizada pela redução do número dos glóbulos vermelhos, por baixos níveis de hemoglobina e pela deterioração neurológica progressiva, podia ser tratada por meio do consumo diário de grandes quantidades de fígado cru. Entretanto, o fator anti-anemia perniciosa – a vitamina B12 – não foi isolado até 1948. A vitamina B12 não é sintetizada por plantas nem por animais, somente por algumas espécies de bactérias. Os herbívoros obtêm vitamina B12 das bactérias que habitam o trato digestivo (na verdade, alguns animais, como o coelho, devem periodicamente comer um pouco de suas fezes para obterem quantidades suficientes dessa substância essencial). Os seres humanos, no entanto, obtêm quase toda vitamina B12
diretamente da dieta, em especial da carne. No intestino, a glicoproteína fator intrínseco, secretada pelo estômago, se liga especificamente à vitamina B12, e o complexo proteína-vitamina é absorvido via um receptor na mucosa intestinal. O complexo dissocia-se, e a vitamina B12 liberada é transportada à corrente sanguínea. Pelo menos três proteínas plasmáticas diferentes, as transcobalaminas, ligam-se à vitamina e facilitam sua captação pelos tecidos. A anemia perniciosa em geral não é uma doença de deficiência da dieta, mas normalmente resulta de uma secreção insuficiente do fator intrínseco. A necessidade humana normal de cobalamina é muito pequena, ~ 3µg ao dia, e o fígado armazena um suprimento dessa vitamina suficiente para 3 a 5 anos. Isso justifica o início tardio da anemia perniciosa e o fato da deficiência dietética de vitamina B12, mesmo entre vegetarianos estritos, ser extremamente rara (Voet, Voet & Pratt, 2000).
Oxidação de ácidos graxos insaturados
Os ácidos graxos insaturados são muito comuns em tecidos animais e vegetais, apresentando suas duplas ligações quase sempre a configuração cis. Depois de remover algumas unidades de dois carbonos (Acetil-CoA) pelo ciclo de Lynen, o ácido graxo insaturado pode dar origem a dois tipos de enoil-CoA, conforme a posição original da dupla ligação em sua molécula: se a dupla ligação for de nº ímpar (ex.: Δ9 do ácido oléico), forma-se uma cis-Δ3-enoil-CoA; se for de nº par (como a Δ12 do ácido linoleico), será formada uma cis-Δ4-enoil-

146
CoA. Para a oxidação dessas acil-CoA insaturadas, são necessárias, além de enzimas da β-oxidação, outras enzimas que as convertam em trans-Δ2-enoil-CoA. O intermediário insaturado da β-oxidação.
Ácidos graxos ramificados ou hidroxilados
Ácidos graxos que possuem ramificações ou hidroxilações são comuns em lipídeos das bactérias, porém pouco frequentes em animais superiores. Nos animais superiores, os ácidos graxos ramificados ocorrem apenas como componentes da cera produzida pelas glândulas sebáceas, e os hidroxilados (com uma OH no carbono α), como componentes de esfingolipídeos do tecido nervoso.
Os ácidos graxos ramificados são importantes constituintes da alimentação (gramíneas) de animais ruminantes e, em consequência, aparecem em componentes da dieta dos seres humanos através das gorduras de origem animal, do leite e de seus derivados, como, por exemplo, o ácido fítânico. Esses ácidos possuem um grupo metil no carbono β, não reconhecido pela acil-CoA desidrogenase, que catalisa a primeira reação da β-oxidação. Essa situação é contornada pela α-oxidação que ocorre nos peroxissomos, iniciando-se com a hidroxilação de carbono α. Segue-se uma oxidação e uma descarboxilação, resultando em um composto que tem o radical metil agora no carbono α e apresenta o carbono β não substituído, podendo ser ativado e oxidado pelo ciclo de Lynen.
Corpos cetônicos
Acetona, Acetoacetato, β-Hidroxibutirato
Na ausência de carboidrato suficiente, quantidades maiores de gordura são usadas para produzir energia, sendo maiores do que o organismo pode processar e restando numa oxidação incompleta, pois os mamíferos não dispõem de via capaz de transformar ácidos graxos, principalmente os da reserva lipídica, em glicose. O acúmulo de intermediários acídicos leva à acidose, ao desequilíbrio sódico e à desidratação. Por um processo conhecido como cetogênese, que ocorre principalmente nas mitocôndrias do fígado, a acetil-CoA é convertida em acetoacetato ou em β-hidroxibutirato. O acetoacetato sofre descarboxilação espontânea, originando acetona. Esses compostos são referidos como corpos cetônicos, e sua síntese é chamada cetogênese. Os corpos cetônicos são combustíveis metabólicos importantes para vários tecidos periféricos, em particular para o coração e para o músculo esquelético.
O O O

147
H3C – C – CH2 – C H3C – C – CH3
O-
Acetacetato Acetona
OH O
H3C – C – CH2 – C
H O-
β - Hidroxibutirato
O cérebro, em circunstâncias normais, utiliza apenas a glicose como fonte de energia (ácidos graxos são incapazes de cruzar a barreira hemato-encefálica), mas, durante um jejum prolongado, os corpos cetônicos tornam-se a principal fonte de combustível metabólico do cérebro. Corpos cetônicos são equivalentes hidrossolúveis dos ácidos graxos. Os corpos cetônicos são liberados na corrente sanguínea, e o acetoacetato e o β-hidroxibutirato são aproveitados como fonte de energia pelos tecidos extra-hepáticos, principalmente coração e músculos esqueléticos. A acetona, por sua vez, é volatilizada pelos pulmões. Os corpos cetônicos são veículos para a transferência de carbonos oxidáveis (originados da Acetil-CoA) do fígado para outros órgãos.
Quando não há oferta de glicose, o organismo lança mão da gliconeogênese que consome oxaloacetato obtido de aminoácidos, principalmente. A oxidação da acetil-CoA pelo ciclo de Krebs fica, então, impedida: a acetil-CoA acumulada condensa-se, formando os corpos cetônicos. É o que ocorre quando há redução drástica da ingestão de carboidratos (jejum ou dieta) ou distúrbios de seu metabolismo (diabetes). Quando a produção ultrapassa o aproveitamento pelos tecidos extra-hepáticos, estabelece-se uma condição denominada Cetose, caracterizada por uma concentração elevada de corpos cetônicos no plasma (cetonemia) e na urina (cetonúria). Um sintoma, característico de indivíduos com Cetose é o odor de acetona de seu hálito. Como os outros dois corpos cetônicos são ácidos, a cetonemia resulta em acidose, isto é, uma diminuição do pH sanguíneo. Em casos de Cetose acentuada, o cérebro obtém uma boa parte de energia de que necessita por oxidação dos corpos cetônicos.

148
Metabolismo do Etanol
Os seres humanos ao ingerirem etanol apresentam absorção rápida. Ocorre a oxidação do etanol a acetaldeído no fígado, por uma álcool desidrogenase citossólica, em uma reação idêntica à última etapa da fermentação alcoólica por leveduras, neste caso em sentido diverso.
H O
CH3-C-OH + NAD+ CH3- C -H + NADH + H+
H
Etanol Acetaldeído
O equilíbrio dessa reação vai favorecer a formação de etanol, porém sua oxidação vai prosseguir por causa da conversão de acetaldeído em acetato, catalisada pela acetaldeído desidrogenase mitocondrial.
O O
CH3 - C - H + H2O + NAD+ CH3 – C – O- + NADH + 2H+
Acetaldeído Acetato
O acetato (como os ácidos graxos) origina acetil-CoA por ação de uma acil-CoA sintetase. Neste ponto, o metabolismo do etanol confunde-se com o metabolismo de carboidratos, lipídios e proteínas, que também originam acetil-CoA. Quando ocorre um consumo de quantidades discretas de etanol, significa que haverá um consumo adicional de calorias, que devem ser somadas àquelas derivadas da ingestão de nutrientes no cômputo das calorias totais da dieta. Porém, quando há ingestão constante de etanol, nem sequer o seu conteúdo calórico é aproveitado pelo organismo. Uma via minoritária, catalisada por um sistema enzimático localizado na membrana do retículo endoplasmático, oxida etanol, utilizando O2, mas sem o concurso da cadeia de transporte de elétrons e, portanto, sem a produção de ATP. No alcoolismo crônico, em que há ingestão de grandes quantidades de etanol, ocorrem inúmeras consequências danosas para o organismo. A oxidação do etanol nas células hepáticas produz altos níveis de NADH no citossol, onde normalmente a concentração NAD+ é muito maior do que a de NADH. A alta concentração de NADH vai deslocar a reação catalisada pela lactato desidrogenase no sentido da formação de lactato, cuja concentração poderá aumentar em até 5 vezes, levando portanto a uma acidose. A constante conversão da piruvato a lactato impossibilita a gliconeogênese, já que todos os amonoácidos glicogênicos devem ser primeiramente convertidos a piruvato; ou seja, em vez de originarem glicose, são transformados em lactato. Muitas vezes, a ingestão de álcool não é acompanhada da ingestão de nutrientes, então, diminuída a reserva de glicogênio, pode ocorrer hipoglicemia e, finalmente, o coma. Uma produção alta

149
acetil-CoA, associada à baixa disponibilidade de glicose, vai ocasionar Cetose. É importante lembrar que concentrações altas de acetaldeído são tóxicas, já que se trata de uma substância extremamente reativa e que se liga covalentemente a proteínas, modificando a sua estrutura e sendo responsável pela ressaca.
Síntese de Ácidos Graxos
Grande parte dos ácidos graxos usada pelo organismo é suprida pela dieta. Carboidratos, proteínas e outras moléculas obtidas da dieta em excesso, podem ser convertidas em ácidos graxos, que são armazenados como triacilgliceróis. Em humanos adultos, a síntese de ácidos graxos ocorre principalmente no fígado e nas glândulas mamária sem lactação e, em menor extensão, no tecido adiposo. O processo incorpora carbonos da acetil-CoA na cadeia de ácido graxo em crescimento, usando adenosina trifosfato (ATP) e Nicotinamida Adenina Dinucleotídeo fosfato reduzido (NADPH). Na via que leva à produção de ácidos graxos, o substrato imediato é a acetil-CoA e o produto final é o ácido palmítico. A síntese ocorre no citossol, para onde deve ser transportada a acetil-CoA formada na mitocôndria, a partir de piruvato. Como a membrana interna da mitocôndria é impermeável a acetil-CoA, os seus carbonos são transportados sob a forma de citrato. Os carboidratos e proteínas (precursores dos ácidos graxos) são degradados a acetil-CoA e oxaloacetato, que sofrem condensação, formando citrato, por ação da primeira enzima do ciclo de Krebs, a citrato sintase. Nas condições consideradas, o citrato não pode ser oxidado pelo ciclo de Krebs em virtude da inibição da isocitrato desidrogenase e é transportado para o citossol pela tricarboxilato translocase, onde é desmembrado em oxaloacetato e acetil-CoA, à custa de ATP, numa reação catalisada pelo citrato liase.
COO COO
CH2 C=O O
HO-C-COO- + ATP 4 + HS-CoA CH2 + H2O –C– ScoA + ADP + P1
CH2 COO
COO
Citrato Oxaloacetato Acetil-CoA
O oxaloacetato é reduzido a malato pela malato desidrogenase do citossol. O malato é substrato da enzima málica. Nesta reação, são produzidos piruvato, que retorna à mitocôndria, e NADPH.
COO- COO-

150
HO –C– H + NADP+ O=C + CO2 + NADPH
CH2 CH3
COO-
Malato Piruvato
O resultado final destas reações é o transporte dos carbonos da acetil-CoA (sob a forma de citrato), com gasto de ATP, da mitocôndria para o citossol, e, ainda, a produção de NADPH. Acetil-CoA e NADPH, ambos no citossol, podem, então, ser utilizados para formar ácidos graxos. A síntese de ácidos graxos consiste na união sequencial de unidades de dois carbonos: a primeira unidade é proveniente de acetil-CoA, e as outras, de malonil-CoA, formada por carboxilação de acetil-CoA. A enzima acetil-CoA carboxilase que catalisa essa reação tem como grupo prostético a biotina.
O O O
H3C – C + ATP + HCO3- C – CH2 – C + ADP + Pi + H+
SCoA -O SCoA
Acetil-CoA Malonil-CoA
A síntese de ácidos graxos é catalisada por um sistema enzimático denominado sintase de ácidos graxos. Também faz parte destas sintases uma pequena proteína não-enzimática, designada proteína carregadora de acila, ou ACP, à qual está sempre ligada a cadeia do ácido graxo em crescimento. O ACP tem como grupo prostético um derivado do ácido pantotênico, a fosfopanteteína, também componente da coenzima A. A sequência das reações de síntese (condensação, redução, desidratação e redução) é inversa à sequência das reações de um ácido graxo pelo ciclo de Lynen (oxidação, hidratação, oxidação, quebra da cadeia carbônica). Os processos diferem quanto às enzimas e coenzimas utilizadas, o compartimento celular onde se processam e o suporte da cadeia carbônica (CoA ou ACP).
As enzimas constituintes da sintase são: acetil-CoA-ACP-transacilase (1), malonil-CoA-ACP-transacilase (2), β-cetoacil-ACP sintase (3), β-cetoacil-ACP redutase (4), β-hidroxiacil-ACP desidratase (5) e enoil-ACP redutase (6). A sintase de ácidos graxos pode ser representada por uma esfera, na qual se destacam o ACP, com sua sulfidrila terminal, e a β-cetoacil-ACP sintase (enzima 3), com o grupo SH de um de seus resíduos de cisteína. Também aparece o primeiro ciclo de síntese, que leva à formação de butiril-ACP. Para que haja o alongamento da cadeia carbônica, o butiril-ACP sintetizado no final da primeira volta sofre a mesma sequência de reações
Carboxilase
acetil-CoA

151
(enzimas 2 a 6), que o acetil-ACP; o radical butiril, é transferido para o grupo SH da enzima 3, como ocorreu com o radical acetil no início da primeira volta, prosseguindo as reações como no primeiro ciclo de síntese. Nos seres humanos, a maioria dos ácidos graxos é produzida pelo fígado a partir de carboidratos da dieta e exportados para os outros tecidos através das lipoproteínas plasmáticas.
CONTEXTUALIZANDO
Corpos Cetônicos como Combustíveis: A Dieta Atkins
A mais conhecida das dietas pobres em carboidratos é a Dieta Atkins. Essa dieta é rica em gordura e proteína e muito pobre em carboidratos (menos do que 20g /dia durante a fase inicial), sendo motivo de controvérsias entre nutricionistas e médicos, devido a seu alto teor de gordura. O interessante é que indivíduos que fazem essa dieta perdem uma quantidade considerável de peso. Estudos clínicos demonstraram que indivíduos obesos perdem mais peso em uma dieta rica em gordura e pobre em carboidratos do que em uma dieta isocalórica com níveis mais altos de carboidratos. É surpreendente também que ocorra uma notável queda nos níveis de triacilgliceróis no sangue desses indivíduos. Até agora, não existem estudos de longo prazo do efeito do perfil lipídico ou da saúde geral dessas pessoas obesas que permanecem nessa dieta. Entretanto, é provável que a dieta Atkins resulte em dramática perda de peso, devido à saciedade causada pelo alto conteúdo de gordura e proteína da dieta, resultando em uma redução de ingesta alimentar. O centro da dieta Atkins é a mobilização de ácidos graxos do tecido adiposo e sua conversão pelo fígado em corpos cetônicos (β-hidroxibutirato, acetoacetato e acetona). A presença de corpos cetônicos na urina é o método prescrito para determinar o estado metabólico durante a dieta, uma vez que mesmo quantidades pequenas de carboidratos na dieta reprimem a síntese de corpos cetônicos, principalmente por inibirem lipólise no tecido adiposo. À medida que a concentração de corpos cetônicos sobe no sangue, uma fração é excretada na urina e uma parte pela respiração. Seria isso responsável pela maior perda de peso observada em indivíduos na dieta Atkins? Para comparação, após 7 dias de jejum, a taxa de excreção urinária diária de acetoacetato e β-hidroxibutirato no homem é de aproximadamente 110 mmol/dia; a taxa de excreção é ainda menor no início do jejum (60 mmol/dia após dois dias de jejum). Esta excreção poderia contribuir para o balanço calórico negativo e perda de peso característicos da dieta de Atkins, embora a perda de energia não exceda 100 kcal/dia.
Em geral, a Cetose ocorre quando a oxidação de glicose é suprimida e o catabolismo de gordura é acelerado. Existem dois tipos de Cetose: a Cetose normal do jejum e a hipercetonemia patológica da cetoacidose diabética.

152
Nenhum outro combustível do sangue humano pode mudar tão drasticamente como os corpos cetônicos, e ainda ser compatível com a vida. Após jejum de uma noite, a concentração de corpos cetônicos é aproximadamente 0,05 mM, mas esta concentração pode subir para 2 mM após 2 dias de jejum e para 7 mM após 40 dias, uma mudança de 140 vezes na concentração. Resultados de pesquisas têm demonstrado que durante um jejum prolongado acetoacetato e β-hidroxibutirato substituem a glicose como o combustível predominante para o cérebro. Isso reduz a necessidade de síntese de glicose a partir de aminoácidos derivados de proteína muscular e do fígado. O tecido muscular consome avidamente corpos cetônicos logo no início do jejum, mas muda para oxidação de ácidos graxos à medida que o jejum progride, economizando assim corpos cetônicos para o metabolismo do cérebro. Portanto, corpos cetônicos são o combustível normal para uma variedade de tecidos e parte de um padrão complexo de metabolismo de combustíveis que ocorre durante o jejum no homem (Devlin, 2007).
ATIVIDADES DE AVALIAÇÃO
1. Esquematizar:
a) a reação catalisada pela lipase do tecido adiposo;
b) as reações de conversão de glicerol a compostos intermediários da via glicolítica;
c) o primeiro passo necessário para a degradação de um ácido graxo (ativação).
2. Citar a localização celular da oxidação dos ácidos graxos e mostrar o papel da carnitina.
3. Citar os compostos formados no fim de cada volta do ciclo de Lynen.
4. Indicar o nº de moléculas de ATP necessárias para degradar uma molécula de ácido graxo.
5. Listar as vitaminas que participam do Ciclo de Lynen.
6. Descrever a regulação do Ciclo de Lynen.
7. Indicar os tecidos que não oxidam ácidos graxos.
8. Fale sobre o metabolismo do etanol indicando os principais passos.
9. Por que a ingestão de etanol em grandes quantidades resulta em problemas?

153
10. Fale sobre o metabolismo da síntese de ácidos graxos.
11. Que são corpos cetônicos? Dê suas estruturas.
12. Como se formam os corpos cetônicos e em quais tecidos do organismo?
13. Defina: cetonúria; cetonemia; acidose; cetose, indicando quando e por que ocorre cada um desses processos.
14. Indique como se processa a síntese de ácidos graxos.
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. No Capítulo 21, do vol. 3, o assunto Metabolismo dos Lipídeos é amplamente discutido.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade III do livro, nos Capítulos 15, 16, 17 e 18 são apresentados tópicos relacionados ao Metabolismo dos Lipídeos com muitas ilustrações, tornando o assunto de fácil compreensão.
Devlin, T. M. Manual de Bioquímica com Correlações Clínicas. São Paulo: Editora Blucher, 2007. Os Capítulos 17 e 18 da Parte IV desse livro abordam tópicos relacionados ao Metabolismo dos Lipídeos.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª ed. 1999. Rio de Janeiro: Editora Guanabara Koogan S. A. O Capítulo 16 da Parte 3 desse livro explana de forma bastante compreensiva o assunto Metabolismo dos Lipídeos.
Pelley, J. W. Bioquímica. Nos Capítulos 10 e 11 são estudados tópicos relativos ao Metabolismo dos Lipídeos de uma forma sucinta e objetiva. Rio de Janeiro: Elsevier, 2007.
Smith, C.; Marks, A. D.; Lieberman, M. Bioquímica Médica Básica de Marks – Uma abordagem clínica. 2ª ed. Porto Alegre: Artmed, 2007. Os Capítulos 32, 33, 34, 35 e 36 abordam tópicos relacionados ao Metabolismo dos Lipídeos de modo detalhado e didático.
Voet, D.; Voet, J. G.; Pratt, C. W. Fundamentos de Bioquímica. Porto Alegre: Artmed, 2000. O Capítulo 19 da Parte IV traz uma discussão bastante abrangente sobre o Metabolismo dos Lipídeos.LEITURA, VÍDEOS E SITES RECOMENDADOS:
underpop.free.fr/b/biologia/metabolismo-de-lipIdios.doc
colesterolgorduraomega.blogspot.com

154
CAPÍTULO 13: METABOLISMO DE AMINOÁCIDOS
OBJETIVOS:
Compreender o papel dos aminoácidos em nosso corpo e como eles são importantes para a manutenção do nosso organismo.
Verificar o papel do nitrogênio proveniente das proteínas.
Entender melhor o ciclo da ureia e associá-la com a excreção do nitrogênio não absorvido pelo nosso organismo.
Conhecer as doenças decorrentes da deficiência do metabolismo dos aminoácidos.
Um adulto saudável ingerindo uma dieta variada está, geralmente, em “balanço de nitrogênio”, ou seja, em um estado em que a quantidade de nitrogênio ingerida a cada dia é equilibrada pela quantidade excretada, resultando em alteração líquida nula no nitrogênio total do corpo. Na condição de um organismo bem alimentado, o nitrogênio excretado vem principalmente do excesso de proteínas ingerido ou da reciclagem (turnover) normal. O turnover de proteínas é definido como a substituição de proteína do corpo por síntese e degradação. Em certas condições, o corpo está em balanço negativo ou positivo de nitrogênio. No balanço negativo de nitrogênio, mais nitrogênio é excretado do que ingerido. Isso ocorre no jejum e em certas doenças. Em um ser humano com idade adulta e dieta adequada, há uma renovação de aproximadamente 400g de proteína por dia.
Uma consequência importante do turnover proteico é restar sempre certa quantidade de aminoácidos não utilizados porque o conjunto de aminoácidos originado das proteínas que estão sendo degradadas nunca é igual ao conjunto de aminoácidos necessários para compor as proteínas que estão sendo sintetizadas. Tendo em vista que não há meios de armazenar os aminoácidos excedentes, eles são oxidados e seu nitrogênio, excretado. Um adulto saudável, com dieta adequada, elimina uma quantidade de nitrogênio correspondente a 100g de proteína aproximadamente. Como 400g de proteínas devem ser renovados neste período, faltam os 100g eliminados, que devem ser repostos pela alimentação. Os aminoácidos presentes nas células animais originam-se, então, das proteínas da dieta (exógenas) e das proteínas
RENOVAÇÃO TURNOVER

155
endógenas e constituem um “pool”: ¼ derivado das proteínas da dieta e ¾ das proteínas endógenas. O “pool” de aminoácidos é usado para síntese de proteínas endógenas e de outras moléculas que contenham nitrogênio. Os aminoácidos são precursores de todos os compostos nitrogenados não-proteicos, que incluem as bases nitrogenadas constituintes dos nucleotídios (dos ácidos nucleicos, coenzimas) e as aminas e seus derivados, como adrenalina, histamina etc.
Aminoácidos Essenciais
A biossíntese de proteínas exige a presença de todos os aminoácidos constituintes. Se um dos vinte aminoácidos estiver ausente ou em pequena quantidade, a biossíntese proteica é inibida. Alguns organismos, como a Escherichia coli, podem sintetizar todos os aminoácidos de que precisam. Outras espécies, incluindo os seres humanos, devem obter alguns aminoácidos dos alimentos. São os chamados aminoácidos essenciais para a nutrição humana (ver Tabela 1). O organismo pode sintetizar alguns desses aminoácidos, mas não em quantidades suficientes para suas necessidades, especialmente no caso de crianças em fase de crescimento. As crianças têm necessidade principalmente de arginina e histidina. Como os aminoácidos não são armazenados (exceto nas proteínas), as fontes alimentares de aminoácidos essenciais são necessárias em intervalos regulares. A falta de proteínas, especialmente uma deficiência prolongada de fontes que contenham aminoácidos essenciais, leva à doença kwashiorkor. O problema dessa doença, muito grave em crianças em fase de crescimento, não é simplesmente a desnutrição, mas a degradação das proteínas do próprio organismo.
Como já foi dito, os seres vivos não são capazes de armazenar aminoácidos nem proteínas e, consequentemente, satisfeitas as necessidades de síntese, os aminoácidos excedentes são degradados. Em um adulto saudável, com dieta adequada, a oxidação de aminoácidos responde por 10 – 15% de suas necessidades energéticas. Como a estrutura dos aminoácidos revela, eles são simplesmente carboidratos ligados a um nitrogênio. Logo, os aminoácidos excedentes são degradados, restando as respectivas cadeias carbônicas e o grupo amino, que é convertido em ureia.
Tabela 1 - Necessidades de aminoácidos no ser humano
Essenciais Não-Essenciais
Arginina Alanina

156
Histidina Asparagina
Isoleucina Aspartato
Leucina Cisteína
Lisina Glutamato
Metionina Glutamina
Fenilalanina Glicina
Treonina Prolina
Triptofano Serina
Valina Tirosina
Fonte: Campbell & Farrel, 2008
Degradação de Aminoácidos
Quando os aminoácidos não são necessários à síntese de outras moléculas contendo nitrogênio, eles são convertidos a carboidratos. A degradação dos aminoácidos, portanto, compreende a remoção e a excreção do grupo amino e a oxidação da cadeia carbônica remanescente (α-cetoácido). Quando o nitrogênio é removido do aminoácido, o carboidrato residual é convertido em piruvato ou em um intermediário do ciclo do ácido cítrico para a produção de energia ou gliconeogênese. Como a amônia é tóxica, uma via é designada para converter o nitrogênio do aminoácido em um composto não tóxico neutro, a ureia, que é eliminada na urina. Em resumo, pode-se dizer então que os aminoácidos, ao se degradarem, têm o grupo amino convertido em uréia, e as vinte cadeias carbônicas resultantes são convertidas em compostos comuns ao metabolismo de carboidratos e lipídios, ou seja, piruvato, acetil-CoA e intermediários do ciclo de Krebs.
Como é Removido o Grupo Amino dos Aminoácidos
O nitrogênio dos aminoácidos se transfere para o ciclo da ureia em três etapas: transaminação, formação da amônia e formação da ureia. O grupo amino da maioria dos aminoácidos é coletado inicialmente como glutamato. O grupo amino de: alanina, arginina, aspartato, asparagina, cisteína, fenilalanina, glutamina, isoleucina, leucina, tirosina, triptofano e valina é retirado por um processo comum, que consiste na transferência deste grupo para o α-cetoglutarato, formando glutamato; a cadeia carbônica do aminoácido é convertida ao α-cetoácido correspondente. É importante lembrar que o glutamato é o principal doador de grupos amino nas reações enquanto o α-cetoglutarato é o principal aceptor.
Aminoácido + α-Cetoglutarato α-Cetoácido + Glutamato

157
Estas reações são catalisadas pelas aminotransferases, também chamadas transaminases (presentes no citossol e na mitocondria e que têm como coenzima piridoxal-fosfato) e transferem o grupo α-amino do aminoácido para o α-cetoglutarato produzindo glutamato. Existem doze tipos de transaminases que catalisam a eliminação do nitrogênio através da formação do glutamato. Duas transaminases clinicamente importantes servem como marcadoras de lesão hepática quando aparecem em altas concentrações no sangue: a aspartato aminotransferase (AST) que catalisa a transaminação reversível do nitrogênio entre aspartato e glutamato; a alanina aminotransferase (ALT), que catalisa a transaminação reversível do nitrogênio entre alanina e piruvato. O piridoxal-fosfato, a forma ativa da vitamina B6, (piridoxina), torna-se necessária como coenzima das aminotransferases.
Na reação geral de transaminação, inicialmente, o grupo amino de um aminoácido é transferido ao piridoxal fosfato, que é convertido à piridoxamina fosfato; a seguir é doado ao α-cetoglutarato, produzindo glutamato. O glutamato é um produto comum às reações de transaminação, constituindo um reservatório temporário de grupos amino, provenientes de diferentes aminoácidos. O aumento da produção de glutamato, (precursor do GABA, um neurotransmissor), gerando um excesso, provoca destruição de neurônios.
Ác. Glutâmico Ác. α-Cetoglutárico
COOH COOH
CH2 CH2
CH2 CH2
CHNH2 C=O
COOH COOH
CH3 CH3
C=O CHNH2
COOH COOH
Ác. Pirúvico Alanina
Figura - Esquema da Reação de Transaminação
O ácido pirúvico pode ser aminado para formar alanina que é transportada para o fígado onde é desaminada e o esqueleto carbônico é reconvertido à glicose. O ciclo da alanina move N do músculo para o fígado sem produzir NH3. Em uma segunda etapa, os grupos amino originam

158
aspartato e/ou amônia. O glutamato formado é consumido em duas reações importantes, uma nova transaminação e uma desaminação. Na remoção do grupo amino do próprio glutamato por transaminação pela ação da aspartato aminotransferase, o grupo amino do glutamato é transferido para o oxaloacetato, formando aspartato.
COO- COO- COO- COO-
H3N+ –C– H + C = O H3N+ –C– H + C = O
CH2 CH2 CH2 CH2
CH2 COO- COO- CH2
COO- COO-
Glutamato Oxaloacetato Aspartato α-Cetoglutarato
A desaminação oxidativa do glutamato em α-cetoglutarato na matriz mitocondrial produz amônia livre. Em outras palavras, o grupo amino do glutamato pode ser liberado como amônia (íon NH4
+, em pH fisiológico); A reação é catalisada pela glutamato desidrogenase (GDH), encontrada principalmente no fígado, e produzindo NADH ou NADPH.
NH3
R – CH – COOH R – C – COOH R – C – COOH
NH2 NH H2O O
α-aminoácido α-iminoácido α-cetoácido
Figura - Desaminação Oxidativa
A reação é reversível sendo, portanto possível incorporar amônia livre em α-cetoglutarato para formar glutamato quando necessário. Os esqueletos de carbono são convertidos em alguns dos mesmos intermediários formados durante o catabolismo da glicose e ácidos graxos. Podem ser levados para os tecidos periféricos, onde entram no ciclo do ácido cítrico para produzir adenosina trifosfato (ATP). Estes fragmentos também podem ser usados nos processos de síntese para fabricar glicose ou gorduras. Aproximadamente 58% da proteína consumida podem ser convertidos em glicose desta maneira. A maioria dos aminoácidos, particularmente a alanina, é potencialmente glicogênica. O piruvato a partir da oxidação da glicose no músculo é aminado para formar a alanina que, por sua vez, é transportada para o fígado onde é desaminada e o esqueleto de C é reconvertido à glicose.
AST
Enzima

159
O ciclo da alanina é uma importante fonte de glicose durante períodos de baixo suprimento exógeno. É também um método de mover N do músculo para o fígado sem a formação de amônia. O grupo amino é liberado na desaminação principalmente como amônia, a qual é usada em processos sintéticos ou carregada para o fígado para conversão em ureia, a forma na qual a maioria dela é excretada. Como a amônia é altamente tóxica, é transportada em combinação com o ácido glutâmico como glutamina.
A amônia é liberada da matriz mitocondrial e serve como um precursor do ciclo da ureia. A ação combinada das aminotransferases e da glutamato desidrogenase resulta na convergência do grupo amino da maioria dos aminoácidos para dois compostos únicos: NH4
+ e aspartato. O aspartato é o segundo depositário do grupo amino dos aminácidos.
COO- COO-
H3N+ –CH + NAD(P)+ + H2O C = O + NAD(P)H + H+ + NH4+
CH2 CH2
CH2 CH2
COO- COO-
Glutamato α-cetoglutarato
Reação catalisada pela glutamato desidrogenase. A glutamato desidrogenase é específica para glutamato.
Reações Especiais para Desaminação de alguns Aminoácidos
Glicina, Histidina, Lisina, Metionina, Prolina, Serina e Treonina, não participam de reações de transaminação. Os grupos amino desses aminoácidos são removidos por reações particulares a cada um deles. Um aspecto comum e importante do metabolismo destes aminoácidos é a forma de remoção do grupo amino que ou é liberado como NH4
+ por reações de desaminação, ou forma glutamato através de transaminação de um intermediário aminado com α-cetoglutarato. Desta forma, os átomos de N destes aminoácidos convergem para os mesmos produtos originados pelo grupo amino dos outros aminoácidos: NH4
+ e glutamato, que pode originar aspartato. Pode-se concluir, portanto que durante a degradação dos 20 aminoácidos, o grupo amino é convertido finalmente em NH4
+ e aspartato, os precursores da ureia. A ureia é sintetizada a partir de NH4
+, aspartato e HCO3-.
Os dois átomos de N presentes na ureia são provenientes de NH4+ e aspartato,
e o átomo de carbono, do bicarbonato. A síntese da ureia é feita no fígado, através do ciclo da ureia.

160
Ciclo da Ureia
A síntese da ureia ocorre através do Ciclo da Ornitina. O CO2 e NH3
(com energia da ATP) se combinam com a ornitina através de uma série de passos para formar a arginina que é então hidrolisada para produzir ureia e ornitina. Sendo assim, uma molécula de ornitina é usada repetidamente na formação da arginina e ureia.
CICLO DA ORNITINA
O
NH4+ + HCO3
- H2NC – OPO3
O
CH2NH3+ C NH2
CH2 NH
CH2 CH2
CHNH3+ CH2
COO- CH2
Ornitina NH2-C-NH2 Arginina CHNH3+
O COO-
Ureia Citrulina
O glutamato formado vai dar origem a aspartato e/ou amônia.
COO- COO- COO- COO-
H3N+ –C– H + C = O H3N+ –C– H + C = O
CH2 CH2 CH2 CH2
CH2 COO- COO- CH2
COO- COO-
Glutamato Oxaloacetato Aspartato α-Cetoglutarato
AAT= Aspartato Amino Transferase
O ciclo da ureia inicia-se na matriz mitocondrial terminando com a formação da ureia no citoplasma. No início da síntese, na matriz mitocondrial, ocorre a formação de carbamoil-fosfato a partir de íons bicarbonato e amônio, com gasto de duas moléculas de ATP. Os íons amônia se ligam com o dióxido
ATP
CARBAMOILFOSFATO
AAT
NO
VATR
ANSA
MIN
AÇÃO

161
de carbono e ATP para produzir carbamoil-fosfato, numa reação catalisada pela carbamoil-fosfato-sintetase.
Em seguida, o carbamoil-fosfato condensa-se com ornitina, numa reação catalisada pela ornitina transcarbamoilase, formando citrulina. Tanto a ornitina como a citrulina têm carreadores específicos de membrana na mitocôndria. No citoplasma, a citrulina e o ácido aspártico se condensam para formar argininossuccinato numa reação catalisada pela argininosuccinato-sintetase. O composto argininossuccinato é clivado em reação catalisada pela argininossuccinase e se decompõe em arginina e fumarato. A arginina é hidrolisada, em reação catalisada pela arginase regenerando ornitina e produzindo ureia, que é transportada para o rim e eliminada pela urina. A ureia é o principal produto de excreção do metabolismo nitrogenado de vertebrados terrestres; aves e reptéis excretam ácido úrico, e, peixes, amônia. A quantidade de ureia excretada por um ser humano adulto é cerca de 30g por dia, mas este valor varia proporcionalmente à quantidade de proteína ingerida. A ureia representa 90% dos compostos nitrogenados excretados; o restante aparece sob a forma de NH4
+, creatinina e ácido úrico. Apesar de NH4+ constituir um
pequeno percentual do N urinário, sua excreção possibilita a eliminação de H+, contribuindo para a manutenção do equilíbrio ácido-base: o conteúdo de NH4
+
da urina aumenta na acidose e diminui na alcalose. Por desaminação ocorre:
COO- Glutamato COO-
H3N+ –CH + NAD(P)+ + H2O C = O + NAD(P)H + H+ + NH4+
CH2 desidrogenase CH2
CH2 CH2
COO- COO-
Glutamato α-Cetoglutarato
A ação combinada das aminotransferases + glutamato desidrogenase originam: +NH4 + aspartato, ou seja:
AMINOÁCIDO α-CETOGLUTARATO ASPARTATOOXALOACETATO
α-CETOGLUTARATOα-CETOÁCIDO GLUTAMATO
T T
GD

162
Compostos nitrogenados excretados pelo homem
Composto Quantidade excretada (g/dia)
Ureia 30
NH4+ 0,7
Creatinina 1,4
Ácido úrico 0,8
A amônia é tóxica para os tecidos animais, especialmente para o cérebro. A conversão da maior parte do NH4
+ em ureia é fundamental para manter baixas as concentrações deste íon nos tecidos. Quando há uma restrição na formação de ureia por disfunção hepática grave – hepatite ou cirrose, por exemplo – a concentração de NH4
+ se eleva, afetando principalmente o cérebro e podendo ocasionar o coma.
Como a amônia é tóxica e a sua conversão em ureia ocorre no fígado, o NH4
+ produzido nos outros tecidos, para ser transportado ao fígado, é incorporado em compostos não-tóxicos e que atravessam membranas com facilidade: glutamina, na maioria dos tecidos extra-hepáticos, e alanina, no músculo. A glutamina e a alanina são, portanto, os transportadores de amônia para o fígado.
Degradação do Esqueleto Carbônico dos Aminoácidos
Quando é removido o grupo amino do aminoácido, resta sua cadeia carbônica, na forma de α-cetoácido. Estes esqueletos de carbono entram no metabolismo intermediário em várias etapas, dependendo se serão convertidos
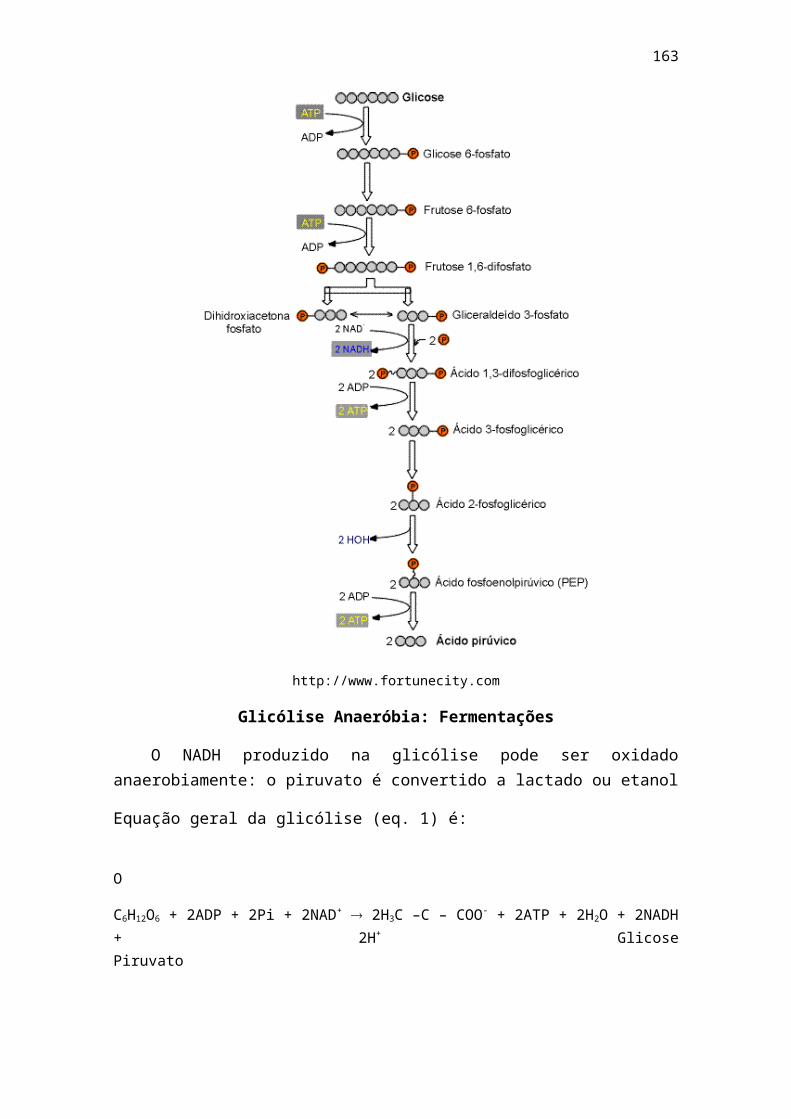
163
em piruvato, acetil-CoA, acetoacetil-CoA ou em intermediários do ciclo do ácido cítrico. Eles irão fornecer substratos para gliconeogênese ou para a produção de corpos cetônicos.
Os aminoácidos cetogênicos são convertidos em acetil-CoA ou acetoacetil-CoA, enquanto os aminoácidos glicogênicos serão convertidos em piruvato ou em intermediários do ciclo do ácido cítrico. Portanto conclui-se que as vinte cadeias carbônicas diferentes não possuem uma via comum de degradação. Seu metabolismo difere, neste aspecto, do metabolismo dos ácidos graxos, todos degradados pelo ciclo de Lynen e dos açúcares, metabolizados pela glicólise. Embora existam vias próprias para a oxidação da cadeia carbônica de cada aminoácido, estas diferentes vias de degradação convergem para a produção de apenas alguns compostos: piruvato, acetil-CoA, ou intermediários do ciclo Krebs (oxaloacetato, α-cetoglutarato, succinil-CoA e fumarato). A partir deste ponto, o metabolismo da cadeia carbônica dos aminoácidos confunde-se com o das cadeias carbônicas de carboidratos ou de ácidos graxos.
O destino final dos α-cetoácidos dependerá do tecido e do estado fisiológico considerados, podendo ser: oxidação pelo ciclo de Krebs, fornecendo energia; utilização pela gliconeogênese, para a produção de glicose; e conversão a triacilgliceróis e armazenamento.
A maioria dos aminoácidos produz piruvato ou intermediários do ciclo de Krebs, precursores da gliconeogênese e são por isso chamados glicogênicos. A leucina origina corpos cetônicos, sendo o único aminoácido exclusivamente cetogênico. Alguns outros aminoácidos como Isoleucina, Fenilalanina, Tirosina, Treonina e Triptofano, são tanto glicogênicos quanto cetogênicos, isto é, são glicocetogênicos.
Os destinos das cadeias carbônicas dos aminoácidos de acordo com o composto formado são:
1. Aminoácidos convertidos a piruvato: Ala,Cys,Gly,Ser,Thr,Trp.
2. Aminoácidos convertidos a oxaloacetato: Asn, Asp.
3. Aminoácidos convertidos a fumarato: Asp, Phe, Tyr.
4. Aminoácidos convertidos a succinil-CoA: Ile,Val, Met e Thr.
5. Aminoácidos convertidos a α-cetoglutarato: Glu, Gln, Pro, Arg, His.
6. Aminoácidos convertidos a acetil-CoA: Phe,Tyr,Trp,Lys,Ile,Thr,Leu.
HIPERTEXTO

164
Doenças Hereditárias do Metabolismo de Aminoácidos
Na fenilcetonúria, a fenilalanina não pode ser convertida em tirosina e origina fenilpiruvato. Há ausência de fenilalanina hidroxilase. A conversão de tirosina a 3,4-diidroxifenilalanina (DOPA) é catalisada por tirosinase, uma oxigenase que contém cobre. As reações que levam à síntese de melanina a partir de DOPA são pouco conhecidas. A tirosina hidroxilase é a enzima ausente no tipo clássico de albinismo. O albinismo está relacionado à incapacidade de sintetizar melanina.
Tabela - Aminoácidos Glicogênicos e Cetogênicos
Glicogênico Cetogênico Glicogênico e Cetogênico
Aspartato Leucina Isoleucina
Asparagina Lisina Fenilalanina
Alanina Triptofano
Glicina Tirosina
Serina
Treonina
Cisteína
Glutamato
Glutamina
Arginina
Prolina
Histidina
Valina
Metionina
Fonte: Campbell & Farrel, 2008
1.Conversão de Ala,Cys,Gly,Ser,Thr,Trp em piruvato
A alanina é convertida diretamente em piruvato através da transaminação, enquanto cisteína e serina necessitam que suas cadeias laterais sejam inicialmente removidas. A glicina é interconvertida em serina, fornecendo uma

165
rota degradativa a piruvato. A treonina é inicialmente convertida em aminoacetona e, depois desaminada em piruvato.
2. Conversão de Asn, Asp em oxaloacetato
A asparaginase remove o nitrogênio amídico da cadeia lateral da asparagina para produzir aspartato, que é convertido em oxalacetato através da transaminação pela AST.
3. Conversão de Asp, Phe, Tyr em fumarato
Aspartato é um dos substratos do ciclo da ureia, em que é convertido a fumarato. A fenilalanina é convertida a tirosina por uma oxidação irreversível, catalisada por fenilalanina hidroxilase. Dos nove carbonos da tirosina, quatro são convertidos a fumarato, quatro a acetoacetato e um a CO2.
4. Conversão de Ile, Val, Met e Thr a succinil-CoA
São inicialmente convertidos a propionil-CoA, que também é produzida na oxidação de ácidos graxos com número ímpar de carbonos. As acil-CoA derivadas da isoleucina e valina são oxidadas por reações semelhantes às da β-oxidação, que convertem valina a propionil-CoA e isoleucina a propionil-CoA e acetil-CoA. A metionina forma α-cetobutirato, que é oxidado a propionil-CoA. A treonina por desaminação catalisada pela treonina desidratase, produz também α-cetobutirato como a metionina.
5. Conversão de Glu, Gln, Pro, Arg, His em α-cetoglutarato
O glutamato converte-se em α-cetoglutarato por transaminação ou por desaminação oxidativa catalisada pela glutamato desidrogenase. A glutamina tem seu grupo amino liberado por ação da glutaminase formando glutamato. A prolina tem todos os seus átomos de carbono aparecendo como glutamato. A arginina ao ser hidrolisada pela arginase (ciclo da ureia, um de seus carbonos aparece na ureia e os outros passam a constituir ornitina, que origina glutamato. Cinco carbonos da histidina produzem glutamato e um carbono é transferido ao tetraidrofolato.
6. Conversão de Phe, Tyr, Trp, Lys, Ile, Thr, Leu em acetil-CoA
A formação de acetil-CoA pode ser direta ou indireta (através de acetoacetato ou acetoacetil-CoA). A fenilalanina, tirosina, triptofano, isoleucina e treonina produzem também compostos precursores de glicose: os aminoácidos glicocetogênicos. Assim, quatro dos carbonos de fenilalanina e tirosina são convertidos a fumarato, três do triptofano à alanina e três da isoleucina a propionil-CoA; a treonina, por uma via alternativa, converte-se a succinil-CoA. A via de degradação da leucina tem passos iniciais comuns à dos outros aminoácidos ramificados, valina e isoleucina, mas os produtos finais são

166
exclusivamente acetoacetato e acetil-CoA. A lisina forma acetoacetil-CoA como o triptofano. O triptofano produz acetoacetil-CoA por uma via que inclui três reações com oxigênio. Um dos intermediários da via de degradação do triptofano é precursor de ácido nicotínico.
CONTEXTUALIZANDO
Kwashiorkor e Marasmo
Nos países desenvolvidos, a desnutrição calórico-proteica é observada mais frequentemente em pacientes hospitalizados com doenças crônicas, ou em indivíduos que sofrem grandes traumas, infecções graves ou grandes cirurgias. Nos países em desenvolvimento, são observados casos onde ocorre ingestão inadequada de proteínas e/ou de energia. Os indivíduos afetados mostram uma variedade de sintomas, incluindo sistema imunitário deprimido, com redução na capacidade de resistir a infecções. Duas formas extremas de desnutrição são o kwashiorkor e o marasmo. Enquanto o marasmo ocorre quando a privação de calorias é relativamente maior do que a redução em proteínas, no kwashiorkor observa-se que a privação de proteína é relativamente maior do que a redução em calorias totais. Ao contrário do marasmo, a grande privação de proteínas está associada à grave perda de proteína visceral. Crianças com kwashiorkor sofrem de perda muscular e diminuição da concentração de proteínas plasmáticas, particularmente albumina. O resultado é um aumento no fluido intersticial, provocando distensão abdominal e edema, o que faz com que a criança pareça gordinha. A perda muscular é causada pela falta de aminoácidos essenciais na dieta; as proteínas existentes devem ser degradadas para produzir tais aminoácidos para a síntese de novas proteínas. A esses problemas pode ser acrescida uma menor habilidade para produzir enzimas digestivas e células novas do epitélio intestinal devido a um decréscimo na disponibilidade de aminoácidos para a síntese de novas proteínas. As vítimas do marasmo não apresentam o edema ou as mudanças nas proteínas plasmáticas observadas no kwashiorkor (Champe, Harvey e Ferrier, 2009; Smith, Marks e Lieberman, 2007).
ATIVIDADES DE AVALIAÇÃO
1. Em relação aos aminoácidos presentes nas células dos mamíferos, indicar:a) suas procedências;b) seus destinos metabólicos;c) se existe uma reserva de aminoácidos ou proteínas.
2. Esquematizar as reações catalisadas pelas seguintes enzimas:- Aspartato Aminotransferase; - Alanina Transferase

167
3. Esquematizar a reação catalisada pela glutamato desidrogenase.4. Citar o principal produto de excreção de nitrogênio no homem.5. Esquematizar a reação de formação de carbamoil-fosfato catalisada por
carbamoil-fosfato sintetase.6. No ciclo da ureia:
a) Indicar a procedência dos átomos de nitrogênio da molécula de ureia.b) Calcular o balanço de ATP.c) Citar o aminoácido proteico sintetizado.
7. Citar o órgão responsável pela produção de ureia.8. A amônia é tóxica para os animais. Mostrar como a amônia produzida nos
tecidos extra-hepáticos é transportada para o fígado.9. Verificar o destino dos esqueletos de carbono dos aminoácidos, seu
catabolismo e indicar aqueles que podem originar glicose.10. Citar as doenças genéticas provenientes de alterações do metabolismo da
tirosina e da fenilalanina.11. Definir aminoácidos essencias e citá-los.12. Qual é o primeiro depositário de grupos amino no metabolismo dos
aminoácidos? E o segundo?
Bibliografia Comentada
Campbell, M. K.; Farrel, S. O. Bioquímica. Vol 3. São Paulo: Thomson Learning - 2008. No Capítulo 23 do vol. 3, o assunto Metabolismo do Nitrogênio é discutido em todos os aspectos de importância próprios do assunto.
Champe, P. C.; Harvey, R. A.; Ferrier, D. R. Bioquímica Ilustrada. 4ª Ed. Porto Alegre: Artmed, 2009. Na Unidade IV do livro nos Capítulos 19, 20 e 21 são apresentados tópicos relacionados ao Metabolismo do Nitrogênio com muitas ilustrações tornando o assunto de fácil compreensão.
Devlin, T. M. Manual de Bioquímica com Correlações Clínicas. São Paulo: Editora Blucher, 2007. O Capítulo 19 da Parte IV desse livro aborda tópicos relacionados ao Metabolismo de Aminoácidos.
Marzzoco, A.; Torres, B. B. Bioquímica Básica. 2ª ed. 1999. Rio de Janeiro: Editora Guanabara Koogan S. A. O Capítulo 17 da Parte 3 desse livro explana de forma bastante compreensiva o assunto Metabolismo de Aminoácidos.
Pelley, J. W. Bioquímica. No Capítulo 12, são estudados tópicos relativos ao Metabolismo de Aminoácido e Heme de uma forma sucinta e objetiva. Rio de Janeiro: Elsevier, 2007.
Smith, C.; Marks, A. D.; Lieberman, M. Bioquímica Médica Básica de Marks – Uma abordagem clínica. 2ª ed. Porto Alegre: Artmed, 2007. Os Capítulos 37,

168
38 e 39 abordam tópicos relacionados ao Metabolismo do Nitrogênio de modo detalhado e didático.
Voet, D.; Voet, J. G.; Pratt, C. W. Fundamentos de Bioquímica. Porto Alegre: Artmed, 2000. O Capítulo 20 da Parte IV traz uma discussão bastante abrangente sobre o Metabolismo dos Aminoácidos.