Ⅱ Ⅲ期药物临床试验项目...
TRANSCRIPT

Ⅱ-Ⅲ期药物临床试验项目设计、实施与常见问题
中山大学肿瘤医院
临床研究部/国家药物临床试验机构洪明晃

学习:
• 《药品注册管理办法》 (局令第28号) 2007.10.1
• 《化学药品注册分类改革工作方案》 (2016年第51号) 2016.3.4
2

一、化学药品注册分类(旧)
1. 未在国内外上市销售的药品:
2. 改变给药途径且尚未在国内外上市销售的制剂。
3. 已在国外上市销售但尚未在国内上市销售的药品:
4. 改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。
5. 改变国内已上市销售药品的剂型,但不改变给药途径的制剂。
6. 已有国家药品标准的原料药或者制剂。

一、化学药品注册分类(旧)
临床试验要求
1. 注册分类1和2(1)统计学要求(2)最低病例数(试验组)要求: I期为20至30例,II期为100例,III期为300例,IV期为2000例
2. 注册分类3和4 人体药代动力学研究和至少 100对随机对照临床试验
多个适应症的,每个主要适应症的病例数不少于60
3. 注册分类5、6:详见原文4

《化学药品注册分类改革工作方案》
1类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。
2类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。
3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。
4类:境内申请人仿制已在境内上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。
5类:境外上市的药品申请在境内上市。

二、治疗用生物制品
1. 临床试验的最低病例数(试验组)要求为:
Ⅰ期:20例,Ⅱ期:100例,Ⅲ期:300例。
2. 注册分类1~12的制品应当按新药要求进行临床试验。
3. 注册分类13~15的制品一般仅需进行Ⅲ期临床试验。
6

三、中药、天然药物
①临床试验的病例数应当符合统计学要求和最低病例数要求
②临床试验的最低病例数(试验组)要求:
Ⅰ期为20~30例,Ⅱ期为100例,Ⅲ期为300例,Ⅳ期为2000例
③属注册分类1、2、4、5、6的新药,以及7类和工艺路线、溶媒等有明显改变的改剂型品种,应当进行Ⅳ期临床试验
④生物利用度试验一般为18~24例;⑤避孕药,…⑥新的中药材代用品,…,临床验证的病例数不少于100对⑦改剂型品种,…,免除或进行不少于100对的临床试验⑧仿制药视情况需要,进行不少于100对的临床试验⑨进口中药、天然药物制剂,…
7

四、关于临床试验分期
II期临床试验:治疗作用初步评价阶段。其目的是初步评价药物对目标适应症患
者的治疗作用和安全性,也包括为III期临床试验研究设计和给药剂量方案的
确定提供依据。此阶段的研究设计可以根据具体的研究目的,采用多种形式
,包括随机盲法对照临床试验。
III期临床试验:治疗作用确证阶段。其目的是进一步验证药物对目标适应症患者
的治疗作用和安全性,评价利益与风险关系,最终为药物注册申请的审查提
供充分的依据。试验一般应为具有足够样本量的随机盲法对照试验。
IV期临床试验:新药上市后应用研究阶段。其目的是考察在广泛使用条件下的药
物的疗效和不良反应,评价在普通或者特殊人群中使用的利益与风险关系以
及改进给药剂量等。
8

第一部分
试验方案的主要内容
9

特别提醒!
• 申办者、研究者、统计专家合作
• 研读CFDA批件
• 遵照相关专业的:– 《指导原则》
– 《临床试验方案设计规范》
• 参考
– SPIRIT– CONSORT
• 问题:忽视批件中特别关注的问题
10

题目
• 研究人群、干预措施、设计等
• 关键词1. 受试者
2. 试验药
3. 对照药
4. 基础治疗(如有)
5. 多中心-?
6. 随机-?
7. 双盲-开放
8. II期-III期-随机对照试验• 题目缩写
11

试验注册、版本号、研究者等
• 注册
– 注册号
• www.Clinicaltrials.org• www.srctn.org• www.chictr.org
• 版本号– 方案定稿日期、版本
• 申办者/赞助者/研究者等– 申办者/CRO/赞助者
– 研究者/SMO– 统计者

研究背景
• 描述研究的问题
• 开展试验的理由
• 对照组选择的理由
• ……

试验目的
II期
• 初步的治疗作用
• 安全性
• 探索III期的最佳给药方案
和受试人群
III期
• 明确治疗效果
• 安全性
• 为上市提供依据
14

试验设计
• 设计类型
– 平行、交叉、析因– 单臂、多阶段设计– 适应性设计
• 分配比例
• 比较类型– 差异性、优效性、等效性、非劣效性
• 问题:
– 缺乏基本的安全性资料,就要上大规模RCT;– 甚至I期、II期方案一起申报
– 为减少例数,把效果或界值定得太宽
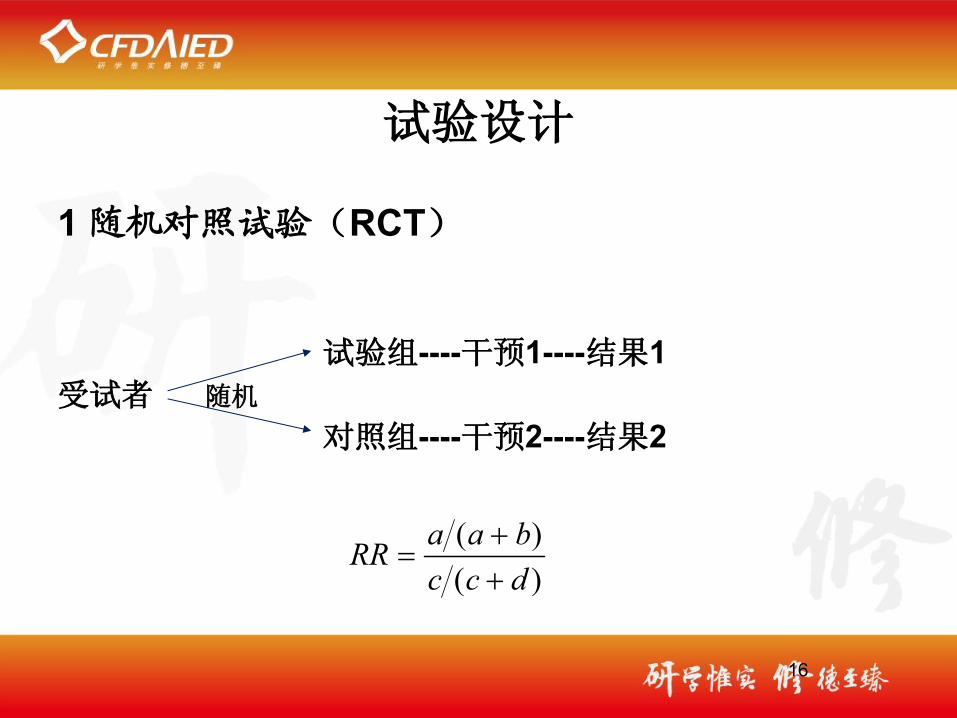
试验设计
1 随机对照试验(RCT)
试验组----干预1----结果1受试者 随机 对照组----干预2----结果2
16
)()(dccbaaRR

试验设计
2 配对设计–自身配对:多见于眼科、皮肤科等的局部治疗
• 部位左右:优点:组间差异小
缺点:全身反应难以鉴别
–异体配对:多为回顾性
• 年龄、性别、分期等“门当户对”
17

试验设计
3 交叉设计 A药 A药受试者 随机 洗脱期
B药 B药
18

试验设计
4 撤药(减量)研究:
减量?%----观察----结果1长期用药者 随机 常规用药----观察----结果2
19

多中心
II期
• 单中心• 如抗肿瘤药物simon 2阶段设计
• 区域
III期• 区域
• 全球
20

多中心临床试验的优缺点
优点:
• 病例收集快;
• 代表性广;
• 集思广益/推进合作
问题:• 合作难
• 管理跟不上
• 不一致性
• 经费不足
不一致的来源:
• 单位:
• 设备:如检验/影像/病理
• 操作: 如手术方式
• 评价:如标准不一
• 人员:如培训、经验
不一致性评价?

减少不一致性的一些做法
• 统一完善的方案/培训
• 中心随机
• 中心实验室/中心疗效评价
– 资质?(卫生部《医疗机构临床实验室管理办法》2006年) – 检测项目?
• 同期开展试验
• 加强监查、稽查
• IDMC
• 问题:资质?等待时间长,延误诊治;医院又另采血一次,且费用
由受试者承担;项目多,加重受试者负担;

合格标准
• 入选标准
– 对受试者,该治疗可能是最佳,且利大于弊的选择
– 不同的疾病,入选标准差别很大
1. 通用入选标准:性别、年龄、签署ICF
2. 疾病特异性:
• 确诊方法、病程、病理类型
• 临床分期或分型、
• 影像学、靶点、
3. 其它特殊要求:如激素水平、预期寿命、PS等
• 问题:过于简单,或条款太多,难以执行
23

合格标准
• 排除标准
– 考虑:安全性、依从性、其它干扰因素
1. 通用排除标准
• 孕妇或哺乳期妇女
1. 其它疾病
2. 器官功能异常
3. 药物相关
• 问题:重复入选标准、太简单
24

治疗方案
用药方案
• 试验药-(开放试验应明确对照组用药方案)–药名、剂量、途径、疗程;包装、标签等
• 基础治疗(如有)
• 合并用药:适应症、用法、日期、结果描述
• 慎用药:什么情况下允许怎么使用
• 禁用药
• 禁用疗法:手术、放疗?
• 后续治疗?
25

治疗方案
• 中止/退出试验– 研究者要求中止/退出
1. 重要不良反应、妊娠
2. 疾病加重
3. 严重违反方案
4. 紧急破盲后
5. 误纳病例
– 受试者坚持退出
• 剂量调整(如抗癌药物)• 问题:
– 中止/暂停/退出/剔除的定义?未规定暂停的时间窗
– 退出后的数据处理(SS、ITT、PP集)及临床处理不明确
26

结局指标
• 主要指标:
– 用于受试者受益、临床意义的判断– 样本估计、统计方法、质量管理、研究结论的依据– 慎用“中间结果”或“替代指标”
• 次要指标
– 安全性指标– 其它重要疗效指标– 生存质量– ……
• 问题:主要指标太多、替代指标不能反映受试者情况、忽视安全性
指标
27

结局指标
• 指标类型/终点事件
1. 临床表现、生存时间等
2. 实验室指标
3. 影像学
4. 病因学、病理学
5. 总体评价:痊愈、显效、好转、无效
6. 受试者报告
28

II期近期指标
替代指标?
例:抗癌药物
• 主要指标
– ORR、PFS• 次要指标
– 安全性– QoL
III期远期指标
受益的客观指标
例:抗癌药物
• 主要指标
– OS、PFS• 次要指标
– 安全性– ORR– QoL
29
结局指标

风险来源
1. 药物相关– 试验用药
– 基础用药/合并用药– 重复/过量用药
1. 试验相关:– 过多的检查/标本采集/访谈/访视– 诊疗延误导致病情加重
– 额外的要求/配合
1. 其它
问题:
– 只关注了试验用药的问题
安全性问题的处理
1. 按医疗常规及时处理
2. 分类:AE/SAE…3. 程度
4. 关联性判断
5. 起止时间
6. 转归
7. 报告
问题:
– 定义不规范
– 如何处理、报告不清晰
30
结局指标
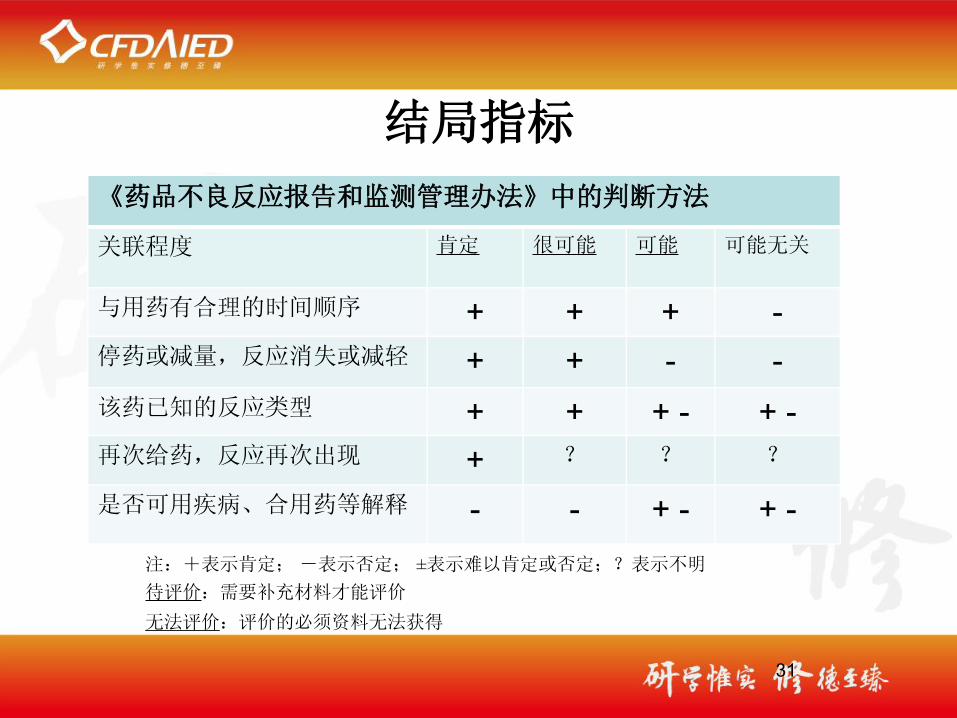
31
《药品不良反应报告和监测管理办法》中的判断方法
关联程度 肯定 很可能 可能 可能无关
与用药有合理的时间顺序 + + + -停药或减量,反应消失或减轻 + + - -该药已知的反应类型 + + + - + -再次给药,反应再次出现 + ? ? ?
是否可用疾病、合用药等解释 - - + - + -注:+表示肯定; -表示否定; ±表示难以肯定或否定;?表示不明待评价:需要补充材料才能评价
无法评价:评价的必须资料无法获得
结局指标

样本量
1. 主要考虑因素
– 设计类型:平行、交叉、析因– 比较类型:优效、等效、非劣效、探索性– 试验目的/主要指标:有效性、安全性– 效应量(effect size)– 检验水准:α 、β
1. 常用软件
– PASS、SAS or R– N-Query、N-Solution
• 问题:比较类型与效果估计缺乏临床依据;
优效或劣效界值未考虑临床意义;安全性方面
未考虑


II期• 统计学样本量计算• 最低要求?(试验组) ≥100例 (80%power检出1例4%的AE
• 对照组例数?
III期• 统计学样本量计算• 最低要求(试验组) ≥ 300例 (80%power检出1例1%的AE)
• 对照组例数?
34
化药分类3和4的,应当进行人体药代动力学研究和至少
100对随机对照临床试验;多个适应症的,每个主要适
应症的病例数不少于60对
样本量

分组
• 分层随机:均衡;分层分析的前提之一
– 分层因素:影响预后最重要的几个因素
• 随机分组– 药物编号顺序发药– 中心随机:网络
• 随机→ 治疗开始:时间窗?
• 问题:
– 中心随机时间长,影响诊治
– 受试者信息泄露
35

对照
II期1. 单臂: 如simon 2阶段设计
2. 空白
3. 安慰剂
或:基础治疗+安慰剂
3. 原研药/标准/阳性
4. 不同剂量
5. 多阶段设计
III期1. 原研药/标准/阳性
2. 安慰剂
3. 基础治疗+安慰剂
36
阳性对照药品的选择一般应按照以下顺序进行:(1)原开发企业的品种;(2)具有明确临床试验数据的同品种;(3)活性成份和给药途径相同,但剂型不同的品种;(4)作用机制相似,适应症相同的其他品种 —《化学药品注册分类及申报资料要求》
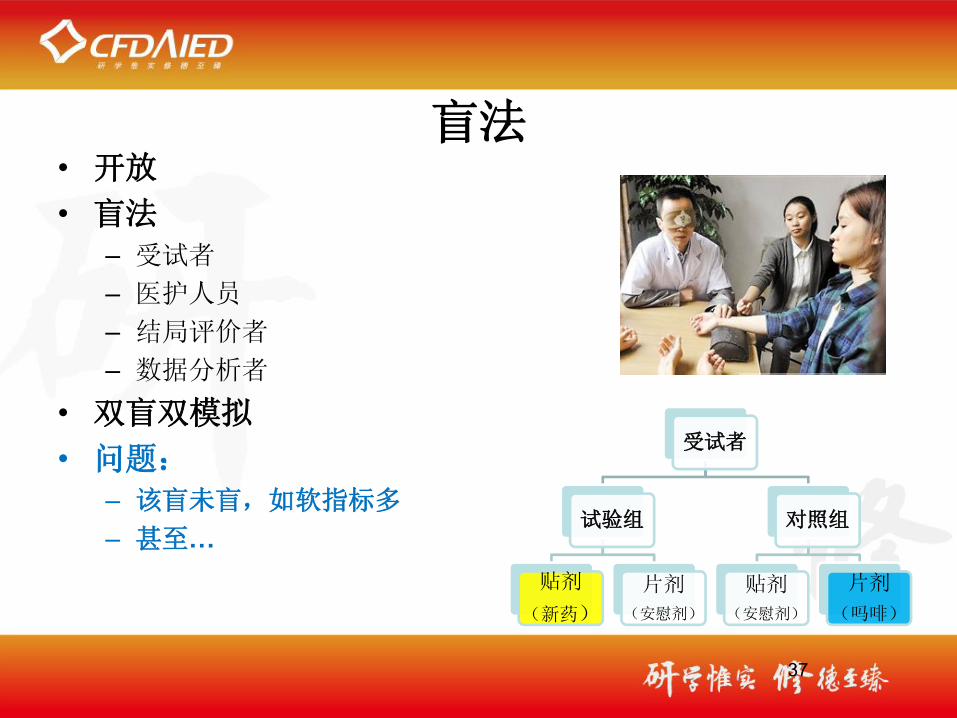
盲法• 开放
• 盲法
– 受试者– 医护人员– 结局评价者– 数据分析者
• 双盲双模拟
• 问题:
– 该盲未盲,如软指标多
– 甚至…
37
受试者
试验组
贴剂
(新药)
片剂
(安慰剂)
对照组
贴剂
(安慰剂)
片剂
(吗啡)

盲法
• 揭盲流程
– 一级揭盲– 二级揭盲
• 紧急揭盲
– 什么情况下?– 谁决定?– 揭盲后受试者的处理?

访视
• 访视安排
• 工作内容
– 间隔时间
– 临床表现/临床检查– 效果/安全性评估– 依从性评估
– 药物发放与回收(门诊受试者)
– 下次访视安排
39

数据采集、录入
• 记录者/谁能记录?– 病历、AE记录表、SAE报告表等– 检测:EKG、血压、肺功能等– 用药日记
• CRF/eCRF):谁能录入/谁签名?• 审核/签字:临床医生/PI• GCP 第二十七条 研究者应保证将数据真实、准确、完整、及时、合法地载入病历和病例报告表

数据管理、统计分析
• 数据管理
– 录入(双重录入?)– 保密及储存方式– 数据核对/数据锁定
• 统计分析– 数据集及其定义– 主要和次要结局指标的分析方法– 分层/亚组分析– 退出病例的分析
• 问题:试验未结束,统计报告已出;点的比较不能反映整体或结局

患者
入选-随机试验组
对照组
中止/暂停
排除
退出 退出
SS、ITT、 SS、ITT、PP
排除
SS
筛选
终点一级揭盲
二级揭盲
总结
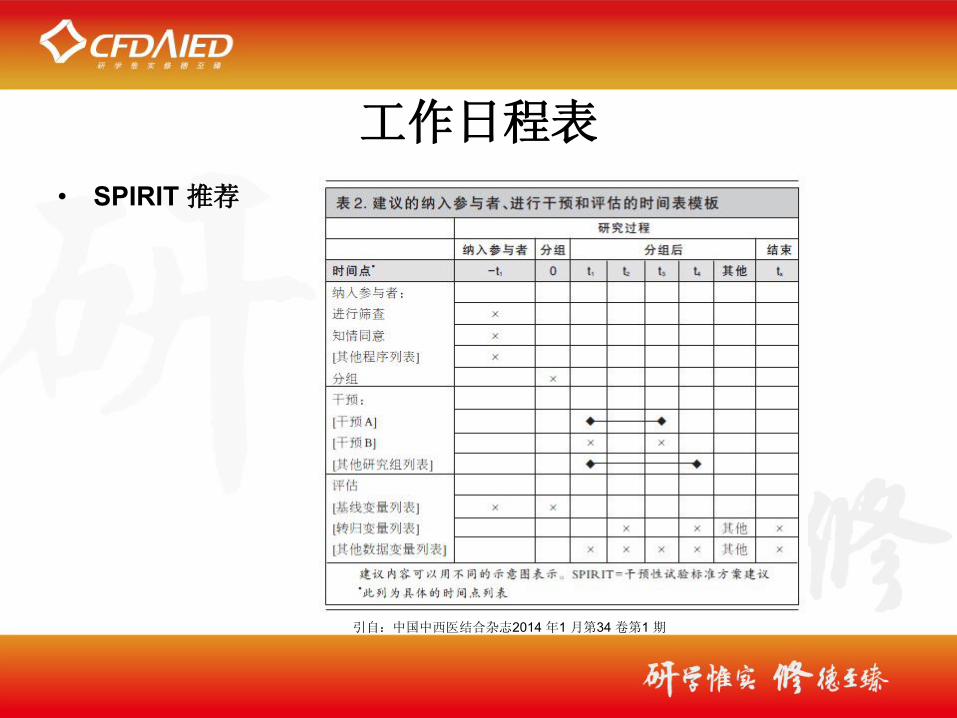
工作日程表
• SPIRIT 推荐
引自:中国中西医结合杂志2014 年1 月第34 卷第1 期

临床专家与统计专家应密切合作
统计专家 临床专家
√
方法方法方法分析
计算
对照假设检验(优效、非劣效、等效)
例数估计数据类型、转换统计方法分析集
ITTFASPPSSSP值
√√
提供参数√√定义
转归?效果、安全
统计学意义 研究结果 临床意义

第二部分
项目实施要点
45
严格执行GCP、SOP、 PROTOCOL就好了!
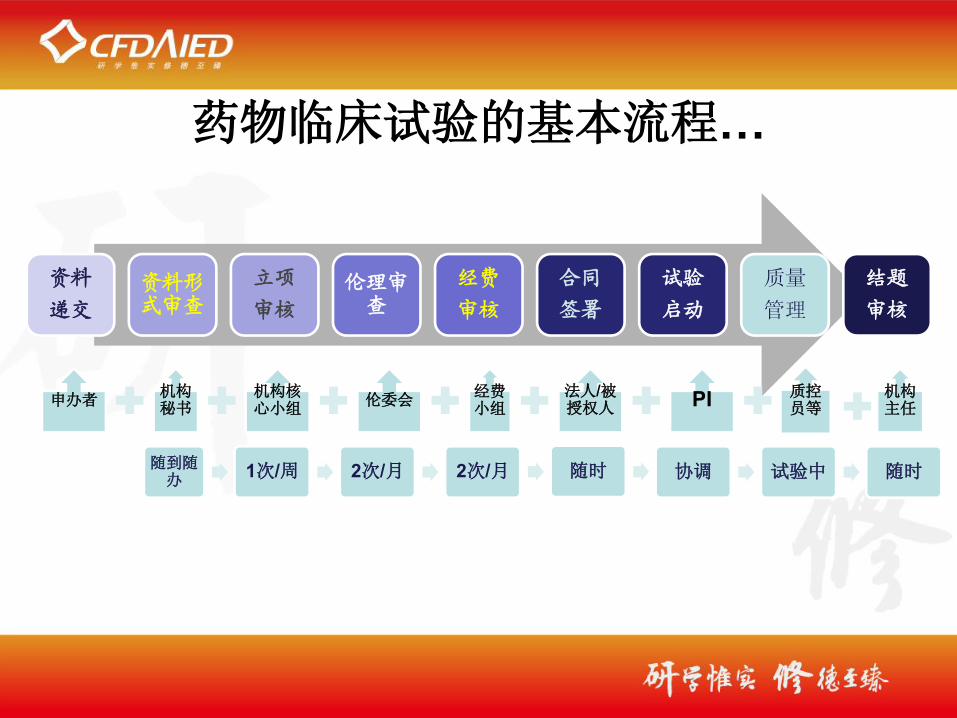
药物临床试验的基本流程…
资料
递交
资料形式审查
立项
审核
伦理审查
经费
审核
合同
签署
试验
启动
质量
管理
结题
审核
申办者机构秘书
机构核心小组
伦委会经费小组
法人/被授权人 PI 质控
员等机构主任
随到随办
1次/周 2次/月 2次/月 随时 协调 试验中 随时

项目实施…
研究者
会议
立项审核
伦理审查
合同签署
启动会
分工授权
筛选
ICF入组 治疗
记录/CRF
AE/SAE 访视 小结

研究者会议
• 会议准备:– 试验方案、CRF、ICF初稿– 必要的参考资料
• 需要解决的问题– 试验方案、CRF、ICF定稿– 研究进程/例数分配/竞争入组– 统一诊断标准/设立中心实验室– 统一疗效评价标准/“独立疗效评价委员会”– 统计专家制订“统计计划书”
– 监查/稽查计划– “项目SOP”?– 成立协调委员会
48

启动
启动会:PI主持,所有研究者、申办者代表参加• 学习有关法规/文件• 熟悉试验方案、SOPs/项目SOP• 医嘱/处方/病程记录模板• 药物的管理
• CRF填写要求• 标本收集与处理
• AE、SAE处理• 监查与质控措施
• PI授权/各司其职• 签到/会议记录• ……
问题:申办者代表不熟悉情况或不能回答问题;研究者缺席;未深入讨论;分工及授权不清晰;
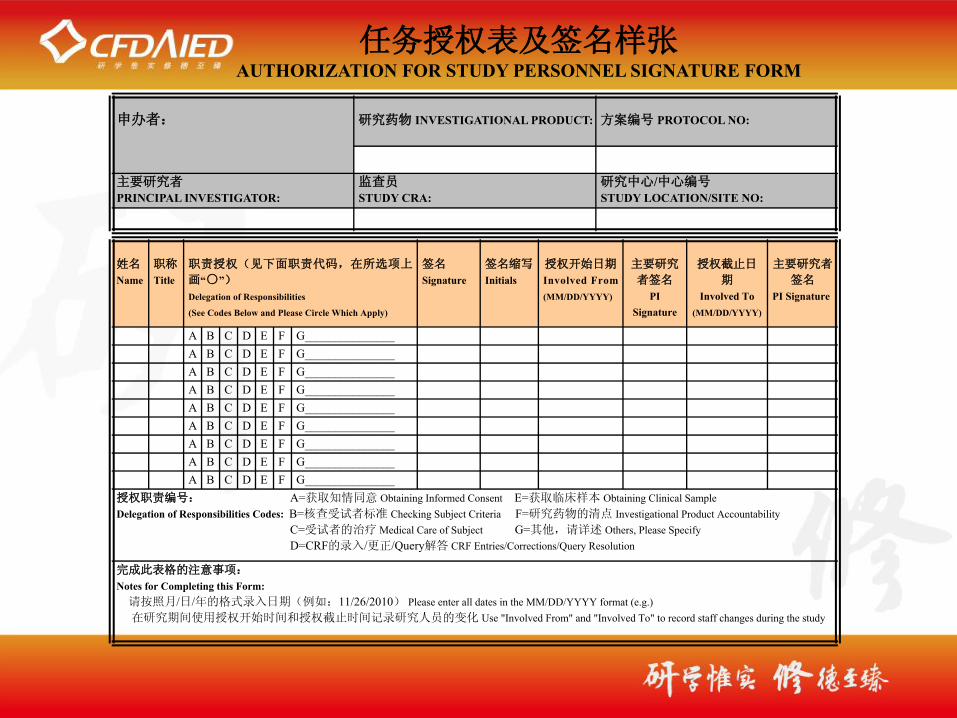
姓名
Name职称
Title职责授权(见下面职责代码,在所选项上
画“○”)Delegation of Responsibilities
(See Codes Below and Please Circle Which Apply)
签名
Signature签名缩写
Initials授权开始日期
Involved From (MM/DD/YYYY)
主要研究
者签名
PI Signature
授权截止日
期
Involved To (MM/DD/YYYY)
主要研究者
签名
PI Signature
A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________A B C D E F G_______________
授权职责编号: A=获取知情同意 Obtaining Informed Consent E=获取临床样本 Obtaining Clinical SampleDelegation of Responsibilities Codes: B=核查受试者标准 Checking Subject Criteria F=研究药物的清点 Investigational Product Accountability C=受试者的治疗 Medical Care of Subject G=其他,请详述 Others, Please Specify D=CRF的录入/更正/Query解答 CRF Entries/Corrections/Query Resolution
完成此表格的注意事项:
Notes for Completing this Form: 请按照月/日/年的格式录入日期(例如:11/26/2010) Please enter all dates in the MM/DD/YYYY format (e.g.) 在研究期间使用授权开始时间和授权截止时间记录研究人员的变化 Use "Involved From" and "Involved To" to record staff changes during the study
任务授权表及签名样张AUTHORIZATION FOR STUDY PERSONNEL SIGNATURE FORM
申办者: 研究药物 INVESTIGATIONAL PRODUCT: 方案编号 PROTOCOL NO:
主要研究者PRINCIPAL INVESTIGATOR:
监查员STUDY CRA:
研究中心/中心编号STUDY LOCATION/SITE NO:
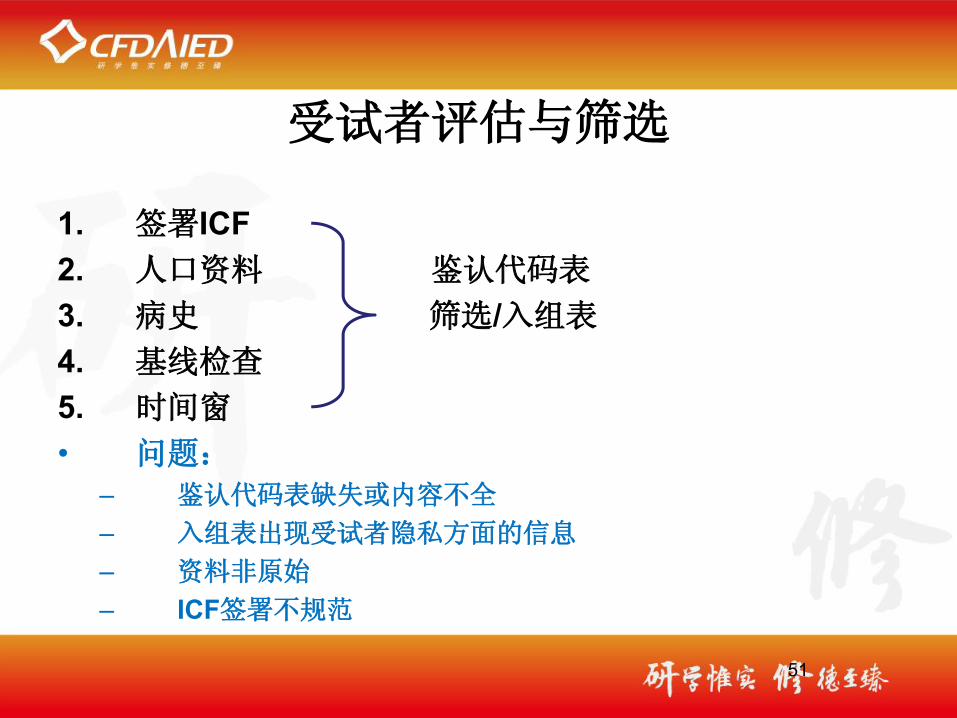
受试者评估与筛选
1. 签署ICF2. 人口资料 鉴认代码表
3. 病史 筛选/入组表
4. 基线检查 5. 时间窗
• 问题:– 鉴认代码表缺失或内容不全
– 入组表出现受试者隐私方面的信息
– 资料非原始
– ICF签署不规范
51

入组
• 随机分组方式、隐蔽?
– 药物编号顺序发药– 中心(央)随机:网络
• 随机→ 治疗开始:时间?
• 受试者管理
– “受试者须知”– 日记卡– QOL表– 访视Check list
52

医嘱/治疗
53
• 住院受试者
– 授权医生:• 医嘱/处方/病程记录(模板)
– 核对/取药/保存– 执行
• 门诊受试者– 授权医生:处方– 取药?
• 问题:
– 无临床相关资质的CRC发药,甚至CRA发药

药物管理
– 中心药房– 中心配制– 科室药柜– 执行:住院、门诊– 药物/日记卡回收/依从性– 退还/销毁
问题:
– 未集中管理、储存条件差
– 相关记录不及时、不准确
– 无相关资质的CRC发药,甚至CRA发药给门诊受试者
– 不按方案要求发药
– 护士未保证受试者一直用同一编号药
– 出现问题未及时报告
– ……54
医嘱/
处方
中心药房 配制 储存 执行 回收 退还 销毁
医嘱/治疗


医嘱/治疗
• 中止
– 其它处理– 恢复试验用药?– 退出
• 退出– 效果、安全性评价、记录– 其它治疗措施、随访
• 问题– 退出受试者的数据未纳入SS、ITT等– 退出受试者未得到合适的临床处理
58

AE/SAE处理
• 执行医院医疗常规/相应的SOP– 住院受试者
研究者和值班护士/医生加强观察、巡视…一旦发现不良事件,当值医护人员进行初步…, 并及时通知研究者;对于程度较重的不良事件,通知PI,…,必要时中止试验…
病情严重…,请求会诊/转诊;必要时报告医务科…如…,应紧急揭盲…,做好相关记录…,判断与药物的因果联系…严重不良事件,应在24小时内,报告…,– 门诊受试者?
• 问题:漏记漏填漏报、尤其是门诊受试者
SAE均被揭盲、紧急揭盲PI不知情、揭盲后受试者未妥善处理
59

AE/SAE记录/报告
1. 程度
3. 处理措施
4. 关联性判断
5. 起止时间
6. 转归、赔偿
7. 报告:SAE、SUSAR→单位、联系方式
• 问题:
– 因果联系判别随意、PI未把好关
– 未按要求及时报告
– 未观察至转归
– 未按方案赔偿
60


效果评估
• 临床医生
– 观察病情– 实验室检查:
• 临床意义判断• 判断CS的应记录AE
– 及时记录
• 受试者报告
• 依从性评价
• 效果、安全性评价
• 问题:
62

CRF的填写
• CRF中的数据来自原始文件并与原始文件一致• 更正时应保持原记录清晰可辩,由更正者签署姓名和时间• CRF/eCRF填写:医生、护士、研究生、CRC?• 审核/签字:临床医生、PI• 问题
– 病情记录不及时– CRF录入慢,缺乏研究护士或CRC– CRF填写后,医生未审核– eCRF:审核?– 溯源?原始文件?

访视
• 访视安排:访视表
– 谁联系/安排;– 谁接诊
• 工作内容
– 间隔时间
– 临床表现/临床检查– 效果/安全性评估– 依从性评估
– 药物发放与回收
– 下次访视安排
• 提高依从性的措施?
64

接受监查/稽查等
• PI重视– 主管医生/研究护士/CRC配合– 准备资料
– 应答/解决问题(按被授权人)
• CRA/稽查员– 通知研究者
– 认真—GCP 第47条– 指出问题/追踪
• 问题:
– CRA不给力,讲进度多讲质量少;稽查更少
– 问题不反馈给研究者
65
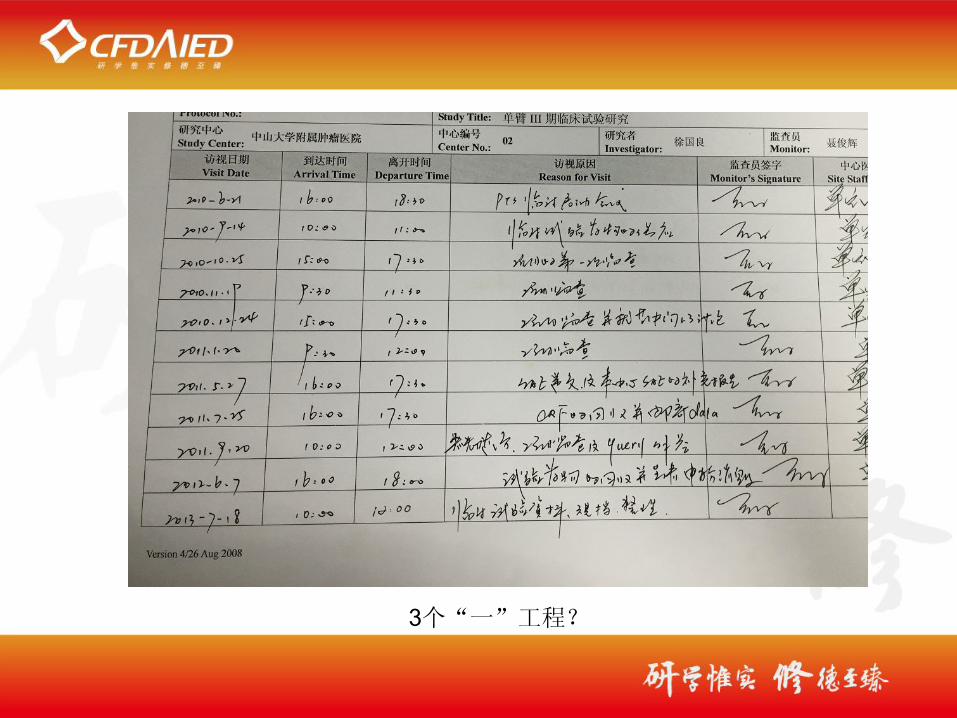
3个“一”工程?

方案修改/项目中止
• 修改
– 方案、ICF;更换研究者等– 伦理审批/备案– 补签ICF
• 中止/终止– PI中止– 机构/伦委会要求中止– 申办方中止– 药监部门责令终止
• 问题:修改频繁、不提交伦委会,未及时补签ICF
1. 伦理委员会未履行职责的;2. 不能有效保证受试者安全的;3. 未按照规定时限报告严重不良事件的;
4. 有证据证明临床试验用药物无效的;
5. 临床试验用药物出现质量问题的;
6. 临床试验中弄虚作假的;7. 其他违反《药物临床试验质量管理规范》的情形。
—《药品注册管理办法》第42条

项目结题(分中心)
• 研究人员:核对、补漏、签字
• 药物管理人员:退还/销毁、记录
• 资料管理人员:收集、整理、归档
• PI:审核、签字• 机构负责人:审核、盖章
• 问题:
• PI只签字,不审核;甚至漏签字
• 试验未完全结束
• 安全性资料不全
• 双盲试验,揭盲后未把受试者的用药情况通知主管医生

数据管理/统计分析
参照《化学药品和生物制品临床试验的生物统计学指导原则》
• 数据管理
– 数据输入:双人,eCRF?– 审核/锁定/存档
• 一级揭盲(双盲试验)
– A组或B组
• 统计分析
• 二级揭盲(1:1双盲试验)
– 试验药或对照药– 尽快把受试者的用药情况通知研究者,保障受试者后续用药安全

总结报告
• 《化学药物临床试验报告的结构与内容技术指导原则》• CONSORT http://www.consort-statement.org/
• 问题:– 试验未完全结束
– PI签名而未严格审核
– 未报告各分中心的重要数据,或数据不符
• 如入组与完成例数、AE与SAE例数
– 结论只根据单个数据的结果
– 未报告各中心的效果、安全性数据,无法比较其差异
– 例数很大的情况,差异有统计学意义,但可能没有临床意义

总结报告
CONSORT Flow Diagram

第三部分
质量管理
72

质量管理贯穿始终……
• 质量涉及多个方面
– 申办方/CRO—责任主体:监查、稽查、IDMC、经费– PI/研究者—项目质量的第1责任人:诊治、安全、数据– 机构/办公室—管理责任:立项、合同、质控、结题– 伦理委员会—项目审查、批准– 药监管理部门:政策、顶层设计;稽查、视察、注册核查、审批
• 722启示录– 质量就是效益!质量就是生命!– 数据真实、完整– 人在!资料在!
90%×90%×90%×90%×90%=59%

监查/稽查
• 监查
– 好的监查员非常重要
– CRA↓,CRC↑,CRB?– GCP 第47条!
• 稽查– 申办者、药监管理部门
– 目前稽查太少
74

视察、现场核查
• INSPECTION、项目现场注册核查– 本省组织,地方保护主义?
– 时间短
– 问题未分级
– 与“审批”存在脱节现象
75

机构内部:项目检查
• 质量管理制度/SOPs• 质控表
• 项目必查/病例抽查/有问题扩大(不能替代CRA!)
• 问题通知书/警告信
• 要求研究者整改/申办者加强监查、稽查等
• 申办者/PI反馈
• 追踪
问题分级与处理
1. 严重问题 → 警告信
2. 重要问题 → 通告书 警告信/
3. 一般问题 → 通告书

机构内部:项目检查
• 进度与质量分析会
• 通报:信息简报、电子邮件等

第四部分
药品上市后的推广
78

药品上市……
• “让优秀的药品尽快送到患者手中”
• 推广会:
研究者应持中立、科学的态度,阐明:– 统计学意义、临床意义– 安全性– 最适合的患者或亚型亚组– 成本-效益– 与其它方案比较的优劣
• 问题
79

临床研究中永恒的追求:
真:真知、真实、真相探究
善:善心、善策、善待患者
美:美德、美行、美好结局